Embed Size (px)
Citation preview

ICAM-1-dependent pathways regulate colonic eosinophilicinflammation
Elizabeth Forbes,* Mark Hulett,† Richard Ahrens,‡ Norbert Wagner,§ Vanessa Smart,*Klaus I. Matthaei,¶ Eric B. Brandt,‡ Lindsay A. Dent,�� Marc E. Rothenberg,‡ Mimi Tang,**Paul. S. Foster,*,†† and Simon P. Hogan‡,1
*Allergy and Inflammation Research Group and ¶Gene Targeting Group, Division of Biochemistry and MolecularBiology, and †Cancer and Vascular Biology Group, Division of Immunology and Genetics, The John Curtin Schoolof Medical Research, Australian National University, Canberra; ††Asthma, Allergy and Inflammation ResearchCentre, School of Biomedical Sciences, University of Newcastle, Australia; ��School of Molecular and BiomedicalScience, University of Adelaide, Australia; **Department of Immunology, Murdoch Childrens Research Institute,Royal Children’s Hospital, Victoria, Australia; §Department of Pediatrics, City Hospital of Dortmund, Germany;and ‡Division of Allergy and Immunology, Department of Pediatrics, Cincinnati Children’s Hospital Medical Center,University of Cincinnati College of Medicine, Ohio
Abstract: Eosinophilic inflammation is a commonfeature of numerous eosinophil-associated gastro-intestinal (GI) diseases. Central to eosinophil mi-gration into the GI tract are the integrin-mediatedinteractions with adhesion molecules. Although themechanisms regulating eosinophil homing into thesmall intestine have begun to be elucidated, theadhesion pathways responsible for eosinophil traf-ficking into the large intestine are unknown. Weinvestigated the role of adhesion pathways in eosin-ophil recruitment into the large intestine duringhomeostasis and disease. First, using a hapten-in-duced colonic injury model, we demonstrate that incontrast to the small intestine, eosinophil recruit-ment into the colon is regulated by a �4�7/mucosaladdressin cell adhesion molecule-1-independentpathway. Characterization of integrin expressionon eosinophils by flow cytometry analysis revealedthat colonic CC chemokine receptor 3� eosino-phils express the intercellular adhesion molecule-1(ICAM-1) counter-receptor integrins �L, �M, and�2. Using ICAM-1-deficient mice and anti-ICAM-1neutralizing antibodies, we show that hapten-in-duced colonic eosinophilic inflammation is criti-cally dependent on ICAM-1. These studies demon-strate that �2-integrin/ICAM-1-dependent path-ways are integral to eosinophil recruitment into thecolon during GI inflammation associated with co-lonic injury. J. Leukoc. Biol. 80: 000–000; 2006.
Key Words: eosinophils � adhesion molecules � gastrointestinal tract
INTRODUCTION
Eosinophil accumulation in the gastrointestinal (GI) tract is acommon feature of numerous eosinophil-associated GI disease(EGID) disorders including food allergy, eosinophilic esoph-agitis (EE), eosinophilic gastroenteritis, allergic colitis, and
inflammatory bowel disease {IBD; ulcerative colitis (UC) andCrohn’s disease [1, 2]}. Although the underlying causes ofEGID are not understood fully, clinical investigations suggestan important role for eosinophils in the etiology of disease.Indeed, a strong correlation has been demonstrated amongclinical symptoms, disease severity, and increased numbers ofthis cell type in the GI tract [1–3]. Furthermore, recentexperimental investigations have provided corroborative evi-dence supporting a role for eosinophils in the pathogenesis ofEGID [4].
Numerous inflammatory mediators have been implicated inregulating eosinophil accumulation, including interleukin(IL)-1, -3, -4, -5, and -13 and granulocyte macrophage-colonystimulating factor and the chemokines regulated on activation,normal T expressed and secreted (RANTES), monocyte che-moattractant protein (MCP)-3, macrophage inflammatory pro-tein-1�, and eotaxin-1, -2, and -3 [5, 6]. Of the mediatorsimplicated in modulating eosinophil accumulation, eotaxin-1appears to be the most important molecule in the regulation ofeosinophil trafficking in the GI tract [7]. Eotaxin-1 is ubiqui-tously expressed in all segments of the GI tract [8]. Further-more, eosinophil levels in the GI tract are reduced significantlyin eotaxin-1-deficient mice as compared with wild-type (WT)mice, and overexpression of eotaxin-1 in the GI tract promotespronounced eosinophilia in the small intestine [9, 10]. Clinicalstudies have demonstrated an association between in-creased expression of CC chemokine ligand 11 (eotaxin-1) inEGID, including cows milk-associated reflux esophagitis andUC [11–15].
The transmigration of leukocytes, such as eosinophils,across the vascular epithelium into mucosal tissues, is regu-lated by coordinated interaction among networks involving
1 Correspondence: Division of Allergy and Immunology, Department ofPediatrics, Cincinnati Children’s Hospital Medical Center, University of Cin-cinnati College of Medicine, Cincinnati, OH 45267. E-mail: [email protected]
Received November 9, 2005; revised March 14, 2006; accepted March 27,2006; doi: 10.1189/jlb.1105643.
0741-5400/06/0080-0001 © Society for Leukocyte Biology Journal of Leukocyte Biology Volume 80, August 2006 1
Uncorrected Version. Published on May 25, 2006 as DOI:10.1189/jlb.1105643
Copyright 2006 by The Society for Leukocyte Biology.

chemokine and cytokine signaling, eosinophil adhesion mole-cules (e.g., selectins and integrins), and integrin receptors[e.g., vascular cell adhesion molecule-1 (VCAM-1), mucosaladdressin cell adhesion molecule-1 (MAdCAM-1), and inter-cellular adhesion molecule 1 (ICAM-1)] expressed on vascularendothelial cells [16, 17]. Integrins are heterodimeric surfacemolecules consisting of an �- and �-chain, and eosinophilsexpress members of the �1 (�4�1 and �6�1)-, �2 (�L�2, �M�2,�X�2, and �D�2)-, and �7 (�4�7)-integrin families [18–22].These various integrin molecules interact selectively with ad-hesion receptors (VCAM-1, MAdCAM-1, ICAM-1, -2, and -3,and fibrinogen) expressed on the vascular endothelium. �4�1
selectively binds to VCAM-1 and fibronectin, and �6�1 is theprimary ligand for the extracellular matrix protein laminin. The�4�7-integrin selectively binds to MAdCAM-1, and lymopho-cyte function-associated antigen-1 (LFA-1; �L�2) and mem-brane-activated complex-1 (MAC-1; �M�2) bind to ICAM-1. Inaddition, LFA-1 (�L�2) can bind ICAM-2 and -3, and �D�2
has been shown to bind VCAM-1 and ICAM-3. Leukocyteintegrin/intercellular adhesion molecule interactions, particu-larly LFA-1/ICAM-1 and very late antigen (VLA)-4/VCAM-1,are regulated by cytokines and chemokines such as IL-1 (� and�), tumor necrosis fator (TNF; � and �), IL-4, and IL-13 [23].IL-1 and TNF stimulate VCAM-1 and ICAM-1 expression on avariety of cell types. By contrast, IL-4 and IL-13 differentiallyenhance the expression of VCAM-1, having little impact onICAM-1. Chemokines alter the activation state and selectivityof adhesion molecules [24, 25]. For example, treatment ofeosinophils with MCP-3, RANTES, or eotaxin-2 switches eo-sinophils from a �1-integrin/VCAM-1-dominant to a �2-inte-grin/ICAM-1-dominant, interacting cell [24].
The specific interaction of cell surface integrins with adhe-sion receptors (VCAM-1, MAdCAM-1, ICAM-1, ICAM-2,ICAM-3, and fibrinogen) facilitates eosinophil migration intovarious tissue compartments during inflammation. For exam-ple, eosinophil recruitment to the site of allergic inflammationin the lung and skin is regulated by a VLA-4 (�4�1-integrin)/VCAM-1-dependent processes [26–30]. Pretreatment of micewith neutralizing monoclonal antibodies (mAb) against �4- or�1-integrin or genetic deletion of VCAM-1 attenuates eosino-phil accumulation in the lung during allergic airways disease[26–30]. In contrast, eotaxin-1-dependent eosinophil recruit-ment to the small intestine of the GI tract is MAdCAM-1/�4�7-integrin-dependent [10].
Recruitment of inflammatory cells into the GI tract is cur-rently believed to occur via an �4�7/MAdCAM-1-dominantinteraction [16]. The �4�7-integrin receptor MAdCAM-1 isexpressed primarily on GI vascular endothelium and is gener-ally absent on nonintestinal venules and most non-GI sites ofinflammation [31]. Blockade of �4�7/MAdCAM-1 interactionsby neutralizing mAb or genetic deletion inhibits T and B celland mast cell recruitment into GI compartments including thesmall intestine, mesenteric lymph nodes, and Peyer’s patches.It is interesting that recent experimental investigations havedemonstrated that leukocyte recruitment into the large intes-tine can occur via a �7-integrin-independent mechanism, sug-gesting that leukocytes use different adhesion systems to infil-trate various GI compartments [32, 33]. We were thereforeinterested in identifying the dominant adhesion complex in-
volved in eosinophil trafficking into the large intestine. This isparticularly important, as there are numerous diseases charac-terized by eosinophil accumulation in the colon (e.g., allergiccolitis and IBD), yet most studies have concentrated on theupper GI tract (e.g., esophagus and small intestine).
In the present study, we demonstrate that colonic eosino-phils express the ICAM-1 ligands [MAC-1 and LFA-1 (�2, �L,and �M)]. Furthermore, using in vivo models of colonic eosin-ophil inflammation, we demonstrate that eosinophil accumula-tion in the colon is regulated by a �2-integrin pathway(ICAM-1) and can occur independently of �4- and �7-integrin-independent pathways. This observation has significant impli-cations for the treatment of disease states characterized bycolonic eosinophilic inflammation.
MATERIALS AND METHODS
MiceBALB/c and C57BL/6 mice (6–8 weeks of age) used in our experiments wereobtained from the Specific Pathogen Free Facility or the Gene TargetingFacility of the John Curtin School of Medical Research (Australian NationalUniversity, Canberra). ICAM-1�/� and L-selectin�/� mice (C57BL/6 back-ground) were kindly provided by Thomas F. Tedder (Duke University MedicalCenter, Durham, NC). CD2-IL-5 transgenic (Tg) �7
�/� mice were generated bycrossing CD2-IL-5Tg (BALB/c) mice into the �7
�/� (C57BL/6) background(kindly provided by Prof. N. Wagner, City Hospital of Dortmund, Germany).The Tg-positive offspring (N1) were subsequently mated with �7
�/� (C57BL/6)to generate CD2 IL-5Tg �7
�/� mice (N2). The (N2) CD2 IL-5Tg �7�/�
(BALB/c�C57BL/6) mice were crossed with �7�/� (C57BL/6) mice, generat-
ing chimeric Tg-positive and -negative �7�/� mice. To generate background-
control mice, the CD2-IL-5Tg mice were crossed with �7�/� (C57BL/6) mice,
and the Tg-positive offspring (N1) were subsequently back-crossed with WTC57BL/6 mice, generating background-matched CD2-IL-5Tg �7
�/� mice,which were treated according to the Australian National University AnimalWelfare guidelines and were housed in an approved containment facility.
Fluorescein-activated cell sorter (FACS) analysisCD2-IL-5Tg mice were killed, spleens from CD2-IL-5Tg mice were removed,and a single-cell suspension was prepared into single-cell suspensions asdescribed previously [34]. To analyze integrin expression on splenic andcolonic eosinophils, cells were incubated with phycoerythrin (PE)-conjugatedanti-�4�7 (Clone DATK32, BD PharMingen, San Diego, CA; 1 �g/106 cells),biotin-conjugated anti-�m (Clone M1/70 15.1, Chemicon Europe CBL, UK),biotin-conjugated anti-�L (Chemicon Europe CBL), PE-conjugated anti-�2-integrin (C71/16; BD PharMingen), Alexa Fluor 647 rat anti-mouse CC che-mokine receptor 3 (CCR3; 83103; BD PharMingen), or isotype-matched con-trol immunoglobulin (Ig; rat IgG biotin, Southern Biotechnology, Birmingham,AL) in phosphate-buffered saline (PBS)/1% fetal calf serum (FCS) on ice for 30min and washed twice in PBS/1% FCS. To visualize binding of the anti-integrin antibodies, cells were incubated with streptavidin-fluorescein isothio-cyanate (FITC; 1:1600 dilution) in PBS/1% FCS on ice for 30 min and washedtwice in PBS/1% FCS. Cells were analyzed by flow cytometry on a FACSad-vantage SE cell sorter (BD Biosciences, San Jose, CA). Eosinophils wereidentified by forward-scatter (FSC) versus side-scatter (SSC) and polarizinglight as described previously [35].
Induction of colonic injuryDextran sulfate sodium (DSS) used for the induction of experimental colitis(ICN Biomedical Inc., Aurora, OH) was supplied as the sodium salt with anaverage molecular weight of 41 kDA. It was used as a supplement in thedrinking water of the mice for 7 days as a 2.5% (w/v) solution.
Disease activity index (DAI)DAI was derived by scoring three major clinical signs (weight loss, diarrhea,and rectal bleeding) [36]. The clinical features were scored separately and then
2 Journal of Leukocyte Biology Volume 80, August 2006 http://www.jleukbio.org

correlated with a histological score. DAI � (body weight change) � (diarrheascore) � (rectal bleeding score).
Body weight
Changes in body weight were calculated as the difference between the pre-dicted body weight and the actual weight on a particular day. The formula forpredicted body weight was derived by simple regression using the body weightdata for the control group. The following formula was used: Y � a � kx, whereY � body weight change (loss or gain), k � daily increase in body weight, x �day, and a � starting body weight.
Diarrhea
The appearance of diarrhea was defined as mucus/fecal material adherent toanal fur. The presence or absence of diarrhea was scored as 1 or 0, respec-tively. The presence or absence of diarrhea was confirmed by examination ofthe colon following completion of the experiment [36]. Mice were killed, andthe colon was excised from the animal. Diarrhea was defined by the absence offecal pellet formation in the colon and the presence of continuous fluid fecalmaterial in the colon.
Rectal bleeding
The appearance of rectal bleeding was defined as diarrhea containing visibleblood or gross rectal bleeding and scored as described for diarrhea.
Histopathological examination
Animals were killed on Day 8, and the colon was excised. The length of thecolon was measured using digmatic calipers (Mitutoyo, Kawasaji, Japan).Tissue specimens were fixed in 4% paraformaldehyde and stained with hema-toxylin and eosin (H/E) and Masson’s trichrome using standard histologicaltechniques. The percentage of colon length with mucosal ulceration wasdetermined by performing morphometric analysis of colon using the ImagePro-Plus 4.5 software package (Media Cybernetics, Inc., Silver Spring, MD). Inbrief, digital images of longitudinal sections (1–2 cm in length) of H/E-stainedcolons were produced. Using the ImageProPlus 4.5 software, the length ofulcerated mucosal lining was divided by the total length of the colonic mucosalsurface, and the value was expressed as a percentage of colon length withmucosal ulceration.
Detection and quantification of eosinophils byimmunohistochemistry
The colon segment of the GI tract was immunostained with antiserum againstmouse major basic protein (MBP) as described previously [8]. Briefly, 5 �msections were quenched with H2O2, blocked with normal goat serum, andstained with a rabbit antimurine eosinophil MBP antiserum (kind gift fromNancy and James Lee, Mayo Clinic, Scottsdale, AZ). The slides were thenwashed and incubated with biotinylated goat anti-rabbit antibody and avidin-peroxidase complex (Vectastain ABC Peroxidase Elite kit, Vector Laborato-ries, Burlingame, CA). The slides were developed by nickel diaminobenzidineand enhanced cobalt chloride to form a black precipitate and counterstainedwith Nuclear Fast Red. Quantification of eosinophils was performed by count-ing the number of immunoreactive cells from 15–25 fields of view (magnifi-cation �40) from at least four to five random sections/mouse. Values wereexpressed as eosinophils per mm2 tissue.
Eosinophil peroxidase (EPO) activity assay
Mice were killed on Day 8, and the colon was excised and flushed with 1 mlPBS solution. The fecal material was vortexed vigorously for 5 min at 4°C andcentrifuged at 10,000 g for 10 min at 4°C. The supernatant was collected andplaced in a sterile Eppendorf tube and stored at –70°C until analysis. EPOactivity was measured in the supernatant of cell-free colon flushes as describedpreviously [37]. This assay is based on the oxidation of o-phenylenediamine(OPD) by EPO in the presence of H2O2. The EPO substrate solution consistedof 12 mM OPD (Sigma Aldrich, St. Louis, MO), 0.005% H2O2, 10 mM HEPES,and 0.22% cetyltrimethylammonium bromide (CTAB). Substrate solution (75�l) was added to cell-free supernatants, which were derived from colon flushes(75 �l) in a 96-well microplate and incubated at room temperature for 15 min
before stopping the reaction with 50 �l cold 8 N sulfuric acid. Absorbance wasmeasured at 490 nm. Standard EPO activity, 100 U/ml, was determined, asEPO activity produced by 1 � 106-purified eosinophils/�l supernatants.Eosinophils were purified from the spleen of CD2-IL-5Tg mice as describedpreviously.
mAb treatment
Mice were injected intraperitoneally (i.p.), daily, with rat anti-mouse integrin�4-chain [200 �l 1 mg/ml Clone PS/2 (IgG2b) mAb in saline], anti-mouseICAM-1 [200 �l 1 mg/ml Clone YN1/1.7.4 (IgG2b) mAb in saline], or rat IgGcontrol antibody (200 �l 1 mg/ml �GL113 mAb in saline) for 4 days. Threehours following the final i.p. injection, mice were killed, the jejunum and colonwere excised and fixed in 4% paraformaldehyde and stained for anti-MBP, andeosinophil levels were quantitiated as described above. In some experiments,mice were injected i.p. daily throughout the 8-day DSS treatment protocol withanti-mouse ICAM-1 [200 �l 1 mg/ml Clone YN1/1.7.4 (IgG2b) mAb in saline]or rat IgG control antibody (200 �l 1 mg/ml �GL113 mAb in saline).
FACS analysis on colonic eosinophils
Colonic injury was induced with 2.5% DSS as described above. On Day 8, thecolon segment of the GI tract was removed and flushed with 20 ml PBS. Thecolon tissue was cut into 1-cm segments and incubated in digestion buffercontaining 48 mg/ml Collagenase A, 24 U/ml Dispase II, and 2.5 mg/ml DNasein RPMI 1640 and incubated for 60 min at 30°C. The tissue was vortexedvigorously every 10 min for 15 s. Following the 60-min incubation, the cellaggregates were dissociated by pipetting and centrifuged at 1200 revolutionsper minute for 10 min at 4°C. The supernatant was decanted, and the cellpellet was resuspended in RPMI 1640 � 10% FCS and filtered through a70-�m filter. The single-cell suspension was prepared by Ficoll densitygradient centrifugation and quantitated by trypan blue exclusion analysis.Colonic cells (1�106) were plated in a 96-well round-bottom plate, and flowcytometry analysis staining for CCR3 and �2-integrin was performed as de-scribed above.
Myeloperoxidase (MPO) activity
MPO activity, a marker of polymorphonuclear neutrophil granules, was as-sessed in colonic luminal contents according to the Bradley method [38]. Micewere killed on Day 8, and the colon was excised and flushed with 1 ml PBSsolution. The fecal material was vortexed vigorously for 5 min at 4°C andcentrifuged at 10,000 g for 10 min at 4°C. The supernatant was collected andplaced in a sterile Eppendorf tube and stored at –70°C until analysis. MPOactivity was measured in the supernatant of cell-free colon flushes as describedpreviously [38]. This assay is based on the oxidation of o-dianisidine dihydro-chloride (oDd) by MPO in the presence of H2O2. The MPO substrate solutionconsisted of 0.167 mg/ml oDd (Sigma Aldrich), 0.005% H2O2, 10 mM HEPES,and 0.22% CTAB. Substrate solution (75 �l) was added to cell-free superna-tants, which were derived from colon flushes (75 �l) in a 96-well microplateand incubated at room temperature for 15 min before stopping the reaction with50 �l cold 3 M sulfuric acid. Absorbance was measured at 450 nm. StandardMPO (human leukocytes, Sigma Aldrich; 50 U/mg protein), 10 U/ml, was usedto generate a standard curve.
Statistical analysis
The significance of differences between the means of experimental groups wasanalyzed using Student’s unpaired t-test. Values were reported as the mean SEM. Differences in mean values were considered significant if P 0.05. Insome experiments, one-way and two-way ANOVA with Bonferroni post-testwere performed as indicated. All calculations were performed using SAS,Version 9.0 (SAS Institute, Cary, NC). To test the hypotheses, that the meanswere equal, one-factor or two-factor ANOVA was used, and the level ofsignificance was at 0.05. In the one-factor analyses, the comparison groupswere the treated groups. If the overall F were statistically significant, Bonfer-roni’s multiple comparison was applied to determine where the differenceswere. In the case of two factors, strain of mice and treatment, if the overall Fwere less than 0.05, Bonferroni’s adjustment was made, dividing the F by thenumber of comparisons made to adjust the P value.
Forbes et al. ICAM-1-mediated colonic eosinophilia 3
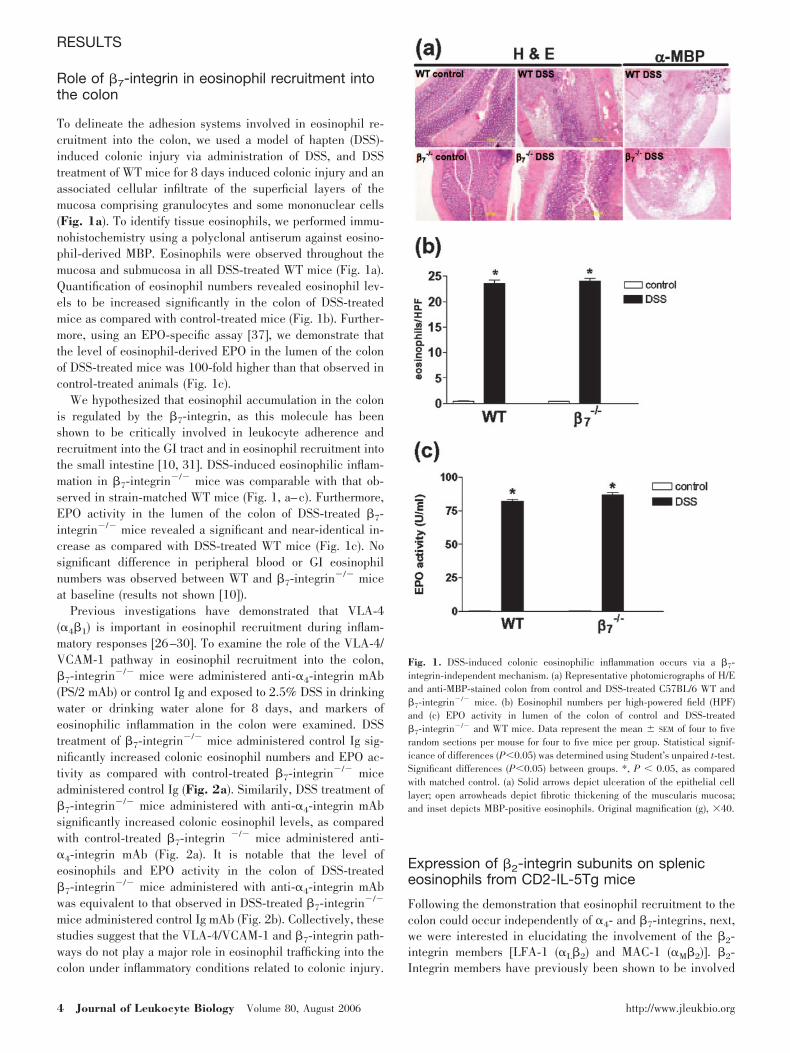
RESULTS
Role of �7-integrin in eosinophil recruitment intothe colon
To delineate the adhesion systems involved in eosinophil re-cruitment into the colon, we used a model of hapten (DSS)-induced colonic injury via administration of DSS, and DSStreatment of WT mice for 8 days induced colonic injury and anassociated cellular infiltrate of the superficial layers of themucosa comprising granulocytes and some mononuclear cells(Fig. 1a). To identify tissue eosinophils, we performed immu-nohistochemistry using a polyclonal antiserum against eosino-phil-derived MBP. Eosinophils were observed throughout themucosa and submucosa in all DSS-treated WT mice (Fig. 1a).Quantification of eosinophil numbers revealed eosinophil lev-els to be increased significantly in the colon of DSS-treatedmice as compared with control-treated mice (Fig. 1b). Further-more, using an EPO-specific assay [37], we demonstrate thatthe level of eosinophil-derived EPO in the lumen of the colonof DSS-treated mice was 100-fold higher than that observed incontrol-treated animals (Fig. 1c).
We hypothesized that eosinophil accumulation in the colonis regulated by the �7-integrin, as this molecule has beenshown to be critically involved in leukocyte adherence andrecruitment into the GI tract and in eosinophil recruitment intothe small intestine [10, 31]. DSS-induced eosinophilic inflam-mation in �7-integrin�/� mice was comparable with that ob-served in strain-matched WT mice (Fig. 1, a–c). Furthermore,EPO activity in the lumen of the colon of DSS-treated �7-integrin�/� mice revealed a significant and near-identical in-crease as compared with DSS-treated WT mice (Fig. 1c). Nosignificant difference in peripheral blood or GI eosinophilnumbers was observed between WT and �7-integrin�/� miceat baseline (results not shown [10]).
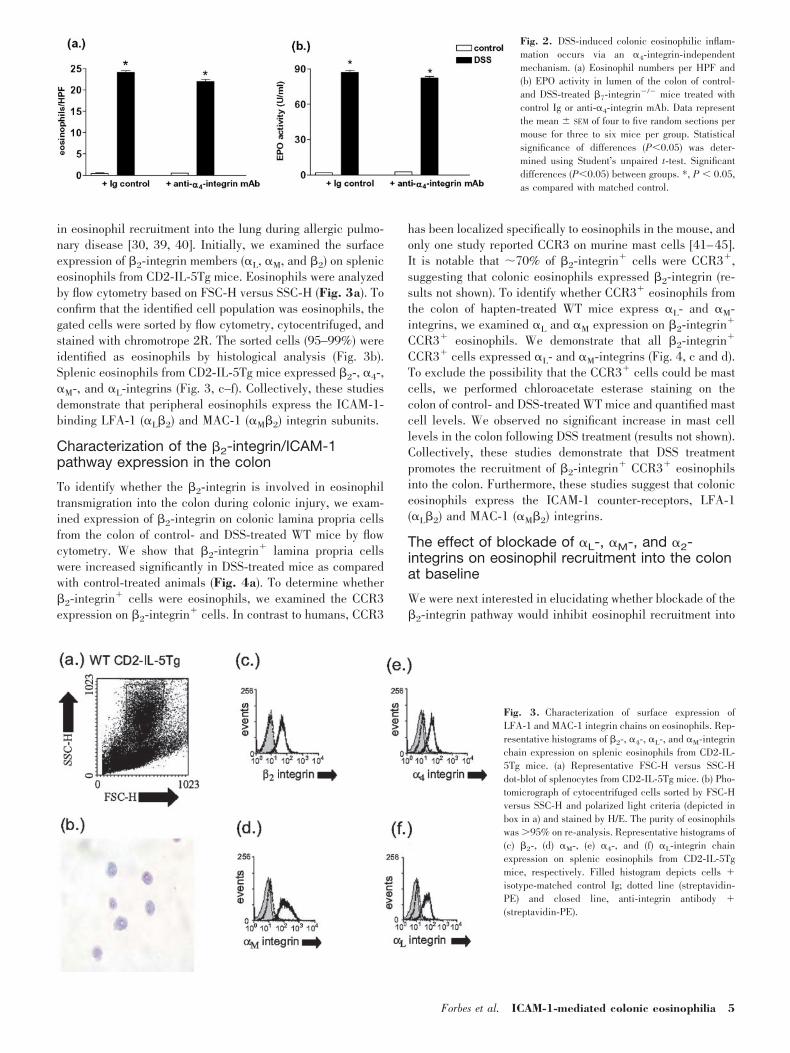
Previous investigations have demonstrated that VLA-4(�4�1) is important in eosinophil recruitment during inflam-matory responses [26–30]. To examine the role of the VLA-4/VCAM-1 pathway in eosinophil recruitment into the colon,�7-integrin�/� mice were administered anti-�4-integrin mAb(PS/2 mAb) or control Ig and exposed to 2.5% DSS in drinkingwater or drinking water alone for 8 days, and markers ofeosinophilic inflammation in the colon were examined. DSStreatment of �7-integrin�/� mice administered control Ig sig-nificantly increased colonic eosinophil numbers and EPO ac-tivity as compared with control-treated �7-integrin�/� miceadministered control Ig (Fig. 2a). Similarily, DSS treatment of�7-integrin�/� mice administered with anti-�4-integrin mAbsignificantly increased colonic eosinophil levels, as comparedwith control-treated �7-integrin �/� mice administered anti-�4-integrin mAb (Fig. 2a). It is notable that the level ofeosinophils and EPO activity in the colon of DSS-treated�7-integrin�/� mice administered with anti-�4-integrin mAbwas equivalent to that observed in DSS-treated �7-integrin�/�
mice administered control Ig mAb (Fig. 2b). Collectively, thesestudies suggest that the VLA-4/VCAM-1 and �7-integrin path-ways do not play a major role in eosinophil trafficking into thecolon under inflammatory conditions related to colonic injury.
Expression of �2-integrin subunits on spleniceosinophils from CD2-IL-5Tg mice
Following the demonstration that eosinophil recruitment to thecolon could occur independently of �4- and �7-integrins, next,we were interested in elucidating the involvement of the �2-integrin members [LFA-1 (�L�2) and MAC-1 (�M�2)]. �2-Integrin members have previously been shown to be involved
Fig. 1. DSS-induced colonic eosinophilic inflammation occurs via a �7-integrin-independent mechanism. (a) Representative photomicrographs of H/Eand anti-MBP-stained colon from control and DSS-treated C57BL/6 WT and�7-integrin�/� mice. (b) Eosinophil numbers per high-powered field (HPF)and (c) EPO activity in lumen of the colon of control and DSS-treated�7-integrin�/� and WT mice. Data represent the mean SEM of four to fiverandom sections per mouse for four to five mice per group. Statistical signif-icance of differences (P0.05) was determined using Student’s unpaired t-test.Significant differences (P0.05) between groups. *, P 0.05, as comparedwith matched control. (a) Solid arrows depict ulceration of the epithelial celllayer; open arrowheads depict fibrotic thickening of the muscularis mucosa;and inset depicts MBP-positive eosinophils. Original magnification (g), �40.
4 Journal of Leukocyte Biology Volume 80, August 2006 http://www.jleukbio.org
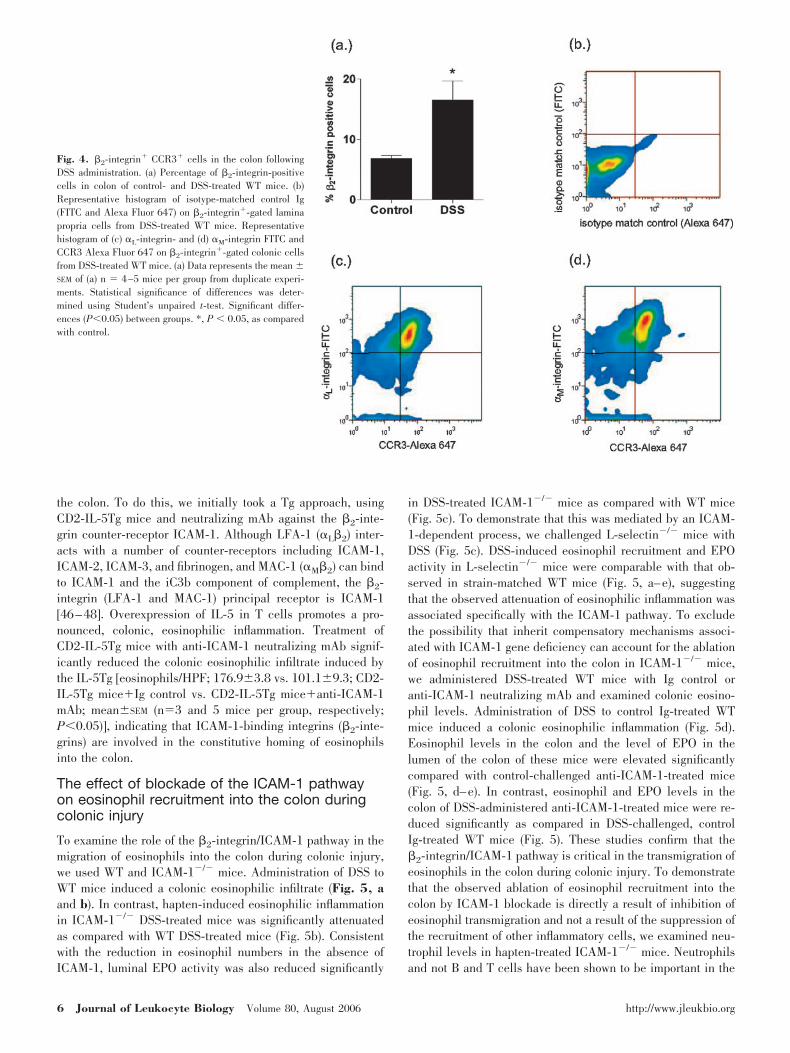
in eosinophil recruitment into the lung during allergic pulmo-nary disease [30, 39, 40]. Initially, we examined the surfaceexpression of �2-integrin members (�L, �M, and �2) on spleniceosinophils from CD2-IL-5Tg mice. Eosinophils were analyzedby flow cytometry based on FSC-H versus SSC-H (Fig. 3a). Toconfirm that the identified cell population was eosinophils, thegated cells were sorted by flow cytometry, cytocentrifuged, andstained with chromotrope 2R. The sorted cells (95–99%) wereidentified as eosinophils by histological analysis (Fig. 3b).Splenic eosinophils from CD2-IL-5Tg mice expressed �2-, �4-,�M-, and �L-integrins (Fig. 3, c–f). Collectively, these studiesdemonstrate that peripheral eosinophils express the ICAM-1-binding LFA-1 (�L�2) and MAC-1 (�M�2) integrin subunits.
Characterization of the �2-integrin/ICAM-1pathway expression in the colon
To identify whether the �2-integrin is involved in eosinophiltransmigration into the colon during colonic injury, we exam-ined expression of �2-integrin on colonic lamina propria cellsfrom the colon of control- and DSS-treated WT mice by flowcytometry. We show that �2-integrin� lamina propria cellswere increased significantly in DSS-treated mice as comparedwith control-treated animals (Fig. 4a). To determine whether�2-integrin� cells were eosinophils, we examined the CCR3expression on �2-integrin� cells. In contrast to humans, CCR3
has been localized specifically to eosinophils in the mouse, andonly one study reported CCR3 on murine mast cells [41–45].It is notable that �70% of �2-integrin� cells were CCR3�,suggesting that colonic eosinophils expressed �2-integrin (re-sults not shown). To identify whether CCR3� eosinophils fromthe colon of hapten-treated WT mice express �L- and �M-integrins, we examined �L and �M expression on �2-integrin�
CCR3� eosinophils. We demonstrate that all �2-integrin�
CCR3� cells expressed �L- and �M-integrins (Fig. 4, c and d).To exclude the possibility that the CCR3� cells could be mastcells, we performed chloroacetate esterase staining on thecolon of control- and DSS-treated WT mice and quantified mastcell levels. We observed no significant increase in mast celllevels in the colon following DSS treatment (results not shown).Collectively, these studies demonstrate that DSS treatmentpromotes the recruitment of �2-integrin� CCR3� eosinophilsinto the colon. Furthermore, these studies suggest that coloniceosinophils express the ICAM-1 counter-receptors, LFA-1(�L�2) and MAC-1 (�M�2) integrins.
The effect of blockade of �L-, �M-, and �2-integrins on eosinophil recruitment into the colonat baseline
We were next interested in elucidating whether blockade of the�2-integrin pathway would inhibit eosinophil recruitment into
Fig. 2. DSS-induced colonic eosinophilic inflam-mation occurs via an �4-integrin-independentmechanism. (a) Eosinophil numbers per HPF and(b) EPO activity in lumen of the colon of control-and DSS-treated �7-integrin�/� mice treated withcontrol Ig or anti-�4-integrin mAb. Data representthe mean SEM of four to five random sections permouse for three to six mice per group. Statisticalsignificance of differences (P0.05) was deter-mined using Student’s unpaired t-test. Significantdifferences (P0.05) between groups. *, P 0.05,as compared with matched control.
Fig. 3. Characterization of surface expression ofLFA-1 and MAC-1 integrin chains on eosinophils. Rep-resentative histograms of �2-, �4-, �L-, and �M-integrinchain expression on splenic eosinophils from CD2-IL-5Tg mice. (a) Representative FSC-H versus SSC-Hdot-blot of splenocytes from CD2-IL-5Tg mice. (b) Pho-tomicrograph of cytocentrifuged cells sorted by FSC-Hversus SSC-H and polarized light criteria (depicted inbox in a) and stained by H/E. The purity of eosinophilswas �95% on re-analysis. Representative histograms of(c) �2-, (d) �M-, (e) �4-, and (f) �L-integrin chainexpression on splenic eosinophils from CD2-IL-5Tgmice, respectively. Filled histogram depicts cells �isotype-matched control Ig; dotted line (streptavidin-PE) and closed line, anti-integrin antibody �(streptavidin-PE).
Forbes et al. ICAM-1-mediated colonic eosinophilia 5
the colon. To do this, we initially took a Tg approach, usingCD2-IL-5Tg mice and neutralizing mAb against the �2-inte-grin counter-receptor ICAM-1. Although LFA-1 (�L�2) inter-acts with a number of counter-receptors including ICAM-1,ICAM-2, ICAM-3, and fibrinogen, and MAC-1 (�M�2) can bindto ICAM-1 and the iC3b component of complement, the �2-integrin (LFA-1 and MAC-1) principal receptor is ICAM-1[46–48]. Overexpression of IL-5 in T cells promotes a pro-nounced, colonic, eosinophilic inflammation. Treatment ofCD2-IL-5Tg mice with anti-ICAM-1 neutralizing mAb signif-icantly reduced the colonic eosinophilic infiltrate induced bythe IL-5Tg [eosinophils/HPF; 176.93.8 vs. 101.19.3; CD2-IL-5Tg mice�Ig control vs. CD2-IL-5Tg mice�anti-ICAM-1mAb; meanSEM (n�3 and 5 mice per group, respectively;P0.05)], indicating that ICAM-1-binding integrins (�2-inte-grins) are involved in the constitutive homing of eosinophilsinto the colon.
The effect of blockade of the ICAM-1 pathwayon eosinophil recruitment into the colon duringcolonic injury
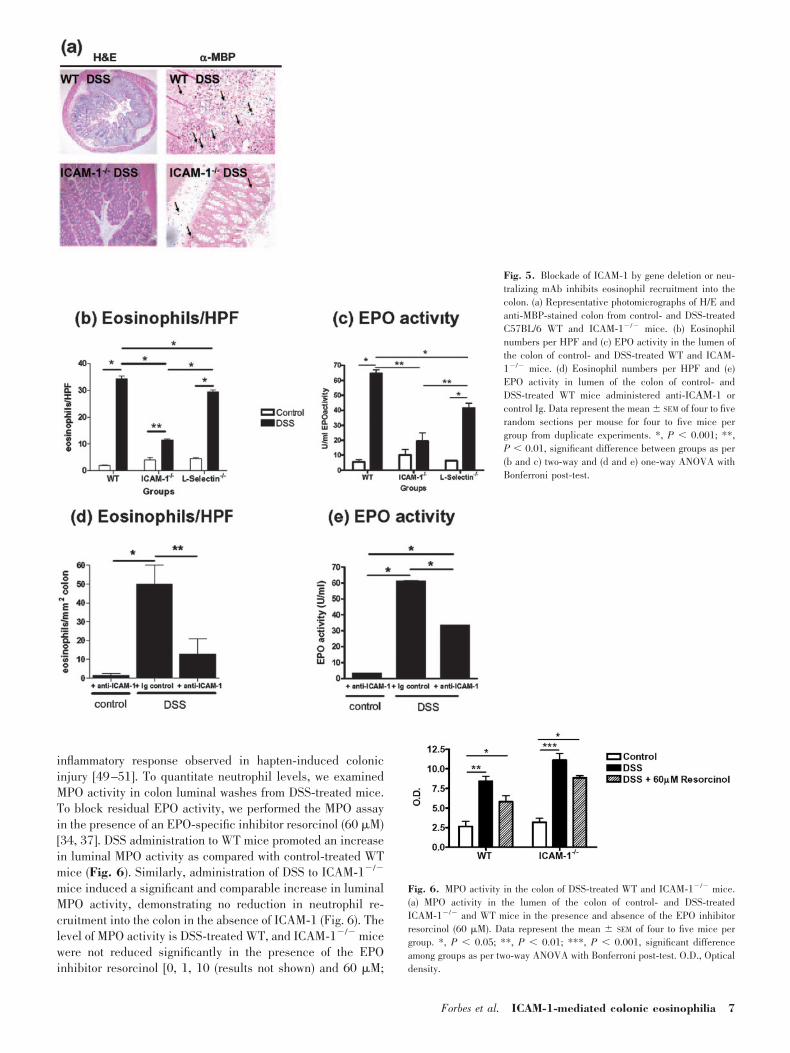
To examine the role of the �2-integrin/ICAM-1 pathway in themigration of eosinophils into the colon during colonic injury,we used WT and ICAM-1�/� mice. Administration of DSS toWT mice induced a colonic eosinophilic infiltrate (Fig. 5, aand b). In contrast, hapten-induced eosinophilic inflammationin ICAM-1�/� DSS-treated mice was significantly attenuatedas compared with WT DSS-treated mice (Fig. 5b). Consistentwith the reduction in eosinophil numbers in the absence ofICAM-1, luminal EPO activity was also reduced significantly
in DSS-treated ICAM-1�/� mice as compared with WT mice(Fig. 5c). To demonstrate that this was mediated by an ICAM-1-dependent process, we challenged L-selectin�/� mice withDSS (Fig. 5c). DSS-induced eosinophil recruitment and EPOactivity in L-selectin�/� mice were comparable with that ob-served in strain-matched WT mice (Fig. 5, a–e), suggestingthat the observed attenuation of eosinophilic inflammation wasassociated specifically with the ICAM-1 pathway. To excludethe possibility that inherit compensatory mechanisms associ-ated with ICAM-1 gene deficiency can account for the ablationof eosinophil recruitment into the colon in ICAM-1�/� mice,we administered DSS-treated WT mice with Ig control oranti-ICAM-1 neutralizing mAb and examined colonic eosino-phil levels. Administration of DSS to control Ig-treated WTmice induced a colonic eosinophilic inflammation (Fig. 5d).Eosinophil levels in the colon and the level of EPO in thelumen of the colon of these mice were elevated significantlycompared with control-challenged anti-ICAM-1-treated mice(Fig. 5, d–e). In contrast, eosinophil and EPO levels in thecolon of DSS-administered anti-ICAM-1-treated mice were re-duced significantly as compared in DSS-challenged, controlIg-treated WT mice (Fig. 5). These studies confirm that the�2-integrin/ICAM-1 pathway is critical in the transmigration ofeosinophils in the colon during colonic injury. To demonstratethat the observed ablation of eosinophil recruitment into thecolon by ICAM-1 blockade is directly a result of inhibition ofeosinophil transmigration and not a result of the suppression ofthe recruitment of other inflammatory cells, we examined neu-trophil levels in hapten-treated ICAM-1�/� mice. Neutrophilsand not B and T cells have been shown to be important in the
Fig. 4. �2-integrin� CCR3� cells in the colon followingDSS administration. (a) Percentage of �2-integrin-positivecells in colon of control- and DSS-treated WT mice. (b)Representative histogram of isotype-matched control Ig(FITC and Alexa Fluor 647) on �2-integrin�-gated laminapropria cells from DSS-treated WT mice. Representativehistogram of (c) �L-integrin- and (d) �M-integrin FITC andCCR3 Alexa Fluor 647 on �2-integrin�-gated colonic cellsfrom DSS-treated WT mice. (a) Data represents the mean SEM of (a) n � 4–5 mice per group from duplicate experi-ments. Statistical significance of differences was deter-mined using Student’s unpaired t-test. Significant differ-ences (P0.05) between groups. *, P 0.05, as comparedwith control.
6 Journal of Leukocyte Biology Volume 80, August 2006 http://www.jleukbio.org
inflammatory response observed in hapten-induced colonicinjury [49–51]. To quantitate neutrophil levels, we examinedMPO activity in colon luminal washes from DSS-treated mice.To block residual EPO activity, we performed the MPO assayin the presence of an EPO-specific inhibitor resorcinol (60 �M)[34, 37]. DSS administration to WT mice promoted an increasein luminal MPO activity as compared with control-treated WTmice (Fig. 6). Similarly, administration of DSS to ICAM-1�/�
mice induced a significant and comparable increase in luminalMPO activity, demonstrating no reduction in neutrophil re-cruitment into the colon in the absence of ICAM-1 (Fig. 6). Thelevel of MPO activity is DSS-treated WT, and ICAM-1�/� micewere not reduced significantly in the presence of the EPOinhibitor resorcinol [0, 1, 10 (results not shown) and 60 �M;
Fig. 5. Blockade of ICAM-1 by gene deletion or neu-tralizing mAb inhibits eosinophil recruitment into thecolon. (a) Representative photomicrographs of H/E andanti-MBP-stained colon from control- and DSS-treatedC57BL/6 WT and ICAM-1�/� mice. (b) Eosinophilnumbers per HPF and (c) EPO activity in the lumen ofthe colon of control- and DSS-treated WT and ICAM-1�/� mice. (d) Eosinophil numbers per HPF and (e)EPO activity in lumen of the colon of control- andDSS-treated WT mice administered anti-IC �-1 orcontrol Ig. Data represent the mean SEM of four to fiverandom sections per mouse for four to five mice pergroup from duplicate experiments. *, P 0.001; **,P 0.01, significant difference between groups as per(b and c) two-way and (d and e) one-way ANOVA withBonferroni post-test.
Fig. 6. MPO activity in the colon of DSS-treated WT and ICAM-1�/� mice.(a) MPO activity in the lumen of the colon of control- and DSS-treatedICAM-1�/� and WT mice in the presence and absence of the EPO inhibitorresorcinol (60 �M). Data represent the mean SEM of four to five mice pergroup. *, P 0.05; **, P 0.01; ***, P 0.001, significant differenceamong groups as per two-way ANOVA with Bonferroni post-test. O.D., Opticaldensity.
Forbes et al. ICAM-1-mediated colonic eosinophilia 7
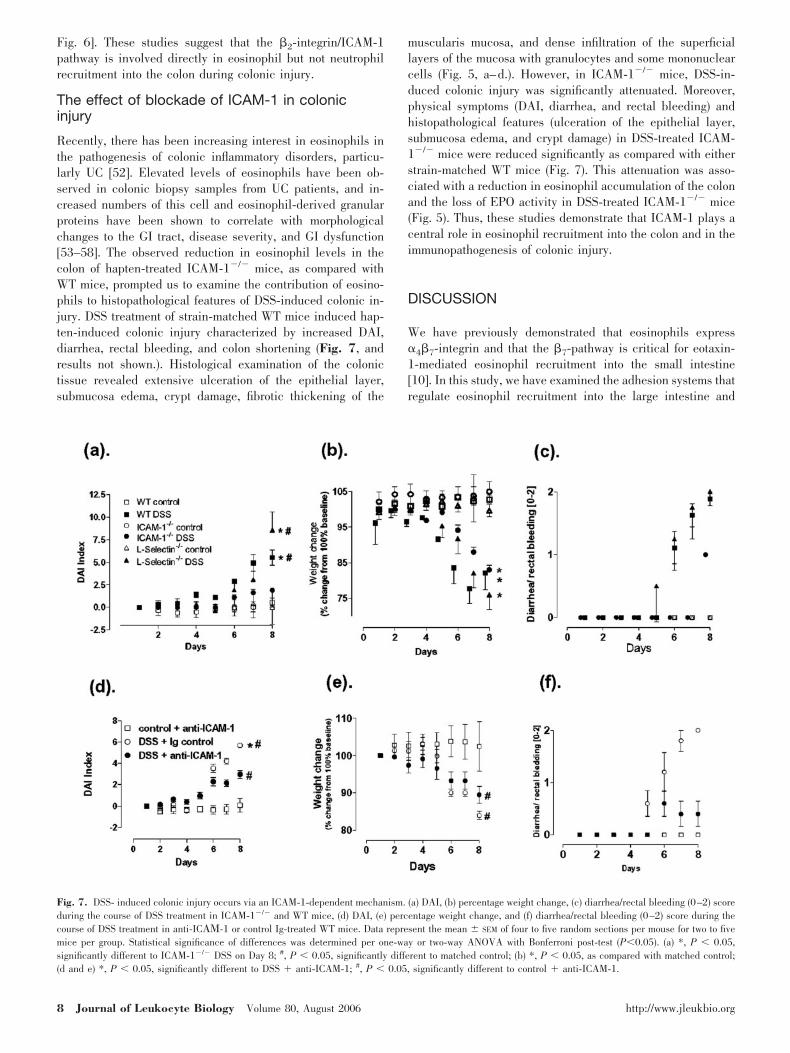
Fig. 6]. These studies suggest that the �2-integrin/ICAM-1pathway is involved directly in eosinophil but not neutrophilrecruitment into the colon during colonic injury.
The effect of blockade of ICAM-1 in colonicinjury
Recently, there has been increasing interest in eosinophils inthe pathogenesis of colonic inflammatory disorders, particu-larly UC [52]. Elevated levels of eosinophils have been ob-served in colonic biopsy samples from UC patients, and in-creased numbers of this cell and eosinophil-derived granularproteins have been shown to correlate with morphologicalchanges to the GI tract, disease severity, and GI dysfunction[53–58]. The observed reduction in eosinophil levels in thecolon of hapten-treated ICAM-1�/� mice, as compared withWT mice, prompted us to examine the contribution of eosino-phils to histopathological features of DSS-induced colonic in-jury. DSS treatment of strain-matched WT mice induced hap-ten-induced colonic injury characterized by increased DAI,diarrhea, rectal bleeding, and colon shortening (Fig. 7, andresults not shown.). Histological examination of the colonictissue revealed extensive ulceration of the epithelial layer,submucosa edema, crypt damage, fibrotic thickening of the
muscularis mucosa, and dense infiltration of the superficiallayers of the mucosa with granulocytes and some mononuclearcells (Fig. 5, a–d.). However, in ICAM-1�/� mice, DSS-in-duced colonic injury was significantly attenuated. Moreover,physical symptoms (DAI, diarrhea, and rectal bleeding) andhistopathological features (ulceration of the epithelial layer,submucosa edema, and crypt damage) in DSS-treated ICAM-1�/� mice were reduced significantly as compared with eitherstrain-matched WT mice (Fig. 7). This attenuation was asso-ciated with a reduction in eosinophil accumulation of the colonand the loss of EPO activity in DSS-treated ICAM-1�/� mice(Fig. 5). Thus, these studies demonstrate that ICAM-1 plays acentral role in eosinophil recruitment into the colon and in theimmunopathogenesis of colonic injury.
DISCUSSION
We have previously demonstrated that eosinophils express�4�7-integrin and that the �7-pathway is critical for eotaxin-1-mediated eosinophil recruitment into the small intestine[10]. In this study, we have examined the adhesion systems thatregulate eosinophil recruitment into the large intestine and
Fig. 7. DSS- induced colonic injury occurs via an ICAM-1-dependent mechanism. (a) DAI, (b) percentage weight change, (c) diarrhea/rectal bleeding (0–2) scoreduring the course of DSS treatment in ICAM-1�/� and WT mice, (d) DAI, (e) percentage weight change, and (f) diarrhea/rectal bleeding (0–2) score during thecourse of DSS treatment in anti-IC �-1 or control Ig-treated WT mice. Data represent the mean SEM of four to five random sections per mouse for two to fivemice per group. Statistical significance of differences was determined per one-way or two-way ANOVA with Bonferroni post-test (P0.05). (a) *, P 0.05,significantly different to ICAM-1�/� DSS on Day 8; #, P 0.05, significantly different to matched control; (b) *, P 0.05, as compared with matched control;(d and e) *, P 0.05, significantly different to DSS � anti-ICAM-1; #, P 0.05, significantly different to control � anti-ICAM-1.
8 Journal of Leukocyte Biology Volume 80, August 2006 http://www.jleukbio.org

have demonstrated that eosinophil recruitment into the colonduring colonic injury occurs via an �4- and �7-integrin-inde-pendent pathway; that murine splenic eosinophils express theligands for ICAM-1 (�2, �L, and �M); eosinophils (CCR3�)isolated from colon of hapten-treated mice express the ICAM-1counter-receptor integrins (�2, �L, and �M); and blockade ofICAM-1 activity by gene knockout or by neutralizing mAbattenuated eosinophil recruitment into the colon. Collectively,these studies suggest that the ICAM-1 adhesion pathway andnot VCAM-1 and �4�7/MAdCAM-1 pathways is important forthe development of colonic eosinophilic inflammation.
Leukocyte recruitment into the GI tract and gut-associatedlymphoid tissue are thought to be critically regulated by �7-integrin [31, 32, 59, 60]. Indeed, the expression of MAdCAM-1and �4�7 expression on leukocytes in the intestinal mucosa isup-regulated significantly in GI diseases, including cows milkallergy and food allergy [61, 62]. Recently, experimental stud-ies have demonstrated that leukocyte recruitment into the largeintestine can occur in a �7-integrin-dependent manner [32, 33,60]. Moreover, no impairment in CD4� T cell and mast cellinfiltration into the colon of �7-integrin�/� mice was observedfollowing helminth infection [32]. Furthermore, the concentra-tion of mast cell progenitors in the large intestine of �7-integrin�/� mice was not reduced as compared with WT mice[60]. We demonstrate that eosinophil recruitment into colonduring inflammatory conditions occurs predominantly via a�7-integrin-independent mechanism, through a �2-integrin/ICAM-1 pathway. The contribution of the �2-integrin pathwayin leukocyte and mast cell recruitment into the colon are notyet fully elucidated.
In our previous investigations, we have demonstrated thateosinophil recruitment into the colon during colonic injury iscritically regulated by the CCR3 ligand, eotaxin-1 [34]. It isnotable that eotaxin-1 up-regulates the expression of ICAM-1-binding integrins �L and �m expression on lymphocytes [63].Furthermore, we have recently demonstrated that in vitro stim-ulation of splenic eosinophils with eotaxin-1 in the presence ofIL-5 up-regulates the surface expression of �L- and �M-inte-grins on eosinophils (results not shown). In light of thesefindings and our demonstration of a role for the �2-integrin/ICAM-1 pathway in eosinophil recruitment, it is tempting tospeculate that eotaxin-1 preferentially promotes �2-integrin/ICAM-1 interactions and eosinophil recruitment into the colon.In different disease states, elevated eosinophil numbers areoften restricted to specific GI compartments. For example, EEis characterized by elevated level of eosinophils restricted tothe esophagus, whereas, eosinophilic colitis and UC are char-acterized by elevated levels of eosinophils in the colon withoutincreased numbers in other GI compartments. Paradoxically,the recruitment of eosinophils into these various GI compart-ments (small and large intestine) is thought to be primarilyregulated by eotaxin-1 [17]. How eotaxin-1 selectively orches-trates the trafficking of eosinophils into specific GI compart-ments remains unclear. It is possible that eotaxin-1 selectivelyup-regulates �L-, �M-, and �2-integrin expression on eosino-phils and the �2-integrin counter-receptor, ICAM-1, on colonicmicrovascular endothelial cells to promote eosinophil infiltra-tion into the colon. Indeed, eotaxin-1 has been shown tostimulate expression of ICAM-1 on microvascular endothelial
cells [64]. However, as we demonstrate that �L-, �M-, �2-integrins are expressed on eosinophils under noninflammatoryconditions, the differential expression of integrins cannot fullyaccount for the eotaxin-1-mediated selectivity for the ICAM-1pathway. An alternate explanation is that chemokines such aseotaxin-1 differentially regulate integrin/adhesion moleculeavidity, promoting �2/ICAM-1-dependent adhesion pathwayinteraction. Weber and colleagues [65] have demonstrated thatRANTES and MCP-3, eotaxin-1 receptor (CCR3) agonists, canselectively down-regulate eosinophil VLA-4/VCAM-1 adhe-sion and promote �2/ICAM-1 adhesion. Furthermore, theyshowed that the differential regulation of adhesion occurredindependently of integrin surface expression and directly in-volved modulation of integrin avidity [65]. We postulate thateotaxin-1-mediated recruitment involves a complex step ofevents involving integrin expression on leukocytes (increased�L, �M, �2), adhesion molecule expression on microvascularendothelium (ICAM-1), and increased integrin and integrinreceptor avidity (LFA-1/ICAM-1, MAC-1/ICAM-1).
The histological presence of eosinophils in the GI mucosa ofpatients with IBD UC has long been recognized; however, thecontribution eosinophils make to disease pathogenesis is stillnot well-understood. Recent clinical investigations of bowelbiopsy specimens from UC patients have shown a correlationamong the eosinophil numbers in the mucosa, the levels ofeosinophil-derived granule proteins (MBP, EPO, eosinophilcationic protein, and eosinophil-derived neurotoxin) in perfu-sion fluid samples, and disease severity [14, 56, 57, 66].Previously, we have demonstrated that eosinophils mediatedisease pathogenesis in experimental colitis through EPO-dependent pathways [34]. EPO catalyzes the oxidation of ha-lides and pseudohalides (Cl–, Br–, and SCN–) with the productsof respiratory burst (O2 and H2O2) to generate cytotoxic oxi-dants. Elevated levels of reactive oxygen species (H2O2) havebeen reported in mucosal tissue samples from patients with UC[67, 68]. Furthermore, H2O2 has been shown to promote anup-regulation of �2-integrin expression on eosinophils [69].
Using L-selectin�/�-deficient mice, we demonstrate thatDSS-induced eosinophil recruitment and EPO activity couldoccur independently of L-selectin. It is notable that eosinophillevels in DSS-treated L-selectin�/�-deficient mice were com-parable with WT. However, although EPO activity levels wereelevated significantly in DSS-treated L-selectin�/�-deficientmice, the EPO activity levels were reduced by �35%, ascompared with DSS-treated WT mice. These studies suggestthat eosinophil release of EPO is at least in part dependent onL-selectin, which binds cell-surface carbohydrates includingsialylated, fucosylated, and sulfated lactosaminoglycans suchas the sialyl LewisX tetrasaccharides. It is notable that liposac-charides of enteric pathogens have been shown to expressLewisX antigens. It is possible that during DSS-induced co-lonic injury, activation of L-selectin via cell-surface carbohy-drates on enteric bacteria promotes eosinophil degranulationand release of EPO. Consistent with this notion, eosinophilgranule proteins have been shown to possess bactericidal ac-tivity [70, 71].
In the present study, we also examined the role of ICAM-1in the recruitment of neutrophils into the colon during hapten-induced colonic injury. To study neutrophil levels, we exam-
Forbes et al. ICAM-1-mediated colonic eosinophilia 9

ined MPO activity, a marker of polymorphonuclear neutrophilgranules in the colon of DSS-treated WT and ICAM-1�/� mice.Consistent with previous investigations, we show that haptentreatment enhances colonic neutrophil levels. Furthermore, weshow no difference in MPO activity between DSS-treated WTand ICAM-1�/� mice. To confirm that contaminating EPOactivity was not contributing to the peroxidase activity, weperformed the assay in the presence of an EPO inhibitor,resorcinol (60 �M), which has been shown to selectively in-hibit EPO activity without neutralizing MPO [37, 72]. Thesestudies suggest that neutrophil recruitment during hapten-induced colonic injury is mediated via ICAM-1-independentmechanisms. Consistent with this observation, Krieglstein andcolleagues [49], using an identical model to what we havedescribed in this investigation, have demonstrated that mono-cyte and neutrophil (measured by MPO activity) accumulationinto the colon is regulated by an �1�1-integrin-dependentpathway [49].
It is notable that although we observed a significant atten-uation in physical symptoms and histopathological features ofdisease in DSS-treated ICAM-1�/� mice as compared withmatched WT mice, we still observed a significant weight-lossover the course of the experimental regime. We cannot fullyaccount for the observed weight-loss in DSS-treated ICAM-1�/� mice. However, although eosinophils and EPO levels aresignificantly attenuated, the level of eosinophils and EPOactivity is two- to threefold higher than that observed at base-line in ICAM-1�/� control mice. It is possible that this resid-ual level of eosinophils and EPO activity may contribute to theweight-loss, or alternatively, weight-loss may be driven byconcurrent pathways, independent of ICAM-1, eosinophils,and EPO.
Clinical and experimental studies have provided evidencefor a role for ICAM-1 in UC [73–76]. ICAM-1 levels areelevated in sonicated colonic tissue samples from UC patients,as compared with control patients, and in the colon of miceexperimental colitis [73]. Furthermore, blockade of ICAM-1function by neutralizing mAb or by antisense oligonucleotideagainst ICAM-1 ameliorates experimental colitis [74–76]. Toour knowledge, this is the first demonstration of a critical rolefor ICAM-1 adhesion pathway in eosinophilic accumulation inthe colon during colonic injury. These findings are particularlyimportant, given that activated eosinophils and eosinophil-derived granular proteins have been linked to the pathogenesisof diseases characterized by colonic injury, such as UC inhumans; that therapeutic approaches targeting ICAM-1, suchas antisense ICAM-1 oligonucleotide (ISIS-2302) therapy, arebeing examined for the treatment of IBD [77–79]; and thatrecent clinical trials examining the use of a humanized anti-body to �4�7 as a therapeutic treatment for IBD, particularlyUC [80], suggest the involvement of adhesion pathways, inde-pendent of �4�7 in the regulation of cellular recruitment in UC.
In conclusion, we have shown that eosinophils expressICAM-1-binding integrins (�2, �L, and �M) and that recruit-ment into the colon occurs via an ICAM-1-dependent and not�7-integrin-dependent pathway. Furthermore, we demonstratea pathogenic role for eosinophils and ICAM-1-mediated adhe-sion pathways in the development of experimental colitis.These studies highlight the importance of eosinophils in GI
diseases and suggest that antagonism of ICAM-1/eosinophilpathways may be a significant, therapeutic approach for thetreatment of UC and allergic colitis.
ACKNOWLEDGMENTS
We thank Anne Prins for excellent technical and histologicalassistance and Nancy and James Lee (Mayo Clinic, Scottsdale,AZ) for providing the anti-MBP mAb. We thank Rachel Akersand Judy Ann Bean for assistance with the statistical analysis.
REFERENCES
1. Sampson, H. A. (1999) Food allergy. Part 1: immunopathogenesis andclinical disorders. J. Allergy Clin. Immunol. 103, 717–728.
2. Furuta, G. T., Ackerman, S. J., Wershil, B. K. (1995) The role of theeosinophil in gastrointestinal diseases. Curr. Opin. Gastroenterol. 11,541–547.
3. Noel, R. J., Putnam, P. E., Collins, M. H., Assa’ad, A. H., Guajardo, J. R.,Jameson, S. C., Rothenberg, M. E. (2004) Clinical and immunopathologiceffects of swallowed fluticasone for eosinophilic esophagitis. Clin. Gastro-enterol. Hepatol. 2, 568–575.
4. Hogan, S. P., Foster, P. S., Rothenberg, M. E. (2002) Experimentalanalysis of eosinophil-associated gastrointestinal diseases. Curr. Opin.Allergy Clin. Immunol. 2, 239–248.
5. Silberstein, D. S. (1995) Eosinophil function in health and disease. Crit.Rev. Oncol. Hematol. 19, 47–77.
6. Rothenberg, M. E. (1998) Eosinophilia. N. Engl. J. Med. 338, 1592–1600.
7. Rothenberg, M. E., Mishra, A., Brandt, E. B., Hogan, S. P. (2001)Gastrointestinal eosinophils in health and disease. Adv. Immunol. 78,291–328.
8. Mishra, A., Hogan, S. P., Lee, J. J., Foster, P. S., Rothenberg, M. E. (1999)Fundamental signals that regulate eosinophil homing to the gastrointesti-nal tract. J. Clin. Invest. 103, 1719–1727.
9. Matthews, A. N., Friend, D. S., Zimmermann, N., Sarafi, M. N., Luster,A. D., Pearlman, E., Wert, S. E., Rothenberg, M. E. (1998) Eotaxin isrequired for the baseline level of tissue eosinophils. Proc. Natl. Acad. Sci.USA 95, 6273–6278.
10. Mishra, A., Hogan, S. P., Brandt, E. B., Wagner, N., Crossman, M. W.,Foster, P. S., Rothenberg, M. E. (2002) Enterocyte expression of theeotaxin and interleukin-5 transgenes induces compartmentalized dysregu-lation of eosinophil trafficking. J. Biol. Chem. 277, 4406–4412.
11. Garcia-Zepeda, E. A., Rothenberg, M. E., Ownbey, R. T., Celestin, J.,Leder, P., Luster, A. D. (1996) Human eotaxin is a specific chemoattrac-tant for eosinophil cells and provides a new mechanism to explain tissueeosinophilia. Nat. Med. 2, 449–456.
12. Butt, A. M., Murch, S. H., Ng, C-L., Kitching, P., Montgomery, S. M.,Phillips, A. D., Walker-Smith, J. A., Thomson, M. A. (2002) Upregulatedeotaxin expression and T cell infiltration in the basal and papillaryepithelium in cows milk associated reflux esophagitis. Arch. Dis. Child.87, 124–130.
13. Mir, A., Minguez, M., Tatay, J., Pascual, I., Pena, A., Sanchiz, V., Almela,P., Mora, F., Benages, A. (2002) Elevated serum eotaxin levels in patientswith inflammatory bowel disease. Am. J. Gastroenterol. 97, 1452–1457.
14. Jeziorska, M., Haboubi, N., Schofield, P., Woolley, D. E. (2001) Distri-bution and activation of eosinophils in inflammatory bowel disease usingan improved immunohistochemical technique. J. Pathol. 194, 484–492.
15. Chen, W., Paulus, B., Shu, D., Wilson, Chadwick, V. (2001) Increasedserum levels of eotaxin in patients with inflammatory bowel disease.Scand. J. Gastroenterol. 36, 515–520.
16. Kunkel, E. J., Butcher, E. C. (2002) Chemokines and the tissue-specificmigration of lymphocytes. Immunity 16, 1–4.
17. Hogan, S. P., Rothenberg, M. E., Forbes, E., Smart, V. E., Matthaei, K. I.,Foster, P. S. (2004) Chemokines in eosinophil-associated gastrointestinaldisorders. Curr. Allergy Asthma Rep. 4, 74–82.
18. Bochner, B. S., Schleimer, R. P. (2001) Mast cells, basophils and eosin-ophils: distinct but overlapping pathways for recruitment. Immunol. Rev.179, 5–15.
10 Journal of Leukocyte Biology Volume 80, August 2006 http://www.jleukbio.org

19. Tachimoto, H., Ebisawa, M., Bochner, B. S. (2002) Cross-talk betweenintegrins and chemokines that influences eosinophil adhesion and migra-tion. Int. Arch. Allergy Immunol. 128 (Suppl. 1), 18–20.
20. Tachimoto, H., Bochner, B. S. (2000) The surface phenotype of humaneosinophils. Chem. Immunol. 76, 45–62.
21. Grayson, M. H., Van der Vieren, M., Sterbinsky, S. A., Michael Gallatin,W., Hoffman, P. A., Staunton, D. E., Bochner, B. S. (1998) ��2 Integrinis expressed on human eosinophils and functions as an alternative ligandfor vascular cell adhesion molecule 1 (VCAM-1). J. Exp. Med. 188,2187–2191.
22. Georas, S. N., McIntyre, B. W., Ebisawa, M., Bednarczyk, J. L., Sterbin-sky, S. A., Schleimer, R. P., Bochner, B. S. (1993) Expression of afunctional laminin receptor (� 6 � 1, very late activation antigen-6) onhuman eosinophils. Blood 82, 2872–2879.
23. Meager, A. (1999) Cytokine regulation of cellular adhesion moleculeexpression in inflammation. Cytokine Growth Factor Rev. 10, 27–39.
24. Tachimoto, H., Burdick, M. M., Hudson, S. A., Kikuchi, M., Konstanto-poulos, K., Bochner, B. S. (2000) CCR3-active chemokines promote rapiddetachment of eosinophils from VCAM-1 in vitro. J. Immunol. 165,2748–2754.
25. Weber, C., Kitayama, J., Springer, T. A. (1996) Differential regulation ofb1 and b2 integrin avidity by chemoattractants in eosinophils. Proc. Natl.Acad. Sci. USA 93, 10939–10944.
26. Weg, V. B., Williams, T. J., Lobb, R. R., Nourshargh, S. (1993) Amonoclonal antibody recognizing very late activation antigen-4 inhibitseosinophil accumulation in vivo. J. Exp. Med. 177, 561–566.
27. Abraham, W. M., Sielczak, M. W., Ahmed, A., Cortes, A., Lauredo, I. T.,Kim, J., Pepinsky, B., Benjamin, C. D., Leone, D. R., Lobb, R. R., et al.(1994) � 4-Integrins mediate antigen-induced late bronchial responsesand prolonged airway hyperresponsiveness in sheep. J. Clin. Invest. 93,776–787.
28. Pretolani, M., Ruffie, C., Lapa e Silva, J. R., Joseph, D., Lobb, R. R.,Vargaftig, B. B. (1994) Antibody to very late activation antigen 4 preventsantigen-induced bronchial hyperreactivity and cellular infiltration in theguinea pig airways. J. Exp. Med. 180, 795–805.
29. Nakajima, H., Sano, H., Nishimura, T., Yoshida, S., Iwamoto, I. (1994)Role of vascular cell adhesion molecule 1/very late activation antigen 4and intercellular adhesion molecule 1/lymphocyte function-associatedantigen 1 interactions in antigen-induced eosinophil and T cell recruit-ment into the tissue. J. Exp. Med. 179, 1145–1154.
30. Gonzalo, J. A., Lloyd, C. M., Kremer, L., Finger, E., Martinez, A. C.,Siegelman, M. H., Cybulsky, M., Gutierrez-Ramos, J. C. (1996) Eosinophilrecruitment to the lung in a murine model of allergic inflammation. Therole of T cells, chemokines, and adhesion receptors. J. Clin. Invest. 98,2332–2345.
31. Butcher, E. C., Williams, M., Youngman, K., Rott, L., Briskin, M. (1999)Lymphocyte trafficking and regional immunity. Adv. Immunol. 72, 209–253.
32. Artis, D., Humphreys, N. E., Potten, C. S., Wagner, N., Muller, W.,McDermott, J. R., Grencis, R. K., Else, K. J. (2000) �7 Integrin-deficientmice: delayed leukocyte recruitment and attenuated protective immunityin the small intestine during enteric helminthic infection. Eur. J. Immu-nol. 30, 1656–1664.
33. Sydora, B. C., Wagner, N., Lohler, J., Takoub, G., Kronenberg, M., Muller,W., Aranda, R. (2002) �7 Integrin expression is not required for thelocalization of T-cells to the intestine and colitis pathogenesis. Clin. Exp.Immunol. 129, 35–42.
34. Forbes, E., Murase, T., Yang, M., Matthaei, K. I., Lee, J. J., Lee, N. A.,Foster, P. S., Hogan, S. P. (2004) Immunopathogenesis of experimentalulcerative colitis is mediated by eosinophil peroxidase. J. Immunol. 172,5664–5675.
35. Shapiro, H. M. (2003) Practical Flow Cytometry, Hoboken, NJ, John Wileyand Sons.
36. Stevceva, L., Pavli, P., Husband, A., Matthaei, K. I., Young, I. G., Doe,W. F. (2000) Eosinophilia is attentuated in experimental colitis induced inIL-5 deficient mice. Genes Immun. 1, 213–218.
37. Schneider, T., Issekutz, A. C. (1996) Quantitation of eosinophil andneutrophil infiltration into rat lung by specific assays for eosinophilperoxidase and myeloperoxidase. Application in a Brown Norway ratmodel of allergic pulmonary inflammation. J. Immunol. Methods 198,1–14.
38. Bradley, P. P., Priebat, D. A., Christensen, R. D., Rothstein, G. (1982)Measurement of cutaneous inflammation: estimation of neutrophil contentwith an enzyme marker. J. Invest. Dermatol. 78, 206–209.
39. Burke-Gaffney, A., Hellewell, P. G. (1998) A CD18/ICAM-1-dependentpathway mediates eosinophil adhesion to human bronchial epithelial cells.Am. J. Respir. Cell Mol. Biol. 19, 408–418.
40. Hakansson, L., Bjornsson, E., Janson, C., Schmekel, B. (1995) Increasedadhesion to vascular cell adhesion molecule-1 and intercellular adhesionmolecule-1 of eosinophils from patients with asthma. J. Allergy Clin.Immunol. 96, 941–950.
41. Senechal, S., Fahy, O., Gentina, T., Vorng, H., Capron, M., Walls, A. F.,McEuen, A. R., Buckley, M. G., Hamid, Q., Wallaert, B., Tonnel, A. B.,Tsicopoulos, A. (2002) CCR3-blocking antibody inhibits allergen-inducedeosinophil recruitment in human skin xenografts from allergic patients.Lab. Invest. 82, 929–939.
42. Humbles, A. A., Lu, B., Friend, D. S., Okinaga, S., Lora, J., Al-Garawi, A.,Martin, T. R., Gerard, N. P., Gerard, C. (2002) The murine CCR3 receptorregulates both the role of eosinophils and mast cells in allergen-inducedairway inflammation and hyperresponsiveness. Proc. Natl. Acad. Sci. USA99, 1479–1484.
43. Gurish, M. F., Humbles, A., Tao, H., Finkelstein, S., Boyce, J. A., Gerard,C., Friend, D. S., Austen, K. F. (2002) CCR3 is required for tissueeosinophilia and larval cytotoxicity after infection with Trichinella spiralis.J. Immunol. 168, 5730–5736.
44. Ma, W., Bryce, P. J., Humbles, A. A., Laouini, D., Yalcindag, A., Alenius,H., Friend, D. S., Oettgen, H. C., Gerard, C., Geha, R. S. (2002) CCR3 isessential for skin eosinophilia and airway hyperresponsiveness in a mu-rine model of allergic skin inflammation. J. Clin. Invest. 109, 621–628.
45. Oliveira, S. H., Lukacs, N. W. (2001) Stem cell factor and IgE-stimulatedmurine mast cells produce chemokines (CCL2, CCL17, CCL22) andexpress chemokine receptors. Inflamm. Res. 50, 168–174.
46. de Fougerolles, A. R., Springer, T. A. (1992) Intercellular adhesionmolecule 3, a third adhesion counter-receptor for lymphocyte function-associated molecule 1 on resting lymphocytes. J. Exp. Med. 175, 185–190.
47. Fawcett, J., Holness, C. L., Needham, L. A., Turley, H., Gatter, K. C.,Mason, D. Y., Simmons, D. L. (1992) Molecular cloning of ICAM-3, a thirdligand for LFA-1, constitutively expressed on resting leukocytes. Nature360, 481–484.
48. Vazeux, R., Hoffman, P. A., Tomita, J. K., Dickinson, E. S., Jasman, R. L.,St John, T., Gallatin, W. M. (1992) Cloning and characterization of a newintercellular adhesion molecule ICAM-R. Nature 360, 485–488.
49. Krieglstein, C. F., Cerwinka, W. H., Sprague, A. G., Laroux, F. S.,Grisham, M. B., Koteliansky, V. E., Senninger, N., Granger, D. N., deFougerolles, A. R. (2002) Collagen-binding integrin �1�1 regulates in-testinal inflammation in experimental colitis. J. Clin. Invest. 110, 1773–1782.
50. Kruidenier, L., van Meeteren, M. E., Kuiper, I., Jaarsma, D., Lamers,C. B., Zijlstra, F. J., Verspaget, H. W. (2003) Attenuated mild colonicinflammation and improved survival from severe DSS-colitis of transgenicCu/Zn-SOD mice. Free Radic. Biol. Med. 34, 753–765.
51. Naito, Y., Takagi, T., Handa, O., Ishikawa, T., Nakagawa, S., Yamaguchi,T., Yoshida, N., Minami, M., Kita, M., Imanishi, J., Yoshikawa, T. (2003)Enhanced intestinal inflammation induced by dextran sulfate sodium intumor necrosis factor-� deficient mice. J. Gastroenterol. Hepatol. 18,560–569.
52. Miner Jr., P. B. (2004) Seeing red in inflammatory bowl disease: theeosinophil at center stage? Inflamm. Bowel Dis. 10, 329–330.
53. Bischoff, S. C., Mayer, J., Nguyen, Q. T., Stolte, M., Manns, M. P. (1999)Immunohistological assessment of intestinal eosinophil activation in pa-tients with eosinophilic gastroenteritis and inflammatory bowel disease.Am. J. Gastroenterol. 94, 3521–3529.
54. Bischoff, S. C., Wedemeyer, J., Herrmann, A., Meier, P. N., Trautwein, C.,Cetin, Y., Maschek, H., Stolte, M., Gebel, M., Manns, M. P. (1996)Quantitative assessment of intestinal eosinophils and mast cells in inflam-matory bowel disease. Histopathology 28, 1–13.
55. Raab, Y., Fredens, K., Gerdin, B., Hallgren, R. (1998) Eosinophil acti-vation in ulcerative colitis: studies on mucosal release and localization ofeosinophil granule constituents. Dig. Dis. Sci. 43, 1061–1070.
56. Carlson, M., Raab, Y., Peterson, C., Hallgren, R., Venge, P. (1999)Increased intraluminal release of eosinophil granule proteins EPO, ECP,EPX, and cytokines in ulcerative colitis and proctitis in segmental per-fusion. Am. J. Gastroenterol. 94, 1876–1883.
57. Saitoh, O., Kojima, K., Sugi, K., Matsuse, R., Uchida, K., Tabata, K.,Nakagawa, K., Kayazawa, M., Hirata, I., Katsu, K. (1999) Fecal eosinophilgranule-derived proteins reflect disease activity in inflammatory boweldisease. Am. J. Gastroenterol. 94, 3513–3520.
58. Carvalho, A. T., Elia, C. C., de Souza, H. S., Elias, P. R., Pontes, E. L.,Lukashok, H. P., de Freitas, F. C., Lapa e Silva, J. R. (2003) Immuno-histochemical study of intestinal eosinophils in inflammatory bowel dis-ease. J. Clin. Gastroenterol. 36, 120–125.
59. Wagner, N., Lohler, J., Kunkel, E. J., Ley, K., Leung, E., Krissansen, G.,Rajewsky, K., Muller, W. (1996) Critical role for �7 integrins in formationof the gut-associated lymphoid tissue. Nature 382, 366–370.
Forbes et al. ICAM-1-mediated colonic eosinophilia 11

60. Gurish, M. F., Tao, H., Abonia, J. P., Arya, A., Friend, D. S., Parker,C. M., Austen, K. F. (2001) Intestinal mast cell progenitors requireCD49d�7 (�4�7 integrin) for tissue-specific homing. J. Exp. Med. 194,1243–1252.
61. Eigenmann, P. A., Tropia, L., Hauser, C. (1999) The mucosal adhesionreceptor �4�7 integrin is selectively increased in lymphocytes stimulatedwith �-lactoglobulin in children allergic to cow’s milk. J. Allergy Clin.Immunol. 103, 931–936.
62. Veres, G., Helin, T., Arato, A., Farkkila, M., Kantele, A., Suomalainen,H., Savilahti, E. (2001) Increased expression of intercellular adhesionmolecule-1 and mucosal adhesion molecule �4�7 integrin in small intes-tinal mucosa of adult patients with food allergy. Clin. Immunol. 99,353–359.
63. Jinquan, T., Quan, S., Feili, G., Larsen, C. G., Thestrup-Pedersen, K.(1999) Eotaxin activates T cells to chemotaxis and adhesion only ifindcued to express CCR3 by IL-2 together with IL-4. J. Immunol. 162,4285–4292.
64. Hohki, G., Terada, N., Hamano, N., Kitaura, M., Nakajima, T., Yoshie, O.,Ikeda, T., Kimura, S., Konno, A. (1997) The effects of eotaxin on thesurface adhesion molecules of endothelial cells and on eosinophil adhe-sion to microvascular endothelial cells. Biochem. Biophys. Res. Commun.241, 136–141.
65. Weber, C., Kitayama, J., Springer, T. A. (1996) Differential regulation ofB1 and B2 integrin avidity by chemoattractants in eosinophils. Proc. Natl.Acad. Sci. USA 93, 10939–10944.
66. Sangfelt, P., Carlson, M., Thorn, M., Loof, L., Raab, Y. (2001) Neutrophiland eosinophil granule proteins as markers of response to local pred-nisolone treatment in distal ulcerative colitis and proctitis. Am. J. Gas-troenterol. 96, 1085–1090.
67. Sands, B. E. (1999) Novel therapies for inflammatory bowel disease.Gastroenterol. Clin. North Am. 28, 323–351.
68. Shanahan, F., Targan, S. R. (1992) Medical treatment of inflammatorybowel disease. Annu. Rev. Med. 43, 125–133.
69. Nagata, M., Yamamoto, H., Shibasaki, M., Sakamoto, Y., Matsuo, H.(2000) Hydrogen peroxide augments eosinophil adhesion via �2 integrin.Immunology 101, 412–418.
70. Carreras, E., Boix, E., Rosenberg, H. F., Cuchillo, C. M., Nogues, M. V.(2003) Both aromatic and cationic residues contribute to the membrane-lytic and bactericidal activity of eosinophil cationic protein. Biochemistry42, 6636–6644.
71. Lehrer, R. I., Szklarek, D., Barton, A., Ganz, T., Hamann, K. J., Gleich,G. J. (1989) Antibacterial properties of eosinophil major basic protein andeosinophil cationic protein. J. Immunol. 142, 4428–4434.
72. Divi, R. L., Doerge, D. R. (1994) Mechanism-based inactivation of lac-toperoxidase and thyroid peroxidase by resorcinol derivatives. Biochem-istry 33, 9668–9674.
73. Vainer, B., Nielsen, O. H. (2000) Changed colnic profile of P-selectin,platelet-endothelial cell adhesion molecule-1 (PECAM-1), intercellularadhesion molecule-1 (ICAM-1), ICAM-2, and ICAM-3 in inflammatorybowel disease. Clin. Exp. Immunol. 121, 242–247.
74. Hamamoto, N., Maemura, K., Hirata, I., Murano, M., Sasaki, S., Katsu,K. (1999) Inhibition of dextran sulphate sodium (DSS)-induced colitisin mice by intracolonically administered antibodies against adhesionmolecules (endothelial leuocyte adhesion molecule (ELAM-1) or inter-cellular adhesion molecule-1 (ICAM-1)). Clin. Exp. Immunol. 117,462– 468.
75. Bennett, C. F., Kornbrust, D., Henry, S., Stecker, K., Howard, R., Cooper,S., Dutson, S., Hall, W., Jacoby, H. I. (1997) An ICAM-1 antisenseoligonucleotide prevents and reverses dextran sulfate sodium-inducedcolitis in mice. J. Pharmacol. Exp. Ther. 280, 988–1000.
76. Kato, S., Hokari, R., Matsuzaki, K., Iwai, A., Kawaguchi, A., Nagao, S.,Miyahara, T., Itoh, K., Ishii, H., Miura, S. (2000) Amelioration of murineexperimental colitis by inhibition of mucosal addressin cell adhesionmolecule-1. J. Pharmacol. Exp. Ther. 295, 183–189.
77. Yacyshyn, B. R., Bowen-Yacyshyn, M. B., Jewell, L., Tami, J. A., Bennett,C. F., Kisner, D. L., Shanahan Jr., W. R. (1998) A placebo-controlled trialof ICAM-1 antisense oligonucleotide in the treatment of Crohn’s disease.Gastroenterology 114, 1133–1142.
78. Yacyshyn, B. R., Barish, C., Goff, J., Dalke, D., Gaspari, M., Yu, R., Tami,J., Dorr, F. A., Sewell, K. L. (2002) Dose ranging pharmacokinetic trial ofhigh-dose alicaforsen (intercellular adhesion molecule-1 antisense oli-godeoxynucleotide) (ISIS 2302) in active Crohn’s disease. Aliment. Phar-macol. Ther. 16, 1761–1770.
79. Yacyshyn, B. R., Chey, W. Y., Goff, J., Salzberg, B., Baerg, R., Buchman,A. L., Tami, J., Yu, R., Gibiansky, E., Shanahan, W. R. (2002) Doubleblind, placebo controlled trial of the remission inducing and steroidsparing properties of an ICAM-1 antisense oligodeoxynucleotide, alicaf-orsen (ISIS 2302), in active steroid dependent Crohn’s disease. Gut 51,30–36.
80. Feagan, B. G., Greenberg, G. R., Wild, G., Fedorak, R. N., Pare, P.,McDonald, J. W., Dube, R., Cohen, A., Steinhart, A. H., Landau, S.,Aguzzi, R. A., Fox, I. H., Vandervoort, M. K. (2005) Treatment ofulcerative colitis with a humanized antibody to the �4�7 integrin. N. Engl.J. Med. 352, 2499–2507.
12 Journal of Leukocyte Biology Volume 80, August 2006 http://www.jleukbio.org





























