Embed Size (px)
Citation preview

of March 2, 2016.This information is current as
HIV-1 ProductionEndogenous IL-32 Controls Cytokine and
Charles A. DinarelloJarod A. Zepp, Milene T. Saavedra, Soo-Hyun Kim and Marcel F. Nold, Claudia A. Nold-Petry, Gregory B. Pott,
http://www.jimmunol.org/content/181/1/557doi: 10.4049/jimmunol.181.1.557
2008; 181:557-565; ;J Immunol
Referenceshttp://www.jimmunol.org/content/181/1/557.full#ref-list-1
, 24 of which you can access for free at: cites 50 articlesThis article
Subscriptionshttp://jimmunol.org/subscriptions
is online at: The Journal of ImmunologyInformation about subscribing to
Permissionshttp://www.aai.org/ji/copyright.htmlSubmit copyright permission requests at:
Email Alertshttp://jimmunol.org/cgi/alerts/etocReceive free email-alerts when new articles cite this article. Sign up at:
Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists All rights reserved.Copyright © 2008 by The American Association of9650 Rockville Pike, Bethesda, MD 20814-3994.The American Association of Immunologists, Inc.,
is published twice each month byThe Journal of Immunology
by guest on March 2, 2016
http://ww
w.jim
munol.org/
Dow
nloaded from
by guest on March 2, 2016
http://ww
w.jim
munol.org/
Dow
nloaded from

Endogenous IL-32 Controls Cytokine and HIV-1 Production1
Marcel F. Nold,*† Claudia A. Nold-Petry,* Gregory B. Pott,* Jarod A. Zepp,*Milene T. Saavedra,* Soo-Hyun Kim,*‡ and Charles A. Dinarello2*
IL-32, a proinflammatory cytokine that activates the p38MAPK and NF-�B pathways, induces other cytokines, for example,IL-1�, IL-6, and TNF-�. This study investigated the role of endogenous IL-32 in HIV-1 infection by reducing IL-32 with smallinterfering (si)RNA in freshly infected PBMC and in the latently infected U1 macrophage cell line. When PBMC were pretreatedwith siRNA to IL-32 (siIL-32), IL-6, IFN-�, and TNF-� were reduced by 57, 51, and 36%, respectively, compared with scrambledsiRNA. Cotransfection of NF-�B and AP-1 reporter constructs with siIL-32 decreased DNA binding of these transcription factorsby 42 and 46%, respectively. Cytokine protein array analysis revealed that the inhibitory activity of siIL-32 primarily targeted Th1and proinflammatory cytokines and chemokines, e.g., MIP-1�/�. Unexpectedly, HIV-1 production (as measured by p24) increased4-fold in these same PBMC when endogenous IL-32 was reduced. Because IFN-� was lower in siIL-32-treated PBMC, we blockedIFN-� bioactivity, which enhanced the augmentation of p24 by siIL-32. Furthermore, siIL-32 reduced the natural ligands ofthe HIV-1 coreceptors CCR5 (MIP-1�/� and RANTES) and CXCR4 (SDF-1). Inhibition of endogenous IL-32 in U1 mac-rophages also increased HIV-1. When rhIL-32� was added to these cells, p24 levels fell by 72%; however, in the samecultures IFN-� increased 4-fold. Blockade of IFN-�/� bioactivity in IL-32�-stimulated U1 cells revealed that IFN-� conveysthe anti-HIV-1 effect of rhIL-32�. In summary, depletion of endogenous IL-32 reduced the levels of Th1 and proinflamma-tory cytokines but paradoxically increased p24, proposing IL-32 as a natural inhibitor of HIV-1. The Journal of Immu-nology, 2008, 181: 557–565.
T he cytokine now termed IL-32 was first reported in 1992as being a cDNA from human NK and T cells stimulatedwith IL-2 and originally called NK cell transcript 4 (NK4)
(1). However, the function(s) of this molecule were not knownuntil 2005, when recombinant human IL-32 was shown to activatethe p38MAPK and the NF-�B signal transduction pathways aswell as induce cytokines and chemokines from human and mousecells, for example, IL-1�, IL-6, TNF-�, and IL-8 (2). IL-32 doesnot share sequence homologies with known cytokine families, butbecause the above-mentioned properties are characteristic ofproinflammatory cytokines, the NK cell transcript 4 was renamedIL-32 (2). There are six isoforms of human IL-32 (2, 3), of whichIL-32� and IL-32� are prominently expressed in human PBMCstimulated with killed Mycobacterium tuberculosis (4). Human IL-32� binds specifically to proteinase-3 with a high affinity (4 nM),but the binding is independent of enzyme activity (5).
In human PBMC and mouse macrophages, IL-32 synergizeswith nucleotide oligomerization domain receptor (NOD)-2 andNOD-1 ligands, respectively, for caspase-1-dependent induction ofIL-1� and IL-6 (6). Furthermore, overexpression of human IL-32�in mouse bone marrow transplantation lead to increased levels ofproinflammatory cytokines, a worsening of collagen-induced ar-thritis, and greater inflammation in sulfonic acid-induced colitis(7). Consistent with these observations, other studies found in-creased expression of IL-32 associated with chronic obstructivepulmonary disease, inflammatory bowel disease (8), and psoriasis(reviewed in Ref. 9). Tissues from patients with rheumatoid ar-thritis exhibit high levels of expression of IL-32 that correlate withdisease severity (10, 11); furthermore, gene array analysis ofmRNA from PBMC determined a greater than 2-fold elevation ofIL-32 expression to be present in 56% of the rheumatoid arthritispatients compared to healthy controls with a statistically signifi-cant overall average increase of 1.9 (12).
Since the introduction of highly active antiretroviral therapy,known as HAART, for HIV-1 infection, morbidity and mortalityhave declined dramatically (13). However, adjuvant interventionswith cytokines or cytokine inhibitors for augmentation of anti-HIV-1 cytotoxic T lymphocyte responses, for example, may havea role in controlling disease progression. IL-32 affects NF-�B andp38MAPK in macrophages and T cells, the target cells of HIV-1.In addition, IL-32-induced IL-1�, TNF-�, IL-6, and chemokines(IL-8 and MIP-2) (2) are each relevant to the pathoimmunology ofHIV-1 (14, 15). We therefore investigated the role of this newcytokine in infection with HIV-1.
To study the role of endogenous IL-32 in acute as well as latentHIV-1 infection, we used a PBMC as well as a U1 cell model,respectively. U1 is a subclone of the promonocytic U937 cell line,which has two copies of HIV-1 integrated into its genome (16).Resembling latently infected cells in vivo, U1 cells release fewHIV-1 virions under resting conditions but synthesize largeamounts of the virus when stimulated by PMA (17), IL-1� (18),
*Department of Medicine, University of Colorado Health Sciences Center, Denver,CO 80262; †Department of Pediatrics, Hospital of the J.W. Goethe University, Frank-furt am Main, Germany; and ‡Department of Biomedical Science and Technology,Konkuk University, Seoul, Republic of Korea
First presented at the 15th Annual Meeting of The International Cytokine Society,October 26–30, 2007, San Francisco, CA and published in Abstract form in Cytokine,Vol. 39, Issue 1, July 2007, page 30.
Received for publication November 5, 2007. Accepted for publication April 3, 2008.
The costs of publication of this article were defrayed in part by the payment of pagecharges. This article must therefore be hereby marked advertisement in accordancewith 18 U.S.C. Section 1734 solely to indicate this fact.1 This work was supported by National Institutes of Health Grant AI 15614 and by theCenter for AIDS Research of the University of Colorado Health Sciences Center (bothto C.A.D.). M.F.N. is supported in part by the Deutsche Forschungsgemeinschaft (No747/1-1).2 Address correspondence and reprint requests to Dr. Charles A. Dinarello, Depart-ment of Medicine, Division of Infectious Diseases, B168, University of ColoradoHealth Sciences Center, 4200 East Ninth Avenue, Denver, CO 80262. E-mail address:[email protected]
Copyright © 2008 by The American Association of Immunologists, Inc. 0022-1767/08/$2.00
The Journal of Immunology
www.jimmunol.org
by guest on March 2, 2016
http://ww
w.jim
munol.org/
Dow
nloaded from

TNF-� (19), or IL-18 (20). U1 cells can thus be used as an in vitromodel resembling latently infected cells, as is the case with chronicHIV infection. PBMC, in contrast, can be rendered susceptible toinfection with HIV-1 in vitro by a conditioning regimen of IL-2and PHA as in Ref. 15, allowing for acute infection. Similar to theacute phase of the HIV infection, freshly infected PBMC produceconsiderable quantities of HIV-1 without exogenous stimulation.
Materials and MethodsReagents
RPMI 1640, PBS, FCS, and penicillin/streptomycin (P/S)3 (50 U/ml and 50�g/ml, respectively) were purchased from Cellgro. LPS (O55:B5), PHA,and PMA were obtained from Sigma-Aldrich. The Dual luciferase kit andconstructs were purchased from Promega. The Nucleofector II electropo-ration device and reagents were obtained from Amaxa. Small interferingRNA (siRNA) to IL-32 (siIL-32) (antisense sequence: 5�-UCAUCAGAGAGGACCUUCGUU-3�) was an ON-TARGETplus duplex suppliedby Thermo Fisher Scientific. Scrambled siRNA (Silencer negative controlno.1) was purchased from Ambion. The lactate dehydrogenase (LDH) de-tection kit was from BioVision. Recombinant human IL-1�, recombinanthuman IL-32�, the IFN-� multisubtype ELISA, and the human cytokineAb arrays (proteome profiles) were purchased from R&D Systems. TheIFN-� multisubtype ELISA was from R&D Systems.
The mAbs to the IFN-� and the IFN-�/� receptors, anti-IFN-�R andanti-IFN-�/�R, as well as to soluble IFN-�/�R were provided by Dr. D.Novick, Weizmann Institute, Rehovot, Israel. The affinity-purified goat anti-human-IL-32 Ab used for Western blotting was prepared with the same Agas that used for the immunization of rabbits. Affinity purification of poly-clonal anti-human IL-32 Abs was described previously (2). All FACS Abs,i.e., CD3, CD4, CD14, CXCR4, and CCR5, were purchased from BDBiosciences/BD Pharmingen.
U1 cells
U1 cells were purchased from American Tissue Culture Collection andcultured in RPMI 1640 with 10% FCS and P/S. For experiments requiringtransfection, cells were counted with trypan blue, centrifuged at 120 � g,and resuspended in prewarmed cell line Nucleofector solution V (Amaxa).Immediately thereafter, siRNA was added and electroporation was per-formed according to manufacturer’s instructions (Amaxa). We tested fourdifferent sequences for siIL-32 and demonstrated all of these to be effectivebefore selecting the most efficient one for further experimentation. Cellswere then transferred to tubes containing 500 �l of serum-free RPMI 1640and incubated at 37°C for 20 min, followed by another transfer to 2 ml ofprewarmed RPMI 1640 with FCS and P/S in 6-well polystyrene plates.After an overnight recovery period, cells were counted with trypan blue,plated into 24-well polystyrene plates at 1 � 106 cells/ml, and either stim-ulated as indicated or left untreated for controls for 21 h. The harvest isdescribed in the next paragraph under the heading PBMC. In addition to theexperiments described, we performed control electroporations withoutsiRNA that did not show significant differences compared with scrambledsiRNA-transfected conditions.
PBMC
These studies were approved by the Colorado Multiple Institutional Re-view Board (Aurora, CO). After informed consent was obtained, PBMCwere isolated from peripheral venous blood of healthy volunteers who hadabstained from taking any medication at least 2 wk before the blood drawas described previously (21). Cells (3 � 106/ml) were suspended in R3medium (RPMI 1640 with 10% FCS, P/S, IL-2 (25 ng/ml), and PHA (3�g/ml). After a 48 h incubation at 37°C in 5% CO2 in polystyrene tissueculture flasks, cells were detached, counted, and examined for viability bytrypan blue exclusion. For experiments requiring transfection, cells werecentrifuged at 120 � g and resuspended in 100 �l of prewarmed human Tcell Nucleofector solution (Amaxa) per condition. Immediately thereafter,siRNA was added and electroporation was performed according to manu-facturer’s instructions (Amaxa). Following the procedure, cells were trans-ferred to tubes containing 500 �l of serum-free human T cell culture me-dium (Amaxa) and incubated at 37°C. At 20 min and 6 h afterelectroporation, the cultures were supplemented with 2 ml of human T cellculture medium with FCS and P/S, as well as IL-2/PHA, respectively, and
allowed to recover overnight. On the next morning, cells were againcounted with trypan blue, transferred to a 15-ml polypropylene tube, andcentrifuged at 400 � g. The pellet was resuspended in 50 �l of RPMI 1640,infected with 500 50% tissue culture-infective doses (TCID50) of T cell-tropic HIV-1 from the AO18A strain per 106 PBMC and incubated for 3 hat 37°C in 5% CO2 as described elsewhere (15). Thereafter, cells (106/ml)were resuspended in fresh R3 medium without PHA and at this time asample of cells was taken and centrifuged and the cell pellet was lysed inlysis buffer (22) for analysis of p24 and cytokines. The remaining infectedPBMC suspension was aliquoted into 24-well flat-bottom polystyreneplates and either stimulated as indicated or remained untreated as controls.After 3 days at 37°C and 5% CO2, supernatants were removed and treatedwith Triton X-100 (1% final concentration). The cells were lysed in lysisbuffer and frozen at �70°C. Before assay, the lysates were clarified bycentrifugation at 20,000 � g for 10 min and the pellet was discarded.
Electrochemiluminescence (ECL) assays
Cytokines and p24 were measured using specific Abs immobilized on mag-netic beads. Biotinylated Abs were mixed with streptavidin-coated beadsand then incubated with the samples. A second cytokine-specific Ab la-beled with ruthenium (Wellstat Diagnostics) was added and the amount ofchemiluminescence was determined by using an Origen analyzer (Bio-Veris). Ab pairs for IL-1�, IL-1�, IL-6, IL-8, and TNF-� were from R&DSystems. Ab pairs for IFN-� were purchased from Fitzgerald IndustriesInternational. The ECL assay for IL-32 has been described previously (2,4). For comparison, Western blotting was performed on the IL-32 proteinstandard used in this assay. According to the results shown in Fig. 1, U1cell lysates contain constitutive IL-32 protein in the 5- to 25-ng range. Thiscontrasts with the ECL readings that measured IL-32 levels in U1 as wellas PBMC samples in the 5- to 500-pg range. One reason for this discrep-ancy may be that during the preparation of the samples for Western blot-ting, boiling may have resulted in the uncovering of epitopes (e.g., by
3 Abbreviations used in this paper: P/S, penicillin/streptomycin; ECL, electrochemi-luminescence; LDH, lactate dehydrogenase; RLU, relative light unit; siIL-32, siRNAto IL-32; siRNA, small interfering RNA; TCID50, 50% tissue culture infective dose.
FIGURE 1. Comparison of the IL-32 ECL assay with Western blotting.A, A serial dilution of the rIL-32� protein used in the ECL assay as thestandard was subjected to Western blot analysis (lanes a–h). In lanes j andk, U1 cell lysates were included for comparison. Lane i is an empty lane.The numbers above the blot indicate the readings of the same samples inthe ECL assay; the numbers below the blot indicate the concentration of thestandard as calculated from the stock preparation. The scientific expres-sions for lanes f, g, and h are for 0.2, 0.04, and 0.008 ng/ml, respectively.One representative of three independently performed experiments isshown. B, The concentrations of the ECL assay (x-axis) are plotted againstthe concentrations calculated from the stock preparation (y-axis) as deter-mined by the Western blot shown in A. The high values of IL-32 in theECL shown on the x-axis were measured in lysates from human endothelialcells stimulated with IL-1� (data not shown).
558 Endogenous IL-32 in HIV
by guest on March 2, 2016
http://ww
w.jim
munol.org/
Dow
nloaded from

deoligomerization and/or unfolding), rendering the protein more readilydetectable. Moreover, the rIL-32 we used as the ECL standard consists ofthe � isoform, whereas cell culture lysates highly probably comprise amixture of IL-32 isoforms. However, as there are no tools available at thistime that can differentiate between these isoforms on the protein level, thisaspect remains to be clarified by a future study. The fact that IL-32 runsslightly higher on the gel than the recombinant protein is highly probablydue to glycosylation.
The biotinylated Ab for p24 determination was provided by L. Lam-brecht, University of Massachussetts, Worchester, MA and the secondaryAb was obtained from the National Institutes of Health AIDS Research andRepository, Germantown, MD. The results from this assay were verified bycomparison to a p24 ELISA from Beckman Coulter.
Western blotting
Twenty-five micrograms of total protein were solubilized in SDS samplebuffer with 2-ME and separated by electrophoresis on 10, 13, or 15%SDS-polyacrylamide gels. Proteins were then transferred to nitrocellulosemembranes (0.2 �m). IL-32 was detected using an affinity-purified goatprimary polyclonal Ab and a HRP-labeled rabbit anti-goat secondary Ab.For visualization, we used the ECL technology by Pierce. Equal loading ofproteins was ascertained by �-actin staining.
Cytokine protein array
Equal volumes of cell culture supernatants were incubated with the pre-coated Proteome Profiler array membrane (R&D Systems) according to themanufacturer’s instructions. Densitometric analysis of the dot blot wasperformed using the AIDA software from Raytest. IL-2 is not shown, as itwas added to the PBMC cultures exogenously.
NF-�B and AP-1 DNA-binding assays
For determination of DNA-bound NF-�B and AP-1, PBMC were simul-taneously transfected with siIL-32 or scrambled siRNA, a firefly luciferase-labeled NF-�B or AP-1 construct, and a Renilla luciferase-labeled con-struct (for control of transfection efficiency). After the recovery period,cells were aliquoted in 24-well plates, stimulated as indicated in Results,and then lysed in a buffer containing 1% Triton X-100, 50 mM Tris (pH7.5), 100 mM NaCl, 50 mM NaF, 40 mM phosphate, and 5 mM EDTA.Firefly and Renilla luciferase activities were analyzed with a Dual lucif-erase kit (Promega), according to the manufacturer’s instructions. Arbitraryunits were calculated for each sample as follows: relative light units (RLU)of target � average RLU all controls/RLU individual control.
FACS analysis
After the 3-day incubation period, PBMC were washed in PBS containing1% BSA. Cells were then stained with Abs directed against CD3, CD4, andCXCR4 or CD14 and CCR5, followed by two more wash steps and fixationin 1% formaldehyde. Fluorescence was measured using a FACSCaliburcytometer (BD Biosciences).
Statistical analysis
Data were analyzed by paired or unpaired Student’s t test and/or by theMann-Whitney rank sum or the Wilcoxon signed rank tests on raw data andare presented either as means of absolute values or percentage of change.Results of the colorimetric LDH assay are given in arbitrary units.
ResultsReduction of endogenous IL-32 by siRNA up-regulates HIV-1 inU1 cells
Under steady-state culture conditions, U1 cells express both IL-32mRNA (data not shown) and protein (see Fig. 2). IL-32 protein isprimarily cell associated in U1 cells. To study the role of endog-enous IL-32 in latent HIV-1 infection, we used specific siRNA toIL-32, which targets each of the IL-32 isoforms as confirmed by aBLAST (basic local alignment search tool) alignment. U1 macro-phages were transfected with increasing concentrations of siIL-32and matching concentrations of scrambled siRNA were used ascontrols. siIL-32 was tested in U1 under both resting and IL-1�-stimulated conditions. As shown in Fig. 2B, constitutive produc-tion of IL-32 protein was dose-dependently reduced in resting cellstransfected with siIL-32 (reductions of 40% at 200 nM and 64% at500 nM; p � 0.05 and �0.01, respectively). These reductions were
also observed by Western blotting of identically treated lysates. Asshown in Fig. 2A, the same concentrations of scrambled siRNA didnot affect IL-32 levels whereas IL-32 was progressively reducedby increasing concentrations of siIL-32. Also shown in Fig. 2B isthe effect of siIL-32 in U1 cells stimulated with IL-1�. Stimulation
FIGURE 2. siIL-32 reduces IL-32 protein levels in U1 cells. ScrambledsiRNA (scr) and siIL-32 (si32) were transfected into U1 cells. For each con-centration of siIL-32, the same concentration of scrambled siRNA was tested.Cells were then either stimulated with IL-1� (10 ng/ml) or left untreated. Afterharvest, IL-32 protein levels in the lysates were determined by Western blot-ting (A) or ECL (B and C). A, Twenty-five microliters of the cell lysates weresubjected to Western blot analysis with staining for IL-32 (upper panel) and�-actin (lower panel). One representative of three independently performedexperiments is shown. B and C, The percentage changes in IL-32 protein levelswere calculated by setting scrambled siRNA at 100% for each concentration(B; range in scrambled siRNA lysates for unstimulated cells, 19–69 pg/ml; forcells stimulated with IL-1�, 18–63 pg/ml) or time point (C). B, Data arepresented as mean � SEM, n � 7; �, p � 0.05; ��, p � 0.01 for siIL-32compared with concentration-matched scrambled siRNA. C, After transfectionwith 500 nM siRNA, U1 cells were harvested at the indicated time points andthe lysates were assayed for IL-32. One representative of four independentlyperformed experiments is shown.
559The Journal of Immunology
by guest on March 2, 2016
http://ww
w.jim
munol.org/
Dow
nloaded from
with IL-1� did not affect the constitutive expression of IL-32 pro-tein levels but the reductions in IL-32 by siIL-32 were comparableto those in resting U1 cells.
The reductions in constitutive IL-32 protein levels by siIL-32were greatest within the first 24 h after transfection (Fig. 2C).Thereafter, IL-32 levels slowly recovered but remained 30% belowbaseline 4 days after electroporation. To exclude the possibilitythat the effects of siIL-32 on endogenous IL-32 protein levels weredue to cell death, we performed cell counts and measured LDHlevels in the supernatants. Both assessments showed no differencesbetween siIL-32 and scrambled siRNA (data not shown).
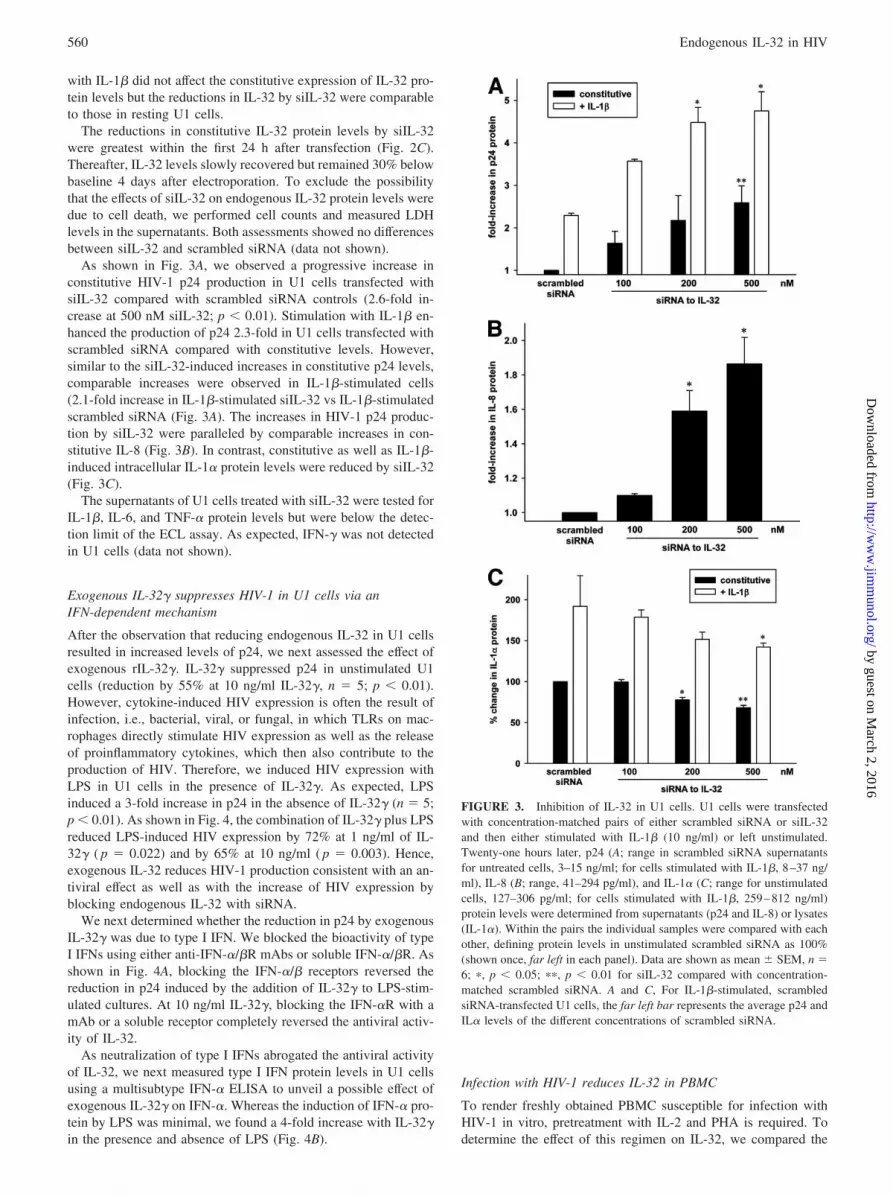
As shown in Fig. 3A, we observed a progressive increase inconstitutive HIV-1 p24 production in U1 cells transfected withsiIL-32 compared with scrambled siRNA controls (2.6-fold in-crease at 500 nM siIL-32; p � 0.01). Stimulation with IL-1� en-hanced the production of p24 2.3-fold in U1 cells transfected withscrambled siRNA compared with constitutive levels. However,similar to the siIL-32-induced increases in constitutive p24 levels,comparable increases were observed in IL-1�-stimulated cells(2.1-fold increase in IL-1�-stimulated siIL-32 vs IL-1�-stimulatedscrambled siRNA (Fig. 3A). The increases in HIV-1 p24 produc-tion by siIL-32 were paralleled by comparable increases in con-stitutive IL-8 (Fig. 3B). In contrast, constitutive as well as IL-1�-induced intracellular IL-1� protein levels were reduced by siIL-32(Fig. 3C).
The supernatants of U1 cells treated with siIL-32 were tested forIL-1�, IL-6, and TNF-� protein levels but were below the detec-tion limit of the ECL assay. As expected, IFN-� was not detectedin U1 cells (data not shown).
Exogenous IL-32� suppresses HIV-1 in U1 cells via anIFN-dependent mechanism
After the observation that reducing endogenous IL-32 in U1 cellsresulted in increased levels of p24, we next assessed the effect ofexogenous rIL-32�. IL-32� suppressed p24 in unstimulated U1cells (reduction by 55% at 10 ng/ml IL-32�, n � 5; p � 0.01).However, cytokine-induced HIV expression is often the result ofinfection, i.e., bacterial, viral, or fungal, in which TLRs on mac-rophages directly stimulate HIV expression as well as the releaseof proinflammatory cytokines, which then also contribute to theproduction of HIV. Therefore, we induced HIV expression withLPS in U1 cells in the presence of IL-32�. As expected, LPSinduced a 3-fold increase in p24 in the absence of IL-32� (n � 5;p � 0.01). As shown in Fig. 4, the combination of IL-32� plus LPSreduced LPS-induced HIV expression by 72% at 1 ng/ml of IL-32� ( p � 0.022) and by 65% at 10 ng/ml ( p � 0.003). Hence,exogenous IL-32 reduces HIV-1 production consistent with an an-tiviral effect as well as with the increase of HIV expression byblocking endogenous IL-32 with siRNA.
We next determined whether the reduction in p24 by exogenousIL-32� was due to type I IFN. We blocked the bioactivity of typeI IFNs using either anti-IFN-�/�R mAbs or soluble IFN-�/�R. Asshown in Fig. 4A, blocking the IFN-�/� receptors reversed thereduction in p24 induced by the addition of IL-32� to LPS-stim-ulated cultures. At 10 ng/ml IL-32�, blocking the IFN-�R with amAb or a soluble receptor completely reversed the antiviral activ-ity of IL-32.
As neutralization of type I IFNs abrogated the antiviral activityof IL-32, we next measured type I IFN protein levels in U1 cellsusing a multisubtype IFN-� ELISA to unveil a possible effect ofexogenous IL-32� on IFN-�. Whereas the induction of IFN-� pro-tein by LPS was minimal, we found a 4-fold increase with IL-32�in the presence and absence of LPS (Fig. 4B).
Infection with HIV-1 reduces IL-32 in PBMC
To render freshly obtained PBMC susceptible for infection withHIV-1 in vitro, pretreatment with IL-2 and PHA is required. Todetermine the effect of this regimen on IL-32, we compared the
FIGURE 3. Inhibition of IL-32 in U1 cells. U1 cells were transfectedwith concentration-matched pairs of either scrambled siRNA or siIL-32and then either stimulated with IL-1� (10 ng/ml) or left unstimulated.Twenty-one hours later, p24 (A; range in scrambled siRNA supernatantsfor untreated cells, 3–15 ng/ml; for cells stimulated with IL-1�, 8–37 ng/ml), IL-8 (B; range, 41–294 pg/ml), and IL-1� (C; range for unstimulatedcells, 127–306 pg/ml; for cells stimulated with IL-1�, 259–812 ng/ml)protein levels were determined from supernatants (p24 and IL-8) or lysates(IL-1�). Within the pairs the individual samples were compared with eachother, defining protein levels in unstimulated scrambled siRNA as 100%(shown once, far left in each panel). Data are shown as mean � SEM, n �6; �, p � 0.05; ��, p � 0.01 for siIL-32 compared with concentration-matched scrambled siRNA. A and C, For IL-1�-stimulated, scrambledsiRNA-transfected U1 cells, the far left bar represents the average p24 andIL� levels of the different concentrations of scrambled siRNA.
560 Endogenous IL-32 in HIV
by guest on March 2, 2016
http://ww
w.jim
munol.org/
Dow
nloaded from
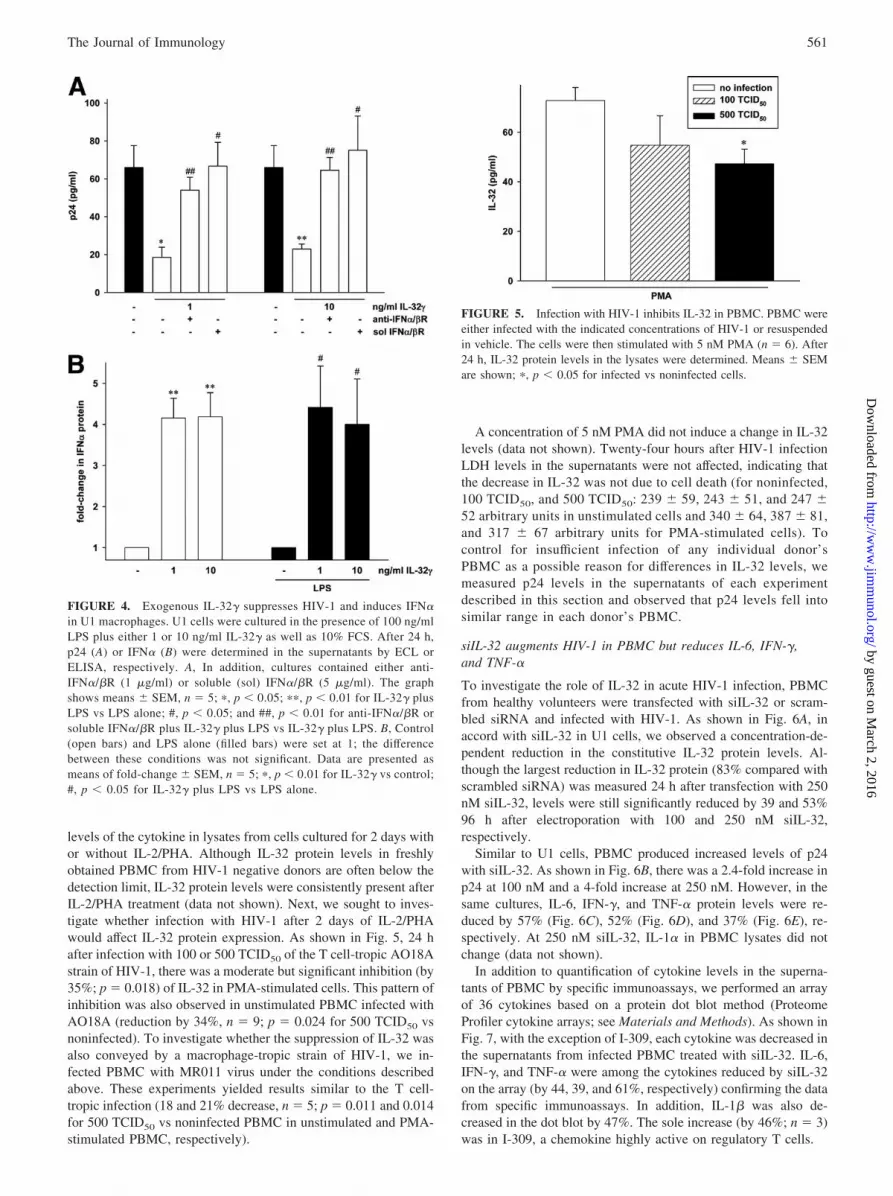
levels of the cytokine in lysates from cells cultured for 2 days withor without IL-2/PHA. Although IL-32 protein levels in freshlyobtained PBMC from HIV-1 negative donors are often below thedetection limit, IL-32 protein levels were consistently present afterIL-2/PHA treatment (data not shown). Next, we sought to inves-tigate whether infection with HIV-1 after 2 days of IL-2/PHAwould affect IL-32 protein expression. As shown in Fig. 5, 24 hafter infection with 100 or 500 TCID50 of the T cell-tropic AO18Astrain of HIV-1, there was a moderate but significant inhibition (by35%; p � 0.018) of IL-32 in PMA-stimulated cells. This pattern ofinhibition was also observed in unstimulated PBMC infected withAO18A (reduction by 34%, n � 9; p � 0.024 for 500 TCID50 vsnoninfected). To investigate whether the suppression of IL-32 wasalso conveyed by a macrophage-tropic strain of HIV-1, we in-fected PBMC with MR011 virus under the conditions describedabove. These experiments yielded results similar to the T cell-tropic infection (18 and 21% decrease, n � 5; p � 0.011 and 0.014for 500 TCID50 vs noninfected PBMC in unstimulated and PMA-stimulated PBMC, respectively).
A concentration of 5 nM PMA did not induce a change in IL-32levels (data not shown). Twenty-four hours after HIV-1 infectionLDH levels in the supernatants were not affected, indicating thatthe decrease in IL-32 was not due to cell death (for noninfected,100 TCID50, and 500 TCID50: 239 � 59, 243 � 51, and 247 �52 arbitrary units in unstimulated cells and 340 � 64, 387 � 81,and 317 � 67 arbitrary units for PMA-stimulated cells). Tocontrol for insufficient infection of any individual donor’sPBMC as a possible reason for differences in IL-32 levels, wemeasured p24 levels in the supernatants of each experimentdescribed in this section and observed that p24 levels fell intosimilar range in each donor’s PBMC.
siIL-32 augments HIV-1 in PBMC but reduces IL-6, IFN-�,and TNF-�
To investigate the role of IL-32 in acute HIV-1 infection, PBMCfrom healthy volunteers were transfected with siIL-32 or scram-bled siRNA and infected with HIV-1. As shown in Fig. 6A, inaccord with siIL-32 in U1 cells, we observed a concentration-de-pendent reduction in the constitutive IL-32 protein levels. Al-though the largest reduction in IL-32 protein (83% compared withscrambled siRNA) was measured 24 h after transfection with 250nM siIL-32, levels were still significantly reduced by 39 and 53%96 h after electroporation with 100 and 250 nM siIL-32,respectively.
Similar to U1 cells, PBMC produced increased levels of p24with siIL-32. As shown in Fig. 6B, there was a 2.4-fold increase inp24 at 100 nM and a 4-fold increase at 250 nM. However, in thesame cultures, IL-6, IFN-�, and TNF-� protein levels were re-duced by 57% (Fig. 6C), 52% (Fig. 6D), and 37% (Fig. 6E), re-spectively. At 250 nM siIL-32, IL-1� in PBMC lysates did notchange (data not shown).
In addition to quantification of cytokine levels in the superna-tants of PBMC by specific immunoassays, we performed an arrayof 36 cytokines based on a protein dot blot method (ProteomeProfiler cytokine arrays; see Materials and Methods). As shown inFig. 7, with the exception of I-309, each cytokine was decreased inthe supernatants from infected PBMC treated with siIL-32. IL-6,IFN-�, and TNF-� were among the cytokines reduced by siIL-32on the array (by 44, 39, and 61%, respectively) confirming the datafrom specific immunoassays. In addition, IL-1� was also de-creased in the dot blot by 47%. The sole increase (by 46%; n � 3)was in I-309, a chemokine highly active on regulatory T cells.
FIGURE 4. Exogenous IL-32� suppresses HIV-1 and induces IFN�in U1 macrophages. U1 cells were cultured in the presence of 100 ng/mlLPS plus either 1 or 10 ng/ml IL-32� as well as 10% FCS. After 24 h,p24 (A) or IFN� (B) were determined in the supernatants by ECL orELISA, respectively. A, In addition, cultures contained either anti-IFN�/�R (1 �g/ml) or soluble (sol) IFN�/�R (5 �g/ml). The graphshows means � SEM, n � 5; �, p � 0.05; ��, p � 0.01 for IL-32� plusLPS vs LPS alone; #, p � 0.05; and ##, p � 0.01 for anti-IFN�/�R orsoluble IFN�/�R plus IL-32� plus LPS vs IL-32� plus LPS. B, Control(open bars) and LPS alone (filled bars) were set at 1; the differencebetween these conditions was not significant. Data are presented asmeans of fold-change � SEM, n � 5; �, p � 0.01 for IL-32� vs control;#, p � 0.05 for IL-32� plus LPS vs LPS alone.
FIGURE 5. Infection with HIV-1 inhibits IL-32 in PBMC. PBMC wereeither infected with the indicated concentrations of HIV-1 or resuspendedin vehicle. The cells were then stimulated with 5 nM PMA (n � 6). After24 h, IL-32 protein levels in the lysates were determined. Means � SEMare shown; �, p � 0.05 for infected vs noninfected cells.
561The Journal of Immunology
by guest on March 2, 2016
http://ww
w.jim
munol.org/
Dow
nloaded from

Flow cytometry analysis revealed that siRNA to IL-32 did notaffect surface expression of CXCR4 and CCR5 on PBMC. In ad-dition, siIL-32 did not induce any changes in the distribution of
CD3�, CD4�, or CD14� cells within the total PBMC population(data not shown).
A reduction in endogenous IL-32 is associated with reducedDNA binding of NF-�B and AP-1 in PBMC
We next investigated the activity levels of NF-�B and AP-1 inPBMC in which endogenous IL-32 was reduced by siRNA. PBMCwere simultaneously transfected with siIL-32 or scrambled siRNAand a luciferase-containing plasmid linked to either AP-1 or NF-�B. As shown in Fig. 8, A and B, significantly lower DNA-bindingactivities of both AP-1 and NF-�B were measured in LPS-stimu-lated cells transfected with siIL-32. The mean reduction in AP-1binding was 46% ( p � 0.04) and that of NF-�B was 42% ( p �0.01).
Modulation of the bioactivity of IFN-� in PMBC culturesaffects p24 production
Having demonstrated that the production of IFN-� was reduced infreshly infected PBMC treated with siIL-32, we considered thatdecreased IFN-� results in a state of reduced endogenous anti-viral activity. In HIV-1-infected PBMC treated with exogenousIL-18, a decrease in p24 production was linked to increasedendogenous IFN-� activity as IL-18 induces IFN-� (21). There-fore, we blocked IFN-� activity using a mAb to the IFN-� re-ceptor (anti-IFN-�R, previously described in Ref. 23). Asshown in Fig. 9, adding exogenous IFN-� to infected PBMCreduced HIV-1 production by 59% at 50 ng/ml. The far rightbars of Fig. 9 shows that anti-IFN-�R increased p24 levels by65%. These studies are consistent with the concept that in
FIGURE 6. Effects of siIL-32 in PBMC. Scrambled siRNA and siIL-32 were transfected into PBMC. IL-32 (A; range for scrambled siRNA-transfected cells at 24 h, 9 –111 pg/ml; at 96 h, 39 –171 pg/ml), p24 (B; range, 1–30 ng/ml), IL-6 (C; range, 3–176 pg/ml), IFN-� (D; range, 495-4321pg/ml), and TNF-� (E; range, 23–168 pg/ml) protein levels in the lysates (IL-32) or supernatants (others) were determined at the time points indicated(in A, time counted from electroporation) or after a 3-day incubation (in B–E, time counted from infection). Each concentration of siIL-32 was pairedwith the same concentration of scrambled siRNA. The percentage changes in protein levels were calculated by defining scrambled siRNA as 100%for each concentration. Data are depicted as mean � SEM, n � 8; �, p � 0.05; ��, p � 0.01 for siIL-32 compared with concentration-matchedscrambled siRNA.
FIGURE 7. siIL-32 vs scrambled siRNA: cytokine expression profile.PBMC were subjected to transfection with either 250 nM siIL-32 or con-centration-matched scrambled siRNA, followed by infection with HIV-1and a 3-day incubation period. Supernatants from three pairs were assayedusing a Proteome Profiler cytokine array (R&D Systems). Mean changesbetween siIL-32 and scrambled siRNA calculated from three indepen-dently performed experiments are shown as means � SEM. A red labelindicates a change of �33%.
562 Endogenous IL-32 in HIV
by guest on March 2, 2016
http://ww
w.jim
munol.org/
Dow
nloaded from

PBMC, endogenous IFN-� provides an antiviral state and thatthe ability of siIL-32 to reduce constitutive IFN-� accounts, inpart, for the increase in p24.
DiscussionSeveral viruses and bacteria are known to skew cytokine pro-duction in host cells to create an environment favorable for theirproliferation. HIV-1 is an example of a virus using this survivalstrategy (24, 25). In the present study, the use of siIL-32 inIL-2/PHA-primed PBMC suggested that IL-32 inhibits HIV-1. In-deed, when constitutive IL-32 was reduced by siIL-32 in both U1macrophages and freshly infected PBMC, HIV-1 production in-creased. In addition, IL-8 levels were also increased by siIL-32.The latter effect may be secondary to the increase in p24, as IL-8is elevated in HIV-positive individuals and known to be inducedby the virion-derived protein R (Vpr) of HIV-1 in various cells (26,27). After having observed the augmentation of HIV-1 induced bysilencing IL-32, we decided to investigate the effects of exogenousstimulation with this cytokine. Indeed, the addition of exogenousrhIL-32� strongly suppressed HIV-1 production in U1 macro-phages (Fig. 4).
In PBMC, the increase in p24 protein associated with loweredlevels of endogenous IL-32 (shown in Fig. 6B) was accompaniedby a reduction in several cytokines; IFN-�, IL-6, and TNF-� pro-tein were decreased by 51, 57, and 36%, respectively (Figs. 6,C–E). The same was observed for IL-1� in U1 cell lysates (re-duction by 32%; Fig. 3C). The extent of this inhibition is remark-able and underscores the importance of IL-32 in the production ofthese cytokines. The decrease in IFN-�, IL-6, TNF-�, and IL-1�was achieved by a knockdown of IL-32 of 83% at 24 h and 53%at 96 h. Moreover, when contrasted with the increase in p24 theseobservations were unexpected, because HIV-1 replication is wellknown to be up-regulated by TNF-� and IL-6 (28, 29) and inhib-ited by blocking these and other cytokines. Hence, the increase inp24 in association with decreased IL-32 is likely independent ofthe reduction in TNF-� and IL-6. The decrease in IFN-�, in con-trast, may explain, in part, the increase in HIV-1 expression, asdiscussed below.
As HIV uses the chemokine receptors CXCR4 and CCR5 ascoreceptors for binding to monocytes/macrophages and T cells(30, 31), we hypothesized that modulation of surface expression ofthese receptors could be a mechanism by which siIL-32 promotesproduction of HIV-1 while reducing the levels of several cyto-kines. However, there were no differences in CXCR4 and CCR5surface expressions between siIL-32- and scrambled siRNA-trans-fected cells.
We also studied the effects of siIL-32-induced decreases in lev-els of endogenous IL-32 on the DNA-binding activities of NF-�Band AP-1. By binding to their respective sites within the long-terminal repeat promoter of HIV-1 (32), both transcription factorsinduce strong boosts of gene expression and release of HIV-1 fromacutely as well as latently infected cells (33). Furthermore, theexpressions of IFN-� (34), IL-6, and TNF-� (35) are, in part,driven by the activity of NF-�B and/or AP-1. IL-32 activatesNF-�B in cell lines (2). In fact, as shown in the present study, inprimary PBMC, siIL-32 reduced DNA binding of NF-�B andAP-1 at the same concentrations which reduced IL-32 and in-creased p24. The reduction in NF-�B and AP-1 binding helps ex-plain the decrease of IL-6, IFN-�, and TNF-� in PBMC and ofIL-1� in U1 cells associated with reduced endogenous IL-32.However, the inhibition of the binding of both transcription factorsfails to provide a sufficient explanation for the paradoxical increasein p24.
On the other hand, diminished levels of IFN-� associated withsiIL-32 and thus a reduction in the antiviral activity within thePBMC cultures may explain the increase in p24 (Fig. 6D). IFN-�is an antiviral cytokine and its range of targets includes HIV-1
FIGURE 8. siIL-32 inhibits DNA binding of NF-�B and AP-1 in PBMC.PBMC were transfected with 250 nM scrambled siRNA or siIL-32, as well aswith either 1 �g of an NF-�B (A) or 2 �g of an AP-1 (B) firefly luciferase-labeled reporter construct and 1 �g of a Renilla luciferase-labeled construct forcontrol of transfection efficiency. After recovery, cells were counted, platedinto 24-well plates, and stimulated with 100 ng/ml LPS for 30 min (NF-�B) or1 h (AP-1). Luciferase activities were then measured in the lysates. Data areshown as mean � SEM, n � 3; �, p � 0.05; ��, p � 0.01 for siIL-32 comparedwith scrambled siRNA.
FIGURE 9. Effect of IFN-� on HIV-1-infected PMBC. PBMC weretransfected with 250 nM siIL-32. After infection, cells were plated andtreated as indicated, followed by a 3-day incubation. Thereafter, p24 pro-tein levels in the supernatants were determined by ECL (range in controlsupernatants, 3–19 ng/ml). Percentage changes in p24 levels are depictedas mean � SEM, n � 7; �, p � 0.05; ��, p � 0.01 for IFN-� or anti-IFN-�R compared with control. The difference between 2 and 10 �g/mlanti-IFN-�R is not significant.
563The Journal of Immunology
by guest on March 2, 2016
http://ww
w.jim
munol.org/
Dow
nloaded from

(36). In PBMC infected with HIV-1 in vitro and stimulated withIL-18, the decrease in p24 was due to an increase in IFN-� (21). Inthe present study, we show that adding exogenous IFN-� to PBMCinfected in vitro suppresses p24 levels and that blocking endoge-nous IFN-� activity increases p24. Thus, the net effect of reducingendogenous IL-32 is in favor of increased HIV-1 production as theantiviral milieu decreases.
Because we were unable to detect IFN-� in U1 cells, it is un-likely that the increase in p24 in these cultures is due to siIL-32-induced decreases in IFN-� levels. Hence, we decided to investi-gate the role of type I IFNs. IFN-�, more specifically at leastsubtype 1a (37), is produced in U937 cells and inhibits HIV-1 (38,39). We detected IFN-� protein in the supernatants of the U1 cellcultures and also observed a 4-fold increase in the presence ofexogenous rIL-32�. Blocking IFN activity with an anti-IFN-�/�Ror neutralizing IFN using soluble IFN-�/� completely reversed theantiviral effect of exogenous IL-32�. We can thus conclude thatthe antiviral activity of IL-32 in latently infected U1 cells is, atleast in part, mediated by IFN-�.
The chemokine receptors CCR5 (40) and/or CXCR4 (41) arecrucial for HIV entry into target cells (31). That discovery laid thefoundation for the development of entry inhibitors that blockCCR5 or CXCR4. For example, maraviroc occupies CCR5, thuspreventing HIV gp120 from binding and using CCR5 as corecep-tor (42). In supernatants from freshly infected PBMC analyzed inthe present study (Fig. 7), siIL-32 resulted in a decrease of theCCR5 ligands MIP-1�, MIP-1�, and RANTES as well as a de-crease of the CXCR4 ligand stromal cell-derived factor-1 (SDF-1),each of which function as an HIV-1 coreceptor. This reduction inthe levels of various chemokines that interfere with HIV-1 fusionis highly relevant, as a greater number of chemokine receptorswould remain unoccupied in cultures transfected with siIL-32,leading to a facilitation of HIV-1 entry and enhanced replication.Hence, the reduction in ligands of the HIV-1 chemokine corecep-tors may be a mechanism by which siIL-32 increases p24. Fur-thermore, the CCR2 ligand MCP-1, which was also reduced bysiIL-32 on the dot blot, was suggested to slow HIV infection bydesensitization of CCR5 and CXCR4 signaling (43, 44).
During the early stages of infection, HIV-1 inhibits the releaseof IFN-�, IL-12, and IL-2, as well as the activation of IL-18 (45,46), thus preventing the mounting of a sufficient Th1 response.Although siRNA-induced reductions in IL-32 were associated witha decrease in nearly all cytokines on the profiler blot, we ob-served a greater decline of Th1-related cytokines and chemo-kines (IL-12 and IFN-� as well as IP-10 and MIP-1��) than thatof Th2 and anti-inflammatory cytokines. Notably, the singleexception from the general inhibition of cytokines by siIL-32was I-309, a chemokine preferentially affecting regulatory Tcells (47). This T cell subset can suppress both Th1 and Th2responses, thus preventing excessive immune reactions and au-toimmunity (48). However, these properties are detrimental inHIV infection as regulatory T cells impair the antiviral activi-ties of HIV-specific T cells (49) and can enhance HIV-1 geneexpression (50). Therefore, IL-32 not only induces the produc-tion of cytokines in general and proinflammatory mediators inparticular but may also play a role in promoting Th1 differen-tiation of T cells and a role in the inhibition of regulatory T cellchemotaxis. Indeed, IFN-� and IL-2 induce IL-32 (2).
Knowledge of the role of endogenous IL-32 in HIV-1 is stilllimited, although it appears that one likely mechanism by whichIL-32 suppresses HIV-1 expression is the induction of IFNs. How-ever, the novel findings we report here seem promising, as theysuggest that this cytokine may have potential to complement andimprove the present control of the disease with highly active an-
tiretroviral therapy (HAART). In general, IL-32 conveys a cyto-kine environment hostile to HIV-1. More specifically, the influ-ence on both types of IFNs as well as on the production ofchemokines that interfere with HIV-1 fusion is likely to be bene-ficial in any stage of the disease, whereas the promotion of a Th1response could be important during the early phase of the infec-tion. In summary, the results of this study suggest IL-32 as anantagonist of HIV-1 and as an amplifier of cytokine responses ingeneral and Th1 and proinflammatory subsets in particular.
AcknowledgmentsWe thank Tania Azam, Alex Bustamante, and Scott Beard for technicalassistance.
DisclosuresThe authors have no financial conflict of interest.
References1. Dahl, C. A., R. P. Schall, H. L. He, and J. S. Cairns. 1992. Identification of a
novel gene expressed in activated natural killer cells and T cells. J. Immunol. 148:597–603.
2. Kim, S. H., S. Y. Han, T. Azam, D. Y. Yoon, and C. A. Dinarello. 2005. Inter-leukin-32: a cytokine and inducer of TNF�. Immunity 22: 131–142.
3. Goda, C., T. Kanaji, S. Kanaji, G. Tanaka, K. Arima, S. Ohno, and K. Izuhara.2006. Involvement of IL-32 in activation-induced cell death in T cells. Int. Im-munol. 18: 233–240.
4. Netea, M. G., T. Azam, E. C. Lewis, L. A. Joosten, M. Wang, D. Langenberg,X. Meng, E. D. Chan, D. Y. Yoon, T. Ottenhoff, et al. 2006. Mycobacteriumtuberculosis induces interleukin-32 production through a caspase- 1/IL-18/inter-feron-�-dependent mechanism. PLoS Med. 3: e277.
5. Novick, D., M. Rubinstein, T. Azam, A. Rabinkov, C. A. Dinarello, andS. H. Kim. 2006. Proteinase 3 is an IL-32 binding protein. Proc. Natl. Acad. Sci.USA 103: 3316–3321.
6. Netea, M. G., T. Azam, G. Ferwerda, S. E. Girardin, M. Walsh, J. S. Park,E. Abraham, J. M. Kim, D. Y. Yoon, C. A. Dinarello, and S. H. Kim. 2005. IL-32synergizes with nucleotide oligomerization domain (NOD) 1 and NOD2 ligandsfor IL-1� and IL-6 production through a caspase 1-dependent mechanism. Proc.Natl. Acad. Sci. USA 102: 16309–16314.
7. Shoda, H., K. Fujio, Y. Yamaguchi, A. Okamoto, T. Sawada, Y. Kochi, andK. Yamamoto. 2006. Interactions between IL-32 and tumor necrosis factor �contribute to the exacerbation of immune-inflammatory diseases. Arthritis Res.Ther. 8: R166.
8. Shioya, M., A. Nishida, Y. Yagi, A. Ogawa, T. Tsujikawa, S. Kim-Mitsuyama,A. Takayanagi, N. Shimizu, Y. Fujiyama, and A. Andoh. 2007. Epithelial over-expression of interleukin-32� in inflammatory bowel disease. Clin. Exp. Immu-nol. 149: 480–486.
9. Dinarello, C. A., and S. H. Kim. 2006. IL-32, a novel cytokine with a possiblerole in disease. Ann. Rheum. Dis. 65 (Suppl. 3): iii61–iii64.
10. Cagnard, N., F. Letourneur, A. Essabbani, V. Devauchelle, S. Mistou, A. Rapinat,C. Decraene, C. Fournier, and G. Chiocchia. 2005. Interleukin-32, CCL2, PF4F1,and GFD10 are the only cytokine/chemokine genes differentially expressed by invitro cultured rheumatoid and osteoarthritis fibroblast-like synoviocytes. Eur. Cy-tokine Network 16: 289–292.
11. Joosten, L. A., M. G. Netea, S. H. Kim, D. Y. Yoon, B. Oppers-Walgreen,T. R. Radstake, P. Barrera, F. A. van de Loo, C. A. Dinarello, andW. B. van den Berg. 2006. IL-32, a proinflammatory cytokine in rheumatoidarthritis. Proc. Natl. Acad. Sci. USA 103: 3298–3303.
12. Edwards, C. J., J. L. Feldman, J. Beech, K. M. Shields, J. A. Stover,W. L. Trepicchio, G. Larsen, B. M. Foxwell, F. M. Brennan, M. Feldmann, andD. D. Pittman. 2007. Molecular profile of peripheral blood mononuclear cellsfrom patients with rheumatoid arthritis. Mol. Med. 13: 40–58.
13. Palella, F. J., Jr., K. M. Delaney, A. C. Moorman, M. O. Loveless, J. Fuhrer,G. A. Satten, D. J. Aschman, and S. D. Holmberg. 1998. Declining morbidity andmortality among patients with advanced human immunodeficiency virus infec-tion: HIV outpatient study investigators. N. Engl. J. Med. 338: 853–860.
14. Poli, G., A. L. Kinter, E. Vicenzi, and A. S. Fauci. 1994. Cytokine regulation ofacute and chronic HIV infection in vitro: from cell lines to primary mononuclearcells. Res. Immunol. 145: 578–582.
15. Shapiro, L., K. A. Heidenreich, M. K. Meintzer, and C. A. Dinarello. 1998. Roleof p38 mitogen-activated protein kinase in HIV type 1 production in vitro. Proc.Natl. Acad. Sci. USA 95: 7422–7426.
16. Folks, T. M., J. Justement, A. Kinter, C. A. Dinarello, and A. S. Fauci. 1987.Cytokine-induced expression of HIV-1 in a chronically infected promonocyte cellline. Science 238: 800–802.
17. Folks, T. M., J. Justement, A. Kinter, S. Schnittman, J. Orenstein, G. Poli, andA. S. Fauci. 1988. Characterization of a promonocyte clone chronically infectedwith HIV and inducible by 13-phorbol-12-myristate acetate. J. Immunol. 140:1117–1122.
18. Poli, G., A. L. Kinter, and A. S. Fauci. 1994. Interleukin 1 induces expression ofthe human immunodeficiency virus alone and in synergy with interleukin 6 inchronically infected U1 cells: inhibition of inductive effects by the interleukin 1receptor antagonist. Proc. Natl. Acad. Sci. USA 91: 108–112.
564 Endogenous IL-32 in HIV
by guest on March 2, 2016
http://ww
w.jim
munol.org/
Dow
nloaded from

19. Poli, G., A. Kinter, J. S. Justement, J. H. Kehrl, P. Bressler, S. Stanley, andA. S. Fauci. 1990. Tumor necrosis factor � functions in an autocrine manner inthe induction of human immunodeficiency virus expression. Proc. Natl. Acad.Sci. USA 87: 782–785.
20. Shapiro, L., A. J. Puren, H. A. Barton, D. Novick, R. L. Peskind, R. Shenkar,Y. Gu, M. S. Su, and C. A. Dinarello. 1998. Interleukin 18 stimulates HIV type1 in monocytic cells. Proc. Natl. Acad. Sci. USA 95: 12550–12555.
21. Choi, H. J., C. A. Dinarello, and L. Shapiro. 2001. Interleukin-18 inhibits humanimmunodeficiency virus type 1 production in peripheral blood mononuclear cells.J. Infect. Dis. 184: 560–568.
22. Petry, C., G. Fritz, J. Pfeilschifter, and A. Huwiler. 2004. Inhibition of Rhomodulates cytokine-induced prostaglandin E2 formation in renal mesangial cells.Biochim. Biophys. Acta 1636: 108–118.
23. Novick, D., D. G. Fischer, Z. Reiter, Z. Eshhar, and M. Rubinstein. 1989. Mono-clonal antibodies to the human interferon-� receptor: blocking of the biologicalactivities of interferon-� and purification of the receptor. J. Interferon Res. 9:315–328.
24. Kedzierska, K., and S. M. Crowe. 2001. Cytokines and HIV-1: interactions andclinical implications. Antiviral Chem. Chemother. 12: 133–150.
25. Butera, S. T. 1993. Cytokine involvement in viral permissiveness and the pro-gression of HIV disease. J. Cell. Biochem. 53: 336–342.
26. Roux, P., C. Alfieri, M. Hrimech, E. A. Cohen, and J. E. Tanner. 2000. Activationof transcription factors NF-�B and NF-IL-6 by human immunodeficiency virustype 1 protein R (Vpr) induces interleukin-8 expression. J. Virol. 74: 4658–4665.
27. Lane, B. R., K. Lore, P. J. Bock, J. Andersson, M. J. Coffey, R. M. Strieter, andD. M. Markovitz. 2001. Interleukin-8 stimulates human immunodeficiency virustype 1 replication and is a potential new target for antiretroviral therapy. J. Virol.75: 8195–8202.
28. Devadas, K., N. J. Hardegen, L. M. Wahl, I. K. Hewlett, K. A. Clouse,K. M. Yamada, and S. Dhawan. 2004. Mechanisms for macrophage-mediatedHIV-1 induction. J. Immunol. 173: 6735–6744.
29. Poli, G., P. Bressler, A. Kinter, E. Duh, W. C. Timmer, A. Rabson,J. S. Justement, S. Stanley, and A. S. Fauci. 1990. Interleukin 6 induces humanimmunodeficiency virus expression in infected monocytic cells alone and in syn-ergy with tumor necrosis factor � by transcriptional and post-transcriptionalmechanisms. J. Exp. Med. 172: 151–158.
30. Ray, N., and R. W. Doms. 2006. HIV-1 coreceptors and their inhibitors. Curr.Top. Microbiol. Immunol. 303: 97–120.
31. Shaheen, F., and R. G. Collman. 2004. Co-receptor antagonists as HIV-1 entryinhibitors. Curr. Opin. Infect. Dis. 17: 7–16.
32. Kulkosky, J., and S. Bray. 2006. HAART-persistent HIV-1 latent reservoirs: theirorigin, mechanisms of stability and potential strategies for eradication. Curr. HIVRes. 4: 199–208.
33. Yang, X., Y. Chen, and D. Gabuzda. 1999. ERK MAP kinase links cytokinesignals to activation of latent HIV-1 infection by stimulating a cooperative in-teraction of AP-1 and NF-�B. J. Biol. Chem. 274: 27981–27988.
34. Akira, S. 2000. The role of IL-18 in innate immunity. Curr. Opin. Immunol. 12:59–63.
35. Palanki, M. S. 2002. Inhibitors of AP-1 and NF-�B mediated transcriptionalactivation: therapeutic potential in autoimmune diseases and structural diversity.Curr. Med. Chem. 9: 219–227.
36. Patterson, B. K., A. Tjernlund, and J. Andersson. 2002. Endogenous inhibitors ofHIV: potent anti-HIV activity of leukemia inhibitory factor. Curr. Mol. Med. 2:713–722.
37. Hussain, M., D. Ni, D. Gill, and M. J. Liao. 2000. IFN-�1a gene is the majorvariant in the North American population. J. Interferon Cytokine Res. 20:763–768.
38. Lallemand, C., P. Lebon, P. Rizza, B. Blanchard, and M. G. Tovey. 1996. Con-stitutive expression of specific interferon isotypes in peripheral blood leukocytesfrom normal individuals and in promonocytic U937 cells. J. Leukocyte Biol. 60:137–146.
39. Locardi, C., C. Petrini, G. Boccoli, U. Testa, C. Dieffenbach, S. Butto, andF. Belardelli. 1990. Increased human immunodeficiency virus (HIV) expressionin chronically infected U937 cells upon in vitro differentiation by hydroxyvitaminD3: roles of interferon and tumor necrosis factor in regulation of HIV production.J. Virol. 64: 5874–5882.
40. Deng, H., R. Liu, W. Ellmeier, S. Choe, D. Unutmaz, M. Burkhart, P. Di Marzio,S. Marmon, R. E. Sutton, C. M. Hill, et al. 1996. Identification of a major co-receptor for primary isolates of HIV-1. Nature 381: 661–666.
41. Feng, Y., C. C. Broder, P. E. Kennedy, and E. A. Berger. 1996. HIV-1 entrycofactor: functional cDNA cloning of a seven-transmembrane, G protein-coupledreceptor. Science 272: 872–877.
42. Reeves, J. D., and A. J. Piefer. 2005. Emerging drug targets for antiretroviraltherapy. Drugs 65: 1747–1766.
43. Genin, P., Y. Mamane, H. Kwon, C. LePage, M. A. Wainberg, and J. Hiscott.1999. Differential regulation of CC chemokine gene expression in human immu-nodeficiency virus-infected myeloid cells. Virology 261: 205–215.
44. Lee, B., B. J. Doranz, S. Rana, Y. Yi, M. Mellado, J. M. Frade, A. C. Martinez,S. J. O’Brien, M. Dean, R. G. Collman, and R. W. Doms. 1998. Influence of theCCR2–V64I polymorphism on human immunodeficiency virus type 1 coreceptoractivity and on chemokine receptor function of CCR2b, CCR3, CCR5, andCXCR4. J. Virol. 72: 7450–7458.
45. Ma, X., and L. J. Montaner. 2000. Proinflammatory response and IL-12 expres-sion in HIV-1 infection. J. Leukocyte Biol. 68: 383–390.
46. Torre, D., and A. Pugliese. 2006. Interleukin-18: a proinflammatory cytokine inHIV-1 infection. Curr. HIV Res. 4: 423–430.
47. Sebastiani, S., C. Albanesi, P. O. De, P. Puddu, A. Cavani, and G. Girolomoni.2002. The role of chemokines in allergic contact dermatitis. Arch. Dermatol. Res.293: 552–559.
48. Romagnani, S. 2006. Regulation of the T cell response. Clin. Exp. Allergy 36:1357–1366.
49. Kinter, A. L., R. Horak, M. Sion, L. Riggin, J. McNally, Y. Lin, R. Jackson,A. O’Shea, G. Roby, C. Kovacs, et al. 2007. CD25� regulatory T cells isolatedfrom HIV-infected individuals suppress the cytolytic and nonlytic antiviral ac-tivity of HIV-specific CD8� T cells in vitro. AIDS Res. Hum. Retroviruses 23:438–450.
50. Holmes, D., G. Knudsen, S. Mackey-Cushman, and L. Su. 2007. FoxP3 enhancesHIV-1 gene expression by modulating NF�B occupancy at the long terminalrepeat in human T cells. J. Biol. Chem. 282: 15973–15980.
565The Journal of Immunology
by guest on March 2, 2016
http://ww
w.jim
munol.org/
Dow
nloaded from





























