Embed Size (px)
Citation preview

HEMOGLOBINOPATIAS

Se ha estimado que aprox. 7% de la población mundial -400 millones de personas-son portadores heterocigotasde estos desórdenes. Es decir que 300.000- 400.000 RN portadores de alguna hemoglobinopatía severa nace cada año. Se ha estimado que hay unos 100.000 pacientes con homocigosispara la beta talasemia vivos en el mundo.
Epidemiologia
Henri WAJCMAN, MD, Ph.D 2005

Cuáles son las razones para la búsqueda de las hemoglobinopatías
Confirmar un diagnóstico sopechado (HbS o talasemia)
Explicar una anormalidad hematológica como anemia, microcitosis, eritrocitosis..
Identificar una anormalidad en fase asintomáticacomo un screening neonatal.
Predecir serias afecciones de síntesis de cadenas de globinas en el feto.
Permitir el correcto asesoramiento genético a los futuros padres. (Talasemia en Chipre, un ejemplo de problema de salud pública).

CLASIFICACIÓN•a) Defectos estructurales de las globinas.
•b) Disminución en la síntesis de globina.
•c) Ambos defectos simultaneamente.

SINDROMES TALASÉMICOS

Definición
Son un grupo heterogeneo de alteraciones hereditarias de la síntesis de hemoglobina, caracterizadas por la disminuciónparcial o total de la producción de unao varias cadenas globínicas y que en su conjunto se conocen comosíndromes talasémicos.

• Defectos estructurales de la globina: hemoglobinopatíasestructurales
• Disminución síntesis globina: talasemia
• Se han identificado hemoglobinopatías con fenotipo talasémico, por ejemplo, la Hb E
Talasemias con alteraciones estructurales, por ejemplo, la HbLepore
• Un cuarto aún más heterogéneo Peristencia Hereditaria Hb F (alteración en la interconversión Hb F a Hb A)



HEMOGLOBINOPATIAS
SINDROMES TALASEMICOS
HEMOGLOBINASVARIANTES
α-TALASEMIAβ-TALASEMIA
HPFHδ-TALASEMIA
Hb SHb C
Constant Spring

Clasificación
o ausencia de sínt. de cadenas alfa talasemias
o ausencia de sínt. de cadenas beta talasemias
o ausencia de sínt. de cad.delta-beta talasemias
o ausencia de sínt. de cad.gamma talasemias
o ausencia de sínt. de cadenas delta talasemias


• Las cadenas α y β de la globina están codificadas por locigenéticos independientes regulados de forma coordinada.
• Los seis tipos de cadenas globínicas que existen (α β γ δ ε y Ζ) se expresan en distintas etapas del desarrollo por lo que se necesitan seis genes estructuralmente diferentes.
• En el ser humano las cadenas globínicas se dividen en dos grupos:
cadenas tipo α (Ζ y α ) cadenas de tipo β (ε, γ, δ y β)
• La síntesis de estas cadenas está regulada por dos flias. de genes organizados en agrupaciones (clusters) localizados en distintos cromosomas.

Genes de de globina

Genes de beta y alfa globina


Estructura Cluster α-globina
• Está formado por un gen embrionario ζ 2, dos genes fetales/adultos α1 y α2 y varios pseudogenes (ψ ζ1, ψ α1, ψ α2) y un gen de función inderterminada θ1
• Telómero- ζ 2- ψ ζ1- ψ α2- ψ α1- α2 - α1- θ1- centrómero
Estructura cluster β-globina
• La flia de genes beta globina se extiende sobre una región de 50 kb a lo largo del brazo corto del cromosoma 11 en la banda p15.5
• está constituida por un gen embrionario (ε) dos genes fetales (Gγy Aγ) dos genes adultos (δ y β) y un seudogen (ψβ)
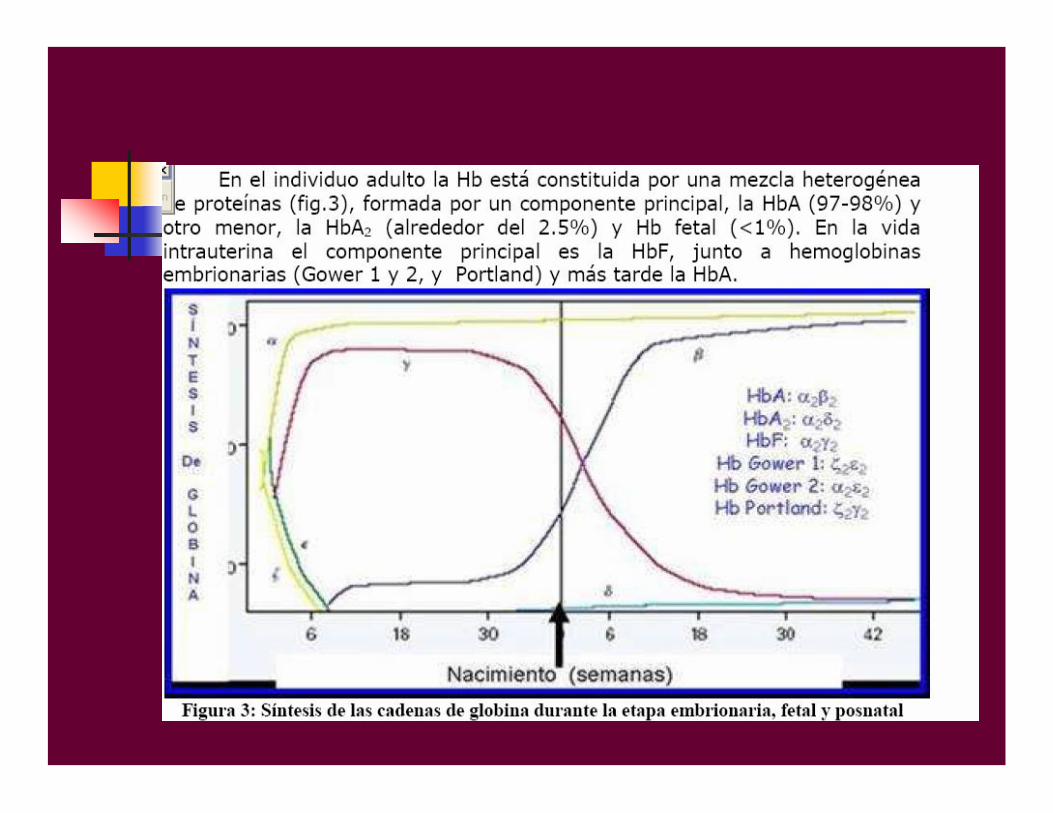

• La herencia de las talasemias muestra un patrón autosómico dominante
α
β
p15.5
p13.3

Beta - talasemia: herencia
Talasemiamenor
Talasemiamayor
Talasemiamenor
Talasemiamenor

Principales tipos de talasemias
•α-talasemia α0 (ausencia total de síntesis)α+ (delecional/no delecional)
(síntesis deficiente o parcial)•β-talasemia β0
β+
con Hb A2 Normal
•δβ-talasemia δβ0
(A γ δβ)0
(δβ)+
• γ-talasemia γ0
•δ-talasemia δ0
δ +

Beta-talasemias
Clasificación
º Th: mutación del gen beta que lleva a ausencia de cadenas beta
+ Th: mutaciones del gen beta que llevan a producir síntesis disminuídas (variables) de cadenas beta

Beta-talasemias
Patología molecular
La mayoría de los defectos génicos responsables de las
b-talasemias es la mutación de un único nucleótido
(mutación puntual) que afecta a uno de los diferentes
procesos moleculares involucrados en la expresión del
gen beta globina; transcripción, procesamiento del pre-
ARNm y traducción.

Genética molecular de beta- th
ߺ-talasemia (completa ausencia de producción de cadena de beta globina) los alelos resultan de mutaciones sin sentido, alteraciones del marco de lectura (frameshift), o (a veces) mutaciones en el empalme (splicing mutations)
ß+-talasemia (producción residual de cadena de beta globina) son producidos por mutaciones en el promotor (caccc o tata box), en la señal de poliadenilación, en las regiones no transcriptas 5' o 3', o por anormalidades en el splicing.

Distribución geográfica de Beta Th- Mutaciones más frecuentes

Fisiopatología de las Beta-ThDÉFICIT DE CADENAS BETA
HB A ó = 0
Microcitosis eHipocromíaVCM y HCM
Exceso de cadenas alfa precipita en el citoplasma de los eritroblastos
Eritropoyesis ineficaz
Hemólisis

Consecuencias Fisiopatológicas
1) Deficiente hemoglobinización microcitosis
disminución de CHCM (hipocromía)La hemoglobinogénesis es un proceso regulado por la concentración de Hbcuando esta alcanza un valor crítico, actúa sobre el núcleo de los precursores eritroides e inhibe el proceso de maduración.Si hay un trastorno en la síntesis de la Hb (ferropenia o talasemia) se tarda más tiempo en alcanzar ese límite crítico aumenta el número de mitosis por cada ciclo y disminuye el tamaño de los eritrocitos (microcitosis)
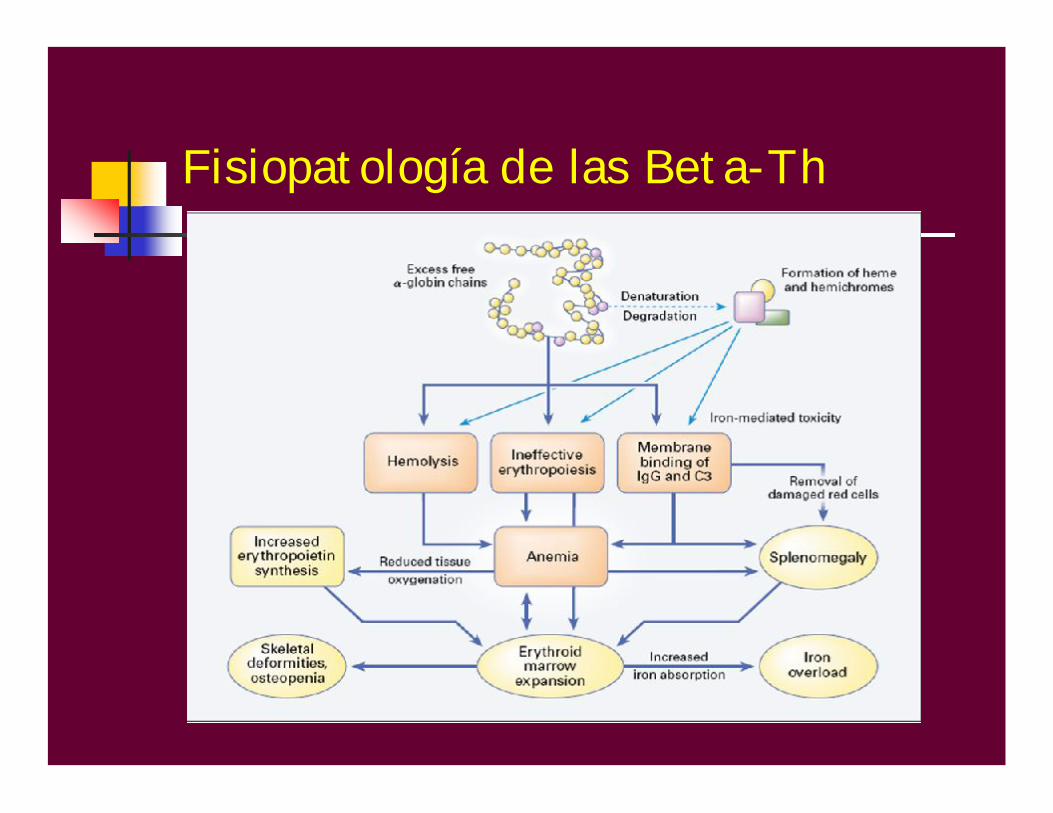
Fisiopatología de las Beta-Th


Talasemia mayor- anemia de Cooley
Diagnóstico clínico:
Bebé entre los 3-6 meses de edadPalidez amarillentaFacies mongoloideAbdomen prominente (gran hepatoesplenomegalia)

Anemia de Cooley
Alteraciones óseas, especialmente en cráneo y
cara, implantación anómala dentaria (facies
mongoloides)
Retraso del desarrollo corporal (hipoxia crónica).
Alteraciones endocrinológicas (sobrecarga férrica
y hemosiderosis)

Anemia de Cooley

Anemia de Cooley

Anemia de Cooley



Diagnóstico de th >
Clínica Hemograma Estudio Convencional de Hem. betaº /betaº : Hb: F 95% - Hb A2 3% beta+/beta+ (Mediterránea): Hb F: 60-90%-
Hb A2 2-5%- Hb A 10- 40% Betaº/beta+: Hb F: 60-90%, Hb A2 :3-5%,
Hb A: 10-40% Estudio familiar: padres con talasemia menor

Diagnóstico de th >
Clínica HemogramaGR: 3300000-3900000/lGB: 18.000/lHB: 3,5- 4 g / dl
Hto: 25%Ret: 8-9%Plaquetas: 315000/l

Estudio convencional de Hbs
Hemograma con IH y morfología de GR. Dosaje de Fe sérico, TIBC, Ferritina sérica Test de drepanocitosis (Hb S) Test de los cuerpos de inclusión (Hb H) Test de inestabilidad Test de Kleihauer y Betke Electroforesis de Hbs en medio ác y alc. Electroforesis de cadenas de globinas Dosaje de Hb álcali resistente (Hb F) Dosaje de hemoglobina A2

Electroforesis de Hbs en medio alcalino

Electroforesis de Hbs en medio ácido y alcalino

Diagnóstico de th >
HemogramaMorfología de los eritrocitos: Marcada anisocitosis con abundantes micro y macrocitos. Marcada poiquilocitosis. Marcada hipocromía. Targets cells, leptocitos. Policromatofilia. Regular cantidad de esquistocitos. Punteado basófilo.
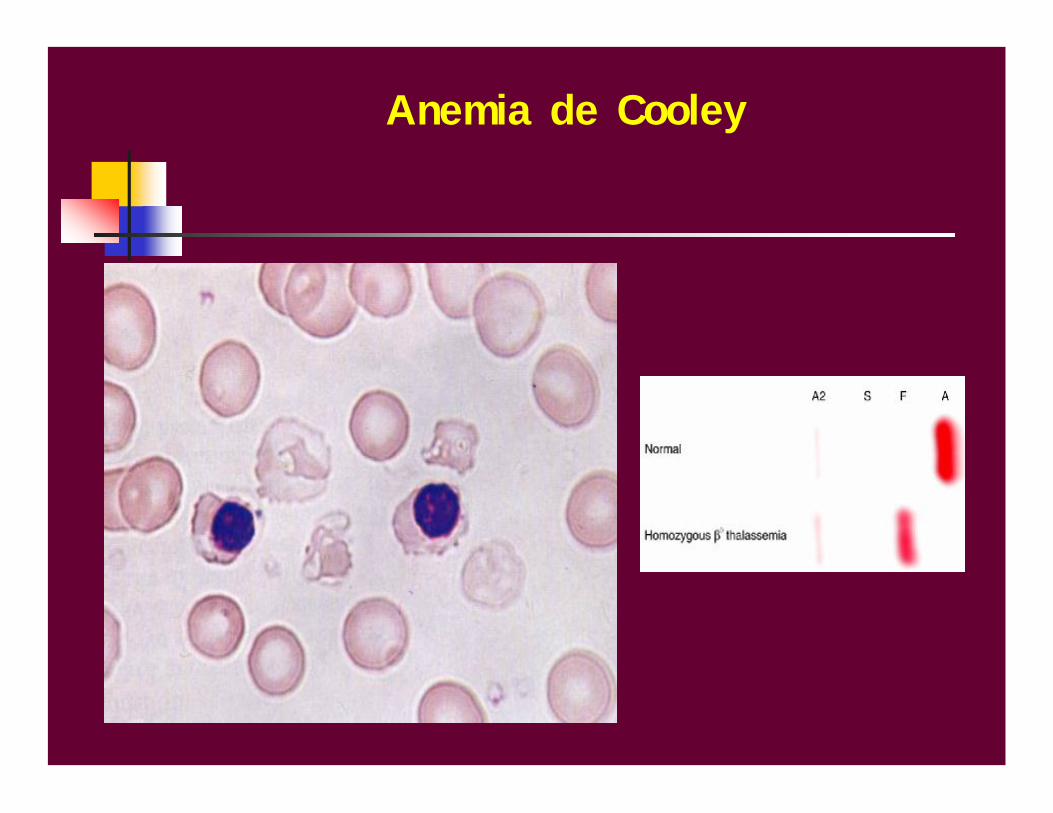
Anemia de Cooley


Diagnóstico molecular
anemia de Cooley
En Rosario y zona de influencia los pacientes son homocigotas o doble heterocigotas para alguna de las siguientes mutaciones:IVS1-1 GA, IVS1-6 TC, IVS1-110 GA, IVS2-l GA , CD 39 CT, IVS2-745 CG, que son las más frecuentes en la zona del Mediterráneo.

Dosaje de Hb FAumentada o disminuída
MÉTODO DE DESNATURALIZACIÓN ALCALINASinger y ChernoffBetkeV.N: menor de 2%Ligeramente aumentada: menor de 5%(HPFH heterocelular),Th menor, embarazoMuy aumentada: 20-50-90% heterocelular (Anemia de Cooley, βδ Th, RN, neonato.Muy aumentada pancelular: HPFH
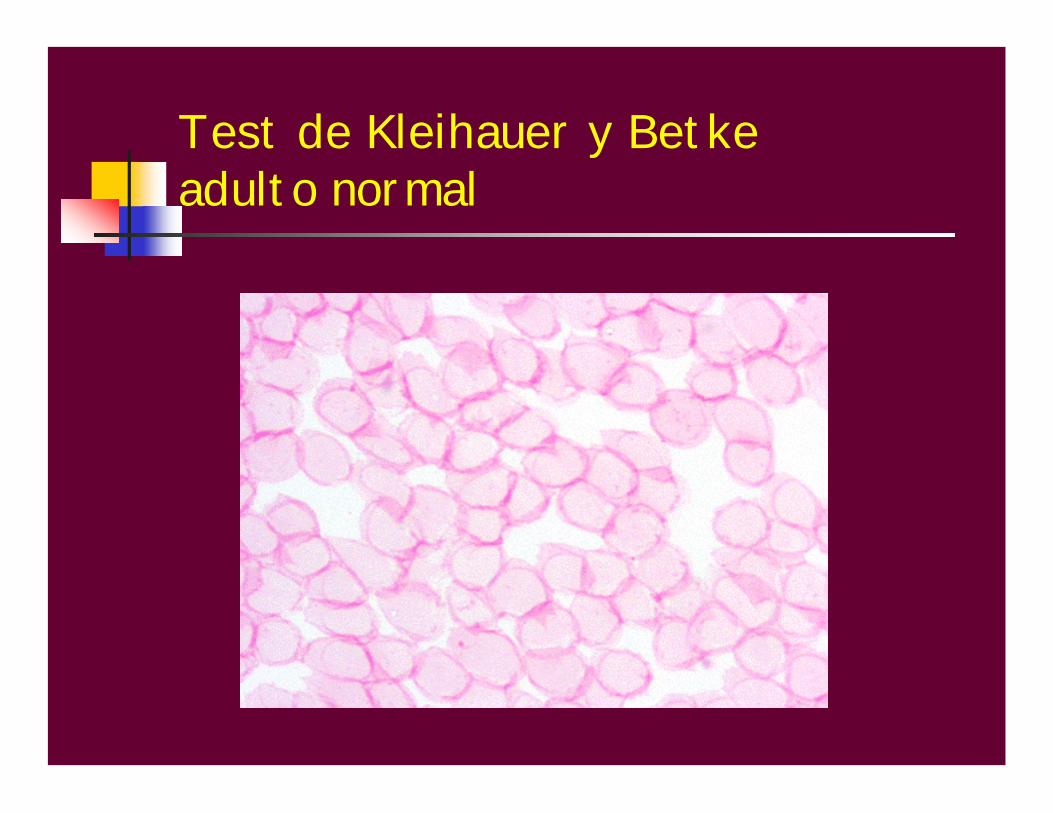
Test de Kleihauer y Betkeadulto normal

Test de kleihauer y Betkeadulto normalHPFH heterocelular

ß-Tal Minorß-Tal MayorMujerHombre
: 11.5-15.3 : 9.1-14
<714.0±0.915.9±1.0Hb g/dl
<2712-2030.2±2.130.9±1.9HCM pg
<79 50-7087.6±5.589.1±5.01VCM fl
PORTADORCOOLEY NormalIndices en sangre
periférica
Indices Hematimétricos en Beta-Talasemia

Talasemia intermediaSe sospecha en individuos que presentan
anemia moderada a una edad mas tardía
y que raramente requieren
transfusiones.
Los individuos con Talasemia Intermedia
tienen riesgo de sobrecarga de hierro
secundaria a absorción intestinal
aumentada como consecuencia de la
eritropoyesis inefectiva.
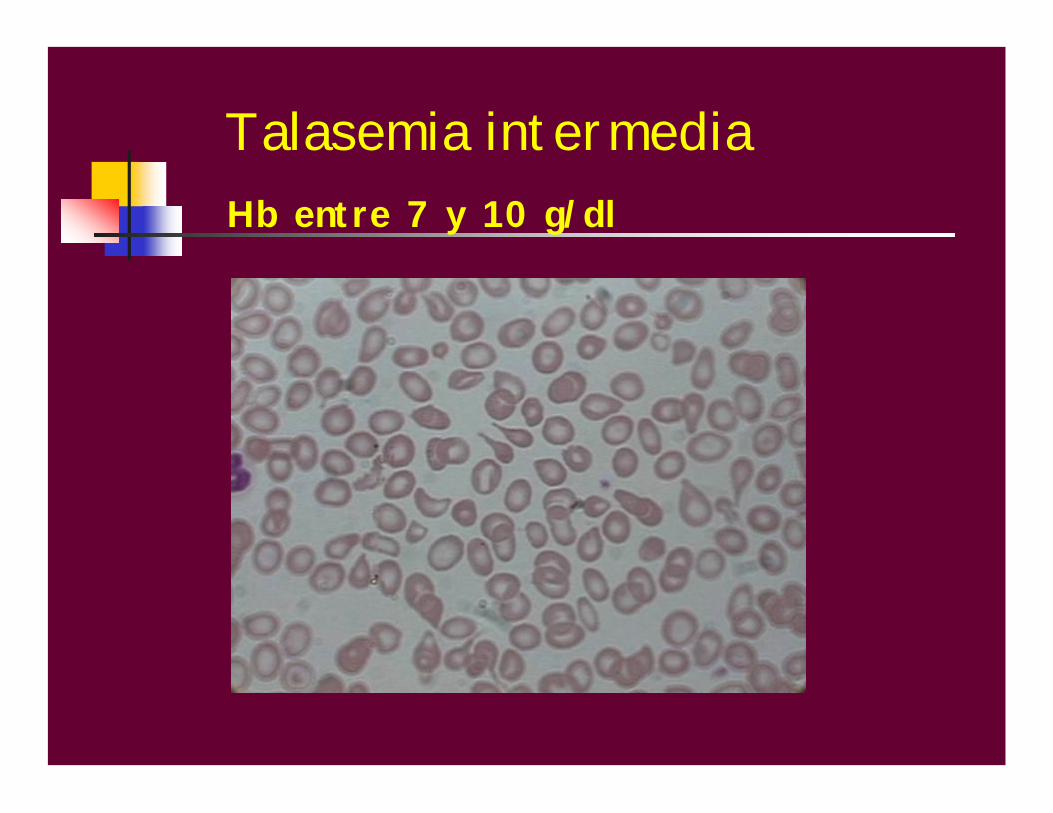
Talasemia intermediaHb entre 7 y 10 g/dl

Talasemia intermedia
Hb: 7-10g /dlVCM disminuídoHCM disminuídaHb A2 aumentada (5%), para las doble heterocigosis u homocigosis de formas leves de beta+ Th, Hb F aumentada: 30-70%Si se trata de talasemias aumenta la Hb F pero no la Hb A2.


Talasemia intermedia
El genotipo puede ser variable:Beta+/beta+ (negros)Betaº/( Lepore)Otros

-Talasemia menor.
Se caracteriza por eritrocitosis microcítica con anemia leve, raravez se aprecia esplenomegalia, sudiagnóstico suele ser casual y facilitado por el empleo de analizadores hematológicos quesuministran de forma sistemáticalos valores de VCM y HCM.
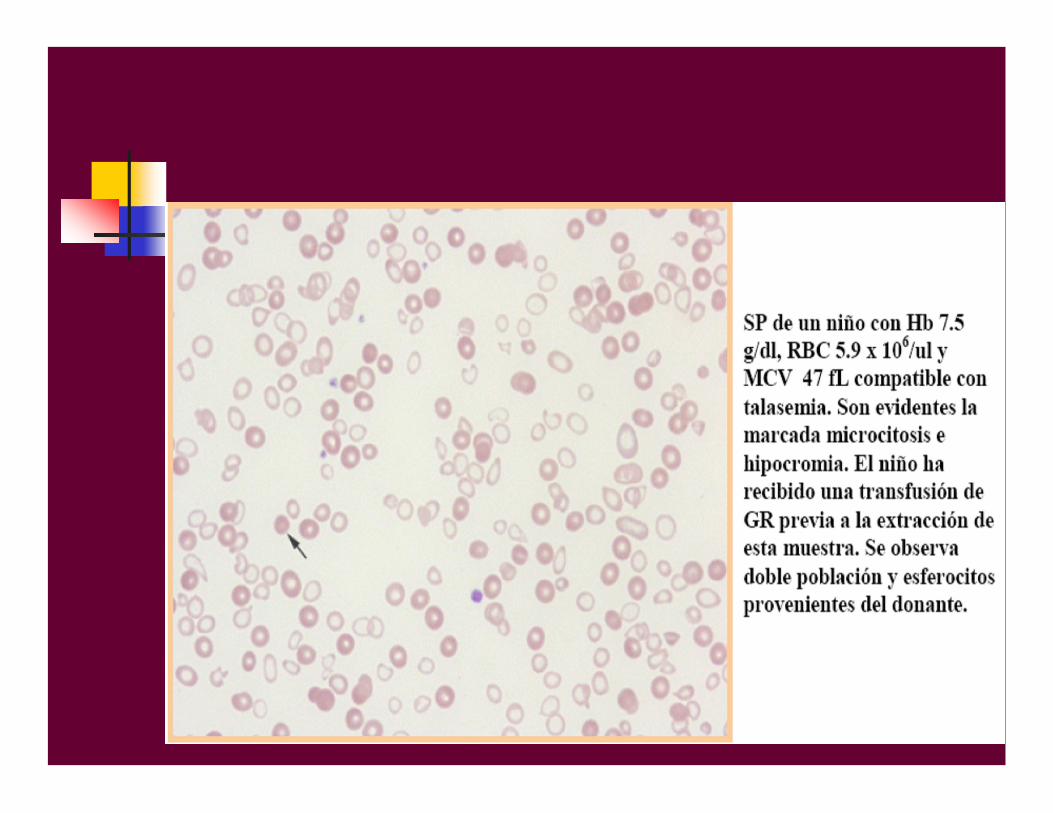

-TALASEMIA MENOR

-talasemia menor

-talasemia menor. diagnósticoº Th:Hb: 8-10 g/dlGR: eritrocitosisVCM y HCM disminuídosHb A2: 4,5-5,5%Hb F: 0,5-4%Hb A: 90-95%

Genetica molecularAnálisis de mutaciones conocidas: las ß-
talasemias pueden ser causadas por mas de 200 mutaciones en el gen beta. Comúnmentelas mutaciones son detectadas porprocedimientos basados en PCR. Los métodosmas usados son reverse dot blot o amplificación primer-específica con un setde primers complementarios a las mutacionesmas frecuentes en la población de la cual el individuo afectado es originario
·

Beta-TalasemiaGenética Molecular: Métodos Clínicos
PCR-ARMSMutación 0 IVS-II-nt 1 (634 bp)
861 pb634 pb
Calle 1: blanco de H2O (control de contaminación).
Calles 2 y 3: individuo normal (A/A) con primers normal y mutado respectivamente.
Calle 4: marcador de peso molecular (100 bp ladder). La banda más brillante corresponde a 500 pb.
Calles 5 y 6: individuo heterocigoto (A/T) con primers normal y mutado respectivamente.

δβ Talasemias
δβ deleción
δβ Lepore-fusión
δβ HPFH

δβ Talasemias
Las delta-beta talasemias heterocigotas presentanun cuadro hematológico y clínico similar a las beta Th heterocigotas.
Presentan Hb F aumentada 5-15% (heterocelular), la Hb A2 está normal-
Las delta-beta talasemias homocigotas o dobleheterocigotas beta-delta/ beta Th presentan un cuadro clínico y hematológico de Th intermedia o mayor con Hb F muy aumentada y Hb A2 disminuída.

Persistencia hereditaria de Hbfetal- HPFH
Es más frecuente en la etnia nergra, sólo el homocigota presenta Hb F de 100% pancelular y VCM disminuído.
El heterocigota puede no presentar anemia pero presenta HB F de alrededor del 30% siempre de distribución pancelular.

δβ talasemias-LeporeLa Hb Lepore corre como la Hb S
En el heterocigota la morfología yelhemograma son similares a la beta Th menor, pero presentan entre 8-15 % de HbLepore, Hb F algo aumentada 5-7%, Hb A2 disminuída.
El homocigota clínica y hematológicamentees una Th mayor, con Hb Lepore de 25-30%, el resto es Hb F.

Alfa talasemias.
La patología molecular de las alfa talasemias está determinada por una serie heterogenea de deleciones de diversa extensión:
º elimina todo el complejo de genes alfa de uno o de los dos cromososmas
+ elimina uno de los genes alfa , parte de los mismos o puede ser una mutación puntual.

-4.2-4.2
-3.7-3.7
- 70 kpb 0 kpb 10 kpb 20 kpb 30 kpb
5’HVR HS-40 2 inter HVR 1 12 1 1 3’HVR
5’ 3’
-5.2-5.2
--MED--MED
--THI--THI
--SEA--SEA
--BO--BO
230 kpb60 kpb
30 kpb35 kpb--DUCH--DUCH
Deleciones causantes de -TalasemiaDeleciones causantes de -Talasemia

- 70 kpb 0 kpb 10 kpb 20 kpb 30 kpb
5’HVR HS-40 2 inter HVR 1 1 2 2 1 1 3’HVR
NcoNcoHphHph CSCST SaudíT Saudí
CAP Señal POLI A1 31 32 99 100 141
5' 3'
CAT ATAbox box
1) Procesamiento del ARNm
Mutaciones Puntuales causantes de -TalasemiaMutaciones Puntuales causantes de -Talasemia
2) Traducción del ARNm
3) Inestabilidad Postranscripcional
Mutaciones en el sitio de SplicingSeñal de Poliadenilación
Mutaciones en el codon de iniciaciónMutaciones en el codon de terminació
Cambio del marco de lecturaMutación sin sentido

Patient: cctgggccgcactgaccctcttctctgcacaactc |||||||||||||||||||||||||||||||||||
Normal sequence: cctgggccgcactgaccctcttctctgcacagctc
Secuenciación: nueva mutación IVS2-142 GEN 2
Secuenciación: nueva mutación IVS2-142 GEN 2

Alfa talasemias.
10.5 / 8.510.5 / 8.5
11.1 / 9.411.1 / 9.4
11.2 / 9.911.2 / 9.9
12.3 / 10.612.3 / 10.6
13.7 / 12.113.7 / 12.1
13.9 / 12.013.9 / 12.0
14.5 / 12.514.5 / 12.5
14.3 / 12.614.3 / 12.6
15.5 / 14.015.5 / 14.0
Hg Hg g/dlg/dl
5.10 / 4.685.10 / 4.68
6.10 / 5.146.10 / 5.14
5.82 / 5.215.82 / 5.21
5.78 / 5.105.78 / 5.10
6.28 / 5.656.28 / 5.65
5.98 / 5.305.98 / 5.30
5.76 / 5.215.76 / 5.21
5.42 / 4.885.42 / 4.88
5.20 / 4.605.20 / 4.60
HtiesHtiesX10X101212/l/l
68.068.0
64.864.8
60.560.5
66.166.1
69.169.1
71.671.6
75.575.5
81.281.2
90 90
VCMVCMflfl
22.322.318.718.7
7.07.019.119.1
10.510.518.918.9
0021.021.0
0021.721.7
0022.922.9
0024.824.8
0026.226.2
003030
Hb HHb H%%
HCMHCMpgpg
GenotipoGenotipo

Alfa talasemias.Deleción alfa 3,7 ó 4,2

Talasemia alfa
Sin alteracionesClínicas ni hematologicas
-a /aaPortador silente
NingunaHbBart 2-20%
Anemia leveO inexistente
-- /aa-a / -a
TalasemiaMenor
HbH 5-20%Trazas deBart
Hb Bart 20 a40%
Anemia Hemolitica crònica
- - / -a Enfermedad por Hb H
HbBart>80%HbHHbPortland
Muerte fetalAnemia grave
- - / - -Hidrops Fetal
Despues del 1° año
RNClínicaGenotipoSìndrome


Enfermedad de la Hb H

Enfermedad de la Hb H


Alfa Talasemias- -/- - Alfa talasemia mayor

Alfa Talasemias- -/- - Alfa talasemia mayor

Alfa TalasemiasDiagnósticoHemograma: cursan sin anemia pero el VCM estásiempre disminuído, aún en la deleción 3,7 heterocigota.La morfología es variable y característica para algunas formas de Th. Sólo las formas -/ - y las - -/ presenten morfología más notoria, sin PB, y a veces es posible detectar un cuerpo de inclusión cada varios campos.En los casos leves es imprescindible efectuar la biología molecular, ya que el estudio de Hb generalmente es normal

1,76 kpb0,5 kpb
1,76 kpb0,5 kpb
1 y 2 control Heterocigosis - 3.7
3 y 8 marcador 100 bp ladder4 y 5 Cordón homocigota - 3.7
6 y 7 Padre - 9 y 10 Madre.
Deleción -3.7Deleción -3.7
ASO-PCR

Alfa Talasemias.-/- homocigota + - -/ heterocigota º
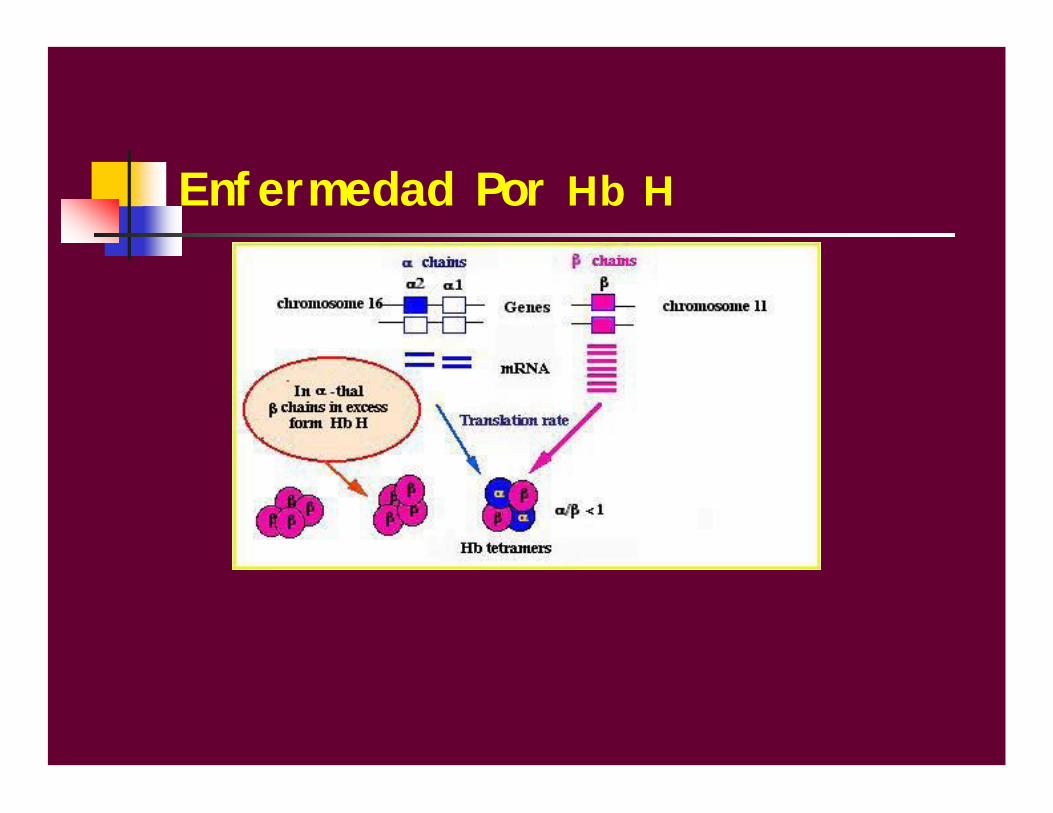
Enfermedad Por Hb H
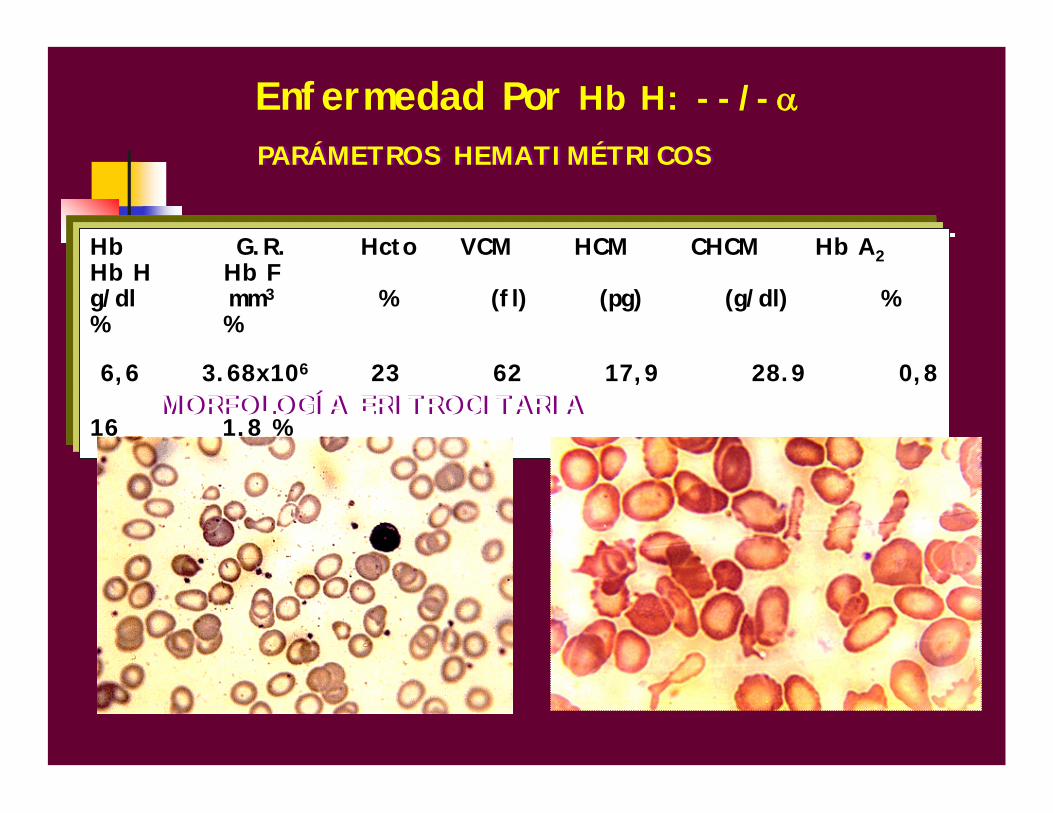
Hb G.R. Hcto VCM HCM CHCM Hb A2Hb H Hb Fg/dl mm3 % (fl) (pg) (g/dl) % % %
6,6 3.68x106 23 62 17,9 28.9 0,8
16 1,8 %MORFOLOGÍA ERITROCITARIAMORFOLOGÍA ERITROCITARIA
PARÁMETROS HEMATIMÉTRICOSPARÁMETROS HEMATIMÉTRICOS
Enfermedad Por Hb H: --/-

ENFERMEDAD POR Hb H

Beta-Talasemia
ASOCIACIÓN DE -TALASEMIA HETEROCIGOTA CON DELECIÓN -3.7
GR
(1012/l)
Hb
(g/dl)
Hto
(%)
VCM
(fl)
HCM
(pg)
CHCM
(g/dl)
4.9 11,2 37 75 22,8 30,3
1 2 3 4 5 6
1,76 KB
039 / -3,7

Beta-Talasemia
ASOCIACIÓN DE -TALASEMIA HETEROCIGOTA CON TRIPLICACIÓN DE GENES ( anti 3,7)
GR
(1012/l)
Hb
(g/dl)
Hto
(%)
VCM
(fl)
HCM
(pg)
CHCM
(g/dl)
(a) 5,7 8,8 28,5 51 15,8 30,9
(b) 5,0 8,6 27 53 17,1 32,2
(a) + II-745/ anti 3,7 (b) 0 39/ anti 3,7

Sindromes talasémicos(pacientes heterocigotas)
Fenotipo (adulto) Genotipo Base molecularVCM A2 F deleción mutación
tal N + 0 + +++ tal N N + 0 +++ + tal N N + 0 + ++ tal N N 0 + -- tal ()0 (A)0 + --tal (LCR )0 + --
(LCR)0
PHHF N N -- +N N ()0 + +


Muñoz Rojas et. al. Protocolo diagnóstico anemias microcíticasMedicine 2008; 10(20):1363-5

HEMATHEMATÓÓLOGOLOGODiagnDiagnóósticostico
Tratamiento y PrevenciTratamiento y Prevencióónn
Banco de SangreBanco de SangreTransfusiTransfusióónnTratamiento con Tratamiento con DesferalDesferaluuotros medicamentos otros medicamentos VacunasVacunas
LaboratorioLaboratorio
Convencional InvestigaciConvencional Investigacióónn
MEDICOS ESPECIALISTASMEDICOS ESPECIALISTAS(Tratamiento y prevenci(Tratamiento y prevencióón)n)EndocrinEndocrinóólogologoCardiCardióólogologoOftalmOftalmóólogologoPsicPsicóólogologo
Apoyo MApoyo MéédicodicoEnfermeras EspecializadasEnfermeras EspecializadasDietistaDietistaDentistaDentista
ABORDAJE MÉDICO (Unidades de Atención a pacientes con Talasemia)
GenetistaGenetista(Asesoramiento Gen(Asesoramiento Genéético)tico) CIBO-GENETICA
2004

MUCHAS GRACIAS





























