Embed Size (px)
Citation preview

á
INSTITUTO POLITÉCNICO NACIONAL
ESCUELA NACIONAL DE CIENCIAS BIOLÓGICAS
LABORATORIO DE BIOFARMACIA
PROYECTO DE TITULACIÓN CURRICULAR
ESTUDIO DEL PERFIL DE DISOLUCIÓN DE PRESENTACIONES COMERCIALES
DE OMEPRAZOL DISPONIBLES EN EL MERCADO MEXICANO
PROYECTO DE TITULACIÓN CURRICULAR
PRESENTA
SANDRA FRANCO FLORES
ASESOR
Dra. En C. SANDRA GARCÍA MEDINA
COASESOR
QFB. JUAN MANUEL JIMÉNEZ VARGAS
2016

AGRADECIMIENTOS
Agradezco a mis padres y hermanos porque siempre están conmigo.
A mi asesora Dra. Sandra García Medina y mi coasesor QFB. Juan Manuel Jiménez
Vargas, ya que tienen el trabajo más importante del mundo, fomentar la educación
generación tras generación, sembrando el conocimiento y formando mejores
ciudadanos.
Mi gratitud al QFB. Miguel Ángel Arellano Ibáñez por su paciencia, control y
tolerancia. Gracias por compartir tu conocimiento.
A mi amiga de toda la vida QFI. Diana Carbajal es un ejemplo a seguir como mujer
y como profesionista; por su empeño y dedicación tanto al trabajo como a su hija y el
compromiso con su familia.
Así mismo, a mis compañeros Claudia Fuentes, América Castañeda y Juan
Rodríguez, porque hicieron que la vida académica fuera más sencilla, nunca
permitieron que alguno de nosotros nos quedáramos en el camino, siempre
estuvieron presentes en cada una de las etapas.
Hay una serie de personas a las que quisiera agradecer su constante apoyo y
ayuda: Carmen Valle, Guadalupe Rodríguez y Ana Silva; ya que contribuyeron a
que el presente trabajo fuera posible.

ÍNDICE
1 INTRODUCCIÓN .................................................................................................. 1
1.1 Medicamentos genéricos en el mercado mexicano ....................................... 1
1.1.1 Medicamento de referencia ......................................................................... 2
1.1.2 Medicamento genérico ................................................................................ 2
1.2 Monografía del Omeprazol ............................................................................. 5
1.2.1 Mecanismo .................................................................................................. 7
1.2.2 Farmacocinética .......................................................................................... 8
1.2.3 Efectos adversos e interacciones farmacológicas ...................................... 9
1.2.4 Usos terapéuticos ..................................................................................... 10
1.3 Disolución ..................................................................................................... 11
1.3.1 Antecedentes ............................................................................................ 11
1.3.2 Definición .................................................................................................. 13
1.3.3 Condiciones para las pruebas de disolución ............................................. 15
1.3.3.1 Aparato 2 de paletas .......................................................................... 15
1.3.3.2 Vaso ................................................................................................... 16
1.3.3.3 Eje transmisor .................................................................................... 16
1.3.3.4 Regulador de velocidad de rotación ................................................... 16
1.3.3.5 Paleta o propela ................................................................................. 17
1.3.3.6 Medio de disolución ............................................................................ 17
1.3.3.7 Agitación ............................................................................................. 19
1.4 Cromatografía de líquidos de alta resolución ............................................... 19
1.4.1 Fundamento .............................................................................................. 20
1.4.2 Partes en un cromatografo de líquidos de alta resolución ........................ 21
1.4.2.1 Sistema de bombeo............................................................................ 21
1.4.2.2 Sistema de inyección.......................................................................... 21
1.4.2.3 Detector .............................................................................................. 21
1.4.2.4 Columna ............................................................................................. 22
1.4.2.5 Registrador de señales ....................................................................... 22
1.4.3 Fase móvil ................................................................................................. 23
1.5 Validación ..................................................................................................... 23
2 JUSTIFICACIÓN ................................................................................................ 28
3 HIPÓTESIS ......................................................................................................... 29

4 OBJETIVO GENERAL ....................................................................................... 29
5 MATERIAL ......................................................................................................... 30
6 MEDICAMENTOS ESTUDIADOS ...................................................................... 31
7 METODOLOGÍA ................................................................................................. 32
7.1 Desarrollo del método analítico .................................................................... 32
7.2 Selección de las condiciones cromatograficas ............................................. 32
7.3 Preparación del medio de disolución ........................................................... 33
7.4 Pruebas de control de calidad ...................................................................... 34
7.4.1 Valoración ................................................................................................. 34
7.4.2 Uniformidad de dosis (Uniformidad de contenido) .................................... 36
7.5 Validación ..................................................................................................... 39
7.5.1 Sistema ..................................................................................................... 39
7.5.1.1 Linealidad del sistema ........................................................................ 39
7.5.1.2 Precisión del sistema.......................................................................... 41
7.5.1.3 Influencia del filtro .............................................................................. 42
7.5.1.4 Estabilidad .......................................................................................... 43
7.5.2 Método ...................................................................................................... 45
7.5.2.1 Linealidad del medicamento referencia y prueba .............................. 45
7.5.2.2 Exactitud del medicamento de referencia y prueba ............................ 48
7.5.2.3 Precisión del medicamento de referencia y prueba ............................ 49
7.5.2.4 Selectividad del medicamento de referencia y prueba ....................... 50
7.6 Perfil de disolución ....................................................................................... 52
8 RESULTADOS ................................................................................................... 55
8.1 Valoración .................................................................................................... 55
8.2 Uniformidad de dosis (uniformidad de contenido) ........................................ 55
8.3 Validación ..................................................................................................... 56
8.4 Perfiles de disolución ................................................................................... 57
9 DISCUSIÓN ........................................................................................................ 60
10 CONCLUSION .................................................................................................... 71
11 ANEXOS ............................................................................................................. 72

11.1 Anexo 1. Aparato 2 de paletas ..................................................................... 72
11.2 ANEXO 2. Validación ................................................................................... 73
11.3 Anexo 3 . Cromatogramas ........................................................................... 83
11.4 Anexo 4. Perfiles de disolución .................................................................... 86
BIBLIOGRAFÍA ...................................................................................................... 100

ÍNDICE DE FIGURAS
Figura 1.Regulación fisiológica y farmacológica de la secreción gástrica 8
Figura 2.Diagrama de flujo del desarrollo del método analítico para la cuantificación
de los fármacos de trabajo (omeprazol) en un perfil de disolución 32
Figura 3.Comparación de perfil de disolución de Losec- A20 contra genérico Elite
Medical 59
Figura 4.Aparato 2, paletas 72
Figura 5.Linealidad del sistema (concentración vs respuesta) 74
Figura 6.Linealidad del método del medicamento de referencia (concentración
nominal contra concentración recuperada) 76
Figura 7. Linealidad del método del medicamento de prueba (concentración
nominal contra concentración recuperada 80
Figura 8. Cromatograma del perfil de disolución en la etapa ácida a los 15 min 83
Figura 9. Cromatograma del perfil de disolución en la etapa ácida a los 30 min 83
Figura 10. Cromatograma del perfil de disolución en la etapa ácida a los 60 min 84
Figura 11.Cromatograma del perfil de disolución en la etapa ácida a los 120 min84
Figura 12. Cromatograma de muestra de omeprazol 85

INDICE DE TABLAS
Tabla 1. Propiedades fisicoquímicas del omeprazol 6
Tabla 2. Equipos e instrumentos, reactivos y materiales utilizados 30
Tabla 3. Medicamentos evaluados 31
Tabla 4. Parámetros del método para el análisis de separación 33
Tabla 5. Preparación de la curva de calibración 40
Tabla 6. Resultado de la valoración 55
Tabla 7. Resultado de la uniformidad de contenido 55
Tabla 8. Resultados de la validación 56
Tabla 9. Resultados de perfiles de disolución 57
Tabla 10. Resultado de f2 58
Tabla 11. Datos de linealidad del sistema 73
Tabla 12. Datos de precisión del sistema 74
Tabla 13. Datos de linealidad del método del medicamento de referencia 75
Tabla 14. Datos de exactitud del medicamento de referencia 76
Tabla 15. Datos de precisión del medicamento de referencia (repetibilidad) 77
Tabla 16. Datos de precisión del medicamento de referencia(reproducibilidad) 77
Tabla 17. Datos de selectividad del medicamento de referencia 78
Tabla 18. Datos de linealidad del método del medicamento de prueba 79
Tabla 19. Datos de exactitud del medicamento de prueba 80
Tabla 20. Datos de precisión del medicamento de prueba (repetibilidad) 81
Tabla 21. Datos de precisión del medicamento de prueba(reproducibilidad) 81
Tabla 22. Datos de selectividad del medicamento de prueba 82
Tabla 23. Datos de la unidad #1 analizadas durante el perfil de disolución de
Losec-A20 86
Tabla 24.Datos de la unidad # 2 analizada durante el perfil de disolución de
Losec-A20. 86

Tabla 25. Datos de la unidad # 3 analizada durante el perfil de disolución de
Losec-A20 87
Tabla 26. Datos de la unidad #4 analizada durante el perfil de disolución de
Losec-A20 87
Tabla 27. Datos de la unidad #5 analizada durante el perfil de disolución de
Losec-A20 88
Tabla 28. Datos de la unidad #6 analizada durante el perfil de disolución de
Losec-A20 88
Tabla 29. Datos de la unidad #7 analizada durante el perfil de disolución de
Losec-A20 89
Tabla 30. Datos de la unidad #8 analizada durante el perfil de disolución de
Losec-A20 89
Tabla 31. Datos de la unidad #9 analizada durante el perfil de disolución de
Losec-A20 90
Tabla 32. Datos de la unidad #10 analizada durante el perfil de disolución de
Losec-A20 90
Tabla 33. Datos de la unidad #11 analizada durante el perfil de disolución de
Losec-A20 91
Tabla 34. Datos de la unidad # 12 analizada durante el perfil de disolución de
Losec-A20 91
Tabla 35. Datos de la unidad #1 analizada durante el perfil de disolución de Medical
Elite 92
Tabla 36. Datos de la unidad #2 analizada durante el perfil de disolución de Medical
Elite 92
Tabla 37. Datos de la unidad #3 analizada durante el perfil de disolución de Medical
Elite 93
Tabla 38. Datos de la unidad #4 analizada durante el perfil de disolución de Medical
Elite 93

Tabla 39. Datos de la unidad #5 analizada durante el perfil de disolución de Medical
Elite 94
Tabla 40. Datos de la unidad # 6 analizada durante el perfil de disolución de Medical
Elite 94
Tabla 41. Datos de la unidad #7 analizada durante el perfil de disolución de Medical
Elite 95
Tabla 42. Datos de la unidad #8 analizada durante el perfil de disolución de Medical
Elite 95
Tabla 43. Datos de la unidad # 9 analizada durante el perfil de disolución de Medical
Elite 96
Tabla 44. Datos de la unidad # 10 analizada durante el perfil de disolución de
Medical Elite 96
Tabla 45. Datos de la unidad #11 analizada durante el perfil de disolución de
Medical Elite 97
Tabla 46. Datos de la unidad #12 analizada durante el perfil de disolución de
Medical Elite 97
Tabla 47. Obtención de porcentaje de omperazol disuelto en el i-ésimo tiempo de
muestreo de Losec-A20 98
Tabla 48. Obtención de porcentaje de omperazol disuelto en el i-ésimo tiempo de
muestreo de Medical Elite 99

SÍMBOLOS Y ACRÓNIMOS
Cuando en el presente proyecto se haga referencia a las siguientes abreviaturas se
entenderá:
% Por ciento ° Grado Celsius CV% Coeficiente de variación CYP Citocromo P450 DCI Denominación Común Internacional f2 Factor de similitud FEUM Farmacopea de los Estados Unidos Mexicanos GERD Enfermedad del Reflujo Gastroesofágico h Hora HPLC Cromatografía de Líquidos de Alta Resolución IUPAC Unión Internacional de Química Pura y Aplicada L Litro Log P Coeficiente de reparto M Molaridad mg Miligramos min Minutos mL Mililitros mm Milímetros N Normalidad ng Nanogramos NOM Norma Oficial Mexicana pH Potencial de hidrógeno PPI Bomba inhibidora de protones rpm Revoluciones por minuto SGF Simular fluido gástrico SIF Simular fluido intestinal uL Microlitros um Micrómetros USP Farmacopea de los Estados Unidos UV Ultravioleta v/v Volumen – volumen

[Escribir el título del documento]
1
1 INTRODUCCIÓN
1.1 Medicamentos genéricos en el mercado mexicano
Todas los medicamentos que están autorizadas para vender en México han
cumplido con los requisitos de calidad, seguridad y eficacia solicitados por la
Secretaria de Salud para el registro sanitario; sin embargo los requisitos para
obtenerlo han cambiado con el avance tecnológico. Conforme la tecnología avanza
para fabricar medicamentos cada vez de mejor calidad, los aspectos técnicos
necesarios para demostrar su calidad farmacéutica, seguridad y eficacia se han
hecho más estrictos. Los estudios que se debían entregar junto con la solicitud de
un registro hace años eran muy pocos, mientras que ahora la autoridad sanitaria es
y debe ser, mucho más exigente. Para facilitar su cumplimiento estos requerimientos
deben ser muy claros y explícitos (Gonzalez, et al., 2005).
Debido a la gran relevancia que tiene la investigación y desarrollo en los
productos farmacéuticos, los laboratorios se clasifican de conformidad con el estado
de propiedad intelectual que guardan los medicamentos que producen (Gonzalez, et
al., 2005):
Empresas que se especializan en desarrollar, fabricar y vender medicamentos
con patente y que son conocidas como empresas innovadoras, y,
Empresas que fundamentalmente fabrican productos que han perdido la
protección de una patente y que son conocidas como de genéricos.
Empresas que participan en ambas actividades.

2
En México, existen varios laboratorios extranjeros que participan tanto en el
segmento de medicamentos de patente como en el de medicamentos genéricos
(Gonzalez, et al., 2005).
La necesidad de abatir los costos del tratamiento médico en los últimos años,
ha dirigido la atención a los genéricos y al papel que éstos pueden jugar en la
disminución de los gastos (Gonzalez, et al., 2005).
En todos los países la entrada al mercado de los genéricos está ligada a la
extinción de la vigencia de una patente de un medicamento innovador. Por lo
anterior es indispensable definir ambos (Brunton, et al., 2012).
1.1.1 Medicamento de referencia
De acuerdo a la Norma Oficial Mexicana (NOM-177, 2013) es un
medicamento indicado por la Secretaría como tal, que cuenta con el registro de
dicha dependencia que se encuentra disponible comercialmente y es seleccionada
conforme a los criterios establecidos por las Normas.
1.1.2 Medicamento genérico
De acuerdo al Decreto del 02 de Enero 2008 (que reforma, adiciona y deroga
diversas disposiciones del Reglamento de Insumos para la Salud) se define como:
especialidad farmacéutica con el mismo fármaco o sustancia activa y forma
farmacéutica, con igual concentración o potencia, que utiliza la misma vía de
administración y que mediante las pruebas reglamentarias requeridas, ha

3
comprobado que sus especificaciones farmacopeicas, perfiles de disolución o su
biodisponibilidad u otros parámetros, según sea el caso, son equivalentes a las del
medicamento de referencia.
Así antes de obtener el registro sanitario, es imperativa la demostración de la
seguridad y eficacia con la investigación preclínica y clínica de los medicamentos.
Los innovadores soportan su eficacia y seguridad por medio de la investigación
básica y clínica necesaria, usualmente costosa y prolongada. La básica de
laboratorio y en animales de experimentación estudia el mecanismo de acción, la
eficacia y los efectos adversos, en especial durante el embarazo y la lactancia, en el
embrión, en el feto y en la descendencia, en la inducción de tumores y en ciertos
casos de interacciones medicamentosas (Gonzalez, et al., 2005).
La clínica analiza el comportamiento del fármaco en el humano, su absorción,
niveles sanguíneos y eliminación, la determinación de la dosis, su eficacia en
estudios preliminares y posteriormente en investigaciones en las que se compara el
medicamento con placebo y otros productos en un grupo mayor de pacientes, así
como la recopilación cuidadosa de los efectos adversos buscados y vigilados
intencionalmente (Gonzalez, et al., 2005).
En cambio los medicamentos genéricos que “copian” a los innovadores no
requieren repetir la investigación ya efectuada en los originales. La forma de
garantizar su eficacia y seguridad es por medio de la realización de pruebas de
intercambiabilidad que demuestren que el genérico se comporta igual que el

4
innovador (una forma más breve y económica que la de la investigación clínica).
Estas pruebas son necesarias para obtener el registro sanitario en la mayoría de los
países. Actualmente se calcula que un porcentaje de medicamentos no cuenta con
esta evidencia (Gonzalez, et al., 2005).
Al darle vigencia al registro sanitario de cinco años, se solicitara para su
prorroga o renovación, las pruebas clínicas o de intercambiabilidad que garanticen
su seguridad a la luz de los avances de la tecnología. La pretensión es que todos los
medicamentos en México posean, en los próximos 5 años, una u otra evidencia de
seguridad y eficacia (Gonzalez, et al., 2005).
En efecto, las pruebas de intercambiabilidad tienen como objetivo
fundamental el demostrar que un medicamento con el mismo principio activo y la
misma forma farmacéutica que un innovador puede sustituir a este sin perjuicio de
su seguridad y de su eficacia. Estas pruebas se realizan por laboratorios “terceros
autorizados”. Los laboratorios farmacéuticos podrían realizar sus propias pruebas
siempre que cumplieran con los mismos requisitos que se les exigen a los terceros
autorizados, previa autorización de la Secretaría de Salud (Gonzalez, et al., 2005).
Para establecer la intercambiabilidad de los medicamentos genéricos es
necesario realizar, de manera científica, pruebas que demuestren que éstos son
equivalentes con respecto al medicamento de referencia dentro de un intervalo
definido. Entre las principales pruebas, están las de biodisponibilidad,

5
bioequivalencia y la comparación de los perfiles de disolución, las cuales están
incluidas en la Norma Oficial Mexicana (NOM-177, 2013).
1.2 Monografía del Omeprazol
El omeprazol es una mezcla racémica de R- y S-isómeros; el S-isómero,
esomeprazol (S-omeprazol), es eliminado con menos rapidez que el R-omeprazol, el
cuál teóricamente proporciona una ventaja terapéutica debido a la semivida mayor
(Brunton, et al., 2012).
El principio activo de las cápsulas de omeprazol 20mg es liberado en un
momento distinto al de la administración, pero no se prolonga el efecto terapéutico
(no hay cambios en ningún otro parámetro terapéutico). Son formas con cubierta
entérica sensible al pH, en las que el principio activo es liberado en una zona
concreta del intestino delgado (Navarra, 2005).
Todas las preparaciones deben protegerse de la luz y conservarse entre 15 a
30°C como máximo (Gomez, et al., 1997).
El omeprazol es químicamente estable y carente de actividad inhibidora a pH
neutro. Sin embargo el compuesto es protonado a pH 5.0, y por debajo de este se
degrada a sus metabolitos el ácido sulfenico y una sulfenamida (Storpirtis &
Rodriguez, 1998).

6
El omeprazol es un polvo blanco o blanquecino, cristalino que funde a 155°C
con descomposición, posee carácter básico débil y es libremente soluble en lípidos,
etanol y metanol, ligeramente soluble en acetona e isopropanol y muy poco soluble
en agua (Tabla 1). La estabilidad de la sustancia está en función del pH, se degrada
rápidamente en medio ácido, pero permanece prácticamente estable en condiciones
alcalinas (Gomez, et al., 1997).
Tabla 1. Propiedades fisicoquímicas del omeprazol
Nombre IUPAC 6-Metoxi-2-[(4-metoxi-3,5-dimetilpiridina-2-il)metilsulfinil]-1H-benzimidazol
Fórmula condensada C17H19N3O3S
Estructura química
Peso molecular 345.4 g
Descripción Polvo blanco, fotosensible
Solubilidad
Ligeramente soluble en agua, soluble en alcohol, alcohol metílico y diclorometano; muy soluble en soluciones alcalinas
PKa 4.0
Log P 2.23 ( octanol / agua)
Clasificación biofarmacéutica (Abdel-
Rahman, et al., 2012)
Baja solubilidad, alta permeabilidad
Estabilidad Sensible a la luz. Conservarse de 15 a 30°C como máximo. Se degrada a pH menores 5.0
(Moffat, et al., 2011)

7
1.2.1 Mecanismo de acción
El omeprazol es clasificado dentro de los inhibidores de la bomba de protones
(PPI, protonpumpinhibitors) son profármacos que exigen activación en un medio
ácido. Después de la absorción hacia la circulación periférica, el profármaco se
difunde hacia las células parietales del estómago y se acumula en los canalículos
secretores de ácido. Ahí, es activado por la formación catalizada por protones de
sulfonamida tetracíclica, lo que atrapa el fármaco de manera que no se puede
difundir de regreso a través de la membrana canalícular. La forma activada se une
luego de manera covalente a los grupos sulfhidrilo de las cisteínas en la H+, K+-
ATPasa, inactivando de manera irreversible la molécula de la bomba. La secreción
de ácido se reanuda sólo después de que se sintetizan nuevas moléculas de la
bomba y se insertan en la membrana luminal, lo que proporciona una supresión
prolongada (de incluso 24 a 48 hrs) en la secreción de ácido, pese a las semividas
plasmáticas más breves (0.5 a 2 h) de los compuestos originales. Puesto que
bloquean el paso final en la producción de ácido, los inhibidores de la bomba de
protones son eficaces para la supresión de ácido independientemente de otros
factores estimulantes (Figura 1) (Brunton, et al., 2012).

8
Figura 1. Regulación fisiológica y farmacológica de la secreción gástrica
(Brunton, et al., 2012).
1.2.2 Farmacocinética
Puesto que se necesita un pH ácido en los canalículos ácidos de la célula
parietal para la activación del fármaco y el alimento estimula la producción de ácido,
estos fármacos en condiciones ideales deberían administrarse aproximadamente 30
minutos antes de las comidas (Brunton, et al., 2012).
Los inhibidores de la bomba de protones, una vez que llegan al intestino
delgado, rápidamente se absorben, se unen en alto grado a la proteína y son
metabolizados considerablemente por las enzimas CYP hepáticas, sobre todo
CYP2C19 y CYP3A4 (Brunton, et al., 2012).
Dado que no todas las bombas o todas las células parietales, están activas en
forma simultánea, la supresión máxima de secreción de ácido exige varias dosis de

9
inhibidores de la bomba de protones. Por ejemplo, es posible tardar dos a cinco días
de tratamiento con una dosis de una vez al día para alcanzar la inhibición de 70% de
las bombas de protones que se observa en un estado de equilibrio dinámico. La
dosis inicial más frecuente reducirá el tiempo en que se logra la inhibición completa
pero no está demostrado que mejore el pronóstico para el paciente. Puesto que la
inhibición de la bomba de protones es irreversible, la secreción de ácido se suprimirá
durante 24 a 48 h, o más, hasta que se sinteticen nuevas bombas de protones y se
incorporen en la membrana luminal de las células parietales (Brunton, et al., 2012).
1.2.3 Efectos adversos e interacciones farmacológicas
Los inhibidores de la bomba de protones por lo regular producen
notablemente pocos efectos adversos. Los efectos adversos más frecuentes son
náuseas, dolor abdominal, estreñimiento, flatulencia y diarrea. Asimismo, se ha
comunicado la presentación de miopatía subaguda, artralgias, cefaleas y
exantemas. Como se mencionó antes, los inhibidores de la bomba de protones son
metabolizados por los CYP hepáticos y por tanto pueden interferir en la
farmacocinética de otros fármacos que se eliminan por esta vía. Los inhibidores de la
bomba de protones interactúan con la warfarina (esomeprazol, lansoprazol,
omeprazol y rabeprazol), el diazepam (esomeprazol y omeprazol) y la ciclosporina
(omeprazol y rabeprazol). Entre los inhibidores de la bomba de protones, sólo el
omeprazol inhibe al CYP2C19 (y por tanto reduce la eliminación de disulfiram,
fenitoína y otros fármacos) y activa la expresión de CYP1A2 (aumentando de esta

10
manera la eliminación de la imipramina, varios antipsicóticos, tacrina y teofilina.
Pruebas indicativas sugieren que el omeprazol puede tener una interacción
adversa con el anticoagulante clopidogrel, al nivel de CYP2C19, para el cual los dos
son sustratos; por consiguiente, el omeprazol puede inhibir la conversión de
clopidogrel a la forma anticoagulante activa (Brunton, et al., 2012).
El tratamiento crónico con omeprazol disminuye la absorción de vitamina B12,
pero no está clara la importancia clínica de este efecto. El empleo crónico de
inhibidores de la bomba de protones se ha relacionado con un incremento del riesgo
de fracturas óseas y un aumento de la susceptibilidad a determinadas infecciones (p.
ej., neumonía intrahospitalaria, infección extrahospitalaria por Clostridium difficile).
La hipergastrinemia es más frecuente y más grave con los inhibidores de la bomba
de protones que con los antagonistas del receptor H2 y las concentraciones de
gastrina > 500 ng/L se presentan en aproximadamente 5 a 10 % de los usuarios con
la administración crónica de omeprazol. Esta hipergastrinemia puede predisponer a
la secreción de ácido gástrico de rebote tras la suspensión de tratamiento y también
favorece la formación de tumores del tubo digestivo (Brunton, et al., 2012).
1.2.4 Usos terapéuticos
Se utilizan los inhibidores de la bomba de protones que se prescriben para
favorecer la cicatrización de las úlceras gástricas y duodenales y tratar la
enfermedad por reflujo gastroesofágico (GERD, gastroesophagealrefluxdisease), lo

11
que comprende esofagitis erosiva, que tiene complicaciones o que no responde al
tratamiento con antagonistas de receptor H2. El omeprazol de venta sin receta está
autorizado para el autotratamiento de la pirosis. Los inhibidores de la bomba de
protones también constituyen la base del tratamiento de los transtornos que cursan
con hipersecreción patológica, entre ellos el síndrome de Zollinger-Ellison. Ademas
todos los inhibidores de la bomba de protones están autorizados para reducir el
riesgo de recidiva de úlcera duodenal relacionada con las infecciones por H. pylori
(Brunton, et al., 2012).
1.3 Disolución
1.3.1 Antecedentes
Es probable que la referencia preliminar a la disolución consista en un artículo
de Noyes y Whitney de 1897 acerca de “The Rate of Solution of Solid Substances in
Their Own Solution” (Velocidad de solución de sustancias sólidas en su propia
solución). Los autores sugerían que la velocidad de disolución de las sustancias
sólidas está determinada por la velocidad de difusión de una capa muy delgada de la
solución saturada que se forma instantáneamente alrededor de la partícula sólida.
Desarrollaron la relación matemática que correlaciona la velocidad de disolución con
el gradiente de solubilidad del sólido. Su ecuación es todavía la fórmula básica que
fundamenta la mayoría de los tratamientos matemáticos modernos del fenómeno de
la disolución (Gennaro, 2003).

12
Sin embargo, los más importantes de estos estudios fueron la aplicación de la
ley de difusión de Fick a la ecuación de Noyes y Whitney por parte de Nerst y
Brunner en 1904 y el desarrollo de la famosa ley de la raíz cúbica de disolución por
parte de Hixson y Crowell en 1931 (Gennaro, 2003).
A mediados del siglo, el énfasis comenzó a desplazarse hacia el examen de
los efectos que el comportamiento de la disolución de drogas tiene sobre la actividad
de los preparados farmacéuticos. Uno de los estudios preliminares con este
propósito fue llevado a cabo por J. Edwards en 1951 en comprimidos de aspirina.
Sobre la base de sus hallazgos informó que “debido a si escasa solubilidad, la
acción analgésica de los comprimidos de aspirina estaría controlada por su
velocidad de disolución en el estómago y en el intestino”. Sin embargo, Edwards no
llevó a cabo ningún estudio in vitro para avalar su postulado (Gennaro, 2003).
Aproximadamente 8 años más tarde, Shenoy y col. demostraron la validez de
la sugerencia de Edwards de la correlación in vitro/in vivo por medio de la
demostración de una relación directa entre la biodisponibilidad de la anfetamina de
comprimidos de liberación sostenida y su velocidad de disolución in vitro. Otros
estudios en especial los de Nelson, Levy y otros, confirmaron más allá de toda duda
el efecto significativo de la conducta de disolución de las drogas sobre sus
actividades farmacológicas. Debido a la importancia de estos hallazgos, las pruebas
de disolución comenzaron a surgir como un tema dominante tanto en el campo
académico farmacéutico como en la industria farmacéutica (Gennaro, 2003).

13
A fines de la década de 1960 las pruebas de disolución se convirtieron en un
requerimiento obligatorio para diversos preparados. Sin embargo, el papel de la
disolución en la absorción de las drogas está lejos de ser comprendido
perfectamente. A pesar del éxito informado de diversos estudios de correlación in
vitro/in vivo, la disolución no es predictor de la eficacia terapéutica. Más bien es una
herramienta cualitativa que puede proporcionar información valiosa acerca de la
biodisponibilidad biológica de una droga así como de la uniformidad entre un lote y
otro. Otra área de dificultad es el hecho de que la exactitud y la precisión del
procedimiento de prueba dependen en gran medida del estricto cumplimiento de
demasiados parámetros sutiles y de detallados controles operativos (Gennaro,
2003).
1.3.2 Definición
La disolución es el proceso por el cual un sólido con características de
solubilidad relativamente buenas entra en solución (Gennaro, 2003).
A pesar de estas desventajas, la disolución se considera hoy en día una de
las pruebas de control de calidad más importantes realizadas en los preparados
farmacéuticos (Gennaro, 2003).
El proceso de absorción de un fármaco contenido en una forma farmacéutica
sólida, después de la administración oral depende, entre otros aspectos, de la
liberación del principio activo del producto y de su disolución o solubilización en las

14
condiciones fisiológicas. Debido a la naturaleza de estos factores, la evaluación de la
velocidad de disolución in vitro puede ser una predicción del comportamiento in vivo,
siempre y cuando el paso limitante para la absorción sea la disolución (Farmacopea,
2014).
Las pruebas de disolución farmacopeicas son pruebas límite puntuales, éstas
únicamente evalúan la cantidad de principio activo disuelto en un tiempo
determinado y el criterio de aceptación es útil para el control de calidad del
medicamento, pero no proporcionan información de la velocidad a la cual el fármaco
se disuelve (Farmacopea, 2014).
En base a esta consideración general, se utilizan las pruebas de disolución in
vitro para las formas de dosificación oral sólidas, como comprimidos y cápsulas, para
evaluar la calidad de los medicamentos lote a lote; guiar el desarrollo de nuevas
formulaciones; y asegurar la calidad y el rendimiento continuados del producto
después de ciertos cambios, tales como cambios en la formulación, el proceso de
fabricación, el sitio de fabricación y el aumento en escala del proceso de fabricación
(FDA, 2015).
Se deberá considerar el conocimiento actual acerca de la solubilidad,
permeabilidad, disolución y farmacocinética de un producto al definir las
especificaciones de las pruebas de disolución para el proceso de aprobación del
fármaco. También se deberá utilizar este conocimiento para asegurar la equivalencia

15
continuada del producto, así como para asegurar la igualdad de estos bajo ciertos
cambios de escala y posteriores a la aprobación (FDA, 2015).
1.3.3 Condiciones para las pruebas de disolución
1.3.3.1 Aparato 2 de paletas
Los métodos de prueba de disolución utilizados más comúnmente son el
método de cesta (Aparato 1) y el método de paleta (Aparato 2). Los métodos de
cesta y paleta son sencillos, robustos, están bien normalizados y se utilizan en todo
el mundo. Estos métodos son lo suficientemente flexibles como para permitir la
realización de pruebas de disolución para una variedad de productos medicinales.
Por este motivo, debería utilizarse los métodos de disolución in vitro descritos en la
Farmacopea Estadounidense (USP), Aparato 1 y Aparato 2, salvo que se pruebe
que no son satisfactorios. Además, la elección del aparato se basa en el
conocimiento que se tenga sobre el diseño de la formulación y los aspectos prácticos
del desempeño de la forma farmacéutica en el sistema de la prueba in vitro (FDA,
2015).
En este caso se usa el aparato dos de paletas que consta de un baño de
agua o en su caso chaquetas de calentamiento y de seis unidades de prueba, donde
cada una está constituida por (Farmacopea, 2014):
Un vaso cilíndrico de fondo semiesférico, con tapa.
Un eje transmisor.

16
Un regulador de velocidad de rotación.
Paleta o propela
1.3.3.2 Vaso
Debe ser de vidrio o de otro material inerte y transparente. De forma cilíndrica
y de fondo semiesférico, de 60 mm a 210 mm de alto y de 98 mm a 106 mm de
diámetro interno, con capacidad para 1 000 mL. La tapa debe estar ajustada para
retardar la evaporación y permitir la inserción de un termómetro, así como la toma de
la muestra. El vaso debe estar firmemente ajustado, sumergido en el baño de agua,
el cual debe mantener la temperatura del medio de disolución a 37°C ± 0.5°C. El
aparato debe permitir la visualización del desarrollo de la prueba (Farmacopea,
2014).
1.3.3.3 Eje transmisor
Debe ser de acero inoxidable tipo 316 y girar suavemente, sin bamboleo, de
9.4 mm a 10.1 mm de diámetro. Debe estar colocado en el centro del vaso, de tal
manera que no quede a más de2.0 mm de cualquier punto del eje vertical del vaso
(Farmacopea, 2014).
1.3.3.4 Regulador de velocidad de rotación
Debe mantener la velocidad constante de acuerdo con lo indicado en la
monografía del producto (Farmacopea, 2014).

17
1.3.3.5 Paleta o propela
Hélice agitadora de 4 mm ± 1 mm de espesor y de 19 mm ± 0.5 mm de alto,
en forma de sección de un círculo de radio de 41.5 mm ± 1.0 mm y cuerdas
paralelas subtendidas de 42 mm ± 1.0 mm y de 74.5 mm± 0.5 mm, quedando la
sección más pequeña hacia abajo. La distancia de la base de la paleta al centro del
círculo imaginario es de 35.8 mm ± 1.0 mm. La línea central de la cuchilla pasa a
través del eje transmisor de tal manera que la sección de 42 mm de la misma quede
perpendicular al eje transmisor al final del mango formando una unidad que puede
estar recubierta con un polímero de fluorocarbono de cualquier otro material inerte.
Durante la prueba se debe mantener una distancia de 25 mm ± 2.0 mm entre la orilla
inferior de la propela y el fondo del vaso. Se puede utilizar un dispositivo de material
no reactivo, para mantener la muestra en el fondo del vaso y evitar que flote
(Farmacopea, 2014).
1.3.3.6 Medio de disolución
En lo posible, las pruebas de disolución se deberán realizar bajo condiciones
fisiológicas. Esto permite la interpretación de los datos de disolución en relación al
rendimiento in vivo del producto. Sin embargo, no hace falta una adherencia estricta
al ambiente gastrointestinal en las pruebas de disolución rutinarias. Las condiciones
de prueba deberán basarse en las características fisicoquímicas de la sustancia

18
medicinal y las condiciones ambientales a las cuales podría estar expuesta la forma
de dosificación tras la administración oral (FDA, 2015).
Por lo general el volumen del medio de disolución es de 500, 900 ó 1000 mL.
Es deseable pero no obligatorio tener condiciones de pila. Se deberá utilizar un
medio acuoso con una gama de pH de 1,2 a 6,8 (la misma concentración iónica de
los tampones de la USP). Para simular el fluido intestinal (SIF), se deberá emplear
un medio de disolución con un pH de 6,8. Se deberá justificar un pH más alto caso
por caso y, por lo general, el pH no deberá excederse de 8,0. Para simular un fluido
gástrico (SGF), se deberá emplear un medio de disolución con un pH de 1,2 sin
enzimas (FDA, 2015).
Se deberá realizar todas las pruebas de disolución para formas de
dosificación de IR a 37±0,5°C. Se puede utilizar el método de cesta y paleta para
realizar las pruebas de disolución bajo condiciones de medios múltiples (p.ej. se
puede realizar la prueba de disolución inicial a un pH de 1,2 y, tras un intervalo
apropiado, se puede agregar una pequeña cantidad de tampón para aumentar el pH
a 6,8 (FDA, 2015).
Ciertos productos y formulaciones medicinales son sensibles al aire disuelto
en el medio de disolución y necesitarán desaireación. Por lo general, las formas de
dosificación en cápsulas tienden a flotar durante las pruebas de disolución con el
método de paleta. En tales casos, se recomienda utilizar varias vueltas de una hélice
de alambre (USP) alrededor de la cápsula (FDA, 2015).

19
1.3.3.7 Agitación
Por lo general, se deberá mantener condiciones de agitación suave durante
las pruebas de disolución para permitir un poder de discriminación máximo y para
detectar productos con un pobre rendimiento in vivo. Utilizando el método de paleta,
es de 50-75 (FDA, 2015).
1.4 Cromatografía de líquidos de alta resolución
En los inicios de la cromatografía de líquidos se utilizaban columnas de vidrio
con diámetros de 1 a 5cm y longitudes de 50 a 500 cm. El relleno de las columnas
consistía en partículas de 150 a 200 micras de diámetro. Más tarde se encontró que
se podía aumentar la eficiencia disminuyendo el tamaño de las partículas de los
rellenos. A finales de los años 60 se logró desarrollar la tecnología adecuada para
producir y utilizar partículas del orden de las 3 las 10 micras. A esta nueva forma de
cromatografía se le llamó cromatografía de líquidos de alta resolución, HPLC por sus
siglas en inglés (High Performance Liquid Cromatography) (Esquivel & Leal, 2004).
El sistema cromatografico de fase reversa fue introducido por Howard y Marlin
en 1950; aquella en la que la fase estacionaria es no polar y la fase móvil es polar,
esta técnica se ha convertido en el tipo de cromatografía más ampliamente utilizada
en HPLC; ya que proporciona retención y selectividad optimas cuando las muestras
tienen un carácter predominantemente alifático o aromático (Esquivel & Leal, 2004).

20
El éxito en a la aplicación de HPLC para un compuesto dado depende de la
combinación correcta de las condiciones de operación, es decir: la preparación de la
muestra, el tipo de la columna, la fase móvil, la longitud y diámetro de la columna, la
velocidad de flujo de la fase móvil, el tipo de detección, el algoritmo de integración,
etc (Farmacopea, 2014).
Para asegurar la efectividad del sistema, es necesario someterlo a una
prueba antes de utilizarse. La esencia de este tipo de pruebas es el concepto de que
el equipo en general, las partes electrónicas, las operaciones analíticas y la muestra,
constituyen un sistema analítico completo el cual puede someterse a una prueba
general de funcionamiento del sistema (Farmacopea, 2014).
1.4.1 Fundamento
La migración diferencial en HPLC es resultado de equilibrio de distribución de
los componentes de una mezcla entre la fase estacionaria y la fase móvil. Dichos
componentes se separan en la columna y al salir de ésta son conducidos por la fase
móvil en el orden en el que emergieron, hacia un detector donde se registra una
respuesta proporcional a su cantidad sus concentraciones y sus tiempos de
retención en la columna. El cromatograma resultante muestra cada compuesto que
sale de la columna en forma de picos simétricos con un tiempo de retención
característico por lo que este tiempo puede emplearse para identificar el compuesto.
Este tiempo de retención (tr) se mide desde el momento de la inyección de la

21
muestra hasta el momento en que aparece el máximo del pico en el cromatograma
(Farmacopea, 2014).
1.4.2 Partes en un cromatografo de líquidos de alta resolución
Esencialmente, un cromatógrafo de líquidos de alta resolución consta de las
siguientes partes (Farmacopea, 2014):
1.4.2.1 Sistema de bombeo
Tiene por objeto impulsar la fase móvil a través de la columna y debe cumplir
ciertas especificaciones como reproducibilidad y precisión, manteniendo un flujo
laminar y de velocidad constante (Farmacopea, 2014).
1.4.2.2 Sistema de inyección
Un factor importante para obtener una buena resolución en la separación es
la adecuada introducción de la muestra en el sistema. La manera ideal de introducir
o inyectar la muestra es en forma de “paquete” pequeño ya que esto ayuda a la
obtención de picos simétricos y angostos. (Farmacopea, 2014)
1.4.2.3 Detector
Puede ser de dos tipos: Tipo 1.- aquellos que miden una propiedad de la fase
móvil, y Tipo 2.- aquellos que miden una propiedad del analito. La selección del
detector estará basada en las propiedades del o los solutos que se desean analizar
(Farmacopea, 2014).

22
1.4.2.4 Columna
Se considera a la columna como la parte fundamental de la cromatografía ya
que es en está, donde se va a llevar a cabo la separación. El material de empaque
seleccionado dependerá básicamente de la separación que se desee hacer. Las
dimensiones de una columna dependerán también del tipo de separación que se
desee hacer. Al aumentar la longitud aumenta el número de platos teóricos y por lo
tanto, se obtiene una mayor resolución aunque en ocasiones es más importante el
tipo de empaque y el tamaño de partícula de éste, ya que al elevar el área de
superficie del empaque, se aumenta la interacción del soluto con la fase
estacionaria. Se debe considerar la influencia de la geometría de la partícula en el
empacamiento de la columna y por tanto en la eficiencia de la separación. Se debe
considerar también la porosidad de la partícula y la influencia que el tamaño del poro
puede tener, sobre todo en separaciones fundamentadas en la diferencia de pesos
moleculares. Otro parámetro importante asociado a la partícula es su tamaño;
generalmente partículas de gran tamaño se emplean en cromatografía preparativa,
en tanto que partículas pequeñas se emplean en separaciones rápidas
(Farmacopea, 2014).
1.4.2.5 Registrador de señales
Al emerger un compuesto ya separado en la columna y pasar por el detector,
la señal que provoca en éste debe ser registrada por un graficador, un integrador o

23
un sistema computarizado de procesamiento de datos. El empleo de una
computadora y del software adecuado puede facilitar el procesamiento de los datos,
desde el algoritmo empleado para la integración, hasta la construcción de curvas de
calibración y cuantificación de los picos. Dichos programas deben cumplir con ciertos
criterios de aseguramiento de la calidad (Farmacopea, 2014).
1.4.3 Fase móvil
En la cromatografía líquida la composición de la fase móvil es una de la
variables que influyen en la separación. Hay una amplia variedad de solventes
usados en HPLC. La fase móvil debe ser (Johnson, 1978):
Pura
No reactiva con el reservorio
Compatible con el detector
Que disuelva la muestra
Baja viscosidad
De preferencia que permita la recuperación de la muestra
Estar disponible comercialmente a un precio razonable
1.5 Validación
La validación de los aparatos y la metodología de disolución deberá incluir la
prueba de aptitud del sistema utilizando calibradores; desaireación, de hacer falta;
validación entre los procedimientos manuales y automatizados; y validación de un

24
paso determinativo (es decir, los métodos analíticos empleados en el análisis
cuantitativo de las muestras de disolución). Esto deberá incluir todos los pasos y
procedimientos apropiados de la validación de los métodos analíticos (FDA, 2015).
Los métodos analíticos utilizados para evaluar la calidad de los productos
farmacéuticos, están sujetos a varios requisitos, de acuerdo con la normatividad
vigente, así como, con otros documentos normativos nacionales e internacionales
(Farmacopea, 2014).
La validación de un método es el proceso que establece, mediante estudios
de laboratorio, que las características de desempeño del método, satisfacen los
requisitos para su aplicación analítica (Farmacopea, 2014).
El proceso de validación de los métodos analíticos puede comprender pero no
está limitado al estudio de las características de desempeño que se describen a
continuación (Farmacopea, 2014):
Especificidad / Selectividad del método
Es la capacidad de un método analítico para obtener una respuesta debida
únicamente al analito de interés y no a otros componentes de la muestra, que
pueden estar presentes (especificidad) o que se pudieran presentar por efectos
ambientales y/o de interacción con los mismos componentes (selectividad) tales
como: impurezas, productos de degradación o componentes de la misma muestra
(Farmacopea, 2014).

25
Exactitud del método
Es la concordancia absoluta entre el resultado obtenido con el método y la
cantidad verdadera del analito presente en la muestra, a una cantidad fija
(Farmacopea, 2014).
Linealidad o intervalo del método
Linealidad es la capacidad de un método analítico para dar resultados que
son directamente proporcionales a la concentración del analito (sin sesgo) dentro de
un intervalo dado. Intervalo es aquel comprendido entre las concentraciones superior
e inferior del analito incluyendo dichas concentraciones) y para el que se ha
demostrado que el analito es cuantificado con un nivel satisfactorio de precisión,
exactitud y linealidad, cuando se aplica el método analítico (Farmacopea, 2014).
Precisión del método
Es el grado de concordancia relativa entre los resultados obtenidos al aplicar
el método analítico, bajo las mismas condiciones analíticas (repetibilidad) o bajo
diferentes condiciones analíticas (reproducibilidad), utilizando una muestra
homogénea. La precisión de un método analítico generalmente se expresa como la
desviación estándar o como el coeficiente de variación (desviación estándar relativa)
(Farmacopea, 2014).
Límite de detección del método

26
Es la cantidad mínima de analito en una muestra que puede ser detectada,
pero no necesariamente cuantificada, bajo las condiciones de aplicación del método.
Así las pruebas limite sólo indican que la cantidad de analito es superior o inferior a
la concentración establecida. El límite de detección (LD) se expresa generalmente
como la concentración indicada en el método analítico (por ejemplo porcentaje, ppm,
partes por billón, etc.) (Farmacopea, 2014).
Límite de cuantificación del método
Es la cantidad mínima de analito en una muestra que puede ser determinada
con exactitud y precisión aceptable, bajo las condiciones de aplicación del método.
Las unidades del límite se expresan como se indica en el método analítico (por
ejemplo, porcentaje, ppm, partes por billón, etc.) (Farmacopea, 2014).
Tolerancia del método
Es el grado de reproducibilidad de los resultados de prueba obtenidos por el
análisis de la misma muestra, bajo una variedad de condiciones tales como:
diferentes laboratorios, analistas, instrumentos, lotes de reactivos, días, etc.
(Farmacopea, 2014).
Robustez del método

27
Capacidad del método analítico de mantener su desempeño al presentarse
variaciones pequeñas pero deliberadas, en las características normales de
operación del método (Farmacopea, 2014).

28
2 JUSTIFICACIÓN
Los medicamentos con la categoría de genéricos, son las especialidades
farmacéuticas que cumplen con las pruebas de intercambiabilidad señaladas por el
Consejo de Salubridad General.
Para establecer la intercambiabilidad de este tipo de medicamentos es
necesario realizar, de manera científica, pruebas que demuestren que estos son
equivalentes con respecto al medicamento de referencia dentro de un intervalo
definido. Una de las pruebas, es la comparación de perfiles de disolución, que está
incluida en la Norma Oficial Mexicana NOM-177-SSA1-2013, que establece las
pruebas y procedimientos para demostrar que un medicamento es intercambiable.
Por lo que, los estudios de los perfiles de disolución permiten comparar la
similitud entre el medicamento establecido como de referencia y el de prueba
(genérico) y esta debe de mantenerse una vez que los medicamentos lleguen al
mercado, independientemente del lote. Por lo que, es de vital importancia realizar
este tipo de estudios con el objetivo de conocer la calidad biofarmaceutica de los
genéricos disponible en el mercado mexicano.

29
3 HIPÓTESIS
Si el medicamento de referencia y el genérico son similares, entonces los
perfiles de disolución obtenidos deben de cumplir con los criterios establecidos por la
NOM-177-SSA1-2013.
4 OBJETIVO GENERAL
Demostrar la intercambiabilidad del omeprazol como medicamento genérico,
mediante un estudio de perfil de disolución.
Objetivo particular
Desarrollar y validar un método analítico por HPLC para cuantificar el
principio activo en los perfiles de disolución.
Realizar el perfil de disolución del medicamento de referencia y el genérico.
Comparar los perfiles de disolución entre las formulaciones.

30
5 MATERIAL
Tabla 2. Equipos e instrumentos, reactivos y materiales utilizados
Equipos, materiales e instrumentos
Matraz volumétrico
5,10,50,100,2000 mL
Micro pipetas transferpette 10-100,
100-1000uL, 0.5 – 5mL
Vasos precipitado
25,50,100,1000mL
Balanza analítica Scientech SM50
Embudo de vidrio Balanza semianalítica ADAM
PW254
Mortero con pistilo Sonicador AS10200AT
Tubos plástico con rosca
Tubos de vidrio con rosca
Termómetro de inmersión parcial
Pipeta pasteur plástico Cronómetro
Gradilla plástico UHPLC Agilent Thecnology 1260
System
Viales ámbar con inserto Aparato 2 de paletas (Ver Anexo 2)
Microtubos Ultracongeladorthermoscientific
88300
Reservorio 250 mL Vortex Genie
Probeta 500 mL Potenciómetro
Espátula
Membrana 0.45um
Reactivos
Ácido clorhídrico concentrado (HCl)
Fosfato de sodio dibásico septahidratado (Na2HPO4.7H2O) 0.235M
Acetonitrilo 99.9 %
Agua grado HPLC
Agua desionizada
Metanol 99.8%
Hidróxido de amonio NH4OH 0.1%
Hidróxido de sodio NaOH 0.25M
Ácido clorhídrico (HCl) 0.1N
.

31
6 MEDICAMENTOS ESTUDIADOS
Tabla 3. Medicamentos evaluados
Característica Medicamento de
referencia Medicamento de
prueba
Denominación Internacional
6-Metoxi-2-[(4-metoxi-3,5-dimetilpiridina-2-
il)metilsulfinil]- 1H-benzimidazol
Denominación Genérica Omeprazol
Denominación Distintiva LosecA-20 Elite Medical
Forma Farmacéutica Cápsula
Contenido 20 mg
Número de Lote YARY L14J1243
Fecha de Caducidad MAY-2016 DIC-2016
Fabricante Astra Zeneca, S.A.
de C.V.
Landsteiner Scientific, S.A. de
C.V.
Cantidad 10 cajas con 7
cápsulas
Caja con 60 cápsulas
Número de Registro Sanitario 245M2003 SSA VI 070M200555AV1

32
7 METODOLOGÍA
7.1 Desarrollo del método analítico
Primero se realizó una investigación bibliográfica de las propiedades
fisicoquímicas, farmacocinéticas, biofarmacéuticas de la molécula.
Posteriormente de acuerdo a la dosis de la presentación farmacéutica, se
estableció una concentración mínima y máxima para probar el rango de trabajo, así
se prepararon soluciones de Omeprazol estándar para construir la curva de
calibración. El desarrollo del método analítico se observa esquematizado en la figura
siguiente.
Figura 2. Diagrama de flujo del desarrollo del método analítico para la cuantificación
de los fármacos de trabajo (Omeprazol) en un perfil de disolución
7.2 Selección de las condiciones cromatograficas
Se establecieron las condiciones cromatográficas de acuerdo a lo establecido
por la FEUM y las características fisicoquímicas del Omeprazol
Investigación bibliográfica
Selección del rango de trabajo
Diseño de la curva de
calibración
Selección de las condiciones
cromatograficas
Pruebas control calidad
Validación Perfil de
disolución

33
Las condiciones cromatográficas empleadas durante la valoración,
uniformidad de contenido, validación y los perfiles de disolución fueron:
Tabla 4. Parámetros del método para el análisis de separación
Módulo
Parámetro
Valores y componentes a utilizar
Bomba Disolvente A Na2HPO4 . 7H2O 10 mM pH 6.8 ± 0.2
Disolvente B Acetonitrilo
Composición del
disolvente
65:35 (A:B)
Tiempo de retención 4.36 min
Inyector automático Inyección 10 uL
Temperatura 4°C
Compartimiento de
las columnas
Columna Columna ZORBAX Eclipse Plus C-18
Rapid Resolution HD 4.6 x 150 mm
Temperatura 40°C
Detector UV 280nm
7.3 Preparación del medio de disolución
Fluido gástrico simulado sin enzima
Se colocó en un matraz volumétrico de 500mL, 7mL de ácido clorhídrico
(HCl) concentrado y se aforó con agua destilada.
Solución de fosfato dibásico de sodio 0.235 M

34
Se pesaron 16.69 g de Na2HPO4 ·7H2O y se disolvieron en agua destilada
tibia con ayuda de un agitador magnético, una vez disueltos, se colocaron en un
matraz volumétrico de 500mL y se llevaron al aforo con agua destilada.
Las curvas preparadas durante la validación fueron realizadas con los medios
de disolución, en la siguiente proporción: Fluido gástrico simulado sin enzima:
Na2HPO4 0,235 M (500:400mL). Esta solución tiene un pH 6.8 ± 0.2, de no ser así
debe se ajustó con NaOH 2 M o HCL 2 M. En adelante esta solución se mencionará
como solución pH 6.8 ± 0.2.
7.4 Pruebas de control de calidad
Se realizan debido a que la calidad de un producto farmacéutico está
directamente relacionada con su efecto terapéutico, debido a la estabilidad y
seguridad del producto.
Las pruebas de valoración y uniformidad de dosis deben realizarse siguiendo
los métodos descritos en la FEUM. Estas pruebas deben realizarse tanto al producto
de prueba como al de referencia.
7.4.1 Valoración
Se realizó la valoración por HPLC, de acuerdo a lo especificado en la
Farmacopea de los Estados Unidos Mexicanos (FEUM) undécima edición.
Preparación de referencia.

35
Se pesó una cantidad de la sustancia de referencia equivalente a 20 mg de
omeprazol. Se transfirió a un matraz volumétrico de 100mL, se disolvió con 20mL de
etanol, se llevó al aforo con solución de metanol NH4OH 0.1% en H2O 50:50 v/v, y se
mezcló. Se transfirió una alícuota de 5mL de esta solución a un matraz volumétrico
de 50mL, se aforó con diluente y se mezcló. Se filtró a través de una membrana de
22um de porosidad. Esta solución contiene 0.2mg/mL de omeprazol.
Preparación de la muestra
Se pesó y mezcló el contenido de no menos de 20 cápsulas, se pesó una
porción de la mezcla equivalente a 20 mg de omeprazol, se transfirió a un matraz
volumétrico de 100mL, se agregaron 5mL de solución de metanol NH4OH 0.1% en
H2O 50:50 v/v y se sometió a la acción de un baño de ultrasonido durante 15 min,
se enfrió y llevó al aforo con solución de metanol NH4OH 0.1% en H2O 50:50 v/v, se
mezcló y filtró a través de una membrana de 22um o equivalente, descartando los
primeros 5mL del filtrado. Se realizó el procedimiento descrito anteriormente a cada
uno de los medicamentos.
Posteriormente se inyectó al cromatografo, por triplicado, volúmenes iguales
(20 uL) de la preparación de referencia, se registraron los picos respuesta y se
calculó el coeficiente de variación en cual no es mayor del 2%, la eficiencia de la
columna no es menor de 20 000 platos teóricos y el factor de coleo es no menor de
0.8 y no mayor de 2. Una vez ajustados los parámetros de operación, se inyectó al
cromatógrafo por separado, volúmenes iguales (20 uL) de la preparación de

36
referencia y de la preparación de la muestra, se obtuvieron sus cromatogramas
correspondientes y se calcularon las áreas bajo los picos. Se calculó la cantidad de
omeprazol en la proporción de la muestra tomada, por medio de la siguiente fórmula:
En donde C es la concentración por mL de la preparación de referencia, D es
el factor de dilución de la muestra, Am es el área bajo el pico obtenida con la
preparación de la muestra y Aref es el área bajo el pico obtenida con la preparación
de referencia.
La valoración para cada producto contiene no menos del 90.0 % y no más del
110.0% de la cantidad de C17 H19N3O3S, indicada en el marbete. (Farmacopea,
2014)
El porcentaje de valoración del medicamento de prueba debe estar dentro de
los límites farmacopeicos y no debe diferir en más del 5% del medicamento de
referencia de acuerdo a la Norma Oficial Mexicana (NOM-177, 2013)
7.4.2 Uniformidad de dosis (Uniformidad de contenido)
Preparación de referencia
Se pesó una cantidad de la sustancia de referencia equivalente a 20 mg de
omeprazol. Se transfirió a un matraz volumétrico de 100 mL, se disolvió con 20 mL
de etanol, se llevó al aforo con solución de metanol NH4OH 0.1% en H2O 50:50 v/v,

37
y se mezcló. Se filtró a través de una membrana de 22um de porosidad. Esta
solución contiene 0.2mg/mL de omeprazol.
Preparación de la muestra
1. Se prepararon tanto para el medicamento de referencia como para el
medicamento de prueba.
2. Se utilizaron 10 capsulas, pesadas e identificadas en la determinación de
peso promedio.
3. Se procedió a trabajar de manera individual cada una de las 10 cápsulas,
tanto del medicamento de prueba como de referencia.
4. Se colocó el contenido de una capsula en un mortero, se maceró y transfirió
a un matraz volumétrico de 100mL, se agregaron 50mL de solución de
metanol NH4OH 0.1% en H2O 50:50 v/v y se sometió a la acción de un baño
de ultrasonido durante 15 min, se enfrió y llevó al aforo con solución de
metanol NH4OH 0.1% en H2O 50:50 v/v, se mezcló y filtro a través de una
membrana de 22um o equivalente, descartando los primeros 5mL del filtrado.
5. Se repitió el paso 4 con las cuatro capsulas restantes para tener.
6. Para cada capsula se obtuvo el porciento de omeprazol en la proporción de
la muestra tomada, por medio de la siguiente fórmula:

38
En donde C es la concentración por mL de la preparación de referencia, D es
el factor de dilución de la muestra, Am es el área bajo el pico obtenida con la
preparación de la muestra y Aref es el área bajo el pico obtenida con la preparación
de referencia.
7. Se calculó el porcentaje de omeprazol por tableta, considerando el peso
promedio para cada producto de acuerdo a la siguientes fórmulas:
%omeprazol x cápsula = (mg omeprazol / capsula) x (100/20)
8. Se determinó el coeficiente de variación (CV) de acuerdo a la ecuación:
Donde el CV es el coeficiente de variación, DE es la desviación estándar y
es el promedio.
9. Se calculó el valor de aceptación de acuerdo a la ecuación:
VA = M – x + k s
En donde M es valor de referencia, x es la media de los contenidos individuales (x1,
x2……..) expresados como el porcentaje de la cantidad declarada, k es la constante
de aceptabilidad y s es la desviación estándar.

39
Se cumple con el requisito de uniformidad de contenido si el valor de
aceptación de las primeras 10 unidades de dosificación es menor o igual al L1%. Si
el valor de aceptación es mayor que L1% analizar las siguientes 20 unidades y
calcular el valor de aceptación. Donde L1 es 15.0 y L2 es 25.0.
7.5 Validación
Se realiza debido a que es la evidencia experimental documentada de que un
procedimiento cumple con el propósito para el que fue diseñado.
Se validó el método analítico para la cuantificación de Omeprazol en el
medio de disolución, en el rango de concentración establecido en el desarrollo del
método. Los parámetros a evaluar fueron los siguientes: linealidad, precisión,
Influencia del filtro, estabilidad del fármaco en el medio de disolución para validación
del sistema. Para la validación del método se evaluó la linealidad, exactitud,
precisión (repetibilidad y reproducibilidad) y selectividad para los medicamentos.
7.5.1 Sistema
7.5.1.1 Linealidad del sistema
El analista debe preparar por lo menos por duplicado 5 niveles de
concentración (intervalo) de la solución de referencia por dilución (a partir de una
misma solución concentrada). El rango de trabajo se determinó tomando en cuenta

40
la dosis del fármaco a estudiar (20 mg), el volumen del medio (900 mL) y la alícuota
a tomar, el cual fue de 2.5 a 30 ug/mL.
Procedimiento:
1. Se pesaron 5 mg de omeprazol USP, y se trasfirieron a un matraz aforado
de 5mL, se aforó con solución de metanol NH4OH 0.1% en H2O 50:50 v/v a
5mL. Esta solución contiene 1000ug/mL.
2. En otro matraz aforado de 5mL se agregaron 500uL de la solución descrita
en el paso (1) y se llevó a aforo con solución de metanol NH4OH 0.1% en
H2O 50:50 v/v Esta solución contiene 100ug/mL.
Preparación de la curva de calibración
1. Para obtener las concentraciones requeridas en la curva se tomaron
alícuotas de la solución estándar como se muestra en la tabla 6.
Tabla 5. Preparación de la curva de calibración
Stock (µg/mL)
Alícuota (mL)
Aforo (mL)
Concentración (µg/mL)
100 0.125 5 2.5
100 0.25 5 5
100 0.5 5 10
100 0.75 5 15
100 1 5 20
100 1.25 5 25
100 1.5 5 30

41
2. Se inyectaron las muestras al cromatógrafo UHPLC /UV bajo las condiciones
cromatográficas mencionadas en la valoración.
3. Con los valores obtenidos se trazó la gráfica la concentración (x) vs área (y),
se incluyó en ella la ecuación, la línea de ajuste y el coeficiente de
correlación
La ecuación para obtener el % de error relativo debido a la regresión es:
Donde A es la ordenada al origen, B es la pendiente y CVy/x es el coeficiente
de variación o error relativo debido a la regresión.
Se cumple con la linealidad del sistema si el coeficiente de correlación (r) es
mayor o igual a 0.99, el error relativo debido a la regresión no es mayor que el 2%
(NOM-177, 2013).
7.5.1.2 Precisión del sistema
EL analista debe determinar la precisión del sistema a partir de los datos de
linealidad del sistema en el cual se debe demostrar que el coeficiente de variación
(CV) del factor respuesta es menor o igual al 2%. El factor respuesta se obtiene del
coeficiente del área entre la concentración. La ecuación para obtener el CV es la
siguiente:

42
Donde el CV es el coeficiente de variación, DE es la desviación estándar y
es el promedio.
7.5.1.3 Influencia del filtro
1. Se pesaron 5 mg de omeprazol USP, y se transfirieron a un matraz aforado
de 5mL, se aforó con solución de metanol NH4OH 0.1% en H2O 50:50 v/v.
Esta solución contiene 1000 ug/mL.
2. En otro matraz aforado de 5mL se agregaron 50uL de la solución descrita en
el paso (1) y se llevó a aforo con solución de metanol NH4OH 0.1% en H2O
50:50 v/v . Esta solución contiene 100 ug/mL.
3. Se realizó por sextuplicado la siguiente dilución. Se tomó una alícuota de
0.5mL, se transfirió a un matraz aforado de 5mL, se aforó con HCl : Fosfato
de sodio dibásico (500: 400mL). Se agitó para homogeneizar la solución.
Esta solución contiene 10ug/mL.
4. Se inyectaron las muestras al cromatógrafo UHPLC /UV bajo las condiciones
cromatográficas mencionadas en la valoración.
5. Se filtraron cada una de las soluciones anteriores desechando los primeros 2
mL.

43
6. Se inyectaron las muestras al cromatógrafo UHPLC /UV bajo las condiciones
cromatográficas mencionadas en la valoración.
7. Se analizaron los resultados
a. Se obtuvo la concentración interpolada
b. Se calculó el porcentaje cuantificado
c. Se obtuvo el promedio del porcentaje para la concentración (10ug/mL).
d. Se calculó la diferencia absoluta de acuerdo a la siguiente fórmula
En donde, di = diferencia absoluta, t = Porcentaje cuantificado de la
solución filtrada 0 = Porcentaje cuantificado de la solución no filtrada.
Se cumple con la influencia del filtro cuando la diferencia absoluta entre el
promedio de los datos de por lo menos seis muestras de solución filtrada y sin filtrar
debe ser igual o menor al 2 % (NOM-177, 2013)
7.5.1.4 Estabilidad
El analista debe establecer la etapa de análisis en la cual se desea evaluar la
estabilidad, además de determinar si en dicha etapa es posible fraccionar (muestras
dependientes) o no (muestras independientes) y las condiciones de almacenaje. Por
lo que se determinó la estabilidad analítica para muestras dependientes, se
procesaron las muestras hasta la etapa preestablecida, se fraccionaron cada una de

44
las preparaciones de acuerdo a las condiciones de interés y para esto último se
realizó investigación bibliográfica en trabajos anteriores evaluando omeprazol
(García, 2008).
1. Se pesaron 5 mg de omeprazol USP, y se transfirieron a un matraz aforado
de 5 mL, se aforo con solución de metanol NH4OH 0.1% en H2O 50:50 v/v a
5 mL. Esta solución contiene 1000ug/mL.
2. En otro matraz aforado de 5mL se agregaron 500uL de la solución descrita
en el paso (1) y se llevó a aforo con solución de metanol NH4OH 0.1% en
H2O 50:50 v/v. Esta solución contiene 100ug/mL
3. Se realizó por triplicado la siguiente dilución. Se tomó una alícuota de 1.1mL
se transfirió a un matraz aforado de 5mL, se aforó con HCl : Fosfato de sodio
dibásico 500: 400. Se agitó para homogeneizar la solución. Esta solución
contiene 22ug/mL.
4. Se dividió la muestra en 2 fracciones:
a. La fracción 1 se inyecto al cromatografo UHPLC/UV bajo las
condiciones cromatográficas mencionadas en la valoración, después
de haberlo preparado (t0)
b. La fracción 2 se guardó en (tubo de plástico) en refrigeración 3 h (t1) y
se inyecto en el cromatografo UHPLC/UV bajo las condiciones
cromatográficas mencionadas en la valoración.
5. Se analizaron los resultados

45
a. Se obtuvo la concentración interpolada
b. Se calculó el porcentaje cuantificado
c. Se obtuvo el promedio de los porcentajes cuantificados para la
concentración evaluada.
d. Se obtuvo la diferencia absoluta para la concentración evaluada, de
acuerdo a la siguiente fórmula:
En donde, di = diferencia absoluta, = Porcentaje cuantificado de la solución
a x tiempo, = Porcentaje cuantificado de la solución a tiempo 0.
7.5.2 Método
7.5.2.1 Linealidad del medicamento referencia y prueba
Preparación del estándar de referencia
1. Se pesaron 5 mg de omeprazol USP, y se transfirieron a un matraz aforado
de 5 mL,se aforó con solución de metanol NH4OH 0.1% en H2O 50:50 v/v a
5 mL. Esta solución contiene 1000ug/mL.
2. En otro matraz aforado de 5mL se agregó 0.5 mL de la solución descrita en
el paso (1) y se llevó al aforo con solución de metanol NH4OH 0.1% en H2O
50:50 v/v . Esta solución contiene 100ug/mL
Preparación de la muestra

46
1. Se pesaron 10 capsulas del producto de prueba. Se calculó el peso
promedio. Se molió el contenido de las cápsulas en un mortero hasta obtener
un polvo fino.
2. Se pesó del polvo obtenido la cantidad equivalente a 20 mg de omeprazol
(considerando el peso promedio) por septuplicado y se transfirió a un matraz
de 100mL, se adicionaron 10mL de solución de metanol NH4OH 0.1% en
H2O 50:50 v/v, se sónico 10 min y se llevó al aforo. Esta solución contiene
200ug/mL.
3. De la solución anterior se tomó una alícuota de 2.5mL y se transfirió a un
matraz de 5mL. Esta solución contiene 100ug/mL.
Solución 1. Se adicionó al primer matraz una alícuota de 0.125 mL de la solución
estándar de referencia, se adicionó 2mL de solución de metanol NH4OH 0.1% en
H2O 50:50 v/v, se sónico 10 min y se aforó con solución pH 6.8 ± 0.2. Se mezcló y
filtro.
Solución 2. Se adicionó al primer matraz una alícuota de 0.25mL de la solución
estándar de referencia, se agregó 2mL de solución de metanol NH4OH 0.1% en H2O
50:50 v/v, se sónico 10 min y se aforó con solución pH 6.8 ± 0.2. Se mezcló y
filtro.
Solución 3. Se adicionó al primer matraz una alícuota de 0.5 mL de la solución
estándar de referencia, se adicionó 2mL de solución de metanol NH4OH 0.1% en

47
H2O 50:50 v/v, se sonicó 10 min y se aforó con solución pH 6.8± 0.2. Se mezcló y
filtró.
Solución 4. Se adicionó al primer matraz una alícuota de 0.75 mL de la solución
estándar de referencia, se agregó 2mL de solución de metanol NH4OH 0.1% en
H2O 50:50 v/v, se sónico 10 min y se aforó solución pH 6.8 ± 0.2. Se mezcló y filtró.
Solución 5.Se adicionó al primer matraz una alícuota de 1 mL de la solución
estándar de referencia, se adicionó 2mL de solución de metanol NH4OH 0.1% en
H2O 50:50 v/v, se sónico 10 min y se aforó con solución pH 6.8 ± 0.2. Se mezcló y
filtró-
Solución 6. Se adicionó al primer matraz una alícuota de 1.25 mL de la solución
estándar de referencia, se adicionó 2mL de solución de metanol NH4OH 0.1% en
H2O 50:50 v/v, se sónico 10 min y se aforó solución pH 6.8 ± 0.2.Se mezcló y filtró.
Solución 7. Se adicionó al primer matraz una alícuota de 1.5 mL de la solución
estándar de referencia, se agregó 2mL de solución de metanol NH4OH 0.1% en H2O
50:50 v/v, se sónico 10 min y se aforó con solución pH 6.8 ± 0.2. Se mezcló y filtro.
4. Se inyectaron las muestras al cromatógrafo UHPLC /UV bajo las condiciones
cromatográficas mencionadas en la valoración.
5. Se realizó un análisis de regresión lineal con los datos de la concentración
nominal contra el área bajo el pico.

48
6. A cada área bajo el pico se restó la ordenada al origen de la regresión
anterior. A esta área bajo el pico se le llamará área bajo el pico corregida.
7. Se realizó un segundo análisis de regresión lineal con el área bajo el pico
corregida y la concentración nominal.
8. Se interpolaron las áreas bajo el pico en la regresión lineal anterior para
obtener la concentración recuperada en ug/mL.
9. Se realizó otra regresión lineal de la concentración recuperada contra la
concentración nominal. Se calculó el coeficiente de correlación (r) y el
coeficiente de variación de la regresión lineal.
Se cumple con la linealidad el método cuando el coeficiente de variación de
la regresión lineal sea menor al 3% y el valor de r es mayor o igual a 0.99 (NOM-177,
2013).
7.5.2.2 Exactitud del medicamento de referencia y prueba
1. Con los datos de concentración recuperada, obtenidos en la linealidad del
método se calculó el porcentaje recuperado.
2. Se obtuvo el promedio de los porcentajes cuantificados para cada
concentración.
3. Se obtuvo la diferencia absoluta para cada concentración de acuerdo a la
siguiente fórmula:

49
Se cumple con la exactitud del medicamento de referencia cuando al calcular
el promedio del porcentaje de la recuperación de los datos de linealidad, este no
varía en más del 3% con respecto a la cantidad nominal en cada punto (NOM-177,
2013).
7.5.2.3 Precisión del medicamento de referencia y prueba
Repetibilidad del medicamento
Con los porcentajes recuperados de la linealidad calcular el coeficiente de
variación del porcentaje recuperado y el coeficiente de variación de cada
concentración y el global.
Se cumple con la precisión del medicamento de referencia (repetibilidad)
cuando al calcular el CV% del porcentaje cuantificado, con los datos de exactitud del
método este es menor o igual al 3% (NOM-177, 2013).
Reproducibilidad del medicamento
Con los datos de linealidad del método de dos analistas se evaluó el efecto
de la precisión del método.
Se calculó el porcentaje recuperado y se determinó el coeficiente recuperado y el
coeficiente de variación global.
Se cumple con la precisión del medicamento de referencia (reproducibilidad)
cuando participan dos o más analistas y se evalúa su efecto en la precisión del

50
método. El CV% global del porcentaje cuantificado, debe ser menor o igual al 3%
(NOM-177, 2013).
7.5.2.4 Selectividad del medicamento de referencia y prueba
Solución de placebo analítico
1. Se pesaron 10 capsulas del producto de prueba. Se calculó el peso
promedio. Se macero el contenido de las cápsulas en un mortero hasta
obtener un polvo fino.
2. Se pesó del polvo obtenido la cantidad equivalente a 20 mg de omeprazol
(considerando el peso promedio) por septuplicado y se transfirió a un matraz
de 100mL, se adicionó 10mLde solución de metanol NH4OH 0.1% en H2O
50:50 v/v, se sónico 10 min y se llevó al aforo. Se mezcló y filtró Esta
solución contiene 200ug/mL.
3. De la solución anterior se tomó una alícuota de 0.5mL y se transfirió a un
matraz de 10mL y se aforó aforar con solución pH 6.8± 0.2. Esta solución
contiene 10ug/mL.
Solución del estándar adicionado
1. Se pesaron 10 capsulas del producto de prueba. Se calculó el peso
promedio. Se maceró el contenido de las cápsulas en un mortero hasta
obtener un polvo fino.

51
2. Se pesó del polvo obtenido la cantidad equivalente a 20 mg de omeprazol
(considerando el peso promedio) por septuplicado y se transfirió a un matraz
de 100mL,se adicionó 10mL de solución de metanol NH4OH 0.1% en H2O
50:50 v/v, se sónico 10 min y se llevó al aforo. Se mezcló y filtro. Esta
solución contiene 200ug/mL.
3. De la solución anterior se tomó una alícuota de 0.5mL y se transfirió a un
matraz de 10mL y se aforó con solución pH 6.8± 0.2. Esta solución contiene
10ug/mL.
4. Al matraz del paso 4, se agregó una alícuota de 1.mL de la solución estándar
de referencia (ver linealidad del método, preparación del estándar de
referencia), se aforó con solución pH 6.8± 0.2, se mezcló.
5. Se inyectaron las muestras al cromatógrafo UHPLC /UV bajo las
condiciones cromatográficas mencionadas en la valoración.
6. Se obtuvo la diferencia entre la respuesta (área bajo el pico) del estándar
adicionado y el placebo analítico.
7. Se interpoló el área bajo el pico resultante de la diferencia en la linealidad del
sistema y se calculó la concentración cuantificada.
8. Se obtuvo el porcentaje cuantificado y se calculó el coeficiente de variación
(CV) y la diferencia absoluta (di), de acuerdo a las siguientes fórmulas

52
En donde, el CV es el coeficiente de variación, DE es la desviación estándar
y es el promedio
En donde, di es la diferencia absoluta y es el promedio.
7.6 Perfil de disolución
El estudio de disolución “in vitro”, que es requerido como criterio de
aceptación en libros oficiales como la Farmacopea Nacional Mexicana, para tabletas
y cápsulas, nos permite predecir los resultados de disponibilidad del principio activo
“in vivo”, ya que la disolución es un proceso previo por el que deben pasar las
capsulas para poder absorberse.
Preparación curva de calibración
El analista debe preparar por lo menos por duplicado 5 niveles de
concentración (intervalo) de la solución de referencia por dilución (a partir de una
misma solución concentrada). Se preparó la curva de calibración de la misma
manera que se preparó en la linealidad del sistema.
Preparación de la muestra
1. Se colocaron las cápsulas en el aparato 2 con 500mL de solución de ácido
clorhídrico 0.1N a 37°C, se aceleró a 100 rpm y se tomaron muestras del
medio (5 mL) a 15, 30, 60 y 120 min. Concluido las dos horas de la fase

53
ácida, se agregó agregará 400 mL de solución de fosfato dibásico de sodio
0.235 M (fase amortiguadora). En caso necesario se ajustó el pH a 6.8 ±
0.05 con solución de hidróxido de sodio 2 N. Se continuó la agitación a 100
rpm, se tomaron 5 mL del medio a los 5, 10, 15, 20, 25, 30 y 45 min.
Inmediatamente los 5 mL se transfirieron a un tubo que contenía una
solución de 1 mL de hidróxido de sodio 0.25 M, se mezcló y filtró a través de
membrana de 1.2 um de porosidad o menor, se protegió de la luz.
2. Se inyectaron en el cromatografo UHPLC/UV bajo las condiciones
cromatográficas mencionadas en la valoración.
3. Calcular la cantidad de omeprazol disuelto al interpolar en la curva de
calibración.
Ningún valor individual excede al 15 por ciento de omeprazol disuelto en la
fase ácida (HCl 0.1N) (Farmacopea, 2014).
En la fase amortiguadora para cápsulas de 10 mg y 20 mg, no menos de 75
por ciento (Q) de omeprazol se disuelve en 30 min (Farmacopea, 2014).
Análisis de datos
La comparación de perfiles de disolución se llevó a cabo empleando un
método modelo independiente. Se aplicó el factor de similitud de 2 puntos (f2). Este
se calculó a partir de la media de los perfiles de disolución de cada uno de los
tiempos de muestreo empleando la siguiente ecuación: (Farmacopea, 2014).

54
Donde n es el número de tiempos de muestreo, Rt es el porcentaje disuelto
promedio en el tiempo t del medicamento sin modificación y Pt corresponde al
porcentaje disuelto promedio en el tiempo t del medicamento con la modificación.
(Farmacopea, 2014)
El valor de f2 varía de 0 a 100. Si el valor es igual o mayor a 50, se considera
que el producto cumple con el factor de similitud en relación al producto sin
modificación. Se utilizará este método siempre y cuando la variabilidad no sea alta.
En caso de que no se cumpla un coeficiente de variación menor al 20% en el primer
tiempo y del 10% en los tiempos subsecuentes, se aplicará el análisis de serie de
tiempos (Farmacopea, 2014).

55
8 RESULTADOS
8.1 Valoración
En la siguiente tabla se presenta los resultados de la valoración del
medicamentos de referencia (Losec A-20) y de prueba (Elite Medical), en esta se
demuestra que los dos productos cumplen con lo especificado en la FEUM 11a ed.
ya que el contenido de porcentaje de principio activo se encuentra en el intervalo de
90.0-110.0% de la cantidad de omeprazol, de valor indicado en el marbete.
Tabla 6. Resultado de la valoración
Medicamento Promedio % de
Omeprazol en las tabletas Diferencia de
promedios
Prueba (Elite Medical) 101.32 3.58%
Referencia (Losec A-20) 97.75
Criterio 90-110% del valor indicado en el marbete
8.2 Uniformidad de dosis (uniformidad de contenido)
De acuerdo a la tabla 7, se demuestra que los dos productos cumplen con lo
especificado en la FEUM undécima edición ya que el valor de aceptación es menor
al L1%.
Tabla 7. Resultado de la uniformidad de contenido
Medicamento de
referencia Medicamento de prueba
Promedio 98.66 101.32
Desviación Estándar 1.88 1.08
Coeficiente de Variación 1.91 1.07
Valor de Aceptación 7.36 2.77
Criterio VA ≤ L1 (15%)

56
8.3 Validación
En la tabla 8 se presentan los diferentes parámetros de desempeño evaluados
durante la validación, por lo que el método analítico desarrollado para cuantificar
omeprazol en un intervalo de 2.5 - 30ug/mL fue validado de acuerdo a los criterios
de la NOM-177-SSA1-2013 y puede ser empleada para evaluar los perfiles de
disolución de omeprazol (cápsulas con capa entérica, 20 mg).
Tabla 8. Resultados de la validación
Sis
tem
a (
Fárm
aco)
PARÁMETRO CRITERIO DE ACEPTACIÓN
RESULTADO
Linealidad (2.5-30ug/mL)
r2>0.99; error debido a la
regresión ≤2% r2 =0.99; CV=0.42%
Precisión CV menor o igual al 2% CV= 0.76%
Influencia del filtro (Robustez)
I di i ≤ 2% I di I = 0.4000%
Estabilidad (3h)
|di| menor o igual al 3% t0(4°C);22ug/mL
|di|=2.92% t1(4°C);22ug/mL
Méto
do (
Med
icam
ento
)
Linealidad del método (2.5-30ug/mL)
r2>0.99; error debido a la
regresión ≤3%
REFERENCIA r2=0.99; CV=1.37%
PRUEBA r2 =0.99; CV=1.49%
Exactitud |di| menor o igual al 3%
en cada punto
REFERENCIA |di|=0.95%, 0.01%, 1.83%, 0.41%, 0.77%, 1.11%, 0.96%
PRUEBA |di|=2.87%, 2.63%,0.97%; 1.13%, 0.57%, 1.29%, 0.90%
Precisión (Repetibilidad)
CV menor o igual al 3% en cada punto
REFERENCIA CV=1.09%,2.47%, 0.98%, 0.21%,1.71%,1.11%,0.97%
PRUEBA CV = 0.39%,2.04%,0.48%, 0.84%,0.69%,0.21%,0.50%
Precisión (Reproducibilidad)
CV menor o igual al 3% REFERENCIA CV= 1.51%
PRUEBA CV= 1.55%
Selectividad CV menor o igual al 3%
REFERENCIA CV= 2.15% |di|= 1.71%
PRUEBA CV=0.78% |di|= 2.03%

57
8.4 Perfiles de disolución
En la tabla 9 se muestran el porciento disuelto de Omeprazol durante la fase
amortiguadora del perfil de disolución, a los 5 minutos en el medicamento de
referencia se disolvió el 67.83%, en cambio en el medicamento de prueba solo se
presentó un 27.76% del fármaco disuelto. Al observar los demás tiempos los valores
obtenidos para el medicamento de prueba se encuentran por debajo a los mostrados
para el medicamento de referencia (figura 3).
En el caso de la fase acida no se detectaron concentraciones del analito en el medio
(ver anexo 3).
Tabla 9. Resultados de perfiles de disolución
Medicamentos
Losec A-20 Elite Medical
Tiempo (minutos)
Promedio de Porcentaje
Disuelto (%)
Coeficiente de
Variación
(%)
Promedio de Porcentaje
Disuelto (%)
Coeficiente de Variación (%)
0 0 0 0 0
5 67.83 17.99 27.76 12.74
10 93.24 4.57 65.43 8.39
15 99.58 3.82 82.11 7.35
20 100.82 3.99 84.30 9.02
25 99.88 2.47 87.33 8.49
30 97.83 3.68 88.81 6.99
45 95.44 3.05 90.00 6.39
Criterio CV menor al 20% en el primer tiempo y del 10% en los tiempos subsecuentes

58
Con respecto a la variabilidad presentada en cada tiempo de muestreo (tabla
9) y de acuerdo a la FEUM undécima edición, el perfil de disolución cumple con el
parámetro del coeficiente de variación menor al 20% en el primer tiempo y del 10%
en los tiempos subsecuentes.
Prueba del factor de similitud
En la tabla 10, se presenta la diferencia entre el medicamento de referencia y
el medicamento de prueba, para establecer el factor de similitud (f2). En esta se
confirma lo puntualizado en la sección anterior, en la mayoría de los tiempos de
muestreo se presenta una diferencia mayor del 10%, obteniéndose un f2 de 33.38
por lo no existe una similitud entre los medicamentos.
Tabla 10. Resultado de f2
Tiempo (minutos)
Promedio de porcentaje disuelto de Losec A-20
Promedio de porcentaje disuelto de
Elite Medical
I Rt – Pt I (Rt-Pt)2
0 0 0 0 0
5 67.83 27.76 40.07 1605.60
10 93.24 65.43 27.81 773.39
15 99.58 82.11 17.47 305.20
20 100-82 84.30 16.52 272.91
25 99.88 87.33 12.55 157.50
30 97.83 88.81 9.02 81.36
45 95.44 90.00 5.44 29.59
f2 33.38
Criterio f2 debe encontrarse en el intervalo 50 a 100
Rt = Promedio del porcentaje disuelto de referencia, Pt = Promedio del porcentaje
disuelto de prueba, f2 = Factor de similitud

59
En la figura 3, se observa el porcentaje del omeprazol con y sin modificación
(prueba y referencia, respectivamente), en esta se observa que a lo largo del perfil el
comportamiento de ambos medicamentos es diferente. Al calcular el factor de
similitud se encontró que no hay similitud entre los perfiles (f2 = 33.38)
Figura 3. Comparación de perfil de disolución de Losec A-20 contra Genérico Elite
Medical

60
9 DISCUSIÓN
De acuerdo a los resultados obtenidos en las pruebas de control de calidad,
es decir valoración y uniformidad de dosis (uniformidad de contenido), se observa
que los resultados de valoración, para los dos medicamentos ( Losec A-20 y Elite
Medical), están dentro del intervalo establecido en la Farmacopea 2014, ya que el
contenido de porcentaje de principio activo se encuentra en el intervalo de 90.0-
110.0% de la cantidad de omeprazol, de valor indicado en el marbete.
Por otro lado para el resultado de uniformidad de dosis se debe mencionar
que el empleo de esta prueba se puede demostrar por los métodos de variación de
masa o el de uniformidad de contenido y este depende de la dosificación y el
criterio de aplicación que se define en el método general correspondiente. La forma
farmacéutica evaluada fue cápsula rígida de 20 mg, por lo tanto de acuerdo a la
Farmacopea, 2014, la prueba que corresponde a este medicamento es uniformidad
de contenido, debido a que la dosis es menor a 25mg y la proporción de fármaco
menor al 25%. En base a los resultados se observa que los dos medicamentos
evaluados (Losec A-20 y Elite Medical) cumplen con lo especificado en la
Farmacopea, ya que el valor de aceptación es menor al L1%.
Estas pruebas son importantes debido a que la calidad representa el conjunto
de características que posee un producto y determina su aceptabilidad. La calidad
de un producto farmacéutico está directamente relacionada con su efecto

61
terapéutico, la estabilidad y seguridad del producto. Y por último uno de los criterios
y requisitos para poder realizar un estudio de intercambiabilidad es que se deben
realizar estas pruebas (valoración y uniformidad de dosis), antes de iniciar el estudio
(Pérez, et al., 2016).
Los parámetros de validación se dividen en los que corresponden al sistema
y los que pertenecen al método; siendo el objetivo de este último, establecer que el
método analítico es adecuado para su uso previsto (Huber, et al., 1999).
La linealidad se define como la habilidad para asegurar que los resultados
obtenidos directamente o por medio de una transformación matemática definida, son
proporcionales a la concentración del analito, dentro de un intervalo determinado
(Biólogos, 2016). El intervalo se definió tomando en cuenta la dosis del fármaco a
estudiar (20 mg), el volumen del medio y la alícuota a tomar, el cual fue de 2.5 a 30
ug/mL, los resultados de linealidad indican que si existe una relación lineal en el
intervalo de concentraciones estudiadas, ya que el valor del coeficiente de
correlación fue de 0.99 con un coeficiente de variación de 0.42; cumpliendo con lo
que establece la NOM-177, 2013 que dice que el valor de r≥ 0.99, CV debido a la
regresión no mayor al 2%, con al menos cinco puntos de concentración a lo largo de
la curva. De igual manera se observa que para la linealidad del método para ambos
medicamentos (referencia y prueba) cumplen con las especificaciones.
La precisión es el grado de concordancia entre resultados analíticos
individuales, cuando el procedimiento se aplica repetidamente a diferentes porciones

62
de una muestra homogénea (Biólogos, 2016). De acuerdo a el resultado obtenido
para la precisión del sistema podemos decir que si hay concordancia entre las
diferentes concentraciones evaluadas y repetidas, es decir; es preciso, ya que el CV
obtenido fue igual a 0.76% y este es menor a la especificación que debe ser menor
o igual al 2% de acuerdo a la NOM-177, 2013. Para la precisión del método esta se
evalúa como repetibilidad y reproducibilidad. El parámetro de repetibilidad se
expresa como se mencionó anteriormente pero bajo las siguientes condiciones: un
solo analista, usando los mismos instrumentos y método. El parámetro de
reproducibilidad se expresa bajo las siguientes condiciones: diferentes laboratorios ó
bajo las variaciones que comúnmente pueden ocurrir dentro del laboratorio,
diferentes días, diferentes analistas, diferentes equipos. De acuerdo a los resultados
obtenidos, ambos medicamentos cumplen con las especificaciones, donde el CV%
no debe ser mayor al 3%, por lo que el método es preciso.
La influencia del filtro, se realiza para comprobar que al usar el filtro para
eliminar los excipientes del medio, este no interviene en la cuantificación del
omeprazol o en otras palabras no se queda retenido el omeprazol en el filtro o se
liberen partículas. Otro objetivo de filtrar la muestra es que no dañe el equipo de
HPLC, principalmente la columna ya que no todos los excipientes de la formulación
son solubles en el medio de disolución. Se probaron filtros acrodiscos de PVDF, de
Nylon y de Celulosa de 0.45um, de los cuales los tres cumplían con la
especificación, sin embargo, debido al alto costo, se usaron membranas de celulosa

63
de 0.45um, las cuales cumplieron con el objetivo de ser inertes, ya que de acuerdo a
los resultados la diferencia absoluta fue menor al 2% de acuerdo a la (NOM-177,
2013).
La estabilidad es la propiedad de una muestra, preparada para su
cuantificación de conservar su integridad fisicoquímica y la del analito, después de
almacenarse durante un tiempo determinado bajo condiciones específicas.
(Biólogos, 2016) La molécula de omeprazol es inestable a pH ácidos, se degrada
bajo la exposición de la luz paulatinamente, por lo tanto durante la manipulación de
este se contemplaron estas condiciones y debido a ello se lograron establecer
medidas para su posterior manipulación en el perfil de disolución. (Oswald, et al.,
2015) Para esta prueba se evaluó una concentración de 22 ug /mL a la temperatura
de 4°C al tiempo 0 min y después de 3 h. de acuerdo a los resultados obtenidos se
cumple con el parámetro de estabilidad debido a que la diferencia absoluta fue
menor al 3% de acuerdo a lo que se especifica en la (NOM-177, 2013).
La exactitud se define como la concordancia entre un valor obtenido
empleando el método y el valor de referencia (Biólogos, 2016). Recordemos que
este parámetro sólo evalúa al método, el cual en este caso es exacto, ya que tanto
el medicamento de prueba como el de referencia cumplen con la especificación ya
que la diferencia absoluta fue menor al 3% en cada punto evaluado.
La selectividad al igual que la exactitud sólo evalúa al método. La selectividad
es la capacidad del método analítico para obtener una respuesta debida únicamente

64
al analito de interés y no a otros componentes de la muestra es decir los aditivos.
Para evaluar este parámetro debido a que no se cuenta con los excipientes se
prepararon dos soluciones, una se denominó solución de placebo analítico la cual
simula la respuesta de los excipientes y otra que se denominó solución del estándar
adicionado la cual contenía la simulación de los excipientes a la misma
concentración que la solución anterior más una concentración conocida del
estándar, debiendo obtener la respuesta del estándar, al restar la respuesta del
excipiente entre las dos preparaciones y repetidamente. De acuerdo a los resultados
se observa que el método es selectivo para ambos medicamentos ya que los valores
del coeficiente de variación y la diferencia absoluta son menores al 3%.
De acuerdo a los resultados de los parámetros anteriormente descritos el
método analítico desarrollado para cuantificar omeprazol en un intervalo de 2.5 -
30ug/mL, validado de acuerdo a los criterios de la NOM-177-SSA1-2013 cumple con
los parámetros de la misma, y puede ser empleada para evaluar los perfiles de
disolución de omeprazol (cápsulas con capa entérica, 20 mg).
En cuanto a los perfiles de disolución, se realizó con seis vasos en cada perfil,
hasta obtener el resultado de las doce unidades, por lo tanto, de manera general las
cápsulas se sometieron a un medio ácido durante en el cual se tomaron muestras
por intervalos de tiempo durante 2 h, en los cuales al analizar las muestras al equipo
de HPLC, no se cuantifico omeprazol (Ver Anexo 3). Estos resultados nos indican
que la capa entérica gastrorresistente cumplió con su función de proteger al principio

65
activo, para ambos medicamentos. Ya que las formas de dosificación de liberación
modificada están diseñadas como sistemas monolíticos o multiparticulados, y su
función es modificar la biodisponibilidad del fármaco y extender la liberación o
controlar el comienzo de la disolución del fármaco en el tracto gastrointestinal. En el
caso de la forma multiparticulada (como es el caso de la forma farmacéutica
estudiada en el presente trabajo), el fármaco se divide en varias subunidades
funcionales, tal como granulos, bolitas o minitabletas (recubierto o no), contenida
dentro de una forma de dosificación final (tableta o cápsula blanda) que se
desintegra rápidamente después de la administración liberando las subunidades en
el tracto gastrointestinal. Esto reduce el riesgo de una alta concentración local del
fármaco y el efecto potencial de irritación localizada de la mucosa gástrica (Tondo, et
al., 2014).
Al termino de las 2 h se agregó un medio de fosfatos en el cual se muestreo
en diferentes intervalos de tiempo durante 45 min, en esta etapa los pellets de
deshicieron y se comenzó a cuantificar omeprazol. Se determinó el CV de las doce
unidades estudiadas de cada medicamento, en los diferentes tiempos de muestreo,
en el primer tiempo de muestreo el CV fue menor al 20% y del 10% en los tiempos
subsecuentes, por lo que se procedió a comparar los perfiles por el método de factor
de similitud (f2). De acuerdo a los resultados obtenidos, se determinó que el
medicamento Genérico Elite Medical no presenta un perfil de disolución similar al de
referencia (el valor de f2 debe encontrarse en el intervalo de 50 a 100). Al tener dos

66
formulaciones, que contienen aditivos diferentes, es posible que la cinética de
disolución se vea afectada (García, 2008). Sin embargo, ambos medicamentos
cumplen con la especificación de la prueba de disolución, de acuerdo a la
Farmacopea 2014 en la fase amortiguadora para cápsulas de 10 mg y 20 mg, el
porciento disuelto de omeprazol debe de ser no menos de 75 por ciento (Q) a los 30
min.
La disolución es considerada una de las pruebas de control de calidad más
importantes en las formas farmacéuticas de dosificación y la validación del método
de disolución es una parte importante de las buenas prácticas de manufactura
(Ameril, et al., 2012).
Cuando no hay una buena correlación se puede deber a varios factores (Pérez, et
al., 2016):
Por ejemplo, las propiedades fisicoquímicas del fármaco ya que hay que
destacar que el omeprazol se comporta de manera inestable a la temperatura
de 37°C pH 6.80 ± 0.02.
Relacionados a la forma de dosificación es decir hay un excipiente que
interviene en la liberación del principio activo.
Y por último relacionados al aparato de disolución y a los parámetros de
prueba (tipo e intensidad de agitación, medio de disolución, pH, temperatura
del medio, lugar de la toma de muestra).

67
De los factores mencionados anteriormente, se considera que el factor que
contribuyó de manera significativa fue el relacionado a la forma de dosificación.
Es evidente que el omeprazol tiene que formularse, para su administración
oral, en una forma adecuada que proteja al fármaco del contacto con el jugo
gástrico (el cual presenta reacción ácida) con el fin de que pueda alcanzar el
intestino delgado sin degradación.
Dadas las peculiaridades del omeprazol y su facilidad para degradarse en los
medios ácidos y neutros, se han desarrollado multitud de formulaciones, para
mejorar el aprovechamiento del mismo por parte del organismo.
Sin embargo, a pesar de los intentos llevados a cabo para obtener una forma
óptima del omeprazol es preciso llevar a cabo procesos largos, engorrosos y muy
caros; en otros casos cuando se intenta trasladar el procedimiento a escala
industrial, se encuentran serios problemas de estabilidad del producto, no pudiendo
conseguirse los resultados pretendidos (Carvajal, et al., 2003).
De manera general las etapas para fabricar un pellet son las siguientes:
Selección y fabricación de esferas de soporte de los pellets con los tamaños
adecuados para la finalidad pretendida
Adhesión del principio activo a las esferas de soporte (núcleos neutros), para
obtener los pellets que contienen al fármaco.

68
Aislamiento de los núcleos activos revistiéndolos con un excipiente protector
inerte para obtener unas esferas o pellets protegidos
Recubrimiento de dichos núcleos activos aislados con una película
gastrorresistente, para obtener los pellets de omeprazol
Cuando vence la patente de un medicamento, es cuando nacen los
genéricos, entonces suponiendo que los dos fabricantes siguen el mismo método
descrito anteriormente, cabe destacar lo siguiente:
Para superar los problemas de estabilidad del omeprazol, el mejor enfoque
parece ser la aplicación de un recubrimiento entérico. En un estudio se desarrollaron
gránulos multicapa revestidos de omeprazol. El sistema consiste en núcleos que
contienen el fármaco, revestidos con una capa de sal, una capa de HPMC y una
capa entérica. El sistema podría mejorar significativamente la velocidad de
disolución y la estabilidad del omeprazol además los niveles de revestimiento tienen
poca influencia en el perfil de disolución del omeprazol. Se comprobó que la
proporción de fármaco liberada fue retrasada significativamente por las capas de
HPMC. La capa de sal podría funcionar para separar y sustituir parte de la capa de
HPMC y disminuir los niveles del HPMC. De esta manera aseguramos una rápida
disolución del omeprazol y la velocidad de disolución es más rápida. Se encontró
también que el almacenamiento a 40°C / 75% HR por seis meses no tiene efecto
en la liberación del fármaco y no hay cambios significativos en la apariencia física
(He, et al., 2009).

69
Muchos factores afectan una eficiente encapsulación del fármaco en las
esferas. Por ejemplo la naturaleza de la droga, la concentración del polímero. La
proporción droga polímero y la velocidad de agitación. Generalmente una baja
concentración del polímero muestra una disminución de la eficiencia en la
encapsulación. (Chandan, et al., 2013)
Otro punto crítico en la obtención de estos pellets y que varía según el
método es en la etapa de la adición del principio activo a las esferas de soporte, se
lleva a cabo a partir de un conjunto de ingredientes convenientemente mezclados en
cantidades adecuadas, es decir el principio activo, un diluyente, un disgregante, un
surfactante, un adherente y un disolvente. En esta etapa el punto crítico es el
disolvente ya que si no se elige de manera conveniente, puede desestabilizar el
omeprazol (Carvajal, et al., 2003).
En otro estudio se mencionan dos métodos de obtención de pellets de
omperazol que producen características muy similares. Uno de ellos fue la extrusión-
esferonización, el cual provocó el bloqueo de la abertura de la matriz de la extrusora
lo que impidió la reutilización inmediata del instrumento, después de la preparación
de un lote. Por el contrario el método de tamizado-esferonización después de varios
lotes no se presentó el problema del método anterior siendo esta la técnica elegida
para la preparación de los pellets (Karim, et al., 2014).

70
Existen otros factores como temperatura, humedad y atomización, que se
deben cuidar durante esta etapa, así como en la etapa del aislamiento de los
núcleos activos.
Es asi como podemos determinar que para ambos medicamentos estudiados
el método de obtención y los aditivos usados intervienen en la prueba de
intercambiabilidad y de ahí deriva el resultado obtenido, lo cual en el humano tiene
como resultado una biodisponibilidad modificada y de esta manera no se alcance el
efecto terapéutico deseado.
Se observa también una ligera disminución del porcentaje disuelto al final de
la prueba para el medicamento de referencia, lo cual se debe a que existe una
degradación del mismo. Todas las preparaciones deben conservarse entre 15 a
30°C como máximo de acuerdo a (Gomez, et al., 1997).

71
10 CONCLUSION
Ambos medicamentos cumplieron con las pruebas de calidad determinando
su aceptabilidad y así continuar con el estudio de intercambiabilidad.
El método analítico desarrollado para cuantificar omeprazol en un intervalo de
2.5 - 30ug/mL, validado de acuerdo a los criterios de la NOM-177-SSA1-2013
cumple con los parámetros de la misma, y puede ser empleada para evaluar
los perfiles de disolución de omeprazol (cápsulas con capa entérica, 20 mg).
En cuanto al perfil de disolución, no se cumplió con la hipótesis planteada
en el presente proyecto ya que el medicamento de referencia y el genérico no
son similares, entonces los perfiles de disolución obtenidos no cumplen con
los criterios establecidos por la NOM-177-SSA1-2013.

72
11 ANEXOS
11.1 Anexo 1. Aparato 2 de paletas
Figura 4. Aparato 2, paletas (Farmacopea, 2014)

73
11.2 ANEXO 2. Validación
Sistema
Tabla 11. Datos de linealidad del sistema
Curva Concentración
(µg/mL) Respuesta m b r Sy/x
1
2.5 29.8
11.85986402 -0.713626062 0.999840863 0.926026681
5 59
10 118.78
15 174.98
20 234.78
25 295.4
30 357.2
2
2.5 29.4
11.94390935 -0.795750708 0.999924454 0.638273855
5 59
10 117.4
15 179
20 237.6
25 299.6
30 356.4
Curva promedio
Concentración (µg/mL)
Respuesta m b r Sy/x
2.5 29.6
11.90188669 -0.754688385 0.999966315 0.426107671
5 59
10 118.09
15 176.99
20 236.19
25 297.5
30 356.8

74
Figura 5. Linealidad del sistema (Concentración vs Respuesta)
Tabla 12. Datos de precisión del sistema
Curva Concentración
(µg/mL) Respuesta
Factor respuesta
1
2.5 29.8 0.08389262
5 59 0.08474576
10 118.78 0.08418926
15 174.98 0.08572408
20 234.78 0.08518613
25 295.4 0.08463101
30 357.2 0.08398656
2
2.5 29.4 0.08503401
5 59 0.08474576
10 117.4 0.08517888
15 179 0.08379888
20 237.6 0.08417508
25 299.6 0.08344459
30 356.4 0.08417508
Promedio 0.08449341
DE 0.0006444
%CV 0.76266073
0
50
100
150
200
250
300
350
400
0 5 10 15 20 25 30 35
Re
spu
est
a
Concentración( µg/mL)

75
Método (Referencia)
Tabla 13. Datos de linealidad del método del medicamento de referencia
Curva Concentración nominal
Respuesta corregida
Concentración Recuperada m b r Sy/x
1
2.5 22.6 2.50
6.54 6.24 0.9998 0.8606
5 38 4.86
10 72.8 10.18
15 103.8 14.92
20 138.2 20.17
25 169.2 24.91
30 202.2 29.96
2
2.5 20 2.55
6.95 2.34 0.9989 2.3355
5 37.6 5.09
10 72.4 10.09
15 106.4 14.98
20 140.4 19.86
25 172 24.41
30 214.6 30.53
3
2.5 21 2.51
6.68 4.27 0.9992 2.0028
5 38 5.05
10 73 10.29
15 104 14.92
20 134.6 19.50
25 170.4 24.85
30 207.4 30.38
Curva promedio
Concentración Promedio de concentración recuperada
m r Sy/x
2.5 2.52
0.9993 0.9996 1.3744795
5 5.00 10 10.18 15 14.94 20 19.84 25 24.72 30 30.29

76
Figura 6. Linealidad del método del medicamento de referencia (Concentración
nominal contra concentración recuperada)
Tabla 14. Datos de exactitud del medicamento de referencia
Cantidad Adicionada
Cantidad Recuperada
%Recuperado Promedio
Recuperado Diferencia Absoluta
2.5
2.50 100.111557
100.951481 -0.95148069 2.55 102.199202
2.51 100.543683
5
4.86 97.1403642
99.9800262 0.01997381 5.09 101.700386
5.05 101.099329
10
10.18 101.769649
101.839232 -1.83923175 10.09 100.875969
10.29 102.872078
15
14.92 99.4399857
99.5836648 0.41633515 14.98 99.8344843
14.92 99.4765245
20
20.17 100.873979
99.2224945 0.7775055 19.86 99.3137422
19.50 97.4797627
25
24.91 99.6553147
98.8892474 1.11075262 24.41 97.6212755
24.85 99.3911518
30
29.96 99.862019
100.963093 -0.96309342 30.53 101.76388
30.38 101.263382
y = 0,9993x + 0,0104 R² = 0,9996
0,00
5,00
10,00
15,00
20,00
25,00
30,00
35,00
0 5 10 15 20 25 30 35 Co
nce
ntr
ació
n r
ecu
pe
rad
a (u
g/m
L)
Concentración nominal (ug/mL)

77
Precisión del medicamento de referencia
Tabla 15. Datos de precisión del medicamento de referencia (repetibilidad)
Precisión (Repetibilidad)
Cantidad nominal
2.5 5 10 15 20 25 30
% Recuperado
100.11 97.14 101.77 99.44 100.87 99.66 99.86
102.20 101.70 100.88 99.83 99.31 97.62 101.76
100.54 101.10 102.87 99.48 97.48 99.39
101.26
Promedio
100.95 99.98 101.84 99.58 99.22 98.89 100.96
Desviacion estandar
1.10194779 2.47751439 0.99987219 0.21798298 1.69894675 1.10601082 0.98584893
%CV
1.09156179 2.47800935 0.98181435 0.21889431 1.71225966 1.11843385 0.97644485
Tabla 16. Datos de precisión del medicamento de referencia (reproducibilidad)
Concentración nominal
Analista
1 2 1 2 1 2
Respuesta corregida
Respuesta corregida
Concentración Recuperada
Concentración Recuperada
% Recuperad
o
% Recuperad
o
2.5 22.6 20 2.50 2.55 100.1116 102.1992
5 38 37.6 4.86 5.09 97.1404 101.7004
10 72.8 72.4 10.18 10.09 101.7696 100.8760
15 103.8 106.4 14.92 14.98 99.4400 99.8345
20 138.2 140.4 20.17 19.86 100.8740 99.3137
25 169.2 172 24.91 24.41 99.6553 97.6213
30 202.2 214.6 29.96 30.53 99.8620 101.7639
Promedio 100.1544
D.E. 1.5182
CV (%) 1.5159

78
Selectividad
Tabla 17. Datos de selectividad del medicamento de referencia
Concentración nominal (ug/mL)
Muestra
Respuesta del
estándar adicionado
Área
Respuesta del Placebo
Analítico Área
Diferencia Absorbancia
Concentración Cuantificada
(ug/mL)
Porcentaje cuantificado
20
1 209.2 72.8 136.4 19.29 96.4500
2 214.6 72.4 142.2 20.12 100.6000
3 211.3 73 138.3 19.56 97.8000
Promedio 98.2833
D.E. 2.1167
CV 2.1537
di 1.7167

79
Método (Prueba)
Tabla 18. Datos de linealidad del método del medicamento de prueba
Curva Concentración nominal
Respuesta Concentración
Recuperada m b r Sy/x
1
2.5 20.6 2.56
8.95 -2.29 0.9996 1.4207
5 43.8 5.15
10 86.2 9.89
15 130.4 14.82
20 177.8 20.11
25 219 24.71
30 268.6 30.25
2
2.5 21 2.57
8.91 -1.88 0.9997 1.3143
5 42.8 5.02
10 86 9.87
15 131.4 14.96
20 178.6 20.26
25 218.2 24.70
30 266.6 30.13
3
2.5 20.4 2.58
9.00 -2.74 0.9992 2.0623
5 44.2 5.23
10 86.8 9.96
15 129.6 14.71
20 177 19.97
25 218.8 24.62
30 271.2 30.44
Curva promedio
Concentración Promedio de respuesta
m r Sy/x
2.5 20.67
8.9532 0.9996 1.4912195
5 43.60
10 86.33
15 130.47
20 177.80
25 218.67
30 268.80

80
Figura 7. Linealidad del método del medicamento de prueba (concentración nominal
contra concentración recuperada
Tabla 19. Datos de exactitud del medicamento de prueba
Cantidad Adicionada
Cantidad Recuperada
%Recuperado Promedio
Recuperado Diferencia absoluta
2.5
2.56 102.457506
102.87411 -2.874109648 2.57 102.887572
2.58 103.277251
5
5.15 103.036844
102.63525 -2.63525005 5.02 100.365148
5.23 104.503758
10
9.89 98.8602981
99.0264553 0.973544663 9.87 98.655116
9.96 99.5639519
15
14.82 98.8079806
98.8680633 1.13193665 14.96 99.7307781
14.71 98.0654313
20
20.11 100.568308
100.572447 -0.572446756 20.26 101.278454
19.97 99.8705787
25
24.71 98.8554509
98.7057919 1.294208145 24.70 98.7960285
24.62 98.4658962
30
30.25 100.839897
100.908641 0.908641351 30.13 100.432424
30.44 101.453604
y = 0,9995x + 0,0074 R² = 0,9996
0,00
5,00
10,00
15,00
20,00
25,00
30,00
35,00
0 10 20 30 40 Co
nce
ntr
ació
n r
ecu
pe
rad
a (u
g/m
L)
Concentración nominal (ug/mL)

81
Precisión del medicamento de prueba
Tabla 20. Datos de precisión del medicamento de prueba (Repetibilidad)
Precisión (Repetibilidad)
Cantidad nominal
2.5 5 10 15 20 25 30
% Recuperado
102.46 103.04 98.86 98.81 100.57 98.86 100.84
102.89 100.37 98.66 99.73 101.28 98.80 100.43
103.28 104.50 99.56 98.07 99.87 98.47 101.45
Promedio
102.87 102.64 99.03 98.87 100.57 98.71 100.91
Desviacion estandar
0.41003817 2.09832857 0.47665694 0.83429757 0.70394667 0.209869472 0.51404915
%CV
0.39858247 2.04445215 0.48134303 0.84384941 0.69993989 0.212621233 0.50942034
Tabla 21. Datos de precisión del medicamento de prueba (Reproducibilidad)
Concentración nominal
Analista
1 2 1 2 1 2
Respuesta corregida
Respuesta corregida
Concentración Recuperada
Concentración Recuperada
% Recuperad
o
% Recuperad
o
2.5 20.6 21 2.56 2.57 102.4575 102.8876
5 43.8 42.8 5.15 5.02 103.0368 100.3651
10 86.2 86 9.89 9.87 98.8603 98.6551
15 130.4 131.4 14.82 14.96 98.8080 99.7308
20 177.8 178.6 20.11 20.26 100.5683 101.2785
25 219 218.2 24.71 24.70 98.8555 98.7960
30 268.6 266.6 30.25 30.13 100.8399 100.4324
Promedio 100.3980
D.E. 1.5621
CV (%) 1.5559

82
Selectividad
Tabla 22. Datos de selectividad del medicamento de prueba
Concentración nominal (ug/mL)
Muestra
Respuesta del
estándar adicionado
Área
Respuesta del Placebo
Analítico Área
Diferencia Absorbancia
Concentración Cuantificada
(ug/mL)
Porcentaje cuantificado
20
1 267.1 86.2 180.9 20.5153 102.5765
2 266.6 86 180.6 20.4817 102.4085
3 265.1 86.8 178.3 20.2235 101.1175
Promedio 102.0342
D.E. 0.7982
CV (%) 0.7823
di -2.0342

83
11.3 Anexo 3 . Cromatogramas
Figura 8. Cromatograma del perfil de disolución en la etapa ácida a los 15 min.
Figura 9. Cromatograma del perfil de disolución en la etapa ácida a los 30 min.

84
Figura 10. Cromatograma del perfil de disolución en la etapa ácida a los 60 min.
Figura 11. Cromatograma del perfil de disolución en la etapa ácida a los 120 min

85
Figura 12. Cromatograma de muestra de omeprazol

86
11.4 Anexo 4. Perfiles de disolución
Tabla 23. Datos de la unidad #1 analizadas durante el perfil de disolución de
Losec-A20
Referencia
Vaso 1
Tiempo Respuesta xi Ei Vi Di %Di
0 0 0 0 0 0
5 149.3 14.2054716 71.0273579 900 12784.92 63.925%
10 224.3 21.260634 106.30317 895 19099.29 95.496%
15 249.3 23.6123548 118.061774 890 21192.33 105.962%
20 253.5 24.0074439 120.03722 885 21541.98 107.710%
25 248.3 23.518286 117.59143 880 21111.52 105.558%
30 249.8 23.6593892 118.296946 875 21234.99 106.175%
45 240.4 22.7751422 113.875711 870 20465.69 102.328%
Tabla 24. Datos de la unidad #2 analizada durante el perfil de disolución de
Losec-A20
Referencia
Vaso 2
Tiempo Respuesta xi Ei Vi Di %Di
0 0 0 0 0 0
5 176.2 16.7359232 83.6796159 900 15062.33 75.312%
10 213.5 20.2446906 101.223453 895 18202.68 91.013%
15 246.3 23.3301483 116.650742 890 20948.74 104.744%
20 241.6 22.8880248 114.440124 885 20557.46 102.787%
25 232.4 22.0225916 110.112958 880 19795.87 98.979%
30 242.8 23.0009074 115.004537 875 20651.90 103.260%
45 231.6 21.9473365 109.736682 870 19735.29 98.676%

87
Tabla 25. Datos de la unidad #3 analizada durante el perfil de disolución de
Losec-A20
Referencia
Vaso 3
Tiempo Respuesta xi Ei Vi Di %Di
0 0 0 0 0 0
5 195.2 18.523231 92.616155 900 16670.91 83.355%
10 220.5 20.9031725 104.515862 895 18800.96 94.005%
15 240.3 22.7657353 113.828677 890 20458.64 102.293%
20 255 24.1485472 120.742736 885 21682.42 108.412%
25 237.6 22.5117495 112.558747 880 20242.04 101.210%
30 227.2 21.5334336 107.667168 875 19386.02 96.930%
45 215.5 20.4328283 102.164141 870 18428.49 92.142%
Tabla 26. Datos de la unidad #4 analizada durante el perfil de disolución de
Losec-A20
Referencia
Vaso 4
Tiempo Respuesta xi Ei Vi Di %Di
0 0 0 0 0 0
5 177.1 16.8205851 84.1029256 900 15138.53 75.693%
10 223 21.1383445 105.691723 895 19002.92 95.015%
15 226.3 21.4487717 107.243858 890 19279.20 96.396%
20 227.9 21.5992818 107.996409 885 19412.40 97.062%
25 227.8 21.5898749 107.949375 880 19404.12 97.021%
30 223.6 21.1947858 105.973929 875 19058.42 95.292%
45 220.4 20.8937656 104.468828 870 18796.53 93.983%

88
Tabla 27. Datos de la unidad #5 analizada durante el perfil de disolución de
Losec- A20
Referencia
Vaso 5
Tiempo Respuesta xi Ei Vi Di %Di
0 0 0 0 0 0
5 124.7 11.8913783 59.4568915 900 10702.24 53.511%
10 221.4 20.9878344 104.939172 895 18843.57 94.218%
15 238 22.549377 112.746885 890 20233.34 101.167%
20 237 22.4553082 112.276541 885 20150.09 100.750%
25 236.3 22.38946 111.9473 880 20092.14 100.461%
30 234.9 22.2577636 111.288818 875 19976.91 99.885%
45 231.4 21.9285227 109.642614 870 19690.47 98.452%
Tabla 28. Datos de la unidad #6 analizada durante el perfil de disolución de
Losec- A20
Referencia
Vaso 6
Tiempo Respuesta xi Ei Vi Di %Di
0 0 0 0 0 0
5 130.2 12.4087569 62.0437844 900 11167.88 55.839%
10 220.8 20.9313931 104.656966 895 18795.64 93.978%
15 231.9 21.9755571 109.877786 890 19724.95 98.625%
20 235 22.2671705 111.335853 885 19983.02 99.915%
25 234.2 22.1919155 110.959577 880 19916.80 99.584%
30 229 21.7027575 108.513788 875 19488.79 97.444%
45 222.5 21.0913101 105.456551 870 18956.83 94.784%

89
Tabla 29. Datos de la unidad #7 analizada durante el perfil de disolución de
Losec- A20
Referencia
Vaso 7
Tiempo Respuesta xi Ei Vi Di %Di
0 0 0 0 0 0
5 125.2 11.7238363 58.6191816 900 10551.45 52.757%
10 210.3 19.4786221 97.3931103 895 17491.99 87.460%
15 229.5 21.2282329 106.141165 890 19049.14 95.246%
20 238.5 22.048363 110.241815 885 19774.95 98.875%
25 245.1 22.6497918 113.248959 880 20304.21 101.521%
30 227.9 21.082432 105.41216 875 18932.77 94.664%
45 225.5 20.8637307 104.318653 870 18742.50 93.713%
Tabla 30. Datos de la unidad #8 analizada durante el perfil de disolución de
Losec- A20
Referencia
Vaso 8
Tiempo Respuesta xi Ei Vi Di %Di
0 0 0 0 0 0
5 185.2 17.1913703 85.9568517 900 15472.23 77.361%
10 216.5 20.0436006 100.218003 895 18024.98 90.125%
15 235.6 21.7840989 108.920495 890 19574.02 97.870%
20 245.8 22.7135797 113.567898 885 20396.61 101.983%
25 245.4 22.6771295 113.385647 880 20364.54 101.823%
30 230.8 21.3466962 106.733481 875 19200.41 96.002%
45 224.5 20.7726051 103.863026 870 18700.95 93.505%

90
Tabla 31. Datos de la unidad #9 analizada durante el perfil de disolución de
Losec- A20
Referencia
Vaso 9
Tiempo Respuesta xi Ei Vi Di %Di
0 0 0 0 0 0
5 185.1 17.1822578 85.9112889 900 15464.03 77.320%
10 250.9 23.1783201 115.8916 895 20830.51 104.153%
15 248.3 22.9413936 114.706968 890 20619.64 103.098%
20 229.8 21.2555706 106.277853 885 19127.69 95.638%
25 235.1 21.7385361 108.692681 880 19552.70 97.763%
30 228.1 21.1006572 105.503286 875 18994.56 94.973%
45 225.6 20.8728432 104.364216 870 18796.36 93.982%
Tabla 32. Datos de la unidad #10 analizada durante el perfil de disolución de
Losec- A20
Referencia
Vaso 10
Tiempo Respuesta xi Ei Vi Di %Di
0 0 0 0 0 0
5 185.2 17.1913703 85.9568517 900 15472.23 77.361%
10 224.9 20.8090553 104.045277 895 18710.06 93.550%
15 226.2 20.9275186 104.637593 890 18815.49 94.077%
20 241.1 22.2852895 111.426448 885 20017.12 100.086%
25 238.2 22.0210254 110.105127 880 19784.57 98.923%
30 236.2 21.8387742 109.193871 875 19625.10 98.125%
45 225.3 20.8455056 104.227528 870 18760.96 93.805%

91
Tabla 33. Datos de la unidad #11 analizada durante el perfil de disolución de
Losec- A20
Referencia
Vaso 11
Tiempo Respuesta xi Ei Vi Di %Di
0 0 0 0 0 0
5 114 10.70323 53.5161499 900 9632.91 48.165%
10 214.6 19.870462 99.35231 895 17837.58 89.188%
15 234.9 21.720311 108.601555 890 19483.95 97.420%
20 242.4 22.4037528 112.018764 885 20088.79 100.444%
25 239.4 22.1303761 110.65188 880 19848.22 99.241%
30 231.8 21.4378218 107.189109 875 19242.23 96.211%
45 230.6 21.3284711 106.642355 870 19147.10 95.735%
Tabla 34. Datos de la unidad #12 analizada durante el perfil de disolución de
Losec- A20
Referencia
Vaso 12
Tiempo Respuesta xi Ei Vi Di %Di
0 0 0 0 0 0
5 175.5 16.3074523 81.5372617 900 14676.71 73.384%
10 218 20.1802889 100.901445 895 18142.90 90.714%
15 236.2 21.8387742 109.193871 890 19618.95 98.095%
20 231.3 21.392259 106.961295 885 19223.78 96.119%
25 232.2 21.474272 107.37136 880 19295.95 96.480%
30 228.4 21.1279948 105.639974 875 18992.96 94.965%
45 226.3 20.9366311 104.683156 870 18826.47 94.132%

92
Medicamento de Prueba (Elite Medical)
Tabla 35. Datos de la unidad #1 analizada durante el perfil de disolución de Medical
Elite
Medicamento de Prueba
Vaso 1
Tiempo Respuesta xi Ei Vi Di %Di
0 0 0 0 0 0
5 60.3 5.05297621 25.264881 900 4547.68 22.738%
10 162.6 13.8080945 69.0404727 895 12383.51 61.918%
15 198 16.8377249 84.1886247 890 15079.88 75.399%
20 244.4 20.8087659 104.04383 885 18594.25 92.971%
25 256.8 21.8699924 109.349962 880 19528.13 97.641%
30 254.4 21.6645937 108.322969 875 19348.41 96.742%
45 261.6 22.2807897 111.403949 870 19884.50 99.422%
Tabla 36. Datos de la unidad #2 analizada durante el perfil de disolución de Medical
Elite
Medicamento de Prueba
Vaso 2
Tiempo Respuesta xi Ei Vi Di %Di
0 0 0 0 0 0
5 86.3 7.27812847 36.3906424 900 6550.32 32.752%
10 150.5 12.7725429 63.8627146 895 11467.82 57.339%
15 241.3 20.5434593 102.717296 890 18383.93 91.920%
20 237.4 20.2096865 101.048432 885 18088.54 90.443%
25 237 20.1754533 100.877267 880 18058.42 90.292%
30 246.1 20.9542566 104.771283 875 18739.87 93.699%
45 246.5 20.9884897 104.942449 870 18769.65 93.848%

93
Tabla 37. Datos de la unidad #3 analizada durante el perfil de disolución de Medical
Elite
Medicamento de Prueba
Vaso 3
Tiempo Respuesta xi Ei Vi Di %Di
0 0 0 0 0 0
5 72.3 6.07996956 30.3998478 900 5471.97 27.360%
10 180.8 15.3657011 76.8285057 895 13782.70 68.914%
15 238 20.2610361 101.305181 890 18139.55 90.698%
20 247.1 21.0398394 105.199197 885 18828.79 94.144%
25 247.9 21.1083056 105.541528 880 18889.04 94.445%
30 252.5 21.5019864 107.509932 875 19233.51 96.168%
45 251.9 21.4506368 107.253184 870 19188.84 95.944%
Tabla 38. Datos de la unidad #4 analizada durante el perfil de disolución de Medical
Elite
Medicamento de Prueba
Vaso 4
Tiempo Respuesta xi Ei Vi Di %Di
0 0 0 0 0 0
5 63.4 5.31828282 26.5914141 900 4786.45 23.932%
10 174.2 14.8008548 74.004274 895 13273.36 66.367%
15 215.5 18.3354236 91.6771179 890 16419.12 82.096%
20 228.2 19.4223249 97.1116244 885 17381.03 86.905%
25 232.7 19.8074474 99.037237 880 17719.94 88.600%
30 227.9 19.39665 96.9832502 875 17360.49 86.802%
45 243.3 20.7146249 103.573124 870 18507.13 92.536%

94
Tabla 39. Datos de la unidad #5 analizada durante el perfil de disolución de Medical
Elite
Medicamento de Prueba
Vaso 5
Tiempo Respuesta xi Ei Vi Di %Di
0 0 0 0 0 0
5 81.6 6.87588941 34.379447 900 6188.30 30.942%
10 193.7 16.469719 82.348595 895 14774.78 73.874%
15 210.3 17.8903931 89.4519657 890 16039.18 80.196%
20 211.8 18.0187673 90.0938365 885 16152.79 80.764%
25 225.3 19.1741348 95.8706741 880 17169.51 85.848%
30 230.8 19.6448401 98.2242006 875 17581.38 87.907%
45 231.5 19.7047481 98.5237403 870 17633.50 88.167%
Tabla 40. Datos de la unidad #6 analizada durante el perfil de disolución de Medical
Elite
Medicamento de Prueba
Vaso 6
Tiempo Respuesta xi Ei Vi Di %Di
0 0 0 0 0 0
5 70.4 5.91736228 29.5868114 900 5325.63 26.628%
10 165.4 14.0477263 70.2386317 895 12602.30 63.012%
15 197.7 16.8120501 84.0602505 890 15062.55 75.313%
20 204.1 17.3597799 86.7988995 885 15547.29 77.736%
25 217.3 18.4894726 92.4473629 880 16541.42 82.707%
30 213.9 18.1984911 90.9924557 875 16286.81 81.434%
45 230.9 19.6533984 98.2669919 870 17552.58 87.763%

95
Tabla 41. Datos de la unidad #7 analizada durante el perfil de disolución de Medical
Elite
Medicamento de Prueba
Vaso 7
Tiempo Respuesta xi Ei Vi Di %Di
0 0 0 0 0 0
5 78.3 6.59346624 32.9673312 900 5934.12 29.671%
10 150.9 12.806776 64.0338801 895 11495.03 57.475%
15 199.6 16.9746574 84.8732869 890 15204.45 76.022%
20 175.4 14.9035541 74.5177706 885 13371.52 66.858%
25 180.5 15.3400263 76.7001315 880 13755.62 68.778%
30 190.4 16.1872958 80.9364791 875 14496.98 75.485%
45 193.1 16.4183693 82.0918466 870 14698.01 76.490%
Tabla 42. Datos de la unidad #8 analizada durante el perfil de disolución de Medical
Elite
Medicamento de Prueba
Vaso 8
Tiempo Respuesta xi Ei Vi Di %Di
0 0 0 0 0 0
5 60 5.02730137 25.1365069 900 4524.57 22.623%
10 168.4 14.3044747 71.5223733 895 12827.64 64.138%
15 237.6 20.226803 101.134015 890 18098.51 90.493%
20 234.5 19.9614964 99.807482 885 17863.72 89.319%
25 245.6 20.9114652 104.557326 880 18699.69 93.498%
30 249.9 21.2794712 106.397356 875 19021.70 95.108%
45 242.4 20.6376004 103.188002 870 18463.27 92.316%

96
Tabla 43. Datos de la unidad # 9 analizada durante el perfil de disolución de Medical
Elite
Medicamento de Prueba
Vaso 9
Tiempo Respuesta xi Ei Vi Di %Di
0 0 0 0 0 0
5 83.9 7.0727298 35.363649 900 6365.46 31.827%
10 188 15.9818971 79.9094857 895 14339.16 71.696%
15 220.8 18.7890123 93.9450616 890 16837.49 84.187%
20 226.4 19.2682759 96.3413794 885 17261.64 86.308%
25 233.7 19.8930302 99.4651509 880 17811.43 89.057%
30 235.8 20.072754 100.36377 875 17968.68 89.843%
45 239.1 20.3551772 101.775886 870 18214.39 91.072%
Tabla 44. Datos de la unidad # 10 analizada durante el perfil de disolución de
Medical Elite
Medicamento de Prueba
Vaso 10
Tiempo Respuesta xi Ei Vi Di %Di
0 0 0 0 0 0
5 80.2 6.75607352 33.7803676 900 6080.47 30.402%
10 180.2 15.3143515 76.5717573 895 13740.12 68.701%
15 213.9 18.1984911 90.9924557 890 16307.01 81.535%
20 223.6 19.0286441 95.1432205 885 17041.69 85.208%
25 234.8 19.9871712 99.9358561 880 17885.20 89.426%
30 231 19.6619567 98.3097833 875 17600.64 88.003%
45 232.5 19.7903308 98.9516542 870 17712.32 88.562%

97
Tabla 45. Datos de la unidad #11 analizada durante el perfil de disolución de
Medical Elite
Medicamento de Prueba
Vaso 11
Tiempo Respuesta xi Ei Vi Di %Di
0 0 0 0 0 0
5 66.9 5.61782255 28.0891127 900 5056.04 25.280%
10 160 13.5855793 67.9278966 895 12187.18 60.936%
15 204.1 17.3597799 86.7988995 890 15546.22 77.731%
20 210.3 17.8903931 89.4519657 885 16015.81 80.079%
25 221.9 18.8831534 94.4157669 880 16889.44 84.447%
30 230.4 19.610607 98.053035 875 17525.96 87.630%
45 231.2 19.6790732 98.3953661 870 17585.53 87.928%
Tabla 46. Datos de la unidad #12 analizada durante el perfil de disolución de
Medical Elite
Medicamento de Prueba
Vaso 12
Tiempo Respuesta xi Ei Vi Di %Di
0 0 0 0 0 0
5 76.6 6.44797551 32.2398776 900 5803.18 29.016%
10 185.7 15.7850568 78.9252838 895 14159.87 70.799%
15 209.3 17.8048104 89.0240518 890 15957.45 79.787%
20 212 18.0358839 90.1794193 885 16161.95 80.810%
25 218.5 18.5921719 92.9608596 880 16651.48 83.257%
30 228.2 19.4223249 97.1116244 875 17377.86 86.889%
45 226.4 19.2682759 96.3413794 870 17243.84 86.219%

98
Tabla 47. Obtención de porcentaje de omperazol disuelto en el i-ésimo tiempo de
muestreo de Losec-A20
0 5 10 15 20 25 30 45
1 0 63.92% 95.50% 105.96% 107.71% 105.56% 106.17% 102.33%
2 0 75.31% 91.01% 104.74% 102.79% 98.98% 103.26% 98.68%
3 0 83.35% 94.00% 102.29% 108.41% 101.21% 96.93% 92.14%
4 0 75.69% 95.01% 96.40% 97.06% 97.02% 95.29% 93.98%
5 0 53.51% 94.22% 101.17% 100.75% 100.46% 99.88% 98.45%
6 0 55.84% 93.98% 98.62% 99.92% 99.58% 97.44% 94.78%
7 0 52.76% 87.46% 95.25% 98.87% 101.52% 94.66% 93.71%
8 0 77.36% 90.12% 97.87% 101.98% 101.82% 96.00% 93.50%
9 0 77.32% 104.15% 103.10% 95.64% 97.76% 94.97% 93.98%
10 0 77.36% 93.55% 94.08% 100.09% 98.92% 98.13% 93.80%
11 0 48.16% 89.19% 97.42% 100.44% 99.24% 96.21% 95.74%
12 0 73.38% 90.71% 98.09% 96.12% 96.48% 94.96% 94.13%
x 0 67.83% 93.24% 99.58% 100.82% 99.88% 97.83% 95.44%
DesEst 0 0.122079 0.042680128 0.038085596 0.040304883 0.024725644 0.036078846 0.029194
%CV 0 17.99736 4.577303667 3.82452095 3.997899855 2.475527794 3.688021392 3.05904
Max 0 83.35% 104.15% 105.96% 108.41% 105.56% 106.17% 102.33%
Min 0 48.16% 87.46% 94.08% 95.64% 96.48% 94.66% 92.14%
Medicamento de Referencia (Losec A-20)
Vaso
Parametros Estadisticos
Tiempos de Muestreo (min)
%Di
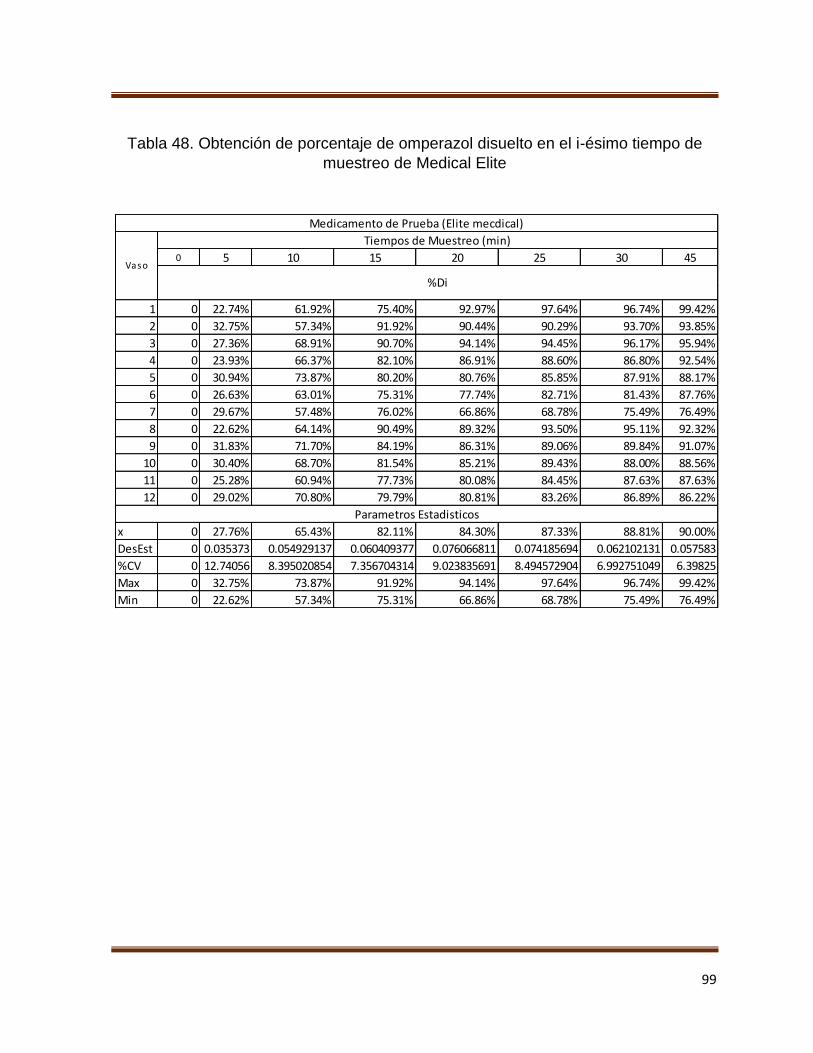
99
Tabla 48. Obtención de porcentaje de omperazol disuelto en el i-ésimo tiempo de
muestreo de Medical Elite
0 5 10 15 20 25 30 45
1 0 22.74% 61.92% 75.40% 92.97% 97.64% 96.74% 99.42%
2 0 32.75% 57.34% 91.92% 90.44% 90.29% 93.70% 93.85%
3 0 27.36% 68.91% 90.70% 94.14% 94.45% 96.17% 95.94%
4 0 23.93% 66.37% 82.10% 86.91% 88.60% 86.80% 92.54%
5 0 30.94% 73.87% 80.20% 80.76% 85.85% 87.91% 88.17%
6 0 26.63% 63.01% 75.31% 77.74% 82.71% 81.43% 87.76%
7 0 29.67% 57.48% 76.02% 66.86% 68.78% 75.49% 76.49%
8 0 22.62% 64.14% 90.49% 89.32% 93.50% 95.11% 92.32%
9 0 31.83% 71.70% 84.19% 86.31% 89.06% 89.84% 91.07%
10 0 30.40% 68.70% 81.54% 85.21% 89.43% 88.00% 88.56%
11 0 25.28% 60.94% 77.73% 80.08% 84.45% 87.63% 87.63%
12 0 29.02% 70.80% 79.79% 80.81% 83.26% 86.89% 86.22%
x 0 27.76% 65.43% 82.11% 84.30% 87.33% 88.81% 90.00%
DesEst 0 0.035373 0.054929137 0.060409377 0.076066811 0.074185694 0.062102131 0.057583
%CV 0 12.74056 8.395020854 7.356704314 9.023835691 8.494572904 6.992751049 6.39825
Max 0 32.75% 73.87% 91.92% 94.14% 97.64% 96.74% 99.42%
Min 0 22.62% 57.34% 75.31% 66.86% 68.78% 75.49% 76.49%
Medicamento de Prueba (Elite mecdical)
Vaso
Tiempos de Muestreo (min)
%Di
Parametros Estadisticos

100
BIBLIOGRAFÍA
Abdel-Rahman, S. M., Amidon, G. L., Vinks, A. A. & Knipp, G. T., 2012. Summary of the National
Institute of Child Health and Human Development-Best Pharmaceuticals for Children Act Pediatric
Formulation Initiatives Workshop-Pediatric Biopharmaceutics Classification System Working Group.
Clinical Therapeutics, p. 518.
Ameril, M. y otros, 2012. The differences between the branded and generic medicines using solid
dosage forms: In-vitro dissolution testing. Results in Pharma Sciences, Volumen 2, pp. 1-8.
Biólogos, C. N. d. Q. F., 2016. Guía de Validación de Métodos Analíticos. México: s.n.
Brunton, L. L., Chabner, B. A. & . Knollmann, B. C., 2012. Las bases farmacológicas de la terapéutica.
México: Mc Graw Hill.
Carvajal, M., Asensio, J. & Sevilla, F., 2003. Procedimiento mejorado para la obtebción de pellets de
omeprazol estables y gastrorresistentes, pellets así obtenidos y aplicaciones. Oficina Española de
Patentes y Marcas, pp. 1-20.
Chandan, M., Rahila, T., Srikanth, M. & Natesh, G., 2013. Design and in-vitro evaluation of stomach
specific floating microspheres of omeprazole. Journal of Drug Delivery & Therapeutics, pp. 31-40.
Douglas A. Skoog, D. M. W. F. H. S. R. C., 2003. Química Analítica. México: Mc Graw Hill.
Esquivel, E. E. & Leal, L. I., 2004. Cromatografía de fase reversa. Morelos: s.n.
Farmacopea, 2014. Farmacopea de los Estados Unidos Mexicanos. México: Secretaria de Salud.
FDA, 2015. U S Food and Drug Administration Home Page. [En línea]
Available at:
http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm200707.htm
[Último acceso: 22 Noviembre 2016].
García, E., 2008. Comparación de perfiles de disolución de productos comerciales de omeprazol en
cápsulas con gránulos de capa entérica de 20 mg. México: Fes Zaragoza Unam
Gennaro, A. R., 2003. Farmacia. Argentina: Médica Panamericana.
Gomez, R., Bafalluy, L., Arranz, C. & Corrales, G., 1997. Utilización terapéutica del omeprazol. Farm
Hosp , pp. 243-256.
Gonzalez, E. P., Frati, A. M. & Enríquez, E. R., 2005. Hacia una política farmacéutica integral para
México. México: Secretaría de Salud.

101
He, W. y otros, 2009. Desing and in Vitro/ in Vivo Evaluation of Multi-layer Film Coated Pellets for
Omeprazole. Chem Pharm Bull, 57(2), pp. 122-128.
Huber, P. y otros, 1999. The SFSTP guide on the validation of cromatographic methods for drug
bioanalysis: from the Whashington Conference to the laboratory. Analytica Chimica Acta 391, pp.
135-148.
Johnson, E., 1978. Basic Liquid Chromatography. Virginia: Varian.
Karim, S., Baie, S., Kah, Y. & Irfan, N., 2014. Development and evaluation of omeprazole pellets
fabricated by sieving-spheronization and extrusión-spheronization process. Pak.J.Pharm. Sci, 27(3),
pp. 425-438.
Moffat, A., David, O. & Brian, W., 2011. Clarke´s Analysis of Drugs and Poisons. Fourth ed. Chicago:
Pharmaceutical Press.
Navarra, 2005. Formas farmacéuticas de liberación modificada y esteroisómeros. Boletín de
Información Farmacoterapéutica, p. 02.
NOM-177, 2013. SSA1, s.l.: Establece las pruebas y procedimientos para demostrar que un
medicamento es intercambiable.
Oswald, C., Kaal, E. & Kagashe, G., 2015. Formulation Development of Generic Omeprazole 20mg
Enteric Coated Tablets. Scientific Research Publishing, Issue 6, pp. 293-301.
Pérez, E. y otros, 2016. Curso Experimental Laboratorio de Biofarmacia. México: IPN.
Storpirtis, S. & Rodriguez, D., 1998. In Vitro Evaluation of Dissolution Properties and Degradation
Products of Omeprazole in Enteric-Coated Pellets. Drug Development and Industrial Pharmacy, pp.
1101-1107.
Tondo, V., Andreazza, I., Otsuka, M. & Murakami, F., 2014. Development of a multiparticulate system
containing enteric-release mini-tablets of omeprazole. Brazilian Journal of Pharmaceutical Sciences,
50(3), pp. 505-511.







![INSTITUTO POLITÉCNICO NACIONAL · Instituto Politécnico Nacional ÍNDICE [Escribir texto] INSTITUTO POLITÉCNICO NACIONAL ESCUELA SUPERIOR DE INGENIERÍA MECÁNICA Y ELÉCTRICA](https://img.dokumen.tips/doc/110x75/5e727d63eb19c64ebc38993a/instituto-politcnico-nacional-instituto-politcnico-nacional-ndice-escribir.jpg)



















