Embed Size (px)
Citation preview

Neuropatológia 2.
Reiniger Lilla

Az előadás felépítése
I. A myelin betegségei
II. Neurodegeneratív betegségek

I. A myelin betegségei
Demyelinizációs
betegségek
• Normális szerkezetű
myelin
• Szerzett
• Okai:
– Immun-mediált
károsodás
– Vírus fertőzések
– Toxikus károsodás
• Sclerosis multiplex
• Egyéb
Dysmyelinizációs
betegségek
• A myelin képzése és
fenntartása zavart
• Öröklött
• Okai:
– Mutációk
• Leukodystrophiák

Sclerosis multiplex (SM)
• „multifokális demyelinizáció”
• Gyakori (1/1000 fő)
• kb. 30-40 éves korban; Nő : férfi = 2 : 1
• Autoimmun betegség
• Klinikum: Shubokban jelentkező idegrendszeri
zavarok (látászavar, végtagzsibbadás/-gyengeség,
járászavar, nyelészavar, szédülés (vertigo), stb.)
• Lefolyás: Relapsus-remisszió vagy krónikus
progresszív

Az SM pathogenezise –
immun mediált myelin károsodás
• Genetikai (HLA-DR2, IL-2 és IL-7 receptorok
génjeinek polimorfizmusai) és környezeti
faktorok
• ↓ tolerancia különféle myelin antigének ellen
• A folyamatot valószínűleg fertőzés indítja el
• CD4+ T-sejtek központi szerepe (TH17 és T
H1
sejtek)
• A myelin vesztés mellett valamennyi axon
és neuron károsodás is megfigyelhető

SM makro-morfológia: Plakkok
A KIR-ben bárhol
N. opticus
Chiasma opticum
Agy
Agytörzs
Kisagy
Gerincvelő

SM mikro-morfológia: Plakkok
Jól körülírt
Éles határú
• Aktív plakkok
• Inaktív plakkok

SM – Aktív plakkok
Akut demyelinizáció
Krónikus aktív plakk
Macrophagok
Lymphocyták
Myelin törmelék
macrophagokban Nincs myelin
törmelék
I. típus: macrophagok, éles határ
II. típus: u.a. + komplement lerakódás
III. típus: kevésbé éles határ,
oligodendrocyta apoptosis
IV. típus: oligodendrocyta vesztés,
nem apoptózissal

SM – Inaktív plakkok
Minimális gyulladás
Astrocytosis
„Csupasz” axonok
NINCS
Myelin
Macrophagok
Jelentős gyulladás

Baló féle koncentrikus sclerosis
Baló József
(1895-1979)

Egyéb szerzett demyelinizációs
betegségek
Immun-mediált
• Posztinfekciózus
(kereszt-reakció myelin
antigénekkel)
– Akut disszeminált
encephalomyelitis (ADEM)
– Akut nekrotizáló
haemorrhagiás
encephalomyelitis
(necrotizáló vasculitis)
• Neuromyelitis optica
(Devic betegség)
– Aquaporin-4 ellenes
autoantitestek (astrocyták)
Nem immun-mediált
• Centrális pontin
myelinolysis
– Hyponatrémia gyors
helyreállítását követően
– Alkoholistáknál vagy súlyos
elektrolit és osmoláris
egyensúlyzavar esetén
• Progresszív multifokális
leukoencephalopathia
– JC vírus

II. Primer neurodegeneratív
betegségek
• Funkcionálisan összekapcsolt idegsejt csoportok
pusztulásával járó betegségek
(szelektív neuronális vulnerabilitás)
• Gyakran – betegségre jellegzetes – kóros
fehérjék halmozódnak fel
• Tünetek az érintett agyi régióktól függően
– Dementia/kognitív hanyatlás (corticalis neuronok)
– Mozgászavarok: hipo- vagy hiperkinetikus
(basalis ganglionok neuronjai)
– Gyengeség (motoneuronok)
– Kevert

Primer neurodegeneratív
betegségek – kóros fehérjék
• Fiziológiásan megtalálható fehérje
konformációja megváltozik
• A kóros konformációjú fehérje aggregálódik és
intra-, ill. extracellulárisan lerakódik
• Pl: Tau, alpha-synuclein, beta-amyloid, TDP-43,
FUS, prion
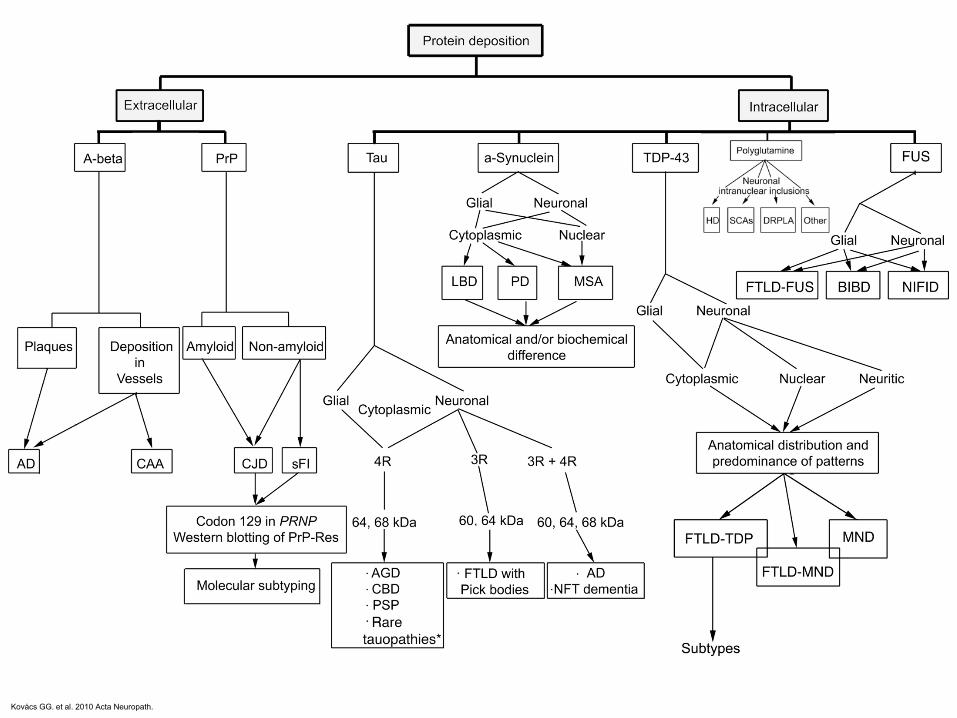
Kovács GG. et al. 2010 Acta Neuropath.

• Prevalencia (gyakoriság)
60-64 év: 1%
85 év: 50%
• Incidencia (új esetek száma) növekszik a korral
70-80 év között: 1-2 eset/100 fő/év
80 év felett: 2-8 eset/100 fő/év
• Kórlefolyás átlagosan 7 év (2-18)
Alzheimer betegség (AD)

Alzheimer betegség (AD)
• Eleinte memória zavar és kognitív zavarok
(pl: elvesznek, új helyzetekkel nem tudnak megbírkózni,
nem tudnak felöltözni stb.)
• Később szótalálási nehézség, pszichiátriai
és viselkedés zavarok, inkontinencia,
akaratlagos mozgások tervezési zavara
• Végül súlyos kognitív hanyatlás, mozgás-
képtelenség, némaság

• Sporadikus (98-99%) – „late onset”
– Számos genetikai és környezeti faktor hatása
együttesen érvényesül
• Familiáris (1-2%) – „early onset”
– Jelentős családi halmozódás (AuD öröklődés)
– Presenilin-1 és -2, ill. amyloid precursor protein
mutációk
– <60 év
Alzheimer betegség (AD)

AD makro-morfológia: Atrófia
Hydrocephalus
ex vacuo
Keskeny gyrusok
Széles sulcusok

Neurofibrilláris kötegek
Hyperfoszforilált Tau
Aβ-plakkok
β-amiloid
Amiloid angiopátia
β-amiloid
AD mikro-morfológia:
Amyloid plakkok és Neurofibrilláris kötegek
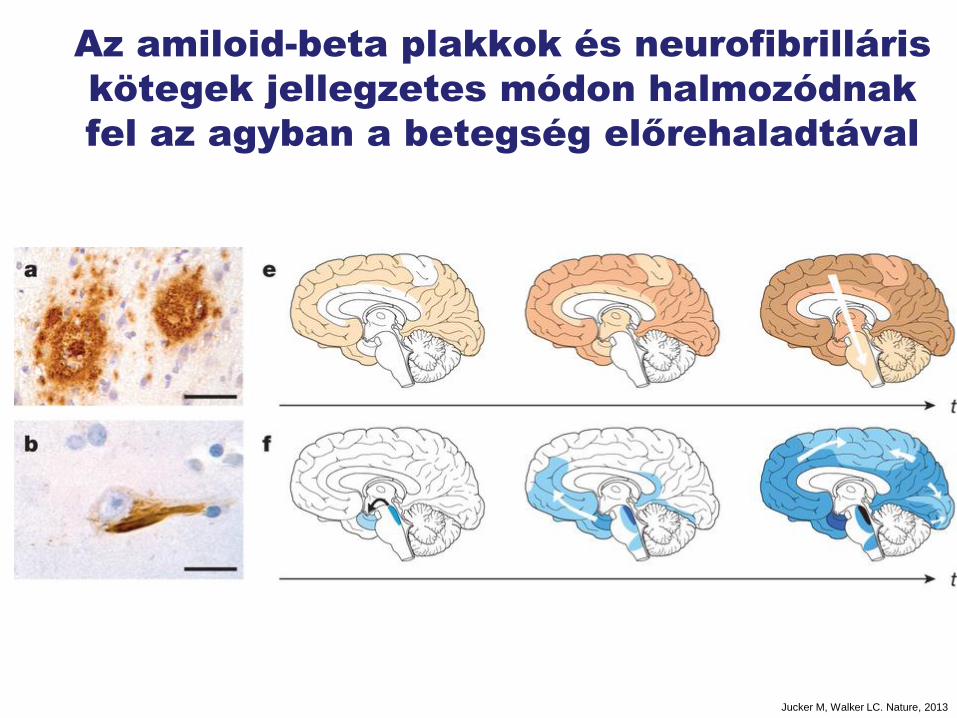
Az amiloid-beta plakkok és neurofibrilláris
kötegek jellegzetes módon halmozódnak
fel az agyban a betegség előrehaladtával
Jucker M, Walker LC. Nature, 2013

Az Aβ-peptid az APP hasításával keletkezik
Az Aβ-oligomerek neurotoxikusak
Túltermelődés
Csökkent lebontás
Fokozott aggregáció

Az Alzheimer betegség pathomechanizmusában szerepet
játszó rizikófaktorok és molekuláris mechanizmusok
Querfurth HW, LaFerla FM. NEJM, 2010

Az Alzheimer betegség kialakulása
szempontjából kockázatot jelentő gének
Barber RC. Scientifica, 2012
APOE4 allél – Aβ aggregációját fokozza és az eltávolítását csökkenti
1 allél – 4x-es rizikó
2 allél – 19x-es rizikó
Nem elegendő az AD kialakulásához

• Gyakori (10-20 új eset/100 000/év)
• 60 év után (40-70 év)
• Mozgászavar jellemzi:
tremor, rigiditás, bradykinesia, instabilitás
• Demencia társulhat hozzá
• Autonóm diszfunkció és személyiségzavar
megelőzi a mozgászavarokat
Parkinson kór (PD)

• Sporadikus
– Számos genetikai és környezeti faktor hatása
együttesen érvényesül
• Familiáris
– AuD öröklődés: α-synuclein mutációk/duplikációk
– AuR öröklődés: Parkin, UCHL-1, LRRK2, PARK7,
PINK1, stb. mutációk
Parkinson kór (PD)

PD makro-morfológia:
SN és LC halvány (depigmentált)
+/- Diffúz corticalis atrófia Normális PD

• Substantia nigra és locus coeroleus neuronjai elpusztulnak
• Lewy testek:
• SN, LC, corticalis neuronokban, híd, nyúltvelő stb.
• α-synuclein, ubiquitin, neurofilament immunpozitívak
• Lewy neurite: demenciával társuló Parkinson kórban
• „Pale” testek
Lewy body Pale body α-synuclein
Corticalis LB Lewy neurite
PD mikro-morfológia:
Neuronvesztés, Lewy testek
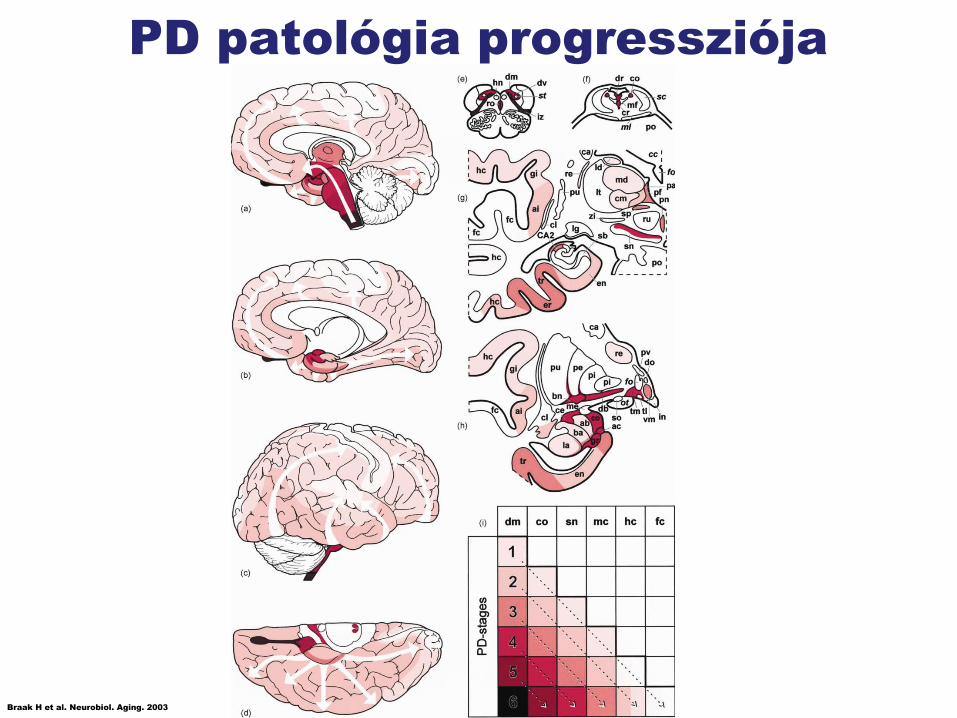
PD patológia progressziója
Braak H et al. Neurobiol. Aging. 2003

• Diffúz Lewy Body betegség
– Demencia dominál, Lewy testek a cortexben és az
agytörzsben
• Multiszisztémás atrófia
– α-synuclein depozitumok oligodendrocytákban
• Progresszív supranucleáris bénulás
– Tau depozitumok a neuronokban és gliasejtekben
• Corticobasalis degeneráció
– Tau depozitumok a neuronokban és gliasejtekben
Parkinsonizmussal járó egyéb
neurodegeneratív betegségek

• AuD öröklődés
• CAG trinucleotid repeat betegség
• Jellegzetes mozgászavarok
– Akaratlan csavaró (choreiform) mozgások, grimaszok
– Ataxia
– Hyperkinesis (vitustánc)
• Személyiség- és hangulatzavarok (fokozott hajlam
az öngyilkosságra), demencia
• Kórlefolyás átlagosan 15 év
Huntington betegség (HD)

Huntington betegség (HD)
4p16.3 – Huntingtin gén
Minél nagyobb az ismétlődések száma, annál fiatalabb korban
kezdődik a betegség (a kórlefolyás hossza nem változik)

(CAG)<36
Az expandált polyglutamin (polyQ) lánc
toxikus funkció nyeréssel jár
Exon 1
Normál allél
(CAG)>36
Mutáns allél
Huntingtin gén

HD – a mutáns huntingtin hatásai
Axonális transzport zavarok
Cytoskeletális abnormitások
Mitokondriális
diszfunkció
Ca2+
homeosztázis
zavarok Megváltozott RNS
metabolizmus
Transzkripciós zavarok
Protein felhalmozódás
Proteolítikus hasítás
Toxikus fragmentumok

HD makro-morfológia:
nucl. caudatus és putamen atrófia
+/- Corticalis atrófia (frontalis) Normális HD
Nucl. caudatus
Putamen
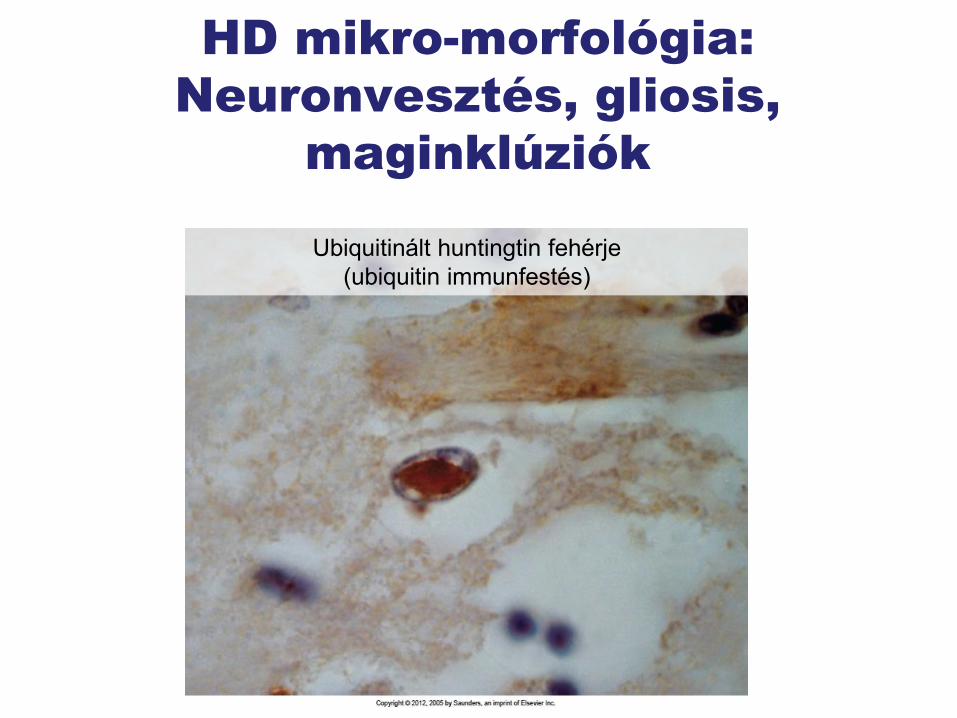
HD mikro-morfológia:
Neuronvesztés, gliosis,
maginklúziók
Ubiquitinált huntingtin fehérje
(ubiquitin immunfestés)

Amyotrophiás Lateral Sclerosis
(ALS)
• Alsó motoneuronok elpusztulnak az agytörzsben és
a gerincvelőben → izomatrófia, gyengeség,
fasciculatio
• Felső motoneuronok elpusztulnak a primer motoros
kéregben → paresis, hyperreflexia, spaszticitás
• 40-70 év között; 1-2/100.000/év
• Sporadikus
• Familiáris – 5-10%, pl. SOD-1, TDP-43, FUS mutáció

ALS – makro: atrófiás és szürke
elülső idegek
Gerincvelő elölről Gerincvelő hátulról

ALS – makro: atrófiás és szürke
elülső idegek (cauda equina)
Elülső idegek Hátsó idegek

ALS – mikro: gerincvelő motoneuron vesztés
gliosissal, következményes izomatrófiával
Bunina testek

ALS – makro: súlyos esetben
motoros cortex atrófia

ALS – mikro: neuronvesztés,
gliosis, spongiosis

Felső motoneuronok pusztulása miatt a corticospinális pálya károsodik
„lateral
sclerosis”
Normál ALS

ALS – TDP-43 pozitív neuronális
és gliális inklúziók

1. Sporadikus prionbetegség
• Sporadikus Creutzfeldt-Jakob kór (sCJD) – ~1/millió/év
• Sporadikus fatális insomnia (sFI)
2. Öröklődő prionbetegségek (IPD) – PRPN gén mutációja
• Örökletes Creutzfeldt-Jakob kór (fCJD)
• Gerstmann–Straussler–Scheinker szindróma (GSS)
• Fatális familiáris insomnia (FFI)
3. Szerzett prionbetegségek – speciális csoportokat érint
• Variáns CJD (vCJD) – fertőzött marha
• Iatrogén CJD – növekedési hormon, cornea, elektróda stb.
• Kuru - kannibalizmus
Prion betegség
TSE – transmissible spongiform
encephalopathies

Prion betegség – közös vonások
• Progresszív demencia
• Egyéb neurológiai és pszichiátriai tünetek
• Fatális
Neuron vesztés
Szivacsos degeneráció
Reaktív gliosis PrPSc
felhalmozódás

Főként α-hélix
szolubilis
proteázok bontják
ismeretlen a funkciója
Sok β-lemezt tartalmaz
nem szolubilis
proteázokra rezisztens
Normális: PrPC
Kóros: PrPSc
Konverzió ?
Prion betegségek
pathomechanizmusa

Konverzió
Öröklődő formák
Mutációk miatt változnak
a fehérje tulajdonságai –
hajlamosabb konverzióra
Szerzett CJD
Exogén PrP már
kóros szerkezetű
sCJD
Öregedéshez kötik
Poszttranszlációs módosulási
folyamatok zavara
„quality control” zavara
A PrPSc
keletkezési mechanizmusai

Prion betegségek pathogenezise

A prion protein gén - PRNP
PRNP

Prion betegség – makro:
atrófiás vagy normális agy

Prion betegség – mikro:
„Spongiform change”
Enyhe Közepes Kifejezett

Granuláris
depozitok
Szinaptikus
depozitok
Erős Gyenge Közepes
Prion betegség – mikro: a PrPSc
lerakódás mintázata sCJD-ben

Plakkok
Peri-
neuronális
hálózat
Erős Gyenge Közepes
Prion betegség – mikro: a PrPSc
lerakódás mintázata sCJD-ben
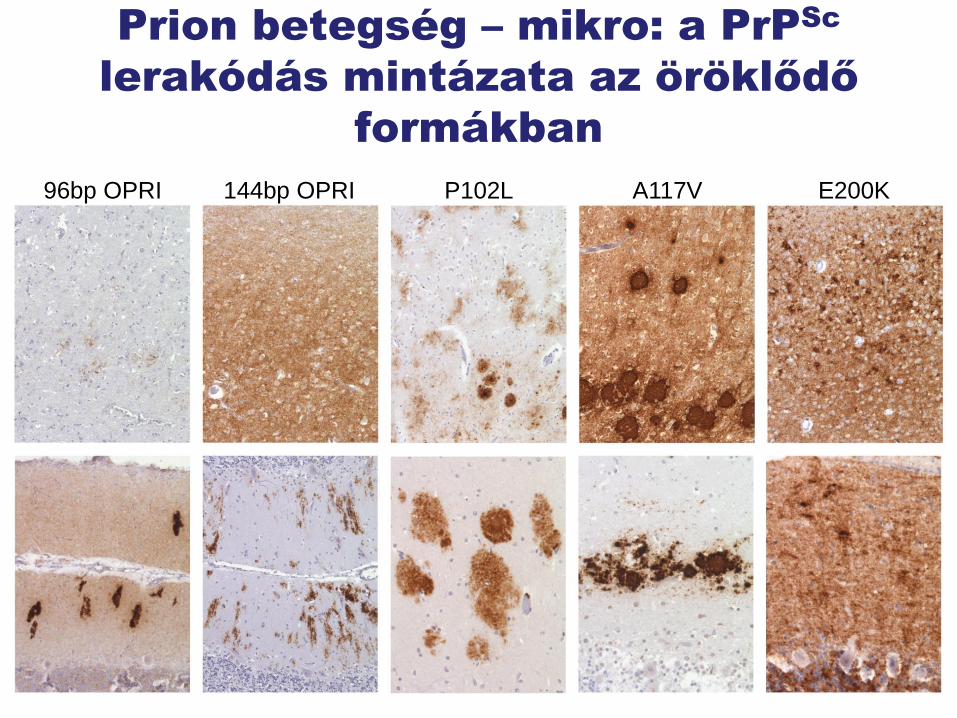
96bp OPRI 144bp OPRI P102L A117V E200K
Prion betegség – mikro: a PrPSc
lerakódás mintázata az öröklődő
formákban

Prion betegség – mikro: vCJD





























