Embed Size (px)
Citation preview

Guillain-Barre Guillain-Barre SyndromeSyndrome

Nerve Roots (acute Nerve Roots (acute polyradiculopathies)polyradiculopathies)
Guillain-Barre SyndromeGuillain-Barre Syndrome Lyme diseaseLyme disease SarcoidosisSarcoidosis HIVHIV Other viruses (CMV, VZV, West Nile)Other viruses (CMV, VZV, West Nile) Cauda Equina SyndromeCauda Equina Syndrome Plexus lesions (brachial plexitis, Plexus lesions (brachial plexitis,
lumbosacral plexopathy)lumbosacral plexopathy)

EpidemiologyEpidemiology
Most common cause of acute flaccid Most common cause of acute flaccid paralysis in Western countriesparalysis in Western countries
Overall incidence 1-2/100,000; up to Overall incidence 1-2/100,000; up to 8.6/100,000 in elderly population8.6/100,000 in elderly population
All age groups can be affected, All age groups can be affected, however more common in elderlyhowever more common in elderly
Bimodal peak, small peak in young Bimodal peak, small peak in young adults and larger peak in elderly; rare adults and larger peak in elderly; rare in infancyin infancy

75% have an antecedent ‘event’ 1-4 75% have an antecedent ‘event’ 1-4 weeks before onset of weakness: weeks before onset of weakness: respiratory (68%), GI (22%), resp and respiratory (68%), GI (22%), resp and GI (10%), surgery (2%), vaccination or GI (10%), surgery (2%), vaccination or pregnancypregnancy
The best documented antecedents The best documented antecedents included infection with C.jejuni, EBV, included infection with C.jejuni, EBV, CMV, HSV, and Mycoplasma. C. jejuni CMV, HSV, and Mycoplasma. C. jejuni is often associated with more severe is often associated with more severe axonal cases and most likely axonal cases and most likely sensitizes the immune system to sensitizes the immune system to shared antigens. shared antigens.

Clinical CourseClinical Course Initial paresthesias in fingers/toes Initial paresthesias in fingers/toes
followed by weaknessfollowed by weakness Weakness rapidly worsens, sensory Weakness rapidly worsens, sensory
loss usually minimalloss usually minimal Usually symmetric, though can be Usually symmetric, though can be
asymmetric initiallyasymmetric initially Classically distal weakness ascending Classically distal weakness ascending
up legs and arms, but proximal up legs and arms, but proximal weakness not uncommon at onsetweakness not uncommon at onset

Cranial nerve involvement (unilateral Cranial nerve involvement (unilateral or bifacial weakness 50%, oculomotor or bifacial weakness 50%, oculomotor paralysis 5%, charachteristic in Miller paralysis 5%, charachteristic in Miller Fischer SyndromeFischer Syndrome
66% reach nadir in 2 weeks, 92% in 3 66% reach nadir in 2 weeks, 92% in 3 weeks; by definition must peak at 4 weeks; by definition must peak at 4 weeksweeks
Brief plateau phase then Brief plateau phase then improvement and gradual resolution improvement and gradual resolution over weeks to monthsover weeks to months

ComplicationsComplications Respiratory weakness/failure (20-30% Respiratory weakness/failure (20-30%
will need intubation at some point will need intubation at some point during admission)during admission)
Autonomic dysfunction in up to 65% Autonomic dysfunction in up to 65% including: arrythmias, hypotension or including: arrythmias, hypotension or hypertension, labile fluctuating BP, hypertension, labile fluctuating BP, Ileus, urinary retentionIleus, urinary retention
Pain in up to 85%: typically back pain, Pain in up to 85%: typically back pain, radiculopathic and musculoskeletal, radiculopathic and musculoskeletal, the straight leg raise test will be the straight leg raise test will be positivepositive

Papilledema (secondary to high CSF Papilledema (secondary to high CSF protein)protein)
Compression neuropathies Compression neuropathies (particularly ulnar and peroneal)(particularly ulnar and peroneal)
DVT/PEDVT/PE SIADH (26%)SIADH (26%) Acute Renal Injury (secondary to IVIG Acute Renal Injury (secondary to IVIG
TX)TX) Hypercalcemia (secondary to Hypercalcemia (secondary to
immobility)immobility) Hepatocelluar dysfunction (30% early Hepatocelluar dysfunction (30% early
on, as high as 60% later in courseon, as high as 60% later in course

Mortality/MorbidityMortality/Morbidity 85% of patients will achieve a full and 85% of patients will achieve a full and
functional recovery within 6-12 months, functional recovery within 6-12 months, maximal recovery at 18 monthsmaximal recovery at 18 months
7-15% of patients will have permanent 7-15% of patients will have permanent neurological sequelae including neurological sequelae including bilateral foot drop, intrinsic hand bilateral foot drop, intrinsic hand muscle wasting, sensory ataxia, muscle wasting, sensory ataxia, dysesthesiadysesthesia
Despite intensive care, 3-8% of Despite intensive care, 3-8% of patients diepatients die
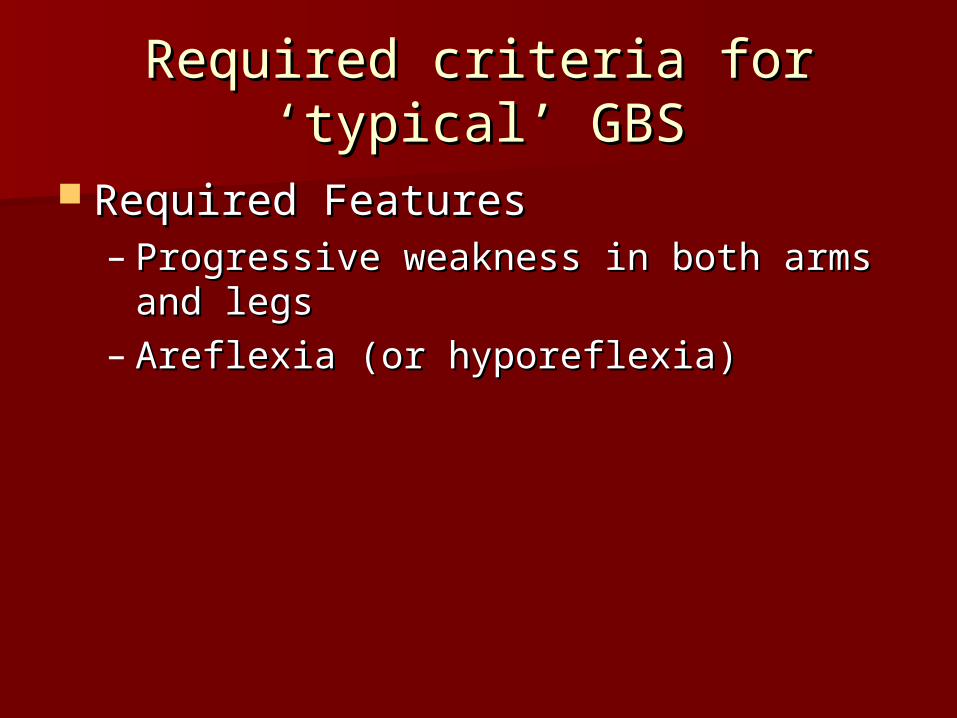
Required criteria for ‘typical’ Required criteria for ‘typical’ GBSGBS
Required FeaturesRequired Features– Progressive weakness in both arms and Progressive weakness in both arms and
legslegs– Areflexia (or hyporeflexia)Areflexia (or hyporeflexia)

Features supportive of Features supportive of DiagnosisDiagnosis
Progression of symptoms over days to 4 Progression of symptoms over days to 4 weeksweeks
Relative symmetricRelative symmetric Mild sensory signs or symptomsMild sensory signs or symptoms CN involvement, especially bilateral facial CN involvement, especially bilateral facial
weaknessweakness Recovery begins 2-4 weeks after Recovery begins 2-4 weeks after
progression ceasesprogression ceases Absence of fever at onsetAbsence of fever at onset Typical CSF and EMG/NCS featuresTypical CSF and EMG/NCS features

PathophysiologyPathophysiology GBS is believed to result from GBS is believed to result from
autoimmune humoral- and cell-mediated autoimmune humoral- and cell-mediated responses to a recent infection or any of responses to a recent infection or any of a long list of medical problems. a long list of medical problems.
Antibodies formed against ganglioside-Antibodies formed against ganglioside-like epitopes in the lipopolysaccharide like epitopes in the lipopolysaccharide (LPS) layer of some infectious agents (LPS) layer of some infectious agents have been shown in both necropsy and have been shown in both necropsy and animal models to cross-react with the animal models to cross-react with the ganglioside surface molecules of ganglioside surface molecules of peripheral nerves. peripheral nerves.

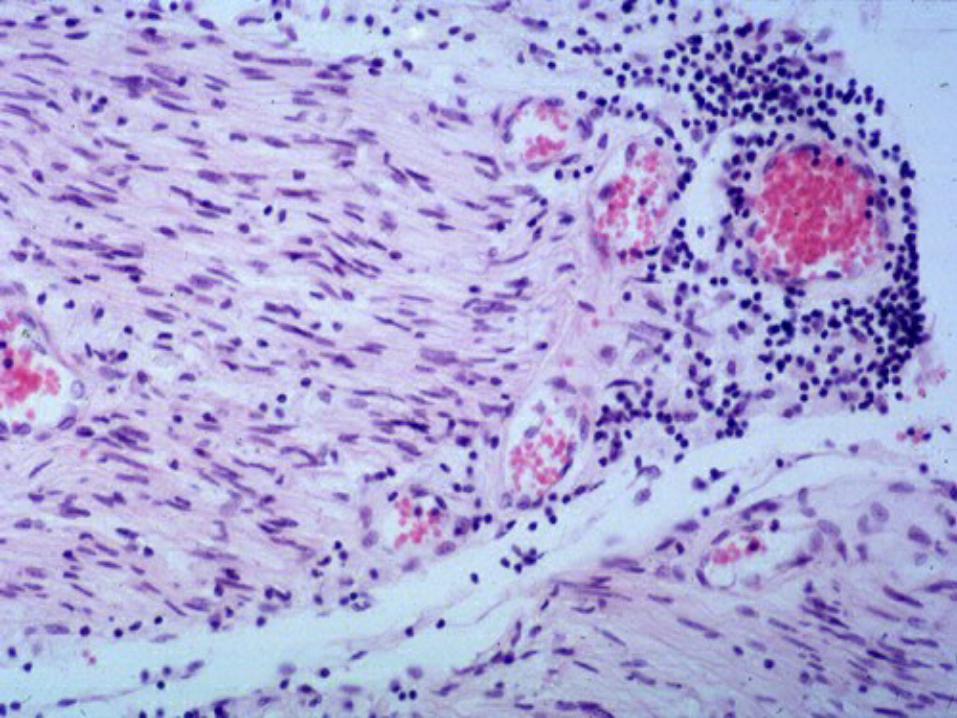
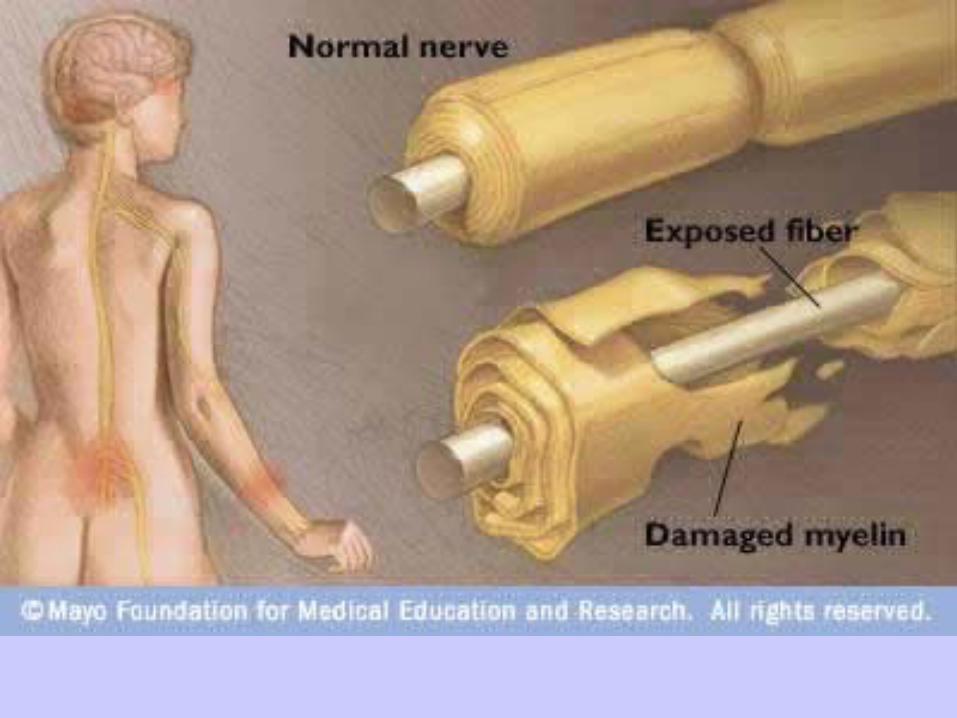
Symptoms generally coincide Symptoms generally coincide pathologically with various patterns pathologically with various patterns of lymphocytic infiltration and of lymphocytic infiltration and macrophage-mediated macrophage-mediated demyelination, depending on the demyelination, depending on the subtype in question. subtype in question.
In a subset of patients, GBS is In a subset of patients, GBS is associated primarily with myelin-associated primarily with myelin-sparing axonal damage resulting sparing axonal damage resulting from a direct cellular immune attack from a direct cellular immune attack on the axon itself. on the axon itself.

Types/VariantsTypes/Variants Acute Inflammatory Demylinating Acute Inflammatory Demylinating
Polyradiculoneuropathy (AIDP)Polyradiculoneuropathy (AIDP) Acute Motor-Sensory Axonal Acute Motor-Sensory Axonal
Neuropathy (AMSAN)Neuropathy (AMSAN) Acute Motor Axonal Neuropathy Acute Motor Axonal Neuropathy
(AMAN)(AMAN) Miller Fisher VariantMiller Fisher Variant Pharyngeal-Cervical-Brachial VariantPharyngeal-Cervical-Brachial Variant Acute PandysautonomiaAcute Pandysautonomia

Acute Inflammatory Acute Inflammatory Demylinating Demylinating
Polyradiculoneuropathy (AIDP)Polyradiculoneuropathy (AIDP) Most common variant, 85% of casesMost common variant, 85% of cases Primarily MotorPrimarily Motor Generally preceded by bacterail or viral Generally preceded by bacterail or viral
infectioninfection Lymphocytic infiltration and macrophage Lymphocytic infiltration and macrophage
mediated demylination of periopheral mediated demylination of periopheral nerves is presentnerves is present
Symptoms generally resolve with Symptoms generally resolve with remylination, max of 4 weeks of progressionremylination, max of 4 weeks of progression

Acute Motor-Sensory Axonal Acute Motor-Sensory Axonal NeuropathyNeuropathy
Motor and sensory involvement with Motor and sensory involvement with severe coursesevere course
Respiratory and bulbar involvementRespiratory and bulbar involvement Primary axonal degenerationPrimary axonal degeneration Patients are typically adults with Patients are typically adults with
both motor and sensory dysfunction both motor and sensory dysfunction with marked muscle wasting and with marked muscle wasting and poor recoverypoor recovery

Acute Motor Axonal Acute Motor Axonal Neuropathy (AMAN)Neuropathy (AMAN)
Motor only with early and severe respiratory Motor only with early and severe respiratory involvement, primary axonal degenerationinvolvement, primary axonal degeneration
More prevalent amongst pediatric age groupsMore prevalent amongst pediatric age groups Up to 75% positive for C. jejuni serology, Up to 75% positive for C. jejuni serology,
often also anti-GM1, anti-GD1a positiveoften also anti-GM1, anti-GD1a positive Hypereflexia is significantly associated with Hypereflexia is significantly associated with
the presence of anti-GM1 antibodiesthe presence of anti-GM1 antibodies Characterized by a rapidly progressive Characterized by a rapidly progressive
weakness, ensuing respiratory failure, and weakness, ensuing respiratory failure, and good recovery.good recovery.

Miller Fisher VariantMiller Fisher Variant Triad: opthalmoplegia, sensory ataxia, Triad: opthalmoplegia, sensory ataxia,
areflexiaareflexia 5% of all cases5% of all cases 96% positive for anti-GQ1b (antiganglioside) 96% positive for anti-GQ1b (antiganglioside)
antibodiesantibodies Patients may also have mild limb weakness, Patients may also have mild limb weakness,
ptosis, facial palsy, or bulbar palsy.ptosis, facial palsy, or bulbar palsy. The ataxia tends to be out of proportion to The ataxia tends to be out of proportion to
the degree of sensory loss.the degree of sensory loss. Recovery generally occurs within 1-3 monthsRecovery generally occurs within 1-3 months

Pharyngeal-Cervical-Brachial Pharyngeal-Cervical-Brachial VariantVariant
Often associated with IgG anti-GT1aOften associated with IgG anti-GT1a Presents with proximal descending Presents with proximal descending
weaknessweakness Must distinguish from botulism and Must distinguish from botulism and
diptheriadiptheria

Acute PandysautonomiaAcute Pandysautonomia
Widespread sympathetic and Widespread sympathetic and parasympathetic failureparasympathetic failure
Very rareVery rare Cardiovascular involvement is Cardiovascular involvement is
common, and dysrhythmias are a common, and dysrhythmias are a significant source of mortality in this significant source of mortality in this form of diseaseform of disease
Recovery is gradual and often Recovery is gradual and often incompleteincomplete

WorkupWorkup Clinical DiagnosisClinical Diagnosis Elevated or rising protein levels on Elevated or rising protein levels on
serial lumbar punctures and 10 or serial lumbar punctures and 10 or fewer mononuclear cells/mm very fewer mononuclear cells/mm very presumptivepresumptive
Normal CSF protein does not rule out Normal CSF protein does not rule out GBS as the level may remain normal in GBS as the level may remain normal in 10% of patients, and a rise in the CSF 10% of patients, and a rise in the CSF protein level may not be observed until protein level may not be observed until 1-2 weeks after the onset of weakness1-2 weeks after the onset of weakness

ImagingImaging MRI is sensitive but nonspecificMRI is sensitive but nonspecific Spinal nerve root enhancement with Spinal nerve root enhancement with
gadolinium is a nonspecific feature seen gadolinium is a nonspecific feature seen in inflammatory conditions and is caused in inflammatory conditions and is caused by disruption of the blood-nerve barrierby disruption of the blood-nerve barrier
Selective anterior nerve root Selective anterior nerve root enhancement appears to be strongly enhancement appears to be strongly suggestive of GBSsuggestive of GBS
The cauda equine nerve roots are The cauda equine nerve roots are enhanced in 83% of patientsenhanced in 83% of patients

Nerve Conduction StudiesNerve Conduction Studies A delay in F waves is present, A delay in F waves is present,
implying nerve root demyelinationimplying nerve root demyelination Compound muscle action potential Compound muscle action potential
(CMAP) amplitude may be decreased(CMAP) amplitude may be decreased Patients may evidence of conduction Patients may evidence of conduction
block or dispersion of responses at block or dispersion of responses at sites of natural nerve compression. sites of natural nerve compression. The extent of decreased action The extent of decreased action potentials correlates with prognosispotentials correlates with prognosis

TreatmentTreatment Only plasmapheresis (the exchange of Only plasmapheresis (the exchange of
patients plasma for albumin) and patients plasma for albumin) and intravenous serum globulin (IVIG) have intravenous serum globulin (IVIG) have proven effectiveproven effective
Both therapies have been shown to shorten Both therapies have been shown to shorten recovery time by as much 50%recovery time by as much 50%
Combining plasma exchange and IVIG Combining plasma exchange and IVIG neither improved outcomes nor shortened neither improved outcomes nor shortened the duration of illnessthe duration of illness
Corticosteriods are ineffective as Corticosteriods are ineffective as monotherapymonotherapy

IVIGIVIG Randomized trials in severe disease Randomized trials in severe disease
show that IVIG started within 4 weeks show that IVIG started within 4 weeks from onset hastens recovery as much from onset hastens recovery as much as pasmapheresisas pasmapheresis
Dosed at 0.4 gram/kg/day IV for 5 DaysDosed at 0.4 gram/kg/day IV for 5 Days Same dose in pediatricsSame dose in pediatrics May increase serum viscosity and May increase serum viscosity and
tromboembolic eventstromboembolic events May increase frequency of migrainesMay increase frequency of migraines May cause aseptic meningitisMay cause aseptic meningitis

Sequela of IVIGSequela of IVIG Increased antiviral and antibacterial Increased antiviral and antibacterial
antibody titers for one monthantibody titers for one month Six-fold increase in ESR lasting two to Six-fold increase in ESR lasting two to
three weeksthree weeks Apparent hyponatremiaApparent hyponatremia