Embed Size (px)
Citation preview

Modelowanie molekularnemetodami chemii kwantowej
Dr hab. Artur MichalakZakład Chemii Teoretycznej
Wydział Chemii UJ
http://www.chemia.uj.edu.pl/~michalak/mmod2007/
Ck08
Wykład 1

Theoretical chemistry
Quantum chemistry
Computational chemistry
Molecular modelling
Molecular informatics(cheminformatics, bioinfiormatics)
Modelowanie molekularneModelowanie molekularneWhat is molecular modelling?
‘Molecular’ clearly implies some
connection with molecules.
The Oxford English Dictionary defines
‘model’ as ‘a simplified or idealized
description of a system or process, often
in mathematical terms, devised to
facilitate calculations and predictions’.
Molecular modelling would therefore
appear to be concerned with ways to
mimic the behaviour of molecules and
molecular systems.

Modelowanie molekularneModelowanie molekularne
- Modelowanie kwantowochemiczne
- Mechanika molekularna
- Dynamika molekularna
- Symulacje Monte-Carlo
- Grafika molekularna

Modelowanie molekularneModelowanie molekularnemetodami chemii kwantowejmetodami chemii kwantowej
Podstawowe zasady obsługi oprogramowania kwantowo-chemicznego; dane do obliczeń
kwantowo-chemicznych; możliwości powszechnie dostępnych systemów obliczeniowych.
Przybliżenie Borna-Oppenheimera; powierzchnia energii potencjalnej; punkty stacjonarne.
Praktyczne aspekty optymalizacji geometrii układów molekularnych; optymalizacja stanów
przejściowych; ścieżki reakcji. Metody wariacyjne; przybliżenie jedno-elektronowe, metoda
Hartree-Focka (HF); metody RHF i UHF; realizacje ab initio i półempiryczne metody HF. Bazy
funkcyjne w obliczeniach ab initio. Orbitale molekularne, gęstość elektronowa, analiza
populacyjna. Sposoby wizualizacji. Wiązanie chemiczne; mapy różnicowe gęstości
elektronowej; orbitale HF i orbitale zlokalizowane; metody lokalizacji orbitali; indeksy rzędów
wiązań. Analiza wibracyjna; drgania normalne. Korelacja elektronowa; metoda mieszania
konfiguracji (CI); perturbacyjna metoda MP. Teoria funkcjonałów gęstości i metoda Kohna-
Shama. Praktyczne aspekty obliczeń DFT; wybór funkcjonału korelacyjno-wymiennego.
Modelowanie dużych układów molekularnych, metody hybrydowe (QMMM). Modelowanie
rozpuszczalnika, metody continuum. Reaktywność chemiczna; jedno- i dwu-reagentowe indeksy
reaktywności; podział energii oddziaływania. Modelowanie reakcji elementarnych
skomplikowanych procesów chemicznych. Wielkości termodynamiczne; energia swobodna
reakcji chemicznych; dynamika molekularna.
Praktyczne zapoznanie z metodami obliczeniowymi chemii kwantowej i ich zastosowaniami w chemii

• Podstawowe idee i metody chemii kwantowej:Funkcja falowa, gęstość elektronowa; równanie Schrodingera; Teoria Funkcjonałów Gęstości (DFT); przyblienie Borna-Oppenheimera, zasada wariacyjna w mechanice kwantowej i w DFT, przyblienie jednoelektronowe; metoda HF; korelacja elektronowa; metody korelacyjne oparte nafunkcji falowej; metoda Kohna-Shama• Dane do obliczeń kwantowo-chemicznych; oprogramowanie; program GAMESS:Geometria czasteczki; macierz Z; bazy funkcyjne w obliczeniach ab initio ; input/output programu GAMESS• Struktura geometryczna układów molekularnych: Optymalizacja geometrii; optymalizacja z wiazami; analiza konformacyjna; problem minimum globalnego• Struktura elektronowa układów molekularnych: Orbitale molekularne, orbitale KS; wiazanie chemiczne; gęstość rónicowa; orbitalezlokalizowane; analiza populacyjna; analiza rzędów wiązań• Analiza wibracyjna; Wielkości termodynamiczne; Reaktywność chemiczna:Analiza wibracyjna; wielkosci termodynamiczne; modelowanie reakcji chemicznych; optymalizacja geometrii stanu przejściowego, IRC; indeksy reaktywności chemicznej, molekularny potencjał elektrostatyczny, funkcja Fukui’ego i teoria orbitali granicznych; jedno- i dwu-reagentowe indeksy reaktywności• Inne zagadnienia:Metody hybrydowe QM/MM; modelowanie wielkich układów; efety rozpuszczalnika; modelowanie w katalizie homo- i heterogenicznej; oddziaływania międzycząsteczkowe, i. in.
Modelowanie molekularne metodami Modelowanie molekularne metodami chch. kwantowej. kwantowejPraktyczne zapoznanie z metodami obliczeniowymi chemii
kwantowej i ich zastosowaniami w chemii

Modelowanie molekularneModelowanie molekularnemetodami chemii kwantowejmetodami chemii kwantowej
Literatura:
Andrew R. Leach „Molecular modelling. Principles
and Applications.”
Prentice Hall, 2001

Modelowanie molekularneModelowanie molekularnemetodami chemii kwantowejmetodami chemii kwantowej
Literatura:
• Frank Jensen, „Introduction to Computational
Chemistry”, Wiley, 1999; second edition 2006• W. Koch, M.C. Holthausen, „A Chemist's Guide to
Density Functional Theory”, Wiley, 2001.
• David C. Young „Computational Chemistry: A Practical
Guide for Applying Techniques to Real-World Problems”,
Wiley 2001
• Encyclopedia of Computational Chemistry. Wiley, 1998.
(wybrane artykuły)
• wybrane artykuły z czasopism chemicznych
Chemia kwantowa (teoria):
• R.F. Nalewajski Podstawy i metody chemii kwantowej. PWN 2001.
• L.Piela, Idee chemi kwantowej. PWN 2001
• A. Szabo, N.L. Ostlund, Modern Quantum Chemistry: Introduction to Advanced
Electronic Structure Theory, Dover, 1989

Chemia kwantowaChemia kwantowa
Fizyka Biologia
Chemia

Quantum ChemistryQuantum Chemistry
= Quantum Mechanics + Chemistry
Who arequantum
mechanics?
Mechanics that repairquantum.

Atomy, cząsteczki, wiązania chemiczneAtomy, cząsteczki, wiązania chemiczne
CC
CC
C
CC
C
C
CC
C
CC
C
CC
C
C
CC
C
N CNC CC
C
C
CCC C
CC
Pd
CC

Atomy, cząsteczki, wiązania chemiczneAtomy, cząsteczki, wiązania chemiczne

jądro
elektron
Atom wodoruAtom wodoru
M = 1 u.
M=1/1840 u.
0,529*10-10 m = 0,529 Å
Model Bohra

jądro
elektron
ok. 10 km
ok. 150 000 000 000 ton
ok. 85 000 000 ton
średnia prędkość elektronuw stanie podstawowym atomu wodoru:ok. 2 000 km/s
Atom wodoruAtom wodoru

Funkcja falowaFunkcja falowa
Funkcja falowa (reprezentacja położeń lub pędów):
Interpretacja probabilistyczna:
N-cząstek, 4N współrzędnych

Funkcja falowaFunkcja falowa
Wartość spodziewana wielkości fizycznej A:
ΨΨ≡ΨΨ= ∫∫ AdQAA ˆˆ... *
ΨΨ≡ΨΨ= ∫∫ HdQHE ˆˆ... *

Hohenberg; Kohn – 1964
Kohn; Sham - 1965
1998 – Walter Kohn – nagroda Nobla
Podstawowa wielkość: gęstość elektronowa, ρ(r)
Teoria Funkcjonałów Gęstości(DFT: Density Functional Theory)
Teoria Funkcjonałów Gęstości(DFT: Density Functional Theory)

I twierdzenie Hohenberga-Kohna
ρ ρ ρ ρ określa jednoznacznie Hamiltonian, a zatem wszystkie wielkości fizyczne układu
E = E[ρρρρ] (dokładna postać funkcjonału - nieznana)
Teoria Funkcjonałów Gęstości(DFT: Density Functional Theory)
Teoria Funkcjonałów Gęstości(DFT: Density Functional Theory)
Podstawowa wielkość: gęstość elektronowa, ρ(r)

Równanie SchrödingeraRównanie SchrödingeraRównanie Schrödingera zależne od czasu:
Stany stacjonarne – N-elektronowa gęstość prawdopodobieństwa nie zależy od czasu:
Funkcja falowa – iloczyn funkcji zależnej od położenia i czasu:
Równanie Schrödingera niezależne od czasu:
Funkcja falowa zawsze zależy od czasu!!!Funkcja falowa zawsze zależy od czasu!!!

Równanie SchrödingeraRównanie Schrödingera
Ψ=Ψ EH
Ψ– funkcja falowaE - energia
H - operator energii (Hamiltonian)

Równanie SchrödingeraRównanie SchrödingeraRozwiązania analityczne – proste układy:
• cząstka swobodna; układ cząstek nieoddziałujących;• cząstka w pudle potencjału;• oscylator harmoniczny; oscylator Morse’a;• rotator sztywny;• atom wodoru;• harmonium (atom dwuelektronowy z elektronami
oddziałującymi z jądrem potencjałem harmonicznym)
Atomy wieloelektronowe i cząsteczki chemiczne
- rozwiązania metodami przybliżonymi

Równanie SchrödingeraRównanie SchrödingeraSeparowalne problemy wielowymiarowe
Jeżeli:
To:

Równanie SchrödingeraRównanie SchrödingeraSeparowalne problemy wielowymiaroweNp. jedno- i dwuwymiarowe pudło potencjału
V=0 a
b V=∞∞∞∞
=
a
xn
an
2cos
2
1 πψ
=
a
yn
ba
xn
a
yxnn yx 2
cos2
1
2cos
2
1,
ππψ

Rotator sztywny:Ruch środka masy:
Hamiltonian ruchu względnego:
Równanie SchrödingeraRównanie Schrödingera

Zagadnienia własne:
Harmoniki sferyczne:
Równanie SchrödingeraRównanie Schrödingera

Harmoniki sferyczneHarmoniki sferyczne: : ss ( l = ( l = 00 ))

Harmoniki sferyczneHarmoniki sferyczne : : pp ( l = 1 )( l = 1 )

Harmoniki sferyczne : Harmoniki sferyczne : dd ( l = 2 )( l = 2 )

Harmoniki sferyczne : Harmoniki sferyczne : ff ( l = 3 )( l = 3 )

Jednostki atomoweJednostki atomowe
Atom wodoru wg. Bohra
l=1a.u.
q=1a.u.
q=-1a.u.m=1a.u.

Atom wodoruAtom wodoru
r
Hamiltonian ruchu względnego:
Zagadnienie własne Hamiltonianu:
),,(
),,(sin
1sin
sin
11
2
2
2
2
222
2
2
2
ϕθ
ϕθϕθθ
θθθ
rE
rr
kZe
rrrr
rrm
Ψ=
Ψ
−
∂
∂+
∂
∂
∂
∂+
∂
∂
∂
∂− h

Atom wodoruAtom wodoru
r
Hamiltonian ruchu względnego:
Zagadnienie własne Hamiltonianu:
Funkcje własne Hamiltonianu:
Ortonormalność:

Atom wodoruAtom wodoru
r
Jednoczesne zagadnienia własne:
),,(),,(ˆ ϕϑψϕϑψ rErh nlmnnlm =
),,()1(),,(ˆ 22 ϕϑψϕϑψ rllrL nlmnlm h+=
),,(),,(ˆ ϕϑψϕϑψ rmrL nlmnlmz h=
),,()1(),,(ˆ 22 ϕϑψϕϑψ rssrS nlmnlm h+=
),,(),,(ˆ ϕϑψϕϑψ rmrS nlmsnlmz h=
n = 1,2,.....
l = 0,1,....,n-1
m = -l,..0,....,l
s = ½
ms = -½, +½

Atom wodoruAtom wodoru
r
Funkcje własne Hamiltonianu:
r
R10
R20
rR30
r
R21
rR31
r
R32
1s
2s
3s
2p
3p 3d

1s
2s3s
OrbitaleOrbitale ss
r
r
r

Orbitale Orbitale 22pp
Orbitale Orbitale 33pp

Orbitale Orbitale 33dd
Orbitale Orbitale 44dd

Atom wodoruAtom wodoru
1s
r
F.falowa stanu podstawowego 1s:
wartośćmaksymalna w pozycji jądra
Sprzeczność?????
Atom wodoru wg. Bohra
R=a0=1a.u.
q=1a.u.Model atomu wodoru Bohra:elektron krąży po orbicie o promieniu
a0 (1 a.u.)

Atom wodoruAtom wodoru
Prawdopodobieństwo napotkania elektronu w odległości rod jądra [w infinitezymalnej warstwie sferycznej wyznaczonej przez dwie kule o promieniach r i (r + dr) ] :
Radialna gęstość prawdopodobieństwa:
r2R2nl(r) , a nie R2
nl(r) !!!

Atom wodoruAtom wodoru
Przykład: stan podstawowy atomu wodoru
Maksimum radialnej gęstości prawdopodobieństwa:
dla Z = 1 rmaks = a0dla Z = 1 rmaks = a0

Atom wodoruAtom wodoru
Radialna gęstośćprawdopodobieństwa:Radialna gęstośćprawdopodobieństwa:
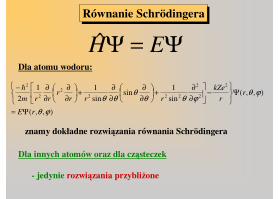
Równanie SchrödingeraRównanie Schrödingera
Ψ=Ψ EHDla atomu wodoru:
),,(
),,(sin
1sin
sin
11
2
2
2
2
222
2
2
2
ϕθ
ϕθϕθθ
θθθ
rE
rr
kZe
rrrr
rrm
Ψ=
Ψ
−
∂
∂+
∂
∂
∂
∂+
∂
∂
∂
∂− h
znamy dokładne rozwiązania równania Schrödingera
Dla innych atomów oraz dla cząsteczek
- jedynie rozwiązania przybliżone

Rozwiązania przybliżone dla atomów i cząsteczek
( ) ( )
( ) ( )∑∑
∑∑ ∑
−+=
=
−
+=
m m
m m N
i
iiq
Ph
CChF
γ δαβαβ
γ δβαβααβ
δαγβαβγδ
δαγβαβγδχχ
|2
1|
|2
1|2ˆ *
∫ ∫= 2121
12
2
*
1
* )()(1
)()()|( drdrrrr
rrklij lkji χχχχ
CDABCCAADCBA lkjilkji δδ)|()|( =
Liczne metody przybliżone chemii kwantowejOparte na funkcji falowej lub gęstości elektronowej

lata 1960lata 1960
Wybitny polski chemik kwantowy prof. Włodzimierz Kołos
pokazał dla cząsteczki H2222 , że dokładność przybliżonych obliczeń kwantowo-chemicznych może być lepsza od doświadczalnej
Prof. Włodzimierz Kołos(1928-1996)

Cząsteczka H2 – kontrowersja Kołosa i Wolniewicza

Cząsteczka H2 – energia jonizacji

Dokładność przybliżonych metod kwantowo-chemicznychDokładność przybliżonych metod kwantowo-chemicznych
Przykład: Odległości C-C w prostych węglowodorach
Etan: H3C-CH3 Etylen: H2C=CH2 Acetylen: HC≡≡≡≡CH
Obliczone: 1,536 ÅEkperyment: 1,540 Å
Różnica: 0,004 Åczyli 0,3%
1,332 Å 1,209 Å1,338 Å 1,203 Å
Różnica: 0,006 Åczyli 0,4%
Różnica: 0,006 Åczyli 0,5%
Różnica ≈≈≈≈0,2 Åczyli ok. 13%
Metoda DFT/BP86/DZP

Struktura elektronowa i własności molekularneStruktura elektronowa i własności molekularne
Akrylonitryl - HOMO Akrylonitryl - LUMO
Akrylan metylu - MEP Akrylan metylu - FF

Cząsteczka benzenu – kontur gęstości elektronowej
Benzen: C6H6
Gęstość elektronowa:ρρρρ(x,y,z)

Cząsteczka benzenu - różnicowa gęstość elektronowa
∑=
−=∆atN
i
at
i
molrrr
1
.. )()()( ρρρ

Etylen – orbitale zlokalizowaneEtylen – orbitale zlokalizowane
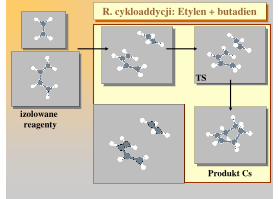
Produkt ostateczny
R. cykloaddycji: Etylen + butadienR. cykloaddycji: Etylen + butadien
izolowane reagenty
TS
Produkt Cs

CC
CC
C
CC
C
C
CC
C
CC
C
CC
C
C
CC
C
N CNC C
C
C
C
CCC C
CC
Pd
CC
Mechanizm polimeryzacji etylenuMechanizm polimeryzacji etylenu

Projektowanie lekówProjektowanie leków
Metody in vivo
Metody in vitro
Metody in sillico

Laboratorium chemika teoretyka
Klaster obliczeniowy „cobalt”, Uniwersytet w Calgary, Kanada, 1998

Metody przyblizone chemii kwantowej– praktyczne narzędzie stosowane w wielu dziedzinach chemii
( ) ( )
( ) ( )∑∑
∑∑ ∑
−+=
=
−
+=
m m
m m N
i
iiq
Ph
CChF
γ δαβαβ
γ δβαβααβ
δαγβαβγδ
δαγβαβγδχχ
|2
1|
|2
1|2ˆ *
∫ ∫= 2121
12
2
*
1
* )()(1
)()()|( drdrrrr
rrklij lkji χχχχ
CDABCCAADCBA lkjilkji δδ)|()|( =

Metody przybliżoneoparte na funkcji falowej lub gęstości elektronowej
Metody przybliżoneoparte na funkcji falowej lub gęstości elektronowej
Ψ Ψ Ψ Ψ = ??????= ??????= ??????= ??????
Ψ=Ψ EHN
O
N
O
O
O
O
O
O
N
N
N
O
N
O
N
OO
O
O
O
O
O O

Przybliżenie Borna-OppenheimeraPrzybliżenie Borna-Oppenheimera

Przybliżenie Borna-Oppenheimera
N elektronów, m – jąder:
Funkcja iloczynowa:
warunki ortonormalności:

Przybliżenie Borna-Oppenheimera
Molekularny Hamiltonian:

Przybliżenie Borna-Oppenheimera
Molekularny Hamiltonian:

Przybliżenie Borna-Oppenheimera
Molekularny Hamiltonian:
Hamiltonian elektronowy:
Nie zawiera energii kinetycznej jąder

Przybliżenie Borna-OppenheimeraRównanie Schrodingera z funkcją iloczynową:
Operator en. kin. jąder działa na f. iloczynową:
Przybliżenie adiabatyczne

Przybliżenie Borna-OppenheimeraRównanie Schrodingera z funkcją iloczynową:
Operator en. kin. jąder działa na f. iloczynową:
Przybliżenie Borna-Oppenheimera

Przybliżenie adiabatyczne i Borna-Oppenheimera
Równanie dynamiki jąder (wycałkowanie po wsp. elektronowych):
Elektronowa powierzchnia energii potencjalnej (PES):
Przybliżenie Borna-Oppenheimera

Przybliżenie Borna-Oppenheimera
Powierzchnia energii potencjalnej:
Eke
RXY

Przybliżenie adiabatyczne i Borna-Oppenheimera
Elektronowa powierzchnia energii potencjalnej (PES):
Dwuetapowe rozwiązanie równania Schrodingera dla molekuły:1) Rozwiązanie równania elektronowego dla wielu geometrii
cząsteczki – wyznaczenie potencjału efektywnego dla ruchu jąder2) Rozwiązanie równania dynamiki jąder poruszajacych się na PES
(w efektywnym potencjale od elektronów)
RXY
Eke

Przybliżenie adiabatyczne i Borna-Oppenheimera
PES- pozwala zdefiniować geometrięcząsteczki
CC
CC
C
CC
C
C
CC
C
CC
C
CC
C
C
CC
C
N CNC C
C
C
C
CCC C
CC
Pd
CC

Przybliżenie adiabatyczne i Borna-Oppenheimera
PES- pozwala zdefiniować geometrięcząsteczki

Przybliżenie adiabatyczne i Borna-Oppenheimera
PES- pozwala zdefiniować geometrięcząsteczki
Punkty charakterystyczne na PES:-minima odpowiadają geometriom równowagowym (substraty, produkty reakcji chemicznych);- punkty siodłowe – stany przejściowe (TS) reakcji chemicznych
TS
TS

Przybliżenie adiabatyczne i Borna-Oppenheimera
Punkty charakterystyczne na PES:-minima odpowiadają geometriom równowagowym (substraty, produkty reakcji chemicznych);- punkty siodłowe – stany przejściowe (TS) reakcji chemicznych
Ścieżki reakcji chemicznej – krzywe na PES łączące substraty i produkty reakcji poprzez odpowiedni TS
TS TS

Przybliżenie adiabatyczne i Borna-Oppenheimera
Punkty charakterystyczne na PES:-minima odpowiadają geometriom równowagowym (substraty, produkty reakcji chemicznych);- punkty siodłowe – stany przejściowe (TS) reakcji chemicznych
Ścieżki reakcji chemicznej – krzywe na PES łączące substraty i produkty reakcji poprzez odpowiedni TS
TS TS

Przybliżenie adiabatyczne i Borna-Oppenheimera
TS TSE
s
substrat
produkt
TS
∆∆∆∆Eakt
Profil energetyczny reakcji

Przybliżenie adiabatyczne i Borna-Oppenheimera
TS TS
Podstawowe wiadomości:1) elektronowa funkcja falowa jest wyznaczana
dla każdej konfiguracji jąder;
2) Struktura reagentów (substratów i produktów) jest wyznaczana w procesie optymalizacji geometrii (poszukiwanie minimów na PES)
3) Struktura stanu przejsciowego jest wyznaczana w procesie optymalizacji TS(poszukiwanie punktów siodłowych na PES)

Metody przybliżoneoparte na funkcji falowej lub gęstości elektronowej
Metody przybliżoneoparte na funkcji falowej lub gęstości elektronowej
ΨΨΨΨel= ??????= ??????= ??????= ??????
Ψ=Ψ EHN
O
N
O
O
O
O
O
O
N
N
N
O
N
O
N
OO
O
O
O
O
O O

Metoda wariacyjnaMetoda wariacyjna
Dowolna funkcja próbna (przybliżona f.f. stanu podstawowego):
Energia stanu próbnego:
ZASADA WARIACYJNA:

II twierdzenie Hohenberga-Kohna
Energia dla dowolnej gęstości próbnej (przybliżenie dla gęstości stanu podstawowego) ρρρρ
dla założonych N oraz v :
Zasada wariacyjna w DFTZasada wariacyjna w DFT
],[)]([)]([ 0 vNENENE ovv =≥ ρρ
Energia stanu podstawowego

Metoda wariacyjnaMetoda wariacyjna
ZASADA WARIACYJNA:
E
E0
E [ φφφφ4 4 4 4 ]]]]E [ φφφφ3 3 3 3 ]]]]
E [ φφφφ2 2 2 2 ]]]]
E [ φφφφ1 1 1 1 ]]]]
np.
Funkcja φφφφ4444 stanowi najlepsze przybliżenie
dla f.f. stanu podstawowego

Metoda wariacyjnaMetoda wariacyjna
ZASADA WARIACYJNA:
E
E0
E [ φφφφ4 4 4 4 ]]]]E [ φφφφ3 3 3 3 ]]]]
E [ φφφφ2 2 2 2 ]]]]
E [ φφφφ1 1 1 1 ]]]]
np.Funkcja φφφφ4444 stanowi
najlepsze przybliżenie dla f.f. stanu podstawowego
Metoda „prób i błędów”- obliczenie funkcjonału energii dla różnych funkcji próbnych

Metoda wariacyjnaMetoda wariacyjnaKlasa funkcji próbnych z parametrami wariacyjnymi:
Funkcjonał energii – funkcja parametrów wariacyjnych:
Szukamy optymalnych wartości parametrów wariacyjnych:
minimalizujących energię:
z układu równań:

Metoda wariacyjnaMetoda wariacyjna

Metoda wariacyjnaMetoda wariacyjnaPrzykład: atom wodoropodobny:
Szukamy funkcji próbnej w postaci:rNer λϕθψ −=),,(
gdzie λλλλ – parametr wariacyjny:
E [ λλλλ ] = ½ λλλλ – λλλλ Z
dE [ λλλλ ] / d λλλλ = λλλλ –Z = 0
daje optymalną wartość:λ λ λ λ = Z
i optymalną postać f. falowej:Zr
Ner−=),,( ϕθψ Eopt = - 0.5 Z2
Rozwiązanie dokładne!

Metoda wariacyjnaMetoda wariacyjnaPrzykład: atom wodoropodobny:
Szukamy funkcji próbnej w postaci:2
),,( rNer λϕθψ −=
daje optymalną wartość:λ λ λ λ = 8 Z2 / (9ππππ)
Eopt = - 0.424 Z2
Energia wyższa od dokładnej!

Metoda wariacyjnaMetoda wariacyjnaPrzykład: atom helu:Szukamy funkcji próbnej w postaci:
Funkcja :
spełnia zagadnienie własne atomu wodoropodobnego:
z efektywnym ładunkiem jądra ζζζζ
Optymalna wartość ζζζζopt = 27/16 < Z
Eopt = -2.8477 a.u.Edokł = -2.9037 a.u.
Epert = -2.75 a.u.
Edokł = -2.9037 a.u.
Epert = -2.75 a.u.

Metoda RitzaMetoda RitzaWariacyjna metoda kombinacji liniowej
Zadana baza:
Poszukiwane współczynniki wariacyjne:

Metoda RitzaMetoda Ritza
Warunek minimum:
Układ równań sekularnych:

Metoda RitzaMetoda RitzaUkład równań sekularnych:
Warunek unormowania:
Wyznacznik sekularny:
Równanie s-tego stopnia na E

Metoda RitzaMetoda RitzaZapis macierzowy:

Metoda RitzaMetoda RitzaKombinacja dwóch funkcji bazy:Całki nakładania:
Elementy macierzy operatora energii:
Równania sekularne:
Wyznacznik sekularny:
Równanie kwadratowe:

Metoda RitzaMetoda Ritza
Przypadek równych poziomów:
Zwykle:
Wyznacznik sekularny – równanie kwadratowe:
Rozwiązania:E
αααα
E1
E2
efekt wiążący
efekt antywiążący

Metoda RitzaMetoda Ritza
Przypadek równych poziomów:Rozwiązania:
E
αααα
E1
E2Cząsteczka H2
kombinacja liniowaorbitali 1s atomów

Metoda RitzaMetoda Ritza
Przypadek równych poziomów:Rozwiązania:
E
αααα
E1
E2Cząsteczka H2
kombinacja liniowaorbitali 1s atomów

Metoda RitzaMetoda Ritza
Przypadek równych poziomów:Rozwiązania:
E
αααα
E1
E2Cząsteczka He2
kombinacja liniowaorbitali 1s atomów

Metoda RitzaMetoda Ritza
Przypadek nierównych poziomów:
Z równania sekularnego:
Modul efektu wiążącego ograniczony nierównością:

Metoda RitzaMetoda Ritza
Przypadek nierównych poziomów:
Z równania sekularnego:
Modul efektu wiążącego ograniczony nierównością:
• efekt energetyczny tym większy, im bliższe poziomy αααα1111, α, α, α, α2 2 2 2
(maksymalny efekt dla αααα1111 = α= α= α= α2 2 2 2 ))))
• efekt energetyczny znika dla ββββ = SSSS = 0 (brak nakładania)

Metoda RitzaMetoda Ritza
Przypadek nierównych poziomów:

Przybliżenie orbitalne(jednoelektronowe)
Przybliżenie orbitalne(jednoelektronowe)
Cząsteczka H2O – orbitale molekularne
Ruch każdego elektronu opisany funkcją zależna od współrzędnych
tylko tego elektronu (funkcją jednoelektronową,
jednoelektronowym orbitalem molekularnym)

Przybliżenie orbitalne(jednoelektronowe)
Przybliżenie orbitalne(jednoelektronowe)
Spinorbital:
część przestrzenna (orbital)
część spinowa

Przybliżenie orbitalne(jednoelektronowe)
Przybliżenie orbitalne(jednoelektronowe)
Spinorbital:
część przestrzenna (orbital)
część spinowa
Przybliżenie orbitalne z restrykcją spinową:
Przybliżenie orbitalne bez restrykcji spinowej:

Przybliżenie orbitalnePrzybliżenie orbitalne
Funkcja wyznacznikowa (wyznacznik Slatera)
Układ N fermionów:
antysymetryczna względem wymiany cząstek

Przybliżenie orbitalnePrzybliżenie orbitalne
Funkcja wyznacznikowa (wyznacznik Slatera)
Układ N fermionów:
Energia – wartość spodziewana Hamiltonianu:
ΨΨ≡ΨΨ= ∫∫ HdQHE ˆˆ... *

Energia elektronowa dla wyznacznika Slatera:
hi,i – całki jednoelektronowe (energia kinetyczna i przyciąganie przez
jądra)
Ji,j - całki kulombowskie (odpychanie międzyelektronowe – część
klasyczna)
Ki,j - całki wymienne (oddziaływanie miedzyelektronowe – część
nieklasyczna)
Przybliżenie orbitalnePrzybliżenie orbitalne

Przybliżenie orbitalnePrzybliżenie orbitalne
Energia elektronowa dla wyznacznika Slatera:
hi,i – całki jednoelektronowe (energia kinetyczna i przyciąganie przez
jądra):

212222*
12
1111*
12 )()(1
)()( drdrrrr
rrJ ψψψψ∫∫=
Przybliżenie orbitalnePrzybliżenie orbitalne
Energia elektronowa dla wyznacznika Slatera:
Ji,j - całki kulombowskie (odpychanie międzyelektronowe – część
klasyczna):

212222*
12
1111*
12 )()(1
)()( drdrrrr
rrJ ψψψψ∫∫=
Ki,j - całki wymienne (odpychanie międzyelektronowe – część
nieklasyczna):
212122*
12
1211*
12 )()(1
)()( drdrrrr
rrK ψψψψ∫∫=
Przybliżenie orbitalnePrzybliżenie orbitalne
Energia elektronowa dla wyznacznika Slatera:
Ji,j - całki kulombowskie (odpychanie międzyelektronowe – część
klasyczna):

Przybliżenie orbitalnePrzybliżenie orbitalne
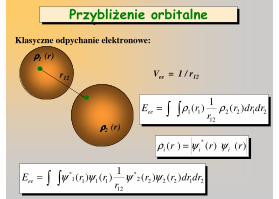
Klasyczne odpychanie elektronowe:
r12Vee = 1 / r12

Przybliżenie orbitalnePrzybliżenie orbitalne
Klasyczne odpychanie elektronowe:
r12Vee = 1 / r12
ρρρρ1 (r)
ρρρρ2 (r)

Przybliżenie orbitalnePrzybliżenie orbitalne
Klasyczne odpychanie elektronowe:
r12Vee = 1 / r12
2122
12
11 )(1
)( drdrrr
rEee ρρ∫∫=
ρρρρ1 (r)
ρρρρ2 (r)
Przybliżenie orbitalnePrzybliżenie orbitalne
Klasyczne odpychanie elektronowe:
r12Vee = 1 / r12
2122
12
11 )(1
)( drdrrr
rEee ρρ∫∫=
ρρρρ1 (r)
ρρρρ2 (r)
)( )()(*
rrr iii ψψρ =
212222*
12
1111* )()(
1)()( drdrrr
rrrEee ψψψψ∫∫=

Przybliżenie orbitalnePrzybliżenie orbitalne
Klasyczne odpychanie elektronowe: całka kulombowska
r12Vee = 1 / r12
2122
12
11 )(1
)( drdrrr
rEee ρρ∫∫=
ρρρρ1 (r)
ρρρρ2 (r))( )()(
*rrr
iiiψψρ =
12212222*
12
1111* )()(
1)()( Jdrdrrr
rrrEee == ∫∫ ψψψψ

212222*
12
1111*
12 )()(1
)()( drdrrrr
rrJ ψψψψ∫∫=
Ki,j - całki wymienne (odpychanie międzyelektronowe – częśćnieklasyczna):
212122*
12
1211*
12 )()(1
)()( drdrrrr
rrK ψψψψ∫∫=
Przybliżenie orbitalnePrzybliżenie orbitalne
Energia elektronowa dla wyznacznika Slatera:
Ji,j - całki kulombowskie (odpychanie międzyelektronowe – część
klasyczna):

Przybliżenie orbitalnePrzybliżenie orbitalneEnergia elektronowa w stanie zamkniętopowłokowym
- suma przyczynków jedno- i dwuelektronowych:
Znajac funkcję falową (orbitale) można policzyć energię jak wyżej.
Jak znaleźć orbitale?

Przybliżenie orbitalnePrzybliżenie orbitalneEnergia elektronowa w stanie zamkniętopowłokowym
- suma przyczynków jedno- i dwuelektronowych:
Znajac funkcję falową (orbitale) można policzyć energię jak wyżej.
Jak znaleźć orbitale?
Z zasady wariacyjnej – orbitale które minimalizują energię:
Metoda Hartree-Focka

Metoda Hartree-FockaMetoda Hartree-FockaRównania HF:
Operator Focka:
Operatory kulombowski i wymienny:

Metoda Hartree-FockaMetoda Hartree-FockaRównania HF:
Operator Focka:
Operatory kulombowski i wymienny – zależą od orbitali :

Metoda Hartree-FockaMetoda Hartree-FockaRównania HF:
Operator Focka:
Operatory kulombowski i wymienny – zależą od orbitali :
Rozwiązanie iteracyjne

Rozwiązanie iteracyjne(SCF – self-consistent-field method,
metoda pola samouzgodnionego)
orbitale→→→→operator Focka →→→→ nowe orbitale →→→→ nowy op. Focka, itd.
dopóki orbitale (i energia) praktycznie się nie zmieniają
Metoda Hartree-FockaMetoda Hartree-FockaRównania HF:

Metoda Hartree-Focka-RoothanaMetoda Hartree-Focka-Roothana
Funkja falowa – wyznacznik Slatera
Orbtale jednoelektronowe – kombinacja liniowa
założonych funkcji bazy
)(...)()(
)1(...)1()1(
!
1
21
21
NNN
N
N
N
ϕϕϕ
ϕϕϕ
MMMM
MMMM=Ψ
∑=
=m
j
jjii c1
)1()1( χϕ
funkcje bazy
Analityczna metoda HF

Metoda Hartree-FockaMetoda Hartree-Focka
Odtwarza do 99 % energii całkowitej
Pozostały 1% jest często bardzo ważny w chemii!
Przybliżenie jednoelektronowe zaniedbuje korelację elektronową

Błędy metody SCFE
ESCF
EHFL (granica Hartree-Focka ‘HF limit’ – nieskończona baza)
ENREL (‘dokładne’ podejście nierelatwistyczne)
Ee (dokładna energia relatywistyczna)
∆εbłąd bazy= EHFL − ESCF < 0
∆Ekorel = ENREL − EHFL < 0 energia korelacji
∆Erel = Ee − ENREL < 0 (poprawka relatywistyczna)
∆Ekorel = −0.0420 ∆Erel = −0.0001>He:
∆Ekorel = −0.4940 ∆Erel = −0.5840< Si:
Metoda Hartree-FockaMetoda Hartree-Focka

Metody korelacyjneMetody korelacyjne
Funkcja falowa jako kombinacja wielu wyznaczników Slatera:Mieszanie konfiguracji (CI, configuration interaction);Metody wielokonfiguracyjne;Metody perturbacyjne (teoria zaburzeń);Metody sprzężonych klastrów;Teoria funkcjonałów gęstości (DFT)
CI, CIS, CID, CISD, etc.
MCSCF,
CASSCF,
CC, CCSD, CCSDT, CCSDTQ, etc.
MP, MP2, MP3, MP4, etc.
DFT, LDA, VWN, BP, PW91,BLYP, PBE, B3LYP, B3PW91, etc.
Nie są oparte na zasadzie wariacyjnej -energia może być niższa od dokładnej!

Wyznacznik KS zbudowany ze spinorbitali
Gestość elektronowa:
Jedno-wyznacznikowa funkcja falowa:
Hipotetyczny układ nieoddziaływających elektronów o gęstości
odpowiadające układowi rzeczywistemu
elektronów odzdiaływajacych
Teoria funkcjonałów gęstości (DFT)Teoria funkcjonałów gęstości (DFT)Metoda Kohna-Shama

Metoda Kohna-Shama
Funkcjonał energii
Exc[ρ] - funkcjonał korelacyjno wymienny (złony nieklasyczne)
Ts[ρ] - energia kinetyczna elektronów niezależnych
Vne[ρ] - przyciąganie elektron-jądro
Jee[ρ] - klasyczne odpychanie międzyelektronowe
zawaira korelacyjną poprawkę do energii kinetycznej
Nieznana dokładna postać EXC
- stosowane przybliżone funkcjonały XC
Teoria funkcjonałów gęstości (DFT)Teoria funkcjonałów gęstości (DFT)

Metoda Kohna-ShamaMetoda Kohna-Shama
Równania KS :
Rozwiązania iteracyjne
ponieważ
Metoda LCAO
baza

Przybliżenie lokalnych gęstości spinowych,
(Local spin density approximation, LSDA)
Kohn-Sham methodKohn-Sham method

Przyczynki korelacyno wymienne:
Funkcjonały XCFunkcjonały XC

LDA/LSDA
Funkcjonały XCFunkcjonały XC

Funkcjonały gradientowe (zawiarają człony zależne od gradientów gęstości)
Perdew-Wang 1986
Becke 1988
Perdew-Wang 1991
Funkcjonały XCFunkcjonały XC

Perdew-Wang 1986 Becke 1988
Perdew-Wang 1991
Lee-Yang-Parr (LYP) Perdew 1986
Gradient functionals (contains terms dependent of density-gradients)
Funkcjonały XCFunkcjonały XC
PW86/88
PW91
BLYP
PBE
Funkcjonały gradientowe (zawiarają człony zależne od gradientów gęstości)
Funkcjonały XCFunkcjonały XC

B3LYP
B3PW91
Funkcjonały hybrydowe
- zawiarają domieszkę dokładnej wymiany (HF)
Metoda HF – dokładna wymiana, brak korelacjiMetoda KS – zawiara wymianę i korelacje, ale oba
przyczynki przybliżone
Funkcjonały XCFunkcjonały XC

Podsumowanie
• przybliżenie Borna-Oppenheimera dla cząsteczek• powierzchnia energii potencjalnej (PES)• przybliżenie jednoelektronowe, przybliżenie orbitalne – metoda HF• DFT – metoda KS (przybliżone funkcjonały XC)• bazy funkcyjne (metoda LCAO) w obu przypadkach - HF, DFT• rozwiązania iteracyjne równań HF i KS
c.d.n.





























