Embed Size (px)
Citation preview

Nanocarriers-encapsulating phytochemicals as potent
therapeutics
in cancer therapy
A thesis submitted in fulfilment of the requirements for
the degree of
Doctor of Philosophy
Rasika Radhakrishnan
School of Science
College of Science, Engineering and Health
RMIT University, Australia

Declaration
I certify that except where due acknowledgement has been made, the work
is that of the author alone; the work has not been submitted previously, in
whole or in part, to qualify for any other academic award; the content of the
thesis is the result of work which has been carried out since the official
commencement date of the approved research program; and, any editorial
work, paid or unpaid, carried out by a third party is acknowledged; and ethics
procedures and guidelines have been followed.
Rasika Radhakrishnan

Acknowledgement
First and foremost, I would like to thank my supervisors Dr. Ravi Shukla,
RMIT, Dr. Sistla Ramakrishna at IICT, Hyderabad and Prof. Suresh
Bhargava in RMIT for giving me the opportunity to work with them for my
PhD.
I would also like to thank RMIT University and IICT, Hyderabad, India for
allowing me the use of their space and services.
It was an utmost pleasure working with Dr. Ravi. He has taught me virtues
beyond science, which are only going to strengthen me for the time to come.
My experience with him has made me realise the importance of upholding
ethics in a workplace, lessons that I will remember throughout my scientific
career. He has also made me understand the qualities that separate a bad
researcher from good ones. I will be forever indebted to you for this.
Dr. Ramakrishna graciously welcomed me into his group despite me not
being from the field. He has high ideals for living and he translates it into his
work. His hard work, resilience and immense patience are qualities I aspire
to have someday. One of the things I most respect him for is that he does not
compromise when it comes to his ideals. I will be grateful to you Sir.
Prof. Suresh is easily one of the most positive influences in my PhD tenure.
Every discussion with him has always had me feeling optimistic, even when
things are low. Continued faith and belief as a supervisor is one of the
greatest gifts that can be given to a student. He has always encouraged me to
believe in myself and never give up. He has exemplary patience and
understanding to shortcomings. Words cannot explain how grateful I am to
you, Prof. Suresh.
Next, I would like to thank my family, my father, mother and brother, each
supporting me in their own different ways, making me the person I am. They
have always been and will continue to be my pillar of strength and support.

I would like to thank my mentors Dr. Hitesh and Dr. Pooja who initiated me
into the field of pharmaceutics and have been with me throughout most of
my PhD. They have been extremely helpful, critical at times but it was
always constructive. My experience with Dr. Pooja has taught me not to
compromise on work even if things are not going your way. She is a strong
woman and I admire her for it.
I would like to thank Mrs. Nadia Zakhartchouk for her help and support in
the cell culture lab. I would also like to thank Dr. Zeyad for his assistance at
MNRF Facility. I would also like to thank Dr. Madhusudana and Dr. Halley
for helping me during animal studies. Dr. Halley was an immense support,
working with me throughout the study.
I would like to thank my colleagues and friends, Arpita, Ram, Vijay, Sudha,
Naresh, Deepthi, Halley, Suma, Tejashri, Salma, Riyaz and Vatsalya, who
had made working in the lab a fun experience. Special mentions to Arpita
and Sudha with whom I could share my thoughts, even the most abstract
ones. A special mention to Dr. Blake Plowman, who has helped me
immensely during drafting of this thesis.
I would also like to thank the lab assistants at IICT, Hyderabad; Laxman,
Chinna and Satish for their help in lab and the animal house.
I would like to honour and remember the precious beings that sacrificed their
lives during my study.
Last, but in no way least I would like to thank God, without Him I would not
be where I am. His continual presence is what keeps me motivated.
Rasika Radhakrishnan

Table of Contents
List of Figures i
List of Tables v
List of Abbreviations vi
Abstract 1
Chapter 1: Introduction
1.1. Cancer 4
1.2. Common causes of cancer 4
1.3. Conventional approaches to combat cancer 4
1.4. Chemotherapy 6
1.5. Problems with current cancer chemotherapy 8
1.6. Nanoparticles used for chemotherapy 10
1.7. Mechanisms for targeting 13
1.8. Rationale 15
1.9. Nanoparticles used in this work 17
1.9.1. Solid lipid nanoparticles 17
1.9.2. Chitosan nanoparticles 22
1.10. Drugs used in this study 25
1.10.1. Epigallocatechin gallate 25
1.10.2. Piperine 27
1.10.3. Mangiferin 27
1.11. Ligands used in this study 28

1.11.1. Bombesin 28
1.11.2. Mannose 29
1.12. Techniques used in this study 30
1.13. Aims and objectives 32
1.14. References 34
Chapter 2: Enhancement in cancer cytotoxicity of EGCG by
encapsulation within solid lipid nanoparticles
2.1. Background 49
2.2. Materials 53
2.3. Methods 53
2.3.1. Aqueous Stability 53
2.3.2. Optimization 54
2.3.3. Preparation of EGCG loaded solid lipid nanoparticles (EGCG-
SLNs)
54
2.3.4. Determination of drug entrapment efficiency 55
2.3.5. Physico-chemical characterization of nanoparticles 55
2.3.5.1. Particle size and surface potential 55
2.3.5.2. FTIR spectroscopy (FTIR) 55
2.3.5.3. Differential scanning calorimetry (DSC) 55
2.3.6. In-vitro drug release 56
2.3.7. In-vitro cytotoxicity studies 56
2.3.8. Colloidal and Serum stability studies 57

2.3.8.1. Colloidal stability of nanoparticles in serum and
physiological conditions
57
2.3.8.2. Electrolyte-induced aggregation 57
2.4. Results 58
2.4.1. Aqueous stability 58
2.4.2. Optimization of different parameters for the blank SLN 60
2.4.3. Influence of formulation parameters 60
2.4.4. Influence of process parameters 61
2.4.5. Encapsulation efficiency of EGCG-SLN 64
2.4.6. Physico-chemical characterization 64
2.4.6.1. FTIR spectroscopy (FTIR) 64
2.4.6.2. Differential scanning calorimetry (DSC) 65
2.4.7. In-vitro drug release studies 67
2.4.8. In-vitro cytotoxicity studies 68
2.4.9. Apoptosis studies 72
2.4.10. Colloidal stability studies 74
2.4.10.1. Stability of NPs in serum and physiological conditions 74
2.4.10.2. Electrolyte induced aggregation 76
2.5. Conclusion 77
2.6. References 78
Chapter 3: Peptide conjugated solid lipid nanoparticles for
breast cancer therapy
3.1. Background 87

3.2. Materials 88
3.3. Methods 89
3.3.1. Preparation of nanoparticles 89
3.3.2. Calculation of Entrapment efficiency 89
3.3.3. Bioconjugation 90
3.3.4. Physicochemical characterization 91
3.3.4.1. Particle size and surface charge 91
3.3.4.2. FTIR analysis 92
3.3.4.3. DSC analysis 92
3.3.5. In-vitro studies 92
3.3.5.1. In-vitro cytotoxicity 92
3.3.5.2. Cellular uptake 93
3.3.5.3. Apoptosis assay 93
3.3.5.4. Migration studies 93
3.3.6. Animal studies 94
3.4. Results 95
3.4.1. Particle size and surface charge 95
3.4.2. FTIR analysis 97
3.4.3. DSC analysis 99
3.4.4. In-vitro studies 99
3.4.4.1. In-vitro cytotoxicity 99
3.4.4.2. Phase contrast microscopy studies 104
3.4.4.3. Cellular uptake studies 107
3.4.4.4. Apoptosis studies 108
3.4.4.5. Migration studies 110

3.4.5. In-vivo studies 111
3.4.6. Conclusions 114
3.4.7. References 116
Chapter 4: Mangiferin and piperine encapsulated
nanoparticles for cancer therapy
4.1. Background 120
A. Mangiferin encapsulated solid lipid nanoparticles for
targeting towards cancer therapy
120
4.2. Materials 122
4.3. Methods 122
4.3.1. Preparation of nanoparticles 122
4.3.2. Determination of entrapment efficiency 123
4.3.3. Bio-conjugation 123
4.3.4. Calculation of conjugation efficiency 124
4.3.5. Physicochemical characterization 124
4.3.5.1. Particle size and surface charge 124
4.3.5.2. FTIR Analysis 124
4.3.6. In-vitro drug release 124
4.4. Results 125
4.4.1. Preparation of nanoparticles 125
4.4.2. Determination of entrapment efficiency 126
4.4.3. Bio-conjugation 127
4.4.4. Calculation of conjugation efficiency 128
4.4.5. Physicochemical characterization 128

4.4.5.1. Particle size and surface charge 128
4.4.5.2. FTIR Analysis 130
4.4.6. In-vitro drug release 131
B. Piperine encapsulated chitosan nanospheres conjugated
with mannose for glycol-targeting towards lectin receptors
overexpressed in cancer
133
4.5. Materials 135
4.6. Methods 135
4.6.1. Preparation of nanoparticles 135
4.6.2. Determination of entrapment efficiency 135
4.6.3. Bio-conjugation 136
4.6.4. Calculation of conjugation efficiency 136
4.6.5. Physico-chemical characterization 136
4.6.5.1. Particle size and surface charge 136
4.6.5.2. FTIR analysis 137
4.7. Results 137
4.7.1. Preparation of nanoparticles 137
4.7.2. Determination of entrapment efficiency 138
4.7.3. Bio-conjugation 139
4.7.4. Calculation of conjugation efficiency 140
4.7.5. Physicochemical characterization 140
4.7.5.1. Particle size and surface charge 140
4.7.5.2. FTIR analysis 142
4.8. Conclusions 143
Chapter 5: Summary and future perspectives

i
Figure
No. LIST OF FIGURES Page
No.
1.1. Different types of nanoparticles used in chemotherapy 11
1.2. Different mechanisms of targeting 14
1.3. Structure of solid lipid nanoparticle 18
1.4. Structure of chitosan polymer 22
1.5. Preparation of chitosan nanoparticles 23
1.6. Structure of epigallocatechin gallate 26
1.7. Structure of piperine 27
1.8. Structure of mangiferin 27
1.9. Structure of bombesin receptor 29
1.10. Structure of bombesin 29
1.11. Structure of D-mannose 30
2.1. Structure of epigallocatechin gallate (EGCG) 52
2.2. UV/Visible spectra of EGCG (50 µg/mL) at different time points in (a)
distilled water (b) 0.1 N HCl (c) normal saline (0.9 %w/v NaCl) (d)
phosphate buffer saline pH 7.4 (e) phosphate buffer pH 5 (f) phosphate
buffer pH 7 (g) phosphate buffer pH 9
59
2.3. Particle size distribution of a) blank solid lipid nanoparticles and b)
EGCG-loaded solid lipid nanoparticles (EGCG-SLN), measured by
dynamic light scattering
62
2.4. Fourier transform infrared (FTIR) spectra of epigallocatechin gallate
(EGCG), blank solid lipid nanoparticles (blank SLN) and EGCG -
loaded solid lipid nanoparticles (EGCG-SLN)
65
2.5. Differential scanning calorimetric analysis of epigallocatechin gallate
(EGCG) and EGCG loaded solid lipid nanoparticles (EGCG-SLN)
66
2.6. In-vitro drug release studies of EGCG loaded solid lipid nanoparticles
(EGCG-SLN) in sodium acetate buffer pH 5.
67
2.7. Percent cell viability of (a) MDA-MB-231 human breast cancer cells
and (b) DU-145 human prostate cancer cells after 24 h of treatment
with pure epigallocatechin gallate (EGCG), blank solid lipid
nanoparticles (blank SLN) and EGCG-loaded SLN (EGCG-SLN)
69

ii
2.8. Phase contrast microscopy images of (a) MDA-MB-231 human breast
cancer cells and (b) DU-145 human prostate cancer cells after 24 h of
treatment with blank solid lipid nanoparticles (blank SLN), pure
epigallocatechin gallate (EGCG), or EGCG-loaded SLN (EGCG-SLN)
equivalent to 20μg/mL EGCG.
71
2.9. Nuclear fragmentation studies: Florescent microscopic images of (a)
MDA-MB-231 human breast cancer cells and (b) human prostate
cancer cells after 24 h of treatment with blank solid lipid
nanoparticles (blank SLN), pure epigallocatechin gallate (EGCG), or
EGCG-loaded SLN (EGCG-SLN) equivalent to 20 μg/mL EGCG
followed by staining with Hoechst 33342
73
2.10. Change in (a) particle size and (b) zeta potential of EGCG-loaded
solid lipid nanoparticles (EGCG-SLN) with respect to time.
75
2.11. Effect of flocculating agent (sucrose sulphate) on the stability of
EGCG loaded solid lipid nanoparticles
76
3.1. Structure of bombesin 81
3.2. Mechanism of EDC-NHS reaction 90
3.3. Particle size distribution of a) blank solid lipid nanoparticles (Blank-
SLN) b) EGCG-loaded solid lipid nanoparticles (EGCG-SLN) and
Bombesin conjugated solid lipid nanoparticles (EB-SLN), measured
by dynamic light scattering
96
3.4. Fourier transform infrared (FTIR) spectra of epigallocatechin gallate
(EGCG), blank solid lipid nanoparticles (Blank SLN), EGCG -loaded
solid lipid nanoparticles (EGCG-SLN) and Bombesin conjugated
solid lipid nanoparticles (EB-SLN)
98
3.5. Differential scanning calorimetric analysis of epigallocatechin gallate
(EGCG) and EGCG loaded solid lipid nanoparticles (EGCG-SLN)
99
3.6. Percent cell viability of MDA-MB-231 human breast cancer after 24
h of treatment with pure EGCG, EGCG-loaded SLN (EGCG-SLN)
and bombesin-conjugated EGCG-SLN (EB-SLN)
102

iii
3.7. Percent cell viability of DU145 prostate cancer cells after 24 h of
treatment with pure EGCG, EGCG-loaded SLN (EGCG-SLN) and
bombesin-conjugated EGCG-SLN (EB-SLN).
102
3.8. Percent cell viability of A549 prostate cancer cells after 24 h of
treatment with pure EGCG, EGCG-loaded SLN (EGCG-SLN) and
bombesin-conjugated EGCG-SLN (EB-SLN).
103
3.9. Percent cell viability of HEK-293 kidney cells after 24 h of treatment
with pure EGCG, EGCG-loaded SLN (EGCG-SLN) and bombesin-
conjugated EGCG-SLN (EB-SLN).
103
3.10. Percent cell viability of L-132 lung cells after 24 h of treatment with
pure EGCG, EGCG-loaded SLN (EGCG-SLN) and bombesin-
conjugated EGCG-SLN (EB-SLN).
104
3.11. Phase contrast microscopy images of MDA-MB-231 human breast
cancer after 24 h and 48 h of treatment with blank solid lipid
nanoparticles (blank SLN), pure epigallocatechin gallate (EGCG),
EGCG-loaded SLN (EGCG-SLN) and bombesin conjugated SLN
(EB-SLN) equivalent to 20 μg/mL EGCG.
106
3.12. Cellular uptake studies: Florescent microscopic images of MDA-MB-
231 human breast cancer cells after 24 h of treatment with Rhodamine
loaded SLN (R-SLN) and bombesin conjugated SLN (RB-SLN).
108
3.13. Nuclear fragmentation studies: Florescent microscopic images of
MDA-MB-231 human breast cancer cells after 24 h of treatment with
blank solid lipid nanoparticles (blank SLN), pure epigallocatechin
gallate (EGCG), EGCG-loaded SLN (EGCG-SLN) and bombesin
conjugated SLN (EB-SLN) equivalent to 20 μg/mL EGCG followed
by staining with Hoechst 33342.
110
3.14. Scratch assay images for MDA-MB-231 human breast cancer cells
after 24 h of treatment with blank solid lipid nanoparticles (blank
SLN), pure epigallocatechin gallate (EGCG), EGCG-loaded SLN
(EGCG-SLN) and bombesin conjugated SLN (EB-SLN) equivalent to
20 μg/mL EGCG along with control cells for reference
111
3.15. Kaplan-Meier plot for survival following administration of pure
epigallocatechin gallate (EGCG), EGCG-loaded SLN (EGCG-SLN)
113

iv
and bombesin conjugated SLN (EB-SLN) formulations with control
mice for reference
3.16 Change in body weight and tumour volume in mice with the number
of days following administration of pure epigallocatechin gallate
(EGCG), EGCG-loaded SLN (EGCG-SLN) and bombesin conjugated
SLN (EB-SLN) formulations with control mice for reference
114
4.1. Structure of Mangiferin 121
4.2. Calibration curve of Mangiferin 126
4.3. Particle Size distribution for blank formulation (Blank-SLN),
Mangiferin-loaded SLN (MgF-SLN) and bombesin-conjugated BM-
SLN formulations measured by dynamic light scattering
129
4.4. Fourier transform infrared (FTIR) spectra for blank formulation
(Blank-SLN), Mangiferin-loaded SLN (MgF-SLN), Mangiferin and
bombesin-conjugated BM-SLN formulations
131
4.5. In vitro drug release studies of Mangiferin-loaded SLN (MgF-SLN)
in sodium acetate buffer pH 5 and phosphate buffer (pH 7.4)
132
4.6. Structure of Piperine 133
4.7. Calibration curve of piperine 139
4.8. Particle Size distribution for blank formulation (B-NP), piperine-
loaded nanoparticle (P-NP) and mannose-conjugated (MP-NP)
formulations measured by dynamic light scattering
142
4.9. Fourier transform infrared (FTIR) spectra for blank formulation (B-
NP), Piperine-loaded chitosan formulation (P-NP), Piperine and
mannose-conjugated chitosan (MP-NP) formulations
143

v
Table
No. LIST OF TABLES Page
No.
2.1 Optimization of formulation parameters for preparation of solid
lipid nanoparticles by double-emulsification method
63
2.2. Optimization of encapsulation of epigallocatechin gallate (EGCG)
in solid lipid nanoparticles
64
2.3. IC50 values (µg/mL) for epigallocatechin gallate (EGCG) and
EGCG-loaded solid lipid nanoparticles (EGCG-SLN) against
MDA-MB-231 human breast cancer cells and DU-145 human
prostate cancer cells after 24 h of treatment
70
3.1. Particle size and surface charge for blank nanoparticles (Blank-
SLN), EGCG-loaded SLN (EGCG-SLN) and bombesin
conjugated SLN (EB-SLN) determined by DLS
96
3.2. IC50 values (µg/mL) for epigallocatechin gallate (EGCG), EGCG-
loaded solid lipid nanoparticles (EGCG-SLN) and bombesin
conjugated SLN (EB-SLN) against MDA-MB-231, DU-145,
A549, HEK-293 and L-132 cells after 24 h of treatment
101
4.1. Optimization of formulation parameters for mangiferin-loaded
solid lipid nanoparticles prepared by single-emulsification method
126
4.2. Optimization of encapsulation of mangiferin and entrapment
efficiency in Mangiferin-loaded SLN (MgF-SLN)
127
4.3 Particle Size distribution, polydispersity and zeta potential values
for blank formulation (Blank-SLN), mangiferin-loaded SLN
(MgF-SLN) and bombesin-conjugated BM-SLN formulations
determined by dynamic light scattering
129
4.4. Optimization of formulation parameters for piperine loaded-
chitosan nanoparticles (P-NP) prepared by ionic gelation method
138
4.5. Optimization of encapsulation of piperine and entrapment
efficiency in piperine loaded chitosan nanoparticles (P-NP)
139
4.6. Particle Size distribution, polydispersity and zeta potential values
for blank formulation (B-NP), piperine-loaded nanoparticle (P-
NP) and mannose-conjugated (MP-NP) formulations determined
by dynamic light scattering
141
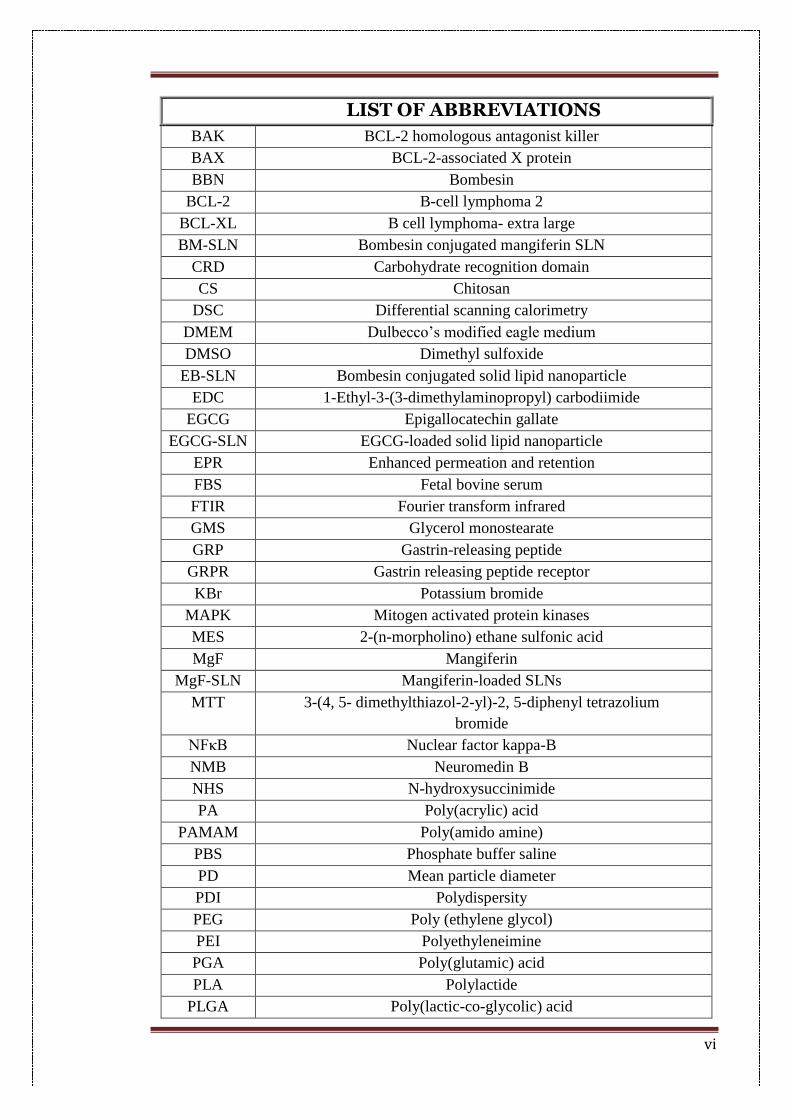
vi
LIST OF ABBREVIATIONS
BAK BCL-2 homologous antagonist killer
BAX BCL-2-associated X protein
BBN Bombesin
BCL-2 B-cell lymphoma 2
BCL-XL B cell lymphoma- extra large
BM-SLN Bombesin conjugated mangiferin SLN
CRD Carbohydrate recognition domain
CS Chitosan
DSC Differential scanning calorimetry
DMEM Dulbecco’s modified eagle medium
DMSO Dimethyl sulfoxide
EB-SLN Bombesin conjugated solid lipid nanoparticle
EDC 1-Ethyl-3-(3-dimethylaminopropyl) carbodiimide
EGCG Epigallocatechin gallate
EGCG-SLN EGCG-loaded solid lipid nanoparticle
EPR Enhanced permeation and retention
FBS Fetal bovine serum
FTIR Fourier transform infrared
GMS Glycerol monostearate
GRP Gastrin-releasing peptide
GRPR Gastrin releasing peptide receptor
KBr Potassium bromide
MAPK Mitogen activated protein kinases
MES 2-(n-morpholino) ethane sulfonic acid
MgF Mangiferin
MgF-SLN Mangiferin-loaded SLNs
MTT
3-(4, 5- dimethylthiazol-2-yl)-2, 5-diphenyl tetrazolium
bromide
NFκB Nuclear factor kappa-B
NMB Neuromedin B
NHS N-hydroxysuccinimide
PA Poly(acrylic) acid
PAMAM Poly(amido amine)
PBS Phosphate buffer saline
PD Mean particle diameter
PDI Polydispersity
PEG Poly (ethylene glycol)
PEI Polyethyleneimine
PGA Poly(glutamic) acid
PLA Polylactide
PLGA Poly(lactic-co-glycolic) acid

vii
PVA Polyvinyl alcohol
RES Reticulo-endothelial system
ROS Reactive oxygen species
RPMI Roswell Park Memorial Institute 1640 medium
SA Stearic acid
SAB Sodium acetate buffer
SD Standard deviation
SFC Supercritical fluid
SLN Solid lipid nanoparticles
TAAs Tumour-associated antigens
VEGF Vascular endothelial growth factor

1
Abstract
Cancer is a disease concerning the abnormal, uncontrolled growth of cells that possess a
potential to invade and injure other parts of the body. Chemotherapy involves
administration of drugs targeted towards the mechanisms responsible for this rapid
division and proliferation. However, most chemotherapy drugs are non-specific, also
causing allied toxicity to normal cells undergoing cell division.
In this work, phytochemicals were used as anti-cancer drugs. These molecules are non-
toxic and biocompatible. However, they also display striking anti-cancer potential, which
was explored in this thesis. In this work, epigallocatechin gallate, piperine and mangiferin
have been explored for their anti-cancer efficacy.
Although phytochemicals are effective agents in cancer chemotherapy, many of them
suffer from disadvantages such as pH instability and limited bioavailability in the body.
To address this problem, nanocarriers were designed to facilitate their increased
bioavailability and stability inside the body. Two nanoparticle matrices were explored in
this work; solid lipid nanoparticles and chitosan nanoparticles. Solid lipid nanoparticles
were used as they are biocompatible, help improve drug stability and show controlled
release. Chitosan nanoparticles are biodegradable, biocompatible and have shown
excellent absorption properties for chemotherapeutic drugs.
Chemotherapy mainly deals with two targeting approaches; active and passive. While
passive targeting relies on the characteristics of the tumour for effective therapy, active
targeting involves attachment of ligands on the nanocarrier for more specific targeting.
Both the targeting approaches have been explored in this work. In addition, two targeting
ligands were used in this work; bombesin and mannose, which are specific for factors
overexpressed in cancer.

2
In summary, this thesis involves the design and fabrication of nanocarriers in order to
increase the anti-cancer efficacy of phytochemicals, utilising different targeting
approaches, matrix components and targeting ligands. The anti-cancer efficacy was
evaluated by in-vitro cytotoxicity studies and in-vivo animal studies. The results validate
the increased anti-cancer efficacy provided by the encapsulation of the drugs within
nanocarrier systems thus offering encouraging, novel possibilities in the field of cancer
therapy.

3

4
1.1. Cancer
Cancer remains one of the world’s most crippling diseases, accounting for about 1 in
every 7 deaths worldwide – more than HIV/AIDS, tuberculosis, and malaria combined[1].
It persists as an unsolved enigma for modern medicine, a cure for which has been sought
since times immemorial. The word 'cancer' is derived from the Greek word for crab
(karkinos), indicating the crab-claw like patterns of veins surrounding the tumour.
Cancerous tissues are characterized by the abnormal growth of cells beyond their usual
boundaries invading adjoining parts of the body and/or spreading to other organs[2].
Lung, prostate, colorectal, stomach and liver cancers are the most common types of
cancer in men, while breast, colorectal, lung, cervix and stomach cancers are most
common among women[3]. Although cancer-related mortality has decreased owing to
better understanding of tumour biology and improved diagnostic devices and treatments,
further advances in this field are necessary to understand the basis of disease progression,
which will in turn lead to better therapy[4].
1.2. Common causes of cancer
Cancer involves the irregular and abnormal growth of cells, perpetuating beyond their
usual lifespan. This diseased condition can be triggered by a number of factors, with the
most well-known being excessive smoking and use of tobacco, exposure to carcinogens,
inadequate diet and physical activity, exposure to radiation, overall genetics, viral and
other infections[3]. However, other factors such as the sex and age of the patient also
influence the development of certain cancers[5].
1.3. Conventional approaches to combat cancer
Current approaches for cancer treatment include surgery, chemotherapy, radiation
therapy, immuno-therapy and gene therapy, as outlined in the following sections:

5
1.3.1. Surgery
Surgery is the preferred method for the excision of solid tumours which are mostly
contained to one area[6]. Excision can be complete or partial (debulking), depending on
the magnitude of expansion of the cancerous tissue, wherein complete removal is
attempted when the tumour is restricted to one area or organ while debulking is carried
out when complete resection might damage an organ or functioning of the body as a
whole[7]. While surgical resection is often a straightforward and plausible line of
treatment, it suffers from the disadvantage of not promising complete removal of
cancerous tissue in cases where metastasis may have taken place, but is yet dormant or
undetected[8]. It also cannot be used for treatment of leukemias.
1.3.2. Radiation therapy
Radiation therapy uses high energy radiations such as gamma rays and X-rays to damage
the DNA in cancerous cells[9]. It is of two types, external beam therapy and internal
radiation therapy. External beam therapy involves the use of a linear accelerator for the
production of X-rays. Internal radiation involves the implantation of a solid or liquid
radioactive source inside the body for the emission of radiations.
1.3.3. Chemotherapy
Chemotherapy involves the administration of an anti-cancer drug (or multiple drugs) as a
part of a regimen for either curative or palliative treatment[10]. It can be used as a
neoadjuvant, wherein chemotherapeutics are administered before radiation therapy or
surgery to reduce the tumour volume or sensitize it for more efficient therapy[8]. It is also
used in tandem with other procedures as an adjuvant in a combinatorial chemotherapeutic
regimen[11].

6
1.3.4. Immunotherapy
Cancer immunotherapy involves stimulating the immune system through the active or
passive targeting against tumour cells[12]. It uses genetically modified cells and viral
particles to stimulate the immune system to destroy cancer cells. Tumour cells express
tumour-associated antigens (TAAs) which can be attacked by the immune system[13].
Active immunotherapy helps the immune system to better detect the TAAs and hence
assist in their removal. In passive immunotherapy, monoclonal antibodies, lymphocytes
and cytokines are used to enhance anti-tumour responses.
1.3.5. Gene therapy
Gene therapy involves the use of genes (in-vitro or in-vivo) to manipulate cancer
cells[14]. However, ineffective delivery of the therapeutic gene to the target cells and
multiple precautions to be taken to ensure the therapeutic gene does not integrate into
unwanted cells, are a few challenges that gene therapy has to circumvent[15].
1.4. Chemotherapy
Chemotherapy is the use of anti-cancer drugs to destroy cancer cells, alone or in
combination with other drugs and sensitizers(As mentioned in Section 1.3)[16]. It mainly
deals with targeting the mechanisms responsible for conferring immortality to cancer
cells. The main types of chemotherapeutic drugs used are as follows[1]:
1.4.1. Alkylating agents
This class includes alkyl group containing synthetic compounds interfering in different
phases of the tumour cell division process, preventing it from further replication. The
alkyl groups react to form irreversible covalent linkages with the various components of
the genetic assembly, such as DNA, RNA or proteins, halting cell division and hence
proliferation[17]. They do suffer from the disadvantage of non-specificity wherein
replicating non-cancerous cells may also be damaged[18]. However, defective DNA

7
repair mechanisms in cancer cells may lead to higher mortality rates in comparison to
normal cells. Cisplatin, oxaliplatin and cyclophosphamide are common examples of
alkylating agents.
1.4.2. Anti-metabolites
Anti-metabolites are modified analogs of purines, pyrimidines, nucleotides and
nucleosides, which are components of the genetic assembly. The similarity of these
analogs to genetic components causes them to be picked up during DNA or RNA strand
synthesis. However irregularity in the strand structure during assimilation of these altered
elements in DNA/RNA strand creates problems in the recoiling and coiling process
during cell division which results in effective cytostatis[19]. Examples include
gemcitabine, 5-fluoro uracil and methotrexate.
1.4.3. Anti-tumour antibiotics
Anti-tumour antibiotics are fermentation products of microbial cultures which are used in
cancer therapeutics. They work by varied mechanisms. Actinomycin and mithromycin
bind to the DNA and inhibit the DNA-dependent RNA synthesis while the anthracyclines
(doxorubicin and daunorubicin) intercalate between base pairs. Bleomycin causes DNA
strand scission whereas mitomycin C carries out DNA alkylation. The common
mechanism of action throughout these agents is their reaction with DNA to interfere in
the DNA replication and division process[20].
1.4.4. Topoisomerase inhibitors
Topoisomerases are enzymes necessary for the supercoiling and relaxation of DNA
strands during the process of cell division. Topoisomerase inhibitors interfere with the
functioning of these enzymes[21,22]. They are classified depending on the enzyme they
inhibit as Topoisomerase I inhibitors, such as irinotecan and topotecan and
Topoisomerase II inhibitors such as etoposide and daunorubicin.

8
1.4.5. Mitotic inhibitors
These agents commonly act by interrupting cell mitosis through the inhibition of
microtubule polymerization which is an indispensable step during cell division[23]. They
are generally derived from plant alkaloids. Common examples include paclitaxel,
docetaxel, vinblastine and vincristine.
1.4.6. Corticosteroids
Corticosteroids are primarily cholesterol-derived hormones of the adrenal cortex. Their
mechanism of action involves multiple signalling processes to induce cell apoptosis in
tumour cells[24]. Common examples include dexamethasone and prednisone.
1.5. Problems with current cancer chemotherapy
Although these enlisted chemotherapeutic drugs have been effective via various
mechanisms against tumour tissue, there are a few drawbacks associated with their use
which are mentioned in the following section.
1.5.1. Non-specificity
Chemotherapy strategies kill cells by targeting important pathways in rapid cell divisions
characteristic of tumourous cells. However, as these therapies lack specificity, they also
affect cellular processes in normal cells undergoing cell division, thus causing allied non-
specific cytotoxicity[25]. Many singular therapies involving one chemotherapeutic drug
therefore have to be co-administered with agents to prevent or alleviate associated non-
specific cytotoxicity[26]. This causes overburdening of the amount of foreign substances
increasing the chances of the reticulo-endothelial system to eliminate the therapeutic
agent[27].
1.5.2. Multi-drug resistance
Development of multi-drug resistance in cancer cells is one of the leading causes for
failure of cancer chemotherapy. Resistance can be effected by numerous mechanisms

9
such as increased drug efflux, activation of DNA repair mechanisms, produce detoxifying
systems and evasion of drug-induced apoptosis[28]. Multi-drug resistance eventually
causes an increase in drug dosages for beneficial therapy as the cells do not take up the
drug in the required amounts. Also, this can increase dose-dependent toxicity at other
sites.
1.5.3. Biological barriers
The human body contains a number of biological and biophysical barriers such as the
blood brain barrier (BBB), cells of the reticulo-endothelial system (RES) and endothelial
cell junctions. The BBB is comprised of tight junctions between epithelial cells, halting
extravasation of chemotherapeutic agents. The BBB also has high specificity in the size
and solubility characteristics of the molecules passing through it. The endothelial cells
and cells of the RES act as immunological barriers by effectively hoarding therapeutic
agents[29]. This causes an inability to deliver therapeutic drugs at the target site and
increased drug dosages.
1.5.4. In-vivo degradation
The metabolic enzymes within the human body, such as proteases, lipases, hydrolases
etc. can rapidly degrade chemotherapy drugs by chemical reactions with their susceptible
reactive groups. This may cause reduction in activity, if the altered functional groups are
important for the therapeutic activity. This causes a lack of significant therapy at the
prescribed dosages and hence, a subsequent increase in drug administration [30].
1.5.5. Poor solubility and stability
Drugs used in cancer treatment are divided into two types based on their solubilities as
hydrophilic and hydrophobic. While hydrophilic drugs are immediately assimilated in the
blood stream if the drug administration route is intravenous, hydrophobic drugs are not
easily soluble and hence require greater therapeutic concentrations to be as effective.

10
Also, certain drugs rapidly deteriorate under certain pH environs[31]. This is a major
challenge as the human body contains many pH conditions for example the physiological
pH of blood is 7.4 while that of a cancer environment is more acidic at around pH 5. This
creates problems in delivering drugs at specific sites.
Nanoparticles have been used as vectors to help protect and deliver anti-cancer drugs by
attempting to circumvent these problems. They are described in greater detail in the
following section.
1.6. Nanoparticles used for chemotherapy
Nanoparticles employed in chemotherapy are typically materials in the size range of 1-
1000 nm, capable of delivering therapeutics in or around the tumour
microenvironment[32]. The small size of nanoparticles allows them to accumulate in
tumour tissues due to the increased permeability and confinement of tumour tissue as a
result of the enhanced permeability and retention (EPR) effect. A size between 10-200
nm is the most preferable size range for anti-cancer drug delivery as particles smaller than
6 nm can be excreted by the kidney, and those larger than 300 nm can be rapidly
recognized and removed by the reticulo-endothelial system (RES)[33]. Therefore,
nanoparticles with this size range are circulated more in the blood after intravenous
administration which provides more opportunity to accumulate in tumour tissues. Over
the years, different types of nanoparticles have been constructed with the view to develop
a fool-proof system for cancer treatment (Refer Fig 1.1.)[34]. Herein, the types of
nanoparticle that have been used for cancer therapy have been discussed.

11
Fig 1.1: Different types of nanoparticles used in chemotherapy (Reproduced with permission from
Masserini et al. 2013)
1.6.1. Polymeric nanoparticles
Polymeric nanoparticles are composed of polymer and co-polymer matrices in the form
of nanospheres and nanovesicles[35]. Nanospheres are matrix systems, wherein the
nanoparticle core is solid and drug molecules may be adsorbed at the sphere surface or
encapsulated within the particle. Nanocapsules are vesicular systems where the entrapped
substances are confined to a cavity consisting of a liquid core (either oil or water)
surrounded by a solid material shell[36]. Commonly used polymeric materials are
poly(lactic-co-glycolic acid) (PLGA), polylactide (PLA), polyethylene glycol (PEG),
polyglutamic acid (PGA), polyethyleneimine (PEI), polyacrylic acid (PA), etc.
1.6.2. Lipid based nanocarriers
Commonly used lipid based nanoparticles include liposomes, nanostructured lipid
carriers and solid lipid nanoparticles. These nanoparticles are favoured more for in-vivo
applications as the lipid matrix is predominantly constructed using physiological lipids

12
which are endogenous to, and hence can be easily degraded within, the cellular milieu
after drug delivery[37]. Commonly used lipid matrix constituents include triglycerides,
fatty acids and phospholipids such as phosphodylcholine and phosphoethanolamine.
1.6.3. Inorganic nanoparticles
Inorganic nanoparticles have demonstrated commendable success in in-vivo and in-vitro
cancer therapies and imaging due to their unique optical-electronic properties. Quantum
dots, carbon nanotubes, gold nanoparticles (spheres, shells, rods and cages), iron oxide
magnetic nanoparticles and ceramic nanoparticles have been employed in tumour
targeting, imaging, photothermal therapy and drug delivery applications[38]. While they
do suffer from several drawbacks such as non-biodegradability and non-biocompatibility,
considerable advances have been made to address their cytotoxicity and give them stealth-
like properties[39].
1.6.4. Carbohydrate-based nanoparticles
Carbohydrate-based nanoparticles have received considerable interest as drug carriers
due to their natural origin and inherent biodegradability and biocompatibility[40].
Commonly used carbohydrates include cyclodextrins, chitosan, amylose, hyaluronic acid
and heparin[41].
1.6.5. Protein-based nanoparticles
Protein-based nanoparticles also display biocompatibility and biodegradability[42].
However, these nanoparticles may show immunogenicity. Abraxane, a human serum
albumin based nanoparticle formulation, has been successfully introduced in the market.
Other proteins explored for nanoparticle synthesis include gelatin, legumin and gliadin.
1.6.6. Dendrimers
Dendrimers are synthetic organic compounds with spherical 3D structural morphology
showing a branched structure. Features like nanoscopic size, narrow polydispersity index,

13
control over molecular structure, the presence of interior cavities and high surface
functionalities makes them attractive candidates for delivery in biological
applications[43]. Commonly used matrix polymers include polypropylene imine,
polyethyleneimine and polyamidoamine (PAMAM). However, they do suffer from
inherent toxicity due to the synthetic nature of the matrix which also makes them more
susceptible to expulsion by the reticulo-endothelial system[44].
1.7. Mechanisms for targeting
Nanoparticle-mediated chemotherapy relies on two types of delivery strategies; active
and passive (Fig. 1.2). Passively-targeted nanoparticle assemblies contain the therapeutic
load but do not have a targeting moiety fabricated in them. This passive targeting relies
on the enhanced permeation and retention (EPR) effect, distinctive of tumour vasculature,
for efficient therapy[45]. The EPR effect accounts for the increased vascular permeability
due to interendothelial gap defects, allowing extravasation of nanoparticles up to 400 nm
in diameter, and also poor lymphatic drainage in the tumour microenvironment aiding the
accumulation of and further release from nanoparticles[46,47].
Passively targeted nanocarriers first reached clinical trials in the mid-1980s, wherein few
liposomes and polymer–protein conjugates based products were marketed in the mid-
1990s[35]. Passive targeting however holds several limitations as the non-specific nature
of the process does not warrant a complete payload delivery at the side of the tumour.
Also certain drugs do not diffuse efficiently inside the tumours and furthermore certain
tumours do not exhibit the EPR effect which makes unmitigated therapy improbable[48].

14
Fig 1.2: Different mechanisms of targeting: Passive tissue targeting achieved by the increased
permeation and retention in tumours. Active cellular targeting (inset) can be achieved by
functionalizing the surface of nanoparticles with ligands that promote cell-specific recognition and
binding. (Reproduced with permission from Peer et al. 2007)
To overcome this limitation, specific molecules can be attached on the surface of the
nanocarrier which are specific to a receptor or protein overexpressed on tumour cells and
can therefore serve as markers for effective ligand binding. These surface modified
nanoparticles will get internalized into the target cells through ligand–receptor
interactions. Targeting ligands such as proteins (antibodies and antibody fragments),
peptides, vitamins, carbohydrates and nucleic acids (aptamers) have been widely explored
for this purpose[49].
However, targeted delivery does have limitations such as the receptor capacity, tumour
volume, cancer cell heterogeneity and differences in overexpression of cancer
markers[50].

15
The density of the surface target receptors plays an important role in determining doses
and dosage regimens. If the density of the targeting receptor is less, any excess targeting
nanocarriers will not have available binding surfaces and thus will be removed[45].
If the targeting agent has a very high affinity for the receptor they can get trapped within
and extravasate in the part of the tumour surrounding the capillaries, and consequently
they do not diffuse throughout the tumour, especially in the case of large solid tumours,
hence recurrence of cancer is more likely, even after sufficient therapeutic dosage
administration[51].
The expression of a particular receptor itself may vary within the same tumour owing to
the high frequency of mutations which are characteristic of cancer cell divisions[52].
Therapies which rely on targeting overexpressed receptors in cancer do not consider the
fact that although these receptors are overexpressed in cancer cells as compared to non-
cancerous cells, the number of non-cancerous cells in the body is far more than the
number of tumorous cells and thus targeting could be considered futile.
With the advantages and disadvantages of the targeting approaches in mind, the work in
this thesis explored both approaches, the rationale for which is explained in the following
section.
1.8. Rationale
Many of the chemotherapy drugs used today target the cellular processes such as
increased cell multiplication which are proven cancer markers as cancerous cells require
a greater amount of energy to account for the increased cell division, metastasis and
angiogenesis[52]. Although these processes are amplified in cancer cells, they are not
exclusive to them alone. Hence, normal cells undergoing mitosis and cell division can
also be affected leading to simultaneous non-specific toxicity and cell death at areas not
intended for action by the drugs[25]. This can in turn lead to several undesirable effects

16
such as hair loss, suppression of bone marrow, multi-drug resistance, neurological
dysfunction and cardiac toxicity[53].
Phytochemicals with potent anti-cancer activity have been used as the drugs of choice in
this thesis. The potential advantage of using phytochemicals is that they have shown
considerable specificity for cancer cells, have a better tolerance within a living system as
they are components of a regular diet and hence will not be harmful in case of non-specific
delivery[54].
Epigallocatechin gallate (EGCG) has shown potent chemopreventive and
chemotherapeutic activity in many cancer cell lines the mechanisms of activity also being
diverse[55–57]. Piperine, one of the most pharmacologically active components of black
pepper, has also shown anti-cancer, anti-inflammatory and anti-oxidant activity[58,59].
Mangiferin from Mangifera indica has also shown a wide range of therapeutic properties
as an anti-diabetic, anti-obesity and anti-cancer agent[60–62].
Two kinds of nanovehicles have been used in this study; solid lipid nanoparticles which
have a matrix made of physiological lipids that are non-toxic. Solid lipid nanoparticles
have a number of advantages including an improvement in drug stability and drug
entrapment, biocompatibility and efficient scale up[63]. Chitosan nanoparticles can also
be easily broken down within the mammalian cell, and thus do not show the accumulation
induced toxicity associated with many other conventional nanoparticle systems such as
the ones with inorganic metals as matrix[64].
Ligands have been employed in two of the projects to confer additional specificity to the
delivery vehicles. One of these ligands is bombesin, which is a tetradecapeptide that is a
natural homolog to the gastrin-releasing peptide receptor which is commonly
overexpressed in many types of cancers such as breast, prostate, ovarian, lung and liver
cancers, and it is therefore an excellent cancer biomarker[65]. The second ligand,

17
mannose, was investigated as a ligand for targeting in cancer as it is a natural homolog to
lectin receptors which are commonly overexpressed in breast, liver, prostate and brain
cancers[66]. A further advantage of ligand conjugation is that it can reduce the toxicity
associated with nanoparticle delivery as the cytotoxic drug will only be delivered to the
cells where it is aimed at and not non-tumorous cells[67].
Prior to investigating their anti-cancer properties, the physico-chemical natures of the
nanoparticle systems were well characterised in order to ensure their suitability for
delivery within living systems. The mean particle size of the nanoparticles was
characterised using dynamic light scattering technique (DLS). To study effective
encapsulation and conjugation differential scanning calorimetry (DSC) and Fourier
transform infrared analysis (FTIR) were used. The in-vitro anti-cancer activity was
checked by assays in cancer cell lines. Uptake and migration assays were carried out to
check the efficiency of the nanoparticle system to be taken up by the cells and suppress
proliferation, respectively.
Thus, different combinations of delivery vehicles (lipid and carbohydrate based),
different approaches for delivery (active and passive) and different targeting ligands
(protein and carbohydrate-based) have been explored in this work.
1.9. Nanoparticles used in this work
1.9.1. Solid lipid nanoparticles (SLNs)

18
Fig 1.3: Structure of an SLN (Reproduced with permission from Khatak et al. 2004.)
Solid lipid nanoparticles (SLNs) are submicron sized colloidal nanocarriers prepared
from physiological lipids which are solid at room temperature[68]. These nanoparticles
have shown an improvement in drug stability and drug entrapment, are biocompatible (as
they are composed of easily biodegradable lipids) and can be efficiently scaled up for
production[69]. The most important advantage of SLNs is that they aid in the controlled,
gradual release of the drug, thus improving the retention time and effectiveness of the
drug while preventing premature degradation[70,71].
SLNs can be prepared as a variety of emulsions. The oil-in-water emulsion (O/W) is
effective to encapsulate hydrophobic drugs that can be dissolved in the inner oil phase
while the water-in-oil emulsion (W/O) emulsion can be used to encapsulate hydrophilic
drugs in the inner aqueous core. In addition, a water-in-oil-in-water (W/O/W) double
emulsion can be used to encapsulate drugs which are sensitive to pH changes. The many
methods for preparation of SLNs are enlisted below:
1.9.1.1. Homogenization
This method of preparation involves the dispersion of the organic phase with the aqueous
phase by the use of strong cavitation forces provided by homogenizers to help in
formation of submicron sized nanoparticles[72]. Homogenisation is of two types:

19
1.9.1.1.1. Hot homogenization
Hot homogenization involves the heating of both the aqueous and organic phases to the
same temperature followed by homogenisation to form a stable emulsion[73]. The
temperature used is higher than the melting point of the lipids used, hence the organic
phase exists as a lipid melt. Also, the higher temperature helps in the formation of smaller
droplets and hence smaller nanoparticle sizes[74].
This method is useful for heat resistant drugs which are not affected by the increase in
temperature during heating. However, care must be taken as the temperature can increase
during homogenisation due to the mechanical stress caused by the cavitation. Hence this
method is not suitable for thermo-labile drugs or those susceptible to structural damage.
1.9.1.1.2. Cold homogenization
This method is employed for encapsulating temperature or shear sensitive drugs where
hot homogenization cannot be used for preparation. The drug is initially dissolved in the
lipid melt to form a uniform organic phase. The melt is then rapidly cooled using liquid
nitrogen to form a solid lipid. Then solid lipid is milled with a mortar to form
microparticles, which are dispersed in an aqueous surfactant solution then into the
aqueous phase followed by homogenization. This method typically gives larger particles
compared to hot homogenization, however the extent of milling of the solid lipid greatly
affects the size of the final nanoparticles.
1.9.1.2. Solvent evaporation and emulsification
In this method, the lipids are initially dissolved in a water-immiscible solvent to form a
continuous organic phase[75]. This organic phase is then homogenized into an aqueous
phase containing surfactants to form a stable emulsion. The formed emulsion is now
subjected to solvent evaporation by stirring, wherein the solvent is removed and the lipids
gradually precipitate into the aqueous phase as nanoparticles. However, if the

20
nanoparticles are to be used for biomedical purposes care must be taken while choosing
the solvents, as there could be additional toxicity induced by the use of toxic solvents[76].
1.9.1.3. Microemulsion
Microemulsions are formed by mixing a hot homogenous lipid phase into a greater
volume of cold aqueous phase[77]. A few factors such as the velocity of addition and the
lipophilicity of the solvents used must be considered for determining particle size. The
faster the distribution in aqueous phase, better the polydispersity of the nanoparticle
suspension. Also, more lipophilic solvents yield bigger particle sizes[78]. This could
again be correlated to the easier distribution in the aqueous phase.
1.9.1.4. Solvent emulsification diffusion technique
This method makes use of a solvent partially miscible with water. Initially, both the water
and the solvent are mixed till the system reaches saturation and are in thermodynamic
equilibrium with each other[79]. Following this, the drugs and the lipids are dissolved in
the water saturated solvent phase. The dissolution is carried out at a higher temperature if
the lipids used have a higher melting temperature. The surfactant phase is made by
saturating the surfactant containing aqueous phase with the solvent. The lipid phase is
now emulsified into the surfactant containing aqueous phase to form a primary emulsion.
After this, more water is added to the system to allow the solvent to diffuse into the
continuous phase and thereby causing the lipid to precipitate in a nanoparticulate form.
The diffused solvent is now eliminated through distillation.
1.9.1.5. Hot melt technique
This method involves melting of the solid lipids to form a hot lipid melt. The drug is
dissolved in lipid melt by vigorous mixing and then emulsified in an aqueous phase that
is heated above the melting temperature of the lipids[80]. The dispersion is then cooled
down to produce nanoparticles.

21
1.9.1.6. Ultrasonication
In this method, the lipid phase is initially melted and the drug is dissolved in this hot
phase[81]. The aqueous phase is then heated to the same temperature and added to the
lipid phase dropwise with ultrasonication. Dispersion occurs due to the strong cavitation
forces supplied by the ultrasonicator. This method does not involve the use of organic
solvents and hence, is preferred for nanoparticle applications due to its minimal toxicity.
1.9.1.7. Solvent injection method
The principle of this method involves lipid precipitation from the dissolved lipid into the
aqueous solution while the lipid is initially dissolved in a water miscible solvent[82]. The
lipid-solvent mixture is then slowly added with an injection needle into the aqueous phase
with or without surfactants. The mixing step is performed while the aqueous phase is
under stirring, to maintain homogeneity and uniform particle size.
1.9.1.8. Supercritical fluid technique
A fluid is called supercritical when its pressure and temperature exceed their critical
value. Beyond the supercritical point, the ability of a fluid to dissolve substances is found
to increase. CO2 has been routinely used as the gas for producing super critical fluid (SFC)
in this method as it is inexpensive, non-inflammable and inert to the drugs. Initially, the
drug is dissolved in the solvent and this drug-solvent mixture is allowed to dissolve in the
SFC. The SFC acts as an anti-solvent, wherein it reduces the solubility of solid in the
solution and therefore the solid precipitates as nanoparticles[83].
1.9.1.9. Double emulsification method
This is a two-step process, wherein in the first step, the drug (hydrophilic) is dissolved in
an aqueous solvent (inner aqueous phase) and then dispersed in a lipid containing an
emulsifier (e.g. lecithin), known as oil phase, to produce a primary emulsion (w/o). A

22
double emulsion (w/o/w) is formed after addition of aqueous solution of the hydrophilic
emulsifier (e.g. Poloxamer, PVA) followed by stirring to evaporate organic solvent[84].
Double emulsification avoids the use of high temperatures to melt the lipid for the
preparation of protein and drug-loaded lipid nanoparticles and there is a scope for surface
modification of the nanoparticles to provide steric stability by using a lipid-PEG
derivative. Steric stabilization significantly improves the stability of these colloidal
systems against pH and electrolyte concentration changes in the gastrointestinal fluids.
1.9.2. Chitosan nanoparticles
Fig 1.4: Structure of chitosan polymer (Reproduced with permission from the Chemistry Glossary)
Chitosan (CS) is a linear copolymer of β-(1–4)-linked D-glucosamine and N-acetyl-D-
glucosamine whose molecular structure comprises a linear backbone linked through
glycosidic bonds[85]. The deacetylated chitosan backbone of glucosamine units has a
high density of basic amine groups which are protonated and thus positively charged in
most physiological fluids[86].
CS is considered hydrophilic, but the percentage of acetylated monomers and their
distribution in the chains has a critical effect on its solubility and conformation in aqueous
media. Because of these molecular features, CS exhibits a pH-dependent behaviour and
interesting biopharmaceutical properties such as muco-adhesiveness and the ability to
open epithelial tight junctions[87].

23
The presence of amino (-NH2) and hydroxyl (-OH) groups in chitosan provides
opportunity for a high degree of chemical modification. Chitosan is an interesting
polymer to prepare nanoparticles, as it is biocompatible, nontoxic, biodegradable and has
shown absorption-enhancing effects[87]. Due to the availability of free amino groups, it
carries a positive charge under physiological conditions and reacts with many negatively
charged surfaces such as the cell membrane. CS is insoluble in water and organic solvents,
however, it is soluble in dilute aqueous acidic solution (pH<6.5), which can convert the
glucosamine units into a soluble form of protonated amine (R–NH3+)[88]. CS is also
known to precipitate in alkaline solutions or with polyanions and form a gel at a lower
pH[89].
Fig 1.5: Preparation of chitosan nanoparticles (Reproduced with permission from Mukhopadhyaya
et al.)

24
Chitosan nanoparticles can be prepared by many methods such as the following:
I. Ionotropic gelation
Ionic cross linking is based on electrostatic interaction to form chitosan nanoparticles
using a negatively charged cross linker such as a triphosphate which can make ionic bonds
with positively charged chitosan molecules, thus forming cross linked polymers. This
method has been widely used to encapsulate proteins and drugs as it avoids the use of
strenuous temperature conditions and organic solvents[90].
II. Covalent conjugation:
Covalent cross linking involves formation of covalent bonds between chitosan chains and
cross linking agents such as glutaraldehydes and polyethylene glycol which helps in the
covalent linking of the chitosan chains[85]. Nanoparticles with a narrow size distribution
have been produced by this method.
III. Precipitation
Precipitation is carried out in two ways. In desolvation, a flocculating agent is added to
an aqueous chitosan solution. The decrease in solubility due to the addition of the
flocculant causes chitosan to precipitate out by the formation of hydrogen bonds. Solvent
emulsification involves the addition of an organic phase containing a drug into the
aqueous chitosan solution. Emulsifiers may be added to reduce the surface tension
between both phases. The interaction of the two phases creates turbulence which results
in of precipitation of chitosan nanoparticles[91].
IV. Radical polymerization
Radical polymerization involves the use of acids and strong conditions such as high
temperatures[92]. Chitosan is dissolved in dilute nitric acid solution and then stirred with
ceric ammonium nitrate, nitric acid and isobutyl cyanoacrylate at 40 °C for 40 minutes in

25
an inert nitrogen environment. This method can be fine-tuned for core and shell properties
by using different monomers.
V. Complex formation
Due to the positive charge on the chitosan chain, it can be used to form ionic complexes
with anionic drugs[93]. Ionic complexes have been formed with retinol encapsulated in
chitosan by this method with a particle size of around 102.6 ± 12.0 nm. Encapsulation of
retinol in chitosan incidentally caused a 1,600-fold increase in its solubility.
VI. Spray drying
This method involves spray drying of chitosan along with another metal oxide that it can
be chelated with. Iron oxide and chitosan nanoparticles have been prepared by this
method[94]. It is a one-step simple method for the preparation of chitosan nanoparticles.
VII. Polyelectrolyte formation
This method involves charge neutralisation of the cationic polymer by use of a negatively
charged anionic component such as a DNA molecule to form a polyelectrolytic
complex[95]. The preparatory method is comparatively mild and requires no
sophisticated instrumentation. A negatively charged anionic solution can be added slowly
to the chitosan dissolved in acetic acid under stirring and slow addition to give chitosan
nanoparticles.
1.10. Drugs used in this study
1.10.1. Epigallocatechin gallate (EGCG)

26
Fig 1.6: Structure of EGCG
IUPAC name: [(2R,3R)-5,7-dihydroxy-2-(3,4,5-trihydroxyphenyl) chroman-3-yl] 3,4,5-
trihydroxybenzoate
Molecular weight: 458.372 g/mol
Solubility: Water, ethanol
Epigallocatechin gallate is one of the most abundant catechins found in green tea. It has
shown anti-diabetic, anti-obesity and anti-cancer potential[96–99]. It has shown to
downregulate mRNA and protein expression of MMP-2 matrix metalloproteinases, which
play an important role in the degradation of the basement membrane, angiogenesis and
metastasis, in MCF-7 breast cancer cell line[100]. It induces prostate cancer cell death by
suppressing androgen receptor acetylation in androgen dependent prostate cancer cell line
LNCaP [101]. EGCG has also shown microRNA targeting potential by upregulating miR-
210 microRNA leading to reduced proliferation and anchorage-independent growth in
H1299 and H460 lung cancer cells[102]. It induces apoptosis in HT-29 colon cancer cells
by upregulation of caspase-3 activity and mitochondrial damage [103]. The structure
consists of three aromatic rings with eight free hydroxyl groups (Fig 1.6). Much of the
beneficial activities of EGCG are linked to its structure. However, the free hydroxyl
groups are susceptible to easy reactions within the biological system by enzymes leading
to an overall loss in activity.

27
1.10.2. Piperine
Fig 1.7: Structure of piperine
IUPAC name: (2E,4E)-5-(1,3-Benzodioxol-5-yl)-1-(1-piperidinyl)-2,4-pentadien-1-on
Molecular weight: 285.338 g/mol
Solubility: Chloroform, ether, acetic acid
Piperine accounts for 5-9% by weight of the components in black pepper. It has been
known as a prominent anti-oxidant, anti-inflammatory and more recently an anti-cancer
agent[104,105].The anti-cancer effect of piperine may be attributed to the inhibition of
NF-κB, c-Fos, ATF-2 and CREB activities, suppression of angiogenesis by inhibiting Akt
phosphorylation, or blockade of the production of pro-inflammatory cytokines and the
activity of matrix metalloproteinases that are likely to promote tumour growth and
metastasis[104].
1.10.3. Mangiferin
Fig 1.8: Structure of mangiferin

28
IUPAC Name: (1S)-1,5-Anhydro-1-(1,3,6,7-tetrahydroxy-9-oxo-9H-xanthen-2-yl)-D-
glucitol
Molecular weight: 422.34 g/mol
Solubility: ethanol, DMSO
Mangiferin, obtained from the bark, leaves and pulp of Mangifera indica, is a polyphenol
used in ancient Indian Ayurvedic and Chinese medicine. The structure consists of a C-
glycosidic derivative attached to a xanthenone. It has shown anti-oxidant, anti-diabetic,
anti-allergic, anti-HIV and anti-cancer activities[60].
Mangiferin exhibits its anti-cancer activity by downregulating inflammation, arresting
cell cycle, reducing proliferation/metastasis, promoting apoptosis in malignant cells and
also providing protection against oxidative stress and DNA damage[106]. A study has
shown that mangiferin reduced the tumour volumes similar to that of conventional
anticancer drugs such as Cisplatin[107]. However, the drug shows low bioavailability
which can be addressed by the use of appropriate delivery vehicles[60].
1.11. Ligands used in the study
1.11.1. Bombesin
Bombesin receptors are a type of G-protein coupled receptors and have 4 subtypes- BB1,
BB2, BB3 and BB4. BB1 is known as the neuromedin B (NMB) receptor because of its
affinity towards mammalian ligand NMB. The BB2 receptor with high affinity to
mammalian gastrin releasing peptide (GRP) is called the GRP receptor[65].
Reubi et al. investigated 161 cancer cell lines, out of which 12 prostate cancer cell lines,
41 breast cancer cell lines, 5 gastrinomas and 3 of 9 small cell lung carcinomas expressed
predominantly GRP receptors; 11 of 24 intestinal, 1 of 26 bronchial, and 1 of 1 thymic
carcinoids had preferentially NMB receptors whereas 9 of 26 bronchial carcinoids, 1 large

29
cell neuroendocrine lung carcinoma, and 4 of 9 small cell lung carcinomas had
preferentially BB3 receptors[108].
Fig 1.9: Structure of Bombesin receptor BB2 (Reproduced with permissions from Atlas of Genetics
and Cytogenetics in Oncology and Haematology)
GRPR is a transmembrane glycoprotein with 7 transmembrane domains consisting of 384
amino acids. Bombesin is a tetradecapeptide originally isolated from the toad Bombina
bombina. GRPR has shown to bind bombesin with very high affinity, internalising via
receptor-mediated endocytosis[109].
Fig 1.10: Structure of bombesin
1.11.2. Mannose
C-type lectins are a group of carbohydrate-binding membrane proteins, commonly
functioning as components in cell adhesion, glycoprotein turnover, pathogen recognition
and apoptosis[12]. Most of them contain an extracellular carbohydrate recognition

30
domain (CRD) that helps in the detection of free carbohydrates or in complexed forms
like glycoproteins or glycolipids. Internalization of carbohydrates takes place through
clathrin-mediated endocytosis. C-type lectins are commonly overexpressed in a variety
of cancers such as liver, lung and ovarian cancers. Mannose is an aldohexose sugar (an
epimer of glucose), which is a common ligand to C-type lectin receptors. Hence, mannose
was used as the ligand for lectin targeting. Glyco-targeting is gaining importance as these
targeting ligands are not foreign to the cellular system and have negligible toxicity, thus
preventing expulsion by the RES system[66].
Fig 1.11: Structure of D-mannose (Reproduced with permission from the Medical Biochemistry)
1.12. Techniques used in the study
A. Dynamic light scattering
Dynamic light scattering technique can help measure particle size as small as 1 nm in
diameter. The sample is illuminated with a laser beam. The scattered light is detected by
a photon detector. Analysis of the fluctuation of the scattered light provides the diffusion
coefficient D which can be used to calculate radius of the particle R by the Stokes-Einstein
Equation:
𝐷 =𝑘𝐵T
6πηR
Where kB is the Boltzmann-constant, T is the temperature and η is the viscosity.
The zeta potential indicates the surface charge on a particle, and therefore provides
information about the electrostatic repulsion with adjacent particles and thus its stability.

31
Theoretically, it is the electric potential at the slipping planes between the Stern layer
surrounding the particle and the diffused layer in contact with the dispersion
medium[110]. Thus, the greater the surface charge, the higher the repulsion with similar
particles, and hence the greater its resistance towards aggregation.
In addition to measuring the particle size DLS provides information about the
polydispersity of the sample. Polydispersity measures the heterogeneity or non-
uniformity of a sample between index values of 0 and 1. The more heterogeneous a
sample is, the greater will be the value of the polydispersity index.
B. Fourier transform infrared (FTIR) analysis
FTIR technique helps in obtaining different vibrational frequencies of molecular
interactions within a sample. When a sample is irradiated with infra-red radiation,
absorbed IR radiation excites molecules into a higher vibrational state. Bonds between
different heteroatoms give rise to distinct vibrational frequencies to give an infrared
spectrum which can be used to determine the nature of chemical interactions between
drug molecules, excipients and matrix of nanoparticles. A detector measures the intensity
of transmitted or absorbed light as a function of its wavelength.
The FTIR spectra are represented as plots of intensity versus wavenumber (which is the
reciprocal of the wavelength and is given in units of cm-1), with the intensity plotted as
the percentage of light transmittance or absorbance at each wavenumber.
C. Differential scanning calorimetry (DSC)
DSC is a thermo-analytical technique which evaluates heat changes during phase
transitions and chemical reactions as a function of temperature. In this technique, the
sample and reference are heated to the same temperature in hermetically sealed pans in
an inert environment. The DSC thermogram is a curve of heat flux versus temperature or

32
time. This curve can be used to calculate enthalpies of transitions obtained by integrating
the peak corresponding to a given transition using the given equation:
ΔH = KA
where ΔH is the enthalpy of transition, K is the calorimetric constant, and A is the area
under the curve.
During phase transitions, energy is either absorbed or released. If it is an endothermic
process such as protein denaturation and reduction reactions more heat must be supplied
to the sample for it to remain at the same temperature as the reference. Therefore, the
enthalpy will be positive. In an exothermic reaction, such as crystallization, less heat
needs to be provided to the sample hence the enthalpy change of that of the sample relative
to the reference is negative. DSC was used in the study for the determination of the phase
transition of the drugs post encapsulation inside the nanoparticles.
1.13. Aims and objectives
Aims of this research work:
The main aim of this thesis work was to develop and evaluate various nanoformulations
for the delivery of phytochemicals to enhance their stability and cytotoxicity.
Major Objectives:
➢ To develop different nanocarriers and nano-carrier combinations for
delivery of phytochemicals as anti-cancer drugs.
➢ To improve therapeutic efficacy of phytochemicals by providing a stable
nanocarrier system for delivery
These research aims were achieved by the use of the following methodogies:
➢ To optimize formulation and process variables for the development of
biocompatible nanoparticles.

33
➢ To characterize nanoformulations using different analytical techniques such
as UV-Visible spectroscopy, dynamic light scattering, Fourier transform infrared
and differential scanning calorimetry techniques.
➢ Bioconjugation of a targeting ligand on the surface of drug loaded
nanoparticles to prepare targeted nanoparticles.
➢ To study the release patterns of encapsulated phytochemicals from
nanoparticle systems.
➢ Evaluation of in vitro cytotoxicity of targeted and non-targeted, drug-
encapsulated nanoparticles in comparison to pure drug.
➢ To study the cellular uptake of nanoparticles by using fluorescent dye-
loaded nanoparticles.
➢ To study the in-vivo efficacy of targeted formulations by using suitable
animal model
➢ To study the in-vivo efficacy of targeted formulations by survival studies
and changes in tumour volume and body weight.

34
1.14. References
[1] American Cancer Society, Cancer Facts & Figures, 2016.
doi:10.3322/caac.20121.
[2] WHO | Cancer, (n.d.). http://www.who.int/mediacentre/factsheets/fs297/en/
(accessed December 29, 2015).
[3] WHO | Cancer, WHO. (2017). http://www.who.int/cancer/en/ (accessed March 25,
2017).
[4] A. Urruticoechea, R. Alemany, J. Balart, A. Villanueva, F. Viñals, G. Capellá,
Recent Advances in Cancer Therapy : An Overview, (2010) 3–10.
[5] Worldwide cancer incidence statistics | Cancer Research UK, (n.d.).
http://www.cancerresearchuk.org/content/worldwide-cancer-incidence-statistics
(accessed January 4, 2016).
[6] Surgery for Cancer- National Cancer Institute, (n.d.).
https://www.cancer.gov/about-cancer/treatment/types/surgery (accessed August 5,
2017).
[7] R. Rami-Porta, C. Wittekind, P. Goldstraw, et International Association for the
Study of Lung Cancer (IASLC) Staging Committee, K. Tsuboshima, N. Tsubota, et al.,
Complete resection in lung cancer surgery: proposed definition., Lung Cancer. 49 (2005)
25–33. doi:10.1016/j.lungcan.2005.01.001.
[8] M.D. Walker, Chemotherapy: adjuvant to surgery and radiation therapy., Semin.
Oncol. 2 (1975) 69–72. http://www.ncbi.nlm.nih.gov/pubmed/137527 (accessed June 13,
2017).
[9] M. Ichel, B. Olla, D. Ionisio, G. Onzalez, P. Adraig, W. Arde, et al., Improved
survival with radiotherapy and goserelin in locally advanced prostate cancer improved
survival in patients with locally advanced prostate cancer treated with radiotherapy and

35
goserelin,337(n.d.).http://www.nejm.org.ezproxy.lib.rmit.edu.au/doi/pdf/10.1056/NEJM
199707313370502 (accessed June 13, 2017).
[10] How Chemotherapy Drugs Work,
(n.d.).https://www.cancer.org/treatment/treatments-and-side-effects/treatment-
types/chemotherapy/how-chemotherapy-drugs-work.html (accessed March 25, 2017).
[11] W. Yu, I. Whang, A. Averbach, D. Chang, P.H. Sugarbaker, Morbidity and
mortality of early postoperative intraperitoneal chemotherapy as adjuvant therapy for
gastric cancer, Am. J. Surg. 64 (1998) 1104–1108.
[12] K. Drickamer, M.E. Taylor, Recent insights into structures and functions of C-type
lectins in the immune system, Curr. Opin. Struct. Biol. 34 (2015) 26–34.
doi:10.1016/j.sbi.2015.06.003.
[13] R.A.B. Wood, A.R. Moossa, The prospective evaluation of tumour-associated
antigens for the early diagnosis of pancreatic cancer, Br. J. Surg. 64 (1977) 718–720.
doi:10.1002/bjs.1800641009.
[14] R. Mulligan, The basic science of gene therapy, Science (80-. ). 260 (1993) 926–
932. doi:10.1126/science.8493530.
[15] D. Cross, J.K. Burmester, Gene therapy for cancer treatment: past, present and
future., Clin. Med. Res. 4 (2006) 218–27. doi:10.3121/CMR.4.3.218.
[16] Chemotherapy- Cancer Council Australia, (n.d.). http://www.cancer.org.au/about-
cancer/treatment/chemotherapy.html (accessed March 25, 2017).
[17] G.P. Wheeler, Studies Related to the Mechanisms of Action of Cytotoxic
Alkylating Agents: A Review, Cancer Res. 22 (1962).
http://cancerres.aacrjournals.org/content/22/6/651.short (accessed June 13, 2017).
[18] R. Saffhill, G. Margison, P. Oconnor, Mechanisms of carcinogenesis induced by
alkylating agents, Biochim. Biophys. Acta - Rev. Cancer. 823 (1985) 111–145.

36
doi:10.1016/0304-419X(85)90009-5.
[19] G.J. Peters, C.L. van der Wilt, C.J.A. van Moorsel, J.R. Kroep, A.M. Bergman, S.P.
Ackland, Basis for effective combination cancer chemotherapy with antimetabolites,
Pharmacol. Ther. 87 (2000) 227–253. doi:10.1016/S0163-7258(00)00086-3.
[20] G.P. Sartiano, W.E. Lynch, W.D. Bullington, Mechanism of action of the
anthracycline anti-tumor antibiotics, doxorubicin, daunomycin and rubidazone:
Preferential inhibition of DNA polymerase .ALPHA.., J. Antibiot. (Tokyo). 32 (1979)
1038–1045. doi:10.7164/antibiotics.32.1038.
[21] Y. Pommier, P. Pourquier, Y. Fan, D. Strumberg, Mechanism of action of
eukaryotic DNA topoisomerase I and drugs targeted to the enzyme, Biochim. Biophys.
Acta - Gene Struct. Expr. 1400 (1998) 83–106. doi:10.1016/S0167-4781(98)00129-8.
[22] D.A. Burden, N. Osheroff, Mechanism of action of eukaryotic topoisomerase II and
drugs targeted to the enzyme, Biochim. Biophys. Acta - Gene Struct. Expr. 1400 (1998)
139–154. doi:10.1016/S0167-4781(98)00132-8.
[23] N. Jiang, X. Wang, Y. Yang, W. Dai, Advances in Mitotic Inhibitors for Cancer
Treatment, Mini-Reviews Med. Chem. 6 (2006) 885–895.
doi:10.2174/138955706777934955.
[24] S. Watanabe, E. Bruera, Corticosteroids as adjuvant analgesics, J. Pain Symptom
Manage. 9 (1994) 442–445. doi:10.1016/0885-3924(94)90200-3.
[25] J.-J. Monsuez, J.-C. Charniot, N. Vignat, J.-Y. Artigou, Cardiac side-effects of
cancer chemotherapy., Int. J. Cardiol. 144 (2010) 3–15. doi:10.1016/j.ijcard.2010.03.003.
[26] V. Pannu, P. Karna, H.K. Sajja, D. Shukla, R. Aneja, Synergistic antimicrotubule
therapy for prostate cancer, Biochem. Pharmacol. 81 (2011) 478–487.
doi:10.1016/j.bcp.2010.11.006.
[27] L.M. Kaminskas, B.J. Boyd, Nanosized Drug Delivery Vectors and the

37
Reticuloendothelial System, in: Springer, Dordrecht, 2011: pp. 155–178.
doi:10.1007/978-94-007-1248-5_6.
[28] J.-P. Gillet, M.M. Gottesman, Mechanisms of Multidrug Resistance in Cancer, in:
Methods Mol. Biol., 2010: pp. 47–76. doi:10.1007/978-1-60761-416-6_4.
[29] M. Ferrari, Frontiers in cancer nanomedicine: directing mass transport through
biological barriers, Trends Biotechnol. 28 (2010) 181–188.
doi:10.1016/j.tibtech.2009.12.007.
[30] Q.Y. Zhu, A. Zhang, D. Tsang, Y. Huang, Z.-Y. Chen, Stability of Green Tea
Catechins, J. Agric. Food Chem. 45 (1997) 4624–4628. doi:10.1021/jf9706080.
[31] H. Singh, R. Bhandari, I.P. Kaur, Encapsulation of Rifampicin in a solid lipid
nanoparticulate system to limit its degradation and interaction with Isoniazid at acidic pH,
Int. J. Pharm. 446 (2013) 106–111. doi:10.1016/j.ijpharm.2013.02.012.
[32] I. Brigger, C. Dubernet, P. Couvreur, Nanoparticles in cancer therapy and
diagnosis, Adv. Drug Deliv. Rev. 54 (2002) 631–651. doi:10.1016/S0169-
409X(02)00044-3.
[33] M. Longmire, P.L. Choyke, H. Kobayashi, Clearance properties of nano-sized
particles and molecules as imaging agents: considerations and caveats, Nanomedicine. 3
(2008) 703–717. doi:10.2217/17435889.3.5.703.
[34] M. Ferrari, Cancer nanotechnology: opportunities and challenges, Nat. Rev.
Cancer. 5 (2005) 161–171. doi:10.1038/nrc1566.
[35] C. Errico, A. Dessy, A. Piras, F. Chiellini, Polymeric Nanoparticles for Targeted
Delivery of Bioactive Agents and Drugs, in: Polym.
Biomater., CRC Press, 2013: pp. 593–616. doi:doi:10.1201/b13757-18.
[36] F. Masood, Polymeric nanoparticles for targeted drug delivery system for cancer
therapy, Mater. Sci. Eng. C. 60 (2016) 569–578. doi:10.1016/j.msec.2015.11.067.

38
[37] K. Manjunath, J.S. Reddy, V. Venkateswarlu, Solid lipid nanoparticles as drug
delivery systems, Methods Find. Exp. Clin. Pharmacol. 27 (2005) 127.
doi:10.1358/mf.2005.27.2.876286.
[38] T.A. Pecorelli, M.M. Dibrell, Z. Li, C.R. Thomas, J.I. Zink, Multifunctional
inorganic nanoparticles for imaging, targeting, and drug delivery, in: S. Achilefu, R.
Raghavachari (Eds.), 2010: p. 75760K. doi:10.1117/12.841168.
[39] K.G. Neoh, E.T. Kang, Functionalization of inorganic nanoparticles with polymers
for stealth biomedical applications, Polym. Chem. 2 (2011) 747–759.
doi:10.1039/C0PY00266F.
[40] M. Marradi, I. Garc?a, S. Penad?s, Carbohydrate-Based Nanoparticles for Potential
Applications in Medicine, in: 2011: pp. 141–173. doi:10.1016/B978-0-12-416020-
0.00004-8.
[41] C.A. Peptu, L. Ochiuz, L. Alupei, C. Peptu, M. Popa, Carbohydrate based
nanoparticles for drug delivery across biological barriers., J. Biomed. Nanotechnol. 10
(2014) 2107–48. http://www.ncbi.nlm.nih.gov/pubmed/25992451 (accessed March 25,
2017).
[42] A. MaHam, Z. Tang, H. Wu, J. Wang, Y. Lin, Protein-Based Nanomedicine
Platforms for Drug Delivery, Small. 5 (2009) 1706–1721. doi:10.1002/smll.200801602.
[43] P. Kesharwani, K. Jain, N.K. Jain, Dendrimer as nanocarrier for drug delivery,
Prog. Polym. Sci. 39 (2014) 268–307. doi:10.1016/j.progpolymsci.2013.07.005.
[44] L.M. Kaminskas, B.J. Boyd, Nanosized Drug Delivery Vectors and the
Reticuloendothelial System, in: A. Prokop (Ed.), Intracell. Deliv. Fundam. Appl.,
Springer Netherlands, Dordrecht, 2011: pp. 155–178. doi:10.1007/978-94-007-1248-5_6.
[45] E. Ruoslahti, S.N. Bhatia, M.J. Sailor, Targeting of drugs and nanoparticles to
tumors, J. Cell Biol. 188 (2010). http://jcb.rupress.org/content/188/6/759 (accessed June

39
13, 2017).
[46] M. Gaumet, A. Vargas, R. Gurny, F. Delie, Nanoparticles for drug delivery: The
need for precision in reporting particle size parameters, Eur. J. Pharm. Biopharm. 69
(2008) 1–9. doi:10.1016/j.ejpb.2007.08.001.
[47] H. Maeda, The enhanced permeability and retention (EPR) effect in tumor
vasculature: the key role of tumor-selective macromolecular drug targeting, Adv. Enzyme
Regul. 41 (2001) 189–207. doi:10.1016/S0065-2571(00)00013-3.
[48] R.K. Jain, Barriers to Drug Delivery in Solid Tumors, Sci. Am. 271 (1994) 58–65.
doi:10.1038/scientificamerican0794-58.
[49] L. Brannon-Peppas, J.O. Blanchette, Nanoparticle and targeted systems for cancer
therapy, Adv. Drug Deliv. Rev. 64 (2012) 206–212. doi:10.1016/j.addr.2012.09.033.
[50] K.S. Shohdy, A.S. Alfaar, Nanoparticles targeting mechanisms in cancer therapy:
current limitations and emerging solutions, Ther. Deliv. 4 (2013) 1197–1209.
doi:10.4155/tde.13.75.
[51] Y. Fukumori, H. Ichikawa, Nanoparticles for cancer therapy and diagnosis, Adv.
Powder Technol. 17 (2006) 1–28. doi:10.1163/156855206775123494.
[52] L.A. Loeb, K.R. Loeb, J.P. Anderson, Multiple mutations and cancer., Proc. Natl.
Acad. Sci. U. S. A. 100 (2003) 776–81. doi:10.1073/pnas.0334858100.
[53] A. Coates, S. Abraham, S.B. Kaye, T. Sowerbutts, C. Frewin, R.M. Fox, et al., On
the receiving end - patient perception of the side-effects of cancer chemotherapy, Eur. J.
Cancer Clin. Oncol. 19 (1983) 203–8. doi:10.1016/0277-5379(83)90418-2.
[54] H. Sakagami, Apoptosis-inducing activity and tumor-specificity of antitumor
agents against oral squamous cell carcinoma, Jpn. Dent. Sci. Rev. 46 (2010) 173–187.
doi:10.1016/j.jdsr.2010.01.004.
[55] C. Braicu, C.D. Gherman, A. Irimie, I. Berindan-Neagoe, Epigallocatechin-3-

40
Gallate (EGCG) Inhibits Cell Proliferation and Migratory Behaviour of Triple Negative
Breast Cancer Cells, J. Nanosci. Nanotechnol. 13 (2013) 632–637.
doi:10.1166/jnn.2013.6882.
[56] Y.D. Jung, M.S. Kim, B.A. Shin, K.O. Chay, B.W. Ahn, W. Liu, et al., EGCG, a
major component of green tea, inhibits tumour growth by inhibiting VEGF induction in
human colon carcinoma cells., Br. J. Cancer. 84 (2001) 844–50.
doi:10.1054/bjoc.2000.1691.
[57] Y. Xu, C.-T. Ho, S.G. Amin, C. Han, F.-L. Chung, Inhibition of Tobacco-specific
Nitrosamine-induced Lung Tumorigenesis in A/J Mice by Green Tea and Its Major
Polyphenol as Antioxidants, Cancer Res. 52 (1992) 3875–3879.
http://cancerres.aacrjournals.org/content/52/14/3875.short (accessed December 30,
2015).
[58] L. Lai, Q. Fu, Y. Liu, K. Jiang, Q. Guo, Q. Chen, et al., Piperine suppresses tumor
growth and metastasis in vitro and in vivo in a 4T1 murine breast cancer model, Acta
Pharmacol. Sin. 33 (2012) 523–530. doi:10.1038/aps.2011.209.
[59] C.R. Pradeep, G. Kuttan, Effect of piperine on the inhibition of lung metastasis
induced B16F-10 melanoma cells in mice, Clin. Exp. Metastasis. 19 (2002) 703–708.
doi:10.1023/A:1021398601388.
[60] P. Rajendran, T. Rengarajan, N. Nandakumar, H. Divya, I. Nishigaki, Mangiferin
in cancer chemoprevention and treatment: pharmacokinetics and molecular targets, J.
Recept. Signal Transduct. 35 (2015) 76–84. doi:10.3109/10799893.2014.931431.
[61] H. Li, J. Huang, B. Yang, T. Xiang, X. Yin, W. Peng, et al., Mangiferin exerts
antitumor activity in breast cancer cells by regulating matrix metalloproteinases,
epithelial to mesenchymal transition, and β-catenin signaling pathway, Toxicol. Appl.
Pharmacol. 272 (2013) 180–190. doi:10.1016/j.taap.2013.05.011.

41
[62] R.K. Khurana, R. Kaur, S. Lohan, K.K. Singh, B. Singh, Mangiferin: a promising
anticancer bioactive, Pharm. Pat. Anal. 5 (2016) 169–181. doi:10.4155/ppa-2016-0003.
[63] A. zur Mühlen, C. Schwarz, W. Mehnert, Solid lipid nanoparticles (SLN) for
controlled drug delivery – Drug release and release mechanism, Eur. J. Pharm. Biopharm.
45 (1998) 149–155. doi:10.1016/S0939-6411(97)00150-1.
[64] S.J. Soenen, P. Rivera-Gil, J.-M. Montenegro, W.J. Parak, S.C. De Smedt, K.
Braeckmans, Cellular toxicity of inorganic nanoparticles: Common aspects and
guidelines for improved nanotoxicity evaluation, Nano Today. 6 (2011) 446–465.
doi:10.1016/j.nantod.2011.08.001.
[65] J. Zhou, J. Chen, M. Mokotoff, R. Zhong, L.D. Shultz, E.D. Ball,
Bombesin/Gastrin-Releasing Peptide Receptor: A Potential Target for Antibody-
Mediated Therapy of Small Cell Lung Cancer, (n.d.).
http://clincancerres.aacrjournals.org/content/clincanres/9/13/4953.full.pdf (accessed
March 25, 2017).
[66] H. Ghazarian, B. Idoni, S.B. Oppenheimer, A glycobiology review: Carbohydrates,
lectins and implications in cancer therapeutics, Acta Histochem. 113 (2011) 236–247.
doi:10.1016/j.acthis.2010.02.004.
[67] M. Srinivasarao, C. V. Galliford, P.S. Low, Principles in the design of ligand-
targeted cancer therapeutics and imaging agents, Nat. Rev. Drug Discov. 14 (2015) 1–17.
doi:10.1038/nrd4519.
[68] W. Mehnert, Solid lipid nanoparticles Production, characterization and
applications, Adv. Drug Deliv. Rev. 47 (2001) 165–196. doi:10.1016/S0169-
409X(01)00105-3.
[69] S. Weber, A. Zimmer, J. Pardeike, Solid Lipid Nanoparticles (SLN) and
Nanostructured Lipid Carriers (NLC) for pulmonary application: a review of the state of

42
the art., Eur. J. Pharm. Biopharm. 86 (2014) 7–22. doi:10.1016/j.ejpb.2013.08.013.
[70] C. Qi, Y. Chen, Q.-Z. Jing, X.-G. Wang, Preparation and characterization of
catalase-loaded solid lipid nanoparticles protecting enzyme against proteolysis., Int. J.
Mol. Sci. 12 (2011) 4282–93. doi:10.3390/ijms12074282.
[71] C. Schwarz, W. Mehnert, J.S. Lucks, R.H. Müller, Solid lipid nanoparticles (SLN)
for controlled drug delivery. I. Production, characterization and sterilization, J. Control.
Release. 30 (1994) 83–96. doi:10.1016/0168-3659(94)90047-7.
[72] A.C. Silva, E. González-Mira, M.L. García, M.A. Egea, J. Fonseca, R. Silva, et al.,
Preparation, characterization and biocompatibility studies on risperidone-loaded solid
lipid nanoparticles (SLN): High pressure homogenization versus ultrasound, Colloids
Surfaces B Biointerfaces. 86 (2011) 158–165. doi:10.1016/j.colsurfb.2011.03.035.
[73] R. Abdullah, N.A. Alhaj, S. Ibrahim, A. Bustamam, Tamoxifen Drug Loading Solid
Lipid Nanoparticles Prepared by Hot High Pressure Homogenization Techniques, Am. J.
Pharmacol. Toxicol. 3 (2008) 219–224.
http://thescipub.com/PDF/ajptsp.2008.219.224.pdf (accessed August 5, 2017).
[74] K. Manjunath, V. Venkateswarlu, Pharmacokinetics, tissue distribution and
bioavailability of clozapine solid lipid nanoparticles after intravenous and intraduodenal
administration, J. Control. Release. 107 (2005) 215–228.
doi:10.1016/j.jconrel.2005.06.006.
[75] D. Liu, S. Jiang, H. Shen, S. Qin, J. Liu, Q. Zhang, et al., Diclofenac sodium-loaded
solid lipid nanoparticles prepared by emulsion/solvent evaporation method, J.
Nanoparticle Res. 13 (2011) 2375–2386. doi:10.1007/s11051-010-9998-y.
[76] A. Sharma, S. V Madhunapantula, G.P. Robertson, Toxicological considerations
when creating nanoparticle-based drugs and drug delivery systems, Expert Opin. Drug
Metab. Toxicol. 8 (2012) 47–69. doi:10.1517/17425255.2012.637916.

43
[77] Z. Rahman, A.S. Zidan, M.A. Khan, Non-destructive methods of characterization
of risperidone solid lipid nanoparticles, Eur. J. Pharm. Biopharm. 76 (2010) 127–137.
doi:10.1016/j.ejpb.2010.05.003.
[78] S. Mukherjee, S. Ray, R.S. Thakur, Solid lipid nanoparticles: a modern formulation
approach in drug delivery system., Indian J. Pharm. Sci. 71 (2009) 349–358.
doi:10.4103/0250-474X.57282.
[79] M. Trotta, F. Debernardi, O. Caputo, Preparation of solid lipid nanoparticles by a
solvent emulsification?diffusion technique, Int. J. Pharm. 257 (2003) 153–160.
doi:10.1016/S0378-5173(03)00135-2.
[80] V. Venkateswarlu, K. Manjunath, Preparation, characterization and in vitro release
kinetics of clozapine solid lipid nanoparticles, J. Control. Release. 95 (2004) 627–638.
doi:10.1016/j.jconrel.2004.01.005.
[81] G. Suresh, K. Manjunath, V. Venkateswarlu, V. Satyanarayana, Preparation,
characterization, and in vitro and in vivo evaluation of lovastatin solid lipid
nanoparticles., AAPS PharmSciTech. 8 (2007) 24. doi:10.1208/pt0801024.
[82] M.A. Schubert, C.C. Müller-Goymann, Solvent injection as a new approach for
manufacturing lipid nanoparticles – evaluation of the method and process parameters,
Eur. J. Pharm. Biopharm. 55 (2003) 125–131. doi:10.1016/S0939-6411(02)00130-3.
[83] P. CHATTOPADHYAY, B. SHEKUNOV, D. YIM, D. CIPOLLA, B. BOYD, S.
FARR, Production of solid lipid nanoparticle suspensions using supercritical fluid
extraction of emulsions (SFEE) for pulmonary delivery using the AERx system?, Adv.
Drug Deliv. Rev. 59 (2007) 444–453. doi:10.1016/j.addr.2007.04.010.
[84] M. Gallarate, M. Trotta, L. Battaglia, D. Chirio, Preparation of solid lipid
nanoparticles from W/O/W emulsions: Preliminary studies on insulin encapsulation, J.
Microencapsul. 26 (2009) 394–402. doi:10.1080/02652040802390156.

44
[85] J.H. Park, G. Saravanakumar, K. Kim, I.C. Kwon, Targeted delivery of low
molecular drugs using chitosan and its derivatives, Adv. Drug Deliv. Rev. 62 (2010) 28–
41. doi:10.1016/j.addr.2009.10.003.
[86] K.A. Janes, M.P. Fresneau, A. Marazuela, A. Fabra, M.J. Alonso, Chitosan
nanoparticles as delivery systems for doxorubicin, J. Control. Release. 73 (2001) 255–
267. doi:10.1016/S0168-3659(01)00294-2.
[87] M. Garcia-Fuentes, M.J. Alonso, Chitosan-based drug nanocarriers: Where do we
stand?, J. Control. Release. 161 (2012) 496–504. doi:10.1016/j.jconrel.2012.03.017.
[88] S. Kunjachan, S. Jose, T. Lammers, Understanding the mechanism of ionic gelation
for synthesis of chitosan nanoparticles using qualitative techniques, Asian J. Pharm. Free
Full Text Artic. from Asian J Pharm. 4 (2014).
http://www.brnsspublicationhub.org/ajp/index.php/ajp/article/view/220 (accessed June
13, 2017).
[89] K.Y. Lee, W.S. Ha, W.H. Park, Blood compatibility and biodegradability of
partially N-acylated chitosan derivatives, Biomaterials. 16 (1995) 1211–1216.
doi:10.1016/0142-9612(95)98126-Y.
[90] W. Fan, W. Yan, Z. Xu, H. Ni, Formation mechanism of monodisperse, low
molecular weight chitosan nanoparticles by ionic gelation technique, Colloids Surfaces B
Biointerfaces. 90 (2012) 21–27. doi:10.1016/j.colsurfb.2011.09.042.
[91] C. Yuwei, W. Jianlong, Preparation and characterization of magnetic chitosan
nanoparticles and its application for Cu(II) removal, Chem. Eng. J. 168 (2011) 286–292.
doi:10.1016/j.cej.2011.01.006.
[92] F. Atyabi, S. Saremi, S.P. Akhlaghi, R. Dinarvand, S.N. Ostad, Thiolated chitosan
nanoparticles for enhancing oral absorption of docetaxel: preparation, in vitro and ex vivo
evaluation, Int. J. Nanomedicine. (2011) 119. doi:10.2147/IJN.S15500.

45
[93] published by Dove Press, NSA-91785-factors-determining-the-stability--size-
distribution--and-ce, (n.d.). doi:10.2147/NSA.S91785.
[94] Z. Zeng, Recent advances of chitosan nanoparticles as drug carriers, Int. J.
Nanomedicine. (2011) 765. doi:10.2147/IJN.S17296.
[95] A. Grenha, Chitosan nanoparticles: a survey of preparation methods, J. Drug
Target. 20 (2012) 291–300. doi:10.3109/1061186X.2011.654121.
[96] S. Wolfram, D. Raederstorff, M. Preller, Y. Wang, S.R. Teixeira, C. Riegger, et al.,
Epigallocatechin Gallate Supplementation Alleviates Diabetes in Rodents, J. Nutr. 136
(2006) 2512–2518. http://jn.nutrition.org/content/136/10/2512.short (accessed December
30, 2015).
[97] M.-S. Lee, C.-T. Kim, Y. Kim, Green tea (-)-epigallocatechin-3-gallate reduces
body weight with regulation of multiple genes expression in adipose tissue of diet-
induced obese mice., Ann. Nutr. Metab. 54 (2009) 151–7. doi:10.1159/000214834.
[98] E. Tedeschi, H. Suzuki, M. Menegazzi, Antiinflammatory Action of EGCG, the
Main Component of Green Tea, through STAT-1 Inhibition, Ann. N. Y. Acad. Sci. 973
(2002) 435–437. doi:10.1111/j.1749-6632.2002.tb04678.x.
[99] Z.P. Chen, J.B. Schell, C.-T. Ho, K.Y. Chen, Green tea epigallocatechin gallate
shows a pronounced growth inhibitory effect on cancerous cells but not on their normal
counterparts, Cancer Lett. 129 (1998) 173–179. doi:10.1016/S0304-3835(98)00108-6.
[100] T. Sen, S. Moulik, A. Dutta, P.R. Choudhury, A. Banerji, S. Das, et al.,
Multifunctional effect of epigallocatechin-3-gallate (EGCG) in downregulation of
gelatinase-A (MMP-2) in human breast cancer cell line MCF-7., Life Sci. 84 (2009) 194–
204. doi:10.1016/j.lfs.2008.11.018.
[101] Y.-H. Lee, J. Kwak, H.-K. Choi, K.-C. Choi, S. Kim, J. Lee, et al., EGCG
suppresses prostate cancer cell growth modulating acetylation of androgen receptor by

46
anti-histone acetyltransferase activity, Int. J. Mol. Med. 30 (2012) 69–74.
http://www.spandidos-publications.com/ijmm/30/1/69/abstract (accessed December 30,
2015).
[102] H. Wang, S. Bian, C.S. Yang, Green tea polyphenol EGCG suppresses lung cancer
cell growth through upregulating miR-210 expression caused by stabilizing HIF-1alpha,
Carcinogenesis. 32 (2011) 1881–1889. doi:10.1093/carcin/bgr218.
[103] C. Chen, G. Shen, V. Hebbar, R. Hu, E.D. Owuor, A.-N.T. Kong, Epigallocatechin-
3-gallate-induced stress signals in HT-29 human colon adenocarcinoma cells.,
Carcinogenesis. 24 (2003) 1369–78. doi:10.1093/carcin/bgg091.
[104] D. Ouyang, L. Zeng, H. Pan, L. Xu, Y. Wang, K. Liu, et al., Piperine inhibits the
proliferation of human prostate cancer cells via induction of cell cycle arrest and
autophagy, Food Chem. Toxicol. 60 (2013) 424–430. doi:10.1016/j.fct.2013.08.007.
[105] J.S. Bang, D.H. Oh, H.M. Choi, B.-J. Sur, S.-J. Lim, J.Y. Kim, et al., Anti-
inflammatory and antiarthritic effects of piperine in human interleukin 1β-stimulated
fibroblast-like synoviocytes and in rat arthritis models, Arthritis Res. Ther. 11 (2009)
R49. doi:10.1186/ar2662.
[106] F. Gold-Smith, A. Fernandez, K. Bishop, Mangiferin and Cancer: Mechanisms of
Action, Nutrients. 8 (2016) 396. doi:10.3390/nu8070396.
[107] African Networks on Ethnomedicines., African journal of traditional,
complementary, and alternative medicines : AJTCAM, [African Networks on
Ethnomedicines], 2004. http://www.bioline.org.br/request?tc11025 (accessed August 6,
2017).
[108] J.C. Reubi, S. Wenger, J. Schmuckli-Maurer, J.-C. Schaer, M. Gugger, Bombesin
Receptor Subtypes in Human Cancers, Clin. Cancer Res. 8 (2002).
http://clincancerres.aacrjournals.org/content/8/4/1139.article-info (accessed June 13,

47
2017).
[109] A.A. Begum, P.M. Moyle, I. Toth, Investigation of bombesin peptide as a targeting
ligand for the gastrin releasing peptide (GRP) receptor, Bioorg. Med. Chem. 24 (2016)
5834–5841. doi:10.1016/j.bmc.2016.09.039.
[110] 2 Origin of charge at interfaces : Structure of the electrical double layer, (n.d.).

48
Enhancement of cancer cytotoxicity of
EGCG by encapsulation within solid
lipid nanoparticles

49
2.1. Background
Tea, obtained from the cured leaves of the tea shrub Camellia sinensis, has been known
as a health promoting beverage since ancient times; its medicinal properties discovered
as early as the 5th century AD[1]. Most of the health benefits associated with drinking
tea including its anti-obesity and anti-oxidant effects have been attributed to its
polyphenolic constituents, especially its catechins including epigallocatechin gallate
(EGCG), (-)-epigallocatechin, (-)-epicatechin gallate and (-)- epicatechin [2–4]. EGCG is
one of the most abundant and active constituents of green tea with extensive therapeutic
properties[5,6]. Research on EGCG has shown that it exhibits a wide array of therapeutic
potencies including anti-inflammatory, anti-diabetic, anti-obesity and anti-cancer
effects[5,7–10]. EGCG has been found effective not only as a chemopreventive but also
as a potent chemotherapeutic agent[11–13]. It has been documented to possess anti-
cancer activity in many cancer cell lines by inhibiting tumorigenesis, proliferation and
angiogenesis[14–16]. Many pathways have been elucidated to explain the anti-cancer
activity of EGCG as described below:
a. Telomerase activity inhibition
Telomeres are long repeating, highly-conserved, end-nucleotide sequences in eukaryotic
cells that help cells to divide without losing any of the crucial genes in the process[17].
During cell replication, the enzyme DNA polymerase uses each strand of the DNA as a
template to synthesize two daughter strands[18].
However, the enzyme is not able to completely replicate the entire strand, and hence there
is a shortening of the end of the strand following each division. Telomeres help in
preserving the important genetic sequences by getting shortened themselves in the
process[17]. Telomerase is an RNA-dependent DNA polymerase (reverse transcriptase)
that helps replenish the units of the telomeres. In most somatic cells, there is no telomerase

50
so the cells cease replicating following a certain number of divisions[19]. Expression of
telomerases is upregulated in a number of cancers thus leading to the maintenance and
the continuous replication of genetic material[20]. EGCG has shown to inhibit telomerase
activity in a number of cancers, thus leading to cell division cessation[21].
b. Gene silencing
DNA hypermethylation is the process of the addition of methyl groups to the DNA strand
by the enzyme DNA methyltransferase. When the DNA subunits at the promoter region
of a gene are hypermethylated, there is no binding of the transcription factors at this
portion leading to gene silencing[22]. Cancer cells are found to induce hypermethylation
at the regions of the DNA that contain pro-apoptotic genes in order to maintain continuous
cell multiplication[23]. EGCG is found to directly inhibit DNA methyltransferase and
also causes reactivation of methylation-silenced genes leading to increased apoptosis
among cancer cell populations [24].
c. Angiogenesis
Angiogenesis is a process of neovascularization wherein new blood vessels are formed
from existing ones. Cancer cells upregulate angiogenesis in order to compensate for the
greater blood supply and nutrients required for the highly escalated rate of cell
division[25]. The process includes disruption of the basement membrane to allow
migration of angiogenic factors which help recruit new vessels. Vascular endothelial
growth factor (VEGF) is one such potent angiogenic factor upregulated in angiogenesis
[26].
EGCG has been reported to inhibit VEGF expression by downregulating expression of
hypoxia-inducible factor-1α which is a VEGF stimulator. Also, the hypoxia and oxidative
stress in cancer cells leads to production of oxygen radicals, which activate transcription
factors such as nuclear factor kappa-B(NFκB), that also cause overexpression of VEGF.

51
EGCG is also found to downregulate expression of NFκB, thus indirectly reducing
VEGF[27].
d. Metastasis
Metastasis is the movement of cancer cells from the primary area of tumour development
to other parts of the body through blood vessels or the lymphatic system. It involves the
degradation of the basement membrane which causes disruption of the membrane wall,
helping dissemination of cancer cells to cause tumour formations elsewhere[28]. EGCG
has been found to inhibit matrix metalloproteinases 2, 3 and 9 which are important in the
degradation of the basement membrane, thus preventing the spread of tumour to other
areas[29].
e. Apoptosis
Apoptosis is a process of programmed cell death that occurs through two pathways-
intrinsic and extrinsic. The extrinsic pathway functions by the activation of death
receptors by binding which triggers a cascade to allow the release of caspases that bring
out events in apoptosis. The intrinsic pathway involves pro-apoptotic proteins such as
BCL-2-associated X protein (BAX) and BCL-2 homologous antagonist killer (BAK) that
triggers activation of caspases. This pathway is regulated by anti-apoptotic proteins such
as B-cell lymphoma 2 (BCL-2) and B cell lymphoma extra-large (BCL-XL), which
antagonize with BAX and BAK. Cancer cells have shown to supress apoptotic pathways
to maintain immortality[30]. EGCG has been shown to promote the expression of pro-
apoptotic proteins BAX and BAK while also consequently inhibiting the expression of
BCL-2 and BCL-XL in many cancer types[31,32].
f. Anti-proliferative activity
Mitogen activated protein kinases (MAPK) are enzymes involved in important steps for
cell survival, proliferation and development of cancer cells[33]. EGCG has shown to

52
inhibit MAPK pathway in many cancer cell types. It has also been shown to inhibit
insulin-like growth factor-I receptor activity which helps to activate the MAPK pathway
and plays an important role in proliferation[34].
Fig 2.1: Structure of epigallocatechin gallate
EGCG’s diverse therapeutic properties are attributed to its polyphenolic structure that
allows electron delocalization on the benzene ring helping quench free radicals[35]. It has
a trihydroxy group substitution on one ring and an esterified gallate group attached to
another. However, it has been found to be unstable in neutral and alkaline conditions;
undergoing rapid degradation involving the deprotonation of the hydroxyl groups[36].
Also, the hydroxyl groups are susceptible to various biotransformation reactions within
the living system such as glucuronidation, methylation and sulphonidation leading to a
loss in biological activity[37]. This instability has also been cited as one of the reasons
for the poor bioavailability of EGCG and other catechins within the biological
system[38]. In this study, the stability of EGCG in solutions of different pH was initially
investigated to determine the pH range where EGCG is stable at room temperature. Then,
EGCG was encapsulated in solid lipid nanoparticles (SLNs) for enhancing the stability
and anti-cancer activity of EGCG. SLN were prepared from biocompatible lipids which
are solid at room and body temperature[39]. They provide multiple advantages over other
conventional nanocarriers such as lipid nanoemulsions and polymeric nanoparticles.
These advantages include an improvement in drug stability, drug entrapment and

53
biocompatibility, as they are composed of easily biodegradable excipients, and offers
efficient scaling up for production[40,41]. Moreover, SLN control the release of a pH-
sensitive drug such as EGCG and therefore, prevent the premature degradation of
encapsulated drug in the biological system[42,43]. Post encapsulation of EGCG in SLN,
the anti-cancer activity of pure EGCG and EGCG-loaded SLN (EGCG-SLN) was
checked by cellular proliferation assays in MDA-MB-231 human breast cancer and DU-
145 human prostate cancer cell lines.
2.2. Materials
Glycerol mono-stearate (GMS) was purchased from Alfa Aesar (Johnson Matthey
Chemicals, Hyderabad, India). EGCG, tristearin (TS), tripalmitin (TP), stearic acid (SA),
Pluronic F68 (P F68), polyvinyl alcohol (PVA), Roswell Park Memorial Institute
(RPMI)-1640 medium, fetal bovine serum (FBS), trypsin–EDTA, phosphate buffered
saline (Ca2+, Mg2+ free), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide
(MTT), dimethyl sulfoxide (DMSO) were purchased from Sigma Aldrich (St. Louis, MO,
USA). Soya lecithin was purchased from HiMedia (Mumbai, India). MDA-MB-231
human breast cancer cells and DU-145 human prostate cancer cells were obtained from
American Type Culture Collection (ATCC, Manassas, USA). Dichloromethane was
obtained from Merck chemicals (Mumbai, India). Tween180 was purchased from sd Fine
Chemicals Limited (Hyderabad, India).
2.3. Methods
2.3.1. Aqueous Stability
The stability of EGCG was determined in different aqueous solutions; 0.1 N HCl (pH
1.5), phosphate buffer saline pH 7.4 (PBS), normal saline and phosphate buffer solutions
of different pH (5, 7.4 and 9). A stock solution of EGCG was prepared (1 mg/mL) and
dilutions were made in the study medium to give a final drug concentration of 50 µg/mL.

54
UV-Visible spectroscopy was used to evaluate changes in the absorption characteristics
of EGCG throughout the study at λmax of EGCG at 274.5 nm. Samples were scanned in
the range of 200-800 nm and spectra were obtained in the various solutions at different
time points of 0, 4, 8, 24, 48 and 96 h using a Jasco-UV650 spectrophotometer.
2.3.2. Optimization
The most important aspect of nanoparticle preparation for biomedical applications is the
regulation of nanoparticle sizes and particle surface charges for obtaining optimum
dimensions as the cell endothelia and tumour microenvironments have stringent
limitations. The synthesis of nanoparticles was optimized with respect to different process
and formulation parameters such as the type of lipid, lipid to co-lipid ratio, surfactant used
and its concentration, amount of co-emulsifier, inner aqueous phase volume and
sonication time.
2.3.3. Preparation of EGCG loaded solid lipid nanoparticles (EGCG-SLNs)
EGCG-SLNs were prepared by emulsion-solvent evaporation method as reported in the
literature[43]. Briefly, 80 mg GMS, 20 mg SA and 20 mg soya lecithin were dissolved
in dichloromethane and sonicated for 3 min. EGCG was dissolved in 500 µL distilled
water and 2 % w/v Pluronic F68 solution was then added to the lipid mixture to form a
water-in-oil (w/o) primary emulsion. The mixture was sonicated for 5 min and then
transferred into 1 % w/v Pluronic F68 solution to form a double emulsion. The emulsion
was stirred for 2 h to evaporate the organic solvent. The nanoparticle suspension was
centrifuged and washed thrice with distilled water to remove any free drug adhering to
the nanoparticle surface. Blank nanoparticles (Blank-SLNs) were prepared in a similar
manner, excluding the drug.
2.3.4. Determination of drug entrapment efficiency
The drug entrapment efficiency (EE) was calculated by the indirect method. The

55
supernatant obtained after centrifugation of the nanoparticle suspension was used for
estimation of the free drug content by UV/Visible spectroscopy at the absorption maxima
of EGCG at 274.5 nm. The percent entrapment efficiency was calculated by the following
formula:
% 𝐸𝐸 =𝑇𝑜𝑡𝑎𝑙 𝑎𝑚𝑜𝑢𝑛𝑡 𝑜𝑓𝐸𝐺𝐶𝐺 𝑎𝑑𝑑𝑒𝑑 − 𝐴𝑚𝑜𝑢𝑛𝑡 𝑜𝑓𝐸𝐺𝐶𝐺 𝑖𝑛 𝑠𝑢𝑝𝑒𝑟𝑛𝑎𝑡𝑎𝑛𝑡
𝑇𝑜𝑡𝑎𝑙 𝑎𝑚𝑜𝑢𝑛𝑡 𝑜𝑓 𝐸𝐺𝐶𝐺 𝑎𝑑𝑑𝑒𝑑×100
2.3.5. Physico-chemical characterization of nanoparticles
2.3.5.1. Particle size and surface potential
The mean particle diameter (PD), polydispersity index (PDI) and surface potential of
Blank-SLN and EGCG-SLN were measured by photon correlation spectroscopy using a
Zetasizer Nano-ZS (Malvern Instrument Ltd., Malvern, UK). Samples were diluted
appropriately and were analysed at 25 °C with a backscattering angle of 173°.
2.3.5.2. FTIR spectroscopy (FTIR)
FTIR analysis of EGCG-SLN and Blank-SLNs was carried out by potassium bromide
(KBr) pellet method, using an IR spectrophotometer (Perkin Elmer, USA). Pellets were
prepared by adding approximately 3 mg of the sample to 100 mg of KBr. Samples were
scanned in the range of 400–4000 cm−1.
2.3.5.3. Differential scanning calorimetry (DSC)
DSC analysis of EGCG-SLN and EGCG were carried out on a DSC1 instrument (Mettler
Toledo, Switzerland). The instrument was calibrated using pure indium. Approximately
5 mg of sample was heated in a hermetically sealed flat-bottom aluminium pan and
scanned from 50 °C to 240 °C at a heat flow rate of 10 °C/min, under a nitrogen
environment. Empty aluminium pans were used as standards.
2.3.6. In-vitro drug release
In-vitro drug release studies were performed by dialysis method in sodium acetate buffer

56
pH 5 (SAB). EGCG-SLN, equivalent to 2 mg of EGCG, was placed in dialysis tubing
and kept in a beaker containing 100 mL of SAB and maintained at 37 °C. At
predetermined time interval, a definite volume was withdrawn and replaced with same
volume of fresh media. The EGCG concentration was determined by measuring the
absorbance at 274.5 nm using a UV/Visible spectrophotometer.
2.3.7. In-vitro cytotoxicity studies
In-vitro cytotoxicity of native EGCG and EGCG-SLN against MDA-MB-231 human
breast cancer cells and DU-145 human prostate cancer cells was evaluated by MTT assay.
MDA-MB-231 human breast cancer cells and DU-145 human prostate cancer cells were
cultured in RPMI-1640 and DMEM medium, respectively, supplemented with 10% FBS,
100 mg mL-1 streptomycin, 100 U mL-1 penicillin, and at 37 °C in a 5% CO2 incubator.
Cells (5 × 103 cells per well) were seeded in a 96-well plate and incubated with different
concentrations of test formulations. After 48 h, the medium was replaced with serum-free
medium containing 0.5 mg mL-1 of MTT reagent and further incubated for 4 h. Then the
medium was removed and formazan crystals generated by live cells were dissolved by
adding 150 µL of DMSO in each well. The absorbance was measured using a microplate
reader at a wavelength of 570 nm. The relative cell viability (%) was determined by
comparing the absorbance with control wells. Data are presented as average ± SD (n =
4).
Cell apoptosis was detected by assessment of nuclear morphology staining with Hoechst
33342. MDA MB-231 and DU145 cell lines were seeded in a 12-well culture plate at a
density of 1 x 105 cells per well, cultured at 37 ºC, 5% CO2 incubator to obtain confluency.
The cells were treated with EGCG and EGCG-SLN formulation at IC-50 concentration
of drug-loaded formulations and incubated for 24 hrs. After incubation, the cells were
washed with ice cold PBS for twice, fixed with 4% paraformaldehyde in PBS (pH 7.4)

57
for 15 min at room temperature, stained with 5 µg/mL Hoechst 33342 in PBS for 15 min
and washed twice with ice cold PBS. The cells were then examined using fluorescence
microscopy.
2.3.8. Colloidal and serum stability studies
2.3.8.1. Colloidal stability of nanoparticles in serum and physiological
conditions
Colloidal stability of EGCG-SLNs with time was tested in PBS, plasma and distilled
water. A stock solution (1 mg/mL) of nanoparticle suspension was prepared in distilled
water. Aliquots of the stock solution were then taken and dispersed in the respective
medium. The samples were stored at room temperature and the particle size,
polydispersity and zeta potential were measured at different time intervals using a
Zetasizer Nano-ZS.
2.3.8.2. Electrolyte-induced aggregation
The effect of electrolytes on the solid lipid nanoparticle formulation was studied by
electrolyte-induced aggregation technique as reported in literature[44]. Sucrose sulphate
solutions were prepared in varying concentrations to study the behaviour of EGCG-SLNs
in the presence of a flocculating agent. Briefly, 16.7 g of sucrose was dissolved in 100
mL double distilled water to give 16.7 % sucrose solution. Sodium sulphate was added to
this native solution to make a 2 M sucrose sulphate solution. Serial dilutions were made
from this stock solution to prepare final concentrations ranging from 0.1 to 2 M.
Nanoparticle suspensions equivalent to 1 mg/mL were added to the serial dilutions to
make a final volume of 3 mL and immediately analysed for particle size, zeta potential,
polydispersity and UV-Visible absorbance.
2.4. Results and discussion
2.4.1. Aqueous stability

58
The stability of EGCG in solutions of different pH was evaluated with UV/Visible
spectroscopy at 274.5 nm which is the absorption maxima for EGCG (Fig.2.2). The
absorption characteristics for EGCG in 0.1 N HCl (pH 1.5), PBS (pH 7.4), normal saline
(0.9 %w/v) and phosphate buffer solutions with three different pH values 5, 7 and 9 were
recorded. This study was carried out to evaluate the behaviour of free EGCG when it is
released in solutions simulating the different environment it encounters when released in
the blood stream. Spectra were recorded in the wavelength range of 200-800 nm up to 96
h to compare the changes in the drug absorption characteristics with changes in the study
environment. EGCG was found to be most stable at highly acidic pH 1.5 in 0.1 N HCl,
showing practically no degradation up to 96 h. In distilled water, the peak intensity for
EGCG is found to change after 4 h but the absorption characteristics remained same till
96 h. At 96 h, the peak is found to disintegrate, which could be due to the formation of
degradation products. In 0.9 % w/v normal saline, the peak distorted at around 4 h, but
complete peak disintegration occurred at 24 h. In PBS, the peak absorption showed a
slight change after 4 h but after 24 h complete peak disintegration was observed. At pH
5, the absorption characteristics remain unchanged up to 24 h after which distortion is
seen at 48 h. At pH 7, peak changes were observed at 8 h but peak distortion occurred at
24 h. EGCG was most unstable at alkaline pH 9. At this pH, changes in the absorption
characteristics are observed immediately after addition and complete disintegration of the
peak was observed after 4 h. These results suggested that EGCG is highly stable at acidic
pH while its stability decreases as the pH tends towards neutral and shows very less or no
stability at highly alkaline conditions.
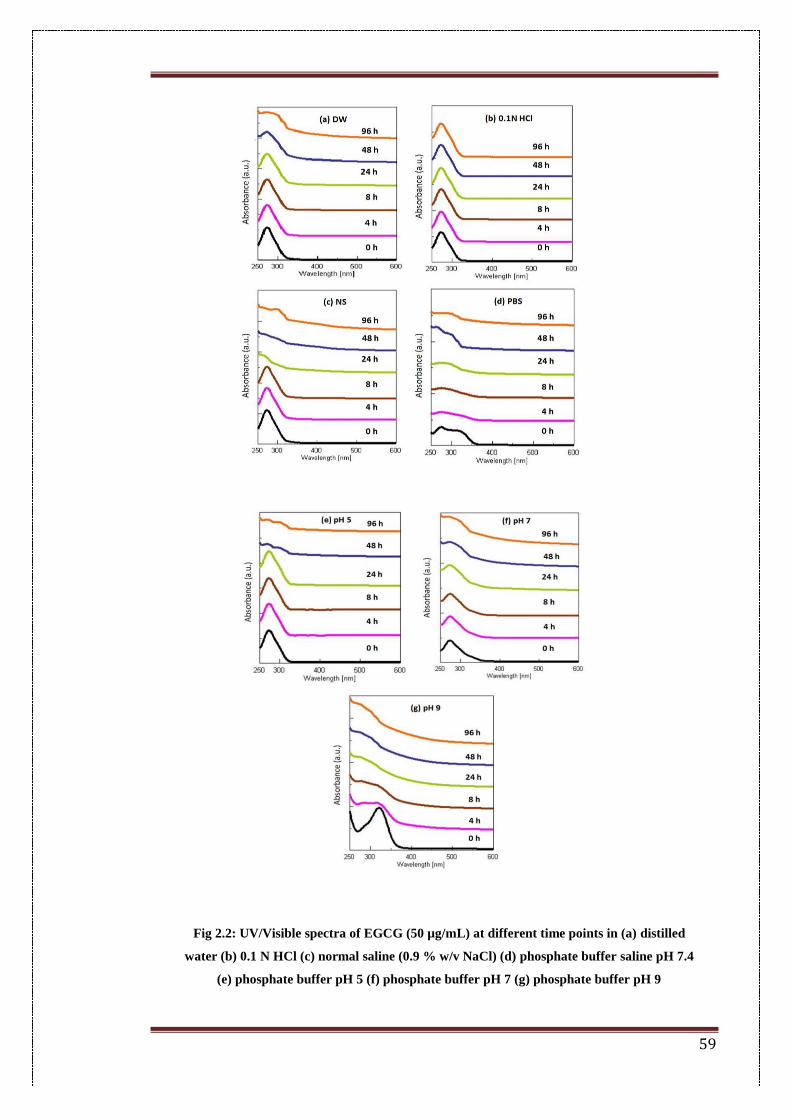
59
Fig 2.2: UV/Visible spectra of EGCG (50 µg/mL) at different time points in (a) distilled
water (b) 0.1 N HCl (c) normal saline (0.9 % w/v NaCl) (d) phosphate buffer saline pH 7.4
(e) phosphate buffer pH 5 (f) phosphate buffer pH 7 (g) phosphate buffer pH 9

60
2.4.2. Optimization of different parameters for the blank SLN
Blank nanoparticles without EGCG encapsulation were prepared by double
emulsification method followed by solvent evaporation. This method shows high
potential for encapsulation of hydrophilic drugs alike. Stearic acid served as charge
modifier as the free carboxylic acid groups of stearic acid renders generation of
electronegative nanoparticles which in-turn can be harnessed for other applications
including ligand conjugation. Formulation and process parameters such as type of lipid,
lipid to co-lipid ratio, type of surfactant and concentration, amount of co-surfactant,
aqueous phase volume, sonication and stirring time were sequentially optimized
2.4.3. Influence of formulation parameters
Lipid crystallinity, lipophilicity, loading capacity, melting point and purity are important
factors to be considered when choosing a lipid for a nanoparticle formulation[45,46].
Tristearin (TS), tripalmitin (TP) and glycerol mono-stearate (GMS) were used for
optimization of particle diameter and surface potential (Table 2.1). The observed particle
size for SLN prepared with TS, TP and GMS were 131.1 ± 2 nm, 133.8 ± 2.7 nm and
121.4 ± 2.6 nm respectively. SLN prepared with GMS not only showed the least particle
size, but also had the least polydispersity. The co-lipid used in SLN preparation was
stearic acid which also acted as a charge modifier. Greater surface potential correlates
with greater stability of nanoparticles as there is a corresponding increase in the
electrostatic repulsive forces with an increase in the charge on each particle[47]. Stearic
acid content was varied between 30%, 20% and 10%w/w of total lipid content. Stearic
acid at 20% w/w of lipid showed a particle diameter of 125.7 ± 2.7 nm and good surface
potential values with least polydispersity. Surfactants stabilize the nanoparticles in the
colloidal state and prevent agglomeration during growth and storage.
The choice of stabilizers is important not only to control particle size and stabilization of

61
dispersions but also to control crystallization and polymorphic transitions[48]. SLN made
with polyvinyl alcohol, Tween® 80 and Pluronic F68 showed a particle diameter of 175
± 3.9 nm, 153.6 ± 3.4 nm and 127.5 ± 2.4 nm, respectively. SLN prepared with Pluronic
F68 showed the smallest particle size compared to the other surfactants. The
concentration of surfactant was varied from 1 to 3% Pluronic F68. SLN prepared with
2% Pluronic solution showed greater uniformity with lesser particle size and hence was
chosen as the surfactant utilized in preparation. Excess lipid molecules employed in the
production form small, stray, unilamellar vesicles which exhibit limited mobility and
therefore they are unable to immediately cover the newly created crystalline interfaces
during lipid recrystallization, leading to aggregation, wherein co-emulsifiers help in
providing additional stability during recrystallization events[49]. The co-emulsifier
employed was soya lecithin (30% phosphatidylcholine) as it has been found to be efficient
in the solvent-evaporation method for preparation of SLN[40]. It was optimized between
10 and 30 mg of which 20 mg soya lecithin showed a higher surface potential with lesser
particle diameter. The inner aqueous phase volume was optimized for particle size.
Increase in the aqueous phase was observed to cause a corresponding increase in the total
particle size. A volume of 400 μL of inner aqueous phase increased the particle size by
around 30 nm i.e. 153 nm compared to that obtained with 200 μL of inner aqueous phase,
which was 115.7 nm.
2.4.4. Influence of process parameters
Sonication helps in the emulsifying process, causing further size reduction of nanoparticle
formulations[50]. Sonication time was optimized for total sonication times of the two
cycles. The optimum sonication time was found to be 8 min for emulsion preparation as
it showed the optimum particle diameter and good surface potential of 118 ± 2.01nm and
-32.2 ± 1.5mV respectively. Solvents are used to dissolve the lipids in the oil phase in
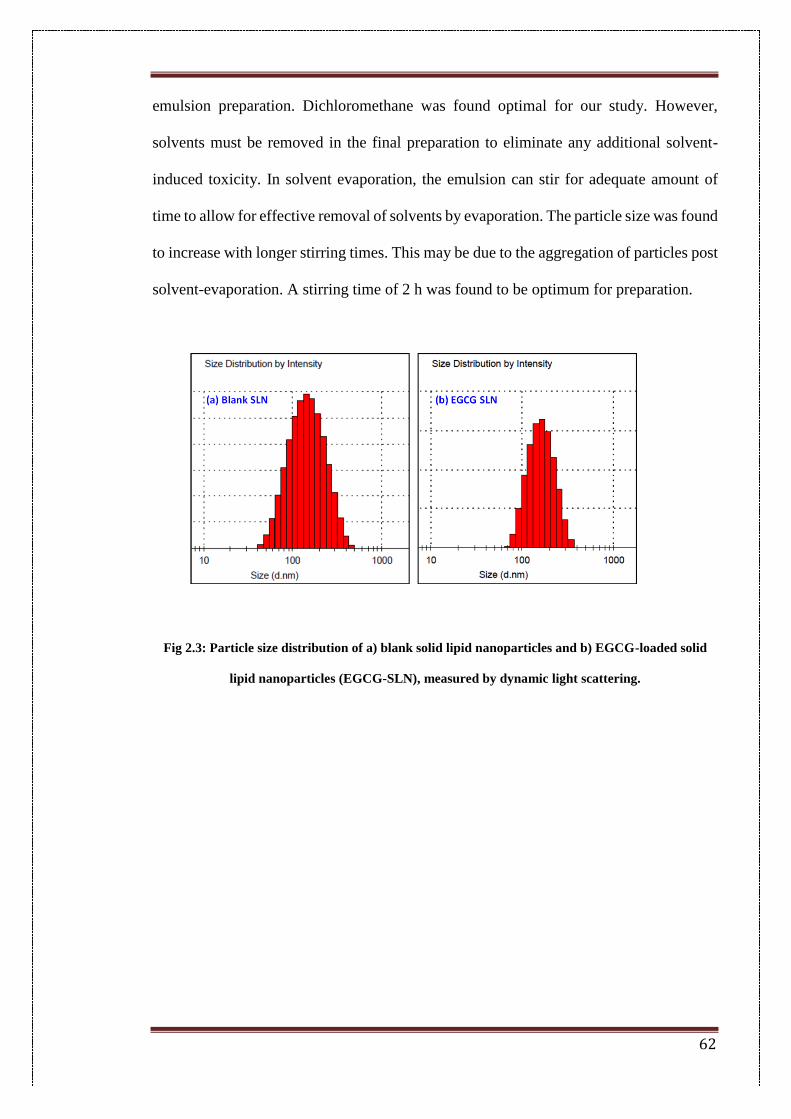
62
emulsion preparation. Dichloromethane was found optimal for our study. However,
solvents must be removed in the final preparation to eliminate any additional solvent-
induced toxicity. In solvent evaporation, the emulsion can stir for adequate amount of
time to allow for effective removal of solvents by evaporation. The particle size was found
to increase with longer stirring times. This may be due to the aggregation of particles post
solvent-evaporation. A stirring time of 2 h was found to be optimum for preparation.
Fig 2.3: Particle size distribution of a) blank solid lipid nanoparticles and b) EGCG-loaded solid
lipid nanoparticles (EGCG-SLN), measured by dynamic light scattering.
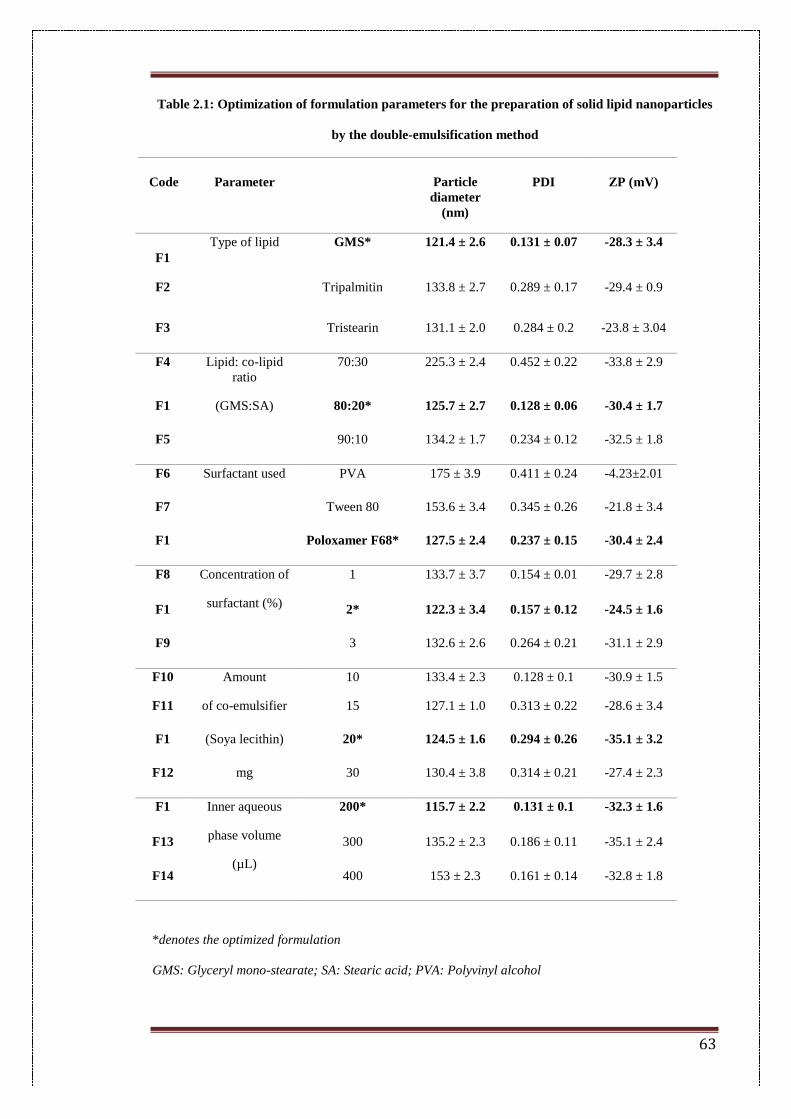
63
Table 2.1: Optimization of formulation parameters for the preparation of solid lipid nanoparticles
by the double-emulsification method
Code Parameter Particle
diameter
(nm)
PDI ZP (mV)
F1
Type of lipid
GMS* 121.4 ± 2.6 0.131 ± 0.07 -28.3 ± 3.4
F2 Tripalmitin 133.8 ± 2.7 0.289 ± 0.17 -29.4 ± 0.9
F3 Tristearin 131.1 ± 2.0 0.284 ± 0.2 -23.8 ± 3.04
F4 Lipid: co-lipid
ratio
70:30 225.3 ± 2.4 0.452 ± 0.22 -33.8 ± 2.9
F1 (GMS:SA) 80:20* 125.7 ± 2.7 0.128 ± 0.06 -30.4 ± 1.7
F5 90:10 134.2 ± 1.7 0.234 ± 0.12 -32.5 ± 1.8
F6 Surfactant used PVA 175 ± 3.9 0.411 ± 0.24 -4.23±2.01
F7 Tween 80 153.6 ± 3.4 0.345 ± 0.26 -21.8 ± 3.4
F1 Poloxamer F68* 127.5 ± 2.4 0.237 ± 0.15 -30.4 ± 2.4
F8 Concentration of
surfactant (%)
1 133.7 ± 3.7 0.154 ± 0.01 -29.7 ± 2.8
F1 2* 122.3 ± 3.4 0.157 ± 0.12 -24.5 ± 1.6
F9 3 132.6 ± 2.6 0.264 ± 0.21 -31.1 ± 2.9
F10 Amount 10 133.4 ± 2.3 0.128 ± 0.1 -30.9 ± 1.5
F11 of co-emulsifier 15 127.1 ± 1.0 0.313 ± 0.22 -28.6 ± 3.4
F1 (Soya lecithin) 20* 124.5 ± 1.6 0.294 ± 0.26 -35.1 ± 3.2
F12 mg 30 130.4 ± 3.8 0.314 ± 0.21 -27.4 ± 2.3
F1 Inner aqueous
phase volume
(µL)
200* 115.7 ± 2.2 0.131 ± 0.1 -32.3 ± 1.6
F13 300 135.2 ± 2.3 0.186 ± 0.11 -35.1 ± 2.4
F14 400 153 ± 2.3 0.161 ± 0.14 -32.8 ± 1.8
*denotes the optimized formulation
GMS: Glyceryl mono-stearate; SA: Stearic acid; PVA: Polyvinyl alcohol

64
2.4.5. Encapsulation efficiency of EGCG-SLN
After optimising the nanoparticles for process and formulation parameters, they were
further optimised for the amount of drug loaded in the nanoparticles. EGCG was
dissolved in the inner aqueous phase of the double emulsion and the rest of the procedure
for preparation remained the same as that of the blank nanoparticles. EGCG-SLNs were
prepared by keeping the lipid concentration constant at 5% w/v and varying the
concentration of EGCG between 3% w/w and 5% w/w with respect to the lipid (Table
2.2). The results showed that an increase in the concentration of the lipid led to an increase
in the drug loading and hence the encapsulation efficiency. SLN with 5% w/w of drug
showed an encapsulation efficiency of 67.2% and particle size of 157 ± 2.5 nm, hence
was chosen as the optimum for future preparations (Fig. 2.3). Higher loading of drug was
not possible due to super-saturation of the drug in the limited inner aqueous volume.
Table 2.2: Optimization of encapsulation of epigallocatechin gallate (EGCG) in solid lipid
nanoparticles
EGCG to lipid
ratio (%w/w)
Particle size
(nm)
Polydispersity
index (PDI)
Zeta potential
(mV)
% EE
3 112.5 ± 3.1 0.14 ± 0.12 -30.1 ± 3.2 89.5 ± 4.7
4 128.2 ± 4.3 0.132 ± 1.21 -31.5 ± 3.7 75.8 ± 2.3
5 157.4 ± 2.5 0.268 ± 0.14 -37.2 ± 2.5 67.2 ± 4.5
PDI: Polydispersity index; ZP: Zeta potential; EE: Encapsulation efficiency
2.4.6. Physico-chemical characterization
2.4.6.1. FTIR analysis
Infrared spectroscopy was used to determine the compatibility of drug with the
components of the nanoparticles (Fig 2.4)[51]. The FTIR spectrum of EGCG shows a
characteristic peak at 3356 cm−1 for the O-H group attached to the aromatic ring, a strong
peak at 1691 cm−1 for the C=O group that links the trihydroxybenzoate group and
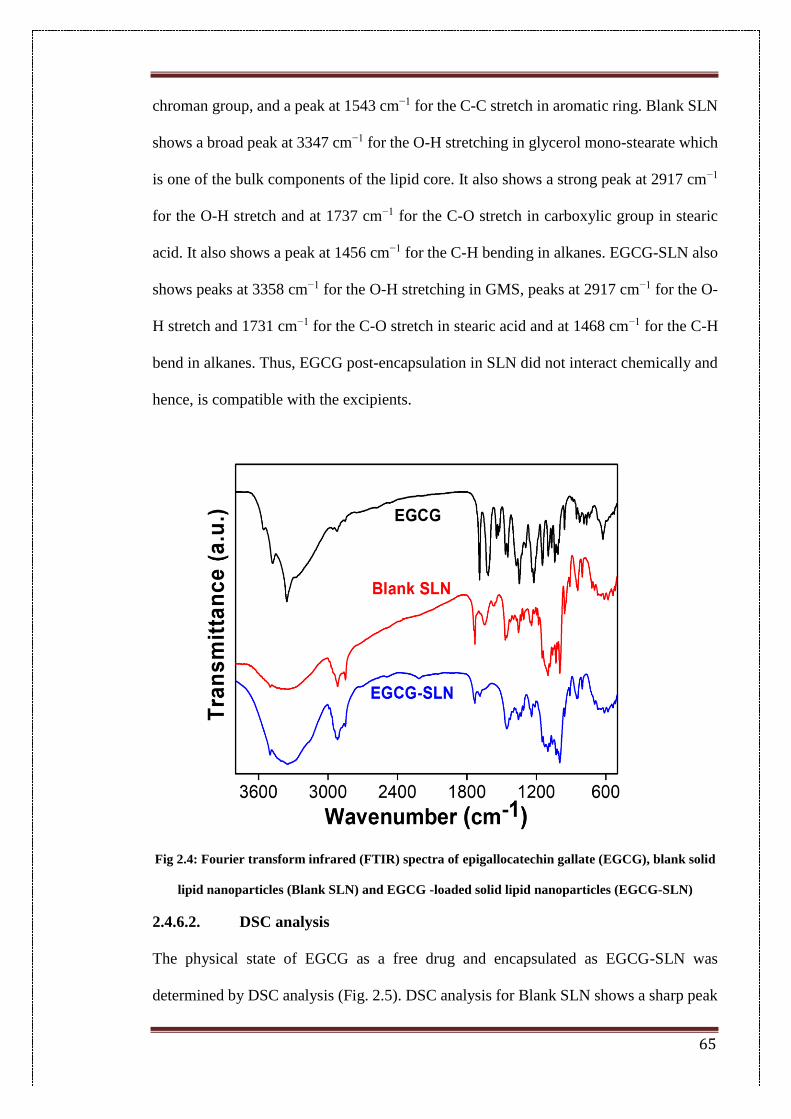
65
chroman group, and a peak at 1543 cm−1 for the C-C stretch in aromatic ring. Blank SLN
shows a broad peak at 3347 cm−1 for the O-H stretching in glycerol mono-stearate which
is one of the bulk components of the lipid core. It also shows a strong peak at 2917 cm−1
for the O-H stretch and at 1737 cm−1 for the C-O stretch in carboxylic group in stearic
acid. It also shows a peak at 1456 cm−1 for the C-H bending in alkanes. EGCG-SLN also
shows peaks at 3358 cm−1 for the O-H stretching in GMS, peaks at 2917 cm−1 for the O-
H stretch and 1731 cm−1 for the C-O stretch in stearic acid and at 1468 cm−1 for the C-H
bend in alkanes. Thus, EGCG post-encapsulation in SLN did not interact chemically and
hence, is compatible with the excipients.
Fig 2.4: Fourier transform infrared (FTIR) spectra of epigallocatechin gallate (EGCG), blank solid
lipid nanoparticles (Blank SLN) and EGCG -loaded solid lipid nanoparticles (EGCG-SLN)
2.4.6.2. DSC analysis
The physical state of EGCG as a free drug and encapsulated as EGCG-SLN was
determined by DSC analysis (Fig. 2.5). DSC analysis for Blank SLN shows a sharp peak
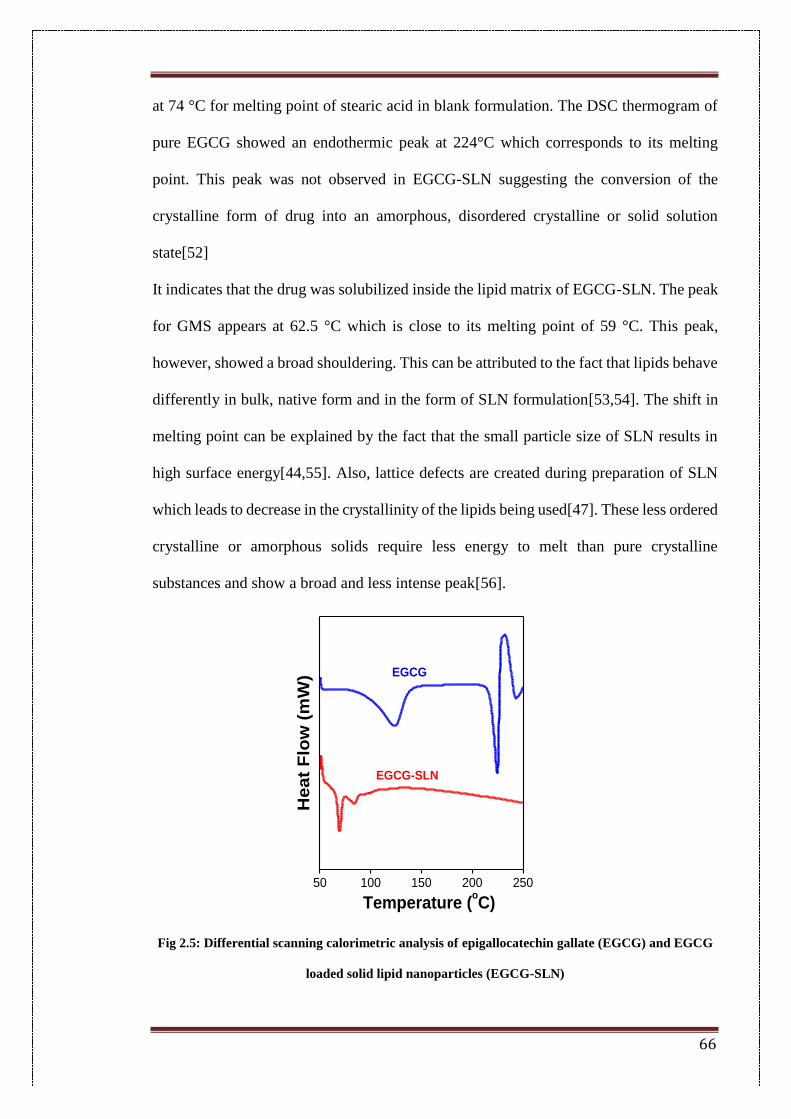
66
at 74 °C for melting point of stearic acid in blank formulation. The DSC thermogram of
pure EGCG showed an endothermic peak at 224°C which corresponds to its melting
point. This peak was not observed in EGCG-SLN suggesting the conversion of the
crystalline form of drug into an amorphous, disordered crystalline or solid solution
state[52]
It indicates that the drug was solubilized inside the lipid matrix of EGCG-SLN. The peak
for GMS appears at 62.5 °C which is close to its melting point of 59 °C. This peak,
however, showed a broad shouldering. This can be attributed to the fact that lipids behave
differently in bulk, native form and in the form of SLN formulation[53,54]. The shift in
melting point can be explained by the fact that the small particle size of SLN results in
high surface energy[44,55]. Also, lattice defects are created during preparation of SLN
which leads to decrease in the crystallinity of the lipids being used[47]. These less ordered
crystalline or amorphous solids require less energy to melt than pure crystalline
substances and show a broad and less intense peak[56].
Fig 2.5: Differential scanning calorimetric analysis of epigallocatechin gallate (EGCG) and EGCG
loaded solid lipid nanoparticles (EGCG-SLN)
50 100 150 200 250
He
at
Flo
w (
mW
)
EGCG-SLN
Temperature (oC)
EGCG
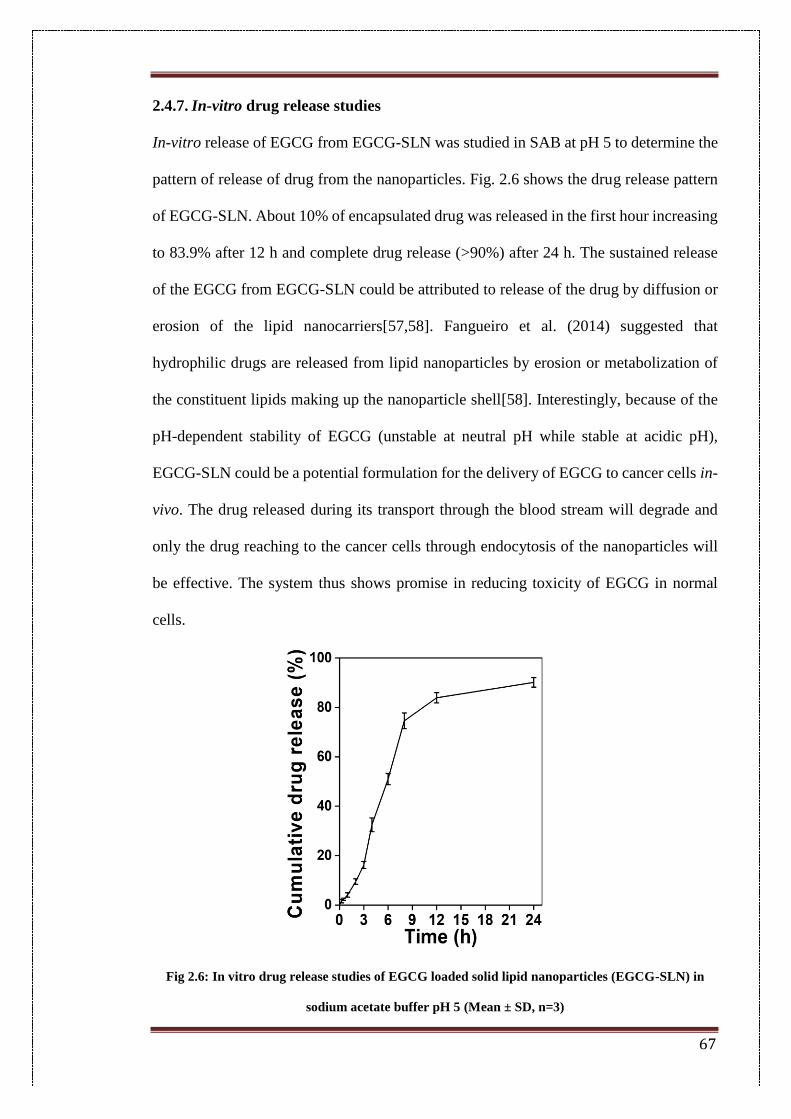
67
2.4.7. In-vitro drug release studies
In-vitro release of EGCG from EGCG-SLN was studied in SAB at pH 5 to determine the
pattern of release of drug from the nanoparticles. Fig. 2.6 shows the drug release pattern
of EGCG-SLN. About 10% of encapsulated drug was released in the first hour increasing
to 83.9% after 12 h and complete drug release (>90%) after 24 h. The sustained release
of the EGCG from EGCG-SLN could be attributed to release of the drug by diffusion or
erosion of the lipid nanocarriers[57,58]. Fangueiro et al. (2014) suggested that
hydrophilic drugs are released from lipid nanoparticles by erosion or metabolization of
the constituent lipids making up the nanoparticle shell[58]. Interestingly, because of the
pH-dependent stability of EGCG (unstable at neutral pH while stable at acidic pH),
EGCG-SLN could be a potential formulation for the delivery of EGCG to cancer cells in-
vivo. The drug released during its transport through the blood stream will degrade and
only the drug reaching to the cancer cells through endocytosis of the nanoparticles will
be effective. The system thus shows promise in reducing toxicity of EGCG in normal
cells.
Fig 2.6: In vitro drug release studies of EGCG loaded solid lipid nanoparticles (EGCG-SLN) in
sodium acetate buffer pH 5 (Mean ± SD, n=3)
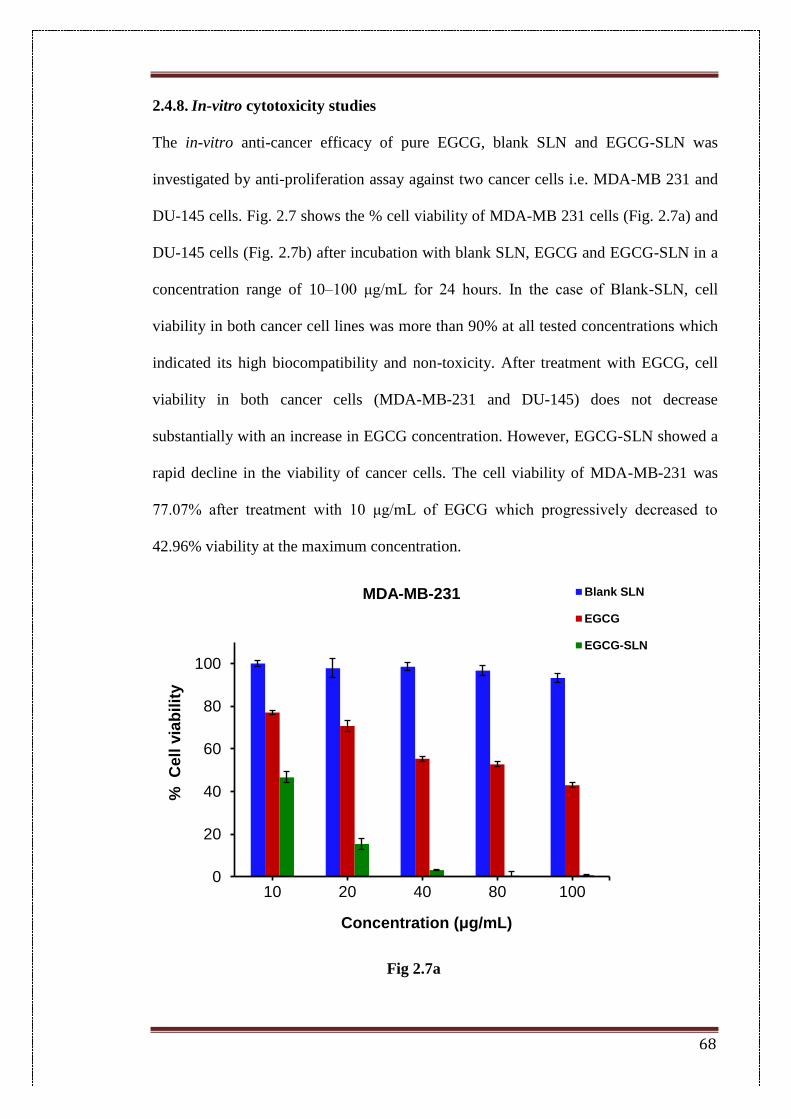
68
2.4.8. In-vitro cytotoxicity studies
The in-vitro anti-cancer efficacy of pure EGCG, blank SLN and EGCG-SLN was
investigated by anti-proliferation assay against two cancer cells i.e. MDA-MB 231 and
DU-145 cells. Fig. 2.7 shows the % cell viability of MDA-MB 231 cells (Fig. 2.7a) and
DU-145 cells (Fig. 2.7b) after incubation with blank SLN, EGCG and EGCG-SLN in a
concentration range of 10–100 μg/mL for 24 hours. In the case of Blank-SLN, cell
viability in both cancer cell lines was more than 90% at all tested concentrations which
indicated its high biocompatibility and non-toxicity. After treatment with EGCG, cell
viability in both cancer cells (MDA-MB-231 and DU-145) does not decrease
substantially with an increase in EGCG concentration. However, EGCG-SLN showed a
rapid decline in the viability of cancer cells. The cell viability of MDA-MB-231 was
77.07% after treatment with 10 μg/mL of EGCG which progressively decreased to
42.96% viability at the maximum concentration.
Fig 2.7a
0
20
40
60
80
100
10 20 40 80 100
%
Cell
via
bilit
y
Concentration (μg/mL)
MDA-MB-231 Blank SLN
EGCG
EGCG-SLN
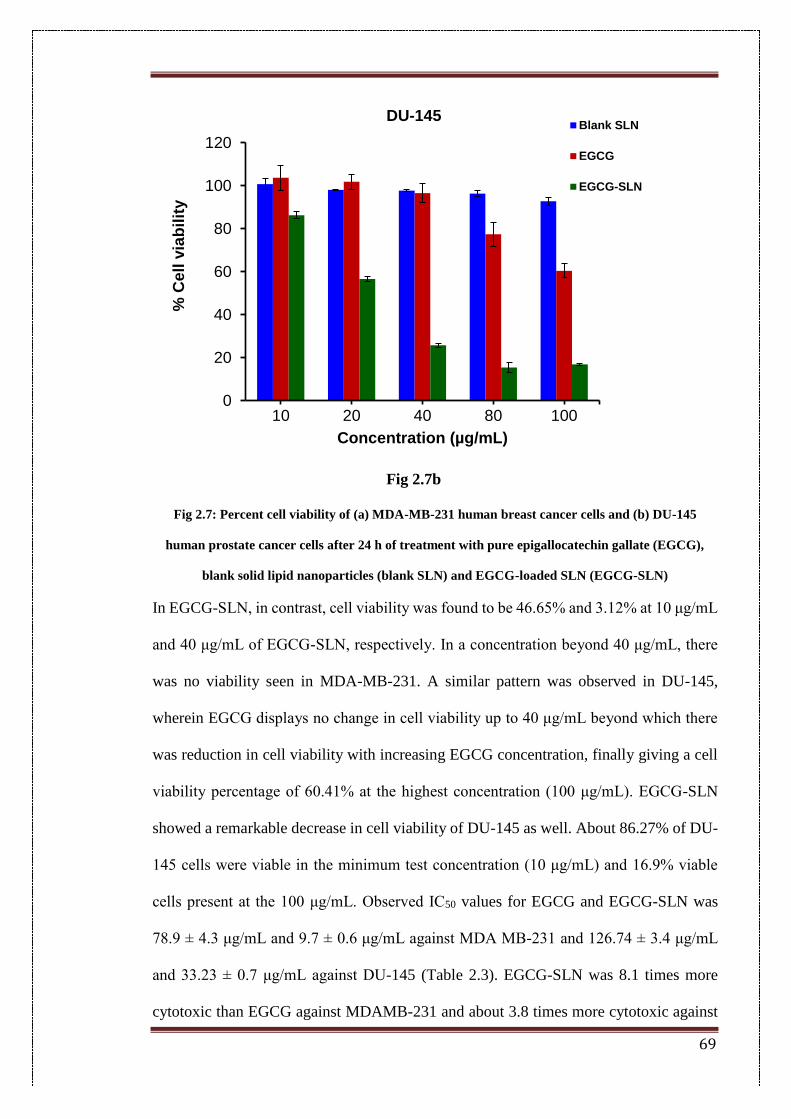
69
Fig 2.7b
Fig 2.7: Percent cell viability of (a) MDA-MB-231 human breast cancer cells and (b) DU-145
human prostate cancer cells after 24 h of treatment with pure epigallocatechin gallate (EGCG),
blank solid lipid nanoparticles (blank SLN) and EGCG-loaded SLN (EGCG-SLN)
In EGCG-SLN, in contrast, cell viability was found to be 46.65% and 3.12% at 10 μg/mL
and 40 μg/mL of EGCG-SLN, respectively. In a concentration beyond 40 μg/mL, there
was no viability seen in MDA-MB-231. A similar pattern was observed in DU-145,
wherein EGCG displays no change in cell viability up to 40 μg/mL beyond which there
was reduction in cell viability with increasing EGCG concentration, finally giving a cell
viability percentage of 60.41% at the highest concentration (100 μg/mL). EGCG-SLN
showed a remarkable decrease in cell viability of DU-145 as well. About 86.27% of DU-
145 cells were viable in the minimum test concentration (10 μg/mL) and 16.9% viable
cells present at the 100 μg/mL. Observed IC50 values for EGCG and EGCG-SLN was
78.9 ± 4.3 μg/mL and 9.7 ± 0.6 μg/mL against MDA MB-231 and 126.74 ± 3.4 μg/mL
and 33.23 ± 0.7 μg/mL against DU-145 (Table 2.3). EGCG-SLN was 8.1 times more
cytotoxic than EGCG against MDAMB-231 and about 3.8 times more cytotoxic against
0
20
40
60
80
100
120
10 20 40 80 100
% C
ell
via
bilit
y
Concentration (µg/mL)
DU-145Blank SLN
EGCG
EGCG-SLN

70
DU-145. This increased cytotoxicity of EGCG-SLN in cancerous cells can be harnessed
in future in-vivo applications as tumours are often characterized by a defective, leaky
vascular architecture because of the poorly regulated nature of tumour angiogenesis and
the interstitial fluid within a tumour is usually inadequately drained by a poorly formed
lymphatic system. All of these factors contribute towards enhanced extravasation and
selective retention of sub-micron particles within tumour tissues[59]. This phenomenon
is called the enhanced permeation and retention (EPR) effect which is can be extensively
used by nanoparticle systems like EGCG-SLN for passive targeting in cancer therapy.
Table 2.3: IC50 values (µg/mL) for epigallocatechin gallate (EGCG) and EGCG-loaded solid lipid
nanoparticles (EGCG-SLN) against MDA-MB-231 human breast cancer cells and DU-145 human
prostate cancer cells after 24 h of treatment
Formulation
MDA-MB-231 DU-145
EGCG 78.9 ± 4.3 126.7 ± 3.4
EGCG-SLN 9.7 ± 0.6 33.2 ± 1.5

71
Fig 2.8a
Fig 2.8b
Fig 2.8: Phase contrast microscopy images of (a) MDA-MB-231 human breast cancer cells and (b)
DU-145 human prostate cancer cells after 24 h of treatment with blank solid lipid nanoparticles
(blank SLN), pure epigallocatechin gallate (EGCG), or EGCG-loaded SLN (EGCG-SLN)
equivalent to 20 μg/mL EGCG.

72
Fig. 2.8 shows phase contrast images of control cells, cells treated with pure EGCG, blank
SLN or EGCG-SLN after 24 h incubation. Fig. 2.8a and 2.8b show MDA-MB 231 and
DU-145 cells treated with the above formulations, respectively. The cell morphology in
MDA-MB 231 and DU-145 control cells is characterized by elongated epithelial cells. A
change in morphology from elongated to spherical and cell shrinkage was visible in both
the cell lines treated with EGCG and EGCG-SLN. However, a greater amount of cell
shrinkage was visible in cells treated with EGCG-SLN suggesting greater cytotoxicity in
the EGCG-SLN formulation compared to pure EGCG in agreement with the results in
Fig. 2.7.
2.4.9. Apoptosis studies
Apoptosis of EGCG-SLN and EGCG in MDA-MB-231 and DU-145 cells was studied
using the nucleus staining dye Hoechst 33342. The cells were treated with EGCG and
EGCG-SLN for 24 h and then stained with Hoechst 33342 (Fig. 2.9). Nuclear
fragmentation and apoptotic bodies were observed at a magnification of 10x with a scale
of 100 µm. Untreated or control cells exhibited spherical and intact nuclei. Cells treated
with EGCG-SLN showed more apoptotic bodies (32.7% in MDA-MB-231 and 24.9% in
DU-145) and nuclear shrinkage compared to the EGCG-treated cells (14.5% in MDA-
MB-231 and 11.8% in DU-145) which are consistent with the results from the anti-
proliferation assay (Section 2.4.7.), thus proving that encapsulation in SLN facilitated
greater anti-cancer efficiency and hence enhanced apoptosis of EGCG.
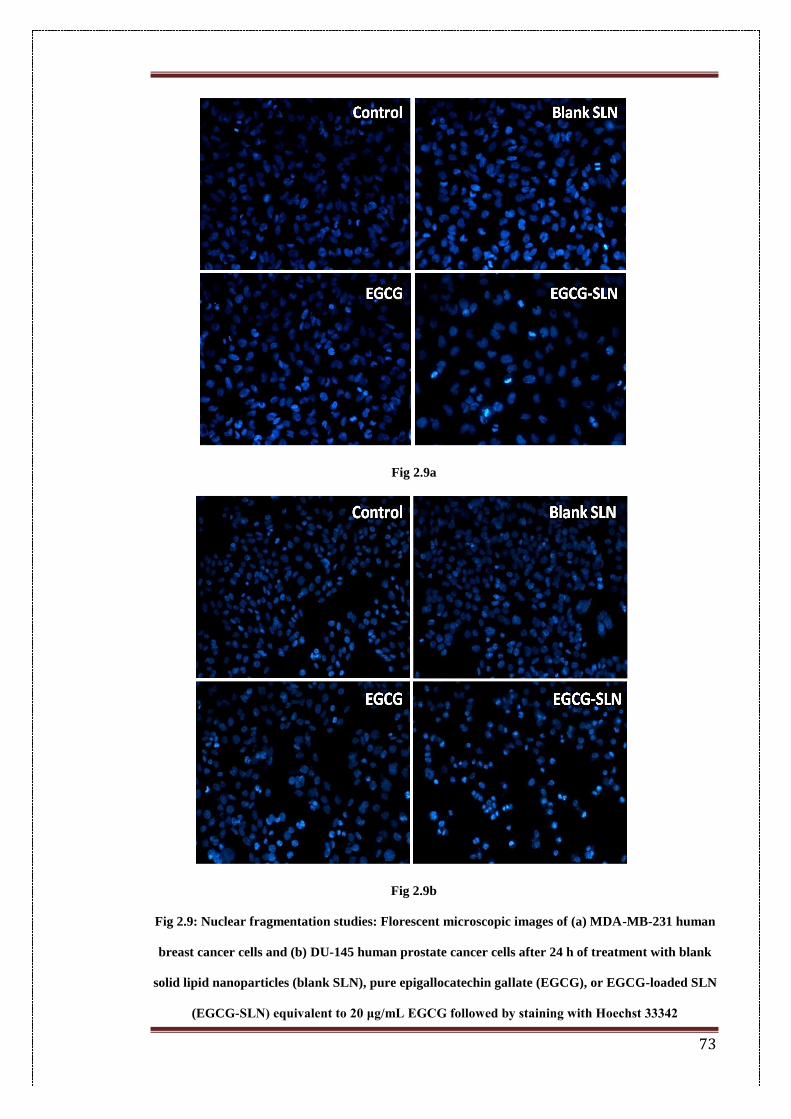
73
Fig 2.9a
Fig 2.9b
Fig 2.9: Nuclear fragmentation studies: Florescent microscopic images of (a) MDA-MB-231 human
breast cancer cells and (b) DU-145 human prostate cancer cells after 24 h of treatment with blank
solid lipid nanoparticles (blank SLN), pure epigallocatechin gallate (EGCG), or EGCG-loaded SLN
(EGCG-SLN) equivalent to 20 μg/mL EGCG followed by staining with Hoechst 33342

74
2.4.10. Colloidal stability studies
2.4.10.1. Stability of NPs in serum and physiological conditions
The colloidal stability of the nanoparticles was determined in distilled water and PBS and
expressed as a change in the particle size and zeta potential with time (Fig. 2.10). After
intravenous administration, the size and charge of the nanoparticles are mainly
determined by the adsorbed serum components[60]. Hence, a similar study was also
carried out in rat serum. Particle size in distilled water remains constant with the size of
112 nm at 0 h and 118.5 nm at 4 h. As expected, an initial increase in size (189.1 nm) in
EGCG-SLN is observed in serum owing to the adsorption of serum proteins, subsequently
falling to 134.3 nm at 2 h, following gradual protein desorption[61].
Particle size increases to 154.3 nm at the end of 8 h and 207 nm at 24 h. Particle size in
PBS increases from 112 nm to 247.15 nm at 2 h, after which it stays constant. Zeta
potential values for EGCG-SLN in distilled water remain constant with negative
potentials of −30.3, −28.4, −32.3, −28.9, −31.3 and −29.5 mV at 0, 1, 2, 4, 8 and 24 h
respectively. In PBS, the surface potential is −16.2, −19.2, −12.3, −10.7, −16.2 and −17.4
mV at 0, 1, 2, 4, 8 and 24 h, respectively. The reduced negative potential of the
nanoparticle suspension may be attributed to electrostatic interaction of opposite ions
present in PBS. Surface potential in serum was −7.01, −7.39, −4.43, −7.64, −6.99 and
−7.17 mV at 0, 1, 2, 4, 8 and 24 h, respectively. The reduced negative potential of EGCG-
SLN after exposing to serum may be due to the cumulative surface charges caused by the
adsorption of serum proteins and other serum components[62].
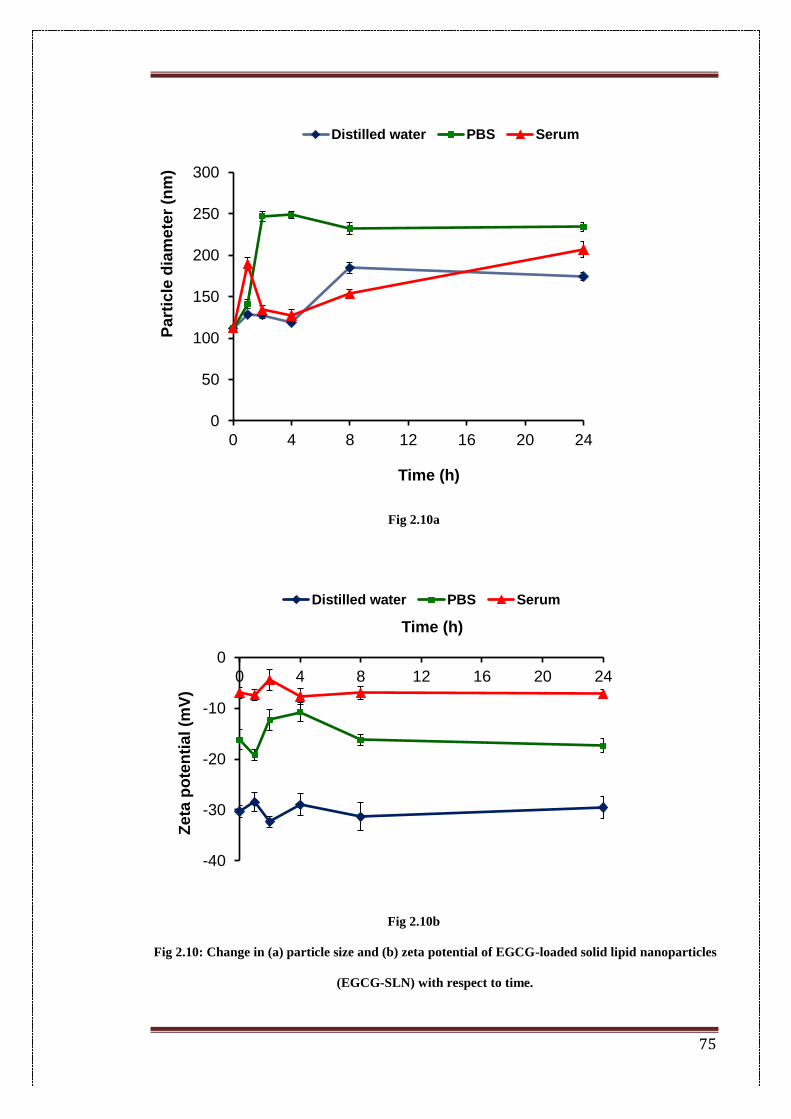
75
Fig 2.10a
Fig 2.10b
Fig 2.10: Change in (a) particle size and (b) zeta potential of EGCG-loaded solid lipid nanoparticles
(EGCG-SLN) with respect to time.
0
50
100
150
200
250
300
0 4 8 12 16 20 24
Pa
rtic
le d
iam
ete
r (n
m)
Time (h)
Distilled water PBS Serum
-40
-30
-20
-10
0
0 4 8 12 16 20 24
Ze
ta p
ote
nti
al
(mV
)
Time (h)
Distilled water PBS Serum
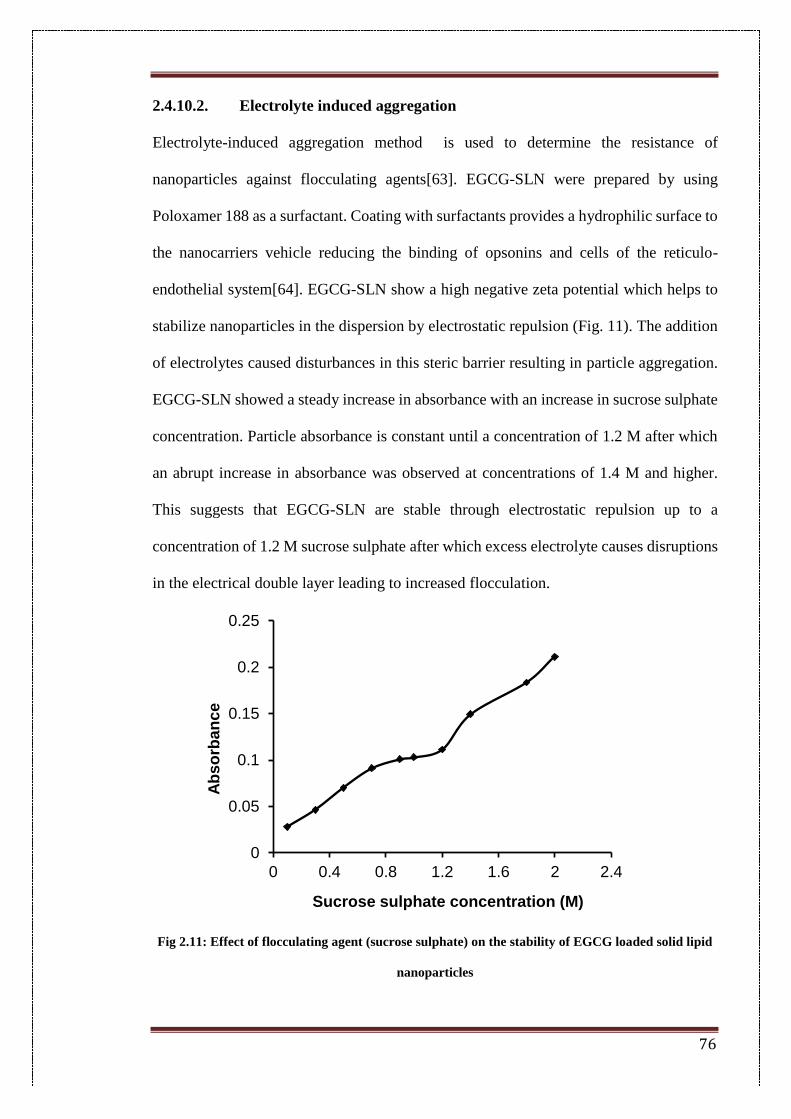
76
2.4.10.2. Electrolyte induced aggregation
Electrolyte-induced aggregation method is used to determine the resistance of
nanoparticles against flocculating agents[63]. EGCG-SLN were prepared by using
Poloxamer 188 as a surfactant. Coating with surfactants provides a hydrophilic surface to
the nanocarriers vehicle reducing the binding of opsonins and cells of the reticulo-
endothelial system[64]. EGCG-SLN show a high negative zeta potential which helps to
stabilize nanoparticles in the dispersion by electrostatic repulsion (Fig. 11). The addition
of electrolytes caused disturbances in this steric barrier resulting in particle aggregation.
EGCG-SLN showed a steady increase in absorbance with an increase in sucrose sulphate
concentration. Particle absorbance is constant until a concentration of 1.2 M after which
an abrupt increase in absorbance was observed at concentrations of 1.4 M and higher.
This suggests that EGCG-SLN are stable through electrostatic repulsion up to a
concentration of 1.2 M sucrose sulphate after which excess electrolyte causes disruptions
in the electrical double layer leading to increased flocculation.
Fig 2.11: Effect of flocculating agent (sucrose sulphate) on the stability of EGCG loaded solid lipid
nanoparticles
0
0.05
0.1
0.15
0.2
0.25
0 0.4 0.8 1.2 1.6 2 2.4
Ab
so
rba
nc
e
Sucrose sulphate concentration (M)

77
2.5. Conclusions
In summary, EGCG loaded solid lipid nanoparticles were formulated to improve the
stability and anti-cancer activity of EGCG. SLNs were prepared by a double-
emulsification method and optimized to provide nanoparticles with a particles size less
than 200 nm and high drug encapsulation efficiency. EGCG-SLN showed significantly
increased stability of encapsulated EGCG which was unstable at physiological conditions
and showed complete degradation in pure form. In-vitro cytotoxicity studies showed that
EGCG-SLN caused an 8.1fold increase in cytotoxicity of EGCG against MDA-MB-231
cells and 3.8 times increase against DU-145 cancer cells. Further, in colloidal stability
studies, EGCG-SLN showed stability in both serum and PBS and high resistance to
electrolyte-induced aggregation. Therefore, EGCG-SLN can be considered a promising
system for delivering EGCG as a potential chemotherapeutic agent.

78
2.6. References
[1] H. Lu, J. Zhang, Y. Yang, X. Yang, B. Xu, W. Yang, et al., Earliest tea as evidence
for one branch of the Silk Road across the Tibetan Plateau, Sci. Rep. 6 (2016) 18955.
doi:10.1038/srep18955.
[2] C. Cabrera, R. Artacho, R. Giménez, Beneficial Effects of Green Tea—A Review,
J. Am. Coll. Nutr. 25 (2006) 79–99. doi:10.1080/07315724.2006.10719518.
[3] C.J. Dufresne, E.R. Farnworth, A review of latest research findings on the health
promotion properties of tea, J. Nutr. Biochem. 12 (2001) 404–421. doi:10.1016/S0955-
2863(01)00155-3.
[4] H. Mukhtar, N. Ahmad, Tea polyphenols: Prevention of cancer and optimizing
health, Am. J. Clin. Nutr. 71 (2000) 1698–1702.
http://ajcn.nutrition.org/content/71/6/1698s.full.pdf (accessed October 1, 2015).
[5] G.-J. Du, Z. Zhang, X.-D. Wen, C. Yu, T. Calway, C.-S. Yuan, et al.,
Epigallocatechin Gallate (EGCG) is the most effective cancer chemopreventive
polyphenol in green tea., Nutrients. 4 (2012) 1679–91. doi:10.3390/nu4111679.
[6] N. Khan, H. Mukhtar, Tea polyphenols for health promotion., Life Sci. 81 (2007)
519–33. doi:10.1016/j.lfs.2007.06.011.
[7] M.E. Cavet, K.L. Harrington, T.R. Vollmer, K.W. Ward, J.-Z. Zhang, Anti-
inflammatory and anti-oxidative effects of the green tea polyphenol epigallocatechin
gallate in human corneal epithelial cells., Mol. Vis. 17 (2011) 533–42.
/pmc/articles/PMC3044696/?report=abstract (accessed December 30, 2015).
[8] S. Klaus, S. Pültz, C. Thöne-Reineke, S. Wolfram, Epigallocatechin gallate
attenuates diet-induced obesity in mice by decreasing energy absorption and increasing
fat oxidation., Int. J. Obes. (Lond). 29 (2005) 615–23. doi:10.1038/sj.ijo.0802926.
[9] M.-S. Lee, C.-T. Kim, Y. Kim, Green tea (-)-epigallocatechin-3-gallate reduces

79
body weight with regulation of multiple genes expression in adipose tissue of diet-
induced obese mice., Ann. Nutr. Metab. 54 (2009) 151–7. doi:10.1159/000214834.
[10] E. Tedeschi, h. suzuki, m. menegazzi, Antiinflammatory Action of EGCG, the
Main Component of Green Tea, through STAT-1 Inhibition, Ann. N. Y. Acad. Sci. 973
(2002) 435–437. doi:10.1111/j.1749-6632.2002.tb04678.x.
[11] A.M. El-Mowafy, M.M. Al-Gayyar, H.A. Salem, M.E. El-Mesery, M.M.
Darweish, Novel chemotherapeutic and renal protective effects for the green tea (EGCG):
role of oxidative stress and inflammatory-cytokine signaling., Phytomedicine. 17 (2010)
1067–75. doi:10.1016/j.phymed.2010.08.004.
[12] M. Shimizu, Y. Shirakami, H. Moriwaki, Targeting receptor tyrosine kinases for
chemoprevention by green tea catechin, EGCG., Int. J. Mol. Sci. 9 (2008) 1034–49.
doi:10.3390/ijms9061034.
[13] H. Tachibana, Molecular basis for cancer chemoprevention by green tea polyphenol
EGCG., Forum Nutr. 61 (2009) 156–69. doi:10.1159/000212748.
[14] C. Braicu, C.D. Gherman, A. Irimie, I. Berindan-Neagoe, Epigallocatechin-3-
Gallate (EGCG) Inhibits Cell Proliferation and Migratory Behaviour of Triple Negative
Breast Cancer Cells, J. Nanosci. Nanotechnol. 13 (2013) 632–637.
doi:10.1166/jnn.2013.6882.
[15] Y.D. Jung, L.M. Ellis, Inhibition of tumour invasion and angiogenesis by
epigallocatechin gallate (EGCG), a major component of green tea, Int. J. Exp. Pathol. 82
(2002) 309–316. doi:10.1046/j.1365-2613.2001.00205.x.
[16] Y. Xu, C.-T. Ho, S.G. Amin, C. Han, F.-L. Chung, Inhibition of Tobacco-specific
Nitrosamine-induced Lung Tumorigenesis in A/J Mice by Green Tea and Its Major
Polyphenol as Antioxidants, Cancer Res. 52 (1992) 3875–3879.
http://cancerres.aacrjournals.org/content/52/14/3875.short (accessed December 30,

80
2015).
[17] D.E. Shippen, Telomeres and telomerases, Curr. Opin. Genet. Dev. 3 (1993) 759–
763. http://ac.els-cdn.com.ezproxy.lib.rmit.edu.au/S0959437X05800954/1-s2.0-
S0959437X05800954-main.pdf?_tid=06936214-5c05-11e7-857f-
00000aab0f6b&acdnat=1498656411_c66add9d63a10db92be00e1f5db484ec (accessed
June 28, 2017).
[18] L.S. Beese, J.R. Kiefer, C. Mao, J.C. Braman, Visualizing DNA replication in a
catalytically active Bacillus DNA polymerase crystal, Nature. 391 (1998) 304–307.
doi:10.1038/34693.
[19] K. Collins, J.R. Mitchell, Telomerase in the human organism, Oncogene. 21 (2002)
564–579. doi:10.1038/sj.onc.1205083.
[20] N.W. Kim, M.A. Piatyszek, K.R. Prowse, C.B. Harley, M.D. West, P.L.C. Ho, et
al., Specific Association of Human Telomerase Activity with Immortal Cells and Cancer,
Science (80-. ). 266 (n.d.) 2011–2015. doi:10.2307/2885288.
[21] J.B. Berletch, C. Liu, W.K. Love, L.G. Andrews, S.K. Katiyar, T.O. Tollefsbol,
Epigenetic and genetic mechanisms contribute to telomerase inhibition by EGCG, J. Cell.
Biochem. 103 (2008) 509–519. doi:10.1002/jcb.21417.
[22] J.G. Herman, S.B. Baylin, Gene Silencing in Cancer in Association with Promoter
Hypermethylation, N. Engl. J. Med. 349 (2003) 2042–2054.
doi:10.1056/NEJMra023075.
[23] S.B. Baylin, DNA methylation and gene silencing in cancer, Nat. Clin. Pract.
Oncol. 2 (2005) S4–S11. doi:10.1038/ncponc0354.
[24] M.Z. Fang, Y. Wang, N. Ai, Z. Hou, Y. Sun, H. Lu, et al., Tea Polyphenol (−)-
Epigallocatechin-3-Gallate Inhibits DNA Methyltransferase and Reactivates
Methylation-Silenced Genes in Cancer Cell Lines, Cancer Res. 63 (2003).

81
http://cancerres.aacrjournals.org/content/63/22/7563.short (accessed June 28, 2017).
[25] J. Folkman, Role of angiogenesis in tumor growth and metastasis, Semin. Oncol.
29 (2002) 15–18. doi:10.1016/S0093-7754(02)70065-1.
[26] N. Ferrara, VEGF and the quest for tumour angiogenesis factors, Nat. Rev. Cancer.
2 (2002) 795–803. doi:10.1038/nrc909.
[27] Y.D. Jung, M.S. Kim, B.A. Shin, K.O. Chay, B.W. Ahn, W. Liu, et al., EGCG, a
major component of green tea, inhibits tumour growth by inhibiting VEGF induction in
human colon carcinoma cells., Br. J. Cancer. 84 (2001) 844–50.
doi:10.1054/bjoc.2000.1691.
[28] I.J. Fidler, Origin and biology of cancer metastasis, Cytometry. 10 (1989) 673–680.
doi:10.1002/cyto.990100602.
[29] S. Garbisa, L. Sartor, S. Biggin, B. Salvato, R. Benelli, A. Albini., Tumor
gelatinases and invasion inhibited by the green tea flavanol epigallocatechin-3-gallate,
Cancer. 91 (2001) 822–832. doi:10.1002/1097-0142(20010215)91:4<822::AID-
CNCR1070>3.0.CO;2-G.
[30] J. Koff, S. Ramachandiran, L. Bernal-Mizrachi, A Time to Kill: Targeting
Apoptosis in Cancer, Int. J. Mol. Sci. 16 (2015) 2942–2955. doi:10.3390/ijms16022942.
[31] J. Sonoda, R. Ikeda, Y. Baba, K. Narumi, A. Kawachi, E. Tomishige, et al.,
Experimental and therapeutic medicine., [Spandidos Pub.], 2014. https://www.spandidos-
publications.com/10.3892/etm.2014.1719 (accessed June 28, 2017).
[32] M. Leone, D. Zhai, S. Sareth, S. Kitada, J.C. Reed, M. Pellecchia, Cancer
Prevention by Tea Polyphenols Is Linked to Their Direct Inhibition of Antiapoptotic Bcl-
2-Family Proteins, Cancer Res. 63 (2003).
http://cancerres.aacrjournals.org/content/63/23/8118.short (accessed June 28, 2017).
[33] J.S. Sebolt-Leopold, R. Herrera, Targeting the mitogen-activated protein kinase

82
cascade to treat cancer, Nat. Rev. Cancer. 4 (2004) 937–947. doi:10.1038/nrc1503.
[34] M. Li, Z. He, S. Ermakova, D. Zheng, F. Tang, Y.-Y. Cho, et al., Direct Inhibition
of Insulin-Like Growth Factor-I Receptor Kinase Activity by (−)−Epigallocatechin-3-
Gallate Regulates Cell Transformation, Cancer Epidemiol. Prev. Biomarkers. 16 (2007).
http://cebp.aacrjournals.org/content/16/3/598.short (accessed June 28, 2017).
[35] J.D. Lambert, R.J. Elias, The antioxidant and pro-oxidant activities of green tea
polyphenols: a role in cancer prevention., Arch. Biochem. Biophys. 501 (2010) 65–72.
doi:10.1016/j.abb.2010.06.013.
[36] Q.Y. Zhu, A. Zhang, D. Tsang, Y. Huang, Z.-Y. Chen, Stability of Green Tea
Catechins, J. Agric. Food Chem. 45 (1997) 4624–4628. doi:10.1021/jf9706080.
[37] S. Sang, J.D. Lambert, C.-T. Ho, C.S. Yang, The chemistry and biotransformation
of tea constituents., Pharmacol. Res. 64 (2011) 87–99. doi:10.1016/j.phrs.2011.02.007.
[38] L. Zhang, Y. Zheng, M.S.S. Chow, Z. Zuo, Investigation of intestinal absorption
and disposition of green tea catechins by Caco-2 monolayer model., Int. J. Pharm. 287
(2004) 1–12. doi:10.1016/j.ijpharm.2004.08.020.
[39] W. Mehnert, Solid lipid nanoparticles Production, characterization and
applications, Adv. Drug Deliv. Rev. 47 (2001) 165–196. doi:10.1016/S0169-
409X(01)00105-3.
[40] D. Pooja, H. Kulhari, L. Tunki, S. Chinde, M. Kuncha, P. Grover, et al.,
Nanomedicines for targeted delivery of etoposide to non-small cell lung cancer using
transferrin functionalized nanoparticles, RSC Adv. 5 (2015) 49122–49131.
doi:10.1039/C5RA03316K.
[41] S. Weber, A. Zimmer, J. Pardeike, Solid Lipid Nanoparticles (SLN) and
Nanostructured Lipid Carriers (NLC) for pulmonary application: a review of the state of
the art., Eur. J. Pharm. Biopharm. 86 (2014) 7–22. doi:10.1016/j.ejpb.2013.08.013.

83
[42] C. Qi, Y. Chen, Q.-Z. Jing, X.-G. Wang, Preparation and characterization of
catalase-loaded solid lipid nanoparticles protecting enzyme against proteolysis., Int. J.
Mol. Sci. 12 (2011) 4282–93. doi:10.3390/ijms12074282.
[43] C. Schwarz, W. Mehnert, J.S. Lucks, R.H. Müller, Solid lipid nanoparticles (SLN)
for controlled drug delivery. I. Production, characterization and sterilization, J. Control.
Release. 30 (1994) 83–96. doi:10.1016/0168-3659(94)90047-7.
[44] K. Vivek, H. Reddy, R.S.R. Murthy, Investigations of the effect of the lipid matrix
on drug entrapment, in vitro release, and physical stability of olanzapine-loaded solid
lipid nanoparticles., AAPS PharmSciTech. (2007). doi:10.1208/pt0804083.
[45] A. Radomska-Soukharev, Stability of lipid excipients in solid lipid nanoparticles.,
Adv. Drug Deliv. Rev. 59 (2007) 411–8. doi:10.1016/j.addr.2007.04.004.
[46] D. Pooja, L. Tunki, H. Kulhari, B.B. Reddy, R. Sistla, Optimization of solid lipid
nanoparticles prepared by a single emulsification-solvent evaporation method., Data Br.
6 (2016) 15–9. doi:10.1016/j.dib.2015.11.038.
[47] C. Freitas, R.H. Müller, Correlation between long-term stability of solid lipid
nanoparticles (SLNTM) and crystallinity of the lipid phase, Eur. J. Pharm. Biopharm. 47
(1999) 125–132. doi:10.1016/S0939-6411(98)00074-5.
[48] T. Helgason, T.S. Awad, K. Kristbergsson, D.J. McClements, J. Weiss, Effect of
surfactant surface coverage on formation of solid lipid nanoparticles (SLN), J. Colloid
Interface Sci. 334 (2009) 75–81. doi:10.1016/j.jcis.2009.03.012.
[49] H. Bunjes, M.H.J. Koch, K. Westesen, Influence of emulsifiers on the
crystallization of solid lipid nanoparticles., J. Pharm. Sci. 92 (2003) 1509–20.
doi:10.1002/jps.10413.
[50] F. Franco, L.A. Pérez-Maqueda, J.L. Pérez-Rodríguez, The effect of ultrasound on
the particle size and structural disorder of a well-ordered kaolinite, J. Colloid Interface

84
Sci. 274 (2004) 107–117. doi:10.1016/j.jcis.2003.12.003.
[51] S. Thomas, D. Rouxel, D. Ponnamma, Spectroscopy of polymer nanocomposites,
n.d.
[52] H. Gleiter, Nanostructured materials: basic concepts and microstructure, Acta
Mater. 48 (2000) 1–29. doi:10.1016/S1359-6454(99)00285-2.
[53] S. Das, W.K. Ng, R.B.H. Tan, Are nanostructured lipid carriers (NLCs) better than
solid lipid nanoparticles (SLNs): development, characterizations and comparative
evaluations of clotrimazole-loaded SLNs and NLCs?, Eur. J. Pharm. Sci. 47 (2012) 139–
51. doi:10.1016/j.ejps.2012.05.010.
[54] A. Kovacevic, S. Savic, G. Vuleta, R.H. Müller, C.M. Keck, Polyhydroxy
surfactants for the formulation of lipid nanoparticles (SLN and NLC): effects on size,
physical stability and particle matrix structure., Int. J. Pharm. 406 (2011) 163–72.
doi:10.1016/j.ijpharm.2010.12.036.
[55] G. Suresh, K. Manjunath, V. Venkateswarlu, V. Satyanarayana, Preparation,
characterization, and in vitro and in vivo evaluation of lovastatin solid lipid
nanoparticles., AAPS PharmSciTech. 8 (2007) 24. doi:10.1208/pt0801024.
[56] A.P. Nayak, W. Tiyaboonchai, S. Patankar, B. Madhusudhan, E.B. Souto,
Curcuminoids-loaded lipid nanoparticles: novel approach towards malaria treatment.,
Colloids Surf. B. Biointerfaces. 81 (2010) 263–73. doi:10.1016/j.colsurfb.2010.07.020.
[57] A. zur Mühlen, C. Schwarz, W. Mehnert, Solid lipid nanoparticles (SLN) for
controlled drug delivery – Drug release and release mechanism, Eur. J. Pharm. Biopharm.
45 (1998) 149–155. doi:10.1016/S0939-6411(97)00150-1.
[58] J.F. Fangueiro, T. Andreani, L. Fernandes, M.L. Garcia, M.A. Egea, A.M. Silva, et
al., Physicochemical characterization of epigallocatechin gallate lipid nanoparticles
(EGCG-LNs) for ocular instillation., Colloids Surf. B. Biointerfaces. 123 (2014) 452–60.

85
doi:10.1016/j.colsurfb.2014.09.042.
[59] K. Greish, Enhanced permeability and retention (EPR) effect for anticancer
nanomedicine drug targeting., Methods Mol. Biol. 624 (2010) 25–37. doi:10.1007/978-
1-60761-609-2_3.
[60] C. Lourenco, Steric stabilization of nanoparticles: Size and surface properties, Int.
J. Pharm. 138 (1996) 1–12. doi:10.1016/0378-5173(96)04486-9.
[61] T. Verrecchia, P. Huve, D. Bazile, M. Veillard, G. Spenlehauer, P. Couvreur,
Adsorption/desorption of human serum albumin at the surface of poly(lactic acid)
nanoparticles prepared by a solvent evaporation process, J Biomed Mater Res. 27 (1993)
1019–1028.
http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Cita
tion&list_uids=8408114.
[62] R. Vinluan, J. Zheng, Serum protein adsorption and excretion pathways of metal
nanoparticles, Nanomedicine. 10 (2015) 2781–2794. doi:10.2217/nnm.15.97.
[63] K. Vivek, H. Reddy, R.S.R. Murthy, Investigations of the effect of the lipid matrix
on drug entrapment, in vitro release, and physical stability of olanzapine-loaded solid
lipid nanoparticles., AAPS PharmSciTech. 8 (2007) E83. doi:10.1208/pt0804083.
[64] V.S. Shenoy, R.P. Gude, R.S.R. Murthy, Paclitaxel-loaded glyceryl palmitostearate
nanoparticles: in vitro release and cytotoxic activity., J. Drug Target. 17 (2009) 304–10.
doi:10.1080/10611860902737938.

86
Peptide conjugated solid lipid
nanoparticles for breast cancer therapy

87
3.1. Background
In 2012, an estimated 14.1 million new cases of cancer occurred worldwide. Four of the
most common cancers occurring worldwide are lung, female breast, bowel and prostate
cancers, together constituting around 40% of all the cancer cases[1]. Breast cancer is the
most frequent cancer in women, with 1.67 million cases diagnosed in 2012, accounting
for one in four of all cancer cases in the world for women[2].
Although many different chemotherapy drugs and drug regimens have been explored for
the treatment of cancer, the disadvantages of non-specificity with cytotoxic drugs have
to be circumvented to reduce non-specific organ damage[3]. The dissipation of the drug
in other areas also results in the concomitant increase in drug dosages, as the intended
organ may not get the total therapeutic dose required[4].
Active targeting involves the attachment of ligands specific to factors overexpressed in
cancer[5] to the nanoparticle. This leads to more accurate delivery as only cancer cells
will receive the cytotoxic drug. Active targeting is therefore being explored in order to
overcome the significant shortcomings associated with traditional chemotherapy. Gastrin
releasing peptide receptor (GRPR) has been observed to be overexpressed in a variety of
cancer types such as prostate, breast, cervical, glioblastomas, small cell lung cancer,
gastric, pancreatic, and colon cancers[6]. It was therefore used as a cancer marker in this
work. GRPR is a glycoprotein receptor (Chapter 1; Fig 1.9) with 7 transmembrane
domains consisting of 384 amino acids[7].
Fig 3.1: Structure of Bombesin

88
Bombesin (BBN) was the targeting ligand used in this study towards GRPR targeting.
BBN is a 14 amino acid peptide with very high affinity to the gastrin releasing peptide
receptor[8]. It was originally isolated from the skin of the toad Bombina bombina.
Bombesin is the amphibian analog to GRPR’s natural ligand mammalian gastrin releasing
peptide (GRP). GRP is a 27 amino acid peptide which shares a homologous C-terminal
seven amino acid long sequence Trp-Ala-Val-Gly-His-Leu-MetNH2 with bombesin[9].
This sequence has been reported to be important for receptor binding and ligand
internalisation via clathrin-mediated endocytosis[9]. Therefore, BBN is taken up by
GRPR with the same mechanism as in its interaction with gastrin-releasing peptide.
In this study, bombesin was conjugated to the EGCG-encapsulated SLN system that was
optimised in Chapter 2 (Please refer to Section 2.3.2.). BBN-conjugated solid lipid
nanoparticles were then explored for their anti-cancer efficacy, examining whether the
addition of a targeting ligand enhances the anti-cancer potential. In-vivo studies on
C57BL6 mice were performed to evaluate changes in tumour volumes and survivability
in comparison to control animals.
3.2. Materials
Glycerol mono-stearate (GMS) was purchased from Alfa Aesar (Johnson Matthey
Chemicals, Hyderabad, India). EGCG, bombesin, Pluronic F68 (P F68), Roswell Park
Memorial Institute (RPMI)-1640 medium, fetal bovine serum (FBS), trypsin–EDTA,
phosphate buffered saline (Ca2+, Mg2+ free), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl
tetrazolium bromide (MTT) and dimethyl sulfoxide (DMSO) were purchased from Sigma
Aldrich (St. Louis, MO, USA). Soya lecithin was purchased from HiMedia (Mumbai,
India). MDA-MB-231 human breast cancer cells, DU-145 (prostate cancer), A549(lung
cancer), HEK-293 (embryonic kidney) and L-132 lung cell lines were obtained from

89
American Type Culture Collection (ATCC, Manassas, USA). Dichloromethane was
obtained from Merck Chemicals (Mumbai, India). Tween 80 was purchased from sd Fine
Chemicals Limited (Hyderabad, India).
3.3. Methods
3.3.1. Preparation of nanoparticles
Solid lipid nanoparticles were prepared by the solvent emulsification-evaporation method
as explained in Chapter 2 (Section.2.3.3.). Briefly, 80 mg glycerol mono-stearate, 20 mg
stearic acid and 20 mg soya lecithin were dissolved in 2 mL of dichloromethane. The
inner aqueous phase containing dissolved EGCG was added to this organic phase
followed by 3 mL of 2% Pluronic F68 solution under sonication at 65% amplitude to
form a primary water-in-oil (w/o) emulsion. This emulsion was added to 7 mL of 2%
Pluronic solution to give a water-in-oil-in-water (w/o/w) emulsion. The nanoparticle
suspension was allowed to stir for 2 hours for evaporation of the solvent and further
centrifuged to precipitate out nanoparticle pellets. The supernatant was used for
evaluation of entrapment efficiency.
3.3.2. Calculation of entrapment efficiency
The drug entrapment efficiency (EE) was calculated by the indirect method as previously
mentioned in Chapter 2 (Section 2.3.3.). Briefly, the supernatant after centrifugation of
the nanoparticle suspension was used for estimation of the free drug content by
UV/Visible spectroscopy at the absorption maxima of EGCG at 274.5 nm. The percent
entrapment efficiency was calculated by the following formula:
% 𝐸𝐸 =𝑇𝑜𝑡𝑎𝑙 𝑎𝑚𝑜𝑢𝑛𝑡 𝑜𝑓 𝐸𝐺𝐶𝐺 𝑎𝑑𝑑𝑒𝑑 − 𝐴𝑚𝑜𝑢𝑛𝑡 𝑜𝑓 𝐸𝐺𝐶𝐺 𝑖𝑛 𝑠𝑢𝑝𝑒𝑟𝑛𝑎𝑡𝑎𝑛𝑡
𝑇𝑜𝑡𝑎𝑙 𝑎𝑚𝑜𝑢𝑛𝑡 𝑜𝑓 𝐸𝐺𝐶𝐺 𝑎𝑑𝑑𝑒𝑑 ×100

90
3.3.3. Bioconjugation
Conjugation, in this work, involves the formation of an amide bond between the peptide
ligand and the lipid excipients of the nanoparticle matrix. Conjugation of bombesin to the
drug loaded nanoparticles was carried out by the EDC-NHS method as described
below[10].
1-Ethyl-3-[3-dimethylaminopropyl] carbodiimide hydrochloride (EDC) is a cross linking
agent used to couple carboxylic acids to primary amines. EDC reacts with a carboxyl
group of the molecule to form an unstable amine-reactive O-acylisourea intermediate.
This could also react with a primary amine to give a stable amide bond, but the instability
of the O-acylisourea intermediate does not allow efficient coupling. If this intermediate
does not encounter an amine, it will hydrolyse and regenerate the carboxyl group.
Addition of sulpho-NHS helps to improve the stability of the intermediate to allow
reaction with the primary amine[11] Sulpho NHS (N-hydroxysulfo succinimide) reacts
with the acylisourea intermediate to form a more stable amine reactive NHS ester, which
can now react with the primary amine leading to the formation of a stable amide bond[11].
Fig 3.2: Mechanism of the EDC-NHS reaction (Reproduced with permission from Pierce et al.)

91
Briefly, the nanoparticle suspension was incubated with 15 mg N-hydroxysuccinimide
(NHS) and 20 mg N-(3-dimethylaminopropyl)-N-ethyl carbodiimide hydrochloride
(EDC). The mixture was allowed to stir for one hour in an inert nitrogen atmosphere.
Bombesin (0.25 mg) was added to this and further allowed to stir for 4 hours. The reaction
was carried out in the presence of MES buffer (pH 6.2). Conjugation of bombesin to the
lipid nanoparticles has been found to be optimum around pH 6.2 based on previous studies
[12].
The nanoparticle dispersion was collected and centrifuged at 10,000 rpm for 10 min and
washed twice with distilled water. The supernatant was collected to estimate the
conjugation efficiency using a standard Bradford protein assay. The amount of bombesin
in the supernatant gives the amount of peptide not conjugated to the nanoparticles. A
calibration curve in the range of 0.5–10 g/mL was prepared using a standard BSA stock
solution (2 mg/mL). The supernatant was diluted appropriately and the absorbance was
measured spectrophotometrically at 595 nm to determine the concentration of bombesin.
3.3.4. Physico-chemical characterization
3.3.4.1. Particle size and surface charge
The particle size and surface charge was determined by a Malvern Zetasizer using the
protocol mentioned in Chapter 2 (Section 2.3.5.1.). Briefly, the mean particle size and
zeta potential of blank nanoparticles (Blank SLN), EGCG-loaded SLNs (EGCG-SLN)
and bombesin conjugated SLNs (EB-SLN) were measured by photon correlation
spectroscopy using a Malvern Zetasizer Nano ZS (Malvern Instrument Ltd., Malvern,
UK). Samples were diluted 10 times with distilled water and analyzed at 25°C with a
backscattering angle of 173°.

92
3.3.4.2. FTIR Analysis
FTIR analysis of blank nanoparticles (Blank SLN), EGCG-loaded SLNs (EGCG-SLN)
and bombesin conjugated SLNs (EB-SLN) was carried by potassium bromide (KBr)
pellet method following the same protocol in Chapter 2 (Section 2.3.5.2.). Samples were
scanned in the range of 400–4000 cm−1.
3.3.4.3. DSC Analysis
DSC analysis of EGCG-SLN and EGCG were carried out on a DSC1 instrument (Mettler
Toledo, Switzerland) using the protocol mentioned previously in Chapter 2 (Section
2.3.5.3.)
3.3.5. In-vitro studies
3.3.5.1. In-vitro cytotoxicity
MDA-MB-231 (breast cancer), DU-145 (prostate cancer), A549(lung cancer), HEK-293
(embryonic kidney) and L-132 lung cell lines were grown in DMEM and RMI medium
supplemented with 10% fetal bovine serum, 100 µg/mL penicillin, 200 µg/mL
streptomycin and 2 mM L-glutamine. The cultures were maintained in a humidified
atmosphere with 5% CO2. Dilutions were made with sterile PBS to get the required
concentration. Formulations were filtered with a 0.22 µm sterile filter before adding to
the wells containing cells.
The cytotoxicity of formulations was determined by MTT assay based on mitochondrial
reduction of yellow MTT tetrazolium dye to a highly coloured blue formazan product
which shows absorption at 570nm[13]. 1 × 104 cells (counted by the Trypan blue
exclusion dye method) in 96 well plates were incubated with EGCG-SLN, EB-SLN and
pure EGCG with a series of concentrations for 48 h at 37 ºC in DMEM with 10% FBS
medium. Then the above media was replaced with 90 µL of fresh serum free media and
10 µL of MTT reagent (5 mg/mL) and the plates were incubated at 37 °C for 4 h. After

93
removing the media, 200 µL of DMSO was added and incubated at 37 °C for a further
10 min. The absorbance at 570 nm was measured using spectrophotometer (Spectramax
Plus, Molecular Devices, USA).
3.3.5.2. Cellular uptake
Rhodamine-B loaded nanoparticles (R-SLN) were prepared and conjugated with BBN
(RB-SLN) for comparative cellular uptake studies. For quantitative studies, 1 × 105 cells
per well were seeded in 12-well plates and allowed to adhere for 24 h. Cells were
incubated with R-SLN and RB-SLN formulations for time intervals of 12 and 24 h. After
removal of the culture media, the cells were washed twice with cold PBS and observed
under a fluorescence microscope.
3.3.5.3. Apoptosis assay
Cell apoptosis was detected by assessment of nuclear morphology staining with Hoechst
33342[14]. MDA-MB-231 cells were seeded in a 12-well culture plate at a density of 1 x
105 cells per well and cultured at 37 ºC in a 5% CO2 incubator to obtain confluency. The
cells were treated with EGCG, EB-SLN and EGCG-SLN formulation at a concentration
of 20 μg/mL of EGCG and incubated for 24 hrs. After incubation the cells were washed
with ice cold PBS twice, fixed with 4% paraformaldehyde in PBS (pH 7.4) for 15 min at
room temperature, stained with 5 µg/mL Hoechst 33342 in PBS for 15 min and washed
twice with ice cold PBS. The cells were then examined under fluorescence microscopy.
3.3.5.4. Migration assay
MDA-MB-231 (5 × 105 cells/mL) were seeded in petri dishes and allowed to grow to
80% confluence. Wounds were created with sterile 250 μL pipette tips. Cells were
incubated with EGCG, EGCG-SLN or EB-SLN, equivalent to 20 μg/mL EGCG. The
zone of wound healing was observed at 0 and 24 h using a bright field microscope. The

94
percentage of wound closure was determined by measuring the wound area using Image
J analysis software.
3.3.6. Animal studies
The animal experimental protocol for this study was approved by the Institutional Animal
Ethics Committee of the CSIR-Indian Institute of Chemical Technology, Hyderabad
(approval no. IICT/PHARM/SRK/FEB/2013/8). All the studies performed on the animals
were in accordance with the guidelines of the Committee for the Purpose of Control and
Supervision of Experiments on Animals (CPCSEA). Male C57/BL6 mice (6–8 weeks)
weighing between 20–25 g were used for the survival studies. Animals were kept in
polypropylene cages under standard laboratory conditions (12:12 h light/ dark cycle) at
24 °C. The animals were housed five per cage and had free access to food and water. The
mouse melanoma cancer cells B16F10 were grown and maintained in RPMI medium,
supplemented with 10% fetal bovine serum, 1% penicillin and streptomycin. Mice were
injected with B16F10 cells (3 × 105 /mouse in 100 µL PBS) subcutaneously into the right
flank via a 30G needle. The control group (PBS), EGCG group (1 mg EGCG/mouse),
EGCG-SLN group (1 mg EGCG equivalent of EGCG-SLN/mouse) and EB-SLN (1 mg
EGCG equivalent of EB-SLN/mouse) were tested in tumour-bearing mice. PBS, EGCG,
EGCG-SLN and EB-SLN were administrated independently via intra-peritoneal injection
every third day from day 14 after tumour implantation. The tumour size and body mass
were measured regularly following the inoculation of tumour cells. Callipers were used
to assess tumour growth by measuring two bisecting diameters in each tumour. The
following equation was used to calculate the tumour volume:

95
Tumour volume (mm3) =(width × length2)
2
Scheme 3.1: Diagrammatic representation of the animal study methodology
3.4. Results
3.4.1. Particle size and surface charge
Particle size and charge are important dimensions when constructing nanovehicles for in-
vivo applications. Nanoparticles within the size range of 6-200 nm show better tissue
penetration and hence more efficient cargo delivery[15]. Surface charge of a particle
determines the stability of the nanoparticles in the biological system. The greater the
magnitude of the surface charge (i.e. strongly positive or strongly negative) the greater
will be the repulsion with similar nanoparticles thus preventing aggregation and helping
to sustain the nanoparticulate dimensions.
Tumour cell Inoculation
•B16F10 mouse melanoma cells (3 x 105/100 µL) injected in each mouse.
•Tumour develops in 12-14 days
Formulation testing
Formulations administered
intraperitoneally at a concentration of 50 mg EGCG/kg body weight
every three days
Tumour regression
and Survival Studies
Tumour volume will be evaluated at periodic
time intervals.
Animals will be kept aside from each group for survival and dosage
will be continued till natural death
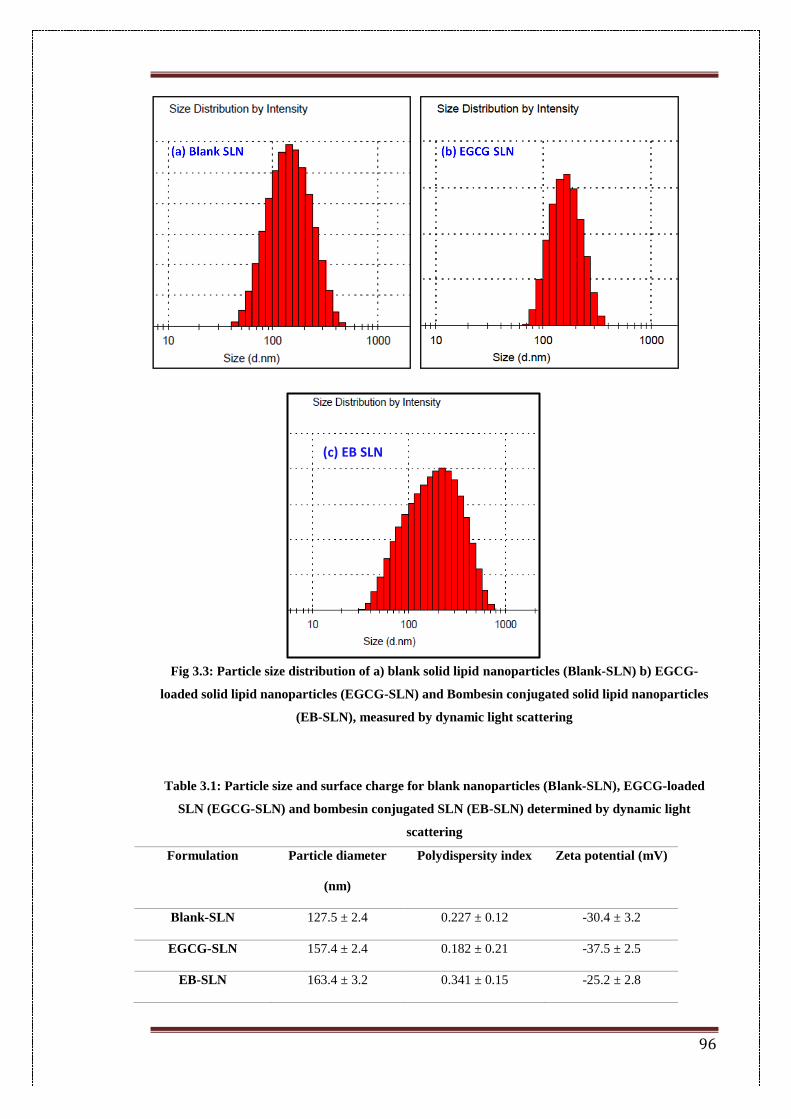
96
Fig 3.3: Particle size distribution of a) blank solid lipid nanoparticles (Blank-SLN) b) EGCG-
loaded solid lipid nanoparticles (EGCG-SLN) and Bombesin conjugated solid lipid nanoparticles
(EB-SLN), measured by dynamic light scattering
Table 3.1: Particle size and surface charge for blank nanoparticles (Blank-SLN), EGCG-loaded
SLN (EGCG-SLN) and bombesin conjugated SLN (EB-SLN) determined by dynamic light
scattering
Formulation Particle diameter
(nm)
Polydispersity index Zeta potential (mV)
Blank-SLN 127.5 ± 2.4 0.227 ± 0.12 -30.4 ± 3.2
EGCG-SLN 157.4 ± 2.4 0.182 ± 0.21 -37.5 ± 2.5
EB-SLN 163.4 ± 3.2 0.341 ± 0.15 -25.2 ± 2.8
(c) EB SLN

97
Blank nanoparticles showed a diameter of around 127.5 ± 2.4 nm with a PD index of
0.227 ± 0.12 and a zeta potential of -30.4 ± 3.2 mV. The negative charges on the
nanoparticles could be attributed to the carboxylate groups of the excipient lipids in the
nanoparticle matrix. EGCG-SLN shows a particle size of around 157.4 ± 2.4 nm, a PD
index of 0.182 ± 0.21 and a surface charge of -37.5 ± 2.5 mV. Bombesin conjugated EB-
SLN shows a particle size of 163.4 ± 3.2 nm with a PD index of 0.341 ± 0.15 and a zeta
potential of -25.2 ± 2.8 mV. The surface potential has reduced from a more negative -
37.5 mV in EGCG-SLN to -25.2 mV in EB-SLN. This reduction in negative potential
could be attributed to the masking of the negatively charged groups on the nanoparticle
matrix. As more peptides get conjugated to the nanoparticle shell, more of the negative
carboxylate groups of the charged lipids are neutralized by the formation of peptide
bonds, thus reducing the total negative potential on the particle.
3.4.2. FTIR analysis
Infrared spectroscopy was used to determine the compatibility of the drug with the
nanoparticle excipients. It also indicates the formation of a chemical bond between the
lipids in the nanoparticle and bombesin (Fig 3.4). The FTIR spectrum of EGCG shows
a characteristic peak at 3356.74 cm−1 for the O-H group attached to the aromatic ring, a
peak at 1691.30 cm−1 for the C-O group that links the trihydroxybenzoate group and
chroman group and a peak at 1543.88 cm−1 for the C-C stretch in the aromatic ring. The
blank SLN shows a broad peak at 3347 cm−1 for O-H stretching in glycerol mono-stearate
which is one of the bulk components of the lipid core. It also shows a strong peak at 2917
cm−1 for the O-H stretch and a peak at 1737.88 cm−1 for the C-O stretch in the carboxylic
group of stearic acid. It also shows a peak at 1456.05 cm−1 for C-H bending in alkanes.
EGCG-SLN also shows peaks at 3358.08 cm−1 for O-H stretching in GMS, peaks at
2917.17 cm−1 for the OH stretch and 1731.24 cm−1 for the C-O stretch in stearic acid and
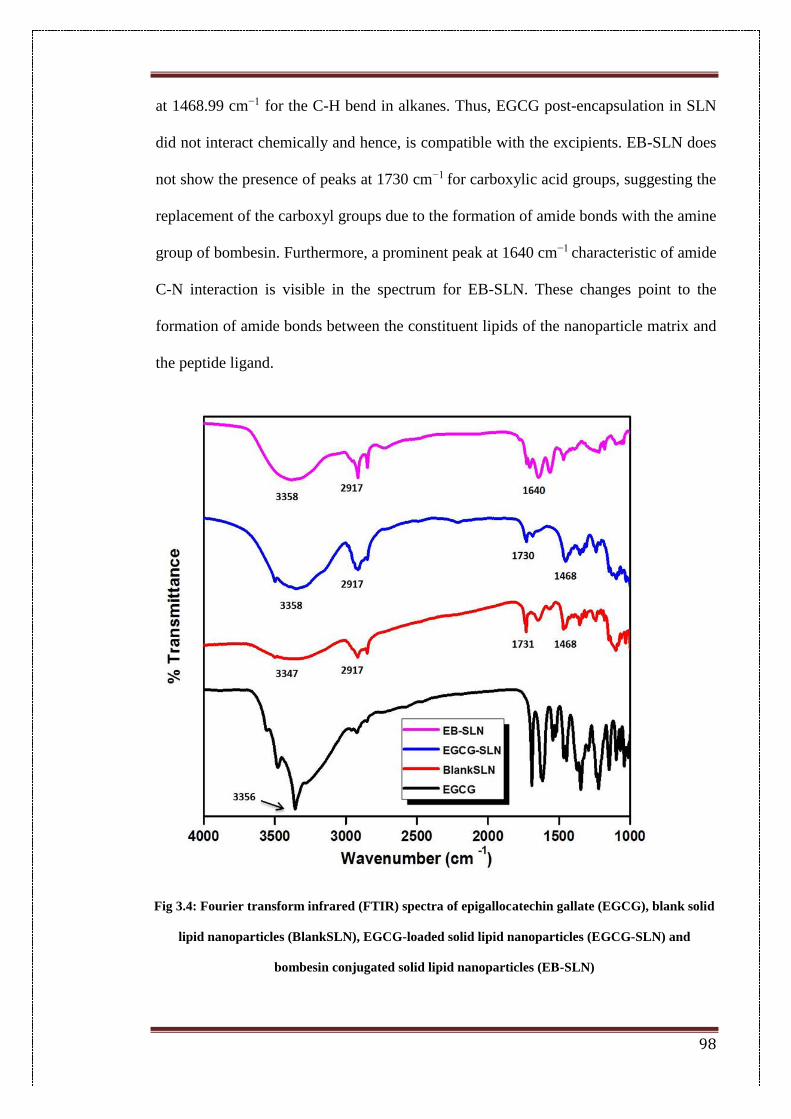
98
at 1468.99 cm−1 for the C-H bend in alkanes. Thus, EGCG post-encapsulation in SLN
did not interact chemically and hence, is compatible with the excipients. EB-SLN does
not show the presence of peaks at 1730 cm−1 for carboxylic acid groups, suggesting the
replacement of the carboxyl groups due to the formation of amide bonds with the amine
group of bombesin. Furthermore, a prominent peak at 1640 cm−1 characteristic of amide
C-N interaction is visible in the spectrum for EB-SLN. These changes point to the
formation of amide bonds between the constituent lipids of the nanoparticle matrix and
the peptide ligand.
Fig 3.4: Fourier transform infrared (FTIR) spectra of epigallocatechin gallate (EGCG), blank solid
lipid nanoparticles (BlankSLN), EGCG-loaded solid lipid nanoparticles (EGCG-SLN) and
bombesin conjugated solid lipid nanoparticles (EB-SLN)
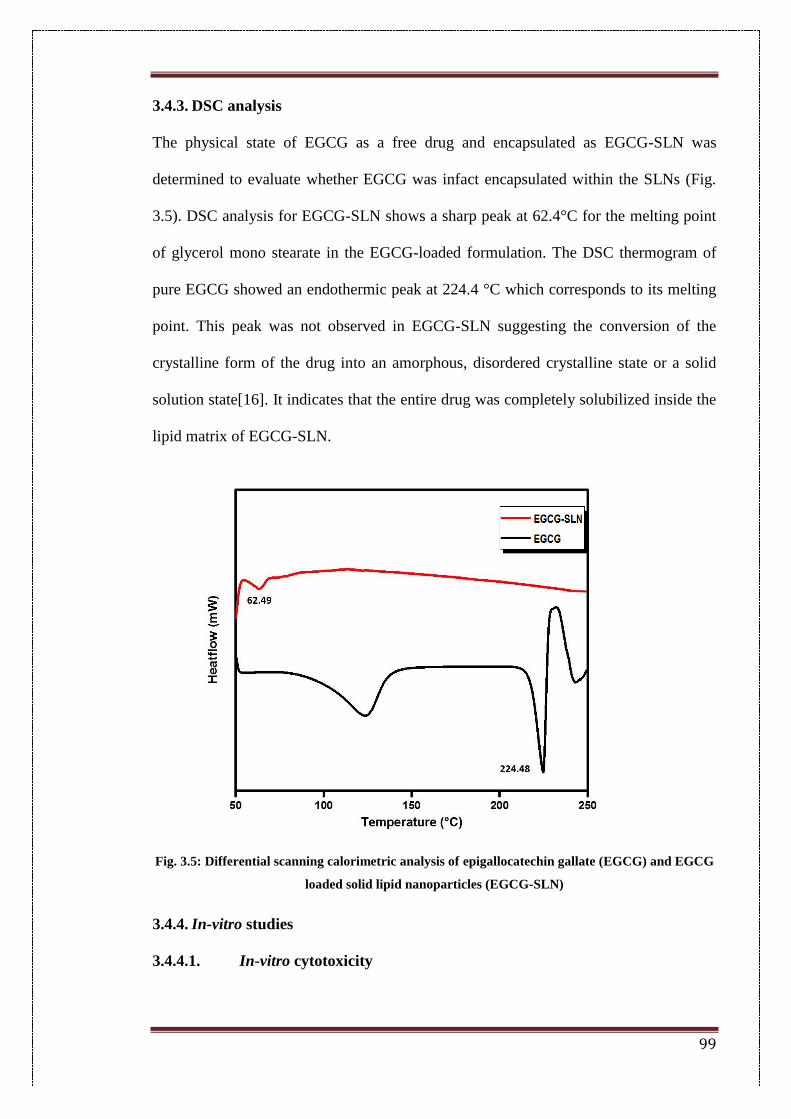
99
3.4.3. DSC analysis
The physical state of EGCG as a free drug and encapsulated as EGCG-SLN was
determined to evaluate whether EGCG was infact encapsulated within the SLNs (Fig.
3.5). DSC analysis for EGCG-SLN shows a sharp peak at 62.4°C for the melting point
of glycerol mono stearate in the EGCG-loaded formulation. The DSC thermogram of
pure EGCG showed an endothermic peak at 224.4 °C which corresponds to its melting
point. This peak was not observed in EGCG-SLN suggesting the conversion of the
crystalline form of the drug into an amorphous, disordered crystalline state or a solid
solution state[16]. It indicates that the entire drug was completely solubilized inside the
lipid matrix of EGCG-SLN.
Fig. 3.5: Differential scanning calorimetric analysis of epigallocatechin gallate (EGCG) and EGCG
loaded solid lipid nanoparticles (EGCG-SLN)
3.4.4. In-vitro studies
3.4.4.1. In-vitro cytotoxicity

100
The in-vitro cytotoxicity of pure EGCG, EGCG-SLN and EB-SLN against MDA-MB-
231, A549, HEK-293 and L132 cell lines was evaluated by MTT assay. The cell viability
of EGCG, EGCG-SLN and EB-SLN at a concentration range of 5-100 µg/mL was
determined in MDA-MB 231 cells. The observed IC50 values for EGCG, EGCG-SLN and
EB-SLN were 78.92 ± 4.3 µg/mL, 9.71 ± 0.6 µg/mL and 5.57 ± 3.2 µg/mL, respectively
(Fig 3.6). The cell viability for EGCG was 42.94% at the highest tested concentration of
100 µg/mL. For the EGCG-SLN formulation the cell viability was found to be 3.02% at
100 µg/mL. Bombesin-conjugated EB-SLN showed a cell viability of 51.77% at the
lowest concentration of 5 µg/mL, reducing drastically to 1.67% at a concentration of 40
µg/mL and showing no cell viability at higher concentrations. The results indicate that the
pure EGCG has low cytotoxicity in MDA-MB-231. However, its activity increased more
than 8 times after encapsulation in SLNs and more than 14 times post conjugation with
bombesin. For DU-145 cell line, the cell viability for maximum concentration of
100µg/mL of EGCG was 60.4%, that for EGCG-SLN was 16.9% whereas for EB-SLN it
was 7.7%. The observed IC50 values for EGCG, EGCG-SLN and EB-SLN were 126.7 ±
3.4 µg/mL, 33.2 ± 1.5 µg/mL and 19.2 ± 2.4 µg/mL respectively. For A549 cell line, the
cell viability for maximum concentration of 100µg/mL of EGCG was 85.1%, that for
EGCG-SLN was 20.4% whereas for EB-SLN it was 14.5%. The observed IC50 values for
EGCG, EGCG-SLN and EB-SLN were224.5 ± 3.9 µg/mL, 27.6 ± 4.5µg/mL and 24.8 ±
3.8 µg/mL respectively. Of the three tested cancer cell lines, EB-SLN showed greater
cytotoxicity in comparison with EGCG-SLN and EGCG in MDA-MB 231 cell line and
hence this cell line was chosen for further studies.
In addition, cytotoxicity evaluation was also carried out in two non-cancer cell lines HEK-
293 embryonic kidney and L132 lung cell lines. An interesting finding in this study was
that EB-SLN was least toxic to these cells in comparison to EGCG-SLN and EGCG. The

101
observed IC50 values in HEK-293 for EGCG, EGCG-SLN and EB-SLN were19.6 ± 4.7
µg/mL, 153.9 ± 2.5 µg/mL and 163.7 ± 4.1 µg/mL respectively. In L-132 cell line it was
13.2 ± 2.3 µg/mL, 182.02 ± 3.4 µg/mL and 252.5 ± 3.7 µg/mL for EGCG, EGCG-SLN
and EB-SLN respectively. These results point to selective specificity of formulation
towards cancer cells as opposed to non-cancerous cells.
Table 3.2: IC50 values (µg/mL) for epigallocatechin gallate (EGCG), EGCG-loaded solid lipid
nanoparticles (EGCG-SLN) and bombesin conjugated SLN (EB-SLN) against MDA-MB-231, DU-
145, A549, HEK-293 and L-132 cells after 24 h of treatment
Formulation
MDA-MB-231 DU-145 A549 HEK-293 L132
EGCG 78.9 ± 4.3 126.7 ± 3.4 224.5 ± 3.9 19.6 ± 4.7 13.2 ± 2.3
EGCG-SLN 9.7 ± 0.6 33.2 ± 1.5 27.6 ± 4.5 153.9 ± 2.5 182.02 ± 3.4
EB-SLN 5.5 ± 3.2 19.2 ± 2.4 24.8 ± 3.8 163.7 ± 4.1 252.5 ± 3.7
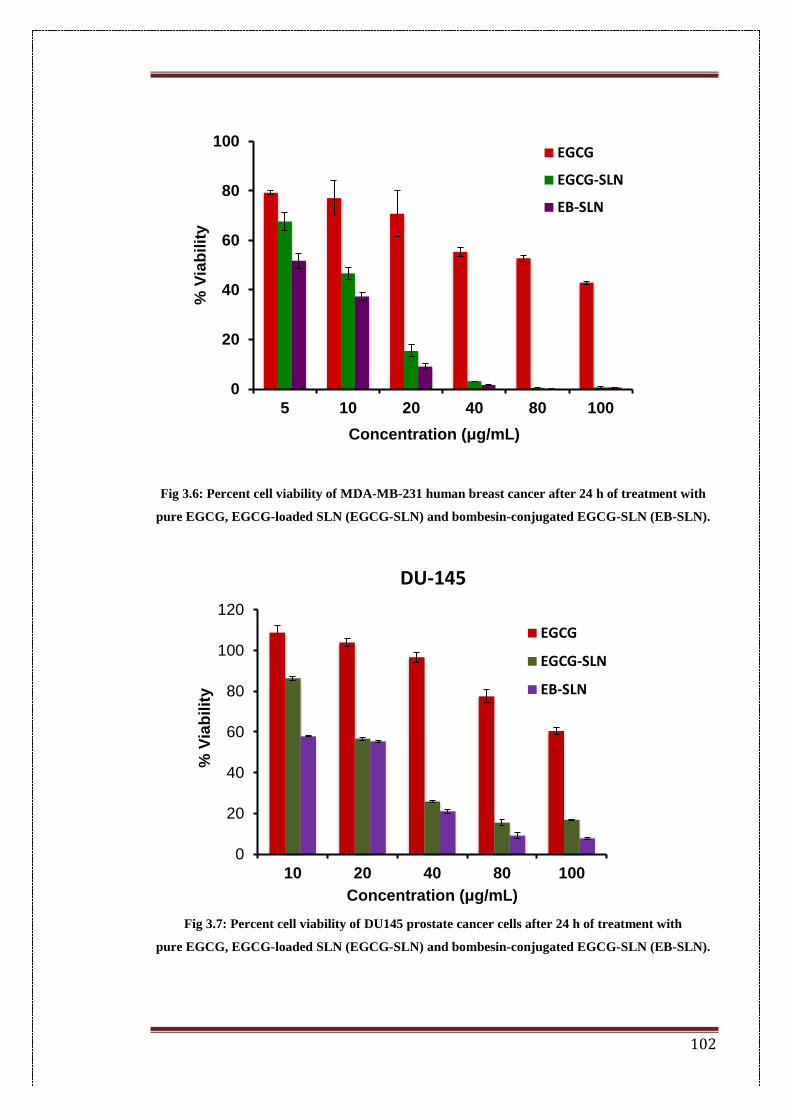
102
Fig 3.6: Percent cell viability of MDA-MB-231 human breast cancer after 24 h of treatment with
pure EGCG, EGCG-loaded SLN (EGCG-SLN) and bombesin-conjugated EGCG-SLN (EB-SLN).
Fig 3.7: Percent cell viability of DU145 prostate cancer cells after 24 h of treatment with
pure EGCG, EGCG-loaded SLN (EGCG-SLN) and bombesin-conjugated EGCG-SLN (EB-SLN).
0
20
40
60
80
100
5 10 20 40 80 100
% V
iab
ilit
y
Concentration (μg/mL)
EGCG
EGCG-SLN
EB-SLN
0
20
40
60
80
100
120
10 20 40 80 100
% V
iab
ilit
y
Concentration (μg/mL)
DU-145
EGCG
EGCG-SLN
EB-SLN
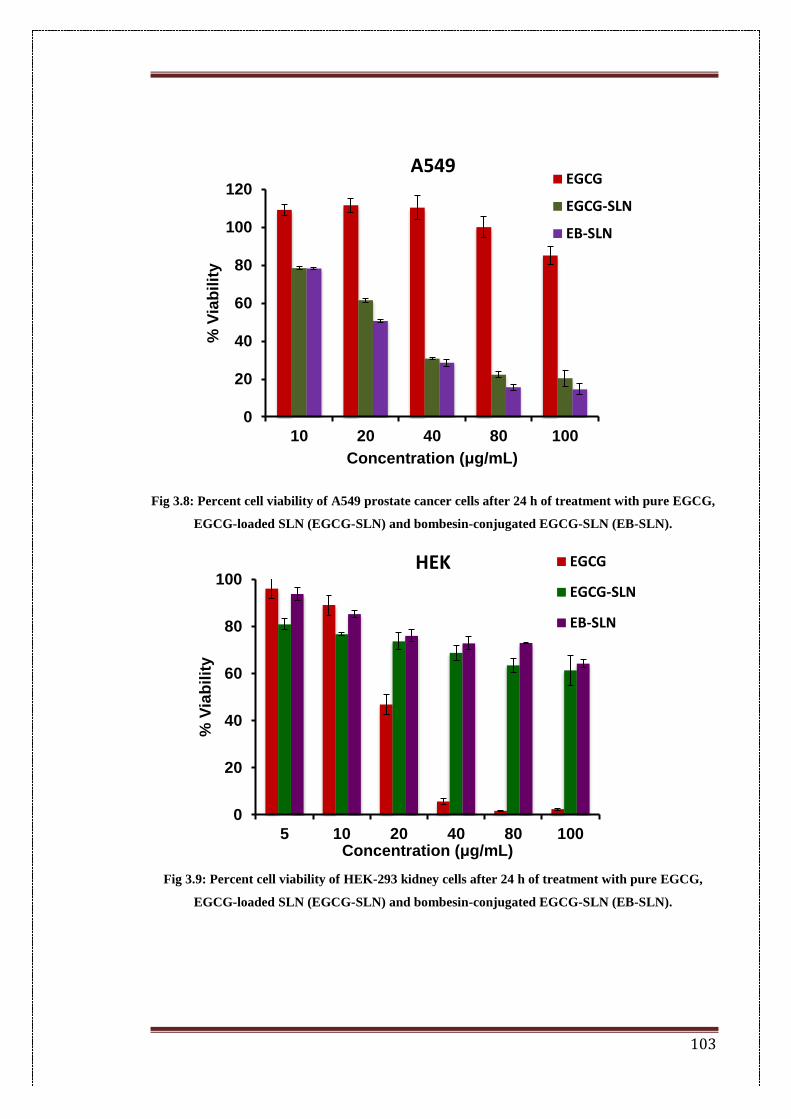
103
Fig 3.8: Percent cell viability of A549 prostate cancer cells after 24 h of treatment with pure EGCG,
EGCG-loaded SLN (EGCG-SLN) and bombesin-conjugated EGCG-SLN (EB-SLN).
Fig 3.9: Percent cell viability of HEK-293 kidney cells after 24 h of treatment with pure EGCG,
EGCG-loaded SLN (EGCG-SLN) and bombesin-conjugated EGCG-SLN (EB-SLN).
0
20
40
60
80
100
120
10 20 40 80 100
% V
iab
ilit
y
Concentration (μg/mL)
A549EGCG
EGCG-SLN
EB-SLN
0
20
40
60
80
100
5 10 20 40 80 100
% V
iab
ilit
y
Concentration (μg/mL)
HEK EGCG
EGCG-SLN
EB-SLN
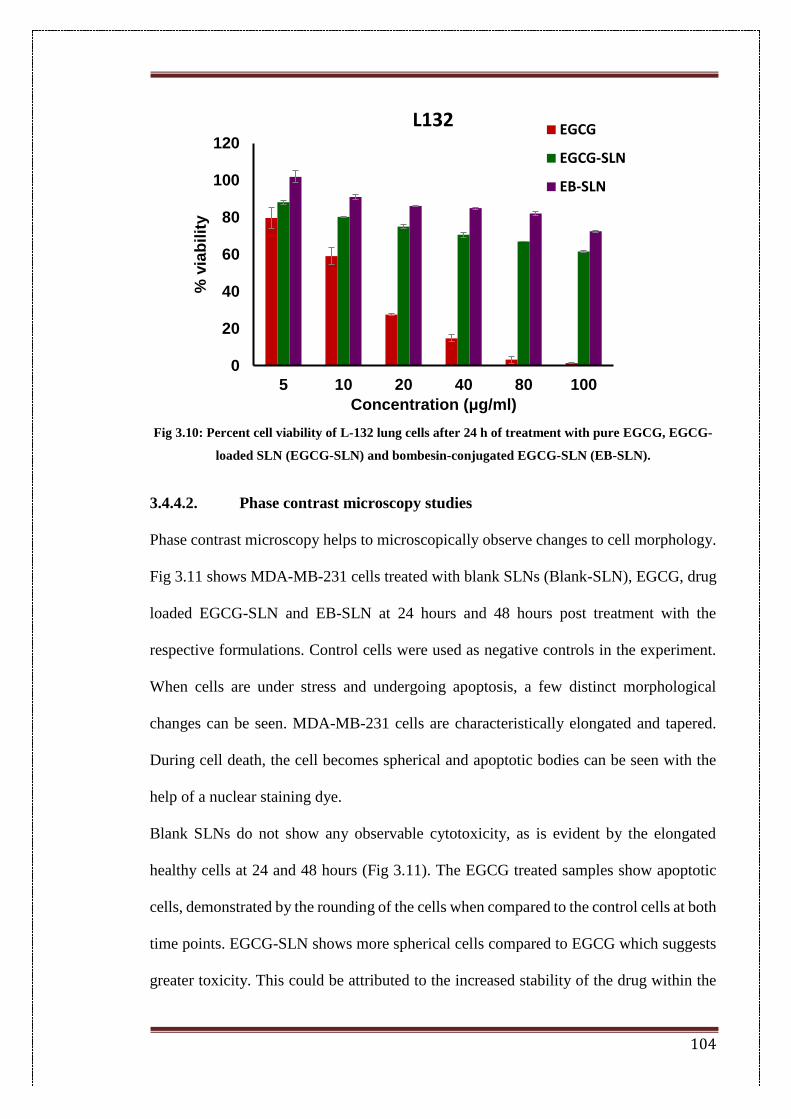
104
Fig 3.10: Percent cell viability of L-132 lung cells after 24 h of treatment with pure EGCG, EGCG-
loaded SLN (EGCG-SLN) and bombesin-conjugated EGCG-SLN (EB-SLN).
3.4.4.2. Phase contrast microscopy studies
Phase contrast microscopy helps to microscopically observe changes to cell morphology.
Fig 3.11 shows MDA-MB-231 cells treated with blank SLNs (Blank-SLN), EGCG, drug
loaded EGCG-SLN and EB-SLN at 24 hours and 48 hours post treatment with the
respective formulations. Control cells were used as negative controls in the experiment.
When cells are under stress and undergoing apoptosis, a few distinct morphological
changes can be seen. MDA-MB-231 cells are characteristically elongated and tapered.
During cell death, the cell becomes spherical and apoptotic bodies can be seen with the
help of a nuclear staining dye.
Blank SLNs do not show any observable cytotoxicity, as is evident by the elongated
healthy cells at 24 and 48 hours (Fig 3.11). The EGCG treated samples show apoptotic
cells, demonstrated by the rounding of the cells when compared to the control cells at both
time points. EGCG-SLN shows more spherical cells compared to EGCG which suggests
greater toxicity. This could be attributed to the increased stability of the drug within the
0
20
40
60
80
100
120
5 10 20 40 80 100
% v
iab
ilit
y
Concentration (μg/ml)
L132EGCG
EGCG-SLN
EB-SLN

105
SLNs and also the sustained release due to encapsulation. EB-SLN shows more apoptotic
cells, with the number of dead or dying cells increasing at 48 hours compared to 24 hours.
This could again be attributed to the sustained release of the drug within SLNs and more
importantly greater uptake due to targeting of bombesin to GRPR receptors. This
enhanced uptake would automatically lead to increased concentrations of drug within the
cell and hence, greater cytotoxicity, in agreement with the observations in Fig 3.6.

106
Fig 3.11: Phase contrast microscopy images of MDA-MB-231 human breast cancer cells after 24 h
and 48 h of treatment with B) pure epigallocatechin gallate (EGCG), C) blank solid lipid
nanoparticles (blank SLN), D) EGCG-loaded SLN (EGCG-SLN) and E) bombesin conjugated SLN
(EB-SLN) equivalent to 20 μg/mL EGCG and A. control cells for reference.
A
B
C
D
E
24 h 48 h

107
3.4.4.3. Cellular uptake studies
Uptake of nanoparticles by cancer cells was performed by loading a fluorescent dye in
the lipid nanoparticle and observing fluorescence through microscopic detection. In this
work Rhodamine B was chosen as the dye due to its hydrophilic nature, as the dye must
be loaded in the same phase as that of the drug. Uptake was studied in the MDA-MB-231
breast cancer cell line. Rhodamine-B loaded SLNs (RB-SLNs) were prepared in the same
method as EGCG-loaded SLNs (Chapter 2, Section 2.3.3.) but with the drug replaced with
Rhodamine-B dye. They were further conjugated with bombesin to give bombesin
conjugated Rhodamine-B nanoparticles (RB-SLNs). An important advantage of targeting
lies in the specificity it provides to the carrier vehicle. This increases the amount of drug
delivered at the target site due to increased uptake via different mechanisms such as
receptor mediated endocytosis; whereas passive targeting merely relies on EPR effect
which does lead to sufficient uptake of nanoparticles but does not guarantee maximum
payload delivery. The use of fluorescence to study uptake is a more visual way to
determine differences in uptake. Fluorescence images were taken at two time points of 12
and 24 hours (Fig 3.12). At 12 hours, visibly more intense red fluorescence is seen in cells
treated with bombesin-conjugated nanoparticles compared with the unconjugated
nanoparticles. At 24 hours, this difference is more pronounced with not only an increase
in the fluorescence in the cells treated with RB-SLN, but there is also visible
intensification of fluorescence in individual cells. These findings point to continual and
increased uptake of nanoparticles in the cancerous cells conjugated with the targeting
ligand bombesin as opposed to the unconjugated formulation.

108
Fig 3.12: Cellular uptake studies: Fluorescent microscopic images of MDA-MB-231 human
breast cancer cells after 24 h of treatment with Rhodamine-loaded SLN (R-SLN) and
bombesin conjugated SLN (RB-SLN)
3.4.4.4. Apoptosis studies
Apoptosis is a process of programmed cell death that occurs after a cell is no longer
needed in the body. It occurs by two pathways, namely extrinsic and intrinsic pathways
(Refer to Section 2.1.e.). In cancer cells, the factors affecting these two pathways are
downregulated causing the cells to continue to proliferate uncontrollably. EGCG has
shown the ability to induce apoptosis, as explained in Chapter 2. Nuclear condensation is
an important process during apoptosis that forms the principle of the nucleus-staining
apoptosis assay. Hoeschst 33342 was the dye used in this study. Hoeschst 33342 is a cell-
permeable, blue fluorescence dye. When this dye binds to DNA it emits blue fluorescence
at 495 nm[14].
To study apoptosis caused by the different formulations, MDA-MB-231 cells were
incubated with blank-SLN, pure EGCG, EGCG-SLN and EB-SLN for 24 hours followed
R-SLN RB-SLN
12 Hrs
24 Hrs

109
by staining with Hoeschst 33342. Fluorescence images in Fig. 3.13 clearly showed the
difference in nuclear staining. Hoeschst 33342 stains all nuclei, hence blue fluorescence
is visible throughout. However, the intensity of fluorescence is more in cells undergoing
apoptosis, owing to nuclear condensation. Control cells and cells treated with blank-SLN
show no intense fluorescence. However, EGCG does show several intense fluorescence
specks. These specks increase in the EGCG-SLN, however the highest intensity is visible
in the EB-SLN formulation treated cells. An explanation for this is that the cells show an
increased uptake of these nanoparticles in comparison to the other samples due to the
presence of the targeting ligand bombesin. This resulted in the successful delivery of
increased concentrations of the cytotoxic EGCG inside the cells, thus causing a greater
extent of apoptosis.

110
Fig. 3.13: Nuclear fragmentation studies: Fluorescent microscopic images of MDA-MB-231 human
breast cancer cells after 24 h of treatment with blank solid lipid nanoparticles (blank SLN), pure
epigallocatechin gallate (EGCG), EGCG-loaded SLN (EGCG-SLN) and bombesin conjugated SLN
(EB-SLN) equivalent to 20 μg/mL EGCG followed by staining with Hoechst 33342.
3.4.4.5. Migration studies
Angiogenesis is the process by which a tumour develops new blood vessels from existing
ones in order to maintain continual growth and nutrition. Therefore, inhibition of
angiogenesis aids in preventing the growth and metastasis of tumours, by essentially
cutting off its nutrition. EGCG has shown anti-angiogenic effects in a number of cell lines
as mentioned in Chapter 2(Section 2.1). However, this effect was found to be increased
when it was encapsulated as EGCG-SLN and further conjugated with bombesin (Fig
3.14). Wound closure is found to more in control cells as compared to EGCG. Wound
EGCG-SLN

111
closure is almost negligible in EGCG-SLN and EB-SLN. Enhanced activity in EGCG-
SLN and EB-SLN could be attributed to the increased stability of the drug within the
solid lipid nanoparticles and hence greater activity. Sustained release of EGCG from the
nanoparticles also aids in continued anti-migratory effect of the drug leading to better
wound closure in EGCG- loaded nanoparticles than pure drug.
Fig 3.14: Scratch assay images for MDA-MB-231 human breast cancer cells after 24 h of treatment
with blank solid lipid nanoparticles (blank SLN), pure epigallocatechin gallate (EGCG), EGCG-
loaded SLN (EGCG-SLN) and and bombesin conjugated SLN (EB-SLN) equivalent to 20 μg/mL
EGCG, along with control cells for reference
3.4.5. In-vivo studies
The in-vivo anticancer efficacy of a therapeutic agent is assessed in terms of prolonging
the quality of life and increasing the survival time. The anti-tumour efficacy of EGCG,
EGCG-SLN or EB-SLN was evaluated on tumour bearing mice upon administration of
multiple doses equivalent to 50 mg/kg EGCG. In-vivo anti-tumour efficacy was evaluated
in a syngeneic C57/BL6 mouse model.
B16F10 cancer cell line was used to develop melanoma in this study. Gastrin-releasing
peptide receptors are overexpressed in many cancers. B16F10 mouse melanoma cell line
0 hr
24 hr

112
shows expression of GRPR receptors and therefore was used for checking the efficacy of
the bombesin conjugated formulation[17]. Mice were first injected subcutaneously with
3 × 105 B16F10 cells on the flank to grow melanoma in- situ. Forty tumour bearing
C57/BL6 mice were randomly divided into 4 groups (n=10) and intraperitoneally
administered EGCG, EGCG-SLN or EB-SLN at a dose equivalent to 50 mg/kg body
weight of EGCG every third day. The control group was administered an equivalent
volume of saline every third day.
The body weight, tumour volume and overall survival of the mice were recorded. The
mean survival time was determined using a Kaplan-Meier survival plot (Fig 3.15). A
mean survival of 41, 48 and 55 days were observed for EGCG, EGCG-SLN and EB-SLN
treated mice, respectively, compared to control mice (37 days) which were administered
with normal saline. There was a significant extension in the life span of mice treated with
EB-SLN.
The changes in body weight and tumour volume of the animals were also recorded during
the whole experiment. The change in body weight is an indicator of systemic toxicity
caused by the formulation[18]. There was rapid loss of body weight in the saline treated
control group in comparison to EGCG formulation treated animals (Fig 3.16). The control
group receiving only saline recorded a loss of 7.01% in total body weight in 30 days
whereas both EGCG-SLN groups showed increase by 1.58% and EB-SLN, 1.06% loss of
body weight, respectively.
The change in tumour volume was also observed and results revealed that tumour shrank
in the mice receiving EGCG formulation. The percent increase in tumour volume in mice
treated with EGCG-SLN and EB-SLN was 3.78% and 0.78% respectively as compared
to mice receiving pure EGCG and control which were 5.45% and 7.97% respectively.
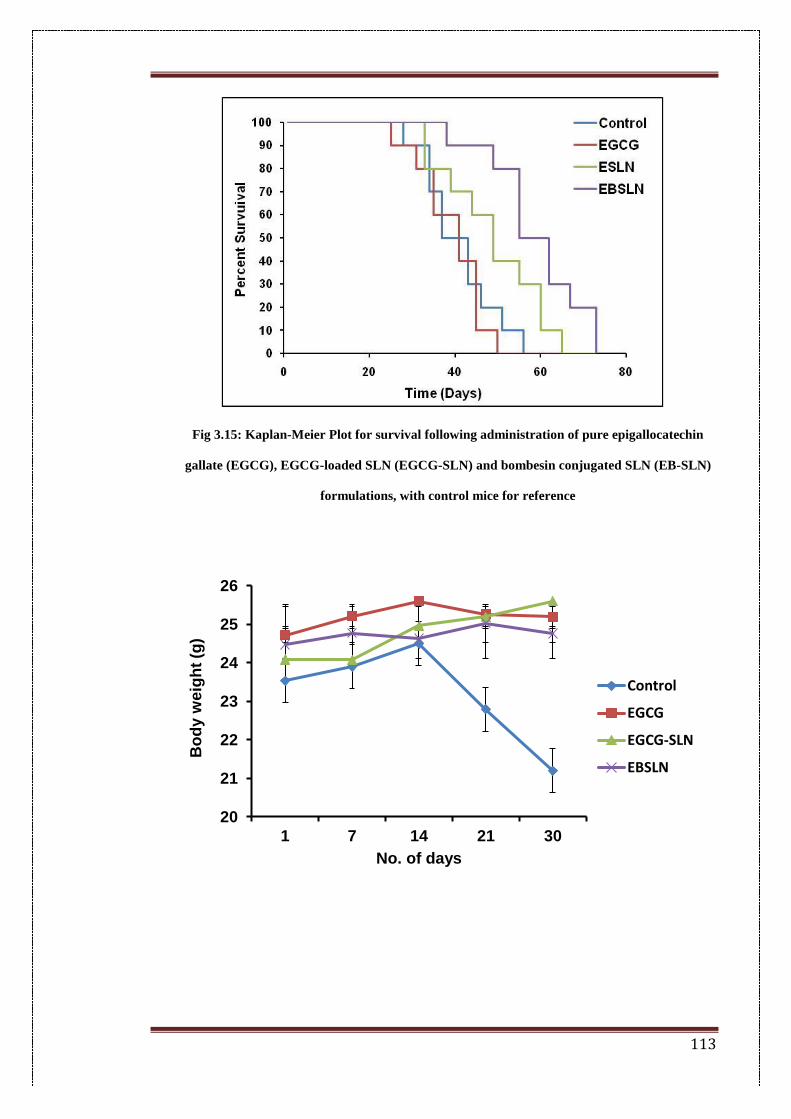
113
Fig 3.15: Kaplan-Meier Plot for survival following administration of pure epigallocatechin
gallate (EGCG), EGCG-loaded SLN (EGCG-SLN) and bombesin conjugated SLN (EB-SLN)
formulations, with control mice for reference
20
21
22
23
24
25
26
1 7 14 21 30
Bo
dy w
eig
ht
(g)
No. of days
Control
EGCG
EGCG-SLN
EBSLN
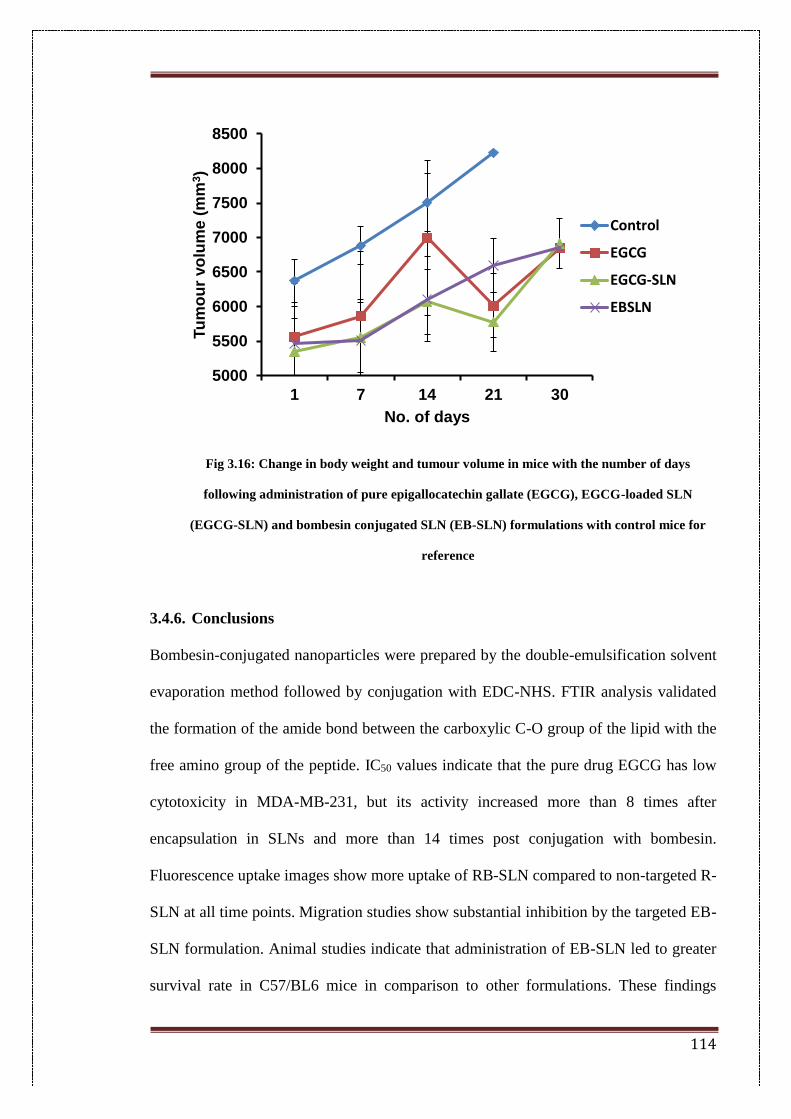
114
Fig 3.16: Change in body weight and tumour volume in mice with the number of days
following administration of pure epigallocatechin gallate (EGCG), EGCG-loaded SLN
(EGCG-SLN) and bombesin conjugated SLN (EB-SLN) formulations with control mice for
reference
3.4.6. Conclusions
Bombesin-conjugated nanoparticles were prepared by the double-emulsification solvent
evaporation method followed by conjugation with EDC-NHS. FTIR analysis validated
the formation of the amide bond between the carboxylic C-O group of the lipid with the
free amino group of the peptide. IC50 values indicate that the pure drug EGCG has low
cytotoxicity in MDA-MB-231, but its activity increased more than 8 times after
encapsulation in SLNs and more than 14 times post conjugation with bombesin.
Fluorescence uptake images show more uptake of RB-SLN compared to non-targeted R-
SLN at all time points. Migration studies show substantial inhibition by the targeted EB-
SLN formulation. Animal studies indicate that administration of EB-SLN led to greater
survival rate in C57/BL6 mice in comparison to other formulations. These findings
5000
5500
6000
6500
7000
7500
8000
8500
1 7 14 21 30
Tu
mo
ur
vo
lum
e (
mm
3)
No. of days
Control
EGCG
EGCG-SLN
EBSLN

115
suggest the merits of attachment of a targeting ligand to the EGCG-encapsulated SLN
system in Chapter 2, further corroborating the potential of EGCG-encapsulated systems
in cancer therapeutics.

116
3.4.7. References
[1] Worldwide cancer statistics | Cancer Research UK, (n.d.).
http://www.cancerresearchuk.org/health-professional/cancer-statistics/worldwide-
cancer#heading-Zero (accessed July 16, 2017).
[2] GLOBOCAN Cancer Fact Sheets: Breast cancer, (n.d.).
http://globocan.iarc.fr/old/FactSheets/cancers/breast-new.asp (accessed July 16, 2017).
[3] J.-J. Monsuez, J.-C. Charniot, N. Vignat, J.-Y. Artigou, Cardiac side-effects of
cancer chemotherapy., Int. J. Cardiol. 144 (2010) 3–15. doi:10.1016/j.ijcard.2010.03.003.
[4] A. Coates, S. Abraham, S.B. Kaye, T. Sowerbutts, C. Frewin, R.M. Fox, et al., On
the receiving end - patient perception of the side-effects of cancer chemotherapy, Eur. J.
Cancer Clin. Oncol. 19 (1983) 203–8. doi:10.1016/0277-5379(83)90418-2.
[5] K.S. Shohdy, A.S. Alfaar, Nanoparticles targeting mechanisms in cancer therapy:
current limitations and emerging solutions, Ther. Deliv. 4 (2013) 1197–1209.
doi:10.4155/tde.13.75.
[6] G. Halmos, J.L. Wittliff, A. V. Schally, Characterization of Bombesin/Gastrin-
releasing Peptide Receptors in Human Breast Cancer and Their Relationship to Steroid
Receptor Expression, Cancer Res. 55 (1995).
http://cancerres.aacrjournals.org/content/55/2/280.short (accessed July 16, 2017).
[7] R. Markwalder, J.C. Reubi, Gastrin-releasing Peptide Receptors in the Human
Prostate: Relation to Neoplastic Transformation, CANCER Res. 59 (1999) 1152–1159.
http://cancerres.aacrjournals.org/content/canres/59/5/1152.full.pdf (accessed July 16,
2017).
[8] J. Zhou, J. Chen, M. Mokotoff, R. Zhong, L.D. Shultz, E.D. Ball,
Bombesin/Gastrin-Releasing Peptide Receptor: A Potential Target for Antibody-
Mediated Therapy of Small Cell Lung Cancer, (n.d.).

117
http://clincancerres.aacrjournals.org/content/clincanres/9/13/4953.full.pdf (accessed
March 25, 2017).
[9] A.A. Begum, P.M. Moyle, I. Toth, Investigation of bombesin peptide as a targeting
ligand for the gastrin releasing peptide (GRP) receptor, Bioorg. Med. Chem. 24 (2016)
5834–5841. doi:10.1016/j.bmc.2016.09.039.
[10] H. Xin, X. Jiang, J. Gu, X. Sha, L. Chen, K. Law, et al., Angiopep-conjugated
poly(ethylene glycol)-co-poly(ε-caprolactone) nanoparticles as dual-targeting drug
delivery system for brain glioma., Biomaterials. 32 (2011) 4293–305.
doi:10.1016/j.biomaterials.2011.02.044.
[11] D. Bartczak, A.G. Kanaras, Preparation of peptide-functionalized gold
nanoparticles using one pot EDC/Sulfo-NHS coupling, Langmuir. 27 (2011) 10119–
10123. doi:10.1021/la2022177.
[12] H. Kulhari, D.P. Kulhari, M.K. Singh, R. Sistla, Colloidal stability and
physicochemical characterization of bombesin conjugated biodegradable nanoparticles,
Colloids Surfaces A Physicochem. Eng. Asp. 443 (2014) 459–466.
doi:10.1016/j.colsurfa.2013.12.011.
[13] J.M. Edmondson, L.S. Armstrong, A.O. Martinez, A rapid and simple MTT-based
spectrophotometric assay for determining drug sensitivity in monolayer cultures, J.
Tissue Cult. Methods. 11 (1988) 15–17. doi:10.1007/BF01404408.
[14] S. Allen, J. Sotos, M.J. Sylte, C.J. Czuprynski, Use of Hoechst 33342 staining to
detect apoptotic changes in bovine mononuclear phagocytes infected with
Mycobacterium avium subsp. paratuberculosis., Clin. Diagn. Lab. Immunol. 8 (2001)
460–464. doi:10.1128/CDLI.8.2.460-464.2001.
[15] M. Gaumet, A. Vargas, R. Gurny, F. Delie, Nanoparticles for drug delivery: The
need for precision in reporting particle size parameters, Eur. J. Pharm. Biopharm. 69

118
(2008) 1–9. doi:10.1016/j.ejpb.2007.08.001.
[16] K. Vivek, H. Reddy, R.S.R. Murthy, Investigations of the effect of the lipid matrix
on drug entrapment, in vitro release, and physical stability of olanzapine-loaded solid
lipid nanoparticles., AAPS PharmSciTech. 8 (2007) E83. doi:10.1208/pt0804083.
[17] J. Fang, Y. Lu, K. Ouyang, G. Wu, H. Zhang, Y. Liu, et al., Specific antibodies
elicited by a novel DNA vaccine targeting gastrin-releasing peptide inhibit murine
melanoma growth in vivo, Clin. Vaccine Immunol. 16 (2009) 1033–1039.
doi:10.1128/CVI.00046-09.
[18] C. Criscitiello, O. Metzger-Filho, K.S. Saini, G. de Castro Jr, M. Diaz, A. La
Gerche, et al., Targeted therapies in breast cancer: are heart and vessels also being
targeted?, Breast Cancer Res. 14 (2012) 209. doi:10.1186/bcr3142.

119
Mangiferin and piperine encapsulated
nanoparticles for cancer therapy

120
4.1. Background
Phytochemicals are plant derived chemical compounds which have been used in ancient
medicine in many parts of the world, with the first literature on their beneficial effects
dating back to 2800 BC[1]. Phytochemicals consist of a wide class of chemical
compounds which includes alkaloids, glycosides, flavonoids, volatile oils, tannins and
resins[2]. They have been known to help in the treatment and prevention of many diseases
such as cardiovascular disease, cancer, osteoporosis and hypercholesterolemia[3].
In this chapter, two phytochemicals have been used to explore anti-cancer efficacy
namely mangiferin and piperine. These formulations were chosen in order to investigate
the use of SLNs with a targeted chemotherapeutic system.
A. Mangiferin encapsulated solid lipid nanoparticles for targeting towards
cancer
Mangiferin (2-b-D-glucopyranosyl-1, 3, 6, 7-tetrahydroxyxanthone) is a C-glucoside
xanthone found in many families such as Anacardiaceae, Bignoniaceae, Moraceae,
Thymelaceae, Aphloiaceae, Malvaceae and Gentianaceae[4]. It is found in higher
concentrations in the root, bark and leaves of Mangifera indica (mango) plant.
Chemically it contains an aromatic ring attached to the C–C bond of a glucose moiety
(Fig. 4.1.), making the molecule polar and water soluble[5].
Its many beneficial medicinal properties such as anti-oxidant, anti-diabetic, analgesic,
anti-inflammatory, anti-hypertensive, immune-modulatory, neuro-protective, anti-viral,
hypo-lipidemic and anti-cancer activities have been reviewed extensively[6–9].

121
Fig 4.1: Structure of mangiferin
Mangiferin has been extensively researched for its anti-cancer properties, exerting anti-
cancer effects by a variety of mechanisms such as anti-inflammatory, anti-proliferative,
apoptotic and immune-modulatory effects[10–12] outlined as follows:
a. Anti-inflammation
Inflammatory processes have been regarded as important contributors in the development
of cancer. Factors such as NFκB (Nuclear factor kappa-B) and interleukins are
overexpressed which are critical mediators of the immune responses that control the
transcription of inflammatory cytokines[13]. Mangiferin downregulates the production of
NFκB by blocking NF-κB and MAPK signalling pathways[14].
b. Proliferation/ metastasis
In normal cells, the rate of cell division and cell death is strictly regulated to prevent
anomalies. In cancer cells, however, the factors responsible for cell death are down-
regulated, causing cells to replicate uncontrollably. Loss of cell adhesion causes cancer
cells to escape from the site of origin and migrate to other sites causing secondary
cancers[15]. Mangiferin has been found to cause induction of miRNA-15b which is a
factor whose upregulation causes cell cycle arrest in cancer cells in the G0/G1 phase,
while simultaneously downregulating matrix metalloproteinase MMP-9, which is an
important factor in the degradation of the basement membrane and metastasis[16].
c. Apoptotic effect

122
Apoptosis is a process of controlled cell death which occurs by two pathways; an extrinsic
pathway which involves activation of death receptors or the intrinsic pathway which
involves the increased permeability of the mitochondria[17]. Mangiferin was found to
activate the intrinsic apoptotic pathway by upregulating caspase-9 cleavage and
activation, which is an initiator caspase that cleaves and activates the executioner caspases
(caspases 3 and 7), leading to apoptosis[18].
d. Immunomodulatory effects
Cancer cells avoid detection by the body’s immune system by the expression of proteins
that dampen the immune response. Mangiferin was found to increase the levels of
immunoglobulins IgM and IgG in animals with benzo(a)pyrene-induced lung
carcinogenesis[19].
In this work, mangiferin was encapsulated in solid lipid nanoparticles to study its anti-
cancer efficacy. The encapsulation was carried out by the single emulsion-solvent
evaporation method. Stearic acid was used as a charge modifier along with glycerol
mono-stearate as the matrix components. Bombesin was conjugated to this system as the
ligand and the release characteristics of the system were studied.
4.2. Materials
Glycerol mono-stearate, stearic acid, Poloxamer F68, mangiferin, chloroform, 1-ethyl-3-
(3-dimethylaminopropyl) carbodiimide (EDC), N-hydroxysuccinimide (NHS) and N-
[tris(hydroxymethyl)methyl]-2-aminoethane sulfonic acid (MES) were used as received.
4.3. Methods
4.3.1. Preparation of nanoparticles
SLNs were prepared by the single solvent-emulsification and evaporation method. The
oil in water emulsion of mangiferin was made by dissolving 5 mg of MgF in 2 mL
chloroform and it was then emulsified slowly in 10 mL of 1.5% Tween 20 surfactant

123
solution. The emulsion was sonicated for 15 mins at 50% amplitude using a probe
sonicator. It was stirred for 3 hours to allow for solvent evaporation and mangiferin-
loaded SLNs (MgF-SLNs) were then pelleted by centrifugation at 15000 rpm for 30
minutes and stored for further work. The supernatant was used for determining the
encapsulation efficiency.
4.3.2. Determination of entrapment efficiency
The entrapment efficiency of solid lipid nanoparticles to encapsulate mangiferin was
calculated by the indirect method. The supernatant collected after centrifugation of MgF-
SLNs was used to determine the concentration of free mangiferin by UV-visible
spectroscopy. A calibration curve of mangiferin was constructed and used for the
calculation of unknown concentrations.
4.3.3. Bioconjugation
MgF-SLNs were conjugated with bombesin using EDC-NHS reaction. Briefly, the
nanoparticle suspension was incubated with 15 mg N-hydroxysuccinimide (NHS) and 20
mg N-(3-dimethylaminopropyl)-N-ethyl carbodiimide hydrochloride (EDC). The
mixture was allowed to stir for an hour for the activation of the carboxylic acid groups of
stearic acid in the SLN matrix. Bombesin (0.25 mg) was then added to this and further
stirred for 4 hours to allow conjugation to take place. The reaction was carried out in the
presence of MES buffer (pH 6.2) as the conjugation of bombesin to lipid nanoparticles
has been found to be optimum around pH 6.2 based on previous studies[20].
The bombesin–conjugated nanoparticles (MB-SLNs) were now centrifuged at 10,000
rpm for 10 minutes to remove unconjugated peptide molecules. The supernatant was
stored for calculation of the unconjugated peptide.
4.3.4. Calculation of conjugation efficiency

124
Unconjugated peptide was calculated by the Bradford assay method for determination of
protein. Briefly, a calibration curve for bombesin was plotted and the unknown
concentration was calculated by spectrophotometric detection at 595nm.
4.3.5. Physico-chemical characterization
4.3.5.1. Particle size and surface charge
Nanoparticles were optimized for particle size and surface charge. The mean particle sizes
and zeta potentials of blank nanoparticles (Blank-SLN), mangiferin-loaded SLNs (MgF-
SLNs) and bombesin conjugated nanoparticles (MB-SLNs) were measured using a
Malvern Zetasizer Nano ZS (Malvern Instrument Ltd., Malvern, UK). Samples were
diluted 10 times with double distilled water and analyzed at 25 ºC with a backscattering
angle of 173º. Surface potentials for the formulations were also determined with the
Zetasizer..
4.3.5.2. FTIR analysis
FTIR analysis of Blank-SLN, MgF-SLN and MB-SLN was carried by using an IR
spectrophotometer (Perkin Elmer, USA). Pellets were prepared by adding approximately
3 mg of the sample to 100 mg of KBr. Samples were scanned in the range of 400–4000
cm−1.
4.3.6. In-vitro drug release
In-vitro drug release studies were performed by dialysis in sodium acetate buffer pH 5
(SAB) and phosphate buffer pH 7.4. MgF-SLNs, equivalent to 2 mg of mangiferin, were
placed in dialysis tubing and kept in a beaker containing 100 mL of SAB and PBS and
maintained at 37 ºC. At predetermined time intervals, 1 mL of buffer was withdrawn and
replaced with same volume of fresh media to maintain sink conditions. The mangiferin
concentration was determined by measuring the absorbance at 342 nm, which is the
absorption maxima for mangiferin, using a UV/Visible spectrophotometer.

125
4.4. Results
4.4.1. Preparation of nanoparticles
Solid lipid nanoparticles were prepared by the emulsification-solvent evaporation method
to form an oil-in-water emulsion. Mangiferin was dissolved in chloroform which was the
inner organic phase. Tween 20 solution was used as the outer aqueous phase.
Emulsification was carried out using a probe sonicator for 15 minutes. The formulation
was optimized for particle diameter and charge for the surfactant used and its
concentration. The lipids used and their concentrations were optimized earlier in Chapter
2 (Section 2.4.2) Surfactants used for optimization included polyvinyl alcohol (PVA),
Tween 20 and Poloxamer F68. Tween 20 gave a particle size of 100.2 ± 3.1 nm, while
PVA and Poloxamer F68 gave a particle size of 132.5 ± 2.2 nm and 125.4 ± 3.6 nm,
respectively. The polydispersity of the Tween 20 formulation (F2) was 0.185 ± 0.2, while
that for PVA and Poloxamer 188 were 0.342 ± 0.2 and 0.379 ± 0.3, respectively. The
particle size and polydispersity of F2 were considerably lower than that of the others and
hence it was chosen to carry out further optimization. The concentration of Tween 20 in
the aqueous phase was optimized between 1, 1.5 and 2% in aqueous solution. The particle
size decreases with an increase in surfactant concentration with 1%, 1.5% and 2% Tween
20 giving particle diameters of 117.7 ± 5.4 nm, 100.2 ± 3.1 nm and 96.5 ± 4.7 nm,
respectively. This could be attributed to the fact that an increase in surfactant
concentration increases particle repulsion hence giving better size and polydispersity
measurements. However, the surface potential is found to decrease at 2% Tween 20
concentration which could lead to a reduction in stability. Thus, 1.5% Tween 20
formulation was chosen as the final formulation.
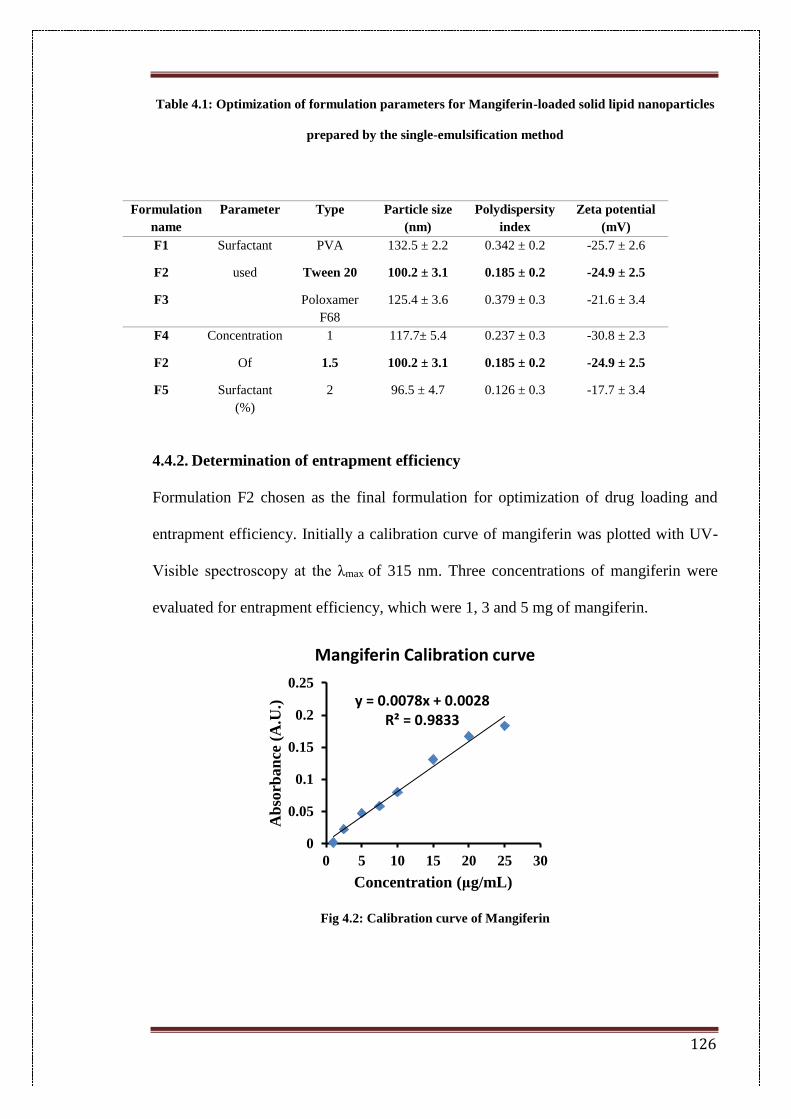
126
Table 4.1: Optimization of formulation parameters for Mangiferin-loaded solid lipid nanoparticles
prepared by the single-emulsification method
4.4.2. Determination of entrapment efficiency
Formulation F2 chosen as the final formulation for optimization of drug loading and
entrapment efficiency. Initially a calibration curve of mangiferin was plotted with UV-
Visible spectroscopy at the λmax of 315 nm. Three concentrations of mangiferin were
evaluated for entrapment efficiency, which were 1, 3 and 5 mg of mangiferin.
Fig 4.2: Calibration curve of Mangiferin
y = 0.0078x + 0.0028R² = 0.9833
0
0.05
0.1
0.15
0.2
0.25
0 5 10 15 20 25 30
Ab
sorb
an
ce (
A.U
.)
Concentration (μg/mL)
Mangiferin Calibration curve
Formulation
name
Parameter Type Particle size
(nm)
Polydispersity
index
Zeta potential
(mV)
F1 Surfactant PVA 132.5 ± 2.2 0.342 ± 0.2 -25.7 ± 2.6
F2 used Tween 20 100.2 ± 3.1 0.185 ± 0.2 -24.9 ± 2.5
F3 Poloxamer
F68
125.4 ± 3.6 0.379 ± 0.3 -21.6 ± 3.4
F4 Concentration 1 117.7± 5.4 0.237 ± 0.3 -30.8 ± 2.3
F2 Of 1.5 100.2 ± 3.1 0.185 ± 0.2 -24.9 ± 2.5
F5 Surfactant
(%)
2 96.5 ± 4.7 0.126 ± 0.3 -17.7 ± 3.4
127
Table 4.2: Optimization of encapsulation of mangiferin and entrapment efficiency in mangiferin-
loaded SLN (MgF-SLN)
Particle sizes for 1, 3 and 5 mg mangiferin loaded formulations were found to be 89.52 ±
3.7 nm, 95.1 ± 4.1 nm and 100.2 ± 3.1 nm, respectively. Particle sizes were found to
increase with an increase in the amount of drug, which could be attributed to an increase
in the amount of drug assimilation between the crystallized lipid matrix therefore causing
an increase on hydrodynamic diameter. Entrapment efficiency for 1, 3 and 5 mg were
found to be 85.6 ± 3.7%, 81.4 ± 2.3% and 75.31 ± 2.3% for the F6, F7 and F2
formulations, respectively (Table 4.2). However, the F2 (5 mg) formulation was chosen
as even with a lesser entrapment efficiecy the total amount of drug loaded will be more
in this case compared to the others (3.765 mg).
4.4.3. Bioconjugation
Bioconjugation of bombesin to MgF-SLN was carried out by the EDC-NHS method as
explained in Chapter 3 (Refer Section 3.3.3.). EDC-NHS reaction helps to activate the
carboxylic groups in the charge modifier lipid (stearic acid) in order to help easier
formation of the amide bond between the carboxylate ion and the amine group of
bombesin. The centrifuged bombesin-conjugated nanoparticle (MB-SLN) formulation
was evaluated for particle size and surface potential. The zeta potential increased from -
24.9 ± 2.5 mV of MgF-SLN formulation to -16.3 ± 2.4 mV in MB-SLN. This could be
Formulatio
n name
Parameter Type Particle size
(nm)
Polydispersity
index
Zeta
potential
(mV)
Entrapmen
t efficiency
(%)
F6 Amount 1 89.52 ± 3.7 0.131 ± 0.2 -24.4 ± 2.1 85.6 ± 3.7
F7 Of drug 3 95.1± 4.2 0.264 ± 0.4 -25.2 ± 2.3 81.4 ± 2.3
F2 (mg) 5 100.2 ± 3.1 0.185 ± 0.2 -24.9 ± 2.5 75.31 ± 2.3

128
attributed to masking of the negative carboxylate groups due to bond formation and
therefore a decrease in the total negative potential on the particle surface.
4.4.4. Calculation of conjugation efficiency
The conjugation efficiency of bombesin (BBN) was calculated by the indirect method.
The amount of unconjugated Bombesin in the supernatant was quantified using the
Bradford assay for protein determination[21]. The conjugation efficiency for 250 µg BBN
was found to be 82.34 ± 2.53%.
4.4.5. Physico-chemical characterization
4.4.5.1. Particle size and surface charge
The particle size and surface charge of (Blank-SLN), MgF-SLN and MB-SLN were
observed by Malvern Zetasizer Nano ZS. The particle size of Blank-SLN, MgF-SLN and
MB-SLN were 96.57 ± 2.8 nm, 100.2 ± 3.1 nm and 121.4 ± 4.2 nm, respectively (Table
4.3). The increase in nanoparticle diameter from Blank SLN to MgF-SLN could be
attributed to the diffusion of the drug between the crystalline lipid matrix therefore
leading to an increase in size[22]. The zeta potential for Blank SLN and MgF-SLN was -
26.9 ± 3.1 mV and -24.9 ± 2.5 mV, respectively. There was not much change in surface
potential suggesting that mangiferin was not adsorbed on the surface and was
encapsulated. The particle size of the MB-SLN increases to 121.4 ± 4.2 nm which could
be due to attachment of the ligand on the particle surface. Also, the zeta potential tends
towards more positive value of -16.3 ± 2.4 mV in MB-SLN. This could be attributed to
masking of the negative carboxylate groups by bond formation with bombesin molecules
on the surface.
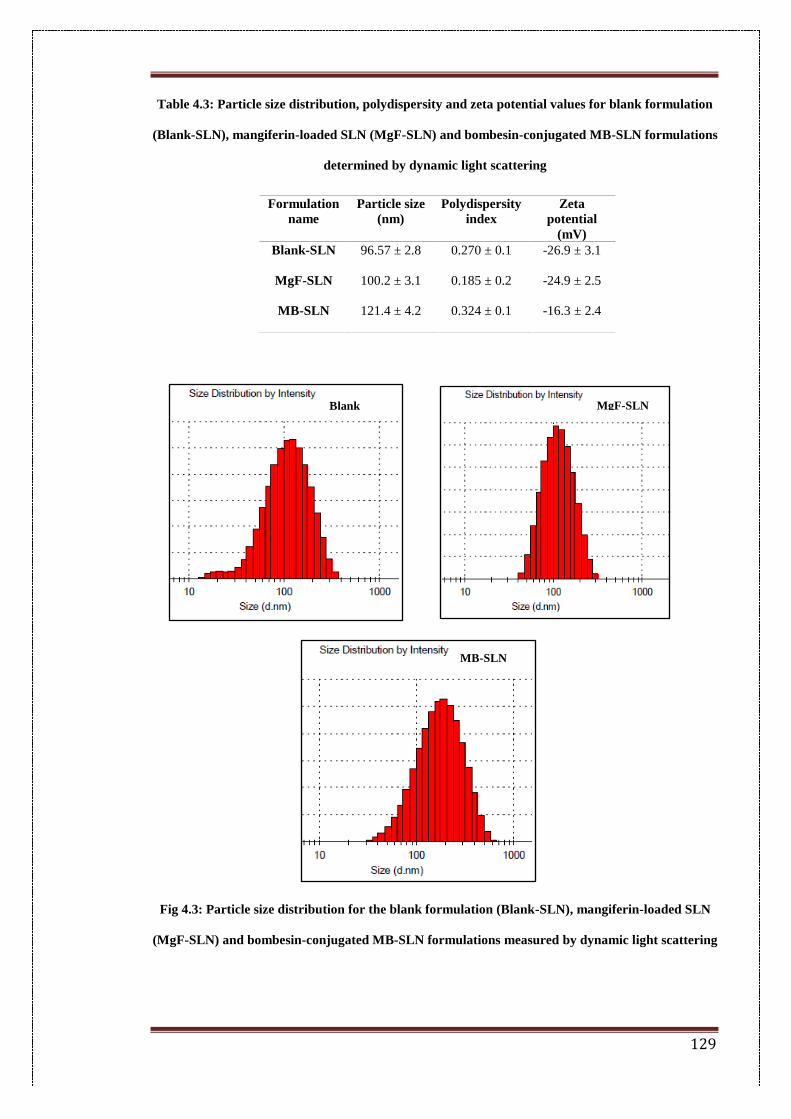
129
Table 4.3: Particle size distribution, polydispersity and zeta potential values for blank formulation
(Blank-SLN), mangiferin-loaded SLN (MgF-SLN) and bombesin-conjugated MB-SLN formulations
determined by dynamic light scattering
Fig 4.3: Particle size distribution for the blank formulation (Blank-SLN), mangiferin-loaded SLN
(MgF-SLN) and bombesin-conjugated MB-SLN formulations measured by dynamic light scattering
Formulation
name
Particle size
(nm)
Polydispersity
index
Zeta
potential
(mV)
Blank-SLN 96.57 ± 2.8 0.270 ± 0.1 -26.9 ± 3.1
MgF-SLN 100.2 ± 3.1 0.185 ± 0.2 -24.9 ± 2.5
MB-SLN 121.4 ± 4.2 0.324 ± 0.1 -16.3 ± 2.4
Blank
SLN
MgF-SLN
MB-SLN

130
4.4.5.2. FTIR analysis
FTIR spectrum of mangiferin shows a peak at 3367 cm-1 indicating presence of secondary
-OH bond (alcoholic OH), a peak at 2938 cm-1 for C-H anti-symmetric stretching and
peaks at 2830 cm-1 and 1649 cm-1 indicating the presence of the C-H and C-O symmetric
stretching. The blank SLN shows a broad peak at 3308 cm-1 for the O-H stretching in
glycerol mono-stearate which is one of the bulk components of the lipid core. It also
shows a peak at 2917 cm-1 for the O-H stretch and at 1730 cm-1 for the C=O stretch in the
carboxylic group in stearic acid. It also shows a peak at 1456 cm-1 strong for the C-H
bending in alkanes. Drug loaded MgF-SLN also shows peaks at 3307 cm-1 for the O-H
stretching in GMS, peaks at 2917 cm-1 for the O-H stretch and 1730 cm-1 for the C=O
stretch in stearic acid. The spectrum for mangiferin was different from the blank-SLN
and MgF-SLN. The spectra for drug loaded MgF-SLN and blank-SLN were similar which
shows that there were no unpleasant interactions between the drug and the lipid
nanoparticle and it was encapsulated efficiently.
Conjugated-SLN (MB-SLN) shows a broad peak at 3390 cm-1 for secondary alcoholic
OH groups from stearic acid. The peak at 2917 cm-1 for the O-H stretching is also visible.
However, peaks at 1645 cm-1 and 1707 cm-1 for amide linkage, which were not visible in
the blank or MgF-SLN are prominent providing evidence for amide bond formation.
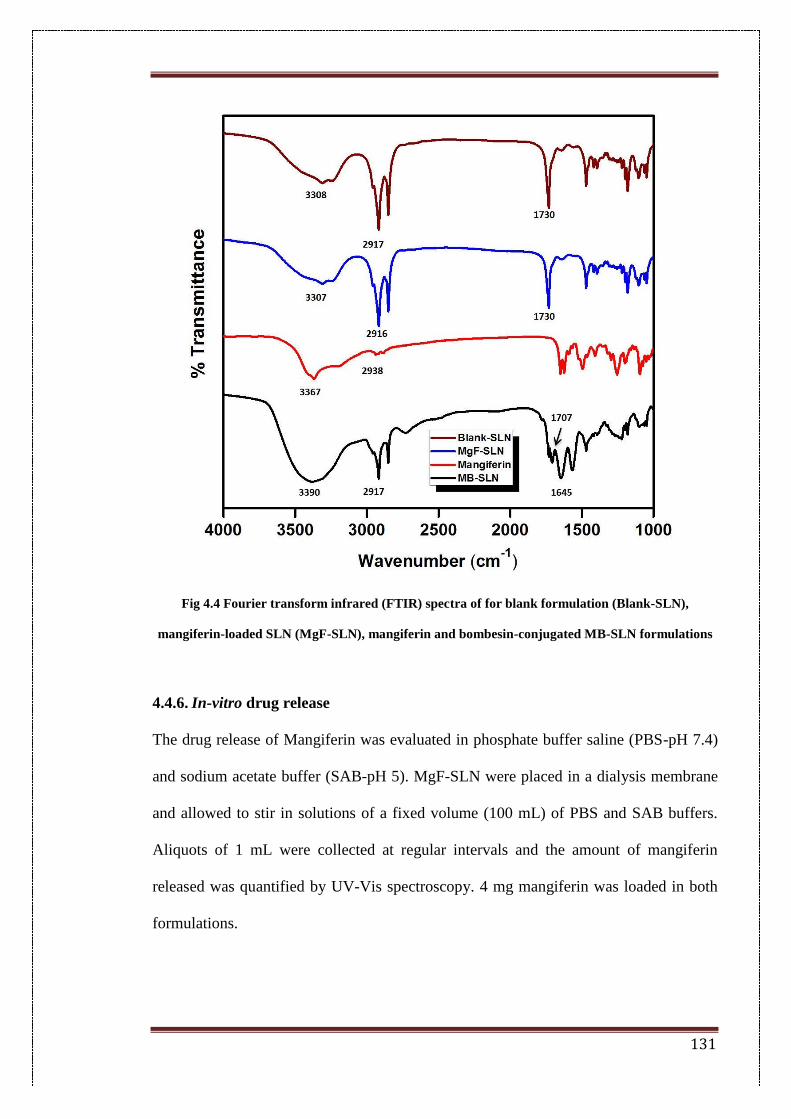
131
Fig 4.4 Fourier transform infrared (FTIR) spectra of for blank formulation (Blank-SLN),
mangiferin-loaded SLN (MgF-SLN), mangiferin and bombesin-conjugated MB-SLN formulations
4.4.6. In-vitro drug release
The drug release of Mangiferin was evaluated in phosphate buffer saline (PBS-pH 7.4)
and sodium acetate buffer (SAB-pH 5). MgF-SLN were placed in a dialysis membrane
and allowed to stir in solutions of a fixed volume (100 mL) of PBS and SAB buffers.
Aliquots of 1 mL were collected at regular intervals and the amount of mangiferin
released was quantified by UV-Vis spectroscopy. 4 mg mangiferin was loaded in both
formulations.
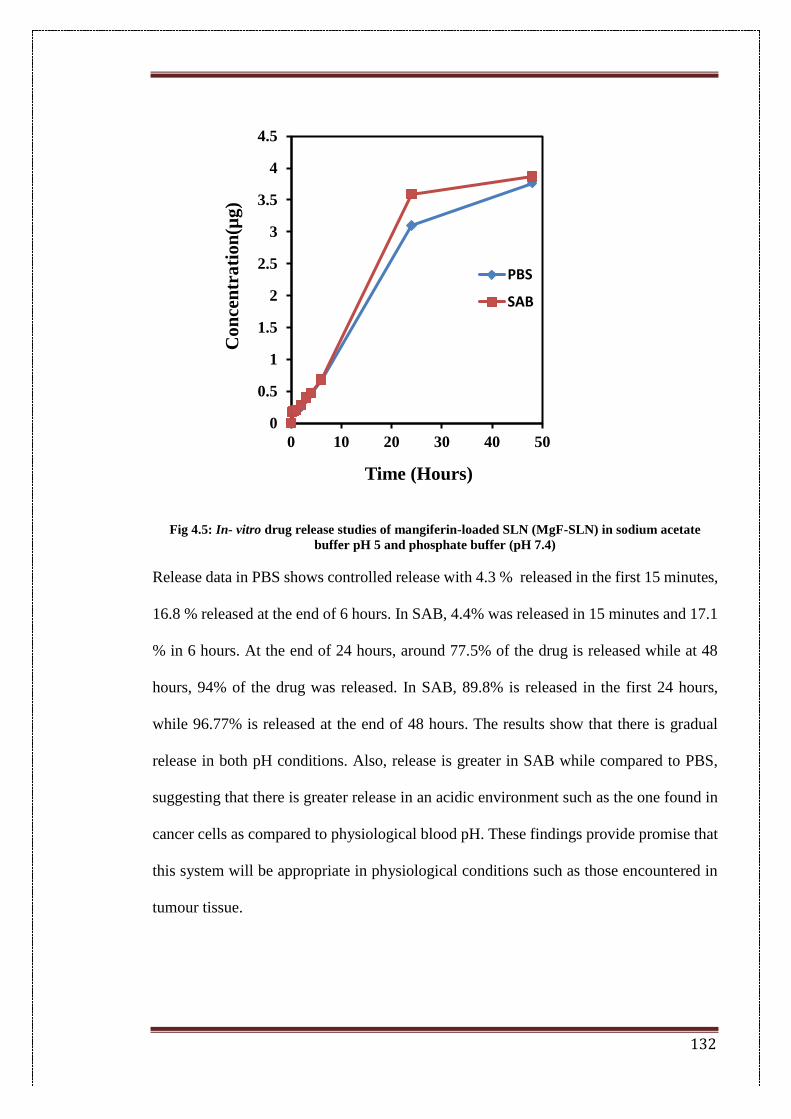
132
Fig 4.5: In- vitro drug release studies of mangiferin-loaded SLN (MgF-SLN) in sodium acetate
buffer pH 5 and phosphate buffer (pH 7.4)
Release data in PBS shows controlled release with 4.3 % released in the first 15 minutes,
16.8 % released at the end of 6 hours. In SAB, 4.4% was released in 15 minutes and 17.1
% in 6 hours. At the end of 24 hours, around 77.5% of the drug is released while at 48
hours, 94% of the drug was released. In SAB, 89.8% is released in the first 24 hours,
while 96.77% is released at the end of 48 hours. The results show that there is gradual
release in both pH conditions. Also, release is greater in SAB while compared to PBS,
suggesting that there is greater release in an acidic environment such as the one found in
cancer cells as compared to physiological blood pH. These findings provide promise that
this system will be appropriate in physiological conditions such as those encountered in
tumour tissue.
0
0.5
1
1.5
2
2.5
3
3.5
4
4.5
0 10 20 30 40 50
Co
nce
ntr
ati
on
(µg
)
Time (Hours)
PBS
SAB

133
B. Piperine encapsulated chitosan nanospheres conjugated with mannose for
glycotargeting towards lectin receptors overexpressed in cancer
Pepper has been used as a spice in southeast Asian and European cuisines and medicine
for a long time. Piperine ((2E,4E)-5-(2H-1,3-Benzodioxol-5-yl)-1-(piperidin-1-yl) penta-
2,4-dien-1-one) is one of the major constituents imparting pungency to pepper. It exhibits
diverse pharmacological activities like anti-oxidant, anti-tumor, anti-inflammatory,
immuno-modulatory, anti-apoptotic and anti-metastatic activities[23].
Fig 4.6: Structure of piperine
a. Anti-oxidant activity
Inflammation leads to the production of free radicals and generates inflammatory
response factors like Interleukin-6 which help in increasing tumour development.
Piperine was found to decrease the amount of superoxide ions (O2–) and hydroxyl radicals
(OH•) which are known to injure surrounding tissues and organs in rats fed with a high
fat diet[24]. It also inhibited the expression of IL-6 and matrix metalloproteinase 13 in rat
arthritis models[25] which are important factors helping in inflammation.
b. Immuno-modulatory effects
Mouse splenocytes were used to evaluate the in-vitro immune-modulation of piperine for
cytokine production, macrophage activation and lymphocyte proliferation. Piperine

134
treated mouse splenocytes demonstrated an increase in the secretion of Th-1 cytokines
(IFN-γ and IL-2), increased macrophage activation and proliferation of T and B cells[26].
c. Apoptotic effect
Apoptosis is often downregulated in cancer cells to maintain continued proliferation. The
apoptotic effect of piperine was studied in prostate cancer cell lines wherein treatment
with piperine induced apoptosis by the activation of caspase-3 and by the cleavage of
PARP-1 proteins in different prostate cancer cells like PC-3, DU-145 and LNCaP prostate
cancer cells[27].
d. Anti-metastatic effect
Metastasis is the development of secondary tumours at places other than the site of origin.
Cells treated with piperine showed a reduction in the expression of cyclin B1 which is
important for cell cycle progression in the 4T1 breast cancer cell line. Piperine
significantly decreased the mRNA expression of MMP-9 and MMP-13 which play an
important role in basement membrane degradation. 4T1 cells are shown to metastasize to
organs like the lung 10th day following inoculation of 1×105 tumour cells. The extent of
metastasis in lungs was found to be less in piperine-treated mice as compared to control
mice as determined by colonogenic assay[28].
Piperine has been extensively used as a bioavailability enhancer for other anti-cancer
drugs, but there is not much research on the anti-cancer activities of piperine alone[29].
In this chapter, piperine was encapsulated in chitosan nanospheres by ionic gelation
method. Mannose was conjugated onto the positively charged amine groups of chitosan
using EDC-NHS reaction. The release and surface characteristics of this system were
studied and propose its potential for further anti-cancer work.
4.5. Materials

135
Chitosan, Tween 20, piperine, 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC),
N-hydroxysuccinimide (NHS), N-[tris(hydroxymethyl)methyl]-2-aminoethane sulfonic
acid (MES) and sodium tripolyphosphate (TPP) were used as received.
4.6. Methods
4.6.1. Preparation of nanoparticles
Piperine encapsulated chitosan nanospheres were made by ionic gelation method as
explained in the first chapter 1(Section B,I). Briefly, chitosan solution was prepared by
adding chitosan to a 1% acetic acid solution (pH 4.8) to yield a solution with a
concentration of 1 mg/mL. The solution was allowed to stir overnight. 10 mL of this
solution was taken in a beaker. Piperine (5 mg) dissolved in 1 mL acetone was added very
slowly dropwise, while stirring. This solution was ultrasonicated with a probe sonicator
at 65% amplitude for 2 minutes. Sodium tripolyphosphate (1 mg/mL) was added
dropwise to this solution with continuous stirring, and the end point was indicated by a
slight turbidity. The above solution was allowed to stir for 3 hours for the evaporation of
the organic solvent and the chitosan encapsulated nanoparticles (P-NP) were collected.
4.6.2. Determination of the entrapment efficiency
The entrapment efficiency was calculated by the indirect method. The supernatant was
collected after centrifugation of the chitosan nanoparticles. The calibration curve of
piperine at known concentrations was constructed using UV-Visible spectroscopy at its
absorption maxima of 342 nm. The amount of unloaded piperine was calculated using the
curve.
4.6.3. Bioconjugation
P-NPs were conjugated with mannose using a protocol mentioned in the literature[30,31].
Briefly, 10 mg of mannose was allowed to undergo a ring opening reaction by stirring in
SAB buffer (pH 5) for 1-2 hours. Mannose solution was added to P-NPs and incubated

136
with 15 mg N-hydroxysuccinimide (NHS) and 20 mg N-(3-dimethylaminopropyl)-N-
ethyl carbodiimide hydrochloride (EDC). The mixture was stirred for 4 hours in an inert
nitrogen environment to allow conjugation to take place.
Mannose–conjugated chitosan nanoparticles (MP-NPs) were then centrifuged at 10,000
rpm for 10 minutes to remove unconjugated mannose molecules. The supernatant was
stored for calculation of the unconjugated peptide.
4.6.4. Calculation of conjugation efficiency
Unconjugated mannose was calculated by the phenol-sulphuric acid mannose detection
method[32]. Briefly, a 5% w/v phenol solution was prepared freshly. 50 µL of unknown
sample along with known concentrations of mannose solution were added into 96 well
plate. 150 µL of concentrated sulphuric acid was pipetted into each well and mixed gently,
followed by the addition of 30 µL of the phenol solution. The plate was covered with
aluminium foil and allowed to sit for 10 minutes at room temperature. The absorbance
was now read at 490 nm.
4.6.5. Physico-chemical characterization
4.6.5.1. Particle size and surface charge
Nanoparticles were optimized for particle size and surface charge. The mean particle size
and zeta potential of blank nanoparticles (B-NP), piperine loaded nanoparticles (P-NPs)
and mannose-conjugated nanoparticles (MP-NPs0 were measured by photon correlation
spectroscopy using a Malvern Zetasizer Nano ZS (Malvern Instrument Ltd., Malvern,
UK). Samples were diluted 10 times with double distilled water and analyzed at 25 °C
with a backscattering angle of 173º.
4.6.5.2. FTIR analysis
FTIR analysis of B-NPs, P-NPs and MP-NPs was carried by using an IR
spectrophotometer (Perkin Elmer, USA). Pellets were prepared by adding approximately

137
3 mg of the sample to 100 mg of KBr. Samples were scanned in an inert atmosphere in
the range of 400–4000 cm−1.
4.7. Results
4.7.1. Preparation of nanoparticles
Piperine-encapsulated chitosan nanoparticles were prepared by ionic gelation method as
explained in Chapter 1. The end of nanoparticle formation was noted as slight turbidity.
The formulation was optimized for sodium tripolyphosphate (TPP) added which was in
the range of 1-3 mg. An amount of 2.5 mg TPP gave a particle size of 163.7 ± 3.4 nm
and zeta potential of 23.4 ± 2.4 mV (Table 4.4). An increase in the amount of TPP was
found to cause a decrease in the hydrodynamic size of the nanoparticles. This can be
hypothesized to the increase in the compactness of the nanoparticle by the increased
degree of cross-linking. A further increase in the amount of TPP (3 mg) did cause a
reduction in particle size. However, the surface potential of the particles could not record
a stable value. Hence, formulation F4 was carried forward for further optimization. The
formulation was also optimized for total volume of chitosan solution. Volumes of 8, 10
and 12 mL gave a particle size of 157.9 ± 2.8 nm, 163.7 ± 3.7 nm and 341.1 ± 4.3 nm,
respectively. Although particle size was lower in formulation F7 (8 mL), the zeta potential
could not be recorded which suggested instability in charges. Formulation F4 (10 mL),
showed adequate particle size and surface potential values and hence, was used for further
studies. This formulation was also optimized for process parameters such as sonication
amplitudes and stirring times but the results were constant. Therefore, the same
parameters as the followed protocol were used for further formulations.
Table 4.4: Optimization of formulation parameters for the piperine loaded chitosan nanoparticles
prepared by the ionic gelation method
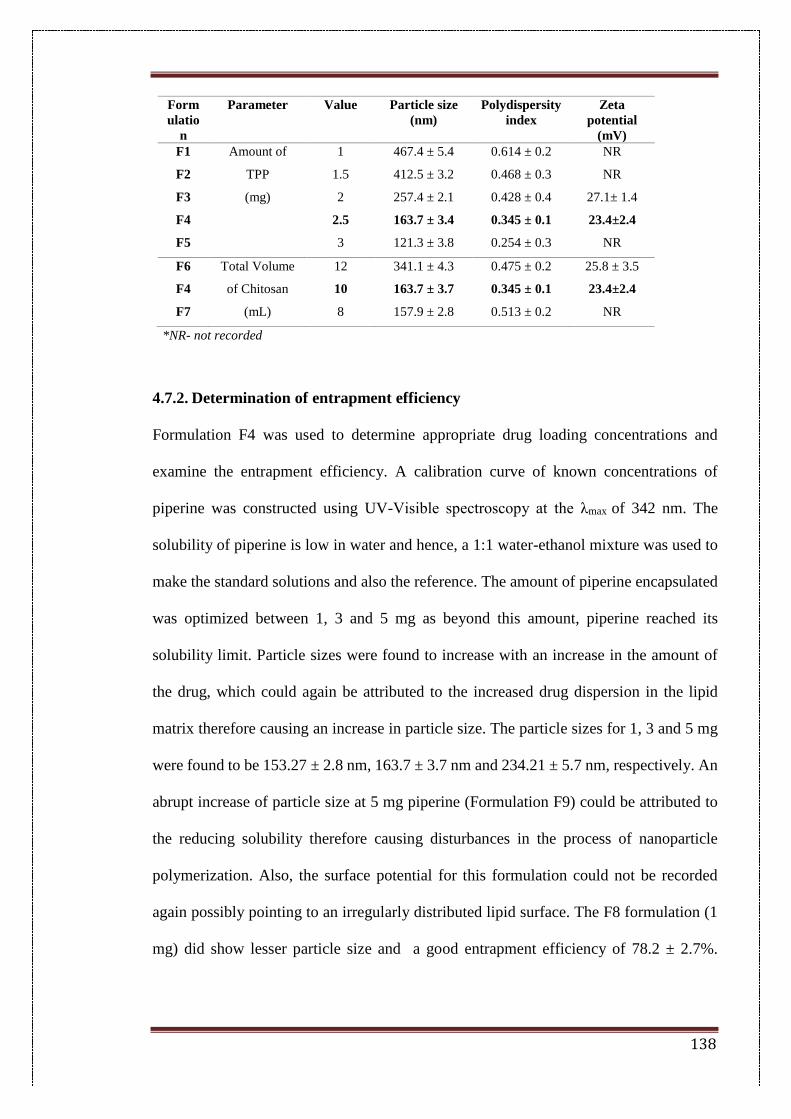
138
Form
ulatio
n
Parameter Value Particle size
(nm)
Polydispersity
index
Zeta
potential
(mV)
F1 Amount of 1 467.4 ± 5.4 0.614 ± 0.2 NR
F2 TPP 1.5 412.5 ± 3.2 0.468 ± 0.3 NR
F3 (mg) 2 257.4 ± 2.1 0.428 ± 0.4 27.1± 1.4
F4
2.5 163.7 ± 3.4 0.345 ± 0.1 23.4±2.4
F5
3 121.3 ± 3.8 0.254 ± 0.3 NR
F6 Total Volume 12 341.1 ± 4.3 0.475 ± 0.2 25.8 ± 3.5
F4 of Chitosan 10 163.7 ± 3.7 0.345 ± 0.1 23.4±2.4
F7 (mL) 8 157.9 ± 2.8 0.513 ± 0.2 NR
*NR- not recorded
4.7.2. Determination of entrapment efficiency
Formulation F4 was used to determine appropriate drug loading concentrations and
examine the entrapment efficiency. A calibration curve of known concentrations of
piperine was constructed using UV-Visible spectroscopy at the λmax of 342 nm. The
solubility of piperine is low in water and hence, a 1:1 water-ethanol mixture was used to
make the standard solutions and also the reference. The amount of piperine encapsulated
was optimized between 1, 3 and 5 mg as beyond this amount, piperine reached its
solubility limit. Particle sizes were found to increase with an increase in the amount of
the drug, which could again be attributed to the increased drug dispersion in the lipid
matrix therefore causing an increase in particle size. The particle sizes for 1, 3 and 5 mg
were found to be 153.27 ± 2.8 nm, 163.7 ± 3.7 nm and 234.21 ± 5.7 nm, respectively. An
abrupt increase of particle size at 5 mg piperine (Formulation F9) could be attributed to
the reducing solubility therefore causing disturbances in the process of nanoparticle
polymerization. Also, the surface potential for this formulation could not be recorded
again possibly pointing to an irregularly distributed lipid surface. The F8 formulation (1
mg) did show lesser particle size and a good entrapment efficiency of 78.2 ± 2.7%.
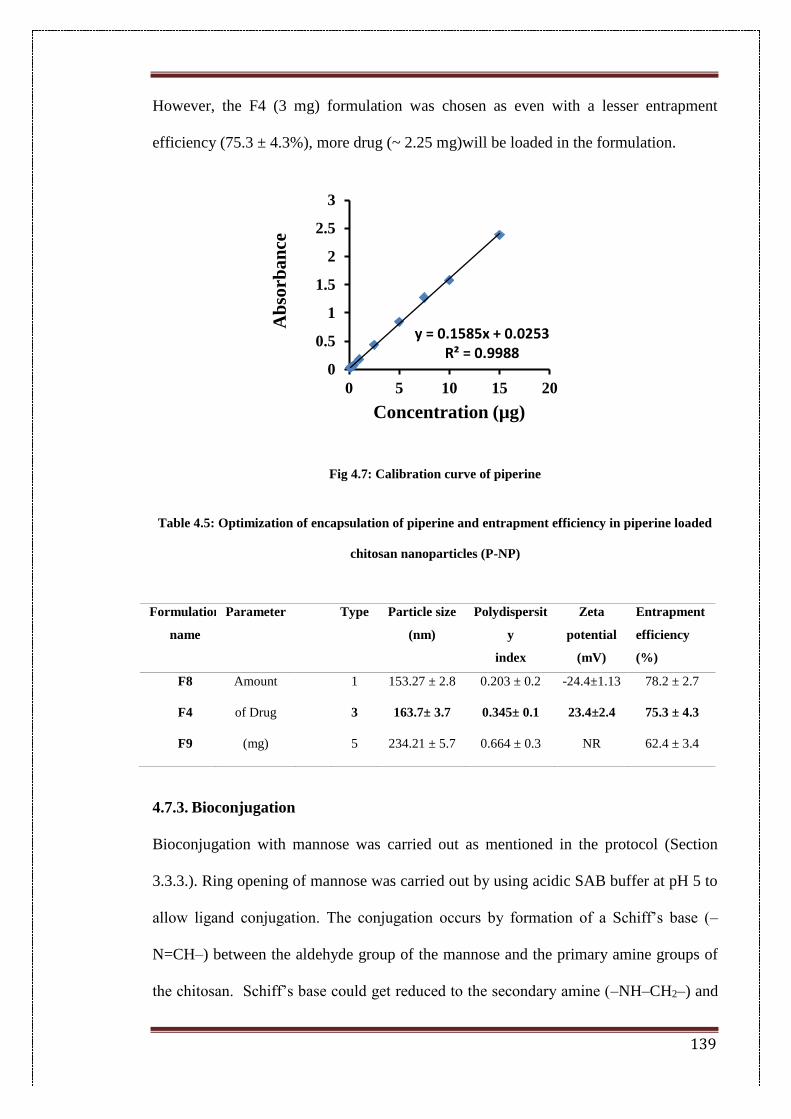
139
However, the F4 (3 mg) formulation was chosen as even with a lesser entrapment
efficiency (75.3 ± 4.3%), more drug (~ 2.25 mg)will be loaded in the formulation.
Fig 4.7: Calibration curve of piperine
Table 4.5: Optimization of encapsulation of piperine and entrapment efficiency in piperine loaded
chitosan nanoparticles (P-NP)
4.7.3. Bioconjugation
Bioconjugation with mannose was carried out as mentioned in the protocol (Section
3.3.3.). Ring opening of mannose was carried out by using acidic SAB buffer at pH 5 to
allow ligand conjugation. The conjugation occurs by formation of a Schiff’s base (–
N=CH–) between the aldehyde group of the mannose and the primary amine groups of
the chitosan. Schiff’s base could get reduced to the secondary amine (–NH–CH2–) and
y = 0.1585x + 0.0253R² = 0.9988
0
0.5
1
1.5
2
2.5
3
0 5 10 15 20
Ab
sorb
an
ce
Concentration (µg)
Formulation
name
Parameter Type Particle size
(nm)
Polydispersit
y
index
Zeta
potential
(mV)
Entrapment
efficiency
(%)
F8 Amount 1 153.27 ± 2.8 0.203 ± 0.2 -24.4±1.13 78.2 ± 2.7
F4 of Drug 3 163.7± 3.7 0.345± 0.1 23.4±2.4 75.3 ± 4.3
F9 (mg) 5 234.21 ± 5.7 0.664 ± 0.3 NR 62.4 ± 3.4

140
remain in equilibrium with the Schiff’s base[31]. The mannose conjugated chitosan
nanoparticles were washed thrice by centrifugation with MilliQ water to remove unbound
mannose. The pellet was stored for further studies.
4.7.4. Calculation of conjugation efficiency
The conjugation efficiency of the mannose to the MgF-SLNs was evaluated by the
phenol-sulphuric acid assay for the detection of sugars. The method works on the
principle of reaction of the aulphonated phenol with the sugars in a solution to give rise
to a yellow orange coloured furfural compound with an absorption maxima at around 490
nm[32]. The supernatant collected post centrifugation of the MP-NPs was used to
determine the unconjugated mannose. The unknown mannose in the supernatant was
determined by using the standard mannose calibration curve. The conjugation efficiency
after using 10 mg of mannose was found to be 92.34 ± 3.31%.
4.7.5. Physico-chemical characterization
4.7.5.1. Particle size and surface charge
The particle size and surface charge of blank nanoparticles (B-NP), piperine-loaded P-
NP and conjugated MP-NP nanoparticles were observed by a Malvern Zetasizer Nano
ZS. The particle size of B-NP, P-NP and MP-NP were 152.9 ± 2.8 nm, 163.7 ± 3.7 nm
and 196.4 ± 4.2 nm, respectively (Table 4.6 and Fig. 4.8). There is an increase of about
10 nm in particle size from B-NP to P-NP which could be attributed to the packing of
piperine molecules between the chitosan polymer matrix therefore leading to increase in
size. The zeta potential for B-NP and P-NP were 24.1 ± 2.3 and 23.4 ± 2.4 mV,
respectively, with the positive potentials due to the amine groups present in chitosan. This
also suggests that there was no change in surface properties suggesting that piperine was
encapsulated and not adsorbed onto the particle surface. The particle size of MP-NP is
196.4 ± 4.2 nm, which is more compared to P-NP (163.7± 3.7). This could be due to the
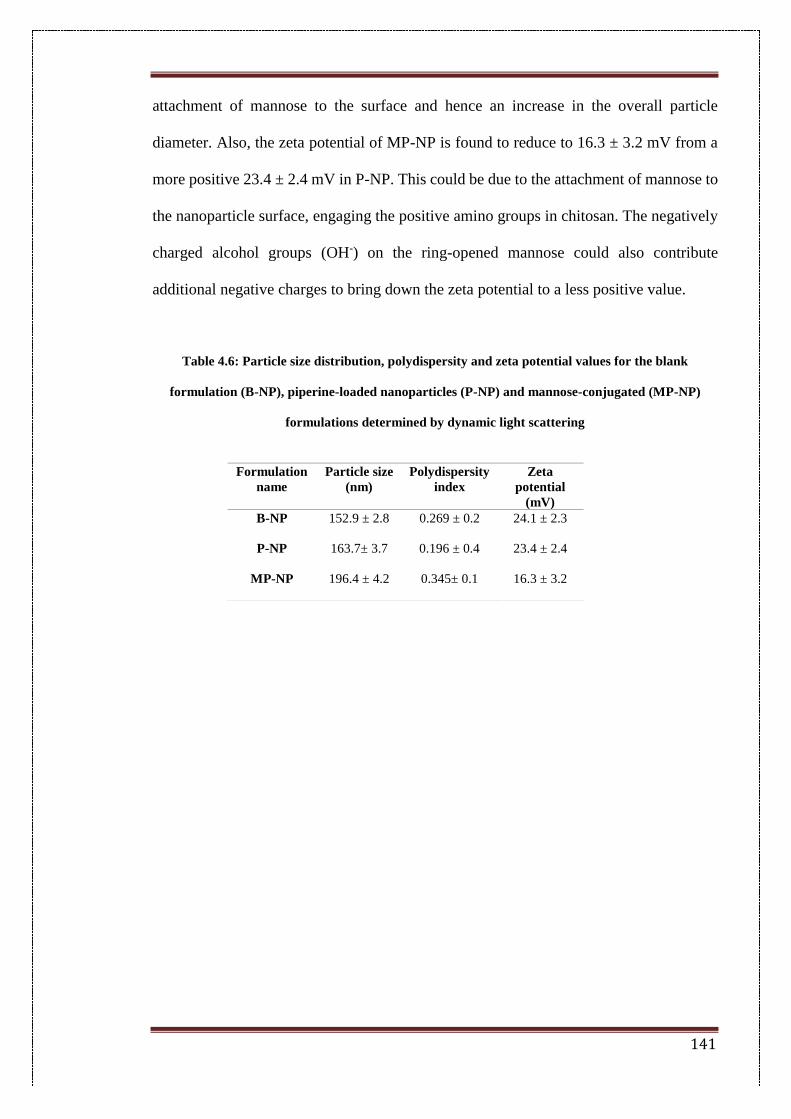
141
attachment of mannose to the surface and hence an increase in the overall particle
diameter. Also, the zeta potential of MP-NP is found to reduce to 16.3 ± 3.2 mV from a
more positive 23.4 ± 2.4 mV in P-NP. This could be due to the attachment of mannose to
the nanoparticle surface, engaging the positive amino groups in chitosan. The negatively
charged alcohol groups (OH-) on the ring-opened mannose could also contribute
additional negative charges to bring down the zeta potential to a less positive value.
Table 4.6: Particle size distribution, polydispersity and zeta potential values for the blank
formulation (B-NP), piperine-loaded nanoparticles (P-NP) and mannose-conjugated (MP-NP)
formulations determined by dynamic light scattering
Formulation
name
Particle size
(nm)
Polydispersity
index
Zeta
potential
(mV)
B-NP 152.9 ± 2.8 0.269 ± 0.2 24.1 ± 2.3
P-NP 163.7± 3.7 0.196 ± 0.4 23.4 ± 2.4
MP-NP 196.4 ± 4.2 0.345± 0.1 16.3 ± 3.2
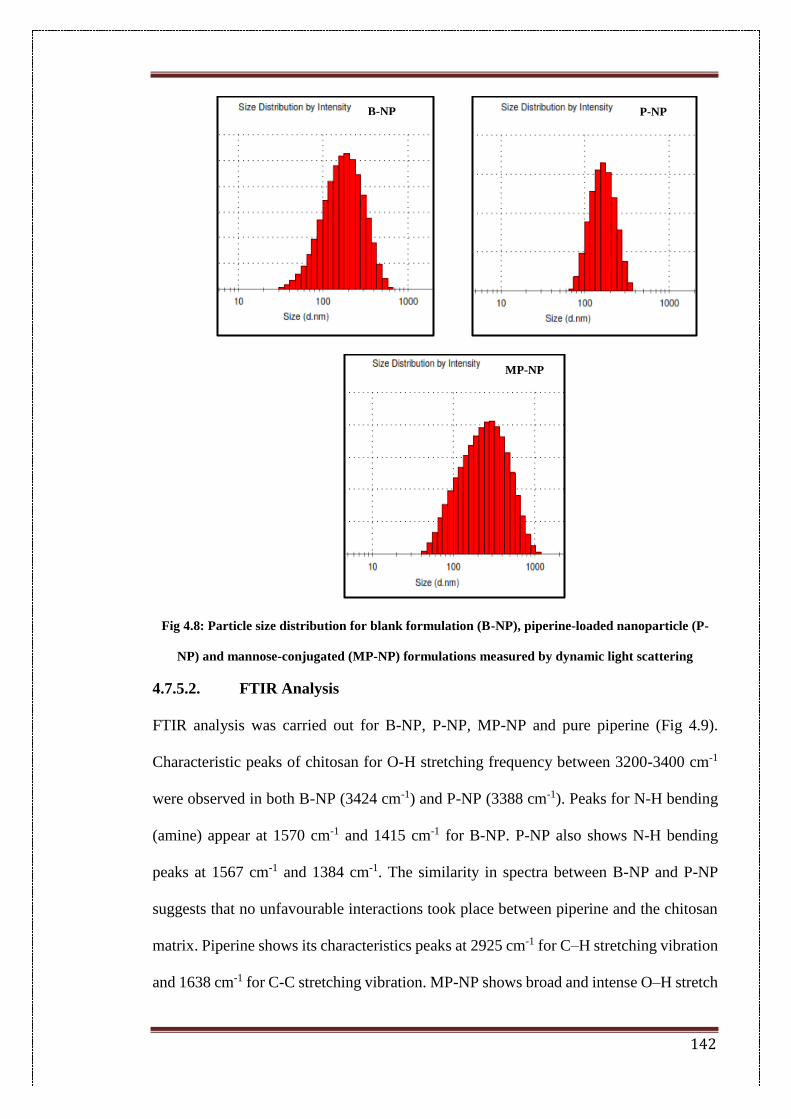
142
Fig 4.8: Particle size distribution for blank formulation (B-NP), piperine-loaded nanoparticle (P-
NP) and mannose-conjugated (MP-NP) formulations measured by dynamic light scattering
4.7.5.2. FTIR Analysis
FTIR analysis was carried out for B-NP, P-NP, MP-NP and pure piperine (Fig 4.9).
Characteristic peaks of chitosan for O-H stretching frequency between 3200-3400 cm-1
were observed in both B-NP (3424 cm-1) and P-NP (3388 cm-1). Peaks for N-H bending
(amine) appear at 1570 cm-1 and 1415 cm-1 for B-NP. P-NP also shows N-H bending
peaks at 1567 cm-1 and 1384 cm-1. The similarity in spectra between B-NP and P-NP
suggests that no unfavourable interactions took place between piperine and the chitosan
matrix. Piperine shows its characteristics peaks at 2925 cm-1 for C–H stretching vibration
and 1638 cm-1 for C-C stretching vibration. MP-NP shows broad and intense O–H stretch
B-NP P-NP
MP-NP
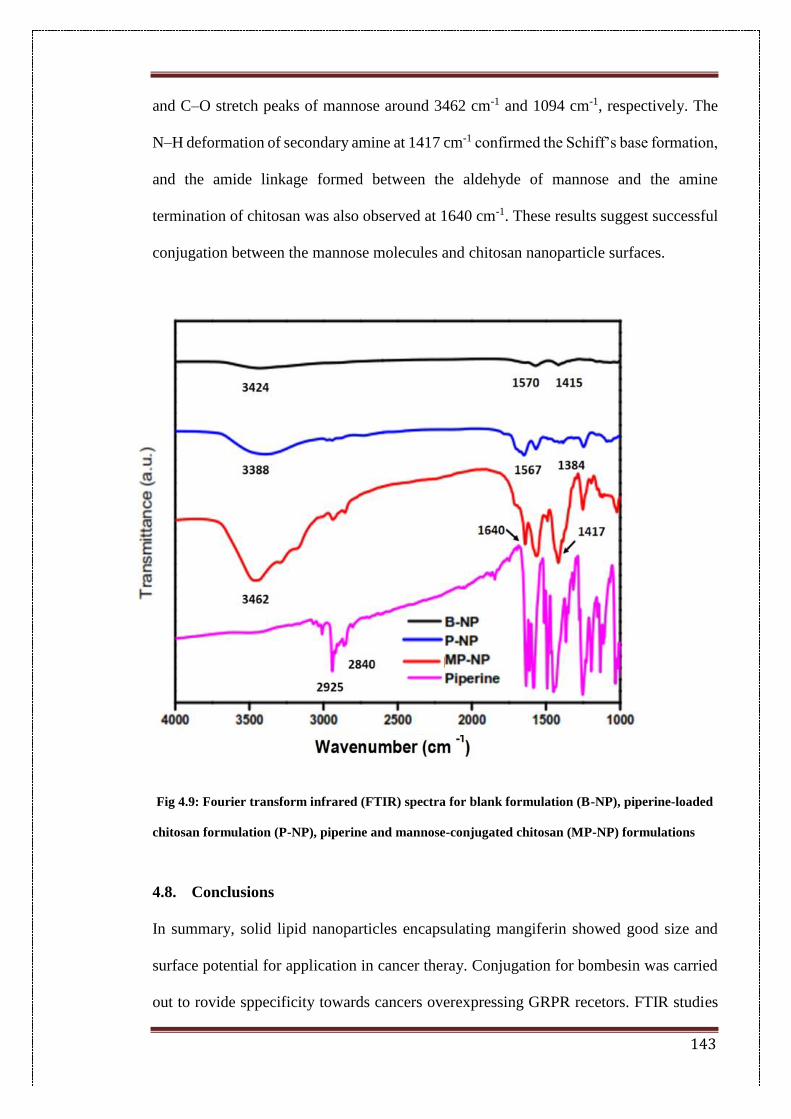
143
and C–O stretch peaks of mannose around 3462 cm-1 and 1094 cm-1, respectively. The
N–H deformation of secondary amine at 1417 cm-1 confirmed the Schiff’s base formation,
and the amide linkage formed between the aldehyde of mannose and the amine
termination of chitosan was also observed at 1640 cm-1. These results suggest successful
conjugation between the mannose molecules and chitosan nanoparticle surfaces.
Fig 4.9: Fourier transform infrared (FTIR) spectra for blank formulation (B-NP), piperine-loaded
chitosan formulation (P-NP), piperine and mannose-conjugated chitosan (MP-NP) formulations
4.8. Conclusions
In summary, solid lipid nanoparticles encapsulating mangiferin showed good size and
surface potential for application in cancer theray. Conjugation for bombesin was carried
out to rovide sppecificity towards cancers overexpressing GRPR recetors. FTIR studies

144
showed formation of amide bond linkages between SLNs and bombesin. Thus, this
system could be a strong candidate for cancer drug delivery. Mannose conjugated
chitosan nanoparticles encapsulating piperine and mangiferin-encapsulated SLNs are
both promising candidates for futher anti-cancer studies based on their physicochemical
properties. These two systems can be used to study in-vitro and in-vivo efficacy in future
studies.

145
4.9. References
[1] H. Wang, T. Oo Khor, L. Shu, Z.-Y. Su, F. Fuentes, J.-H. Lee, et al., Plants vs.
Cancer: A Review on Natural Phytochemicals in Preventing and Treating Cancers and
Their Druggability, Anticancer. Agents Med. Chem. 12 (2012) 1281–1305.
doi:10.2174/187152012803833026.
[2] N.M. Meybodi, A.M. Mortazavian, A.B. Monfared, F.A. Meybodi, Phytochemicals
in Cancer Prevention : A Review of the Evidence, 10 (2017). doi:10.17795/ijcp-
7219.Review.
[3] D.M. Tham, C.D. Gardner, W.L. Haskell, Potential Health Benefits of Dietary
Phytoestrogens: A Review of the Clinical, Epidemiological, and Mechanistic Evidence1,
Http://Dx.Doi.Org/10.1210/Jcem.83.7.4752. 83 (2011) 2223–2235.
doi:10.1210/jc.83.7.2223.
[4] A. Matkowski, P. Kus, E. Goralska, D. Wozniak, Mangiferin – a Bioactive
Xanthonoid, not only from Mango and not just Antioxidant, Mini Rev. Med. Chem. 13
(2013) 439–455. doi:10.2174/138955713804999838.
[5] R.H. Mirza, N. Chi, Y. Chi, Therapeutic Potential of the Natural Product
Mangiferin in Metabolic Syndrome, J. Nutr. Ther. 2 (2013) 74–79.
[6] R.K. Khurana, R. Kaur, S. Lohan, K.K. Singh, B. Singh, Mangiferin: a promising
anticancer bioactive, Pharm. Pat. Anal. 5 (2016) 169–181. doi:10.4155/ppa-2016-0003.
[7] S. Saha, P. Sadhukhan, P.C. Sil, Mangiferin: A xanthonoid with multipotent anti-
inflammatory potential, BioFactors. 42 (2016) 459–474. doi:10.1002/biof.1292.
[8] L. He, X. Peng, J. Zhu, X. Chen, H. Liu, C. Tang, et al., Mangiferin attenuate sepsis-
induced acute kidney injury via antioxidant and anti-inflammatory effects., Am. J.
Nephrol. 40 (2014) 441–50. doi:10.1159/000369220.
[9] P. Rajendran, T. Rengarajan, N. Nandakumar, H. Divya, I. Nishigaki, Mangiferin

146
in cancer chemoprevention and treatment: pharmacokinetics and molecular targets, J.
Recept. Signal Transduct. 35 (2015) 76–84. doi:10.3109/10799893.2014.931431.
[10] H. Li, J. Huang, B. Yang, T. Xiang, X. Yin, W. Peng, et al., Mangiferin exerts
antitumor activity in breast cancer cells by regulating matrix metalloproteinases,
epithelial to mesenchymal transition, and β-catenin signaling pathway, Toxicol. Appl.
Pharmacol. 272 (2013) 180–190. doi:10.1016/j.taap.2013.05.011.
[11] J. Das, J. Ghosh, A. Roy, P.C. Sil, Mangiferin exerts hepatoprotective activity
against D-galactosamine induced acute toxicity and oxidative/nitrosative stress via Nrf2–
NFκB pathways, Toxicol. Appl. Pharmacol. 260 (2012) 35–47.
doi:10.1016/j.taap.2012.01.015.
[12] D. Du Plessis-Stoman, J. Du Preez, M. Van de Venter, Combination treatment with
oxaliplatin and mangiferin causes increased apoptosis and downregulation of nfκb in
cancer cell lines, African J. Tradit. Complement. Altern. Med. 8 (2011).
doi:10.4314/ajtcam.v8i2.63206.
[13] X. Dolcet, D. Llobet, J. Pallares, X. Matias-Guiu, NF-kB in development and
progression of human cancer, Virchows Arch. 446 (2005) 475–482. doi:10.1007/s00428-
005-1264-9.
[14] W. Dou, J. Zhang, G. Ren, L. Ding, A. Sun, C. Deng, et al., Mangiferin attenuates
the symptoms of dextran sulfate sodium-induced colitis in mice via NF-κB and MAPK
signaling inactivation, Int. Immunopharmacol. 23 (2014) 170–178.
doi:10.1016/j.intimp.2014.08.025.
[15] I.J. Fidler, Origin and biology of cancer metastasis, Cytometry. 10 (1989) 673–680.
doi:10.1002/cyto.990100602.
[16] J. Xiao, L. Liu, Z. Zhong, C. Xiao, J. Zhang, Mangiferin regulates proliferation and
apoptosis in glioma cells by induction of microRNA-15b and inhibition of MMP-9

147
expression, Oncol. Rep. 33 (2015) 2815–2820. doi:10.3892/or.2015.3919.
[17] S. Elmore, Apoptosis: A Review of Programmed Cell Death, Toxicol. Pathol. 35
(2007) 495–516. doi:10.1080/01926230701320337.
[18] M. Cuccioloni, L. Bonfili, M. Mozzicafreddo, V. Cecarini, S. Scuri, M. Cocchioni,
et al., Mangiferin blocks proliferation and induces apoptosis of breast cancer cells via
suppression of the mevalonate pathway and by proteasome inhibition, Food Funct. 7
(2016) 4299–4309. doi:10.1039/C6FO01037G.
[19] P. Rajendran, T. Jayakumar, I. Nishigaki, G. Ekambaram, Y. Nishigaki, J.
Vetriselvi, et al., Immunomodulatory Effect of Mangiferin in Experimental Animals with
Benzo(a)Pyrene-induced Lung Carcinogenesis., Int. J. Biomed. Sci. 9 (2013) 68–74.
http://www.ncbi.nlm.nih.gov/pubmed/23847456 (accessed August 15, 2017).
[20] H. Kulhari, D.P. Kulhari, M.K. Singh, R. Sistla, Colloidal stability and
physicochemical characterization of bombesin conjugated biodegradable nanoparticles,
Colloids Surfaces A Physicochem. Eng. Asp. 443 (2014) 459–466.
doi:10.1016/j.colsurfa.2013.12.011.
[21] J.M. Walker, Handbook Edited by, 2002. doi:10.1385/1592591698.
[22] H. Patel, D.R. Panchal, U. Patel, T. Brahmbhatt, M. Suthar, Matrix Type Drug
Delivery System : A Review, J. Pharm. Sci. Biosci. Res. 1 (2011) 143–151.
[23] Z.A. Damanhouri, A. Ahmad, A Review on Therapeutic Potential of Piper nigrum
L. (Black Pepper): The King of Spices, Med. Aromat. Plants. 3 (2014) 1–6.
doi:10.4172/2167-0412.1000161.
[24] R.S. Vijayakumar, D. Surya, N. Nalini, Antioxidant efficacy of black pepper (
Piper nigrum L.) and piperine in rats with high fat diet induced oxidative stress, Redox
Rep. 9 (2004) 105–110. doi:10.1179/135100004225004742.
[25] J.S. Bang, D.H. Oh, H.M. Choi, B.-J. Sur, S.-J. Lim, J.Y. Kim, et al., Anti-

148
inflammatory and antiarthritic effects of piperine in human interleukin 1β-stimulated
fibroblast-like synoviocytes and in rat arthritis models, Arthritis Res. Ther. 11 (2009)
R49. doi:10.1186/ar2662.
[26] E.. Sunila, G. Kuttan, Immunomodulatory and antitumor activity of Piper longum
Linn. and piperine, J. Ethnopharmacol. 90 (2004) 339–346.
doi:10.1016/j.jep.2003.10.016.
[27] A. Samykutty, A.V. Shetty, G. Dakshinamoorthy, M.M. Bartik, G.L. Johnson, B.
Webb, et al., Piperine, a Bioactive Component of Pepper Spice Exerts Therapeutic Effects
on Androgen Dependent and Androgen Independent Prostate Cancer Cells, PLoS One. 8
(2013) e65889. doi:10.1371/journal.pone.0065889.
[28] L. Lai, Q. Fu, Y. Liu, K. Jiang, Q. Guo, Q. Chen, et al., Piperine suppresses tumor
growth and metastasis in vitro and in vivo in a 4T1 murine breast cancer model, Acta
Pharmacol. Sin. 33 (2012) 523–530. doi:10.1038/aps.2011.209.
[29] C.R. Pradeep, G. Kuttan, Effect of piperine on the inhibition of lung metastasis
induced B16F-10 melanoma cells in mice, Clin. Exp. Metastasis. 19 (2002) 703–708.
doi:10.1023/A:1021398601388.
[30] A. Jain, A. Agarwal, S. Majumder, N. Lariya, A. Khaya, H. Agrawal, et al.,
Mannosylated solid lipid nanoparticles as vectors for site-specific delivery of an anti-
cancer drug, J. Control. Release. 148 (2010) 359–367. doi:10.1016/j.jconrel.2010.09.003.
[31] N. Nimje, A. Agarwal, G.K. Saraogi, N. Lariya, G. Rai, H. Agrawal, et al.,
Mannosylated nanoparticulate carriers of rifabutin for alveolar targeting, J. Drug Target.
17 (2009) 777–787. doi:10.3109/10611860903115308.
[32] P. Rao, T.N. Pattabiraman, Further studies on the mechanism of phenol-sulfuric
acid reaction with furaldehyde derivatives, Anal. Biochem. 189 (1990) 178–181.
doi:10.1016/0003-2697(90)90103-G.

149
Summary and future perspectives

150
Cancer remains one of the world’s most debilitating diseases with 8.8 million deaths
worldwide in 2015, with the number of new cases expected to rise by 70% in the next
two decades[1].
Drug delivery in cancer mainly uses two types of approaches; passive and active
targeting[2]. Passive targeting relies on the distinct properties of tumour tissue (such as
enhanced permeation and retention effect) for delivery of the therapeutic payload.
Increased interendothelial gaps in tumour blood vessels causes increased penetration of
nanoparticles up to a size of 400 nm, with defective lymphatic drainage leading to
improved retention and, therefore, more delivery of cytotoxic drugs at tumour sites[3].
The rationale of this thesis is the preparation of nanoparticle assemblies for cancer therapy
with reduced non-specific toxicity. Many conventional model drugs for cancers lead to
non-specific associated toxicities at other sites, creating undesirable side effects[4].
Phytochemicals were used as cancer therapeutics in this thesis, which are plant-derived,
naturally occurring compounds. Epigallocatechin gallate (EGCG), mangiferin and
piperine are derived from green tea, mango and pepper, respectively. All of these
compounds have shown anti-cancer potential in-vitro[5–7]. These compounds have been
part of the regular human diet for centuries, and hence have mechanisms in place for their
effective metabolism, thus leading to reduced injury to non-targeted sites.
Two types of nanoparticle vehicles have been explored in this thesis viz. solid lipid
nanoparticles and chitosan nanoparticles. Solid lipid nanoparticles are made of a lipid
matrix which is solid at room temperature. The solid core gives added protection to the
encapsulated drugs. Also, these lipids can be broken down within the cells by metabolic
enzymes such as lipases. This helps to circumvent accumulation-induced toxicity which
is one of the major drawbacks associated with nanoparticle delivery[8]. Solid lipid
nanoparticles have also been known to show controlled release of the drugs from the

151
matrix and thus, improved bioavailability and sustained efficacy[9]. Chitosan
nanoparticles have a matrix of covalently cross-linked chitosan molecules. Chitosan is a
naturally occurring polysaccharide extracted from the shells of crabs. The cross linked
polymer also helps in effective encapsulation and increased bioavailability of the
encapsulated drugs within the biological system[10]. These nanoparticles can also be
easily degraded within the biological system and thus do not lead to accumulation[11].
Both the targeting approaches (active and passive) were explored in this study. Passive
targeting was used in Chapter 2 wherein EGCG was encapsulated in SLNs and studied
the effect of encapsulation on the anti-cancer efficacy. Other targeting ligands have been
used in the following chapters, including bombesin which is a tetra-decapeptide isolated
from the toad Bombina bombina. Bombesin is an amphibian analogue of the mammalian
gastrin releasing peptide which is a natural ligand to gastrin releasing peptide receptors,
overexpressed in a variety of cancers and hence used as a tumour marker[12]. The
mechanism of nanoparticle uptake through GRPR binding is via receptor mediated
endocytosis. Mannose was used as a ligand to target lectin receptors which are commonly
overexpressed in lung, liver, breast and brain tumours. Uptake of nanoparticles in this
case is also via receptor mediated or clathrin-mediated endocytosis.
EGCG is the most abundant catechin found in green tea. It has been explored in recent
times for its anti-cancer activity[13]. The structure of EGCG however causes it to be
easily degraded within the cell[14]. It has 8 free hydroxyl groups which can be easily
acted upon by cellular enzymes, causing it to be glucuronidated, sulphonated or amidated
therefore causing a loss in activity[14].
The stability of EGCG with different pH conditions using buffer solutions was initially
studied. EGCG was found to be most stable in acidic pH with stability increasing as the
solution becomes increasingly acidic. However, stability was found to reduce as we move

152
towards the neutral pH and further is found to degrade almost instantly in basic solutions.
Therefore, to combat stability issues EGCG was encapsulated in solid lipid nanoparticles.
Furthermore, the physico-chemical parameters such as particle size and surface charge of
the ECG-encapsulated SLNs were studied. Surface functionalities was studied using
FTIR spectroscopy to study any changes in the SLN surface post encapsulation. Spectra
showed encapsulation of EGCG as opposed to adsorption. Also, DSC studies were carried
out to further test whether EGCG was encapsulated within SLNs. The crystalline melting
peak of EGCG was not seen in the EGCG-encapsulated in SLNs, which therefore
confirms that EGCG was not present in its bulk state. Drug release studies were
performed to determine the release characteristics of EGCG from the SLNs. The results
indicated sustained release in acidic pH which could demonstrate favourable release in
acidic tumour tissue. Colloidal stability of the nanoparticles was studied in different
physiological mediums such as PBS, DW and serum to check the behaviour of the
nanoparticles in biological mimicking environments, wherein EGCG-SLN showed
stability in both serum and PBS and high resistance to electrolyte-induced aggregation.
Finally, in-vitro cytotoxicity was employed to assess the change in anti-cancer efficacy
post encapsulation. EGCG-SLN showed an 8-fold increase in cytotoxicity in the breast
cancer cell line and a 4-fold increase in cytotoxicity in the prostate cancer cell line as
opposed to pure EGCG. It also showed enhanced apoptosis as opposed to the free drug.
EGCG-SLN also showed more apoptosis in both cell lines as opposed to pure EGCG.
Thus, we can conclude that the increase in anti-cancer efficacy was obtained owing to the
encapsulation of EGCG within SLNs.
Bombesin-targeted solid lipid nanoparticles were prepared by the double-emulsification
solvent evaporation method as shown in Chapter1 followed by conjugation with the
peptide by EDC-NHS reaction. Bombesin was used as a ligand to target the gastrin-

153
releasing peptide receptor which is overexpressed in breast cancer[15]. FTIR analysis
validated the formation of an amide bond between the carboxylic COO- of the lipid with
the free amino group of the peptide. In-vitro studies in breast cancer cell line MDA-MB-
231 as it shows increased expression of GRP receptors were carried out. IC50 values
indicate that the pure drug EGCG has low cytotoxicity in MDA-MB 231, but its activity
increased more than 8 times after encapsulation in SLNs and more than 14 times post
conjugation with bombesin. Fluorescence uptake images show more uptake in RB-SLN
compared to non-targeted R-SLN at all time points. Migration studies show substantial
inhibition by the targeted EB-SLN formulation. Survival studies on C57BL6 mice
showed greater survival in mice administered with the targeted formulation compared to
the non-targeted ones, which was in turn more than mice administered with only EGCG.
Reductions in tumour volumes in mice administered with targeted formulations were also
observed. This study shows the potential of bombesin conjugated formulation in efficient
breast cancer therapy.
Mangiferin and piperine are two phytochemicals which have been used in ancient
medicine for their medicinal properties including their anti-cancer activities. Although
their mechanisms of action have been widely explored and documented, the number of
formulations that have been tried with these phytochemicals is comparatively low. In this
work, two novel systems for the delivery of these phytochemicals have been explored.
Mangiferin was encapsulated in solid lipid nanoparticles by a single-emulsification
method and further conjugated with bombesin, which is an efficient ligand for targeting
that was explored in Chapter 3. In this work, the formulation was optimized for particle
size and surface potential and checked for its physicochemical characteristics. Fourier
transform infrared analysis showed successful conjugation between the stearic acid
carboxylic group in SLNs and amine group in bombesin. For piperine, a chitosan

154
formulation was tested to optimise a nano-formulation with a carbohydrate matrix.
Mannose was conjugated onto this nanoparticle for targeting towards lectin receptors.
Zetasizer measurements showed size dimensions of <160 nm and surface charge values
of approximately 24mV. FTIR showed successful conjugation and there were no
incompatibilities between piperine and the excipients used for the nanoparticle system.
Both systems show good promise for further studies representing exciting candidates for
further in-vitro and in-vivo studies.
Future prospective in this work:
EGCG-SLN formulation showed 8 times more cytotoxicity than EGCG in breast
cancer cell lines MDA-MB 231 and 4 times more than EGCG in DU-145 prostate cancer
cell lines. In-vivo studies such as biodistribution and pharmacokinetics can be carried out
to further study the efficacy of encapsulated system compared to free EGCG.
A surprising observation was the differential cytotoxicities of the EGCG
formulations in cancerous and non-cancerous cell lines. EGCG showed more cytotoxity
in non-cancerous cell lines as compared to cancerous cell lines. These findings suggest
differential specificity of the conjugated system which could be a superior advantage if
sufficiently validated. More work remains to be done in this respect. Work on in-vivo
studies with this system such as pharmacokinetic studies and tissue distribution can be
carried out to determine in-vivo efficacy.
Mannose conjugated chitosan nanoparticles encapsulating piperine and mangiferin-
encapsulated SLNs conjugated with bombesin are both promising candidates for futher
anti-cancer studies based on their physicochemical properties. These two systems can be
used to study in-vitro cancer efficacy by cytotoxicity, uptake and apoptosis assays. In-
vivo studies such as pharmacookinetics can be carried out to determine distribution of
these nanoparticle system in-vivo.

155
Key knowledge additions provided by this thesis:
➢ EGCG-loaded SLNs helped improve the stability of a labile polyphenol such as
EGCG which has pH dependent stability issues. The increase in in-vitro efficacy could
be attributed to the additional stability imparted by the lipid core and also the slow release
from the SLN.
➢ EGCG-loaded SLNs conjugated with bombesin showed greater efficacy in-vitro as
well as in-vivo when compared to non-targeted nanoparticles. In-vitro studies have shown
specificity to cancerous cell lines in comparison to non-cancerous cells. Survival studies
showed greater survival of mice injected with targeted formulations.
➢ Piperine and mangiferin remain to be explored as potent chemotherapeutics in a
targeted system. SLN formulation with mangiferin and chitosan formulation with
piperine were optimized for size and charge parameters. The conjugation reactions
with bombesin and mannose were validated by FTIR analysis. Thus, two novel
nanoparticle systems have been optimized for particle size, surface conjugation and
surface potential in this thesis.
Publications from this thesis:
1. Published article:
Radhakrishnan, Rasika, et al. "Encapsulation of biophenolic phytochemical EGCG within
lipid nanoparticles enhances its stability and cytotoxicity against cancer." Chemistry and
physics of lipids 198 (2016): 51-60.
2. Article under preparation:
Bombesin-conjugated solid lipid nanoparticles for targeted breast cancer therapy- Work
represented in Chapter 3.

156
References
[1] WHO | Cancer, WHO. (2017). http://www.who.int/cancer/en/ (accessed March 25,
2017).
[2] L. Brannon-Peppas, J.O. Blanchette, Nanoparticle and targeted systems for cancer
therapy, Adv. Drug Deliv. Rev. 64 (2012) 206–212. doi:10.1016/j.addr.2012.09.033.
[3] Y. Fukumori, H. Ichikawa, Nanoparticles for cancer therapy and diagnosis, Adv.
Powder Technol. 17 (2006) 1–28. doi:10.1163/156855206775123494.
[4] J.-J. Monsuez, J.-C. Charniot, N. Vignat, J.-Y. Artigou, Cardiac side-effects of
cancer chemotherapy., Int. J. Cardiol. 144 (2010) 3–15. doi:10.1016/j.ijcard.2010.03.003.
[5] T. Sen, S. Moulik, A. Dutta, P.R. Choudhury, A. Banerji, S. Das, et al.,
Multifunctional effect of epigallocatechin-3-gallate (EGCG) in downregulation of
gelatinase-A (MMP-2) in human breast cancer cell line MCF-7., Life Sci. 84 (2009) 194–
204. doi:10.1016/j.lfs.2008.11.018.
[6] D. Ouyang, L. Zeng, H. Pan, L. Xu, Y. Wang, K. Liu, et al., Piperine inhibits the
proliferation of human prostate cancer cells via induction of cell cycle arrest and
autophagy, Food Chem. Toxicol. 60 (2013) 424–430. doi:10.1016/j.fct.2013.08.007.
[7] M. Cuccioloni, L. Bonfili, M. Mozzicafreddo, V. Cecarini, S. Scuri, M. Cocchioni,
et al., Mangiferin blocks proliferation and induces apoptosis of breast cancer cells via
suppression of the mevalonate pathway and by proteasome inhibition, Food Funct. 7
(2016) 4299–4309. doi:10.1039/C6FO01037G.
[8] Solid Lipid Nanoparticles ( SLN ) and Nanostructured Lipid Carriers ( NLC ):
Occlusive Effect and Penetration Enhancement Ability, (2015) 62–72.
file:///C:/Users/Rasika/Downloads/JCDSA_2015052814334704.pdf (accessed October
5, 2015).
[9] K. Manjunath, J.S. Reddy, V. Venkateswarlu, Solid lipid nanoparticles as drug

157
delivery systems, Methods Find. Exp. Clin. Pharmacol. 27 (2005) 127.
doi:10.1358/mf.2005.27.2.876286.
[10] K.Y. Lee, W.S. Ha, W.H. Park, Blood compatibility and biodegradability of
partially N-acylated chitosan derivatives, Biomaterials. 16 (1995) 1211–1216.
doi:10.1016/0142-9612(95)98126-Y.
[11] G. Suresh, K. Manjunath, V. Venkateswarlu, V. Satyanarayana, Preparation,
characterization, and in vitro and in vivo evaluation of lovastatin solid lipid
nanoparticles., AAPS PharmSciTech. 8 (2007) 24. doi:10.1208/pt0801024.
[12] R. Markwalder, J.C. Reubi, Gastrin-releasing Peptide Receptors in the Human
Prostate: Relation to Neoplastic Transformation, CANCER Res. 59 (1999) 1152–1159.
http://cancerres.aacrjournals.org/content/canres/59/5/1152.full.pdf (accessed July 16,
2017).
[13] Y.D. Jung, M.S. Kim, B.A. Shin, K.O. Chay, B.W. Ahn, W. Liu, et al., EGCG, a
major component of green tea, inhibits tumour growth by inhibiting VEGF induction in
human colon carcinoma cells., Br. J. Cancer. 84 (2001) 844–50.
doi:10.1054/bjoc.2000.1691.
[14] B. Li, W. Du, J. Jin, Q. Du, Preservation of (-)-epigallocatechin-3-gallate
antioxidant properties loaded in heat treated β-lactoglobulin nanoparticles., J. Agric.
Food Chem. 60 (2012) 3477–84. doi:10.1021/jf300307t.
[15] J. Zhou, J. Chen, M. Mokotoff, R. Zhong, L.D. Shultz, E.D. Ball,
Bombesin/Gastrin-Releasing Peptide Receptor: A Potential Target for Antibody-
Mediated Therapy of Small Cell Lung Cancer, (n.d.).
http://clincancerres.aacrjournals.org/content/clincanres/9/13/4953.full.pdf (accessed
March 25, 2017).




























