Embed Size (px)
Citation preview

REPÚBLICA BOLIVARIANA DE VENEZUELALA UNIVERSIDAD DEL ZULIA
FACULTAD DE MEDICINAPROGRAMA DE GENETICA CLINICA
Dra. Ana BrachoPediatría - Genetista
HERENCIA AUTOSÓMICA DOMINANTE

HERENCIA AUTOSÓMICA DOMINANTE

HERENCIA AUTOSÓMICA DOMINANTE
Factores que modifican las proporciones mendelianas
Del patrón de herencia autosómico dominante
Penetración Incompleta
Expresividad
Edad de aparición
Mutaciones Nuevas
Mosaicismo germinal
Pleiotropia
Heterogeneidad genética
Impronta genomica
Anticipación
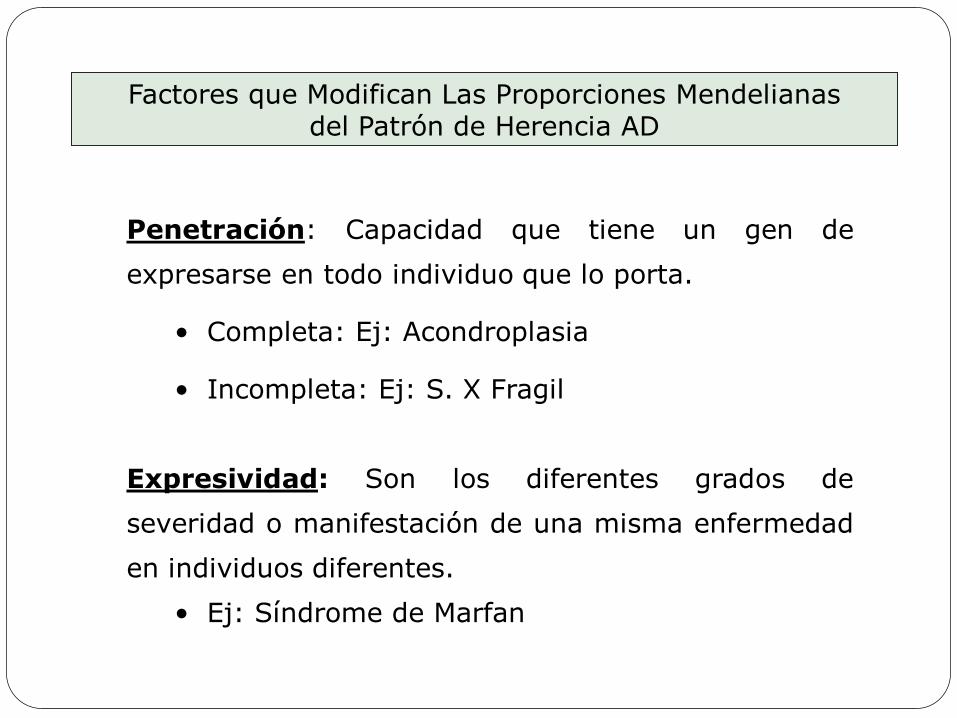
Penetración: Capacidad que tiene un gen de
expresarse en todo individuo que lo porta.
• Completa: Ej: Acondroplasia
• Incompleta: Ej: S. X Fragil
Expresividad: Son los diferentes grados de
severidad o manifestación de una misma enfermedad
en individuos diferentes.
• Ej: Síndrome de Marfan
Factores que Modifican Las Proporciones Mendelianas del Patrón de Herencia AD

Edad de Aparición:
Existen enfermedades hereditarias presentes al
momento del nacimiento (congénitas).
• Ej: acondroplasia
Existen otras cuya edad de aparición es tardía
• Ej: Enfermedad de Huntington
Factores que Modifican Las Proporciones Mendelianas del Patrón de Herencia AD

Mutación:
Cambios transmisibles en el material genético.
En sentido amplio es el origen de un nuevo tipo
hereditario.
Mosaicismo germinal:
Durante el desarrollo embrionario de uno de los
progenitores ocurre una mutación que afecta una
parte o toda la línea germinal, individuos normales
pero con una mutación germinal
Factores que Modifican Las Proporciones Mendelianas del Patrón de Herencia AD

Pleiotropia:
La mutación en un solo gen afecta a múltiples
características fenotípicas.
• Ej: Neurofibromatosis
Heterogeneidad genética:
Diferentes genes mutantes pueden producir un
cuadro clínico similar, pero mostrar diferentes
formas de herencia
• Ej: Sordera profunda congénita
Factores que Modifican Las Proporciones Mendelianas del Patrón de Herencia AD

Impronta genomica:
El mecanismo por el cual la actividad de los genes
homólogos en una persona difieren, dependiendo
del sexo del progenitor que lo transmite. Esto
ocurre durante etapas muy tempranas del
desarrollo embrionario.
• Ej: Síndrome de Prader Willi y síndrome de
Angelman (15 q11.2-11.3)
Factores que Modifican Las Proporciones Mendelianas del Patrón de Herencia AD

Anticipación:
Algunas enfermedades genéticas aparecen a
edades mas tempranas y son mas severas en
las siguientes generaciones dependiendo del
progenitor que la transmita
• Ej: Distrofia Miotonica
Factores que Modifican Las Proporciones Mendelianas del Patrón de Herencia AD

Herencia: autosómica dominante
Es el mas frecuente
enanismo de miembros
cortos
Penetración: completa
ACONDROPLASIA

Mapeado: 4 pl6.3
Proteína: Receptor 3 del factor de crecimiento del fibroblasto
80% mutaciones nuevas
Incidencia: 1 x 26.000 a 40.000 nacidos vivos
Ocurre en todas las razas y en ambos sexos
ACONDROPLASIA

Clínica:
Acortamiento de segmentos proximales de los miembros (rizomelia)
Tórax costillas cortas
Pliegues redundantes en miembros
Macrocefalia
Inteligencia normal
ACONDROPLASIA

ACONDROPLASIA
Frente amplia y prominente
Raíz nasal deprimida
Narinas antevertidas
Abdomen prominente
Limitación extensión del codo
Manos y pies cortos
Dedos triangulares (mano en tridente)

ACONDROPLASIA
Genus varum
Cifosis lumbar en la infancia
por hipotonía
Luego lordosis lumbar

ACONDROPLASIA
Diagnostico:
Clínico y radiologico.
Rx de cráneo: Huesos faciales ↓. Hidrocefalia. Base
del cráneo con foramen mágnum pequeño.
Rx Tórax: Costillas cortas. Cuerpos vertebralesplanos. Estrechamiento interpedicular región lumbar
Rx Pelvis: alas iliacas cuadradas, escotadura ciáticamayor ensanchada.
Huesos largos: metáfisis ensanchadas
Acortamiento de falanges proximales y medias.

ACONDROPLASIA

Diagnostico prenatal
Ecografía fetal:
Relación diámetro biparietal/longitud del fémur
Análisis del ADN: Identificación de la mutación
ACONDROPLASIA

Evolución:
Enanismo
Hidrocefalia
Infecciones respiratorias
Estrechez canal espinal: tetraplejia
Muerte súbita por compresión del tallo cerebral (6-24
años) Cardiacas (25–54 años)
ACONDROPLASIA

TRATAMIENTO
Prevenir complicaciones
Vigilancia ortopédica
Vigilar compromiso respiratorio
Alargamiento óseo
Embarazadas acondroplásicas:
Cesárea con anestesia general
ACONDROPLASIA



SÍNDROME DE MARFÁN
DEFINICION:
Es una condición hereditaria que afectael tejido conectivo y por ende órganos ysistemas, incluyendo el sistemaesquelético, cardiovascular, el sistemanervioso, ojos y la piel, entre otros.
INCIDENCIA:
• 1:5.000 personas padece de este síndrome en losEstados Unidos
• Afecta tanto a hombres como a mujeres y niños.

MECANISMO DE TRANSMISION:
Se hereda como un rasgo autosómico dominante. El30% de los casos son "esporádicos".
UBICACIÓN GÉNICA:
Cromosoma 15q. El gen fibrilina-1 codifica unaproteína denominada fibrilina.
SÍNDROME DE MARFÁN

SÍNDROME DE MARFÁN
Esqueleto:
Las personas con el síndrome de Marfanson muy altas, delgadas, y tienen lasarticulaciones muy laxas o flexibles.
Ojos:
Más del 50% experimentan dislocacióndel cristalino. El desprendimiento de laretina es una complicación seria. Puedeobservarse además, miopía, glaucoma ycataratas.
ÓRGANOS Y SISTEMAS DEL CUERPO MAS AFECTADOS

SÍNDROME DE MARFÁN
Sistema cardiovascular:
La mayoría de las personas con el síndrome de Marfan
tienen anormalidades en el corazón y los vasos
sanguíneos. Las paredes de la aorta pueden debilitarse y
estirarse debido a deficiencias en el tejido conectivo,
produciendo dilatación de la aorta. En algunos casos
presentan prolapso valvular.

Sistema nervioso:
A medida que las personas con Marfan envejecen, la
duramadre se debilita y se estira. Esta dilatación de la
membrana puede causar desde presión de las vértebras en
la parte baja de la columna vertebral así como desgaste de
los huesos alrededor de la médula espinal, hasta molestias
leves como dolor abdominal y dolor, adormecimiento o
debilitamiento de las piernas.
SÍNDROME DE MARFÁN

SÍNDROME DE MARFÁN
Piel:
Pueden desarrollar estrías en la piel, las cualesocurren sin que haya cambios de peso. Además tienenun mayor riesgo de desarrollar hernia abdominal oinguinal.
Pulmones:
Pueden tener trastornos de larespiración tales como ronquidos oapnea del sueño.

SÍNDROME DE MARFÁN
ÓRGANOS Y SISTEMAS DEL CUERPO MAS AFECTADOS
Pectus
excavatum

SÍNDROME DE MARFÁN
DIAGNÓSTICO:
• Criterios diagnósticos: 4 mayores ó 2 mayores y 2menores
• Se pueden llevar a cabo los siguientes exámenes:
• Ecocardiograma
• Examen de ojos
• Prueba para la mutación de la fibrilina-1 (en algunosindividuos)

SÍNDROME DE MARFÁN
1.- ESQUELETICOS.
• Pectus excavatum o carinatum.
• Reducción en la relación segmento superior-segmento inferior o
en relación con la brazada/talla (mayor de 1,05).
• Signo de la muñeca (Walter Murdoch).
• Escoliosis mayor de 20 grados o espondilolistesis.
• Rotación medial del maleolo medio que causa pie plano.
• Protusion acetabular de cualquier grado por radiografía.
CRITERIOS MAYORES

2.- OCULARES
• Ectopia lentis.
3.- CARDIOVASCULARES
• Dilatación de la aorta ascendente que involucre los
senos de valsalva
• Disección de la aorta descendente.
4.- DURAMADRE
• Ectopia dural lumbosacra por TAC o RMN
SÍNDROME DE MARFÁN
CRITERIOS MAYORES

CRITERIOS MAYORES
5.- HISTORIA FAMILIAR
• Padre, un hijo, un hermano que cumpla los siguientes
criterios:
- Mutación del gen FBN1.
- Haplotipo alrededor del gen FBN1, heredado conocido
asociado al síndrome de Marfan en la familia (análisis
de ligamiento)
SÍNDROME DE MARFÁN

CRITERIOS MENORES
1.- ESQUELETICOS
• Pectus excavatun de moderada intensidad
• Hipermovilidad articular
• Paladar alto con apiñamiento dentario
• Apariencia facial (Dolicocefalia, hipoplasia malar, enoftalmos,
retrognatia, hendiduras palpebrales dirigidas hacia abajo)
SÍNDROME DE MARFÁN

CRITERIOS MENORES
2.- CARDIOVASCULARES
• Prolapso de válvula mitral MVP con o sin regurgitación mitral
• Dilatación de la arteria pulmonar antes de los 40 años sin
causa obvia
• Calcificación del anillo mitral antes de los 40 años
• Dilatación o disección de la aorta toráxica o abdominal por
debajo de los 50 años
SÍNDROME DE MARFÁN

CRITERIOS MENORES
3.- OCULARES
• Cornea anormalmente adelgazada
• Aumento de la longitud axial del globo ocular (ecograma)
• Iris Hipoplásico del músculo ciliar que causan disminución
en la miosis
4- PULMONARES
• Neumotorax espontáneo
• Ampolla apical (Rx)
5.- PIEL
• Estrías atróficas sin causa obvia
• Hernias recurrentes
SÍNDROME DE MARFÁN



TRATAMIENTO:
• No existe un tratamiento curativo único para estaafección.
• Los medicamentos que disminuyen la frecuencia cardiaca(beta bloqueadores) pueden ayudar a prevenir el estrés enesta arteria.
• Evitar las actividades atléticas competitivas y los deportesde contacto.
SÍNDROME DE MARFÁN

TRATAMIENTO:
• Cada año, se debe realizar un ecocardiograma.
• Antes de cualquier tratamiento odontológico: debenrecibir antibióticos para prevenir una endocarditis.
• El embarazo en personas con el síndrome de Marfan
debe ser vigilado muy de cerca debido al incremento
del estrés sobre el corazón y la aorta.
SÍNDROME DE MARFÁN


Neurofibromatosis Tipo I
AD
17q11.2
NF1 Neurofibromina Gen clonado 1991
Incidencia
1 : 3.500 n v
Neurofibromatosis Tipo I

Neurofibromatosis Tipo I
Neurofibromatosis tipo 1
Enfermedad de Von Recklinghausen
Mutaciones de novo 50%
Trastorno genético más común del sistema nervioso

Neurofibromatosis Tipo I
Criterio consenso de Instituto de Salud (NIH)
• Manchas café con leche: > de 5 mm en prepuberes y más de
15 mm en postpuberes
• Neurofibromas de cualquier tipo o 1 neurofibroma y 2 o más del
neurofibroma plexiforme
• Pecas axilares o inguinales
• Glioma del nervio óptico
• Nódulos de Lisch (hamartomas del iris) En < de 6 años y en
numero de 2 o más
• Displasia del esfenoides o enrarecimiento de la corteza del
hueso con o sin pseudoartrosis
• Pariente de Primer grado con NF1

Neurofibromatosis Tipo I
Neurofibroma plexiforme
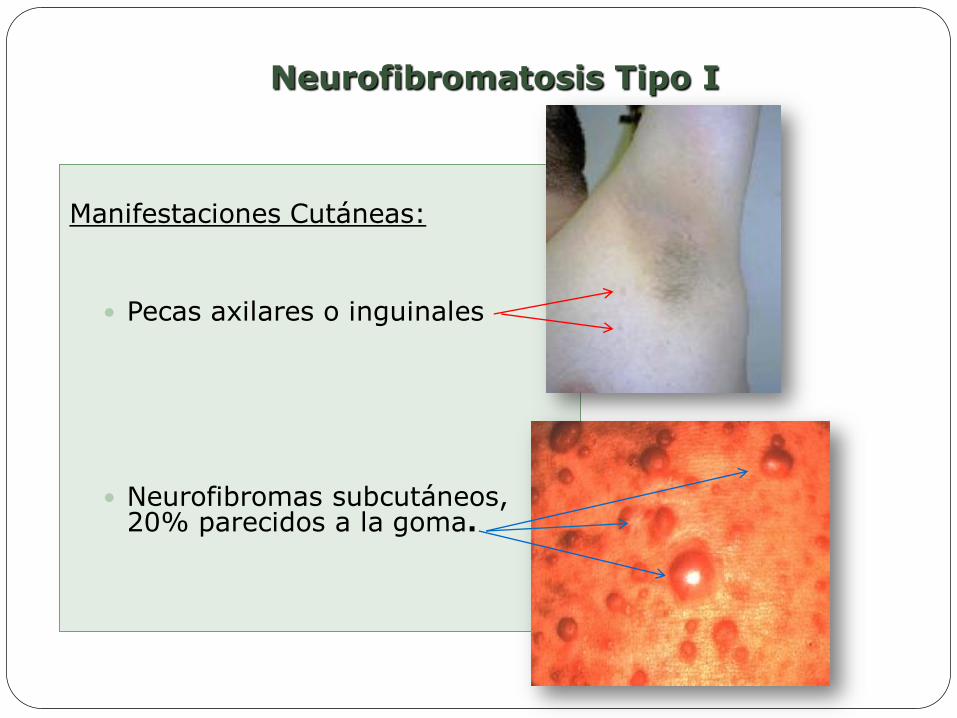
Neurofibromatosis Tipo I
Manifestaciones Cutáneas:
Pecas axilares o inguinales
Neurofibromas subcutáneos, 20% parecidos a la goma.

Neurofibromatosis Tipo I
Manifestaciones Neurológicas
• Gliomas ópticos 15% → ceguera
• Astrocitomas
• Schwanomas vestibulares (neuroma acústico)
• Ependimomas
• Meningiomas

Neurofibromatosis Tipo I
Manifestaciones Oculares
Nódulos de Lisch(hamartomas del iris)

Neurofibromatosis Tipo I
Otras manifestaciones
- Neurofibrosarcoma - Glaucoma congénito
- Rabdomiosarcoma - Feocromacitoma
- Tumor de Wilms - Leucemia no linfocitica
- Neurofibroma visceral - Hidrocefalia
- Tratornos del aprendizaje - Hipertensión
- Pubertad precoz
- Deficiencia de la hormona del crecimiento
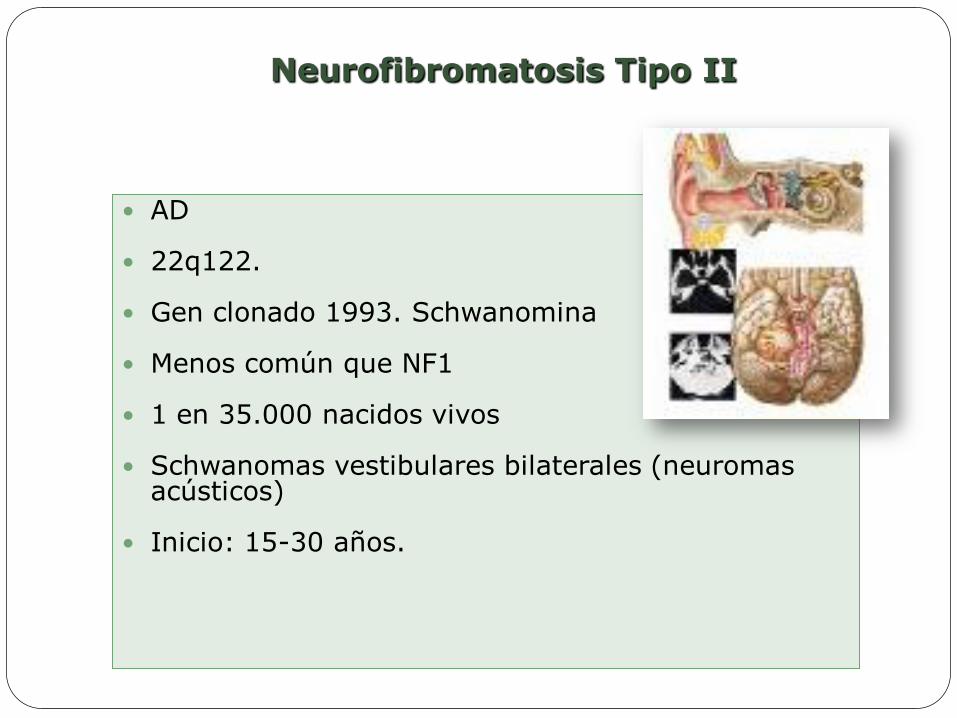
AD
22q122.
Gen clonado 1993. Schwanomina
Menos común que NF1
1 en 35.000 nacidos vivos
Schwanomas vestibulares bilaterales (neuromas acústicos)
Inicio: 15-30 años.
Neurofibromatosis Tipo II

Neurofibromatosis Tipo II
Criterio consenso del Instituto de salud (NIH)
• Schwanomas vestibulares bilaterales
(neuromas acústicos) en TC o RM
• Un pariente primer grado con NF2 más un Schwanoma
vestibular unilateral antes de los 30 años
• Cualquiera 2 de los siguientes:
*neurofibroma *meningioma *glioma *schwanoma
u *opacidad subcapsular posterior

Neurofibromatosis Tipo II
Manifestaciones Cutáneas
• Neurofibromas menos común que NF1
• Neurofibromas Plexiforme son inusuales
• Pocas manchas café con leche
• 8% de NF2 tienen más de 3 manchas
• Pecas axilares o inguinales no están presentes
• Schwanomas planos o esféricos, cutáneos y subcutáneos
en nervios periféricos.
• Schwanomas nodulares subcutáneos en nervios periféricos
de los miembros y del tronco

Neurofibromatosis Tipo II
Manifestaciones Neurológicas
• Schwanomas vestibulares mas frecuente, pueden ocurrir en
cualquiera de los nervios craneales a excepción del nervio
olfatorio.
• La presión en el complejo vestibulococlear y del nervio facial
→ sordera, desequilibrio y debilidad facial
• Evitar nadar debajo del agua, desorientación subacuática
• Otros: meningiomas intracraneales y espinales, astrocitomas,
y ependimomas.

George Huntington (1851-1916)

Enfermedad de Huntington
Enfermedad neurodegenerativa hereditaria, caracterizada pormovimientos corporales anormales, demencia y problemaspsiquiátricos.
Mecanismo de transmisión: AD
Afecta ambos sexos por igual.
Edad de aparición: entre 30 - 50 años de edad,excepcionalmente en la primera infancia o tan tardíamentecomo a los 70 años.
Base molecular: expansión del trinucleótido CAG (36 a121 copias) en el exon 1 del cromosoma 4 (4p16.3) quecodifica para una proteína llamada huntingtina

Características:
• Cambios en la personalidad, depresión, cambios del
carácter.
• Marcha inestable, movimientos involuntarios.
• Problemas con el juicio.
• Dificultad al tragar.
•Apariencia de intoxicación alcohólica.
Enfermedad de Huntington

Enfermedad de Huntington
Sintomas:
Cambios de comportamiento
• irritabilidad, malhumor• inquietud, impaciencia • comportamientos antisociales • psicosis, paranoia, alucinaciones
Demencia progresiva
• pérdida de la memoria • pérdida del juicio • cambios en el lenguaje • pérdida de otras funciones (capacidad para calcular, etc.) • cambios de personalidad • desorientación o confusión

Enfermedad de Huntington
Otros
• Movimientos faciales, muecas
• Necesidad de girar la cabeza para desplazar la mirada
• Marcha inestable
• Desarrollo progresivo de movimientos anormales
• Deterioro del lenguaje
• Ansiedad, estrés y tensión
• Dificultad para deglutir
En los niños:
• Movimientos lentos • Temblor • Rigidez

Enfermedad de Huntington
Diagnóstico
• TAC de cráneo: puede evidenciar atrofia delcerebro, especialmente de estructuras profundas(caudado) u otras anomalías.
• IRM de cráneo
• Tomografía por emisión de positrones (isótopos) delcerebro (PET)
• Puede haber disponibilidad de estudios conmarcadores de ADN (estudios genéticos que indican latendencia a desarrollar este trastorno)

Enfermedad de Huntington
Tratamiento
En la actualidad solo permite aliviar algunas manifestacionespero no retrasa ni la aparición ni la progresión de los síntomas.
MEDICAMENTOS
Butirofenonas y neurolépticos: antagonistas de los receptoresdopaminérgicos del cuerpo estriado (control de la corea)
Tetrabenazina y reserpina: inhibidores del almacenamiento oliberación de la dopamina.

Enfermedad de Huntington
Tratamiento
Para la demencia: No existe tratamiento eficaz, los fármacos antipsicóticos y antidepresivos son de utilidad, además tienen un efecto parcial sobre la corea y la agitación. Ej. Clorpromazina y haloperidol.
Se ha experimentado con la cirugía estereostática pero es poco útil e insatisfactoria.
Actualmente se investiga la búsqueda de drogas que puedan prevenir la formación de inclusiones in vivo por mecanismos diversos.

Enfermedad de Huntington
CONSEJO GENÉTICO
La detección de los portadores de la enfermedad en fasespresintomáticas es ahora posible gracias a las técnicas debiología molecular.
Se han creado comités en los que participan psicologos,médicos, juristas y expertos en ética médica con el fin deasesorar en cada caso sobre la conveniencia de poner enpráctica dichas técnicas.
“ Se necesita un equipo multidisciplinario ”

Enfermedad de Huntington
CURSO CLÍNICO Y PRONÓSTICO
La Enfermedad de Huntington sigue un curso progresivo,es más rápido en las formas de inicio juvenil.
En fases avanzadas los pacientes pierden la capacidadfísica y mental para el cuidado personal, la marcha sehace imposible, el tragar dificultoso y al final existe unademencia grave.
La muerte se produce por complicaciones derivadas de ladebilidad general en 15 años con límites entre 10 y 25años.

Craneosinostosis
Fusión prematura de las suturas que restringe el crecimiento del cráneo, con crecimiento asimétrico del mismo. No se conoce la causa de esta fusión prematura.
Craneosinostosis

Craneosinostosis
Síndromes que involucran craneosinostosis:
• Síndrome de Apert (acrocéfalosindactilia)
• Síndrome de Carpenter (deformidad del cráneo
en forma de cruce en trébol)
• Enfermedad de Crouzon (disostosis craneofacial)
• Síndrome de Saethre-Chotzen
• Síndrome de Pfeiffer

Se caracteriza por el cierre precoz de las suturas
craneales: coronal, sagital, escamosa y lamdoidea, lo que
ocasiona un crecimiento asimétrico de la cabeza.
Adicionalmente sindactilia de los dedos 2, 3 y 4 de
ambas manos, dando la apariencia de manopla o cuchara,
defectos que pueden observarse en los pies.
Acrocéfalosindactilia
Síndrome de Apert

Síndrome de Apert
Mecanismo de transmisión:
• Autosómico dominante• Mutaciones esporádicas
Cromosoma: 10q26
Gen: F6FR2 que codifica una proteína llamada Factor decrecimiento fibroblástico
Mutaciones en el gen: P253R = sindactiliaS252W = paladar hendido
Prevalencia: 1:200.000 hasta 15:1.000.000aumenta con el incremento en la edad paterna

Acrocéfalosindactilia
Manifestaciones Clínica
Apariencia típica craneofacial: cara plana y alargadacon ensanchamiento de la región temporal yaplanamiento occipital
- Craneosinostosis: • Acrocefalia• Hipertensión intracraneal• Retraso mental• Edema de papila, ceguera y pérdida de la
visión por atrofia óptica.
- Sindactilia:• 2, 3 y 4 de manos y pies

Acrocéfalosindactilia
- Rasgos faciales inusuales:
• hipertelorismo
• hipoplasia del tercio medio facial
• exoftalmos
- Macroglosia
- Maloclusión dental
- Paladar ojival fisura palatina

Acrocéfalosindactilia
Diagnóstico:
• Clínico:
• Imagen:

Acrocéfalosindactilia
Tratamiento
Quirúrgico
Antes de los 6 meses de edad dirigido a:
• Descomprimir el espacio intracraneal
• Mejorar la función respiratoria
• Permitir el desarrollo normal de las distintas áreas cerebrales
Alrededor de los 6 años dirigido a:
• Mejorar el aspecto físico del niño
• Sobre todo cara y manos































