Embed Size (px)
Citation preview

424 www.postepybiochemii.pl
Jolanta Barańska*
Instytut Biologii Doświadczalnej im. Marcele-go Nenckiego PAN, Warszawa
*Instytut Biologii Doświadczalnej im. Marcelego Nenckiego PAN, ul. Pasteura 3, 02-093 Warszawa; tel. (22) 58 92 300, e-mail: [email protected]
Artykuł otrzymano 12 września 2014 r.Artykuł zaakceptowano 16 września 2014 r.
Słowa kluczowe: receptory nukleotydowe, P1, P2X, P2Y
Wykaz skrótów: αβMeATP — α,β-metyleno-ATP; AC — cyklaza adenylanowa; Akt — ki-naza białkowa B/kinaza Akt; Ap4A — dia-denozyno-5,5-P1,P4-tetrafosforan; BzATP - 2’,3’-O-(4-benzoilobenzoil)-ATP; cAMP — cy-kliczny AMP; DAG — diacyloglicerol; ERK1/ERK2 (ang. extracellular signal regulated kinases) — kinazy regulowane sygnałem zewnątrzko-mórkowym; GPCR (ang. G-protein-coupled-re-ceptors) — receptory metabotropowe sprzężo-ne z białkiem G; IP3 — trisfosfoinozytol; MAP (ang. mitogen activated protein) — białko akty-wowane przez czynniki mitogenne; IUPHAR (ang. International Union of Basic and Clinical Pharmacology) — Międzynarodowa Unia Far-makologiczna; 2MeSADP — 2-methylotio--ADP; 2MeSATP — 2-methylotio-ATP; PI3K — kinaza 3-fosfatydyloinozytolu; PIP2 — fos-fatydyloinozytolo-(4,5)-bisfosforan; PLC — fosfolipaza C; PKA, PKC — kinazy białkowe; PPADS — 6-azofenylo-2’,4’-disulfonowy fosfo-ran pirydoksalu
Receptory nukleotydowe — budowa i funkcje, historia i perspektywy
STRESZCZENIE
Receptory nukleotydowe zostały odkryte w latach 70. ubiegłego wieku przez brytyjskiego badacza Geoffreya Burnstocka. Pierwotnie nosiły one nazwę „receptorów purynergicz-
nych”. Kiedy wykazano, że w ich aktywacji biorą udział zarówno nukleotydy purynowe jak i piramidynowe nazwę zmieniono na „receptory nukleotydowe” i podzielono na dwie grupy: P1 i P2. Receptory P1 to receptory, których agonistą jest nukleozyd, adenozyna, pod-czas gdy receptory P2, dalej podzielone na podgrupy P2X i P2Y są aktywowane przez nu-kleotydy. Receptory P2X to receptory jonotropowe, tworzące kanał w błonie komórkowej, aktywowane przez ATP. Receptory P2Y to receptory metabotropowe, związane z białkami G (podobnie jak P1), aktywowane przez ATP, ADP, UTP, UDP i cukrowe pochodne UDP. W ar-tykule opisano historię odkrycia i omówiono strukturę i funkcje poszczególnych receptorów nukleotydowych. Zwrócono uwagę na ich znaczącą rolę w regulacji rozlicznych procesów zachodzących w organizmie i związane z tym możliwości terapeutyczne. Zaproponowano kierunki perspektywiczne przyszłych badań, którymi byłaby charakterystyka dotychczas nierozpoznanych receptorów sierocych oraz terapia wybiórcza, celowana na określony na-rząd i receptor.
WPROWADZENIE
Nukleotydy są związkami powszechnie występującymi w świecie żywym. Odgrywają kluczową rolę we wszystkich podstawowych procesach zachodzą-cych wewnątrz komórek, regulują aktywność wielu enzymów, biorą udział w syntezie kwasów nukleinowych. Najwszechstronniej przebadany nukleotyd, ATP, jest obecny w stężeniu 1–10 mM we wszystkich komórkach zwierząt, ro-ślin i mikroorganizmów. Nukleotyd ten jest stale produkowany i zużywany. Człowiek w ciągu doby zużywa mniej więcej tyle ATP ile sam waży. W całym świecie żywym ATP służy jako paliwo, uniwersalny nośnik energii napędzający reakcje biologiczne komórek, a przez to umożliwiający funkcjonowanie i życie organizmów. Jednak ATP i inne nukleotydy i nukleozydy pełnią jeszcze inną, również bardzo ważną funkcję. Mianowicie są cząsteczkami sygnałowymi, uży-wanymi w przekazywaniu informacji ze środowiska zewnętrznego do wnętrza komórki, służą również w komunikacji międzykomórkowej [1,2].
Teza o podwójnej roli ATP i innych nukleotydów początkowo spotykała się z ogromnym niedowierzaniem. Sądzono, że ATP pełni wyłącznie funkcje we-wnątrzkomórkowego źródła i nośnika energii. Jednak poczynając od lat 70. ubie-głego wieku dokonano wielu odkryć wyjaśniających, w jaki sposób nukleotyd ten jest uwalniany do środowiska zewnątrzkomórkowego, i jak pod wpływem ektoenzymów ulega degradacji, prowadzącej kolejno do powstania ostateczne-go produktu tych przemian, adenozyny. Wykazano, że ATP i inne związki ade-ninowe, znajdując się poza komórką, oddziałują ze specyficznymi receptorami usytuowanymi, podobnie jak ektoenzymy, na zewnętrznej powierzchni błony plazmatycznej komórek [2-4].
Receptory te zostały początkowo nazwane „receptorami purynergicznymi” i podzielone na te, na które działa adenozyna i te, które rozpoznają ATP i ADP. Gdy wykazano, że na receptory te działają również nukleotydy pirymidynowe, takie jak UTP i UDP, dokonano weryfikacji nazwy całej klasy. Obecnie noszą one nazwę „receptorów nukleotydowych” [5,6], choć ich pierwotna nazwa jest stale spotykana w literaturze.
Receptory nukleotydowe są wszechobecne, oddziałują z nukleotydami uwal-nianymi aktywnie do środowiska zarówno w warunkach normy, jak i w pro-cesach patologicznych. To oddziaływanie powoduje aktywację receptorów i przekazanie określonych sygnałów do wnętrza komórki, modulując i regulując rozliczne funkcje organizmu, jak skurcze mięśni, czynności serca, czy przekaź-nictwo nerwowe [7]. Porównując sygnały staramy się je poznać i wpływać na ich

Postępy Biochemii 60 (4) 2014 425
zmianę, zmierzając w celach terapeutycznych do poprawy ludzkiego zdrowia [8-10].
HISTORIA BADAŃ RECEPTORÓW NUKLEOTYDOWYCH
Historia badań receptorów nukleotydowych rozpoczęła się ponad 80 lat temu. Za jej początek należy uznać odkry-cie ATP, które przypadło na rok 1929. Było ono udziałem dwóch grup badawczych, niemieckiej i amerykańskiej. Nie-miecki chemik, Lohmann pracujący w Heidelbergu oraz Fi-ske i jego student Subbarow, pracujący w Harvard Medical School, niemal jednocześnie pokazali, że skurcz komórek mięśniowych zależy od dostępności cząsteczki występują-cej w ekstraktach mięśni. Wykazali, że cząsteczka ta skła-da się z łańcucha trzech grup fosforanowych połączonych z adenozyną, czyli purynową zasadą adeniny związaną z cukrem, rybozą [11,12]. Badania z lat późniejszych potwier-dziły budowę tej cząsteczki jako adenozynotrifosforanu (ATP) i w połowie lat czterdziestych ostatecznie ustaliły jego strukturę.
W tym samym czasie, w którym nastąpiło odkrycie ATP, Drury i Szent-Gyȍrgyi przedstawili wyniki swoich badań pokazujące działanie adenozyny i AMP, otrzymanych z mięśnia sercowego, na pracę tego organu [13]. Wykazali, że podanie dożylne śwince morskiej tych związków wywołu-je rozszerzenie naczyń krwionośnych i obniżenie ciśnienia krwi [13]. Kontynuując powyższą tematykę, w 1934 r., Gil-lespie zwrócił uwagę na rolę struktury związków adenino-wych w wywoływanych przez nie efektach fizjologicznych [14]. Gillespie pokazał, że kolejne usuwanie grup fosfora-nowych z podawanych królikowi czy kotu związków ade-ninowych wywołuje coraz bardziej postępujące obniżanie ciśnienia krwi. Zgodnie z tą obserwacją podanie ATP pod-wyższało ciśnienie, podczas gdy adenozyna i AMP takich efektów nie wywoływały. Ponadto, ATP powodowało wie-lokrotnie silniejszy niż adenozyna skurcz jelit [14]. Wyniki te były pierwszymi wskazującymi, że ATP i adenozyna wy-wołują odmienne efekty fizjologiczne i sugerowały istnienie specyficznych receptorów, różnych dla różnych nukleoty-dów. W owym czasie nikt jednak nie przypuszczał, że ATP może odgrywać jakąś rolę poza komórką.
Musiało upłynąć wiele lat, aby okazało się, że ATP może działać na zewnątrz komórki, pełniąc funkcje zewnątrzko-mórkowego przekaźnika informacji. Aż do lat 70. zeszłego wieku uważano bowiem, że ATP pełni wyłącznie rolę czą-steczki przenoszącej energię wewnątrz komórki, nośnika energii w procesach wewnątrzkomórkowych wymagają-cych i zużywających energię. Taką funkcję ATP w układach biologicznych przedstawił w 1941 r. Lippman [15] i za to odkrycie został 12 lat później uhonorowany Nagrodą No-bla. ATP uznano za najważniejszą, uniwersalną cząsteczkę, źródło łatwo dostępnej chemicznej energii we wszystkich żywych komórkach [16,17]. W wyniku tych odkryć badania nukleotydów w ciągu wielu lat koncentrowały się jedynie na ich wewnątrzkomórkowych funkcjach energetycznych.
Jak powszechnie wiadomo, ATP jest syntetyzowany w komórkach eukariotycznych w procesie glikolizy oraz oksy-dacyjnej fosforylacji. Tak otrzymany ATP ulega hydrolizie do ADP i fosforanu w wielu reakcjach biochemicznych wy-
magających energii. Toteż stężenie ATP wewnątrz komórki jest wypadkową szybkości syntezy i hydrolizy tego nukle-otydu i jest ściśle związane ze stanem metabolicznym da-nej komórki. Może zatem być różne w różnych komórkach, ale też i w tej samej komórce. Jest jednak zawsze wysokie, w cytoplazmie komórek ssaków wartość ta wynosi około 3–5 mM. W odróżnieniu, stężenie ATP w środowisku ze-wnętrznym komórek w stanie spoczynku jest bardzo niskie i utrzymuje się na poziomie 1–10 nM [18,19]. Tak niskie stę-żenie wydawało się racjonalne, bowiem wielkość i ładunek cząsteczki ATP nie pozwala jej na przekroczenie dwuwar-stwy lipidowej błony plazmatycznej w procesie prostej dy-fuzji [20,21]. Sądzono, że jedynie w wyniku mechanicznego uszkodzenia tkanek i komórek, ATP i inne nukleotydy wy-dostają się z cytoplazmy i pojawiają w strumieniu krwi [22]. Istotnie, nagły wzrost stężenia zewnątrzkomórkowego ATP obserwowano we krwi rannych żołnierzy zabieranych z pól bitwy w czasie II wojny światowej [22,23].
Bardzo niskie stężenie zewnątrzkomórkowego ATP sta-nowiło argument popierający tezę, że nukleotyd ten działa jedynie wewnątrz komórek. Dane literaturowe wskazują-ce na jego uwalnianie traktowano sceptycznie. Jednak w ciągu ostatnich lat dokonano wielu odkryć wskazujących, że ATP może i jest uwalniany do środowiska zewnątrzko-mórkowego zarówno przez komórki o typowych właści-wościach wydzielniczych, jak i komórki niesekrecyjne. I tak wykazano, że w neuronach, komórkach chromochłonnych i groniastych komórkach trzustki, ATP jest pakowane do pęcherzyków wydzielniczych: pęcherzyków synaptycz-nych czy granul chromochłonnych i w takiej postaci trans-portowane poza komórkę [23-26]. ATP odkryto również w środowisku zewnątrzkomórkowym komórek niesekre-cyjnych, takich jak komórki nabłonkowe [27,28], śródbłon-kowe [29,30], mięśni gładkich, fibroblastów i limfocytów [31], hepatocytów [32] czy wielu transformowanych linii komórkowych. Wydzielanie ATP z tego typu komórek było wywoływane przez stymulację mechaniczną (stres mechaniczny), działanie związków farmakologicznych (np. trypsyny, trombiny, epinefryny) czy stres fizjologiczny (np. niedotlenienie) [23,30,31]. W neuronach obwodowego i ośrodkowego układu nerwowego, ATP zmagazynowany w pęcherzykach synaptycznych jest uwalniany w wyniku depolaryzacji błony [23]. Wiele rodzajów komórek uwalnia ATP oraz adenozynę za pośrednictwem specyficznych bia-łek transportujących. Wydaje się, że taką rolę w uwalnianiu nukleotydów z komórek śródbłonka, w procesie regulują-cym światło naczyń krwionośnych, mogą odgrywać ATPa-zy z rodziny ABC (ang. ATP-binding casette proteins) [23,30]. W komórkach astrocytów hodowanych in vitro, Coco i wsp. [33], opisali mechanizm wydzielania ATP, jako proces za-leżnej od jonów wapnia egzocytozy. Uważa się, że nie tylko ATP, lecz również inne nukleotydy (np. UTP) oraz cukrowe pochodne nukleotydów (np. UDP-glukoza) są uwalniane z komórek na drodze transportu pęcherzykowego w podob-nym procesie [34]. Należy dodać, że wydzielane z komórek do środowiska zewnątrzkomórkowego nukleotydy ulegają szybko degradacji, jak również fosforylacji przez specyficz-ne ektoenzymy obecne na zewnętrznej powierzchni błon plazmatycznych [3,4,18,35,36]. W ten sposób uwolniony do przestrzeni zewnątrzkomórkowej ATP jest hydrolizowany kolejno do ADP, AMP i adenozyny, a UDP fosforylowany

426 www.postepybiochemii.pl
do UTP [34]. Powstałe związki mogą teraz oddziaływać ze specyficznymi błonowymi receptorami obecnymi na po-wierzchni tej samej lub sąsiedniej komórki. Podwyższone stężenie zewnątrzkomórkowych nukleotydów jest więc zja-wiskiem przejściowym, ma miejsce po zadziałaniu określo-nych czynników stymulujących i jest niezwykle precyzyjnie regulowane.
Obecność receptorów dla ATP została po raz pierwszy za-sugerowana przez Burnstocka na początku lat 70. ubiegłego wieku [37]. Była ona wynikiem jego wcześniejszych badań prowadzonych w latach 60. na Uniwersytecie w Melbourne, w których stwierdził, że skurcz jelit czy pęcherza moczowe-go zachodzi w procesie, w którym nie biorą udziału prze-kaźniki nerwowe, acetylocholina i noradrenalina, a bierze udział ATP. Wynik ten był zgodny z wcześniejszymi obser-wacjami Holton [38], która wykazała uwalnianie cząsteczek ATP z nerwów czuciowych. W środowisku naukowym ist-niało jednak tak silne przekonanie o nadrzędnej, wewnątrz-komórkowej funkcji ATP jako nośnika energii, że uzyska-nie pewności i opublikowanie wyników świadczących o ATP jako działającym zewnątrzkomórkowo przekaźniku nerwowym zajęło Burnstockowi około dziesięciu lat. I tak, wreszcie w 1972 r., Burnstock w Pharmacological Review [37] przedstawił rezultaty swoich 10-letnich doświadczeń, z których niezbicie wynikało, że istotnie ATP jest wydzielany z nerwów, które nazwał „purynergicznymi”. Wnioskował, że skoro ATP jest uwalniany do szczeliny synaptycznej, to należy przypuszczać, że komórki postsynaptyczne odbiera-jące taki sygnał powinny posiadać receptory dla tego nu-kleotydu znajdujące się na powierzchni komórki [37]. Zało-żenie to zostało poparte dowodami farmakologicznymi; w 1976 r., Burnstock zdefiniował białka pełniące taką funkcję i nazwał je „receptorami purynergicznymi” [39]. Dwa lata później, opierając się na danych farmakologicznych i bio-chemicznych, dokonał podziału tych receptorów na dwie klasy: P1, odpowiadające na adenozynę i P2, odpowiadają-ce na ATP i ADP [40]. Dalsze badania prowadzone w latach 80., również w pracowni Burnstocka, doprowadziły do ak-tualnego do dziś podziału receptorów P2 na dwa podtypy: P2X i P2Y [41]. Następnie, w latach 90. ubiegłego wieku, dzięki wprowadzeniu metod burzliwie rozwijającej się bio-logii molekularnej, w tym analizy sekwencji klonowanych receptorów, wyizolowano i określono strukturę molekular-ną receptorów P2X i P2Y i poznano szlaki przekazywanych przez nie wewnątrzkomórkowych sygnałów [1,2,24,42].
Badania lat 90. wykazały również, że nie tylko nukle-otydy purynowe, lecz także pirymidynowe, takie jak UTP i UDP, posiadają swoje receptory w wielu komórkach [5,6,43]. Stwierdzono, że receptory te mają podobną charakterysty-kę farmakologiczną i molekularną jak receptory P2. Z tego powodu, zgodnie z zaleceniem Komitetu do Spraw Nazew-nictwa Receptorów i Klasyfikacji Leków Międzynarodowej Unii Farmakologicznej (IUPHAR, ang. International Union of Basic and Clinical Pharmacology) dokonano zmiany nazwy „receptory purynergiczne” na „receptory nukleotydowe” [44,45]. Jednak podana przez Burnstocka pierwotna nazwa „receptory purynergiczne” jest stale obecna w literaturze.
W latach następnych, już w XXI wieku, nastąpił lawino-wy rozwój tej dziedziny wiedzy [2]. Identyfikacja recepto-
rów nukleotydowych pozwoliła ustalić ich lokalizację na komórkach różnych tkanek i organizmów i dostarczyła dowodów wskazujących, że reprezentują one wielką ro-dzinę, szeroko rozpowszechnioną w całym świecie żywym [7]. Obecnie nikt już nie wątpi, że ATP, inne nukleotydy, a także adenozyna pełnią istotną rolę jako zewnątrzkomór-kowe przekaźniki informacji oddziałujące ze specyficznymi receptorami, a Burnstock jest słusznie uważany nie tylko za odkrywcę, lecz także „duchowego ojca” tych receptorów.
EKTOENZYMY I PODZIAŁ RECEPTORÓW NUKLEOTYDOWYCH
Jak już wspomniano, wydzielane nukleotydy nie są sta-bilne w środowisku zewnątrzkomórkowym i ulegają degra-dacji katalizowanej przez ektoenzymy związane z błoną ko-mórkową. Nazwą „ektoenzymy nukleotydowe” określamy enzymy błony plazmatycznej, które po zewnętrznej stronie błony specyficznie metabolizują nukleotydy docierające do nich z przestrzeni zewnątrzkomórkowej [3,4].
Do najbardziej znanych grup ektoenzymów należą: E--NTPDazy (ang. ecto-nucleotidase 5’-triphosphate diphospho-hydrolases), E-NPPazy (ang. ecto-phosphodiesterase/nucleotide pyrophosphatase), ekto-5’-nukleotydaza oraz alkaliczna fos-fataza [18,35,36].
Rodzina E-NTPDaz (klasyfikowana poprzednio jako ro-dzina E-ATPaz) charakteryzuje się zdolnością do hydrolizy tri- i dinukleotydów. U ssaków wyróżniamy, co najmniej 8 sklonowanych i scharakteryzowanych enzymów należą-cych do tej grupy (NTPDazy1-8). Enzymy te to integralne białka błony plazmatycznej składające się z dwóch domen transbłonowych o krótkich odpowiednio N- i C-końcach znajdujących się od strony cytosolowej. Po stronie zewnątrz- komórkowej domeny transbłonowe połączone są dużą pętlą utworzoną przez łańcuch aminokwasów zawiera-jący zachowane w ewolucji regiony wrażliwe na apyrazę [3,36]. Wśród NTPDaz należy wyróżnić NTPDazę1 (znaną również jako ekto-apyraza i ekto-ATP-difosfohydrolaza). Enzym ten przeprowadza dwie reakcje, hydrolizuje tri- i difosforany nukleozydów prowadząc do powstania mono-fosforanów nukleozydów. NTPDaza1 działa zatem zarów-no na ATP jak i na ADP przeprowadzając reakcję: ATP → AMP + 2Pi, oraz reakcję: ADP → AMP + Pi, w obu przypad-kach prowadząc do powstania AMP. Natomiast NTPDaza2 selektywnie hydrolizuje trifosforany nukleozydów do di-fosforanów nukleozydów (ATP → ADP + Pi) [35]. Z kolei NTPDaza3 hydrolizuje zarówno trifosforany nukleozydów jak i difosforany nukleozydów (ATP → ADP + Pi, ADP → AMP + Pi), z preferencją 3:1 dla trifosforanów, a NTPDaza8 charakteryzuje się szerokim spektrum działania [18,34,36].
Rodzina ekto-pirofosfataz/fosfodiesteraz (E-NPP) za-wiera trzy białka (E-NPP1, E-NPP2 oraz E-NPP3), kodo-wane przez różne geny. W budowie ich można wyróżnić krótki koniec aminowy od strony cytosolowej błony, jedną domenę transbłonową oraz długą zewnątrzkomórkową domenę zawierającą miejsce katalityczne enzymu. Enzymy te charakteryzują się szeroką specyficznością substratową. Mogą hydrolizować zarówno tri- i difosforany nukleozy-dów do monofosforanów nukleozydów, używając jako sub-

Postępy Biochemii 60 (4) 2014 427
stratów nie tylko ATP, lecz również UTP, GTP, ADP i UDP, a także hydrolizować cykliczny AMP (cAMP) do AMP, jak i kwasy nukleinowe oraz cukrowe pochodne nukleotydów (UDP-glukoza) [34,36].
Ekto-5’-nukleotydaza to enzym, który konwertuje mo-nofosforany nukleozydów do nukleozydów i ortofosfora-nu (AMP → Adenozyna + Pi). A więc w wyniku degradacji AMP (powstającego w przestrzeni zewnątrzkomórkowej w wyniku hydrolizy ATP i ADP), enzym ten ma udział w aku-mulacji zewnątrzkomórkowej adenozyny. Natomiast alka-liczna fosfataza hydrolizuje niespecyficznie każdy z powyż-szych etapów, działając zarówno na ATP, jak i ADP i AMP, prowadząc również do powstania adenozyny [3,4,18,34-36].
Znane są także interkonwertujące właściwości enzymów takich jak mono- i di-kinazy nukleozydów. Zewnątrzko-mórkowe difosfokinazy nukleozydowe, NPDK (ang. nuc-leoside diphosphokinase) katalizują bowiem dwukierunkową reakcję transfosforylacji, zachodzącą według wzoru: ATP + UDP ↔ ADP + UTP. W komórkach nowotworów pocho-dzenia glejowego (gwiaździaka 1321N1 oraz glejaka C6) NDPK bardzo szybko tworzą ATP z endogennego ADP w obecności odpowiedniego stężenia UTP w środowisku ze-wnątrzkomórkowym [18,34].
Tak więc, w krótszym lub dłuższym cyklu przemian en-zymatycznych, z uwolnionego do przestrzeni zewnątrzko-mórkowej ATP może w wyniku hydrolizy powstać zarów-no ADP jak i adenozyna oraz w wyniku reakcji transfosfory-lacji nie tylko UTP, lecz również ATP [18,34,36].
Tak powstałe związki mogą teraz specyficznie oddziały-wać z błonowymi receptorami wrażliwymi na zewnątrzko-mórkowe nukleotydy: ATP, ADP, UTP, UDP, cukrowe po-chodne UDP oraz adenozynę. Zasadniczy podział recepto-rów nukleotydowych opiera się właśnie na tych kryteriach, specyficzności receptorów wobec określonych, działających na nie agonistów.
Zgodnie z tymi kryteriami receptory nukleotydowe dzie-limy na dwie duże klasy: receptory P1 aktywowane przez adenozynę i receptory P2 wykazujące powinowactwo do ATP, ADP, UTP, UDP oraz do cukrowych pochodnych UDP. Ze względu na różnice w budowie oraz charaktery-stykę farmakologiczną i molekularną receptory P2 dzielimy dalej na receptory P2X i P2Y. Receptory P2X są receptorami jonotropowymi, błonowymi kanałami jonowymi, natomiast receptory P2Y, podobnie jak P1, to receptory metabotropo-we, związane z trójpodjednostkowymi białkami G [1,2,9,39-42].
Rycina 1 przedstawia podział receptorów nukleotydo-wych. Opierając się na danych farmakologicznych i bioche-micznych, jak również na analizie sekwencji klonowanych receptorów, receptory P1 podzielono na 4 podtypy (A1, A2A, A2B, A3), a receptory P2Y na 8 podtypów (P2Y1, P2Y2, P2Y4, P2Y6, P2Y11, P2Y12, P2Y13, P2Y14). Konsekwencją pobudzenia tych receptorów jest aktywacja różnych szlaków sygnało-wych przebiegających wewnątrz komórki. Mechanizm ich działania przedstawia schematycznie rycina 1. Natomiast, wśród jonotropowych receptorów P2X wyróżniamy 7 pod-typów (P2X1-7), a ich charakterystyka i mechanizm działania zostaną omówione w dalszych częściach tekstu.
RECEPTORY P1 — METABOTROPOWE, AKTYWOWANE PRZEZ ADENOZYNĘ
Adenozyna stanowi podstawową „cegiełkę budulcową” wielu biologicznie czynnych związków, takich jak ATP, di-nukleotyd nikotynoamidoadeninowy (NAD+) czy kwasy nukleinowe. Powstając w komórce w wyniku przekształceń S-adenozylometioniny i S-adenozylohomocysteiny może jednak nie ulegać dalszym przekształceniom, a jako taka być transportowana poza komórkę przez działające dwukierun-kowo białko transportujące (ENT, ang. equilibrative nucleosi-de transporter), za pośrednictwem którego wewnątrz- i ze-wnątrzkomórkowe stężenia tej substancji są ze sobą ściśle powiązane [46-48]. Należy dodać, że adenozyna może nie
tylko być transportowana z powrotem do komór-ki poprzez owe specyficzne białko transportują-ce, lecz także wnikać do niej przez prostą dyfu-zję, procesy, które nie mają miejsca w przypadku ATP i ADP [20,21,46]. A więc, pierwszy ze spo-sobów, dzięki któremu adenozyna może pojawić się na zewnątrz komórki to transport. Drugi, to jej powstanie w wyniku omówionej już uprzednio degradacji uwolnionego z komórki ATP w proce-sie: ATP→ADP→AMP→adenozyna, katalizowa-nym przez ektonukleotydazy znajdujące się na zewnętrznej powierzchni błony plazmatycznej [4,18,36]. Uwolniona z komórki, czy też utworzo-na pozakomórkowo adenozyna może teraz od-działywać ze specyficznymi receptorami błony plazmatycznej, wywierając wpływ na wiele pro-cesów związanych z funkcjonowaniem układu krążenia, układu wewnątrzwydzielniczego, od-pornościowego czy wreszcie nerwowego [48-51].
W płynach ustrojowych stężenie adenozyny w warunkach spoczynkowych wynosi 30-200 nM i
Rycina 1. Schematyczny podział receptorów nukleotydowych z zaznaczeniem ich głównych funkcji. Rycina pokazuje podtypy receptorów P1 (A1,A2A,A2B,A3) aktywowanych przez adenozy-nę. Receptory P2 dzielimy na P2X i P2Y. Podtypy receptorów P2Y (P2Y1,P2Y2, P2Y4, P2Y6, P2Y11, P2Y12, P2Y13, P2Y14) są aktywowane przez ATP, ADP, UTP, UDP i cukrowe pochodne UDP, a podtypy receptorów P2X1-7 (niepokazane na rycinie, omówione w tekście) są aktywowane przez ATP. Podtypy receptorów P1 i P2Y współdziałają z trójpodjednostkowymi białkami G i indukują różne szlaki sygnalizacyjne wewnątrz komórki. AC — cyklaza adenylanowa; cAMP — cykliczny-AMP; [Ca2+] — stężenie jonów wapnia; PLC — fosfolipaza C; ↑ — wzrost ilości; ↓ — spadek ilości.
428 www.postepybiochemii.pl
tak jak w przypadku ATP, jest ono zbyt niskie, aby aktywo-wać receptory błony plazmatycznej [52]. Jednak po pobu-dzeniu stężenie adenozyny gwałtownie wzrasta. W sytuacji stresowej, np. przy niedokrwistości (ischemii), wartości te mogą wzrastać nawet stukrotnie. Po pobudzeniu komórki i uwolnieniu ATP, moment pojawienia się wzrostu zewną-trzkomórkowego stężenia adenozyny jest zawsze wtórny, opóźniony w stosunku do obserwowanego na początku skoku wzrostu stężenia ATP [48,53]. Przy tej wzajemnej relacji: ATP — adenozyna, pobudzające działanie ATP jest zazwyczaj kompensowane przeciwstawnym działaniem adenozyny. W układzie nerwowym, adenozyna nie będąc magazynowana w pęcherzykach synaptycznych, ani uwal-niana w wyniku depolaryzacji, nie jest traktowana jako „prawdziwy” neuroprzekaźnik. Natomiast, ze względu na wspomnianą powyżej relację z ATP, jest uważana za istotny neuromodulator [48,50].
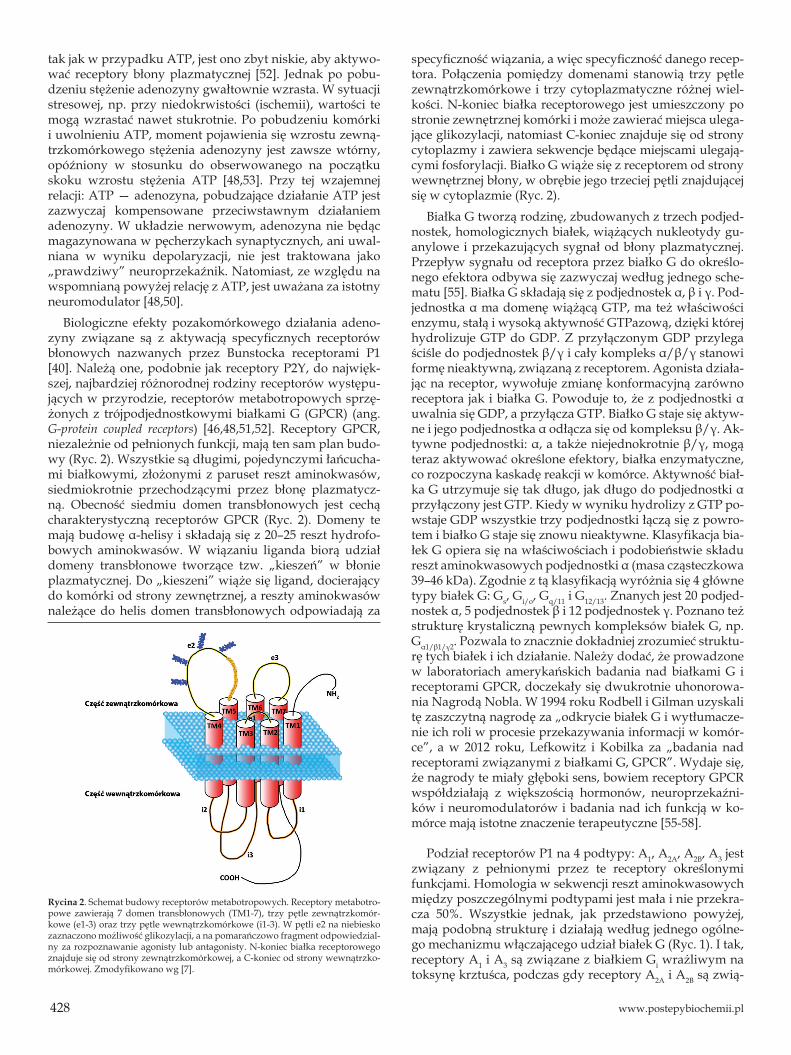
Biologiczne efekty pozakomórkowego działania adeno-zyny związane są z aktywacją specyficznych receptorów błonowych nazwanych przez Bunstocka receptorami P1 [40]. Należą one, podobnie jak receptory P2Y, do najwięk-szej, najbardziej różnorodnej rodziny receptorów występu-jących w przyrodzie, receptorów metabotropowych sprzę-żonych z trójpodjednostkowymi białkami G (GPCR) (ang. G-protein coupled receptors) [46,48,51,52]. Receptory GPCR, niezależnie od pełnionych funkcji, mają ten sam plan budo-wy (Ryc. 2). Wszystkie są długimi, pojedynczymi łańcucha-mi białkowymi, złożonymi z paruset reszt aminokwasów, siedmiokrotnie przechodzącymi przez błonę plazmatycz-ną. Obecność siedmiu domen transbłonowych jest cechą charakterystyczną receptorów GPCR (Ryc. 2). Domeny te mają budowę α-helisy i składają się z 20–25 reszt hydrofo-bowych aminokwasów. W wiązaniu liganda biorą udział domeny transbłonowe tworzące tzw. „kieszeń” w błonie plazmatycznej. Do „kieszeni” wiąże się ligand, docierający do komórki od strony zewnętrznej, a reszty aminokwasów należące do helis domen transbłonowych odpowiadają za
specyficzność wiązania, a więc specyficzność danego recep-tora. Połączenia pomiędzy domenami stanowią trzy pętle zewnątrzkomórkowe i trzy cytoplazmatyczne różnej wiel-kości. N-koniec białka receptorowego jest umieszczony po stronie zewnętrznej komórki i może zawierać miejsca ulega-jące glikozylacji, natomiast C-koniec znajduje się od strony cytoplazmy i zawiera sekwencje będące miejscami ulegają-cymi fosforylacji. Białko G wiąże się z receptorem od strony wewnętrznej błony, w obrębie jego trzeciej pętli znajdującej się w cytoplazmie (Ryc. 2).
Białka G tworzą rodzinę, zbudowanych z trzech podjed-nostek, homologicznych białek, wiążących nukleotydy gu-anylowe i przekazujących sygnał od błony plazmatycznej. Przepływ sygnału od receptora przez białko G do określo-nego efektora odbywa się zazwyczaj według jednego sche-matu [55]. Białka G składają się z podjednostek α, β i γ. Pod-jednostka α ma domenę wiążącą GTP, ma też właściwości enzymu, stałą i wysoką aktywność GTPazową, dzięki której hydrolizuje GTP do GDP. Z przyłączonym GDP przylega ściśle do podjednostek β/γ i cały kompleks α/β/γ stanowi formę nieaktywną, związaną z receptorem. Agonista działa-jąc na receptor, wywołuje zmianę konformacyjną zarówno receptora jak i białka G. Powoduje to, że z podjednostki α uwalnia się GDP, a przyłącza GTP. Białko G staje się aktyw-ne i jego podjednostka α odłącza się od kompleksu β/γ. Ak-tywne podjednostki: α, a także niejednokrotnie β/γ, mogą teraz aktywować określone efektory, białka enzymatyczne, co rozpoczyna kaskadę reakcji w komórce. Aktywność biał-ka G utrzymuje się tak długo, jak długo do podjednostki α przyłączony jest GTP. Kiedy w wyniku hydrolizy z GTP po-wstaje GDP wszystkie trzy podjednostki łączą się z powro-tem i białko G staje się znowu nieaktywne. Klasyfikacja bia-łek G opiera się na właściwościach i podobieństwie składu reszt aminokwasowych podjednostki α (masa cząsteczkowa 39–46 kDa). Zgodnie z tą klasyfikacją wyróżnia się 4 główne typy białek G: Gs, Gi/o, Gq/11 i G12/13. Znanych jest 20 podjed-nostek α, 5 podjednostek β i 12 podjednostek γ. Poznano też strukturę krystaliczną pewnych kompleksów białek G, np. Gα1/β1/γ2. Pozwala to znacznie dokładniej zrozumieć struktu-rę tych białek i ich działanie. Należy dodać, że prowadzone w laboratoriach amerykańskich badania nad białkami G i receptorami GPCR, doczekały się dwukrotnie uhonorowa-nia Nagrodą Nobla. W 1994 roku Rodbell i Gilman uzyskali tę zaszczytną nagrodę za „odkrycie białek G i wytłumacze-nie ich roli w procesie przekazywania informacji w komór-ce”, a w 2012 roku, Lefkowitz i Kobilka za „badania nad receptorami związanymi z białkami G, GPCR”. Wydaje się, że nagrody te miały głęboki sens, bowiem receptory GPCR współdziałają z większością hormonów, neuroprzekaźni-ków i neuromodulatorów i badania nad ich funkcją w ko-mórce mają istotne znaczenie terapeutyczne [55-58].
Podział receptorów P1 na 4 podtypy: A1, A2A, A2B, A3 jest związany z pełnionymi przez te receptory określonymi funkcjami. Homologia w sekwencji reszt aminokwasowych między poszczególnymi podtypami jest mała i nie przekra-cza 50%. Wszystkie jednak, jak przedstawiono powyżej, mają podobną strukturę i działają według jednego ogólne-go mechanizmu włączającego udział białek G (Ryc. 1). I tak, receptory A1 i A3 są związane z białkiem Gi wrażliwym na toksynę krztuśca, podczas gdy receptory A2A i A2B są zwią-
Rycina 2. Schemat budowy receptorów metabotropowych. Receptory metabotro-powe zawierają 7 domen transbłonowych (TM1-7), trzy pętle zewnątrzkomór-kowe (e1-3) oraz trzy pętle wewnątrzkomórkowe (i1-3). W pętli e2 na niebiesko zaznaczono możliwość glikozylacji, a na pomarańczowo fragment odpowiedzial-ny za rozpoznawanie agonisty lub antagonisty. N-koniec białka receptorowego znajduje się od strony zewnątrzkomórkowej, a C-koniec od strony wewnątrzko-mórkowej. Zmodyfikowano wg [7].

Postępy Biochemii 60 (4) 2014 429
zane z białkiem Gs wrażliwym na toksynę cholery [52]. Biał-ka Gi hamują, a białka Gs stymulują (w obu przypadkach ich podjednostki α) aktywność cyklazy adenylanowej (AC). A więc, w wyniku aktywacji przez adenozynę, receptory prze-kazują sygnał na określone białka G, które działając na dany efektor, w tym przypadku cyklazę adenylanową, powodują albo wzrost, albo zmniejszenie poziomu cyklicznego AMP (cAMP) w komórce (Ryc. 1). Ponadto receptory A1 i A3 są również związane z białkiem Gq/G11, którego aktywacja prowadzi do stymulacji aktywności fosfolipazy C i w efek-cie kaskady następujących po sobie reakcji do zwiększenia stężenia wolnych jonów wapnia w komórce [59]. Ponadto, receptor A1, dominujący w sercu i w centralnym układzie nerwowym, może przez włączenie białka Go (również ha-mującego cyklazę adenylanową) oddziaływać bezpośred-nio na kanał K+ w błonie plazmatycznej i przez jego otwar-cie i wypływ K+ z komórki prowadzić do hiperpolaryzacji błony, stanowiąc podstawę antyepileptycznego działania adenozyny [48,60]. Z kolei, receptor A3 może również akty-wować kaskadę kinaz MAP, a także z udziałem białka G12/13 aktywować małe, jednopodjednostkowe białko G, białko RhoA (nie pokazano na Ryc. 1) [48].
Receptory A1 i A2A charakteryzuje wysokie powinowac-two do adenozyny, co powoduje możliwość ich aktywacji przez jej fizjologiczne stężenia, występujące po pobudzeniu komórki w warunkach normalnych. Natomiast receptory A2B i A3 charakteryzują się niskim powinowactwem, toteż jedynie wysokie stężenia adenozyny są zdolne do ich ak-tywacji. W warunkach niedotlenienia (hipoksji), występu-jących w ischemii, stanach zapalnych, czy wnętrzu litych guzów nowotworowych, następuje znaczne zwiększenie stężenia adenozyny, jej akumulacja. Jest to spowodowane zatrzymaniem syntezy ATP przez nieaktywną w tych wa-runkach cytosolową kinazę adenozyny i wskazuje na rolę receptorów P1 w powyższych stanach patologicznych [48]. Receptory adenozyny są szeroko rozpowszechnione w świecie żywym, często w jednej komórce występuje więcej niż jeden ich podtyp. Wytłumaczeniem tego zjawiska może być właśnie odpowiedź na różne stężenia adenozyny. I tak, w płodowych komórkach miocytów serca kurczaka wystę-pują wspólnie receptory A2A i A2B. Oba, będąc związane z białkiem Gs generują zwiększenie poziomu cAMP i odgry-wają rolę w procesie skurczu. Za proces ten w warunkach fizjologicznych odpowiada jednak jedynie receptor A2A, na-tomiast przy niedokrwistości (ischemii) za proces ten odpo-wiada receptor A2B [52]. Z kolei, w neutrofilach, niskie stę-żenia adenozyny aktywują receptor A1 związany z białkiem Gi, podczas gdy wysokie działają na receptor A2B związany z białkiem Gs, niwelując efekty wywołane przez receptor A1 [61].
W układzie limfatycznym receptory adenozynowe P1 odgrywają istotną rolę w mechanizmie selekcji limfocytów T w trakcie ich dojrzewania w grasicy. Likwidacja autoreak-tywnych klonów odbywa się na drodze apoptozy induko-wanej przez adenozynę [51]. W układzie limfatycznym wy-stępuje szeroko rozpowszechniony receptor A2A. Występuje on także w mózgu, w obszarach charakteryzujących się wy-soką reprezentacją receptorów dopaminowych. Takim ob-szarem bogatym w receptory D2 jest prążkowie (striatum). W prążkowiu wzajemne działanie receptorów A2A i D2 jest
antagonistyczne, bowiem A2A poprzez białko Gs pozytyw-nie oddziałuje na cyklazę adenylanową, a D2 są związane z białkiem Gi i hamują aktywność tego enzymu. Uważa się, że zmiany w funkcjonalnym współdziałaniu tych recepto-rów mogą odgrywać rolę w rozwoju choroby Parkinsona. W chorobie tej następuje drastyczne obniżenie poziomu do-paminy. Wydaje się, że zastosowanie selektywnych antago-nistów receptora A2A mogłoby stanowić pewną nową formę terapii powstrzymującą rozwój choroby Parkinsona [62].
Receptor A3 poznano stosunkowo najpóźniej. Jest on pierwszym podtypem receptora P1, który został sklono-wany przed charakterystyką farmakologiczną [48]. Choć obecnie struktura jego jest już poznana, to właściwości tego receptora są stale trudne do dokładnego określenia ze względu na niską homologię międzygatunkową i różny profil farmaceutyczny, np. różną odpowiedź na działanie antagonistów, a nawet różne rozmieszczenie tkankowe [63]. Wysoka ekspresja mRNA białka receptorowego A3 wystę-puje w płucach, mózgu, sercu czy wątrobie, a bardzo wy-soka jest charakterystyczna dla różnych linii komórek no-wotworowych. Poziom białka receptorowego A3 jest w tych komórkach tak wysoki, że Gessi i wsp. [63], zaproponowali nawet uznanie go za marker nowotworzenia. Receptor A3 jest związany z białkiem Gi i Gq, a więc zmniejsza poziom cAMP i powoduje wzrost stężenia Ca2+ w komórce (Ryc. 1). Ponadto, przez podjednostkę β/γ białka Gi stymuluje kaskadę MAP kinaz, co w efekcie końcowym prowadzi do indukcji czynników transkrypcyjnych w jądrze i wpływa na ekspresję genów. W liniach komórkowych czerniaka, chło-niaka i komórek raka prostaty in vitro, receptor A3 działa na białko Gi. Wynikiem zahamowania syntezy cAMP jest redukcja aktywności kinazy białkowej A i kinazy białko-wej B, zwanej także Akt [48]. Z drugiej strony, aktywacja białka Gq prowadzi do stymulacji aktywności fosfolipazy C (PLC) i wzrostu stężenia Ca2+. Wydłużony w czasie sygnał wapniowy i zahamowanie aktywności powyższych kinaz prowadzi do zahamowania proliferacji komórek i włącze-nia szlaku prowadzącego do apoptozy. To proapoptotyczne działanie receptora A3 sugeruje możliwość używania agoni-stów tego receptora w chemioterapii przeciwko nowotwo-rowym liniom komórkowym [63]. Natomiast w przypadku litych guzów nowotworowych działanie receptora A3 jest odmienne, bowiem stymulując kaskadę szlaku MAP kinaz działa w kierunku wzmożenia proliferacji; w tym przypad-ku użyteczni w terapii mogliby być antagoniści tego recep-tora [48,63]. Konkludując, receptor A3 może zarówno sty-mulować, jak i hamować proliferację nowotworów, a jego wielostronne działanie jest od kilku lat przedmiotem inten-sywnych badań [48].
RECEPTORY P2Y — METABOTROPOWE, AKTYWOWANE PRZEZ NUKLEOTYDY
Receptory P2Y reprezentują najliczniejszą grupę wśród wszystkich receptorów nukleotydowych. Należy do nich 8 podtypów: P2Y1, P2Y2, P2Y4, P2Y6, P2Y11, P2Y12, P2Y13 i P2Y14, aktywowanych przez nukleotydy purynowe i pirymidyno-we: ATP, ADP, UTP, UDP i cukrowe pochodne UDP. Braku-jące numery receptorów P2Y odpowiadają receptorom nie w pełni scharakteryzowanym, tzw. „sierocym”, czy znale-zionym u kręgowców, ale niemającym jak dotąd odpowied-

430 www.postepybiochemii.pl
ników u ssaków [64]. Wszystkie należą do wielkiej rodziny receptorów metabotropowych GPCR, a więc charakteryzują się obecnością 7 domen transbłonowych i przekazywaniem sygnałów do wnętrza komórki przez współdziałające z re-ceptorami trójpodjednostkowe (α/β/γ) białka G. Wiązanie liganda ma miejsce w utworzonej przez domeny TM3, TM6 i TM7 „kieszeni” w błonie plazmatycznej. Za ten proces odpowiadają dodatnio naładowane reszty aminokwasów obecne w domenach tworzących kieszeń, oddziałujące elek-trostatycznie z grupami fosforanowymi nukleotydów do-cierającymi do komórki od strony zewnętrznej. Regiony te są zachowane w ewolucji, mimo, że homologia między po-szczególnymi podtypami receptorów P2Y wynosi od 21% do 57%, wskazując na ich różnorodność [5,6,42,45,64,65].
Filogenetycznie i strukturalnie podtypy receptorów P2Y dzielimy na dwie grupy, co znajduje swoje odbicie w drze-wie filogenetycznym [64]. Do pierwszej należą receptory P2Y1, P2Y2, P2Y4, P2Y6 i P2Y11, do drugiej P2Y12, P2Y13 i P2Y14. O ich rozróżnieniu decydują reszty aminokwasów w dome-nie TM6 i TM7. Dla wiązania agonistów istotny jest wystę-pujący w domenie TM6 u wszystkich receptorów P2Y mo-tyw: H-X-X-R/K. Natomiast w domenie TM7 u podtypów receptorów P2Y należących do pierwszej grupy występuje motyw: Y-Q/K-X-X-R. Receptory drugiej grupy, receptory P2Y12, P2Y13 i P2Y14, takiego motywu nie posiadają, charak-teryzuje je natomiast występujący w domenie TM7 motyw: K-E-X-X-L [64]. Ponadto funkcjonalnie, receptory pierwszej grupy współpracują z białkami G typu Gq/11 (pewien wy-jątek stanowi receptor P2Y11, o czym szerzej będzie mowa później), a drugiej z białkami typu Gi/o (Ryc. 1). Przedsta-wiony powyżej podział ma charakter filogenetyczno-struk-turalno-funkcjonalny. Natomiast od strony farmakologicz-nej receptory P2Y1-P2Y13 charakteryzują się różną wrażli-wością na nukleotydy adeninowe i urydynowe i możemy je podzielić na te, które odpowiadają na ATP i ADP, te, które odpowiadają na UTP i UDP, oraz te, które charakteryzują się tzw. selektywnością mieszaną [64]. Receptor P2Y14
to stosunkowo niedawno sklonowane białko receptorowe, uważane do 2003 r. za tzw. receptor „sierocy” [64]. Receptor P2Y14 należy do drugiej grupy receptorów P2Y i farmakolo-gicznie odpowiada na cukrowe pochodne UDP [64].
Wszystkie receptory zakwalifikowane do pierwszej gru-py: P2Y1, P2Y2, P2Y4, P2Y6 i P2Y11 są związane z białkami Gq/11 [51,57]. Ich stymulacja poprzez białko Gq powoduje aktywację PLC (Ryc. 1). Jest to fosfolipaza C typu β, działa-jąca specyficznie na znajdujący się w błonie plazmatycznej fosfolipid inozytolowy, fosfatydyloinozytolo-(4,5)-bisfosfo-ran (PIP2) [65]. W związku z tym, w powyższym szlaku sy-gnalizacyjnym sekwencja zdarzeń jest następująca: agonista działa na receptor sprzężony z białkiem Gq. Podjednostka α tego białka aktywuje fosfolipazę C typu β. Zaktywowany enzym działa hydrolitycznie na fosfolipid PIP2. W wyniku hydrolizy powstają dwa wtórne przekaźniki informacji: trisfosfoinozytol (IP3) i diacyloglicerol (DAG). DAG pozo-staje w błonie plazmatycznej i aktywuje kinazę białkową C (PKC); DAG jest uważany za naturalny aktywator tej kina-zy [57]. Aktywna PKC może wywierać hamujący efekt na receptor oraz na PLC, hamując hydrolizę PIP2 i wygasza-jąc sygnał wapniowy [66]. Aktywacja PKC prowadzi też do zwiększenia aktywności fosfolipazy D (PLD) oraz szlaku
kinaz MAP [65]. Natomiast IP3, cząsteczka inozytolu z 3 resztami fosforanowymi w pozycji 1,4,5 pierścienia inozy-tolowego, jest dobrze rozpuszczalna w wodzie i dyfunduje z błony plazmatycznej do siateczki śródplazmatycznej. Sia-teczka śródplazmatyczna jest głównym magazynem jonów wapnia w komórce zwierzęcej. IP3 łączy się w błonie siatecz-ki ze specyficznym receptorem IP3, będącym kanałem jono-wym. Po związaniu IP3 kanał otwiera się i zmagazynowane jony Ca2+ zostają uwolnione do cytosolu [67]. Opróżnienie magazynów siateczki śródplazmatycznej stanowi sygnał do otwarcia specyficznego, niezależnego od napięcia, kanału w błonie plazmatycznej i napływu dodatkowej porcji Ca2+ z przestrzeni międzykomórkowej, w tzw. procesie pojemno-ściowego napływu jonów wapnia (ang. capacitative calcium entry) [67,68]. A więc efektem końcowym aktywacji recepto-ra związanego z białkiem Gq jest zwiększenie poziomu wol-nych jonów wapnia w cytosolu, co schematycznie przedsta-wia rycina 1 i rycina 3.
Stężenie Ca2+ w cytosolu jest niskie i w komórce niepo-budzonej wynosi około 50-100 nM, podczas gdy w płynach ustrojowych jest to wartość dziesięć tysięcy razy większa (1-2 mM). Po pobudzeniu komórki poziom Ca2+ wzrasta dziesięciokrotnie (1 µM) i dlatego zmiana stężenia tego jonu jest tak ważna, kontrolująca wiele życiowych procesów, jak wzrost, podział, różnicowanie, procesy apoptozy, zapłod-nienie i śmierć [68]. Należy dodać, że PIP2 może ulegać nie tylko hydrolizie, lecz także fosforylacji, prowadzącej do powstania fosfatydyloinozytolo-(3,4,5)-trisfosforanu (PIP3). Ten fosfolipid nie ulega hydrolizie przez PLCβ, a rekrutu-je do błony plazmatycznej kinazę białkową B, zwaną także Akt i wiążąc się z nią powoduje jej aktywację [57,65]. Kinaza ta pełni istotne funkcje prożyciowe, zapobiegające apopto-zie; zahamowanie jej aktywności ma działanie przeciwne,
Rycina 3. Działanie nukleotydów na wybrane receptory P2 i generowane przez nie szlaki sygnalizacyjne. ATP działa na receptory P2X (składające się z 3 podjed-nostek tworzących kanał) i razem z UTP aktywuje receptor P2Y2. Ektoenzymy hy-drolizują ATP do ADP i adenozyny, która działa na receptory P1. ADP stymuluje zarówno receptor P2Y1 jak i receptor P2Y12. Receptory P2Y1 i P2Y2 poprzez białko Gq aktywują PLC, która hydrolizuje PIP2 do DAG i IP3. DAG aktywuje PKC, a IP3 dyfunduje do siateczki śródplazmatycznej (ER, ang. endoplasmic reticulum) i wiążąc się ze specyficznym receptorem uwalnia jony Ca2+ do cytosolu. Recep-tor P2Y12, poprzez białko Gi hamuje aktywność AC i zmniejsza poziom cAMP w komórce, hamując aktywność PKA. Objaśnienie używanych skrótów w tekście, zmodyfikowano wg [65].
Postępy Biochemii 60 (4) 2014 431
pro-apoptotyczne, co już sygnalizowano poprzednio oma-wiając działanie receptora A3.
Szlak sygnałowy, prowadzący do mobilizacji Ca2+ w ko-mórce jest typowy dla receptorów P2Y1, P2Y2, P2Y4 i P2Y6. Do pierwszej grupy należy jednak również receptor P2Y11. Ten receptor stanowi wyjątek, bowiem może być związany nie tylko z białkiem Gq/11, lecz także z białkiem Gs (Ryc. 1). W konsekwencji oddziałuje pozytywnie na dwa szlaki sy-gnałowe aktywując zarówno PLCβ jak i cyklazę adenylano-wą (AC), prowadząc do mobilizacji Ca2+ i powodując zwięk-szenie poziomu cAMP w komórce [5]. Z kolei, receptory P2Y12, P2Y13 i P2Y14, należące do drugiej grupy, zmniejszają poziom cAMP w komórce, hamując aktywność AC. Te re-ceptory są związane z białkiem Gi/o (Ryc. 1).
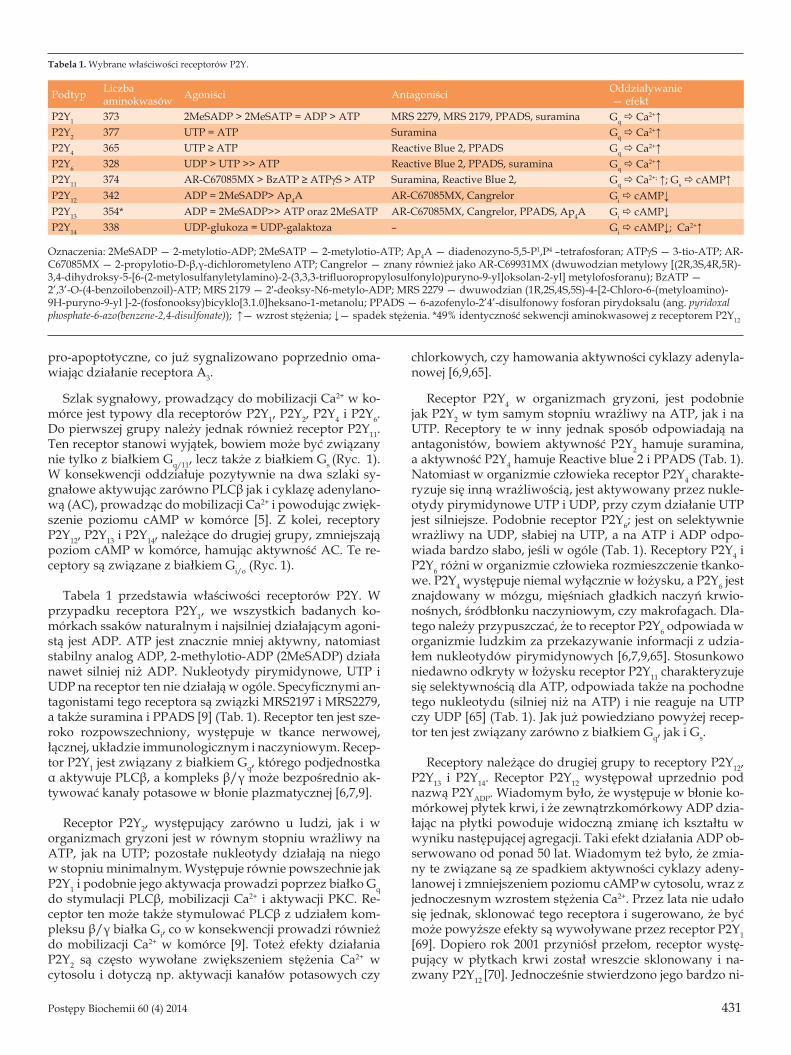
Tabela 1 przedstawia właściwości receptorów P2Y. W przypadku receptora P2Y1, we wszystkich badanych ko-mórkach ssaków naturalnym i najsilniej działającym agoni-stą jest ADP. ATP jest znacznie mniej aktywny, natomiast stabilny analog ADP, 2-methylotio-ADP (2MeSADP) działa nawet silniej niż ADP. Nukleotydy pirymidynowe, UTP i UDP na receptor ten nie działają w ogóle. Specyficznymi an-tagonistami tego receptora są związki MRS2197 i MRS2279, a także suramina i PPADS [9] (Tab. 1). Receptor ten jest sze-roko rozpowszechniony, występuje w tkance nerwowej, łącznej, układzie immunologicznym i naczyniowym. Recep-tor P2Y1 jest związany z białkiem Gq, którego podjednostka α aktywuje PLCβ, a kompleks β/γ może bezpośrednio ak-tywować kanały potasowe w błonie plazmatycznej [6,7,9].
Receptor P2Y2, występujący zarówno u ludzi, jak i w organizmach gryzoni jest w równym stopniu wrażliwy na ATP, jak na UTP; pozostałe nukleotydy działają na niego w stopniu minimalnym. Występuje równie powszechnie jak P2Y1 i podobnie jego aktywacja prowadzi poprzez białko Gq do stymulacji PLCβ, mobilizacji Ca2+ i aktywacji PKC. Re-ceptor ten może także stymulować PLCβ z udziałem kom-pleksu β/γ białka Gi, co w konsekwencji prowadzi również do mobilizacji Ca2+ w komórce [9]. Toteż efekty działania P2Y2 są często wywołane zwiększeniem stężenia Ca2+ w cytosolu i dotyczą np. aktywacji kanałów potasowych czy
chlorkowych, czy hamowania aktywności cyklazy adenyla-nowej [6,9,65].
Receptor P2Y4 w organizmach gryzoni, jest podobnie jak P2Y2 w tym samym stopniu wrażliwy na ATP, jak i na UTP. Receptory te w inny jednak sposób odpowiadają na antagonistów, bowiem aktywność P2Y2 hamuje suramina, a aktywność P2Y4 hamuje Reactive blue 2 i PPADS (Tab. 1). Natomiast w organizmie człowieka receptor P2Y4 charakte-ryzuje się inną wrażliwością, jest aktywowany przez nukle-otydy pirymidynowe UTP i UDP, przy czym działanie UTP jest silniejsze. Podobnie receptor P2Y6; jest on selektywnie wrażliwy na UDP, słabiej na UTP, a na ATP i ADP odpo-wiada bardzo słabo, jeśli w ogóle (Tab. 1). Receptory P2Y4 i P2Y6 różni w organizmie człowieka rozmieszczenie tkanko-we. P2Y4 występuje niemal wyłącznie w łożysku, a P2Y6 jest znajdowany w mózgu, mięśniach gładkich naczyń krwio-nośnych, śródbłonku naczyniowym, czy makrofagach. Dla-tego należy przypuszczać, że to receptor P2Y6 odpowiada w organizmie ludzkim za przekazywanie informacji z udzia-łem nukleotydów pirymidynowych [6,7,9,65]. Stosunkowo niedawno odkryty w łożysku receptor P2Y11 charakteryzuje się selektywnością dla ATP, odpowiada także na pochodne tego nukleotydu (silniej niż na ATP) i nie reaguje na UTP czy UDP [65] (Tab. 1). Jak już powiedziano powyżej recep-tor ten jest związany zarówno z białkiem Gq, jak i Gs.
Receptory należące do drugiej grupy to receptory P2Y12, P2Y13 i P2Y14. Receptor P2Y12 występował uprzednio pod nazwą P2YADP. Wiadomym było, że występuje w błonie ko-mórkowej płytek krwi, i że zewnątrzkomórkowy ADP dzia-łając na płytki powoduje widoczną zmianę ich kształtu w wyniku następującej agregacji. Taki efekt działania ADP ob-serwowano od ponad 50 lat. Wiadomym też było, że zmia-ny te związane są ze spadkiem aktywności cyklazy adeny-lanowej i zmniejszeniem poziomu cAMP w cytosolu, wraz z jednoczesnym wzrostem stężenia Ca2+. Przez lata nie udało się jednak, sklonować tego receptora i sugerowano, że być może powyższe efekty są wywoływane przez receptor P2Y1 [69]. Dopiero rok 2001 przyniósł przełom, receptor wystę-pujący w płytkach krwi został wreszcie sklonowany i na-zwany P2Y12 [70]. Jednocześnie stwierdzono jego bardzo ni-
Tabela 1. Wybrane właściwości receptorów P2Y.
Podtyp Liczbaaminokwasów Agoniści Antagoniści Oddziaływanie
— efektP2Y1 373 2MeSADP > 2MeSATP = ADP > ATP MRS 2279, MRS 2179, PPADS, suramina Gq ð Ca2+↑P2Y2 377 UTP = ATP Suramina Gq ð Ca2+↑P2Y4 365 UTP ≥ ATP Reactive Blue 2, PPADS Gq ð Ca2+↑P2Y6 328 UDP > UTP >> ATP Reactive Blue 2, PPADS, suramina Gq ð Ca2+↑P2Y11 374 AR-C67085MX > BzATP ≥ ATPγS > ATP Suramina, Reactive Blue 2, Gq ð Ca2+; ↑; Gs ð cAMP↑P2Y12 342 ADP = 2MeSADP> Ap4A AR-C67085MX, Cangrelor Gi ð cAMP↓P2Y13 354* ADP = 2MeSADP>> ATP oraz 2MeSATP AR-C67085MX, Cangrelor, PPADS, Ap4A Gi ð cAMP↓P2Y14 338 UDP-glukoza = UDP-galaktoza – Gi ð cAMP↓; Ca2+↑
Oznaczenia: 2MeSADP — 2-metylotio-ADP; 2MeSATP — 2-metylotio-ATP; Ap4A — diadenozyno-5,5-P1,P4 –tetrafosforan; ATPgS — 3-tio-ATP; AR-C67085MX — 2-propylotio-D-β,γ-dichlorometyleno ATP; Cangrelor — znany również jako AR-C69931MX (dwuwodzian metylowy [(2R,3S,4R,5R)-3,4-dihydroksy-5-[6-(2-metylosulfanyletylamino)-2-(3,3,3-trifluoropropylosulfonylo)puryno-9-yl]oksolan-2-yl] metylofosforanu); BzATP — 2’,3’-O-(4-benzoilobenzoil)-ATP; MRS 2179 — 2′-deoksy-N6-metylo-ADP; MRS 2279 — dwuwodzian (1R,2S,4S,5S)-4-[2-Chloro-6-(metyloamino)-9H-puryno-9-yl ]-2-(fosfonooksy)bicyklo[3.1.0]heksano-1-metanolu; PPADS — 6-azofenylo-2’4’-disulfonowy fosforan pirydoksalu (ang. pyridoxal phosphate-6-azo(benzene-2,4-disulfonate)); ↑— wzrost stężenia; ↓— spadek stężenia. *49% identyczność sekwencji aminokwasowej z receptorem P2Y12

432 www.postepybiochemii.pl
ską, wynoszącą jedynie 24% homologię z receptorem P2Y1, co automatycznie wykluczało poprzednie przypuszczenia, choć dalsze badania wykazały daleko idącą zbieżność lo-kalizacji obu tych receptorów. Dalsze badania potwierdziły także wyłączny udział receptora P2Y12 w hamowaniu pod wpływem ADP aktywności cyklazy adenylanowej w płyt-kach krwi [70] oraz w komórkach glejaka C6 [71]. Natomiast za proces mobilizacji Ca2+ obserwowany podczas agregacji płytek krwi odpowiada receptor P2Y1 [70]. Odkrycie recep-tora P2Y12 ułatwiło sklonowanie jego bliskiego homologa (48% identyczności), receptora P2Y13. Te dwa receptory są podobne filogenetycznie i strukturalnie, obydwa są zwią-zane z białkiem Gi/o wrażliwym na toksynę krztuśca (PTX, ang. pertusis toxin), są selektywnie wrażliwe na ADP i 2Me-SADP, blokowane przez pochodne ARC, a do ich rozróż-nienia mogą służyć określone syntetyczne związki pełniące rolę agonistów i antagonistów (Tab. 1). I tak, PPADS jest an-tagonistą P2Y13, ale nie działa na P2Y12, a z kolei Ap4A działa jako agonista receptora P2Y12, ale jest antagonistą receptora P2Y13 [65] (Tab. 1). Należy dodać, że receptor P2Y12 prowa-dzi także szlak sygnałowy, którym jest aktywacja szlaku MAP kinaz (ERK1/ERK2) oraz kinaz PI3K/Akt, przy czym procesy te są niezależne od Ca2+ i PKC [72].
Z białkiem Gi/o, jak powiedziano powyżej, jest rów-nież związany receptor P2Y14 (Ryc. 1). Receptor ten został sklonowany w 2000 r., ale jego dokładniejsza charaktery-styka i nadanie nazwy miało miejsce ostatecznie w roku 2003 [64]. Receptor ten wykazuje 45% homologii z recep-torem P2Y12 i jest aktywowany przez monosacharydowe pochodne nukleotydów, UDP-glukozę, UDP-galaktozę, UDP-glukoronian i UDP-N-acetyl-glukozaminę, podczas gdy ATP, ADP i UTP nie wywierają na niego wpływu [65] (Tab. 1). Receptor ten przez podjednostkę α białka Gi/o hamuje aktywność AC, ale ponadto, przez kompleks podjednostek β/γ może dodatkowo, w pewnych komór-kach aktywować PLCβ i powodować mobilizację Ca2+ w komórce [65,73]. Podobna aktywacja PLCβ przez podjed-nostki β/γ białka Gi/o występuje też w przypadku innych receptorów P2Y, a wśród nich, jak już wspomniano, re-ceptora P2Y2 [9]. Obecność receptora P2Y14 stwierdzono w łożysku, tkance tłuszczowej, sercu, mózgu, a także komórkach gleju (astrocytach i mikrogleju) i komórkach glejaka C6 [73]. W komórkach astrocytów i mikrogleju jego poziom jest regulowany przez czynniki prozapalne, co może sugerować udział receptora P2Y14 w przekazy-waniu sygnału między układem immunologicznym, a nerwowym. Ostatnio wykazano, że UDP może być także aktywnym agonistą receptora P2Y14, przy czym daleko posunięta zbieżność występowania tego receptora wraz z receptorem P2Y6 w tkankach organizmów ludzi i gryzoni sugeruje możliwość wspólnej odpowiedzi obu recepto-rów na UDP [74]. W takim przypadku P2Y14 hamowałby zależną od białka Gi cyklazę adenylanową, a P2Y6 sty-mulowałby poprzez białko Gq aktywność fosfolipazy C, zwiększając poziom Ca2+ w komórce [74]. Należy dodać, że receptory P2Y mogą także aktywować małe białka G, a także oddziaływać z integrynami biorąc aktywny udział w migracji komórek [65]. Natomiast uwalniany do prze-strzeni zewnątrzkomórkowej ATP może brać udział w komunikacji międzykomórkowej typu: astrocyt-astrocyt czy astrocyt-neuron [75].
Rycina 3 częściowo obrazuje przekazane dotychczas in-formacje dotyczące receptorów P2Y. Zewnątrzkomórkowe ATP i UTP działają równocennie na receptor P2Y2. W wy-niku działania ektoenzymów, ADP powstający z ATP dzia-ła zarówno na receptor P2Y1 jak i P2Y12. Receptory P2Y1 i P2Y2 są związane z białkiem Gq i ich aktywacja powoduje mobilizację Ca2+ w komórce, podczas gdy P2Y12 zmniejsza poziom cAMP. A więc receptory P2Y1 i P2Y2, aktywowane odpowiednio przez ADP i ATP lub UTP współdziałają ze sobą w kierunku zwiększenia stężenia jonów Ca2+ w komór-ce. Natomiast w przypadku ADP, działającego jednocześnie na P2Y1 i P2Y12, odpowiedzi różnych komórek są różne. W płytkach krwi aktywacja obu tych receptorów przez ADP wywołuje wspólnie proces agregacji, jest to więc przykład pozytywnej współpracy. W komórkach glejaka C6 od-powiedź jest inna. Należy dodać, że w zdrowych komór-kach astrocytów czy neuronów, receptor P2Y1 dominuje, podczas gdy w komórkach nowotworowych pochodzenia neuronalnego nie jest znajdowany [76]. Komórki glejaka C6 są komórkami nowotworowymi pochodzenia glejowego. Charakteryzują się zwiększoną ekspresją mRNA białka re-ceptorowego P2Y12, a receptor P2Y1, choć jest w nich obecny, jest wysoce niestabilny. W niesprzyjających warunkach fi-zjologicznych, np. przy długotrwałej hodowli komórek w środowisku niezawierającym surowicy, ekspresja mRNA białka receptorowego P2Y1 zanika i na ADP „odpowiada” wyłącznie receptor P2Y12. Można sądzić, że powodem tego jest tendencja komórek nowotworowych do wzmożonej proliferacji, bowiem w komórkach glejaka C6 receptor P2Y12 moduluje aktywność szlaku MAP kinaz (ERK1/ERK2) i szlaku kinaz PI3K/Akt działających stymulująco na pro-liferację i zwiększających inwazyjność, podczas gdy P2Y1 działa hamująco na aktywność kinaz PI3K/Akt [19,72]. W warunkach fizjologicznie niesprzyjających niska ekspresja mRNA białka receptorowego P2Y1, a wysoka P2Y12 sprzyja zatem procesom przeżywania, a więc w przypadku komó-rek glejaka C6 działanie receptorów P2Y1 i P2Y12 jest prze-ciwstawne [19,65,72]. Reasumując, różnorodność szlaków sygnałowych uruchamianych przez receptory P2Y i ich wzajemne współzależności generują w różnych komórkach różne odpowiedzi zależne od różnych stanów fizjologicz-nych, co wskazuje na bogactwo wywoływanych przez nie efektów biologicznych.
RECEPTORY P2X — JONOTROPOWE, AKTYWOWANE PRZEZ ATP
Receptory P2X są błonowymi kanałami jonowymi otwie-ranymi przez przyłączenie ATP lub jego syntetycznych po-chodnych. A więc, otwieranie kanału następuje przez wią-zanie liganda, a nie przez zmianę napięcia błony. Dlatego kanały te zostały zakwalifikowane jako receptory, receptory jonotropowe. Jak już sama nazwa wskazuje są one odmien-ne od receptorów metabotropowych, których przedstawi-cielami były opisane poprzednio receptory P1 i P2Y. Przede wszystkim nie są monomerami, występują jako wielkoczą-steczkowe oligomeryczne białka, w skład których wchodzi kilka podjednostek otaczających centralnie zlokalizowa-ny kanał. Receptory jonotropowe są zarówno homo- jak i heterooligomerami. Funkcjonalny receptor P2X składa się zazwyczaj z trzech podjednostek okalających i tworzących

Postępy Biochemii 60 (4) 2014 433
kanał (Ryc. 3). Dotychczas sklonowano siedem podtypów receptorów P2X: P2X1-7, a homologia sekwencji ich reszt aminokwasowych mieści się w granicach 30-50% [6,77,78].
Schemat budowy monomeru/podjednostki receptora P2X przedstawia rycina 4. Jak widać, monomer zawiera w swojej cząsteczce dwie domeny transbłonowe, a końce C- i N- białka receptorowego są zlokalizowane po stronie cyto-solowej. Bardzo duża część zewnątrzkomórkowa tworząca pętlę łączącą domeny składa się z 280 reszt hydrofilowych aminokwasów, podczas gdy białko receptorowe zawiera od 379 do 595 reszt aminokwasów. W skład pętli wchodzą liczne miejsca glikozylacji, a także 10 reszt cysteinowych tworzących mostki dwusiarczkowe; jest to cecha charakte-rystyczna receptorów P2X. Sekwencja aminokwasów w pę-tli jest zachowana ewolucyjnie, podczas gdy domeny trans-błonowe o strukturze α-helisy mogą się od siebie znacznie różnić. W tworzenie części kanałowej receptora, otwierają-cej i zamykającej kanał jonowy jest zaangażowana domena TM1. ATP wiąże się do regionu pętli zewnątrzkomórkowej przyległej do domen w bliskim sąsiedztwie z błoną pla-zmatyczną. Hydrofobowy region H5 pętli moduluje kanał przez przyłączenie kationów Mg2+, Zn2+, Ca2+ i Cu2+ (Ryc. 4) [6,77,78].
Związanie liganda i aktywacja receptora przez ATP (ale nie przez UTP) prowadzi do otwarcia kanału przepuszczal-nego selektywnie dla jonów Ca2+, Na+ i K+. Jony Ca2+ i Na+ napływają do komórki ze środowiska zewnątrzkomórko-wego, a jony K+ wypływają z komórki zgodnie z gradientem stężeń. Przepływ tych jonów powoduje depolaryzację błony i może dodatkowo prowadzić do otwarcia zależnych od na-pięcia kanałów wapniowych i powodować dalszy napływ Ca2+ do komórki. Zatem, w wyniku aktywacji receptorów P2X otwarcie kanałów tych receptorów powoduje wzrost stężenia cytosolowego Ca2+ i redukcję potencjału błony ko-
mórkowej. Te dwa parametry są miarą aktywności danego receptora P2X [65,77-80].
Receptory jonotropowe, bezpośrednio związane z ka-nałem jonowym to receptory dla szybkich neuroprzekaź-ników, biorące udział w szybkiej transmisji sygnałowej. Toteż głównym miejscem ich lokalizacji jest błona komórek neuronalnych, ale też komórek mięśni, a więc komórek po-budliwych. Zasada ta dotyczy także receptorów P2X, choć występują również w komórkach niepobudliwych, takich jak np. komórki tuczne, granulocyty, limfocyty, monocyty, komórki skóry, trzustki, a także komórki glejowe czy gleja-ki, w tym komórki glejaka C6 [51,65,77-80].
ATP jest selektywnym ligandem jonotropowych recepto-rów P2X, ale jak wiadomo oddziałuje również z receptora-mi metabotropowymi P2Y, powodując w obu przypadkach wzrost stężenia jonów Ca2+ w cytosolu komórki. Rozróż-nienie obu typów receptorów jest możliwe dzięki różnej szybkości działania receptorów i odpowiedzi komórki po ich stymulacji. W receptorach jonotropowych efekt działa-nia agonisty osiąga szczyt w ciągu kilku milisekund i rów-nie szybko zanika, podczas gdy receptory metabotropowe są wielokrotnie wolniejsze, działają przez aktywację białek G i generowanie wtórnych przekaźników informacji, toteż odpowiedź komórki na ich stymulację jest obserwowana w minutach. Podobnie jest w przypadku receptorów P2Y związanych z białkiem Gq. Takim przykładem może być ak-tywowany przez ATP czy UTP receptor P2Y2, gdzie po sto-sunkowo szybkim wyrzucie jonów Ca2+ z siateczki śródpla-zmatycznej podwyższony poziom Ca2+ w komórce utrzy-muje się przez kilka minut. Dla doświadczonego badacza odpowiedzi tego typu nie sposób pomylić z nagłą, przemi-jającą odpowiedzią wapniową obserwowaną po pobudze-niu przez ATP receptorów jonotropowych P2X [65,79,80]. Aktywność receptora jonotropowego można także mierzyć przez pomiar prądu jonowego przewodzonego przez poje-dynczy kanał stosując metodę „patch-clamp” [77,78].
Podtypy receptorów P2X występują dość powszechnie w układzie nerwowym, gdzie odpowiadają za transmisję sygnałową i w mięśniach gładkich, szczególnie w układzie naczyniowym oraz pęcherzu moczowym, gdzie odpowia-dają za skurcz. Jednak w kilku przypadkach można mówić o ich dużej specyficzności. Receptor P2X1 jest jedynym re-ceptorem P2X występującym w komórkach mięśni gładkich naczyń krwionośnym, a P2X3 występuje w niektórych ner-wach czuciowych. Receptor P2X3 znajdujący się na zakoń-czeniach nerwów czuciowych w skórze uczestniczy w reak-cji na ból i dotyk. Bierze też udział w indukcji przewlekłego bólu [8,9]. Receptor P2X5 jest znajdowany w obwodowym układzie nerwowym i rdzeniu kręgowym, a nie występuje w mózgu [77,78]. Receptor ten odgrywa istotną rolę w pro-cesie różnicowania mioblastów w komórkach satelitarnych C2C12 [80]. Z kolei receptor P2X7 występuje zarówno w ko-mórkach neuronalnych i komórkach mięśni szkieletowych jak i w niektórych komórkach układu immunologicznego, komórkach tucznych, granulocytach, monocytach i limfo-cytach, a także komórkach nabłonka, śródbłonka czy w ko-mórkach glejowych [65,77,78].
Rycina 4. Schemat budowy receptorów P2X. Podjednostka receptora zawiera dwie domeny transbłonowe (TM1,2). Duża pętla zewnątrzkomórkowa zawiera mostki dwusiarczkowe zaznaczone jako S-S, miejsca glikozylacji zaznaczone nie-bieskim gwiazdkami i hydrofobowy region H5, modulujący kanał przez wiąza-nie kationów. Końce N- i C- znajdują się od strony cytosolowej błony komórko-wej (zmodyfikowano wg. [5]).

434 www.postepybiochemii.pl
Poszczególne podtypy receptorów P2X różnią się mię-dzy sobą nie tylko rozmieszczeniem tkankowym, lecz także wrażliwością na agonistów, antagonistów oraz podatno-ścią na desensytyzację. Jak już powiedziano, naturalnym agonistą wszystkich receptorów P2X jest ATP, różni je jed-nak wrażliwość na syntetyczne analogi ATP, takie jak α,β-metyleno-ATP (α,βMeATP) oraz 2-metylotio-ATP (2Me-SATP) [51,65,77,78]. Z różną też wrażliwością reagują na antagonistów, którymi dla wszystkich podtypów, za wyjąt-kiem receptora P2X7 jest PPADS i suramina, jednak należy przypomnieć, że zarówno suramina, jak i PPADS są także antagonistami receptorów P2Y (Tab. 1). Specyficznym anta-gonistą receptora P2X7 jest Briliant blue G [78,79]. Receptor P2X7 różni się od pozostałych podtypów także odpowiedzią na agonistów.
Receptor P2X7 jest receptorem atypowym, jego właści-wości odbiegają od pozostałych receptorów P2X. Główną różnicę stanowi stosunkowo niskie powinowactwo do ATP; receptor P2X7 do swojej aktywacji wymaga milimolowych stężeń tego nukleotydu, o rząd wielkości większych niż inne receptory. Dopiero tak wysokie stężenie otwiera kanał i powoduje szybki napływ Ca2+ i Na+ do komórki i wypływ K+, tak jak ma to miejsce w przypadku pozostałych recepto-rów P2X. Następną różnicą jest odpowiedź receptora P2X7 na syntetycznego agonistę, którym dla tego receptora jest analog ATP, 2’,3’-O-(4-benzoilobenzoil)-ATP (BzATP). Re-ceptor P2X7 jest od 10 do 30 razy bardziej czuły na BzATP niż na ATP. Należy dodać, że wysokie, 1mM stężenie ATP wymagane do aktywacji receptora P2X7 może zmieniać wa-runki pomiaru poziomu jonów Ca2+ w cytosolu komórki, obniżając pH środowiska zewnątrzkomórkowego przez silnie ujemny ładunek tego nukleotydu. Dlatego do badań zazwyczaj używa się BzATP, w odpowiednio 10-krotnie niższych stężeniach. Jednak ten syntetyczny analog ATP, do niedawna stosowany jako selektywny agonista recep-tora P2X7, okazał się skuteczny w aktywacji innych recep-torów P2X, mianowicie receptorów P2X1 i P2X3 [77,78]. Co więcej, także receptorów P2Y [79]. Badania prowadzone na komórkach glejaka C6 wykazały, wbrew poprzednim da-nym literaturowym, że odpowiedzi wapniowe generowane przez działanie BzATP były wywołane przez aktywację re-ceptora P2Y2, a nie P2X7, wskazując, wraz z innymi danymi doświadczalnymi, że w tych komórkach receptor P2X7 nie występuje lub jest niefunkcjonalny [79].
Kolejną wyjątkową właściwością receptora P2X7 (przy powtarzającej się, czy przedłużonej ekspozycji komórek na wysokie, milimolowe stężenia ATP) jest zamiana kanałów jonowych specyficznych dla kationów, na duże, nieselek-tywne transbłonowe pory. Stosunkowo niedawno pojawiły się sugestie, że przy otwieraniu błonowych porów genero-wanych przez aktywację receptora P2X7 biorą także udział inne białka, panneksyny-1 (ang. pannexin-1) [81]. Pory te odpowiadają za masowy napływ jonów Ca2+ do komórki, ale też są zdolne do przepuszczania większych kationów or-ganicznych, cząsteczek naładowanych dodatnio o masie na-wet do 1 kDa, a także niektórych barwników fluoroscencyj-nych [6,65,77-79]. W komórkach układu immunologicznego aktywacja i tworzenie porów przez receptor P2X7 powoduje zwiększenie wydzielania z komórek makrofagów interleu-
kiny IL-1β [81]. Przy różnych rodzajach uszkodzeń cen-tralnego układu nerwowego, wysoki poziom uwalnianego ATP aktywując receptory P2X7 powoduje śmierć neuronów. Właściwości receptorów P2X7 powodują, że w określonych warunkach receptor P2X7 może być także odpowiedzialny za apoptotyczną śmierć komórki, w tym komórki nowotwo-rowej [51,65,77,78]. Sygnałem włączającym proces progra-mowanej śmierci jest zwiększenie wewnątrzkomórkowego poziomu jonów Ca2+ dzięki otwarciu kanałów receptoro-wych, czy utworzeniu porów. Te właściwości receptorów P2X, a w szczególności receptora P2X7 powodują, że recep-tory te mogą pełnić istotną funkcję w procesie zwalczania nowotworów.
KONKLUZJE I PERSPEKTYWY
ATP i inne nukleotydy są w świecie żywym wszech-obecne. Także szeroko rozpowszechnione są aktywowane przez nie receptory. Receptory nukleotydowe występują nie tylko u kręgowców; receptory ATP są znajdywane w orga-nizmach prostych bezkręgowców, takich jak pierwotniaki, ameby, czy śluzowce. Znaleziono je również u roślin. U ślu-zowca Dictostelium discoideum receptory aktywowane przez ATP są podobne do ludzkich receptorów P2X. W tych orga-nizmach regulują one napływ i wypływ wody do komórki. Wskazuje to na pojawienie się informacyjnych funkcji nu-kleotydów już na wczesnych etapach ewolucji [82].
To szerokie rozpowszechnienie nukleotydów musi po-niekąd powodować, że znaczna liczba receptorów, na które działają te związki jest stale niedostatecznie opisana. Recep-tory te to tzw. „receptory sieroce” (ang. orphan receptors). Zespół, który ma największe osiągnięcia w badaniach nad takimi receptorami to zespół kierowany przez Marię Pia Abbracchio z Uniwersytetu w Mediolanie we Włoszech. Między innymi, dzięki pracom tego zespołu udała się peł-na charakterystyka i poznanie właściwości receptora P2Y14, który przed zatwierdzeniem przez IUPHAR, nosił nazwy GPR105 czy KIAA0001 [64]. Jak już wspomniano, także re-ceptor P2Y12 został stosunkowo niedawno poznany i opisa-ny, co pozwoliło następnie na identyfikację receptora P2Y13, który jako receptor sierocy nosił uprzednio nazwy GPR86, GPR94 czy SP174 [64]. W pracowni Abbracchio zakończono prace nad charakterystyką następnego sierocego receptora, receptora GPR17, wiążącego nie tylko cukrowe pochodne UDP, lecz również leukotrieny. Stosunkowo niedawno opi-sano również receptory sieroce GPR91 i GPR99, pokrewne receptorom nukleotydowym. Receptory te, choć homolo-giczne z receptorem P2Y1, nie rozpoznają jednak nukleoty-dów, a współdziałają z intermediatami cyklu Krebsa [83]. Należy dodać, że w spisach IUPHAR znajdują się wszystkie receptory, w tym sieroce. Badania nad receptorami sierocy-mi to najprawdopodobniej jeden z dwóch głównych kierun-ków perspektywicznych, jakie będą się rozwijać w tej dzie-dzinie wiedzy w okresie najbliższych lat.
Drugim takim kierunkiem perspektywicznym są ba-dania o implikacjach klinicznych. Stosowane obecnie i nieustannie udoskonalane leki są ukierunkowane na etap odbierania przez receptory sygnałów i przekazywanie ich dalej na określone enzymy generujące biologicznie czynne cząsteczki wewnątrz komórki. Dlatego poznanie właści-

Postępy Biochemii 60 (4) 2014 435
wości receptorów ma ważne znaczenie terapeutyczne. Do-brym przykładem działań tego typu są badania dotyczące receptora P2Y12. Określenie jego właściwości okazało się mieć istotne znaczenie kliniczne w leczeniu chorób serco-wo-naczyniowych. I tak, przy uszkodzeniu komórek lub po podaniu ich stresowi, uwolniony z nich czynnie czy bier-nie ATP, degradowany do ADP, rozpoczyna proces gojenia ran aktywując receptory P2Y12 znajdujące się na płytkach krwi. Zaktywowany receptor wywołuje agregację płytek tworząc tzw. „czop płytkowy” zatykający uszkodzenie. Jednak ten sam mechanizm prowadzi do powstania zakrze-pów w naczyniach krwionośnych, powodując zawały lub udary, groźne dla życia. Okazało się, że stosowany w tego typu zaburzeniach lek, klopidogrel, zatrzymuje ten proces hamując działanie receptora P2Y12, uniemożliwiając tworze-nie zakrzepów [7,8,70,82]. Trwają badania nad poznaniem szeregu innych lekarstw, które modulując aktywności re-ceptorów działałyby terapeutycznie na rozmaite funkcje fizjologiczne narządów i tkanek [57]. Dla tego typu działań konieczne jest poznanie i porównanie funkcji receptorów, w tym receptorów nukleotydowych, w normie i patologii. Takie właśnie podejście reprezentują autorzy licznych arty-kułów publikowanych w obecnym zeszycie „Postępów Bio-chemii”. Należy dodać, że także wydany w bieżącym roku numer pierwszy czasopisma „Purinergic Signalling” jest poświęcony w całości porównaniu receptorów nukleotydo-wych działających w zdrowiu i w chorobie w układzie tra-wiennym, moczowym, rozrodczym, endokrynnym, a także wątrobie [84]. Wszystkie artykuły przeglądowe w tym nu-merze „Purinergic Signalling” są autorstwa lub współau-torstwa odkrywcy receptorów nukleotydowych, Geoffreya Burnstocka.
Konkludując, odkryte w końcu lat siedemdziesiątych ubiegłego wieku receptory nukleotydowe są stale w cen-trum zainteresowań badaczy. Jest to między innymi zwią-zane z powszechnością ich występowania i coraz większą wiedzą wskazująca na ich istotną rolę w regulacji licznych procesów fizjologicznych. Dwa wymienione powyżej per-spektywiczne kierunki badań, w jakich prawdopodobnie będzie dalej rozwijać się ta gałąź wiedzy, wzajemnie się przenikają. Badania nad sierocymi receptorami mają bo-wiem w zamyśle znalezienie takich, których właściwości pozwolą na ściśle ukierunkowane i selektywne działania kliniczne. Działania te, ograniczając lub stymulując funkcje określonych receptorów, nie powinny wpływać na funkcje innych receptorów, na inne komórki i inne narządy. Bada-nia takie są już prowadzone. Można mieć nadzieję, że w naj-bliższej przyszłości nowe generacje leków, oparte na wie-dzy o molekularnych właściwościach receptorów, pozwolą na ograniczenie wielu chorób dręczących dziś ludzkość.
PIŚMIENNICTWO1. Burnstock G (1990) Overview – purinergic mechanisms, W: Dubyak
GR, Fedan JA (red), Biological actions of extracellular ATP. New York Academy of Sciences, New York 603: 31-45
2. Burnstock G (2006) Purinegic signalling – an overview. Novartis Found Symp 276: 20-48; discussion 48-57, 275-281
3. Zimmermann H, Braun N (1999) Ecto-nucleotidases – molecular struc-tures, catalytic properties and functional roles in the nervous system. Prog Brain Res 120: 371-385
4. Ziganshin AU, Hoyle C, Burnstock G (1994) Ecto-enzymes and metab-olism of extracellular ATP. Drug Dev Res 32: 134-146
5. Ralevic V, Burnstock G (1998) Receptors for purines and pyrimidines. Pharmacol Rev 50: 413-492
6. Burnstock G (2007) Purine and pyrimidine receptors. Cell and Mol Life Sci 64: 1471-1483
7. Burnstock G, Knight GE (2004) Cellular distribution and functions of P2 receptor subtypes in different systems. Int Rev Cytol 240: 301-304
8. Burnstock G (2006) Pathophysiology and therapeutic potential of pu-rinergic signaling. Pharmacol Rev 58: 58-86
9. Burnstock G (2007) Physiology and pathophysiology of purinergic neurotransmission. Physiol Rev 87: 659-797
10. Deli T, Csernoch L (2008) Extracellular ATP and cancer: an overview with special reference to P2 purinergic receptors. Pathol Oncol Res 14: 219-231
11. Lohmann K (1929) Ȕber die Pyrophosphatfraction in Muskel. Natur-wissenschaften 17: 624-625
12. Fiske CH, Subbarow Y (1929) Phosphorus compounds of muscle and liver. Science 70: 381-382
13. Drury AN, Szent-Gyȍrgyi A (1929) The physiological activity of ade-nine compounds with special reference to their action upon the mam-malian heart. J Physiol 68: 213-237
14. Gillespie JH (1934) The biological significance of the linkages in ade-nosine triphosphoric acid. J Physiol 80: 345-349
15. Lippman F (1941) Metabolic generation and utilization of phosphate bond energy. Enzymology 1: 99
16. Kalckar HM (1969) Biological phosphorylations: Development of con-cepts. Englewood Cliffs, NJ: Prentice-Hall
17. Kalckar HM (1991) 50 years of biological research – from oxidative phosphorylation to energy requiring transport regulation. Ann Rev Biochem 60: 1-37
18. Lazarowski ER, Boucher RC, Harden TK (2000) Constitutive release of ATP and evidence for major contribution of ecto-nucleotide pyrophos-phatase and nucleoside diphosphokinase to extracellular nucleotide concentration. J Biol Chem 275: 31061-31068
19. Krzemiński P, Supłat D, Czajkowski R, Pomorski P, Barańska J (2007) Expression and functional characterization of P2Y1 and P2Y12 nucle-otide receptors in long-term serum-deprived glioma C6 cells. FEBS J 274: 1970-1982
20. Glynn IM (1968) Membrane adenosine triphosphate and cation trans-port. Br Med Bull 24: 165-169
21. Chaudry IH (1982) Does ATP cross the plasma membrane. Yale J Biol Med 55: 1-10
22. Green HN, Stoner HB (1950) Biological actions of the adenine nucle-otides. Lewis, London
23. Bodin P, Burnstock G (2001) Purinergic signalling: ATP release. Neu-rochem Res 26: 959-969
24. Burnstock G (1997) The past, present and future of purine nucleotides as signalling molecules. Neuropharmacol 36: 1127-1139
25. Evans RJ, Derkach V, Surprenant A (1992) ATP mediates fast synaptic transmission in mammalian neurons. Nature 357: 503-505
26. Sorensen CE, Novak I (2001) Visualization of ATP release in pancre-atic acini in response to cholinergic stimulus. Use of fluorescent probes and confocal microscopy. J Biol Chem 276: 3295-32932
27. Grygorczyk R, Hanrahan JW (1997) CFTR-independent ATP release from epithelial cells triggered by mechanical stimuli. Am J Physiol 272: C1058-1066
28. Guyot A, Hanrahan JW (2002) ATP release from human airway epi-thelial cells studied using a capillary cell culture system. J Physiol 545: 199-206
29. Milner P, Bodin P, Loesch A, Burnstock G (1990) Rapid release of en-dothelin and ATP from isolated aortic endothelial cells exposed to in-creased flow. Biochem Biophys Res Commun 170: 649-656
30. Burnstock G (1999) Release of vasoactive substances from endothelial cells by shear stress and purinegic mechanosensory transduction. J Anat 194: 335-342

436 www.postepybiochemii.pl
31. Lazarowski ER, Boucher RC, Harden TK (2003) Mechanisms of release of nucleotides and integration of their action as P2X- and P2Y-receptor activating molecules. Mol Pharmacol 64: 785-795
32. Frenchak AP, Lewis MA, Kresge C, Sathe M, Bugde A, Luby-Phelps K, Antich PP, Fitz JG (2010) Initiation of purinergic signaling by exocy-tosis of ATP-containing vesicles in liver epithelium. J Biol Chem 285: 8138-8147
33. Coco S, Calegari F, Pravettoni E, Pozzi D, Taverna E, Rosa P, Matteoli M, Verderio C (2003) Storage and release of ATP from astrocytes in culture. J Biol Chem 278: 1354-1362
34. Lazarowski E (2006) Regulated release of nucleotides and UDP sugars from astrocytoma cells. Novartis Found Symp 276: 76-84; discussion: 84-90, 107-112, 275-281
35. Zimmermann H (2000) Extracellular metabolism of ATP and other nucleotides. Naunyn Schmiedebergs Arch Pharmacol 362: 299-309
36. Zimmermann H (2006) Ectonucleotidases in the nervous system. No-vartis Found Symp 276: 113-128; discussion: 128-130, 275-281
37. Bunstock G (1972) Purinergic nerves. Pharmacol Rev 24: 509-58138. Holton P (1959) The liberation of adenosine triphosphate on antidro-
mic stimulation of sensory nerves. J Physiol 145: 494-50439. Bunstock G (1976) Purinergic receptors. J Theor Biol 62: 491-50340. Burnstock G (1978) A basis for distinguishing two types of purynergic
receptor, W: Straub RW, Boils L (red), Cell membrane receptors for drugs and hormones: a multidisciplinary approach. Raven Press, New York, str. 107-118
41. Burnstock G, Kennedy C (1985) Is there a basis for distinguishing two types of P2-purinoceptor? Gen Phamacol 16: 433-440
42. Abbracchio MP, Burnstock G (1994) Purinoceptors: are there families of P2X and P2Y purinoceptors? Pharmacol Ther 64: 445-475
43. O’Connor SE, Dainty IA, Leff P (1991) Further subclassification of ATP receptors based on agonist studies. Tends Pharmacol Sci 12: 137-141
44. Fredholm BB, Abbracchio MP, Burnstock G, Daly JW, Harden TK, Jacobson KA, Leff P, Williams M (1994) International Union of Phar-macology. VI. Nomenclature and classification of purinoceptors. Phar-macol Rev 46: 143-156
45. Fredholm BB, Abbracchio MP, Burnstock G, Dubyak GR, Harden TK, Jacobson KA, Schwabe U, Williams M (1997) Towards a revised no-menclature for P1 and P2 receptors. Trends Pharmacol Sci 18: 79-82
46. Stiles GL (1992) Adenosine receptors. J Biol Chem 267: 6451-645447. Khakh BS, Kennedy C (1998) Adenosine and ATP: progress in their
receptors structures and functions. Trends Pharmacol Sci 19: 39-4148. Ceruti S, Abbracchio MP (2013) Adenosine signaling in glioma cells,
W: Barańska J (red), Glioma signaling. Adv Exp Med Biol 986: 13-31, Springer, Dordrecht, Heidelbeg, New York, London
49. Belardinelli L (1993) Adenosine system in heart. Drug Dev Res 28: 263-267
50. Romanowska M, Komoszyński M (2002) Adenozyna – neuroprzekaź-nik i neuromodulator w centralnym układzie nerwowym. Postepy Biochem 48: 230-238
51. Czajkowski R, Sabała P, Barańska J (2004) Receptory nukleotydowe, W: Nowak JZ, Zawilska JB (red), Receptory i mechanizmy przekazy-wania sygnału. PWN, Warszawa, str. 438-450
52. Fredholm BB, Ijzerman AP, Jacobson KA, Linden J, Mȕller CE (2011) International Union of Basic and Clinical Pharmacology. LXXXI. No-menclature and classification of adenosine receptors - an update. Phar-macol Rev 63: 1-34
53. Lazarowski ER, Sesma JI, Seminario-Vidal L, Kreda SM (2011) Molec-ular mechanisms of purine and pyrimidine nucleotide release. Adv Pharmacol 61: 221-261
54. Klotz KN (2000) Adenosine receptors and their ligands. Naunyn Schmiedebergs Arch Pharmacol 362: 382-391
55. Barańska J (1994) Białka G - nagroda Nobla 1994. Post Biol Kom 21: 479-488
56. Kobilka BK, Deupl X (2007) Conformation complexity of G-pro-tein-coupled receptors. Trends Pharmacol Sci 28: 397-406
57. Barańska J, Nalepa I (2010) Wewnątrzkomórkowe szlaki sygnalizacyj-ne wczoraj i dziś: zaburzenia w sygnalizacji a stany chorobowe, W: Koroniak H, Barciszewski J (red), Na pograniczu chemii i biologii. Wyd Nauk UAM, Poznań, str. 51-67
58. Latek D, Modzelewska A, Traskowski B, Palczewski K, Filipek S (2012) G protein-coupled receptors - recent advances. Acta Biochim Polon 59: 515-529
59. Abbracchio NP, Brambilla R, Ceruti S, Kim HO, von Lubitz DKJE, Jacobson KA, Cattabeni F (1995) G protein-dependent activation of phospholipase C by adenosine A3 receptor in at brain. Mol Pharmacol 48: 1038-1045
60. Fredholm BB (2010) Adenosine receptors as drug targets. Exp Cell Res 316: 1284-1288
61. Fields RD, Burnstock G (2006) Purinergic signalling in neuron-glia in-teractions. Nat Rev Neurosci 7: 423-436
62. Jacobson KA, Gao ZG (2006) Adenosine receptors as therapeutic tar-gets. Nature Rev Drug Discov 5: 247-264
63. Gessi S, Merighi S, Varani K, Leung E, Mac Lennan S, Borea PA (2008) The A3 adenosine receptors: an enigmatic player in cell biology. Phar-macol Ther 117: 123-140
64. Abbracchio MP, Boeynaems J-M, Barnard EA, Boyer JL, Kennedy C, Miras-Portugal MT, King BF, Gachet C, Jacobson KA, Weisman GA, Burnstock G (2003) Characterization of the UDP-glucose receptor (re--named here the P2Y14 receptor) adds diversity to the P2Y receptor fa-mily. Trends Pharmacol Sci 24: 52-55
65. Wypych D, Barańska J (2013) Cross-talk in nucleotide signaling in glio-ma C6 cells, W: Barańska J (red), Glioma signaling. Adv Exp Med Biol 986: 61-81
66. Czajkowski R, Barańska J (1999) Sphingosine and phorbol ester modu-late protein kinase C activity and modify ATP-evoked calcium mobili-zation in glioma C6 cells. Biochem Biophys Res Commun 260: 614-618
67. Berridge MJ (1993) Inositol trisphosphate and calcium signaling. Na-ture 361: 315-325
68. Barańska J, Przybyłek K, Sabała P (1999) Capacitative calcium entry. Glioma C6 as a model of nonexcitable cells. Pol J Pharmacol 51: 153-162
69. Sabała P, Czajkowski R, Przybyłek K, Kalita K, Kaczmarek L, Barań-ska J (2001) Two subtypes of G protein-coupled nucleotides receptors, P2Y(1) and P2Y(2) are involved in calcium signaling in glioma C6 cells. Br J Pharmacol 132: 393-402
70. Hollopeter J, Jantzen HM, Vincent D, Li G, England L, Ramakrishnan V, Yang RB, Nurden P, Nurden A, Julius D, Conley PB (2001) Identifi-cation of the platelet ADP receptor targeted by antithrombotic drugs. Nature 409: 202-207
71. Czajkowski R, Lei L, Sabała P, Barańska J (2002) ADP-evoked pho-spholipase C stimulation and adenylyl cyclase inhibition in glioma C6 cells occur through two didtinct nucleotide receptors, P2Y(1) and P2Y(12). FEBS Lett 513: 179-183
72. Czajkowski R, Banachewicz W, Ilnytska O, Drobot LB, Barańska J (2004) Differential effects of P2Y1 and P2Y12 nucleotide receptors on ERK1/ERK2 and phosphatidylinositol-3-kinase. Br J Pharmacol 141: 497-507
73. Krzemiński P, Pomorski P, Barańska J (2008) The P2Y14 receptor acti-vity in glioma C6 cells. Eur J Pharmacol 594: 49-54
74. Harden TK, Sesma JI, Fricks IP, Lazarowski ER (2010) Signalling and pharmacological properties of P2Y14 receptor. Acta Physiol (Oxf) 199: 149-160
75. Bezzi P, Volterra A (2001) A neuron-glia signaling network in the acti-ve brain. Curr Opin Neurobiol 11: 387-394
76. Sak K, Illes P (2005) Neuronal and glial cell lines as model systems for studying P2Y receptor pharmacology. Neurochem Int 47: 401-412
77. North RA, Surprenant A (2000) Pharmacology of cloned jonotropic P2X receptors. Annu Rev Pharmacol Toxicol 40: 563-580
78. North RA (2002) Molecular physiology of P2X receptors. Physiol Rev 82: 1016-1067
79. Supłat-Wypych D, Dygas A, Barańska J (2010) 2’,3’-0-(4-Benzoylben-zoyl)-ATP-mediated calcium signaling in rat glioma C6: role of the P2Y(2) nucleotide receptor. Purinergic Signal 6: 317-325

Postępy Biochemii 60 (4) 2014 437
Nucleotide receptors — structure and function, history and perspectivesJolanta Barańska*
Nencki Institute of Experimental Biology, 3 Pasteur St., 02-093 Warsaw, Poland
*e-mail: [email protected]
Key words: P1 receptors, P2X and P2Y receptors
ABSTRACTFirst nucleotide receptors were discovered by Geoffrey Burnstock in 70ties of the last century, as a purinoreceptors activated by ATP. It was further found that they may be activated both by purine and pyrimidine nucleotides and their name was changed to nucleotide receptors. They are divided into two fsamilies: P1, activated by adenosine and P2, activated by nucleotides which are further divided into P2X and P2Y subfamilies. P2X are ionotropic receptors activated by ATP, P2Y (as the P1) are metabotropic receptors coupled with protein G. P2Y receptors are activated by ATP, ADP, UTP, UDP and UDP-sugar derivatives. This review describes early history of extracellular nucleotide signaling studies and presents current knowledge of the particular nucleotide receptors subtypes. The article also describes the structure and functional roles of these receptors and speculates about future research and therapeutic directions in this field.
80. Banachewicz W, Supłat D, Krzemiński P, Pomorski P, Barańska J (2005) P2 nucleotide receptors on C2C12 satellite cells. Purinergic Sig-nal 1: 249-257
81. Pelegrin P, Surprenant A (2006) Pannexin-1 mediates large pore for-mation and interleukin-1β release by the ATP-gated P2X7 receptor. EMBO J 25: 5071-5082
82. Khakh BS, Burnstock G (2010) Podwójne życie ATP. Świat Nauki 1: 38-45
83. He W, Miao FJ-P, Lin DC-H, Schwandner RT, Wang Z, Gao J, Chen J-L, Tian H, Ling L (2004) Citric acid cycle intermediates as ligands for orphan G-protein-coupled receptors. Nature 429: 188-193
84. Special Issue: Purinergic signalling in visceral organs in health and disease (2014) Purinergic Signal 10: 1-231
















![Fryzjer 514[01]Z1.01 u01]_z1.01_u.pdf · 4.1. Budowa i funkcje skóry 4.1.1. Materiał nauczania Budowa szczegółowa i funkcje skóry Skóra jest narządem pokrywającym całe ciało,](https://img.dokumen.tips/doc/110x75/5ed10253fbddcf49463f0a4d/fryzjer-51401z101-u-01z101updf-41-budowa-i-funkcje-skry-411-materia.jpg)












