Embed Size (px)
Citation preview

Published Ahead of Print 23 November 2011. 2012, 86(3):1758. DOI: 10.1128/JVI.05666-11. J. Virol.
Covadonga AlonsoRaquel Muñoz-Moreno, Miguel A. Cuesta-Geijo and Jose I. Quetglas, Bruno Hernáez, Inmaculada Galindo, African Swine Fever Virus InfectionBiosynthetic Pathway Intermediates in Small Rho GTPases and Cholesterol
http://jvi.asm.org/content/86/3/1758Updated information and services can be found at:
These include:
REFERENCEShttp://jvi.asm.org/content/86/3/1758#ref-list-1at:
This article cites 66 articles, 35 of which can be accessed free
CONTENT ALERTS more»articles cite this article),
Receive: RSS Feeds, eTOCs, free email alerts (when new
http://jvi.asm.org/site/misc/reprints.xhtmlInformation about commercial reprint orders: http://journals.asm.org/site/subscriptions/To subscribe to to another ASM Journal go to:
on January 12, 2012 by guesthttp://jvi.asm
.org/D
ownloaded from

Small Rho GTPases and Cholesterol Biosynthetic PathwayIntermediates in African Swine Fever Virus Infection
Jose I. Quetglas,* Bruno Hernáez, Inmaculada Galindo, Raquel Muñoz-Moreno, Miguel A. Cuesta-Geijo, and Covadonga Alonso
Departamento de Biotecnología–Instituto Nacional de Investigación y Tecnología Agraria y Alimentaria, INIA, Madrid, Spain
The integrity of the cholesterol biosynthesis pathway is required for efficient African swine fever virus (ASFV) infection. Incor-poration of prenyl groups into Rho GTPases plays a key role in several stages of ASFV infection, since both geranylgeranyl andfarnesyl pyrophosphates are required at different infection steps. We found that Rho GTPase inhibition impaired virus morpho-genesis and resulted in an abnormal viral factory size with the accumulation of envelope precursors and immature virions. Fur-thermore, abundant defective virions reached the plasma membrane, and filopodia formation in exocytosis was abrogated. Rac1was activated at early ASFV infection stages, coincident with microtubule acetylation, a process that stabilizes microtubules forvirus transport. Rac1 inhibition did not affect the viral entry step itself but impaired subsequent virus production. We foundthat specific Rac1 inhibition impaired viral induced microtubule acetylation and viral intracellular transport. These findingshighlight that viral infection is the result of a carefully orchestrated modulation of Rho family GTPase activity within the hostcell; this modulation results critical for virus morphogenesis and in turn, triggers cytoskeleton remodeling, such as microtubulestabilization for viral transport during early infection.
The members of Rho family of small GTPases are essential keyregulators of diverse critical cellular functions, including cyto-
skeleton dynamics, cell cycle progression, migration, the genera-tion of reactive oxygen species, and gene expression (16, 29, 35,53). Like the majority of Ras superfamily proteins, most RhoGTPases function as molecular switches and cycle between an ac-tive GTP-bound form and an inactive GDP-bound one. Two typesof regulatory proteins control this cycling: guanine nucleotide ex-change factors (GEFs) that promote activation of these proteinsduring signal transduction by exchanging of GDP for GTP mole-cules and, in contrast, GTPase-activating proteins (GAPs) thatpromote the hydrolysis of the bound GTP molecules, thus allow-ing the transfer of the GTPase back to the inactive state (7, 9). Inaddition, Rho GTPases must often undergo posttranslational pre-nylation to become functionally active (43). Thus, their localiza-tion, insertion into membranes, and protein-protein interactionsrequire covalent incorporation into the carboxy terminus of eitherfarnesyl pyrophosphate (FPP) or geranylgeranyl pyrophosphate(GGPP) (62). Since these prenyl groups are derived from meval-onic acid, which is also the starting material for the cholesterolbiosynthesis, statins have been widely used to inhibit the prenyla-tion of Ras-related proteins, particularly the Rho GTPase subfam-ily (24, 28, 41).
Given the control that the most studied Rho GTPase members(RhoA, Rac1, and Cdc42) exert over cytoskeleton dynamics, ves-icle trafficking, and signaling pathways, it has been hypothesizedthat they make a major contribution to viral entry, replication,and morphogenesis. In this regard, Rac1/Cdc42 regulates actindynamics and architecture during macropinocytotic entry of di-verse large DNA viruses, such as vaccinia virus (42, 45) and ade-novirus (40). In addition, during entry into host cells, herpes sim-plex virus 1 (HSV-1) activates Rac1 and Cdc42, which results inthe induction of filopodia and lamellipodia in epithelial cells andfibroblasts (33). Rho GTPases are also implicated in microtubuleregulation during capsid trafficking of Kaposi’s sarcoma-associated herpesvirus (48). Recently, it has been shown that vac-cinia virus F11L protein interacts directly with RhoA to inhibit its
downstream signaling (61). This F11L-mediated inhibition ofRhoA signaling has been proposed to be required for an efficientvirus release from infected cells (4) and also for stimulating virus-induced cell motility (4, 12, 66) and the spreading of infection.RhoA signaling is required for respiratory syncytial virus replica-tion and morphogenesis (26). Moreover, the expression of activeRac1 is increased after hepatitis B virus replication (59).
African swine fever virus (ASFV) is the causative agent of asevere and highly lethal hemorrhagic disease that affects domesticpigs. This large icosahedral and enveloped DNA virus is the onlyknown member of the family Asfarviridae (17). It enters host cellsby clathrin- and dynamin-dependent endocytosis after attach-ment to a still unknown cell receptor(s) and requires later fusionbetween the viral envelope and endosome membrane to deliverDNA into cytoplasm (31, 60). This fusion event requires the char-acteristic acidic pH of the endosomal environment and also thepresence of cholesterol at the plasma membrane of the target cell(6, 22). Like many other viruses, during the early stages of infec-tion ASFV interacts with the microtubule cytoskeleton and re-quires retrograde dynein-based transport to constitute the peri-nuclear virus factory (3, 30), where DNA replication and assemblyoccur. This specialized site, close to the microtubule organizingcenter, contains mostly viral DNA, most of the viral proteins, im-mature and mature virions, and also abundant virus-inducedmembranes. Microtubule motors have also been proposed to beinvolved in at least three other events that occur in the ASFVreplication cycle, namely, vimentin rearrangement into a cage that
Received 13 July 2011 Accepted 8 November 2011
Published ahead of print 23 November 2011
Address correspondence to C. Alonso, [email protected].
* Present address: Division of Gene Therapy, School of Medicine, Center forApplied Medical Research, University of Navarre, Pamplona, Spain.
Copyright © 2012, American Society for Microbiology. All Rights Reserved.
doi:10.1128/JVI.05666-11
1758 jvi.asm.org 0022-538X/12/$12.00 Journal of Virology p. 1758–1767
on January 12, 2012 by guesthttp://jvi.asm
.org/D
ownloaded from

finally surrounds the viral factory (57), the recruitment of virus-targeted membranes to the virus factory (54), and the transport offully assembled virions to the plasma membrane before their re-lease from an infected cell by budding (36).
Here we explored, for the first time, the relevance of the RhoGTPase subfamily during ASFV infection. We found that a gen-eral inhibition of Rho GTPases leads to a decrease in viral yieldsand deficient viral morphogenesis. Most importantly, since Rac1resulted activated early during infection and coincident with anincrease in microtubule acetylation, the data obtained suggest theinvolvement of Rac1-mediated signaling on ASFV intracellulartransport.
MATERIALS AND METHODSCell culture, viruses, and plasmids. Vero cells were obtained from theEuropean Collection of Cell Cultures (ECACC) and maintained in Dul-becco modified Eagle medium supplemented with 5% fetal bovine serumat 37°C and 5% CO2. The tissue culture-adapted ASFV strain BA71V wasused in all experiments (18). Where indicated, a recombinant virus ex-pressing viral protein p54 fused to the green fluorescence protein (GFP)was used (B54GFP-2 [32]) to identify viral factories by confocal micros-copy. Cell infections and titrations were carried out as previously de-scribed. When indicated, we used highly purified ASFV as described else-where (10).
Plasmids (pcDNA3) encoding wild-type (wt), dominant-negative(17N), and constitutively active (Q61L) forms of Rac1 fused to GFP werekindly provided by Ole Gjoerup (58). Constructs expressing the glutathi-one S-transferase (GST)-Rac1/Cdc42 binding domain of Pak1 (GST-PBD) were a generous gift from Keith Burridge (5). Transient expressionin Vero cells was achieved by transfection using Fugene HD transfectionreagent (Roche) and 2 �g of DNA/106 cells (ratio 1:6) according to themanufacturer’s instructions.
Antibodies and other reagents. Antibodies against Cdc42 and Rac1were obtained from BD Transduction Laboratories. Monoclonal antibod-ies against �-tubulin and acetylated �-tubulin were purchased fromSigma. Jose M. Escribano provided the monoclonal antibody against p30.The monoclonal antibody against p72 clone 18BG3 was purchased fromIngenasa. Rabbit polyclonal serum against ASFV protein pE120R was ob-tained after immunization with recombinant protein. Anti-rabbit andanti-mouse antibodies conjugated to horseradish peroxidase were pur-chased from GE Healthcare. Alexa Fluor 488- and Alexa Fluor 594-conjugated anti-mouse IgG antibodies were from Molecular Probes.
Triton X-100, lovastatin (Mevinolin), farnesyl pyrophosphate (FPP),geranylgeranyl pyrophosphate (GGPP), L-mevalonate, cholesterol,�-actin polyclonal antibody, and Hoechst 33258 were purchased fromSigma. Clostridium difficile toxin B (CdTB), FTI-277 (farnesyltransferaseinhibitor [FTase] inhibitor), GGTI-286 (geranylgeranyltransferase inhib-itor [GGTI]), NSC23766 (Rac1 inhibitor), and Fluorsave were obtainedfrom Calbiochem.
Cytotoxicity assay. The cytotoxicity elicited by inhibitors was ana-lyzed by using a lactate dehydrogenase cytotoxicity assay kit (Promega)and by trypan blue exclusion.
Drug treatments and infections. Vero cells were treated with a rangeof concentrations (1 to 3 �M) of lovastatin (Lov) for 24 or 48 h at 37°C.Add-back of intermediate metabolites was done at the following concen-trations: 200 �M mevalonate (Mev), 5 �M GGPP, 5 �M FPP, or 5 �g ofcholesterol/ml, together with 1 to 3 �M Lov. In all cases, at 24 h after ASFVinfection (BA71V isolate, 0.5 PFU/cell), cells and supernatants were col-lected to determine virus progeny production by plaque assay, as previ-ously described (25). In a second set of experiments, the cells were incu-bated with a range of concentrations of GGTI-286 (10 to 30 �M), FTI-277(10 to 30 �M), or CdTB (25 to 100 ng/ml). These chemical inhibitors wereadded either 2 h before virus inoculation and removed after viral adsorp-tion or maintained throughout infection, or they were added at 3 h postin-
fection (hpi) and maintained until the end of the experiment. In order toevaluate the infectivity of ASFV in the presence of these drugs, we infectedpretreated cells (2 h before virus inoculation for GGTI-286, FTI-277, orCdTB or 24 and 48 h before virus inoculation when using Lov) and ana-lyzed the number of infected cells at 3 hpi by immunofluorescence detec-tion of viral early protein p30 (see the discussion of immunofluorescenceanalysis). Where indicated, cells were infected at high multiplicity of in-fection (MOI), i.e., �20 PFU/cell.
GTPase activation assays. Serum-starved Vero cells (60% conflu-ence), infected or mock infected with ASFV at an MOI of 5 PFU/cell, werewashed with phosphate-buffered saline (PBS) at a range of times postin-fection and lysed in a buffer containing 10% glycerol, 50 mM Tris-HCl(pH 7.4), 100 mM NaCl, 1% NP-40, 2 mM MgCl2, and protease inhibi-tors.
Bacterially expressed and purified GST-PBD (p21-binding domain ofPak1) interacts with activated GTP-bound Rac1 and Cdc42 was used topull-down GTP-bound forms of Rac1 and Cdc42, as described previously(15, 55). Equal amounts of total protein from cell lysates were incubatedfor 1 h at 4°C with glutathione-Sepharose 4B beads (GE Healthcare) pre-viously conjugated to GST-PBD. After extensive washing, bound proteinswere analyzed by Western blotting with monoclonal antibodies againstRac1 or Cdc42. Previously, lysates were probed with the same antibodiesto detect total Rac1 and Cdc42 in the samples. To determine the levels ofactive GTPases, the intensities of the corresponding bands were measuredby densitometry (Tina 2.0) and normalized to values obtained with mock-infected cells.
RhoA, Rac1, and Cdc42 activation was also measured using RhoA-,Rac1-, and Cdc42-specific G-Lisa activation kits (Cytoskeleton, Inc.) ac-cording to the manufacturer’s instructions. Briefly, Vero cells were seededin 25-cm2 flasks for the indicated confluence and serum starved for 24 hprior to BA71V infection (5 PFU/cell). Mock-infected and infected cellswere harvested in lysis buffer at the indicated times postinfection. G-Lisakits used 96-well plates coated with the binding domain of the corre-sponding GTPase effector protein. GTPase-GDP was removed duringwashing steps, and GTPase-GTP was detected with specific antibodies,followed by absorbance at 490 nm.
Detection and quantitation of the ASFV genome. DNA from infectedor mock-infected Vero cells with ASFV 0.5 PFU/cell was extracted andpurified with a DNeasy blood and tissue kit (Qiagen) at 16 hpi. Detectionand quantitation of the ASFV genome was achieved by quantitative real-time PCR using specific oligonucleotides and a TaqMan probe, as previ-ously described (38). The amplification reaction was performed in aRotor-Gene RG3000 (Corbett Research) as follows: 1 cycle at 94°C for 10min, then 45 cycles at 94°C for 15 s, and finally 45 cycles at 58°C for 1 min.Positive and negative amplification controls (DNA purified from ASFVvirions and DNA from mock-infected cells, respectively) were included inthe assay, and duplicates from each sample were analyzed.
Western blot analysis. After estimation of total protein in samples bythe Bradford method, 30 �g of protein was resolved by SDS-PAGE andtransferred to nitrocellulose membranes (Bio-Rad). Membranes wereprobed for 1 h at room temperature with the corresponding primaryantibodies in PBS containing 0.05% Tween 20 (PBS-T; Sigma) at thefollowing dilutions: monoclonal antibody against p30 (1:500), monoclo-nal antibody anti-p72 (1:5000), mouse monoclonal antibody anti-Rac1(1:1,000), mouse monoclonal antibody anti-Cdc42 (1:250), mousemonoclonal antibodies anti-�-tubulin (1:4,000), and anti-acetylated�-tubulin (1:2,000). As a protein loading control, we included the detec-tion of actin with polyclonal rabbit anti-�-actin serum diluted 1:250. Af-ter an extensive washing with PBS-T, membranes were incubated withanti-mouse IgG antibody (1:5,000) or anti-rabbit IgG (1:4,000), both con-jugated to horseradish peroxidase. Finally, bands were developed by usingan enhanced chemiluminescence reaction with a Western blot detectionreagent (GE Healthcare).
Immunofluorescence analysis. Vero cells (60% confluence) weregrown on glass coverslips and then infected with BA71V or B54GFP-2. In
Role of Rho GTPases during ASFV Infection
February 2012 Volume 86 Number 3 jvi.asm.org 1759
on January 12, 2012 by guesthttp://jvi.asm
.org/D
ownloaded from
infectivity assays, at 3 hpi, the cells were rinsed with PBS, fixed with PBS–3.8% paraformaldehyde for 10 min, and permeabilized with 0.2% PBS–Triton X-100 for 15 min. Infected cells at this time were identified bydetection of early ASFV protein with monoclonal anti-p30 antibody (1:200). For the viral factory analysis, cells infected with B54GFP-2 werefixed at 16 hpi. Internalized virions in pCDNA-3-transfected cells weredetected at 1 hpi with monoclonal antiviral major capsid protein p72(1:1,000). Rabbit polyclonal serum against pE120R was diluted 1:500. Thesecondary antibodies used were an anti-mouse IgG antibody conjugatedto either Alexa Fluor 488 or Alexa Fluor 549. Nuclei and also DNA in virusfactories were identified by staining with Hoechst 33258. Finally, cover-slips were mounted onto slides using Fluorsave reagent.
Confocal microscopy was carried out in a Leica confocal microscopeTCS SP2-AOBS equipped with �63 and �100 objective lenses. The digitalimages were processed with Adobe Photoshop 8.0.
Electron microscopy. For conventional Epon section analysis, Verocells, preincubated or not with toxin B, were infected with ASFV at 1PFU/cell and fixed at 16 hpi with 2.5% glutaraldehyde (Sigma) in PBS for30 min at room temperature. Postfixation was carried out with 1% OsO4
in PBS at room temperature for 90 min. The samples were then dehy-drated with acetone and embedded in Epon according to standardprocedures. Samples were examined at 80 kV in a JEOL JEM 10-10electron microscope. The digital images were processed with AdobePhotoshop 8.0.
Statistical analysis. All error terms are expressed as standard devia-tions (SD). Prism software (GraphPad Software, Inc., San Diego, CA) wasused for the statistical analysis. A one-way analysis of variance test, fol-lowed by Bonferroni’s multiple-comparison test, was used to comparedifferent experimental groups. P values of �0.05 were considered statis-tically significant.
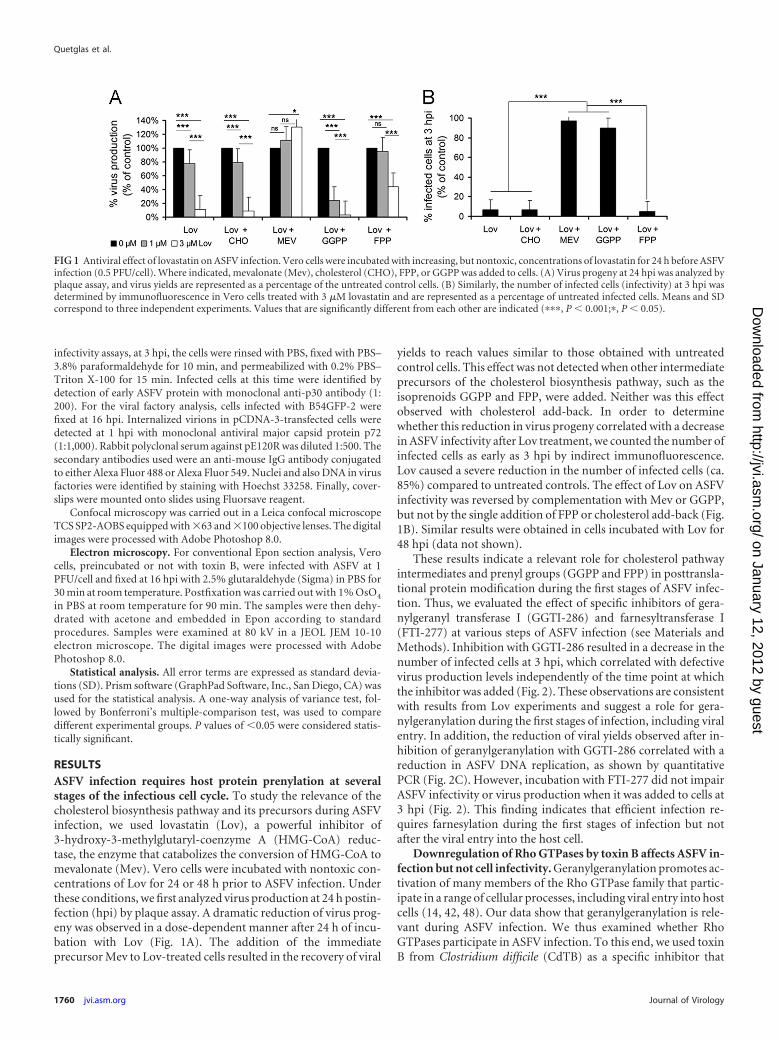
RESULTSASFV infection requires host protein prenylation at severalstages of the infectious cell cycle. To study the relevance of thecholesterol biosynthesis pathway and its precursors during ASFVinfection, we used lovastatin (Lov), a powerful inhibitor of3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reduc-tase, the enzyme that catabolizes the conversion of HMG-CoA tomevalonate (Mev). Vero cells were incubated with nontoxic con-centrations of Lov for 24 or 48 h prior to ASFV infection. Underthese conditions, we first analyzed virus production at 24 h postin-fection (hpi) by plaque assay. A dramatic reduction of virus prog-eny was observed in a dose-dependent manner after 24 h of incu-bation with Lov (Fig. 1A). The addition of the immediateprecursor Mev to Lov-treated cells resulted in the recovery of viral
yields to reach values similar to those obtained with untreatedcontrol cells. This effect was not detected when other intermediateprecursors of the cholesterol biosynthesis pathway, such as theisoprenoids GGPP and FPP, were added. Neither was this effectobserved with cholesterol add-back. In order to determinewhether this reduction in virus progeny correlated with a decreasein ASFV infectivity after Lov treatment, we counted the number ofinfected cells as early as 3 hpi by indirect immunofluorescence.Lov caused a severe reduction in the number of infected cells (ca.85%) compared to untreated controls. The effect of Lov on ASFVinfectivity was reversed by complementation with Mev or GGPP,but not by the single addition of FPP or cholesterol add-back (Fig.1B). Similar results were obtained in cells incubated with Lov for48 hpi (data not shown).
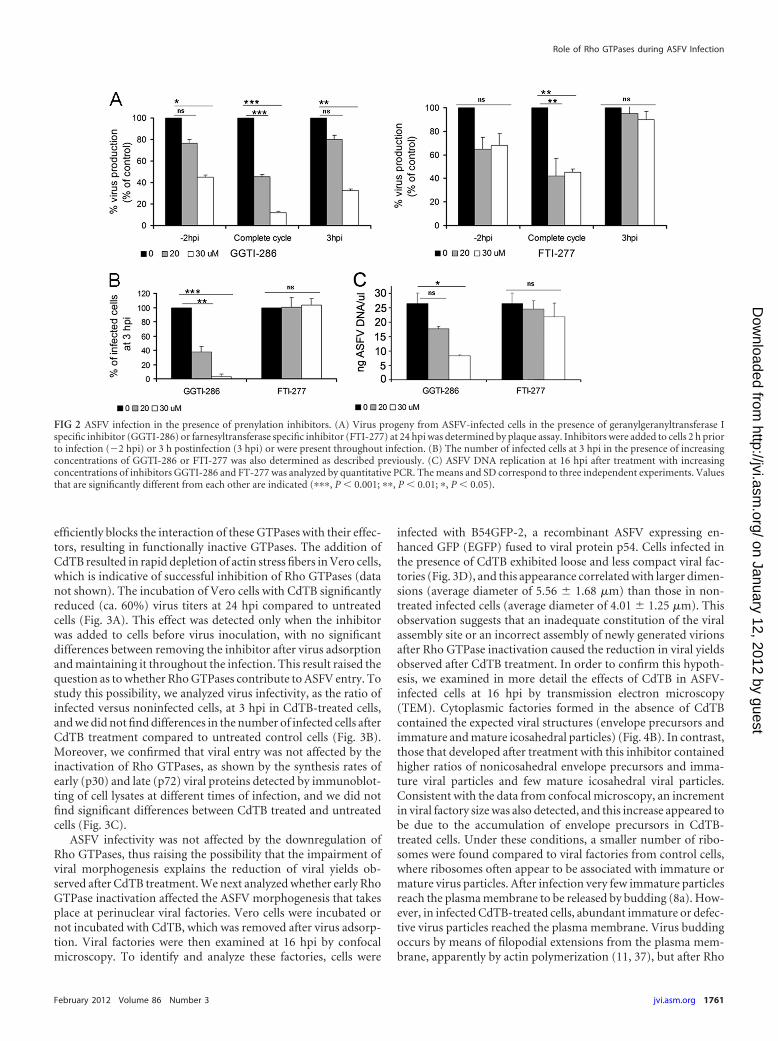
These results indicate a relevant role for cholesterol pathwayintermediates and prenyl groups (GGPP and FPP) in posttransla-tional protein modification during the first stages of ASFV infec-tion. Thus, we evaluated the effect of specific inhibitors of gera-nylgeranyl transferase I (GGTI-286) and farnesyltransferase I(FTI-277) at various steps of ASFV infection (see Materials andMethods). Inhibition with GGTI-286 resulted in a decrease in thenumber of infected cells at 3 hpi, which correlated with defectivevirus production levels independently of the time point at whichthe inhibitor was added (Fig. 2). These observations are consistentwith results from Lov experiments and suggest a role for gera-nylgeranylation during the first stages of infection, including viralentry. In addition, the reduction of viral yields observed after in-hibition of geranylgeranylation with GGTI-286 correlated with areduction in ASFV DNA replication, as shown by quantitativePCR (Fig. 2C). However, incubation with FTI-277 did not impairASFV infectivity or virus production when it was added to cells at3 hpi (Fig. 2). This finding indicates that efficient infection re-quires farnesylation during the first stages of infection but notafter the viral entry into the host cell.
Downregulation of Rho GTPases by toxin B affects ASFV in-fection but not cell infectivity. Geranylgeranylation promotes ac-tivation of many members of the Rho GTPase family that partic-ipate in a range of cellular processes, including viral entry into hostcells (14, 42, 48). Our data show that geranylgeranylation is rele-vant during ASFV infection. We thus examined whether RhoGTPases participate in ASFV infection. To this end, we used toxinB from Clostridium difficile (CdTB) as a specific inhibitor that
FIG 1 Antiviral effect of lovastatin on ASFV infection. Vero cells were incubated with increasing, but nontoxic, concentrations of lovastatin for 24 h before ASFVinfection (0.5 PFU/cell). Where indicated, mevalonate (Mev), cholesterol (CHO), FPP, or GGPP was added to cells. (A) Virus progeny at 24 hpi was analyzed byplaque assay, and virus yields are represented as a percentage of the untreated control cells. (B) Similarly, the number of infected cells (infectivity) at 3 hpi wasdetermined by immunofluorescence in Vero cells treated with 3 �M lovastatin and are represented as a percentage of untreated infected cells. Means and SDcorrespond to three independent experiments. Values that are significantly different from each other are indicated (���, P � 0.001;�, P � 0.05).
Quetglas et al.
1760 jvi.asm.org Journal of Virology
on January 12, 2012 by guesthttp://jvi.asm
.org/D
ownloaded from
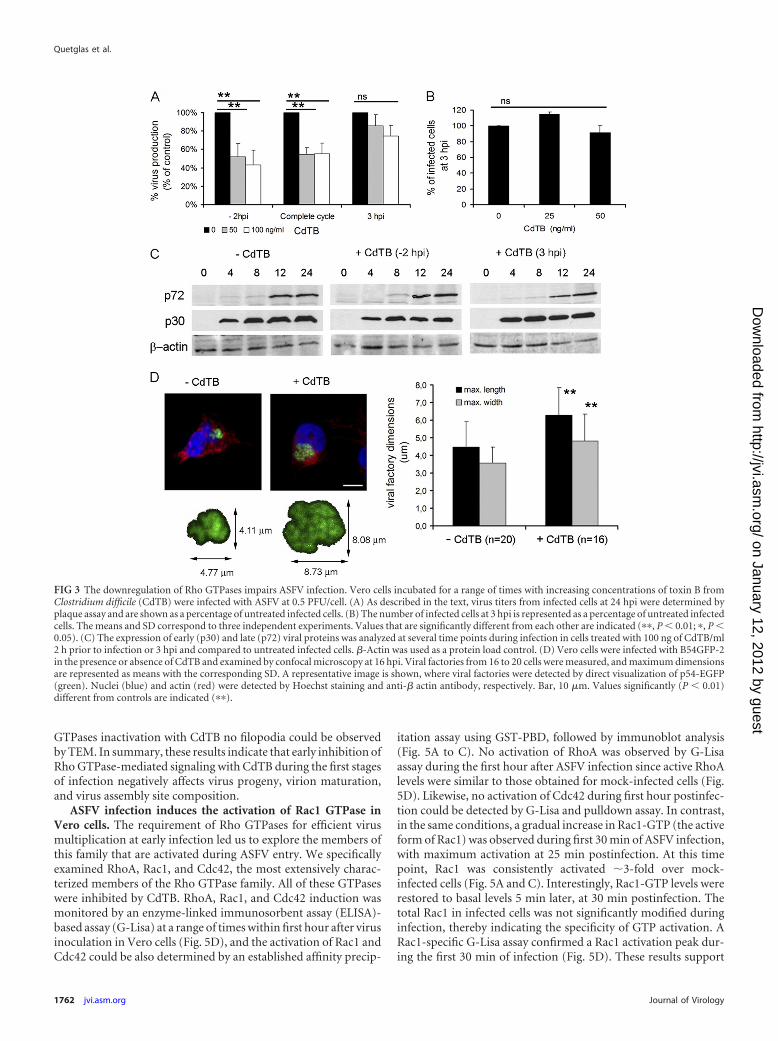
efficiently blocks the interaction of these GTPases with their effec-tors, resulting in functionally inactive GTPases. The addition ofCdTB resulted in rapid depletion of actin stress fibers in Vero cells,which is indicative of successful inhibition of Rho GTPases (datanot shown). The incubation of Vero cells with CdTB significantlyreduced (ca. 60%) virus titers at 24 hpi compared to untreatedcells (Fig. 3A). This effect was detected only when the inhibitorwas added to cells before virus inoculation, with no significantdifferences between removing the inhibitor after virus adsorptionand maintaining it throughout the infection. This result raised thequestion as to whether Rho GTPases contribute to ASFV entry. Tostudy this possibility, we analyzed virus infectivity, as the ratio ofinfected versus noninfected cells, at 3 hpi in CdTB-treated cells,and we did not find differences in the number of infected cells afterCdTB treatment compared to untreated control cells (Fig. 3B).Moreover, we confirmed that viral entry was not affected by theinactivation of Rho GTPases, as shown by the synthesis rates ofearly (p30) and late (p72) viral proteins detected by immunoblot-ting of cell lysates at different times of infection, and we did notfind significant differences between CdTB treated and untreatedcells (Fig. 3C).
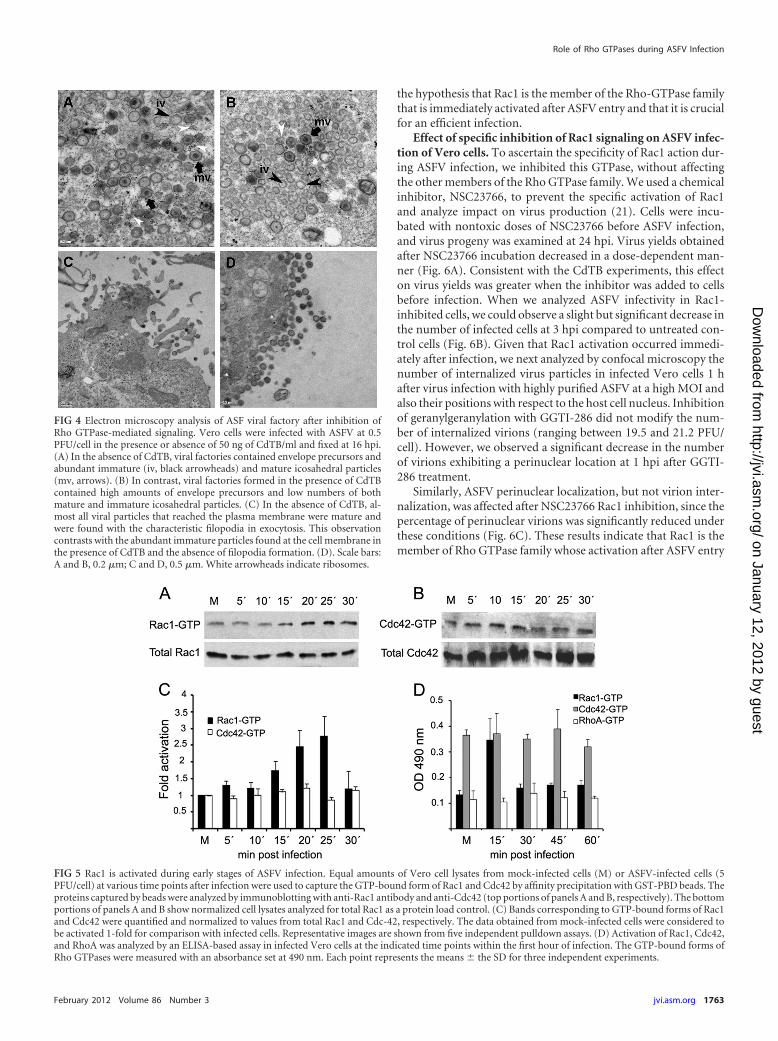
ASFV infectivity was not affected by the downregulation ofRho GTPases, thus raising the possibility that the impairment ofviral morphogenesis explains the reduction of viral yields ob-served after CdTB treatment. We next analyzed whether early RhoGTPase inactivation affected the ASFV morphogenesis that takesplace at perinuclear viral factories. Vero cells were incubated ornot incubated with CdTB, which was removed after virus adsorp-tion. Viral factories were then examined at 16 hpi by confocalmicroscopy. To identify and analyze these factories, cells were
infected with B54GFP-2, a recombinant ASFV expressing en-hanced GFP (EGFP) fused to viral protein p54. Cells infected inthe presence of CdTB exhibited loose and less compact viral fac-tories (Fig. 3D), and this appearance correlated with larger dimen-sions (average diameter of 5.56 � 1.68 �m) than those in non-treated infected cells (average diameter of 4.01 � 1.25 �m). Thisobservation suggests that an inadequate constitution of the viralassembly site or an incorrect assembly of newly generated virionsafter Rho GTPase inactivation caused the reduction in viral yieldsobserved after CdTB treatment. In order to confirm this hypoth-esis, we examined in more detail the effects of CdTB in ASFV-infected cells at 16 hpi by transmission electron microscopy(TEM). Cytoplasmic factories formed in the absence of CdTBcontained the expected viral structures (envelope precursors andimmature and mature icosahedral particles) (Fig. 4B). In contrast,those that developed after treatment with this inhibitor containedhigher ratios of nonicosahedral envelope precursors and imma-ture viral particles and few mature icosahedral viral particles.Consistent with the data from confocal microscopy, an incrementin viral factory size was also detected, and this increase appeared tobe due to the accumulation of envelope precursors in CdTB-treated cells. Under these conditions, a smaller number of ribo-somes were found compared to viral factories from control cells,where ribosomes often appear to be associated with immature ormature virus particles. After infection very few immature particlesreach the plasma membrane to be released by budding (8a). How-ever, in infected CdTB-treated cells, abundant immature or defec-tive virus particles reached the plasma membrane. Virus buddingoccurs by means of filopodial extensions from the plasma mem-brane, apparently by actin polymerization (11, 37), but after Rho
FIG 2 ASFV infection in the presence of prenylation inhibitors. (A) Virus progeny from ASFV-infected cells in the presence of geranylgeranyltransferase Ispecific inhibitor (GGTI-286) or farnesyltransferase specific inhibitor (FTI-277) at 24 hpi was determined by plaque assay. Inhibitors were added to cells 2 h priorto infection (�2 hpi) or 3 h postinfection (3 hpi) or were present throughout infection. (B) The number of infected cells at 3 hpi in the presence of increasingconcentrations of GGTI-286 or FTI-277 was also determined as described previously. (C) ASFV DNA replication at 16 hpi after treatment with increasingconcentrations of inhibitors GGTI-286 and FT-277 was analyzed by quantitative PCR. The means and SD correspond to three independent experiments. Valuesthat are significantly different from each other are indicated (���, P � 0.001; ��, P � 0.01; �, P � 0.05).
Role of Rho GTPases during ASFV Infection
February 2012 Volume 86 Number 3 jvi.asm.org 1761
on January 12, 2012 by guesthttp://jvi.asm
.org/D
ownloaded from
GTPases inactivation with CdTB no filopodia could be observedby TEM. In summary, these results indicate that early inhibition ofRho GTPase-mediated signaling with CdTB during the first stagesof infection negatively affects virus progeny, virion maturation,and virus assembly site composition.
ASFV infection induces the activation of Rac1 GTPase inVero cells. The requirement of Rho GTPases for efficient virusmultiplication at early infection led us to explore the members ofthis family that are activated during ASFV entry. We specificallyexamined RhoA, Rac1, and Cdc42, the most extensively charac-terized members of the Rho GTPase family. All of these GTPaseswere inhibited by CdTB. RhoA, Rac1, and Cdc42 induction wasmonitored by an enzyme-linked immunosorbent assay (ELISA)-based assay (G-Lisa) at a range of times within first hour after virusinoculation in Vero cells (Fig. 5D), and the activation of Rac1 andCdc42 could be also determined by an established affinity precip-
itation assay using GST-PBD, followed by immunoblot analysis(Fig. 5A to C). No activation of RhoA was observed by G-Lisaassay during the first hour after ASFV infection since active RhoAlevels were similar to those obtained for mock-infected cells (Fig.5D). Likewise, no activation of Cdc42 during first hour postinfec-tion could be detected by G-Lisa and pulldown assay. In contrast,in the same conditions, a gradual increase in Rac1-GTP (the activeform of Rac1) was observed during first 30 min of ASFV infection,with maximum activation at 25 min postinfection. At this timepoint, Rac1 was consistently activated �3-fold over mock-infected cells (Fig. 5A and C). Interestingly, Rac1-GTP levels wererestored to basal levels 5 min later, at 30 min postinfection. Thetotal Rac1 in infected cells was not significantly modified duringinfection, thereby indicating the specificity of GTP activation. ARac1-specific G-Lisa assay confirmed a Rac1 activation peak dur-ing the first 30 min of infection (Fig. 5D). These results support
FIG 3 The downregulation of Rho GTPases impairs ASFV infection. Vero cells incubated for a range of times with increasing concentrations of toxin B fromClostridium difficile (CdTB) were infected with ASFV at 0.5 PFU/cell. (A) As described in the text, virus titers from infected cells at 24 hpi were determined byplaque assay and are shown as a percentage of untreated infected cells. (B) The number of infected cells at 3 hpi is represented as a percentage of untreated infectedcells. The means and SD correspond to three independent experiments. Values that are significantly different from each other are indicated (��, P � 0.01; �, P �0.05). (C) The expression of early (p30) and late (p72) viral proteins was analyzed at several time points during infection in cells treated with 100 ng of CdTB/ml2 h prior to infection or 3 hpi and compared to untreated infected cells. �-Actin was used as a protein load control. (D) Vero cells were infected with B54GFP-2in the presence or absence of CdTB and examined by confocal microscopy at 16 hpi. Viral factories from 16 to 20 cells were measured, and maximum dimensionsare represented as means with the corresponding SD. A representative image is shown, where viral factories were detected by direct visualization of p54-EGFP(green). Nuclei (blue) and actin (red) were detected by Hoechst staining and anti-� actin antibody, respectively. Bar, 10 �m. Values significantly (P � 0.01)different from controls are indicated (��).
Quetglas et al.
1762 jvi.asm.org Journal of Virology
on January 12, 2012 by guesthttp://jvi.asm
.org/D
ownloaded from
the hypothesis that Rac1 is the member of the Rho-GTPase familythat is immediately activated after ASFV entry and that it is crucialfor an efficient infection.
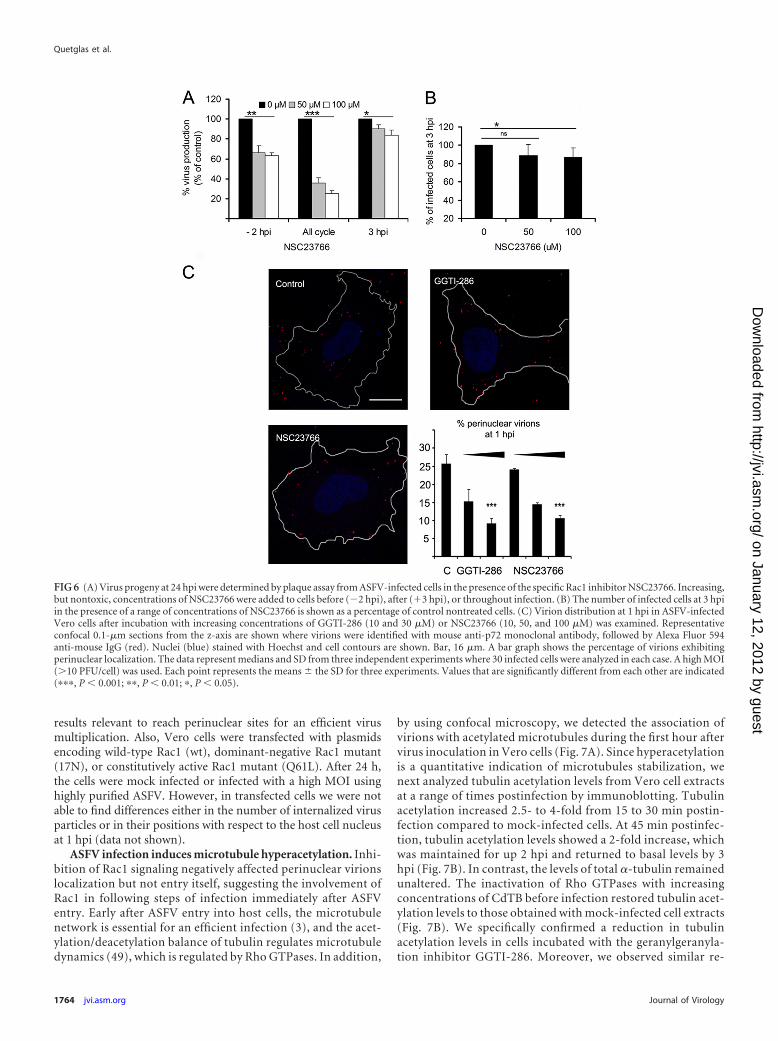
Effect of specific inhibition of Rac1 signaling on ASFV infec-tion of Vero cells. To ascertain the specificity of Rac1 action dur-ing ASFV infection, we inhibited this GTPase, without affectingthe other members of the Rho GTPase family. We used a chemicalinhibitor, NSC23766, to prevent the specific activation of Rac1and analyze impact on virus production (21). Cells were incu-bated with nontoxic doses of NSC23766 before ASFV infection,and virus progeny was examined at 24 hpi. Virus yields obtainedafter NSC23766 incubation decreased in a dose-dependent man-ner (Fig. 6A). Consistent with the CdTB experiments, this effecton virus yields was greater when the inhibitor was added to cellsbefore infection. When we analyzed ASFV infectivity in Rac1-inhibited cells, we could observe a slight but significant decrease inthe number of infected cells at 3 hpi compared to untreated con-trol cells (Fig. 6B). Given that Rac1 activation occurred immedi-ately after infection, we next analyzed by confocal microscopy thenumber of internalized virus particles in infected Vero cells 1 hafter virus infection with highly purified ASFV at a high MOI andalso their positions with respect to the host cell nucleus. Inhibitionof geranylgeranylation with GGTI-286 did not modify the num-ber of internalized virions (ranging between 19.5 and 21.2 PFU/cell). However, we observed a significant decrease in the numberof virions exhibiting a perinuclear location at 1 hpi after GGTI-286 treatment.
Similarly, ASFV perinuclear localization, but not virion inter-nalization, was affected after NSC23766 Rac1 inhibition, since thepercentage of perinuclear virions was significantly reduced underthese conditions (Fig. 6C). These results indicate that Rac1 is themember of Rho GTPase family whose activation after ASFV entry
FIG 4 Electron microscopy analysis of ASF viral factory after inhibition ofRho GTPase-mediated signaling. Vero cells were infected with ASFV at 0.5PFU/cell in the presence or absence of 50 ng of CdTB/ml and fixed at 16 hpi.(A) In the absence of CdTB, viral factories contained envelope precursors andabundant immature (iv, black arrowheads) and mature icosahedral particles(mv, arrows). (B) In contrast, viral factories formed in the presence of CdTBcontained high amounts of envelope precursors and low numbers of bothmature and immature icosahedral particles. (C) In the absence of CdTB, al-most all viral particles that reached the plasma membrane were mature andwere found with the characteristic filopodia in exocytosis. This observationcontrasts with the abundant immature particles found at the cell membrane inthe presence of CdTB and the absence of filopodia formation. (D). Scale bars:A and B, 0.2 �m; C and D, 0.5 �m. White arrowheads indicate ribosomes.
FIG 5 Rac1 is activated during early stages of ASFV infection. Equal amounts of Vero cell lysates from mock-infected cells (M) or ASFV-infected cells (5PFU/cell) at various time points after infection were used to capture the GTP-bound form of Rac1 and Cdc42 by affinity precipitation with GST-PBD beads. Theproteins captured by beads were analyzed by immunoblotting with anti-Rac1 antibody and anti-Cdc42 (top portions of panels A and B, respectively). The bottomportions of panels A and B show normalized cell lysates analyzed for total Rac1 as a protein load control. (C) Bands corresponding to GTP-bound forms of Rac1and Cdc42 were quantified and normalized to values from total Rac1 and Cdc-42, respectively. The data obtained from mock-infected cells were considered tobe activated 1-fold for comparison with infected cells. Representative images are shown from five independent pulldown assays. (D) Activation of Rac1, Cdc42,and RhoA was analyzed by an ELISA-based assay in infected Vero cells at the indicated time points within the first hour of infection. The GTP-bound forms ofRho GTPases were measured with an absorbance set at 490 nm. Each point represents the means � the SD for three independent experiments.
Role of Rho GTPases during ASFV Infection
February 2012 Volume 86 Number 3 jvi.asm.org 1763
on January 12, 2012 by guesthttp://jvi.asm
.org/D
ownloaded from
results relevant to reach perinuclear sites for an efficient virusmultiplication. Also, Vero cells were transfected with plasmidsencoding wild-type Rac1 (wt), dominant-negative Rac1 mutant(17N), or constitutively active Rac1 mutant (Q61L). After 24 h,the cells were mock infected or infected with a high MOI usinghighly purified ASFV. However, in transfected cells we were notable to find differences either in the number of internalized virusparticles or in their positions with respect to the host cell nucleusat 1 hpi (data not shown).
ASFV infection induces microtubule hyperacetylation. Inhi-bition of Rac1 signaling negatively affected perinuclear virionslocalization but not entry itself, suggesting the involvement ofRac1 in following steps of infection immediately after ASFVentry. Early after ASFV entry into host cells, the microtubulenetwork is essential for an efficient infection (3), and the acet-ylation/deacetylation balance of tubulin regulates microtubuledynamics (49), which is regulated by Rho GTPases. In addition,
by using confocal microscopy, we detected the association ofvirions with acetylated microtubules during the first hour aftervirus inoculation in Vero cells (Fig. 7A). Since hyperacetylationis a quantitative indication of microtubules stabilization, wenext analyzed tubulin acetylation levels from Vero cell extractsat a range of times postinfection by immunoblotting. Tubulinacetylation increased 2.5- to 4-fold from 15 to 30 min postin-fection compared to mock-infected cells. At 45 min postinfec-tion, tubulin acetylation levels showed a 2-fold increase, whichwas maintained for up 2 hpi and returned to basal levels by 3hpi (Fig. 7B). In contrast, the levels of total �-tubulin remainedunaltered. The inactivation of Rho GTPases with increasingconcentrations of CdTB before infection restored tubulin acet-ylation levels to those obtained with mock-infected cell extracts(Fig. 7B). We specifically confirmed a reduction in tubulinacetylation levels in cells incubated with the geranylgeranyla-tion inhibitor GGTI-286. Moreover, we observed similar re-
FIG 6 (A) Virus progeny at 24 hpi were determined by plaque assay from ASFV-infected cells in the presence of the specific Rac1 inhibitor NSC23766. Increasing,but nontoxic, concentrations of NSC23766 were added to cells before (�2 hpi), after (�3 hpi), or throughout infection. (B) The number of infected cells at 3 hpiin the presence of a range of concentrations of NSC23766 is shown as a percentage of control nontreated cells. (C) Virion distribution at 1 hpi in ASFV-infectedVero cells after incubation with increasing concentrations of GGTI-286 (10 and 30 �M) or NSC23766 (10, 50, and 100 �M) was examined. Representativeconfocal 0.1-�m sections from the z-axis are shown where virions were identified with mouse anti-p72 monoclonal antibody, followed by Alexa Fluor 594anti-mouse IgG (red). Nuclei (blue) stained with Hoechst and cell contours are shown. Bar, 16 �m. A bar graph shows the percentage of virions exhibitingperinuclear localization. The data represent medians and SD from three independent experiments where 30 infected cells were analyzed in each case. A high MOI(�10 PFU/cell) was used. Each point represents the means � the SD for three experiments. Values that are significantly different from each other are indicated(���, P � 0.001; ��, P � 0.01; �, P � 0.05).
Quetglas et al.
1764 jvi.asm.org Journal of Virology
on January 12, 2012 by guesthttp://jvi.asm
.org/D
ownloaded from
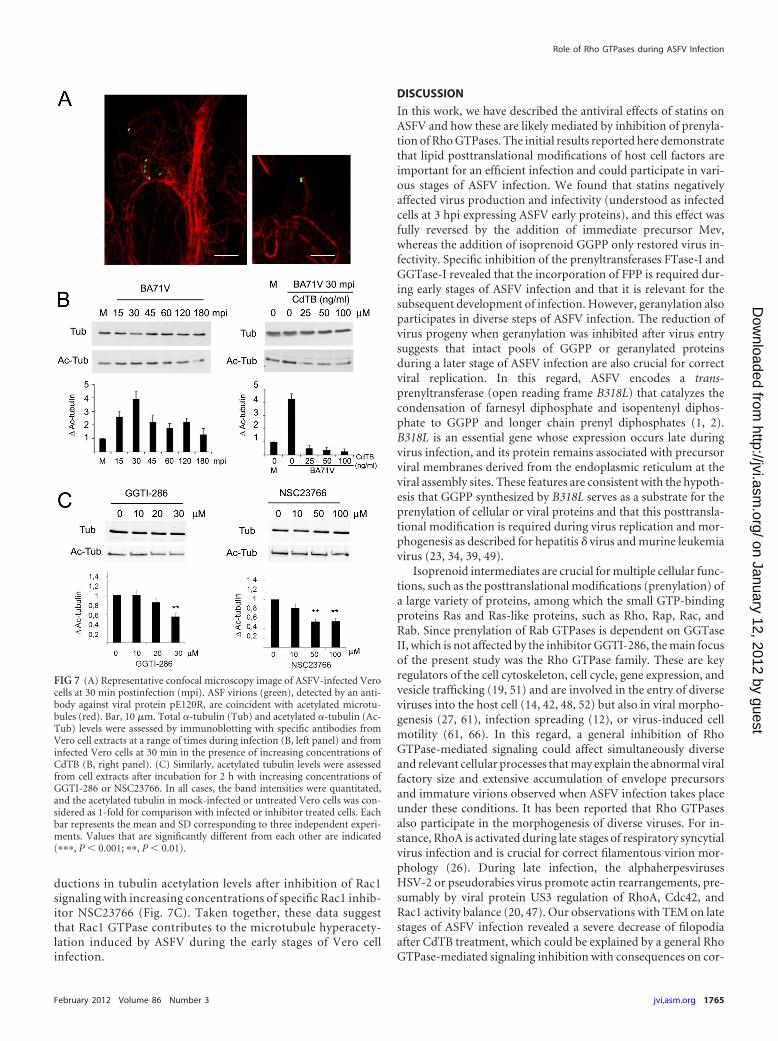
ductions in tubulin acetylation levels after inhibition of Rac1signaling with increasing concentrations of specific Rac1 inhib-itor NSC23766 (Fig. 7C). Taken together, these data suggestthat Rac1 GTPase contributes to the microtubule hyperacety-lation induced by ASFV during the early stages of Vero cellinfection.
DISCUSSION
In this work, we have described the antiviral effects of statins onASFV and how these are likely mediated by inhibition of prenyla-tion of Rho GTPases. The initial results reported here demonstratethat lipid posttranslational modifications of host cell factors areimportant for an efficient infection and could participate in vari-ous stages of ASFV infection. We found that statins negativelyaffected virus production and infectivity (understood as infectedcells at 3 hpi expressing ASFV early proteins), and this effect wasfully reversed by the addition of immediate precursor Mev,whereas the addition of isoprenoid GGPP only restored virus in-fectivity. Specific inhibition of the prenyltransferases FTase-I andGGTase-I revealed that the incorporation of FPP is required dur-ing early stages of ASFV infection and that it is relevant for thesubsequent development of infection. However, geranylation alsoparticipates in diverse steps of ASFV infection. The reduction ofvirus progeny when geranylation was inhibited after virus entrysuggests that intact pools of GGPP or geranylated proteinsduring a later stage of ASFV infection are also crucial for correctviral replication. In this regard, ASFV encodes a trans-prenyltransferase (open reading frame B318L) that catalyzes thecondensation of farnesyl diphosphate and isopentenyl diphos-phate to GGPP and longer chain prenyl diphosphates (1, 2).B318L is an essential gene whose expression occurs late duringvirus infection, and its protein remains associated with precursorviral membranes derived from the endoplasmic reticulum at theviral assembly sites. These features are consistent with the hypoth-esis that GGPP synthesized by B318L serves as a substrate for theprenylation of cellular or viral proteins and that this posttransla-tional modification is required during virus replication and mor-phogenesis as described for hepatitis � virus and murine leukemiavirus (23, 34, 39, 49).
Isoprenoid intermediates are crucial for multiple cellular func-tions, such as the posttranslational modifications (prenylation) ofa large variety of proteins, among which the small GTP-bindingproteins Ras and Ras-like proteins, such as Rho, Rap, Rac, andRab. Since prenylation of Rab GTPases is dependent on GGTaseII, which is not affected by the inhibitor GGTI-286, the main focusof the present study was the Rho GTPase family. These are keyregulators of the cell cytoskeleton, cell cycle, gene expression, andvesicle trafficking (19, 51) and are involved in the entry of diverseviruses into the host cell (14, 42, 48, 52) but also in viral morpho-genesis (27, 61), infection spreading (12), or virus-induced cellmotility (61, 66). In this regard, a general inhibition of RhoGTPase-mediated signaling could affect simultaneously diverseand relevant cellular processes that may explain the abnormal viralfactory size and extensive accumulation of envelope precursorsand immature virions observed when ASFV infection takes placeunder these conditions. It has been reported that Rho GTPasesalso participate in the morphogenesis of diverse viruses. For in-stance, RhoA is activated during late stages of respiratory syncytialvirus infection and is crucial for correct filamentous virion mor-phology (26). During late infection, the alphaherpesvirusesHSV-2 or pseudorabies virus promote actin rearrangements, pre-sumably by viral protein US3 regulation of RhoA, Cdc42, andRac1 activity balance (20, 47). Our observations with TEM on latestages of ASFV infection revealed a severe decrease of filopodiaafter CdTB treatment, which could be explained by a general RhoGTPase-mediated signaling inhibition with consequences on cor-
FIG 7 (A) Representative confocal microscopy image of ASFV-infected Verocells at 30 min postinfection (mpi). ASF virions (green), detected by an anti-body against viral protein pE120R, are coincident with acetylated microtu-bules (red). Bar, 10 �m. Total �-tubulin (Tub) and acetylated �-tubulin (Ac-Tub) levels were assessed by immunoblotting with specific antibodies fromVero cell extracts at a range of times during infection (B, left panel) and frominfected Vero cells at 30 min in the presence of increasing concentrations ofCdTB (B, right panel). (C) Similarly, acetylated tubulin levels were assessedfrom cell extracts after incubation for 2 h with increasing concentrations ofGGTI-286 or NSC23766. In all cases, the band intensities were quantitated,and the acetylated tubulin in mock-infected or untreated Vero cells was con-sidered as 1-fold for comparison with infected or inhibitor treated cells. Eachbar represents the mean and SD corresponding to three independent experi-ments. Values that are significantly different from each other are indicated(���, P � 0.001; ��, P � 0.01).
Role of Rho GTPases during ASFV Infection
February 2012 Volume 86 Number 3 jvi.asm.org 1765
on January 12, 2012 by guesthttp://jvi.asm
.org/D
ownloaded from

tical actin barrier regulation. However, and given that a high num-ber of immature virus particles are accumulated at the cell surfaceafter CdTB treatment, we cannot rule out the possibility that ma-ture ASFV particles were required for stimulating filopodia for-mation, as reported for vaccinia virus-induced actin tails that re-semble filopodia (reviewed in reference 56).
Our data with CdTB indicated that cellular signaling pathwaysregulated by Rho GTPases are activated during early stages ofASFV infection; however, its inactivation did not affect virus in-fectivity. Moreover, virus internalization was not affected bygeranylgeranylation inhibition, thus demonstrating that RhoGTPases are not essential for ASFV entry itself in Vero cells.
Among the most extensively characterized members of the RhoGTPase family, we have demonstrated that Rac1 is activated dur-ing the first 30 min of ASFV infection in Vero cells. Surprisingly,Cdc42, which may act as an upstream activator of Rac1, was notinduced by ASFV infection, as deduced from pull-down and alsoELISA-based assays. A similar situation, Rac1 but not Cdc42 acti-vation, was also recently described during hepatitis B virus infec-tion (59). Rac1 regulates a wide variety of cellular functions (8)and participates in the entry of the following viruses into the hostcell: vaccinia virus (44), dengue virus (65), herpes simplex virus 1(33, 50), group B coxsackieviruses (13), and hepatitis B virus (59),among others. Most of these viruses exploit macropinocytosis insome way in order to gain access to the host cell by means ofmembrane ruffling or blebbing, which requires Rac1 activation(45). However, this entry strategy does not appear to be followedby ASFV, which enters the host cell by dynamin- and clathrin-dependent endocytosis (31), since specific Rac1 inhibition byNSC23766 before virus addition did not affect virus internaliza-tion, thus indicating the absence of deficiencies in ASFV endocy-tosis.
Thus, Rac1 may participate in further steps of early infection,such as intracellular transport to replication sites. Indeed, micro-tubule network integrity during the initial stages of ASFV infec-tion is critical for successful infection (3). Our results demonstratethat ASFV infection promotes early microtubule hyperacetyla-tion, a hallmark of the preferential stabilization of these structures.This process may be regulated by Rho GTPases, since their inhibi-tion prevented microtubule hyperacetylation. Adenovirus andKaposi’s sarcoma-associated herpesvirus also induce microtubulehyperacetylation during early infection of human foreskin fibro-blast (HFF) cells by a RhoA- and Rac1-dependent mechanism (48,64). However, in A549 cells, microtubule hyperacetylation in-duced by incoming adenovirus is mediated exclusively by a Rac1-dependent mechanism (63), as we observed for ASFV. We deter-mined that maximal Rac1 activity during early ASFV infection iscoincident with hyperacetylation of microtubules (around 30 minpostinfection), and tubulin acetylation levels were reduced afterinhibition of Rac1 signaling with increasing concentrations of spe-cific inhibitor.
Moreover, our results showed that specific inhibition of Rac1-mediated signaling with NSC23766 impaired virion perinuclearlocalization but not viral entry itself. Similar results were obtainedwith geranylgeranyl inhibitor which is concordant with a Rac1-mediated signaling inhibition by GGTI-286, given that Rac1 un-dergoes geranylgeranylation at its C terminus, and this posttrans-lational modification has been previously associated with anincrease in Rac1 GTP binding and activation (46). However, wedid not find differences in Rac1 dominant-negative mutant trans-
fected cells affecting either the number of internalized virions ortheir localization to perinuclear areas. A possible explanation isthat the inhibition of Rac1 in Vero cells was not complete in thiscase and a minimal proportion of acetylated microtubules mightbe sufficient to facilitate the start of ASFV infection.
Our data suggest that Rac1 modulates the intracellular trans-port of ASFV by inducing microtubule acetylation. In this regard,microtubules and associated molecular motors have been previ-ously shown to be critical for ASFV trafficking from entry to rep-lication and assembly sites (3), and these results open up the pos-sibility that Rho GTPases could constitute an early target forstatins during ASFV infection, relevant for the development ofnovel strategies to the eradication of African swine fever.
ACKNOWLEDGMENTS
We thank Ole Gjoerup for the plasmids encoding wild-type, dominant-negative, and constitutively active forms of Rac1 fused to GFP. The con-struct expressing the GST-Rac1/Cdc42 binding domain of Pak1 was agenerous gift from Keith Burridge.
This study was supported by grants from Consolider ProgramCSD2006-00007 and by AGL2009-09209 from the Spanish Ministry ofScience and Innovation.
REFERENCES1. Alejo A, Andres G, Vinuela E, Salas ML. 1999. The African swine fever
virus prenyltransferase is an integral membrane trans-geranylgeranyl-diphosphate synthase. J. Biol. Chem. 274:18033–18039.
2. Alejo A, Yanez RJ, Rodriguez JM, Vinuela E, Salas ML. 1997. Africanswine fever virus trans-prenyltransferase. J. Biol. Chem. 272:9417–9423.
3. Alonso C, et al. 2001. African swine fever virus protein p54 interacts withthe microtubular motor complex through direct binding to light-chaindynein. J. Virol. 75:9819 –9827.
4. Arakawa Y, Cordeiro JV, Way M. 2007. F11L-mediated inhibition ofRhoA-mDia signaling stimulates microtubule dynamics during vacciniavirus infection. Cell Host Microbe 1:213–226.
5. Bagrodia S, Taylor SJ, Jordon KA, Van Aelst L, Cerione RA. 1998. Anovel regulator of p21-activated kinases. J. Biol. Chem. 273:23633–23636.
6. Bernardes C, Antonio A, Pedroso de Lima MC, Valdeira ML. 1998.Cholesterol affects African swine fever virus infection. Biochim. Biophys.Acta 1393:19 –25.
7. Bos JL, Rehmann H, Wittinghofer A. 2007. GEFs and GAPs: criticalelements in the control of small G proteins. Cell 129:865– 877.
8. Bosco EE, Mulloy JC, Zheng Y. 2009. Rac1 GTPase: a “Rac” of all trades.Cell. Mol. Life Sci. 66:370 –374.
8a.Brookes SM, Dixon LK, Parkhouse RM. 1996. Assembly of African swinefever virus: quantitative ultrastructural analysis in vitro and in vivo.Virol-ogy 224:84 –92.
9. Bustelo XR, Sauzeau V, Berenjeno IM. 2007. GTP-binding proteins ofthe Rho/Rac family: regulation, effectors and functions in vivo. Bioessays29:356 –370.
10. Carrascosa AL, del Val M, Santaren JF, Vinuela E. 1985. Purification andproperties of African swine fever virus. J. Virol. 54:337–344.
11. Carvalho ZG, De Matos AP, Rodrigues-Pousada C. 1988. Association ofAfrican swine fever virus with the cytoskeleton. Virus Res. 11:175–192.
12. Cordeiro JV, et al. 2009. F11-mediated inhibition of RhoA signalingenhances the spread of vaccinia virus in vitro and in vivo in an intranasalmouse model of infection. PLoS One 4:e8506.
13. Coyne CB, Shen L, Turner JR, Bergelson JM. 2007. Coxsackievirus entryacross epithelial tight junctions requires occludin and the small GTPasesRab34 and Rab5. Cell Host Microbe 2:181–192.
14. del Real G, et al. 2004. Statins inhibit HIV-1 infection by down-regulatingRho activity. J. Exp. Med. 200:541–547.
15. de Rooij J, Bos JL. 1997. Minimal Ras-binding domain of Raf1 can beused as an activation-specific probe for Ras. Oncogene 14:623– 625.
16. Diebold BA, Fowler B, Lu J, Dinauer MC, Bokoch GM. 2004. Antago-nistic cross-talk between Rac and Cdc42 GTPases regulates generation ofreactive oxygen species. J. Biol. Chem. 279:28136 –28142.
17. Dixon LK, et al. 2005. Asfarviridae, p 135–143. InFauquet CM, et al. (ed),
Quetglas et al.
1766 jvi.asm.org Journal of Virology
on January 12, 2012 by guesthttp://jvi.asm
.org/D
ownloaded from

Virus taxonomy: eighth report of the International Committee on Taxon-omy of Viruses. Elsevier/Academic Press, London, England.
18. Enjuanes L, Carrascosa AL, Moreno MA, Vinuela E. 1976. Titration ofAfrican swine fever (ASF) virus. J. Gen. Virol. 32:471– 477.
19. Etienne-Manneville S, Hall A. 2002. Rho GTPases in cell biology. Nature420:629 – 635.
20. Favoreel HW, Van Minnebruggen G, Adriaensen D, Nauwynck HJ.2005. Cytoskeletal rearrangements and cell extensions induced by the US3kinase of an alphaherpesvirus are associated with enhanced spread. Proc.Natl. Acad. Sci. U. S. A. 102:8990 – 8995.
21. Gao Y, Dickerson JB, Guo F, Zheng J, Zheng Y. 2004. Rational designand characterization of a Rac GTPase-specific small molecule inhibitor.Proc. Natl. Acad. Sci. U. S. A. 101:7618 –7623.
22. Geraldes A, Valdeira ML. 1985. Effect of chloroquine on African swinefever virus infection. J. Gen. Virol. 66(Pt 5):1145–1148.
23. Glenn JS, Watson JA, Havel CM, White JM. 1992. Identification of aprenylation site in delta virus large antigen. Science 256:1331–1333.
24. Goldstein JL, Brown MS. 1990. Regulation of the mevalonate pathway.Nature 343:425– 430.
25. Gomez-Puertas P, et al. 1995. Improvement of African swine fever virusneutralization assay using recombinant viruses expressing chromogenicmarker genes. J. Virol. Methods 55:271–279.
26. Gower TL, et al. 2005. RhoA signaling is required for respiratory syncytialvirus-induced syncytium formation and filamentous virion morphology.J. Virol. 79:5326 –5336.
27. Gower TL, Peeples ME, Collins PL, Graham BS. 2001. RhoA is activatedduring respiratory syncytial virus infection. Virology 283:188 –196.
28. Guijarro C, et al. 1998. 3-Hydroxy-3-methylglutaryl coenzyme a reduc-tase and isoprenylation inhibitors induce apoptosis of vascular smoothmuscle cells in culture. Circ. Res. 83:490 –500.
29. Heasman SJ, Ridley AJ. 2008. Mammalian Rho GTPases: new insightsinto their functions from in vivo studies. Nat. Rev. Mol. Cell. Biol.9:690 –701.
30. Heath CM, Windsor M, Wileman T. 2001. Aggresomes resemble sitesspecialized for virus assembly. J. Cell Biol. 153:449 – 455.
31. Hernaez B, Alonso C. 2010. Dynamin- and clathrin-dependent endocy-tosis in African swine fever virus entry. J. Virol. 84:2100 –2109.
32. Hernaez B, Escribano JM, Alonso C. 2006. Visualization of the Africanswine fever virus infection in living cells by incorporation into the virusparticle of green fluorescent protein-p54 membrane protein chimera.Virology 350:1–14.
33. Hoppe S, et al. 2006. Early herpes simplex virus type 1 infection is depen-dent on regulated Rac1/Cdc42 signaling in epithelial MDCKII cells. J. Gen.Virol. 87:3483–3494.
34. Hwang SB, Lai MM. 1993. Isoprenylation mediates direct protein-protein interactions between hepatitis large delta antigen and hepatitis Bvirus surface antigen. J. Virol. 67:7659 –7662.
35. Jaffe AB, Hall A. 2005. Rho GTPases: biochemistry and biology. Annu.Rev. Cell Dev. Biol. 21:247–269.
36. Jouvenet N, Monaghan P, Way M, Wileman T. 2004. Transport ofAfrican swine fever virus from assembly sites to the plasma membrane isdependent on microtubules and conventional kinesin. J. Virol. 78:7990 – 8001.
37. Jouvenet N, et al. 2006. African swine fever virus induces filopodia-likeprojections at the plasma membrane. Cell Microbiol. 8:1803–1811.
38. King DP, et al. 2003. Development of a TaqMan PCR assay with internalamplification control for the detection of African swine fever virus. J.Virol. Methods 107:53– 61.
39. Lee CZ, Chen PJ, Lai MM, Chen DS. 1994. Isoprenylation of largehepatitis delta antigen is necessary but not sufficient for hepatitis deltavirus assembly. Virology 199:169 –175.
40. Li E, Stupack D, Bokoch GM, Nemerow GR. 1998. Adenovirus endo-cytosis requires actin cytoskeleton reorganization mediated by Rho familyGTPases. J. Virol. 72:8806 – 8812.
41. Liao JK, Laufs U. 2005. Pleiotropic effects of statins. Annu. Rev. Pharma-col. Toxicol. 45:89 –118.
42. Locker JK, et al. 2000. Entry of the two infectious forms of vaccinia virusat the plasma membrane is signaling-dependent for the IMV but not theEEV. Mol. Biol. Cell 11:2497–2511.
43. McTaggart SJ. 2006. Isoprenylated proteins. Cell. Mol. Life Sci. 63:255–267.
44. Mercer J, Helenius A. 2010. Apoptotic mimicry: phosphatidylserine-mediated macropinocytosis of vaccinia virus. Ann. N. Y. Acad. Sci. 1209:49 –55.
45. Mercer J, Helenius A. 2009. Virus entry by macropinocytosis. Nat. CellBiol. 11:510 –520.
46. Michaelson D, et al. 2001. Differential localization of Rho GTPases in livecells: regulation by hypervariable regions and RhoGDI binding. J. CellBiol. 152:111–126.
47. Murata T, Goshima F, Daikoku T, Takakuwa H, Nishiyama Y. 2000.Expression of herpes simplex virus type 2 US3 affects the Cdc42/Rac path-way and attenuates c-Jun N-terminal kinase activation. Genes Cells5:1017–1027.
48. Naranatt PP, Krishnan HH, Smith MS, Chandran B. 2005. Kaposi’ssarcoma-associated herpesvirus modulates microtubule dynamics viaRhoA-GTP-diaphanous 2 signaling and utilizes the dynein motors to de-liver its DNA to the nucleus. J. Virol. 79:1191–1206.
49. Overmeyer JH, Maltese WA. 1992. Isoprenoid requirement for intracel-lular transport and processing of murine leukemia virus envelope protein.J. Biol. Chem. 267:22686 –22692.
50. Petermann P, Haase I, Knebel-Morsdorf D. 2009. Impact of Rac1 andCdc42 signaling during early herpes simplex virus type 1 infection of ke-ratinocytes. J. Virol. 83:9759 –9772.
51. Qualmann B, Mellor H. 2003. Regulation of endocytic traffic by RhoGTPases. Biochem. J. 371:233–241.
52. Quinn K, et al. 2009. Rho GTPases modulate entry of Ebola virus andvesicular stomatitis virus pseudotyped vectors. J. Virol. 83:10176 –10186.
53. Ridley AJ. 2001. Rho family proteins: coordinating cell responses. TrendsCell Biol. 11:471– 477.
54. Rodriguez JM, Garcia-Escudero R, Salas ML, Andres G. 2004. Africanswine fever virus structural protein p54 is essential for the recruitment ofenvelope precursors to assembly sites. J. Virol. 78:4299 – 4313.
55. Sander EE, et al. 1998. Matrix-dependent Tiam1/Rac signaling in epithe-lial cells promotes either cell-cell adhesion or cell migration and is regu-lated by phosphatidylinositol 3-kinase. J. Cell Biol. 143:1385–1398.
56. Smith GL, Vanderplasschen A, Law M. 2002. The formation and func-tion of extracellular enveloped vaccinia virus. J. Gen. Virol. 83:2915–2931.
57. Stefanovic S, Windsor M, Nagata KI, Inagaki M, Wileman T. 2005.Vimentin rearrangement during African swine fever virus infection in-volves retrograde transport along microtubules and phosphorylation ofvimentin by calcium calmodulin kinase II. J. Virol. 79:11766 –11775.
58. Subauste MC, et al. 2000. Rho family proteins modulate rapid apoptosisinduced by cytotoxic T lymphocytes and Fas. J. Biol. Chem. 275:9725–9733.
59. Tan TL, et al. 2008. Rac1 GTPase is activated by hepatitis B virusreplication: involvement of HBX. Biochim. Biophys. Acta 1783:360 –374.
60. Valdeira ML, Bernardes C, Cruz B, Geraldes A. 1998. Entry of Africanswine fever virus into Vero cells and uncoating. Vet. Microbiol. 60:131–140.
61. Valderrama F, Cordeiro JV, Schleich S, Frischknecht F, Way M. 2006.Vaccinia virus-induced cell motility requires F11L-mediated inhibition ofRhoA signaling. Science 311:377–381.
62. Van Aelst L, D’Souza-Schorey C. 1997. Rho GTPases and signalingnetworks. Genes Dev. 11:2295–2322.
63. Warren JC, Cassimeris L. 2007. The contributions of microtubule stabil-ity and dynamic instability to adenovirus nuclear localization efficiency.Cell Motil. Cytoskeleton 64:675– 689.
64. Warren JC, Rutkowski A, Cassimeris L. 2006. Infection with replication-deficient adenovirus induces changes in the dynamic instability of host cellmicrotubules. Mol. Biol. Cell 17:3557–3568.
65. Zamudio-Meza H, Castillo-Alvarez A, Gonzalez-Bonilla C, Meza I.2009. Cross-talk between Rac1 and Cdc42 GTPases regulates formation offilopodia required for dengue virus type 2 entry into HMEC-1 cells. J. Gen.Virol. 90:2902–2911.
66. Zwilling J, Sliva K, Schwantes A, Schnierle B, Sutter G. 2010. FunctionalF11L and K1L genes in modified vaccinia virus Ankara restore virus-induced cell motility but not growth in human and murine cells. Virology404:231–239.
Role of Rho GTPases during ASFV Infection
February 2012 Volume 86 Number 3 jvi.asm.org 1767
on January 12, 2012 by guesthttp://jvi.asm
.org/D
ownloaded from





























