Embed Size (px)
Citation preview

ORIGINAL PAPER
dsDNA modified carbon nanofiber—solidified pasteelectrodes: probing Ni(II)—dsDNA interactions
Adriana Ferancová & Liza Rassaei & Frank Marken &
Jan Labuda & Mika Sillanpää
Received: 26 February 2010 /Accepted: 21 May 2010 /Published online: 30 June 2010# Springer-Verlag 2010
Abstract In order to develop a renewable electrodesurface, carbon nanofibers (CNF) were embedded intosolidified paste electrodes using a composite of paraffinwax and paraffin oil. A range of different compositions wassurveyed and the optimal composition of the paste forelectroanalysis was found to be 43% of CNF, 41% ofparaffin wax, and 16% of paraffin oil. The electrochemicalproperties of the novel composite electrode were investi-gated using cyclic voltammetry and electrochemical im-pedance spectroscopy and compared to those of similargraphite—solidified paste electrodes. The carbon nano-fibers enhance the activity of the surface of the electrodeand provide a good substrate for the adsorption andvoltammetric detection of dsDNA. Responses of dsDNAbases and Ni2+ ions accumulated from ammonium buffer
pH 8.5 (with a Langmuirian binding constant of 105 mol−1 L)were investigated and a limit of detection of 7 nmol L−1
(at 3σ) was obtained using “nucleation stripping voltamme-try”. Interferences by other metal cations are examined anddiscussed.
Keywords Carbon nanofibers . Solidified paste electrode .
DNA biosensor . Nucleation stripping voltammetry .
Nickel—DNA interaction
Introduction
Metal cations have the ability to bind covalently or non-covalently to parts of DNA such as structural phosphates orelectron-donor atoms in nucleotide bases [1]. For example,Hg(I) preferably binds to the DNA bases whereas Co(II), Ni(II), Cd(II), and Pb(II) interact with both phosphate groups aswell as DNA bases [2]. It has been suggested that in selectedcases the binding interaction between DNA and metalcations can be exploited and used for analytical purposes [3].
Electrochemical DNA-based sensors employ the immo-bilized DNA as a “recognition layer” and the electrode asan electrochemical transducer. Today, DNA-based sensorsare frequently used for the assessment of health-riskchemicals which interact with DNA and which may causetoxic effects of damage to DNA [4, 5]. AmperometricDNA-based electrodes with stationary mercury-films wereinvestigated for the determination of heavy metals such asFe(III), Cd(II), Cu(II), and Pb(II) in biological [6], food,and water [7, 8] samples. Due to the nanomolar detectionlimits, it was concluded that such DNA electrodes can bepotentially useful in practical analysis and environmentalcontrol [9]. Gold electrodes were modified with single-stranded DNA to prepare very sensitive electrochemical
A. Ferancová :M. SillanpääLaboratory of Applied Environmental Chemistry, Departmentof Environmental Sciences, University of Eastern Finland,Patteristonkatu 1,50100 Mikkeli, Finland
A. Ferancová (*) : J. LabudaInstitute of Analytical Chemistry,Slovak University of Technology in Bratislava,Radlinskeho 9,81237 Bratislava, Slovakiae-mail: [email protected]: [email protected]
L. Rassaei : F. MarkenDepartment of Chemistry, University of Bath,Bath BA2 7AY, UK
M. SillanpääLUT Faculty of Technology,Lappeenranta University of Technology,Patteristonkatu 1,50100 Mikkeli, Finland
Microchim Acta (2010) 170:155–164DOI 10.1007/s00604-010-0388-z

sensors for Cd(II) with an accumulation step at open circuitpotential [10]. Glassy carbon electrodes modified withDNA were used for the investigation of Pb(II), Ni(II), Cd(II), and Pd(II) [11, 12]. It was suggested that due to highaffinity these metal cations may form covalent bondswith nitrogen bases associated with characteristic changesin differential pulse voltammograms for guanine andadenine. The interaction of Ni(II) with DNA moleculewas investigated recently [11, 12] and this system hasbeen chosen in the present study to demonstrate thedevelopment of a novel carbon nanofiber—solidified pasteelectrode methodology.
Carbon nanofibers (CNFs) are known to possessbeneficial chemical and physical properties such as goodelectrical conductivity and a high active surface area.Electrochemical properties of carbon nanofibers immobi-lized bare at the surface of glassy carbon electrodes as wellas embedded into a different composite matrices werestudied using voltammetric methods [13–15]. It wasreported that such electrodes provide low electrical resis-tance and a low capacitive background. Casting of carbonnanofiber dispersions in dimethylformamide onto electrodesurfaces was used to modify glassy carbon and goldsubstrates applied in the electrochemical oxidation ofNADH [16]. A carbon paste electrode surface modifiedwith carbon nanofibers [17] was used for the voltammetricdetermination of dopamine, ascorbic acid, and uric acid.
Carbon nanofiber surfaces were successfully used as asupport for DNA immobilization. DNA was directlycovalently attached to carboxylic acid sites [18] orindirectly with gold nanoparticle anker [19, 20]. DNAadsorption under potential control has been reported [21] aswell as the covalent immobilization of the DNA probes forthe preparation of DNA hybridization sensors [22–25].Peaks for guanine (G) and for adenine (A) oxidation wereinvestigated after accumulation of single-stranded DNA(ssDNA) and double-stranded DNA (dsDNA) at the surfaceof carbon nanofiber paste electrodes prepared with siliconeoil [26]. Nano-arrays of carbon nanofibers were function-alized with oligonucleotides and used for highly sensitiveDNA detection based on hybridization [27].
In this study, a novel carbon nanofiber—solidified pasteelectrode (CNFPE) is prepared using a composite ofparaffin wax and paraffin oil as binders. The optimalcomposition of the paste is studied (avoiding oil contam-ination but retaining the reproducibility of paste surfaces)and the resulting electrodes are characterized using cyclicvoltammetry, differential pulse voltammetry, and electro-chemical impedance spectroscopy. CNFPEs with optimalcomposition are then modified with dsDNA using electro-chemically controlled adsorption. Differential pulse voltam-metric responses of guanine and adenine oxidation are usedfor the evaluation of dsDNA immobilization. The dsDNA-
based sensor was then tested for the trace determination ofnickel by “nucleation stripping voltammetry”.
Experimental
Apparatus and reagents
Voltammetric and impedance measurements were per-formed with an Autolab potentiostat (PGSTAT12) andsoftware GPES (Metrohm Autolab B.V., Netherlands,www.metrohm-autolab.com). Electrochemical impedancespectroscopic measurements were carried out under opencircuit conditions and over a 10 kHz to 0.1 Hz frequencyrange with 10 mV amplitude. A three-electrode system wasused consisting of working paste electrode with geometricsurface area 3.1 mm2, a Ag/AgCl reference electrode (Ag/AgCl 3 mol L−1 KCl) and a Pt counter electrode. Thetemperature during experiments was 20±2°C.
Carbon nanofibers (Pyrograph III PR-PS) with BET(Brunauer, Emmett, and Teller) surface area 50–60 m2/g, adiameter of ca. 70–200 nm, and length of 50–100 μm wereobtained from Pyrograf Product Inc. (USA, pyrografproduct.com). Graphite powder extra pure was obtained from Merck(www.merck-chemicals.com). Paraffin wax (melting point73–80°C) was obtained from Sigma (www.sigmaaldrich.com) and paraffin oil from Fluka (www.sigmaaldrich.com).Calf-thymus dsDNA was obtained from Sigma (www.sigmaaldrich.com). Stock solution with 5 mg mL−1 wasprepared in a solution containing 1×10−2 mol L−1 Tris-HCland 1×10−3 mol L−1 EDTA adjusted to pH 8.0. NiSO4 wasobtained from Merck (www.merck-chemicals.com). Stocksolutions were prepared in deionized water. A stock solutionof Ru(NH3)6Cl3 (Sigma, www.sigmaaldrich.com) was pre-pared in 0.1 mol L−1 KCl (Merck, www.merck-chemicals.com). Na2HPO4 (Riedel-de Haёn, www.riedeldehaen.com)and NaH2PO4 (Merck, www.merck-chemicals.com) wereused for the preparation of 0.1 mol L−1 phosphate buffersolutions. Acetic acid and sodium acetate (both Merck,www.merck-chemicals.com) were used for the preparation of0.2 mol L−1 acetate buffer solutions. Ammonium hydroxide(Fluka, www.sigmaaldrich.com) and ammonium chloride(Merck, www.merck-chemicals.com) were used for thepreparation of 0.01 mol L−1 ammonium buffer solutions.
Preparation of carbon nanofiber paste electrodes (CNFPE)and graphite paste electrodes (GPE)
CNFPEs were prepared by thoroughly mixing of carbonnanofibers, paraffin oil, and liquefied paraffin wax invarious ratios (see Table 1) in a mortar at a temperature ofca. 80°C until the paste was homogeneous. A Teflonelectrode body was filled with the CNF paste. Solidified
156 A. Ferancová et al.
carbon nanofiber paste electrodes were formed after coolingdown to ambient temperature. The surface of the pasteelectrode was renewed by polishing on a wet filter paper.The GPEs were prepared employing the same procedure.
Preparation of dsDNA modified electrodes
The polished CNFPE (or GPE) was first potentiostaticallyactivated using a potential of 1.7 V vs. Ag/AgCl for 120 sin 0.1 mol L−1 phosphate buffer at pH 7.0. The activatedelectrode was then washed with deionized water anddsDNA was potentiostatically adsorbed from a50 μg mL−1 solution in 0.2 mol L−1 acetate buffer atpH 5.0 with an applied potential of 0.5 V vs. Ag/AgClduring 300 s under stirring. The resulting sensor electrodewas thoroughly rinsed with deionized water before use.
Accumulation and differential pulse voltammetricdetermination of Ni(II)
Ni(II) cations were first potentiostatically accumulated atthe DNA modified electrode from a solution in ammoniabuffer at pH 8.5 at −0.2 V vs. Ag/AgCl for 300 s. Theelectrode was then rinsed with deionized water andtransferred into the blank 0.01 mol L−1 ammonia bufferand a differential pulse voltammetry scan was recorded inthe potential range from −0.2 V to −1.4 V with the scan rateof 10 mV s−1. After each measurement the CNFPE (orGPE) electrode surface was renewed by polishing on a wetfilter paper, washing with deionized water and performingnew modification by DNA.
Results and discussion
Electrochemical characterization of carbon nanofiber pasteelectrode
In order to adjust and optimize the viscosity and electro-chemical properties of the paraffin paste electrode, amixture of paraffin oil and a high melting paraffin wax
was employed. A survey of electrode property versus pastecomposition was conducted by investigating the cyclicvoltammetry responses for the one-electron reduction of 1×10−3 mol L−1 [Ru(NH3)6]
3+ in aqueous 0.1 mol L−1 KCl.Electrodes were studied with varying content of carbonnanofibers (CNF), paraffin wax (PW), and paraffin oil (PO)(see Table 1). Peak currents and the peak potential dataobtained at a scan rate of 50 mV s−1 are summarized.
The peak-to-peak separation ΔEp (see Table 1) is relatedto the availability of active carbon at the electrode surfaceand it can be observed that for a higher content of the oilcomponent in the paste ΔEp and other electrochemicalproperties for the electrode deteriorate. It is likely thatexcess paraffin oil in the paste partially blocks the access of[Ru(NH3)6]
3+ to the CNF surface and thereby is inhibitingelectron transfer. A decreasing content of paraffin oil down toca. 16% improved the reversibility of the redox process. Forexample, in the case of electrode 3 with 47% paraffin oil thepeak potential separation observed was 153 mV. In contrast tothis, in case of electrode 7 (16% paraffin oil) the peak potentialseparation was 68 mV, which is closer to the ideal value ofΔEp ¼ 2:218� RT
nF ¼ 56 mV [28] (R is the gas constant, T isthe absolute temperature, F denotes the Faraday constant,and n is the number of electrons transferred per moleculereacting at the electrode surface). The most well-definedpeak currents were observed for the electrode 7 (CNFPE7)and therefore the composition 43% carbon nanofiber—41%paraffin wax—16% paraffin oil was employed further forvoltammetric experiments.
The cathodic peak current for the reduction of [Ru(NH3)6]
3+ depends linearly on υ1/2 consistent with a processdominated by planar diffusion to the paste electrodesurface. A decrease in linearity for this dependence wasobserved with increasing paraffin oil content in the paste (i.e. correlation coefficient was 0.999 for electrode 7 and0.941 for the electrode 2). Because of their large intrinsicsurface area, carbon nanofibers as a modifier of the pasteelectrode often enlarge the real electrode area or result inporosity. In contrast, inhomogeneous paste electrodes withnon-conducting paste binder components can also cause theelectroactive area of the electrode to be smaller than that
Electrode No. Composition / % (w / w) Ic / μA Ia / μA ΔEp/ mV
CNF PW PO
1 45 55 – −3.85 3.56 100
2 35 – 65 −2.05 1.98 207
3 45 8 47 −3.72 1.99 153
4 47 15 38 −4.19 3.34 118
5 38 32 30 −4.48 3.38 118
6 38 42 20 −4.12 3.37 95
7 43 41 16 −6.04 6.50 68
Table 1 Composition andcyclic voltammetric character-ization of carbon nanofiber—paste electrodes in an aqueoussolution of 1×10−3 mol L−1 [Ru(NH3)6]
3+ in 0.1 mol L−1 KCl(scan rate 50 mV s−1)
dsDNA modified carbon nanofiber—solidified paste electrodes: probing Ni(II)—dsDNA interactions 157
expected based on the geometric area [29]. The effectivesurface area (based on diffusion) for carbon nanofiber pasteelectrodes was evaluated using Randles-Ševčik equation[30] (equation 1).
Ip ¼ 0:446 FAc
ffiffiffiffiffiffiffiffiffi
FDv
RT
r
ð1ÞIn this expression Ip is the peak current (A), D is
diffusion coefficient (8.06×10−10 m2 s−1 [31]), v is scan rate(V s−1), A is the electrode area, and c is the concentration of[Ru(NH3)6]
3+, R, T and F have their usual denotation. Forexperimental conditions employed in Table 1, a theoreticalpeak current of 5.3 μA is predicted. This value is in goodagreement with the experimentally observed peak currents.Therefore these paste electrodes are homogeneous andaccessible.
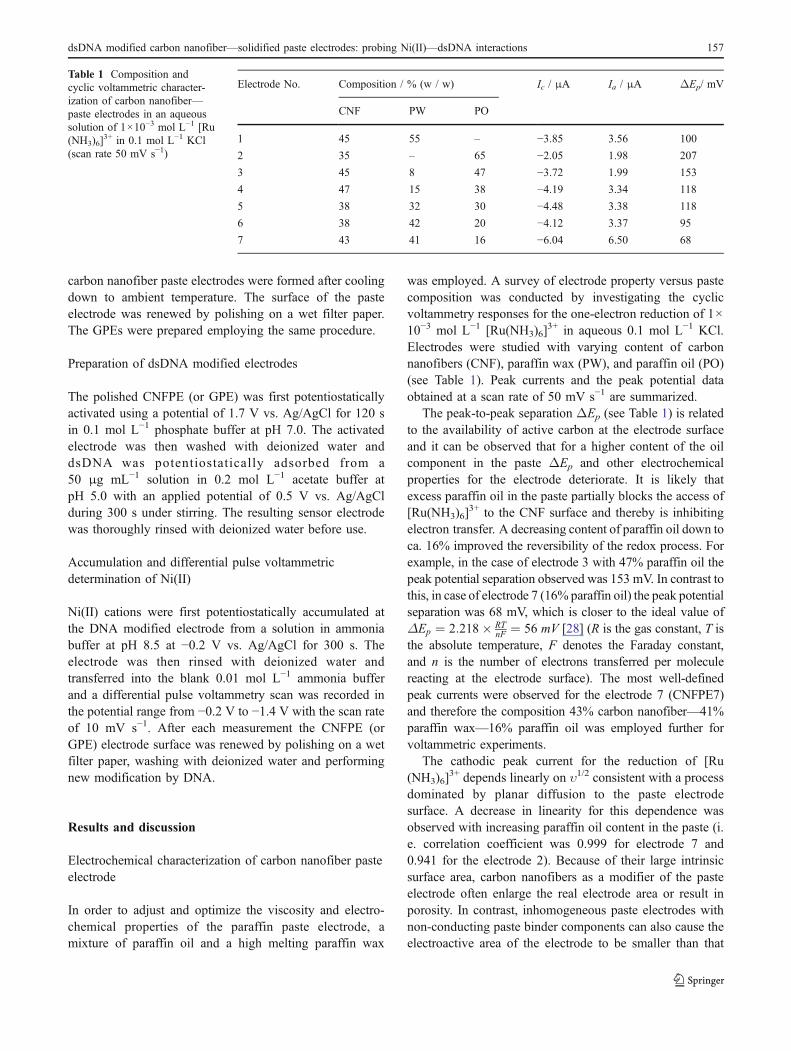
In order to examine the beneficial effects of carbonnanofibers in the electrode material, a graphite powderpaste electrode (GPE) was prepared with a paste composi-tion close to that employed in the electrode 7. A widepotential window of approximately 3 V in aqueous0.1 mol L−1 KCl was observed in case of carbon nanofiberelectrode 7. In contrast, for the GPE the potential windowwas only ca. 2 V. Figure 1 shows typical cyclic voltamme-try responses obtained in (a) blank 0.1 mol L−1 KClsolution as well as in (b) a solution of 1×10−3 mol L−1 [Ru(NH3)6]
3+ for both GPE and carbon nanofiber electrode 7.It can be seen that the carbon nanofiber electrode exhibits alower capacitive background current in comparison tothe GPE. Well-defined redox peaks for the reduction of[Ru(NH3)6]
3+ were obtained at both electrodes. An increas-ing of ΔEp as well as decreasing of peak current wasobserved in the case of GPE indicating lower reversibilityand/or a more coarse heterogeneity of the electrode surface.
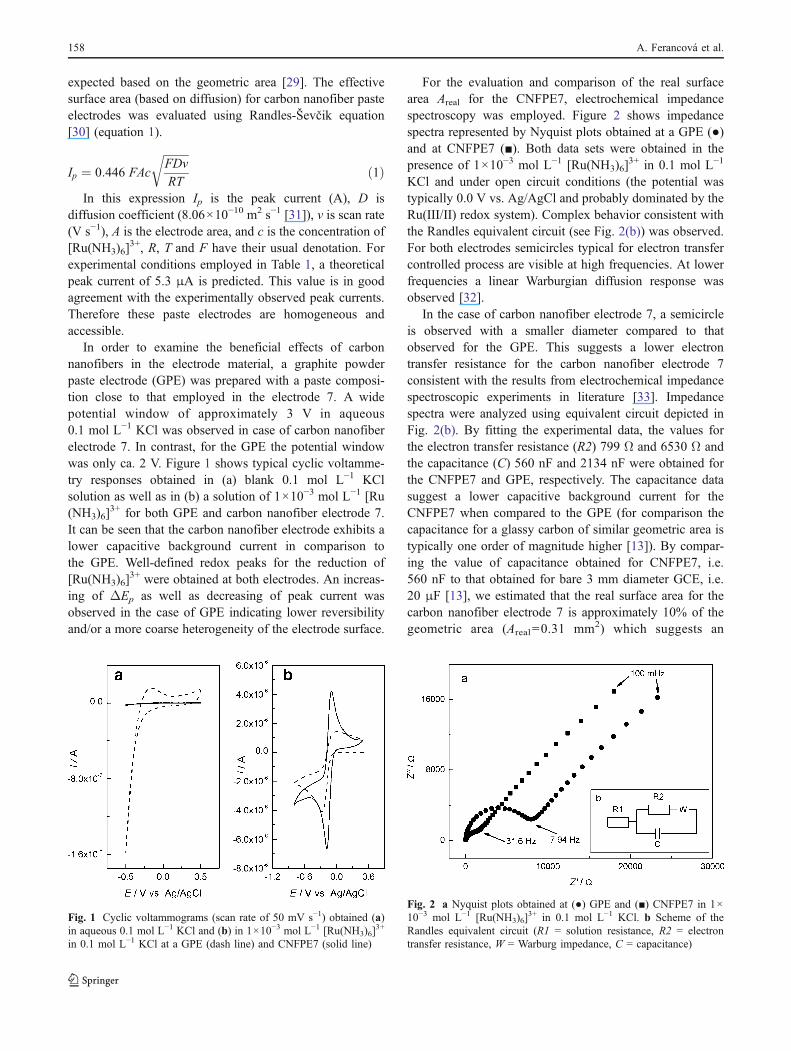
For the evaluation and comparison of the real surfacearea Areal for the CNFPE7, electrochemical impedancespectroscopy was employed. Figure 2 shows impedancespectra represented by Nyquist plots obtained at a GPE (●)and at CNFPE7 (■). Both data sets were obtained in thepresence of 1×10−3 mol L−1 [Ru(NH3)6]
3+ in 0.1 mol L−1
KCl and under open circuit conditions (the potential wastypically 0.0 V vs. Ag/AgCl and probably dominated by theRu(III/II) redox system). Complex behavior consistent withthe Randles equivalent circuit (see Fig. 2(b)) was observed.For both electrodes semicircles typical for electron transfercontrolled process are visible at high frequencies. At lowerfrequencies a linear Warburgian diffusion response wasobserved [32].
In the case of carbon nanofiber electrode 7, a semicircleis observed with a smaller diameter compared to thatobserved for the GPE. This suggests a lower electrontransfer resistance for the carbon nanofiber electrode 7consistent with the results from electrochemical impedancespectroscopic experiments in literature [33]. Impedancespectra were analyzed using equivalent circuit depicted inFig. 2(b). By fitting the experimental data, the values forthe electron transfer resistance (R2) 799 Ω and 6530 Ω andthe capacitance (C) 560 nF and 2134 nF were obtained forthe CNFPE7 and GPE, respectively. The capacitance datasuggest a lower capacitive background current for theCNFPE7 when compared to the GPE (for comparison thecapacitance for a glassy carbon of similar geometric area istypically one order of magnitude higher [13]). By compar-ing the value of capacitance obtained for CNFPE7, i.e.560 nF to that obtained for bare 3 mm diameter GCE, i.e.20 μF [13], we estimated that the real surface area for thecarbon nanofiber electrode 7 is approximately 10% of thegeometric area (Areal=0.31 mm2) which suggests an
Fig. 1 Cyclic voltammograms (scan rate of 50 mV s−1) obtained (a)in aqueous 0.1 mol L−1 KCl and (b) in 1×10−3 mol L−1 [Ru(NH3)6]
3+
in 0.1 mol L−1 KCl at a GPE (dash line) and CNFPE7 (solid line)
Fig. 2 a Nyquist plots obtained at (●) GPE and (■) CNFPE7 in 1×10−3 mol L−1 [Ru(NH3)6]
3+ in 0.1 mol L−1 KCl. b Scheme of theRandles equivalent circuit (R1 = solution resistance, R2 = electrontransfer resistance, W = Warburg impedance, C = capacitance)
158 A. Ferancová et al.
improved Faradaic to background current ratio and poten-tial for application in electroanalytical protocols.
Electrochemical behavior of dsDNA adsorbed onto carbonnanofiber paste electrode
Next, the surface modification of the carbon nanofiberelectrode 7 with dsDNA was studied. The potentiostaticpretreatment of conventional carbon paste electrodes athigh positive potential is known to be important foreffective adsorption of nucleic acids [34]. Pedano et al.[35] studied the effect of pretreatment conditions on DNAadsorption at carbon nanotube modified paste electrodes.They reported that during electrochemical pretreatment thedensity of oxygenated groups is increased which leads toimproved adsorption and improved electro-oxidation of theDNA bases guanine (G) and adenine (A). Positive effects ofpotentiostatic pretreatment on dsDNA response were alsoreported for highly ordered pyrolytic graphite electrode[36], glassy carbon electrode [37], and screen-printedcarbon electrode [38].
When dsDNA was adsorbed onto an untreated CNFPE7,no voltammetric response other than the background wasobserved. In contrast, after adsorption of dsDNA at thepotentiostatically pretreated CNFPE7, the oxidation signals ofboth, G and A, were observed. Several electrode pretreatmentpotentials (i.e. +0.5 V, +1.3 V, +1.7 V, and +1.8 V vs. Ag/AgCl) were tested for the anodic/electrooxidative pretreat-ment of CNFPE7. The clearest voltammetric responses for Gand A were obtained after the pretreatment of the carbonnanofiber electrode 7 at a potential of +1.7 V vs. Ag/AgCl for120 s.
Next, the influence of the buffer solution used forpretreatment and dsDNA adsorption was studied. For thispurpose, 0.2 mol L−1 acetate buffer pH 5.0 and 0.1 mol L−1
phosphate buffer pH 7.0 were tested. Improved currentresponse for G and A oxidation were obtained when thepretreatment was performed in 0.1 mol L−1 phosphatebuffer pH 7.0 and when dsDNA adsorption was performedin 0.2 mol L−1 acetate buffer pH 5.0. Different adsorptionpotentials, e.g. +0.2 V or +0.5 V vs. Ag/AgCl applied forthe immobilization of dsDNA, were reported in theliterature [35, 39]. In this study, good results were obtainedusing potential of +0.5 V vs. Ag/AgCl and with anadsorption time of 300 s. Differential pulse voltammetryat dsDNA modified electrodes was employed after transferinto clean 0.1 mol L−1 phosphate buffer pH 7.0 (see Fig. 3).Compared to literature reports, the use of the new electrodebased on carbon nanofibers in a solidified paste allowedimproved voltammetric signals to be obtained.
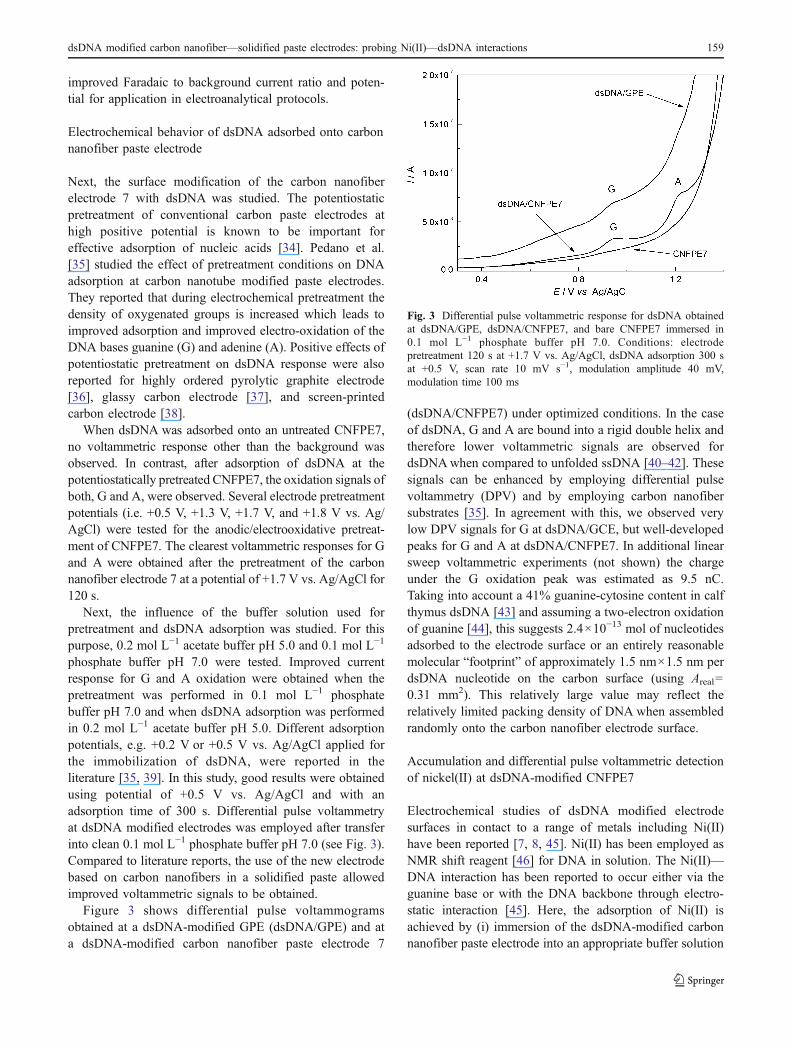
Figure 3 shows differential pulse voltammogramsobtained at a dsDNA-modified GPE (dsDNA/GPE) and ata dsDNA-modified carbon nanofiber paste electrode 7
(dsDNA/CNFPE7) under optimized conditions. In the caseof dsDNA, G and A are bound into a rigid double helix andtherefore lower voltammetric signals are observed fordsDNAwhen compared to unfolded ssDNA [40–42]. Thesesignals can be enhanced by employing differential pulsevoltammetry (DPV) and by employing carbon nanofibersubstrates [35]. In agreement with this, we observed verylow DPV signals for G at dsDNA/GCE, but well-developedpeaks for G and A at dsDNA/CNFPE7. In additional linearsweep voltammetric experiments (not shown) the chargeunder the G oxidation peak was estimated as 9.5 nC.Taking into account a 41% guanine-cytosine content in calfthymus dsDNA [43] and assuming a two-electron oxidationof guanine [44], this suggests 2.4×10−13 mol of nucleotidesadsorbed to the electrode surface or an entirely reasonablemolecular “footprint” of approximately 1.5 nm×1.5 nm perdsDNA nucleotide on the carbon surface (using Areal=0.31 mm2). This relatively large value may reflect therelatively limited packing density of DNA when assembledrandomly onto the carbon nanofiber electrode surface.
Accumulation and differential pulse voltammetric detectionof nickel(II) at dsDNA-modified CNFPE7
Electrochemical studies of dsDNA modified electrodesurfaces in contact to a range of metals including Ni(II)have been reported [7, 8, 45]. Ni(II) has been employed asNMR shift reagent [46] for DNA in solution. The Ni(II)—DNA interaction has been reported to occur either via theguanine base or with the DNA backbone through electro-static interaction [45]. Here, the adsorption of Ni(II) isachieved by (i) immersion of the dsDNA-modified carbonnanofiber paste electrode into an appropriate buffer solution
Fig. 3 Differential pulse voltammetric response for dsDNA obtainedat dsDNA/GPE, dsDNA/CNFPE7, and bare CNFPE7 immersed in0.1 mol L−1 phosphate buffer pH 7.0. Conditions: electrodepretreatment 120 s at +1.7 V vs. Ag/AgCl, dsDNA adsorption 300 sat +0.5 V, scan rate 10 mV s−1, modulation amplitude 40 mV,modulation time 100 ms
dsDNA modified carbon nanofiber—solidified paste electrodes: probing Ni(II)—dsDNA interactions 159
followed by (ii) voltammetry in a separate buffer solution.The highest voltammetric responses for nickel wereobserved when Ni(II) cations were adsorbed from ammo-nium buffer pH 8.5. Under these conditions Ni(II) is presentas [Ni(NH3)6]
2+ cation and both electrostatic interaction
with phosphate in the DNA backbone as well as interactionwith guanine base via coordination with N7 and O6 atomsappear possible [1, 47]. Differential pulse voltammetrysignals were recorded after transfer of the electrode into ablank ammonium buffer solution. The current peak
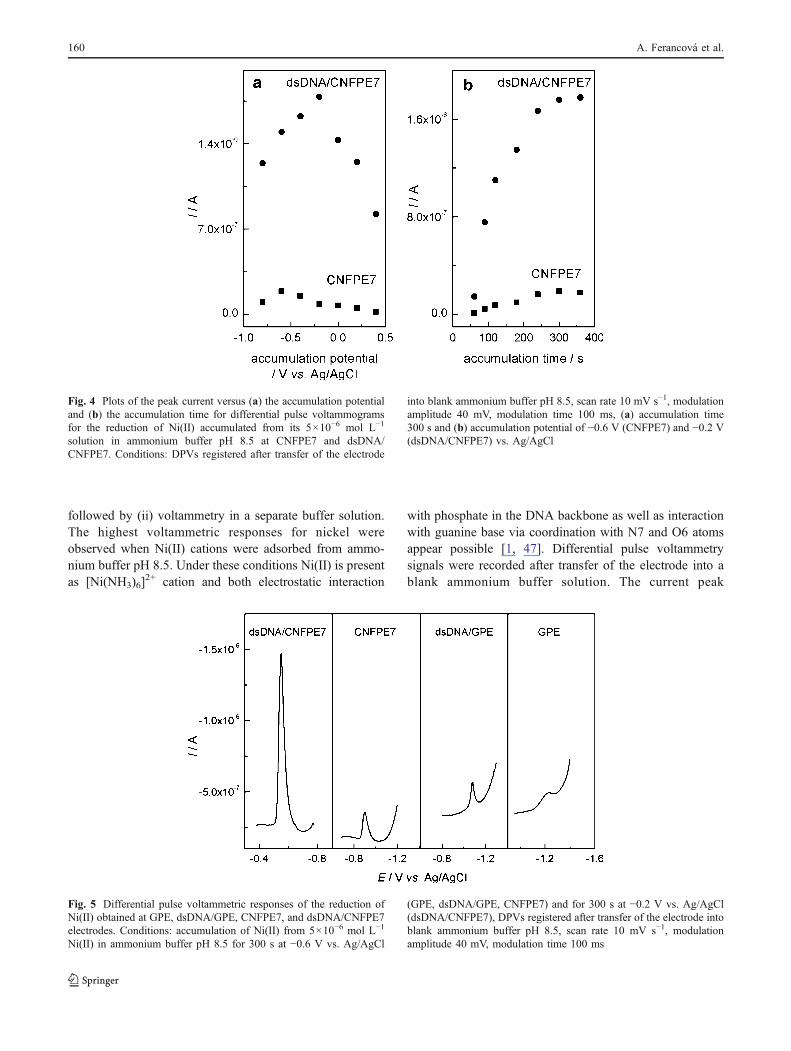
Fig. 4 Plots of the peak current versus (a) the accumulation potentialand (b) the accumulation time for differential pulse voltammogramsfor the reduction of Ni(II) accumulated from its 5×10−6 mol L−1
solution in ammonium buffer pH 8.5 at CNFPE7 and dsDNA/CNFPE7. Conditions: DPVs registered after transfer of the electrode
into blank ammonium buffer pH 8.5, scan rate 10 mV s−1, modulationamplitude 40 mV, modulation time 100 ms, (a) accumulation time300 s and (b) accumulation potential of −0.6 V (CNFPE7) and −0.2 V(dsDNA/CNFPE7) vs. Ag/AgCl
Fig. 5 Differential pulse voltammetric responses of the reduction ofNi(II) obtained at GPE, dsDNA/GPE, CNFPE7, and dsDNA/CNFPE7electrodes. Conditions: accumulation of Ni(II) from 5×10−6 mol L−1
Ni(II) in ammonium buffer pH 8.5 for 300 s at −0.6 V vs. Ag/AgCl
(GPE, dsDNA/GPE, CNFPE7) and for 300 s at −0.2 V vs. Ag/AgCl(dsDNA/CNFPE7), DPVs registered after transfer of the electrode intoblank ammonium buffer pH 8.5, scan rate 10 mV s−1, modulationamplitude 40 mV, modulation time 100 ms
160 A. Ferancová et al.
observed under these conditions corresponds to the reduc-tion of adsorbed Ni(II) to nickel metal which is associatedwith a nucleation process and therefore sharp and substratesensitive.
The influence of accumulation potential and accumula-tion time on differential pulse voltammetry peak currentsfor Ni(II) reduction were investigated (see Fig. 4) in thepresence and in the absence of immobilized dsDNA. In theabsence of dsDNA, the highest differential pulse voltam-metry signal for Ni(II) reduction at a carbon nanofiber pasteelectrode (electrode 7) was obtained when Ni(II) wasaccumulated at a potential of −0.6 V vs. Ag/AgCl for300 s (Fig. 4). In the presence of dsDNA, the optimalaccumulation potential was −0.2 V vs. Ag/AgCl (see Fig. 4(a)) and an accumulation time of 300 s was necessary toreach the saturation (see Fig. 4(b)). In the presence of thedsDNA, the considerable shift in both the accumulationpotential and in the peak potential in the DPV response (seeFig. 5) are observed. The adsorption of Ni(II) and theformation of nickel metal appear both to be stronglyaffected by the presence of dsDNA at the electrode surface.
Figure 5 shows a comparison of differential pulsevoltammetry responses for Ni(II) reduction obtained withand without dsDNA and at GPE or CNFPE7 underoptimized conditions. The Ni(II/0) peak potential is shiftedto less negative potentials in the presence of dsDNA (peakpotential −1.21 V and −1.07 V vs. Ag/AgCl at GPE and atdsDNA/GPE, peak potential −0.92 V and −0.55 V vs. Ag/AgCl at CNFPE7 and at dsDNA/CNFPE7). This substantialshift is likely to be associated with a lower overpotential forthe formation of nickel metal nuclei from Ni(II) in thepresence of dsDNA. The magnitude of the DPV signal forNi(II) at dsDNA/CNFPE7 was an order of magnitude higherthan that obtained at the CNFPE7 electrode (see Fig. 5)indicating the presence of binding sites on the dsDNA.
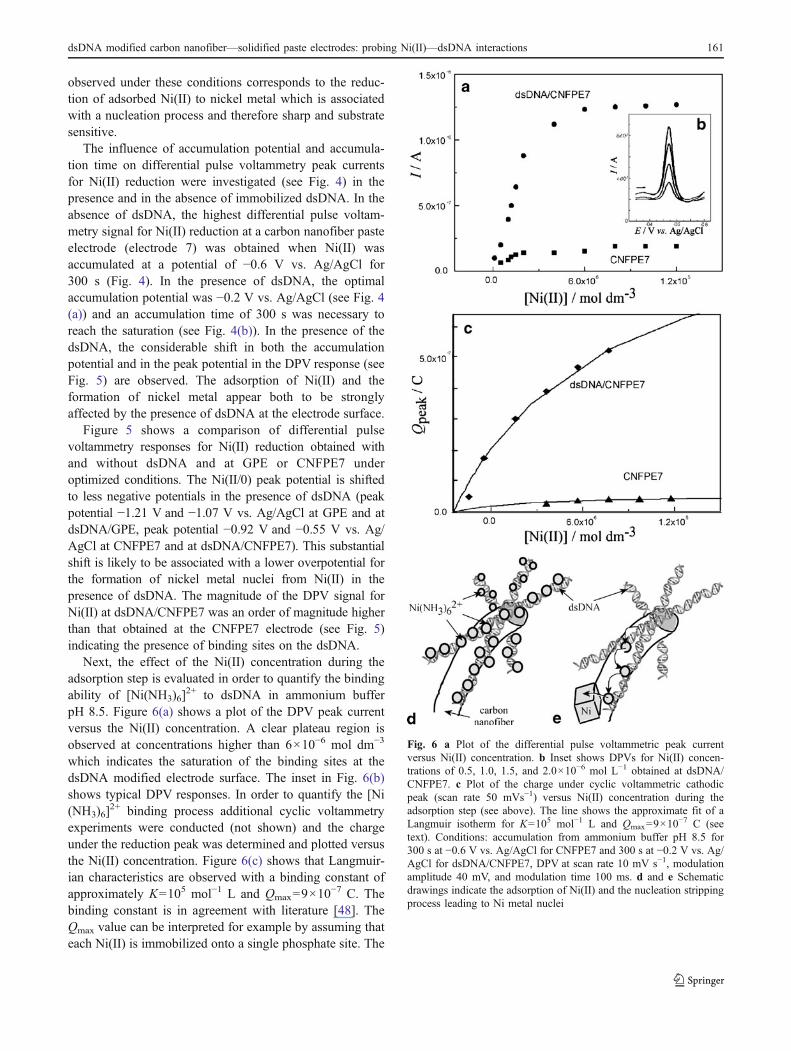
Next, the effect of the Ni(II) concentration during theadsorption step is evaluated in order to quantify the bindingability of [Ni(NH3)6]
2+ to dsDNA in ammonium bufferpH 8.5. Figure 6(a) shows a plot of the DPV peak currentversus the Ni(II) concentration. A clear plateau region isobserved at concentrations higher than 6×10−6 mol dm−3
which indicates the saturation of the binding sites at thedsDNA modified electrode surface. The inset in Fig. 6(b)shows typical DPV responses. In order to quantify the [Ni(NH3)6]
2+ binding process additional cyclic voltammetryexperiments were conducted (not shown) and the chargeunder the reduction peak was determined and plotted versusthe Ni(II) concentration. Figure 6(c) shows that Langmuir-ian characteristics are observed with a binding constant ofapproximately K=105 mol−1 L and Qmax=9×10
−7 C. Thebinding constant is in agreement with literature [48]. TheQmax value can be interpreted for example by assuming thateach Ni(II) is immobilized onto a single phosphate site. The
Fig. 6 a Plot of the differential pulse voltammetric peak currentversus Ni(II) concentration. b Inset shows DPVs for Ni(II) concen-trations of 0.5, 1.0, 1.5, and 2.0×10−6 mol L−1 obtained at dsDNA/CNFPE7. c Plot of the charge under cyclic voltammetric cathodicpeak (scan rate 50 mVs−1) versus Ni(II) concentration during theadsorption step (see above). The line shows the approximate fit of aLangmuir isotherm for K=105 mol−1 L and Qmax=9×10
−7 C (seetext). Conditions: accumulation from ammonium buffer pH 8.5 for300 s at −0.6 V vs. Ag/AgCl for CNFPE7 and 300 s at −0.2 V vs. Ag/AgCl for dsDNA/CNFPE7, DPV at scan rate 10 mV s−1, modulationamplitude 40 mV, and modulation time 100 ms. d and e Schematicdrawings indicate the adsorption of Ni(II) and the nucleation strippingprocess leading to Ni metal nuclei
dsDNA modified carbon nanofiber—solidified paste electrodes: probing Ni(II)—dsDNA interactions 161
two-electron Ni(II/0) reduction is therefore consistent withthe presence of 5×10−12 mol phosphate or DNA nucleo-tides. It is interesting to note that this value is a factor 20higher compared to that evaluated based on the directguanine oxidation response (vide supra). The directelectrochemical oxidation of dsDNA appears to occur onlyclose to the electrode surface. However, the Ni(II) accumu-lation method provides a measure of the total dsDNAamount even when dsDNA strands are in several nanometerdistance from the electrode surface. This process describedhere as “nucleation stripping voltammetry” is schematicallydepicted in Fig. 6d and 6e. The Ni(II) adsorbed ontodsDNA is sufficiently mobile to allow nucleation of Nimetal nuclei while being “stripped off” the dsDNA.
For analytical purposes [49, 50] the repeatability of theDPV measurements (expressed as R.S.D. value) wasdetermined for two concentrations of Ni(II) (with n=5)with 6% for 1×10−6 mol L−1 and 8% for 5×10−7 mol L−1.The developed analytical procedure was applied for thenucleation determination of Ni(II) at the surface of dsDNA/CNFPE7 sensor. Data in Fig. 6(a) show a comparison of thecalibration curves obtained at dsDNA/CNFPE7 andCNFPE7. From the initially approximately linear depen-dence a significantly higher slope was observed at dsDNA/CNFPE7 in comparison to CNFPE7. This indicates a highersensitivity because of the higher pre-concentration abilityintroduced by the dsDNA layer. The DPV limit of detectionat the dsDNA/CNFPE7 electrode was determined (3σ) as7×10−9 mol L−1
Interference study for the detection of nickel(II) at dsDNA-modified CNFPE7
DNA is known to interact with many metal cations [1] andtherefore effects of possible interferences have to beconsidered. Other metal cations can compete with Ni(II)in the formation of complexes with dsDNA. It was reportedthat mono-valent alkali metal cations prefer the interactionwith AT rich region of minor groove, while divalent
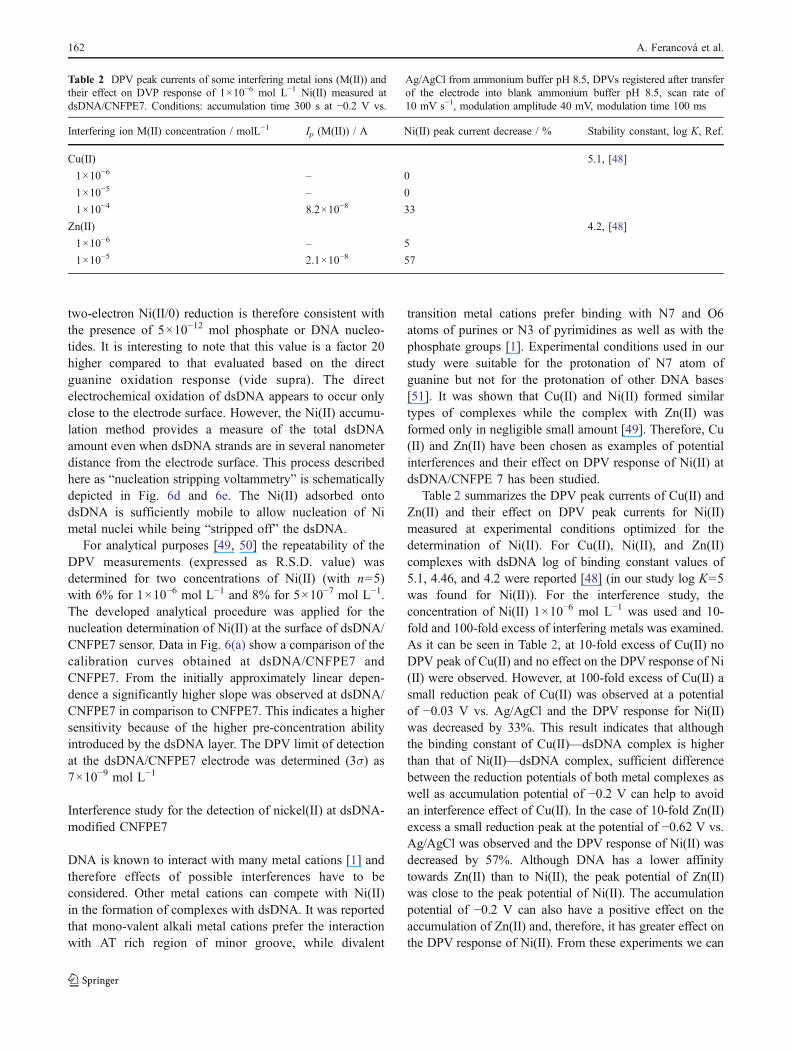
transition metal cations prefer binding with N7 and O6atoms of purines or N3 of pyrimidines as well as with thephosphate groups [1]. Experimental conditions used in ourstudy were suitable for the protonation of N7 atom ofguanine but not for the protonation of other DNA bases[51]. It was shown that Cu(II) and Ni(II) formed similartypes of complexes while the complex with Zn(II) wasformed only in negligible small amount [49]. Therefore, Cu(II) and Zn(II) have been chosen as examples of potentialinterferences and their effect on DPV response of Ni(II) atdsDNA/CNFPE 7 has been studied.
Table 2 summarizes the DPV peak currents of Cu(II) andZn(II) and their effect on DPV peak currents for Ni(II)measured at experimental conditions optimized for thedetermination of Ni(II). For Cu(II), Ni(II), and Zn(II)complexes with dsDNA log of binding constant values of5.1, 4.46, and 4.2 were reported [48] (in our study log K=5was found for Ni(II)). For the interference study, theconcentration of Ni(II) 1×10−6 mol L−1 was used and 10-fold and 100-fold excess of interfering metals was examined.As it can be seen in Table 2, at 10-fold excess of Cu(II) noDPV peak of Cu(II) and no effect on the DPV response of Ni(II) were observed. However, at 100-fold excess of Cu(II) asmall reduction peak of Cu(II) was observed at a potentialof −0.03 V vs. Ag/AgCl and the DPV response for Ni(II)was decreased by 33%. This result indicates that althoughthe binding constant of Cu(II)—dsDNA complex is higherthan that of Ni(II)—dsDNA complex, sufficient differencebetween the reduction potentials of both metal complexes aswell as accumulation potential of −0.2 V can help to avoidan interference effect of Cu(II). In the case of 10-fold Zn(II)excess a small reduction peak at the potential of −0.62 V vs.Ag/AgCl was observed and the DPV response of Ni(II) wasdecreased by 57%. Although DNA has a lower affinitytowards Zn(II) than to Ni(II), the peak potential of Zn(II)was close to the peak potential of Ni(II). The accumulationpotential of −0.2 V can also have a positive effect on theaccumulation of Zn(II) and, therefore, it has greater effect onthe DPV response of Ni(II). From these experiments we can
Table 2 DPV peak currents of some interfering metal ions (M(II)) and their effect on DVP response of 1×10−6 mol L−1 Ni(II) measured at dsDNA/CNFPE7. Conditions: accumulation time 300 s at −0.2 V vs. Ag/AgCl from ammonium buffer pH 8.5, DPVs registered after transfer of the electrodeinto blank ammonium buffer pH 8.5, scan rate of 10 mV s−1, modulation amplitude 40 mV, modulation time 100 ms
Interfering ion M(II) concentration / molL−1 Ip (M(II)) / A Ni(II) peak current decrease / % Stability constant, log K, Ref.
Cu(II) 5.1, [48]
1×10−6 – 0
1×10−5 – 0
1×10−4 8.2×10−8 33
Zn(II) 4.2, [48]
1×10−6 – 5
1×10−5 2.1×10−8 57
Table 2 DPV peak currents of some interfering metal ions (M(II)) andtheir effect on DVP response of 1×10−6 mol L−1 Ni(II) measured atdsDNA/CNFPE7. Conditions: accumulation time 300 s at −0.2 V vs.
Ag/AgCl from ammonium buffer pH 8.5, DPVs registered after transferof the electrode into blank ammonium buffer pH 8.5, scan rate of10 mV s−1, modulation amplitude 40 mV, modulation time 100 ms
162 A. Ferancová et al.

summarize that the effect of interfering metal cations can beto some extent minimized by using a carefully optimizedaccumulation potential.
Conclusions
(1) A new type of solidified composite paste electrodebased on a mixture of paraffin oil and paraffin wax hasbeen constructed. Carbon nanofiber paste compositeelectrodes were prepared using an optimized compo-sition of the mixture of paraffin wax and paraffin oil(43% of CNF, 41% of PW, and 16% of PO). Theelectrodes were characterized using electrochemicalmethods and then employed for the surface modifica-tion by dsDNA.
(2) It was shown that dsDNA modified carbon nano-fiber—solidified paste electrodes can be advanta-geously used for the differential pulse voltammetricdetermination of metal cations such as Ni(II)accumulated into the calf thymus dsDNA layer.
(3) Direct guanine moiety oxidation has been employed toevaluate the amount of dsDNA in “electrochemicallyactive” contact with the carbon nanofiber electrodesurface. The Ni(II) adsorption process has beenemployed to estimate the total dsDNA amountadsorbed at the carbon nanofiber electrode. The totaldsDNA amount was estimated to be 20-times highercompared to the directly oxidizable dsDNA.
(4) A “nucleation stripping voltammetry” method hasbeen developed and the binding of Ni(II) to dsDNAinvestigated. A high sensitivity for the analytical Ni(II)determination was found in ammonium buffer pH 8.5with 7×10−9 mol L−1 limit of detection.
Acknowledgements This research project has been supported by aMarie Curie Transfer of Knowledge Fellowship of the EuropeanCommunity’s Sixth Framework Programme under contract numberMTKD-CT-2006-042637. L.R. also thanks the EPSRC (EP/F025726/1)for financial support. A.F. and J.L. thank also the Scientific Grant Agencyof the Slovak Republic (VEGA 1/0852/08).
References
1. Anastassopoulou J (2003) Metal—DNA interactions. J Mol Struct651–653:19–26
2. Freisinger E, Sigel RKO (2007) From nucleotides to ribozymes—A comparison of their metal ion binding properties. CoordinChem Rev 251:1834–1851
3. Wang H, Kim YM, Liu HP, Zhu Z, Bamrungsap S, Tan WH(2009) Engineering a unimolecular DNA-catalytic probe forsingle lead ion monitoring. J Am Chem Soc 131:8221–8226
4. Saenger W (1984) Principles of nucleic acid structure. Springer-Verlag, New York
5. Ovádeková R, Labuda J (2007) DNA binding interactions,structural damage and protection investigated by electrochemicalDNA biosensors. In: Adam V, Kizek K (eds) Utilizing of bio-electrochemical and mathematical methods in biological research.Research Signpost, India, pp 173–201
6. Babkina SS, Ulakhovich NA, Moiseeva EN, Filyushina EE (2003)Preconcentration of heavy metals on immobilized deoxyribonu-cleic acid incorporated into a biosensor for determining metals inbiological samples. J Anal Chem 58:651
7. Babkina SS, Ulakhovich NA (2004) Amperometric biosensorbased on denatured DNA for the study of heavy metals complex-ing with DNA and their determination in biological, water andfood samples. Bioelectrochemistry 63:261–265
8. Babkina SS, Paleček E, Jelen F, Fojta M (2005) Electrochemicalsensors based on stationary electrodes and immobilized DNA orits fragments and the assessment of their analytical potentials. JAnal Chem 60:567–572
9. Teles FRR, Fonseca LP (2008) Trends in DNA biosensors.TRAC-Trend Anal Chem 7:606–623
10. Wong ELS, Chow E, Gooding JJ (2007) The electrochemicaldetection of cadmium using surface-immobilized DNA. Electro-chem Commun 9:845–849
11. Oliveira SCB, Corduneanu O, Oliveira-Brett AM (2008) Insitu evaluation of heavy metal—DNA interactions using anelectrochemical DNA biosensor. Bioelectrochemistry 72:53–58
12. Chiorcea-Paquim AM, Corduneanu O, Oliveira SCB, DiculescuVC, Oliveira-Brett AM (2009) Electrochemical and AFM evalu-ation of hazard compounds—DNA interaction. Electrochim Acta54:1978–1985
13. Marken F, Gerrard ML, Mellor IM, Mortimer RJ, Madden CE,Fletcher S, Holt K, Foord JS, Dahm RH, Page F (2001)Voltammetry at carbon nanofiber electrodes. Electrochem Com-mun 3:177–180
14. Van Dijk N, Fletcher S, Madden CE, Marken F (2001) Nano-composite electrodes made of carbon nanofibers and black wax.Anodic stripping voltammetry of zinc and lead. Analyst126:1878–1881
15. Rassaei L, Sillanpää M, Bonné MJ, Marken F (2007) Carbonnanofiber-polystyrene composite electrodes for electroanalyticalprocesses. Electroanalysis 19:1461–1466
16. Pérez B, del Valle M, Alegret S, Merkoçi A (2007) Carbonnanofiber vs. carbon microparticles as modifiers of glassy carbonand gold electrodes applied in electrochemical sensing of NADH.Talanta 74:398–404
17. Liu Y, Huang J, Hou H, You T (2008) Simultaneous determinationof dopamine, ascorbic acid and uric acid with electrospun carbonnanofibers modified electrode. Electrochem Commun 10:1431–1434
18. Fletcher BL, McKnight TE, Melechko AV, Simpson ML, DoktyczMJ (2006) Biochemical functionalization of vertically alignedcarbon nanofibres. Nanotechnology 17:2032–2039
19. Peckys DB, de Jonge N, Simpson ML, McKnight TE (2008) End-specific strategies of attachment of long double stranded DNAonto gold-coated nanofiber arrays. Nanotechnology 19:435301–435309
20. Peckys DB, Melechko AV, Simpson ML, McKnight TE (2009)Immobilization and release strategies for DNA delivery usingcarbon nanofiber arrays and self-assembled monolayers. Nano-technology 20:145304–145311
21. Lin X, Jiang X, Lu L (2005) DNA deposition on carbonelectrodes under controlled dc potentials. Biosens Bioelectron20:1709–1717
22. Wang J, Lin Y (2008) Functionalized carbon nanotubes andnanofibers for biosensing applications. TRAC-Trend Anal Chem27:619–626
dsDNA modified carbon nanofiber—solidified paste electrodes: probing Ni(II)—dsDNA interactions 163

23. Lee CS, Baker SE, Marcus MS, Yang W, Eriksson MA, HamersRJ (2004) Electrically addressable biomolecular functionalizationof carbon nanotube and carbon nanofiber electrodes. Nano Lett4:1713–1716
24. Baker SE, Tse KY, Hindin E, Nichols BM, Clare TL, HamersRJ (2005) Covalent functionalization for biomolecular recog-nition on vertically aligned carbon nanofibers. Chem Mater17:4971–1978
25. Arumugam PU, Chen H, Siddiqui S, Weinrich JAP, Jejelowo A,Li J, Meyyappan A (2009) Wafer-scale fabrication of patternedcarbon nanofiber nanoelectrode arrays: a route for development ofmultiplexed, ultrasensitive disposable biosensors. Biosens Bio-electron 24:2818–2824
26. Pruneanu S, Ali Z, Watson G, Hu SQ, Lupu D, Biris AR, OlenicL, Mihailescu G (2006) Investigation of electrochemical proper-ties of carbon nanofibers prepared by CCVD method. Part SciTechnol 24:311–320
27. Koehne JE, Chen H, Cassell A, Liu GY, Li J, Meyyappan M(2009) Arrays of carbon nanofibers as a platform for biosensing atthe molecular level and for tissue engineering and implantation.Biomed Mater Eng 19:35–43
28. Fisher AC (1996) Electrode dynamics. Oxford University Press,Oxford
29. Barsan MM, Pinto EM, Florescu M, Brett CMA (2009)Development and characterization of a new conducting carboncomposite electrode. Anal Chim Acta 635:71–78
30. Compton RG, Banks CE (2007) Understanding voltammetry.World Scientific, London
31. Fanjul-Bolado P, Hernández-Santos D, Lamas-Ardisana PJ,Martín-Pernía A, Costa-García A (2008) Electrochemical charac-terization of screen-printed and conventional carbon paste electro-des. Electrochim Acta 53:3635–3642
32. Katz E, Willner I (2003) Probing biomolecular interactions atconductive and semiconductive surfaces by impedance spectros-copy: routes to impedimetric immunosensors, DNA-sensors, andenzyme biosensors. Electroanalysis 15:913–947
33. Wu L, Zhang X, Ju H (2007) Detection of NADH and ethanolbased on catalytic activity of soluble carbon nanofiber with lowoverpotential. Anal Chem 79:453–458
34. Wang J, Cai X, Tian B, Shiraishi H (1996) Microfabricated thick-film electrochemical sensor for nucleic acid determination.Analyst 121:965–969
35. Pedano ML, Rivas GA (2004) Adsorption and electrooxidation ofnucleic acids at carbon nanotubes paste electrodes. ElectrochemCommun 6:10–16
36. Wu L, Zhou J, Luo J, Lin Z (2000) Oxidation and adsorption ofdeoxyribonucleic acid at highly ordered pyrolytic graphiteelectrode. Electrochim Acta 45:2923–2927
37. Pedano ML, Rivas GA (2003) Immobilization of DNA on glassycarbon electrodes for the development of affinity biosensors.Biosens Bioelectron 18:269–277
38. Marrazza G, Chianella I, Mascini M (1999) Disposable DNAelectrochemical biosensors for environmental monitoring. AnalChim Acta 387:297–307
39. Wang J, Cai X, Rivas G, Shiraishi H (1996) Stripping potentio-metric transduction of DNA hybridization processes. Anal ChimActa 326:141–147
40. Özcan A, Şahin Y, Özsöz M, Turan S (2007) Electrochemicaloxidation of ds-DNA on polypyrrole nanofiber Modified pencilgraphite electrode. Electroanalysis 19:2208–2216
41. Jelen F, Fojta M, Paleček E (1997) Voltammetry of native double-stranded, denatured and degraded DNAs. J Electroanal Chem427:49–56
42. Zabost E, Nowicka AM, Donten M, Stojek Z (2009) Substantialdifference between temperature dependencies of dsDNA predena-turation process obtained by voltammetry and spectroscopy. PhysChem Chem Phys 11:8933–8938
43. Novak RL, Dohnal J (1974) DNA-snapback peptides. NucleicAcids Res 1:753–759
44. Oliveira-Brett AM, Diculescu V, Piedade JAP (2002) Electro-chemical oxidation mechanism of guanine and adenine using aglassy carbon microelectrode. Bioelectrochemistry 55:61–62
45. Alvarez JLM, Calzón JAG, Fonseca JML (1999) Catalyticreduction of metal ions by adsorbed ligands in differential pulsevoltammetry: evaluation of catalytic effects from the non-catalysed reduction peaks. Electrochim Acta 45:477–482
46. Sletten E, Froystein NA (2009) Sequence-selective binding oftransition metal complexes to DNA. In: Hadjiliadis N, Sletten E(eds)Metal complex—DNA interactions.Wiley, NewYork, pp 15–29
47. Abrescia NGA, Huybh-Dinh T, Subirana JA (2002) Nickel-guanine interactions in DNA: crystal structure of nickel-d[CGTGTACACG]2. J Biol Inorg Chem 7:195–199
48. Bregadze V, Gelagutashvili E, Tsakadze K (2009) Thermodynam-ic models of metal ion—DNA interactions. In: Hadjiliadis N,Sletten E (eds) Metal complex—DNA interactions. Wiley, NewYork, pp 31–54
49. Virkutyte J, Sillanpää M (2006) Chemical evaluation of potablewater in Qinghai Province, China: Human health aspects. EnvironInt 32:80–86
50. Huang X, Sillanpää M, Duo B, Gjessing ET (2008) Water qualityin the Tibetan Plateau: metal contents of four selected rivers.Environ Pollut 156:270–277
51. Mukherjee G, Ghosh T (1995) Metal ion interaction withPenicillins-Part VII: mixed-ligand complex formation of cobalt(II), nickel(II), copper(II), and zinc(II) with Ampicillin andnucleic bases. J Inorg Biochem 59:827–833
164 A. Ferancová et al.




























