Embed Size (px)
Citation preview

The FASEB Journal • Research Communication
Thrombospondin 1 binding to calreticulin-LRP1 signalsresistance to anoikis
Manuel A. Pallero,*,1 Carrie A. Elzie,*,1,2 Jiping Chen,* Deane F. Mosher,†
and Joanne E. Murphy-Ullrich*,3
*Department of Pathology, Division of Molecular and Cellular Pathology and the Center forMetabolic Bone Disease and the BioMatrix Engineering and Regenerative Medicine Center,University of Alabama at Birmingham, Birmingham, Alabama, USA; and †Department of Medicine,University of Wisconsin, Madison, Wisconsin, USA
ABSTRACT Anoikis, apoptotic cell death due to lossof cell adhesion, is critical for regulation of tissuehomeostasis in tissue remodeling. Fibrogenesis is asso-ciated with reduced fibroblast apoptosis. The matricel-lular protein thrombospondin 1 (TSP1) regulates celladhesion and motility during tissue remodeling and infibrogenesis. The N-terminal domain of TSP1 binds tothe calreticulin-LRP1 receptor co-complex to signaldown-regulation of cell adhesion and increased cellmotility through focal adhesion disassembly. TSP1 sig-naling through calreticulin-LRP1 activates cell survivalsignals such as PI3-kinase. Therefore, we tested thehypothesis that TSP1 supports cell survival under ad-hesion-independent conditions to facilitate tissue re-modeling. Here, we show that platelet TSP1, its N-terminal domain (NoC1) as a recombinant protein, or apeptide comprising the calreticulin-LRP1 binding site[amino acids 17–35 (hep I)] in the N-terminal domainpromotes fibroblast survival under anchorage-indepen-dent conditions. TSP1 activates Akt and decreasesapoptotic signaling through caspase 3 and PARP1 insuspended fibroblasts. Inhibition of PI3K/Akt activityblocks TSP1-mediated anchorage-independent survival.Fibroblasts lacking LRP1 or expressing calreticulinlacking the TSP1 binding site do not respond to TSP1with anchorage-independent survival. These data definea novel role for TSP1 signaling through the calreticulin/LRP1 co-complex in tissue remodeling and fibroticresponses through stimulation of anoikis resistance.—Pallero, M. A., Elzie, C. A., Chen, J., Mosher, D. F.,Murphy-Ullrich, J. E. Thrombospondin 1 binding to cal-reticulin-LRP1 signals resistance to anoikis. FASEB J. 22,3968–3979 (2008)
Key Words: apoptosis � fibroblasts � cell adhesion � focal adhe-sions � Akt
Signaling through the extracellular matrix (ECM)controls cell proliferation, differentiation, and survival.Adhesion to the ECM mediates survival primarilythrough integrin-mediated signals, which initiate down-stream pathways, including focal adhesion kinase(FAK) and phosphoinositide 3-kinase (PI3K) -depen-dent Akt activation, to suppress apoptosis (1, 2). Re-
modeling of the ECM during development, woundhealing, fibrosis, and tumorigenesis alters these signals.Lack of signaling from the ECM or inappropriatecell-matrix contacts induces anoikis, programmed celldeath, in most adherent cell types (3, 4). Anoikis iscritical for regulation of tissue homeostasis duringdevelopment, tissue remodeling, and in resolution ofwound-healing responses (5–7). In contrast, resistanceto anoikis in tumor cells increases survival time in theabsence of matrix (8, 9). Anoikis resistance is alsoinvolved in pathological events, including prolongedmyofibroblast survival in lung fibrosis and other fibro-proliferative diseases, such as arthritis and atheroscle-rosis (9–11). Therapeutic modulation of anoikis sus-ceptibility or resistance can be utilized to increasesurvival of transplanted islet cells, improve graft viabil-ity, and improve seeding of tissue engineered scaffoldsby increasing mesenchymal stem cell survival (12, 13).Thus, understanding the mechanisms that regulateanoikis is important for understanding critical physio-logical and pathological processes.
Thrombospondin 1 (TSP1) is a matricellular proteinreleased from platelet �-granules and secreted by mostcell types in response to stress and injury (14, 15). TSP1plays important roles in cell adhesion and motility,growth factor regulation, nitric oxide signaling, andremodeling of the ECM through both direct and indi-rect, protease-mediated effects (14, 16, 17). TSP1 hasparadoxical functions due to signaling through multi-ple receptor binding sites located in different domainsof the polypeptide (14, 18). It is well established thatTSP1 can induce apoptosis of endothelial cells viainteractions of the type 1 repeats of TSP1 with one of itsreceptors, CD36 (19, 20). Binding of the C-terminaldomain of TSP1 to CD47 stimulates caspase-indepen-dent apoptosis in promyelocytic leukemia NB4 cells
1 These authors contributed equally to this work.2 Current address: Biological Sciences Department, U-5214
MRB III, Vanderbilt University, Nashville, TN 37235, USA.3 Correspondence: Department of Pathology, VH 668 1530
3rd Ave., South, Birmingham, AL 35294-0019, USA. E-mail:[email protected]
doi: 10.1096/fj.07-104802
3968 0892-6638/08/0022-3968 © FASEB

(21), initiates mechanosensitive apoptosis of fibro-blasts, and mediates apoptosis of endothelial cells in-duced by proatherogenic flow conditions (22, 23).TSP1 signaling of cell death through CD47, however,may be context and/or cell type specific, because theCD47 binding TSP1 4N1K peptide reduced ceramide-stimulated apoptosis of thyroid epithelial cells (24).
In contrast to these generally proapoptotic actions ofthe type 1 repeats and the C-terminal region of TSP1,the N-terminal domain of TSP1 has been shown tostimulate endothelial cell proliferation and angiogene-sis through both syndecan 4 and integrins �3�1 and�9�1 (25–28). Proteases released by inflammatory cellscan cleave the N-terminal domain of TSP1 with releaseinto endothelial cell-conditioned media (29, 30). Cleav-age of the N-terminal domain by ADAMTS (a disinte-grin-like and metalloprotease with thrombospondintype 1 motifs) results in N-terminal domain localizationto wounds (31). In addition, apoptotic macrophagesrelease the N-terminal domain following cleavage by achymotrypsin-like serine protease (32). These data sug-gest that the N-terminal domain of TSP1 might haveunique functions, either in the context of the entireTSP1 molecule or as a truncated protein, distinct fromthose of the C-terminal regions of TSP1.
Previously, we established that TSP1 signals focaladhesion disassembly in adherent, spread cells, a statereferred to as intermediate adhesion (33, 34). Thisaction of TSP1 is mediated by a sequence located in theN-terminal domain (aa 17–35) and can be mimicked bya peptide (hep I) comprising this sequence or by theN-terminal domain (35). Although the N-terminal do-main recognizes multiple receptors (18, 28), the actionof the hep I sequence is mediated by binding to cellsurface calreticulin (CRT), which signals in associationwith LRP1 (36–38). The structure of the N-terminaldomain suggests that the key basic residues in the aa17–35 span are accessible for binding (39). TSP1 bind-ing to CRT stimulates a G�i-dependent activation ofFAK, ERK, and PI3K (40). These signals converge todown-regulate Rho, stimulating cytoskeletal reorganiza-tion with loss of focal adhesions and an increase in bothrandom and directed cell migration (41, 42). TSP1binding to CRT/LRP1 in cooperation with binding toCD47 on T cells also stimulates autocrine T cell adhe-sion and motility (43).
On the basis of our findings that the hep I sequenceof TSP1 stimulates cellular deadhesion and increasedmotility in the presence of concomitant increases in cellsurvival signals (41, 42, 44), we hypothesized that TSP1signaling through the CRT-LRP1 receptor might pro-tect cells from apoptotic cell death during down-regu-lation of cell adhesion, i.e., anoikis. We now report thatTSP1 signaling through binding of the hep I sequenceto the CRT-LRP1 receptor complex increases cell sur-vival of fibroblasts under adhesion-independent condi-tions by down-regulating apoptotic signaling and stim-ulating Akt activity. Anoikis resistance in fibroblasts ismediated not only by platelet TSP1, but also by thetrimeric N-terminal domain as a recombinant protein
and the hep I peptide mimetic of the CRT-bindingsequence. These findings suggest a novel function forTSP1 in modulating cell survival during conditions ofintermediate cell adhesion, such as ECM turnover andtissue remodeling in response to injury.
MATERIALS AND METHODS
Antibodies and other reagents
Rabbit anti-Akt (#9272), mouse monoclonal anti-phosphoAktS473 (#4051), rabbit anti-PARP1 (#9542) antibody, and rab-bit antibody to cleaved and uncleaved caspase 3 (#9664) werepurchased from Cell Signaling Technology (Beverly, MA,USA). Rabbit anti-�-tubulin antibody (SC9104) and rabbitanti-hemagglutinin (#805) immunoglobulin G (IgG) werefrom Santa Cruz Biotechnology (Santa Cruz, CA, USA).Nonimmune rabbit IgG and goat anti-mouse and anti-rabbitantibodies conjugated to horseradish peroxidase (HRP) wereobtained from Jackson ImmunoResearch Laboratories (WestGrove, PA, USA). AlexaFluor 488 conjugated goat anti-rabbitIgG (A11008) was from Molecular Probes/Invitrogen (Carls-bad, CA, USA). Poly (2-hydroxyethyl methacrylate) (poly-HEMA), soybean trypsin inhibitor, and insulin were purchasedfrom Sigma Chemical (St. Louis, MO, USA). Wortmannin wasfrom Kamiya Biomedical Company (Seattle, WA, USA).LY294002, U0126, and PD98059 were purchased from Promega(Madison, WI, USA).
Proteins and peptides
TSP1 was isolated from human platelets purchased fromthe American Red Cross and purified as described previ-ously, using heparin affinity and gel filtration chromatog-raphy (35). Peptides hep I (ELTGAARKGSGRRLVKGPD),modified hep I (ELTGAARAGSGRRLVAGPD), CRT19.36(RWIESKHKSDFGKFVLSS), and CRT20.30A (RWIESAAASDKFGLAAASS), a control for CRT19.36, were purchased fromAnaSpec, Inc. (San Jose, CA, USA) (36). All peptides are�95% pure by HPLC and mass spectrometry. Recombinanttrimeric N-terminal heparin binding domain of TSP1, NoC1,and the trimeric construct of TSP1 lacking the N-terminaldomain (delN1) were purified from conditioned medium ofHigh-Five cells infected with recombinant baculovirus (28, 45,46). Molar concentrations of the trimeric proteins TSP1 andNoC1 and delN1 were calculated on the basis of absorbanceat 280 nm and the molecular mass of the monomeric sub-units. cDNA for GST-CRT was a gift from Dr. Marek Michalak(University of Alberta, Edmonton, AB, Canada). TenascinFNIII A-D was a gift from Dr. Harold Erickson (Duke Univer-sity, Durham, NC, USA).
Cell culture and generation of CRT mouse embryonicfibroblasts cell lines
Wild-type (K41) and CRT(�/�) null (K42) mouse embryonicfibroblasts (MEFs) were a gift from Dr. Marek Michalak (Uni-versity of Alberta, Edmonton, AB, Canada) (47). LRP1-deficient(PEA13 ATCC-CRL-2216) MEFs were purchased from AmericanType Culture Collection (Manassas, VA, USA). Cells were main-tained in Dulbecco modified Eagle medium (DMEM), supple-mented with 10% fetal bovine serum, and routinely tested formycoplasma.
Stable cell lines expressing either wild-type CRT (K42CRT)or mutated CRT lacking aa 19–36 (K42CRT�19.36) weregenerated. Plasmid pcDNA3-CRT-HAQ [CRT with a hemag-
3969TSP1/HEP I INDUCES ANOIKIS RESISTANCE

glutinin (HA) tag] was a gift from Dr. Marek Michalak(University of Alberta, Edmonton, AB, Canada), andpcDNA3-CRT-HAQ-�19–36 was generated using the Quick-Change site-directed mutagenesis kit (Stratagene, La Jolla, CA,USA) (36). Wild-type or mutant CRT cDNAs were subclonedinto a vector containing the zeocine selective marker, pcDNA4-Topo/His-myc-A (Invitrogen). Plasmids pcDNA3-CRT-HAQand pCDNA3-CRT-HAQ-�19–36 were double-digested withXbaI and HindIII, and the resulting 1.2-kb fragments wereisolated and replaced the 1.2 kb XbaI-HindIII DNA fragmentof pcDNA4-Topo/His-myc-A to generate plasmids pcDNA4-wtCRTand pcDNA4-CRT�19–36, respectively. The wild-typeand mutant CRT sequences were confirmed by automateddideoxynucleotide sequencing [University of Alabama at Bir-mingham (UAB) Center for AIDS Research DNA SequencingCore]. DNA was purified with the EndoFree Plasmid Maxi Kit(Qiagen, Valencia, CA, USA). K42 MEFs were seeded at adensity of 500,000/100 mm plate and grown in regular mediaovernight. Cells were transfected with 6 �g of DNA (pcDNA4-wtCRT or pcDNA4-CRT�19–36) using the TransIT reagent(Mirus, Madison, WI, USA) in RPMI media. Forty-eight hoursafter transfection, cells were split 1:40 or 1:80 into mediacontaining 400 �g/ml Zeocin (Invitrogen) or until individualcolonies appeared. Colonies were picked, amplified, andscreened for HA and CRT expression by immunoblotting.Clones with levels of CRT expression similar to wild-type K41MEFs were selected. Surface expression of CRT (wild-typeand mutated) was also confirmed by flow cytometry onnonpermeabilized cells (Supplemental Fig. 1). Cells werethen further analyzed for their ability to undergo focaladhesion disassembly in the presence of hep I and TSP1(Supplemental Fig. 2). Cells were cultured from passages3–21 in DMEM with 4.5 g/L glucose, 2 mM glutamine, and10% FBS.
Anoikis conditions
Wells were coated with poly-HEMA to prevent adhesion andspreading (4, 48). Poly-HEMA at 120 �g/ml was dissolved in95% ethanol by shaking at 37°C, and 100 �l was added toeach well of a 24-well plate, followed by overnight incubationto allow evaporation of ethanol. Control wells were coatedwith FBS. Cells were harvested with 0.05% trypsin. Trypsin wasinhibited with 10% FBS in DMEM and cells were rinsed twicein DMEM. Cells (30,000 cells/well in 500 �l) were added toeither poly-HEMA or serum-coated wells and incubated at37°C, 5% CO2 with DMEM, TSP1, hep I, modified hep I,NoC1, or insulin, as indicated in the figure legends. Mainte-nance of suspended cells in poly-HEMA-coated wells wasmonitored by phase microscopy. Suspended cells were col-lected by aspiration and then lysed in 2� Laemmli buffer forimmunoblotting. Adherent cells from serum-coated wellswere harvested in Laemmli buffer and pooled with anynonattached cells recovered by centrifugation.
Cell viability assay
Cells were trypsinized, trypsin was neutralized with 1 �g/mlsoybean trypsin inhibitor, and cells were resuspended inserum-free DMEM. Cells (25,000) serum-free DMEM wereadded to poly-HEMA-coated wells and maintained for timesindicated. Control cells were plated on serum-coated wells inserum-free medium. Cell viability was measured with CellTiterBlue (Promega, Madison, WI). CellTiterBlue reagent(100 �l) was added to each well and incubated at 37°C for 3 h.Fluorescence was measured with a plate reader (excitation530/emission 580). Samples were assayed in triplicate in atleast 3 separate experiments.
Fluorescein isothiocyanate (FITC) -annexin V flowcytometry for apoptosis
K41 MEFs were plated on either poly-HEMA or serum-coatedwells as described in the anoikis assay. Attached cells wereincubated in serum-free DMEM and suspended cells (105
cells/condition) were incubated with either 32.5 nM TSP1, 30nM NoC1, 30 nM delN1, 1 �M hep I, 1 �M modified hep I,500 nM insulin, or serum-free DMEM for 5.5 h at 37°C with5% CO2. Cells were washed and then tested using theAnnexin V-FITC apoptosis detection kit (Calbiochem/Nova-biochem/Novagen, San Diego, CA, USA; PF032). Cell suspen-sions (20,000 cells/condition) were evaluated for FITC-an-nexin V binding and for propidium iodide staining at theUAB Center for AIDS Research Flow Cytometry Core Facilityusing a Becton-Dickinson FACS Aria Cell sorter with BDFACSDiva 6.0 software (Becton Dickinson, Franklin Lakes,NJ, USA). Samples were evaluated in triplicate.
Caspase 3 activity assay
Caspase-3 activity in K41 MEF cell lysates was measured usinga caspase-3 activity kit (Becton-Dickinson-Pharmingen, SanJose, CA). K41 MEFs (150,000 cells) were plated in triplicatefor each condition in poly-HEMA-coated-wells in serum-freemedia with treatments for 6 h. Adherent cells were plated onserum-coated wells in serum-free media. Cells were lysed, andlysates were incubated in a 96-well plate in HEPES buffer with5 �l of caspase-3 fluorogenic substrate (Ac-DEVD-AMC) for1 h at 37°C in the dark. Fluorescence was measured with aplate reader (excitation 360/emission 460).
Caspase 3 and PARP1 immunoblotting
MEFs (150,000 cells) were plated in poly-HEMA-coated wellsof a 6-well plate in serum-free DMEM with treatments asindicated in the figure legends. Adherent cells were plated onserum-coated wells in serum-free DMEM. Following treat-ments, suspended cells were removed and lysed in reducingLaemmli buffer with a protease inhibitor cocktail (SigmaP2714). Adherent cells from serum-coated wells were har-vested in Laemmli buffer. Samples were sonicated on ice,boiled for 6 min, and centrifuged at 10,000 rpm to pelletinsoluble material. Equal volumes of lysates were separatedon a 4–15% gradient SDS-PAGE. Proteins were transferred tonitrocellulose membranes for 1–1.5 h at 100 V at 4°C.Membranes were blocked with 1% casein (1 h at roomtemperature) and then probed overnight at 4°C with rabbitanti-cleaved caspase 3 (1:1000) or rabbit anti-PARP1 poly-clonal antibody (1:1000), washed with TBS-T (10 mM Tris-HCl, 100 mM NaCl, and 0.1% Tween 20), probed with goatanti-rabbit IgG-HRP (1:10,000) at room temperature for 1 h,and washed with TBS-T. Blots were developed using WesternLightning Chemiluminescence Reagent Plus (Perkin Elmer,Wellesley, MA, USA). Membranes were stripped with ReBlotStrong (Chemicon, Temecula, CA, USA) at 1:10 in water for15 min at room temperature and then washed twice withTBS-T. Membranes were reprobed with rabbit anti-�-tubulinantibody (1:1000) to normalize for protein loading.
To assess phosphorylated and total Akt, cells on poly-HEMA were incubated with treatments for 10 min, or cells onserum-coated wells were incubated for 30 min and then lysedin 2� Laemmli buffer with both protease and phosphataseinhibitor cocktails (Sigma P2714, P2850). Samples were pro-cessed for immunoblotting as described above. Membraneswere probed with rabbit anti-phosphoAkt S473 rabbit mono-clonal antibody (1:1000). Blots were developed using WesternLightning Chemiluminescence Reagent Plus (Perkin Elmer).
3970 Vol. 22 November 2008 PALLERO ET AL.The FASEB Journal
Membranes were stripped with ReBlot Strong and thenprobed overnight at 4°C with rabbit anti-Akt polyclonalantibody (1:1000). Bands were visualized as described above.All experiments were performed on at least 3 separateoccasions.
Focal adhesion assays
The presence or absence of focal adhesions in spread cellswas scored on K41, K42, K42 CRT, and K42 CRT�19.36 MEFsusing interference reflection microscopy by established meth-ods, as described previously (35).
Analysis of cell surface CRT�19.36 and CRTby flow cytometry
K42 CRT(�/�) MEFS, K42 MEFs stably transfected with eitherHA-tagged CRT (K42 CRT), or HA-tagged CRT with aa 19–36deleted (K42 CRT�19.36) were assessed for cell surface CRTexpression by flow cytometric analysis of the HA tag on nonper-meabilized cells. Cells cultured until 60–70% confluent wereharvested with 0.05% trypsin (Sigma), and trypsin was inacti-vated with DMEM with 10% FBS. Cells were pelleted by centrif-ugation for 2 min at 1200 rpm, resuspended in phenol red-freeDMEM, and rinsed twice. Single-cell suspensions were passedthrough a strain cap tube (352235; B-D Falcon, Canaan, CT,USA) and then pelleted by centrifugation. Cells were resus-pended in 1% cold paraformaldehyde and fixed for 10 min at4°C and then washed twice in serum-free phenol red-freeDMEM. Cell suspensions (300,000–500,000 cells) were incu-bated with 2–4 �g/ml rabbit anti-HA or nonimmune rabbit IgGfor 1 h at 4°C. Cells were washed twice with phenol red-freeDMEM and then incubated with Alexa Fluor 488-labeled goatanti-rabbit IgG (1:250) for 45–60 min at 4°C. Cells were washedtwice in phenol red-free DMEM and resuspended to 170,000cells/ml in 300 �l phenol red-free DMEM with 1 �M propidiumiodide. �10,000 cells/condition were evaluated by flow cytom-etry.
Statistical analysis
Statistical significance where indicated was determined using1-way ANOVA. Results were considered to be significant at P 0.01, as indicated.
RESULTS
The N-terminal domain of TSP1 protects MEFsfrom anoikis
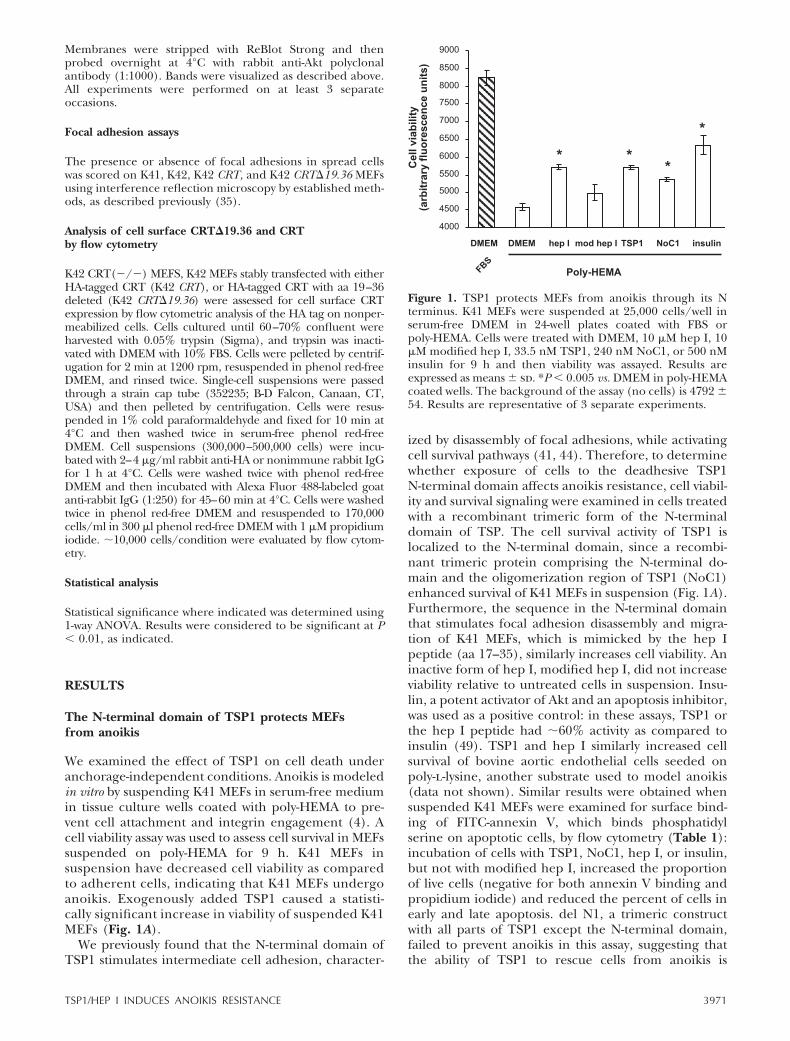
We examined the effect of TSP1 on cell death underanchorage-independent conditions. Anoikis is modeledin vitro by suspending K41 MEFs in serum-free mediumin tissue culture wells coated with poly-HEMA to pre-vent cell attachment and integrin engagement (4). Acell viability assay was used to assess cell survival in MEFssuspended on poly-HEMA for 9 h. K41 MEFs insuspension have decreased cell viability as comparedto adherent cells, indicating that K41 MEFs undergoanoikis. Exogenously added TSP1 caused a statisti-cally significant increase in viability of suspended K41MEFs (Fig. 1A).
We previously found that the N-terminal domain ofTSP1 stimulates intermediate cell adhesion, character-
ized by disassembly of focal adhesions, while activatingcell survival pathways (41, 44). Therefore, to determinewhether exposure of cells to the deadhesive TSP1N-terminal domain affects anoikis resistance, cell viabil-ity and survival signaling were examined in cells treatedwith a recombinant trimeric form of the N-terminaldomain of TSP. The cell survival activity of TSP1 islocalized to the N-terminal domain, since a recombi-nant trimeric protein comprising the N-terminal do-main and the oligomerization region of TSP1 (NoC1)enhanced survival of K41 MEFs in suspension (Fig. 1A).Furthermore, the sequence in the N-terminal domainthat stimulates focal adhesion disassembly and migra-tion of K41 MEFs, which is mimicked by the hep Ipeptide (aa 17–35), similarly increases cell viability. Aninactive form of hep I, modified hep I, did not increaseviability relative to untreated cells in suspension. Insu-lin, a potent activator of Akt and an apoptosis inhibitor,was used as a positive control: in these assays, TSP1 orthe hep I peptide had �60% activity as compared toinsulin (49). TSP1 and hep I similarly increased cellsurvival of bovine aortic endothelial cells seeded onpoly-l-lysine, another substrate used to model anoikis(data not shown). Similar results were obtained whensuspended K41 MEFs were examined for surface bind-ing of FITC-annexin V, which binds phosphatidylserine on apoptotic cells, by flow cytometry (Table 1):incubation of cells with TSP1, NoC1, hep I, or insulin,but not with modified hep I, increased the proportionof live cells (negative for both annexin V binding andpropidium iodide) and reduced the percent of cells inearly and late apoptosis. del N1, a trimeric constructwith all parts of TSP1 except the N-terminal domain,failed to prevent anoikis in this assay, suggesting thatthe ability of TSP1 to rescue cells from anoikis is
Figure 1. TSP1 protects MEFs from anoikis through its Nterminus. K41 MEFs were suspended at 25,000 cells/well inserum-free DMEM in 24-well plates coated with FBS orpoly-HEMA. Cells were treated with DMEM, 10 �M hep I, 10�M modified hep I, 33.5 nM TSP1, 240 nM NoC1, or 500 nMinsulin for 9 h and then viability was assayed. Results areexpressed as means sd. *P 0.005 vs. DMEM in poly-HEMAcoated wells. The background of the assay (no cells) is 4792 54. Results are representative of 3 separate experiments.
3971TSP1/HEP I INDUCES ANOIKIS RESISTANCE
restricted to the N-terminal domain (Table 1) (46).The increase in viability of cells in suspension is not dueto increased proliferation, because studies from this labshowed that hep I does not stimulate proliferation ofadherent K41 MEFs (unpublished results).
Protection against anoikis by TSP1 and hep I is dosedependent. TSP1 at 67.5 nM or greater and hep I atconcentrations equal to or greater than 1 �M wereeffective. However, control peptide (modified hep I)did not have any significant protective effect (data notshown).
TSP1 inhibits apoptotic signaling of cellsin suspension
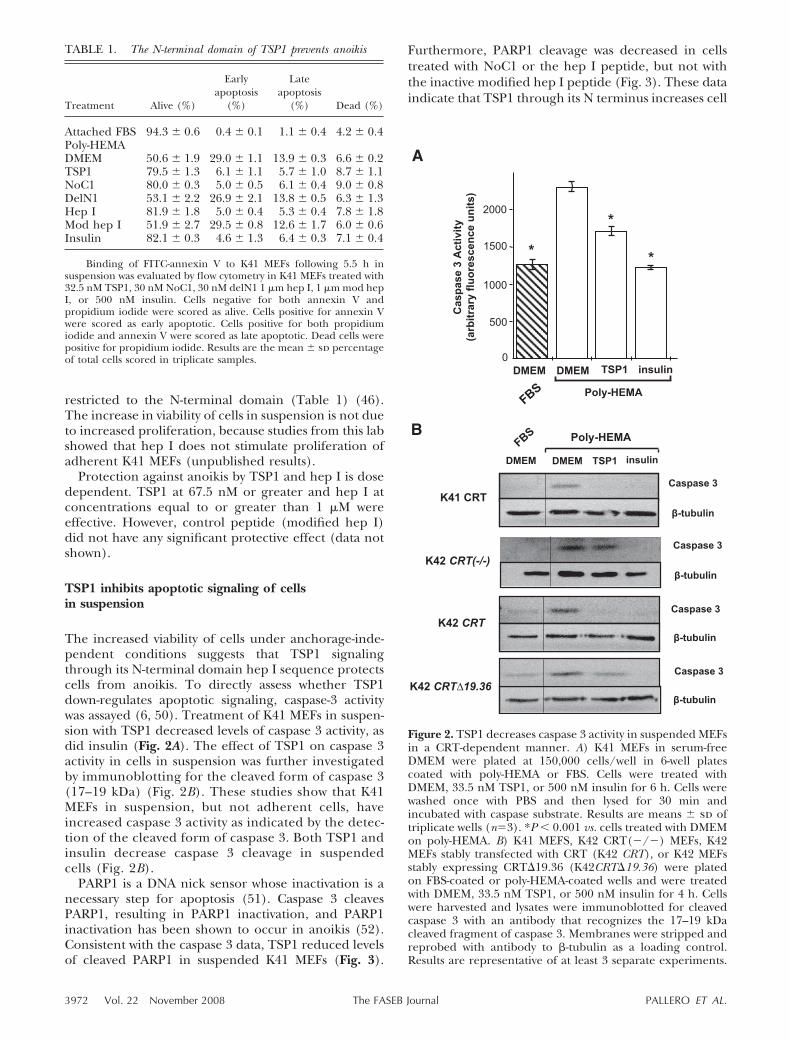
The increased viability of cells under anchorage-inde-pendent conditions suggests that TSP1 signalingthrough its N-terminal domain hep I sequence protectscells from anoikis. To directly assess whether TSP1down-regulates apoptotic signaling, caspase-3 activitywas assayed (6, 50). Treatment of K41 MEFs in suspen-sion with TSP1 decreased levels of caspase 3 activity, asdid insulin (Fig. 2A). The effect of TSP1 on caspase 3activity in cells in suspension was further investigatedby immunoblotting for the cleaved form of caspase 3(17–19 kDa) (Fig. 2B). These studies show that K41MEFs in suspension, but not adherent cells, haveincreased caspase 3 activity as indicated by the detec-tion of the cleaved form of caspase 3. Both TSP1 andinsulin decrease caspase 3 cleavage in suspendedcells (Fig. 2B).
PARP1 is a DNA nick sensor whose inactivation is anecessary step for apoptosis (51). Caspase 3 cleavesPARP1, resulting in PARP1 inactivation, and PARP1inactivation has been shown to occur in anoikis (52).Consistent with the caspase 3 data, TSP1 reduced levelsof cleaved PARP1 in suspended K41 MEFs (Fig. 3).
Furthermore, PARP1 cleavage was decreased in cellstreated with NoC1 or the hep I peptide, but not withthe inactive modified hep I peptide (Fig. 3). These dataindicate that TSP1 through its N terminus increases cell
Figure 2. TSP1 decreases caspase 3 activity in suspended MEFsin a CRT-dependent manner. A) K41 MEFs in serum-freeDMEM were plated at 150,000 cells/well in 6-well platescoated with poly-HEMA or FBS. Cells were treated withDMEM, 33.5 nM TSP1, or 500 nM insulin for 6 h. Cells werewashed once with PBS and then lysed for 30 min andincubated with caspase substrate. Results are means sd oftriplicate wells (n�3). *P 0.001 vs. cells treated with DMEMon poly-HEMA. B) K41 MEFS, K42 CRT(�/�) MEFs, K42MEFs stably transfected with CRT (K42 CRT), or K42 MEFsstably expressing CRT�19.36 (K42CRT�19.36) were platedon FBS-coated or poly-HEMA-coated wells and were treatedwith DMEM, 33.5 nM TSP1, or 500 nM insulin for 4 h. Cellswere harvested and lysates were immunoblotted for cleavedcaspase 3 with an antibody that recognizes the 17–19 kDacleaved fragment of caspase 3. Membranes were stripped andreprobed with antibody to �-tubulin as a loading control.Results are representative of at least 3 separate experiments.
TABLE 1. The N-terminal domain of TSP1 prevents anoikis
Treatment Alive (%)
Earlyapoptosis
(%)
Lateapoptosis
(%) Dead (%)
Attached FBS 94.3 0.6 0.4 0.1 1.1 0.4 4.2 0.4Poly-HEMADMEM 50.6 1.9 29.0 1.1 13.9 0.3 6.6 0.2TSP1 79.5 1.3 6.1 1.1 5.7 1.0 8.7 1.1NoC1 80.0 0.3 5.0 0.5 6.1 0.4 9.0 0.8DelN1 53.1 2.2 26.9 2.1 13.8 0.5 6.3 1.3Hep I 81.9 1.8 5.0 0.4 5.3 0.4 7.8 1.8Mod hep I 51.9 2.7 29.5 0.8 12.6 1.7 6.0 0.6Insulin 82.1 0.3 4.6 1.3 6.4 0.3 7.1 0.4
Binding of FITC-annexin V to K41 MEFs following 5.5 h insuspension was evaluated by flow cytometry in K41 MEFs treated with32.5 nM TSP1, 30 nM NoC1, 30 nM delN1 1 �m hep I, 1 �m mod hepI, or 500 nM insulin. Cells negative for both annexin V andpropidium iodide were scored as alive. Cells positive for annexin Vwere scored as early apoptotic. Cells positive for both propidiumiodide and annexin V were scored as late apoptotic. Dead cells werepositive for propidium iodide. Results are the mean sd percentageof total cells scored in triplicate samples.
3972 Vol. 22 November 2008 PALLERO ET AL.The FASEB Journal
viability in K41 MEFs undergoing anoikis by inhibitingapoptotic signaling.
TSP1 binding to CRT regulates survival signaling
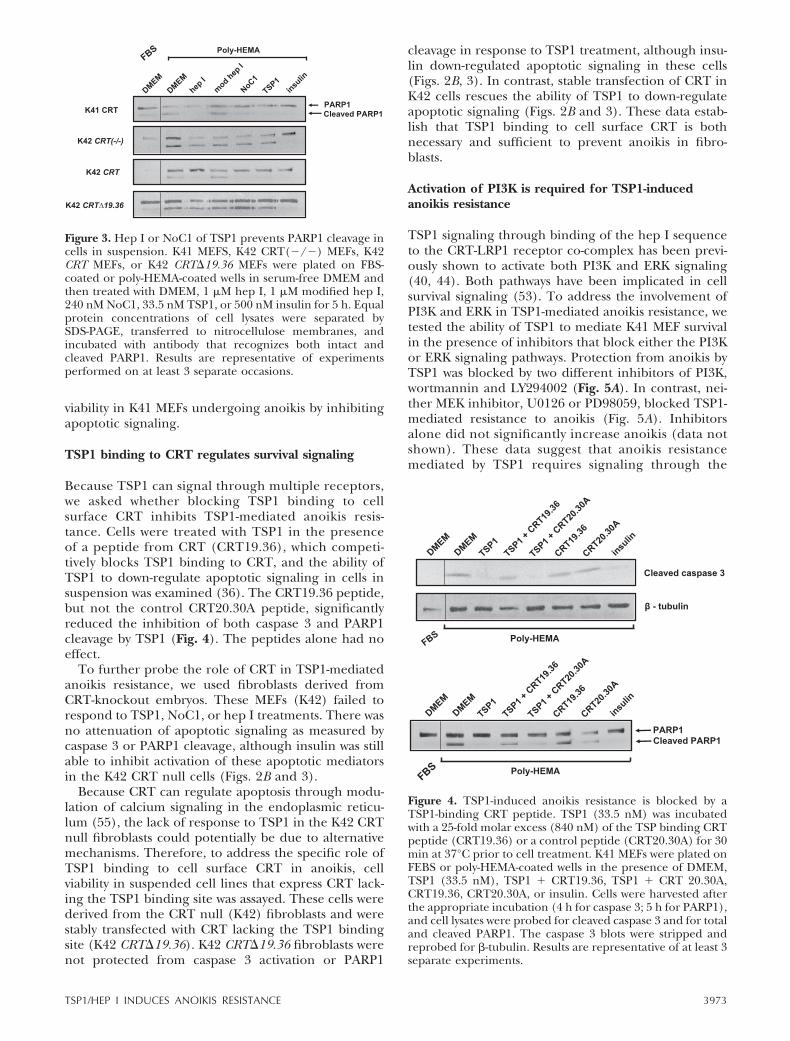
Because TSP1 can signal through multiple receptors,we asked whether blocking TSP1 binding to cellsurface CRT inhibits TSP1-mediated anoikis resis-tance. Cells were treated with TSP1 in the presenceof a peptide from CRT (CRT19.36), which competi-tively blocks TSP1 binding to CRT, and the ability ofTSP1 to down-regulate apoptotic signaling in cells insuspension was examined (36). The CRT19.36 peptide,but not the control CRT20.30A peptide, significantlyreduced the inhibition of both caspase 3 and PARP1cleavage by TSP1 (Fig. 4). The peptides alone had noeffect.
To further probe the role of CRT in TSP1-mediatedanoikis resistance, we used fibroblasts derived fromCRT-knockout embryos. These MEFs (K42) failed torespond to TSP1, NoC1, or hep I treatments. There wasno attenuation of apoptotic signaling as measured bycaspase 3 or PARP1 cleavage, although insulin was stillable to inhibit activation of these apoptotic mediatorsin the K42 CRT null cells (Figs. 2B and 3).
Because CRT can regulate apoptosis through modu-lation of calcium signaling in the endoplasmic reticu-lum (55), the lack of response to TSP1 in the K42 CRTnull fibroblasts could potentially be due to alternativemechanisms. Therefore, to address the specific role ofTSP1 binding to cell surface CRT in anoikis, cellviability in suspended cell lines that express CRT lack-ing the TSP1 binding site was assayed. These cells werederived from the CRT null (K42) fibroblasts and werestably transfected with CRT lacking the TSP1 bindingsite (K42 CRT�19.36). K42 CRT�19.36 fibroblasts werenot protected from caspase 3 activation or PARP1
cleavage in response to TSP1 treatment, although insu-lin down-regulated apoptotic signaling in these cells(Figs. 2B, 3). In contrast, stable transfection of CRT inK42 cells rescues the ability of TSP1 to down-regulateapoptotic signaling (Figs. 2B and 3). These data estab-lish that TSP1 binding to cell surface CRT is bothnecessary and sufficient to prevent anoikis in fibro-blasts.
Activation of PI3K is required for TSP1-inducedanoikis resistance
TSP1 signaling through binding of the hep I sequenceto the CRT-LRP1 receptor co-complex has been previ-ously shown to activate both PI3K and ERK signaling(40, 44). Both pathways have been implicated in cellsurvival signaling (53). To address the involvement ofPI3K and ERK in TSP1-mediated anoikis resistance, wetested the ability of TSP1 to mediate K41 MEF survivalin the presence of inhibitors that block either the PI3Kor ERK signaling pathways. Protection from anoikis byTSP1 was blocked by two different inhibitors of PI3K,wortmannin and LY294002 (Fig. 5A). In contrast, nei-ther MEK inhibitor, U0126 or PD98059, blocked TSP1-mediated resistance to anoikis (Fig. 5A). Inhibitorsalone did not significantly increase anoikis (data notshown). These data suggest that anoikis resistancemediated by TSP1 requires signaling through the
Figure 4. TSP1-induced anoikis resistance is blocked by aTSP1-binding CRT peptide. TSP1 (33.5 nM) was incubatedwith a 25-fold molar excess (840 nM) of the TSP binding CRTpeptide (CRT19.36) or a control peptide (CRT20.30A) for 30min at 37°C prior to cell treatment. K41 MEFs were plated onFEBS or poly-HEMA-coated wells in the presence of DMEM,TSP1 (33.5 nM), TSP1 � CRT19.36, TSP1 � CRT 20.30A,CRT19.36, CRT20.30A, or insulin. Cells were harvested afterthe appropriate incubation (4 h for caspase 3; 5 h for PARP1),and cell lysates were probed for cleaved caspase 3 and for totaland cleaved PARP1. The caspase 3 blots were stripped andreprobed for �-tubulin. Results are representative of at least 3separate experiments.
Figure 3. Hep I or NoC1 of TSP1 prevents PARP1 cleavage incells in suspension. K41 MEFS, K42 CRT(�/�) MEFs, K42CRT MEFs, or K42 CRT�19.36 MEFs were plated on FBS-coated or poly-HEMA-coated wells in serum-free DMEM andthen treated with DMEM, 1 �M hep I, 1 �M modified hep I,240 nM NoC1, 33.5 nM TSP1, or 500 nM insulin for 5 h. Equalprotein concentrations of cell lysates were separated bySDS-PAGE, transferred to nitrocellulose membranes, andincubated with antibody that recognizes both intact andcleaved PARP1. Results are representative of experimentsperformed on at least 3 separate occasions.
3973TSP1/HEP I INDUCES ANOIKIS RESISTANCE
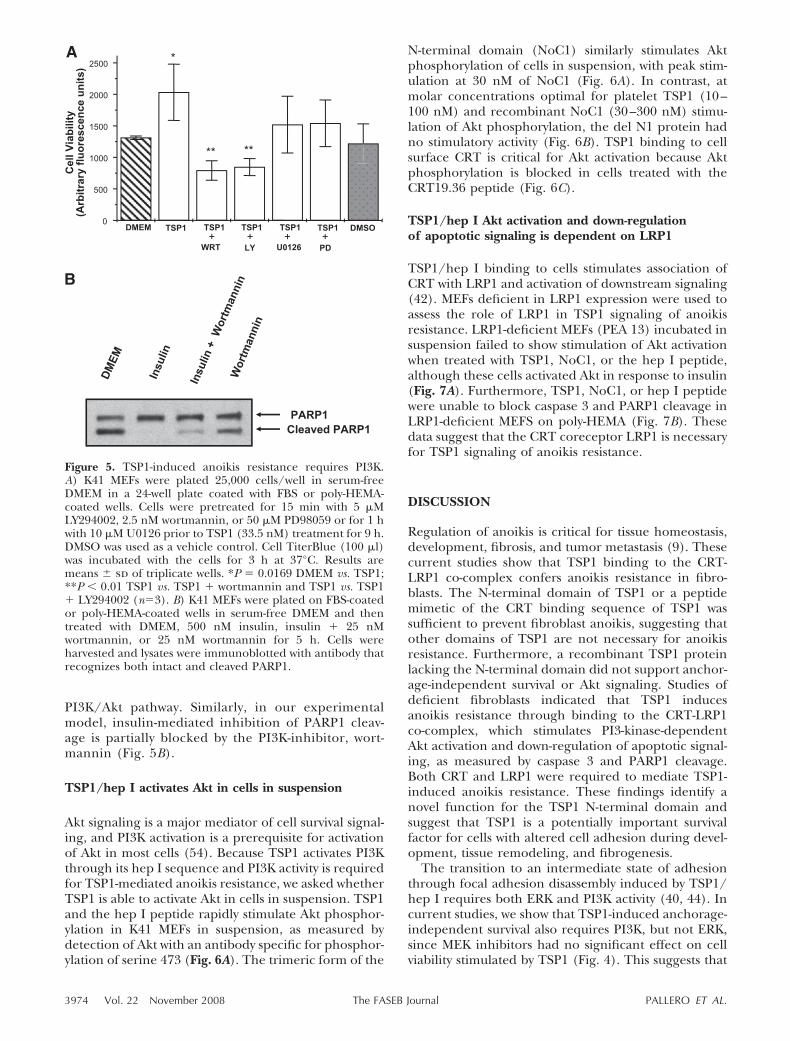
PI3K/Akt pathway. Similarly, in our experimentalmodel, insulin-mediated inhibition of PARP1 cleav-age is partially blocked by the PI3K-inhibitor, wort-mannin (Fig. 5B).
TSP1/hep I activates Akt in cells in suspension
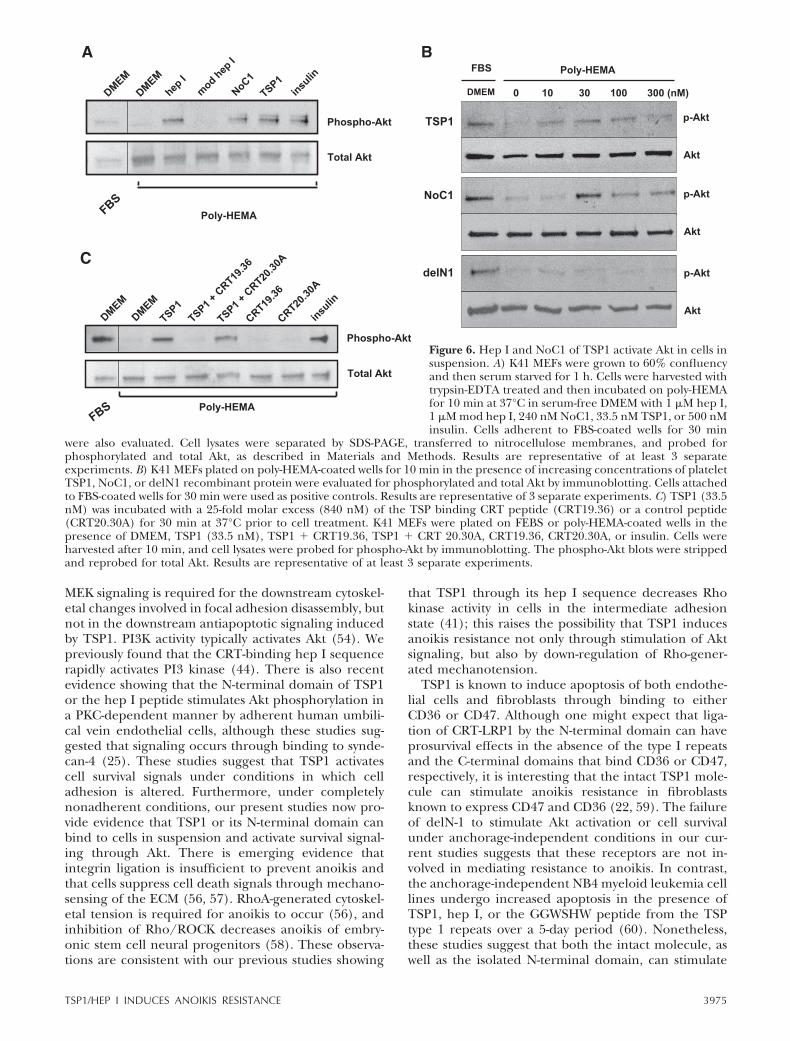
Akt signaling is a major mediator of cell survival signal-ing, and PI3K activation is a prerequisite for activationof Akt in most cells (54). Because TSP1 activates PI3Kthrough its hep I sequence and PI3K activity is requiredfor TSP1-mediated anoikis resistance, we asked whetherTSP1 is able to activate Akt in cells in suspension. TSP1and the hep I peptide rapidly stimulate Akt phosphor-ylation in K41 MEFs in suspension, as measured bydetection of Akt with an antibody specific for phosphor-ylation of serine 473 (Fig. 6A). The trimeric form of the
N-terminal domain (NoC1) similarly stimulates Aktphosphorylation of cells in suspension, with peak stim-ulation at 30 nM of NoC1 (Fig. 6A). In contrast, atmolar concentrations optimal for platelet TSP1 (10–100 nM) and recombinant NoC1 (30–300 nM) stimu-lation of Akt phosphorylation, the del N1 protein hadno stimulatory activity (Fig. 6B). TSP1 binding to cellsurface CRT is critical for Akt activation because Aktphosphorylation is blocked in cells treated with theCRT19.36 peptide (Fig. 6C).
TSP1/hep I Akt activation and down-regulationof apoptotic signaling is dependent on LRP1
TSP1/hep I binding to cells stimulates association ofCRT with LRP1 and activation of downstream signaling(42). MEFs deficient in LRP1 expression were used toassess the role of LRP1 in TSP1 signaling of anoikisresistance. LRP1-deficient MEFs (PEA 13) incubated insuspension failed to show stimulation of Akt activationwhen treated with TSP1, NoC1, or the hep I peptide,although these cells activated Akt in response to insulin(Fig. 7A). Furthermore, TSP1, NoC1, or hep I peptidewere unable to block caspase 3 and PARP1 cleavage inLRP1-deficient MEFS on poly-HEMA (Fig. 7B). Thesedata suggest that the CRT coreceptor LRP1 is necessaryfor TSP1 signaling of anoikis resistance.
DISCUSSION
Regulation of anoikis is critical for tissue homeostasis,development, fibrosis, and tumor metastasis (9). Thesecurrent studies show that TSP1 binding to the CRT-LRP1 co-complex confers anoikis resistance in fibro-blasts. The N-terminal domain of TSP1 or a peptidemimetic of the CRT binding sequence of TSP1 wassufficient to prevent fibroblast anoikis, suggesting thatother domains of TSP1 are not necessary for anoikisresistance. Furthermore, a recombinant TSP1 proteinlacking the N-terminal domain did not support anchor-age-independent survival or Akt signaling. Studies ofdeficient fibroblasts indicated that TSP1 inducesanoikis resistance through binding to the CRT-LRP1co-complex, which stimulates PI3-kinase-dependentAkt activation and down-regulation of apoptotic signal-ing, as measured by caspase 3 and PARP1 cleavage.Both CRT and LRP1 were required to mediate TSP1-induced anoikis resistance. These findings identify anovel function for the TSP1 N-terminal domain andsuggest that TSP1 is a potentially important survivalfactor for cells with altered cell adhesion during devel-opment, tissue remodeling, and fibrogenesis.
The transition to an intermediate state of adhesionthrough focal adhesion disassembly induced by TSP1/hep I requires both ERK and PI3K activity (40, 44). Incurrent studies, we show that TSP1-induced anchorage-independent survival also requires PI3K, but not ERK,since MEK inhibitors had no significant effect on cellviability stimulated by TSP1 (Fig. 4). This suggests that
Figure 5. TSP1-induced anoikis resistance requires PI3K.A) K41 MEFs were plated 25,000 cells/well in serum-freeDMEM in a 24-well plate coated with FBS or poly-HEMA-coated wells. Cells were pretreated for 15 min with 5 �MLY294002, 2.5 nM wortmannin, or 50 �M PD98059 or for 1 hwith 10 �M U0126 prior to TSP1 (33.5 nM) treatment for 9 h.DMSO was used as a vehicle control. Cell TiterBlue (100 �l)was incubated with the cells for 3 h at 37°C. Results aremeans sd of triplicate wells. *P � 0.0169 DMEM vs. TSP1;**P 0.01 TSP1 vs. TSP1 � wortmannin and TSP1 vs. TSP1� LY294002 (n�3). B) K41 MEFs were plated on FBS-coatedor poly-HEMA-coated wells in serum-free DMEM and thentreated with DMEM, 500 nM insulin, insulin � 25 nMwortmannin, or 25 nM wortmannin for 5 h. Cells wereharvested and lysates were immunoblotted with antibody thatrecognizes both intact and cleaved PARP1.
3974 Vol. 22 November 2008 PALLERO ET AL.The FASEB Journal
MEK signaling is required for the downstream cytoskel-etal changes involved in focal adhesion disassembly, butnot in the downstream antiapoptotic signaling inducedby TSP1. PI3K activity typically activates Akt (54). Wepreviously found that the CRT-binding hep I sequencerapidly activates PI3 kinase (44). There is also recentevidence showing that the N-terminal domain of TSP1or the hep I peptide stimulates Akt phosphorylation ina PKC-dependent manner by adherent human umbili-cal vein endothelial cells, although these studies sug-gested that signaling occurs through binding to synde-can-4 (25). These studies suggest that TSP1 activatescell survival signals under conditions in which celladhesion is altered. Furthermore, under completelynonadherent conditions, our present studies now pro-vide evidence that TSP1 or its N-terminal domain canbind to cells in suspension and activate survival signal-ing through Akt. There is emerging evidence thatintegrin ligation is insufficient to prevent anoikis andthat cells suppress cell death signals through mechano-sensing of the ECM (56, 57). RhoA-generated cytoskel-etal tension is required for anoikis to occur (56), andinhibition of Rho/ROCK decreases anoikis of embry-onic stem cell neural progenitors (58). These observa-tions are consistent with our previous studies showing
that TSP1 through its hep I sequence decreases Rhokinase activity in cells in the intermediate adhesionstate (41); this raises the possibility that TSP1 inducesanoikis resistance not only through stimulation of Aktsignaling, but also by down-regulation of Rho-gener-ated mechanotension.
TSP1 is known to induce apoptosis of both endothe-lial cells and fibroblasts through binding to eitherCD36 or CD47. Although one might expect that liga-tion of CRT-LRP1 by the N-terminal domain can haveprosurvival effects in the absence of the type I repeatsand the C-terminal domains that bind CD36 or CD47,respectively, it is interesting that the intact TSP1 mole-cule can stimulate anoikis resistance in fibroblastsknown to express CD47 and CD36 (22, 59). The failureof delN-1 to stimulate Akt activation or cell survivalunder anchorage-independent conditions in our cur-rent studies suggests that these receptors are not in-volved in mediating resistance to anoikis. In contrast,the anchorage-independent NB4 myeloid leukemia celllines undergo increased apoptosis in the presence ofTSP1, hep I, or the GGWSHW peptide from the TSPtype 1 repeats over a 5-day period (60). Nonetheless,these studies suggest that both the intact molecule, aswell as the isolated N-terminal domain, can stimulate
Figure 6. Hep I and NoC1 of TSP1 activate Akt in cells insuspension. A) K41 MEFs were grown to 60% confluencyand then serum starved for 1 h. Cells were harvested withtrypsin-EDTA treated and then incubated on poly-HEMAfor 10 min at 37°C in serum-free DMEM with 1 �M hep I,1 �M mod hep I, 240 nM NoC1, 33.5 nM TSP1, or 500 nMinsulin. Cells adherent to FBS-coated wells for 30 min
were also evaluated. Cell lysates were separated by SDS-PAGE, transferred to nitrocellulose membranes, and probed forphosphorylated and total Akt, as described in Materials and Methods. Results are representative of at least 3 separateexperiments. B) K41 MEFs plated on poly-HEMA-coated wells for 10 min in the presence of increasing concentrations of plateletTSP1, NoC1, or delN1 recombinant protein were evaluated for phosphorylated and total Akt by immunoblotting. Cells attachedto FBS-coated wells for 30 min were used as positive controls. Results are representative of 3 separate experiments. C) TSP1 (33.5nM) was incubated with a 25-fold molar excess (840 nM) of the TSP binding CRT peptide (CRT19.36) or a control peptide(CRT20.30A) for 30 min at 37°C prior to cell treatment. K41 MEFs were plated on FEBS or poly-HEMA-coated wells in thepresence of DMEM, TSP1 (33.5 nM), TSP1 � CRT19.36, TSP1 � CRT 20.30A, CRT19.36, CRT20.30A, or insulin. Cells wereharvested after 10 min, and cell lysates were probed for phospho-Akt by immunoblotting. The phospho-Akt blots were strippedand reprobed for total Akt. Results are representative of at least 3 separate experiments.
3975TSP1/HEP I INDUCES ANOIKIS RESISTANCE

survival of adhesion-dependent cells under adhesion-independent conditions.
CRT is a calcium-binding endoplasmic reticulumchaperone that is also present on the cell surface andhas multiple roles in regulating cell adhesion, calciumsignaling, clearance of apoptotic cells, and immunoge-nicity of tumor cells (61–66). Recent evidence showsthat soluble forms of CRT enhance cell migration andwound healing (61). Like TSP1, CRT has complex rolesin apoptosis. CRT has the potential to influence apop-tosis both positively and negatively via modulation ofintracellular calcium stores (67–69). Overexpression ofCRT promotes apoptosis during cardiac differentiationby negatively regulating Akt activation through up-regulation of the serine/threonine phosphatase PP2A(70). In contrast, our results show that TSP1 binding toCRT on the cell surface is critical for activation of Aktand anchorage-independent survival. This is likely in-dependent of CRT’s role in calcium homeostasis, sinceK42 MEFs expressing CRT lacking the TSP1 bindingsite (K42 CRT�19.36) are nonresponsive to TSP1-induced anoikis resistance. Although the mechanism ofCRT translocation from the endoplasmic reticulum to
the cell surface has not been defined, it is well estab-lished that CRT expression on the cell surface isup-regulated by conditions of cellular stress, as well asby apoptotic inducers such as anthracyclines and byalterations in endoplasmic reticulum calcium levels(62, 71, 72). We also showed that CRT expressioninduced by stable transfection of fibroblasts is sufficientto induce CRT surface expression (Supplemental Fig.1). Purified CRT binds to cell death receptors CD40Ligand, TRAIL, and Fas ligand (73). Furthermore, cellsurface CRT is a receptor for an apoptogenic bacterialwall peptidoglycan peptide and cell surface CRT com-plexes with tumor necrosis factor (TNF) receptor1-TRADD to mediate apoptosis on peptide binding(67). CRT expression on apoptotic cells mediates theclearance of these cells by LRP1 (63, 74). An anti-inflammatory protein, adiponectin binds CRT, whichacts in concert with LRP1 to mediate clearance ofapoptotic cells (75). Interestingly, TSP1 was recentlyidentified as a binding protein for adiponectin (76). Itwill be interesting to determine how TSP1 binding toCRT affects CRT interactions with these inflammatoryand cell death mediators. These data show that cellsurface CRT is important for regulation of apoptoticsignaling and cell clearance; furthermore, cell surfaceCRT appears capable of interacting with multiple fac-tors that can stimulate differing outcomes, suggestingthat CRT can act as a cell survival switch, depending onthe stimulating ligand and its binding partners at themembrane.
TSP1 is highly expressed during embryogenesis, butits expression in the adult is limited to areas of activetissue remodeling, such as during wound healing andin fibrotic tissues (17, 77, 78). TSP1 stimulates cellmigration into wounds, and it can affect deposition ofthe extracellular matrix through modulation of collag-ens, matrix metalloproteases, and TGF-� activation(79–81). A study by Lee et al. (31) also demonstratesthe presence of the N-terminal TSP1 fragment inwounds. During early wound repair, fibroblasts becomemigratory and might require survival signals generatedby TSP1-binding to the CRT-LRP1 co-complex as asubstitute for integrin-dependent signals during exten-sive extracellular matrix remodeling: this scenariowould be consistent with the delayed induction ofgranulation tissue in dermal wounds of TSP1 null mice(79, 81). Conversely, fibroblast apoptosis is critical forresolution of granulation tissue, and persistent fibro-blast survival is associated with fibrosis (11, 82, 83). Thiswould be consistent with down-regulation of TSP1expression during the resolving stages of wound heal-ing (79). TSP1 is frequently elevated in many fibroticconditions, and it is possible that anoikis resistancesignaling through TSP1 contributes to fibrotic progres-sion.
TSP1 belongs to a family of matricellular proteins,including TSP2, SPARC, hevin, tenascin-C and os-teopontin, which are expressed at high levels duringdevelopment and in response to tissue injury or remod-eling. Matricellular proteins are generally deadhesive,
Figure 7. LRP1 is required for mediating Akt signaling andfor inhibiting apoptotic signaling by TSP1, NoC1, or hep I.A) LRP1 deficient PEA13 MEFs plated on either poly-HEMAcoated or FBS-coated wells were treated with DMEM, 33.5 nMTSP1, 240 nM NoC1, 1 �M hep I, 1 �M modified hep I, or 500nM insulin for 10 min. Cell lysates were separated by SDS-PAGE, transferred to nitrocellulose membranes, and probedfor phosphorylated and total Akt, as described in Materialsand Methods. Results are representative of at least 3 separateexperiments. B) LRP1-deficient PEA13 MEFs plated on eitherpoly-HEMA-coated or FBS-coated wells were treated withDMEM, 1 �M hep I, 240 nM NoC1, 33.5 nM TSP1, or 500 nMinsulin for either 4 h (caspase 3) or 5 h (PARP1). Cells wereharvested and lysates were immunoblotted for cleavedcaspase 3 with an antibody that recognizes uncleaved caspase3 and the 17–19 kDa cleaved fragment of caspase 3 or withantibody that recognizes both intact and cleaved PARP1.
3976 Vol. 22 November 2008 PALLERO ET AL.The FASEB Journal

inducing an intermediate state of adhesion (34). Thus,it is interesting to speculate that induction of cellsurvival signals and anoikis resistance could be a com-mon phenotype induced by other matricellular pro-teins to promote cell survival in the context ofdecreased cell adhesion during tissue remodeling. Con-sistent with this idea, SPARC and tenascin-C mediatesurvival of gliomas and human chondrosarcoma cells,respectively, through activation of Akt (84, 85). Tenas-cin-C treated neuronal cell suspensions had increasednumbers of surviving neurons after transplantation(86). Results from our lab also show that tenascin FNIIIA-D increases anchorage-independent MEF survival(data not shown). Osteopontin activates Akt in a PI3-kinase dependent manner through CD44 (87): anchor-age-independent growth of breast cancer cells wasspecifically inhibited with an antibody against os-teopontin and a splice variant of osteopontin enhancedanoikis resistance (88). The urokinase/uPAR complex,which also mediates intermediate adhesion, similarlyinduces anoikis resistance (89–91). In turn, activationof Akt appears to regulate the expression of variousmatricellular proteins, including tenascin-C, osteopon-tin, and TSP1, creating a possible positive feedbackloop (92–94).
In summary, these studies have identified a novelfunction for the N-terminal CRT-binding sequence ofTSP1 in mediating anoikis resistance in fibroblasts. Thestudies suggest additional mechanisms by which TSP1regulates wound healing and fibrotic responses.
The authors gratefully acknowledge Marek Michalak (Uni-versity of Alberta, Edmonton, AB, Canada) for providing theCRT constructs, K41 and K42 CRT(�/�) MEFs. We alsothank Doug Annis (University of Wisconsin, Madison, WI,USA) for assistance with the preparation of recombinantTSP1 proteins. We thank Silvia Goicoechea (University ofNorth Carolina, Chapel Hill, NC, USA) for making the CRTdeletion construct. We appreciate the assistance of Mariya T.Sweetwyne in preparation of the figures. This work wassupported by a comprehensive training grant in bone biologyand disease (T32AR47512–01A1) to C.A.E., a training grantin oral biology (T32DE014300) to J.C., and a NationalInstitutes of Health (NIH) grant (HL079644) to J.E.M.U. Thisinvestigation was conducted in a facility constructed withsupport from Research Facilities Improvement Program grantC06 RR 15490 from the National Center for Research Re-sources, NIH.
REFERENCES
1. Xia, H., Nho, R. S., Kahm, J., Kleidon, J., and Henke, C. A.(2004) Focal adhesion kinase is upstream of phosphatidylinosi-tol 3-kinase/Akt in regulating fibroblast survival in response tocontraction of type I collagen matrices via a beta 1 integrinviability signaling pathway. J. Biol. Chem. 279, 33024–33034
2. Yamaki, N., Negishi, M., and Katoh, H. (2007) RhoG regulatesanoikis through a phosphatidylinositol 3-kinase-dependentmechanism. Exp. Cell. Res. 313, 2821–2832
3. Meredith, J. E., Jr., Fazeli, B., and Schwartz, M. A. (1993) Theextracellular matrix as a cell survival factor. Mol. Biol. Cell 4,953–961
4. Frisch, S. M., and Francis, H. (1994) Disruption of epithelialcell-matrix interactions induces apoptosis. J. Cell Biol. 124,619–626
5. Gilmore, A. P. (2005) Anoikis. Cell Death Differ. 12 (Suppl. 2),1473–1477
6. Grossmann, J., Walther, K., Artinger, M., Kiessling, S., andScholmerich, J. (2001) Apoptotic signaling during initiation ofdetachment-induced apoptosis (“anoikis”) of primary humanintestinal epithelial cells. Cell Growth Differ. 12, 147–155
7. Boudreau, N., Sympson, C. J., Werb, Z., and Bissell, M. J. (1995)Suppression of ICE and apoptosis in mammary epithelial cellsby extracellular matrix. Science 267, 891–893
8. Liotta, L. A., and Kohn, E. (2004) Anoikis: cancer and thehomeless cell. Nature 430, 973–974
9. Valentijn, A. J., Zouq, N., and Gilmore, A. P. (2004) Anoikis.Biochem. Soc. Trans. 32, 421–425
10. Michel, J. B. (2003) Anoikis in the cardiovascular system: knownand unknown extracellular mediators. Arterioscler. Thromb. Vasc.Biol. 23, 2146–2154
11. Horowitz, J. C., Rogers, D. S., Sharma, V., Vittal, R., White, E. S.,Cui, Z., and Thannickal, V. J. (2007) Combinatorial activation ofFAK and AKT by transforming growth factor-beta1 confers ananoikis-resistant phenotype to myofibroblasts. Cell. Signal. 19,761–771
12. Pinkse, G. G., Voorhoeve, M. P., Noteborn, M., Terpstra, O. T.,Bruijn, J. A., and De Heer, E. (2004) Hepatocyte survivaldepends on beta1-integrin-mediated attachment of hepatocytesto hepatic extracellular matrix. Liver Int. 24, 218–226
13. Zvibel, I., Smets, F., and Soriano, H. (2002) Anoikis: roadblockto cell transplantation? Cell Transplant. 11, 621–630
14. Adams, J. C., and Lawler, J. (2004) The thrombospondins. Int.J. Biochem. Cell Biol. 36, 961–968
15. Bornstein, P. (2001) Thrombospondins as matricellular modu-lators of cell function. J. Clin. Invest. 107, 929–934
16. Isenberg, J. S., Jia, Y., Fukuyama, J., Switzer, C. H., Wink, D. A.,and Roberts, D. D. (2007) Thrombospondin-1 inhibits nitricoxide signaling via CD36 by inhibiting myristic acid uptake.J. Biol. Chem. 282, 15404–15415
17. Murphy-Ullrich, J. E., and Poczatek, M. (2000) Activation oflatent TGF-beta by thrombospondin-1: mechanisms and physi-ology. Cytokine Growth Factor Rev. 11, 59–69
18. Elzie, C. A., and Murphy-Ullrich, J. E. (2004) The N-terminus ofthrombospondin: the domain stands apart. Int. J. Biochem. CellBiol. 36, 1090–1101
19. Dawson, D. W., Pearce, S. F., Zhong, R., Silverstein, R. L.,Frazier, W. A., and Bouck, N. P. (1997) CD36 mediates the Invitro inhibitory effects of thrombospondin-1 on endothelialcells. J. Cell Biol. 138, 707–717
20. Good, D. J., Polverini, P. J., Rastinejad, F., Le Beau, M. M.,Lemons, R. S., Frazier, W. A., and Bouck, N. P. (1990) A tumorsuppressor-dependent inhibitor of angiogenesis is immunolog-ically and functionally indistinguishable from a fragment ofthrombospondin. Proc. Natl. Acad. Sci. U. S. A. 87, 6624–6628
21. Saumet, A., Slimane, M. B., Lanotte, M., Lawler, J., and Duber-nard, V. (2005) Type 3 repeat/C-terminal domain of throm-bospondin-1 triggers caspase-independent cell death throughCD47/alphavbeta3 in promyelocytic leukemia NB4 cells. Blood106, 658–667
22. Graf, R., Freyberg, M., Kaiser, D., and Friedl, P. (2002) Mech-anosensitive induction of apoptosis in fibroblasts is regulated bythrombospondin-1 and integrin associated protein (CD47).Apoptosis 7, 493–498
23. Freyberg, M. A., Kaiser, D., Graf, R., Buttenbender, J., andFriedl, P. (2001) Proatherogenic flow conditions initiate endo-thelial apoptosis via thrombospondin-1 and the integrin-associ-ated protein. Biochem. Biophys. Res. Commun. 286, 141–149
24. Rath, G. M., Schneider, C., Dedieu, S., Sartelet, H., Morjani, H.,Martiny, L., and El Btaouri, H. (2006) Thrombospondin-1C-terminal-derived peptide protects thyroid cells from ceram-ide-induced apoptosis through the adenylyl cyclase pathway. Int.J. Biochem. Cell Biol. 38, 2219–2228
25. Nunes, S. S., Outeiro-Bernstein, M. A., Juliano, L., Vardiero, F.,Nader, H. B., Woods, A., Legrand, C., and Morandi, V. (2008)Syndecan-4 contributes to endothelial tubulogenesis throughinteractions with two motifs inside the pro-angiogenic N-termi-nal domain of thrombospondin-1. J. Cell. Physiol. 214, 828–837
26. Ferrari do Outeiro-Bernstein, M. A., Nunes, S. S., Andrade,A. C., Alves, T. R., Legrand, C., and Morandi, V. (2002) Arecombinant NH(2)-terminal heparin-binding domain of theadhesive glycoprotein, thrombospondin-1, promotes endothe-
3977TSP1/HEP I INDUCES ANOIKIS RESISTANCE

lial tube formation and cell survival: a possible role for synde-can-4 proteoglycan. Matrix Biol. 21, 311–324
27. Chandrasekaran, L., He, C. Z., Al-Barazi, H., Krutzsch, H. C.,Iruela-Arispe, M. L., and Roberts, D. D. (2000) Cell contact-dependent activation of alpha3beta1 integrin modulates endo-thelial cell responses to thrombospondin-1. Mol. Biol. Cell 11,2885–2900
28. Staniszewska, I., Zaveri, S., Del Valle, L., Oliva, I., Rothman, V. L.,Croul, S. E., Roberts, D. D., Mosher, D. F., Tuszynski, G. P., andMarcinkiewicz, C. (2007) Interaction of alpha9beta1 integrin withthrombospondin-1 promotes angiogenesis. Circ. Res. 100, 1308–1316
29. Bonnefoy, A., and Legrand, C. (2000) Proteolysis of subendo-thelial adhesive glycoproteins (fibronectin, thrombospondin,and von Willebrand factor) by plasmin, leukocyte cathepsin G,and elastase. Thromb. Res. 98, 323–332
30. Rabhi-Sabile, S., Pidard, D., Lawler, J., Renesto, P., Chignard, M.,and Legrand, C. (1996) Proteolysis of thrombospondin duringcathepsin-G-induced platelet aggregation: functional role of the165-kDa carboxy-terminal fragment. FEBS Lett. 386, 82–86
31. Lee, N. V., Sato, M., Annis, D. S., Loo, J. A., Wu, L., Mosher,D. F., and Iruela-Arispe, M. L. (2006) ADAMTS1 mediates therelease of antiangiogenic polypeptides from TSP1 and 2. EMBOJ. 25, 5270–5283
32. Krispin, A., Bledi, Y., Atallah, M., Trahtemberg, U., Verbovetski,I., Nahari, E., Zelig, O., Linial, M., and Mevorach, D. (2006)Apoptotic cell thrombospondin-1 and heparin-binding domainlead to dendritic-cell phagocytic and tolerizing states. Blood 108,3580–3589
33. Murphy-Ullrich, J. E., and Hook, M. (1989) Thrombospondinmodulates focal adhesions in endothelial cells. J. Cell Biol. 109,1309–1319
34. Murphy-Ullrich, J. E. (2001) The de-adhesive activity of matri-cellular proteins: is intermediate cell adhesion an adaptivestate? J. Clin. Invest. 107, 785–790
35. Murphy-Ullrich, J. E., Gurusiddappa, S., Frazier, W. A., andHook, M. (1993) Heparin-binding peptides from thrombospon-dins 1 and 2 contain focal adhesion-labilizing activity. J. Biol.Chem. 268, 26784–26789
36. Goicoechea, S., Pallero, M. A., Eggleton, P., Michalak, M., andMurphy-Ullrich, J. E. (2002) The anti-adhesive activity of throm-bospondin is mediated by the N-terminal domain of cell surfacecalreticulin. J. Biol. Chem. 277, 37219–37228
37. Goicoechea, S., Orr, A. W., Pallero, M. A., Eggleton, P., andMurphy-Ullrich, J. E. (2000) Thrombospondin mediates focaladhesion disassembly through interactions with cell surfacecalreticulin. J. Biol. Chem. 275, 36358–36368
38. Orr, A. W., Pedraza, C. E., Pallero, M. A., Elzie, C. A., Goico-echea, S., Strickland, D. K., and Murphy-Ullrich, J. E. (2003)Low-density lipoprotein receptor-related protein is a calreticulincoreceptor that signals focal adhesion disassembly. J. Cell Biol.161, 1179–1189
39. Tan, K., Duquette, M., Liu, J. H., Zhang, R., Joachimiak, A.,Wang, J. H., and Lawler, J. (2006) The structures of thethrombospondin-1 N-terminal domain and its complex with asynthetic pentameric heparin. Structure 14, 33–42
40. Orr, A. W., Pallero, M. A., and Murphy-Ullrich, J. E. (2002)Thrombospondin stimulates focal adhesion disassemblythrough Gi- and phosphoinositide 3-kinase-dependent ERKactivation. J. Biol. Chem. 277, 20453–20460
41. Orr, A. W., Pallero, M. A., Xiong, W. C., and Murphy-Ullrich,J. E. (2004) Thrombospondin induces RhoA inactivationthrough FAK-dependent signaling to stimulate focal adhesiondisassembly. J. Biol. Chem. 279, 48983–48992
42. Orr, A. W., Elzie, C. A., Kucik, D. F., and Murphy-Ullrich, J. E.(2003) Thrombospondin signaling through the calreticulin/LDL receptor-related protein co-complex stimulates randomand directed cell migration. J. Cell Sci. 116, 2917–2927
43. Li, S. S., Forslow, A., and Sundqvist, K. G. (2005) Autocrineregulation of T cell motility by calreticulin-thrombospondin-1interaction. J. Immunol. 174, 654–661
44. Greenwood, J. A., Pallero, M. A., Theibert, A. B., and Murphy-Ullrich, J. E. (1998) Thrombospondin signaling of focal adhe-sion disassembly requires activation of phosphoinositide 3-ki-nase. J. Biol. Chem. 273, 1755–1763
45. Sottile, J., Selegue, J., and Mosher, D. F. (1991) Synthesis oftruncated amino-terminal trimers of thrombospondin. Biochem-istry 30, 6556–6562
46. Anilkumar, N., Annis, D. S., Mosher, D. F., and Adams, J. C.(2002) Trimeric assembly of the C-terminal region of throm-bospondin-1 or thrombospondin-2 is necessary for cell spread-ing and fascin spike organisation. J. Cell Sci. 115, 2357–2366
47. Nakamura, K., Zuppini, A., Arnaudeau, S., Lynch, J., Ahsan, I.,Krause, R., Papp, S., De Smedt, H., Parys, J. B., Muller-Esterl, W.,Lew, D. P., Krause, K. H., Demaurex, N., Opas, M., andMichalak, M. (2001) Functional specialization of calreticulindomains. J. Cell Biol. 154, 961–972
48. Folkman, J., and Moscona, A. (1978) Role of cell shape ingrowth control. Nature 273, 345–349
49. Lawlor, M. A., and Alessi, D. R. (2001) PKB/Akt: a key mediatorof cell proliferation, survival and insulin responses? J. Cell Sci.114, 2903–2910
50. Rajeswari, J., and Pande, G. (2006) Direct association betweencaspase 3 and alpha5beta1 integrin and its role during anoikis ofrat fibroblasts. Cell Biol. Int. 30, 963–969
51. Soldani, C., and Scovassi, A. I. (2002) Poly(ADP-ribose) poly-merase-1 cleavage during apoptosis: an update. Apoptosis 7,321–328
52. Hou, Y., Wong, E., Martin, J., Schoenlein, P. V., Dostmann,W. R., and Browning, D. D. (2006) A role for cyclic-GMPdependent protein kinase in anoikis. Cell. Signal. 18, 882–888
53. Frisch, S. M., and Screaton, R. A. (2001) Anoikis mechanisms.Curr. Opin. Cell Biol. 13, 555–562
54. Andjelkovic, M., Jakubowicz, T., Cron, P., Ming, X. F., Han,J. W., and Hemmings, B. A. (1996) Activation and phosphory-lation of a pleckstrin homology domain containing proteinkinase (RAC-PK/PKB) promoted by serum and protein phos-phatase inhibitors. Proc. Natl. Acad. Sci. U. S. A. 93, 5699–5704
55. Arnaudeau, S., Frieden, M., Nakamura, K., Castelbou, C.,Michalak, M., and Demaurex, N. (2002) Calreticulin differ-entially modulates calcium uptake and release in the endo-plasmic reticulum and mitochondria. J. Biol. Chem. 277,46696 – 46705
56. Ma, Z., Myers, D. P., Wu, R. F., Nwariaku, F. E., and Terada, L. S.(2007) p66Shc mediates anoikis through RhoA. J. Cell Biol. 179,23–31
57. Zahir, N., and Weaver, V. M. (2004) Death in the thirddimension: apoptosis regulation and tissue architecture. Curr.Opin. Genet. Dev. 14, 71–80
58. Koyanagi, M., Takahashi, J., Arakawa, Y., Doi, D., Fukuda, H.,Hayashi, H., Narumiya, S., and Hashimoto, N. (2008) Inhibitionof the Rho/ROCK pathway reduces apoptosis during transplan-tation of embryonic stem cell-derived neural precursors. J Neu-rosci. Res. 86, 270–280
59. Ring, A., Le Lay, S., Pohl, J., Verkade, P., and Stremmel, W.(2006) Caveolin-1 is required for fatty acid translocase (FAT/CD36) localization and function at the plasma membrane ofmouse embryonic fibroblasts. Biochim. Biophys. Acta. 1761, 416–423
60. Bruel, A., Touhami-Carrier, M., Thomaidis, A., and Legrand, C.(2005) Thrombospondin-1 (TSP-1) and TSP-1-derived heparin-binding peptides induce promyelocytic leukemia cell differen-tiation and apoptosis. Anticancer Res. 25, 757–764
61. Gold, L. I., Rahman, M., Blechman, K. M., Greives, M. R.,Churgin, S., Michaels, J., Callaghan, M. J., Cardwell, N. L.,Pollins, A. C., Michalak, M., Siebert, J. W., Levine, J. P., Gurtner,G. C., Nanney, L. B., Galiano, R. D., and Cadacio, C. L. (2006)Overview of the role for calreticulin in the enhancement ofwound healing through multiple biological effects. J. Investig.Dermatol. Symp. Proc. 11, 57–65
62. Obeid, M., Tesniere, A., Ghiringhelli, F., Fimia, G. M., Apetoh,L., Perfettini, J. L., Castedo, M., Mignot, G., Panaretakis, T.,Casares, N., Metivier, D., Larochette, N., van Endert, P., Cicco-santi, F., Piacentini, M., Zitvogel, L., and Kroemer, G. (2007)Calreticulin exposure dictates the immunogenicity of cancercell death. Nat. Med. 13, 54–61
63. Gardai, S. J., McPhillips, K. A., Frasch, S. C., Janssen, W. J.,Starefeldt, A., Murphy-Ullrich, J. E., Bratton, D. L., Oldenborg,P. A., Michalak, M., and Henson, P. M. (2005) Cell-surfacecalreticulin initiates clearance of viable or apoptotic cellsthrough trans-activation of LRP on the phagocyte. Cell 123,321–334
64. Groenendyk, J., Lynch, J., and Michalak, M. (2004) Calreticulin,Ca2�, and calcineurin signaling from the endoplasmic reticu-lum. Mol. Cells 17, 383–389
3978 Vol. 22 November 2008 PALLERO ET AL.The FASEB Journal

65. Eggleton, P., and Llewellyn, D. H. (1999) Pathophysiologicalroles of calreticulin in autoimmune disease. Scand. J. Immunol.49, 466–473
66. Szabo, E., Papp, S., and Opas, M. (2007) Differential calreticulinexpression affects focal contacts via the calmodulin/CaMK IIpathway. J. Cell. Physiol. 213, 269–277
67. Chen, D., Texada, D. E., Duggan, C., Liang, C., Reden, T. B.,Kooragayala, L. M., and Langford, M. P. (2005) Surface calre-ticulin mediates muramyl dipeptide-induced apoptosis in RK13cells. J. Biol. Chem. 280, 22425–22436
68. Vanoverberghe, K., Vanden Abeele, F., Mariot, P., Lepage, G.,Roudbaraki, M., Bonnal, J. L., Mauroy, B., Shuba, Y., Skryma, R.,and Prevarskaya, N. (2004) Ca2� homeostasis and apoptoticresistance of neuroendocrine-differentiated prostate cancercells. Cell Death Differ. 11, 321–330
69. Mesaeli, N., and Phillipson, C. (2004) Impaired p53 expression,function, and nuclear localization in calreticulin-deficient cells.Mol. Biol. Cell 15, 1862–1870
70. Kageyama, K., Ihara, Y., Goto, S., Urata, Y., Toda, G., Yano, K.,and Kondo, T. (2002) Overexpression of calreticulin modulatesprotein kinase B/Akt signaling to promote apoptosis duringcardiac differentiation of cardiomyoblast H9c2 cells. J. Biol.Chem. 277, 19255–19264
71. Conway, E. M., Liu, L., Nowakowski, B., Steiner-Mosonyi, M.,Ribeiro, S. P., and Michalak, M. (1995) Heat shock-sensitiveexpression of calreticulin. In vitro and in vivo up-regulation.J. Biol. Chem. 270, 17011–17016
72. Tufi, R., Panaretakis, T., Bianchi, K., Criollo, A., Fazi, B., DiSano, F., Tesniere, A., Kepp, O., Paterlini-Brechot, P., Zitvogel,L., Piacentini, M., Szabadkai, G., and Kroemer, G. (2008)Reduction of endoplasmic reticulum Ca(2�) levels favorsplasma membrane surface exposure of calreticulin. Cell DeathDiffer. 15, 274–282
73. Duus, K., Pagh, R. T., Holmskov, U., Hojrup, P., Skov, S., andHouen, G. (2007) Interaction of calreticulin with CD40 ligand,TRAIL and Fas ligand. Scand. J. Immunol. 66, 501–507
74. Ogden, C. A., deCathelineau, A., Hoffmann, P. R., Bratton, D.,Ghebrehiwet, B., Fadok, V. A., and Henson, P. M. (2001) C1qand mannose binding lectin engagement of cell surface calre-ticulin and CD91 initiates macropinocytosis and uptake ofapoptotic cells. J. Exp. Med. 194, 781–795
75. Takemura, Y., Ouchi, N., Shibata, R., Aprahamian, T., Kirber,M. T., Summer, R. S., Kihara, S., and Walsh, K. (2007) Adi-ponectin modulates inflammatory reactions via calreticulin re-ceptor-dependent clearance of early apoptotic bodies. J. Clin.Invest. 117, 375–386
76. Wang, Y., Xu, L. Y., Lam, K. S., Lu, G., Cooper, G. J., and Xu, A.(2006) Proteomic characterization of human serum proteinsassociated with the fat-derived hormone adiponectin. Proteomics6, 3862–3870
77. O’Shea, K. S., and Dixit, V. M. (1988) Unique distribution of theextracellular matrix component thrombospondin in the devel-oping mouse embryo. J. Cell Biol. 107, 2737–2748
78. Raugi, G. J., Olerud, J. E., and Gown, A. M. (1987) Throm-bospondin in early human wound tissue. J. Invest. Dermatol. 89,551–554
79. Agah, A., Kyriakides, T. R., Lawler, J., and Bornstein, P. (2002)The lack of thrombospondin-1 (TSP1) dictates the course ofwound healing in double-TSP1/TSP2-null mice. Am. J. Pathol.161, 831–839
80. DiPietro, L. A., Nissen, N. N., Gamelli, R. L., Koch, A. E., Pyle,J. M., and Polverini, P. J. (1996) Thrombospondin 1 synthesisand function in wound repair. Am. J. Pathol. 148, 1851–1860
81. Nor, J. E., DiPietro, L., Murphy-Ullrich, J. E., Hynes, R. O.,Lawler, J., Polverini, P. J. (2005) Activation of latent TGF-�1 byThrombospondin-1 is a major component of wound repair. OralBiosci. Med. 2, 153–161
82. Aarabi, S., Bhatt, K. A., Shi, Y., Paterno, J., Chang, E. I., Loh,S. A., Holmes, J. W., Longaker, M. T., Yee, H., and Gurtner,G. C. (2007) Mechanical load initiates hypertrophic scar forma-tion through decreased cellular apoptosis. FASEB J. 21, 3250–3261
83. Rai, N. K., Tripathi, K., Sharma, D., and Shukla, V. K. (2005)Apoptosis: a basic physiologic process in wound healing. Int. J.Low. Extrem. Wounds 4, 138–144
84. Shi, Q., Bao, S., Maxwell, J. A., Reese, E. D., Friedman, H. S.,Bigner, D. D., Wang, X. F., and Rich, J. N. (2004) Secretedprotein acidic, rich in cysteine (SPARC), mediates cellularsurvival of gliomas through AKT activation. J. Biol. Chem. 279,52200–52209
85. Jang, J. H., and Chung, C. P. (2005) Tenascin-C promotes cellsurvival by activation of Akt in human chondrosarcoma cell.Cancer Lett. 229, 101–105
86. Marchionini, D. M., Collier, T. J., Camargo, M., McGuire, S.,Pitzer, M., and Sortwell, C. E. (2003) Interference with anoikis-induced cell death of dopamine neurons: implications foraugmenting embryonic graft survival in a rat model of Parkin-son’s disease. J. Comp. Neurol. 464, 172–179
87. Lin, Y. H., and Yang-Yen, H. F. (2001) The osteopontin-CD44survival signal involves activation of the phosphatidylinositol3-kinase/Akt signaling pathway. J. Biol. Chem. 276, 46024–46030
88. He, B., Mirza, M., and Weber, G. F. (2006) An osteopontinsplice variant induces anchorage independence in humanbreast cancer cells. Oncogene 25, 2192–2202
89. Alfano, D., Iaccarino, I., and Stoppelli, M. P. (2006) Urokinasesignaling through its receptor protects against anoikis by in-creasing BCL-xL expression levels. J. Biol. Chem. 281, 17758–17767
90. Hasanuzzaman, M., Kutner, R., Agha-Mohammadi, S., Reiser, J.,and Sehgal, I. (2007) A doxycycline-inducible urokinase recep-tor (uPAR) upregulates uPAR activities including resistance toanoikis in human prostate cancer cell lines. Mol. Cancer 6, 34
91. Czekay, R. P., Aertgeerts, K., Curriden, S. A., and Loskutoff, D. J.(2003) Plasminogen activator inhibitor-1 detaches cells fromextracellular matrices by inactivating integrins. J. Cell Biol. 160,781–791
92. Jinnin, M., Ihn, H., Asano, Y., Yamane, K., Trojanowska, M., andTamaki, K. (2006) Upregulation of tenascin-C expression byIL-13 in human dermal fibroblasts via the phosphoinositide3-kinase/Akt and the protein kinase C signaling pathways.J. Invest. Dermatol. 126, 551–560
93. Chen, J., Somanath, P. R., Razorenova, O., Chen, W. S., Hay, N.,Bornstein, P., and Byzova, T. V. (2005) Akt1 regulates patholog-ical angiogenesis, vascular maturation and permeability in vivo.Nat. Med. 11, 1188–1196
94. Zhang, G., Huang, Z., Shi, R., Lin, Y., and Hao, B. (2006)Osteopontin regulation by protein kinase B (Akt) in HepG2cells. Exp. Oncol. 28, 36–39
Received for publication March 7, 2008.Accepted for publication July 2, 2008.
3979TSP1/HEP I INDUCES ANOIKIS RESISTANCE





























