Embed Size (px)
Citation preview

© 2010 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim 1261
Biotechnol. J. 2010, 5, 1261–1276 DOI 10.1002/biot.201000183 www.biotechnology-journal.com
1 Introduction to Caenorhabditis elegansbiology
The roundworm Caenorhabditis elegans was select-ed as a simple metazoan model in the early 1960sby Sydney Brenner for pursuing research in devel-opmental biology and neurobiology. Since its intro-duction as a model organism, C. elegans has beenwidely used to investigate important biologicalprocesses most of which have remained essential-ly unchanged during evolution [1]. C. elegans is a
free-living, non-parasitic nematode, with a life cy-cle of 3.5 days at 20°C and a lifespan of about2–3 weeks under suitable living conditions. Theadult is about 1 mm in length and 80 µm in diame-ter; it feeds on bacteria such as Escherichia coli inliquid medium or on agar plates, and can be easilycultivated in large numbers. C. elegans has fivepairs of autosomes and one pair of sex chromo-somes. It has two sexes, hermaphrodites and males;the ratio of sex chromosomes to autosomes deter-mines its sex. A single hermaphrodite produces~300 progeny by self-fertilization, and more if itmates with males, which arise occasionally at a fre-quency of 0.1%.Wild-type individuals consist of 959somatic cells, 302 of which are neurons.The animalbody is transparent so it is easy to track cells andfollow cell lineages and biological processes [2].The genome of C. elegans, comprising about a hun-dred million base pairs, is completely sequencedand surprisingly similar to that of humans; it is es-timated that 60–80% of the genes have a humancounterpart [3]. Viable mutant strains, strains thatoverexpress a gene or lack a gene function can beefficiently generated and the resulting phenotypescan be rapidly identified [4]. Comprehensive infor-mation concerning gene structure, expression pat-terns, protein-protein interactions, mutant or RNAinterference (RNAi) phenotypes and microarray
Review
Modeling human diseases in Caenorhabditis elegans
Maria Markaki and Nektarios Tavernarakis
Institute of Molecular Biology and Biotechnology, Foundation for Research and Technology – Hellas, Heraklion, Crete, Greece
Genes linked to human diseases often function in evolutionarily conserved pathways, which canbe readily dissected in simple model organisms. Because of its short lifespan and well-known bi-ology, coupled with a completely sequenced genome that shares extensive homology with that ofmammals, Caenorhabditis elegans is one of the most versatile and powerful model organisms. Re-search in C. elegans has been instrumental for the elucidation of molecular pathways implicated inmany human diseases. In this review, we introduce C. elegans as a model organism for biomedicalresearch and we survey recent relevant findings that shed light on the basic molecular determi-nants of human disease pathophysiology. The nematode holds promise of providing clear leadstowards the identification of potential targets for the development of new therapeutic interventionsagainst human diseases.
Keywords: Ageing · Cell death · Model organisms · Neurodegeneration · Protein aggregation
Correspondence: Dr. Nektarios Tavernarakis, Institute of Molecular Biology and Biotechnology, Foundation for Research and Technology –Hellas, N. Plastira 100, Vassilika Vouton, PO Box 1385, Heraklion 70013,Crete, GreeceE-mail: [email protected]: +30-2810-391067
Abbreviations: Aββ, β-amyloid peptide; AD, Alzheimer’s disease; ALS, amy-otrophic lateral sclerosis; APP, amyloid precursor protein; αα-syn, α-synu-clein; CHIP, C terminus of Hsc70 interacting protein; DA, dopaminergic;DMD, Duchenne muscular dystrophy; DR, dietary restriction; ER, endo-plasmic reticulum; Htt, Huntingtin; HD, Huntington’s disease;LRRK2, leucine-rich repeat kinase 2; miRNAs, microRNAs; MSP, majorsperm protein; OPMD, oculopharyngeal muscular dystrophy; PD, Parkin-son’s disease; SMA, spinal muscular atrophy; SMN, survival motor neurongene; SOD1, Cu/Zn superoxide dismutase 1; YFP, yellow fluorescent protein
Received 31 August 2010Revised 22 October 2010Accepted 25 October 2010

BiotechnologyJournal Biotechnol. J. 2010, 5, 1261–1276
1262 © 2010 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim
data, is available in Wormbase, the online resourcefor nematode-related information (http://www.wormbase.org/) [5]. Nematode strains can bestored indefinitely in liquid nitrogen allowing largemutant collections and public mutant repositoriesto be set up. C. elegans is amenable to unbiased for-ward and reverse genetic screens and is particular-ly susceptible to gene inactivation by RNAi [6, 7].Due to its value as a research tool, a large set ofmethods that include advanced high-resolutionimaging techniques have been developed.
C. elegans has emerged as a powerful experi-mental system to study the molecular and cellularaspects of human disease in vivo. It has been esti-mated that about 42% of the human disease geneshave an ortholog in the genome of C. elegans, in-cluding those genes associated with Alzheimer’sdisease (AD), juvenile Parkinson’s disease (PD),spinal muscular atrophy (SMA), hereditary non-polyposis colon cancer, and many others age-relat-ed disorders [8–10]. Modeling a human disease in asimple invertebrate, such as C. elegans, allows thedissection of complex molecular pathways intotheir component parts, thus providing a meaning-ful insight into the pathogenesis of a complex dis-ease phenotype. Here, we survey nematode modelsof human disease, highlighting recent discoveriesthat shed light on the molecular mechanisms un-derlying disease pathogenesis.
2 C. elegans models of neurodegenerativedisorders
Neurodegeneration has a profound effect on hu-man health, yet the mechanisms underlying neu-ronal injury and death remain unclear. C. elegansmodels of neuronal dysfunction have been estab-lished for a number of neurodegenerative diseases,including AD, PD and polyglutamine-expansiondisorders.These models typically involve the trans-genic expression of human disease genes.
2.1 Alzheimer’s disease
AD is a progressive neurological disease that re-sults in the irreversible loss of neurons particular-ly in the neocortex. The brain of AD-affected pa-tients is characterized by the accumulation of in-tracellular neurofibrillary tangles (NFTs) com-posed of the microtubule-associated protein tauthat stabilizes microtubules when phosphorylated[11] and of extracellular senile plaques primarilycomposed of β-amyloid peptide (Aβ) [12].Com-pelling evidence supports the causative role ofAβ1–42, which derives from the proteolysis of amy-
loid precursor protein (APP) by β- and γ- secretase,in AD. Similarly, mutations in the tau gene lead tofamiliar tauopathies.
Autosomal dominant mutations in APP are cor-related with rare cases of early-onset AD. Deter-mining the in vivo functions of APP is difficult be-cause mammals have an APP gene family contain-ing two APP-related genes. C. elegans has a singleAPP-related gene, apl-1 mapped on the X chromo-some. The apl-1 gene is expressed in multiple celltypes and is necessary for many developmentalprocesses, including proper molting and morpho-genesis. Loss of apl-1 causes larval lethality, whichcan be rescued by neuronal expression of the ex-tracellular domain of APL-1. Similarly, the overex-pression of APL-1 causes defects in movement,brood size and larval viability of transgenic nema-todes. The apl-1 overexpression-induced lethalityis partially rescued by the reduced activity of sel-12, a C. elegans homologue of the human γ-secre-tase gene component presenilin 1, suggesting thatSEL-12, like mammalian PS1, regulates the activi-ty of APL-1 either directly or indirectly [13].
C. elegans AD models were developed that ex-press Aβ variants under the control of male-specif-ic, body wall muscle-specific and neuronal-specif-ic promoters. Only transgenic lines carrying theunc-54/Aβ1–42 minigene accumulate intracellularAβ, and show a progressive paralysis beginning inyoung adulthood. Co-immunoprecipitation studiescoupled with mass spectrometry reveal that chap-erone-related proteins (two human HSP70, threeHSP-16 homologous to αB-crystallin and a putativenegative regulator of HSP70 function) interact withAβ in vivo. This interaction is considered as part ofa cellular protective response since the overex-pression of either HSP-16.2 or a human HSP70partially suppresses the Aβ-induced paralysis inworms [14]. Heat shock treatment reduces Aβoligomeric protein and delays paralysis in trans-genic Aβ worms, probably through the activation ofheat shock proteins regulated by the heat shocktranscription factor 1 (HSF-1) [15]. HSF-1 has beencoupled to normal ageing and age-related diseases[16]. Previous studies showed that constituents ofGinkgo biloba leaf extract (EGb 761 and ginkgo-lides) delay β-amyloid-induced paralysis in trans-genic C. elegans [17].This effect was not additive toprotective heat shock, suggesting a shared mecha-nism of action by the two treatments [15]. Microar-ray analysis in a transgenic worm strain that accu-rately expresses a temperature-inducible Aβ42 inbody wall muscle, reveals that the gene encodingthe arsenite-inducible protein (aip-1) is up-regu-lated as part of a cellular protective response.Worms overexpressing AIP-1 show decreased ac-

© 2010 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim 1263
cumulation of Aβ42 peptide and attenuated paraly-sis.The AIP-1 human homologue, AIRAPL, but notAIRAP when expressed under the control of myo-3 promoter also confers protection against Aβ toxi-city [18]. Both AIP-1 and AIRAPL contain a pre-dicted farnesylation site known to be critical for theAIP-1-mediated longevity in C. elegans [19]. AIP-1and AIRAPL enhance at least partially general pro-tein turnover by acting as positive regulators ofproteosomal function, and thus protect against Aβ-induced toxicity [18].The effects of tetracycline andits analogues doxycycline and minocycline on Aβ42-induced toxicity were assayed [20] using the wormsexpressing a temperature-inducible Aβ1–42 trans-gene [21]. Tetracyclines successfully protectedtransgenic worms from the Aβ insult by reducingthe concentration of oligomers considered to be re-sponsible for the toxic phenotype. These effectswere specific, dose-dependent and not linked toany antibiotic activity. Furthermore, tetracyclinesprotect Aβ-expressing nematodes from oxidativestress by reducing the superoxide production, sug-gesting a potential use of these drugs in targetingAβ aggregates.
2.2 Tauopathies
Abnormal phosphorylation, aggregation and ulti-mately functional alteration of the main micro-tubule-associated protein, tau, is the major cause ofneurodegenerative disorders known as tauopa-thies [22, 23].Tau aggregates are found in AD, Pick’sdisease, corticobasal degeneration, Down’s syn-drome and frontotemporal dementia with parkin-sonism chromosome 17 type (FTDP-17) [24]. Themost common tauopathy is AD. To analyze the roleof tau modification during AD progression, humantau and a pseudohyperphosphorylated tau (PHPtau; in which ten serine/threonine residues werechanged to glutamates), which mimics AD-relevanttau modification, were brought under the control ofa pan neuronal (a 3.4 kb upstream of the geneF25B3.3) promoter. Transgenic worms expressingeither tau or PHP tau developed a phenotype of un-coordinated (Unc) locomotion, characteristic of avariety of C. elegans nervous system defects. How-ever, only PHP tau-expressing worms showed a de-fective pattern of motor neuron development, sug-gesting that PHP tau may better mimic the stabledisease-like tau modification that interferes withmechanisms of axonal outgrowth and path finding[25]. To identify specific modifiers for the tau-in-duced pathological phenotype, a genome-wideRNAi screen was conducted using transgenicworms expressing mutations in tau (FTDP-17 tau)driven by the aex-3 pan neuronal promoter. Sixty
enhancer genes modified only the tau-induced Uncphenotype. These genes normally function in pro-tein phosphorylation, protein folding, stress re-sponse, nucleic acid function, proteolysis and neu-rotransmission [26]. In addition, random mutation-al screens on these transgenic animals resulted inthe identification of two novel tauopathy-associat-ed genes, sut-1 and sut-2 (suppressor of tauopathy)[27, 28]. SUT-1 requires the activity of UNC-34,which plays a role in the correct neuronal cell mi-gration and axonal guidance. SUT-2 encodes aCCCH zinc finger protein, primarily expressed inthe nucleus of neuronal cells, which binds ZYG-12,a protein of the HOOK family. In turn, ZYG-12 actsas a linker connecting membrane compartmentswith the microtubule cytoskeleton protein.
2.3 Polyglutamine-expansion disorders
Numerous human neurodegenerative disorders in-cluding Huntington’s disease (HD), several spin-ocerebellar ataxias and Kennedy’s disease [29, 30]are associated with misfolded and aggregation-prone proteins that interfere with protein home-ostasis, leading to cellular dysfunction. Polygluta-mine diseases are caused by a CAG triplet expan-sion in the coding regions of seemingly unrelatedgenes, resulting in a pathogenic protein with ab-normally expanded track of glutamines (Fig. 1) [31].
To address the mechanisms underlying the im-pact of aggregation-prone proteins on cellularfunction, transgenic C. elegans lines expressing dif-ferent lengths of polyQ-YFP (yellow fluorescentprotein) or CFP (cyan fluorescent protein) from in-tegrated arrays in muscle (polyQm) [32] or neu-ronal (polyQn) [33] cells have been utilized in ge-netic screens. These polyglutamine aggregationmodels show polyQ-length-dependent aggregationand toxicity. C. elegans polyQm or polyQn strainswere crossed with animals expressing tempera-ture-sensitive mutations considered to be sensitiveindicators of a disruption in protein homeostasis.These experiments have shown that chronic ex-pression of an aggregation-prone protein inter-feres with the function of multiple structurally andfunctionally unrelated proteins in a polyQ-length-dependent manner. PolyQ expansions uncover thefolding defect of proteins harboring mildly destabi-lizing temperature-sensitive mutations, which inturn modify polyQ aggregation and toxicity and en-hance the disruption of cellular folding homeosta-sis. Thus, mild folding mutations in the geneticbackground can modify the toxic phenotypes ofpolyglutamine diseases [34].
Protein aggregation and cellular dysfunctioncan occur at a threshold of approximately 40 gluta-
Biotechnol. J. 2010, 5, 1261–1276 www.biotechnology-journal.com

BiotechnologyJournal Biotechnol. J. 2010, 5, 1261–1276
1264 © 2010 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim
mine residues. However, the threshold for polyQaggregation and toxicity is dynamic and age de-pendent in C. elegans. At 3 days of age or less, onlyworms expressing Q40 or greater exhibit aggre-gates, while in older animals aggregates appeareven in Q33- and Q35-expressing worms [32].
A link between molecular determinants of age-ing and aggregation-mediated proteotoxicity wasestablished in C. elegans. Polyglutamine toxicitywas dependent on the activity of daf-16, a forkheadtranscription factor of the FOXO family functioningdownstream of age-1 (a phosphoinositide-3 ki-nase) in the insulin-like signaling pathway [35].Work on polyQ suppression shows that TOR-2, theworm ortholog of the mammalian torsinA, is an en-doplasmic reticulum (ER)-associated protein withchaperone activity that suppresses polyglutamineaggregation in C. elegans [36].
HD is the most frequent autosomal dominantinherited polyQ disorder caused by an expansion ofa CAG trinucleotide sequence in the huntingtin(Htt) protein. Normal Huntington alleles encode upto 37 CAG repeats, whereas HD-affected individu-als have more than 40.
Previous work has shown that ubiquilin is an in-teractor of presenilin, a protein associated with AD.Ubiquilin proteins contain multiple ubiquitin-re-lated domains and are linked to the ubiquitin-pro-teasome system of protein degradation as boththeir ubiquitin-like domain and the ubiquitin-as-sociated domain have been shown to bind the pro-teosomal subunit S5a [37]. The role of ubiquilin inHD pathogenesis was investigated using C. elegans
expressing GFP-Htt-polyQ fusion proteins in bodywall muscle. Transgenic worms display a polyQ-length-dependent motility defect that is sup-pressed by ubiquilin overexpression through anmRFP-tagged ubiquilin fusion protein. Conversely,knockdown by RNAi of C. elegans ubiquilin geneexacerbated the motility defect associated with theexpression of GFP-Htt (Q55) fusion protein. Thesefindings show that, similar to AD, ubiquilin protectsagainst polyglutamine-induced toxicity, and theysuggest a general role for ubiquilin in regulating anumber of different neurodegenerative disorders[38]. The motility defect that these C. elegans HDmodels display is reduced by the knockdown of thegene encoding the dynamin-related protein 1(Drp-1), which controls mitochondrial fission. Thisresult establishes a link between HD pathogenesisand mitochondrial dysfunction [39]. A role for in-sulin-like signaling pathway in the amelioration ofHD-associated proteotoxicity has been reported[16].
Two different C. elegans models of human dis-ease were used to explore the effects of dietary re-striction (DR) on polyglutamine-associated age-re-lated diseases.An HD model in which 35 glutamineresidues were fused to YFP (Q35YFP) and ex-pressed in the body wall muscles [32], and a trans-genic model expressing a 42-amino acid Aβ (Aβ42)under the control of the unc-54 promoter, wereused in this investigation [40]. The results demon-strate that DR suppresses age-associated paralysisin these worms.To test if DR acts as a general sup-pressor of proteotoxicity, the effects of food depri-
polyQ Aberrant proteolysis aggregates inclusions proteasome
protein degradationpathways
Q>30misfolded protein
Degraded proteins
lysosomepathways
mitochondrial dysfunction
Figure 1. Pathogenesis of polyglutamine diseases. Polyglutamine expansion disorders are caused by abnormally expanded tracks of polyglutamine residues(polyQ) in seemingly unrelated proteins. In many cases, abnormal proteolytic cleavage of the affected proteins could result in toxic folding intermediates,oligomers and aggregates, which ultimately form intracellular inclusions. Damaged proteins cannot be degraded through the ubiquitin (proteosomal) andlysosomal degradation pathways. Thus, the accumulation of unwanted proteins may lead to neurodegeneration. Mitochondrial dysfunction has also beenlinked to polyglutamine-expansion proteotoxicity.

© 2010 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim 1265
vation on transgenic worms expressing an aggre-gation-prone form of GFP (GFP-degron) were alsoexamined. It was shown that both environmentaland genetic (represented by the RNAi inhibition ofeat-2 and the knockdown of rab-10, sams-1 or drr-1 genes that act downstream of food consumption)models of DR confer protection against proteotox-icity in C. elegans. Compelling evidence was pre-sented that the general protective effect of DR ismediated by the activity of the heat shock tran-scription factor, hsf-1 [41].
2.4 Parkinson’s disease
PD is the second most common neurodegenerativedisorder characterized by involuntary movementsincluding bradykinesia and difficulty in balanceand muscle rigidity due to the progressive loss ofdopaminergic (DA) neurons from the substantianigra. At the cellular level, PD is characterized bycytoplasmic protein inclusions called Lewy bodies,which are the pathological feature of PD and otherneurodegenerative disorders known as synucle-opathies [42]. α-Synuclein (α-syn), a major con-stituent of Lewy bodies, is causatively related to PDsince missense mutations in the α-syn gene or du-plications and triplications of the α-syn locus causefamiliar forms of PD [43–45]. Sporadic PD affecting1–4% of those over 65 years of age appears to be un-related to mutations or multiplications of the α-synlocus, and implicates a combination of environ-mental factors. How α-syn inclusions are formedand how these are linked to disease are poorly un-derstood. Synucleopathies have been associatedwith mitochondria dysfunction, defective au-tophagy, impairment of the ubiquitin-proteasomesystem, production of reactive oxygen species(ROS), and stress within the ER [46].
The worm genome includes orthologs of all es-tablished familiar PD genes (Parkin, DJ-1, PINK1,UCHL-1, LRRK2, PARK9 and NURR1) except the α-syn. A transgene bearing Rab1 guanosine triphos-phate, a GTPase involved in transport from the ERto Golgi, under the control of the dopamine trans-porter (DAT-1) gene promoter was used to drive theexpression of Rab1 along with α-syn in DA neuronsin a worm PD model. Human α-syn overexpressionin C. elegans leads to the formation of misfolded α-syn aggregates that increase with age. Coexpres-sion of Rab1 significantly rescued the neurodegen-eration caused by the expression of α-syn alone.Considering that Rab1 plays an essential role inER-Golgi vesicle trafficking and its enhanced ex-pression protects against α-syn pathology, thesecombined results indicate that the accumulation ofα-syn inhibits the ER-Golgi trafficking [47]. Once
coexpressed,TOR-2 [48] ameliorated the formationof these aggregates by associating with α-syn inLewy bodies [49].
Worms expressing both α-syn::GFP and TOR-2in body wall muscles were used in a large-scaleRNAi screen for modifiers of α-syn misfolding andneurodegeneration. In all, 868 candidate genes po-tentially associated with existing PD genes andpathways were targeted for knockdown; 20 geneproducts that enhance misfolding of human α-synupon depletion were isolated. The neuroprotectivegenes, F32A6.3 (the C. elegans ortholog of VPS41, aconserved vesicular protein, essential for lysoso-mal biogenesis), W08D2.5 (the worm ortholog ofPARK9), and F55A4.1 (ortholog of Sec22p, a wellcharacterized vesicular trafficking protein inyeast), were among those identified.The identifica-tion of C. elegans ATG7 as a neuroprotective geneindicates the significant role of autophagy andlysosomal function in preventing the DA neuronloss caused by excess α-syn [48]. To further char-acterize the processes involved in inclusion bodyformation, a genome-wide RNAi screen was per-formed in transgenic worms expressing the α-synuclein-YFP chimera driven by the unc-54 pro-moter. The screen identified 80 suppressors, ofwhich 49 have an established human ortholog.Most of the genes are involved in protein qualitycontrol and vesicle trafficking between the ER-Golgi as well as other vesicular compartments.Moreover, this screen uncovered several regulatorsof lifespan (the NAD+-dependent protein deacety-lase sir-2.1, the sphingolipid synthase lagr-1) asmodifiers of inclusion formation, suggesting a linkbetween the molecular mechanisms of ageing andα-syn pathology [50].
Mutations in GTPase (R1441C/G) and kinase(G2019S) domains of leucine-rich repeat kinase(LRRK2) are the most frequent cause of autosomaldominant and idiopathic PD [51, 52]. Expression ofhuman LRRK2 in C. elegans neurons modifies theresponse to mitochondrial inhibition, inferred bythe increased survival of worms exposed torotenone or paraquat, which are agents that impairmitochondrial function. These findings demon-strate the power of the nematode as a model tostudy the possible gain-of-function phenotype ac-quired by LRRK2 mutations. lrk-1 is the sole C. el-egans ortholog of LRRK2. lrk-1 and LRRK2 seemsto share similar functions since LRRK2 comple-ments the sensitization to rotenone caused by thelrk-1 knockdown. These results indicate a role forLRRK2 and its worm ortholog in regulating mito-chondrial dysfunction [53]. R1441C/G and G2019Smutations increase kinase activity of LRRK2 in vit-ro [54, 55]. The role of LRRK2 in PD pathogenesis
Biotechnol. J. 2010, 5, 1261–1276 www.biotechnology-journal.com

BiotechnologyJournal Biotechnol. J. 2010, 5, 1261–1276
1266 © 2010 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim
was investigated in vivo using transgenic C. ele-gans overexpressing human LRRK2 wild type,R1441C/G and G2019S mutations and a LRRK2-in-active mutation K1347A in DA neurons. All trans-genic worms show an age-dependent degenerationof DA neurons, locomotor dysfunction, behavioralabnormalities and dopamine deficiency. In con-trast, loss of endogenous LRK-1 and the GTP-binding defective mutation, K1347A, amelioratedLRRK2-associated phenotypes [56].
2.5 Amyotrophic lateral sclerosis
Amyotrophic lateral sclerosis (ALS), the most com-mon motor neuron disorder, is characterized bydysfunction or death of motor neurons in the mo-tor cortex, brain stem and spinal cord. Approxi-mately 10% of ALS cases are familiar with an auto-somal dominant inheritance [57] and ~20% of fa-miliar ALS (FALS) are associated with mutations inthe gene coding for Cu/Zn superoxide dismutase(SOD1) [58], which catalyzes the conversion ofhighly reactive superoxide anions into hydrogenperoxide [59]. While it is accepted that toxic gain-of-function mutations in SOD1 gene evoke the dis-ease phenotype, the mechanisms of toxicity remainunclear.
Transgenic C. elegans expressing FALS-relatedmutant (A4V, G37R, G93A) human SOD1 are moresensitive to paraquat-induced oxidative stress thancontrol worms expressing wild-type human SOD1.Moreover, the transgenic worms that harbor gain-of-function alleles show an aberrant accumulationand aggregate formation of mutant proteins underoxidative stress. Thus, oxidative damage duringageing might cause FALS by inhibiting degradationand enhancing aggregate formation of mutantSOD1 proteins [60].
G85R, an ALS-associated human mutant SOD1,causes a severe locomotor defect in transgenic C.elegans when expressed pan-neuronally under thecontrol of the synaptobrevin (snb-1) gene promot-er. Paralyzed animals have soluble oligomers andinsoluble aggregates in neurons. The movementdefect results from the dysfunction of synapses, theconnections between neurons and between neu-rons and muscles. An RNAi screen identified mo-lecular chaperones as critical modifiers of proteinaggregation-mediated toxicity [61]. Expression ofthree distinct SOD1 mutations (G85R, G93A, 127X)in the body muscle cells of wild-type (N2) wormscauses only mild cellular toxicity. However, whenSOD1 mutants are expressed on the background ofdestabilized protein polymorphisms, their toxicityis enhanced. These results suggest that the geneticbackground may intensify the specific toxic pheno-
types of misfolded and aggregation-prone proteinsthat characterize conformational diseases [62].
A recent study identified the gene ALS8 as anovel cause of ALS [63]. Mutations in ALS8, as inSOD, result in a wide range of defects that vary inthe age of onset, the speed of progression and themotor neurons affected. This variation may be dueto genetic modifiers, redundancy or environmentalcontribution. ALS8 codes for VAMP (synapto-brevin)-associated protein B (VAPB), which isclosely related to VAPA, a protein associated withthe cytoplasmic face of the ER and the Golgi appa-ratus [64–66]. Many species have VAP proteinscharacterized by the major sperm protein domain(MSP) of about 125 residues at their N-terminalend [67], a central region of coiled-form motif anda hydrophobic C terminus, which acts as a mem-brane anchor. The MSP domain is named from itssimilarity to C. elegans MSPs, the most abundantproteins in the worm sperm [68].
MSP functions in oocyte maturation and sheathcontraction in C. elegans [69]. After secretion, ex-tracellular MSP directly binds to the VAB-1 Eph re-ceptor and other yet-to-be-identified receptors onoocyte and ovarian cell surfaces [70].
The biological function of VAPs and the mecha-nisms underlying their impact in ALS pathogenesisis not well understood. To define the role of VAPs,C. elegans was used together with other model sys-tems. C. elegans has a single VAP homolog, theVPR-1, and VAP MSP domains share a 25% identi-ty in their primary sequences with MSPs. Injectionof recombinant VPR-1 MSP stimulated oocyte mat-uration and sheath contraction in the gonads ofworms lacking MSP and sperm, suggesting that C.elegans MSP and VAP MSP domains have evolu-tionary conserved extracellular signaling activity.Microinjection of Drosophila VAP (dVAP) and hu-man VAP also induced both responses. VAPs havebeen shown to regulate Eph receptor signaling invivo. MSP domains of VAP proteins are cleaved andprovide ligands for Eph receptors. Mutations inVAPs may result in a failure to secrete the MSP do-main, accumulation of inclusions in the ER and anunfolded protein response. These findings provideinsight into the disease mechanism [71].
2.6 Spinal muscular atrophy
SMA, the most common genetic cause of infantmortality, is an autosomal recessive neuromusculardisorder. SMA patients suffer from degeneration oflower motor neurons in the anterior horn of thespinal cord leading to atrophy of the correspondingmuscles [72]. The survival motor neuron gene(SMN) has been causatively linked with SMA, but

© 2010 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim 1267
the mechanisms underlying the disease pathogen-esis are poorly understood. Given that SMN iswidely expressed both within and outside the nerv-ous system [73], the specific motor neuron defectthat the SMA patients face may be rather linked tothe gene function [74]. C. elegans smn-1 gene hasbeen identified as the ortholog of human SMN [75]with a widespread expression in various tissues in-cluding the nervous system and body wall muscles.The mutant smn-1 (ok355) harbors a deletion allelethat removes most of the smn-1, including thetranslational start codon. Having reduced SMN-1levels throughout the organism, smn-1 (ok355) isused for modeling aspects of SMA. Mutant animalsproceed through embryogenesis due to maternallycontributed SMN-1, but showed late larval arrest,decreased lifespan, defects in motility and pharyn-geal pumping. Neuronal, but not muscle-directedexpression of full-length smn-1 partially rescuesthe smn-1 (ok355) phenotype [76]. A genome-wideRNAi screen and behavioral studies in the loss offunction smn-1 (ok355) mutant strain provide pre-liminary evidence that the TGF-β/BMP signalingpathway plays a role in the SMN-mediated neuro-muscular pathology in C. elegans [77].
3 Stroke – Excitotoxicity
Necrotic cell death underlies the pathology of nu-merous human degenerative conditions [78]. In C.elegans, non-apoptotic cell death (necrosis) is trig-gered by specific genetic lesions or environmentalconditions such as energy depletion, toxin expo-sure or extreme temperatures. Severe deprivationof energy resources can rapidly develop during is-chemic or hypoglycemic episodes. Prolonged hy-poxia, a condition of oxygen deprivation that takesplace in ischemic episodes and stroke, also inducescell death in C. elegans [79]. The most thoroughlycharacterized necrosis-induced mutations in thenematode are gain-of-function mutations in spe-cific ion channel genes, such as degenerin genesdeg-1 and mec-4, the acetylcholine receptor chan-nel subunit gene deg-3 and the Gs protein a-sub-unit gene gsa-1, which result in necrotic cell deathof neurons expressing the mutant proteins [80].Execution of necrotic cell death requires the activ-ity of calcium-regulated CLP-1 and TRA-3 calpainproteases and ASP-3 ASP-4 aspartyl proteases.Perturbation of intracellular concentrations of cal-cium, either by extracellular calcium influx or byrelease of ER stores, activates calpain proteases,which in turn engage executioner aspartyl proteas-es leading to cell destruction [81]. In mammalianmodels of excitotoxicity, calpains increase Ca2+
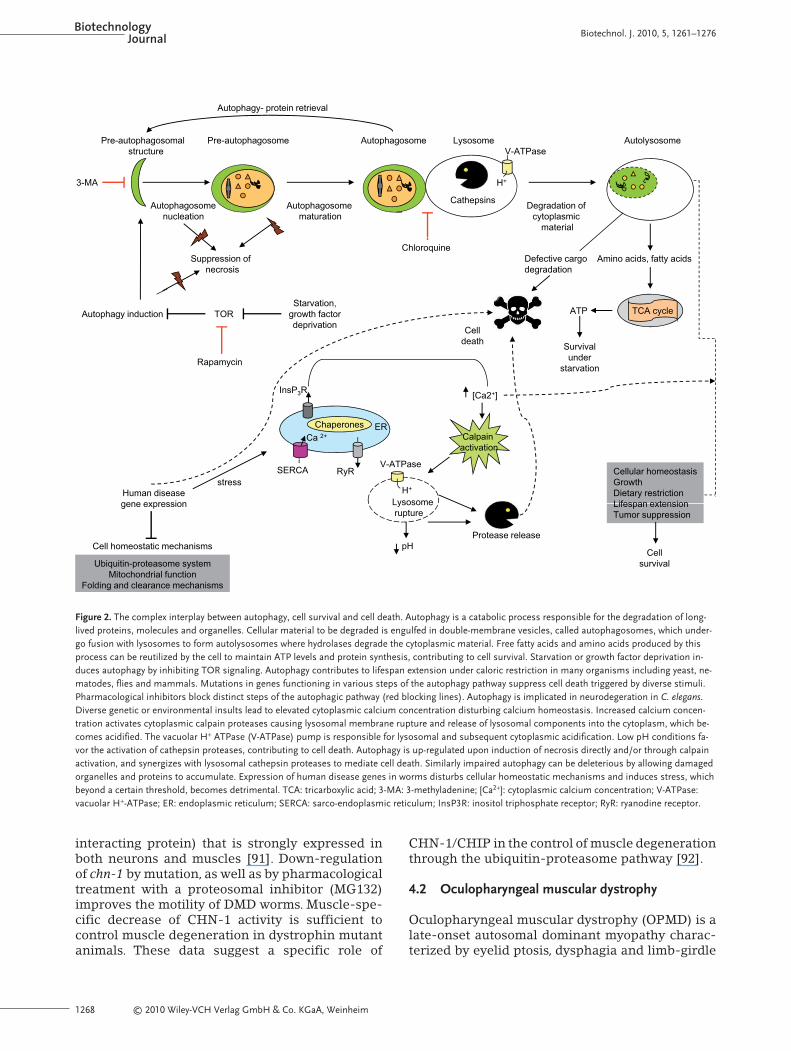
concentration by mediating the cleavage of specif-ic glutamate receptors [82]. Aspartyl proteases areeither cytoplasmic or lysosomal, being releasedinto the cytoplasm as a result of lysosomal rupture.Acidification of the cytoplasm due to the action ofhydrolytic enzymes liberated during the lysosomalrupture requires the vacuolar H+-V-ATPase (V-AT-Pases), a proton pump that acidifies endocytic com-partments. Similar mechanisms may underlie theextreme acidosis that is observed during stroke inhumans [83]. Evidence suggests that lysosomescontribute to execution of necrosis. Suppression oflysosome biogenesis and of lysosome-mediated cy-toplasmic acidification, which develops duringnecrosis and is required for cell death, affectsnecrosis in C. elegans [84]. A working model fornecrotic cell death and cellular defense pathwayssuch as autophagy that may underlie diverse as-pects of development, tissue homeostasis and dis-ease pathogenesis in C. elegans is outlined in Fig. 2.
4 Muscle-associated disorders
Dystrophin is a huge cytosolic protein that links theintracellular F-actin filaments to the members ofthe dystrophin-glycoprotein complex (DGC) [85,86]. Genetic defects in the complex componentscause a variety of pathological conditions, includ-ing muscular dystrophy, cardiomyopathy and va-sospasm. Determining the molecular function ofthe complex will increase our understanding ofmuscle diseases pathogenesis. C. elegans has a dy-strophin complex (DAPC) and mutations in itscomponents result in head bending and hypercon-traction [87, 88]. Several lines of evidence coupledefects in the DAPC with calcium homeostasisdisruption through a mechanism involving theISLO-1, which mediates the interaction of SLO-1(the ortholog of the mammalian BK channel) withDAPC. A defect in either DAPC or ISLO-1 impactsSLO-1 channel localization resulting in musclehyperactivity [89].
4.1 Duchenne muscular dystrophy
Duchenne muscular dystrophy (DMD) is one of themost severe X-linked childhood diseases, charac-terized by progressive muscle wasting and weak-ness due to mutations in the dystrophin gene [90].A C. elegans DMD model carrying mutations in dys-1 and hlh-1, the worm orthologs of the human dys-trophin and MyoD, respectively, was used to inves-tigate the effects of chn-1 on muscle degeneration.chn-1 encodes the homololog of the human E3/E4ubiquitylation enzyme CHIP (C terminus of Hsc-70
Biotechnol. J. 2010, 5, 1261–1276 www.biotechnology-journal.com
BiotechnologyJournal Biotechnol. J. 2010, 5, 1261–1276
1268 © 2010 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim
interacting protein) that is strongly expressed inboth neurons and muscles [91]. Down-regulationof chn-1 by mutation, as well as by pharmacologicaltreatment with a proteosomal inhibitor (MG132)improves the motility of DMD worms. Muscle-spe-cific decrease of CHN-1 activity is sufficient tocontrol muscle degeneration in dystrophin mutantanimals. These data suggest a specific role of
CHN-1/CHIP in the control of muscle degenerationthrough the ubiquitin-proteasome pathway [92].
4.2 Oculopharyngeal muscular dystrophy
Oculopharyngeal muscular dystrophy (OPMD) is alate-onset autosomal dominant myopathy charac-terized by eyelid ptosis, dysphagia and limb-girdle
Pre-autophagosomal t t
AutolysosomePre-autophagosome Autophagosome LysosomeV ATP
Autophagy- protein retrieval
structure
3-MA
V-ATPase
H+
Degradation of cytoplasmic
material
Autophagosome nucleation
Autophagosome maturation
Cathepsins
Defective cargo degradation
Suppression of necrosis
Amino acids, fatty acidsChloroquine
TCA cycleATPAutophagy induction TORStarvation,
growth factordeprivation
Survival under
starvation
Cell death
Rapamycin
deprivation
starvation
InsP3R [Ca2+]
Chaperones
Ca 2+ER
Calpain activation
Human disease gene expression
stressSERCA RyR
LysosomeH+
V-ATPaseCellular homeostasisGrowthDietary restrictionLifespan extensiongene expression
Cell homeostatic mechanismsProtease release
Lysosomerupture
pH
Lifespan extensionTumor suppression
Ubiquitin-proteasome systemMitochondrial function
Folding and clearance mechanisms
Cell homeostatic mechanismsCell
survival
pH
Figure 2. The complex interplay between autophagy, cell survival and cell death. Autophagy is a catabolic process responsible for the degradation of long-lived proteins, molecules and organelles. Cellular material to be degraded is engulfed in double-membrane vesicles, called autophagosomes, which under-go fusion with lysosomes to form autolysosomes where hydrolases degrade the cytoplasmic material. Free fatty acids and amino acids produced by thisprocess can be reutilized by the cell to maintain ATP levels and protein synthesis, contributing to cell survival. Starvation or growth factor deprivation in-duces autophagy by inhibiting TOR signaling. Autophagy contributes to lifespan extension under caloric restriction in many organisms including yeast, ne-matodes, flies and mammals. Mutations in genes functioning in various steps of the autophagy pathway suppress cell death triggered by diverse stimuli.Pharmacological inhibitors block distinct steps of the autophagic pathway (red blocking lines). Autophagy is implicated in neurodegeration in C. elegans.Diverse genetic or environmental insults lead to elevated cytoplasmic calcium concentration disturbing calcium homeostasis. Increased calcium concen-tration activates cytoplasmic calpain proteases causing lysosomal membrane rupture and release of lysosomal components into the cytoplasm, which be-comes acidified. The vacuolar H+ ATPase (V-ATPase) pump is responsible for lysosomal and subsequent cytoplasmic acidification. Low pH conditions fa-vor the activation of cathepsin proteases, contributing to cell death. Autophagy is up-regulated upon induction of necrosis directly and/or through calpainactivation, and synergizes with lysosomal cathepsin proteases to mediate cell death. Similarly impaired autophagy can be deleterious by allowing damagedorganelles and proteins to accumulate. Expression of human disease genes in worms disturbs cellular homeostatic mechanisms and induces stress, whichbeyond a certain threshold, becomes detrimental. TCA: tricarboxylic acid; 3-MA: 3-methyladenine; [Ca2+]: cytoplasmic calcium concentration; V-ATPase:vacuolar H+-ATPase; ER: endoplasmic reticulum; SERCA: sarco-endoplasmic reticulum; InsP3R: inositol triphosphate receptor; RyR: ryanodine receptor.

© 2010 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim 1269
muscle weakness [93]. A short polyalanine (GCG-triplet) stretch of 10–13 alanines or more in thegene encoding polyalanine-binding protein 1(PABPN1) on chromosome 14q causes gain-of-function mutations leading to misfolding, aggre-gate formation and dysfunction of the ubiquitinproteasome system [94–96]. However, the patho-genesis of disease remains unclear.
In a C. elegans OPMD model, loss-of-functionmutations in sir-2.1/SIRT1 and daf-16 protecttransgenic worms from the muscle decline and ab-normal motility induced by expression of mutant(13 alanines) PAPBN1 protein [97].
5 Cancer
Cancer is a major cause of human mortality andtypically evolves as a multistep process from muta-tions in key cellular genes.Although C. elegans doesnot form tumors per se, other mutant phenotypesare linked to worm homologues of human cancergenes, which often function at distinct steps withinevolutionarily conserved pathways. Thus, identify-ing C. elegans genes that disrupt processes relevantto cancer could contribute importantly to our un-derstanding of the cancer gene functions.
Several studies show that c-Met, a receptor ty-rosine kinase (RTK) is overexpressed and ampli-fied in many human cancers [98–102]. Mutations inthe c-Met juxtamembrane domain as well asgermline mutations have been identified in smallcell lung cancer [103] and in non-small cell lungcancer [104] cases, respectively.
Transgenic worms expressing wild-type c-Metor the frequent human lung cancer specific c-Metmutants (R988C and T1010I) were generated to in-vestigate cancer gene function in a whole organismcontext. Mutant transgenic worms show locomo-tion defects, low fecundity and vulval hyperplasia,phenotypes that were intensified upon exposure tonicotine [105].
The RTK/Ras/Raf/MAPK signaling pathway ishighly conserved and interfaces with many aspectsof animal development. Work in C. elegans hasgreatly contributed in our understanding of thispathway and its regulation [106]. In C. elegans, Rascontrols vulval induction and its hyperactivationinterferes with vulval cell differentiation, leading toa multivulval phenotype [107].Vulval developmentprovides a model system to understand cell prolif-eration and differentiation, deregulation of whichis causatively linked to human tumors.
Recently, the role of microRNAs (miRNAs) inhuman cancer has been extensively studied. miR-NAs are small non-protein coding RNA molecules
of ~21–23 nucleotides, which control gene expres-sion at the post-transcriptional level acting as de-velopmental timing regulators [108]. Many of the112 miRNAs identified so far in C. elegans are con-served across phylogeny. Compelling evidence sug-gests that miRNAs are involved in the regulation ofageing and metabolism and provides insight intotheir function as tumor suppressor genes or onco-genes.
Several miRNAs act in the Notch and Ras sig-naling pathways [109–111]. miR-84, a let-7 familymiRNA, targets let-60, a C. elegans ortholog ofHRAS, KRAS and NRAS, the human ras oncogenes.All of the human ras genes have complementarysites for human let-7 family miRNAs in their3’UTRs, suggesting a similar mode of regulation tothat of the worm let-60 [110]. Gain-of-function mu-tations in let-60 result in a multivulval phenotypethat is partially suppressed by mir-84 overexpres-sion [112]. Accumulating evidence suggests thatlet-7 acts as a tumor suppressor gene. Similarly,miR-61, which targets vav-1, a homologue of theVav oncogene family in vertebrates [113], may in-fluence cancer through the regulation of Notch sig-naling [111].
The role of apoptosis in cancer treatment sensi-tivity is well established. Apoptosis is a geneticallyregulated cell death mechanism that is required fornormal development and homeostasis. Defects inkey apoptotic proteins can interfere with tumorige-nesis and chemotherapy [114]. Therefore, under-standing the events that affect DNA damage-in-duced apoptosis at the molecular level is importantfor cancer research.The apoptotic molecular path-way is highly conserved [115].
In C. elegans, germline apoptosis limits thenumber of oocytes competing for nutrients in thegonad. It can also be activated in response to path-ogenic microorganism invasion or DNA-damagingagents and genotoxic stress [116]. The p53/CEP-1,CED-9 (Bcl-2), CED-4 (ApafI) and CED-3 (caspase)proteins function in the checkpoint machinery ofDNA damage-induced germline apoptosis. A re-cent study revealed that the hypoxia-inducibletranscription factor (HIF-1) antagonizes the func-tion of CEP-1 in DNA-damage induced apoptosis.HIF-1 inhibits apoptosis by transcriptionally up-regulating the tyrosinase family member TYR-2 inthe ASJ neurons. TYR-2 is then secreted and, onentering the gonad, it blocks apoptosis.These find-ings yield insight into a novel pathway throughwhich two single neurons regulate apoptotic fate inanother tissue. Ultimately, this pathway links hy-poxia to apoptosis. Knockdown of the TYR-2 ho-mologue TRP2 in human melanoma cells increasesbasal and cisplatin-induced p53 apoptosis, indicat-
Biotechnol. J. 2010, 5, 1261–1276 www.biotechnology-journal.com

BiotechnologyJournal Biotechnol. J. 2010, 5, 1261–1276
1270 © 2010 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim
ing an evolutionary conserved function. TYR-2would be a candidate target in new therapeuticstrategies [117].
6 Metabolic diseases
The O-GlcNAc signaling pathway, one of the mostevolutionary ancient pathways, couples the nutri-ent-dependent synthesis of UDP-GlcNAc to O-GlcNAc modification of Ser/Thr residues of a largenumber of nuclear and cytoplasmic protein targets.Accumulating data suggest the involvement of O-GlcNAc cycling in age-associated human diseasessuch as insulin resistance, X-linked dystoniaparkinsonism, cancer and disease of the immunesystem.
Two highly conserved enzymes are responsiblefor the cycling of O-GlcNAc on Ser/Thr residues oftarget proteins: glysosyltransferase, OGT, and theO-GlcNAcase, which mediate the addition and theremoval of O-GlcNAc, respectively. The identifica-tion of the enzymes involved in O-GlcNAc cyclingled to the detection of their interacting partners.Two-hybrid screens reveal diverse proteins associ-ated with vesicle and organelle movements in neu-rons, mitochondrial movement, or factors involvedin the control of gene expression that interact withOGT. In C. elegans, two-hybrid analysis indicatesthe interaction of OGT-1 with the MAP kinase,PMK-1. The highly conserved hexosamine biosyn-thetic pathway is upstream of O-GlcNAc cycling.Accumulating findings suggested a link betweenthe hexosamine biosynthetic pathway, O-GlcNAccycling and mammalian insulin resistance [118].
C elegans strains with putative null alleles ofogt-1 (ortholog of O-GlcNAc transferase) or oga-1(ortholog of O-GlcNAcase) provide versatile plat-forms for studying the role of O-GlcNAc in cellularsignaling and metabolism. Nuclear transport oftranscription factors is normal in homozygous ogt-1(ok430) [119] and oga-1(ok1207) knockout ani-mals [120], yet the ogt-1(ok430) mutant completelylacks any detectable O-GlcNAc on nuclear poreproteins, whereas the oga-1(ok1207) accumulatedO-GlcNAc. Both mutant strains show increasedglycogen and trehalose levels and decreased lipidstorage.The link of human OGT and OGA to insulinresistance prompted the examination of the impactof ogt-1 and oga-1 deletions on the insulin- like-signaling pathway controlling nutrient storage,dauer formation, longevity and stress responses inC. elegans. oga-1(ok1207) knockout enhances dauerformation induced by a temperature-sensitive al-lele of the insulin-like receptor daf-2 (e1370) [120]under conditions in which the ogt-1(ok430) null al-
lele lowers dauer formation [119]. C. elegans is thefirst animal with viable and fertile knockout allelesof OGA or OGT, which provide geneticallyamenable models of non-insulin-dependent dia-betes. Use of O-GlcNAc cycling mutants in high-throughput RNAi screening revealed the interac-tion of ogt-1 with essential components of impor-tant signaling pathways such as β-catenin. In mam-mals, β-catenin triggers key developmentalswitches including the formation of T cells andB cells in adaptive immunity [121].
Accumulating evidence suggests that the en-zymes of O-GlcNAc cycling may play a role in mod-ulating transcription [122, 123]. The impact of nullalleles of O-GlcNAc cycling genes on chromatinstructure and gene expression can be globally ex-amined in C. elegans. Whole-genome chromatinimmunoprecipitation on- chip tiling arrays re-vealed over 800 promoters in which O-GlcNAc cy-cling occurs, including miRNA genes and multi-gene operons. Transcriptional profiling of O-Glc-NAc knockout mutants confirmed the deregulationof numerous genes associated with insulin-like sig-naling, ageing, stress response, innate immunityand carbohydrate/lipid metabolism [124]. Thesefindings suggest that altered O-GlcNAc cycling hasimportant implications in the diseases associatedwith ageing, including obesity, type II diabetes mel-litus, cardiovascular diseases, cancer and neurode-generative diseases.
In C. elegans, insulin and its signaling pathwayare implicated in the regulation of fat storage andreproduction and in modulating lifespan. daf-2, thehomologue of the mammalian insulin receptor, ispart of a signal transduction cascade homologousto the insulin pathway in mammals. DAF-2 knock-out induces dauer formation and extends lifespan.Restoration of DAF-2 function specifically in neu-rons is sufficient to restore lifespan and reproduc-tion of daf-2 knockouts to wild-type levels. Grow-ing evidence suggests that impaired insulin signal-ing in the central nervous system plays a funda-mental role in the pathogenesis of commonmetabolic diseases, including obesity and type IIdiabetes [125].
C. elegans provides a versatile platform for dis-secting the molecular mechanisms underlying glu-cose-mediated impact on cellular and mitochondr-ial function in diabetes. An intracellular glucoseconcentration of 14 mmol/L, which is within therange observed in poorly controlled diabetes pa-tients, results in methylglyoxal-derived modifica-tions of mitochondrial proteins and increase inROS formation, thus reducing lifespan in C. ele-gans. These effects are independent of osmoticpressure and the pathways activated by caloric re-
© 2010 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim 1271
Biotechnol. J. 2010, 5, 1261–1276 www.biotechnology-journal.com
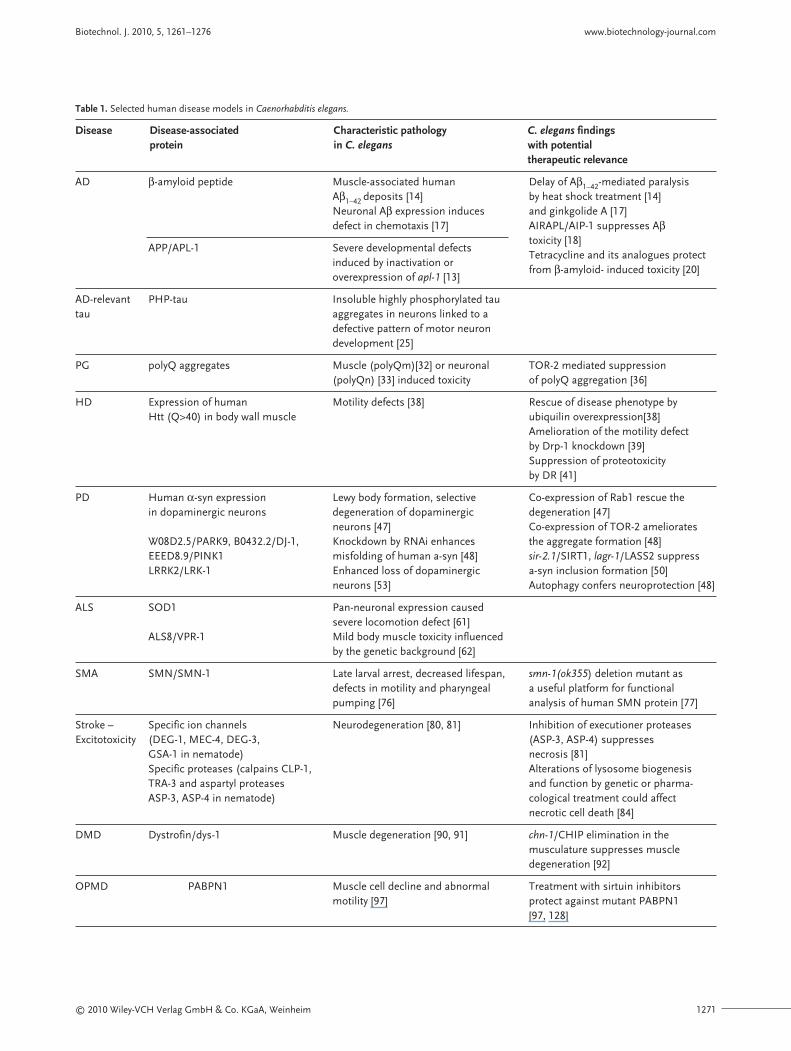
Table 1. Selected human disease models in Caenorhabditis elegans.
Disease Disease-associated Characteristic pathology C. elegans findings protein in C. elegans with potential
therapeutic relevance
AD β-amyloid peptide Muscle-associated human Delay of Aβ1–42-mediated paralysis Aβ1–42 deposits [14] by heat shock treatment [14]Neuronal Aβ expression induces and ginkgolide A [17]defect in chemotaxis [17] AIRAPL/AIP-1 suppresses Aβ
APP/APL-1 Severe developmental defects toxicity [18]
induced by inactivation orTetracycline and its analogues protect
overexpression of apl-1 [13]from β-amyloid- induced toxicity [20]
AD-relevant PHP-tau Insoluble highly phosphorylated tau tau aggregates in neurons linked to a
defective pattern of motor neuron development [25]
PG polyQ aggregates Muscle (polyQm)[32] or neuronal TOR-2 mediated suppression (polyQn) [33] induced toxicity of polyQ aggregation [36]
HD Expression of human Motility defects [38] Rescue of disease phenotype by Htt (Q>40) in body wall muscle ubiquilin overexpression[38]
Amelioration of the motility defect by Drp-1 knockdown [39]Suppression of proteotoxicity by DR [41]
PD Human α-syn expression Lewy body formation, selective Co-expression of Rab1 rescue the in dopaminergic neurons degeneration of dopaminergic degeneration [47]
neurons [47] Co-expression of TOR-2 ameliorates W08D2.5/PARK9, B0432.2/DJ-1, Knockdown by RNAi enhances the aggregate formation [48]EEED8.9/PINK1 misfolding of human a-syn [48] sir-2.1/SIRT1, lagr-1/LASS2 suppress LRRK2/LRK-1 Enhanced loss of dopaminergic a-syn inclusion formation [50]
neurons [53] Autophagy confers neuroprotection [48]
ALS SOD1 Pan-neuronal expression caused severe locomotion defect [61]
ALS8/VPR-1 Mild body muscle toxicity influenced by the genetic background [62]
SMA SMN/SMN-1 Late larval arrest, decreased lifespan, smn-1(ok355) deletion mutant as defects in motility and pharyngeal a useful platform for functional pumping [76] analysis of human SMN protein [77]
Stroke – Specific ion channels Neurodegeneration [80, 81] Inhibition of executioner proteases Excitotoxicity (DEG-1, MEC-4, DEG-3, (ASP-3, ASP-4) suppresses
GSA-1 in nematode) necrosis [81]Specific proteases (calpains CLP-1, Alterations of lysosome biogenesis TRA-3 and aspartyl proteases and function by genetic or pharma-ASP-3, ASP-4 in nematode) cological treatment could affect
necrotic cell death [84]
DMD Dystrofin/dys-1 Muscle degeneration [90, 91] chn-1/CHIP elimination in the musculature suppresses muscle degeneration [92]
OPMD PABPN1 Muscle cell decline and abnormal Treatment with sirtuin inhibitors motility [97] protect against mutant PABPN1
[97, 128]

BiotechnologyJournal Biotechnol. J. 2010, 5, 1261–1276
1272 © 2010 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim
striction in eat-2 mutants and the insulin-like re-ceptor daf-2/daf-16 pathway. Conversely, high glu-cose-mediated lifespan reduction in C. elegans isdependent on glyoxalase-1-controlled mitochon-drial complexes II and III. Further studies shoulddelineate the relevance of these mechanisms de-scribed in C. elegans to the situation in diabetic pa-tients [126].
miRNAs have also been implicated in the regu-lation of glucose and lipid metabolism, and hencein the initiation and progression of diabetes and itsspecific complications. miRNAs that play a criticalrole in the pathogenesis of the disease may serve aspotential biomarkers for prognosis and diagnosisof diabetes. Furthermore, the altered expressionprofiles of miRNAs in the pancreas and insulin-targeted tissues in diabetic patients could provideinsights for treating this complex metabolic disease[127].
7 Concluding remarks
Accumulating evidence demonstrates that most ofthe important pathways such as insulin signaling,Ras/Notch signaling, p53, and many miRNAs haveremained essentially unchanged during evolution.Many human diseases, including cancer, originatefrom mutations in genes functioning in pathwaysconserved in C. elegans. Although nematode dis-ease models might not perfectly recapitulate thepathophysiology of human disease, the small,
transparent worm enables the use of powerful ge-netic and molecular approaches to dissect the tox-icity mechanisms and determine the in vivo factorsthat exacerbate or cure pathological conditions.Table 1 summarizes a selection of human diseasemodels in C. elegans. Besides being an excellentmodel for drug target identification, C. elegans isamenable to high-throughput screens for rapidand inexpensive drug evaluation before embarkingon expensive and elaborate animal models.
N.T. is supported by grants from the European Re-search Council (ERC), the European Commission 6thand 7th Framework programs and the EuropeanMolecular Biology Organization (EMBO).
The authors have declared no conflict of interest.
8 References[1] Brenner, S.,The genetics of Caenorhabditis elegans. Genet-
ics 1974, 77, 71–94.[2] Sulston, J. E., Schierenberg, E., White, J. G., Thomson, J. N.,
The embryonic cell lineage of the nematode Caenorhabdi-tis elegans. Dev. Biol. 1983, 100, 64–119.
[3] Harris,T.W., Chen, N., Cunningham, F.,Tello-Ruiz, M. et al.,WormBase: a multi-species resource for nematode biologyand genomics. Nucleic Acids Res. 2004, 32, D411–417.
[4] Hariharan, I. K., Haber, D. A., Yeast, flies, worms, and fishin the study of human disease. N. Engl. J. Med. 2003, 348,2457–2463.
Table 1. Continued
Disease Disease-associated Characteristic pathology C. elegans findings protein in C. elegans with potential
therapeutic relevance
Cancer c-Met Locomotion defects, low fecundity and vulval hyperplasia [105]Anti-apoptotic factor, antagonizes p53/CEP-1, interferes with tumor progression and therapy [117]
let-60/ras oncogenes Multivulval phenotype [109–111]vav-1/Vavcep-1/p53 Apoptotic defects interfere with
tumorigenesis and resistance to chemotherapeutic drugs [114]
HIFa/HIF-1 Anti-apoptotic factor, antagonizes Target TYR-2/TRP2 [117]p53/cep-1, interferes with tumor progression and therapy [117]
Metabolic ogt-1, oga-1 Null mutations cause alterations in Overexpression or inhibition of diseases carbohydrate and lipid metabolism specific miRNAs may be a future (insulin resistance, [119, 120, 124] tool for targeting diabetes [127]type II diabetes)

© 2010 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim 1273
[5] Chen, N., Harris, T. W., Antoshechkin, I., Bastiani, C. et al.,WormBase: a comprehensive data resource for Caenor-habditis biology and genomics. Nucleic Acids Res. 2005, 33,D383–389.
[6] Kamath, R. S., Ahringer, J., Genome-wide RNAi screeningin Caenorhabditis elegans. Methods 2003, 30, 313–321.
[7] Fire, A., Xu, S., Montgomery, M. K., Kostas, S. A. et al., Po-tent and specific genetic interference by double-strandedRNA in Caenorhabditis elegans. Nature 1998, 391, 806–811.
[8] Baumeister, R., Ge, L.,The worm in us – Caenorhabditis el-egans as a model of human disease. Trends Biotechnol.2002, 20, 147–148.
[9] Poulin, G., Nandakumar, R., Ahringer, J., Genome-wideRNAi screens in Caenorhabditis elegans: impact on cancerresearch. Oncogene 2004, 23, 8340–8345.
[10] Kenyon, C.,The plasticity of aging: insights from long-livedmutants. Cell 2005, 120, 449–460.
[11] Kosik, K. S., Joachim, C. L., Selkoe, D. J., Microtubule-asso-ciated protein tau (tau) is a major antigenic component ofpaired helical filaments in Alzheimer disease. Proc. Natl.Acad. Sci. USA 1986, 83, 4044–4048.
[12] Glenner, G. G., Wong, C. W., Alzheimer’s disease: initial re-port of the purification and characterization of a novelcerebrovascular amyloid protein. Biochem. Biophys. Res.Commun. 1984, 120, 885–890.
[13] Hornsten, A., Lieberthal, J., Fadia, S., Malins, R. et al., APL-1, a Caenorhabditis elegans protein related to the humanbeta-amyloid precursor protein, is essential for viability.Proc. Natl. Acad. Sci. USA 2007, 104, 1971–1976.
[14] Link, C. D., C. elegans models of age-associated neurode-generative diseases: lessons from transgenic worm modelsof Alzheimer’s disease. Exp. Gerontol. 2006, 41, 1007–1013.
[15] Wu, Y., Cao, Z., Klein, W. L., Luo, Y., Heat shock treatmentreduces beta amyloid toxicity in vivo by diminishingoligomers. Neurobiol. Aging 31, 1055–1058.
[16] Hsu, A. L., Murphy, C. T., Kenyon, C., Regulation of agingand age-related disease by DAF-16 and heat-shock factor.Science 2003, 300, 1142–1145.
[17] Wu,Y.,Wu, Z., Butko, P., Christen,Y. et al.,Amyloid-beta-in-duced pathological behaviors are suppressed by Ginkgobiloba extract EGb 761 and ginkgolides in transgenicCaenorhabditis elegans. J. Neurosci. 2006, 26, 13102–13113.
[18] Hassan,W. M., Merin, D.A., Fonte,V., Link, C. D.,AIP-1 ame-liorates beta-amyloid peptide toxicity in a Caenorhabditiselegans Alzheimer’s disease model. Hum. Mol. Genet. 2009,18, 2739–2747.
[19] Yun, C., Stanhill, A., Yang, Y., Zhang, Y. et al., Proteasomaladaptation to environmental stress links resistance to pro-teotoxicity with longevity in Caenorhabditis elegans. Proc.Natl. Acad. Sci. USA 2008, 105, 7094–7099.
[20] Diomede, L., Cassata, G., Fiordaliso, F., Salio, M. et al.,Tetra-cycline and its analogues protect Caenorhabditis elegansfrom beta amyloid-induced toxicity by targeting oligomers.Neurobiol. Dis. 2010, 40, 424–431.
[21] Link, C. D., Invertebrate models of Alzheimer’s disease.Genes Brain Behav. 2005, 4, 147–156.
[22] Goedert, M., Jakes, R., Mutations causing neurodegenera-tive tauopathies. Biochim. Biophys. Acta 2005, 1739,240–250.
[23] Iqbal, K.,Alonso Adel, C., Chen, S., Chohan, M. O. et al.,Taupathology in Alzheimer disease and other tauopathies.Biochim. Biophys. Acta 2005, 1739, 198–210.
[24] Lee,V. M., Goedert, M., Trojanowski, J. Q., Neurodegenera-tive tauopathies. Annu. Rev. Neurosci. 2001, 24, 1121–1159.
[25] Brandt, R., Gergou, A., Wacker, I., Fath, T., Hutter, H., ACaenorhabditis elegans model of tau hyperphosphoryla-tion: induction of developmental defects by transgenicoverexpression of Alzheimer’s disease-like modified tau.Neurobiol. Aging 2009, 30, 22–33.
[26] Kraemer, B. C., Burgess, J. K., Chen, J. H., Thomas, J. H.,Schellenberg, G. D., Molecular pathways that influence hu-man tau-induced pathology in Caenorhabditis elegans.Hum. Mol. Genet. 2006, 15, 1483–1496.
[27] Kraemer, B. C., Schellenberg, G. D., SUT-1 enables tau-in-duced neurotoxicity in C. elegans. Hum. Mol. Genet. 2007,16, 1959–1971.
[28] Guthrie, C. R., Schellenberg, G. D., Kraemer, B. C., SUT-2potentiates tau-induced neurotoxicity in Caenorhabditiselegans. Hum. Mol. Genet. 2009, 18, 1825–1838.
[29] Orr, H. T., Beyond the Qs in the polyglutamine diseases.Genes Dev. 2001, 15, 925–932.
[30] Ross, C.A., Polyglutamine pathogenesis: emergence of uni-fying mechanisms for Huntington’s disease and relateddisorders. Neuron 2002, 35, 819–822.
[31] Orr, H. T., Zoghbi, H. Y., Trinucleotide repeat disorders.Annu. Rev. Neurosci. 2007, 30, 575–621.
Biotechnol. J. 2010, 5, 1261–1276 www.biotechnology-journal.com
Nektarios Tavernarakis is a Research
Director at the Institute of Molecular
Biology and Biotechnology, and Profes-
sor of Molecular Systems Biology at the
Medical School of the University of
Crete in Heraklion, Greece. He earned
his PhD at the University of Crete,
studying gene expression in yeast, and
trained in Caenorhabditis elegans biolo-
gy at Rutgers University, New Jersey,
USA. His research focuses on neuronal function and dysfunction. His
main interests are the molecular mechanisms of necrotic cell death
and neurodegeneration, the molecular mechanisms of sensory trans-
duction and integration by the nervous system, the interplay between
cellular metabolism and ageing, and the development of novel genetic
tools for C. elegans research. He is the recipient of a European Re-
search Council (ERC) Advanced Investigator grant award, a European
Molecular Biology Organization (EMBO) Young Investigator award,
the Alexander von Humboldt Foundation, Friedrich Wilhelm Bessel re-
search award, and is member of EMBO.
Maria Markaki is a staff scientist in the
Caenorhabditis elegans molecular genet-
ics laboratory at the Institute of Molec-
ular Biology and Biotechnology, in Her-
aklion, Crete, Greece. She earned her
PhD degree at the University of Crete
studying the ecophysiology of Oxalis
pes-caprae L. Her main interests are the
molecular mechanisms that underlie
the effects of autophagy on ageing.

BiotechnologyJournal Biotechnol. J. 2010, 5, 1261–1276
1274 © 2010 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim
[32] Morley, J. F., Brignull, H. R.,Weyers, J. J., Morimoto, R. I.,Thethreshold for polyglutamine-expansion protein aggrega-tion and cellular toxicity is dynamic and influenced by ag-ing in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA2002, 99, 10417–10422.
[33] Brignull, H. R., Moore, F. E., Tang, S. J., Morimoto, R. I.,Polyglutamine proteins at the pathogenic threshold dis-play neuron-specific aggregation in a pan-neuronalCaenorhabditis elegans model. J. Neurosci. 2006, 26, 7597–7606.
[34] Gidalevitz, T., Ben-Zvi, A., Ho, K. H., Brignull, H. R., Mori-moto, R. I., Progressive disruption of cellular protein fold-ing in models of polyglutamine diseases. Science 2006, 311,1471–1474.
[35] Guarente, L., Kenyon, C., Genetic pathways that regulateageing in model organisms. Nature 2000, 408, 255–262.
[36] Caldwell, G. A., Cao, S., Sexton, E. G., Gelwix, C. C. et al.,Suppression of polyglutamine-induced protein aggrega-tion in Caenorhabditis elegans by torsin proteins. Hum.Mol. Genet. 2003, 12, 307–319.
[37] Walters, K. J., Kleijnen, M. F., Goh, A. M., Wagner, G., How-ley, P. M., Structural studies of the interaction betweenubiquitin family proteins and proteasome subunit S5a.Biochemistry 2002, 41, 1767–1777.
[38] Wang, H., Lim, P. J.,Yin, C., Rieckher, M. et al., Suppressionof polyglutamine-induced toxicity in cell and animal mod-els of Huntington’s disease by ubiquilin. Hum. Mol. Genet.2006, 15, 1025–1041.
[39] Wang, H., Lim, P. J., Karbowski, M., Monteiro, M. J., Effectsof overexpression of huntingtin proteins on mitochondri-al integrity. Hum. Mol. Genet. 2009, 18, 737–752.
[40] Link, C. D., Expression of human beta-amyloid peptide intransgenic Caenorhabditis elegans. Proc. Natl. Acad. Sci.USA 1995, 92, 9368–9372.
[41] Steinkraus, K. A., Smith, E. D., Davis, C., Carr, D. et al., Di-etary restriction suppresses proteotoxicity and enhanceslongevity by an hsf-1-dependent mechanism in Caenor-habditis elegans. Aging Cell 2008, 7, 394–404.
[42] Vila, M., Przedborski, S., Genetic clues to the pathogenesisof Parkinson’s disease. Nat. Med. 2004, 10 Suppl, S58–62.
[43] Polymeropoulos, M. H., Lavedan, C., Leroy, E., Ide, S. E. etal., Mutation in the alpha-synuclein gene identified infamilies with Parkinson’s disease. Science 1997, 276, 2045–2047.
[44] Singleton, A. B., Farrer, M., Johnson, J., Singleton, A. et al.,Alpha-synuclein locus triplication causes Parkinson’s dis-ease. Science 2003, 302, 841.
[45] Chartier-Harlin, M. C., Kachergus, J., Roumier, C., Mouroux,V. et al.,Alpha-synuclein locus duplication as a cause of fa-milial Parkinson’s disease. Lancet 2004, 364, 1167–1169.
[46] Dauer, W., Przedborski, S., Parkinson’s disease: mecha-nisms and models. Neuron 2003, 39, 889–909.
[47] Cooper, A. A., Gitler, A. D., Cashikar, A., Haynes, C. M. et al.,Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescuesneuron loss in Parkinson’s models. Science 2006, 313,324–328.
[48] Hamamichi, S., Rivas, R. N., Knight, A. L., Cao, S. et al., Hy-pothesis-based RNAi screening identifies neuroprotectivegenes in a Parkinson’s disease model. Proc. Natl. Acad. Sci.USA 2008, 105, 728–733.
[49] Shashidharan, P., Good, P. F., Hsu, A., Perl, D. P. et al.,Torsi-nA accumulation in Lewy bodies in sporadic Parkinson’sdisease. Brain Res. 2000, 877, 379–381.
[50] van Ham,T. J.,Thijssen, K. L., Breitling, R., Hofstra, R. M. etal., C. elegans model identifies genetic modifiers of alpha-synuclein inclusion formation during aging. PLoS Genet.2008, 4, e1000027.
[51] Paisan-Ruiz, C., Jain, S., Evans, E. W., Gilks, W. P. et al.,Cloning of the gene containing mutations that causePARK8-linked Parkinson’s disease. Neuron 2004, 44, 595–600.
[52] Zimprich,A., Biskup, S., Leitner, P., Lichtner, P. et al., Muta-tions in LRRK2 cause autosomal-dominant parkinsonismwith pleomorphic pathology. Neuron 2004, 44, 601–607.
[53] Saha, S., Guillily, M. D., Ferree, A., Lanceta, J. et al., LRRK2modulates vulnerability to mitochondrial dysfunction inCaenorhabditis elegans. J. Neurosci. 2009, 29, 9210–9218.
[54] West, A. B., Moore, D. J., Biskup, S., Bugayenko, A. et al.,Parkinson’s disease-associated mutations in leucine-richrepeat kinase 2 augment kinase activity. Proc. Natl. Acad.Sci. USA 2005, 102, 16842–16847.
[55] Gloeckner, C. J., Kinkl, N., Schumacher,A., Braun, R. J. et al.,The Parkinson disease causing LRRK2 mutation I2020T isassociated with increased kinase activity. Hum. Mol. Genet.2006, 15, 223–232.
[56] Yao, C., El Khoury, R., Wang, W., Byrd, T. A. et al., LRRK2-mediated neurodegeneration and dysfunction ofdopaminergic neurons in a Caenorhabditis elegans modelof Parkinson’s disease. Neurobiol. Dis. 2010, 40, 73–81.
[57] Mulder, D. W., Kurland, L. T., Offord, K. P., Beard, C. M., Fa-milial adult motor neuron disease: amyotrophic lateralsclerosis. Neurology 1986, 36, 511–517.
[58] Cudkowicz, M. E., McKenna-Yasek, D., Sapp, P. E., Chin,W.et al., Epidemiology of mutations in superoxide dismutasein amyotrophic lateral sclerosis. Ann. Neurol. 1997, 41,210–221.
[59] Hart, P. J., Pathogenic superoxide dismutase structure, fold-ing, aggregation and turnover. Curr. Opin. Chem. Biol. 2006,10, 131–138.
[60] Oeda,T., Shimohama, S., Kitagawa, N., Kohno, R. et al., Ox-idative stress causes abnormal accumulation of familialamyotrophic lateral sclerosis-related mutant SOD1 intransgenic Caenorhabditis elegans. Hum. Mol. Genet. 2001,10, 2013–2023.
[61] Wang, J., Farr, G. W., Hall, D. H., Li, F. et al., An ALS-linkedmutant SOD1 produces a locomotor defect associated withaggregation and synaptic dysfunction when expressed inneurons of Caenorhabditis elegans. PLoS Genet. 2009, 5,e1000350.
[62] Gidalevitz, T., Krupinski, T., Garcia, S., Morimoto, R. I.,Destabilizing protein polymorphisms in the genetic back-ground direct phenotypic expression of mutant SOD1 tox-icity. PLoS Genet. 2009, 5, e1000399.
[63] Nishimura,A. L., Mitne-Neto, M., Silva, H. C., Richieri-Cos-ta, A. et al., A mutation in the vesicle-trafficking proteinVAPB causes late-onset spinal muscular atrophy and amy-otrophic lateral sclerosis. Am. J. Hum. Genet. 2004, 75, 822–831.
[64] Soussan, L., Burakov, D., Daniels, M. P., Toister-Achituv, M.et al., ERG30, a VAP-33-related protein, functions in pro-tein transport mediated by COPI vesicles. J. Cell Biol. 1999,146, 301–311.
[65] Skehel, P. A., Fabian-Fine, R., Kandel, E. R., Mouse VAP33is associated with the endoplasmic reticulum and micro-tubules. Proc. Natl. Acad. Sci. USA 2000, 97, 1101–1106.

© 2010 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim 1275
[66] Kaiser, S. E., Brickner, J. H., Reilein, A. R., Fenn, T. D. et al.,Structural basis of FFAT motif-mediated ER targeting.Structure 2005, 13, 1035–1045.
[67] Nishimura,Y., Hayashi, M., Inada, H.,Tanaka,T., Molecularcloning and characterization of mammalian homologuesof vesicle-associated membrane protein-associated(VAMP-associated) proteins. Biochem. Biophys. Res. Com-mun. 1999, 254, 21–26.
[68] Bottino, D., Mogilner, A., Roberts, T., Stewart, M., Oster, G.,How nematode sperm crawl. J. Cell Sci. 2002, 115, 367–384.
[69] Miller, M. A., Nguyen,V. Q., Lee, M. H., Kosinski, M. et al., Asperm cytoskeletal protein that signals oocyte meioticmaturation and ovulation. Science 2001, 291, 2144–2147.
[70] Miller, M. A., Ruest, P. J., Kosinski, M., Hanks, S. K., Green-stein, D., An Eph receptor sperm-sensing control mecha-nism for oocyte meiotic maturation in Caenorhabditis ele-gans. Genes Dev. 2003, 17, 187–200.
[71] Tsuda, H., Han, S. M., Yang, Y., Tong, C. et al., The amy-otrophic lateral sclerosis 8 protein VAPB is cleaved, se-creted, and acts as a ligand for Eph receptors. Cell 2008,133, 963–977.
[72] Ogino, S.,Wilson, R. B., Genetic testing and risk assessmentfor spinal muscular atrophy (SMA). Hum. Genet. 2002, 111,477–500.
[73] Lefebvre, S., Burglen, L., Reboullet, S., Clermont, O. et al.,Identification and characterization of a spinal muscularatrophy-determining gene. Cell 1995, 80, 155–165.
[74] Briese, M., Esmaeili, B., Sattelle, D. B., Is spinal muscularatrophy the result of defects in motor neuron processes?Bioessays 2005, 27, 946–957.
[75] Miguel-Aliaga, I., Culetto, E., Walker, D. S., Baylis, H. A. etal., The Caenorhabditis elegans orthologue of the humangene responsible for spinal muscular atrophy is a mater-nal product critical for germline maturation and embryon-ic viability. Hum. Mol. Genet. 1999, 8, 2133–2143.
[76] Briese, M., Esmaeili, B., Fraboulet, S., Burt, E. C. et al., Dele-tion of smn-1, the Caenorhabditis elegans ortholog of thespinal muscular atrophy gene, results in locomotor dys-function and reduced lifespan. Hum. Mol. Genet. 2009, 18,97–104.
[77] Dimitriadi, M., Hart, A. C., Neurodegenerative disorders:Insights from the nematode Caenorhabditis elegans. Neu-robiol. Dis. 2010, 40, 4–11.
[78] Syntichaki, P., Tavernarakis, N., Death by necrosis. Uncon-trollable catastrophe, or is there order behind the chaos?EMBO Rep. 2002, 3, 604–609.
[79] Scott, B.A.,Avidan, M. S., Crowder, C. M., Regulation of hy-poxic death in C. elegans by the insulin/IGF receptor ho-molog DAF-2. Science 2002, 296, 2388–2391.
[80] Syntichaki, P., Tavernarakis, N., The biochemistry of neu-ronal necrosis: rogue biology? Nat. Rev. Neurosci. 2003, 4,672–684.
[81] Syntichaki, P., Xu, K., Driscoll, M.,Tavernarakis, N., Specif-ic aspartyl and calpain proteases are required for neu-rodegeneration in C. elegans. Nature 2002, 419, 939–944.
[82] Xu,W.,Wong,T. P., Chery, N., Gaertner,T. et al., Calpain-me-diated mGluR1alpha truncation: a key step in excitotoxic-ity. Neuron 2007, 53, 399–412.
[83] Syntichaki, P., Samara, C., Tavernarakis, N., The vacuolarH+ -ATPase mediates intracellular acidification requiredfor neurodegeneration in C. elegans. Curr. Biol. 2005, 15,1249–1254.
[84] Artal-Sanz, M., Samara, C., Syntichaki, P.,Tavernarakis, N.,Lysosomal biogenesis and function is critical for necrotic
cell death in Caenorhabditis elegans. J. Cell Biol. 2006, 173,231–239.
[85] Ervasti, J. M., Campbell, K. P., Membrane organization ofthe dystrophin-glycoprotein complex. Cell 1991, 66,1121–1131.
[86] Ervasti, J. M., Campbell, K. P.,A role for the dystrophin-gly-coprotein complex as a transmembrane linker betweenlaminin and actin. J. Cell Biol. 1993, 122, 809–823.
[87] Gieseler, K., Bessou, C., Segalat, L., Dystrobrevin- and dy-strophin-like mutants display similar phenotypes in thenematode Caenorhabditis elegans. Neurogenetics 1999, 2,87–90.
[88] Kim, H., Rogers, M. J., Richmond, J. E., McIntire, S. L., SNF-6 is an acetylcholine transporter interacting with the dys-trophin complex in Caenorhabditis elegans. Nature 2004,430, 891–896.
[89] Kim, H., Pierce-Shimomura, J.T., Oh, H. J., Johnson, B. E. etal., The dystrophin complex controls bk channel localiza-tion and muscle activity in Caenorhabditis elegans. PLoSGenet. 2009, 5, e1000780.
[90] Bulfield, G., Siller, W. G., Wight, P. A., Moore, K. J., X chro-mosome-linked muscular dystrophy (mdx) in the mouse.Proc. Natl. Acad. Sci. USA 1984, 81, 1189–1192.
[91] Ballinger, C. A., Connell, P., Wu, Y., Hu, Z. et al., Identifica-tion of CHIP, a novel tetratricopeptide repeat-containingprotein that interacts with heat shock proteins and nega-tively regulates chaperone functions. Mol. Cell. Biol. 1999,19, 4535–4545.
[92] Nyamsuren, O., Faggionato, D., Loch, W., Schulze, E.,Baumeister, R., A mutation in CHN-1/CHIP suppressesmuscle degeneration in Caenorhabditis elegans. Dev. Biol.2007, 312, 193–202.
[93] Brais, B., Oculopharyngeal muscular dystrophy: a late-on-set polyalanine disease. Cytogenet. Genome Res. 2003, 100,252–260.
[94] Wang, Q., Mosser, D. D., Bag, J., Induction of HSP70 expres-sion and recruitment of HSC70 and HSP70 in the nucleusreduce aggregation of a polyalanine expansion mutant ofPABPN1 in HeLa cells. Hum. Mol. Genet. 2005, 14, 3673–3684.
[95] Klein, A. F., Ebihara, M., Alexander, C., Dicaire, M. J. et al.,PABPN1 polyalanine tract deletion and long expansionsmodify its aggregation pattern and expression. Exp. CellRes. 2008, 314, 1652–1666.
[96] Abu-Baker, A., Messaed, C., Laganiere, J., Gaspar, C. et al.,Involvement of the ubiquitin-proteasome pathway andmolecular chaperones in oculopharyngeal muscular dys-trophy. Hum. Mol. Genet. 2003, 12, 2609–2623.
[97] Catoire, H., Pasco, M.Y.,Abu-Baker,A., Holbert, S. et al., Sir-tuin inhibition protects from the polyalanine musculardystrophy protein PABPN1. Hum. Mol. Genet. 2008, 17,2108–2117.
[98] Di Renzo, M. F., Olivero, M., Katsaros, D., Crepaldi, T. et al.,Overexpression of the Met/HGF receptor in ovarian can-cer. Int. J. Cancer 1994, 58, 658–662.
[99] Di Renzo, M. F., Olivero, M., Giacomini, A., Porte, H. et al.,Overexpression and amplification of the met/HGF recep-tor gene during the progression of colorectal cancer. Clin.Cancer Res. 1995, 1, 147–154.
[100] Ferracini, R., Di Renzo, M. F., Scotlandi, K., Baldini, N. et al.,The Met/HGF receptor is over-expressed in human os-teosarcomas and is activated by either a paracrine or anautocrine circuit. Oncogene 1995, 10, 739–749.
Biotechnol. J. 2010, 5, 1261–1276 www.biotechnology-journal.com

BiotechnologyJournal Biotechnol. J. 2010, 5, 1261–1276
1276 © 2010 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim
[101] Hellman,A., Zlotorynski, E., Scherer, S.W., Cheung, J. et al.,A role for common fragile site induction in amplification ofhuman oncogenes. Cancer Cell 2002, 1, 89–97.
[102] Maulik, G., Kijima, T., Ma, P. C., Ghosh, S. K. et al., Modula-tion of the c-Met/hepatocyte growth factor pathway insmall cell lung cancer. Clin. Cancer Res. 2002, 8, 620–627.
[103] Ma, P. C., Kijima,T., Maulik, G., Fox, E. A. et al., c-MET mu-tational analysis in small cell lung cancer: novel jux-tamembrane domain mutations regulating cytoskeletalfunctions. Cancer Res. 2003, 63, 6272–6281.
[104] Ma, P. C., Jagadeeswaran, R., Jagadeesh, S.,Tretiakova, M. S.et al., Functional expression and mutations of c-Met andits therapeutic inhibition with SU11274 and small inter-fering RNA in non-small cell lung cancer. Cancer Res. 2005,65, 1479–1488.
[105] Siddiqui, S. S., Loganathan, S., Krishnaswamy, S., Faoro, L.et al., C. elegans as a model organism for in vivo screeningin cancer: effects of human c-Met in lung cancer affect C.elegans vulva phenotypes. Cancer Biol. Ther. 2008, 7, 856–863.
[106] Tan, P. B., Kim, S. K., Signaling specificity: the RTK/RAS/MAP kinase pathway in metazoans. Trends Genet. 1999, 15,145–149.
[107] Moghal, N., Sternberg, P. W., The epidermal growth factorsystem in Caenorhabditis elegans. Exp. Cell Res. 2003, 284,150–159.
[108] Nilsen, T. W., Mechanisms of microRNA-mediated generegulation in animal cells. Trends Genet. 2007, 23, 243–249.
[109] Sundaram, M. V., The love-hate relationship between Rasand Notch. Genes Dev. 2005, 19, 1825–1839.
[110] Johnson, S. M., Grosshans, H., Shingara, J., Byrom, M. et al.,RAS is regulated by the let-7 microRNA family. Cell 2005,120, 635–647.
[111] Yoo, A. S., Greenwald, I., LIN-12/Notch activation leads tomicroRNA-mediated down-regulation of Vav in C. elegans.Science 2005, 310, 1330–1333.
[112] Esquela-Kerscher,A., Johnson, S. M., Bai, L., Saito, K. et al.,Post-embryonic expression of C. elegans microRNAs be-longing to the lin-4 and let-7 families in the hypodermisand the reproductive system. Dev. Dyn. 2005, 234, 868–877.
[113] Tybulewicz, V. L., Vav-family proteins in T-cell signalling.Curr. Opin. Immunol. 2005, 17, 267–274.
[114] Johnstone, R.W., Ruefli, A. A., Lowe, S.W., Apoptosis: a linkbetween cancer genetics and chemotherapy. Cell 2002, 108,153–164.
[115] Ellis, H. M., Horvitz, H. R., Genetic control of programmedcell death in the nematode C. elegans. Cell 1986, 44, 817–829.
[116] van Haaften, G., Romeijn, R., Pothof, J., Koole, W. et al.,Identification of conserved pathways of DNA-damage re-
sponse and radiation protection by genome-wide RNAi.Curr. Biol. 2006, 16, 1344–1350.
[117] Sendoel, A., Kohler, I., Fellmann, C., Lowe, S. W., Hengart-ner, M. O., HIF-1 antagonizes p53-mediated apoptosisthrough a secreted neuronal tyrosinase. Nature 2010, 465,577–583.
[118] Hanover, J. A., Krause, M. W., Love, D. C., The hexosaminesignaling pathway: O-GlcNAc cycling in feast or famine.Biochim. Biophys. Acta 1800, 80–95.
[119] Hanover, J.A., Forsythe, M. E., Hennessey, P.T., Brodigan,T.M. et al., A Caenorhabditis elegans model of insulin resist-ance: altered macronutrient storage and dauer formationin an OGT-1 knockout. Proc. Natl. Acad. Sci. USA 2005, 102,11266–11271.
[120] Forsythe, M. E., Love, D. C., Lazarus, B. D., Kim, E. J. et al.,Caenorhabditis elegans ortholog of a diabetes susceptibili-ty locus: oga-1 (O-GlcNAcase) knockout impacts O-Glc-NAc cycling, metabolism, and dauer. Proc. Natl. Acad. Sci.USA 2006, 103, 11952–11957.
[121] Hanover, J. A., Love, D. C., Prinz, W. A., Calmodulin-drivennuclear entry: trigger for sex determination and terminaldifferentiation. J. Biol. Chem. 2009, 284, 12593–12597.
[122] Sinclair, D. A., Syrzycka, M., Macauley, M. S., Rastgardani,T. et al., Drosophila O-GlcNAc transferase (OGT) is encod-ed by the Polycomb group (PcG) gene, super sex combs(sxc). Proc. Natl. Acad. Sci. USA 2009, 106, 13427–13432.
[123] Gambetta, M. C., Oktaba, K., Muller, J., Essential role of theglycosyltransferase sxc/Ogt in polycomb repression. Sci-ence 2009, 325, 93–96.
[124] Love, D. C., Ghosh, S., Mondoux, M. A., Fukushige, T. et al.,Dynamic O-GlcNAc cycling at promoters of Caenorhabdi-tis elegans genes regulating longevity, stress, and immuni-ty. Proc. Natl. Acad. Sci. USA 107, 7413–7418.
[125] Porte, D. Jr., Baskin, D. G., Schwartz, M. W., Insulin signal-ing in the central nervous system: a critical role in meta-bolic homeostasis and disease from C. elegans to humans.Diabetes 2005, 54, 1264–1276.
[126] Schlotterer, A., Kukudov, G., Bozorgmehr, F., Hutter, H. etal., C. elegans as model for the study of high glucose- me-diated life span reduction. Diabetes 2009, 58, 2450–2456.
[127] Pandey, A. K., Agarwal, P., Kaur, K., Datta, M., MicroRNAsin diabetes: tiny players in big disease. Cell Physiol. Bio-chem. 2009, 23, 221–232.
[128] Pasco, M. Y., Catoire, H., Parker, J. A., Brais, B. et al., Cross-talk between canonical Wnt signaling and the sirtuin-FoxO longevity pathway to protect against muscularpathology induced by mutant PABPN1 expression in C. el-egans. Neurobiol. Dis. 38, 425–433.





























