Embed Size (px)
Citation preview

www.elsevier.com/locate/yviro
Virology 334 (20
Potent inhibition of HIV-1 entry by (s4dU)35
Andras Horvatha,b, Szilvia T}ookesa,b, Tracy Hartmanc,1, Karen Watsonc,1, Jim A. Turpinc,2,
Robert W. Buckheit Jr.c,1, Zsolt Sebestyenb,d,e, Janos Szfll}oosib,d,e,Ilona Benk}oof, Thomas J. Bardosg, Joseph A. Dunnh, Laszlo Fesqsa,b,
Ferenc D. Tothi, Janos Aradia,b,TaDepartment of Biochemistry and Molecular Biology, University of Debrecen, H-4012 Debrecen, Nagyerdei krt. 98, HungarybResearch Center for Molecular Medicine, Medical and Health Science Center, Faculty of Medicine, University of Debrecen,
H-4012 Debrecen, Nagyerdei krt. 98, HungarycSouthern Research Institute, Infectious Disease Research, Frederick, MD 21701, USA
dDepartment of Biophysics, University of Debrecen, H-4012 Debrecen, Nagyerdei krt. 98, HungaryeCell Biophysics Research Group of the Hungarian Academy of Sciences, University of Debrecen, H-4012 Debrecen, Nagyerdei krt. 98, Hungary
fDepartment of Pharmacology and Pharmacotherapy, University of Debrecen, H-4012 Debrecen, Nagyerdei krt. 98, HungarygDepartment of Chemistry, State University of New York, Buffalo, NY 14260, USA
hOmniPharm Inc. 255 Great Arrow Avenue Buffalo, NY 14207, USAiDepartment of Medical Microbiology, University of Debrecen, H-4012 Debrecen, Nagyerdei krt. 98, Hungary
Received 22 November 2004; returned to author for revision 4 January 2005; accepted 26 January 2005
Abstract
We have previously reported the potent in vitro HIV-1 anti-reverse transcriptase activity of a 35-mer of 4-thio-deoxyuridylate [(s4dU)35].
In efforts to define its activity in a more physiological system, studies were carried out to determine the stage of viral infection that this
compound mediates its anti-viral effect. Results of the studies reported herein show that (s4dU)35 is nontoxic and is capable of inhibiting both
single and multi-drug resistant HIV strains (IC50: 0.8–25.4 Ag/ml) in vitro. Besides its previously reported anti-RT activity, (s4dU)35 mediated
its antiviral action by preventing virus attachment (IC50: 0.002–0.003 Ag/ml), and was stable in vitro and slowly degraded by DNAses.
Competition studies and fluorescence resonance energy transfer (FRET) experiments indicated that (s4dU)35 preferentially binds to CD4
receptors, but not to CD48. Confocal laser scanning microscopy (CLSM) studies showed that (s4dU)35 did not penetrate into the cells and
colocalized with cell surface thioredoxin. Our studies identify (s4dU)35 as a potential novel HIVentry inhibitor that may have utility as either
a systemic antiretroviral or as a preventing agent for HIV transmission.
D 2005 Elsevier Inc. All rights reserved.
Keywords: HIV-entry; Inhibition; Oligonucleotide; Redox process; CD4
0042-6822/$ - see front matter D 2005 Elsevier Inc. All rights reserved.
doi:10.1016/j.virol.2005.01.033
* Corresponding author. Department of Biochemistry and Molecular
Biology, University of Debrecen, H-4012 Debrecen, POB. 6. Hungary.
Fax: +36 52 314 989.
E-mail address: [email protected] (J. Aradi).1 Current address: Genelogic, 18761 N. Frederick Avenue, Gaithersburg,
MD 20879, USA.2 Current address: Henry M. Jackson Foundation, 6700 B Rockledge
Drive, Bethesda, MD 20892, USA.
Introduction
The development and discovery of inhibitors of human
immunodeficiency virus type 1 (HIV-1), the etiological
agent for HIV/AIDS, has been dominated by inhibitors of
the HIV enzymes reverse transcriptase (RT) and protease
(De Clercq, 1993; Richman, 2001). However, as issues of
patient compliance, cost and access to therapies and the
emergence of single- and multi-drug resistant strains have
come to the forefront of HIV/AIDS therapy, the need for
05) 214–223

A. Horvath et al. / Virology 334 (2005) 214–223 215
more potent and novel antivirals as well as new antiviral
targets has arisen. Part of the need for new antiviral targets
has been met by the identification of many of the processes
involved in HIV entry into cells (Cooley and Lewin, 2003;
Dimitrov, 1997; Doms and Moore, 2000; Eckert and Kim,
2001; Kilby and Eron, 2003; LaBranche et al., 2001).
The HIV-1 envelope protein is a trimer of gp120–gp41
heterodimers on the virion surface. The multi-step HIVentry
process is initiated by the highly specific interaction
between gp120 HIV envelope glycoprotein and the CD4
molecule, its primary receptor. This interaction induces
conformational alterations in gp120, enabling it to sub-
sequently interact with the co-receptor (CCR5 or CXCR4)
(Eckert and Kim, 2001; LaBranche et al., 2001). Following
binding of gp120 to CD4 and a chemokine receptor, a
further conformational change occurs in the gp120–gp41
oligomer that leads to insertion of the hydrophobic N-
terminal peptide sequence of gp41 into the membrane of the
host cell bringing about the fusion process. Recently,
Enfuvirtide (T20), an inhibitor of gp41 and virus entry,
has been approved for use in combination with other anti-
HIV medications, providing access to a new class of drugs
as antiviral targets for the treatment of HIV/AIDS (Kilby
and Eron, 2003).
In addition to the above conformational changes, a
chemical process, that is, redox changes of cell surface
proteins and the viral envelope glycoprotein (gp120) are
required for successful entry of HIV-1 into cells. It was
recently shown that the 2nd domain (D2) disulfide bond of
CD4 is redox-active and the redox process is controlled by
cell surface thioredoxin, secreted by the T cell (Hogg, 2003;
Matthias and Hogg, 2003, Matthias et al., 2002). Protein-
disulfide isomerase (PDI) is another cell surface protein that
is known to catalyze redox changes, and thus, may also be
involved in the control of cell surface redox chemistry
required for HIV-1 entry. It was reported that purified PDI
can cleave disulfide bonds in recombinant gp120 and two of
the nine disulfides of gp120 are reduced during interaction
with the lymphocyte surface on CD4+ cells (Barbouche et
al., 2003; Gallina et al., 2002) and anti-PDI agents
effectively block CXCR4 Env-mediated fusion (Markovic
et al., 2004). Localization of CD4 receptors in cholesterol
and glycosphingolipid-rich lipid rafts, present in the external
leaflet of the plasma membrane, also appear indispensable
for successful HIV-1 entry (del Real et al., 2002; Popik et
al., 2002), although these data remain a subject of debate
(Percherancier et al., 2003). Further studies are needed to
elucidate the detailed functional connection between the
well-established conformational changes and the disulfide
exchange processes involved in the entry of HIV-1 into
target cells. However, these two findings, the cell surface
reducing power, that is, thioredoxin and perhaps PDI, as
well as the localization of the CD4 receptors in lipid rafts,
together may represent new information which can be
exploited as potential targets in the inhibition of HIV-1
entry.
Although oligonucleotides with demonstrated activity
against various viral structural and enzyme targets have
been reported, no oligonucleotide drug has yet been
approved for routine therapeutic use against HIV infection.
Both antisense and triple-helix forming oligonucleotides
exert their inhibitory activities by sequence-specific nucleic
acid interactions (Agrawal et al., 1989; Inagawa et al., 2002;
Kinchington et al., 1992; Tsukahara et al., 1997). In vitro
oligodeoxycytidine containing a phosphorothioate backbone
binds to CD4, blocking the interaction between gp120 and
CD4. In addition, it has also been shown to inhibit
polymerase activity of purified RT enzyme (Majumdar et
al., 1989; Matsukura et al., 1987; Stein et al., 1991).
Guanine-rich oligonucleotides with phosphorothioate inter-
nucleotide linkages also exert anti-HIV activity (Buckheit et
al., 1994; Jing and Hogan, 1998; Kuwasaki et al., 2003;
Ojwang et al., 1995). We have previously shown that 5-
mercaptopyrimidine-containing oligo- and polynucleotides
are potent inhibitors of RT enzyme activity and HIV-1
replication in vitro (Bardos et al., 1992; T}ookes and Aradi,
1995). In the studies described herein, we have conducted
further detailed studies aimed of the characterization of the
precise stage of the viral life cycle which is the target of the
anti-HIV activity of a 35-mer homo-oligonucleotide com-
posed of 4-thio-deoxyuridylates, (s4dU)35.
While this 35-mer oligonucleotide was previously shown
to be a potent inhibitor of cell-free HIV-1 RT and telomerase
in vitro (Szatmari et al., 2000; T}ookes and Aradi, 1996),
results of the studies reported herein utilizing a more
physiological assay for the measurement of anti-viral
activity provide data that supports the view that this novel
oligo-compound (s4dU)35 primarily mediates its anti-viral
effect by inhibiting virus entry.
Results
Antiviral activity of (s4dU)35
(s4dU)35 was assessed for inhibition of HIV-1 replication
in a standard cell-based assay (Buckheit et al., 1993; Rice
et al., 1997; Weislow et al., 1989) using both wild-type and
viruses expressing mutations known to confer resistance to
nucleoside (NRTI) and non-nucleoside (NNRTI) reverse
transcriptase inhibitors (Buckheit et al., 1995a, 1995b).
Inhibitory concentration 50% (IC50s) ranged from 0.8 to
25.4 Ag/ml (Table 1). Minor 0.9- to 3.3-fold variations in
(s4dU)35 IC50s were noted for non-nucleoside reverse
transcriptase inhibitor (NNRTI) and nucleoside reverse
transcriptase inhibitor (NRTI) resistant viruses compared
to corresponding wild type strains (NL4-3 and IIIB); but
these changes are not statistically significant in the bioassay.
There was no detectable evidence of cytotoxicity (morpho-
logically or by formazan dye reduction) when doses as high
as 100 Ag/ml of (s4dU)35 were utilized in these assays. The
results presented in Table 1 are representative of multiple
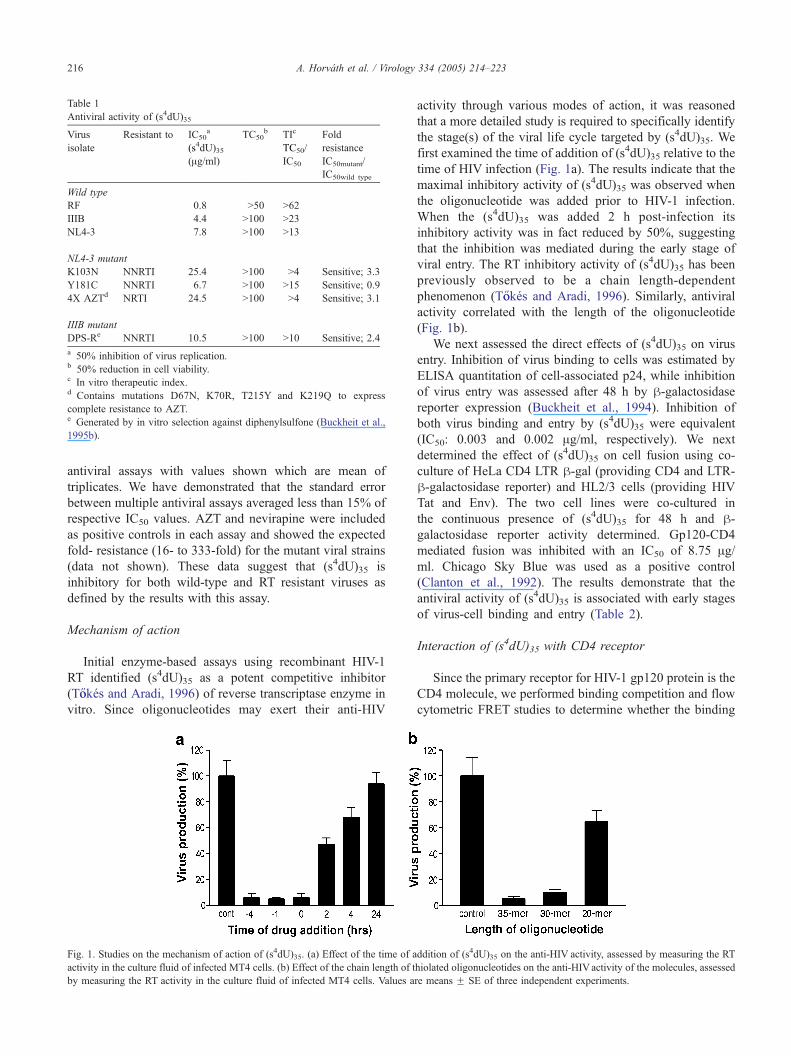
Table 1
Antiviral activity of (s4dU)35
Virus
isolate
Resistant to IC50a
(s4dU)35(Ag/ml)
TC50b TIc
TC50/
IC50
Fold
resistance
IC50mutant/
IC50wild type
Wild type
RF 0.8 N50 N62
IIIB 4.4 N100 N23
NL4-3 7.8 N100 N13
NL4-3 mutant
K103N NNRTI 25.4 N100 N4 Sensitive; 3.3
Y181C NNRTI 6.7 N100 N15 Sensitive; 0.9
4X AZTd NRTI 24.5 N100 N4 Sensitive; 3.1
IIIB mutant
DPS-Re NNRTI 10.5 N100 N10 Sensitive; 2.4
a 50% inhibition of virus replication.b 50% reduction in cell viability.c In vitro therapeutic index.d Contains mutations D67N, K70R, T215Y and K219Q to express
complete resistance to AZT.e Generated by in vitro selection against diphenylsulfone (Buckheit et al.,
1995b).
A. Horvath et al. / Virology 334 (2005) 214–223216
antiviral assays with values shown which are mean of
triplicates. We have demonstrated that the standard error
between multiple antiviral assays averaged less than 15% of
respective IC50 values. AZT and nevirapine were included
as positive controls in each assay and showed the expected
fold- resistance (16- to 333-fold) for the mutant viral strains
(data not shown). These data suggest that (s4dU)35 is
inhibitory for both wild-type and RT resistant viruses as
defined by the results with this assay.
Mechanism of action
Initial enzyme-based assays using recombinant HIV-1
RT identified (s4dU)35 as a potent competitive inhibitor
(T}ookes and Aradi, 1996) of reverse transcriptase enzyme in
vitro. Since oligonucleotides may exert their anti-HIV
Fig. 1. Studies on the mechanism of action of (s4dU)35. (a) Effect of the time of a
activity in the culture fluid of infected MT4 cells. (b) Effect of the chain length of t
by measuring the RT activity in the culture fluid of infected MT4 cells. Values a
activity through various modes of action, it was reasoned
that a more detailed study is required to specifically identify
the stage(s) of the viral life cycle targeted by (s4dU)35. We
first examined the time of addition of (s4dU)35 relative to the
time of HIV infection (Fig. 1a). The results indicate that the
maximal inhibitory activity of (s4dU)35 was observed when
the oligonucleotide was added prior to HIV-1 infection.
When the (s4dU)35 was added 2 h post-infection its
inhibitory activity was in fact reduced by 50%, suggesting
that the inhibition was mediated during the early stage of
viral entry. The RT inhibitory activity of (s4dU)35 has been
previously observed to be a chain length-dependent
phenomenon (T}ookes and Aradi, 1996). Similarly, antiviral
activity correlated with the length of the oligonucleotide
(Fig. 1b).
We next assessed the direct effects of (s4dU)35 on virus
entry. Inhibition of virus binding to cells was estimated by
ELISA quantitation of cell-associated p24, while inhibition
of virus entry was assessed after 48 h by h-galactosidasereporter expression (Buckheit et al., 1994). Inhibition of
both virus binding and entry by (s4dU)35 were equivalent
(IC50: 0.003 and 0.002 Ag/ml, respectively). We next
determined the effect of (s4dU)35 on cell fusion using co-
culture of HeLa CD4 LTR h-gal (providing CD4 and LTR-
h-galactosidase reporter) and HL2/3 cells (providing HIV
Tat and Env). The two cell lines were co-cultured in
the continuous presence of (s4dU)35 for 48 h and h-galactosidase reporter activity determined. Gp120-CD4
mediated fusion was inhibited with an IC50 of 8.75 Ag/ml. Chicago Sky Blue was used as a positive control
(Clanton et al., 1992). The results demonstrate that the
antiviral activity of (s4dU)35 is associated with early stages
of virus-cell binding and entry (Table 2).
Interaction of (s4dU)35 with CD4 receptor
Since the primary receptor for HIV-1 gp120 protein is the
CD4 molecule, we performed binding competition and flow
cytometric FRET studies to determine whether the binding
ddition of (s4dU)35 on the anti-HIV activity, assessed by measuring the RT
hiolated oligonucleotides on the anti-HIVactivity of the molecules, assessed
re means F SE of three independent experiments.
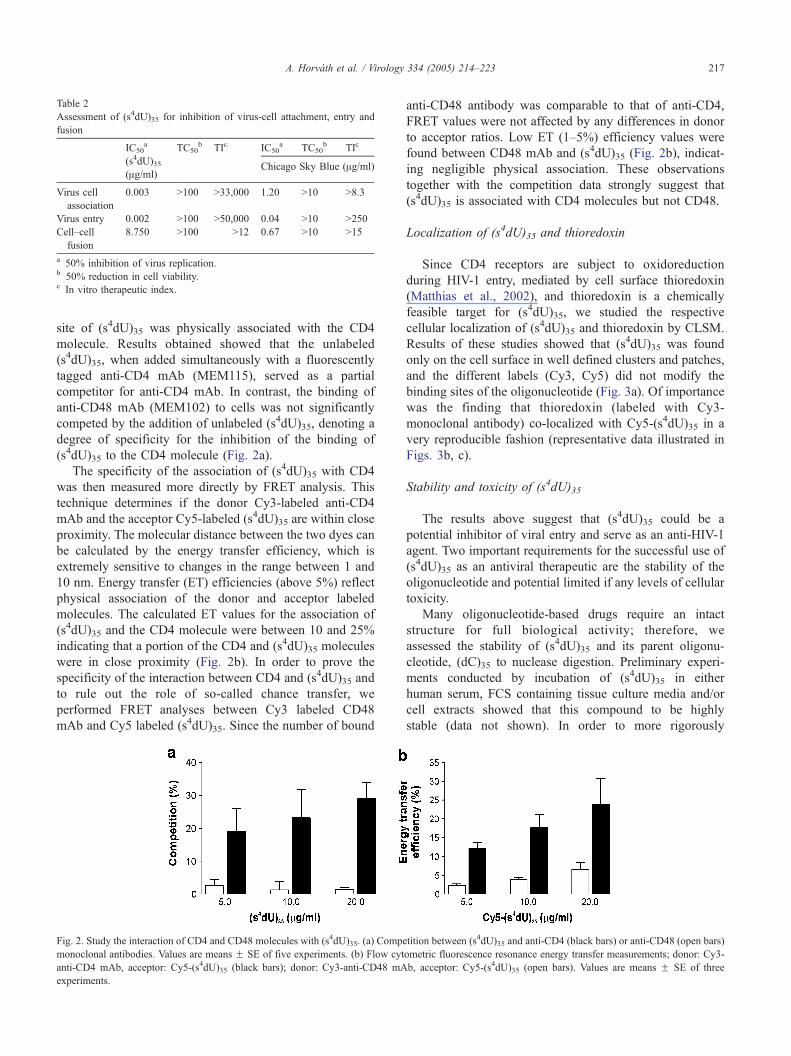
Table 2
Assessment of (s4dU)35 for inhibition of virus-cell attachment, entry and
fusion
IC50a TC50
b TIc IC50a TC50
b TIc
(s4dU)35(Ag/ml)
Chicago Sky Blue (Ag/ml)
Virus cell
association
0.003 N100 N33,000 1.20 N10 N8.3
Virus entry 0.002 N100 N50,000 0.04 N10 N250
Cell–cell
fusion
8.750 N100 N12 0.67 N10 N15
a 50% inhibition of virus replication.b 50% reduction in cell viability.c In vitro therapeutic index.
A. Horvath et al. / Virology 334 (2005) 214–223 217
site of (s4dU)35 was physically associated with the CD4
molecule. Results obtained showed that the unlabeled
(s4dU)35, when added simultaneously with a fluorescently
tagged anti-CD4 mAb (MEM115), served as a partial
competitor for anti-CD4 mAb. In contrast, the binding of
anti-CD48 mAb (MEM102) to cells was not significantly
competed by the addition of unlabeled (s4dU)35, denoting a
degree of specificity for the inhibition of the binding of
(s4dU)35 to the CD4 molecule (Fig. 2a).
The specificity of the association of (s4dU)35 with CD4
was then measured more directly by FRET analysis. This
technique determines if the donor Cy3-labeled anti-CD4
mAb and the acceptor Cy5-labeled (s4dU)35 are within close
proximity. The molecular distance between the two dyes can
be calculated by the energy transfer efficiency, which is
extremely sensitive to changes in the range between 1 and
10 nm. Energy transfer (ET) efficiencies (above 5%) reflect
physical association of the donor and acceptor labeled
molecules. The calculated ET values for the association of
(s4dU)35 and the CD4 molecule were between 10 and 25%
indicating that a portion of the CD4 and (s4dU)35 molecules
were in close proximity (Fig. 2b). In order to prove the
specificity of the interaction between CD4 and (s4dU)35 and
to rule out the role of so-called chance transfer, we
performed FRET analyses between Cy3 labeled CD48
mAb and Cy5 labeled (s4dU)35. Since the number of bound
Fig. 2. Study the interaction of CD4 and CD48 molecules with (s4dU)35. (a) Compe
monoclonal antibodies. Values are means F SE of five experiments. (b) Flow cyt
anti-CD4 mAb, acceptor: Cy5-(s4dU)35 (black bars); donor: Cy3-anti-CD48 mA
experiments.
anti-CD48 antibody was comparable to that of anti-CD4,
FRET values were not affected by any differences in donor
to acceptor ratios. Low ET (1–5%) efficiency values were
found between CD48 mAb and (s4dU)35 (Fig. 2b), indicat-
ing negligible physical association. These observations
together with the competition data strongly suggest that
(s4dU)35 is associated with CD4 molecules but not CD48.
Localization of (s4dU)35 and thioredoxin
Since CD4 receptors are subject to oxidoreduction
during HIV-1 entry, mediated by cell surface thioredoxin
(Matthias et al., 2002), and thioredoxin is a chemically
feasible target for (s4dU)35, we studied the respective
cellular localization of (s4dU)35 and thioredoxin by CLSM.
Results of these studies showed that (s4dU)35 was found
only on the cell surface in well defined clusters and patches,
and the different labels (Cy3, Cy5) did not modify the
binding sites of the oligonucleotide (Fig. 3a). Of importance
was the finding that thioredoxin (labeled with Cy3-
monoclonal antibody) co-localized with Cy5-(s4dU)35 in a
very reproducible fashion (representative data illustrated in
Figs. 3b, c).
Stability and toxicity of (s4dU)35
The results above suggest that (s4dU)35 could be a
potential inhibitor of viral entry and serve as an anti-HIV-1
agent. Two important requirements for the successful use of
(s4dU)35 as an antiviral therapeutic are the stability of the
oligonucleotide and potential limited if any levels of cellular
toxicity.
Many oligonucleotide-based drugs require an intact
structure for full biological activity; therefore, we
assessed the stability of (s4dU)35 and its parent oligonu-
cleotide, (dC)35 to nuclease digestion. Preliminary experi-
ments conducted by incubation of (s4dU)35 in either
human serum, FCS containing tissue culture media and/or
cell extracts showed that this compound to be highly
stable (data not shown). In order to more rigorously
tition between (s4dU)35 and anti-CD4 (black bars) or anti-CD48 (open bars)
ometric fluorescence resonance energy transfer measurements; donor: Cy3-
b, acceptor: Cy5-(s4dU)35 (open bars). Values are means F SE of three
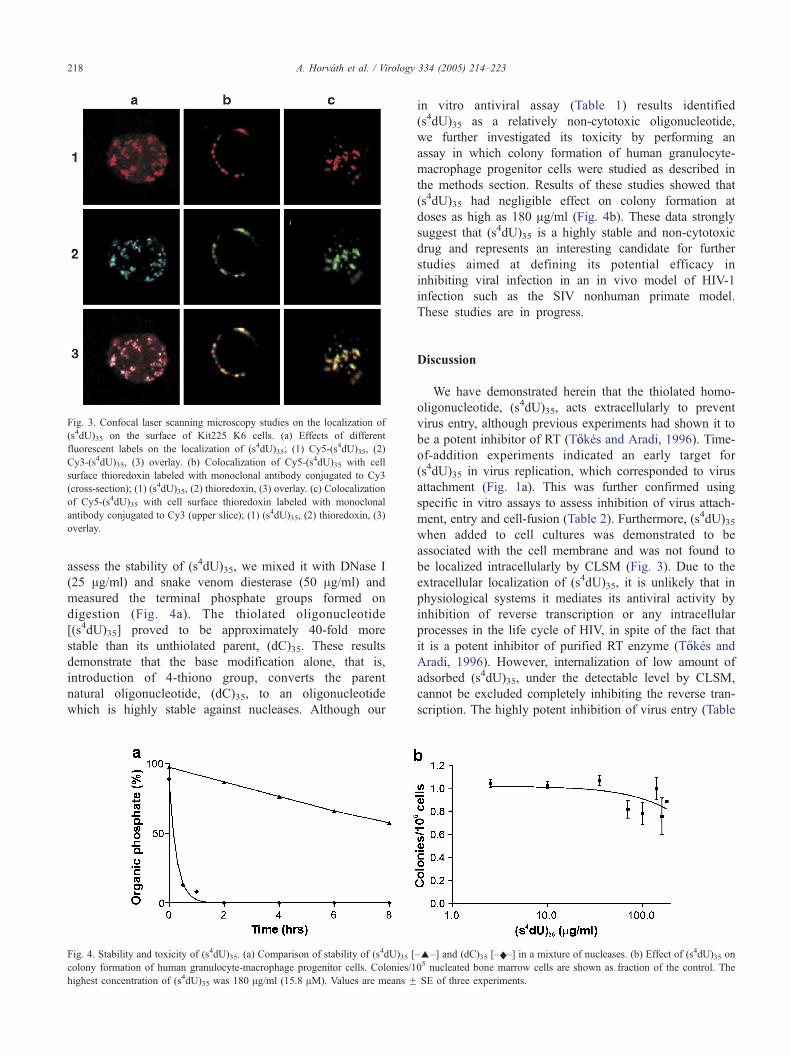
Fig. 3. Confocal laser scanning microscopy studies on the localization of
(s4dU)35 on the surface of Kit225 K6 cells. (a) Effects of different
fluorescent labels on the localization of (s4dU)35; (1) Cy5-(s4dU)35, (2)
Cy3-(s4dU)35, (3) overlay. (b) Colocalization of Cy5-(s4dU)35 with cell
surface thioredoxin labeled with monoclonal antibody conjugated to Cy3
(cross-section); (1) (s4dU)35, (2) thioredoxin, (3) overlay. (c) Colocalization
of Cy5-(s4dU)35 with cell surface thioredoxin labeled with monoclonal
antibody conjugated to Cy3 (upper slice); (1) (s4dU)35, (2) thioredoxin, (3)
overlay.
A. Horvath et al. / Virology 334 (2005) 214–223218
assess the stability of (s4dU)35, we mixed it with DNase I
(25 Ag/ml) and snake venom diesterase (50 Ag/ml) and
measured the terminal phosphate groups formed on
digestion (Fig. 4a). The thiolated oligonucleotide
[(s4dU)35] proved to be approximately 40-fold more
stable than its unthiolated parent, (dC)35. These results
demonstrate that the base modification alone, that is,
introduction of 4-thiono group, converts the parent
natural oligonucleotide, (dC)35, to an oligonucleotide
which is highly stable against nucleases. Although our
Fig. 4. Stability and toxicity of (s4dU)35. (a) Comparison of stability of (s4dU)35 [
colony formation of human granulocyte-macrophage progenitor cells. Colonies/1
highest concentration of (s4dU)35 was 180 Ag/ml (15.8 AM). Values are means F
in vitro antiviral assay (Table 1) results identified
(s4dU)35 as a relatively non-cytotoxic oligonucleotide,
we further investigated its toxicity by performing an
assay in which colony formation of human granulocyte-
macrophage progenitor cells were studied as described in
the methods section. Results of these studies showed that
(s4dU)35 had negligible effect on colony formation at
doses as high as 180 Ag/ml (Fig. 4b). These data strongly
suggest that (s4dU)35 is a highly stable and non-cytotoxic
drug and represents an interesting candidate for further
studies aimed at defining its potential efficacy in
inhibiting viral infection in an in vivo model of HIV-1
infection such as the SIV nonhuman primate model.
These studies are in progress.
Discussion
We have demonstrated herein that the thiolated homo-
oligonucleotide, (s4dU)35, acts extracellularly to prevent
virus entry, although previous experiments had shown it to
be a potent inhibitor of RT (T}ookes and Aradi, 1996). Time-
of-addition experiments indicated an early target for
(s4dU)35 in virus replication, which corresponded to virus
attachment (Fig. 1a). This was further confirmed using
specific in vitro assays to assess inhibition of virus attach-
ment, entry and cell-fusion (Table 2). Furthermore, (s4dU)35when added to cell cultures was demonstrated to be
associated with the cell membrane and was not found to
be localized intracellularly by CLSM (Fig. 3). Due to the
extracellular localization of (s4dU)35, it is unlikely that in
physiological systems it mediates its antiviral activity by
inhibition of reverse transcription or any intracellular
processes in the life cycle of HIV, in spite of the fact that
it is a potent inhibitor of purified RT enzyme (T}ookes and
Aradi, 1996). However, internalization of low amount of
adsorbed (s4dU)35, under the detectable level by CLSM,
cannot be excluded completely inhibiting the reverse tran-
scription. The highly potent inhibition of virus entry (Table
–E–] and (dC)35 [–x–] in a mixture of nucleases. (b) Effect of (s4dU)35 on
05 nucleated bone marrow cells are shown as fraction of the control. The
SE of three experiments.

A. Horvath et al. / Virology 334 (2005) 214–223 219
2) indicates that the inhibition of reverse transcription
cannot be the main mechanism of the anti-HIV action of
(s4dU)35.
(s4dU)35 inhibited the in vitro replication of various
laboratory strains of HIV, including resistant mutants to
NRTI and NNRTI (Table 1), with IC50s in the 0.8 to 25.4
Ag/ml range (0.07-2.23 AM), which is better or comparable
to the activity of other reported oligonucleotide based anti-
HIV agents (Agrawal et al., 1989; Bardos et al., 1992;
Buckheit et al., 1994; Inagawa et al., 2002; Jing and Hogan,
1998; Kinchington et al., 1992; Kuwasaki et al., 2003;
Matsukura et al., 1987; Ojwang et al., 1995; Tsukahara et
al., 1997; Yamaguchi et al., 1997). (s4dU)35 was also shown
to be noncytotoxic at concentrations 50 to 1000 times its
IC50 and exhibited negligible effect on colony formation of
granulocyte-macrophage progenitor cells (Fig. 4b). These
experimental data identify (s4dU)35 as a nontoxic anti-HIV
agent, acting as an entry inhibitor.
The potent inhibition of virus entry (IC50 = 0.002 Ag/ml)
and the inhibition of the virus replication at much higher
concentrations, as both of them are documented herein
(Tables 1 and 2), is an apparent contradiction. However,
similar observations have also been reported in the literature
(Chang and Bewley, 2002). It is potentially possible that the
reasons for this differential outcome could be due to the fact
that whereas the assay for entry determines the effect of one
round of infection, the antiviral assay measures multiple
rounds of infections.
Chemically, at least three features of (s4dU)35 could be
important for its antiviral activity. First: the 4-thiono group
increases the hydrophobic character of the base (T}ookes andAradi, 1996). Second: the 4-thiono group has a propensity
toward tautomeric conversion (Simuth et al., 1970) to form
reactive -SH groups at position 4. Third: the 4-thiolation
converts the natural oligonucleotide to a highly nuclease
resistant molecule (Fig. 4a). The dependence of antiviral
activity on the length of the thiolated oligonucleotides
indicates that the presence of the 4-thiono or 4-mercapto
group alone were not sufficient to mediate inhibition of
virus entry. Chain-length dependency further suggests an
aptamer-like effect or a size dependent affinity for the
antiviral target(s) requiring hydrophobic and/or ionic inter-
action. A similar chain length-dependent effect was
observed for other oligonucleotide-based entry inhibitors
(Bardos et al., 1992; Matsukura et al., 1987).
A significant difference between (s4dU)35 and many
other previously reported oligonucleotides with anti-HIV
activity is that (s4dU)35 does not penetrate into the cells.
This feature may be partly responsible for its low toxicity.
On the other hand, the nontoxic nature of (s4dU)35 could be
due to the fact that its biological effect is mediated at the
cell surface with specificity for CD4 and perhaps other yet
to be identified proteins involved in HIV-1 entry. It should
be noted that 4-thiouracil is a naturally occurring base
(Lipsett, 1965), and that disulfide linkage can be formed
between two 4-thiouridines (Shefter and Kalman, 1968),
and poly-4-thiouridylic acid could support the synthesis
(Fiser et al., 1974) of polyphenylalanine as an artificial
mRNA.
Since the 4-thiono group has a propensity toward
tautomeric conversion (Simuth et al., 1970) to form reactive
-SH groups at position 4, intervention by (s4dU)35 during
the cell surface disulfide exchange processes is chemically
feasible and if it precedes the cell-virus interaction could be
the bases for its antiviral activity.
Redox change within the second domain of CD4
molecule is required for successful HIV-1 entry, mediated
by cell surface thioredoxin (Matthias et al., 2002). Therefore
we first studied the interaction between of (s4dU)35 and CD4
and thioredoxin. The results of CD4/(s4dU)35 and CD48/
(s4dU)35 competition studies together with the data of FRET
analysis strongly suggest that (s4dU)35 is associated with
CD4 molecules but not with CD48. On addition, the CLSM
study (Fig. 3) suggests that (s4dU)35 may interact with cell
surface thioredoxin, too. These proteins, with essential roles
in the entry process, carry free -SH groups. Our results and
the physico-chemical nature of -SH groups are consistent
with the formation of disulfide linkage between -SH
carrying proteins and thiolated oligonucleotide. While this
is conceptually possible, it is important to specifically
demonstrate that this indeed occurs. Thus, further studies are
needed to determine the nature of these interactions and to
show whether any covalent interactions could occur
between (s4dU)35, CD4, thioredoxin and other proteins,
subjects of redox reactions and involved in HIV entry. It has
to be noted that another redox process between PDI and
gp120 is also required for HIV-1 entry (Barbouche et al.,
2003; Gallina et al., 2002) which perhaps may serve as
additional targets for -SH interacting agents.
In summary, (s4dU)35 is a thiolated oligonucleotide with
low in vitro cytotoxicity and potent inhibition of HIV entry.
The results presented herein further demonstrate that the
entry process of HIV can be specifically interdicted with
low cellular toxicity, identifying (s4dU)35 as a new member
of entry inhibitors, with a potential as either a systemic
inhibitor of HIV replication or for use in prevention of HIV
transmission.
Materials and methods
Synthesis and characterization of (s4dU)35
We synthesized (s4dU)35 according to our earlier
developed method (T}ookes and Aradi, 1996), by conversion
of (dC)35 to (s4dU)35. This method is based on published
procedures of the synthesis of 4-thio-uridylate containing
oligonucleotides (Miura et al., 1973) and of the conversion
of polycytidylic acid to poly-4-thiouridylic acid (Hochberg
and Keren-Zur, 1974).
For preparation of (s4dU)35 the starting material, chro-
matography-purified (dC)35, was purchased from IDT

A. Horvath et al. / Virology 334 (2005) 214–223220
(Coralville, IA). The thiolated oligonucleotide was isolated
by two precipitations with 2 volumes of alcohol from a
solution containing 0.3 M Na-acetate (pH 5.4). The
precipitates were washed twice with 80% alcohol, dissolved
in water and purified in 6 mg portion by ion exchange
chromatography (Horvath and Aradi, 2005). Most of the
batches proved to be homogenous by ion exchange
chromatography; therefore, the purification by chromato-
graphy could have been omitted.
The ratio of thiolated and unmodified bases in the final
product was determined after complete degradation of the
thiolated oligonucleotide to nucleosides (4-thio-deoxyuri-
dine and deoxycytidine) by phosphodiesterase and phos-
phatase followed by HPLC analysis, as we have described
earlier (T}ookes and Aradi, 1996).
The homogeneity of (s4dU)35 was further proved by
denaturing gel electrophoresis; 0.5, 1.0, 2.0, 4.0 Agoligonucleotide was applied to denaturing polyacrylamide
gel, and visualized by silver staining (PlusDNARAmersham-Pharmacia).
The (s4dU)35 used in this study was homogenous in
length and 98.64% of the bases were thiolated and 1.36%
were unmodified. The theoretical molecular mass of
(s4dU)35-Na salt is 11,403 kDa.
The fluorescently labeled (s4dU)35 was synthesized
using an automated oligonucleotide synthesizer (Pharma-
cia Gene-Assembler Plus) by standard phosphoramidite
chemistry in our laboratory. The 4-thio-deoxyuridine
phosphoramidite and Cy3 or Cy5 phosphoramidite were
purchased from Glen Research (Sterling, VA) and
Amersham-Pharmacia, respectively. Crude products were
purified by ion-exchange chromatography (Amersham-
Pharmacia Akta Purifier, on a RESOURCE Q column)
using Na-perchlorate as an eluent (Horvath and Aradi,
2005).
Cells and viruses
The CEM-SS, HL2/3 and HeLa CD4 LTR h-gal cell lineswere obtained from the NIH AIDS Research and Reference
Reagent Program (Bethesda, MD). CEM-SS cells were
maintained in RPMI 1640 medium supplemented with 10%
Fetal Calf Serum (FCS), 2 mM l-glutamine, and antibiotics.
HL2/3 and HeLa CD4 LTR h-gal were maintained in
DMEM 10% FCS, 2 mM l-glutamine, and antibiotics.
Kit225 K6 human T lymphoma cells were cultured in
RPMI 1640 medium supplemented with 10% FCS and
antibiotics in the presence of human recombinant IL-2.
Human immunodeficiency virus type 1 (HIV-1) strains
RF, IIIB and the molecular clones of NL4-3 were
obtained from the NIH AIDS Research and Reference
Reagent Program (Buckheit et al., 1995a). 4X AZT
contains mutations at D67N, K70R, T215Y and K219Q to
express complete resistance to AZT. DSP-R, a viral strain
resistant to NNRTIs, was generated as described earlier
(Buckheit et al., 1995b).
Antiviral assay based on RT activity measurement
Antiviral activity in the indicated experiments was
determined by measuring the RT activity in the culture
fluid of infected MT-4 cells after 96 h of infection (Hoffman
et al., 1985). The protocol is summarized briefly below.
To determine the anti-HIV activity of thiolated oligo-
nucleotides with various chain-length, 2.5 � 105 MT-4
cells were treated with the oligonucleotide inhibitors (in
300 Al) for 1 h before infection then infection was made at
a MOI (multiplicity of infection) of 0.01 with a virus stock
of HIV-1IIIB (100 Al) and after 1 h 600 Al of complete
tissue culture medium was added to get the final volume of
1 ml. Eventually, the treated and infected cells were grown
at 37 8C in RPMI 1640 medium, supplemented with 10%
heat-inactivated FCS, 2 mM l-glutamine and antibiotics.
The RT activity was determined 96 h of postinfection as
described (Hoffman et al., 1985). The indicated oligonu-
cleotide concentration was present in the 300 Al cell
culture during the pre-incubation of the cells with (s4dU)n.
The inhibitors and unabsorbed viruses were not removed
after the infection. Uninfected and untreated samples
(negative control), infected and untreated samples (positive
control) were included in each experiment.
In the time-of-addition assay, the above described
protocol was used, except that the time of infection varied
relative to the addition of (s4dU)35.
Antiviral assay by determination of cytoprotection
The HIV-inhibitory activity of the compounds on CEM-
SS cells was evaluated as described previously (Buckheit et
al.,l 1993; Rice et al., 1997; Weislow et al., 1989) in a
microtiter anti-HIV assay which measures the ability of the
compound to inhibit HIV-1 induced cell killing. Zidovudine
(AZT, Sigma Chemical Co., St. Louis, MO) or nevirapine
were used as positive controls.
HIV attachment, binding and inhibition of cell–cell fusion
assays
Attachment and fusion assays were performed as we
described earlier (Buckheit et al., 1994) with minor
modifications. Briefly, the virus attachment and entry assays
were performed following the interaction of HIV-1IIIB with
HeLa CD4 LTR h-gal cells for 1 h at 37 8C, washing to
remove unbound virus and immediately quantitating cell-
associated p24 (virus binding) by ELISA (Coulter) or
culturing the cells for an additional 48 h and assessing
h-galactosidase reporter expression (virus entry). The h-galactosidase enzyme expression was detected by Tropix
Gal-screen (Tropix, Bedford, MA).
Inhibition of fusion was quantitated by coculture of HL2/
3 (expressing HIV Tat and Env proteins) and HeLa CD4
LTR h-gal cells for 48 h in the presence of drug and
determining h-galactosidase reporter expression measured

A. Horvath et al. / Virology 334 (2005) 214–223 221
by Tropix Gal-screen. Chicago Sky Blue (CSB), a poly-
sulfonic acid dye inhibitor of HIV attachment and fusion
was used as a positive control for all mechanism assays
(Clanton et al., 1992).
Methods for study the interaction between CD4 receptor
and (s4dU)35
The MEM115 monoclonal antibody (mAb) against CD4
molecule and MEM102 mAb against CD48 molecule was a
kind gift from Vaclav Horejsi (Institute of Molecular
Genetics, Prague, Czech Republic).
For labeling of the antibodies with fluorescent probes,
purified Abs were conjugated with succinimidyl ester of
Alexa 546 (Molecular Probes, Eugene, OR) or with
sulfoindo-cyanin succinimidyl bifunctional ester derivative
of Cy3 and Cy5 (Amersham, Braunschweig, Germany) as
we described earlier (Matko et al., 2002).
For labeling of cell-surface antigens, about 106 cells in 50
Al of PBS were incubated with fluorescently tagged label-
ing molecules for 30 min on ice in the dark. The applied
antibody concentration (50–100 Ag/ml) saturated all avail-
able binding sites. The excess mAb was removed by washing
the cells twice in PBS. Antibodies were centrifuged at
100,000 � g for 30 min before use to remove aggregated
molecules. Flow cytometric measurements were carried out
using a Becton Dickinson FACScalibur flow cytometer ac-
cording to our earlier published method (Matko et al., 2002).
For competition assays, cells were incubated with the
mixture of fluorescently conjugated mAb and unlabeled
(s4dU)35. Competition was calculated from the fractional
decrease of the fluorescence of the antibody in the presence of
the (s4dU)35.
For FRET measurements, cells were labeled simulta-
neously with the mixture of donor Cy3-labeled mAb and
with acceptor Cy5-labeled (s4dU)35. Four fluorescent
intensities were detected. Three were excited at 488 nm
and detected at 530 F 30 nm, 585 F 42 nm and above
670 nm, respectively, and the fourth was excited at 635 nm
and detected at 661 F 16 nm. Correction factors for the
spectral overlap between the different fluorescence channels
were obtained from data measured on unlabeled and single-
labeled cells. Forward and side angle light scatterings were
used for gating out debris and dead cells. We calculated the
energy transfer efficiency on a cell-by-cell basis from the
four fluorescence intensities on 10,000 cells using list mode
data (Sebestyen et al., 2002).
The calculated energy transfer efficiency was expressed
as the ratio of the number of excited donor molecules
tunneling their excitation energy to the acceptor and the
number of all excited molecules.
Confocal laser scanning microscopy analysis
The CLSM studies were performed as we have described
earlier (Sebestyen et al., 2002). Briefly, monoclonal anti-
body against cell surface thioredoxin (Pharmingen) was
labeled with sulfoindo-cyanin succinimidyl ester derivative
of Cy3 (Amersham). For labeling of cell-surface, about 106
cells in 50 Al of PBS were incubated with Cy3 and Cy5
tagged molecules for 30 min on ice, in the dark, the cells
were washed twice before analysis. The microscope was a
Zeiss, LSM 510 model.
DNase stability of (s4dU)35
Briefly, DNase stability was determined in a final volume
of 500 Al. The final reaction mixture contained 100 mMTris–
HCl (pH 8.0), 5 mMMgCl2, 1,250 Ag/ml (s4dU)35, 50 Ag/ml
phosphodiesterase I (0.16 U/mg, Sigma), 25 Ag/ml deoxy-
ribonuclease I (3000 Kunitz U/mg, Sigma) and 0.025%
NaN3. The reaction mixture was incubated at 37 8C, 40 Alaliquots were removed at the times indicated, and then the
terminal phosphate hydrolyzed by alkaline phosphatase (E.
coli A19, Amersham) and determined (Tokes and Aradi,
1996).
Toxicity of (s4dU)35 on human bone marrow
granulocyte-macrophage progenitor cells
The effect of (s4dU)35 on the colony formation of human
granulocyte-macrophage progenitor cells was determined
using published methods (Benk}oo et al., 1999). Management
of patients conformed to the Helsinki Declaration. The
protocol was approved by the Regional and Institutional
Ethics Committee of University of Debrecen, Hungary
(DEOEC RKEB/IKEB-2055/2003).
Human bone marrow samples, derived from adults,
were aspirated for diagnostic purposes and included in
these experiments only if subsequent morphological
examination failed to find any major abnormality (from
three patients). The mononuclear cell fraction was
separated by Ficoll-Iodamide gradient centrifugation
(1.077 g/ml). Methyl cellulose cultures were made by
using inocula of 2 � 105 bone marrow cells/ml in Petri
dishes and were grown in McCoy’s 5A modified medium
(GIBCO, Grand Island, NY) supplemented with amino
acids, Na pyruvate, NaHCO3 and antibiotics as described
(Benk}oo et al., 1999), using 1.2% Methyl cellulose
(Methocel, Fluka Chemie AG, Buchs). Mononuclear bone
marrow cells were grown in the presence of (s4dU)35(highest concentration: 180 Ag/ml, 15.8 AM) incubated
for 14 days at 37 8C at 100% relative humidity in an
atmosphere containing 5% CO2. Colonies, defined as
groups of at least 50 cells, were counted under a
dissecting microscope at the end of the incubation period.
Acknowledgments
This work was supported by a Hungarian Science Foun-
dation (OTKA) grant T 038163 to J.A., grant T 043061 to J.S.

A. Horvath et al. / Virology 334 (2005) 214–223222
References
Agrawal, S., Ikeuchi, T., Sun, D., Sarin, P.S., Konopka, A., Maizel, J.,
Zamecnik, P.C., 1989. Inhibition of human immunodeficiency virus in
early infected and chronically infected cells by antisense oligodeoxy-
nucleotides and their phosphorothioate analogues. Proc. Natl. Acad.
Sci. U.S.A. 86, 7790–7794.
Barbouche, R., Miquelis, R., Jones, I.M., Fenouillet, E., 2003. Protein-
disulfide isomerase-mediated reduction of two disulfide bonds of HIV
envelope glycoprotein 120 occurs post-CXCR4 binding and is required
for fusion. J. Biol. Chem. 278, 3131–3136.
Bardos, T.J., Schinazi, R.F., Ling, K.H., Heider, A.R., 1992. Structure-
activity relationships and mode of action of 5-mercapto-substituted
oligo- and polynucleotides as antitemplates inhibiting replication of
human immunodeficiency virus type 1. Antimicrob. Agents Chemother.
36, 108–114.
Benk}oo, I., Hernadi, F., Megyeri, A., Kiss, A., Somogyi, G., Tegyey, Z.,
Kraicsovits, F., Kovacs, P., 1999. Comparison of the toxicity of
fluconazole and other azole antifungal drugs to murine and human
granulocyte-macrophage progenitor cells in vitro. J. Antimicrob.
Chemother. 43, 675–681.
Buckheit, Jr., R.W., Hollingshead, M.G., Germany-Decker, J., White, E.L.,
McMahon, J.B., Allen, L.B., Ross, L.J., Decker, W.D., Westbrook, L.,
Shannon, W.M., Weislow, O., Bader, J.B., Boyd, M.R., 1993.
Thiazolobenzimidazole: biological and biochemical anti-retroviral
activity of a new nonnucleoside reverse transcriptase inhibitor. Antiviral
Res. 21, 247–265.
Buckheit, Jr., R.W., Roberson, J.L., Lackman-Smith, C., Wyatt, J.R.,
Vickers, T.A., Ecker, D.J., 1994. Potent and specific inhibition of
HIV envelope-mediated cell fusion and virus binding by G quartet-
forming oligonucleotide (ISIS 5320). AIDS Res. Hum. Retroviruses
10, 1497–1506.
Buckheit, Jr., R.W., Fliakas-Boltz, V., Decker, W.D., Roberson, J.L., Stup,
T.L., Pyle, C.A., White, E.L., McMahon, J.B., Currens, M.J., Boyd,
M.R., Bader, J.P., 1995a. Comparative anti-HIV evaluation of diverse
HIV-1-specific reverse transcriptase inhibitor-resistant virus isolates
demonstrates the existence of distinct phenotypic subgroups. Antiviral
Res. 26, 117–132.
Buckheit Jr., R.W., Kinjerski, T.L., Fliakas-Boltz, V., Russell, J.D., Stup,
T.L., Pallansch, L.A., Brouwer, W.G., Dao, D.C., Harrison, W.A.,
Schultz, R.J., Bader, J.P., Stringner, Y.S., 1995b. Structure-activity and
cross-resistance evaluation of a series of human immunodeficiency
virus type-1-specific compounds related to oxathiin carboxanilide.
Antimicrob. Agents Chemother. 39, 2718–2727.
Chang, L.C., Bewley, C.A., 2002. Potent inhibition of HIV-1 fusion by
cyanovirin-N requires only a single high affinity carbohydrate binding
site: characterization of low affinity carbohydrate binding site knockout
mutants. J. Mol. Biol. 318, 1–8.
Clanton, D.J., Moran, R.A., McMahon, J.B., Weislow, O.S., Buckheit Jr.,
R.W., Hollingshead, M.G., Ciminale, V., Pavlakis, B.K., Bader, J.P.,
1992. Sulfonic acid dyes: inhibition of the human immunodeficiency
virus and mechanism of action. J. Acquired Immune Defic. Syndr. 5,
771–781.
Cooley, L.A., Lewin, S.R., 2003. HIV-1 cell entry and advances in viral
entry inhibitor therapy. J. Clin. Virol. 26, 121–132.
De Clercq, E., 1993. HIV-1-specific RT inhibitors: highly selective
inhibitors of human immunodeficiency virus type 1 that are
specifically targeted at the viral reverse transcriptase. Med. Res. Rev.
13, 229–258.
Del Real, G., Jimenez-Baranda, S., Lacalle, R.A., Mira, E., Lucas, P.,
Gomez-Mouton, C., Carrera, A.C., Martınez-A., C., Manes, S., 2002.
Blocking of HIV-1 infection by targeting CD4 to nonraft membrane
domains. J. Exp. Med. 196, 293–301.
Dimitrov, D.S., 1997. How do viruses enter cells? The HIV coreceptors
teach us a lesson of complexity. Cell 91, 721–730.
Doms, R.W., Moore, J.P., 2000. HIV-1 membrane fusion: targets of
opportunity. J. Cell Biol. 151, F9–F13.
Eckert, D.M., Kim, P.S., 2001. Mechanisms of viral membrane fusion and
its inhibition. Annu. Rev. Biochem. 70, 777–810.
Fiser, I., Scheit, K.H., Stfffler, G., Kuechler, E., 1974. Identification of
protein S1 at the messenger RNA binding site of the Escherichia coli
ribosome. Biochem. Biophys. Res. Commun. 60, 1112–1118.
Gallina, A., Hanley, T.M., Mandel, R., Trahey, M., Broder, C.C.,
Viglianti, G.A., Ryser, H.J.-P., 2002. Inhibitors of protein-disulfide
isomerase prevent cleavage of disulfide bonds in receptor-bound
glycoprotein 120 and prevent HIV-1 entry. J. Biol. Chem. 277,
50579–50588.
Hochberg, A.A., Keren-Zur, M., 1974. The synthesis of poly-4-thiouridylic
acid and its effect on protein synthesis in a cell free system derived from
rat liver. Nucleic Acids Res. 1, 1619–1630.
Hoffman, A.D., Banapour, B., Levy, J.A., 1985. Characterization of the
AIDS-associated retrovirus reverse transcriptase and optimal conditions
for its detection in virions. Virology 147, 326–335.
Hogg, P.J., 2003. Disulfide bonds as switches for protein function. Trends
Biochem. Sci. 28, 210–214.
Horvath, A., Aradi, J., 2005. Advantages of sodium-perchlorate solution as
mobile phase for purification of synthetic oligonucleotides by anion-
exchange chromatography. Anal. Biochem. 338, 341–343.
Inagawa, T., Nakashima, H., Karwowski, B., Guga, P., Stec, W.J., Takeuchi,
H., Takaku, H., 2002. Inhibition of human immunodeficiency virus type
1 replication by P-stereodefined oligo(nucleoside phosphorothioate)s in a
long-term infection model. FEBS Lett. 528, 48–52.
Jing, N., Hogan, M.E., 1998. Structure-activity of tetrad-forming oligonu-
cleotides as a potent anti-HIV therapeutic drug. J. Biol. Chem. 273,
34992–34999.
Kilby, J.M., Eron, J.J., 2003. Novel therapies based on mechanism of HIV-1
cell entry. N. Engl. J. Med. 348, 2228–2238.
Kinchington, D., Galpin, S., Jaroszewski, J.W., Ghosh, K., Subasinghe, C.,
Cohen, J.S., 1992. A comparison of gag, pol and rev antisense
oligodeoxynucleotides as inhibitors of HIV-1. Antiviral Res. 17, 53–62.
Kuwasaki, T., Hatta, M., Takeuchi, H., Hiroshi, T., 2003. Inhibition of
human immunodeficiency virus 1 replication in vitro by a self-stabilized
oligonucleotide with 2V-O-methyl-guanosine-uridine quadruplex motifs.
J. Antimicrob. Chemother. 51, 813–819.
LaBranche, C.C., Galasso, G., Moore, J.P., Bolognesi, D.P., Hirsch, M.S.,
Hammer, S.M., 2001. HIV fusion and its inhibition. Antiviral Res. 50,
95–115.
Lipsett, M.N., 1965. The isolation of 4-thiouridylic acid from the soluble
ribonucleic acid of Escherichia coli. J. Biol. Chem. 240, 3975–3978.
Majumdar, C., Stein, C.A., Cohen, J.S., Broder, S., Wilson, S.H., 1989.
Stepwise mechanism of HIV reverse transcriptase: primer function of
phosphorothioate oligodeoxynucleotide. Biochemistry 28, 1340–1346.
Markovic, I., Stantchev, T.Z., Fields, K.H., Tiffany, L.J., Tomic, M., Weiss,
C.D., Broder, C.C., Strebel, K., Clouse, K.A., 2004. Thiol/disulfide
exchange is a pre-requisite for CXCR4-tropic HIV-1 envelope-mediated
T-cell fusion during viral entry. Blood 103, 1586–1594.
Matko, J., Bodnar, A., Vereb, G., Bene, L., Vamosi, G., Szentesi, G.,
Szfllfsi, J., Gaspar Jr., R., Horejsi, V., Waldmann, T.A., Damjanovich,
S., 2002. GPI-microdomains (membrane rafts) and signaling of the
multi-chain interleukin-2 receptor in human lymphoma/leukemia T cell
lines. Eur. J. Biochem. 269, 1199–1208.
Matsukura, M., Shinozuka, K., Zon, G., Mitsuya, H., Reitz, M., Cohen,
J.S., Broder, S., 1987. Phosphorothioate analogs of oligodeoxynucleo-
tides: inhibitors of replication and cytopathic effects of human
immunodeficiency virus. Proc. Natl. Acad. Sci. U.S.A. 84, 7706–7710.
Matthias, L.J., Hogg, P.J., 2003. Redox control on the cell surface:
implication for HIV-1 entry. Antioxid. Redox Signal. 5, 133–138.
Matthias, L.J., Yam, P.T.W., Jiang, X.-M., Vandegraaff, N., Li, P.,
Poumbourios, P., Donoghue, N., Hogg, P.J., 2002. Disulfide exchange
in domain 2 of CD4 is required for entry of HIV-1. Nat. Immunol. 3,
727–732.
Miura, K., Shiga, M., Ueda, T., 1973. Nucleosides and nucleotides: VI.
Preparation of diribonucleoside monophosphates containing 4-thiour-
idine. J. Biochem. 73, 1279–1284.

A. Horvath et al. / Virology 334 (2005) 214–223 223
Ojwang, J.O., Buckheit, R.W., Pommier, Y., Mazumder, A., De Vreese, K.,
Este, J.A., Reymen, D., Pallansch, L.A., Lackman-Smith, C., Wallace,
T.A., De Clercq, E., McGrath, M.S., Rando, R.F., 1995. T30177, an
oligonucleotide stabilized by an intramolecular guanosine octet, is a
potent inhibitor of laboratory strains and clinical isolates of human
immunodeficiency virus type 1. Antimicrob. Agents Chemother. 39,
2426–2435.
Percherancier, Y., Lagane, B., Planchenault, T., Staropoli, I., Altmeyer, R.,
Virelizier, J.-L., Arenzana-Seisdedos, F., Hoessli, D.C., Bachelerie, F.,
2003. HIV-1 entry into T-cells is not dependent on CD4 and CCR5
localization to sphingolipid-enriched, detergent-resistant, raft membrane
domains. J. Biol. Chem. 278, 3153–3161.
Popik, W., Alce, T.M., Au, W.-C., 2002. Human immunodeficiency virus
type 1 uses lipid raft-colocalized CD4 and chemokine receptors for
productive entry into CD4+ T cells. J. Virol. 76, 4709–4722.
Rice, W.G., Turpin, J.A., Huang, M., Clanton, D., Buckheit Jr., R.W.,
Civell, D.G., Wallqvist, A., McDonnell, N.B., DeGuzman, R.N.,
Summers, M.F., Zalkow, L., Bader, J.P., Haugwitz, R.D., Sausville,
E.A., 1997. Azodicarbonamide inhibits HIV-1 replication by targeting
the nucleocapsid protein. Nat. Med. 3, 341–345.
Richman, D.D., 2001. HIV chemotherapy. Nature 410, 995–1001.
Sebestyen, Z., Nagy, P., Horvath, G., Vamosi, G., Debets, R., Gratama, J.W.,
Alexander, D.R., Szfll}oosi, J., 2002. Long wavelength fluorophores and
cell-by-cell correction for autofluorescence significantly improves the
accuracy of flow cytometric energy transfer measurements on a dual-laser
benchtop flow cytometer. Cytometry 48, 124–135.
Shefter, E., Kalman, T.I., 1968. The molecular structure of the disulfide of
the t-RNA constituent, 4-thiouridine. Biochem. Biophys. Res. Com-
mun. 32, 878–884.
Simuth, J., Scheit, K.H., Gottschalk, E.M., 1970. The enzymatic synthesis
of poly 4-thiouridylic acid by polynucleotide phosphorylase from
Escherichia coli. Biochim. Biophys. Acta 204, 371–380.
Stein, C.A., Neckers, L.M., Nair, B.C., Mumbauer, S., Hoke, G., Pal,
R., 1991. Phosphorpthioate oligodeoxycytidine interferes with
binding HIV-1 gp120 to CD4. J. Acquired Immune Defic. Syndr. 4,
686–693.
Szatmari, I., T}ookes, S., Dunn, C.B., Bardos, T.J., Aradi, J., 2000.
Modified telomeric repeat amplification protocol: a quantitative
radioactive assay for telomerase without using electrophoresis. Anal.
Biochem. 282, 80–88.
T}ookes, S., Aradi, J., 1995. Inhibition of moloney murine leukemia virus
reverse transcriptase by partially 5-thiolated polyuridylic acid. Biochim.
Biophys. Acta 1261, 115–120.
T}ookes, S., Aradi, J., 1996. (s4dU)35: a novel highly potent oligonucleotide
inhibitor of the human immunodeficiency virus type 1 reverse
transcriptase. FEBS Lett. 396, 43–46.
Tsukahara, S., Suzuki, J., Hiratou, T., Takai, K., Koyanagi, Y., Yamamoto,
N., Takaku, H., 1997. Inhibition of HIV-1 replication by triple-helix-
forming phosphorothioate oligonucleotides targeted to the polypurine
tract. Biochem. Biophys. Res. Commun. 233, 742–747.
Weislow, O.S., Kiser, R., Fine, D.L., Bader, J., Shoemaker, R.H., Boyd,
M.R., 1989. New soluble-formazan assay for HIV-1 cytopathic effects:
application to high-flux screening of synthetic and natural products for
AIDS-antiviral activity. J. Natl. Cancer. Inst. 81, 577–586.
Yamaguchi, K., Papp, B., Zhang, D., Ali, A.N., Agrawal, S., Byrn, R.A.,
1997. The multiple inhibitory mechanisms of GEM 91R, a gag
antisense phosphorothioate oligonucleotide, from human immunodefi-
ciency virus type 1. AIDS Res. Hum. Retroviruses 13, 545–554.





























