Embed Size (px)
Citation preview

REVIEW
IgG glycosylation analysis
Carolin Huhn, Maurice H. J. Selman, L. Renee Ruhaak, André M. Deelderand Manfred Wuhrer
Biomolecular Mass Spectrometry Unit, Department of Parasitology,Leiden University Medical Center, Leiden, The Netherlands
A multitude of monoclonal IgG antibodies directed against a variety of therapeutic targets iscurrently being developed and produced by biotechnological companies. The biological activity ofIgGs is modulated by the N-glycans attached to the fragment crystallizable (Fc) part. For example,lack of core-fucoses on these N-glycans may lead to a drastic enhancement of antibody-mediatedcellular cytotoxicity. Moreover, sialylation of Fc N-glycans determines the immunosuppressiveproperties of polyclonal IgG from human blood, which stimulates research into Fc glycosylationof human plasma IgG in various disease settings. This review presents and evaluates the differ-ent approaches which are used for IgG glycosylation analysis: N-glycans may be enzymatically orchemically released from purified IgG, prior to chromatographic or mass spectrometric analysis.Moreover, IgGs may be treated with endoproteinases such as trypsin, followed by glycosylationanalysis at the glycopeptide level, which is generally accomplished by HPLC with ESI-MS.Alternatively, intact IgGs or fragments thereof obtained by enzymatic cleavages in the hingeregion and by reduction may be analyzed by a large number of analytical techniques, includingMS and chromatography or CE.
Received: September 6, 2008Revised: October 13, 2008
Accepted: October 13, 2008
Keywords:
Antibody / Glycopeptides / Glycoprotein / Mass spectrometry / Post-translationalmodification
882 Proteomics 2009, 9, 882–913
1 Introduction
Two major application fields for IgG glycosylation analysishave evolved: IgGs in the human circulation as part of thehumoral immune response, and biotechnologically pro-duced mAb for therapeutic purposes. IgGs are present inhigh abundance in the human circulation (concentrationapproximately 10 mg/mL in human plasma) and occur infour different subclasses (IgG1, IgG2, IgG3, and IgG4). Nextto the enormous variety in the sequence of the antigen-binding region of plasma IgG, human IgGs are known sincemore than two decades to vary in the N-glycosylation at thehighly conserved N-glycosylation site of the fragment crys-tallizable (Fc) part. As a general pattern, IgG Fc N-glycans arepredominantly biantennary complex-type structures, most ofthem being core-fucosylated (Fig. 1A). Some of the glycanscarry a bisecting N-acetylglucosamine, and a small portionmay carry a sialic acid residue on the antennae. Notably, thelargest variation has been reported regarding galactosylation:a varying portion of the IgG Fc glycoforms is truncated. The
Correspondence: Dr. Manfred Wuhrer, Biomolecular Mass Spec-trometry Unit, Department of Parasitology, Leiden UniversityMedical Center, P.O. Box 9600, 2300 RC Leiden, The NetherlandsE-mail: [email protected]: 131-71-526-6907
Abbreviations: 2-AA, 2-aminobenzoic acid; 2-AB, 2-aminobenz-amide; 3-AA, 3-Aminobenzoic acid; ADCC, antibody-dependentcellular cytotoxicity; APTS, 8-aminopyrene-1,3,6-trisulfonate;CIEX, cation exchange chromatography; Endo H, endoglycosi-dase H; FA, formic acid; Fab, fragment antigen binding; Fc, frag-ment cristallizable; Fuc, fucose; G0, biantennary N-glycan with-out galactose residues; G1, biantennary N-glycan with one galac-tose; G2, biantennary N-glycan with two galactoses; Glu-C,
endoproteinase Glu-C and V8 protease; GlcNAc, N-acetyl gluco-samine; Hex, hexose; HexNAc, N-acetylhexosamine; HILIC,
hydrophilic interaction LC; IVIG, intravenous Ig; LIF, laser-induced fluorescence; Lys-C, lysylendopeptidase; PA, 2-amino-pyridine; PGC, porous graphitic carbon; PNGase A, glycoami-dase A; PNGase F, glycoamidase F; RA, rheumatoid arthritis;SLE, systemic lupus erythematosus; ZIC-HILIC, zwitterionic-typehydrophilic interaction chromatography
DOI 10.1002/pmic.200800715
© 2009 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com
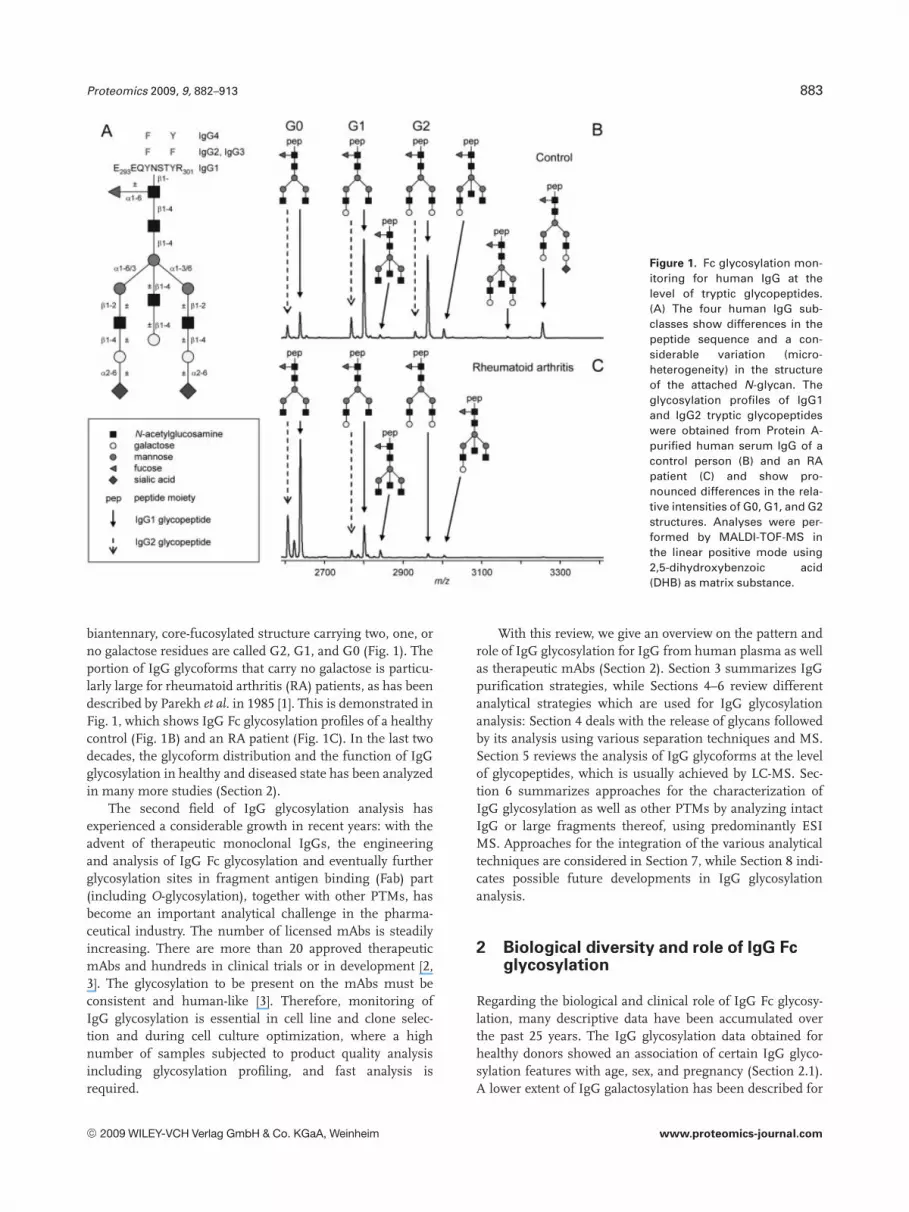
Proteomics 2009, 9, 882–913 883
Figure 1. Fc glycosylation mon-itoring for human IgG at thelevel of tryptic glycopeptides.(A) The four human IgG sub-classes show differences in thepeptide sequence and a con-siderable variation (micro-heterogeneity) in the structureof the attached N-glycan. Theglycosylation profiles of IgG1and IgG2 tryptic glycopeptideswere obtained from Protein A-purified human serum IgG of acontrol person (B) and an RApatient (C) and show pro-nounced differences in the rela-tive intensities of G0, G1, and G2structures. Analyses were per-formed by MALDI-TOF-MS inthe linear positive mode using2,5-dihydroxybenzoic acid(DHB) as matrix substance.
biantennary, core-fucosylated structure carrying two, one, orno galactose residues are called G2, G1, and G0 (Fig. 1). Theportion of IgG glycoforms that carry no galactose is particu-larly large for rheumatoid arthritis (RA) patients, as has beendescribed by Parekh et al. in 1985 [1]. This is demonstrated inFig. 1, which shows IgG Fc glycosylation profiles of a healthycontrol (Fig. 1B) and an RA patient (Fig. 1C). In the last twodecades, the glycoform distribution and the function of IgGglycosylation in healthy and diseased state has been analyzedin many more studies (Section 2).
The second field of IgG glycosylation analysis hasexperienced a considerable growth in recent years: with theadvent of therapeutic monoclonal IgGs, the engineeringand analysis of IgG Fc glycosylation and eventually furtherglycosylation sites in fragment antigen binding (Fab) part(including O-glycosylation), together with other PTMs, hasbecome an important analytical challenge in the pharma-ceutical industry. The number of licensed mAbs is steadilyincreasing. There are more than 20 approved therapeuticmAbs and hundreds in clinical trials or in development [2,3]. The glycosylation to be present on the mAbs must beconsistent and human-like [3]. Therefore, monitoring ofIgG glycosylation is essential in cell line and clone selec-tion and during cell culture optimization, where a highnumber of samples subjected to product quality analysisincluding glycosylation profiling, and fast analysis isrequired.
With this review, we give an overview on the pattern androle of IgG glycosylation for IgG from human plasma as wellas therapeutic mAbs (Section 2). Section 3 summarizes IgGpurification strategies, while Sections 4–6 review differentanalytical strategies which are used for IgG glycosylationanalysis: Section 4 deals with the release of glycans followedby its analysis using various separation techniques and MS.Section 5 reviews the analysis of IgG glycoforms at the levelof glycopeptides, which is usually achieved by LC-MS. Sec-tion 6 summarizes approaches for the characterization ofIgG glycosylation as well as other PTMs by analyzing intactIgG or large fragments thereof, using predominantly ESIMS. Approaches for the integration of the various analyticaltechniques are considered in Section 7, while Section 8 indi-cates possible future developments in IgG glycosylationanalysis.
2 Biological diversity and role of IgG Fcglycosylation
Regarding the biological and clinical role of IgG Fc glycosy-lation, many descriptive data have been accumulated overthe past 25 years. The IgG glycosylation data obtained forhealthy donors showed an association of certain IgG glyco-sylation features with age, sex, and pregnancy (Section 2.1).A lower extent of IgG galactosylation has been described for
© 2009 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

884 C. Huhn et al. Proteomics 2009, 9, 882–913
various diseases (Section 2.2). Many of these data wereacquired already one or two decades ago. Analytical ap-proaches used in most of these studies involved IgG purifi-cation, N-glycan release by PNGase F (also known as N-gly-cosidase F, glycoamidase F and N-glycanase) or hydrazino-lysis, radioactive or fluorescent labeling of the glycans,followed by their chromatographic analysis, often in con-junction with exoglycosidase treatment (see Table 1). High-end analytical techniques, as they are presented in thisreview, have hardly been implemented for these applications,though this is expected to happen within the coming years.
Another set of data on the biological role of IgG Fc gly-cosylation has predominantly been obtained in in vitro stud-ies using mAbs. The production of mAbs in differentexpression systems resulted in distinct glycosylation fea-tures, which have been shown to influence antibody-de-pendent cellular cytotoxicity (ADCC) via a modulation of theinteraction between IgGs and Fcg-receptors (Section 2.3).Finally, recent mechanistic studies of intravenous Ig (IVIG)indicated an immunosuppressive role of IgG which dependson the degree of sialylation on the Fc-linked N-glycans (Sec-tion 2.4).
2.1 IgG glycosylation in healthy human populations
Three publications have analyzed the glycosylation of IgG inhuman blood within reasonably large human populations.The specific approaches and findings of these three studiesare presented and summarized.
A first publication on the age-dependency of IgG glyco-sylation was from Parekh et al. [4] who chose the followingapproach: the degree of galactosylation of IgG N-glycans wasanalyzed for a group of 151 healthy individuals of both sexesvarying in age from 1 to 70 years. The authors did not dis-tinguish between Fc and Fab glycosylation and focusedspecifically on galactosylation: all heterogeneity in terms ofattached sialic acids, N-acetylglucosamine, and fucose waseliminated by performing corresponding exoglycosidasetreatments prior to oligosaccharide analysis, leaving galacto-sylation as the only feature to be analyzed. Parekh et al. [4]found that the mean incidence of N-glycans on IgG with bothouter arms terminating in N-acetylglucosamine is higherthan 30% on average at birth, reaches a minimum of ap-proximately 20% for persons of 25 years of age and thenincreases continuously to reach approximately 40% by 70years of age. The mean incidence of monogalactosylated N-glycans remains remarkably constant at approximately 40%over the whole age range, while the digalactosylated N-gly-cans show a pattern which is complementary to the parabolicage-dependency curve of the nongalactosylated N-glycans.No sex-specific differences where found. Notably, much ofthe biological variation observed in this study could not beexplained by sex and age heterogeneity.
Approximately 10 years later two IgG glycosylation stud-ies in healthy individuals were published by Japanese re-search teams. Yamada et al. [5] enzymatically released N-gly-
cans from human serum IgG of 176 female and 227 maleindividuals ranging in age from 0 to 84 years. N-glycans werelabeled with 2-aminopyridine (PA), desialylated, and profiledby RP-HPLC with fluorescence detection, allowing the regis-tration of 12 different glycan structures. Yamada et al. [5]confirmed the age-dependency of galactosylation [4] for bothsexes. In addition, a difference in the degree of galactosyla-tion was found between males and females at age of approx-imately 25 years: women showed on average lower levels ofagalactosyl glycoforms than men. Moreover, the occurrenceof bisecting N-acetylglucosamine showed an age-dependen-cy: bisecting N-acetyl glucosamine (GlcNAc) is generallyincreasing with age and seems to reach a plateau of an aver-age of 18% at an age of 50 years.
Corroborating the findings of Yamada et al. [5], Shikata etal. [6] released N-glycans by hydrazinolysis from humanserum IgG of a small cohort consisting of 43 female and 37male individuals ranging in age from 18 to 73 years. N-gly-cans were labeled with p-aminobenzoic acid ethyl ester(ABEE) and analyzed for their degree of sialylation by anionexchange chromatography. Glycans were profiled by RP-HPLC with fluorescence detection after desialylation, allow-ing the registration of 12 glycan structures, demonstratingthe age-dependency of galactosylation only for females. Forthe males a slight trend toward lower galactosylation athigher age was observed. However, this tendency was notfound to be significant with the small number of male indi-viduals included in the study due to the considerable biolog-ical variability. The decreasing incidence of monosialylatedN-glycans with age might be explained by the lower inci-dence of galactosylated antennae (acceptor structures) athigher lifetime. The incidence of bisecting GlcNAc wasfound to increase with age for both sexes.
A first indication for a particularly high degree of IgGgalactosylation during pregnancy was published by Pekel-haring et al. [7]. In the following, Rook et al. [8] showed forboth healthy woman and an RA patient a low degree of IgG-G0 during pregnancy with particularly low values in the thirdtrimester. Moreover, for a group of seven patients theyshowed that the incidence of IgG-G0 tends to increase fromthe third trimester of pregnancy to 6 wk post partum [8]. Thiswas recently confirmed in a pregnant RA patient for the IgGsubclasses IgG1, IgG2, and IgG4 [9]. In addition, glycosyla-tion of IgG from the fetal circulation was analyzed and com-pared to maternal IgG. Interestingly the incidence of IgG-G0was found to be approximately 25% higher in maternalserum as compared to fetal umbilical vein serum, whichmight indicate a glycosylation-sensitive transport of mater-nal IgG via the placenta to the fetal circulation with a selec-tion for highly galactosylated glycoforms [10].
In conclusion, glycosylation of IgG varies considerablybetween individuals of the same sex and age. The differencesbetween sexes are minor compared to the influence of life-time. There is a clear age-dependency of IgG N-glycangalactosylation and decoration with bisecting GlcNAc.Galactosylation increases after birth up to age 25 and
© 2009 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

Proteomics 2009, 9, 882–913 885
decreases again with higher lifetime. This decrease in galac-tosylation seems to be accompanied by a decrease in sialyla-tion. Moreover, pregnancy is associated with a temporalincrease in IgG galactosylation.
2.2 IgG glycosylation in diseases
In 1985, researchers from Oxford and Tokyo showed anincrease in IgG-G0 for patients with RA and osteoarthritis(OA) [1], and many more reports have followed describingsimilar IgG glycosylation features for other autoimmunediseases, infectious diseases, and cancer. An overview of keypublications on diseases with an elevated incidence of G0 onIgG is given in Table 1. Low galactosylation of IgG is clearly acommon feature in various diseases. In RA, IgG-G0 has beenshown to correlate with clinical parameters like jointdestruction and disease activity and may serve as an indicatorfor the disease course [11]. Interestingly, remission of arthri-tis is often observed during pregnancy, which is associatedwith a decrease of IgG-G0 [8]. A pathogenic role of IgG-G0autoantibodies has been demonstrated by Rademacher et al.[12] for murine collagen-induced arthritis: the treatment ofan autoantibody-containing IgG preparation with b-galacto-sidase to increase the level of IgG-G0 glycoforms resulted ina more efficient induction of arthritis than the untreated IgGpreparation. In the following, a role has been suggested forthe mannose-binding protein in complement activation byIgG-G0 glycoforms in RA [13]. More research will be needed,however, to establish the mechanisms of pathogenicity ofIgG-G0 in RA and related diseases.
A few reports have shown changes in IgG galactosylationduring a humoral immune response: Lastra et al. [14] haveshown in mice that during a humoral immune response IgGswith a low degree of galactosylation are produced, whereas,when IgG titers were falling antibodies became more highlygalactosylated. Kaneko et al. [15], in contrast, found a specificdecrease of IgG sialylation during humoral immune responsein a mouse nephropathy model. Future studies will hopefullyshed light on the regulation and dynamics of IgG glycosyla-tion in human humoral immune response.
2.3 ADCC
IgG Fc glycosylation is crucial for high-affinity interactionwith Fc receptors, which can be demonstrated by enzymati-cally removing parts of the glycan structures. After cleavageof the chitobiose core of the N-glycan by endoglycosidaseEndo S from Streptococcus pyogenes (leaving a single N-acet-ylglucosamine on the Fc portion) [24], IgG binding to the Fcgreceptor changes and the binding to activated monocytesdecreases. Endo S was found to have a protective effect in amouse model of idiopathic thrombocytopenic purpura (ITP)and is a potential therapeutic agent against antibody-medi-ated autoimmune diseases [25]. Notably, Fc N-glycans influ-ence the binding to Fcg receptors by determining the con-formational structure of the Fc moiety: fully galactosylated N-glycans are positioned between the Fc polypeptide chains,resulting in an open Fc conformation. Removal of N-glycanresulted in a closed conformation, which is unfavorable formost IgG–Fcg receptor interactions [26].
Table 1. Diseases for which elevated IgG-G0 and/or lowered IgG galactosylation have been reported
Disease Study cohort Analytical method Reference
RA 19 patients Glycan release and chromatography [1]RA 127 patients Lectin binding assay [11]RA 29 patients Lectin binding assay [16]Adult onset RA 53 patients Glycan release and chromatography [17]Juvenile onset RA 27 patients Glycan release and chromatography [17]Juvenile onset RA 9 patients Lectin binding assay [16]Osteoarthritis 13 patients Glycan release and chromatography [1]Osteoarthritis 13 patients Lectin binding assay [16]Crohn’s disease 9 patients Glycan release and chromatography [18]Crohn’s disease 17 patients Lectin binding assay [16]SLE with Sjörgen’s syndrome 10 patients Glycan release and chromatography [18]SLE 12 patients Lectin binding assay [16, 19]Ankylosing spondylitis 12 patients Lectin binding assay [16]Ulcerative colitis 32 patients Immunoassay with anti-GlcNAc mAb [20]Ulcerative colitis 16 patients Lectin binding assay [16]Infective endocarditis 12 patients Lectin binding assay [16]Tuberculosis 21 patients Glycan release and chromatography [18]Liver fibrosis and cirrhosis in hepatitis C infection ca. 200 patients Lectin binding assay (AAL; alauria aurantia lectin)a) [21]HIV infection 28 patients Lectin binding assay; glycan release and MS [22]Ovarian cancer 3 patients Glycan release and chromatography [23]
a) Specific analysis of anti-a-Gal antibodies.
© 2009 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

886 C. Huhn et al. Proteomics 2009, 9, 882–913
Next to the removal of most of the Fc N-glycans by Endo Streatment, more subtle changes in the IgG Fc glycosylationpattern have likewise been shown to have a profound influ-ence on the interaction of the IgG Fc portion with Fcgreceptors, as achieved by choosing different expression sys-tems [27] and by modifying the glycosylation using exogly-cosidases [28, 29].
Many reports have shown that the presence or absenceof core-fucose is the most important feature with respect toADCC of IgG1, while the degree of sialylation, antennagalactosylation, and the presence of bisecting N-acet-ylglucosamine do not seem to have a strong influence [28–31]. The importance of these findings on the influence of FcN-glycosylation features – particularly core-fucosylation –for the development of new therapeutic antibodies has beenstressed by Mori et al. [32]: ADCC of IgG1 therapeutic anti-bodies is dramatically enhanced by a reduction in core-fucosylation, and next generation therapeutic antibodieswith improved efficacy are expected to be nonfucosylated.Notably, we have recently obtained indications that thepathogenic antiplatelet IgGs of various women with humanfetal-maternal alloimmune thrombocytopenia are largelylacking core-fucose [33]. The pathogenic IgGs which arelacking core-fucose are expected to be very efficient inmediating effector functions toward the platelets of thefetus, and the glycosylation patterns may, therefore, repre-sent an important determinant of disease severeness.Hence, achieving high ADCC by expression of noncorefucosylated, antigen-specific IgGs may actually be a conceptshared between therapeutic mAbs and pathogenic alloim-mune antibodies. Future studies will have to show if IgGsubpopulations lacking core-fucose play a major role inother immune responses to, e.g., vaccines, infectious agents,and autoantigens.
2.4 IVIG
Glycosylation of IgG has recently received much attentiondue to the findings of Ravetch and coworkers [15, 34], whoanalyzed the protective effect of IVIG in a mouse arthritismodel and could demonstrate that only the Fc parts exhibitthe immunosuppressive function. Moreover, using bothneuraminidase treatment and enrichment of sialylated spe-cies by lectin affinity chromatography, they showed thatsialylated IgGs as well as sialylated IgG Fc moieties aremuch more potent in preventing pathology [15]. Recently,2,6-sialylated N-glycan antennae on the Fc moieties wereidentified in a mouse model to be the motifs binding to yetunidentified receptors on macrophages, thereby inducingthe expression of inhibitory IgG Fc receptors on effectormacrophages [34]. Identification of the IVIG receptor to-gether with the refinement of its binding properties to IgGFc moieties and derived recombinant drug candidates isexpected to provide a way of efficient treatment of auto-immune disorders [34, 35]. Hitherto IVIG has been used onthe basis of its anti-inflammatory properties at high doses
(above 1 g/kg) for the treatment of various autoimmunediseases such as RA, ITP, and systemic lupus erythemato-sus (SLE) [36].
3 IgG purification
Parekh et al. [1] were the first to analyze IgG glycosylationin great detail. To purify the IgGs, ammonium sulfateprecipitation and anion-exchange chromatography wasused, based on a protein fractionation protocol which isconsiderably older [37]. Adaptations of this method are stillin use, primarily for the purification and subsequent gly-can release of IgG from human serum or plasma [6, 38–40]. The most popular technique is, however, affinity-puri-fication using immobilized Protein A (e.g., [41–44]) andProtein G (e.g., [5, 45–47]): these bacterial proteins bind toIgGs of various species and are commercially available inimmobilized form (e.g., Sepharose beads and magneticbeads). IgGs may be loaded at neutral pH, elution isachieved at low pH. Generally, Protein A and Protein Gcolumns are reusable for many purification cycles. The useof Protein A and Protein G is particularly popular for thepurification and subsequent glycosylation analysis ofrecombinant mAbs. For the purification of IgG from hu-man serum, both Protein A and Protein G may be applied,with the important difference that Protein G will bind allfour human IgG subclasses, while Protein A will onlyretain IgG1, IgG2, and IgG4, but not IgG3 [9, 48]. Addi-tional sample preparation steps like buffer exchange ordesalting may be necessary to get the IgG samples purifiedfor downstream analytical methods [49].
For pharmacokinetic studies in animals, mAbs have tobe purified from animal plasma. For this purpose, immo-bilized goat antihuman Ig Fcg antibodies have beenapplied [50]. Alternatively, the target antigen of the mAbmay be immobilized and used for capturing the mAb [51],or anti-idiotypic mAbs may be used to specifically capturethe mAb of interest from animal serum.
An alternative approach for IgG purification fromplasma or serum is the use of SDS-PAGE, followed by the in-gel N-glycan release or generation of glycopeptides [52].Moreover, the simultaneous glycosylation analysis of majorhuman serum glycoproteins after albumin depletion hasrecently been demonstrated by RP-HPLC-MS/MS at the gly-copeptide level, covering the glycosylation profiles of 23 N-glycosylation sites of serum glycoproteins including the Fc-part of the different IgG subclasses [53].
4 Analysis of released N-glycans
Starting from purified IgG, glycans are released using eitherenzymatic or chemical cleavage and subsequently purified.Glycans are often subjected to derivatization, followed by asecond purification step. Analysis of oligosaccharides can be
© 2009 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

Proteomics 2009, 9, 882–913 887
performed directly by MS or by coupling MS or fluorescencedetection to separation techniques such as HPLC, CE, CGE,and IC.
Most mAbs are only glycosylated at the conserved N-gly-cosylation site on the Fc portions of the two heavy chains.When these mAbs are sufficiently purified, assignment ofreleased glycans to the Fc portions is possible. For mAbswhich are also glycosylated in the Fab part, specific proteoly-tic cleavage and peptide purification have to be performedprior to glycan release in order to allow the assignment of theglycans to specific portions of the molecule (e.g., [43, 45, 54]).Likewise, the analysis of released glycans of polyclonal IgGdoes only allow the assignment of the glycans to Fc and Fabportions when combined with appropriate purificationmethods for IgG [45, 48]. Analysis of released glycans is per-formed for different purposes: (i) glycan release is theapproach of choice for in-depth structural characterizationincluding linkage information [43, 49]. (ii) Glycan profiles oflarge numbers of IgG samples may be analyzed after N-gly-can release [5, 46].
4.1 Glycan release
Glycans are released from purified IgG using either enzy-matic cleavage or hydrazinolysis. Several enzymes have beenused for N-glycan release, including PNGase F [43, 46, 49,54–68], PNGase A (also known as N-glycosidase A and gly-coamidase A) [3, 5, 45, 69–72], and endoglycosidase H (EndoH) [3, 62]. PNGase F releases all asparagine-linked oligo-saccharides with high yields, unless they are a1–3 core-fuco-sylated. This core-modification is not observed in mammals,but is common in plants. MAbs expressed in plants are,therefore, resistant to deglycosylation with PNGase F. Gly-cans with a1–3 core-fucosylation may instead be releasedusing PNGase A. Notably, an efficient N-glycan release withPNGase A can only be achieved at the glycopeptide level,while PNGase F is capable of deglycosylating intact IgG.Using Endo H, only high-mannose-like N-glycans can bereleased, making Endo H a highly specific glycosidase [3, 62].Prior to enzymatic glycan release, antibodies are generallydenatured to minimize steric hindrance of PNGase F. Dena-turation may be achieved with SDS, the reducing agent b-mercaptoethanol, or a combination of SDS and b-mercap-toethanol [49, 60, 61]. Similarly, antibodies may be treatedwith proteases such as trypsin and chymotrypsin to facilitateN-glycan release [73, 74]. These procedures are, however,time consuming and do not seem to be necessary for IgGconstant region N-glycans as good results are also obtainedwithout any pretreatment [65, 75].
As mentioned above, a second method for release of gly-cans is chemical hydrazinolysis [6, 38–40, 47, 57, 76, 77].With this technique, O-glycans can be released in addition toN-glycans. For this approach, the IgG preparation needs tobe free of water. Moreover, hydrazinolysis is a rather harshmethod leading to the loss of N-acetyl groups that are presenton N-acetylglucosamine residues of the N-glycans, making
re-N-acetylation an obligatory step after hydrazinolysis.Importantly, working with anhydrous hydrazine requiresspecial safety precautions, and any contact with water has tobe avoided.
Several approaches aim at the separation of antibodyfragments prior to N-glycan release, to allow the unambig-uous assignment of glycans to the Fc and Fab moieties. Hol-land et al. treated IgG with pepsin under slightly acidic con-ditions (pH 4.5). While the Fc part was cleaved into peptides,the Fab part stayed intact (see also Section 6.1.1). Upondigestion, size exclusion ultrafiltration using 10 kDa cut-offallowed recovery of Fc glycopeptides in the flow-through,while Fab was retained on the filter [45]. Alternatively, IgGmay be treated with papain which cleaves in the hinge region[3, 43, 70, 72] (see also Section 6.1.1). In one case, papaincleavage was followed by anion exchange purification, andessentially pure Fab fragments were found in the flow-through, while Fc-portions showed some cleavage hetero-geneity and were recovered in the eluate [3]. Papain digests(see also Section 6.1.1) may be alternatively applied to Pro-tein A columns which however – next to the Fc portions –will retain any undigested antibody [43, 72]. Finally, Fab andFc fragments can be separated by SDS-PAGE, which may befollowed by in-gel release of N-glycans, resulting in relativelypure N-glycan samples [46, 54, 64].
4.2 Purification of released N-glycans
Several strategies for purification of oligosaccharides prior tolabeling are available. Proteins but not peptides may be pre-cipitated either by addition of ethanol or application of heat[43, 56, 59–61, 63]. Even though this procedure is very fast,other impurities like detergents and salts are not removed.Furthermore, heating may induce the loss of sialic acids.Alternatively, released oligosaccharides can be purified usinggel-filtration on Biogel P-2 or P-4 columns [3, 45, 63, 66, 69,72]. Salts and detergents will be removed, as well as intactproteins. Since gel filtration is based on size, this method isnot applicable for oligosaccharide purification from peptidemixtures, as peptides and oligosaccharides have similarmasses and will coelute. Moreover, cation exchange columnscan be used for the removal of salts, detergents, peptides, andproteins, as most peptides and proteins are retained on cati-on exchangers [3, 46, 70]. Various SPE methods includinghydrophilic interaction LC (HILIC), RP, and porous graphi-tic carbon (PGC) chromatography have recently been report-ed for the purification of oligosaccharides [55, 57, 68]. Since1998, PGC is widely used in glycan analysis and was shownto quantitatively recover pure oligosaccharides from complexsamples [78]. Usually peptides and proteins strongly adsorbto PGC and do not coelute with the glycans at 25% ACN [55].When using RP-SPE cartridges, native glycans are collectedin the flow-through [57, 79], while peptides and proteins canbe eluted using more stringent eluates. Yu et al. [68] reportedthe use of a HILIC-based 96-well SPE plate for purification ofoligosaccharides. It is a disadvantage of HILIC that glycans
© 2009 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

888 C. Huhn et al. Proteomics 2009, 9, 882–913
have to be dissolved in high concentrations of organics,which may induce glycan precipitation. All three SPE meth-ods are widely used in glycan analysis, and can easily beapplied in a 96-well format for high-throughput analyses.
To date, most methods for enzymatic N-glycan releaseare performed in solution. Additionally, a method usingprotein immobilization on PVDF membranes (also availablein the 96 well format) has been described which facilitatesremoval of detergents and therefore bypasses the purifica-tion step prior to analysis [80, 81]. Due to the low capacity(,10 pmol of (glyco-)protein binding per well) only smallquantities of glycans can be retrieved [82].
4.3 Removal of sialic acids
To decrease the complexity of the oligosaccharide mixtureand thus allow for an easier analytical strategy, sialic acidscan be removed from the glycans. This can either be per-formed using acid hydrolysis or sialidase treatment. Acidhydrolysis results in efficient desialylation and is usuallyperformed prior to the release of the N-glycans [3, 69, 73].Takegawa et al. [66], in contrast, performed acid hydrolysisafter labeling and purification, followed by immediate glycananalysis. Enzymatic desialylation is generally performed di-rectly before or in between analyses as no further purifica-tion is necessary. It is commonly applied prior to RP-HPLCleading to a simplified peak pattern [5]. In other cases, enzy-matic desialylation is part of extensive exoglycosidase treat-ment for identification purposes [43, 49, 57, 60].
4.4 Analytical strategies
Three major strategies are used for analyzing oligosaccha-rides from antibodies. A first approach includes labeling ofthe reducing end of released glycans followed by LC, ionchromatography, or CE. These separation techniques may becombined with fluorescence detection or MS. Second, nativereducing-end N-glycans may be analyzed by MALDI-TOF-MS or high-pH anion exchange chromatography with elec-trochemical detection. Third, permethylation of all hydroxylgroups may be performed, followed by mass spectrometricanalysis using either MALDI or ESI-MS. With these meth-ods, the composition of the glycans can be established. Inorder to obtain information regarding linkages, branching,and epimers, several studies have applied multiple exoglyco-sidases [43, 46, 49, 54, 57, 60, 64, 66].
4.4.1 Analysis of reductively aminated glycans
The use of different labels for mono- and oligosaccharideanalysis has recently been reviewed [83, 84]. Reducing-endlabeling of oligosaccharides with a fluorescent or UV-absorbing tag allows glycan detection and quantification incombination with various separation systems. The labelinginvolves reductive amination with several types of aromaticamines. It can be performed both under nonaqueous [49, 55,
61] and aqueous conditions [43, 56, 59, 85]. Nonaqueouslabeling with 2-aminobenzoic acid (2-AA) and 2-amino-benzamide (2-AB) was optimized by Bigge et al. [86], whilelabeling with 2-AA under aqueous conditions was estab-lished by Anumula [84, 87].
To label glycans from IgG, mainly three fluorescentlabels have been used, which are 2-AA, 2-AB, and PA. Allthese labels have their own features making them compati-ble with different kinds of purification strategies and ana-lytical methods.
4.4.1.1 PA
Labeling with PA has been used extensively for both in-depthanalysis and profiling of IgG oligosaccharides. Upon label-ing, excess label is removed using gel filtration [3, 69, 73],resulting in pure oligosaccharide samples. Takahashi et al.[73] used PA labeling followed by RP-HPLC-FLU analysisusing an ODS column for the in-depth analysis of N-glycansattached to IgG and additional NMR analysis of glycan frac-tions. Using similar procedures, IgG glycosylation was pro-filed for mAbs and in population studies [5, 45, 69, 70, 72].Recently, Takegawa et al. [66] introduced an RP-LC-MSmethod with sonic spray ionization for the in-depth analysisof IgG glycosylation, revealing galactosylated bisectingGlcNAc as a novel glycosylation feature on a low abundanceN-glycan of IgG. The presence of this modification on IgG N-glycans has recently been confirmed [57] (see Section 4.4.2).The use of a zwitterionic-type hydrophilic interaction chro-matography (ZIC-HILIC) column with sulfobetaine groupsfor the separation of desialylated, PA-labeled N-glycans fromIgG has recently been reported [67]. With this method [67]isomeric glycans of Hex4HexNAc4Fuc1 and Hex4HexNAc5-
Fuc1 could be baseline separated, and the separation seemsto be somewhat superior to that obtained using anotherHILIC system with 2-AB-labeled glycans (TSK amide-80 col-umn; see below) [23, 46]. Recently, Takegawa et al. [88]showed that sialylated PA-glycans can be successfully sepa-rated on ZIC-HILIC, yet this technique has not been appliedto IgG.
4.4.1.2 2-AB
2-AB is a neutral label which is frequently used in glycananalysis. Either HILIC-SPE or paper chromatography is usedfor the removal of excess label [46, 81]. For the analysis oflarger sample sets of IgG glycans, 2-AB labeled glycans aremost frequently profiled without desialylation by HILICusing TSK amide-80 columns (Tosoh Biosciences) [23, 46,81]. The group of Pauline Rudd has established a database(http://glycobase.nibrt.ie/) for the assignment of glycanstructures based on HILIC retention properties, which aredetermined for native glycans as well as for glycans treatedwith exoglycosidases [81, 89]. This technology is widely usedfor the analysis of antibody glycosylation [23, 46, 54, 81, 89,90]. Watson et al. [38] chose to first analyze the sialylation
© 2009 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

Proteomics 2009, 9, 882–913 889
status of 2-AB labeled N-glycans by weak anion exchangechromatography, followed by desialylation of the N-glycansand profiling using HILIC (glycosep N column). Using thelatter method, the degree of sialylation can be determinedvery accurately, but the procedure is rather time-consuming.
Recently, Chen and Flynn [55] developed an RP-HPLCmethod of 160 min run time with on-line fluorescence andMS detection for the in-depth analysis of 2-AB labeled N-glycans derived from IgG. They observed the elution of sia-lylated N-glycans in the front part of the chromatogram, fol-lowed by oligomannosidic N-glycans, with neutral complex-type N-glycans eluting at the end of the chromatogram. Thisis in contrast to results obtained with HILIC separation,where the size of the glycan has the largest influence on theretention time, with neutral complex-type glycans, sialylatedglycans, and oligomannosidic glycans eluting in overlappingregions [91].
4.4.1.3 2-AA
2-AA has a wide range of applications within the field of gly-can analysis. Due to the negative charge of this tag, CE-separations can easily be obtained with 2-AA-labeled glycans.Moreover, 2-AA-labeled glycans can be analyzed by negative-mode as well as positive-mode MALDI-TOF-MS. Negative-mode MALDI-TOF-MS and MALDI-Q-TOF-MS of 2-AA-labeled glycans is particularly attractive, as it allows theregistration of both neutral and sialylated glycans in thesame experiment [43, 92]. Moreover, 2-AA-labeled glycanscan be analyzed by both HILIC [92] and nano-RP-HPLC-ESI-MS. Excess label can be removed using Oasis HLB cartridgesin the NP-mode [43, 60]. Recently, several methods weredeveloped for the fast, high-throughput purification of 2-AAlabeled N-glycans from glycoproteins, including IgG [93] andwhole plasma glycoproteins [92], on the basis of HILIC sta-tionary phases DPA-6S or microcrystalline cellulose, respec-tively.
Kamoda et al. developed a CE method with laser-inducedfluorescence detection (LIF) for the separation of 2-AA-labeled glycans, which separates N-glycans without desialy-lation at high resolution in 24 min. Using this method iso-meric glycans of Hex4HexNAc4Fuc1 could be separated, andgood quantification was obtained. For the structural assign-ment of the N-glycans, peaks were fractionated using HILICwith fluorescence detection. Fractions were subsequentlyanalyzed by MALDI-TOF-MS [60, 94].
Qian et al. [43] recently performed a detailed analysis of 2-AA labeled N-glycans from a mAb. Using MALDI-Qq-TOF MSand MS/MS analysis, the composition of the N-glycans wasdetermined. For in-depth characterization, HILIC with fluo-rescence detection was performed with peak fractionation,followed by treatment with various exoglycosidases andMALDI-TOF mass spectrometric detection. Using this time-consuming method, anomericity and epimericity of the 2-AAlabeled N-glycans was revealed, resulting in an extensive char-acterization of the oligosaccharide content of the antibody.
4.4.1.4 Other labels
3-Aminobenzoic acid (3-AA)-labeled N-glycans from IgGwere profiled by CE-LIF [59]. Excess label was removed usingSEC. Compared to 8-aminopyrene-1,3,6-trisulfonate (APTS)labeled N-glycans (see below), 3-AA-labeled glycans showed aslightly reduce retention, while analysis times are slightlyincreased [59]. Nevertheless, 3-AA has several advantagescompared to APTS: high quality grade 3-AA is easily avail-able, inexpensive and the conditions for the labeling reactionare less stringent. Furthermore, 3-AA is compatible withboth HILIC and RP-HPLC, similar to the 2-AA label. Notably,the method used for the analysis of 2-AA labeled N-glycansand 3-AA labeled N-glycans by CE-LIF gave similar results[59, 60].
APTS is widely used for N-glycan analysis with CE cou-pled to LIF detection due to the short separation times andgood sensitivity that can be achieved. Excess label has to beremoved using ultrafiltration or gel filtration. In studies onthe glycosylation of a mAb, it was shown that with run timesof around 10 min structural isomers could be separated [49,59]. Even though good separation can be achieved, no directstructural information can be obtained regarding themigrating APTS-labeled N-glycans. Therefore, a capillary gelelectrophoresis-MS technology with on-line LIF detectionhas been developed [56]. Due to the modifications whichwere necessary for compatibility with MS detection, thelength of the run had to be doubled in order to obtain rea-sonable resolution. With this technology, previously uni-dentified minor glycans were detected, as well as isomericglycans which are comigrating with other components and,therefore, require mass spectrometric detection for dis-crimination. Recently, Zhuang et al. [95] reported on the useof a microfluidic device for the electrophoretic analysis ofAPTS labeled N-glycans from plasma proteins. With this de-vice very good separations were achieved in only 3 min.
A method for quick and high-throughput analysis ofAPTS labeled N-linked oligosaccharides by a DNA-sequencerwas reported several years ago [82, 96]. This method wouldappear to be useful for the fast profiling of many N-glycansamples from antibodies [89]. However, this approach is notsuitable for in-depth analysis as no MS data are generated.
ABEE is a UV-absorbing label that has been used forprofiling of N-glycans from IgG. Upon labeling, excess labelcan be removed using simple and straightforward liquid–liquid extraction, which is a fast procedure. Profiling is thenachieved by anion exchange chromatography to determinethe sialic acid content, followed by desialylation and sub-sequent profiling using RP-HPLC with UV detection [6, 39,40].
4.4.2 Analysis of nonlabeled and reduced glycans
When labeling is omitted and native N-glycans are analyzed,the total sample preparation often involves only a few steps.The panel of separation methods available for native glycans
© 2009 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

890 C. Huhn et al. Proteomics 2009, 9, 882–913
is however rather restricted. Majid et al. [63] purified thereleased N-glycans by gel filtration followed by high-pHanion exchange chromatography with pulsed amperometricdetection (HPAEC-PAD) [63], but obtained only poor resolu-tion. Nevertheless, HPAEC-PAD is generally a very goodchoice for glycan analysis, with analysis times convenientlybeing around 30 min. The use of MS for the analysis ofnative N-glycans is a second option. In general, MS allowsrapid and highly sensitive profiling of N-glycans. UsingMALDI-TOF-MS in both positive and negative ionizationmode together with exoglycosidase treatment, profiles fromIgG-derived N-glycans can be obtained [49, 64], revealinginformation on the glycan composition together with theirstructural characterization. For example, Yu et al. reportedthe use of a HILIC-SPE plate for the purification of native N-glycans from IgG followed by MALDI-Q-TOF-MS/MS anal-ysis. Even though mass spectrometric methods for the anal-ysis of native glycans comprise generally fast and highlysensitive procedures, no information can be obtainedregarding linkages, epimericity, and anomericity.
In a study by Harvey et al. [57], in-depth analysis of aspecific glycan from IgG was performed using HILIC frac-tionation of native N-glycans followed by MALDI-TOF anal-ysis for the rapid analysis of fractions and subsequent nega-tive-mode ESI-Q-TOF MS/MS structural analysis (Fig. 2).Using this approach, several new structures could be detect-ed which shared the unusual motif of a b1,4-linked galactoseon the bisecting GlcNAc (Fig. 2C), in accordance with anearlier report by Takegawa et al. [66].
Graphitized carbon-HPLC-MS of reduced glycans is aparticularly useful technique for assigning detailed oligo-saccharide structures based on the comparison of molecularmass and retention time with a large set of oligosaccharidestandards [97]. This method has recently been applied for thein-depth analysis of N-glycans from both serum IgG andrecombinant mAbs [52]. It allows to distinguish between a2–
3-linked and a2–6-linked sialic acid residues as well as be-tween b1–4-linked and b1–3-linked galactose [52]. Moreover,these substitutions can be assigned to the upper (a6-linkedmannose) or lower antenna (a3-linked mannose) [52]. Anal-ysis of released glycans by graphitized carbon-HPLC-MSallows the differentiation between glycans containing N-acetylneuraminic acid and N-glycolylneuraminic acids: whilethe chromatographic properties of N-glycans exhibiting thisstructural difference were very similar, they could easily bedifferentiated due to the difference in mass [97].
4.4.3 Analysis of permethylated glycans
In a comparative study, many laboratories investigated theglycosylation of IgG with a variety of methods, including themass spectrometric analysis of permethylated N-glycans [48](Fig. 3). Permethylation stabilizes the sialic acids by convert-ing the carboxylic groups into methyl esters, thus preventingsialic acid loss during mass spectrometric analysis. Per-methylated oligosaccharides can easily be purified usingliquid-liquid extraction. Mass spectrometric detection of per-methylated glycans is very sensitive, both with MALDI andESI. MALDI-TOF-MS of permethylated glycans is particu-larly suitable for relative quantification, as reproducible gly-can profiles can be generated. In the future, internal stan-dards may be used based on one of the recently introducedisotope labeling techniques which should make MALDI-TOF-MS [98] and MALDI-Fourier transform ICR (FTICR)-MS [99] of permethylated glycans suitable for absolutequantification of IgG glycoforms. MS/MS of sodium adductsof permethylated glycans is particularly suitable for in-depthanalysis of glycans [100], as it provides information on link-age and branching and easily allows to differentiate betweenterminal and internal structural elements [101]. ESI of per-methylated glycans has only recently been hyphenated withgraphitized carbon HPLC [102], which – combined with
Figure 2. Negative ion MS/MSspectra of the [M 1 H2PO4]
2 ionsfrom the isomeric triantennaryglycans: (A) triantennary glycanfrom human a1-acid glycopro-tein; (B) triantennary glycanfrom CEACAM 1; and (C) trian-tennary glycan from IgG. Emptycircle, mannose; filled square,GlcNAc; empty diamond, galac-tose; diamond with dot, fucose.Linkage positions are shown bythe angle of the lines connectingthe monosaccharide symbols() = 1–2, / = 1–3, } = 1–4, \ = 1–6);full line = b-bond; broken line =a-bond. Reproduced with per-mission from [57].
© 2009 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

Proteomics 2009, 9, 882–913 891
Figure 3. MALDI-TOF-MS spec-trum of permethylated oligo-saccharides from human IgG[48].
the efforts for miniaturizing and streamlining the per-methylation procedure [103] – could represent an interestingstrategy for the detailed analysis of IgG N-glycans at a rea-sonable throughput.
5 Analysis of glycopeptides
Mass spectrometric analysis of glycoproteins in general andIgG in particular at the level of glycopeptides are generatedwith endoproteinases offers the possibility to determine gly-can attachment sites and evaluate glycosylation site micro-heterogeneity. Usually these analyses are performed on pu-rified proteins [104–106]. However, adequate assignment of apeptide sequence to a certain protein may already beachieved for peptide stretches of ten amino acids length,offering the possibility to investigate and assign PTMs,including glycosylation, at the level of complex proteindigests [53]. Notably, enrichment of the obtained glycopep-tides can be useful, in order to avoid the suppression of gly-copeptide ionization by strongly ionizing coexisting pep-tides. In this chapter, we give an introduction to proteolyticcleavage of glycoproteins with the focus on IgG (Section 5.1)and subsequently we address often applied techniques forIgG glycopeptide analysis (Section 5.2).
5.1 Proteolytic cleavage
Various specific and nonspecific proteolytic enzymes areavailable for the digestion of glycoproteins, including tryp-
sin, lysylendopeptidase (Lys-C), endoproteinase Glu-C (V8protease), and pronase. In general, completeness of proteo-lytic cleavage is improved by the addition of denaturingagents, such as guanidine hydrochloride, urea and ACN. Inaddition, reduction of cysteine disulfide bonds by reducingagents such as DTT and b-mercaptoethanol furtherimproves accessibility of proteolytic cleavage sites (see alsoSection 6.1.7). Since not all proteolytic enzymes retain theirfull activity under reducing conditions, and in order to avoidreformation of disulfide bonds during (glyco-)peptide stor-age and analysis, sulfhydryl groups are often blocked withalkylating agents, such as iodoacetamide, iodoacetic acid,and ethyleneimine. To ensure full activity of the proteolyticenzyme, the reduction/alkylation buffer is usually diluted orreplaced by a digestion buffer using dialysis and gel filtra-tion which may be followed by lyophilization of the desaltedsample [53, 107, 108]. In order to simplify IgG analysis,heavy and light chains resulting from the reduction andalkylation can be separated by RP-HPLC or SDS-PAGEprior enzymatic digestion. This approach is often applied instudies characterizing recombinant mAb glycosylation [64,105, 109].
An improvement of the sample-processing using pro-teolytic cleavage reactions is often achieved by the use ofenzymes which are immobilized on beads, monoliths, oron capillary walls, offering the advantage of enzyme re-us-ability, automation, and complete digestion in very shorttimes with a resulting peptide mixture lacking contamina-tion with protease and its autoproteolysis fragments [110–114].
© 2009 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

892 C. Huhn et al. Proteomics 2009, 9, 882–913
5.1.1 Trypsin
Trypsin is by far the most widely applied proteolytic enzymein proteomics and glycoproteomics. Trypsin is a serine pro-tease, cleaving peptides at the carboxyl side of lysine and ar-ginine, unless the following amino acid is a proline. IgG Fcglycopeptides resulting from tryptic digestion are often easyto ionize by ESI and MALDI. Trypsin digestion is typicallyperformed overnight at 377C in a 10–100 mM reaction buffer(Tris-HCl or ammonium bicarbonate, pH,8) using a 1:100–1:20 protease/protein ratio [9, 53, 64, 67, 112, 115–118]. Toimprove trypsin digestion enzyme mixtures [117] or dena-turing agents such as guanidine–HCl or urea with con-centrations up to 2 M are sometimes applied [64, 104, 112].Interestingly, addition of ACN with concentrations up to80% was shown to improve enzymatic activity and digestionefficiency for a protein standard mixture [119, 120], thoughthis has not been shown hitherto for IgG.
Samples prepared by trypsin digestion have successfullybeen used for the identification and characterization of theglycosylation of mAbs [64, 118, 121] and serum IgG [9, 52,53, 116, 122]. Due to the occurrence of tryptic Fc glycopep-tides with identical peptide moieties for the IgG2 and IgG3subclasses, the analysis of a proteolytic digest of total serumIgG will not allow the specific assignment of glycopeptides tothese subclasses [9] (Fig. 1). Therefore, serum IgG was frac-tionated into a pool of IgG1, IgG2, and IgG4 by Protein Acapturing, followed by the capturing of IgG 3 from the flow-through using Protein G beads. Analysis of the trypsinizedeluates by RP-LC allowed the unambiguous assignment ofthe observed tryptic glycopeptides to specific IgG subclasses[9, 33, 123].
5.1.2 Lysyl endopeptidase
Lysyl endopeptidase is an alkaline protease, specificallycleaving peptides at the carboxyl side of lysine. As for IgG,lysyl endopeptidase has been reported to specifically cleavethe core hinge portion at low protease/protein ratios andshort digestion times (see Section 6.1.6). In contrast, an al-most complete digest is achieved by overnight digestion at377C using 1:200–1:20 protease/protein ratios. Althoughlysyl endopeptidase is more tolerable to higher guanidine–HCl or urea concentrations than trypsin, digestion is typi-cally performed using the same reaction buffers [50, 107,124, 125]. Lysyl endopeptidase has been successfully appliedfor LC-MS glycopeptide mapping of several mAb, includingmouse IgG2b and humanized IgG1 and IgG4 [50, 107, 124–126].
5.1.3 Endoproteinase Glu-C
Endoproteinase Glu-C is a serine protease, specifically cleav-ing peptide bonds at the carboxyl-terminal side of glutamicacid. Depending on the pH of the digestion buffer, peptidebonds are additionally cleaved at the carboxyl-terminal side
of aspartic acid with a 100–3000-fold lower rate [127]. Ingeneral, specificity for glutamic acid is obtained if digestionis performed in ammonium acetate buffer at pH , 4 [127].Endoproteinase Glu-C treatment of human IgG has beenshown to result in the complete degradation of the heavychains, while the light chains were affected only partially[128]. Endoproteinase Glu-C digestion of human IgGallowed the characterization and quantification of Fcg glyco-sylation of plasma purified IgG1 and IgG4 by LC-ESI-MSand LC-ESI-MS/MS [129]. It was suggested that the de-scribed procedure was additionally applicable for IgG2 andIgG3 glycosylation characterization and quantification.
5.1.4 Pronase
Pronase is a commercially available mixture of endo- andexopeptidases, nonspecifically cleaving proteins into theirindividual amino acids. Glycosylation generally leads to asteric hindrance of the proteinases, which results in glyco-peptides of several amino acids. Using this approach, thedetermination of N-linked glycosylation sites was possiblefor several pure model proteins, including ribonuclease B,chicken ovalbumin, horse radish peroxidase, a1-acid glyco-protein, and k-casein [106, 130–133]. For IgG, pronase hasbeen applied to elucidate glycan structural and heterogeneityinformation [134–137].
5.2 MS of glycopeptides
Mass spectrometric detection of glycopeptides offers thepossibility to determine glycosylation profiles in a site-spe-cific manner. Traditionally, glycopeptides were ionized byfast atom bombardment (FAB) and laser desorption (LD).Within the past two decades, the softer ionization techniquesESI and MALDI came in place, providing a higher sensitivityand making intact ions of high-molecular mass compoundsaccessible (see also Section 6.2.3).
MALDI of tryptic IgG glycopeptides typically results insingly charged [M 1 H]1 ions [64, 67, 104, 115–117, 138],while ESI often results in doubly charged [M 1 2H]21 [9, 53,108, 139] and/or triply charged [M 1 3H]31 ions [9, 42, 52].However, quadruply charged [M 1 4H]41 ions due to onemissed cleavage (see figure 2c in [52]) and doubly chargedsodiated [M 1 H 1 Na]21 ions [115] of tryptic IgG glycopep-tides have also been reported with ESI. ESI of larger IgGglycopeptides typically obtained by lysyl endopeptidase orendoproteinase Glu-C cleavage often result in triply charged[M 1 3H]31 ions [107, 129]. The ESI conditions markedlyinfluence the charge state distribution, with harsher iontransfer conditions leading to lower charge states.
Ionization of glycopeptides can be suppressed by highlyabundant coexisting peptide ions. In addition, salts present inthe original sample or introduced during reduction, alkyla-tion, and enzymatic cleavage can suppress glycopeptide ioni-zation, especially with ESI. Finally, overlapping isotope clus-ters from coexisting peptides or missed cleavages can com-
© 2009 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

Proteomics 2009, 9, 882–913 893
plicate glycopeptide mass determination and identification.Therefore, proper sample pretreatment (enrichment, effi-cient enzymatic cleavage, desalting) prior to ionization isimportant. Sample purification and preseparation of (glyco-)peptides can be achieved using coupled techniques such asLC-ESI-MS. Alternatively, samples can be fractionated anddesalted by preparative LC or SPE, followed by MALDI-MS.
5.2.1 RP-HPLC-ESI-MS
One of the most widely applied analytical techniques for gly-copeptide analysis involves direct analysis of proteolyticdigests by RP-HPLC coupled to ESI-MS. RP-HPLC separationof proteolytic IgG digests is mostly performed on C18 analyti-cal columns of various dimensions. As an alternative to RP-HPLC, graphitized carbon chromatography can be applied[140]. In general, a water/ACN gradient with increasing ACNconcentrations, typically containing approximately 0.1%acidic mobile phase additive such as TFA or formic acid (FA) isused in RP-HPLC-MS [9, 53, 107, 108, 141]. TFA is a com-monly used mobile phase additive due to its positive effect onanalyte retention and peak concentration [142]. However, TFAcan form gas-phase ion pairs with positively charged analyteions potentially increasing ESI suppression [142, 143]. For IgGglycosylation analysis, FA is therefore applied more often asacidic mobile phase additive. In addition, to ensure methodstability in terms of separation, ionization, and detection, moststudies have applied micro-LC [108, 141, 144] or capillary-LC[52, 53, 105, 107] coupled to ESI-MS, while nano-LC is lessfrequently used [9, 33, 42, 123, 139, 145].
ESI-MS/MS spectra of LC separated glycopeptides can behighlighted on the basis of glycan specific oxonium ions ofm/z 204 [HexNAc 1 H]1, 366 [Hex1HexNAc1 1 H]1, 528[Hex2HexNAc1 1 H]1, and 657 [Hex1HexNAc1NANA1 1 H]1
(N-acetyl neuraminic acid (NANA)) obtained on CID or byscanning for specific neutral losses of terminal mono-saccharides such as fucose, N-acetylglucosamine, and sialicacid [53, 115, 132]. Moreover, multiply charged IgG glycopep-tide ions as generated by ESI allow the performance of CIDand electron transfer dissociation (ETD) experiments, withthe latter resulting in peptide sequence information [9]. Gly-copeptides with a common peptide moiety generally eluteclose to each other. Interestingly, some RP-HPLC columnsshow an enhanced retention of sialylated glycopeptides whencompared to glycopeptides with neutral glycan chains [9],while other RP-HPLC columns do not exhibit this pro-nounced difference in retention times [52]. To ensure themeasurement of intact glycopeptides, in-source decay, whichoften results in the loss of sialic acid or the cleavage of theGlcNAcb1–2Man linkage, has to be ruled out by choosing theion transfer voltages carefully [52]. This allows the acquisitionof Fc glycosylation profiles by RP-HPLC-ESI-MS [50, 52, 53,126], as shown, e.g., for IgG1 from two RA patients and twohealthy individuals (Fig. 4) [9].
MS of glycopeptides allows the determination of glycanstructure and attachment site in a single experiment. For
example, analysis of lysyl endopeptidase-digested mouseantidansyl mAb IgG2b (RCM-IgG2b) by an automated LC-MS(/MS) mode, allowed assignment of 12 different types ofO-linked oligosaccharides to the heavy chain threonine 221in the hinge region [107] (Figs. 5A and B). Moreover, threemajor types of N-linked oligosaccharides could be assignedto the heavy chain asparagine 297 from endoproteinase Glu-C digested RCM-IgG2b (Fig. 5C).
Accurate MS measurement of glycopeptide amounts incomplex samples is complicated by ionization variability. UVdetection, in contrast, often allows proper quantification inpeptide mapping by RP-HPLC [109]. Due to the unspecificnature of UV detection, quantification is only possible whencompounds are at least partially resolved into individual LCpeaks. Monitoring LC separations with in-line UV followedby ESI-MS characterization and identification combines thequantitative strength of UV with the profiling strength ofMS. Additionally, an extra quality control step is offered todetermine digestion completeness, separation quality, andsample identity.
As for IgG glycosylation, quantification of non-glycosylated, deglycosylated, and total N-glycosylated speciesin tryptic digests of Herceptin IgG1 was possible by RP-UPLC-UV-MS [146]. Nonglycosylated and deglycosylatedpeptides were separated into individual LC peaks. The totalN-glycosylated species were determined by the analysis of thetryptic digests before and after PNGase-A treatment. Addi-tionally, IgG1 purified from human polyclonal IgG was ana-lyzed, showing the presence of a peptide coeluting with thedeglycosylated peptides. This contamination preventedproper quantification on the basis of UV only, pointing outthe necessity of combination with the more tedious andtime-consuming MS detection.
5.2.2 HILIC-ESI-MS
HILIC coupled to ESI-MS is another powerful technique forthe separation and analysis of glycopeptides. While retentionin RP-HPLC is primarily mediated by hydrophobic interac-tions of the peptide moiety, retention in HILIC is mainlyachieved via hydrophilic interactions between the stationaryphase and the glycan moieties of the glycopeptides, whichresults in the (partial) separation of glycopeptides from pep-tides. Retention will generally increase with the size of theglycan moiety. Therefore, separation of glycopeptides can beachieved according to the degree of sialylation [147]. ZIC-HILIC separation of tryptic IgG1 and IgG2 glycopeptidescontaining the same glycan moiety showed that the presenceof more hydrophilic amino acids in the peptide chain affectsglycopeptide retention [67]. IgG1 tryptic Fc glycopeptideswhich contain two tyrosine residues showed more retentionthan IgG2 tryptic Fc glycopeptides which instead contain twophenylalanine residues, thus structurally only differing bytwo aromatic hydroxyl groups. Moreover, with ZIC-HILIC itwas possible to separate IgG glycopeptides with identicalpeptide moieties containing isomeric glycan structures. For
© 2009 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

894 C. Huhn et al. Proteomics 2009, 9, 882–913
Figure 4. Comparison of IgG1 glycosylation profiles. Glycopeptides with neutral (left) and acidic (right) glycan chains elute in differentpositions of the chromatogram. C1, control 1; C2, control 2; RA1, rheumatoid arthritis patient 1; RA2, rheumatoid arthritis patient 2. pep,peptide moiety; glycan code as in Fig. 1. As for monogalactosylation and sialylation, these residues have not been assigned to a specificantenna. Moreover, features like the fucosylated trimannosyl core were assigned based on literature data. Taken from [9] with permis-sion.
example, G1 glycopeptides of both IgG1 and IgG2 contain-ing an upper antenna galactose were efficiently separatedfrom those containing a lower antenna galactose [67].
Detailed glycan structural information can be obtainedby sequential exoglycosidase treatments of antibody glyco-peptides followed by LC-MS. For example, sequential exo-glycosidase treatments of endoproteinase Lys-C digestedhumanized anti-Ab IgG1 mAb showed that the major N-linked oligosaccharide structures in the Fc region were core-fucosylated biantennary oligosaccharides with none or one b-linked N-acetylgalactosamine, zero or one a-linked galactoseand one or two N-glycolylneuraminic acid residues [50]. Inaddition, typical core-fucosylated, biantennary N-glycanswith one or two galactose residues were observed in the Fcregion.
5.2.3 MALDI-MS
IgG glycosylation is often studied by MALDI-MS. Generally,MALDI is less sensitive to ion suppression than ESI. Never-theless, direct glycopeptide analysis from proteolytic digests
is often prohibited by the presence of nonvolatile salts,detergents, and nonglycosylated peptides. In addition,occurrence of overlapping isotope clusters from coexistingpeptides or missed cleavages can further complicate glyco-peptide identification and mass determination. Therefore,IgG glycopeptides are often enriched and desalted by RP-HPLC fractionation [64, 104, 116, 138], HILIC affinity chro-matography [68, 104, 117], or lectin-based affinity chroma-tography [104]. MALDI sources are most often coupled toTOF-MS, but also to IT-MS, FTICR-MS, and Orbitrap MS,with the MALDI-TOF-MS combination being the most rele-vant. MALDI-TOF-MS analysis of tryptic IgG glycopeptidesoverlaid, mixed, or sandwiched with 2,5-dihydroxybenzoicacid (DHB) and/or CHCA can be performed in linear posi-tive, linear negative, and/or reflectron positive mode. Due tothe soft nature of MALDI ionization most IgG glycopeptideions remain intact. Sialylated glycopeptides, however, whenanalyzed by positive-ion MALDI-TOF-MS with delayedextraction in the reflectron mode, often show massive desia-lylation due to in-source decay and metastable decay causedby laser-induced decomposition and/or collisional fragmen-
© 2009 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

Proteomics 2009, 9, 882–913 895
Figure 5. LC-ESI-IT-MS/MS product ion spectra of digested RCM-IgG2b. (A) Doubly charged ion (m/z 1167.5) of peptideG341LVRAPQVYILPPPAEQLSRK361. Peptides were generated bylysyl endopeptidase digestion. (B) Doubly charged molecular ion(m/z 1489.0) of the O-glycopeptide K212LEPSGPISTINPCPPCK229.Peptides were generated by lysyl endopeptidase digestion, andthe sugar is linked to Thr-221. (C) Triply charged ion (m/z 1472.2)of N-glycopeptide D295YNSTIRVVSTLPIQHQDWMSGKE318. Pep-tides were generated by endoproteinase Glu-C digestion, and thesugar is linked to Asn-297H. Taken from [107] with permission.
tation during ion acceleration. In contrast, detection of sialy-lated glycopeptides can be achieved by MALDI-TOF-MS inthe linear mode [104, 117], since the lack of an extractiondelay strongly reduces in-source decay of the sialylated gly-copeptides. Moreover, any metastable decay products result-ing after the acceleration steps are not resolved in linearmode.
Additional structural information can be obtained byCID if MALDI is coupled to tandem mass spectrometers,such as QIT/TOF, QTOF, and TOF/TOF mass spectro-meters. For example, CID of [M 1 H]1 glycopeptide ions byMALDI-QIT/TOF allowed glycan identification and se-quencing [117]. In addition, fragment ions corresponding tothe peptide with attached N-acetylglucosamine were formed,which could be isolated and subjected to CID providing pep-tide sequence and glycan attachment site information inMS3. Notably, MALDI-TOF/TOF-MS as well as MALDI-QTOF-MS analyses of glycopeptides often directly providedetailed peptide sequence information. In contrast, production spectra obtained by automatic LC-MS, LC-MS/MS oftenlack sufficient information, as exemplified for a glycatedpeptide [148].
6 Analysis of intact IgGs, subunits, heavychains, and light chains
A major reason for analyzing intact IgG is the minimalsample preparation necessary. After purification, the IgG isdirectly accessible for analysis. Moreover, MS of intact IgGprovides an integrated analysis of a large number of PTMs.Besides the glycosylation in the Fc part and in the variableregions of the light and heavy chain these include the con-version of N-terminal glutamine of the heavy chain into pyr-oglutamate, complete or partial absence of C-terminal lysinethrough the action of carboxypeptidases, oxidation onmethionine residues, deamidation and formation of succin-imide from aspartic acids, cysteinylation, dimer formation,and peptide bond cleavage, e.g., by acid hydrolysis, which canalso be induced by analytical conditions (e.g., acidic condi-tions at elevated temperature in LC [41]). This approach is,however, not feasible with polyclonal IgG such as total serumIgG, because of the enormous heterogeneity of the variableregions which prohibits mass spectrometric detection. Theanalysis of IgG or its fragments is thus mainly applied for thecharacterization and quality control of biotechnologicallyproduced mAbs.
A major challenge in analyzing intact monoclonal IgG isits size [149]. Minor modifications present in the molecule,e.g., N-terminal pyroglutamate, cannot be resolved by MS ofintact IgG, and most of the microheterogeneity based onglycosylation differences has a too small effect to be resolvedby SDS-PAGE. In order to simplify the analytical task, smal-ler fragments of the antibody are generated enzymatically:The analysis of IgG fragments such as the Fc and Fab partsas well as light and heavy chains reduces the complexity of
© 2009 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

896 C. Huhn et al. Proteomics 2009, 9, 882–913
the sample and provides more detailed information on thetype and site of modifications.
In this section, we give an introduction to the generationof antibody fragments and summarize the techniques for theanalysis of the heterogeneity of intact IgG and IgG fragmentswith a focus on glycosylation.
6.1 Fragment generation
In general, two strategies for the formation of fragments ofthe intact antibody are applied: (i) cleavage in the hingeregion using enzymes, mainly papain and pepsin, and (ii)disruption of disulfide bonds between light and heavychains. Figure 6 shows possible fragments.
6.1.1 Cleavage in the hinge region
Cleavage in the hinge region is done enzymatically. Papainis applied in most studies, and pepsin, ficin, IdeS, and Lys-Chave also been used. A list of different enzymes and theirspecificity is given in [151]. Optimized conditions are need-ed for each type of antibody due to variations in the cleav-age patterns of papain and pepsin with different IgG sub-classes and IgG derived from different species. In addition,the broad substrate specificity of these two proteases givesrise to heterogeneous fragments if the optimal digestionconditions are not properly met and controlled [152]. Mon-itoring of the fragmentation and purification processes canbe done by a large number of analytical techniques such asSDS-PAGE, IEF, cation exchange chromatography (CIEX),and CE [153].
6.1.2 Pepsin
Pepsin prefers aromatic and hydrophobic residues like Pheand Leu for cleavage, but has a rather broad specificity. When
Figure 6. Intact IgG and fragments generated thereof. Taken from[150] with permission.
applied to IgG under strictly controlled conditions, an F(ab0)2
part as well as an Fc moiety are formed, with the two poly-peptide chains of the Fc moiety connected via disulfidebonding (see Fig. 6). Due to the broad specificity of pepsin,some heterogeneity of the C-terminal F(ab0)2 is oftenobserved due to additional cleavages [154]. Kelly et al. [155]used agarose-bound pepsin in a 100 mM citrate bufferpH 3.5 for the digestion, followed by filtration as a first puri-fication step. Further purification was achieved by dialysisusing a 10 kDa cut-off membrane. Ashton et al. [153] used asodium acetate buffer (100 mM, pH 4.3) and soluble pepsinwith an enzyme to substrate ratio of 1:100 w/w. The resultingF(ab0)2 fragments were purified by ultrafiltration.
6.1.3 Papain
Papain cleavage of IgG generates two Fab fragments consist-ing of the light chain and the N-terminal section of the heavychain (see Fig. 6). The additionally produced Fc fragmentcomprises the disulfide-linked C-terminal sections of theheavy chains [156]. Papain has been used in a large number ofpublications dealing with the glycosylation analysis of IgGantibodies [41, 50, 70, 116, 126, 149, 152, 153, 156–158]. A pHof 6–7 is normally chosen for papain cleavage, which is routi-nely obtained using a phosphate or a Tris buffer containinglow concentrations of EDTA (1–4 mM) for papain stabiliza-tion. Papain is a thiol protease, requiring reducing agents(usually cysteine) for its activation. Lim et al. [41] stated thatpapain digestion in absence of cysteine was not reproducibleand may result in the formation of F(ab0)2 fragments. In mostcases, cysteine is present for activation of papain at low con-centration (1–10 mM) to ensure papain activation but to avoidfurther reduction of disulfide bonds between heavy and lightchain of the antibody due to its reducing action [152]. Thereaction is usually performed at 377C with typical incubationtimes of 1–8 h. Purification steps may include ultrafiltration,CIEX, and Protein A affinity chromatography. The fragmentsolution may directly be analyzed by LC-MS which accom-plishes both matrix removal and (partial) separation of thefragments. Further techniques to analyze the cleavage prod-ucts are IEF, SDS-PAGE, and MS (see Section 6.2).
6.1.4 Ficin
Ficin is similar to papain and produces either Fab or F(ab0)2
fragments, cleaving at one or several sites in the hingeregion. The incubation is accomplished in the presence ofmainly cysteine. In a study by Kelly et al. [155], a mAb wassuccessfully digested with pepsin, whereas another mAbunderwent additional cleavage resulting in peptide forma-tion.
6.1.5 IdeS
IdeS (streptococcal Ig-degrading enzyme), which has beendescribed only recently [151, 159] and is now commercially
© 2009 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

Proteomics 2009, 9, 882–913 897
available (“FabRICATOR”), is a streptococcal cysteine pro-teinase with structural homology to enzymes of the papainfamily. It was shown to have a unique specificity for IgG,which is cleaved in the hinge region with high specificityresulting in F(ab0)2 and two Fc fragments. Von Pawel-Ram-mingen et al. [159] showed that IdeS does not act on other Igclasses such as IgM, IgA, IgD, and IgE.
6.1.6 Lysyl endopeptidase
Limited proteolysis can be achieved when incubating IgGwith endoproteinase Lys-C at an enzyme/substrate ratio ofabout 1:500 in Tris-HCl at about pH 8 as described by Ren etal. [160] and Gadgil et al. [161]. The reaction is terminatedafter 30 min by shifting the pH to 4.5 or by adding DTT [162],then yielding the light and heavy chains of Fc and Fab. Ingeneral, when using Lys-C, care has to be taken not to inducepeptide formation [126].
6.1.7 Cleavage of disulfide bonds
Another possibility for fragment generation is the reductionof the disulfide bonds present between the heavy and thelight chain. Reduction may be applied to intact IgG or to Fcand Fab fragments obtained by enzymatic digestion [41, 42,153, 157] (see also Fig. 6).
DTT is most commonly used for reduction [41, 64, 70,108, 116, 126, 153, 157, 160, 162–166]. Alternatively,dithioerythritol (DTE) [149] and mercaptoethanolamine [155]can be used. Frequently, the reduction is accomplishedunder denaturing conditions using, e.g., 7.5 M guanidinehydrochloride in Tris buffer at a pH of around 7 [42, 108, 116,126, 160, 165]. In order to avoid rebinding of the cysteinethiol groups during the following analytical steps, alkylationreagents such as iodoacetamide [42, 64, 108, 116, 126, 149,153, 160, 166] and iodoacetic acid [162] are frequently added.Reduction can also be performed during analysis: IEF underdenaturing and reducing conditions was used for a fastanalysis of the isoform pattern of heavy and light chainwithout the need for sample workup [154].
6.2 Analytical techniques
Many different techniques are used for the analysis of intactIgG, as summarized in Table 1. The separation principlesinclude size, charge, polarity, affinity, and mass. The infor-mation that can be obtained largely differs depending onwhich technique is used. Many of the more classical tech-niques deliver only a limited amount of information and arenot sufficient for a detailed characterization of mAbs andtheir fragments. However, they are still widely used to obtainpreliminary information on molecular mass, to judge theoutcome of enzymatic reactions and to assess the sites ofheterogeneity. Many of them can also be used for purificationand fractionation purposes prior to MS analysis. In addition,they can be used for a fast monitoring during the optimiza-
tion of manufacturing processes. More detailed informationcan be obtained when chemical reactions on the PTMs areincluded such as removal of glycans by PNGase F [64, 70,108, 126, 140, 153, 158], desialylation [50, 158, 159], exogly-cosidase treatment on specific glycan substructures [64], ortreatment with carboxypeptidase B for the identification of C-terminal lysine residues [158, 173].
According to Table 2 the dominant separation proceduresare SDS-PAGE, (c)IEF and CIEX, and RP-HPLC. The latter isincreasingly used with on-line MS providing detailed struc-tural information. Most often, several of the techniquessummarized in Table 2 are combined in order to get a morecomprehensive understanding of the PTMs of the IgG and tocharacterize the mAb.
In the following, we will review the analytical techniquesregarding their efficiency and the information depth that canbe obtained. Due to the large number of analytical tech-niques in use, we will focus on selected publications.
6.2.1 Electromigrative separation techniques
6.2.1.1 SDS-PAGE (2-D) gel electrophoresis
SDS-PAGE and 2-DE in gels enable a separation according tosize and pI value. Both methods are widely applied inlaboratories and enable simple and fast measurements.Nowadays they are often used for fractionation prior to MSdue to the high sensitivity achievable with state-of-the-artmass spectrometric equipment. SDS-PAGE allows a roughestimate of the molecular mass. A positive bias in the esti-mation of the molecular mass of glycoproteins was observed,which is due to a lower SDS binding in the presence of car-bohydrates [149]. In general, the microheterogeneity presenton a glycoprotein is partly resolved in SDS-PAGE, but oftenbroad bands are obtained due to the insufficient mass reso-lution [126]. In 2-DE, series of spots are observed due to dif-ferences in glycosylation and particularly sialylation, asseparation is based both on size and pI [184, 185]. Gel bandsand spots may be excised, and proteins may be cleavedenzymatically with subsequent molecular characterizationby MS [166].
Gel electrophoresis with LIF detection after labeling themAb of interest or its fragments via succinimidyl esterreaction (5-carboxytetramethylrhodamine succinimidylester) has been described [166, 169]. Heavy and light chainas well as fragments thereof could easily be separated fromeach other, in general with high resolution. Peak assign-ment was critical and accomplished using chemical meth-ods, e.g., partial reduction with the observation of changesin signal heights. The methodology allowed, among others,impurity detection, and the separation of various fragmentssuch as F(ab0)2, Fab, Fc, and heavy as well as light chain.Shifts in migration times after removal of carbohydrateswith PNGase F were observed for the heavy chain, Fc andF(ab0)2.
© 2009 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

898 C. Huhn et al. Proteomics 2009, 9, 882–913
Table 2. Methods used for the analysis of intact IgG or its fragments
Analyticalmethod
Principle Information/use Major advantagesand shortcomings
References
SDS-PAGE Size Estimation of purity and molecular mass 1 General equipment, cheap, fast1 High-throughput possible2 Limited mass resolution2 Limited information
[116, 126, 149, 150,159, 166–172]
SEC Size Desalting, purification,and estimation of MW
1 Major isoform separation possible1 Orthogonal to RP-HPLC and HILIC2 Limited mass resolution
[126, 140, 156, 173]
CIEX Charge Number of charge variants andcharge heterogeneity,including chemical modificationsinformation on type of PTM;site specificity; N- and C-terminalmodifications
1 Cheap, fast1 High resolution and specificity1 Information on, e.g., deamidation2 Detailed information only when
chemical reactions included
[126, 156, 158, 172,174]
IEF [41, 126, 154, 158,172, 174, 175]
CE [153, 176, 177]
RP-HPLC Polarity Purification (desalting and matrixremoval), fractionation, separationof fragment species; Information onPTMs such as oxidation, N- andC-terminal modifications
1 High resolution1 Separation of major isoforms2 Risk of hydrolysis
[50, 64, 126, 126,150, 165, 168,173, 178]
Affinity columns(CE and LC)
Affinity Protein A and Protein G for separationof Fab and Fc purification andseparation of Fc, Fab and F(ab0)2
1 Fast, easy to handle2 Very limited information
[150, 157, 171]
Immunoblotting/immunoassay
Lectin affinity for glycosylation analysison intact or fragment level,assessment of glycosylation sites;differentiation between O-/N-glyco-sylation, detection of sialic acids
6 Limited information onO-/N-glycosylation onnonpeptide level
[116, 149, 155]
MALDI Mass Molecular mass, heterogeneity in mass,relative ratio of glycoforms,high number of possible PTMsaccessible, sequence confirmation,estimation of relative concentrationsof glycoforms
1 Detailed information, fast2 Only high mass differences on
intact level accessible6 Fragment generation for reduced
complexity2 Laborious sample purification
[64, 116, 126, 153,179, 180]
ESI-MS Mass [42, 64, 70, 116, 126,149, 153, 154,161, 180, 181]
SEC-MS Size 1 mass Compared to MS alone, coupling to LCand SEC provides additionalconfidence in signal assignmentbased on migration times and mass
1 Reduced sample preparation1 On-line desalting and matrix removal1 On-line separation of fragments1 Pure spectra with less adducts2 Optimization necessary2 Expensive equipment and
trained personnel2 Risk of on-column hydrolysis
[165]LC-MS Polarity 1 mass [41, 42, 50, 108, 126,
140, 150, 152,154, 160–164,173, 178, 180,182, 183]
6.2.1.2 cIEF/IEF
cIEF is widely applied for the analysis of IgG. The method ischeap and easy to handle, and the necessary equipment ispresent in many laboratories working with proteins. In termsof optimization, the ampholytes must be chosen to fit to the pIvalues of the IgG isoforms to be detected. Applications forprotein and glycoprotein analysis in general are given in [185–187]. For cIEF but also for slab gel IEF, care has to be taken notto precipitate the proteins enriched in narrow pI-bands [182].
At first, using appropriate markers, the pI of the IgG canbe determined. The impressive resolving power based on
subtle differences in the pI of different isoforms enables adetailed look on the charge heterogeneity of IgG molecules[126, 158, 175]. pI differences are due to the presence of sialicacids, deamidation, N-terminal lysine variation, C-terminalpyroglutamate formation, and succinimide formation. LikeCIEX, cIEF is able to make deamidation on asparagine andglutamine residues visible at the level of intact protein orfragments thereof. Lim et al. [41] analyzed a therapeutic mAbwith two glycosylation sites revealing a high heterogeneity asvisible in the IEF-electropherogram of the intact antibody(Fig. 7). Using papain digestion, it was nicely shown that thecharge heterogeneity was present in the Fab part. By LC-MS
© 2009 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

Proteomics 2009, 9, 882–913 899
the heterogeneity was shown to be largely due to differencesin sialylation of the Fab N-glycan.
Additional information may be obtained by combininganalytical techniques with enzymatic reactions that address aspecific PTM.
Figure 8 shows an example, were cIEF was used to ana-lyze the heterogeneity of light and heavy chains of the F(ab0)2
fragment of a murine mAb. Analysis was performed beforeand after incubation with neuraminidase to remove sialicacid residues. The two peaks assigned to the Fab part of theheavy chain collapsed after neuraminidase treatment, whichindicated partial sialylation of the Fab part of the heavy chain.
IgG glycosylation analysis by IEF has been restricted tothe analysis of sialoforms [154] (Fig. 8). In addition, C-ter-minal variants of IgG differing in terminal lysine residuescan be well separated by cIEF [158], and carboxypeptidase Bcan be applied to identify the presence of this PTM.
6.2.1.3 CE
There is a large number of applications of CE in glycoproteinanalysis [186]. Glycoform separation was achieved for ery-thropoietin (EPO) [188, 189] and transferrin [190] withseparation of sialoforms. Additionally, erythropoietin iso-forms differing in the glycan composition can be further
separated and analyzed with MS [191, 192]. Regarding IgGanalysis, there are publications on the separation of IgGsubclasses (IgG1, IgG2, and IgG3) [176], the on-line pre-concentration of IgG on monolithic devices [177] and on theuse of CE for the control of enzymatic reactions [153]. Arecent CE-LIF analysis of IgG did not show separation ofglycoforms (Application Note Picometrics AN 066).
6.2.2 Chromatographic separation techniques
6.2.2.1 CIEX
Similar to cIEF, CIEX is used to assess the charge hetero-geneity of IgG isoforms. The use of this method seems tobe straightforward with a high potential of separation,while especially N-terminal and C-terminal modificationscan be analyzed. C-terminal lysine variants were separatedby CIEX [126, 156, 158] and identified by, e.g., treatmentwith carboxypeptidase. The presence of sialic acid residuescan be determined by reanalyzing a sample after sialidasetreatment [14]. The separation of Fc and Fab fragmentsreveals site-specific information on the presence of PTMs,e.g., for C- and N-terminal variants [156]. CIEX was alsoused for purification purposes [126].
Figure 7. Imaged cIEF electropherograms of a mAb showing the pI values of the intact mAb (A) and of the Fab and Fc fragments aftertreatment with papain (B). Taken from [41] with permission.
© 2009 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

900 C. Huhn et al. Proteomics 2009, 9, 882–913
Figure 8. cIEF under reducing conditions with EOF mobilization. (A) Isoform pattern of heavy chain (HC) and light chain (LC) of F(ab0)2. (B)Isoform pattern after neuraminidase treatment. The more acidic isoform of the heavy chain HC2 is largely reduced due to the removal ofsialic acids, while HC1 increases. The peak areas of the light chain isoforms remain unaltered. Taken from [154] with permission.
6.2.2.2 RP-HPLC
RP-HPLC is extensively used for the analysis of glycopro-teins, including IgG. The most often applied stationaryphases include C4, C8, and C18 RP materials. Dillon et al.[108, 173, 178] demonstrated the effect of column tempera-ture, solvent strength, and pH on IgG separation. Heavy andlight chain together with additional N-terminal variants werewell resolved and analyzed by RP-HPLC-UV [108].
When MS is used for detection, the optimization of the ionpairing reagent in the mobile phase might be necessary to ob-tain a good peak shape as well as a high sensitivity [126, 164,173, 178]. TFA is most often used (see Section 5.2.1), resultingin excellent peak shapes. However, ionization efficiency isgenerally decreased with TFA, thus other systems with FA,acetic acid, and heptafluorobutyric acid have been proposed inorder to obtain a compromise between sensitivity and peakshape. Care has to be taken when analyzing sialylated glyco-proteins by LC, because the loss of sialic acids is possible underacidic conditions at elevated temperature [41]. Further hydro-lysis can occur, e.g., cleavage between aspartic acid and prolineresidues on the heavy chain [108]. A separation of glycoformsis generally not achieved, thus most of the work on glycosyla-tion analysis has been done with MS detection (both on-lineand off-line; see Section 6.2.4).
6.2.2.3 SEC
SEC has been used for the analysis of the heavy and lightchains of mAbs as well as for preparative purposes [126, 174].
The fragments were well separated and could be identifiedwith off-line ESI-MS. Moreover, SEC can readily be coupledon-line to ESI-MS (see Section 6.2.4).
6.2.2.4 Affinity chromatography
Lectin binding assays have been used to analyze the glycosy-lation of IgG from human serum (see Section 2). Immobi-lized lectins are used for glycoprotein purification ratherthan for analytical purposes. Lectins with high specificity forcertain oligosaccharide structures can be utilized to selec-tively concentrate specific glycoproteins or glycoformsthereof [193, 194].
Moreover, Protein G and Protein A columns are used topurify IgG (Section 3) and to separate larger fragments [116,126, 149, 171]. Papain digestion was characterized usingHPLC with affinity columns based on Protein A monolithswhich separated (Fab0)2 from the Fc fragment. An affinitycolumn placed in-line with RP-HPLC for capturing and pu-rification of a mAb has been presented by Battersby et al.[150].
6.2.3 MS
MALDI and ESI are the two ionization techniques used inMS of intact IgG. When analyzing intact IgG as well as otherhigh-molecular mass proteins [195, 196], the resolution ofthe mass spectrum is not sufficient to allow detailed charac-terization, as minor modifications such as deamidation orpyroglutamate formation are not resolved. The relatively
© 2009 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

Proteomics 2009, 9, 882–913 901
large mass differences between various glycoforms (e.g.,162 Da mass increment of a Hex residue) allow the identifi-cation of glycoforms at the level of the intact protein, both forMALDI and ESI. In general, however, the mass spectromet-ric analysis of IgG fragments often gives a more detailedpicture of glycosylation and additional modifications thanthe analysis of intact IgG.
6.2.3.1 MALDI
While the high molecular mass of IgG can hinder the reso-lution of isoforms with low mass differences by MALDI-TOF-MS, differences in the overall galactosylation of the twoFc N-glycans are usually fully resolved (i.e., G0, G1, and G2glycoforms). This information on the relative ratios of glycanstructures is important for cell culture monitoring.
In order to assess the molecular mass of the proteinbackbone (including nonglyco PTMs), a deglycosylation stepis often included (e.g., [153]). MALDI-TOF-MS of IgG beforeand after deglycosylation directly reveals the overall glycancomposition. Additional stepwise release of glycan sub-structures using exoglycosidases allows a more precisedescription of the glycan moiety. More detailed analysisusing MALDI is carried out at the level of IgG fragments [64,153]. The reduced overall molecular mass allows the assess-ment of glycosylation sites as well as the analysis of otherPTMs, which are not clearly visible at the level of intact IgG[196]. It has to be noted that the glycosylation analysis of sia-loforms with MALDI is difficult due to the lability of the sia-lic acid linkages and the resulting (partial) loss of this residueduring MALDI-TOF-MS analysis, and due to the low ioniza-tion efficiency of sialylated glycoforms.
The major drawback of MALDI-TOF-MS is that one hasto work with carefully purified samples. Effective desaltinghas to be carried out to avoid adduct formation, which wouldotherwise further complicate the mass spectra. Differentpurification strategies have been applied (see below), includ-ing fractionation of antibody samples by LC [126]. An ad-vantage of MALDI-TOF-MS is that one instrument can beused for the analysis of intact IgG, its fragments but also forglycans and peptides, so that a full characterization of a mAbis possible without the need for additional equipment [179](Section 7).
6.2.3.2 ESI-MS
ESI-MS spectra of proteins show multiply charged ions, anddata are often represented after deconvolution, for which pa-rameters have to be set properly to avoid signal artifacts [42,195]. As in the case of MALDI-MS, a thorough sample puri-fication is necessary to avoid adduct formation and to obtainhigh-quality spectra [196] (see below). Notably, glycoproteinsare usually ionized less efficiently by ESI than their deglyco-sylated counterparts due to the potential protonation sitesthat are covered by glycans and due to the fact that desolva-
tion phenomena are less effective when glycans are attachedto the protein [186, 196].
Similar to MALDI-MS, direct infusion ESI-MS measure-ments on the intact IgG are used to determine the overallmolecular compositions of the sum of glycans attached,which may be performed by comparing glycosylated anddeglycosylated species [70], assuming that the ionizationefficiency is similar for all isoforms, as has been demon-strated by Wan et al. [164]. A comparison of ESI-MS for theanalysis of IgG fragment glycosylation with HPLC analysisof the released glycans gave very similar results, showingthat ESI-MS can yield glycoform profiles (also relative pro-portions of glycoforms) including semiquantitative detectionof sialylation [70].
Additional information on PTMs can be obtained whenlight and heavy chain [70, 149, 180, 181] or Fc and Fab frag-ments [70] are analyzed: Jiskoot et al. [149] analyzed the Fcsubunit of a mAb and observed two major and several minormultiply charged components in a relatively complex spec-trum already at this fragment level. The complexity was at-tributed to a second glycosylation site additional to the com-mon N-glycosylation in the CH2 domain. Further analysisusing lectin binding revealed the presence of an O-glycosy-lation. Deamidation was suggested to be the reason for a6 Da mass difference (experimental vs. theory) [149] but noproof on the basis of additional analytical techniques waspresented.
6.2.3.3 Sample preparation for ESI and MALDI
As mentioned before, sample purification is a major issue,when direct infusion ESI-MS or MALDI-MS measurementsare to be performed. Several strategies have been describedwhich are often similar for the purification of intact IgG andits fragments. Fractionation by LC or SEC and also CIEX hasbeen used by several groups [126, 158, 180]. This alsoincludes the preseparation of Fc and Fab fragments by affin-ity columns (see Section 6.2.2). The advantages here aresimilar to the on-line coupling except that the sample prepa-ration is more labor intensive. Ultrafiltration [149, 153, 180]and dialysis [70, 149, 180] are commonly applied. Desaltingis also carried out using ion exchange columns [64]. Excisinggel bands is a further approach, which represents an effi-cient, but labor-intensive purification step [180].
6.2.4 Coupling of separation techniques to MS
A large number of papers demonstrate the need for fastanalytical techniques with minimized sample preparation,high robustness, and good repeatability providing generalinformation on PTMs to allow the monitoring of biotechno-logical antibody production and a fast quality control. Allthese tasks are met by separation techniques used in con-junction with MS. Separation techniques may be used for theefficient removal of salt and matrix components from intactIgG or fragments thereof. In addition, (partial) separation of
© 2009 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

902 C. Huhn et al. Proteomics 2009, 9, 882–913
fragments and their isoforms may be obtained. This stronglyreduces the complexity of the mass spectra when severalPTMs are present on the IgG molecule.
6.2.4.1 SEC-MS
Brady et al. [165] have applied SEC-MS to obtain the molec-ular mass of mAbs. The SEC column allowed an efficientdesalting and was shown to be faster and more sensitive thancommon RP-HPLC analyses. An advantage of this method-ology is that the elution profiles of different mAbs are simi-lar, making method optimization unnecessary when analyz-ing different mAbs. The use of FA buffers enhanced thesensitivity compared to TFA. Next to intact mAb, heavy andlight chains were analyzed, though the latter two were notseparated from each other. The method proved to be highlytolerable with regard to matrix salt composition.
6.2.4.2 LC-MS
The use of LC-MS in glycoprotein analysis is well estab-lished. The large number of papers dealing with the analysisof glycoforms present on IgG (see Table 2) demonstrates theusefulness of this method. A growing number of laboratoriesare equipped with this technology and its application in IgGanalysis will surely increase.
As with SEC, the use of LC-MS for the analysis of intactIgG allows an efficient and automated desalting process.Sample preparation steps such as dialysis or ultrafiltrationare normally not necessary. In addition, matrix components,e.g., other proteins or peptides as well as cell debris from cellculture samples can eventually be separated before detection.However, LC methods have hitherto not achieved a separa-tion of IgG glycoforms. Therefore, MS offers a second di-mension that allows the identification of the glycoforms. Anexample is given in Fig. 9 [42]: the first ESI-MS spectrumshows the charge envelope of an IgG1 analyzed by LC-MS.
Figure 9B gives a close-up view of the 531 charge state,where the glycoforms were assigned by calculation using themolecular mass of the mAb protein part. Deconvolutionnicely shows six pairs of glycoforms (Fig. 9C). In case of theisobaric G0F/G2F and G1F/G1F, discrimination is not pos-sible at the level of intact IgG. In this example, no furtherPTMs were observed, so that the assignment of mass signalsto glycoforms was straightforward. In other cases, highercomplexity by additional PTMs is often addressed by analyz-ing Fab and Fc fragments [152, 160, 161] or heavy and lightchain [108, 140, 160, 163–165, 178] using LC-MS. Notably,while some reports obtain only partial chromatographic res-olution of the fragments [152, 165], others succeed to sepa-rate the major fragments (e.g., [108, 140, 160, 161, 164, 178]).In the latter case, pure spectra of the fragments are obtainedand the glycoforms can easily be assigned.
Additional PTMs on the IgG fragments can lead to fur-ther signals in the LC. Rehder et al. [108], for example, ana-lyzed light and heavy chains of a reduced mAb. Beside gly-
Figure 9. ESI-TOF MS spectra of the intact IgG1 by LC-MS analy-sis. (A) ESI-MS raw spectrum showing a series of multiplycharged molecular ions. (B) ESI mass spectrum for the 531
charged ion of the intact IgG1 with glycoform labels. (C) Decon-voluted MS spectrum showing the overview of the glycosylationheterogeneity of the mAb. Taken from [42] with permission.
cosylation comprising the unusual Man5 structure, theydetected variants of the heavy chain containing N-terminalglutamine and pyroglutamic acid. These isoforms were par-tially separated by LC and identified using MS.
The versatility of RP-HPLC-MS was demonstrated byRen et al. [160], who applied RP-HPLC-MS to determine
© 2009 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

Proteomics 2009, 9, 882–913 903
chemical modifications on a mAb with an unpaired cysteineon each of the heavy chains in the Fab domain, analyzing theintact and reduced glycoprotein as well as fragments gener-ated by limited proteolysis via Lys-C. The use of a diphenylcolumn together with the thorough optimization of the LCconditions enabled (i) the separation of the cysteinylatedfrom the noncysteinylated intact mAb. (ii) The separation ofthe heavy and light chain after reduction was achieved,showing additional small peaks corresponding to methio-nine oxidation products of 5% relative abundance. (iii) Di-sulfide bond connectivity was also analyzed using this meth-od: Lys-C limited proteolysis was applied instead of digestionwith papain or pepsin. This treatment caused a single cleav-age, C-terminal to a lysine residue in the hinge region, whilemaintaining the disulfide structure of the Fc and Fabdomains. Differences in migration times as well as differ-ences in the molecular mass of 2–4 Da demonstrated theability of this LC-MS method to distinguish disulfide bondconnectivity. Additional chemical methods were applied inorder to prove that the registered mass difference was notdue to deamidation or even insufficient mass precision.
Gadgil et al. [161] also used LC-MS for the characteriza-tion of another recombinant monoclonal IgG1 antibody.Within one LC-MS run of Fc and Fab fragments generated bylimited proteolysis with Lys-C, many modifications includinggalactosylation, cysteinylation, C-terminal lysine variants,oxidation, and a possible succinimide formation were ana-lyzed. The good chromatographic separation of differentisoforms helped to largely reduce the complexity of the massspectra. The LC-MS separation of IgG1 or IgG2 isoforms wasoptimized using high column temperature, low pH, solventswith high elutropic strength, and columns with long alkylchains [178]. Mass data reflected a common glycosylationprofile of a biantennary carbohydrate attached to the Fc do-main. Multiple mass peaks for each LC signal were shown tobe due to the truncation of terminal galactose residues. Inaddition, conformational isoforms formed by partial reduc-tion of disulfide bonds were separated by LC.
The ease of automation is an additional aspect to men-tion here: Wan et al. [164] monitored galactosylation levels inthe SIM mode using RP-HPLC-MS via monitoring the 381
ions for G0, G1, and G2 during recombinant antibody pro-duction. To summarize, a detailed picture of the PTMs ofIgG can be obtained by LC-MS without the need of excessivesample preparation and purification.
6.3 Applications to IgG microheterogeneity analysis
6.3.1 Additional O-glycosylation
The presence of O-glycans on 30% of the IgG2 expressed inChinese hamster ovary (CHO) cells was demonstrated by LC-MS of the Fc subunit together with a lectin binding assay[160], and by LC-MS of the light chain, peptide mapping, N-terminal sequencing, and a-mannosidase treatment [163].
Furthermore, Masuda et al. [181] detected 14% O-glycosyla-tion in the hinge region of an IgG2 mAb by ESI-MS.
6.3.2 Second N-glycosylation site
The analysis of IgG fragments allows site-specific determi-nation of glycosylation. Chicken serum IgG contains twopotential N-glycosylation sites located on CH2 and CH3domains. Lectin binding assays showed that the Fc part con-tained high-mannose-type oligosaccharides (staining withCon A) whereas the variable part of Fab contained complex-type N-glycans (staining with Ricinus communis agglutinin-I(RCA-I)) [116].
An additional N-glycosylation site has also been de-scribed for a recombinant mAb from NSO-cells [64]. It waspresent on the heavy chain and contained nonsialylatedbiantennary fucosylated structures. The analytical strategyincluded ESI-MS and MALDI-MS after fractionation withLC, oligosaccharide analysis with MALDI-MS after enzy-matic glycan release from the light and heavy chains excisedfrom SDS-PAGE bands, MALDI-MS of tryptic peptides, andsequencing of the amino acid heavy and light chain.
A mAb from CHO cell lines was analyzed to show anadditional N-glycosylation on the heavy chain [41]. Aftertreating the mAb with papain and DTT, the analyses werecarried out using LC-MS and a cIEF method (see also Fig. 7).The chromatographic run enabled the separation of the gly-cosylated Fc heavy chain fragment from the glycosylated Fabheavy chain fragment.
6.3.3 Glycan pairing on the Fc part
In order to analyze the glycan pairing, Masuda et al. [157] an-alyzed the Fc fragments with and without the interchain S–Sbridges present (Fc and Fc/2) using LC-MS and glycan analy-sis. The Fc/2 fragment showed two major and two minor sig-nals in the mass spectrum with compositions of ((Hex)3(Hex-NAc)3(Fuc), (Hex)3(HexNAc)4(Fuc), (Hex)4(HexNAc)4(Fuc),and (Hex)5(HexNAc)4(Fuc), corresponding to G0-GlcNac, G0,G1, and G2) as calculated from the mass differences to themolecular mass of the mAb backbone. The ratios of glycans inthe single Fc/2 parts were compared to those determined forthe Fc fragment. Various combinations of glycans, both ho-mologous and heterologous, were observed. The ratios weresignificantly different from what could be expected on thebasis of random pairing. Pairing tended to be preferably ho-mologous, which might result in symmetric folding of the twopolypeptide chains of the Fc part.
6.3.4 Presence of disialic acids
The analysis of disialic acid moieties present on the lightchains of IgG from mouse serum, presumably on an O-gly-can was performed by Yasukawa et al. [171, 197]. IgG lightchains were purified from mouse serum and confirmed tocontain disialic acids by three methods including immuno-
© 2009 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

904 C. Huhn et al. Proteomics 2009, 9, 882–913
staining with an antibody specific for the a2,8-linkage, anionexchange chromatography after labeling with 1,2-diamino-4,5-methylenedioxybenzene (DMB) and a fluorimetric C7/C9
analysis.
6.3.5 Disulfide bond connectivity
Reduction of disulfide bonds leads to free sulfhydryl groupsand is a common chemical degradation observable for mAbs,possibly leading to cross-linkages in the protein. Ren et al.[160] used LC-MS for the determination of the disulfide bondconnectivity after limited proteolysis with Lys-C maintainingthe disulfide structure of the Fc and Fab domains. The chro-matogram showed several minor signals besides the ones forFc and Fab, partly due to methionine oxidation (16 Da massshift) but also due to single or double disulfide bond reduc-tion (2 and 4 Da). Additional analysis after blocking the freethiol groups was necessary to confirm this assumption.
6.3.6 Cysteinylation
Ren et al. [160] applied RP-LC-MS to determine chemicalmodifications on a mAb with an unpaired cysteine on each ofthe heavy chains in the Fab domain, analyzing the intact andreduced glycoprotein as well as fragments generated by lim-ited proteolysis via Lys-C. The use of a diphenyl column to-gether with thorough optimization of the LC conditionsenabled the separation of the cysteinylated from the non-cysteinylated intact mAb, where the mass spectra revealedglycoforms of, e.g., G0/G0 and G0/G1, overlaid by the het-erogeneity in cysteinylation. LC-MS was also used to deter-mine cysteinylation of free cysteine in the Fab region of arecombinant IgG1 mAb [161]. Cysteinylation on the Fabfragment caused a 119 Da mass difference.
6.3.7 C-Terminal lysine clipping and N-terminal
pyroglutamate formation
The mass spectrum of the reduced Fc part of a mAb clearlyshowed the presence of C-terminal lysine with a mass dif-ference of 128 Da, with the relative abundance being de-pendent on the cell culture conditions [41]. C- and N-termi-nal modifications were analyzed using cIEF and cIEX [158,174], showing the advantages of separations based on chargefor these modifications. N-terminal modifications, here pyr-oglutamate formation, were determined by Moorhouse et al.[156] analyzing Fc and Fab fragments from papain digestionusing a validated cIEX method. Pyroglutamate was presentat 0, 1, (with two different variants) or 2 termini, and theresulting four isoforms were well separated by cIEX. C-ter-minal variants were also detected on the Fc part showing 0–2lysines. Identification of the signals was achieved with addi-tional methods such as MS and tryptic digestion. Alter-natively, C- and N-terminal modifications were analyzed byLC-MS [161].
6.3.8 Methionine oxidation
Methionine oxidation to sulfoxide can be an artifact of sam-ple preparation, but can also occur in vivo [199]. Methionineoxidation on the heavy chain was analyzed using LC-MS[160]. The optimized LC method allowed the separation ofisoforms differing in the number of methionine oxidationsas reflected in the mass differences of 16 Da, accounting foran overall methionine oxidation of about 5%. A mass differ-ence of 16 Da was also observed by Gadgil et al. [161] and anoxidation was presumed.
6.3.9 Further modifications
Succinimide formation was assumed for the Fab fragment ofa mAb due to an observed mass shift of 17 Da [161]. Glyca-tion analysis has been achieved by MALDI-MS [200] and LC-MS [41, 183], and nitration analysis by LC-MS [162]. Deami-dation analysis was carried out for the heavy and light chainusing CIEX [174].
7 Integration of analytical strategies
In order to fully characterize IgG derived as mAbs from bio-technological synthesis, the above-described methods have tobe combined to get a detailed picture on the amino acidsequence, but also on the large variety of PTMs present. Incase of glycosylation analysis this means that all levels ofanalysis have to be implemented: the analysis of intact IgGand its fragments (Section 6) gives preliminary informationon the glycosylation as well as other PTMs of the mAb. Thein-depth characterization of a mAb requires the analysis ofreleased carbohydrates (Section 4) and the analysis of trypticpeptides (Section 5). Strategies for an in-depth characteriza-tion of mAbs are manifold [186]. Next to methods for in-depth characterization, other approaches allow the monitor-ing of antibody structural features during production andquality control. In this section, we want to present somestrategies applied to IgG characterization extracted fromsome selected publications in order to show the complexanalytical schemes necessary.
The analysis of mAbs from different cell lines hasrecently been presented by Wagner-Rousset et al. [140]. Theyused three levels of analysis: (i) the analysis of intact andreduced mAbs was accomplished by LC-ESI-TOF-MS. Theauthors stated that the analysis of mAb heavy and light chainafter LC separation revealed mass spectra of higher resolu-tion due to the lower molecular mass of the fragments com-pared to the intact antibody. In addition, the complexity isreduced because only one glycosylation site remains permolecule. The assignment of the major glycoforms was pos-sible with a high accuracy. (ii) The analysis of the structuresof the released N-glycan pool by ESI-TOF-MS was achievedusing direct infusion and MS/MS analysis. The common N-glycans such as G0, G1, and G2 were identified and their
© 2009 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

Proteomics 2009, 9, 882–913 905
fragmentation patterns discussed. Two further minor glycanstructures were reported as isobars of G2. (iii) The third levelof glycoprotein analysis was introduced as peptide mappingwith a novel nano-LC-Chip-MS/MS approach using a gra-phitized carbon column, showing some advantages of thiscolumn over RP material. Peptides and glycopeptides fromthe heavy chain of the mAb were generated using trypsin.Nine glycopeptides were detected and identified via theirMS/MS spectra. The peptide mapping provided informationon the position of the N-glycan (here Asn297), the peptideamino acid sequence and the microheterogeneity of the N-linked glycan [140].
The glycosylation analysis directly from SDS-PAGEseparated proteins or protein fragments was introduced byKüster et al. [166]. The bands of interest were excised andpurified by washing with NaHCO3 buffer or just dried. Theresulting gel pieces containing the analyte were then sub-jected to in situ digestion by PNGase F or trypsin. Trypsin waseither applied to the glycoprotein itself or to the deglycosy-lated proteins. Extracted glycans were purified and (i) thesialic acids esterified with methyliodide to facilitate MALDImeasurements (better stability and better ionization in thepositive mode) or (ii) subjected to LC analysis after fluores-cent labeling with 2-AB. In addition, exoglycosidases wereapplied to analyze the terminal glycan structures. The ad-vantage of this strategy is its high sensitivity as 50–100 pmolglycoprotein applied to gels are sufficient for glycan profilingand sequencing. Samples are easily purified by SDS-PAGE.The whole analysis is possible within three days, revealinginformation on specific glycoproteins also from proteinmixtures. The analysis of both intact proteins as well as theirfragments (in case of IgG light and heavy chain) is possiblewith information on the glycosylation sites.
A somewhat similar approach was presented by Lattovaet al. [179], who used an on-target method for the investiga-tion of oligosaccharides and glycosylation sites in glycopep-tides and standard glycoproteins using MALDI. No sampleclean-up steps were necessary between analytical steps, andenzymatic reactions were accomplished on the MALDI tar-get. The method was applied to different glycoproteins andtryptic peptides from IgG and mouse serum. In contrast tothe use of gel electrophoresis prior to the analysis [166], apreliminary sample preparation, like HPLC fractionationwas necessary for mouse sera. Another strategy for a com-prehensive characterization of IgG from chicken serum waspresented by Suzuki and Lee [116]. Glycans were released byPNGase F from a trypsin/chymotrypsin digest of IgG andpurified with a Dowex column and a Carbograph cartridge.Reductive amination was carried out using PA. The glycanswere identified using HPLC and MALDI-TOF-MS, revealingthe presence of (i) high-mannose type oligosaccharides and(ii) biantennary complex-type oligosaccharides, with sialyla-tion present in some cases. Site specific localization of theglycans was achieved at the level of Fc and Fab fragmentsobtained by papain digestion, which were separated on aDEAE-Sepharose column and subjected to immunoblotting,
SDS-PAGE and lectin blotting, staining with Con A (only Fcfragment with high mannose glycans was positive), andRCA-I (only Fab fractions with complex glycans were posi-tive). Furthermore, the exact glycosylation sites as well asglycans present were derived via the mass spectrometricanalysis of glycopeptides. Another strategy [180] usedMALDI for the determination of the molecular mass of anintact mAb, a tryptic LC-MS map for the determination ofglycosylation sites and glycosylation composition andEdman degradation for the analysis of peptide sequences.The approach revealed C- and N-terminal variants, methio-nine oxidations on the basis of MS/MS data and one N-gly-cosylation site per heavy chain. No O-glycosylation wasreported. The glycosylation profiles were determined byHPAEC of released oligosaccharides. Their sequences andlinkages were assessed by MALDI-MS and controlled diges-tions with specific exoglycosidases.
The glycosylation profiles of two different myelomaIgGs were compared by Mimura et al. [70] at the level of Fcfragments, heavy chains, and light chains using ESI-MS.In order to analyze the glycosylation, the molecular massof Fc was analyzed before and after deglycosylation,revealing G0, G1, and G2 glycans with different degrees offucosylation for the two IgG preparations analyzed. Quan-titative HPLC analysis of oligosaccharides released fromIgG tryptic peptides gave comparable results. A similar setof data was obtained for the heavy chain. For one IgGsample, additional glycosylation on the light chain wasobserved. The Fab glycans could only be released by EndoF2 and not by PNGase F. The glycans of the light chainwere estimated to be highly sialylated biantennary oligo-saccharides in agreement with the results from releasedglycan analysis. Regarding the good correlation of glycananalysis and MS results, the authors concluded that ESI-MS is a valuable tool to obtain preliminary glycoform pro-files for IgG.
In conclusion, these publications show that a full char-acterization of IgG derived from cell cultures requires thecombination of many analytical techniques. In all cases, MSis included to obtain detailed information on the glycosyla-tion present. Expensive equipment and trained personnel isthus required.
8 Outlook
The glycosylation analysis of IgG from human serum hasuntil now mainly been descriptive, showing as major fea-tures the lifetime dependency of the galactosylation of the FcN-glycan and the association of IgG G0-glycoforms with var-ious diseases. Research from the group of Ravetch has addeda functional facet to IgG glycosylation, as they described animmunosuppressive property of IgG which is largely de-pendent on sialylation of the Fc N-glycans [15]. This obser-vation is expected to stimulate research on the expressionand regulation of sialic acid on Fc N-glycans for different IgG
© 2009 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

906 C. Huhn et al. Proteomics 2009, 9, 882–913
populations in health and disease. Moreover, the degree offucosylation of the Fc N-glycan of human serum IgG willreceive more attention in future studies, because the lack ofcore-fucose strikingly enhances the interaction of IgGs withFc-receptors, as has been shown for various mAbs. Differ-ences in core-fucosylation might, therefore, strongly influ-ence the ADCC activity of human serum IgGs during ahumoral immune response, both in the defense againstinfectious agents and in autoimmune diseases. An addition-al level of complexity has recently been indicated by lookingat the Fc glycosylation of antigen-specific IgGs in infection[21] and autoimmunity [123], showing that IgG glycosylationdiffers for various IgG subpopulations from human serum.Many more studies will be necessary, however, to elucidatethe mechanisms regulating IgG Fc glycosylation in humansand animal models.
Regarding the analytical techniques, the requirement formore detailed characterizations of IgG glycosylation (sialyla-tion and fucosylation) will lead to an increase of the applica-tion of mass spectrometric techniques also in this clinical/biological field. While mass spectrometric analysis of intactserum IgGs will not be an option due to the polyclonal na-ture of most human immune responses, both analysis ofoligosaccharides and glycopeptides as well as IgG fragments(mainly the Fc part) will have their place in future studies.Due to the higher degree of automation and the reducedamount of sample preparation necessary, coupled tech-niques of separation and mass spectrometric detection willdominate the methodology, such as LC-MS of (glyco-)pep-tides but also graphitized carbon-HPLC-MS for oligo-saccharide analysis allowing the differentiation of structuralisomers (Section 4.4.2). For pharmacokinetic studies, glyco-peptide analysis is the method of choice offering both highsensitivity and specificity in both the glycan structureassignment and the identification of the mAb.
The other, considerably larger field of IgG glycosylationanalysis, which deals with the characterization of recombi-nantly expressed mAbs, is expected to grow within the nextyears. New expression systems are expected to gain impor-tance, next to the currently predominantly used mammaliancell lines. These may include genetically modified mamma-lian cell lines which show altered IgG glycosylation profileswith, e.g., lowered core-fucosylation [201], or plant [71, 202,203] and yeast [204, 205] expression systems with a thor-oughly refurbished glycosylation pathway, established inorder to avoid potentially antigenic glycan structures and gettoward a humanized glycosylation. A full characterization ofthese antibodies, including the amino acid structure and thepresence of PTMs is necessary to avoid immunological reac-tions to nonhuman-like glycosylation. All levels of glycopro-teins analysis, intact IgG and its major fragments, the analy-sis of glycopeptides and of released glycans have to beapplied. Importantly, MS of the released glycans can providevery detailed structural information and will, therefore, con-tinue to play a key role (Section 4). Recently, Chung et al.[206] have highlighted the complications caused by galac-
tose-a1,3-galactose epitopes on a therapeutic antibody, whichinduces anaphylaxis in some of the patients, often withinminutes after the first drug application. Anaphylaxis is pre-sumably mediated by pre-existing anti-a-Gal IgE antibodies.
Next to in-depth analysis, quality control during devel-opment and production will further stimulate the develop-ment of analytical techniques with higher throughput. Manyanalytical steps including sample preparation, separation,and detection are expected to be automated, which will helpto increase capacity and robustness. Another aspect that isgaining importance is the shortening of the total analysistime: feedback information from IgG glycosylation analysiscan be used to regulate the production process in large-scalecell culture. Two methods which are suitable to address theseanalytical challenges are the analysis of released glycans byHILIC-HPLC with fluorescence detection or by capillary gelelectrophoresis using DNA sequencer equipment [81, 89].For both methods, sample preparation may be shortened andstreamlined.
To summarize, the analytical techniques applied for IgGanalysis will further develop toward a more detailed charac-terization of the glycosylation and possible further PTMs butalso toward fast, automated high-throughput method forglycosylation monitoring.
This work was supported by IOP Grant IGE05007 and by afellowship within the Postdoc-Programme of the German Aca-demic Exchange Service (DAAD).
The authors have declared no conflict of interest.
9 References
[1] Parekh, R. B., Dwek, R. A., Sutton, B. J., Fernandes, D. L. et al.,Association of rheumatoid arthritis and primary osteoar-thritis with changes in the glycosylation pattern of totalserum IgG. Nature 1985, 316, 452–457.
[2] Wang, W., Singh, S., Zeng, D. L., King, K., Nema, S., Antibodystructure, instability, and formulation. J. Pharm. Sci. 2007, 96,1–26.
[3] Jefferis, R., Glycosylation of recombinant antibody ther-apeutics. Biotechnol. Prog. 2005, 21, 11–16.
[4] Parekh, R., Roitt, I., Isenberg, D., Dwek, R., Rademacher, T.,Age-related galactosylation of the N-linked oligosaccharidesof human serum IgG. J. Exp. Med. 1988, 167, 1731–1736.
[5] Yamada, E., Tsukamoto, Y., Sasaki, R., Yagyu, K., Takahashi,N., Structural changes of immunoglobulin G oligosacchar-ides with age in healthy human serum. Glycoconj. J. 1997, 14,401–405.
[6] Shikata, K., Yasuda, T., Takeuchi, F., Konishi, T. et al., Struc-tural changes in the oligosaccharide moiety of human IgGwith aging. Glycoconj. J. 1998, 15, 683–689.
[7] Pekelharing, J. M., Hepp, E., Kamerling, J. P., Gerwig, G. J.,Leijnse, B., Alterations in carbohydrate composition of serum
© 2009 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

Proteomics 2009, 9, 882–913 907
IgG from patients with rheumatoid arthritis and from preg-nant women. Ann. Rheum. Dis. 1988, 47, 91–95.
[8] Rook, G. A. W., Steele, J., Brealey, R., Whyte, A. et al., Chan-ges in IgG glycoform levels are associated with remission ofarthritis during pregnancy. J. Autoimmun. 1991, 4, 779–794.
[9] Wuhrer, M., Stam, J. C., van de Geijn, F. E., Koeleman, C. A.et al., Glycosylation profiling of immunoglobulin G (IgG)subclasses from human serum. Proteomics 2007, 7, 4070–4081.
[10] Williams, P. J., Arkwright, P. D., Rudd, P, Scragg, I. G. et al.,Short communication: Selective placental transport ofmaternal IgG to the fetus. Placenta 1995, 16, 749–756.
[11] van Zeben, D., Rook, G. A., Hazes, J. M., Zwinderman, A. H.et al., Early agalactosylation of IgG is associated with a moreprogressive disease course in patients with rheumatoidarthritis: Results of a follow-up study. Br. J. Rheumatol.1994, 33, 36–43.
[12] Rademacher, T. W., Williams, P., Dwek, R. A., Agalactosylglycoforms of IgG autoantibodies are pathogenic. Proc.Natl. Acad. Sci. USA 1994, 91, 6123–6127.
[13] Malhotra, R., Wormald, M. R., Rudd, P. M., Fischer, P. B. et al.,Glycosylation changes of IgG associated with rheumatoidarthritis can activate complement via the mannose-bindingprotein. Nat. Med. 1995, 1, 237–243.
[14] Lastra, G. C., Thompson, S. J., Lemonidis, A. S., Elson, C. J.,Changes in the galactose content of IgG during humoralimmune responses. Autoimmunity 1998, 28, 25–30.
[15] Kaneko, Y., Nimmerjahn, F., Ravetch, J. V., Anti-inflamma-tory activity of immunoglobulin G resulting from Fc sialyla-tion. Science 2006, 313, 670–673.
[16] Bond, A., Alavi, A., Axford, J. S., Bourke, B. E. et al., Adetailed lectin analysis of IgG glycosylation, demonstratingdisease specific changes in terminal galactose and N-acet-ylglucosamine. J. Autoimmun. 1997, 10, 77–85.
[17] Parekh, R. B., Roitt, I. M., Isenberg, D. A., Dwek, R. A. et al.,Galactosylation of IgG associated oligosaccharides: Reduc-tion in patients with adult and juvenile onset rheumatoidarthritis and relation to disease activity. Lancet 1988, 331,966–969.
[18] Parekh, R., Isenberg, D., Rook, G., Roitt, I. et al., A compara-tive analysis of disease-associated changes in the galacto-sylation of serum IgG. J. Autoimmun. 1989, 2, 101–114.
[19] Bond, A., Alavi, A., Axford, J. S., Youinou, P., Hay, F. C., Therelationship between exposed galactose and N-acet-ylglucosamine residues on IgG in rheumatoid arthritis (RA),juvenile chronic arthritis (JCA) and Sjögren’s syndrome(SS). Clin. Exp. Immunol. 1996, 105, 99–103.
[20] Dube, R., Rook, G. A., Steele, J., Brealey, R. et al., Agalacto-syl IgG in inflammatory bowel disease: Correlation with C-reactive protein. Gut 1990, 31, 431–434.
[21] Mehta, A. S., Long, R. E., Comunale, M. A., Wang, M. et al.,Increased levels of galactose-deficient anti-Gal immunoglo-bulin G in the sera of hepatitis C virus-infected individualswith fibrosis and cirrhosis. J. Virol. 2008, 82, 1259–1270.
[22] Moore, J. S., Wu, X., Kulhavy, R., Tomana, M. et al.,Increased levels of galactose-deficient IgG in sera of HIV-1-infected individuals. AIDS 2005, 19, 381–389.
[23] Saldova, R., Royle, L., Radcliffe, C. M., Hamid, U. M. et al.,Ovarian cancer is associated with changes in glycosylation
in both acute-phase proteins and IgG. Glycobiology 2007,17, 1321–1332.
[24] Allhorn, M., Olin, A. I., Nimmerjahn, F., Collin, M., HumanIgG/Fc gamma R interactions are modulated by strepto-coccal IgG glycan hydrolysis. PLoS ONE 2008, 3, e1413.
[25] Collin, M., Shannon, O., Björck, L., IgG glycan hydrolysis bya bacterial enzyme as a therapy against autoimmune condi-tions. Proc. Natl. Acad. Sci. USA 2008, 105, 4265–4270.
[26] Krapp, S., Mimura, Y., Jefferis, R., Huber, R., Sondermann, P.,Structural analysis of human IgG-Fc glycoforms reveals acorrelation between glycosylation and structural integrity. J.Mol. Biol. 2003, 325, 979–989.
[27] Kanda, Y., Yamada, T., Mori, K., Okazaki, A. et al., Compar-ison of biological activity among nonfucosylated ther-apeutic IgG1 antibodies with three different N-linked Fc oli-gosaccharides: The high-mannose, hybrid, and complextypes. Glycobiology 2007, 17, 104–118.
[28] Mimura, Y., Church, S., Ghirlando, R., Ashton, P. R. et al., Theinfluence of glycosylation on the thermal stability andeffector function expression of human IgG1-Fc: Properties ofa series of truncated glycoforms. Mol. Immunol. 2000, 37,697–706.
[29] Mimura, Y., Sondermann, P., Ghirlando, R., Lund, J. et al.,Role of oligosaccharide residues of IgG1-Fc in Fc gammaRIIb binding. J. Biol. Chem. 2001, 276, 45539–45547.
[30] Shields, R. L., Lai, J., Keck, R., O’Connell, L. Y. et al., Lack offucose on human IgG1 N-linked oligosaccharide improvesbinding to human Fcgamma RIII and antibody-dependentcellular toxicity. J. Biol. Chem. 2002, 277, 26733–26740.
[31] Iida, S., Misaka, H., Inoue, M., Shibata, M. et al., Non-fucosylated therapeutic IgG1 antibody can evade the inhibi-tory effect of serum immunoglobulin G on antibody-de-pendent cellular cytotoxicity through its high binding to Fcgamma RIIIa. Clin. Cancer Res. 2006, 12, 2879–2887.
[32] Mori, K., Iida, S., Yamane-Ohnuki, N., Kanda, Y. et al., Non-fucosylated therapeutic antibodies: The next generation oftherapeutic antibodies. Cytotechnology 2007, 55, 109–114.
[33] Wuhrer, M., Porcelijn, L., Kapur, R., Koeleman, C. A. M. et al.,Regulated glycosylation patterns of IgG during alloimmuneresponses against human platelet antigens. J. ProteomeRes. 2008, in press.
[34] Anthony, R. M., Nimmerjahn, F., Ashline, D. J., Reinhold, V.N. et al., Recapitulation of IVIG anti-inflammatory activitywith a recombinant IgG Fc. Science 2008, 320, 373–376.
[35] Nimmerjahn, F., Ravetch, J. V., The antiinflammatory activityof IgG: The intravenous IgG paradox. J. Exp. Med. 2007, 204,11–15.
[36] Katz, U., Achiron, A., Sherer, Y., Shoenfeld, Y., Safety ofintravenous immunoglobulin (IVIG) therapy. Autoimmun.Rev. 2007, 6, 257–259.
[37] Prahl, J. W., Porter, R. R., Allotype-related sequence varia-tion of the heavy chain of rabbit immunoglobulin G. Bio-chem. J. 1968, 107, 753–763.
[38] Watson, M., Rudd, P. M., Bland, M., Dwek, R. A., Axford, J. S.,Sugar printing rheumatic diseases: A potential method fordisease differentiation using immunoglobulin G oligo-saccharides. Arthritis Rheum. 1999, 42, 1682–1690.
[39] Kuroda, Y., Shikata, K., Takeuchi, F., Akazawa, T. et al.,Structural alterations in outer arms of IgG oligosaccharides
© 2009 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

908 C. Huhn et al. Proteomics 2009, 9, 882–913
in patients with Werner syndrome. Exp. Gerontol. 2007, 42,545–553.
[40] Kuroda, Y., Nakata, M., Makino, A., Matsumoto, A. et al.,Structural studies on IgG oligosaccharides of patients withprimary Sjögren’s syndrome. Glycoconj. J. 2002, 19, 23–31.
[41] Lim, A., Reed-Bogan, A., Harmon, B. J., Glycosylation pro-filing of a therapeutic recombinant monoclonal antibodywith two N-linked glycosylation sites using liquid chroma-tography coupled to a hybrid quadrupole time-of-flightmass spectrometer. Anal. Biochem. 2008, 375, 163–172.
[42] Olivova, P., Chen, W., Chakraborty, A. B., Gebler, J. C.,Determination of N-glycosylation sites and site hetero-geneity in a monoclonal antibody by electrospray quadru-pole ion-mobility time-of-flight mass spectrometry. RapidCommun. Mass Spectrom. 2008, 22, 29–40.
[43] Qian, J., Liu, T., Yang, L., Daus, A., Crowley, R., Zhou, Q.,Structural characterization of N-linked oligosaccharides onmonoclonal antibody cetuximab by the combination oforthogonal matrix-assisted laser desorption/ionizationhybrid quadrupole-quadrupole time-of-flight tandem massspectrometry and sequential enzymatic digestion. Anal.Biochem. 2007, 364, 8–18.
[44] Schuster, M., Jost, W., Mudde, G. C., Wiederkum, S. et al., Invivo glyco-engineered antibody with improved lytic poten-tial produced by an innovative nonmammalian expressionsystem. Biotechnol. J. 2007, 2, 700–708.
[45] Holland, M., Yagi, H., Takahashi, N., Kato, K. et al., Differ-ential glycosylation of polyclonal IgG, IgG-Fc and IgG-Fabisolated from the sera of patients with ANCA-associatedsystemic vasculitis. Biochim. Biophys. Acta 2006, 1760, 669–677.
[46] Omtvedt, L. A., Royle, L., Husby, G., Sletten, K. et al., Glycananalysis of monoclonal antibodies secreted in depositiondisorders indicates that subsets of plasma cells differentiallyprocess IgG glycans. Arthritis Rheum. 2006, 54, 3433–3440.
[47] Youings, A., Chang, S. C., Dwek, R. A., Scragg, I. G., Site-specific glycosylation of human immunoglobulin G isaltered in four rheumatoid arthritis patients. Biochem. J.1996, 314, 621–630.
[48] Wada, Y., Azadi, P., Costello, C. E., Dell, A. et al., Comparisonof the methods for profiling glycoprotein glycans-HUPOHuman Disease Glycomics/Proteome Initiative multi-insti-tutional study. Glycobiology 2007, 17, 411–422.
[49] Mechref, Y., Muzikar, J., Novotny, M. V., Comprehensiveassessment of N-glycans derived from a murine monoclonalantibody: A case for multimethodological approach. Elec-trophoresis 2005, 26, 2034–2046.
[50] Huang, L., Biolsi, S., Bales, K. R., Kuchibhotla, U., Impact ofvariable domain glycosylation on antibody clearance: AnLC/MS characterization. Anal. Biochem. 2006, 349, 197–207.
[51] Millward, T. A., Heitzmann, M., Bill, K., Längle, U. et al.,Effect of constant and variable domain glycosylation onpharmacokinetics of therapeutic antibodies in mice. Biolog-icals 2008, 36, 41–47.
[52] Stadlmann, J., Pabst, M., Kolarich, D., Kunert, R., Altmann,F., Analysis of immunoglobulin glycosylation by LC-ESI-MSof glycopeptides and oligosaccharides. Proteomics 2008, 8,2858–2871.
[53] Harazono, A., Kawasaki, N., Itoh, S., Hashii, N. et al., Simul-taneous glycosylation analysis of human serum glycopro-
teins by high-performance liquid chromatography/tandemmass spectrometry. J. Chromatogr. B 2008, 869, 20–30.
[54] Jacquemin, M., Radcliffe, C. M., Lavend’homme, R., Wor-mald, M. R. et al., Variable region heavy chain glycosylationdetermines the anticoagulant activity of a factor VIII anti-body. J. Thromb. Haemost. 2006, 4, 1047–1055.
[55] Chen, X., Flynn, G. C., Analysis of N-glycans from recombi-nant immunoglobulin G by on-line reversed-phase high-performance liquid chromatography/mass spectrometry.Anal. Biochem. 2007, 370, 147–161.
[56] Gennaro, L. A., Salas-Solano, O., On-line CE-LIF-MS tech-nology for the direct characterization of N-linked glycansfrom therapeutic antibodies. Anal. Chem. 2008, 80, 3838–3845.
[57] Harvey, D. J., Crispin, M., Scanlan, C., Singer, B. B. et al.,Differentiation between isomeric triantennary N-linked gly-cans by negative ion tandem mass spectrometry and con-firmation of glycans containing galactose attached to thebisecting (beta1–4-GlcNAc) residue in N-glycans from IgG.Rapid Commun. Mass Spectrom. 2008, 22, 1047–1052.
[58] Hodoniczky, J., Zheng, Y. Z., James, D. C., Control of recom-binant monoclonal antibody effector functions by Fc N-gly-can remodeling in vitro. Biotechnol. Prog. 2005, 21, 1644–1652.
[59] Kamoda, S., Nomura, C., Kinoshita, M., Nishiura, S. et al.,Profiling analysis of oligosaccharides in antibody pharma-ceuticals by capillary electrophoresis. J. Chromatogr. A2004, 1050, 211–216.
[60] Kamoda, S., Ishikawa, R., Kakehi, K., Capillary electrophore-sis with laser-induced fluorescence detection for detailedstudies on N-linked oligosaccharide profile of therapeuticrecombinant monoclonal antibodies. J. Chromatogr. A2006, 1133, 332–339.
[61] Kanoh, Y., Mashiko, T., Danbara, M., Takayama, Y. et al.,Analysis of the oligosaccharide chain of human serumimmunoglobulin G in patients with localized or metastaticcancer. Oncology 2004, 66, 365–370.
[62] Leibiger, H., Wustner, D., Stigler, R. D., Marx, U., Variabledomain-linked oligosaccharides of a human monoclonalIgG: Structure and influence on antigen binding. Biochem.J. 1999, 338, 529–538.
[63] Majid, F. A., Butler, M., Al Rubeai, M., Glycosylation of animmunoglobulin produced from a murine hybridoma cellline: The effect of culture mode and the anti-apoptotic gene,bcl-2. Biotechnol. Bioeng. 2007, 97, 156–169.
[64] Fernández, L. E. M., Kalume, D. E., Calvo, L., Fernández, M.M. et al., Characterization of a recombinant monoclonalantibody by mass spectrometry combined with liquid chro-matography. J. Chromatogr. B 2001, 752, 247–261.
[65] Saba, J. A., Kunkel, J. P., Jan, D. C., Ens, W. E. et al., A studyof immunoglobulin G glycosylation in monoclonal andpolyclonal species by electrospray and matrix-assisted laserdesorption/ionization mass spectrometry. Anal. Biochem.2002, 305, 16–31.
[66] Takegawa, Y., Deguchi, K., Nakagawa, H., Nishimura, S.,Structural analysis of an N-glycan with “beta1–4 bisectingbranch” from human serum IgG by negative-ion MSn spec-tral matching and exoglycosidase digestion. Anal. Chem.2005, 77, 6062–6068.
[67] Takegawa, Y., Deguchi, K., Keira, T., Ito, H. et al., Separationof isomeric 2-aminopyridine derivatized N-glycans and N-
© 2009 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

Proteomics 2009, 9, 882–913 909
glycopeptides of human serum immunoglobulin G by usinga zwitterionic type of hydrophilic-interaction chromatogra-phy. J. Chromatogr. A 2006, 1113, 177–181.
[68] Yu, Y. Q., Gilar, M., Kaska, J., Gebler, J. C., A rapid samplepreparation method for mass spectrometric characterizationof N-linked glycans. Rapid Commun. Mass Spectrom. 2005,19, 2331–2336.
[69] Homma, H., Tozawa, K., Yasui, T., Itoh, Y. et al., Abnormalglycosylation of serum IgG in patients with IgA nephro-pathy. Clin. Exp. Nephrol. 2006, 10, 180–185.
[70] Mimura, Y., Ashton, P. R., Takahashi, N., Harvey, D. J., Jef-feris, R., Contrasting glycosylation profiles between Fab andFc of a human IgG protein studied by electrospray ionizationmass spectrometry. J. Immunol. Methods 2007, 326, 116–126.
[71] Rouwendal, G. J., Wuhrer, M., Florack, D. E., Koeleman, C. A.et al., Efficient introduction of a bisecting GlcNAc residue intobacco N-glycans by expression of the gene encoding hu-man N-acetylglucosaminyltransferase III. Glycobiology2007, 17, 334–344.
[72] Yamaguchi, Y., Nishimura, M., Nagano, M., Yagi, H. et al.,Glycoform-dependent conformational alteration of the Fcregion of human immunoglobulin G1 as revealed by NMRspectroscopy. Biochim. Biophys. Acta 2006, 1760, 693–700.
[73] Takahashi, N., Ishii, I., Ishihara, H., Mori, M. et al., Compara-tive structural study of the N-linked oligosaccharides of hu-man normal and pathological immunoglobulin G. Bio-chemistry 1987, 26, 1137–1144.
[74] Takegawa, Y., Deguchi, K., Ito, S., Yoshioka, S. et al., Simul-taneous analysis of 2-aminopyridine-derivatized neutral andsialylated oligosaccharides from human serum in the nega-tive-ion mode by sonic spray ionization ion trap mass spec-trometry. Anal. Chem. 2005, 77, 2097–2106.
[75] Kunkel, J. P., Jan, D. C., Butler, M., Jamieson, J. C., Compar-isons of the glycosylation of a monoclonal antibody pro-duced under nominally identical cell culture conditions intwo different bioreactors. Biotechnol. Prog. 2000, 16, 462–470.
[76] Taniguchi, T., Mizuochi, T., Beale, M., Dwek, R. A. et al.,Structures of the sugar chains of rabbit immunoglobulin G:Occurrence of asparagine-linked sugar chains in Fab frag-ment. Biochemistry 1985, 24, 5551–5557.
[77] Harada, H., Kamei, M., Tokumoto, Y., Yui, S. et al., System-atic fractionation of oligosaccharides of human immu-noglobulin G by serial affinity chromatography on immobi-lized lectin columns. Anal. Biochem. 1987, 164, 374–381.
[78] Packer, N. H., Lawson, M. A., Jardine, D. R., Redmond, J. W.,A general approach to desalting oligosaccharides releasedfrom glycoproteins. Glycoconj. J. 1998, 15, 737–747.
[79] Yuen, C. T., Gee, C. K., Jones, C., High-performance liquidchromatographic profiling of fluorescent labelled N-glycanson glycoproteins. Biomed. Chromatogr. 2002, 16, 247–254.
[80] Papac, D. I., Briggs, J. B., Chin, E. T., Jones, A. J., A high-throughput microscale method to release N-linked oligo-saccharides from glycoproteins for matrix-assisted laserdesorption/ionization time-of-flight mass spectrometricanalysis. Glycobiology 1998, 8, 445–454.
[81] Royle, L., Campbell, M. P., Radcliffe, C. M., White, D. M. et al.,HPLC-based analysis of serum N-glycans on a 96-well plateplatform with dedicated database software. Anal. Biochem.2008, 376, 1–12.
[82] Laroy, W., Contreras, R., Callewaert, N., Glycome mappingon DNA sequencing equipment. Nat. Protoc. 2006, 1, 397–405.
[83] Shilova, N. V., Bovin, N. V., Fluorescent labels for analysis ofmono- and oligosaccharides. Russ. J. Bioorg. Chem. 2003,29, 309–324.
[84] Anumula, K. R., Advances in fluorescence derivatizationmethods for high-performance liquid chromatographicanalysis of glycoprotein carbohydrates. Anal. Biochem.2006, 350, 1–23.
[85] Kamoda, S., Nakano, M., Ishikawa, R., Suzuki, S., Kakehi, K.,Rapid and sensitive screening of N-glycans as 9-fluo-renylmethyl derivatives by high-performance liquid chroma-tography: A method which can recover free oligosaccha-rides after analysis. J. Proteome Res. 2005, 4, 146–152.
[86] Bigge, J. C., Patel, T. P., Bruce, J. A., Goulding, P. N. et al.,Nonselective and efficient fluorescent labeling of glycansusing 2-amino benzamide and anthranilic acid. Anal. Bio-chem. 1995, 230, 229–238.
[87] Anumula, K. R., Quantitative determination of mono-saccharides in glycoproteins by high-performance liquidchromatography with highly sensitive fluorescence detec-tion. Anal. Biochem. 1994, 220, 275–283.
[88] Takegawa, Y., Deguchi, K., Ito, H., Keira, T. et al., Simpleseparation of isomeric sialylated N-glycopeptides by a zwit-terionic type of hydrophilic interaction chromatography. J.Sep. Sci. 2006, 29, 2533–2540.
[89] Domann, P. J., Pardos-Pardos, A. C., Fernandes, D. L., Spen-cer, D. I. et al., Separation-based glycoprofiling approachesusing fluorescent labels. Proteomics 2007, 7, 70–76.
[90] Wormald, M. R., Rudd, P. M., Harvey, D. J., Chang, S. C. et al.,Variations in oligosaccharide-protein interactions in immu-noglobulin G determine the site-specific glycosylation pro-files and modulate the dynamic motion of the Fc oligo-saccharides. Biochemistry 1997, 36, 1370–1380.
[91] Guile, G. R., Rudd, P. M., Wing, D. R., Prime, S. B., Dwek, R.A., A rapid high-resolution high-performance liquid chro-matographic method for separating glycan mixtures andanalyzing oligosaccharide profiles. Anal. Biochem. 1996,240, 210–226.
[92] Ruhaak, L. R., Huhn, C., Waterreus, W. J., de Boer, A. R. et al.,Hydrophilic interaction chromatography-based high-throughput sample preparation method for N-glycan analy-sis from total human plasma glycoproteins. Anal. Chem.2008, 80, 6119–6126.
[93] Prater, B. D., Anumula, K. R., Hutchins, J. T., Automatedsample preparation facilitated by PhyNexus MEA purifica-tion system for oligosaccharide mapping of glycoproteins.Anal. Biochem. 2007, 369, 202–209.
[94] Kamoda, S., Kakehi, K., Evaluation of glycosylation forquality assurance of antibody pharmaceuticals by capillaryelectrophoresis. Electrophoresis 2008, 29, 3595–3604.
[95] Zhuang, Z., Starkey, J. A., Mechref, Y., Novotny, M. V.,Jacobson, S. C., Electrophoretic analysis of N-glycans onmicrofluidic devices. Anal. Chem. 2007, 79, 7170–7175.
[96] Callewaert, N., Contreras, R., Mitnik-Gankin, L., Carey, L. etal., Total serum protein N-glycome profiling on a capillaryelectrophoresis-microfluidics platform. Electrophoresis2004, 25, 3128–3131.
[97] Pabst, M., Bondili, J. S., Stadlmann, J., Mach, L., Altmann, F.,Mass 1 retention time = structure: A strategy for the analy-
© 2009 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

910 C. Huhn et al. Proteomics 2009, 9, 882–913
sis of N-glycans by carbon LC-ESI-MS and its application tofibrin N-glycans. Anal. Chem. 2007, 79, 5051–5057.
[98] Kang, P., Mechref, Y., Kyselova, Z., Goetz, J. A., Novotny, M.V., Comparative glycomic mapping through quantitativepermethylation and stable-isotope labeling. Anal. Chem.2007, 79, 6064–6073.
[99] Atwood, J. A. III, Cheng, L., Alvarez-Manilla, G., Warren, N.L. et al., Quantitation by isobaric labeling: Applications toglycomics. J. Proteome Res. 2008, 7, 367–374.
[100] Prien, J. M., Huysentruyt, L. C., Ashline, D. J., Lapadula, A.J. et al., Differentiating N-linked glycan structural isomersin metastatic and nonmetastatic tumor cells using sequen-tial mass spectrometry. Glycobiology 2008, 18, 353–366.
[101] Ashline, D. J., Lapadula, A. J., Liu, Y. H., Lin, M. et al., Car-bohydrate structural isomers analyzed by sequential massspectrometry. Anal. Chem. 2007, 79, 3830–3842.
[102] Costello, C. E., Contado-Miller, J. M., Cipollo, J. F., A gly-comics platform for the analysis of permethylated oligo-saccharide alditols. J. Am. Soc. Mass Spectrom. 2007, 18,1799–1812.
[103] Kang, P., Mechref, Y., Novotny, M. V., High-throughputsolid-phase permethylation of glycans prior to mass spec-trometry. Rapid Commun. Mass Spectrom. 2008, 22, 721–734.
[104] Zhang, Y., Go, E. P., Desaire, H., Maximizing coverage ofglycosylation heterogeneity in MALDI-MS analysis of gly-coproteins with up to 27 glycosylation sites. Anal. Chem.2008, 80, 3144–3158.
[105] Schähs, M., Strasser, R., Stadlmann, J., Kunert, R. et al.,Production of a monoclonal antibody in plants with ahumanized N-glycosylation pattern. Plant Biotechnol. J.2007, 5, 657–663.
[106] Yu, Y. Q., Fournier, J., Gilar, M., Gebler, J. C., Identificationof N-linked glycosylation sites using glycoprotein diges-tion with pronase prior to MALDI tandem time-of-flightmass spectrometry. Anal. Chem. 2007, 79, 1731–1738.
[107] Hirayama, K., Yuji, R., Yamada, N., Kato, K. et al., Completeand rapid peptide and glycopeptide mapping of mousemonoclonal antibody by LC/MS/MS using ion trap massspectrometry. Anal. Chem. 1998, 70, 2718–2725.
[108] Rehder, D. S., Dillon, T. M., Pipes, G. D., Bondarenko, P. V.,Reversed-phase liquid chromatography/mass spectrome-try analysis of reduced monoclonal antibodies in pharma-ceutics. J. Chromatogr. A 2006, 1102, 164–175.
[109] Bongers, J., Cummings, J. J., Ebert, M. B., Federici, M. M.et al., Validation of a peptide mapping method for a ther-apeutic monoclonal antibody: What could we possiblylearn about a method we have run 100 times? J. Pharm.Biomed. Anal. 2000, 21, 1099–1128.
[110] Janecki, D. J., Broshears, W. C., Reilly, J. P., Photo-immobilization of proteins for affinity capture combinedwith MALDI TOF MS analysis. Anal. Chem. 2004, 76, 6643–6650.
[111] Jmeian, Y., El Rassi, Z., Tandem affinity monolithic micro-columns with immobilized protein A, protein G0, and anti-bodies for depletion of high abundance proteins fromserum samples: Integrated microcolumn-based fluidicsystem for simultaneous depletion and tryptic digestion. J.Proteome Res. 2007, 6, 947–954.
[112] Korecka, L., Bilkova, Z., Holeapek, M., Kralovsky, J. et al.,Utilization of newly developed immobilized enzyme reac-
tors for preparation and study of immunoglobulin G frag-ments. J. Chromatogr. B 2004, 808, 15–24.
[113] Stigter, E. C., de Jong, G. J., van Bennekom, W. P., Devel-opment of an open-tubular trypsin reactor for on-linedigestion of proteins. Anal. Bioanal. Chem. 2007, 389,1967–1977.
[114] Stigter, E. C., de Jong, G. J., van Bennekom, W. P., Pepsinimmobilized in dextran-modified fused-silica capillaries foron-line protein digestion and peptide mapping. Anal.Chim. Acta 2008, 619, 231–238.
[115] Ito, H., Takegawa, Y., Deguchi, K., Nagai, S. et al., Directstructural assignment of neutral and sialylated N-glycansof glycopeptides using collision-induced dissociation MSnspectral matching. Rapid Commun. Mass Spectrom. 2006,20, 3557–3565.
[116] Suzuki, N., Lee, Y. C., Site-specific N-glycosylation ofchicken serum IgG. Glycobiology 2004, 14, 275–292.
[117] Wada, Y., Tajiri, M., Yoshida, S., Hydrophilic affinity isola-tion and MALDI multiple-stage tandem mass spectrometryof glycopeptides for glycoproteomics. Anal. Chem. 2004,76, 6560–6565.
[118] Weitzman, S., Portmore, J., Immunoglobulin glycopep-tides from an IgG2b-producing mouse myeloma cell lineand from variant cell lines. J. Immunol. 1981, 127, 2095–2101.
[119] Strader, M. B., Tabb, D. L., Hervey, W. J., Pan, C., Hurst, G.B., Efficient and specific trypsin digestion of microgram tonanogram quantities of proteins in organic-aqueous sol-vent systems. Anal. Chem. 2006, 78, 125–134.
[120] Hervey, W. J., Strader, M. B., Hurst, G. B., Comparison ofdigestion protocols for microgram quantities of enrichedprotein samples. J. Proteome Res. 2007, 6, 3054–3061.
[121] Rousseaux, J., Rousseaux-Prevost, R., Bazin, H., Isolationand characterization of a VH domain fragment frommonoclonal rat IgG of IgG1 and IgG2a subclasses. Mol.Immunol. 1989, 26, 27–32.
[122] Suzuki, N., Khoo, K. H., Chen, C. M., Chen, H. C., Lee, Y. C.,N-glycan structures of pigeon IgG: A major serum glyco-protein containing Galalpha1-4 Gal termini. J. Biol. Chem.2003, 278, 46293–46306.
[123] Scherer, H. U., Wang, J., Toes, R. E. M., van der Woude, D.et al., Immunoglobulin 1 (IgG1) Fc-glycosylation profilingof anti-citrullinated peptide antibodies from human serum.Prot. Clin. Appl. 2009, 3, 106–115.
[124] Kim, H., Yamaguchi, Y., Masuda, K., Matsunaga, C. et al., O-glycosylation in hinge region of mouse immunoglobulinG2b. J. Biol. Chem. 1994, 269, 12345–12350.
[125] Sheeley, D. M., Merrill, B. M., Taylor, L. C., Characterizationof monoclonal antibody glycosylation: Comparison ofexpression systems and identification of terminal alpha-linked galactose. Anal. Biochem. 1997, 247, 102–110.
[126] Beck, A., Bussat, M. C., Zorn, N., Robillard, V. et al., Char-acterization by liquid chromatography combined withmass spectrometry of monoclonal anti-IGF-1 receptorantibodies produced in CHO and NS0 cells. J. Chromatogr.B 2005, 819, 203–218.
[127] Rousseaux, J., Rousseaux-Prevost, R., Bazin, H., Biserte,G., Proteolysis of rat IgG subclasses by Staphylococcusaureus V8 proteinase. Biochim. Biophys. Acta 1983, 748,205–212.
© 2009 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

Proteomics 2009, 9, 882–913 911
[128] Prokesova, L., Potuznikova, B., Potempa, J., Zikan, J. et al.,Cleavage of human immunoglobulins by serine proteinasefrom Staphylococcus aureus. Immunol. Lett. 1992, 31, 259–265.
[129] Martens, L., Immunomics: Entwicklung einer ESI-MSbasierten methodik zur differenzierung und quantifizierungvon humanen immunglobulinen und ihrer glykosylierung.PhD Thesis, Koeln, 2003, pp. 1–185. http://kups.ub.uni-koeln.de/volltexte/2004/1056/pdf/Dissertation_Lejon_Martens2.pdf.
[130] An, H. J., Peavy, T. R., Hedrick, J. L., Lebrilla, C. B., Deter-mination of N-glycosylation sites and site heterogeneity inglycoproteins. Anal. Chem. 2003, 75, 5628–5637.
[131] Temporini, C., Perani, E., Calleri, E., Dolcini, L. et al., Pro-nase-immobilized enzyme reactor: An approach for auto-mation in glycoprotein analysis by LC/LC-ESI/MSn. Anal.Chem. 2007, 79, 355–363.
[132] Wuhrer, M., Koeleman, C. A., Hokke, C. H., Deelder, A. M.,Protein glycosylation analyzed by normal-phase nano-liquid chromatography-mass spectrometry of glycopep-tides. Anal. Chem. 2005, 77, 886–894.
[133] Clowers, B. H., Dodds, E. D., Seipert, R. R., Lebrilla, C. B.,Site determination of protein glycosylation based ondigestion with immobilized nonspecific proteases andFourier transform ion cyclotron resonance mass spec-trometry. J. Proteome Res. 2007, 6, 4032–4040.
[134] Rothman, R. J., Warren, L., Vliegenthart, J. F., Hard, K. J.,Clonal analysis of the glycosylation of immunoglobulin Gsecreted by murine hybridomas. Biochemistry 1989, 28,1377–1384.
[135] Tai, T., Ito, S., Yamashita, K., Muramatsu, T., Kobata, A.,Asparagine-linked oligosaccharide chains of IgG: A revisedstructure. Biochem. Biophys. Res. Commun. 1975, 65, 968–974.
[136] Cheron, A., Fournet, B., Spik, G., Montreuil, J., Completestructure of glycopeptides isolated from IgG immunoglo-bulins of cow colostrum. Biochimie 1976, 58, 927–942.
[137] Lee, S. O., Connolly, J. M., Ramirez-Soto, D., Poretz, R. D.,The polypeptide of immunoglobulin G influences itsgalactosylation in vivo. J. Biol. Chem. 1990, 265, 5833–5839.
[138] Kroon, D. J., Freedy, J., Burinsky, D. J., Sharma, B., Rapidprofiling of carbohydrate glycoforms in monoclonal anti-bodies using MALDI/TOF mass spectrometry. J. Pharm.Biomed. Anal. 1995, 13, 1049–1054.
[139] Perdivara, I., Deterding, L., Moise, A., Tomer, K. B., Przy-bylski, M., Determination of primary structure and micro-heterogeneity of a beta-amyloid plaque-specific antibodyusing high-performance LC-tandem mass spectrometry.Anal. Bioanal. Chem. 2008, 391, 325–336.
[140] Wagner-Rousset, E., Bednarczyk, A., Bussat, M. C., Colas,O. et al., The way forward, enhanced characterization oftherapeutic antibody glycosylation: Comparison of threelevel mass spectrometry-based strategies. J. Chromatogr.B 2008, 872, 23–37.
[141] Huang, L., Lu, J., Wroblewski, V. J., Beals, J. M., Riggin, R.M., In vivo deamidation characterization of monoclonalantibody by LC/MS/MS. Anal. Chem. 2005, 77, 1432–1439.
[142] Chakraborty, A. B., Berger, S. J., Optimization of reversed-phase peptide liquid chromatography ultraviolet mass
spectrometry analyses using an automated blendingmethodology. J. Biomol. Tech. 2005, 16, 327–335.
[143] Shou, W. Z., Naidong, W., Simple means to alleviate sen-sitivity loss by trifluoroacetic acid (TFA) mobile phases inthe hydrophilic interaction chromatography-electrospraytandem mass spectrometric (HILIC-ESI/MS/MS) bioana-lysis of basic compounds. J. Chromatogr. B 2005, 825, 186–192.
[144] Huddleston, M. J., Bean, M. F., Carr, S. A., Collisional frag-mentation of glycopeptides by electrospray ionization LC/MS and LC/MS/MS: Methods for selective detection of gly-copeptides in protein digests. Anal. Chem. 1993, 65, 877–884.
[145] Kilgore, B. R., Lucka, A. W., Patel, R., Andrien, B. A., Jr.,Dhume, S. T., Comparability and monitoring immunogenicN-linked oligosaccharides from recombinant monoclonalantibodies from two different cell lines using HPLC withfluorescence detection and mass spectrometry. MethodsMol. Biol. 2008, 446, 333–346.
[146] Karnoup, A. S., Kuppannan, K., Young, S. A., A novel HPLC-UV-MS method for quantitative analysis of protein glyco-sylation. J. Chromatogr. B 2007, 859, 178–191.
[147] Boersema, P. J., Mohammed, S., Heck, A. J., Hydrophilicinteraction liquid chromatography (HILIC) in proteomics.Anal. Bioanal. Chem. 2008, 391, 151–159.
[148] Zhang, B., Yang, Y., Yuk, I., Pai, R. et al., Unveiling a glyca-tion hot spot in a recombinant humanized monoclonalantibody. Anal. Chem. 2008, 80, 2379–2390.
[149] Jiskoot, W., van de, W. G., Martin, J. M., Green, B. N. et al.,Application of electrospray mass spectrometry (ES-MS) forthe analysis of monoclonal antibody Fc subunits. Pharm.Res. 1992, 9, 945–951.
[150] Battersby, J. E., Snedecor, B., Chen, C., Champion, K. M. etal., Affinity-reversed-phase liquid chromatography assayto quantitate recombinant antibodies and antibody frag-ments in fermentation broth. J. Chromatogr. A 2001, 927,61–76.
[151] Ryan, M. H., Petrone, D., Nemeth, J. F., Barnathan, E. et al.,Proteolysis of purified IgGs by human and bacterialenzymes in vitro and the detection of specific proteolyticfragments of endogenous IgG in rheumatoid synovialfluid. Mol. Immunol. 2008, 45, 1837–1846.
[152] Adamczyk, M., Gebler, J. C., Wu, J., Papain digestion ofdifferent mouse IgG subclasses as studied by electrospraymass spectrometry. J. Immunol. Methods 2000, 237, 95–104.
[153] Ashton, D. S., Beddell, C. R., Cooper, D. J., Craig, S. J. et al.,Mass spectrometry of the humanized monoclonal antibodyCAMPATH 1H. Anal. Chem. 1995, 67, 835–842.
[154] Hagmann, M. L., Kionka, C., Schreiner, M., Schwer, C.,Characterization of the F(ab0)2 fragment of a murinemonoclonal antibody using capillary isoelectric focusingand electrospray ionization mass spectrometry. J. Chro-matogr. A 1998, 816, 49–58.
[155] Kelly, L. S., Kozak, M., Walker, T., Pierce, M., Puett, D., Lectinimmunoassays using antibody fragments to detect glyco-forms of human chorionic gonadotropin secreted by chor-iocarcinoma cells. Anal. Biochem. 2005, 338, 253–262.
[156] Moorhouse, K. G., Nashabeh, W., Deveney, J., Bjork, N. S.et al., Validation of an HPLC method for the analysis of thecharge heterogeneity of the recombinant monoclonal anti-
© 2009 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

912 C. Huhn et al. Proteomics 2009, 9, 882–913
body IDEC-C2B8 after papain digestion. J. Pharm. Biomed.Anal. 1997, 16, 593–603.
[157] Masuda, K., Yamaguchi, Y., Kato, K., Takahashi, N. et al.,Pairing of oligosaccharides in the Fc region of immu-noglobulin G. FEBS Lett. 2000, 473, 349–357.
[158] Santora, L. C., Krull, I. S., Grant, K., Characterization ofrecombinant human monoclonal tissue necrosis factor-alpha antibody using cation-exchange HPLC and capillaryisoelectric focusing. Anal. Biochem. 1999, 275, 98–108.
[159] von Pawel-Rammingen, U., Johansson, B. P., Bjorck, L.,IdeS, a novel streptococcal cysteine proteinase with uniquespecificity for immunoglobulin G. EMBO J. 2002, 21, 1607–1615.
[160] Ren, D., Pipes, G., Xiao, G., Kleemann, G. R. et al.,Reversed-phase liquid chromatography-mass spectrome-try of site-specific chemical modifications in intact immu-noglobulin molecules and their fragments. J. Chromatogr.A 2008, 1179, 198–204.
[161] Gadgil, H. S., Bondarenko, P. V., Pipes, G. D., Dillon, T. M. etal., Identification of cysteinylation of a free cysteine in theFab region of a recombinant monoclonal IgG1 antibodyusing Lys-C limited proteolysis coupled with LC/MS analy-sis. Anal. Biochem. 2006, 355, 165–174.
[162] Liu, H., Gaza-Bulseco, G., Chumsae, C., Radziejewski, C. H.,Mass spectrometry analysis of in vitro nitration of arecombinant human IgG1 monoclonal antibody. RapidCommun. Mass Spectrom. 2008, 22, 1–10.
[163] Martinez, T., Pace, D., Brady, L., Gerhart, M., Balland, A.,Characterization of a novel modification on IgG2 lightchain. Evidence for the presence of O-linked mannosyla-tion. J. Chromatogr. A 2007, 1156, 183–187.
[164] Wan, H. Z., Kaneshiro, S., Frenz, J., Cacia, J., Rapid methodfor monitoring galactosylation levels during recombinantantibody production by electrospray mass spectrometrywith selective-ion monitoring. J. Chromatogr. A 2001, 913,437–446.
[165] Brady, L. J., Valliere-Douglass, J., Martinez, T., Balland, A.,Molecular mass analysis of antibodies by on-line SEC-MS.J. Am. Soc. Mass Spectrom. 2008, 19, 502–509.
[166] Küster, B., Wheeler, S. F., Hunter, A. P., Dwek, R. A., Harvey,D. J., Sequencing of N-linked oligosaccharides directlyfrom protein gels: In-gel deglycosylation followed bymatrix-assisted laser desorption/ionization mass spec-trometry and normal-phase high-performance liquid chro-matography. Anal. Biochem. 1997, 250, 82–101.
[167] Yamaguchi, Y., Kim, H., Kato, K., Masuda, K. et al., Proteo-lytic fragmentation with high specificity of mouse immu-noglobulin G. Mapping of proteolytic cleavage sites in thehinge region. J. Immunol. Methods 1995, 181, 259–267.
[168] Mariani, M., Camagna, M., Tarditi, L., Seccamani, E., A newenzymatic method to obtain high-yield F(ab)2 suitable forclinical use from mouse IgGl 1. Mol. Immunol. 1991, 28, 69–77.
[169] Hunt, G., Nashabeh, W., Capillary electrophoresis sodiumdodecyl sulfate nongel sieving analysis of a therapeuticrecombinant monoclonal antibody: A biotechnology per-spective. Anal. Chem. 1999, 71, 2390–2397.
[170] Lausch, R., Scheper, T., Reif, O-W., Schlösser, J. et al., Rapidcapillary gel electrophoresis of proteins. J. Chromatogr. A1993, 654, 190–195.
[171] Yasukawa, Z., Sato, C., Sano, K., Ogawa, H., Kitajima, K.,Identification of disialic acid-containing glycoproteins inmouse serum: A novel modification of immunoglobulinlight chains, vitronectin, and plasminogen. Glycobiology2006, 16, 651–665.
[172] Harris, R. J., Kabakoff, B., Macchi, F. D., Shen, F. J. et al.,Identification of multiple sources of charge heterogeneityin a recombinant antibody. J. Chromatogr. B 2001, 752,233–245.
[173] Dillon, T. M., Bondarenko, P. V., Speed, R. M., Developmentof an analytical reversed-phase high-performance liquidchromatography-electrospray ionization mass spectrome-try method for characterization of recombinant antibodies.J. Chromatogr. A 2004, 1053, 299–305.
[174] Perkins, M., Theiler, R., Lunte, S., Jeschke, M., Determina-tion of the origin of charge heterogeneity in a murinemonoclonal antibody. Pharm. Res. 2000, 17, 1110–1117.
[175] Tang, S., Nesta, D. P., Maneri, L. R., Anumula, K. R., Amethod for routine analysis of recombinant immunoglo-bulins (rIgGs) by capillary isoelectric focusing (cIEF). J.Pharm. Biomed. Anal. 1999, 19, 569–583.
[176] Kelly, J. A., Lee, C. S., On-line post-capillary affinity detec-tion of immunoglobulin G subclasses and monoclonalantibody variants for capillary electrophoresis. J. Chroma-togr. A 1997, 790, 207–214.
[177] Armenta, J. M., Gu, B., Humble, P. H., Thulin, C. D., Lee, M.L., Design and evaluation of a coupled monolithic pre-concentrator-capillary zone electrophoresis system for theextraction of immunoglobulin G from human serum. J.Chromatogr. A 2005, 1097, 171–178.
[178] Dillon, T. M., Bondarenko, P. V., Rehder, D. S., Pipes, G. D. etal., Optimization of a reversed-phase high-performanceliquid chromatography/mass spectrometry method forcharacterizing recombinant antibody heterogeneity andstability. J. Chromatogr. A 2006, 1120, 112–120.
[179] Lattova, E., Chen, V. C., Varma, S., Bezabeh, T., Perreault,H., Matrix-assisted laser desorption/ionization on-targetmethod for the investigation of oligosaccharides and gly-cosylation sites in glycopeptides and glycoproteins. RapidCommun. Mass Spectrom. 2007, 21, 1644–1650.
[180] Roberts, G. D., Johnson, W. P., Burman, S., Anumula, K. R.,Carr, S. A., An integrated strategy for structural characteri-zation of the protein and carbohydrate components ofmonoclonal antibodies: Application to anti-respiratorysyncytial virus MAb. Anal. Chem. 1995, 67, 3613–3625.
[181] Masuda, K., Yamaguchi, Y., Kato, K., Kim, H. H. et al., Post-translational modifications of immunoglobulin G: A mouseIgG variant that lacks the entire CH1 domain. Mol. Immu-nol. 1999, 36, 993–1003.
[182] Ahrer, K., Jungbauer, A., Chromatographic and electro-phoretic characterization of protein variants. J. Chroma-togr. B Biomed. Sci. Appl. 2006, 841, 110–122.
[183] Gadgil, H. S., Bondarenko, P. V., Pipes, G., Rehmer, D. et al.,The LC/MS analysis of glycation of IgG molecules insucrose containing formulations. J. Pharm. Sci. 2007, 96,2607–2621.
[184] Görg, A., Weiss, W., Dunn, M. J., Current two-dimensionalelectrophoresis technology for proteomics. Proteomics2004, 4, 3665–3685.
[185] Dolník, V., Capillary electrophoresis of proteins 2003–2005.Electrophoresis 2005, 27, 126–141.
© 2009 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

Proteomics 2009, 9, 882–913 913
[186] Mechref, Y., Novotny, M. V., Structural investigations ofglycoconjugates at high sensitivity. Chem. Rev. 2002, 102,321–369.
[187] Kilár, F., Recent applications of capillary isoelectric focus-ing. Electrophoresis 2003, 24, 3908–3916.
[188] Tran, A. D., Park, S., Lisi, P. J., Huynh, O. T. et al., Separationof carbohydrate-mediated microheterogeneity of recombi-nant human erythropoietin by free solution capillary elec-trophoresis: Effects of pH, buffer type and organic addi-tives. J. Chromatogr. A 1991, 542, 459–471.
[189] Watson, E., Yao, F., Capillary electrophoretic separation ofhuman recombinant erythropoietin (r-HuEPO) glycoforms.Anal. Biochem. 1993, 210, 389–393.
[190] Sanz-Nebot, V., Balaguer, E., Benavente, F., Neusüß, C.,Barbosa, J., Characterization of transferrin glycoforms inhuman serum by CE-UV and CE-ESI-MS. Electrophoresis2006, 28, 1949–1957.
[191] Balaguer, E., Demelbauer, U., Pelzing, M., Sanz-Nebot, V.,Neusüß, C., Glycoform characterization of erythropoietincombining glycan and intact protein analysis by capillaryelectrophoresis - electrospray-time-of-flight mass spec-trometry. Electrophoresis 2006, 27, 2638–2650.
[192] Balaguer, E., Neusüß, C., Glycoprotein characterizationcombining intact protein and glycan analysis by capillaryelectrophoresis-electrospray ionization-mass spectrome-try. Anal. Chem. 2006, 78, 5384–5393.
[193] Madera, M., Mechref, Y., Novotny, M. V., Combining lectinmicrocolumns with high-resolution separation techniquesfor enrichment of glycoproteins and glycopeptides. Anal.Chem. 2005, 77, 4081–4090.
[194] Madera, M., Mechref, Y., Klouckova, I., Novotny, M. V.,Semiautomated high-sensitivity profiling of human bloodserum glycoproteins through lectin preconcentration andmultidimensional chromatography/tandem mass spec-trometry. J. Proteome Res. 2006, 5, 2348–2363.
[195] Gadgil, H. S., Pipes, G. D., Dillon, T. M., Treuheit, M. J.,Bondarenko, P. V., Improving mass accuracy of high per-formance liquid chromatography/electrospray ionizationtime-of-flight mass spectrometry of intact antibodies. J.Am. Soc. Mass Spectrom. 2006, 17, 867–872.
[196] Geyer, H., Geyer, R., Strategies for analysis of glycoproteinglycosylation. Biochim. Biophys. Acta 2006, 1764, 1853–1869.
[197] Yasukawa, Z., Sato, C., Kitajima, K., Inflammation-depend-ent changes in a-2,3-, a2,6- and a2,8-salic acid glycotopeson serum glycoproteins in mice. Glycobiology 2005, 15,827–837.
[198] Squier, T. C., Bigelow, D. J., Protein oxidation and age-de-pendent alterations in calcium homeostasis. Front Biosci.2000, 5, D504–D526.
[199] Davies, M. J., The oxidative environment and proteindamage. Biochim. Biophys. Acta 2005, 1703, 93–109.
[200] Lapolla, A., Fedele, D., Aronica, R., Garbeglio, M. et al.,Evaluation of IgG glycation levels by matrix-assisted laserdesorption/ionization mass spectrometry. Rapid Commun.Mass Spectrom. 1997, 11, 1342–1346.
[201] Umana, P., Jean-Mairet, J., Moudry, R., Amstutz, H., Bailey,J. E., Engineered glycoforms of an antineuroblastoma IgG1with optimized antibody-dependent cellular cytotoxic ac-tivity. Nat. Biotechnol. 1999, 17, 176–180.
[202] Cox, K. M., Sterling, J. D., Regan, J. T., Gasdaska, J. R. et al.,Glycan optimization of a human monoclonal antibody inthe aquatic plant Lemna minor. Nat. Biotechnol. 2006, 24,1591–1597.
[203] Bakker, H., Rouwendal, G. J., Karnoup, A. S., Florack, D. E.et al., An antibody produced in tobacco expressing ahybrid beta-1,4-galactosyltransferase is essentially devoidof plant carbohydrate epitopes. Proc. Natl. Acad. Sci. USA2006, 103, 7577–7582.
[204] Hamilton, S. R., Gerngross, T. U., Glycosylation engineer-ing in yeast: The advent of fully humanized yeast. Curr.Opin. Biotechnol. 2007, 18, 387–392.
[205] Li, H., Sethuraman, N., Stadheim, T. A., Zha, D. et al., Opti-mization of humanized IgGs in glycoengineered Pichiapastoris. Nat. Biotechnol. 2006, 24, 210–215.
[206] Chung, C. H., Mirakhur, B., Chan, E., Le, Q. T. et al., Cetux-imab-induced anaphylaxis and IgE specific for galactose-alpha-1,3-galactose. N. Engl. J. Med. 2008, 358, 1109–1117.
© 2009 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com





























