Embed Size (px)
Citation preview

Evaluación de la costo-efectividad de la prueba de secuenciación completa del gen CFTR por técnica Sanger para portadores
asintomáticos en población colombiana de primer, segundo y tercer grado de
consanguinidad con historia familiar de fibrosis quística.
Ernesto Andrade Cerquera
Universidad Nacional de Colombia
Facultad de Medicina Instituto de
Investigaciones Clínicas
Bogotá, Colombia
2017

Evaluación de la costo-efectividad de la prueba de secuenciación completa del gen CFTR por técnica Sanger para portadores
asintomáticos en población colombiana de primer, segundo y tercer grado de
consanguinidad con historia familiar de fibrosis quística.
Ernesto Andrade Cerquera
Tesis presentada como requisito parcial para optar al título de:
Magister en Epidemiologia Clínica
Director
PhD©. Jorge Augusto Díaz Rojas
Línea de investigación de Evaluación de Tecnologías
Grupo de investigación: Evaluación de Tecnologías y Políticas de Salud
Universidad Nacional de Colombia
Facultad de Medicina Instituto de
Investigaciones Clínicas
Bogotá, Colombia
2017

III
Dedicado
A:
Dios, por darme vida y salud para con esfuerzo y dedicación poder dar este
siguiente paso en mi vida académica y poder seguir compartiendo junto a
mi familia y demás seres queridos que me acompañan. A mis padres con quienes
tengo la gran oportunidad de seguir compartiendo muchos momentos de vida
y quienes me han apoyado constantemente en mi vida personal y académica.
A mis profesores de especialización y maestría, especialmente a los profes
Jorge Augusto Díaz Rojas y Heidi Eliana Mateus Arbeláez quienes me asesoraron
en el proceso de idealización y construcción de este proyecto de investigación.

IV
Resumen
Introducción: La fibrosis quística es una enfermedad autosómica recesiva, con
incidencia estimada en Colombia de 1/5000 nacidos vivos (1). Se estima a nivel mundial
que 1/25 individuos son portadores sanos (2). Las pruebas genéticas de portador
permiten identificar a parejas en riesgo con 25% de probabilidad en tener hijos con la
enfermedad.
Metodología: Se realizó una búsqueda sistemática en bases electrónicas especializadas
en salud, incluyendo literatura gris y panel de expertos. Se evaluó las características
operativas de la prueba diagnóstica de secuenciación para portadores asintomáticos de
mutaciones en fibrosis quística, y los diferentes modelos de árbol de decisiones en
estudios de costo-efectividad. Se aplicó Quadas2 modificada y QHES para pruebas
diagnósticas y estudios económicos, respectivamente. Se adaptó un modelo de árbol de
decisiones al contexto clínico del país, teniendo como unidad de análisis la prevención
de futuras concepciones mediante la prueba con asesoría genética, por riesgo de
presentar fibrosis quística y la razón de costo efectividad incremental, aplicada en
familiares asintomáticos de primer, segundo y tercer grado de consanguinidad al caso
índice con diagnostico de fibrosis quística en Colombia. Los costos de la enfermedad
fueron obtenidos del reporte de alto costo del Ministerio de Salud y Protección Social de
Colombia. Los costos de la prueba genética fueron referenciados por laboratorios
nacionales. Se realizó un análisis de sensibilidad determinístico y probabilístico bajo la
perspectiva del tercer pagador con horizonte temporal a un año.

V
Resultados: Se obtuvo un ICER de $ 17.082.833,90 pesos colombianos, que equivale al
costo incremental por obtener un 10,89 % más de probabilidad de evitar el nacimiento de
un niño enfermo con fibrosis quística por pareja tamizada. Se evidenció que al aplicar el
PIB per cápita como disposición a pagar; esta tecnología diagnostica resulta ser costo-
efectiva por tener un ICER entre 1 y 3 PIB per cápita.
Conclusiones: La prueba genética de detección de portador para fibrosis quística es
costo efectiva dependiendo del umbral de disponibilidad a pagar aplicado, teniendo en
cuenta los supuestos, los escenarios clínicos y las limitaciones establecidas en el
modelo.
Palabras clave: Fibrosis quística, asesoramiento genético, pruebas genéticas,
evaluación de costo efectividad.

VI
Abstract
Introduction: Cystic fibrosis is an autosomal recessive disease, with an estimated
incidence in Colombia of 1/5000 live births (1). It is estimated worldwide that 1/25
individuals are healthy carriers (2). Genetic carrier screening allow the identification of
couples at risk with a 25% chance of having children with the disease.
Methodology: A systematic search was carried out in electronic databases specialized in
health, including gray literature and panel of experts. We evaluated the operative
characteristics of the diagnostic test of sequencing for asymptomatic carriers of mutations in
cystic fibrosis, and the different decision tree models in cost-effectiveness studies.
Modified Quadas2 and QHES were applied for diagnostic tests and economic studies,
respectively. A decision tree model was adapted to the clinical context of the country,
having as a unit of analysis the prevention of future conceptions through the test with
genetic counseling, for the risk of presenting cystic fibrosis and the cost-effectiveness
ratio, applied in asymptomatic relatives of first, second and third degree of consanguinity
to the index case with diagnosis of cystic fibrosis in Colombia. The costs of the disease
were obtained from the report of high cost of the Ministry of Health and Social Protection
of Colombia. The costs of genetic testing were referenced by national laboratories. A
deterministic and probabilistic sensitivity analysis was performed under the perspective of
the third payer with a one-year time horizon.
Results: An ICER of $ 17,082,833.90 Colombian pesos was obtained, which is equivalent
to the incremental cost of obtaining 10.89% more probability of avoiding the birth of a sick
child with cystic fibrosis by screening couple. It was evidenced that when applying GDP
per capita as a willingness to pay; this diagnostic technology proves to be cost-effective to
have an ICER between 1 and 3 GDP per capita.
Conclusions: The genetic test of carrier detection for cystic fibrosis is cost effective
depending on the availability threshold to pay applied, taking into account the
assumptions, clinical scenarios and limitations established in the model.
Keywords: Cystic fibrosis, genetic counseling, genetic testing, cost-effectiveness
evaluation.

1
Contenido
Pág. Resumen………………………………………………………………………………..………..IV
Lista de figuras…………………………………………………………………………...……. .3
Lista de tablas...…………………………………………………………………………..……...4
Lista de abreviaturas…………………………..………………………………………………..5
Introducción.......................................................................................................................6
Justificación.......................................................................................................................8
1. Capitulo 1 ……………………………………………………………..………………..10
1.1 Objetivos...........................................................................................................10
1.1.1 Objetivo general.....................................................................................10
1.1.2 Objetivos específicos……..…………………………………………………10
1.2 Marco teórico……...……………………………………………….………………..11
1.2.1 Epidemiología………………………………………………….………………..11 1.2.2 Fisiopatología………………………………………………...….….…………..14 1.2.3 Diagnostico…………………………………………………...….….…………..15 1.2.4 Pronostico……………………………………………………...….….………....20 1.2.5 Estudio de portadores…………………………..…………...…………………21 1.2.6 Prueba de secuenciación…………………………..………...………………..25 1.2.7 Tamizaje versus test para portador de fibrosis quística………………..….28 1.2.8 Tratamiento….….……………….……………………………..………………..31 1.2.9 Costo de la enfermedad……………...………………………..………...…….32
1.2.10 Costo del test de portador………………………………………...…..…….32
2. Capitulo 2 ……………………………………………………………………………..34 2.1 Marco Metodológico…………………………….………………………………..34
2.1.1 Documentación de estudios diagnósticos y de costo efectividad………....35
2.1.2 Población Objetivo…………….…………………………..…………………....36 2.1.3 Perspectiva de análisis…………….……………………….………………….36

2
2.1.4 Horizonte temporal………….………………………………………………….36 2.1.5 Identificación de las tecnologías sanitarias a comparar……………………36 2.1.6 Estimación de los costos asociados…………………………………….……37 2.1.7 Medidas de resultado en salud…………………………………………….….37 2.1.8 Tasa de descuento………………………………………………………….….37 2.1.9 Modelación……………………………….………………………………….......37 2.1.10 Análisis de sensibilidad…………………………………………………….…38 2.1.11 Consideraciones Éticas………………………………………………….……38 2.1.12 Supuestos……...……………………………………………………………..38
3. Capitulo 3 ……………………………………………………………………………..40 3.1 Resultados……………………………………………………………….………..40
3.1.1 Revisión sistemática de estudios de costo efectividad para identificación de portadores de mutaciones de fibrosis quística…………40 3.1.2 Valoración de los costos de la prueba diagnóstica para portadores y del costo de un paciente con fibrosis quística durante un año…………..54 3.1.3 Evaluación de la sensibilidad y especificidad de la prueba diagnóstica……………………………………………………………………..57
3.1.4 Resultados del modelo……………………..….………………………………69
4. Discusión……………………….…….………………………….……………………...77
5. Conclusiones…………………………………………………………………………...82
Anexos
A. Consecuencias clínicas y económicas según escenario de probabilidad de aceptación de asesoría genética……………………………...83
B. Operacionalización de variables……………..…………………….………................84 C. Reporte de búsqueda electrónica de estudios de costo efectividad
para detección de portadores de fibrosis quística…………………….....................86 D. Reporte de búsqueda electrónica de estudios de prueba de
secuenciación para detección de portadores de fibrosis quística………………...94 E. Árbol de decisiones aplicado al modelo……..……….……………………………..106
Bibliografía…………………………………………………………………………................107

3
Lista de figuras
Figura 1 Patrón de herencia autosómica recesiva de la FQ………………………………..16
Figura 2 PRISMA……………………………………………………………...………………..41
Figura 3 Plano de análisis de costo-efectividad………………………….…………………69
Figura 4 Diagrama de tornado (ICER)……………………….……………..………………..70
Figura 5 Análisis de sensibilidad sobre Conyuge1pos y Conyuge2pos (Beneficio
neto, Disponibilidad a pagar= 3.3227902E7)……………………………………..…..………73
Figura 6 Análisis de sensibilidad probabilístico, simulación de Monte Carlo,
gráfico de dispersión ICER. 1 PIB per cápita……………...…….……………………………74
Figura 7 Análisis de sensibilidad probabilístico, simulación de Monte Carlo,
gráfico de dispersión ICER. 2 PIB per cápita…………………………………………………75
Figura 8 Análisis de sensibilidad probabilístico, simulación de Monte Carlo,
gráfico de dispersión ICER. 3 PIB per cápita……………………………………...………….75
Figura 9 Curva de aceptabilidad de costo-efectividad………………………………………76

4
Lista de tablas
Tabla 1. Riesgo estimado para población de alto riesgo……………………………………17
Tabla 2. Claves diagnosticas según edad…………………………………………………….19
Tabla 3. Detección de portadores de fibrosis quística antes y después del test por panel
de 25 mutaciones…………………………………………..……………………………………22
Tabla 4. Revisión de los estudios económicos de test de portador para fibrosis quística
incluidos en el análisis…………………………………………………………………………..44
Tabla 5. Probabilidades, costos y resultados de estudios incluidos……………………….49
Tabla 6. Costos de la prueba de portador por secuenciación completa del gen CFTR…54
Tabla 7. Costos de tratamiento en pacientes con fibrosis quística según reporte de alto
costo 2012, Colombia……………………………………………………………………………55
Tabla 8. Distribución y prevalencia de pacientes con fibrosis quística en Colombia,
2012……………………………………………………………………………………………….56
Tabla 9. Parámetros de costos aplicados al modelo………………………………..……....57
Tabla 10. Riesgo de tener un niño afectado después del test con proporción
de detección de portador del 90%.....................................................................................58
Tabla 11. Probabilidad de tener un niño afectado por fibrosis quística antes y
después del test de portador……………………………………………………………………59
Tabla 12. Estudios no incluidos en el análisis de características operativas…………….61
Tabla 13. Estudios incluidos en el análisis de características operativas………………..63
Tabla 14. Resultados QUADAS 2………………………………………………………..……67
Tabla 15. Probabilidades aplicadas al modelo……………………………………………....68
Tabla 16. Reporte del diagrama de tornado………………………………………………….71
Tabla 17. Análisis de sensibilidad de dos vías, probabilidad de portador de
mutación en cónyuge 1 y 2……………………………………………………………………..72

5
Lista de abreviaturas
CFTR Regulador de la conductancia transmembranal de fibrosis quística
ACOG Colegio Americano de Ginecólogos y Obstetras
VEF Volumen espiratorio forzado
PCR Reacción en cadena polimerasa
ASO Hibridización de oligonucleótido específica
SSCP Polimorfismo conformacional simple
DGGE Electroforesis denaturación de gradiente en gel
ACMG Colegio Americano de Genética Médica
OMS Organización mundial de la Salud
OPS Organización panamericana de la Salud
OLA Ensayo de ligación de oligonucleótido
ARMS Sistema de amplificación refractaria de mutaciones
DHPLC Desnaturalización liquida de alta resolución cromatografica
MLPA Prueba de amplificación dependiente de ligación múltiple
CAP Colegio Americano de Patología
MGL Laboratorio de genética molecular
ICER Razón de costo efectividad incremental
PIB Producto Interno Bruto
SISPRO Sistema integral de información de la Protección Social

6
Introducción
La fibrosis quística es una enfermedad genética de carácter autosómico recesivo
clasificada como una enfermedad huérfana de alto costo. En Colombia presenta una
incidencia estimada de 1/5000 nacidos vivos (1). Existe variabilidad en el riesgo de
probabilidad de portador de mutación según el origen étnico sin presentar antecedente
familiar de la enfermedad. En familiares del caso índice de fibrosis quística, esta
probabilidad de portador de mutación aumenta según su grado de consanguinidad.
El riesgo de ser portador de mutación del gen CFTR se detecta a través de pruebas
genéticas que estudian de manera parcial o total los componentes del gen. Esta
enfermedad disminuye la sobrevida alcanzando un promedio de 37 años de edad, con un
adecuado manejo multidisciplinario en centros especializados (21).
En Colombia son pocos los estudios que contribuyen a establecer la epidemiologia y la
estadística de las mutaciones más frecuentes en pacientes afectados con la enfermedad
y en portadores asintomáticos, ya que no es frecuente que se analice la totalidad del gen
CFTR (1, 3, 13). No existe un panel de mutaciones específico para Colombia que
mantenga unas características operativas altas y pueda reducir los costos de la prueba
genética. Tampoco se han realizado en el país estudios de análisis de costo efectividad
de pruebas genéticas que permitan la detección de portadores de mutaciones en fibrosis
quística para la prevención de futuras concepciones en riesgo de presentar la
enfermedad.
En el presente estudio se establece la siguiente pregunta de investigación con base en la
estrategia PICO:
• Población: hombres y mujeres en edad fértil asintomáticos en primer, segundo
y tercer grado de consanguinidad a un caso índice con diagnóstico de
fibrosis quística.

7
• Intervención: prueba diagnóstica de secuenciación del gen CFTR con asesoría
genética.
• Comparación: no hacer prueba, ya que no se realiza de rutina en la atención
clínica.
• Desenlace: razón de costo efectividad incremental en términos de
prevención de futuras concepciones mediante asesoría genética por riesgo de
presentar fibrosis quística.
• Tipo de estudio: evaluación económica de tecnologías sanitarias con análisis de
costo-efectividad basado en un modelo de toma de decisiones desde la
perspectiva del tercer pagador.
• Horizonte temporal: 1 año.
Cuál es la costo-efectividad de la prueba diagnóstica de secuenciación del gen CFTR
comparado con no realizarla, para personas asintomáticas en edad fértil con
antecedente familiar de fibrosis quística en Colombia?

8
Justificación
La fibrosis quística es considerada la enfermedad genética más prevalente del grupo de
las autosómicas recesivas, estimándose una incidencia mundial alta en caucásicos de
aprox. 1 de cada 2.500 nacidos vivos (3). En Colombia se estima una incidencia
aproximada de la enfermedad en uno de cada 5000 nacidos vivos.
En los adolescentes portadores debería de ofrecerse la oportunidad de discutir su
estado genético y sus posibles implicaciones reproductivas con consejería
genética. Los padres y familiares pueden ser informados de su estado genético por
pruebas moleculares con la posibilidad de requerir diagnóstico prenatal en embarazos
futuros (4).
Para los padres portadores, existe un riesgo del 25% de transmitir ambos alelos
defectuosos a sus hijos desarrollando la enfermedad y un 50% de riesgo de tener hijos
portadores sanos. Existen pruebas genéticas para identificar a personas portadoras
de estas mutaciones; permitiendo ofrecer asesoría genética en técnicas
reproductivas a familiares de primer, segundo y tercer grado de consanguinidad y a sus
respectivos cónyuges, con antecedente familiar de diagnóstico de fibrosis quística.
El costo de un paciente con FQ se estima entre $ 10.000 y 40.000 dólares por año en
costos médicos directos y $ 9.000 dólares en costos secundarios (5); mientras que la
prueba de secuenciación completa del gen CFTR tiene un costo estimado entre $ 1.000
y $ 2.000 dólares en Colombia, considerándose una posible medida costo efectiva a
ser evaluada e implementada en el sistema de salud colombiano.

9
En Colombia se aplican unos criterios de priorización enfocados en la “Metodología de
ponderación de criterios para la selección de tecnologías en salud a evaluar”, teniendo
en cuenta la carga de la enfermedad, el perfil epidemiológico, la situación de salud
según el plan decenal de salud publica, las recomendaciones de guías de práctica
clínica colombianas, la intervención de primera línea de atención o uso cotidiano, los
grupos poblacionales vulnerables y la frecuencia de recobro de la tecnología; estos
criterios corresponden a una disposición del Ministerio de Salud y Protección Social (6).
Para el caso de fibrosis quística se cumplen algunos de estos criterios, los cuales
podrían tenerse en cuenta al evaluar la costo-efectividad de la prueba diagnóstica de
portador y asesoría genética para familiares de pacientes con fibrosis quística.
Es importante mencionar que en Colombia aún no se han realizado estudios de costo
efectividad de la prueba de secuenciación ni se encuentra incluido en e l s is tema de
salud colombiano, mientras que otros países, como Australia y Países Bajos (48, 52)
es incluido en la atención clínica y se ha avanzado en la aplicación de esta prueba
diagnóstica con repercusiones a futuro. Por eso es cuestionable evaluarla en el
contexto económico y clínico colombiano para poder considerarla dentro de la
atención en salud.

10
Capitulo 1
1.1 Objetivos
1.1.1 Objetivo general
Determinar la razón de costo-efectividad de la prueba diagnóstica de secuenciación del
gen CFTR para portadores asintomáticos en edad fértil familiares de primer, segundo
y tercer grado de consanguinidad al caso índice con diagnostico de fibrosis quística en
términos de prevención de futuras concepciones por riesgo de presentar fibrosis
quística mediante la asesoría genética bajo la perspectiva del tercer pagador.
1.1.2 Objetivos específicos
• Buscar y valorar la calidad de la evidencia de las características operativas de la
prueba diagnóstica de secuenciación del gen CFTR para portadores
asintomáticos de fibrosis quística con antecedente familiar.
• Adaptar o crear un modelo económico a p l i c a b l e al contexto clínico
colombiano, a través de un árbol de decisiones para la detección de portadores
asintomáticos de fibrosis quística con antecedente familiar.
• Medir, valorar y estimar los costos directos de la prueba diagnóstica de
secuenciación y el costo anual de atención de un paciente con fibrosis
quística desde la perspectiva del tercer pagador.

11
1.2 Marco teórico
Su primera identificación clínica surgió a partir de 1934 con Andersen y Fanconi et al. (7,
8). En 1985 se identificó y localizó la mutación responsable de la fibrosis quística en el
brazo largo del cromosoma 7 humano y cuatro años después gracias a equipos
multidisciplinarios principalmente Tsui y Collins (9) se reportó el gen responsable de la
enfermedad, identificado con la mutación más frecuente, la DF508, presentando
deleción de tres pares de bases que codifican un único aminoácido, la fenilalanina. Es
considerada como una enfermedad genética de transmisión autosómica recesiva que
compromete el gen CFTR, regulador de la conductancia transmembrana de la fibrosis
quística (FQ), localizado en el cromosoma 7 y compuesto por 27 exones (10).
Las mutaciones presentan variabilidad según el grupo étnico y área geográfica. En la
actualidad se describen 2008 mutaciones, desde la no producción de proteína CFTR
hasta la regulación defectuosa del canal de cloro y reducción de la síntesis de ARN
mensajero, generando variabilidad clínica (11).
1.2.1 Epidemiología
La incidencia mundial de Fibrosis quística es variable; en Europa se da en 1 de cada
2000 a 3000 nacidos, en África 1 de cada 7056 nacidos, en Norteamérica 1 de cada 3500
nacidos, en Latinoamérica varía entre 1 de cada 3900 a 8500 nacidos, en Medio Oriente
1 de cada 2560 a 15876 nacidos, en Asia 1 de cada 40000 a 100000 nacidos. Esta gran
variabilidad depende de la consanguinidad y del origen étnico, de mayor incidencia en
raza caucásica; 1 de cada 2500 recién nacidos con gran variabilidad regional por
heterogeneidad étnica, siendo más frecuente a nivel mundial la mutación DF508 (12).
Según estudios realizados en Latinoamérica, se han encontrado incidencias de la
enfermedad en 1 de cada 8000 recién nacidos en México y en 1 de cada 3862 recién
nacidos en Cuba (13).

12
Según el Colegio Americano de Ginecólogos y Obstetras (ACOG) 2011 (14), la incidencia
de la enfermedad es de 1 en 2500 individuos blancos no hispánicos. La frecuencia de
portadores sanos heterocigotos en la población caucásica se ha estimado en 1 de cada
25 personas (3, 13).
Keyeux y col, (13, 15) en Colombia, analizaron la presencia de mutaciones del gen
CFTR por reacción en cadena polimerasa de 92 pacientes diagnosticados con fibrosis
quística y 130 pacientes control sin la enfermedad y sin ningún antecedente familiar de
riesgo, encontrando que de los 130 analizados dos eran portadores de la mutación
DF508 heterocigoto con una frecuencia de portadores de 1 en 65. De los 92 pacientes
con fibrosis quística analizados se descartaron 16 por tener antecedente directo de
pariente en primer grado afectado con la enfermedad; en los pacientes restantes se
encontró un 48% de frecuencia mutacional DF508 y una alta heterogeneidad regional y
de haplotipos. Algunas otras mutaciones encontradas fueron la c.1811+1.6kbA>G (6.5%),
p.G542X (3.8%), p.S549R (2.2%), p.W1282X (1.1%) and p.R1162X (1.1%),
desconociendo el resto de mutaciones predominantes en Colombia.
En el 2000 Restrepo y Cols. (16), analizaron 96 pacientes latinoamericanos con fibrosis
quística por diagnostico clínico y test de sudor; 45 mexicanos, 24 colombianos y 27
venezolanos. Se analizaron por técnica de reacción en cadena polimerasa PCR con un
panel de 16 mutaciones. La eficacia de esta técnica fue reportada en 47.9% a nivel
general y de 45.8% específicamente para Colombia. La mutación más común fue la
DF508 en 39.6%, seguida de la G542X y N1303k, 4,7 % y 1 % respectivamente. Las
mutaciones no detectadas con este panel fue del 52.1%.
En el 2010 Vásquez y Cols. (17), realizaron un estudio de corte transversal evaluando
un total de 128 pacientes colombianos con fibrosis quística, encontrando una ligera
prevalencia de la enfermedad en mujeres del 53.9%, en su mayoría provenientes de
Bogotá 57%, con edades entre los 2 y 25 años, contando que solo el 14,8 % era mayor
de 18 años, con principales manifestaciones de síntomas respiratorios y falla en el
crecimiento. Las bacterias más frecuentes en cultivo de esputo fueron el stafilococcus
aureus y Pseudomona aureginosa, con 57 % y 39.8% respectivamente. Según el VEF 1,
el 32,7% de los pacientes no tenían perdida de la función pulmonar, mientras 23%

13
presentaban obstrucción leve, 25% moderada y 19% severa. El 35.9% de estos
pacientes presentaban talla baja para la edad y 29.7% peso bajo para la edad. Las
complicaciones más frecuentes fueron sinusitis e hipertensión pulmonar con 26.6% y
compromiso hepatobiliar e hipoxemia con 20.3%. El análisis genético fue realizado en 66
pacientes (52%) encontrando que la mutación más frecuente fue la DF508 y la
621+1G>T con 54.5 y 10.6% respectivamente, evaluado con el panel de 32 mutaciones.
La edad de diagnostico inicial fue de 3,6 años mostrando demora en el diagnostico,
realizado a los 6 meses de edad en otros países.
Jay y col en 2006 (3) realizaron un análisis de frecuencia de portador mutacional DF508
en Colombia a través de la técnica PCR heteroduplex por agrupamiento en 400 personas
asintomáticas, comparada con la técnica de heteroduplex simple. La sensibilidad de la
técnica fue de 92.5% y la especificidad de 100% en la heteroduplex por agrupamientos
de hasta 10 muestras, y del 100% de sensibilidad y especificidad para heteroduplex
simple. Se reportaron 10 casos positivos para mutación DF508 tanto en la técnica simple
como en la de agrupamiento de 5 muestras mostrando una frecuencia de portadores de
1 en 40, 7 casos positivos para la técnica por agrupamiento de 10 muestras y 0 casos
para agrupamiento de 20 muestras.
Pérez y col (18), realizaron en el 2007 un análisis estadístico de los diferentes estudios
en Centroamérica y Suramérica para la detección de distribución de mutaciones del gen
CFTR a través de diferentes técnicas diagnosticas tales como reacción en cadena
polimerasa (PCR), digestión enzimática, hibridización de oligonucleótido especifica
(ASO), DGGE Electroforesis denaturación de gradiente en gel, SSCP Polimorfismo
conformacional simple y análisis heteroduplex. Se incluyeron estudios de Argentina,
Brasil, Chile, Colombia, México, Ecuador, Venezuela, Costa Rica, Cuba y Uruguay. De
4354 cromosomas analizados, 89 mutaciones fueron encontradas, con una proporción de
detección del 62.79%. La DF508 fue la mutación más frecuente con 46.69% en promedio
con variabilidad según la región, seguidas de la p.G542X y p.N1303K, 5,08% y 1,65%
respectivamente. Para Colombia con un panel de las 7 mutaciones más frecuentes, se
estima un poder de detección de mutación del 56%.

14
Silva y col (1), en el 2007 determinaron en Colombia la presencia de la mutación
F508del mediante reacción en cadena de la polimerasa (PCR) y análisis de heteroduplex
en 2608 personas sanas sin evidencia clínica ni historia familiar de fibrosis quística. Se
determinó una frecuencia de portadores de 1/84 para Colombia; 31 fueron heterocigotos
para la deleción de tres bases en el exón 10. Se estimo una incidencia de la enfermedad
de 1 recién nacido afectado por cada 5025 recién nacidos.
Un estudio piloto realizado el 2011 en Bogotá, encontró una incidencia de 1 caso de
fibrosis quística por cada 8297 recién nacidos, observando un subdiagnóstico y
subregistro con respecto al número de pacientes registrados con la enfermedad (19).
1.2.2 Fisiopatología
La glándula sudorípara normal produce un líquido isotónico, gracias a su contenido de
cloro (Cl-) y sodio (Na+). Cuando pasa por el conducto excretor, que es impermeable al
agua, se produce la entrada de Na+ en la célula, a través de la membrana apical y
gracias a la existencia de un gradiente electroquímico favorable este ion abandona la
célula a través de la membrana baso-lateral, intercambiándose por potasio (K+). En la
bomba Na+- K+-ATPasa, el K+ entra en la célula por los canales para este ion que existen
en la membrana basolateral. El Na+ intracelular establece un gradiente favorable para
que penetre el Cl-, tanto por los canales apicales de CFTR como por los basolaterales.
En la FQ la pérdida de función de la CFTR hace que el Cl- no pueda entrar en la célula y
por lo tanto tampoco lo haga el Na+, dando lugar a un aumento de estos iones en el
sudor (20).
Existe una alteración de la regulación del canal de cloro activado por AMP cíclico,
localizado en la región apical de células epiteliales secretoras, generando disfunción de
canales de cloro y sodio (10).

15
El CFTR es un gen que codifica un canal de membrana celular esencial en el transporte
de iones. Al alterarse su estructura las secreciones tienden a aumentarse y a volverse
viscosas. Este canal es responsable del transporte de cloruro y regula los canales de
cloro, bicarbonato, sodio y calcio.
El gradiente electroquímico lo produce la bomba Na+-K+ ATPasa. En la vía aérea el
sodio entra a la célula desde el canal y para mantener el equilibrio debe reabsorberse Cl-
por la vía paracelular. A nivel de la membrana apical se ubican dos canales para el Cl- ,
el CFTR y el canal alternativo regulado por Ca+ intracelular. En la Fibrosis quística la
permeabilidad para el Cl- se altera. La proteína CFTR se expresa en niveles más altos en
glándulas submucosas. A nivel de páncreas el CFTR se expresa en el ápice de células
epiteliales de sus conductos donde actúan el Na+, Cl- y el bicarbonato (1).
Se aumenta el cloruro de sodio en el sudor y la viscosidad de las secreciones
glandulares con riesgo de obstrucción de los canales excretores, ocasionando perdida de
la función ciliar en vía aérea y perdida de la función glandular con riesgo de colonización
por Pseudomona aeuriginosa.
1.2.3 Diagnóstico
El diagnostico clínico se manifiesta con al menos tres determinaciones positivas de
electrolitos en sudor asociada con uno de los siguientes criterios; íleo meconial, historia
familiar de fibrosis quística, insuficiencia pancreática exocrina, enfermedad pulmonar
crónica, azoospermia obstructiva, síndrome de pérdida de sal, entre otras variaciones
clínicas por la gran cantidad de mutaciones infrecuentes (10).
Por la variabilidad y polimorfismos en el gen CFTR, existen una forma clásica de fibrosis
quística y una amplia sintomatología secundaria a la FQ, tales como ausencia congénita
de vasos deferentes, bronquiectasia diseminada, panbronquiolitis difusa, enfisema
pulmonar, asma, pancreatitis crónica e hipertripsinemia neonatal (12).

16
En la prueba de electrolitos en sudor, se estimula la sudoración por iontoforesis con
pilocarpina; se recoge el sudor y se determina la concentración de electrolitos. Es
positivo si el cloro es > 60mEq/L, y negativo < 40mEq/L, entre 40 y 60 se considera
dudoso, sin embargo existen diagnósticos diferenciales tales como hipotiroidismo e
insuficiencia suprarrenal, entre otros. Hay exámenes complementarios al diagnostico
de la fibrosis quística; el cultivo del tracto respiratorio para patógenos relacionados,
pruebas de función pulmonar, radiografía de tórax, TAC de tórax, TAC de senos
paranasales, evaluación de la función pancreática y el recuento de esperma (21).
Al ser una enfermedad genética autosómica recesiva, una pareja portadora tiene
probabilidad del 25% de tener un hijo afectado, el cual debe poseer la mutación en los
dos alelos de el gen CFTR; si ellas son diferentes el paciente se denomina heterocigoto
compuesto. Ante la presencia de una mutación que afecte solo un alelo, se considera
como un paciente portador de mutación CFTR asintomático (22).
Figura 1 Patrón de herencia autosomica recesiva de la FQ. Fuente: Tomado de Goetzinger 2010. (23)

17
Tabla 1 Riesgo estimado para población de alto riesgo. Fuente: Tomado de Gregg 2002. (22)
Riesgo sin
tamizaje
Riesgo individual Riesgo para el
Embarazo sin
tamizaje
Judio Ashkenazi 1/29 1/29* 1/29* 1/4: 1/3364
Caucásico 1/29 129* 1/29* 1/4: 1/3364
Riesgo con
tamizaje
(negativo)
Riesgo individual
(negativo)
Pareja negativa Uno negativo,
compañero no
testeado
Judío Ashkenazi 1/930 1/930* 1/930* 1/4:
1/3459600
1/930* 1/29* 1/4:
1/107880
Caucásico 1/140 1/140* 1/140* 1/4:
1/78400
1/140* 1/29* 1/4:
1/16240
Riesgo con
tamizaje (positivo)
Uno positivo,
compañero negativo
Uno positivo,
compañero no
testeado
Pareja positiva
Judio Ashkenazi 1* 1/930* 1/4: 1/3720 1* 1/29* 1/4: 1/116 1* 1* 1/4: ¼
Caucásico 1* 1/140* 1/4: 1/560 1* 1/29* 1/4: 1/116 1* 1* 1/4: ¼
Las variantes detectadas en la secuenciación de ADN son validadas en la base de datos
de la CFTR.org database.; las nuevas son reportadas para interpretación clínica posterior
(21). Su diagnostico clínico es variable; en 80-90% de los casos presenta insuficiencia
pancreática exocrina y enfermedad pulmonar progresiva (10).

18
Es una enfermedad crónica y progresiva, con características clínicas típicas de
compromiso respiratorio y gastrointestinal, tales como tos productiva persistente, coriza,
estornudos, disnea, hemoptisis, hipocratismo digital y neumonía recurrente secundaria a
Pseudomona aeruginosa, Staphylococcus aureus y Hemophilius influenzae, mostrando
un patrón obstructivo restrictivo en las pruebas de función pulmonar con hiperinflación y
bronquiectasias en la radiografía de tórax. A nivel gastrointestinal se inicia con intestino
ecogénico e íleo meconial, intususcepción, obstrucción intestinal recurrente,
malabsorción, esteatorrea, prolapso rectal, hepatopatía, déficit de vitamina A y E,
malaabsorción intestinal, retardo de crecimiento, desmineralización ósea, compromiso
dérmico, visual y distrofia neuronal. En pacientes de mayor edad puede debutar con
diabetes mellitus secundaria a la fibrosis quística y déficit pancreático. Puede presentarse
azoospermia por ausencia, atrofia o fibrosis de vasos deferentes. Muchas de estas
manifestaciones clínicas están relacionadas con el tipo de mutación presente en el
paciente debido a la heterogeneidad génica, logrando predecirse manifestaciones
clínicas acorde a la mutación detectada, conocida como correlación genotipo-fenotipo
(22).
Las mutaciones de fibrosis quística pueden generar cuatro respuestas clínicas, tales
como fibrosis quística, desordenes relacionados con el CFTR, mutaciones con
consecuencia clínica desconocida y mutaciones con relevancia clínica incierta o no
probada. Entre las mutaciones presentes, suele haber un cambio en la secuencia de
aminoácido que afecta severamente la síntesis o la función del CFTR, o se introduce una
señal de terminación prematura como inserción, deleción o alteración de los dos primeros
o últimos nucleótidos con deleción de uno o más exones (24).

19
Tabla 2 Claves diagnosticas según edad. Fuente: Tomado de Guía de práctica clínica FQ 2014 Colombia.
Grupo de
edad
Presentación frecuente Menos frecuente
Antenatal Vellosidades coriónicas
Intestino ecogénico en
Ecografías
Íleo meconial perforado
Neonatal Tamización neonatal, íleo
Meconial, obstrucción intestinal
Con perforación o sin ella
Y peritonitis.
Atresia intestinal , ictericia
Obstructiva, deficiencia de
Vitaminas liposolubles.
Lactantes y
niños
Pequeños
Síntomas respiratorios persistentes (tos,
sibilancias, neumonía, infiltrados en
radiografía de tórax, bronquiectasias,
sudor salado, falla de crecimiento,
esteatorrea, diarrea, distensión abdominal,
aumento de apetito, colestasis.
Prolapso rectal, trastorno de
deshidratación, anemia,
edema e hipoproteinemia.
Niños
mayores y
adultos
Síntomas respiratorios recurrentes, tos
crónica, broncorrea, hipocratismo digital,
pólipos nasales, sinusitis, infertilidad
masculina, obstrucción intestinal distal,
diabetes.
Pancreatitis aguda, crónica,
enfermedad hepática,
malabsorción, deshidratación,
trastornos hidroelectrolíticos.

20
Para un seguimiento clínico existe el Score de Shwachman y Kulczycki que evalúa la
actividad general, el aparato respiratorio, el estado nutricional y la radiografía de tórax
con una puntuación ideal de 100 para establecer el estado clínico de un paciente con
fibrosis quística. A nivel radiológico y de tomografía de tórax se puede valorar la
presencia de atrapamiento aéreo, sombras lineales, lesiones nodulo-quisticas y
consolidaciones segmentarias o lobares (10).
Las características fenotípicas de la fibrosis quística incluyen la enfermedad
sinopulmonar crónica, las alteraciones gastrointestinales y nutricionales, los síndromes
por perdida de sal en sudor y la ausencia congénita bilateral de conductos deferentes.
La edad promedio de diagnostico de la enfermedad está entre los 6 a 8 meses y en más
del 50% de los casos son diagnosticados antes de cumplir el año de edad. Entre la
segunda y tercera década de la vida puede cursar desde un cuadro atípico hasta la
muerte debido principalmente a complicaciones pulmonares. La edad promedio de
sobrevida para 1995 era de 30.1 años pero ha aumentado en los últimos años sin cura
total de la enfermedad a pesar de los avances en el tratamiento (25). En el 2004 la
Asociación Colombiana de Neumología pediátrica encontró que el diagnóstico se había
realizado tardíamente, a los 3,6 años en promedio (21).
1.2.4 Pronostico
El promedio de edad de supervivencia en un paciente con FQ es de 37 años presentando
falla respiratoria como principal causa de muerte, aunque aproximadamente un 15%
tienen un curso moderado de la enfermedad y alcanzan un promedio de vida de 56 años
(14).
Es considerada una enfermedad de alto costo derivada principalmente de las
prescripciones ambulatorias y de atención hospitalaria. Por ser una enfermedad
multisistémica requiere manejo multidisciplinario en programas de rehabilitación,

21
ejercicio, soporte nutricional, trabajo social, psicología y medicina especializada debido al
impacto sobre la salud y calidad de vida del paciente y su familia (21).
1.2.5 Estudio de portadores
Según la guía de práctica clínica colombiana de FQ del 2014 se recomienda realizar
estudios moleculares a los familiares directos de pacientes con fibrosis quística en edad
fértil porque disminuye la incidencia de la enfermedad, siempre y cuando vayan
acompañados de asesoría genética para optar diferentes estrategias de reproducción. El
asesoramiento genético lo debe realizar un medico genetista o consejero genético,
explicando al paciente y a su familia la historia natural de la enfermedad, la etiología, los
riesgos de recurrencia en la familia, tratamientos disponibles y opciones reproductivas
respetando la decisión del paciente.
Las opciones reproductivas para parejas portadoras incluyen diagnostico prenatal con
posible terminación del embarazo, la donación de oocito o esperma, la adopción o el
diagnostico genético preimplantacional (23).
Los paneles de mutación buscan identificar mutaciones comunes de cada región, pero
depende de la epidemiologia genética con respecto al gen CFTR, ya que el análisis
completo del gen es laborioso y costoso. El panel de 25 mutaciones recomendado por
el Colegio Americano de Genética Médica (ACMG) detecta 83.7 % de las mutaciones del
gen CFTR pero difiere según las diferentes regiones (12).
El ACMG y el Colegio Americano de Ginecólogos y Obstetras (ACOG) recomendaron en
el 2001 el test prenatal/preconcepcional para portador de Fibrosis Quística cuando uno o
ambos miembros de una pareja fueran Caucásicos, o tuvieran historia familiar de
fibrosis quística o alguno de la pareja tuviera la enfermedad. El objetivo propuesto fue
que el test de tamizaje identificara parejas en riesgo de tener un niño con fibrosis
quística clásica. Con un resultado negativo del test de portador de 25 mutaciones se
reduce mas no se elimina el riesgo de ser un portador de fibrosis quística. Este test

22
incluye mutaciones que al menos alcancen una frecuencia de 0.1%, pero no se tuvo en
cuenta la posibilidad de aumentar su sensibilidad al incluir muchas más mutaciones
presentes (26).
En 2005 la ACOG y ACMG reportaron la proporción de portador de mutación para FQ
antes del test en 1 de cada 25 personas para Caucásicos no hispanos con una
proporción de detección del 88% usando el panel de 25 mutaciones, que dos años
después se redujo a 23 mutaciones por no alcanzar el 0.1% de frecuencia en dos de
ellas. Según la ACOG y ACMG el análisis completo del gen CFTR por secuenciación es
reservado solo para pacientes con fibrosis quística, o con historia familiar de fibrosis
quística, o con infertilidad masculina con ausencia bilateral de vasos deferentes o test de
tamizaje de recién nacido positivo, pero no sería apropiado como tamizaje de rutina para
portador poblacional sin ningún antecedente familiar (14, 26).
La proporción de detección de portador con el test de 25 mutaciones es de 94% para
judíos Ashkenazi, 88% para blancos no hispánicos, 72 % para blancos hispánicos; con
riesgo de portador antes del test de 1/24, 1/25 y 1/58 respectivamente (14).
Tabla 3 Detección de portadores de fibrosis quística antes y después del test por panel de 25 mutaciones.
Fuente: Tomado de ACOG 2011 (14).
Grupo étnico o
racial
Detección
(%)
Riesgo de
portador
Antes del test
Riesgo de portador
Después de un test
negativo
Judíos Ashkenazi 94 1/24 1/380
Blancos no
hispánicos
88 1/25 1/200
Blancos hispánicos 72 1/58 1/200
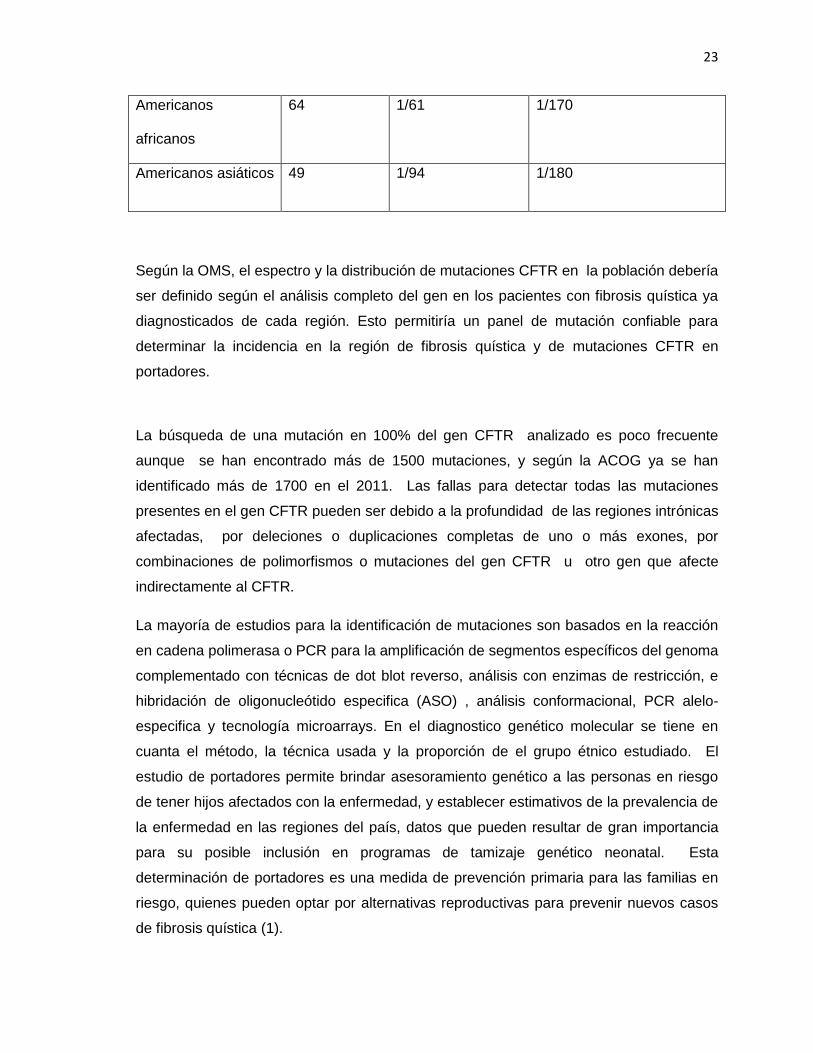
23
Americanos
africanos
64 1/61 1/170
Americanos asiáticos 49 1/94 1/180
Según la OMS, el espectro y la distribución de mutaciones CFTR en la población debería
ser definido según el análisis completo del gen en los pacientes con fibrosis quística ya
diagnosticados de cada región. Esto permitiría un panel de mutación confiable para
determinar la incidencia en la región de fibrosis quística y de mutaciones CFTR en
portadores.
La búsqueda de una mutación en 100% del gen CFTR analizado es poco frecuente
aunque se han encontrado más de 1500 mutaciones, y según la ACOG ya se han
identificado más de 1700 en el 2011. Las fallas para detectar todas las mutaciones
presentes en el gen CFTR pueden ser debido a la profundidad de las regiones intrónicas
afectadas, por deleciones o duplicaciones completas de uno o más exones, por
combinaciones de polimorfismos o mutaciones del gen CFTR u otro gen que afecte
indirectamente al CFTR.
La mayoría de estudios para la identificación de mutaciones son basados en la reacción
en cadena polimerasa o PCR para la amplificación de segmentos específicos del genoma
complementado con técnicas de dot blot reverso, análisis con enzimas de restricción, e
hibridación de oligonucleótido especifica (ASO) , análisis conformacional, PCR alelo-
especifica y tecnología microarrays. En el diagnostico genético molecular se tiene en
cuanta el método, la técnica usada y la proporción de el grupo étnico estudiado. El
estudio de portadores permite brindar asesoramiento genético a las personas en riesgo
de tener hijos afectados con la enfermedad, y establecer estimativos de la prevalencia de
la enfermedad en las regiones del país, datos que pueden resultar de gran importancia
para su posible inclusión en programas de tamizaje genético neonatal. Esta
determinación de portadores es una medida de prevención primaria para las familias en
riesgo, quienes pueden optar por alternativas reproductivas para prevenir nuevos casos
de fibrosis quística (1).

24
Existen diferentes métodos para la identificación de mutaciones en fibrosis quística, por
un lado métodos para mutaciones conocidas o establecidas tales como el análisis de
heteroduplex para DF508 micro inserciones y deleciones, el análisis de restricción
enzimática útil para mutaciones puntuales especificas, la hibridización reversa dot blot
para detectar hasta 20 mutaciones, al igual que el ARMS (Sistema de amplificación
refractaria de mutaciones) y el OLA (ensayo de ligación de oligonucleótidos) entre otros
que detectan hasta 32 y 36 mutaciones. De igual manera existen métodos para la
identificación de mutaciones desconocidas, entre estos el DGGE (electroforesis en gel
con gradiente de desnaturalización), el DHPLC (desnaturalización liquida de alta
resolución cromatográfica), el MLPA (Prueba de amplificación dependiente de ligación
múltiple) que logran altas sensibilidades mayor al 95%. La más aplicada para su uso en
este grupo es la secuenciación del gen que logra una sensibilidad cercana al 100% con
muy pocas limitaciones. Aun con todos estos métodos entre el 1 y 5 % de alelos
permanecen indeterminados en pacientes con presentaciones de fibrosis quística clásica
y aun más en pacientes con presentaciones atípicas. No todas las mutaciones
encontradas están relacionadas fenotípicamente con caso típico de fibrosis quística; las
mutaciones en fibrosis quística se pueden agrupar en; 1) mutaciones que causan fibrosis
quística clásica, 2) mutaciones que resultan en enfermedades relacionadas con el CFTR,
3) mutaciones con consecuencias no clínicas y 4) mutaciones de relevancia clínica
incierta. Esto se debe tener en cuenta a la hora de evaluar los portadores de mutaciones
en especial las mutaciones del grupo 1 y 4, por tal motivo es importante la consejería
genética para analizar la relación genotipo/fenotipo de los hallazgos encontrados.
Debido a la alta heterogeneidad locus de este gen, los paneles de detección de mutación
para CFTR varían en su proporción de detección por diferencias étnicas y geográficas
debido a que la mayoría de estos paneles son realizados según la frecuencia de
mutaciones encontradas en Estados Unidos y Europa. Cada región debería de tener su
propio panel de mutaciones que causen fibrosis quística sobre una frecuencia mutacional
del 1%. Para pacientes con fibrosis quística diagnosticada clínicamente se deben buscar
las mutaciones más frecuentes y se confirma la enfermedad con la presencia de los dos
alelos afectados por mutación, si hay solo una o ninguna mutación, se extiende la
búsqueda por secuenciación a todo el gen y se puede determinar que posiblemente no

25
es un caso de fibrosis quística sino de portador. Se rastrea igualmente a los padres del
caso índice de fibrosis quística y rara vez se obtienen mutaciones de novo, aunque se
deben descartar también casos de no paternidad y error en la toma de muestras (27).
En el 2004 Watson y col (28), revisaron el panel de mutación de portadores para fibrosis
quística propuesto en el 2001 por el colegio americano de genética médica teniendo en
cuenta que se descartaban las mutaciones < 0.1 % en frecuencia. El análisis se basó en
42737 cromosomas de pacientes con Fibrosis quística provistos por la Fundación para
Fibrosis Quística de los Estados Unidos. Se encontró que algunas mutaciones que fueron
<0.1% ahora son más frecuentes y viceversa sin ser removidas o añadidas al panel
propuesto de 25 mutaciones. Se evidencio que la frecuencia de las mutaciones difiere
con relación a su etnicidad, por ejemplo el DF508 tiene una proporción de 72.4% en
Caucásicos no hispanos, 54.38% en Caucásicos hispanos y 31.4% en judíos. Se
recomienda por eso tener en cuenta la distribución étnica y geográfica de las mutaciones
al momento de realizar un tamizaje para portadores.
Monoghan y col (29) en el 2004, realizaron un análisis genético de 2189 muestras de
origen Americano africano con el panel de 25 mutaciones referido por la ACMG a través
de heteroduplex, ensayo de ligación de oligonucleótido entre otros métodos. Se
encontraron 33 portadores de Fibrosis quística con una proporción de detección del 66%,
concluyendo que para aumentar esa proporción de detección debe aumentar el número
de mutaciones analizadas según cada región.
1.2.6 Prueba de Secuenciación
El método enzimático de Sanger conocido también como técnica de terminación de
cadena de Sanger o técnica del didesoxi requiere 1) de una plantilla o secuencia simple
de ADN a copiar, 2) de un primer o secuencia corta de ADN con 20 nucleótidos aprox.
que permite ser el punto de inicio para sintetizar una secuencia, 3) de una enzima DNA
polimerasa para sintetizar cadenas de ADN con una terminación especifica; generando
fragmentos de ADN de todos los tamaños posibles distinguidos por un marcaje o
terminador especifico. La ADN polimerasa trabaja con ADN de cadena sencilla llamado

26
“templado” y sintetiza la hebra complementaria a partir de un iniciador o “primer” en
dirección 5´ a 3´. Los terminadores son nucleótidos que no tienen un grupo hidroxilo en
su extremo 3´ para obtener una terminación específica ddNTP dideoxinucleótidos
trifosfato evitando que la cadena de ADN sintetizada continúe extendiéndose, ya que la
ADN polimerasa no puede enlazar otros nucleótidos al ddNTP. La estrategia de Sanger
hace 4 reacciones diferentes de síntesis de ADN utilizando un ddNTP distinto en cada
tubo para cada nucleótido. Con la mezcla de un nucleótido normal (dNTP) y su
terminador (ddNTP) se generan fragmentos complementarios de diferentes tamaños que
terminan en el mismo nucleótido, estos fragmentos se separan en un gel de
electroforesis con cuatro carriles distintos determinando la secuencia del templado. El
método de secuenciación de Sanger ha ido mejorando gracias a la técnica de PCR
(reacción en cadena polimerasa) para amplificar segmentos de ADN seguido del método
de electroforesis capilar que permite identificar el orden de los nucleótidos del ADN con
una terminación específica, además del descubrimiento de enzimas resistentes al calor
como la enzima termoestable polimerasa Taq que cortan el ADN en secuencias
específicas, el marcaje de ADN (ddNTP) con fluoróforos para identificar los fragmentos
de ADN sintetizados y el desarrollo de mejores técnicas de secuenciación con técnicas
de clonación de ADN. Gracias al marcaje de ADN con fluoróforos ya no se requieren de
cuatro tubos para cada reacción ni de cuatro carriles ya que en uno solo se pueden
identificar los terminales ddNTP marcados fluorescentes distintos en su base (30).
La secuenciación es útil cuando las mutaciones están dispersas en la totalidad del gen a
estudiar, o cuando los genes no han sido suficientemente estudiados para determinar los
puntos claves. El comité de la ACMG recomienda que el panel de mutación pan-étnico
incluya todas las mutaciones causantes de fibrosis quística con una frecuencia alélica de
≥ 0.1% en la población estadounidense en general. Los estudios han sido derivados de
análisis con población estadounidense, por tanto los paneles de mutación varían en la
proporción de detección en diferentes regiones y grupos étnicos. Los pacientes
tamizados con estos paneles tienen un riesgo residual de ser portador de mutaciones
desconocidas o no testeadas, que aumenta cuando son familiares de primer, segundo o
tercer grado de consanguinidad (31).

27
El estudio de cascada de portadores para los familiares de pacientes afectados con
fibrosis quística (primer grado) se recomienda que sea preconcepcional ofreciendo
asesoría genética y opciones reproductivas con el fin de disminuir la incidencia de la
enfermedad.
En Colombia aun no se ha implementado de rutina clínica el tamizaje neonatal para
fibrosis quística que permita un diagnostico precoz y un mejor desenlace clínico en el
paciente afectado; de igual manera tampoco se realiza el estudio genético a través de
secuenciación de ADN, el cual permitiría un mejor asesoramiento genético y lograría
establecer un panel especifico para población colombiana con el fin de detectar a un
menor costo las familias portadoras en riesgo por mutación del gen (21).
En la práctica clínica se procedería a buscar en los pacientes y en sus familias las
mutaciones más frecuentes por rastreo sin necesidad de realizar directamente la
secuenciación completa o la búsqueda completa de mutaciones en los 27 exones que
componen el gen. En Colombia debido a que no se ha estudiado ampliamente la
prevalencia de mutaciones para CFTR, no es posible determinar por ahora un panel de
detección; por eso es útil realizar la secuenciación completa del gen en pacientes con
fibrosis quística y en sus parientes con el fin de detectar la prevalencia de mutaciones y
poder implementar a futuro un panel especifico con alta proporción de detección para el
país.
Se recomienda la secuenciación completa del gen para Colombia porque en caso de que
un test expandido salga negativo sería conveniente realizar el análisis completo del gen
teniendo en cuenta que las recomendaciones del panel de 23 mutaciones aplican en
población estadunidense. En Colombia no se han realizado análisis completo del gen y
no se conoce la distribución epidemiológica de las mutaciones más frecuentes por tanto
la sensibilidad de un panel sería distinta a la referenciada en otras poblaciones. Se debe
evaluar si el paciente ya ha sido tamizado con anterioridad para no repetir exámenes, y
mejorar los resultados en costo efectividad.

28
1.2.7 Tamizaje versus test para portador de FQ
El tamizaje de portador se refiere a la detección de personas portadoras de la mutación
sin tener riesgo previo por antecedente familiar de la enfermedad, o sea que se tamiza a
toda la población de manera rutinaria sin importar antecedentes, diferente al termino test
de portador el cual se realiza en personas que tengan riesgo por antecedente familiar de
la enfermedad.
En Australia nacen alrededor de 70 bebes afectados con fibrosis quística cada año; de
los cuales en más del 90% sus padres no tenían ningún antecedente familiar de fibrosis
quística (32).
Massie y col (33) en Victoria Australia analizaron entre el 2006 y el 2008 a 3200
pacientes; entre ellos 3000 mujeres preconcepcionales y prenatales menores de 14
semanas de gestación y 200 hombres con un panel de 12 mutaciones con la técnica
PCR multiplex con sensibilidad aproximada de 83.5% para la población de Victoria. De
estos pacientes, 100 parejas fueron tamizadas en paralelo. Del total de analizados, se
identificaron 106 portadores obteniendo una frecuencia de 1 en 30 pacientes. Las parejas
de los 106 portadores fueron testeadas, logrando identificar 9 parejas portadoras en
riesgo del 25% de tener un hijo con fibrosis quística. De estas parejas, 6 estaban
embarazadas y se sometieron al diagnóstico prenatal por muestra de vellosidad
coriónica; cuatro fetos resultaron sanos, tres de ellos portadores de solo una mutación.
Los dos fetos restantes fueron afectados homocigotos con la mutación F508del, con la
decisión de terminar la gestación. Las tres parejas restantes no estaban en gestación y
decidieron someterse a diagnostico genético preimplantacional.
En el 2009 Christie y col (34) en Inglaterra, analizaron retrospectivamente la presencia de
portadores de mutaciones de fibrosis quística desde el 2003 al 2007 en 1000 individuos.
De esta población, algunas estaban en embarazo de menos de 14 semanas de
gestación y 83% constituidas como pareja. Si un compañero era identificado con
mutación DF508, se le realizaba un tamizaje expandido de 28 mutaciones a su pareja. La
técnica de tamizaje empleada fue la PCR multiplex ARMS con sensibilidad de 82% para
individuos de Reino Unido. De los 1000 individuos testeados, 730 no tenían historia
familiar de fibrosis quística; 27 de ellos fueron portadores del DF508 con una frecuencia

29
de portador de 1 en 27. De los 27 portadores, 11 eran mujeres en embarazo y 16 eran
individuos en plan preconcepcional. Dos parejas resultaron portadoras de DF508, una sin
embarazo la cual decidió ingresar al programa de fertilización in vitro sin éxito, ya que se
detecto en la mujer la presencia de dos alelos mutacionales para fibrosis quística atípica.
La otra pareja se encontraba en la sexta semana de gestación y por medio de
diagnostico prenatal se obtuvo un feto femenino no afectado portador DF508. De los 270
individuos restantes tamizados con historia familiar de fibrosis quística, 126 resultaron
portadores con una frecuencia de portador de 1 en 2. Dos parejas fueron identificadas
con alto riesgo de tener un hijo con fibrosis quística sin estar en embarazo actual. Seis
meses después, una de las parejas decidió quedar en gestación y por diagnostico
prenatal se determino un feto afectado con Fibrosis quística por dos alelos mutados,
decidiendo terminar con la gestación. La otra pareja decidió no tener hijos por el alto
riesgo presentado ya que la mujer resulto tener fibrosis quística y el hombre portador
DF508.
Picci y col (35) en 2010 reportaron en Italia los resultados de un programa de tamizaje
de portares de fibrosis quística en un periodo de 10 años desde 1996 a 2006, tamizando
a 57999 sujetos sin historia familiar previa ni signos clínicos de fibrosis quística de los
cuales 25104 eran parejas y 7791 eran solteros, fueron excluidos 1783 sujetos por
presentar historia familiar de fibrosis quística. Se analizó la presencia de las 47
mutaciones más frecuentes en Italia, con la técnica de amplificación PCR multiplex
seguido de análisis especifico de alelo de oligonucleótido usando linfocitos de sangre
periférica. Los sujetos negativos fueron analizados con denaturación y electroforesis en
gel evaluando todos los 27 exones y las zonas limítrofes. Los sujetos con sospecha de
resultados erróneos fueron evaluados con secuenciación completa del gen usando el ABI
PRISM 3100. Entre todos los individuos tamizados se identificaron 1879 portadores de
fibrosis quística con una frecuencia de portador de 1 en 31 personas. Se identifico la
DF508 como la mutación más frecuente con 42.6%. Diez individuos resultaron afectados
siendo heterocigotos compuestos en donde 7 de ellos presentaban problemas de
infertilidad y 5 ausencia bilateral congénita de conductos deferentes. De las 25104
parejas, 24181 resultaron negativas al tamizaje, 815 individuos de cada pareja fueron
portadores y 108 parejas fueron ambos portadores para mutaciones de fibrosis quística.
De estas parejas en riesgo, 89 estaban en embarazo en curso sometiéndose a

30
diagnostico prenatal encontrando 20 fetos no portadores de mutaciones (22%), 47
portadores sanos con una mutación en uno de los dos alelos (53%) y 22 con mutación en
ambos alelos (25%).
Rohlfs y cols. (36), en el 2011 reportaron la frecuencia de 98 mutaciones para 364890
individuos sin historia familiar de fibrosis quística analizados entre el 2005 y 2008 en
Estados Unidos. El 93.1% fueron mujeres, con múltiple etnicidad y de diferentes países
de origen usando la técnica por hibridización y fluorescencia. Se encontró una frecuencia
de detección de portador de 1 en 38 individuos y los caucásicos fue la etnicidad más
frecuente con una proporción del 43%. La mutación más frecuente fue la DF 508 con
57.7%. Las mutaciones no incluidas en el panel de 23 mutaciones sumaron el 13 % de la
totalidad de individuos analizados y 9.3% de los caucásicos presentaron 44 mutaciones
distintas al panel básico de 23 mutaciones. Fueron encontradas 87 de las 98 mutaciones
ofrecidas y en 36 individuos se encontró la presencia de dos mutaciones sin tener
síntomas clásicos de fibrosis quística.
Dugueperoux y col (37) en el 2013 analizaron en Francia en dirección retrospectiva el
test de cascada de portador en padres y parientes de niños portadores de mutaciones
identificados por tamizaje prenatal de 1991 a 2010. El test fue ofrecido a los padres para
identificar si compartían la mutación con su hijo, y con sus familiares estableciendo una
proporción de identificación del 90% para mutación familiar y del 95% con escaneo
completo de exones. De 200.378 recién nacidos tamizados, 202 fueron identificados
como portadores de mutaciones de fibrosis quística, tres de ellos se excluyeron porque
fueron dados en adopción. De 199 portadores sanos identificados, 106 fueron mujeres y
21 mutaciones genéticas fueron identificadas. Las 5 mutaciones más frecuentes
sumaban el 90.5%. Los 199 portadores correspondieron a 195 familias, de las cuales 47
no se sometieron a test familiar, las 148 familias restantes se sometieron a 374 test
individuales. De los padres testeados, se identifico 141 parejas y 6 mujeres madres. Solo
un caso evidencio que ninguno de los padres compartía la mutación presente en su hijo,
por lo que fue excluido por sospecha de exclusión parental. Entre las 141 parejas y 6
mujeres, se evidencio 148 portadores padres en donde al menos uno de ellos tenía la
misma mutación en su hijo. De estos, 3 parejas estaban en riesgo de 1 a 4 en tener un
hijo con FQ. De los 69 familiares restantes tamizados se encontraron mutaciones en 35

31
de ellos. De igual manera se tamizaron 15 compañeros sexuales de los parientes
portadores de los que se detectaron 2 de ellos como portadores, encontrando a 2
parejas en riesgo de 1 a 4 de tener hijos con FQ. De 38 tíos y tías testeados 23 fueron
portadores, de 14 hermanos testeados 7 fueron portadores, de 6 abuelos testeados 3
fueron portadores, de 4 primos testeados 1 fue portador, y de otros 7 siguientes
familiares testeados 1 fue portador. Con este estudio se logro identificar a 138 portadores
y 5 parejas en riesgo de 1 a 4 en tener un niño con FQ. Cuatro parejas se sometieron a
diagnostico prenatal y se identifico un embarazo afectado.
1.2.8 Tratamiento
El tratamiento de los pacientes con FQ debe ser realizado en unidades especializadas,
que permitan a un equipo interdisciplinario interactuar para abordar la infección
respiratoria y evitar el deterioro de la función respiratoria. Se debe facilitar la eliminación
de secreciones y una adecuada nutrición. La fisioterapia respiratoria permite drenajes
bronquiales, permitiendo mantener despejadas las vías aéreas. El ejercicio y la actividad
física facilita la eliminación de secreciones de la vía aérea.
Un paciente con fibrosis quística debe ser monitorizado con cultivo de esputo, hisopado
faríngeo o de tos cada 2 a 3 meses. Se emplean esquemas antibióticos contra
Pseudomona aeruginosa y Staphylococcus aureus sin recomendación profiláctica ya que
no previenen exacerbaciones. El TACAR de tórax es útil para diagnostico y seguimiento
de un paciente con fibrosis quística, al igual que las pruebas de función pulmonar
mediante la curva flujo volumen en mayores de 5 años. La espirometría debe hacerse
como seguimiento cada 3 a 6 meses para diagnostico y monitoreo de exacerbaciones
pulmonares. No se recomienda repetir TACAR de tórax en menos de 2 a 3 años por el
riesgo de radiación. En pacientes con fibrosis quística se recomienda realizar con
periodicidad anual un examen físico completo, medición de aminotransferasas y
ecografía hepatobiliar. Toda exacerbación pulmonar requiere cultivo de secreciones y un
manejo empírico inicial con respecto al último cultivo realizado, por lo general el
tratamiento antibiótico intravenoso dura 14 días pero depende de cada paciente. En
infecciones crónicas por Pseudomona aeruginosa se recomienda el manejo por periodos
de 6 meses con azitromicina oral en mayores de 6 años para disminuir exacerbaciones y
mejorar la función pulmonar. No se recomienda el uso de esteroides inhalados ni por vía

32
oral a largo plazo por efectos secundarios y poca mejoría pulmonar. La dornasa alfa aun
es útil como medicamento mucolítico por efecto enzimático directo sobre el DNA
extracelular de secreciones viscosas a pesar de su alto costo. También es útil la solución
salina hipertónica al 7% para disminuir la viscosidad de las secreciones y reducir las
exacerbaciones pulmonares además de su bajo costo.
Se recomienda en pacientes con fibrosis quística terapia respiratoria permanente
independiente de presencia o no de síntomas respiratorios. Las enzimas pancreáticas
permiten compensar la insuficiencia pancreática de la enfermedad y mejoran el estado
nutricional de los pacientes, sin embargo no se recomiendan usarlas concomitantemente
junto con inhibidores de secreción de acido gástrico. En pacientes mayores de 10 años,
se recomienda una prueba anual de tolerancia oral a la glucosa para tamización y
diagnostico temprano de la diabetes relacionada a la fibrosis quística (21).
1.2.9 Costo de la enfermedad
El Instituto Nacional de Salud de los Estados Unidos estimó para 1997 el costo de vida
de un paciente con fibrosis quística alcanzando los 800.000 dólares. Liu y col para 1996
estimó el costo anual del cuidado médico para un paciente entre 13.300 y 43.000 dólares
(25). La guía de práctica clínica colombiana del 2014 estimó el costo directo de un grupo
de 53 pacientes con fibrosis quística seguidos de forma ambulatoria por tres meses en la
ciudad de Bogotá, con un costo mensual promedio de $ 10.973.301,32 (21).
1.2.10 Costo del test de portador
Se considera que el costo del panel de DNA para portador de fibrosis quística en
Colombia es de $ 1.770.500 y el de secuenciación completa del gen CFTR es de
$3.014.720 aproximadamente. La guía Colombiana recomienda desarrollar paneles de
mutación específicos para la población colombiana que logren detectar un 80% de los
afectados, reduciendo el costo de los estudios moleculares (21).

33
Chandrasekharan y col (38), reportaron los costos para el 2008 del test de portador para
secuenciación completa del gen que va desde $ 1.200 dólares hasta $ 2.586 dólares
para entidades con o sin ánimo de lucro como las Universidades de Utah, Harvard, Johns
Hopkins, laboratorios Genzyme, Prevention entre otros.
Esta evaluación económica se hace necesaria debido a que no ha sido realizada en el
contexto colombiano y con su aporte permitiría un gran beneficio en salud y un impacto
en costos y presupuestos del sistema de salud desde el ámbito preventivo.

34
Capitulo 2
2.1 Marco Metodológico
Se realizó una búsqueda sistemática bajo la pregunta de investigación en formato
PICO evaluando si es costo-efectivo realizar o no la prueba de secuenciación del gen
CFTR para la detección de portadores asintomáticos en edad fértil en primer, segundo
y tercer grado de consanguinidad a un caso índice familiar con diagnostico de fibrosis
quística. Esta búsqueda incluyó la identificación, selección y apreciación de la calidad
de la evidencia, por medio del análisis y consulta de bases de datos científicas
robustas y especializadas en el área de la salud, estableciendo previamente la
estrategia de búsqueda ajustada a cada una de ellas; de igual manera se tuvo en
cuenta la literatura gris, eventos de divulgación científica y paneles de expertos
para ampliar el campo de búsqueda. Se evaluó la sensibilidad y especificidad de la
prueba diagnóstica y se tuvo en cuenta modelos de costo efectividad de árboles de
decisiones publicados en la literatura.
La evidencia fue evaluada según los estudios encontrados: para revisiones
sistemáticas el AMSTAR, para ensayos clínicos aleatorizados la herramienta
Cochrane para evaluación de riesgo de sesgos, para pruebas diagnosticas el
Quadas2, para cohortes el Newcastle Ottawa Scale (NOS) y para estudios económicos
el QHES (39, 40). El diseño realizado a través de un modelo de árbol de decisiones
tuvo en cuenta como unidad de análisis la prevención de futuras concepciones
mediante asesoría genética por riesgo de presentar fibrosis quística y la razón de costo
efectividad incremental. La mejor evidencia disponible fue tenida en cuenta para la
adaptación del modelo, medición, estimación y valoración de desenlaces y de costos
directos de las pruebas diagnósticas y los costos de atención anual para un paciente
con fibrosis quística; estos últimos obtenidos de los sistemas de información de la
cuenta de alto costo del Ministerio de Salud y Protección Social.

35
Al modelo adaptado se le realizó un análisis de sensibilidad determinístico y
probabilístico, y su perspectiva fue la del tercer pagador (Entidades prestadoras de
salud). Esta investigación se orientó por la “Guía metodológica para la
actualización integral del Plan Obligatorio de Salud del Sistema General de Seguridad
Social en Salud” (42) y la “Guía metodológica para la elaboración de guías de
atención integral en el sistema general de seguridad social en salud colombiano”
(41).
2.1.1 Documentación de estudios diagnósticos y de costo efectividad
Con el objetivo de documentar la efectividad y los diferentes modelos económicos de la
prueba, se buscaron estudios diagnósticos y económicos de costo efectividad:
• MEDLINE®, Ovid platform (1990 - presente).
• MEDLINE® In-Process & Other Non-Indexed Citations, Ovid platform (1990-
presente).
• MEDLINE® Daily Update, Ovid platform (1990 - presente).
• NHS Economic Evaluation Database (1990 - presente).
• HuGE Net (Human Genomic Epidemiology Network) (1990 - presente).
• EMBASE (1990 - presente).
• Cochrane Database of Systematic Reviews, Ovid platform (2005 -
presente).
• LILACS, IAHx interface (1990 - presente).
• Health Technology Assessment, Ovid platform (1990 - presente).
• Genetests.org
• Genetsickkids.org

36
Cada búsqueda incluyó la siguiente información: base de datos, motor de búsqueda,
fecha de búsqueda, rango de fecha de búsqueda y estrategia de búsqueda. Las
búsquedas se realizaron sin restricción de idioma y fecha. Posteriormente se realizó el
tamizaje, selección y recuperación de la literatura pertinente.
2.1.2 Población Objetivo
Hombres y mujeres en edad fértil asintomáticos en primer, segundo y tercer grado de
consanguinidad a un caso índice con diagnóstico de fibrosis quística.
2.1.3 Perspectiva de análisis
Tercer pagador.
2.1.4 Horizonte temporal
Se consideró un horizonte temporal de 1 año, el cual permitió incluir los costos directos
relevantes asociados a la fibrosis quística.
2.1.5 Identificación de las tecnologías sanitarias a comparar
Hacer o no hacer la prueba de secuenciación del gen CFTR para portadores
asintomáticos de mutaciones de fibrosis quística con antecedente familiar.

37
2.1.6 Estimación de los costos asociados
Se utilizaron los costos directos reportados por las EPS a la cuenta de alto costo
recolectada en el SISPRO por el ministerio de salud y protección social para el año
2012. Los costos fueron actualizados al valor presente aplicando el IPC (Índice del
Precio al Consumidor) en salud de Colombia para el año 2013, 2014, 2015 y 2016. No
se tuvo en cuenta los costos indirectos ni costos por pérdida de productividad por no
ser el objetivo de la presente investigación además del difícil cálculo en el contexto del
país. Los costos de la prueba genética de secuenciación del gen CFTR por técnica
Sanger fueron obtenidos de laboratorios genéticos nacionales y de referencias
colombianas.
2.1.7 Medidas de resultado en salud
La principal medida de efectividad de la prueba genética de secuenciación del gen
CFTR fue la prevención de futuras concepciones asociada a la asesoría genética por
riesgo de presentar fibrosis quística y la razón de costo efectividad incremental.
2.1.8 Tasa de descuento
No se aplica por tener un horizonte temporal a 1 año.
2.1.9 Modelación
Se construyo un modelo de árbol de decisiones con el programa TreeAge Pro
Healthcare 2015. El modelo se ajustó con los costos pertinentes para cada brazo del
árbol de decisiones y las probabilidades de los eventos clínicos.

38
Cálculo de los valores:
El cálculo de los valores esperados (promedios) de los costos y de la efectividad de
cada estrategia, se efectuó hacia atrás “Roll back” con el valor del costo y de la
efectividad de cada rama, ajustándolos por sus respectivas probabilidades.
2 . 1 . 1 0 Análisis de sensibilidad
Se realizó un análisis de sensibilidad deterministico y probabilístico con los
parámetros de costos y de las probabilidades incorporadas en el modelo para evaluar la
incertidumbre.
2.1.11 Consideraciones Éticas
Este proyecto fue clasificado como una investigación con mínimo riesgo según la
Resolución 8430/93 art.11 (43).
El modelo de costo efectividad en este caso se consideró como un estudio secundario
que utilizó estudios primarios y modelos de árboles de decisiones, recolectando la
evidencia disponible. Por ser un estudio secundario, no requirió solicitud de
consentimiento informado.
2.1.12 Supuestos
Debe ser ofrecido a parejas de manera voluntaria, ofreciendo toda la consejería
genética necesaria a quienes son portadores en riesgo para tomar la decisión a través
de un consentimiento informado que tenga en cuenta la sensibilidad y especificidad del
test, la probabilidad del 25% de tener hijos enfermos y del 50% de tener portadores
sanos, y la descripción detallada de la severidad de la enfermedad, su expectativa de
vida y costos asociados.

39
Es decisión de la pareja tener o no el hijo con la enfermedad, teniendo en cuenta que
no es incompatible con la vida pero que presenta un contexto social y económico
complejo por el grado de severidad y por su expectativa de vida reducida ya que no
hay curación de la misma.
Se expone que no es conveniente ofrecerlo a mujeres en gestación por el riesgo de
aborto voluntario o asociado al diagnostico prenatal al tomar muestra de vellosidad
coriónica. Además del aumento del costo asociado, su resultado clínico no está ligado a
un aborto obligatorio y la pareja puede decidir continuar con el embarazo a pesar del
riesgo.
Los supuestos aplicados fueron:
• Todas las parejas en riesgo por algún antecedente familiar en primer segundo o
tercer grado aceptan someterse a la prueba de secuenciación del gen CFTR.
• El 100% de las parejas en riesgo por el resultado positivo de la prueba de
secuenciación aceptan las recomendaciones dadas en la consejería genética.
• Las parejas asintomáticas portadoras de mutaciones tienen el 25% de
probabilidad de tener hijos con la enfermedad y 50% de hijos portadores
asintomáticos de la mutación.
• Las parejas tamizadas recordaran su estado de portador anotado en su historia
clínica y no se repetirán nuevamente sus exámenes.

40
Capitulo 3
3.1 Resultados
3.1.1 Revisión sistemática de estudios de costo-efectividad para identificación de portadores de mutaciones de fibrosis quística.
Fueron identificados 573 artículos, de los cuales 3 eran duplicados. Con revisión de titulo
y abstract, se excluyeron 560 artículos. Al final se sometieron 13 estudios (44-56) que
cumplieron con criterios de inclusión al ser evaluaciones económicas completas
relevantes para su revisión, sin exclusión de idioma ni de tiempo de publicación para
incluir en el análisis, revisando de igual manera las referencias de los artículos
seleccionados. Entre los estudios incluidos, 4 fueron realizados en Estados Unidos, 2 en
Reino Unido, 3 en Países Bajos, 3 en Australia y 1 en México. Para nuestro objetivo solo
se incluyó estudios de costo efectividad, aunque dos tenían también análisis de costo
beneficio (49, 50) y uno análisis de costo utilidad (50). Seis de los estudios tenían como
intervención un tamizaje preconcepcional añadido al tamizaje prenatal (46, 48, 49, 50, 54,
56), cinco exclusivamente en tamizaje prenatal (44, 45, 47, 51, 55) y dos exclusivos con
tamizaje preconcepcional (52, 53). La perspectiva más usada fue la del tercer pagador
con nueve de los 13 estudios incluidos, seguida de la perspectiva de la sociedad. La
tasa de descuento aplicada estuvo entre un rango de 3.5 % a 5%.

41
Figura 2 PRISMA Fuente: propia.
La unidad de análisis principal aplicada fue la del costo por pareja portadora detectada
seguida de costo por nacimiento evitado con fibrosis quística. De igual manera el
resultado de cada estudio fue reportado consecuente con su unidad de análisis ya que no
todos los estudios aplicaron el RCEI (Razón de costo efectividad incremental) como
medida final. Diez estudios aplicaron el modelo de análisis de decisiones.
Hubo heterogeneidad en los supuestos aplicados tales como número de gestaciones,
probabilidad de aceptación del test genético, probabilidad de olvido de su estado de
portador y la mayoría utilizó el método univariado para el análisis de sensibilidad. Todos
los estudios aplicaron como características operativas la sensibilidad y especificidad del
test de portador y del test prenatal, según la intervención planteada.

42
En todos los estudios seleccionados, la intervención fue comparada con no realizarla,
teniendo en cuenta que la mayoría de países no habían establecido aun un programa de
tamizaje genético para portador de fibrosis quística. No se encontró en ningún estudio la
aplicación de la estrategia de test de portador en cascada para fibrosis quística. Los
estudios encontrados usaron la prevalencia de portador para población caucásica en
general, lo que indica que no se tuvo en cuenta exclusivamente la probabilidad de ser
portador teniendo antecedente familiar de fibrosis quística. Todos los estudios utilizaron
el supuesto de una cohorte de pacientes para ser aplicada las probabilidades dadas al
modelo de análisis de decisiones. La prevalencia de portador más usada fue de 1/25
pacientes seguida de 1/30, que son las de mayor aplicación para raza caucásica. Los
resultados obtenidos en cada estudio son heterogéneos debido a variación en moneda
aplicada, año, tasa de descuento y supuestos que difieren en relación al sistema de salud
de cada país, además de la percepción de aceptación de un test genético y de la
decisión de abortar un feto con alto riesgo de fibrosis quística. La probabilidad de elegir
aborto terapéutico se presento entre el 30 % y el 100%.
En la estrategia preconcepcional se encontró el supuesto que hasta 25% de los
pacientes estaban dispuestos a abstenerse de tener hijos en caso de que la pareja fuera
portadora. Ningún estudio reportó la posibilidad de diagnóstico genético
preimplantacional en el análisis de decisiones. Solo se presento en el estudio más
reciente la posibilidad y el costo de la técnica IVF.
Se evidenció que la expectativa de vida más usada para un paciente con fibrosis quística
fue de 30 años aunque uno de los estudios usó una expectativa de 50 años. Se evidenció
que la estrategia más costo efectiva fue la del tamizaje preconcepcional asociada al test
prenatal, mientras que la preconcepcional aislada no garantizaba costo efectividad dada
por los supuestos que debía cumplir, como la probabilidad de aceptación o no de la
población a optar por otras opciones de consejería genética: abstenerse a la gestación,
adoptar un niño o el diagnostico genético preimplantacional.

43
La mejor estrategia de tamizaje genético fue la secuencial en la cual primero se realiza
el test a un integrante de la pareja y si resulta positivo se realiza el test a su
compañero(a); si ambos son positivos se considera pareja en riesgo, pero si resulta
negativo alguno de ellos su reporte final es sin riesgo, la ventaja es que se da un
diagnostico de portador de manera individual. Esta estrategia resultó ser mejor que la
estrategia paralela o simultanea, en la se realiza el examen diagnostico al mismo tiempo
a los dos integrantes, dando su reporte como pareja con o sin riesgo.

44
Tabla 4 Revisión de los estudios económicos de test de portador para fibrosis quística incluidos en el análisis. Fuente: propia.
Referencia País Intervención Perspectiva T.
descue
nto
U. de
análisis
Evaluación
/Modelo
Supuestos A.De sensibilidad
Lieu (44) EU Tamizaje en
prenatal.
Estrategia
secuencial.
Tercer
pagador
5% a
costos.
Nacimien
to
evitado
con FQ.
Costo por
embaraz
o de alto
riesgo
detectad
o.
ACE/AD Cohorte 1 millón de
mujeres, sin
antecedente de FQ,
en embarazo.
Univariado y bivariado.
Aceptación del test de
portador.
S del test portador
Prob. de elegir aborto
terapéutico.
Costo de test portador.
Costo de vida FQ.
Asch (45) EU Tamizaje en
prenatal.
Estrategia
paralela y
secuencial.
Sociedad 4% a
costos.
Nacimien
to
evitado
con FQ.
ACE/AD Prob. de no
paternidad. (0.1 -
0.5)
Estrategia sin
pérdidas.
Prob. de no continuar
el embarazo de alto
riesgo.
Test estándar vs test
expandido.
Bivariado
Costo de test expandido y su
sensibilidad.
Costo de test básico y
expandido.
Probabilidad de no paternidad
y de no continuar el embarazo
de alto riesgo.
Más de una gestación.
Morris (46) RU Tamizaje
preconcepcion
al y en
prenatal.
Estrategia
paralela y
Tercer
pagador
Ningun
a
Nacimien
to
evitado
con FQ
Costo por
ACE/Simulac
ión.
Solo una gestación. Univariado
Aceptación del test de
portador.
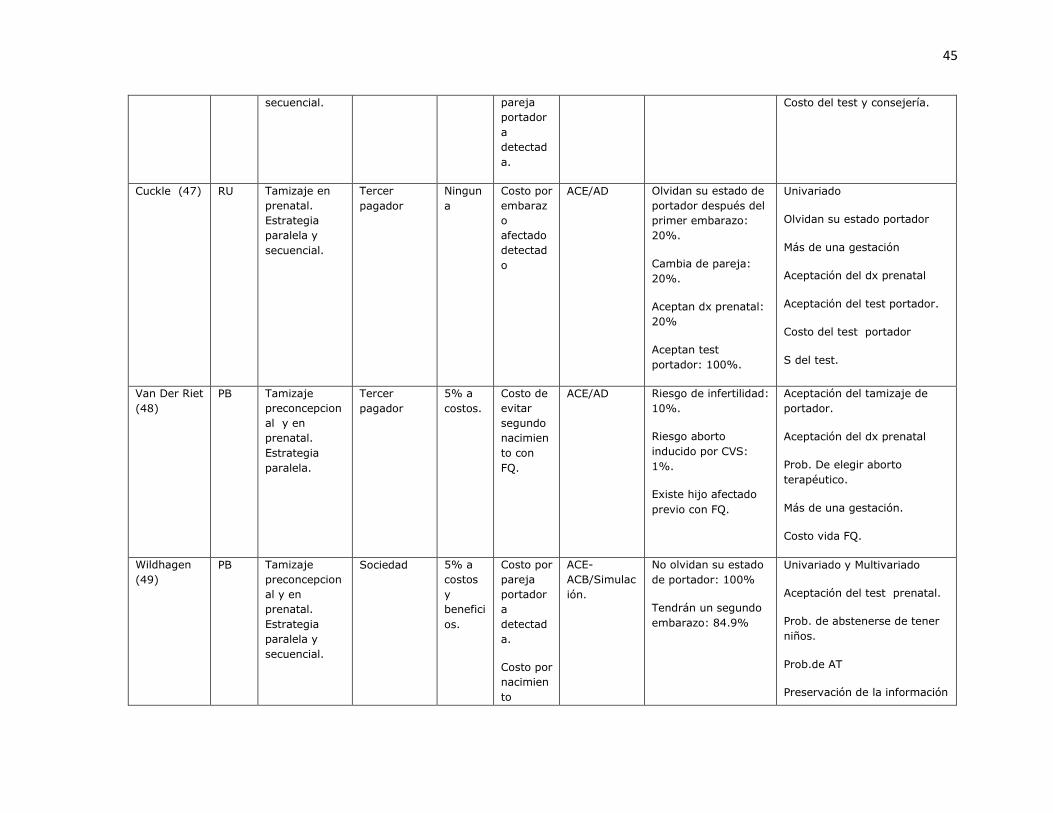
45
secuencial. pareja
portador
a
detectad
a.
Costo del test y consejería.
Cuckle (47) RU Tamizaje en
prenatal.
Estrategia
paralela y
secuencial.
Tercer
pagador
Ningun
a
Costo por
embaraz
o
afectado
detectad
o
ACE/AD Olvidan su estado de
portador después del
primer embarazo:
20%.
Cambia de pareja:
20%.
Aceptan dx prenatal:
20%
Aceptan test
portador: 100%.
Univariado
Olvidan su estado portador
Más de una gestación
Aceptación del dx prenatal
Aceptación del test portador.
Costo del test portador
S del test.
Van Der Riet
(48)
PB Tamizaje
preconcepcion
al y en
prenatal.
Estrategia
paralela.
Tercer
pagador
5% a
costos.
Costo de
evitar
segundo
nacimien
to con
FQ.
ACE/AD Riesgo de infertilidad:
10%.
Riesgo aborto
inducido por CVS:
1%.
Existe hijo afectado
previo con FQ.
Aceptación del tamizaje de
portador.
Aceptación del dx prenatal
Prob. De elegir aborto
terapéutico.
Más de una gestación.
Costo vida FQ.
Wildhagen
(49)
PB Tamizaje
preconcepcion
al y en
prenatal.
Estrategia
paralela y
secuencial.
Sociedad 5% a
costos
y
benefici
os.
Costo por
pareja
portador
a
detectad
a.
Costo por
nacimien
to
ACE-
ACB/Simulac
ión.
No olvidan su estado
de portador: 100%
Tendrán un segundo
embarazo: 84.9%
Univariado y Multivariado
Aceptación del test prenatal.
Prob. de abstenerse de tener
niños.
Prob.de AT
Preservación de la información

46
evitado
con FQ.
de portador.
Aceptación del test portador.
Costo test portador
Rowley
(50)
EU Tamizaje
preconcepcion
al y en
prenatal.
Estrategia
secuencial.
Sociedad 3% a
costos
y
benefici
os
Costo por
nacimien
to
evitado
con FQ.
ACE-ACB-
ACU/AD
Una gestación por
pareja.
Que no haya
antecedente familiar
de FQ.
90% de test
portador se realizó en
gestantes.
Test de dx prenatal
para resultados
positivos de portador
en la pareja.
Univariado
Aceptación test portador.
S test portador.
Costo vida FQ.
Aceptación del test prenatal.
Prob. de AT
Costo test portador.
Doyle (51) Mex Tamizaje
prenatal.
Estrategia
secuencial.
Tercer
pagador
Ningun
a
Costo por
nacimien
to
evitado
con FQ.
ACE/AD No hubo falsos
positivos ni para la
enfermedad ni para
portadores.
Los falsos negativos
se consideraron bajo
la sensibilidad de la
prueba.
Igual etnicidad en las
parejas.
No se consideró la no
paternidad, la no
disponibilidad del
hombre a la prueba,
ni cambios de pareja
futuros. La prueba se
realiza en la primera
Bivariado
Costo del test
S de test
Aceptación de AC y AT
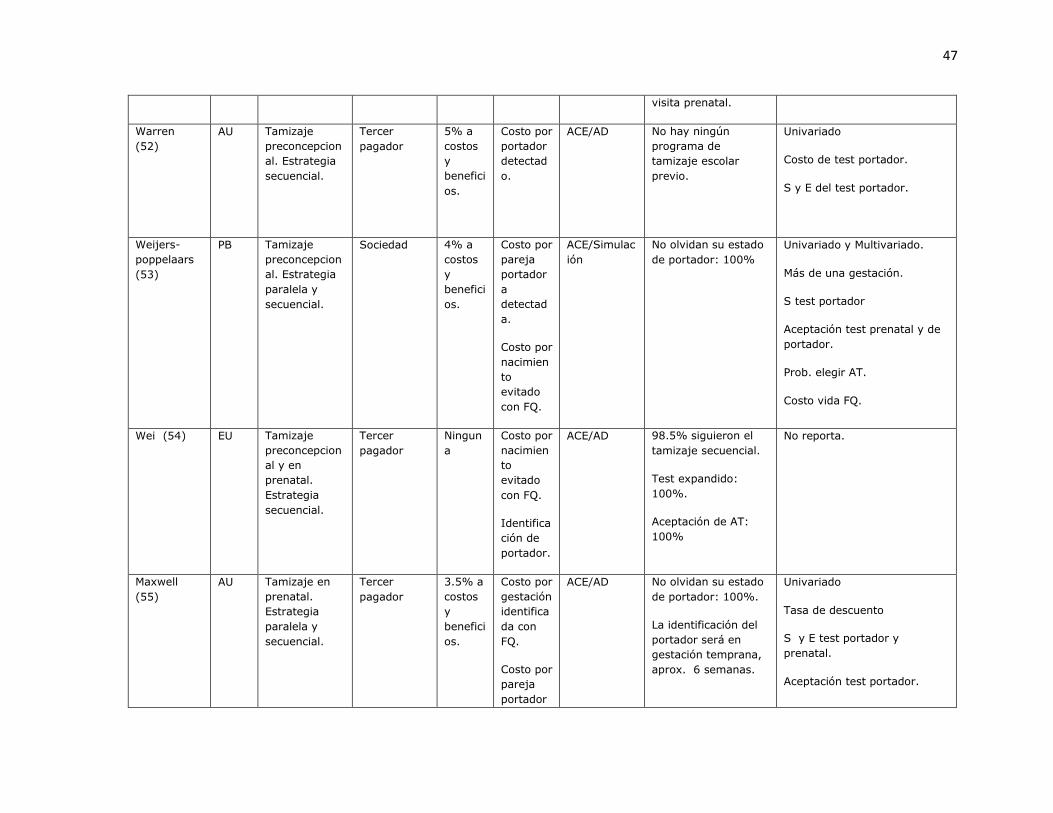
47
visita prenatal.
Warren
(52)
AU Tamizaje
preconcepcion
al. Estrategia
secuencial.
Tercer
pagador
5% a
costos
y
benefici
os.
Costo por
portador
detectad
o.
ACE/AD No hay ningún
programa de
tamizaje escolar
previo.
Univariado
Costo de test portador.
S y E del test portador.
Weijers-
poppelaars
(53)
PB Tamizaje
preconcepcion
al. Estrategia
paralela y
secuencial.
Sociedad 4% a
costos
y
benefici
os.
Costo por
pareja
portador
a
detectad
a.
Costo por
nacimien
to
evitado
con FQ.
ACE/Simulac
ión
No olvidan su estado
de portador: 100%
Univariado y Multivariado.
Más de una gestación.
S test portador
Aceptación test prenatal y de
portador.
Prob. elegir AT.
Costo vida FQ.
Wei (54) EU Tamizaje
preconcepcion
al y en
prenatal.
Estrategia
secuencial.
Tercer
pagador
Ningun
a
Costo por
nacimien
to
evitado
con FQ.
Identifica
ción de
portador.
ACE/AD 98.5% siguieron el
tamizaje secuencial.
Test expandido:
100%.
Aceptación de AT:
100%
No reporta.
Maxwell
(55)
AU Tamizaje en
prenatal.
Estrategia
paralela y
secuencial.
Tercer
pagador
3.5% a
costos
y
benefici
os.
Costo por
gestación
identifica
da con
FQ.
Costo por
pareja
portador
ACE/AD No olvidan su estado
de portador: 100%.
La identificación del
portador será en
gestación temprana,
aprox. 6 semanas.
Univariado
Tasa de descuento
S y E test portador y
prenatal.
Aceptación test portador.

48
a
identifica
da.
Aceptación test prenatal.
Prob. AT.
Más de una gestación.
Costo vida FQ.
Norman
(56)
AU Tamizaje
preconcepcion
al y en
prenatal.
Estrategia
secuencial.
Tercer
pagador
5% a
costo
de vida
FQ y
benefici
os.
Costo por
nacimien
to
evitado
con FQ.
ACE/AD Parejas estables.
No olvidan su estado
de portador: 100%
Univariado
S y E de test portador
Costo IVF
Más de una gestación.
Aceptación de test portador y
prenatal.
Costo vida FQ.
Costo test portador.

49
Tabla 5 Probabilidades, costos y resultados de estudios incluidos. Fuente: propia.
Referencia Probabilidades Costos Resultados
Lieu 1994 Aceptación de test de portador
materno: 78%.
S test portador en sangre: 85%.
E test portador: 99.9%
Aceptación de test prenatal: 80%
Prevalencia portador FQ: 1/25
Prob. aborto por dx prenatal:
CVS: 0.013
AC: 0.005
Atención aborto espontaneo: $
1000
Prob. elegir Aborto terapéutico:
30%.
Expectativa de vida: 29 años.
Dólar 1993
Costo test portador: $
100
Cita consejería: $170
CVS: $1.175
AC: $1.200
AT: $400
Costo vida FQ: $ 243.650
Costo por nacimiento evitado de
FQ: $ 1.411.000
Costo por identificación de
embarazo de alto riesgo: $82.000
Cohorte de 1 millón de mujeres
embarazadas:
Sin tamizaje: 400 niños con FQ
Con tamizaje: 350 niños con FQ
50 niños sin FQ
No costo efectivo.
Asch 1995 S test básico : 85% E: 99,5%
S test expandido: 90% E: 99%
Prevalencia portador FQ: 1/25
Prob. elegir AT: 100%
S test AC: 99.5 % E: 99.9 %
Prob. aborto por AC: 0.5%
Dólar 1995
Costo test básico: $ 50
Costo test expandido: $
100
Cita consejería: $ 26
AC: $ 200
Costo vida FQ: $ 351.278
Costo nacimiento evitado con FQ:
Estrategia secuencial o individual:
$ 367.000 (146 nacimientos
evitados con FQ).
Estrategia paralela o pareja: $
594.000 (187 nacimientos
evitados con FQ).
No costo efectivo
Morris 1995 S test portador bucal: 85%
Prevalencia portador FQ: 1/25
Aceptación de test portador
secuencial: 72-99%
Libra esterlina 1994
Costo test portador: £ 40
Cita consejería: £ 30
No toman en cuenta el
costo de vida de FQ.
Estrategia en pareja:
Costo por pareja portadora
detectada: £ 35.700
Costo por nacimiento evitado de
FQ: £ 142.900
Estrategia secuencial:
Costo por pareja portadora
detectada: £ 36.600

50
Costo por nacimiento evitado de
FQ: £ 146.500
No costo efectivo
Cuckle 1995 Aceptación test prenatal: 20%
Prevalencia portador FQ: 1/25
S test básico: 70-85%
S test expandido: 80-95%
Aceptación del test portador: 55-
95%
Libra esterlina 1995
Costo test básico: £ 16
Costo test expandido: £
33
Cita consejería: £ 25
Costo test prenatal: £
200
Costo por embarazo afectado
detectado:
Test Secuencial: £ 40.000 – £
90.000
Test en pareja: £ 46.000 - £
104.000.
No costo efectivo
Van Der
Riet 1997
S test portador: 100%
S dx prenatal: 90%
Aceptación AT: 99%
Prevalencia portador FQ: 1/25
Dólar 1994.
Costo test portador:
$1.200
Costo vida FQ: $
545.968
AT: $ 668
Costo de evitar segundo
nacimiento con FQ: $ -62.621
Costo de evitar nacimiento con FQ
en parientes: $ +42 - $ 2.075.
Costo efectivo a partir del segundo
embarazo.
Wildhagen
1998
Aceptación test prenatal : 62-
91%
Aceptación test portador: 4- 87%
Prevalencia portador FQ: 1/30
Prob. De elegir AT: 80%
Prob. Aborto por dx prenatal:
0.75%
Libra esterlina
Costo test básico: £ 8,25
Costo test expandido: £
33
Cita consejería: £ 23,24
Costo dx prenatal: £
1.106,72
Costo AT: £ 192,51
Costo aborto iatrogénico:
£ 60,30
Costo vida FQ: £ 238.634
Prenatal:
Costo por pareja portadora
detectada:
Test secuencial: £ 58.000
Test en pareja: £ 70.000
Costo por nacimiento evitado:
Test secuencial: £ 177.000 ( 18
nacimientos evitados)
Test en pareja: £ 213.000 ( 21
nacimientos evitados)
Preconcepcional:
Costo por pareja portadora
detectada:
Test secuencial: £ 69.000
Test en pareja: £ 80.000
Costo por nacimiento evitado:
Test secuencial: £ 223.000 ( 10
nacimientos evitados)
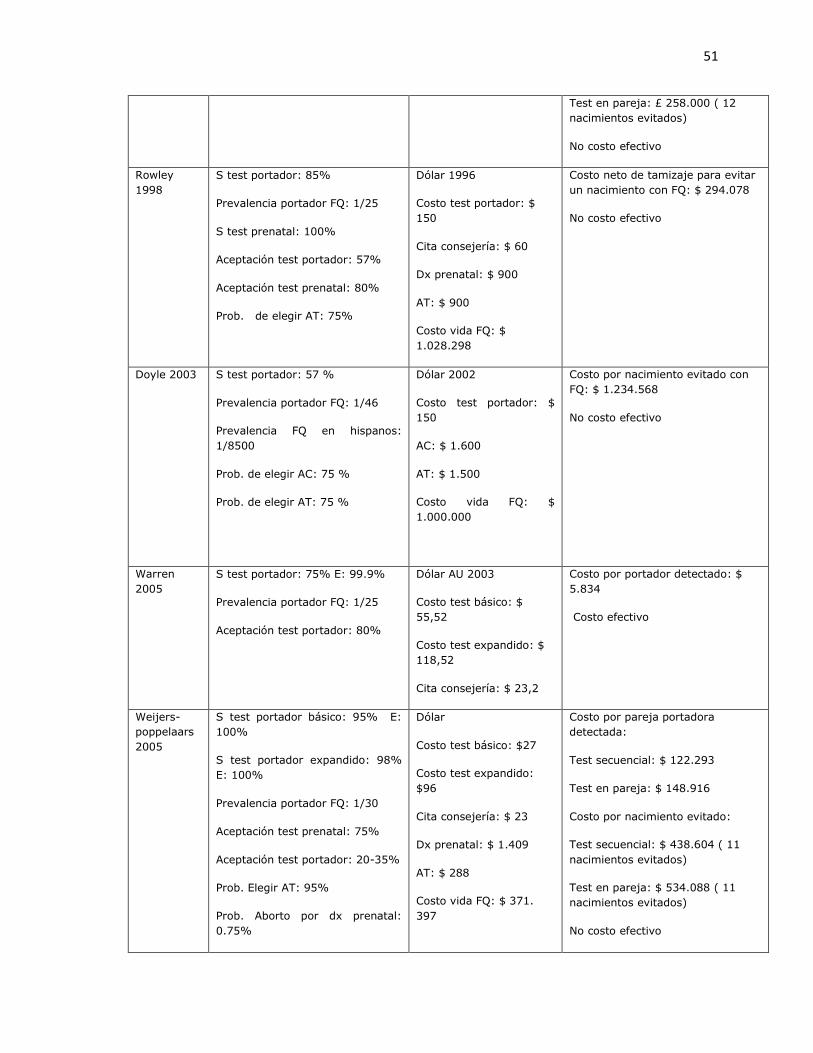
51
Test en pareja: £ 258.000 ( 12
nacimientos evitados)
No costo efectivo
Rowley
1998
S test portador: 85%
Prevalencia portador FQ: 1/25
S test prenatal: 100%
Aceptación test portador: 57%
Aceptación test prenatal: 80%
Prob. de elegir AT: 75%
Dólar 1996
Costo test portador: $
150
Cita consejería: $ 60
Dx prenatal: $ 900
AT: $ 900
Costo vida FQ: $
1.028.298
Costo neto de tamizaje para evitar
un nacimiento con FQ: $ 294.078
No costo efectivo
Doyle 2003 S test portador: 57 %
Prevalencia portador FQ: 1/46
Prevalencia FQ en hispanos:
1/8500
Prob. de elegir AC: 75 %
Prob. de elegir AT: 75 %
Dólar 2002
Costo test portador: $
150
AC: $ 1.600
AT: $ 1.500
Costo vida FQ: $
1.000.000
Costo por nacimiento evitado con
FQ: $ 1.234.568
No costo efectivo
Warren
2005
S test portador: 75% E: 99.9%
Prevalencia portador FQ: 1/25
Aceptación test portador: 80%
Dólar AU 2003
Costo test básico: $
55,52
Costo test expandido: $
118,52
Cita consejería: $ 23,2
Costo por portador detectado: $
5.834
Costo efectivo
Weijers-
poppelaars
2005
S test portador básico: 95% E:
100%
S test portador expandido: 98%
E: 100%
Prevalencia portador FQ: 1/30
Aceptación test prenatal: 75%
Aceptación test portador: 20-35%
Prob. Elegir AT: 95%
Prob. Aborto por dx prenatal:
0.75%
Dólar
Costo test básico: $27
Costo test expandido:
$96
Cita consejería: $ 23
Dx prenatal: $ 1.409
AT: $ 288
Costo vida FQ: $ 371.
397
Costo por pareja portadora
detectada:
Test secuencial: $ 122.293
Test en pareja: $ 148.916
Costo por nacimiento evitado:
Test secuencial: $ 438.604 ( 11
nacimientos evitados)
Test en pareja: $ 534.088 ( 11
nacimientos evitados)
No costo efectivo

52
Prob. abstenerse de quedar en
gestación: 25%
Wei 2007 Prevalencia portador FQ: 1/25
Aceptación test portador: 100%
Aceptación test prenatal: 83.3%
Dólar
Costo test portador: $ 50
Cita consejería: $ 175
CVS: $ 450
AC: $ 500
Costo por nacimiento evitado con
FQ: $ 334.000
No costo efectivo
Maxwell
2010
S test portador: 80% E: 100%.
S test prenatal: 100%.
Aceptación test portador: 60%
Aceptación test prenatal: 90%
Prob. Aborto test prenatal: 1%
Prob de elegir AT: 90%
Expect. Vida: 50 años.
Prevalencia portador FQ: 1/25
Dólar AU 2008
Costo test portador: $
116,77
Cita consejería: $ 119
AC/CVS: $ 93
AT: $ 1.836 dólares.
Costo vida FQ: $ 508.370
Estrategia secuencial:
Costo por pareja portadora
identificada: $ 139.538
Costo por gestación identificada
con FQ: $ 695.258
Estrategia simultanea:
Costo por pareja portadora
identificada: $ 159.611
Costo por gestación identificada
con FQ: $ 795.272
No costo efectivo
Norman
2012
S test portador: 90% E: 100%
S CVS: 100% E: 100%
Prevalencia portador FQ: 1/25
Aceptación test portador: 20%
Aceptación test prenatal: 80%
Prob. De elegir AT: 90%
Prob. Aborto por CVS: 1.3%
Dólar AU 2010
Costo test portador: $
116,77
AT: $ 1.708
CVS $ 115,20
Costo un ciclo IVF $ $
7.500
Costo vida FQ: $ 336.000
Costo por nacimiento evitado con
FQ: $ 150.000
No costo efectivo
Morris (46), Van Der Riet (48) y Wei (54) no especificaron fuente de financiación, ni
estado de conflicto de interés en la realización de los estudios económicos.

53
Esta revisión sistemática permitió encontrar que la técnica de análisis de decisión
seleccionada más frecuente en la literatura científica mundial para la realización de
evaluaciones económicas de tipo costo-efectividad del test genético de portador para
fibrosis quística, fue un árbol de decisiones en orden secuencial.
Se encontró marcada heterogeneidad en la metodología aplicada que llevó a que los
resultados no fueran comparables y se concluyera que existen diferentes enfoques de
este test genético; según características operativas de la prueba diagnóstica, moneda
aplicada, supuestos y probabilidades que deben ajustarse a las necesidades y
características de cada país. No se encontró ningún estudio económico realizado en
Suramérica ni en Colombia lo que planteó la necesidad de realizarlo según supuestos y
costos colombianos bajo un modelo de análisis de decisiones.
Radhakrishnan et al. (57), en 2008 realizaron una revisión de evaluación económica para
test de fibrosis quística donde encontraron 14 estudios desde 1990 a 2006 reportando
también como análisis primario el costo por nacimiento con fibrosis quística evitado y el
costo por identificación de pareja portadora afectada; entre otros hallazgos que
concuerdan con esta revisión sistemática.
La evidencia sugiere que la estrategia de tamizaje preconcepcional puede ser exitosa,
pero depende de la aceptación por parte de la pareja y de un excelente abordaje en
consejería genética para tener alta probabilidad de aceptar opciones costo efectivas
como la de no tener gestaciones entre parejas en riesgo, adoptar o la donación de
gametos (IVF); mientras que las opciones de tamizaje prenatal y de diagnostico genético
preimplantacional tienen riesgo de aborto y un costo más elevado para el sistema de
salud. Además se debe considerar que no hay una cultura de consulta preconcepcional,
las valoraciones prenatales suelen iniciarse de forma tardía y tampoco hay un panel
especifico ni un estudio de mutaciones prevalentes para ser aplicado en Colombia con el
fin de reducir los costos de estas pruebas genéticas, mejorando su sensibilidad y
especificidad.

54
3.1.2 Valoración de los costos de la prueba diagnóstica para portadores y del costo de un paciente con fibrosis quística durante un año.
Se realizo cotización nacional de la prueba diagnóstica de secuenciación del gen CFTR
para el 2016 a dos laboratorios genéticos de referencia nacional. Además se incluyó el
costo de la secuenciación completa del gen reportada en la guía de práctica clínica
colombiana para fibrosis quística del 2014, actualizando su costo a valor presente 2016.
Tabla 6 Costos de la prueba de portador por secuenciación completa del gen CFTR. Fuente: propia.
Costos prueba
(pesos colombianos)
Referencia
$ 4.234.000
Genetix –Centro de investigación en
Genética humana y reproductiva.
$ 3.474.000 Genética molecular de Colombia
$ 3.014.720 actualizado a
$ 3.432.904
GPC FQ 2014 Colombia. Actualizado
a 2016.
Se solicitó permiso para acceder a la cuenta de alto costo de la base de datos SISPRO
del ministerio de salud y protección social de Colombia. Los datos recolectados
corresponden al registro único para enfermedades de alto costo reportado por las EPS´s
del país al Ministerio de Salud realizado entre el 1 de enero al 31 de diciembre de 2012
según la Resolución 3681 de 2013 (58), la cual define los contenidos y requerimientos
técnicos de la información a reportar por una única vez, a la cuenta de alto costo para el
censo de pacientes con enfermedades huérfanas, incluyendo costos de tratamiento y
procedimientos en salud.

55
Se encontró un total de 424 pacientes diagnosticados con fibrosis quística distribuidos
por departamentos, con mayor frecuencia en Bogotá D.C. y Antioquia.
Tabla 7 Costos de tratamiento en pacientes con fibrosis quística según reporte de alto costo 2012,
Colombia. Fuente: Cuenta de alto costo.
Edad por
rango años
N° pacientes
Costo No POS
Costo POS
Costo total 2012
Costo total 2016
Costo por paciente
0 - antes de 1 4 24.097.861 22.581.268 46.679.129 57.435.025 14.358.756
01 a 05 114 1.637.016.175 924.178.738 2.561.194.913 3.151.350.465 27.643.425
06 a 09 78 2.864.399.025 337.885.343 3.202.284.368 3.940.161.009 50.514.885
10 a 14 74 1.439.692.099 345.718.270 1.785.410.369 2.196.808.126 29.686.596
15 a 18 60 1.120.522.443 795.598.808 1.916.121.251 2.357.637.665 39.293.961
19 a 26 58 1.663.902.245 543.049.243 2.206.951.488 2.715.481.576 46.818.648
27 a 44 22 411.111.916 74.430.535 485.542.451 597.422.094 27.155.550
45 a 59 9 50.862.315 22.796.807 73.659.122 90.631.801 10.070.200
60 y más 5 7.368.224 115.649.977 123.018.201 151.364.296 30.272.859
Total 424 9.218.972.304 3.181.888.989 12.400.861.293 15.258.292.059 Promedio
35.986.538
Se registró el costo total Pos y no Pos para el año 2012 en pesos colombianos el cual fue
actualizado al valor presente 2016 aplicando el IPC en salud de Colombia; para el 2013:
0,0444; 2014: 0,0346; 2015: 0,053 y 2016: 0,0814. Se encontró en dicho registro, el costo
individual y promedio por rango de edades evidenciando una mayor frecuencia de
pacientes entre 1 a 5 años y 6 a 9 años. El costo promedio por paciente

56
independientemente de la edad y severidad de la enfermedad fue de $ 35.986.538, con
una desviación estándar de $ 21.530.047. Con esta información se determinó un costo
mínimo de $ 14.456.491 y un costo máximo de $ 57.516.585 pesos colombianos. Esta
alta variabilidad en los costos se debe a la diferencia de los estadios clínicos que puede
presentar la fibrosis quística; según el número de consultas, de hospitalizaciones o la
cantidad de tratamiento necesario.
Tabla 8 Distribución y prevalencia de pacientes con fibrosis quística en Colombia, 2012. Fuente: Cuenta
de alto costo.
Departamento N°
pacientes
Antioquia 85
Atlántico 29
Bogotá, D.C. 141
Bolívar 26
Boyacá 8
Caldas 6
Cauca 5
Cesar 7
Córdoba 1
Cundinamarca 15
Huila 6
La Guajira 1
Magdalena 6
Meta 3
Nariño 5
Norte de Santander 10
Quindío 3
Risaralda 12
Santander 5
Sucre 3
Tolima 12
Valle del Cauca 35
Total Colombia 424

57
Tabla 9 Parámetros de costos aplicados al modelo. Fuente: propia.
3.1.3 Evaluación de la sensibilidad y especificidad de la prueba diagnóstica.
Para evaluar las características operativas de las pruebas genéticas en fibrosis quística,
existe un test de competencia que intenta ser una medida externa de calidad de un
laboratorio clínico genético. El Colegio Americano de Patología (CAP) es uno de los
participantes en la elaboración y aplicación del test, el cual permite evaluar los errores
preanalíticos tales como el procesamiento de la muestra o extracción del ácido nucleico y
errores postanalíticos como la interpretación y el error de reporte de las mutaciones
encontradas.
En el 2010 se desarrollo el método basado en una encuesta educacional de
secuenciación (SEC) dirigida por el comité genético molecular del CAP y el Colegio
Americano de Genética y Genómica Médica (ACMG) implementada en Estados Unidos y
posteriormente en Europa. Este test permite interpretar y reconocer los hallazgos de los
genes secuenciados, valorando su capacidad analítica e interpretación clínica de los
hallazgos usando la secuenciación Sanger. Para el 2013 se complemento el test con el

58
SEC-1 que valora no solo la interpretación clínica y analítica sino también el componente
técnico de la secuenciación Sanger. Para el caso de la fibrosis quística existen dos
modalidades de evaluación de competencias de la técnica de secuenciación Sanger, la
MGL 2 y la MGL 5 (laboratorio de genética molecular) (59).
La sensibilidad analítica corresponde a la proporción de resultados positivos
correctamente reportados de muestras que contienen una mutación en que la prueba de
laboratorio está diseñada para detectarla. Las mutaciones no detectadas son reportadas
como falsos negativos y puede ocurrir durante la fase analítica como en la mezcla de
muestras y en la falla de la reacción por expiración de los reactivos usados. También
puede ocurrir durante la fase pre o post analítica como en el error de clasificación, el
error en la entrada de datos, lectura, registros de resultados o incorrecta interpretación.
La especificidad analítica corresponde a la proporción de resultados negativos
correctamente reportados por el laboratorio cuando ninguna mutación es detectable. Los
resultados falsos positivos pueden presentarse en la fase analítica, pre o post analítica
por contaminación o reacciones no específicas (60).
Tabla 10 Riesgo de tener un niño afectado después del test con proporción de detección de portador del
90%. Fuente: Tomado de Roberts 2003 (61).
Resultado Riesgo de FQ
Acción
Pareja portadora 1 en 4 Consejería genética Opciones reproductivas Preimplantación
Pariente portador, pareja negativa
1 en 1000 Seguridad acerca del bajo riesgo No test en embarazo Buscar descendencia para fibrosis quística
Pariente negativo, pareja portadora
1 en 2000 Re test para asegurar que no hubo confusión No test en embarazo Buscar descendencia para fibrosis quística
Pareja negativa 1 en 250000 Fuerte seguridad

59
Tabla 11 Probabilidad de tener un niño afectado por fibrosis quística antes y después del test de
portador. Fuente: Tomado de Roberts 2003 (61).
Parentesco a un paciente Con fibrosis quística
Probabilidad de Ser un portador
Riesgo de tener hijo con FQ Con una pareja no testeada
Padres 100% 1 en 4
Persona con FQ 100% 1 en 50
Niño de una mujer con FQ 100% 1 en 100
Hermanos 2 en 3 1 en 150
Tíos 1 en 2 1 en 200
Abuelos 1 en 2 1 en 200
Primo 1 grado 1 en 4 1 en 400
Primo 2 grado 1 en 8 1 en 800
No familiar 1 en 25 1 en 2500
En la tabla 10 se muestra la probabilidad genética del riesgo de tener un hijo afectado
según la presencia de mutación en la pareja, encontrándose un 25% de probabilidad de
tener hijos con fibrosis quística cuando ambos cónyuges son portadores de mutación. La
tabla 11 muestra la probabilidad de ser portador de mutación según el parentesco a un
paciente con fibrosis quística, evidenciando que los familiares en primer grado de
consanguinidad tienen mayor probabilidad (66.6%) de ser portadores de mutaciones que
los familiares de segundo (50%) y tercer grado (25%). Los padres del paciente
diagnosticado con fibrosis quística se consideran portadores con probabilidad del 100%
por tanto no aplica para el objetivo de este estudio. Para personas sin antecedentes de
fibrosis quística en población caucásica se considera una probabilidad de portador de
mutación de 1/25 personas (4%) pero para estudios reportados en Colombia la
probabilidad de portador más acertada corresponde a 1/65 personas (1.5%) (13).
Teniendo en cuenta esta información se considera para el presente estudio realizar el
análisis de costo efectividad en familiares de primer grado de consanguinidad por tener la
probabilidad de portador más alta (66.6%). A través de un análisis de sensibilidad se
evaluará la costo efectividad para familiares en segundo y tercer grado.

60
Al realizar la búsqueda sistemática de los estudios que reportan las características
operativas de la prueba diagnóstica de portador para fibrosis quística o prueba genética
para pacientes con diagnostico clínico de fibrosis quística, se encontraron nueve
artículos, de los cuales cuatro de ellos se seleccionaron para el análisis completo y la
revisión con la herramienta de evaluación de calidad QUADAS 2 para estudios
diagnósticos.

61
Tabla 12 Estudios no incluidos en el análisis de características operativas. Fuente: propia.
Estudio Tipo de estudio
Objetivo Técnica Resultados Observación
Bombieri 2001 (62)
Diagnostico Análisis de 113 pacientes con fibrosis quística
Secuenciación de DNA para exones 9 y 13, polimorfismo de conformación de RNA de filamento único (RNA SSCP) para exones de 1 al 12 y electroforesis en gel con gradiente de desnaturalización (DGGE) para exones 14 a 24.
22 mutaciones diferentes identificadas en 203 de 225 cromosomas tamizados. (90.2% de detección).
No aplica secuenciación a la totalidad del gen CFTR, utiliza otras técnicas distintas a la secuenciación. No aplicaría para obtener sensibilidad y especificidad en el estudio. No hay un gold estándar especificado.
Palomaki 2004 (63)
Diagnostico
Valoración según etnicidad a partir de estudios previos principalmente de estados unidos.
Panel de 25 mutaciones valoradas por etnicidad a partir de estudios previos correlacionados con la epidemiologia genética de cada región.
Reportada según etnicidad. Se reporta la frecuencia de mutación genética según origen étnico.
No aplica para el presente estudio, usa el panel de 25 mutaciones, sin especificar la técnica genética, no hay una valoración completa del gen y varía según la etnicidad. Colombia tiene origen multiétnico por lo cual no podría clasificarse solo como caucásicos no hispánicos. Evalúan la proporción de detección del panel de 25 mutaciones según etnicidad pero no se reporta la validez analítica de la técnica usada.
Watson 2004 (28)
Revisión genética ACMG
Panel de 25 mutaciones según origen étnico.
No se especifica técnica genética, panel de 25 mutaciones.
Según origen étnico, una proporción de detección dada.
No aplica para el estudio, panel de 25 mutaciones, multietnicidad, no especifican técnica genética. No hay secuenciación completa del gen. No hay gold estándar
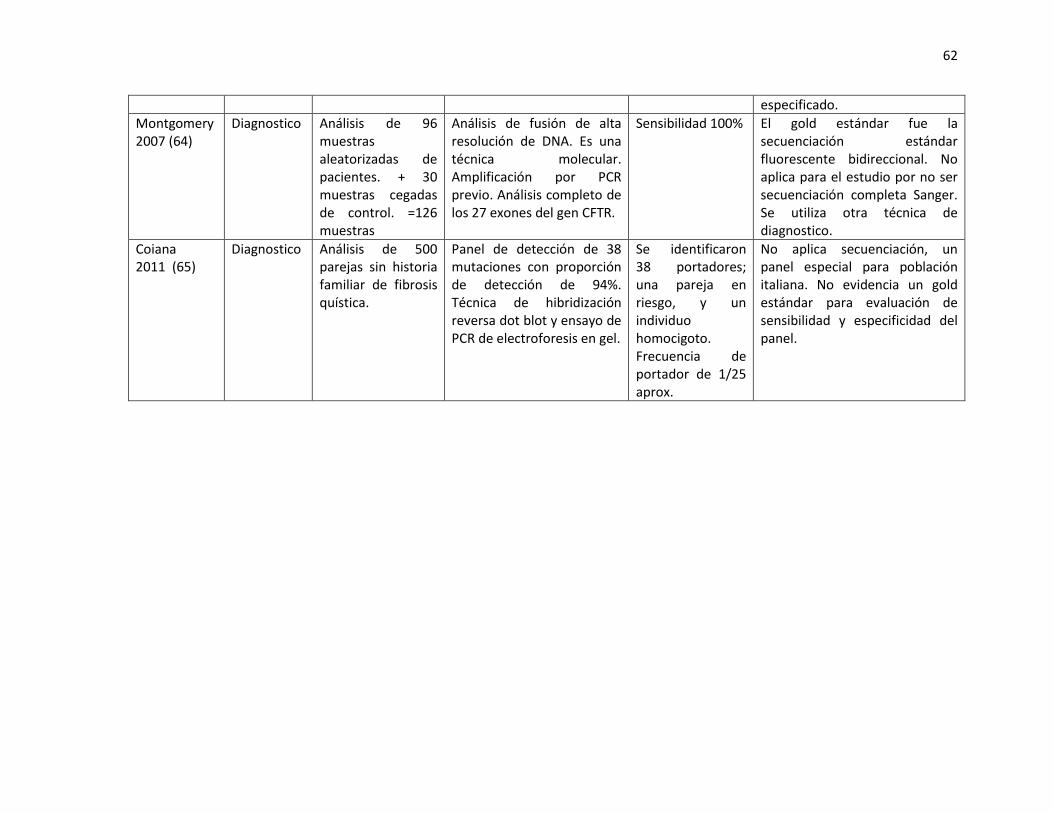
62
especificado.
Montgomery 2007 (64)
Diagnostico Análisis de 96 muestras aleatorizadas de pacientes. + 30 muestras cegadas de control. =126 muestras
Análisis de fusión de alta resolución de DNA. Es una técnica molecular. Amplificación por PCR previo. Análisis completo de los 27 exones del gen CFTR.
Sensibilidad 100% El gold estándar fue la secuenciación estándar fluorescente bidireccional. No aplica para el estudio por no ser secuenciación completa Sanger. Se utiliza otra técnica de diagnostico.
Coiana 2011 (65)
Diagnostico Análisis de 500 parejas sin historia familiar de fibrosis quística.
Panel de detección de 38 mutaciones con proporción de detección de 94%. Técnica de hibridización reversa dot blot y ensayo de PCR de electroforesis en gel.
Se identificaron 38 portadores; una pareja en riesgo, y un individuo homocigoto. Frecuencia de portador de 1/25 aprox.
No aplica secuenciación, un panel especial para población italiana. No evidencia un gold estándar para evaluación de sensibilidad y especificidad del panel.

63
Tabla 13 Estudios incluidos en el análisis de características operativas. Fuente: propia.
Estudio Tipo de estudio
Objetivo Técnica Resultados Observación
Palomaki 2003 (60)
Diagnostico Se evalúan a través del
test de competencia
externa MGL 2 y 5
preparado por la
CAP/ACMG, que consiste
en DNA purificado de
líneas celulares
establecidas distribuidas a
los laboratorios
participantes y aplicadas 2
veces al año.
Total de 2198 alelos testeados desde 1996 hasta 2001, reportan falsos positivos, falsos negativos, respuestas correctas, al comparar con el test de competencia externa CAP/ACMG MGL.
Análisis de muestras de laboratorio de EU e internacionales de 1996 al 2001. No especifican técnicas aplicadas al diagnostico.
Sensibilidad global : 97.9% IC 95% (96.8-98.7) Especificidad global: 98.4% IC 95% (96.8-98.7)
Evalúan laboratorios principalmente de estados unidos. El Gold estándar es la
evaluación que hacen los
laboratorios del
CAP/ACMG teniendo en
cuenta el DNA purificado
de líneas celulares
establecidas. No se
especifican las técnicas
genéticas usadas por los
diferentes laboratorios, ni
el numero de laboratorios
participantes. Se reportan
el número de alelos
evaluados de muestras de
Estados Unidos sin
reconocer la etnicidad. Se
aplica la búsqueda sobre
el panel de 23 mutaciones
recomendado por el
ACMG/ACOG.

64
Strom 2003 (66)
Diagnostico DNA de 17 pacientes
sintomáticos con FQ, con
mutaciones conocidas,
fueron codificados,
anonimizados, y enviados
para análisis con controles
de DNA. También fueron
analizados 8 pacientes
posteriormente enviados
por sus médicos por
presentar pruebas clínicas
positivas como la de
sudor, al examen fueron
confirmados siete como
positivos por
secuenciación completa.
Ensayo diagnostico para
secuenciación completa
del gen, detectando 991
de 1004 mutaciones
detectadas al 2002, con
técnicas de PCR y ABI
PRISM Big Dye y ABI
Prism 3100 Genetic
Analyzer, todo el proceso
es automatizado.
El laboratorio fue cegado de todas las muestras y de las mutaciones esperadas.
Todas las
mutaciones fueron
correctamente
identificadas y los
controles normales
también fueron
correctamente
identificados.
98.7% de detección.
Usan la técnica de Sanger en población con diagnostico clínico de fibrosis quística.
No especifican perdidas,
no especifican gold
estándar usado.
La clínica podría actuar como gold estándar en la detección de mutaciones.
Pratt 2009 (67)
Diagnostico 15 muestras de líneas
celulares de DNA
obtenidas del “Coriell Cell
repositorie”. Se analizaron
también las 23
mutaciones del panel
recomendado por la
ACOG. Se realizo cultivo y
criopreservación de las
muestras.
Seis laboratorios
genéticos clínicos
voluntarios participaron
en el estudio,
provenientes de EU.
Tienen certificación CLIA
y son acreditados por el
colegio de patólogos
americanos (CAP).
Se usaron 6 métodos
diagnósticos “Luminex
tag-it, asuragen
Sensibilidad y especificidad del 100%
Usan diferentes técnicas
entre ellas la
secuenciación. El
protocolo está bien
definido, hubo
cegamiento de los alelos y
mutaciones a los
laboratorios participantes.
Ningún falso positivo o
resultado discordante fue
reportado entre los
laboratorios, obteniendo

65
signature, laboratory
developed test, third
wave inplex, OLA y
secuenciación”.
La secuenciación fue
realizada por dos
laboratorios usando el
ABI 3100 y el ABI 3730
realizada bidireccional
con un control normal.
sensibilidad y
especificidad de 100% con
la secuenciación de las
mutaciones cegadas.
No se especifica si las muestras de los 15 alelos provienen de pacientes con diagnostico clínico de fibrosis quística o solamente portadores asintomáticos.
Lyon/ Palomaqui 2015 (68) CAP-ACMG
Diagnostico El CAP evalúa por test de competencias MGL 2 y MGL 5 aplicadas dos veces al año con muestras de ADN analizadas por laboratorios de Estados Unidos e internacionales. Ambas encuestas se basan principalmente en la detección de las 23 mutaciones recomendadas por el panel ACMG/ACOG.
Las muestras del CAP/ACMG consisten en DNA purificado de líneas celulares establecidas por el Coriell Repositories. Estas líneas celulares han sido validadas por el programa previo GeT-RM (Pratt 2009) y por laboratorios del CAP/ACMG. La valoración de la competencia se basa en la exactitud del genotipo identificado y de la interpretación asociada al significado clínico.
Del 2003 al 2013 participaron 322 laboratorios clínicos de USA y 35 laboratorios internacionales. 2008-2013: USA S: 99.3% IC 95% (98.8-99.5) E: 99.6 % IC(99.3-99.8) INTERNACIONAL S: 96 % IC 95% (92.9-97.8) E: 100% IC (99.9-100)
Utiliza la valoración de diferentes técnicas de prueba de portador basada principalmente del panel de 23 mutaciones. Tiene un gold estándar que sería el comité de CAP-ACMG quienes revisan y analizan la exactitud diagnostica. Se evalúa la sensibilidad y especificidad por test de competencias incluidos laboratorios internacionales. No se reporta la clínica o el antecedente medico ni la etnicidad de las

66
También fueron analizadas mutaciones diferentes a las incluidas en el panel de 23 mutaciones. Las técnicas más comunes fueron OLA, ASO, PCR, incluidas secuenciación Sanger y nueva generación.
muestras analizadas.
CAP: Colegio de Patólogos Americanos, ACMG: Colegio Americano de Genética y Genómica Médica, EU: Estados Unidos, OLA: ensayo de ligación de
oligonucleótido, MGL: laboratorio de genética molecular, ASO: oligonucleótido especifico de alelo, PCR: reacción en cadena polimerasa. CLIA:
correcciones de mejora para laboratorios clínicos.

67
Tabla 14 Resultados QUADAS 2. Fuente: propia.
Estudio Riesgo de Sesgo Aplicabilidad
Selección
de
pacientes
Prueba
índice
Prueba de
referencia
Flujo y
cronograma
Selección
de
pacientes
Prueba
índice
Prueba de
referencia
Palomaki
2003
Strom
2003
Pratt
2009
Lyon
2015
Bajo Riesgo Alto Riesgo Riesgo poco claro
Los resultados de QUADAS 2 se interpretan de manera subjetiva. En la presente
investigación se tiene en cuenta que las pruebas MGL 2 y 5 son test de competencias
diagnosticas especificas para detectar mutaciones de fibrosis quística, en el cual se
envían muestras de laboratorio purificadas con mutaciones conocidas revisadas por el
comité CAP. Se desconoce por tanto la selección de los pacientes, si son o no
portadores o si tienen la condición de fibrosis quística. Dentro de la prueba de
competencia que envía el comité, solicitan que cada laboratorio interprete clínicamente la
mutación o mutaciones encontradas, y esto podría definirse como enmascaramiento
simple a los laboratorios, lo cual disminuye el riesgo de sesgos de selección y de
diagnostico.

68
Se encuentra que el estudio de Lyon 2015 es el de menor riesgo de sesgo y de alta
calidad para ser incluido en el análisis ya que reporta de manera diferencial las
características analíticas de laboratorios internacionales no estadounidenses y reportan
las técnicas de diagnostico aplicadas a las muestras, entre ellas la de secuenciación
Sanger, aunque solo se incluyen en las muestras enviadas las 23 mutaciones más
frecuentes recomendadas por el panel de detección ACOG/ACMG.
Tabla 15 Probabilidades aplicadas al modelo. Fuente: propia.

69
3.1.4 Resultados del modelo de costo efectividad de aplicar versus no aplicar la prueba genética en portadores asintomáticos.
Figura 3 Plano de análisis de costo efectividad. Fuente: Propia.
Al realizar el análisis de costo efectividad en el programa TreeAge Pro 2015 se obtuvo un
ICER de $ 17.082.833,90 pesos colombianos, evidenciando que este es el costo
incremental por obtener un 10,89 % más de probabilidad de evitar el nacimiento de un
niño enfermo con fibrosis quística por pareja tamizada. La estrategia de no hacer la
prueba genética tiene una efectividad basal de 89,11 % de tener niños sanos por cada
gestación, dado por características de herencia autosomica recesiva propias de la
enfermedad. Al realizar la prueba genética junto a la consejería, se logra obtener el 100%
de efectividad, evitando nacimientos con fibrosis quística. De este análisis se puede
inferir que; de 100 gestaciones de parejas en riesgo sin realizar la prueba genética; 89
nacimientos serán sanos y 11 estarán afectados con fibrosis quística. Con la nueva
estrategia, de 100 parejas en riesgo a quienes se les ofrece la prueba con su respectiva
consejería genética, se evitarían 11 futuras concepciones con fibrosis quística. En el
plano de costo efectividad (Figura 3) se encontró que son estrategias localizadas en el

70
cuadrante I mostrando que hacer la prueba genética es más efectiva pero también más
costosa en relación a la estrategia de no realizarla.
Figura 4 Diagrama de tornado (ICER). Fuente: Propia.
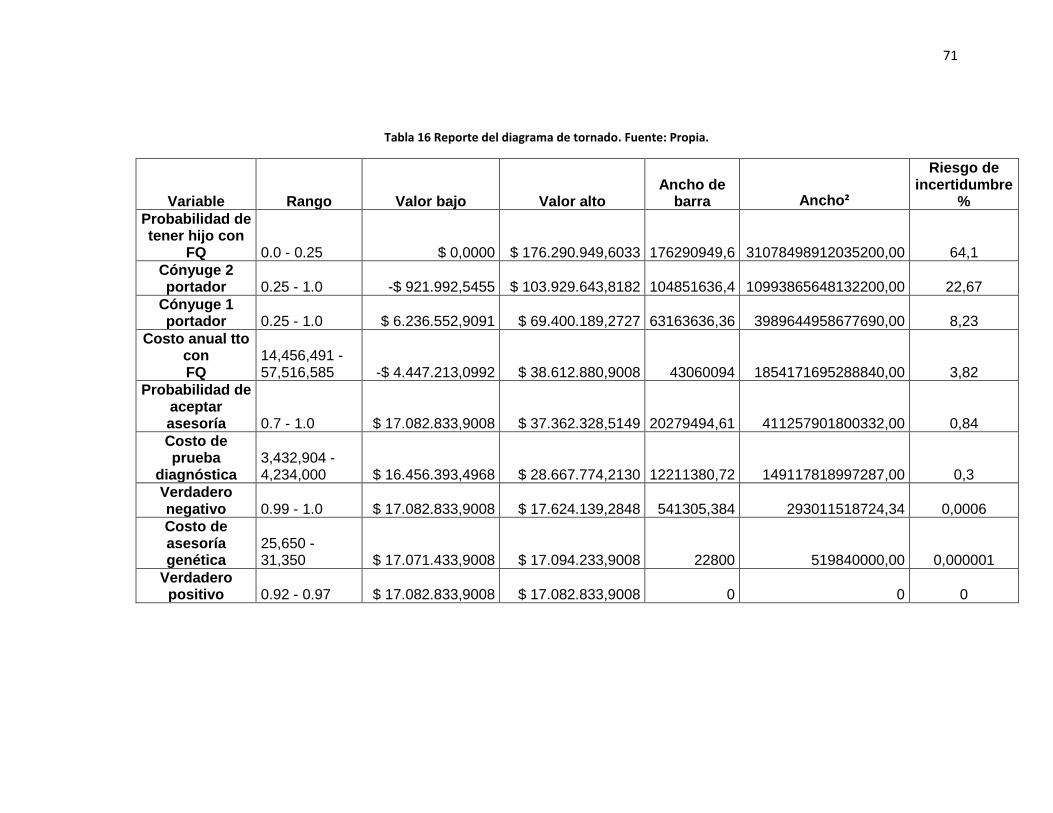
71
Tabla 16 Reporte del diagrama de tornado. Fuente: Propia.
Variable Rango Valor bajo Valor alto Ancho de
barra Ancho²
Riesgo de incertidumbre
%
Probabilidad de tener hijo con
FQ 0.0 - 0.25 $ 0,0000 $ 176.290.949,6033 176290949,6 31078498912035200,00 64,1
Cónyuge 2 portador 0.25 - 1.0 -$ 921.992,5455 $ 103.929.643,8182 104851636,4 10993865648132200,00 22,67
Cónyuge 1 portador 0.25 - 1.0 $ 6.236.552,9091 $ 69.400.189,2727 63163636,36 3989644958677690,00 8,23
Costo anual tto con FQ
14,456,491 - 57,516,585 -$ 4.447.213,0992 $ 38.612.880,9008 43060094 1854171695288840,00 3,82
Probabilidad de aceptar asesoría 0.7 - 1.0 $ 17.082.833,9008 $ 37.362.328,5149 20279494,61 411257901800332,00 0,84
Costo de prueba
diagnóstica 3,432,904 - 4,234,000 $ 16.456.393,4968 $ 28.667.774,2130 12211380,72 149117818997287,00 0,3
Verdadero negativo 0.99 - 1.0 $ 17.082.833,9008 $ 17.624.139,2848 541305,384 293011518724,34 0,0006
Costo de asesoría genética
25,650 - 31,350 $ 17.071.433,9008 $ 17.094.233,9008 22800 519840000,00 0,000001
Verdadero positivo 0.92 - 0.97 $ 17.082.833,9008 $ 17.082.833,9008 0 0 0

72
Al realizar el análisis de sensibilidad univariado representado en el diagrama de tornado
(figura 4) encontramos que las variables con mayor probabilidad de riesgo de
incertidumbre son: hijo con FQ, cónyuge 1 y 2 positivos y costo del tratamiento; lo cual
indica que al cambiar cada una de estas variables de manera independiente entre los
rangos establecidos, influye directamente en el resultado siendo menor o mayor al ICER
obtenido con los datos basales del modelo. La variable hijo con FQ refiere que por ser
una enfermedad autosómica recesiva existe el riesgo hasta del 25 % de tener hijos con
fibrosis quística con ambos padres portadores de una mutación. Con relación a las
variables costo de tratamiento y cónyuge 2 positivo, se encuentra que el ICER llega a ser
negativo representando ahorro con los límites superiores de dichas variables. El resto de
variables no representan una proporción de incertidumbre considerable a tener en cuenta
en el análisis.
Tabla 17 Análisis de sensibilidad de dos vías, probabilidad de portador de mutación en cónyuge 1 y 2. Fuente: Propia.
Prob. Cónyuge 1 sea portador
Prob. Cónyuge 2 sea portador
0.25 0.437
0.25
Hacer prueba
Costo
Efectividad
No hacer prueba
Costo
Efectividad
ICER
$ 4.344.281,25
1
$ 562.289,66
0.98
$ 189.099.579,5
$ 4.996.992,19
1
$ 984.006,90
0.97
$ 133.766.176,3
0.437
Hacer prueba
Costo
Efectividad
No hacer prueba
Costo
Efectividad
ICER
$ 4.345.617,19
1
$ 984.006,90
0.97
$ 112.053.676,3
$ 4.999.330,08
1
$ 1.722.012,07
0.95
$ 65.546.360,2
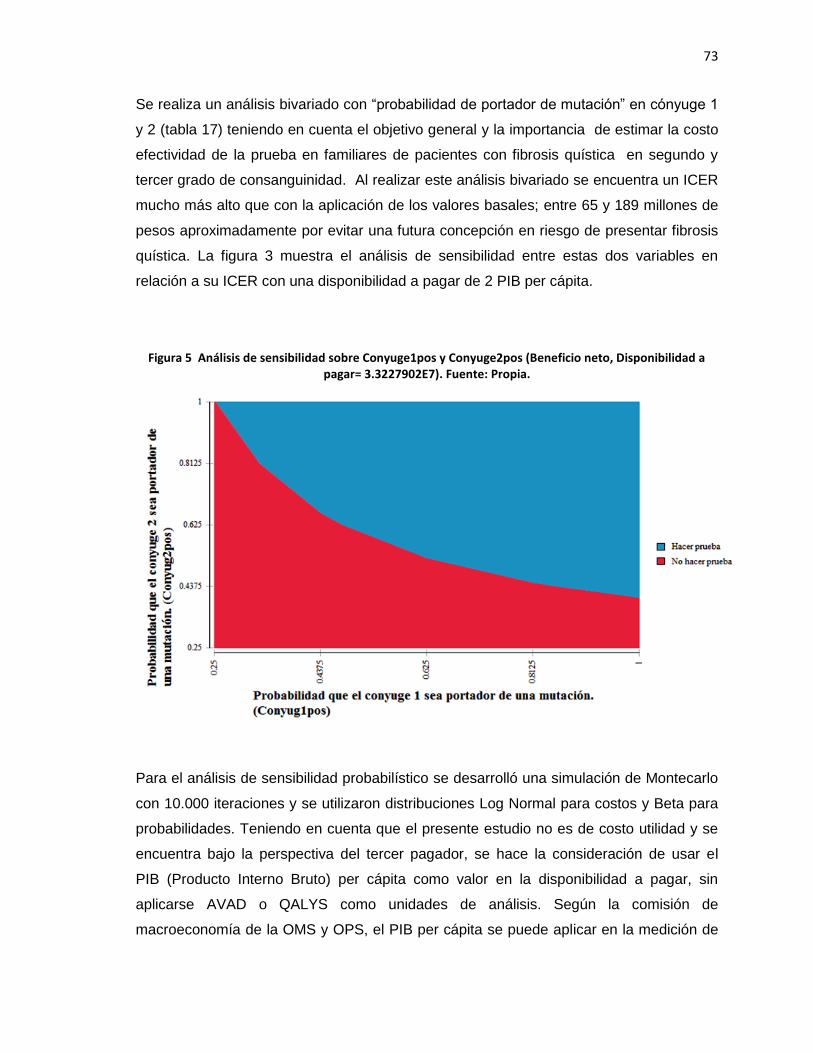
73
Se realiza un análisis bivariado con “probabilidad de portador de mutación” en cónyuge 1
y 2 (tabla 17) teniendo en cuenta el objetivo general y la importancia de estimar la costo
efectividad de la prueba en familiares de pacientes con fibrosis quística en segundo y
tercer grado de consanguinidad. Al realizar este análisis bivariado se encuentra un ICER
mucho más alto que con la aplicación de los valores basales; entre 65 y 189 millones de
pesos aproximadamente por evitar una futura concepción en riesgo de presentar fibrosis
quística. La figura 3 muestra el análisis de sensibilidad entre estas dos variables en
relación a su ICER con una disponibilidad a pagar de 2 PIB per cápita.
Figura 5 Análisis de sensibilidad sobre Conyuge1pos y Conyuge2pos (Beneficio neto, Disponibilidad a pagar= 3.3227902E7). Fuente: Propia.
Para el análisis de sensibilidad probabilístico se desarrolló una simulación de Montecarlo
con 10.000 iteraciones y se utilizaron distribuciones Log Normal para costos y Beta para
probabilidades. Teniendo en cuenta que el presente estudio no es de costo utilidad y se
encuentra bajo la perspectiva del tercer pagador, se hace la consideración de usar el
PIB (Producto Interno Bruto) per cápita como valor en la disponibilidad a pagar, sin
aplicarse AVAD o QALYS como unidades de análisis. Según la comisión de
macroeconomía de la OMS y OPS, el PIB per cápita se puede aplicar en la medición de
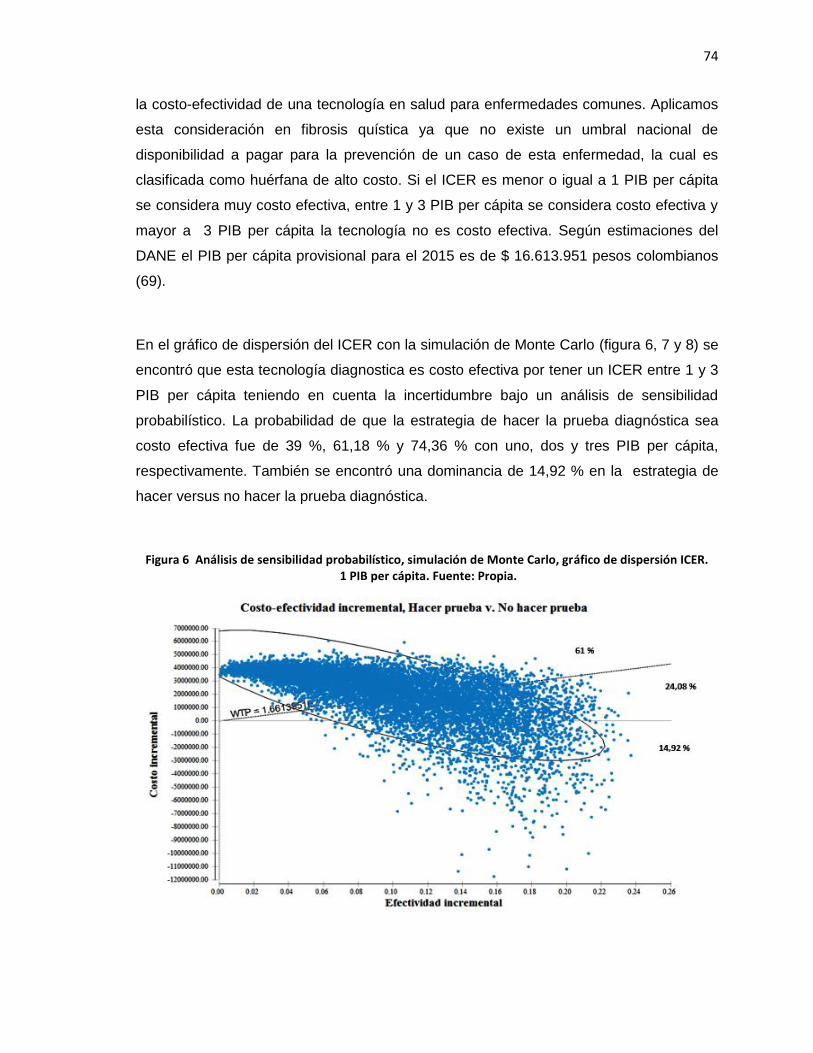
74
la costo-efectividad de una tecnología en salud para enfermedades comunes. Aplicamos
esta consideración en fibrosis quística ya que no existe un umbral nacional de
disponibilidad a pagar para la prevención de un caso de esta enfermedad, la cual es
clasificada como huérfana de alto costo. Si el ICER es menor o igual a 1 PIB per cápita
se considera muy costo efectiva, entre 1 y 3 PIB per cápita se considera costo efectiva y
mayor a 3 PIB per cápita la tecnología no es costo efectiva. Según estimaciones del
DANE el PIB per cápita provisional para el 2015 es de $ 16.613.951 pesos colombianos
(69).
En el gráfico de dispersión del ICER con la simulación de Monte Carlo (figura 6, 7 y 8) se
encontró que esta tecnología diagnostica es costo efectiva por tener un ICER entre 1 y 3
PIB per cápita teniendo en cuenta la incertidumbre bajo un análisis de sensibilidad
probabilístico. La probabilidad de que la estrategia de hacer la prueba diagnóstica sea
costo efectiva fue de 39 %, 61,18 % y 74,36 % con uno, dos y tres PIB per cápita,
respectivamente. También se encontró una dominancia de 14,92 % en la estrategia de
hacer versus no hacer la prueba diagnóstica.
Figura 6 Análisis de sensibilidad probabilístico, simulación de Monte Carlo, gráfico de dispersión ICER. 1 PIB per cápita. Fuente: Propia.

75
Figura 7 Análisis de sensibilidad probabilístico, simulación de Monte Carlo, gráfico de dispersión ICER. 2 PIB per cápita. Fuente: Propia.
Figura 8 Análisis de sensibilidad probabilístico, simulación de Monte Carlo, grafico de dispersión ICER. 3 PIB per cápita. Fuente: Propia.

76
Figura 9 Curva de aceptabilidad de costo efectividad. Fuente: Propia.
La curva de aceptabilidad de costo efectividad (figura 9) muestra que con una
disponibilidad a pagar de 25 millones de pesos la probabilidad de que la estrategia de
hacer la prueba genética sea costo efectiva es del 50%. El resultado del ICER de $
17.082.833,90 pesos colombianos muestra que la probabilidad de que la estrategia de
hacer la prueba genética sea costo efectiva es del 36% aproximadamente.
Adicionalmente se evidencia que la probabilidad de costo-efectividad de hacer la prueba
genética con un IC 60% está entre un rango de disponibilidad a pagar de $ 4.000.000 y $
60.000.000 pesos.

77
4. Discusión
Este estudio planteó la evaluación de costo efectividad con un modelo de árbol de
decisiones; el hacer o no la prueba genética de detección de portadores de mutaciones,
en familiares asintomáticos en edad fértil de pacientes con fibrosis quística en primer,
segundo y tercer grado de consanguinidad. Se desarrolló bajo la perspectiva del tercer
pagador, con un horizonte de seguimiento a un año, con costos directos propios del país
y probabilidades encontradas en la literatura. Se logró determinar el costo incremental de
obtener un 10,89 % más de probabilidad de evitar el nacimiento de un niño enfermo con
fibrosis quística con un ICER de $ 17.082.833,90 pesos colombianos en familiares de
primer grado de consanguinidad. Para familiares de segundo y tercer grado de
consanguinidad se encontró un ICER de $ 65.546.360,2 y $ 189.099.579,5 pesos
obtenido de un análisis de sensibilidad bivariado mostrando un costo incremental muy
superior a los de primer grado. Al evaluar la incertidumbre del modelo en un análisis de
sensibilidad univariado se encontró que las variables hijo con FQ, cónyuge 1 y 2 positivos
y costo del tratamiento representan la mayor probabilidad de riesgo de incertidumbre en
el modelo.
Para el análisis de sensibilidad probabilístico se utilizó el PIB per cápita como valor de
disponibilidad a pagar, teniendo en cuenta que no existe un umbral para fibrosis quística
considerada como una enfermedad huérfana de alto costo. Se obtuvo en este análisis la
probabilidad que con la estrategia de hacer la prueba diagnóstica, presente una costo
efectiva de 39 %, 61,18 % y 74,36 % con uno, dos y tres PIB per cápita, respectivamente.
La curva de aceptabilidad mostró que con una disponibilidad a pagar de 25 millones de
pesos hay una probabilidad del 50% de que la estrategia de hacer la prueba genética sea
costo efectividad.
La evidencia muestra que puede existir variabilidad entre la aceptación o no del test
genético para portadores de fibrosis quística y de las recomendaciones dadas durante la
asesoría genética; donde se incluyen opciones tales como evitar tener hijos o adoptar;
las cuales no representarían costos adicionales. Otras opciones reproductivas como el
diagnostico prenatal, la donación de oocito o esperma y el diagnóstico genético
preimplantacional implicarían costos adicionales, los cuales no fueron contemplados en

78
el modelo, teniendo en cuenta la autonomía reproductiva de la pareja que son quienes
deciden entre abortar o no en caso que el producto este afectado con la enfermedad.
Es importante tener en cuenta que la presente investigación se desarrolló en un
escenario ideal; en el cual el 100% de las parejas aceptan usar la prueba de portador
para fibrosis quística y las recomendaciones dadas durante la asesoría genética. Ante
este fuerte supuesto planteado, se evaluó el desenlace dependiendo del escenario
clínico según la probabilidad de aceptación de las recomendaciones durante asesoría
genética, mostrando que se logra evitar al menos un caso de fibrosis quística con un 10
% de aceptación de la asesoría genética por cada 100 parejas en riesgo por ser
portadoras de mutación. De igual manera se logró determinar el costo total de los 11
casos de fibrosis quística por cada 100 parejas en riesgo en caso de no hacerse la
prueba, con un total de $ 13.315.019.060 pesos, según la esperanza de vida promedio
de 37 años para Colombia asumiendo el costo anual promedio como constante (Ver
Anexo A).
Watson y col en 1992 (70), analizaron a 88 portadores de mutaciones sin antecedente
familiar de fibrosis quística, evaluando sus actitudes reproductivas y psicológicas. Un
55% de los analizados no tenían hijos y 45 % tenían al menos un hijo. 67 % estaban
sorprendidos por los resultados, 25% experimentaron ansiedad, 22% preocupación y 7 %
depresión. 42% de los encuestados decidieron no querer tener más hijos, 49% estaban
indecisos. 79% de los encuestados estuvieron dispuestos a acceder al diagnostico
prenatal y 20% a abortar en caso de producto afectado.
Callanan et al (71) en 1999, testearon 296 estadounidenses, familiares de pacientes con
fibrosis quística. Se reportaron 52 parejas con al menos una mutación. Al preguntar sobre
su deseo de paridad, el 48% decidieron no planear hijos a futuro, 26% estaban
planeando tener hijos y 7,4% estaban indecisos.
Lafayette y col en 1999 (72), encuestaron y evaluaron a 81 familiares portadores de
pacientes con fibrosis quística, encontrando que en 42% de los casos tendrían menos
hijos de los planeados, 10% preferían no tener hijos y 5% usarían inseminación artificial.

79
En 70% de los casos se cambiaría su plan familiar y estarían dispuestos a usar el
diagnostico prenatal, solo 7% estuvieron dispuestos a abortar en caso que el producto
tuviera la enfermedad.
Braekeleer et al (73) en 2000, encuestaron 825 personas de Quebec, Canadá, entre
familiares con y sin hijos afectados con fibrosis quística, pacientes, doctores y
enfermeras. Entre los resultados, 34 de 56 parejas con un niño diagnosticado con fibrosis
quística, decidieron no tener más hijos, las 22 parejas restantes decidieron tener al
menos un segundo hijo. Del total de parejas incluidas, un 70,3 % decidieron no tener más
hijos. Solo 12,7% de las parejas utilizaron el diagnostico prenatal.
Henneman y col en 2002 (74), encuestaron nueve parejas de Netherlands identificadas
como portadores mutacionales para fibrosis quística con antecedente familiar de la
enfermedad. Cuatro parejas ya estaban en gestación, reportando angustia y ansiedad
antes y después del test. Tres parejas continuaron con sus planes reproductivos, 2
tuvieron problemas de fertilidad y 4 pensaron en no tener más hijos. Ocho de las nueve
parejas usaron el diagnostico prenatal como opción gestacional, obteniendo 17
embarazos viables y 6 abortos terapéuticos. Estas decisiones reproductivas son más
influenciadas luego de presentar un niño afectado con la enfermedad, debido a la poca
implementación del test de manera preconcepcional.
Ioannou y col (75) en 2014 realizaron una revisión sistemática de estudios que evaluaban
consecuencias psicológicas y de preferencia con relación al uso del test en familiares con
antecedente de fibrosis quística. Fueron seleccionados 85 estudios en los que se
encontró un 40% de aplicación del test a nivel prenatal y 30,6% a nivel preconcepcional.
El 89% de mujeres no gestantes y el 98% de gestantes prefirieron que el test fuera
ofrecido a nivel preconcepcional. El rango de uso del test para seguimiento
preconcepcional se registró entre 2 y 96 %. En 50 a 94% de los estudios se encontró que
la población prefiere recibir asesoría pretest en la consulta general.

80
Ioannou et al (76) en 2015, analizaron 8 parejas de un programa de tamizaje para
portadores de fibrosis quística, de las cuales 6 decidieron realizarse diagnostico prenatal,
encontrándose 2 productos afectados con la enfermedad. Tres parejas decidieron no
tener más hijos.
Dugueperoux y cols. (77) en 2016 analizaron en Francia retrospectivamente por 25 años,
la probabilidad del uso de la prueba de portador de mutaciones para fibrosis quística;
encontrando 40,3% de uso de la prueba, ofrecida a 459 personas mayores de 18 años
familiares provenientes de 40 pacientes con fibrosis quística, de las cuales 185 familiares
aceptaron ser tamizados. Esta estrategia permitió la detección de 5 parejas portadoras
en riesgo de tener hijos con la enfermedad.
Como todo modelo, el presente estudio de costo efectividad también tiene limitaciones,
las cuales son: A) Nuestro modelo no incluye valoraciones desde la perspectiva social,
dejando por fuera los costos indirectos. B) Por tener un modelo de horizonte temporal a 1
año, no permite evaluar resultados posteriores, como el caso de tener más de una
gestación o al tener en cuenta la esperanza de vida asociada a los costos de atención en
salud, con lo cual mejoraría el resultado de costo efectividad. C) Las probabilidades
aplicadas en el modelo se obtienen de estudios internacionales que evalúan las
características operativas de la prueba genética con población principalmente
estadounidense y europea, limitando la valoración de las características operativas de la
prueba en la población colombiana. D) No existe un umbral de disponibilidad a pagar
para la prevención de casos de fibrosis quística en Colombia, lo cual deja a
consideración del sistema de salud y del tomador de decisiones identificar un umbral de
disponibilidad a pagar para la prevención de un caso de fibrosis quística, que podría
variar la clasificación de la costo efectividad de tecnologías en salud para enfermedades
de alto costo. De igual manera, el umbral de disponibilidad a pagar para la prevención de
casos de fibrosis quística asociado a la esperanza de vida de la enfermedad podría
mejorar el resultado de costo efectividad del modelo. E) Se tiene en cuenta que el costo
de la prueba aplicado es para secuenciación completa del gen CFTR, ya que no hay
estudios epidemiológicos de distribución genética completa de mutaciones para fibrosis
quística en el país, que permita aplicar un panel de mutaciones con alta sensibilidad y
especificidad para la población colombiana. Si se tuviera un panel de mutaciones

81
específico para ser aplicado en Colombia, los costos de la prueba genética bajarían,
mejorando su costo efectividad. F) El PIB per cápita como umbral de disponibilidad a
pagar podría sobrevalorar o subvalorar la costo-efectividad del modelo.
El presente estudio de costo efectividad puede ser aplicado a población colombiana
teniendo en cuenta que el modelo de árbol de decisiones fue adaptado a la atención
clínica del país, sin embargo, no hay una cultura de consulta preconcepcional para
asegurar que el 100% de los familiares accedan a la prueba genética. Para mejorar esta
limitación se requiere de la búsqueda activa en las familias de los pacientes
diagnosticados con fibrosis quística. De igual manera se debe tener en cuenta que no se
incluyó la perspectiva de la sociedad influyendo en el resultado de costo efectividad. Las
probabilidades de las características operativas de la prueba genética fueron
identificadas de estudios diagnósticos internacionales con heterogeneidad en la etnicidad
de los pacientes involucrados, lo cual podría limitar la aplicación de un panel de mutación
en nuestra población.

82
5. Conclusiones
• Se evidencia costo efectividad en la prueba genética de secuenciación para
portadores de fibrosis quística según disponibilidad a pagar de 1 a 3 PIB per
cápita por parte del tercer pagador.
• Para familiares en segundo y tercer grado de consanguinidad se presenta un
ICER mayor y su costo efectividad es dependiente de la disponibilidad a pagar.
• No se encontraron estudios de costo efectividad para prueba genética en
portadores de fibrosis quística ni en Colombia ni en Suramérica.
• No existe un umbral de disponibilidad a pagar para la prevención de casos en
enfermedades de alto costo, como la fibrosis quística.
• Falta investigación en la distribución epidemiológica de las mutaciones más
frecuentes de fibrosis quística en pacientes afectados y familiares portadores, que
permitan establecer un panel de mutaciones con alta sensibilidad y especificidad
para la población colombiana.
Conflicto de intereses: Ninguno
Financiación: Universidad Nacional de Colombia. Código Hermes 20532.

83
Anexos
Anexo A. Consecuencias clínicas y económicas según escenario de probabilidad de aceptación de asesoría genética por cada 100 parejas tamizadas en riesgo por ser portadores de mutaciones. Fuente: Propia.
Aceptación de
Asesoría genética
%
N° de Niños con FQ
Haciendo la Prueba
+ asesoría genética
N° de Niños con FQ
Sin Hacer la Prueba genética
(no incluye asesoría genética)
Total niños sanos
de 100
parejas
Costo según N° de
Niños FQ al hacer Prueba +
asesoría genética.
($ 35.986.538 promedio año)
$
Costo según niños
afectados (costo
enfermedad anual
constante por esperanza de
vida de 37 años)
$
100 0 10,89 100 0 0
90 1,003 10,89 98,99 35.986.538 1.331.501.906
80 2,007 10,89 97,99 71.973.076 2.663.003.812
70 3,0105 10,89 96,98 107.959.614 3.994.505.718
60 4,014 10,89 95,98 143.946.152 5.326.007.624
50 5,0175 10,89 94,98 179.932.690 6.657.509.530
40 6,021 10,89 93,97 215.919.228 7.989.011.436
30 7,0245 10,89 92,97 251.905.766 9.320.513.342
20 8,028 10,89 91,97 287.892.304 10.652.015.248
10 9,0315 10,89 90,96 323.878.842 11.983.517.154
0 10,035 10,89 89,9 359.865.380 13.315.019.060

84
Anexo B. Operacionalización de variables. Fuente: propia.
VARIABLE DEFINICION INDICADOR CATEGORIA
NIVEL DE MEDICION
INDICE
Probabilidad de portador de mutación en primer grado familiar de FQ
Probabilidad o proporción de tener
una mutación de fibrosis quística
teniendo en cuenta la consanguinidad
familiar.
En probabilidad o porcentaje
Cuantitativa continua
Probabilidad
Porcentaje
Sensibilidad de la prueba de
secuenciación gen CFTR
Proporción de resultados positivos
correctamente reportados con
mutación detectada, son los verdaderos
positivos detectados con la prueba.
En probabilidad o porcentaje
Cuantitativa continua
Probabilidad
Porcentaje
Especificidad de la prueba de
secuenciación gen CFTR
Proporción de resultados negativos
correctamente reportados sin
mutación detectada, son los verdaderos
negativos detectados con la prueba.
En probabilidad o porcentaje
Cuantitativa continua
Probabilidad
Porcentaje
Aceptación de asesoría genética
Probabilidad de que la pareja en riesgo de tener una gestación con fibrosis quística
acepte las recomendaciones
dadas en la asesoría genética.
En probabilidad o porcentaje
Cuantitativa continua
Probabilidad
Porcentaje
Riesgo de tener un hijo con
fibrosis quística
Probabilidad determinada
genéticamente por ser una enfermedad
autosómica recesiva, en tener una gestación
con fibrosis quística
En riesgo o sin riesgo Cualitativa Nominal
Porcentaje

85
Costo de
asesoría genética
Costo en pesos colombianos de brindar asesoría
genética a la pareja en riesgo por tener ambos
mutaciones del gen CFTR.
Peso colombiano
Cuantitativa continua
Promedio
Costo de prueba de secuenciación
gen CFTR
Costo en pesos colombianos de la
prueba genética de secuenciación
completa del gen CFTR por técnica Sanger.
Peso colombiano Cuantitativa continua
Promedio
Costo de tratamiento por
paciente con fibrosis quística
anual
Costo en pesos colombianos del
tratamiento anual por paciente con
diagnostico de fibrosis quística.
Peso colombiano Cuantitativa continua
Promedio

86
Anexo C. Reporte de búsqueda electrónica de estudios de costo efectividad para detección de portadores de fibrosis quística.
Reporte de búsqueda electrónica#1
Tipo de búsqueda Nueva
Base de datos Medline - NHS Economic Evaluation Database
Health Technology Assessment
Plataforma Ovid Medline ®
Fecha de búsqueda 24/02/2015
Fecha de actualización (auto alerta) Indefinida
Rango de fecha de búsqueda Medline 1946 hasta marzo 2015 NHS 1995 hasta marzo 2015 HTA 2001 hasta marzo 2015
Restricciones de lenguaje Ninguna
Otros límites Tipo de estudio: Estudios de costo efectividad
Estrategia de búsqueda (resultados) 1 exp Cystic fibrosis/ (28874)
2 cystic.tw. (84819)
3 fibrosis.tw. (123441)
4 2 and 3 (34623)
5 (cyst$ adj3 fibros$).tw. (34429)
6 (Gen adj3 CFTR adj3 mutations).tw. (0)
7 CFTR.tw. (6911)
8 (CFTR adj3 mutatio$).tw. (1652)
9 (Transmembrane adj3 conductance adj3 regulator adj3
gen$).tw. (1099)
10 transmembrane.tw. (76636)
11 conductance.tw. (50264)
12 regulator.tw. (98453)
13 gen$.tw. (4024096)
14 10 and 11 and 12 and 13 (2646)
15 exp Mucoviscidosis/ (28874)
16 Mucoviscidosis.tw. (1394)
17 (Gen adj3 CFTR).tw. (1)
18 1 or 4 or 5 or 6 or 7 or 8 or 9 or 14 or 15 or 16 or 17 (41239)
19 exp genetic testing/ (27160)
20 (genetic adj3 testing).tw. (12237)
21 genetic.tw. (582672)
22 testing.tw. (364471)
23 21 and 22 (28448)
24 exp genetic screening/ (27272)
25 (genetic adj3 screening).tw. (4980)
26 (carrier adj3 screening).tw. (860)
27 carrier.tw. (81707)

87
28 (carrier adj3 testing).tw. (543)
29 (screening adj3 before adj3 conception).tw. (4)
30 (screening adj3 conception).tw. (23)
31 conception.tw. (22640)
32 (PCR adj3 analys$).tw. (44162)
33 PCR.tw. (350363)
34 (DNA adj3 test$).tw. (10331)
35 DNA.tw. (796671)
36 (preconceptional adj3 carrier adj3 screening).tw. (18)
37 (preconceptional adj3 screening).tw. (47)
38 preconceptional.tw. (690)
39 (Preconceptional adj3 carrier adj3 testing).tw. (3)
40 (Preconceptional adj3 testing).tw. (8)
41 (cascade adj3 testing).tw. (121)
42 exp Multiplex polymerase chain reaction/ (1777)
43 (Multiplex adj3 polymerase adj3 chain adj3 reaction).tw.
(2716)
44 multiplex.tw. (23277)
45 polymerase.tw. (243968)
46 44 and 45 (4507)
47 exp Multiplex ligation dependent probe amplification/ (1777)
48 (Multiplex adj3 ligation adj3 dependent adj3 probe adj3
amplification).tw. (1739)
49 (Molecular adj3 gen$ adj3 testing).tw. (770)
50 (Molecular adj3 gen$ adj3 testing).tw. (770)
51 (Pan-ethnic adj3 panel).tw. (2)
52 pan-ethi$.tw. (0)
53 (mutation adj3 panel).tw. (125)
54 panel.tw. (85582)
55 mutation.tw. (261219)
56 54 and 55 (2427)
57 (expanded adj3 mutation adj3 panels).tw. (0)
58 (23-mutation adj3 panel).tw. (2)
59 23-muta$.tw. (206)
60 (standard adj3 panel).tw. (391)
61 (Standard adj3 23-muta$ adj3 panel).tw. (0)
62 19 or 20 or 23 or 24 or 25 or 26 or 27 or 28 or 29 or 30 or 31
or 32 or 33 or 34 or 35 or 36 or 37 or 38 or 39 or
40 or 41 or 42 or 43 or 46 or 47 or 48 or 49 or 50 or 51 or 52 or 53
or 56 or 57 or 58 or 59 or 60 or 61 (1190426)
63 cost.tw. (277966)
64 exp cost effectiveness/ (73235)
65 effectiveness.tw. (297809)
66 (cost adj3 effectiveness).tw. (51451)

88
67 exp cost of illness/ (19047)
68 (cost adj3 illness).tw. (1916)
69 63 or 64 or 65 or 66 or 67 or 68 (548985)
70 18 and 62 and 69 (226)
# de referencias identificadas 226
# de referencias sin duplicados 226
Reporte de búsqueda electrónica#2
Tipo de búsqueda
Nueva
Base de datos
▪ EMBASE
Plataforma Embase.com
Fecha de búsqueda
21/03/2015
Fecha de actualización (auto alerta)
Indefinida
Rango de fecha de búsqueda
1966 - hasta marzo 2015
Restricciones de lenguaje
Ninguna
Otros límites Tipo de estudio: Estudios de costo efectividad
Estrategia de búsqueda (resultados)
1. cystic fibrosis'/exp/ (51,095) 2. cystic:ab,ti/ (108,025) 3. fibrosis:ab,ti/ (172,471) 4. (cyst* NEAR/3 fibros*):ab,ti/ (45,425) 5. (gen NEAR/3 cftr NEAR/3 mutations):ab,ti/ (0) 6. cftr:ab,ti/ (9,959) 7. (cftr NEAR/3 mutatio*):ab,ti/ (2,688) 8. (transmembrane NEAR/3 conductance NEAR/3 regulator NEAR/3 gen*):ab,ti/ (1,301) 9. transmembrane:ab,ti/ (85,426) 10. conductance:ab,ti/ (53,563) 11. regulator:ab,ti/ (118,902) 12. gen*:ab,ti/ (4,817,232) 13. 'mucoviscidosis'/exp/ (51,095) 14. mucoviscidosis:ab,ti/ (1,537) 15. (gen NEAR/3 cftr):ab,ti/ (11) 16. #2 AND #3/ (45,818) 17. #9 AND #10 AND #11 AND #12/ (3,104) 18. #1 OR #4 OR #5 OR #6 OR #7 OR #8 OR #13 OR #14 OR #15 OR #16 OR #17/ (61,375) 19. 'genetic testing'/exp/ (49,124) 20. (genetic NEAR/3 testing):ab,ti/ (17,434) 21. genetic:ab,ti/ (692,706) 22. testing:ab,ti/ (474,611) 23. 'genetic screening'/exp/ (49,124) 24. (genetic NEAR/3 screening):ab,ti/ (6,645)

89
25. screening:ab,ti/ (455,927) 26. (carrier NEAR/3 screening):ab,ti/ (1,008) 27. carrier:ab,ti/ (93,322) 28. (carrier NEAR/3 testing):ab,ti/ (658) 29. (screening NEAR/3 before NEAR/3 conception):ab,ti/ (4) 30. (screening NEAR/3 conception):ab,ti/ (37) 31. conception:ab,ti/ (28,661) 32. (pcr NEAR/3 analys*):ab,ti/ (56,106) 33. pcr:ab,ti/ (470,614) 34. (dna NEAR/3 test*):ab,ti/ (13,025) 35. dna:ab,ti/ (896,691) 36. (preconceptional NEAR/3 carrier NEAR/3 screening):ab,ti/ (20) 37. (preconceptional NEAR/3 screening):ab,ti/ (57) 38. preconceptional:ab,ti/ (911) 39. (preconceptional NEAR/3 carrier NEAR/3 testing):ab,ti/ (3) 40. (preconceptional NEAR/3 testing):ab,ti/ (11) 41. (cascade NEAR/3 testing):ab,ti/ (174) 42. 'multiplex polymerase chain reaction'/exp/ (7410) 43. (multiplex NEAR/3 polymerase NEAR/3 chain NEAR/3 reaction):ab,ti/ (2,999) 44. multiplex:ab,ti/ (32,246) 45. polymerase:ab,ti/ (265,486) 46. 'multiplex ligation dependent probe amplification'/exp/ (3,505) 47. (multiplex NEAR/3 ligation NEAR/3 dependent NEAR/3 probe NEAR/3 amplification):ab,ti/
(2,441) 48. ligation:ab,ti/ (70,522) 49. (molecular NEAR/3 gen* NEAR/3 testing):ab,ti/ (1180) 50. ('pan ethnic' NEAR/3 panel):ab,ti/ (2) 51. (mutation NEAR/3 panel):ab,ti/ (296) 52. panel:ab,ti/ (114,462) 53. mutation:ab,ti/ (319,343) 54. (expanded NEAR/3 mutation NEAR/3 panels):ab,ti/ (0) 55. expanded:ab,ti/ (93,608) 56. ('23 mutation' NEAR/3 panel):ab,ti/ (5) 57. (standard NEAR/3 panel):ab,ti/ (612) 58. stand*:ab,ti/ (1,238,540) 59. #21 AND #22/ (39,655) 60. #21 AND #25/ (33,477) 61. #25 AND #27/ (4,748) 62. #25 AND #31/ (728) 63. #22 AND #35/ (24,901) 64. #25 AND #27 AND #38/ (38) 65. #22 AND #27 AND #38/ (17) 66. #44 AND #45/ (5070) 67. #44 AND #48/ (2,918) 68. #52 AND #53/ (4,783) 69. #52 AND #53 AND #55/ (116) 70. #52 AND #58/ (12,174) 71. #52 AND #53 AND #58/ (471) 72. cost:ab,ti/ (340,464) 73. 'cost effectiveness'/exp/ (104,081) 74. effectiveness:ab,ti/ (362,989)

90
75. (cost NEAR/3 effectiveness):ab,ti/ (52,522) 76. 'cost of illness'/exp/ (14,518) 77. (cost NEAR/3 illness):ab,ti/ (1,848) 78. #72 AND #76/ (5740) 79. #73 OR #75 OR #76 OR #77 OR #78/ (132,408) 80. #19 OR #20 OR #23 OR #24 OR #26 OR #28 OR #29 OR #30 OR #32 OR #33 OR #34
OR #36 OR #37 OR #38 OR #39 OR #40 OR #41 OR #42 OR #43 OR #46 OR #47 OR #49 OR #50 OR #51 OR #54 OR #56 OR #57 OR #59 OR #60 OR #61 OR #62 OR #63 OR #64 OR #65 OR #66 OR #67 OR #68 OR #69 OR #70 OR #71/ (599,903)
81. #18 AND #79 AND #80/ (103)
82. #18 AND #79 AND #80 AND [embase]/lim/ (90)
# de referencias identificadas
90
# de referencias sin duplicados
88
Reporte de búsqueda electrónica#3
Tipo de búsqueda Nueva
Base de datos The Cochrane Library (CLIB)
Plataforma EBM Reviews - Cochrane Database of Systematic Reviews
Fecha de búsqueda 21/03/2015
Fecha de actualización (auto alerta) Indefinida
Rango de fecha de búsqueda 1995- hasta marzo 2015
Restricciones de lenguaje Ninguna
Otros límites Estudios de costo efectividad
Estrategia de búsqueda (resultados) 1. Cystic fibrosis/exp {Including Limited Related Terms}/ (175)
2. cystic.tw./ (405) 3. fibrosis.tw./ (596) 4. (cyst$ adj3 fibros$).tw./ (300) 5. (Gen adj3 CFTR adj3 mutations).tw./ (0) 6. CFTR.tw./ (28) 7. (CFTR adj3 mutatio$).tw./ (10) 8. (Transmembrane adj3 conductance adj3 regulator adj3
gen$).tw./ (8) 9. transmembrane.tw./ (48) 10. conductance.tw./ (97) 11. regulator.tw./ (68) 12. gen$.tw./ (8175) 13. Mucoviscidosis/exp {Including Limited Related Terms}/
(175)

91
14. Mucoviscidosis.tw. (1) 15. (Gen adj3 CFTR).tw./ (0) 16. 2 and 3/ (303) 17. 9 and 10 and 11 and 12/ (19) 18. 1 or 4 or 5 or 6 or 7 or 8 or 13 or 14 or 15 or 16 or 17/
(325) 19. genetic testing/exp {Including Limited Related Terms}/
(31) 20. (genetic adj3 testing).tw./ (84) 21. genetic.tw./ (923) 22. testing.tw./ (2386) 23. genetic screening/exp {Including Limited Related Terms}/
(22) 24. (genetic adj3 screening).tw./ (17) 25. screening.tw./ (3030) 26. (carrier adj3 screening).tw./ (3) 27. carrier.tw./ (157) 28. (carrier adj3 testing).tw./ (3) 29. (screening adj3 before adj3 conception).tw./ (5) 30. (screening adj3 conception).tw/ (5) 31. conception.tw./ (489) 32. (PCR adj3 analys$).tw./ (21) 33. PCR.tw./ (171) 34. (DNA adj3 test$).tw./ (45) 35. DNA.tw./ (445) 36. (preconceptional adj3 carrier adj3 screening).tw./ (0) 37. (preconceptional adj3 screening).tw./ (1) 38. preconceptional.tw./ (5) 39. (Preconceptional adj3 carrier adj3 testing).tw./ (0) 40. (Preconceptional adj3 testing).tw./ (1) 41. (cascade adj3 testing).tw./ (1) 42. Multiplex polymerase chain reaction/exp {Including
Limited Related Terms}/ (1) 43. (Multiplex adj3 polymerase adj3 chain adj3 reaction).tw./
(2) 44. multiplex.tw./ (16) 45. polymerase.tw./ (204) 46. Multiplex ligation dependent probe amplification/exp
{Including Limited Related Terms}/ (0) 47. (Multiplex adj3 ligation adj3 dependent adj3 probe adj3
amplification).tw./ (1) 48. ligation.tw./ (145) 49. (Molecular adj3 gen$ adj3 testing).tw./ (6) 50. (Pan-ethnic adj3 panel).tw./ (0) 51. pan-ethi$.tw./ (0) 52. (mutation adj3 panel).tw./ (0) 53. panel.tw./ (642) 54. mutation.tw./ (169) 55. (expanded adj3 mutation adj3 panels).tw./ (0) 56. expanded.tw./ (944) 57. (23-mutation adj3 panel).tw./ (0)

92
58. 23-muta$.tw./ (0) 59. (standard adj3 panel).tw./ (1) 60. stand$.tw./ (7854) 61. (Standard adj3 23-muta$ adj3 panel).tw./ (0) 62. 21 and 22/ (368) 63. 21 and 25/ (403) 64. 25 and 27/ (67) 65. 25 and 31/ (179) 66. 22 and 35/ (163) 67. 25 and 27 and 38/ (1) 68. 22 and 27 and 38/ (1) 69. 44 and 45/ (7) 70. 44 and 48/ (1) 71. 52 and 53/ (0) 72. 52 and 53 and 55/ (0) 73. 52 and 58/ (0) 74. 52 and 53 and 58/ (0) 75. cost.tw./ (3647) 76. cost effectiveness/exp {Including Limited Related Terms}/
(191) 77. effectiveness.tw./ (5622) 78. (cost adj3 effectiveness).tw./ (1449) 79. cost of illness/exp {Including Limited Related Terms}/
(312) 80. (cost adj3 illness).tw./ (26) 81. 75 or 76 or 77 or 78 or 79 or 80/ (6372) 82. or/19-74/ (8253) 83. 18 and 81 and 82/ (255)
# de referencias identificadas 255
# de referencias sin duplicados 255
Reporte de búsqueda electrónica#4
Tipo de búsqueda Nueva
Base de datos LILACS
Plataforma LILACS BVS
Fecha de búsqueda 21/03/2015
Fecha de actualización (auto alerta) Indefinida
Rango de fecha de búsqueda 1982- hasta marzo 2015
Restricciones de lenguaje Ninguna
Otros límites Estudios de costo efectividad
Estrategia de búsqueda (resultados) (Cystic fibrosis OR Gen CFTR mutations OR Transmembrane
conductance regulator gene OR Mucoviscidosis OR Gen CFTR)
AND (Genetic testing OR Genetic screening OR Carrier testing
OR Screening before conception OR PCR analysis OR DNA
testing OR Preconceptional carrier screening OR Preconceptional
carrier testing OR Cascade testing OR Multiplex polymerase chain

93
reaction OR Multiplex ligation dependent probe amplification OR
Molecular genetic testing OR Pan-ethnic panel OR Mutation
panels OR Expanded mutation panels OR 23-mutation panel OR
Standard panel OR Standard 23-mutation panel) AND (cost
effectiveness OR cost of illness).
# de referencias identificadas 0

94
Anexo D. Reporte de búsqueda electrónica de estudios de prueba de secuenciación para detección de portadores de fibrosis quística.
Reporte de búsqueda electrónica#1 Tipo de búsqueda Nueva Base de datos Medline
Plataforma Ovid Medline ® Fecha de búsqueda 21/12/2015 Fecha de actualización (auto alerta) Indefinida Rango de fecha de búsqueda Medline 1946 hasta el presente
Restricciones de lenguaje Ninguna Otros límites Tipo de estudio: Estudios de pruebas diagnosticas Estrategia de búsqueda (resultados) 1 exp Cystic fibrosis/ (30868)
2 cystic.tw. (90901)
3 fibrosis.tw. (135860)
4 (cyst$ adj3 fibros$).tw. (37102)
5 (Gen adj3 CFTR adj3 mutations).tw. (0)
6 CFTR.tw. (7312)
7 (CFTR adj3 mutatio$).tw. (1758)
8 (Transmembrane adj3 conductance adj3 regulator adj3
gen$).tw. (1180)
9 transmembrane.tw. (82306)
10 conductance.tw. (53200)
11 regulator.tw. (111256)
12 gen$.tw. (4368407)
13 exp Mucoviscidosis/ (30868)
14 Mucoviscidosis.tw. (1420)
15 (Gen adj3 CFTR).tw. (1)
16 1 or 2 or 3 or 4 or 5 or 6 or 7 or 8 or 9 or 10 or 11 or 12
or 13 or 14 or 15 (4649806)
17 exp Sequence Analysis, DNA/ (180247)
18 Sequence Analysis.tw. (56747)
19 (Sequence adj3 Analysis).mp. [mp=title, abstract,
original title, name of substance word, subject heading word,
keyword heading word, protocol supplementary concept word,
rare disease supplementary concept word, unique identifier]
(263913)
20 exp Massively-Parallel Sequencing/ (9797)
21 Massively-Parallel Sequencing.tw. (1143)
22 (Massively-Parallel adj3 Sequencing).tw. (1633)
23 exp High-Throughput Nucleotide Sequencing/ (9797)
24 High-Throughput Nucleotide Sequencing.tw. (11)
25 (High-Throughput adj3 Nucleotide adj3 Sequencing).tw.

95
(16)
26 Next generation sequencing.tw. (10717)
27 (Next adj3 generation adj3 sequencing).tw. (11656)
28 exp genetic testing/ (29907)
29 (genetic adj3 testing).tw. (13565)
30 exp genetic screening/(29907)
31 (genetic adj3 screening).tw. (5339)
32 (carrier adj3 screening).tw. (909)
33 (carrier adj3 testing).tw. (571)
34 DNA testing.tw. (2109)
35 (DNA adj3 testing).tw. (3254)
36 (preconceptional adj3 carrier adj3 screening).tw. (17)
37 (Preconceptional adj3 testing).tw. (8)
38 Cascade testing.tw. (96)
39 (Cascade adj3 testing).tw. (138)
40 mutation panels.tw. (25)
41 (mutation adj3 panels).tw. (31)
42 expanded.tw. (82120)
43 40 or 42 (82144)
44 41 or 42 (82150)
45 17 or 18 or 19 or 20 or 21 or 22 or 23 or 24 or 25 or 26
or 27 or 28 or 29 or 30 or 31 or 32 or 33 or 34 or 35 or 36 or
37 or 38 or 39 or 40 or 41 or 43 or 44 (441387)
46 16 and 45 (322291)
47 exp Sensitivity/ and Specificity/ (303252)
48 Sensitivity.tw. (604781)
49 Specificity.tw. (362549)
50 exp Predictive Value of Tests/ (162386)
51 Predictive Value of Tests.tw. (50)
52 exp Diagnostic Tests/ (7961)
53 Diagnostic Tests.tw. (17393)
54 47 or 48 or 49 or 50 or 51 or 52 or 53 (1124937)
55 46 and 54 (24469)
56 limit 55 to (humans and "diagnosis (maximizes
specificity)") (5319)
# de referencias identificadas 5
# de referencias sin duplicados 5
Reporte de búsqueda electrónica#2 Tipo de búsqueda
Nueva
Base de datos ▪ Health Technology Assessment Plataforma Ovid Medline ® Fecha de búsqueda
21/12/2015

96
Fecha de actualización (auto alerta)
Indefinida
Rango de fecha de búsqueda
2001 - hasta el presente
Restricciones de lenguaje
Ninguna
Otros límites Tipo de estudio: Estudios de pruebas diagnosticas Estrategia de búsqueda (resultados)
1. exp Cystic fibrosis/ (51)
2. cystic.tw. (64)
3. fibrosis.tw. (110)
4. (cyst$ adj3 fibros$).tw. (59)
5. (Gen adj3 CFTR adj3 mutations).tw. (0)
6. CTRF.tw. (0)
7. (CTRF adj3 mutatio$).tw. (0)
8. (Transmembrane adj3 conductance adj3 regulator adj3 gen$).tw. (0)
9. transmembrane.tw. (7)
10. conductance.tw. (4)
11. regulator.tw. (4)
12. gen$.tw. (1855)
13. exp Mucoviscidosis/ (51)
14. Mucoviscidosis.tw. (0)
15. (Gen adj3 CFTR).tw. (0)
16. 1 or 2 or 3 or 4 or 5 or 6 or 7 or 8 or 9 or 10 or 11 or 12 or 13 or 14 or 15 (1952)
17. exp Sequence Analysis, DNA/ (63)
18. Sequence Analysis.tw. (37)
19. (Sequence adj3 Analysis).mp. [mp=title, text, subject heading word] (44)
20. exp Massively-Parallel Sequencing/ (0)
21. Massively-Parallel Sequencing.tw. (3)

97
22. (Massively-Parallel adj3 Sequencing).tw. (5)
23. exp High-Throughput Nucleotide Sequencing/ (0)
24. High-Throughput Nucleotide Sequencing.tw. (2)
25. (High-Throughput adj3 Nucleotide adj3 Sequencing).tw. (2)
26. Next generation sequencing.tw. (5)
27. (Next adj3 generation adj3 sequencing).tw. (6)
28. exp genetic testing/ (0)
29. (genetic adj3 testing).tw. (171)
30. exp genetic screening/ (68)
31. (genetic adj3 screening).tw. (77)
32. (carrier adj3 screening).tw. (6)
33. (carrier adj3 testing).tw. (4)
34. DNA testing.tw. (15)
35. (DNA adj3 testing).tw. (16)
36. (preconceptional adj3 carrier adj3 screening).tw. (0)
37. (Preconceptional adj3 testing).tw. (0)
38. Cascade testing.tw. (1)
39. (Cascade adj3 testing).tw. (1)
40. mutation panels.tw. (0)
41. (mutation adj3 panels).tw. (0)
42. expanded.tw.(25)
43. 40 or 42 (25) 44. 41 or 42 (25) 45. 17 or 18 or 19 or 20 or 21 or 22 or 23 or 24 or 25 or 26 or 27 or 28 or 29 or 30 or 31 or 32
or 33 or 34 or 35 or 36 or 37 or 38 or 39 or 40 or 41 or 43 or 44 (335)
46. 16 and 45 (292) 47. exp Sensitivity/ and "Specificity"/ (94)
48. Sensitivity.tw. (372)

98
49. Specificity.tw. (251)
50. exp Predictive Value of Tests/ (163)
51. Predictive Value of Tests.tw. (163)
52. exp Diagnostic Tests/ (36)
53. Diagnostic Tests.tw. (102)
54. 47 or 48 or 49 or 50 or 51 or 52 or 53 (604)
55. 46 and 54 (23) 56. limit 55 to humans (17)
# de referencias identificadas
1
# de referencias sin duplicados
1
Reporte de búsqueda electrónica#3 Tipo de búsqueda Nueva Base de datos Cochrane
Plataforma OVID-EBM Reviews
Fecha de búsqueda 21/12/2015 Fecha de actualización (auto alerta) Indefinida Rango de fecha de búsqueda 1995- hasta el presente Restricciones de lenguaje Ninguna Otros límites Estudios de pruebas diagnosticas. Estrategia de búsqueda (resultados) 1. exp Cystic fibrosis/ (919)
2. cystic.tw.(3300)
3. fibrosis.tw. (5515)
4. (cyst$ adj3 fibros$).tw. (2951)
5. (Gen adj3 CFTR adj3 mutations).tw. (0)
6. CTRF.tw. (0)
7. (CTRF adj3 mutatio$).tw. (0)

99
8. (Transmembrane adj3 conductance adj3 regulator adj3
gen$).tw. (15)
9. transmembrane.tw. (202)
10. conductance.tw. (1295)
11. regulator.tw. (551)
12. gen$.tw. (96907)
13. exp Cystic Fibrosis/ (919)
14. Mucoviscidosis.tw. (31)
15. (Gen adj3 CFTR).tw. (0)
16. 1 or 2 or 3 or 4 or 5 or 6 or 7 or 8 or 9 or 10 or 11 or 12 or
13 or 14 or 15 (103506)
17. exp Sequence Analysis, DNA/ (282)
18. Sequence Analysis.tw. (96)
19. (Sequence adj3 Analysis).mp. [mp=title, original title,
abstract, mesh headings, heading words, keyword] (558)
20. exp Massively-Parallel Sequencing/ (7)
21. Massively-Parallel Sequencing.tw. (5) 22. (Massively-Parallel adj3 Sequencing).tw. (6) 23. exp High-Throughput Nucleotide Sequencing/ (7) 24. High-Throughput Nucleotide Sequencing.tw. (0)
25. (High-Throughput adj3 Nucleotide adj3 Sequencing).tw.(0)
26. Next generation sequencing.tw. (51) 27. (Next adj3 generation adj3 sequencing).tw. (52) 28. exp genetic testing/ (292) 29. (genetic adj3 testing).tw. (219) 30. exp genetic screening/ (292)
31. (genetic adj3 screening).tw. (92)
32. (carrier adj3 screening).tw. (14) 33. (carrier adj3 testing).tw. (11) 34. DNA testing.tw. (87)
35. (DNA adj3 testing).tw. (114) 36. (preconceptional adj3 carrier adj3 screening).tw. (1) 37. (Preconceptional adj3 testing).tw. (0)

100
38. Cascade testing.tw. (0)
39. (Cascade adj3 testing).tw. (1) 40. mutation panels.tw. (0) 41. (mutation adj3 panels).tw. (0) 42. expanded.tw. (2006)
43. 40 or 42 (2006) 44. 41 or 42 (2006) 45. 17 or 18 or 19 or 20 or 21 or 22 or 23 or 24 or 25 or 26 or
27 or 28 or 29 or 30 or 31 or 32 or 33 or 34 or 35 or 36 or
37 or 38 or 39 or 40 or 41 or 43 or 44 (3316) 46. 16 and 45 (1377) 47. exp "Sensitivity and Specificity"/ and "Specificity"/ (7619) 48. Sensitivity.tw. (22392) 49. Specificity.tw. (6762) 50. exp Predictive Value of Tests/ (5319)
51. Predictive Value of Tests.tw. (8) 52. exp Diagnostic Tests, Routine/ (137)
53. Diagnostic Tests.tw. (369)
54. 47 or 48 or 49 or 50 or 51 or 52 or 53 (32880)
55. 46 and 54 (149)
# de referencias identificadas 0
# de referencias sin duplicados 0
Reporte de búsqueda electrónica#4 Tipo de búsqueda Nueva Base de datos NHS Economic Evaluation Database
Plataforma Ovid Medline ®
Fecha de búsqueda 21/12/2015 Fecha de actualización (auto alerta) Indefinida Rango de fecha de búsqueda 1995- hasta el presente Restricciones de lenguaje Ninguna Otros límites Estudios de pruebas diagnosticas. Estrategia de búsqueda (resultados) 1. exp Cystic fibrosis/ (36)
2. cystic.tw. (78)
3. fibrosis.tw. (105)
4. (cyst$ adj3 fibros$).tw. (61)
5. (Gen adj3 CFTR adj3 mutations).tw. (0)

101
6. CTRF.tw. (0)
7. (CTRF adj3 mutatio$).tw. (0)
8. (Transmembrane adj3 conductance adj3
regulator adj3 gen$).tw. (0)
9. transmembrane.tw. (2)
10. conductance.tw. (2)
11. regulator.tw. (2)
12. gen$.tw. (8526)
13. exp Mucoviscidosis/ (36)
14. Mucoviscidosis.tw. (0)
15. (Gen adj3 CFTR).tw. (0)
16. 1 or 2 or 3 or 4 or 5 or 6 or 7 or 8 or 9 or 10
or 11 or 12 or 13 or 14 or 15 (8557)
17. exp Sequence Analysis, DNA/ (19)
18. Sequence Analysis.tw. (11)
19. (Sequence adj3 Analysis).mp. [mp=title,
text, subject heading word] (14)
20. exp Massively-Parallel Sequencing/ (0)
21. Massively-Parallel Sequencing.tw. (0)
22. (Massively-Parallel adj3 Sequencing).tw. (0)
23. exp High-Throughput Nucleotide
Sequencing/(0)
24. High-Throughput Nucleotide
Sequencing.tw.(0)
25. (High-Throughput adj3 Nucleotide adj3
Sequencing).tw. (0)
26. Next generation sequencing.tw.(0)
27. (Next adj3 generation adj3 sequencing).tw.
(1)
28. exp genetic testing/ (0)

102
29. (genetic adj3 testing).tw.(121)
30. exp genetic screening/ (44)
31. (genetic adj3 screening).tw. (63)
32. (carrier adj3 screening).tw. (11)
33. (carrier adj3 testing).tw. (4)
34. DNA testing.tw. (48)
35. (DNA adj3 testing).tw.(53)
36. (preconceptional adj3 carrier adj3
screening).tw.(1)
37. (Preconceptional adj3 testing).tw.(0)
38. Cascade testing.tw. (1)
39. (Cascade adj3 testing).tw. (2)
40. mutation panels.tw. (0)
41. (mutation adj3 panels).tw.(0)
42. expanded.tw.(112)
43. 40 or 42 (112)
44. 41 or 42 (112) 45. 17 or 18 or 19 or 20 or 21 or 22 or 23 or 24
or 25 or 26 or 27 or 28 or 29 or 30 or 31 or
32 or 33 or 34 or 35 or 36 or 37 or 38 or 39
or 40 or 41 or 43 or 44 (324)
46. 16 and 45 (286) 47. exp Sensitivity/ and "Specificity"/(1427)
48. Sensitivity.tw.(9389)
49. Specificity.tw.(1990)
50. exp Predictive Value of Tests/ (275)
51. Predictive Value of Tests.tw.(275)
52. exp Diagnostic Tests/(79)
53. Diagnostic Tests.tw.(615)
54. 47 or 48 or 49 or 50 or 51 or 52 or 53 (9502)
55. 46 and 54 (239)

103
56. limit 55 to humans (234)
# de referencias identificadas 0
# de referencias sin duplicados 0
Reporte de búsqueda electrónica#5 Tipo de búsqueda Nueva Base de datos LILACS
Plataforma LILACS BVS
Fecha de búsqueda 21/12/2015 Fecha de actualización (auto alerta) Indefinida Rango de fecha de búsqueda 1982- hasta el presente Restricciones de lenguaje Ninguna Otros límites Estudios de pruebas diagnosticas Estrategia de búsqueda (resultados) • (Cystic fibrosis OR Gen CFTR mutations OR
Transmembrane conductance regulator gene OR Mucoviscidosis OR Gen CFTR) AND (Sequence Analysis, DNA OR Massively-Parallel Sequencing OR High-Throughput Nucleotide Sequencing OR Next generation sequencing OR Genetic testing OR Carrier screening OR Carrier testing OR DNA testing OR Preconceptional carrier testing OR Cascade testing OR Mutation panels OR Expanded mutation panels) AND
(Sensitivity and Specificity OR Diagnostic Tests
OR Predictive Value of Tests).
# de referencias identificadas 0
Reporte de búsqueda electrónica#6
Tipo de búsqueda Nueva
Base de datos ▪ EMBASE
Plataforma Embase.com
Fecha de búsqueda 21/12/2015
Fecha de actualización (auto alerta)
Indefinida
Rango de fecha de búsqueda
1966 - hasta el presente
Restricciones de lenguaje
Ninguna
Otros límites Tipo de estudio: Estudios de pruebas diagnosticas
Estrategia de búsqueda (resultados)
83. 'cystic fibrosis'/exp OR 'cystic fibrosis' (66821) 84. cystic:ab,ti (116667) 85. fibrosis:ab,ti (191207)

104
86. (cyst* NEAR/3 fibros*):ab,ti (49690) 87. (gen NEAR/3 cftr NEAR/3 mutations):ab,ti (0) 88. ctrf:ab,ti (13) 89. (ctrf NEAR/3 mutatio*):ab,ti (0) 90. (transmembrane NEAR/3 conductance NEAR/3 regulator
NEAR/3 gen*):ab,ti (1383) 91. transmembrane:ab,ti (89727) 92. conductance:ab,ti (56104) 93. regulator:ab,ti (129476) 94. gen*:ab,ti (5,160,927) 95. 'mucoviscidosis'/exp (55703) 96. mucoviscidosis:ab,ti (1572) 97. (gen NEAR/3 cftr):ab,ti (12) 98. #1 OR #2 OR #3 OR #4 OR #5 OR #6 OR #7 OR #8 OR #9
OR #10 OR #11 OR #12 OR #13 OR #14 OR #15 (5508893) 99. 'sequence analysis dna'/exp (159700) 100. sequence AND analysis:ab,ti (397261) 101. 'massively-parallel sequencing' (1766) 102. 'massively parallel' AND sequencing:ab,ti (2622) 103. ('massively parallel' NEAR/3 sequencing):ab,ti (2291) 104. 'high-throughput nucleotide sequencing'/exp (8728) 105. 'high throughput' AND nucleotide AND sequencing:ab,ti
(4072) 106. ('high
throughput' NEAR/3 nucleotide NEAR/3 sequencing):ab,ti (17)
107. next AND generation AND sequencing:ab,ti (18939) 108. (next NEAR/3 generation NEAR/3 sequencing):ab,ti
(16855) 109. 'genetic testing'/exp (54005) 110. 'genetic screening'/exp (54005) 111. (genetic NEAR/3 screening):ab,ti (7400) 112. (carrier NEAR/3 screening):ab,ti (1092) 113. (carrier NEAR/3 testing):ab,ti (696) 114. dna AND testing:ab,ti (38761) 115. (dna NEAR/3 testing):ab,ti (4383) 116. (preconceptional NEAR/3 carrier NEAR/3 screening):ab,ti
(21) 117. (preconceptional NEAR/3 testing):ab,ti (12) 118. cascade AND testing:ab,ti (1502) 119. (cascade NEAR/3 testing):ab,ti (213) 120. mutation AND panels:ab,ti (1174) 121. (mutation NEAR/3 panels):ab,ti (59) 122. expanded:ab,ti (101737) 123. #38 AND #40 (58) 124. #39 AND #40 (3) 125. #17 OR #18 OR #19 OR #20 OR #21 OR #22 OR #23
OR #24 OR #25 OR #26 OR #27 OR #28 OR #29 OR #30 OR #31 OR #32 OR #33 OR #34 OR #35 OR #36 OR #37 OR #38 OR #39 OR #41 OR #42 (596182)
126. #16 AND #43 (444088)

105
127. 'sensitivity' AND 'specificity' (343408) 128. sensitivity:ab,ti (731196) 129. specificity:ab,ti(433614) 130. 'predictive value of tests'/exp (81739) 131. predictive AND value AND of AND tests:ab,ti (16131) 132. 'diagnostic tests' (22952) 133. #45 OR #46 OR #47 OR #48 OR #49 OR #50 (1125144) 134. #44 AND #51 (33066)
135. #44 AND #51 AND [humans]/lim (16613) # de referencias identificadas
2
# de referencias sin duplicados
2
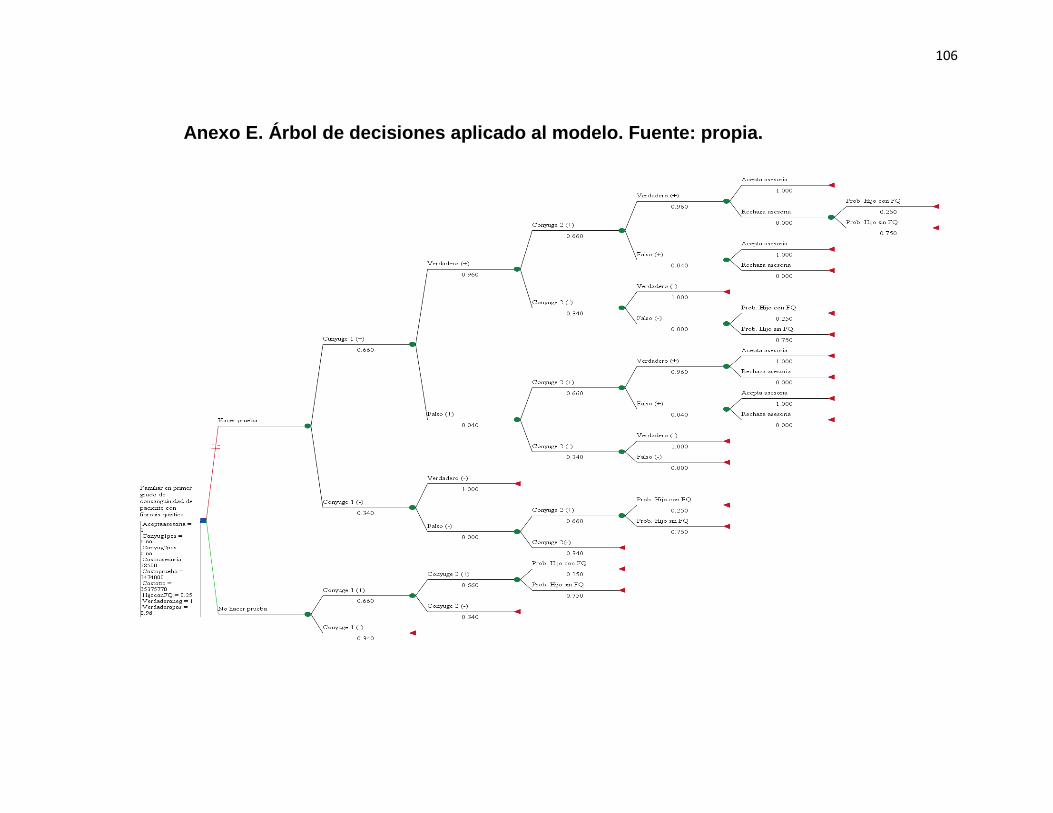
106
Anexo E. Árbol de decisiones aplicado al modelo. Fuente: propia.

107
Bibliografía
1. Silva Marín MS, Determinación de la frecuencia de portadores de la mutación
DF508 en la población colombiana (Tesis de maestría en Genética Humana).
Universidad del Rosario; 2007.
2. Orozco L, Chávez M, Saldaña Y, Velázquez R, et al. Fibrosis quística: la frontera
del conocimiento molecular y sus aplicaciones clínicas. Rev. Invest Clin 2006; 58
(2): 139-152.
3. Jay M. L, Mateus H, Fonseca D, Restrepo M. C, Keyeux G. PCR-
heteroduplex por agrupamiento: implementación de un método de identificación
de portadores de la mutación más común causal de Fibrosis quística en
Colombia. Colomb Med 2006; 37: 176-182.
4. Dugueperoux I, Pierre M, et al. Cascade testing in families of carriers identified
through newborn screening in Western Brittany (France). Journal of Cystic
Fibrosis 2013; 12: 338 - 344.
5. National Institutes of Health Consensus Development Conference Statement on
Genetic Testing for Cystic Fibrosis. Genetic Testing for Cystic Fibrosis Arch intern
med 1999; 159: 1529-1539.
6. Ministerio de Salud y Protección Social. Metodología de ponderación de criterios
para seleccionar las tecnologías en salud a evaluar, 2014. Bogota, Colombia.

108
7. Andersen DH. Cystic fibrosis of the pancreas and its relation to celiac disease: A
clinical and pathological study. Am J Dis Child 1938; 56: 344-349.
8. Fanconi G, Vehlinger E, Knauer C. Das Coeliakie-Syndrom bei angeborenem
zystischem Pancreas fibromatose und Bronchiektasien. Wien Med Wschr 1936;
86: 753-756.
9. Tsui LC, Buchwald M, Barker D, Braman JC, Knowlton R, Schrumm JW, et al.
Cystic fibrosis locus defined by a genetically linked polymorphic DNA marker.
Science 1985; 23: 1054-1057.
10. Ortigosa L. Fibrosis quística. Aspectos diagnósticos. Colomb Med 2007; 38 (Supl
1): 41-49.
11. CFTR1, Cystic Fibrosis Mutation Database; 1989 (Actualizado 2010). Revisado
en mayo 2016. Disponible en:
http://www.genet.sickkids.on.ca/cftr/StatisticsPage.html.
12. The molecular genetic epidemiology of cystic fibrosis. World Health Organization
(WHO). 1983.
13. Keyeux G, Sánchez D, Garavito P, Stand I, Rodas C, Bienvenu T, et al. Estudios
moleculares en pacientes colombianos con fibrosis quística. Acta Médica
Colombiana 1997; 22(4): 167-173.
14. Update on carrier screening for cystic fibrosis. Committee Opinion No 486.
American College of Obstetricians and Gynecologists. Obstet Gynecol 2011; 117:
1028 - 1031.
15. Keyeux G, Rodas C, Bienvenu T, Garavito P, Vidaud D, Sanchez D, et al. CFTR
mutations in patients from Colombia: implications for local and regional molecular
diagnosis programs. Hum Mutat 2003; 644: 1-7.

109
16. Restrepo CM, Pineda L, Rojas-Martínez A, Gutiérrez CA, Morales A, Gómez Y, et
al. CFTR mutations in three Latin American countries. Am J Med Genet 2000;
91: 277–279.
17. Vásquez C, Aristizával R, Daza W. Fibrosis Quística en Colombia. Rev Chil
Neumol Ped 2010; 5: 44-50.
18. Pérez M, Luna M, Pivetta H. O, Keyeux G. CFTR gene analysis in Latin American
CF patients: Heterogeneous origin and distribution of mutations across the
continent. Journal of Cystic Fibrosis 2007; 6: 194-208.
19. Mateus H, Amado P. Tamizaje neonatal para fibrosis quística en una muestra de
la ciudad de Bogotá. Bogotá: Universidad del Rosario; 2011.
20. Schwlebert E, Egan M, Hwang T, Fulmer S, Allen S, Cutting G, et al. CFTR
Regulates Outtwardly Rectifying Chloride Channels through an Autocrine
mechanism Involving ATP. Cell 1995; 81: 1063-73.
21. Ministerio de Salud y Protección Social, Colciencias. Grupo de trabajo Guía de
práctica clínica para la prevención, diagnostico, tratamiento y rehabilitación de
fibrosis quística. Bogotá, Colombia 2014.
22. Gregg A, Simpson J. L. Genetic screening for cystic fibrosis. Obstet Gynecol Clin
N Am 2002; 29: 329– 340.
23. Goetzinger R. K, Cahill G. A. An Update on Cystic Fibrosis Screening. Clin Lab
Med 2010; 30: 533-543.
24. Culling B, Ogle R. Genetic Counselling Issues in Cystic Fibrosis. Paediatric
Respiratory Reviews 2010; 11: 75-79.
25. Doyle M. N, Gardner O. M. Prenatal cystic fibrosis screening in Mexican
Americans: An economic analysis. Am J Obstet Gynecol 2003; 189: 769-774.

110
26. Update on carrier screening for cystic fibrosis. Committee Opinion No 325.
American College of Obstetricians and Gynecologists. Obstet Gynecol 2005; 106:
1465 - 1468.
27. Dequeker E, Stuhrmann M, Morris M, Casals T, Castellani C, Claustres M, et al
Best practice guidelines for molecular genetic diagnosis of cystic fibrosis and
CFTR-related disorders – updated European recommendations, European Journal
of Human Genetics 2009; 17: 51–65.
28. Watson S. M, Cutting R. G, Desnick J. R, Driscoll A. D, Klinger K, Mennut M et al.
Cystic fibrosis population carrier screening: 2004 revision of American College of
Medical Genetics mutation panel. Genetics IN Medicine 2004; 6 (5).
29. Monaghan G. K, Bluhm D, Phillips M, Feldman L. G. Preconception and prenatal
cystic fibrosis carrier screening of African Americans reveals unanticipated
frequencies for specific mutations. Genet Med 2004: 6 (3):141-144.
30. Necochea C. R, Canul T. J. Secuenciación de ácidos nucleicos. Proyecto de
investigación. Instituto de Biotecnología. UNAM. Cuernavaca; 2004.
31. Grody W, Cutting R. G, Klinger W. K, Richards S. C, Watson S. M, Desnick J. R.
Laboratory standards and guidelines for population based cystic fibrosis carrier
screening. ACMG statement 2001; 3.
32. Massie J, Forbes R, duSart D, Bankier A, Delatycki B. M. Community-wide
screening for cystic fibrosis carriers could replace newborn screening for the
diagnosis of cystic fibrosis. Journal of Paediatrics and Child Health 2007; 43: 721-
723.
33. Massie J, Petrou V, Forbes R, Curnow L, Ioannou L, Dusart D. Population-based
carrier screening for cystic fibrosis in Victoria: The first three years experience.
Australian and New Zealand Journal of Obstetrics and Gynaecology 2009; 49:
484-489.

111
34. Christie M. L, Ingrey J. A, Turner M. G, Proos L. A, Watts E. G. Outcomes of a
cystic fibrosis carrier testing clinic for couples. MJA 2009; 191: 499-501.
35. Picci L, Cameran M, Marangon O, Marzenta D, Ferrari S, Frigo C. A, et al. A 10-
year large-scale cystic fibrosis carrier screening in the Italian population. Journal
of Cystic Fibrosis 2010; 9: 29-35.
36. Rohlfs M. E, Zhou Z, Heim A. R, Nagan N, Rosenblum S. L, Flynn K. Cystic
Fibrosis Carrier Testing in an Ethnically Diverse US Population. Clinical Chemistry
2011; 57 (6): 841-848.
37. Duguépéroux I, Audrézet P. M, Parent P, Bellanger A. S, Roussey M, Férec C.
Cascade testing in families of carriers identified through newborn screening in
Western Brittany (France). Journal of Cystic Fibrosis 2013; 12: 338-344.
38. Chandrasekharan S, Heaney C, James T, Conover C, Cook-Deegan R. Impact of
Gene Patents and Licensing Practices on Access to Genetic Testing for Cystic
Fibrosis: Patents and Licensing for Cystic Fibrosis Testing. Genet Med 2010;
12(4): 194-211.
39. Whiting P, Rutjes A, Westwood M, Mallett S, et al. QUADAS-2 Group. QUADAS-2:
A Revised Tool for the Quality Assessment of Diagnostic Accuracy Studies.
Annals of Internal Medicine 2011; 155 (8).
40. Chiou C, Hay J, Wallace J, et al. Development and Validation of a Grading System
for the Quality of Cost-Effectiveness Studies. MEDICAL CARE 2003; 41 (1): 32–
44.
41. Ministerio de la Protección Social, Colciencias, Centro de Estudios e Investigación
en Salud de la Fundación Santa Fe de Bogotá, Escuela de Salud Pública de la
Universidad de Harvard. Guía Metodológica para el desarrollo de Guías de
Atención Integral en el Sistema General de Seguridad Social en Salud
Colombiano. Bogotá, Colombia 2010.

112
42. Metodología para la actualización integral del plan obligatorio de salud del sistema
general de seguridad social en salud. Bogotá, Colombia 2011.
43. Ministerio de Salud. Resolución 8430 de 1993. Bogotá, Colombia.
44. Lieu TA, Watson SE, Washington AE. The cost-effectiveness of prenatal carrier
screening for cystic fibrosis. Obstet Gynecol 1994; 84: 903-912.
45. Asch D, Hershey J, DeKay M, Pauly M, Patton J, Jedrziewski K, et al. Carrier
screening for cystic fibrosis: costs and clinical outcomes. Med Decis Making 1998;
18: 202-212.
46. Morris JK, Oppenheimer PM. Cost comparison of different methods of screening
for cystic fibrosis. J Med Screen 1995; 2: 22-27.
47. Cuckle HS, Richardson GA, Sheldon TA, Quirke P. Cost effectiveness of antenatal
screening for cystic fibrosis. BMJ 1995; 311: 1460-1464.
48. Van der Riet A, Van Hout B, Rutten F. Cost effectiveness of DNA diagnosis for
four monogenic diseases. J Med Genet 1997; 34: 741-745.
49. Wildhagen MF, Hilderink HB, Verzijl JG, Verheij JB, Kooij L, Tijmstra T, et al.
Costs, effects, and savings of screening for cystic fibrosis gene carriers. J
Epidemiol Community Health 1998; 52: 459–467.
50. Rowley PT, Loader S, Kaplan RM. Prenatal screening for cystic fibrosis carriers:
an economic evaluation. Am J Hum Genet 1998; 63: 1160–1174.
51. Doyle M. N, Gardner O. M. Prenatal cystic fibrosis screening in Mexican
Americans: An economic analysis. Am J Obstet Gynecol 2003; 189: 769-774.

113
52. Warren E, Anderson R, Proos AL, Burnett LB, Barlow-Stewart K, Hall J. Cost-
effectiveness of a school-based tay-sachs and cystic fibrosis genetic carrier
screening program. Genet Med 2005; 7: 484- 494.
53. Weijers-Poppelaars FA, Wildhagen MF, Henneman L, Cornel MC, Kate LP.
Preconception cystic fibrosis carrier screening: costs and consequences. Genet
Test 2005; 9:158–166.
54. Wei S, Quigg M. H, Monaghan K. G. Is Cystic Fibrosis Carrier Screening Cost
Effective?. Community Genet 2007; 10: 103–109.
55. Maxwell S, Brameld K, Youngs L, Geelhoed E, O’leary P. Informing policy for the
Australian context – Costs, outcomes and cost savings of prenatal carrier
screening for cystic fibrosis. Australian and New Zealand Journal of Obstetrics and
Gynaecology 2010; 50: 51–59.
56. Norman R, Van Gool K, Hall J, Delatycki M, Massie J. Cost-effectiveness of carrier
screening for cystic fibrosis in Australia. Journal of Cystic Fibrosis 2012; 11: 281-
287.
57. Radhakrishnan M, van Gool K, Hall J, Delatycki M, Massie J. Economic evaluation
of cystic fibrosis screening: a review of the literature. Health Policy 2008; 85(2):
133-147.
58. Ministerio de Salud. Resolución 3681 de 2013. Bogotá, Colombia.
59. Schrijver I, Aziz N, Jennings J. L, Richards S. C, Voelkerding V. K, Weckzzxx E K.
Methods-Based Proficiency Testing in Molecular Genetic Pathology. The Journal
of Molecular Diagnostics 2014; 16: 283-287.
60. Palomaki E. G, Bradley A. L, Richards S. C, Haddow E. J. Analytic validity of
cystic fibrosis testing: A preliminary estimate.Genet Med 2003; 5(1): 15-20.

114
61. Roberts T, Schwarz J. M, Kerr-Liddell R, Hinks L. J, Super M. Cascade carrier-
testing in cystic fibrosis. Paediatric Respiratory reviews 2003; 4: 293-298.
62. Bombieri C, Pignatti F. P. CF mutation testing in Italy. Genetic Testing 2001; 5:
229-233.
63. Palomaki E. G, FitzSimmons C. S, Haddow E. J. Clinical sensitivity of prenatal
screening for cystic fibrosis via CFTR carrier testing in a United States panethnic
population. Genet Med 2004; 6(5): 405-414.
64. Montgomery J, Wittwer T. C, Kent O. J, Zhou L. Scanning the Cystic Fibrosis
Transmembrane Conductance Regulator Gene Using High-Resolution DNA
Melting Analysis. Clinical Chemistry 2007; 53(11): 1891-1898.
65. Coiana A, Faa V, Carta D, Puddu R, Cao A, Rosatelli C. M. Preconceptional
identification of cystic fibrosis carriers in the Sardinian population: A pilot
screening program. Journal of Cystic Fibrosis 2011; 10: 207-211.
66. Strom M. C, Huang D, Chen C, Buller A, Peng M, Quan F, et al. Extensive
sequencing of the cystic fibrosis transmembrane regulator gene: Assay validation
and unexpected benefits of developing a comprehensive test. Genet Med 2003;
5(1): 9-14.
67. Pratt M. V, Caggana M, Bridges C, Buller M. A, DiAntonio L, Highsmith E. W.
Development of Genomic Reference Materials for Cystic Fibrosis Genetic Testing.
J Mol Diagn 2009; 11: 186-193.
68. Lyon E, Schrijver I, Weck E. K, Gonzalez F. A, Richards S, Palomaki E. G.
Molecular genetic testing for cystic fibrosis: laboratory performance on the College
of American Pathologists external proficiency surveys. Genetics in medicine 2015;
17(3): 219-225.

115
69. Banco de la Republica de Colombia. Dirección de Síntesis y Cuentas Nacionales,
estudios económicos - Cuentas financieras. Bogotá, Colombia. Metodología año
base 2005. Disponible en: http://www.banrep.gov.co/es/pib.
70. Watson E. K, Maynall E. S, Lamb J, Chapple J, Williamson R. Psychological and
social consequences of community carrier screening programme for cystic
fibrosis. Lancet 1992; 340: 217-220.
71. Callanan P. N, Cheuvront J. B, Sorenson R. J. CF Carrier testing in a high risk
population: Anxiety, risk perceptions, and reproductive plans of carrier by "non-
carrier'' couples. Genetics in Medicine 1999; 1(7): 323-327.
72. Lafayette D, Abuelo D, Passero A. M, Tantravahi U. Attitudes Toward Cystic
Fibrosis Carrier and Prenatal Testing and Utilization of Carrier Testing Among
Relatives of Individuals with Cystic Fibrosis. Journal of Genetic Counseling 1999;
8(1): 17-36.
73. Braekeleer D. M, Bellis G, Rault G, Allard C, Milot M, Simard F. Reproductive
attitudes of couples having a child with cystic fibrosis in Saguenay–Lac-Saint-Jean
(Quebec, Canada). Annales de Génétique 2000; 43: 93-97.
74. Henneman L, Kooij L, Bouman K, Kate P. L. Personal Experiences of Cystic
Fibrosis (CF) Carrier Couples Prospectively Identified in CF Families. American
Journal of Medical Genetics 2002; 110: 324-331.
75. Ioannou L, McClaren J. B, Massie J, Lewis S, Metcalfe A. S, Forrest L, et al.
Population-based carrier screening for cystic fibrosis: a systematic review of 23
years of research. Genetics in medicine 2014; 16(3): 207-216.

116
76. Ioannou L, Delatycki B. M, Massie J, Hodgson J, Lewis S. “Suddenly Having two
Positive People who are Carriers is a Whole New Thing”- Experiences of Couples
Both Identified as Carriers of Cystic Fibrosis Through a Population-Based Carrier
Screening Program in Australia. J Genet Counsel 2015; 24: 987-1000.
77. Duguépéroux I, L'Hostis C, Audrézet MP, Rault G, Frachon I, Bernard R, et al.
Highlighting the impact of cascade carrier testing in cystic fibrosis families. Journal
of Cystic Fibrosis 2016; 15: 452-459.





























