Embed Size (px)
Citation preview

Polyhedron 79 (2014) 161–169
Contents lists available at ScienceDirect
Polyhedron
journal homepage: www.elsevier .com/locate /poly
Design, structural and spectroscopic elucidation, and the in vitrobiological activities of new triorganotin dithiocarbamates – Part II
http://dx.doi.org/10.1016/j.poly.2014.05.0010277-5387/� 2014 Elsevier Ltd. All rights reserved.
⇑ Corresponding author. Tel.: +55 31 3409 5744; fax: +55 31 3409 5720.E-mail address: [email protected] (G.M. de Lima).
I.P. Ferreira a, G.M. de Lima a,⇑, E.B. Paniago a, W.R. Rocha a, J.A. Takahashi a, C.B. Pinheiro b, J.D. Ardisson c
a Departamento de Química, Universidade Federal de Minas Gerais, UFMG, Avenida Antônio Carlos 6627, Belo Horizonte, MG CEP 31270-901, Brazilb Departamento de Física, Universidade Federal de Minas Gerais, UFMG, Avenida Antônio Carlos 6627, Belo Horizonte, MG CEP 31270-901, Brazilc Centro de Desenvolvimento em Tecnologia Nuclear, CDTN/CNEN, Avenida Antônio Carlos 6627, Belo Horizonte, MG CEP 31270-901, Brazil
a r t i c l e i n f o
Article history:Received 7 March 2014Accepted 5 May 2014Available online 13 May 2014
Keywords:Biological activityOrganotin dithiocarbamateStructural determinationSARAspergillosis
a b s t r a c t
The two novel dithiocarbamate salts, [Na{S2CNR(R1)}] (i), [Na{S2CNR(R2)}] (ii), R = methyl, R1 = CH2
CH(OMe)2, R2 = 2-methyl-1,3-dioxolane, previously synthesized by us, have been used in chemical reac-tions with triorganotin halides. Hence, five new complexes: [SnPh3{S2CNR(R1)}] (1), [SnCy3{S2CNR(R1)}](2), [SnMe3{S2CNR(R2)}] (3), [SnPh3{S2CNR(R2)}] (4) and [SnCy3{S2CNR(R2)}] (5), [R = methyl, R1 = CH2
CH(OMe)2, and R2 = 2-methyl-1,3-dioxolane], have been isolated. All compounds were authenticated interms of infrared, 1H and 13C NMR, and the complexes were also characterized using 119Sn NMR, 119SnMössbauer and X-ray crystallography, in the case of complexes (1), (4) and (5). The biological activityof all derivatives has been screened in terms of IC90 (lmol L�1) and IC50 (lmol L�1) against Aspergillusflavus, Aspergillus niger, Aspergillus parasiticus and Penicillium citrinum, and the results correlated well witha performed study of structure–activity relationship (SAR). Complexes (1) and (4) displayed nanomolarinhibition concentration in terms of IC50.
� 2014 Elsevier Ltd. All rights reserved.
1. Introduction
The dithiolates involves ligands such as dithiocarmates, R2NCS2�,
dithiocarboxylates (R-CS2�), xanthates, ROCS2
�, dithiophosphates,(RO)2PS2
�, which in view of their important applications are stillunder investigation [1]. Apart from their ability to stabilise metalcations in a variety of oxidation states, well documented in thefield of coordination chemistry [2,3], their pharmaceutical proper-ties are worth mentioning. They are used to remove excess of cop-per due to Wilson’s disease [4], they are also able to reduce thenephrotoxicity of platinum-based drugs for chemotherapy [5]and in addition they are employed in the clinical treatment of alco-holism [6], or of other diseases [7]. Apart from that they find appli-cations in the vulcanization of rubber [8], preparation of pesticides[9], and as precursors for the production of metal sulfide nanopar-ticles [10].
It is also significant the wide range of applications and potentialuse of organotin derivatives among other metal-derivatives, asdiverse as in agriculture, biology, catalysis, or organic synthesis[11]. As both organotins and dithiocarbamates interact with livingcells, perhaps an enhanced biological activity can be observed bybonding them together. Many works have described not only the
preparation and characterization of related complexes [12–18]but also their action against tumours [19,20], fungi, bacteria, andother microorganisms [21], as well as other applications [22,23].The number and nature of the organic groups bonded to the tincentre influences the toxicity towards microorganisms, which, ingeneral, decreases in the order R3SnX > R2SnX2 > RSnX3. However,the order of toxicity depends on the microorganism, and variesfrom strain to strain [24]. It has been proposed that toxicity inthe R3Sn series correlates with total molecule surface (TSA) andhence n-propyl-, n-butyl-, n-pentyl-, phenyl-, and cyclohexyl-substituted tin should be more toxic than ethyl- and methyltin.Moreover, if the toxic effects are intra-cellular, following transportthrough the cell membrane, a correlation should exist betweentoxicity and lipophilicity [25]. Besides preparing new organotin–dithiocarbamates, investigating their potential applications [26]and screening their activity in the presence of some parasites[27] we have been interested in the mechanism of action of suchcomplexes in biological media. The effect of organotin–dithiocar-bamate and –carboxylate complexes on the cellular activity ofsome variety of Candida albicans revealed that there are no changesin DNA integrity or in the mitochondria function. However, allcomplexes reduced the ergosterol biosynthesis. Special techniquesused for morphological investigations such as scanning electronmicroscopy (SEM) and transmission electron microscopy (TEM)suggested that the organotin complexes act on the cell membrane,

Fig. 1. Structure of the complex [SnPh3{S2CNMe(R1)}] (1), [R1 = CH2CH(OMe)2].
Fig. 2. Structure of the complex [SnPh3{S2CNMe(R2)}] (4) and [SnCy3{S2CNMe(R2)}](5), [2-methyl-1,3-dioxolane].
162 I.P. Ferreira et al. / Polyhedron 79 (2014) 161–169
in view of the observed cytoplasm leakage and strong deteriorationof the cellular membrane [28,29]. In order to proceed with suchinvestigations we have been interested in attaching less simpledithiocarbamate (DTC) ligands to metal cations in order to evaluatetheir biocide responses as a consequence of the new bonding,chemical or structural properties [30–33]. In the last 2 years wehave been exploring ligands such as [S2CNMe(R0)]�, {R0 = CH2
CH(OMe)2 (i) or = 2-methyl-1,3-dioxolane (ii)} synthesized by us,resulting in a series of published papers describing the outcomes[34–37]. Herein we report the second part of the work involvingligands (i) and (ii) and organotin fragments, the characterizationand the crystallographic authentication of a series of triorganotindithiocarbamates (1)–(5). The biological activity of the complexeshave been screened in the presence of Aspergillus flavus, Aspergillusniger, Aspergillus parasiticus and Penicillium citrinum.
2. Experimental
2.1. Chemistry
2.1.1. Materials and instrumentsAll starting materials were purchased from Aldrich, Merck or
Synth and used as received. NMR spectra were recorded at400 MHz using a Bruker DPX-400 spectrometer equipped with an89 mm wide-bore magnet. 1H and 13C{1H} shifts are reportedrelative to SiMe4 and 119Sn shifts relative to SnMe4. The infraredspectra were recorded with samples pressed as CsI pellets on aPerkin–Elmer GX FT-IR spectrometer in the range of 4000–200 cm�1.Carbon, hydrogen and nitrogen analysis were performed on aPerkin–Elmer PE-2400 CHN-analysis using tin sample-tubes.119Sn Mössbauer spectra were run in standard equipment at liquidnitrogen temperature using a BaSnO3 source kept at room temper-ature. Intensity data for the X-ray study were collected at 293(2) Kon a Xcalibur, Atlas, Gemini, Mo Ka (k = 0.7107 Å). Data collection,reduction and cell refinement were performed using the CRYSALIS RED
program [38]. The structures were solved and refined employingthe SHELXS-97 [39]. Further details are given in Table 1. All non-Hatoms were refined anisotropic. The H atoms were refined withfixed individual displacement parameters [Uiso (H)Z1.2 Ueq (C)]using the SHELXL riding model. The ORTEP-3 program for Windows[40] was used in the preparation of Figs. 1–3, sketched employingthe MERCURY program [41].
2.2. The structure–activity relationship (SAR)
Geometry optimization and frequency calculations of all com-pounds studied in this work were performed at the Density Func-tional Theory level [42], employing the hybrid B3LYP [43,44]exchange-correlation functional and using the 6-31G(d) all elec-tron basis set [45,46] for all atoms. The tin atom was treated usingthe LANL2DZ pseudo-potential for the core electrons and its asso-ciated double-zeta basis set for the valence electrons [47]. All prop-erties involved in the SAR studies were obtained for the optimized
Table 1Selected 1H, 13C and 119Sn NMR an Infrared spectroscopic data for compounds (i), (ii) and[SnPh3{S2CNR(R2)}] (4) and [SnCy3{S2CNR(R2)}] (5), [R = methyl, R1 = CH2CH(OMe)2, and R
Compound 1H/d (NCH2) 13C/d (NCH2)
(i) 4.39–4.42 57.8[SnPh3{S2CNR(R1)}] 3.32–3.39 59.8[SnCy3{S2CNR(R1)}] 3.99–4.02 59.4
(ii) 4.49–4.51 57.3[SnMe3{S2CNR(R2)}] 4.11–4.13 59.6[SnPh3{S2CNR(R2)}] 3.98–4.01 60.3[SnCy3{S2CNR(R2)}] 4.11–4.13 59.6
structures. The quantum mechanical calculations were performedusing the GAUSSIAN 03 program [48]. The molecular surface area,molecular volume and octanol-water partition coefficients (LogP)of all species were calculated using the MARVINVIEW program [49].
2.3. Syntheses
2.3.1. Synthesis of [SnPh3{S2CN(Me)R}] {R = CH2CH(OMe)2} (1)To a round-bottom flask (125 mL) containing [SnPh3Cl] (3.53 g,
9.2 mmol) in methanol (25 mL) was added [Na{S2CN(Me)CH2
CH(OMe)2}], (2 g, 9.2 mmol). After 24 h at room temperature and
complexes [SnPh3{S2CNR(R1)}] (1), [SnCy3{S2CNR(R1)}] (2), [SnMe3{S2CNR(R2)}] (3),2 = 2-methyl-1,3-dioxolane].
13C/d (CS2) mðN—CS2Þ masymðCS2Þ
214.2 1474 966197.57 1492 993201.0 1484 989
214.4 1473 982201.1 1488 991198.5 1495 996201.1 1482 993

Fig. 3. Structure of the complex [SnCy3{S2CNMe(R2)}] (5), [R2 = 2-methyl-1,3-dioxolane].
I.P. Ferreira et al. / Polyhedron 79 (2014) 161–169 163
the white precipitate was filtered and washed with hot water andethanol. The solid obtained was re-crystallized in a mixture of CH2
Cl2/toluene (4:1) rendering X-ray quality crystals. Yield 71%. Mp89.1–91 �C. IR. (cm�1, KBr): 1492 (mN–CS); 993 {masym(C–S)}; 626{msym(C–S)}; 397 {masym(S–Sn)}; 375 {msym(S–Sn)}. 1H NMR (d, CDCl3):7.89–7.31 (Sn–C6H5); 4.70 (CH); 3.87–3.84 (OCH3) and (NCH3);3.39–3.32 (NCH2). 13C NMR (d, CDCl3): 197.6 (CS2); 142–127.5(Sn–C6H5); 102.1 (CH); 59.8 (NCH2); 54.9 (OCH3); 45.9 (NCH3).119Sn NMR (d, CDCl3): �181.8 {1J(119Sn–13C) = 605 Hz,1J(119Sn–1H) = 62.3 Hz}. d (mm s�1): 1.41; D (mm s�1): 2.78. Car-bon, hydrogen and nitrogen analyses for C24H27NO2S2Sn, respec-tively. Found (calc.) 52.43 (52.96); 4.91 (5.00); 2.49 (2.57).
2.3.2. Synthesis of [SnCy3{S2CN(Me)R}] {R = CH2CH(OMe)2} (2)Prepared in a similar manner using ethanol (20 mL), [SnCy3Cl]
(1.85 g, 4.6 mmol) and [Na{S2CN(Me)CH2CH(OMe)2}] (1 g,4.6 mmol). Yield 50%. Mp 57–58 �C. IR. (cm�1, KBr): 1484 (mN–CS);989 {masym(C–S)}; 626 {msym(C–S)}; 412 {masym(S–Sn)}; 385 {msym(S–Sn)}.1H NMR (d, CDCl3): 4.82–4.76 (CH); 4.02–3.99 (NCH2); 3.51–3.44(OCH3) and (NCH3); 2.01–1.29 (Sn–C6H11). 13C NMR (d, CDCl3):200.4 (CS2); 102.1 (CH); 59.4 (NCH2); 55.26 (OCH3); 45.7 (NCH3);34.7–27.02 (Sn–C6H11). 119Sn NMR (d, CDCl3): �23.7{1J(119Sn–13C) =341 Hz}. d (mm s�1): 1.41; D (mm s�1): 2.78.Carbon, hydrogen and nitrogen analyses for C24H45NO2S2Sn,respectively. Found (calc.) 51.17 (51.26); 7.94 (8.06); 2.38 (2.49).
2.3.3. Synthesis of [SnMe3{S2CN(Me)R}] {R = 2-methyl-1,3-dioxolane}(3)
To a round-bottom flask (125 mL) containing [SnMe3Cl] (1 g,5 mmol) in methanol (15 mL) was added [Na{S2CN(Me)R}], R = 2-methyl-1,3-dioxolane) (1.08 g, 5 mmol). After stirring for 24 h atroom temperature the white precipitate was filtered and washedwith hot water and ethanol. The solid obtained was re-crystallizedin a mixture of CH2Cl2/ethanol (4:1) rendering X-ray quality crys-tals. Yield 90%. Mp 129–130 �C. IR. (cm�1, KBr): 1488 (mN–CS); 991{masym(C–S)}; 609 {msym(C–S)}. 1H NMR (d, CDCl3): 5.27–5.22 (OCHO);4.13–4.11 (NCH2); 4.00–4.00–3.85 (OCH2); 3.54 (NCH3); 1.46 (Sn–CH3). 13C NMR (d, CDCl3): 201.1 (CS2); 101.6 (OCHO); 64.9(OCH2); 59.6 (NCH2); 45.1 (NCH3); 15.1–15.0 (Sn–CH3). 119SnNMR (d, CDCl3): �334.8 {1J(119Sn–13C) = 608 Hz, 1J(119Sn–1H) =83.3 Hz}. d (mm s�1): 1.43; D (mm s�1): 3.01. Carbon, hydrogenand nitrogen analyses for C9H19NO2S2Sn, respectively. Found (calc.)31.86 (30.39); 5.36 (5.38).
2.3.4. Synthesis of [SnPh3{S2CN(Me)R}] (R = 2-methyl-1,3-dioxolane)(4)
Synthesized accordingly using [SnPh3Cl] {3.6 g, 9.3 mmol) and[Na{S2CN(Me)R}] (R = 2-methyl-1,3-dioxolane) {2.0 g, 9.3 mmol).Yield 73%. Mp 111.5–112.8 C. IR. (cm�1, KBr): 1495 (mN–CS); 996{masym(C–S)}; 609 {msym(C–S)}; 473 {masym(S–Sn)}; 342 {msym(S–Sn)}. 1HNMR (d, CDCl3): 7.73–7.35 (Sn–C6H5); 5.23–5.19 (OCHO); 3.94–3.79 (OCH2); 4.01–3.98 (NCH2); 3.47 (NCH3). 13C NMR (d, CDCl3):198.5 (CS2); 142.1–128.5 (Sn–C6H5); 101.1 (OCHO); 64.9 (OCH2);60.3 (NCH2); 45.51 (NCH3). 119Sn NMR (d, CDCl3): �181.8{1J(119Sn–13C) = 603 Hz, 1J(119Sn–1H) = 62.3 Hz}. d (mm s�1): 1.41;D (mm s�1): 2.78. Carbon, hydrogen and nitrogen analyses for C24
H25NO2S2Sn, respectively. Found (calc.) 53.02 (53.16); 4.56 (4.65);2.62 (2.58).
2.3.5. Synthesis of [SnCy3{S2CN(Me)R}] (R = 2-methyl-1,3-dioxolane)(5)
Synthesized accordingly using [SnCy3Cl] {1.0 g, 2.5 mmol) and[Na{S2CN(Me)R}] (R = 2-methyl-1,3-dioxolane) {0.53 g, 2.5 mmol).Yield 63%. Mp 103.9–105.9 �C. IR. (cm�1, KBr): 1482 (mN–CS); 993{masym(C–S)}; 598 {msym(C–S)}. 1H NMR (d, CDCl3): 5.27–5.22 (OCHO);4.13–4.11 (NCH2); 4.00–3.85 (OCH2); 3.54 (NCH3); 1.96–1.29 (Sn–C6H11). 13C NMR (d, CDCl3): 201.1 (CS2); 101.6 (OCHO); 64.9(OCH2); 59.6 (NCH2); 45.1 (NCH3); 34.7–27.0 (Sn–C6H11). 119SnNMR (d, CDCl3): �25.4 {1J(119Sn–13C) = 328 Hz}. d (mm s�1): 1.41;D (mm s�1): 2.78. Carbon, hydrogen and nitrogen analyses for C24-
H43NO2S2Sn, respectively. Found (calc.) 51.35 (51.44); 7.61 (7.73);2.40 (2.50).
2.4. Biological tests
2.4.1. Filamentous fungiA. flavus (CCT 4952) was obtained from Tropical Collection Cul-
ture, S.P. (Brazil). A. niger (NRRL 3), A. parasiticus (ATCC 15517), andP. citrinum (ATCC 756) were obtained from ARS Culture Collection(NRRL, USA), American Type Culture Collection (ATCC, USA) andBiotechnology and Bioassays Laboratory (LABB, MG, Brazil). Theywere kept in potato dextrose agar (PDA) under refrigeration at 7�C. The tests were performed in Broth Heart Infusion (BHI) med-ium. The fungal spores were counted in a Neubauer chamber. Dilu-tions were carried to achieve the required final concentration ofspores of 5.0 � 103 spores mL�1.
3. Results and discussions
3.1. Chemistry
[Na{S2CNR(R1)}] (i), [Na{S2CNR(R2)}] (ii), [R = methyl, R1 = CH2
CH(OMe)2, R2 = 2-methyl-1,3-dioxolane], have been prepared ascolorless solids, with yields higher than 50%, by the appropriateamine reaction with CS2 in alkaline media. The synthesis of com-plexes (1)–(5) have been performed by the chemical reaction ofSnR3Cl with (i) or (ii), Scheme 1.
All complexes have been isolated as colorless, air stable andcrystalline solids. Their purity has been attested in terms of thesatisfactory melting points and C, H and N analysis.
3.2. Infrared results
The signals related to the N–CS2, C–S and Sn–C vibration modeswere used in the infrared study. The asymmetric (masym) and sym-metric (msym) C–S vibration of [Na{S2CNR(R1)}] (i), [Na{S2CNR(R2)}](ii), [R = methyl, R1 = CH2CH(OMe)2, R2 = 2-methyl-1,3-dioxolane]have been observed at 966, 613 cm�1, and 982, 606 cm�1, respec-tively. The (masym) C–S stretching vibration moved to higher

Scheme 1. Synthesis of complexes [SnPh3{S2CNR(R1)}] (1), [SnCy3{S2CNR(R1)}] (2), [SnMe3{S2CNR(R2)}] (3), [SnPh3{S2CNR(R2)}] (4) and [SnCy3{S2CNR(R2)}] (5), [R = methyl,R1 = CH2CH(OMe)2, and R2 = 2-methyl-1,3-dioxolane].
164 I.P. Ferreira et al. / Polyhedron 79 (2014) 161–169
frequency, 995–979 cm�1 upon complexation, demonstratingmostly a symmetrical bidentate coordination mode. On the otherhand little variations have been detected in the (msym) C–S bands,640–553 cm�1, in (1)–(5) in comparison to the ligand salts [50].
It is observed an increasing in the N–CS2 bond strength, sincethe vibration frequencies move from 1473 cm�1 (i) and1474 cm�1 (ii) to 1482–1495 cm�1 in (1)–(5), indicating anincreasing in the N–CS2 double bond character.
3.3. NMR results
The 1H NMR experiments revealed the dithiocarbamate coordi-nation to the organotin fragment, in view of the obtained patternsfor complexes (1)–(5). All complexes exhibited an upfield shift forthe NCH2 hydrogens in comparison to the ligands, as well as thecorrect hydrogen integrations, Table 1.
The main interest in the 13C NMR spectra concerns the reso-nance of the N13CS2 group, and that of the Ca, bonded to the tinatom. The d (13CS2) signal in the ligands moves to lower frequencyin complexes (1)–(5). A closer relation between the 13C chemicalshift, d, and mðN—CS2Þ has been observed. Higher values of the stretch-ing frequency of N–C bond indicates a higher double bond charac-ter. It effects an intense p-electron circulation at the N–C contact,and therefore decreases the net magnetic field experienced bythe N13CS2, since it locates in the diamagnetic zone of the molecule[51]. Consequently lower values of chemical shift (d) are observed,Table 1.
The 119Sn chemical shifts for penta-, or hexa- or hepta-coordinated organotin dithiocarbamates have been found in theliterature in the range of �150 to �250 ppm, �300 to �500 ppmand �700 to �800, respectively [52]. We have observed the
Table 2119Sn NMR parameters and results and C–Sn–C angle obtained by X-ray crystallography fo[SnPh3{S2CNR(R2)}] (4) and [SnCy3{S2CNR(R2)}] (5), [R = methyl, R1 = CH2CH(OMe)2, and R
Complexa 119Sn NMR/d 1J(11
[SnPh3{S2CNR(R1)}] (1) �181.75 605[SnCy3{S2CNR(R1)}] (2) �23.73 341[SnMe3{S2CNR(R2)}] (3) �334.82 608[SnPh3{S2CNR(R2)}] (4) �181.76 603[SnCy3{S2CNR(R2)}] (5) �25.38 328
a CDCl3 as solvent.b Obtained using |1J(119Sn–13C)| = [(15.56 ± 0.84)(h)–(1160 ± 101)].c Obtained using |1J(119Sn–13C)| = [(9.99 ± 0.73)(h)–(746 ± 100)].d Obtained using |1J(119Sn–13C)| = [11.4(h)–875].e The average of the C–Sn–C angles obtained by X-ray crystallography.
following resonances in the 119Sn NMR experiments 181.8,�23.7, �334.8, �181.8 and �25.4 ppm, Table 2, for [SnPh3
{S2CNR(R1)}] (1), [SnCy3{S2CNR(R1)}] (2), [SnMe3{S2CNR(R2)}] (3),[SnPh3{S2CNR(R2)}] (4), [SnCy3{S2CNR(R2)}] (5), accordingly, whereR = methyl, R1 = CH2CH(OMe)2, and R2 = 2-methyl-1,3-dioxolane.More informative are the coupling constant 1J (119Sn, 13C) and 2J(119Sn, 1H), which allowed an evaluation of the C–Sn–C angle ofthe organotin fragment. The literature suggests that 1J and 2J arevery sensitive to variation in the coordination number. Empiricalequations express a mathematical relationship between h and 1J(119Sn, 13C) coupling constants for organotin complexes [52–55]:
j1Jð119Sn� 13CÞj ¼ 11:4ðhÞ� 875 for methyl-possessing derivatives; ð1Þ
j1Jð119Sn� 13CÞj ¼ ½ð15:56� 0:84ÞðhÞ � ð1160
� 101Þ� for phenyl-containing derivatives ð2Þ
and
j1Jð119Sn� 13CÞj ¼ ½ð9:99� 0:73ÞðhÞ � ð746
� 100Þ� for butyl-containing complexes: ð3Þ
The interpretation of chemical shifts and coupling constants insolution is generally based on crystal structure data (X-ray), there-fore subject to uncertainties arising from solvation and dynamiceffects. Eqs. (1)–(3) provide, with reasonable accuracy, means ofdetermining the C–Sn–C angle in solution by measuring J couplingparameters. The comparison of this angle and those measured by
r complexes [SnPh3{S2CNR(R1)}] (1), [SnCy3{S2CNR(R1)}] (2), [SnMe3{S2CNR(R2)}] (3),2 = 2-methyl-1,3-dioxolane].
9Sn–13C) (Hz) C–Sn–C (�) C–Sn–C(�)e
114.5(av.)b 108.49(av.)108.7(av.)c –130.1d –114.3(av.)b 108.49(av.)107.4(av.)d 111.04(av.)

I.P. Ferreira et al. / Polyhedron 79 (2014) 161–169 165
X-ray crystallography, discussed in Section 3.5, suggests little var-iation of the solution- and in the solid-state structure of complexes(1), (4) and (5).
3.4. Mössbauer spectroscopic results
The 119Sn-Mössbauer experiments were performed in order todetermine the differences in the Sn nuclei on going from organotinstarting materials to their respective complexes. The isomer shiftparameters (d/mm s�1) indicates the oxidation state and the pres-ence of s electron density at the tin nuclei, therefore it is a goodindication of alterations of the s contribution to the frontier molec-ular orbitals formation in the complexes. The structure and the cor-responding 119Sn parameters of the organotin precursors used inthis work are required: [SnPh3Cl] {tetrahedral, d = 1.34 mm s�1
and D = 2.46 mm s�1}, [56]; [SnCy3Cl] {Cy = cyclohexyl, distortedtrigonal bipiramid, d = 1.51 mm s�1 and D = 2.95 mm s�1}, [57];[SnMe3Cl] {distorted trigonal bipiramid, d = 1.47 mm s�1 andD = 3.32 mm s�1}, [58]. Upon complexation we have observed thefollowing parameters for the complexes [SnPh3{S2CNR(R1)}] (1),d = 1.28 mm s�1 and D = 1.76 mm s�1; [SnCy3{S2CNR(R1)}] (2),d = 1.50 mm s�1 and D = 2.20 mm s�1; [SnMe3{S2CNR(R2)}] (3),d = 1.46 mm s�1 and D = 2.95 mm s�1; [SnPh3{S2CNR(R2)}] (4),d = 1.25 mm s�1 and D = 1.74 mm s�1; and for [SnCy3{S2CNR(R2)}](5), d = 1.51 mm s�1 and D = 2.24 mm s�1. The isomer shift, d,reduced a little on going from tetrahedral [SnPh3Cl] to complexes(1) and (4), suggesting a decrease in the s electron density contri-bution to the frontier orbitals in these complexes. So it is consistentwith a distorted bipyramid geometry for (1) and (4), as determinedby X-ray experiments. In addition a distorted trigonal bipyramidarrangement around the tin cation is observed in [SnCy3Cl] atlow temperature [59]. We have detected a modest isomer shift var-iation comparing [SnCy3Cl] with (2) and (5), so the geometry ispossibly preserved after complexation. Again the crystallographicdetermination agrees with the 119Sn Mössbauer results. On theother hand the isomer shift obtained for (3), 1.46 mm s�1, is veryclose to that of the starting material SnMe3Cl, 1.47 mm s�1, justify-ing a trigonal bipyramid as well. The non-zero value of the quadru-polar splitting parameters (D/mm s�1) indicates deviation from aspherical charge distribution at the metallic cation. For complexes(1), (2), (4) and (5) we have obtained quadrupolar splitting at 1.76,2.20, 1.74 and 2.24 mm s�1, smaller than those of [SnPh3Cl],246 mm s�1 and [SnCy3Cl] 2.95 mm s�1, which agrees with a dis-torted trigonal bipyramid with two ligands in cis position, asrevealed by the crystallographic results obtained for (1), (4) and(5). The D observed for (3), 2.95 mm s�1 agrees with the proposedgeometry.
3.5. X-ray crystallographic results
Biological performance of organotin complexes relies upon sub-tle structural arrangements in solution or in the solid state, sostructural authentication plays an key role in the investigation ofthe biocide activity. The structures of complexes (1), (4) and (5)have been authenticated by X-ray crystallography, Table 3.
In complexes (1), (4) and (5), the tin atom is asymmetricallycoordinated by a dithiocarbamate ligand and three carbon atomsfrom the organic substituents, phenyl or cyclohexyl, sketching adistorted trigonal bipyramid, Figs. 1–3.
The equatorial position is occupied by two cis Ph or Cy groups,and one of the sulfur atoms. The Sn–C bonds at the equatorial posi-tion in (1), (4) and (5), respectively: Sn1–C7, 2.139(2), Sn1–C19,2.129(2) Å; Sn1–C7, 2.137(3), Sn1–C13, 2.128(3) Å; and Sn1–C7,2.159(3), Sn1–C13, 2.169(3) Å are shorter than the axial Sn–Cbonds, Sn1–C13, 2.157(2) in (1); Sn1–C19, 2.158(3) Å in (4); Sn1–C19, 2.179(3) in (5), accordingly. The Sn–S are even more asym-
metric, the apical Sn–S contacts, 3.0780(7) in (1), 3.0183(9) in (4)and 3.2160(11) Å in (5) are longer than those at the equatorialposition, 2.4687(6) in (1), 2.4751(8) in (4) and 2.4697(10) Å in(5). These features agree with other results of the literature, asnicely described in a recent review [1]. Despite the longer Sn–Sbond in complex (5) it is still smaller than the sum of the Vander Waals radii of Sn and S (4.0 Å) [60], suggesting a strong cova-lent character, Table 4.
This crystallographic arrangement goes along with Bent’s rulethat explain some deviations of the VSEPR theory [61]. The molec-ular orbitals are normally obtained by equivalent contribution ofthe atomic orbitals (LCAO) with appropriate symmetry and energy.However organotin complexes tend to deviate from this rule. Theorbital s contributes more for the LCAO at the equatorial coordina-tion, that involve spxpy orbitals, than to axial chemical bonds, thatcomprise pzdz2 wave functions. The Sn–C and Sn–S of the apicallocation is much longer in the cyclohexyl derivative [SnCy3{S2-
CNR(R2)}] (5), than in the others, suggesting as well the influenceof steric factors.
The average of the C–Sn–C angles in (1), (4) and (5), 108.49(av.),108.49(av.) and 111.04(av.), obtained by X-ray experiments, com-pare closely to the angles obtained by using the 1J(119Sn–13C) cou-pling constants, 114.5�, 114.3� and 107.4� for the respectivecomplexes, suggesting little structural variations between the sol-ids and their solutions, Table 2. Therefore the solid state structuresmight be kept in solution.
Intermolecular interactions in complex (5) can be neglected,however they are important the 3D crystal packing in complex(1) and (4). The following contact are observed in (1):S2� � �S2 = 3.584 Å (symmetry operator 2 � x, 2 � y, 2 � z), C2–H2B� � �O2 = 2.639 Å, (symmetry operator 1 � x, 1 � y, 2 � z), C24–H24� � �p(C17) = 2.894 Å (symmetry operator 2 � x, 2 � y, 1 � z),C21–H21� � �p(C18) = 2.816 Å (symmetry operator 1 � x, 2 � y,1 � z) e C15–H15� � �p(C12) = 2.883 Å (symmetry operator 2 � x,1 � y, 1 � z). In (5) we can observe the following intermolecularinteractions C10–H10� � �p(C20) = 2.840 Å (symmetry operators1 + x, y, z), C2–H2A� � �p(C18) = 2.734 Å (symmetry operators 1 � x,1 � y, 1 � z) e C21–H21� � �p(C9) = 2.866 Å (symmetry operators�0.5 + x, 0.5 � y, 0.5 + z).
3.6. Biocide assay results
Aspergillosis has been in focus lately due to the growing numberof deaths in consequence of fungal infections in individual immu-nocompromised either from the action of immunosuppressivedrugs, diseases, cancer or AIDS, etc, [62]. Voriconazole and liposo-mal amphotericin B are the first choice for the clinical treatment ofaggressive Aspergillosis, however they can be as dangerous as thefungus for the patient, particularly to their kidneys and liver [63].Other optional drugs are amphotericin B, caspofungin, flucytosineor itraconazole, however, in view of the increasing cases of resis-tant infections, more powerful and less toxic drugs are required[64]. Metal-containing drugs alone or in combination with otherin used in clinics might represent a way to overcome resistance.
In this work the biocide assays were performed in terms ofinhibitory concentrations, which are more consistent and reliable.A pre-screening against A. flavus, A. niger, A. parasiticus andP. citrinum, have been performed with complexes (1)–(5) in aconcentration of 250 lg mL�1, according to a pre-establishedprotocol [65]. Experiments of IC90 and IC50 were only performedfor those complexes with 100% inhibition growth of the studiedmicroorganism in this concentration of 250 lg mL�1.
The sodium salts (i) and (ii) were inactive towards the studiedfungi. On the other hand, complexes (1)–(5) were quite activeagainst A. flavus, A. niger, A. parasiticus and P. citrinum. Complexes[SnPh3{S2CNR(R1)}] (1) and [SnPh3{S2CNR(R2)}] (4), [R = methyl,
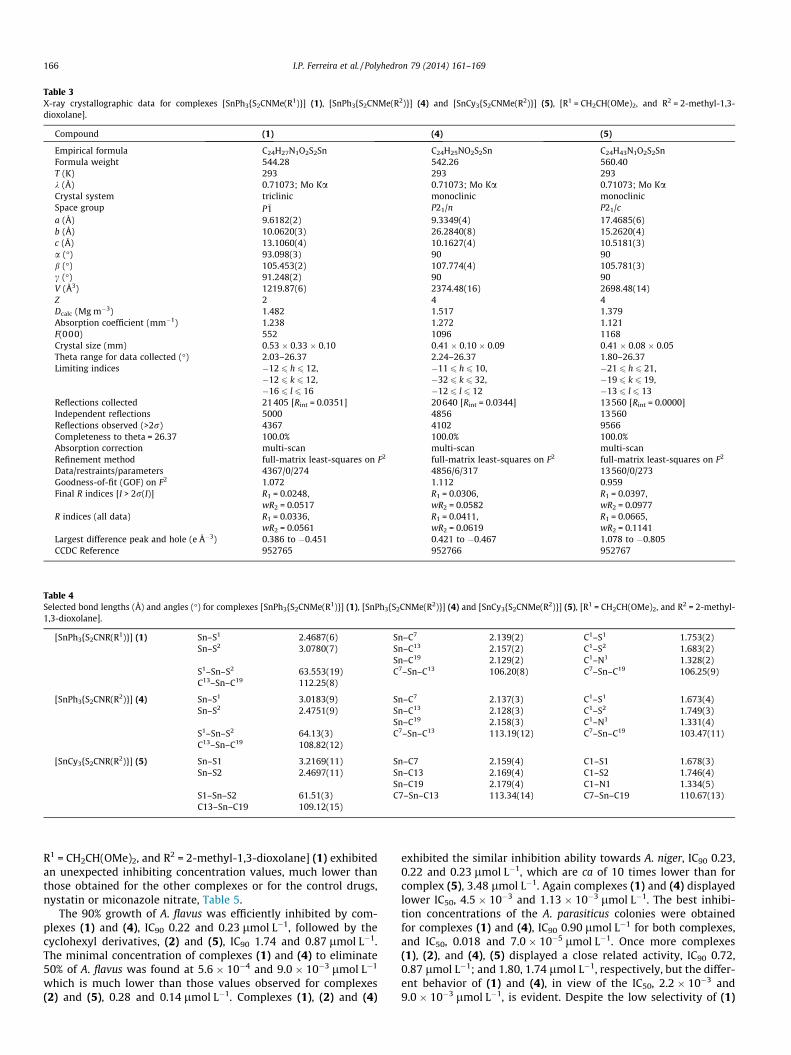
Table 3X-ray crystallographic data for complexes [SnPh3{S2CNMe(R1)}] (1), [SnPh3{S2CNMe(R2)}] (4) and [SnCy3{S2CNMe(R2)}] (5), [R1 = CH2CH(OMe)2, and R2 = 2-methyl-1,3-dioxolane].
Compound (1) (4) (5)
Empirical formula C24H27N1O2S2Sn C24H25NO2S2Sn C24H43N1O2S2SnFormula weight 544.28 542.26 560.40T (K) 293 293 293k (Å) 0.71073; Mo Ka 0.71073; Mo Ka 0.71073; Mo KaCrystal system triclinic monoclinic monoclinicSpace group P�1 P21/n P21/ca (Å) 9.6182(2) 9.3349(4) 17.4685(6)b (Å) 10.0620(3) 26.2840(8) 15.2620(4)c (Å) 13.1060(4) 10.1627(4) 10.5181(3)a (�) 93.098(3) 90 90b (�) 105.453(2) 107.774(4) 105.781(3)c (�) 91.248(2) 90 90V (Å3) 1219.87(6) 2374.48(16) 2698.48(14)Z 2 4 4Dcalc (Mg m�3) 1.482 1.517 1.379Absorption coefficient (mm�1) 1.238 1.272 1.121F(000) 552 1096 1168Crystal size (mm) 0.53 � 0.33 � 0.10 0.41 � 0.10 � 0.09 0.41 � 0.08 � 0.05Theta range for data collected (�) 2.03–26.37 2.24–26.37 1.80–26.37Limiting indices �12 6 h 6 12, �11 6 h 6 10, �21 6 h 6 21,
�12 6 k 6 12, �32 6 k 6 32, �19 6 k 6 19,�16 6 l 6 16 �12 6 l 6 12 �13 6 l 6 13
Reflections collected 21405 [Rint = 0.0351] 20640 [Rint = 0.0344] 13560 [Rint = 0.0000]Independent reflections 5000 4856 13560Reflections observed (>2r) 4367 4102 9566Completeness to theta = 26.37 100.0% 100.0% 100.0%Absorption correction multi-scan multi-scan multi-scanRefinement method full-matrix least-squares on F2 full-matrix least-squares on F2 full-matrix least-squares on F2
Data/restraints/parameters 4367/0/274 4856/6/317 13560/0/273Goodness-of-fit (GOF) on F2 1.072 1.112 0.959Final R indices [I > 2r(I)] R1 = 0.0248,
wR2 = 0.0517R1 = 0.0306,wR2 = 0.0582
R1 = 0.0397,wR2 = 0.0977
R indices (all data) R1 = 0.0336,wR2 = 0.0561
R1 = 0.0411,wR2 = 0.0619
R1 = 0.0665,wR2 = 0.1141
Largest difference peak and hole (e �3) 0.386 to �0.451 0.421 to �0.467 1.078 to �0.805CCDC Reference 952765 952766 952767
Table 4Selected bond lengths (Å) and angles (�) for complexes [SnPh3{S2CNMe(R1)}] (1), [SnPh3{S2CNMe(R2)}] (4) and [SnCy3{S2CNMe(R2)}] (5), [R1 = CH2CH(OMe)2, and R2 = 2-methyl-1,3-dioxolane].
[SnPh3{S2CNR(R1)}] (1) Sn–S1 2.4687(6) Sn–C7 2.139(2) C1–S1 1.753(2)Sn–S2 3.0780(7) Sn–C13 2.157(2) C1–S2 1.683(2)
Sn–C19 2.129(2) C1–N1 1.328(2)S1–Sn–S2 63.553(19) C7–Sn–C13 106.20(8) C7–Sn–C19 106.25(9)C13–Sn–C19 112.25(8)
[SnPh3{S2CNR(R2)}] (4) Sn–S1 3.0183(9) Sn–C7 2.137(3) C1–S1 1.673(4)Sn–S2 2.4751(9) Sn–C13 2.128(3) C1–S2 1.749(3)
Sn–C19 2.158(3) C1–N1 1.331(4)S1–Sn–S2 64.13(3) C7–Sn–C13 113.19(12) C7–Sn–C19 103.47(11)C13–Sn–C19 108.82(12)
[SnCy3{S2CNR(R2)}] (5) Sn–S1 3.2169(11) Sn–C7 2.159(4) C1–S1 1.678(3)Sn–S2 2.4697(11) Sn–C13 2.169(4) C1–S2 1.746(4)
Sn–C19 2.179(4) C1–N1 1.334(5)S1–Sn–S2 61.51(3) C7–Sn–C13 113.34(14) C7–Sn–C19 110.67(13)C13–Sn–C19 109.12(15)
166 I.P. Ferreira et al. / Polyhedron 79 (2014) 161–169
R1 = CH2CH(OMe)2, and R2 = 2-methyl-1,3-dioxolane] (1) exhibitedan unexpected inhibiting concentration values, much lower thanthose obtained for the other complexes or for the control drugs,nystatin or miconazole nitrate, Table 5.
The 90% growth of A. flavus was efficiently inhibited by com-plexes (1) and (4), IC90 0.22 and 0.23 lmol L�1, followed by thecyclohexyl derivatives, (2) and (5), IC90 1.74 and 0.87 lmol L�1.The minimal concentration of complexes (1) and (4) to eliminate50% of A. flavus was found at 5.6 � 10�4 and 9.0 � 10�3 lmol L�1
which is much lower than those values observed for complexes(2) and (5), 0.28 and 0.14 lmol L�1. Complexes (1), (2) and (4)
exhibited the similar inhibition ability towards A. niger, IC90 0.23,0.22 and 0.23 lmol L�1, which are ca of 10 times lower than forcomplex (5), 3.48 lmol L�1. Again complexes (1) and (4) displayedlower IC50, 4.5 � 10�3 and 1.13 � 10�3 lmol L�1. The best inhibi-tion concentrations of the A. parasiticus colonies were obtainedfor complexes (1) and (4), IC90 0.90 lmol L�1 for both complexes,and IC50, 0.018 and 7.0 � 10�5 lmol L�1. Once more complexes(1), (2), and (4), (5) displayed a close related activity, IC90 0.72,0.87 lmol L�1; and 1.80, 1.74 lmol L�1, respectively, but the differ-ent behavior of (1) and (4), in view of the IC50, 2.2 � 10�3 and9.0 � 10�3 lmol L�1, is evident. Despite the low selectivity of (1)
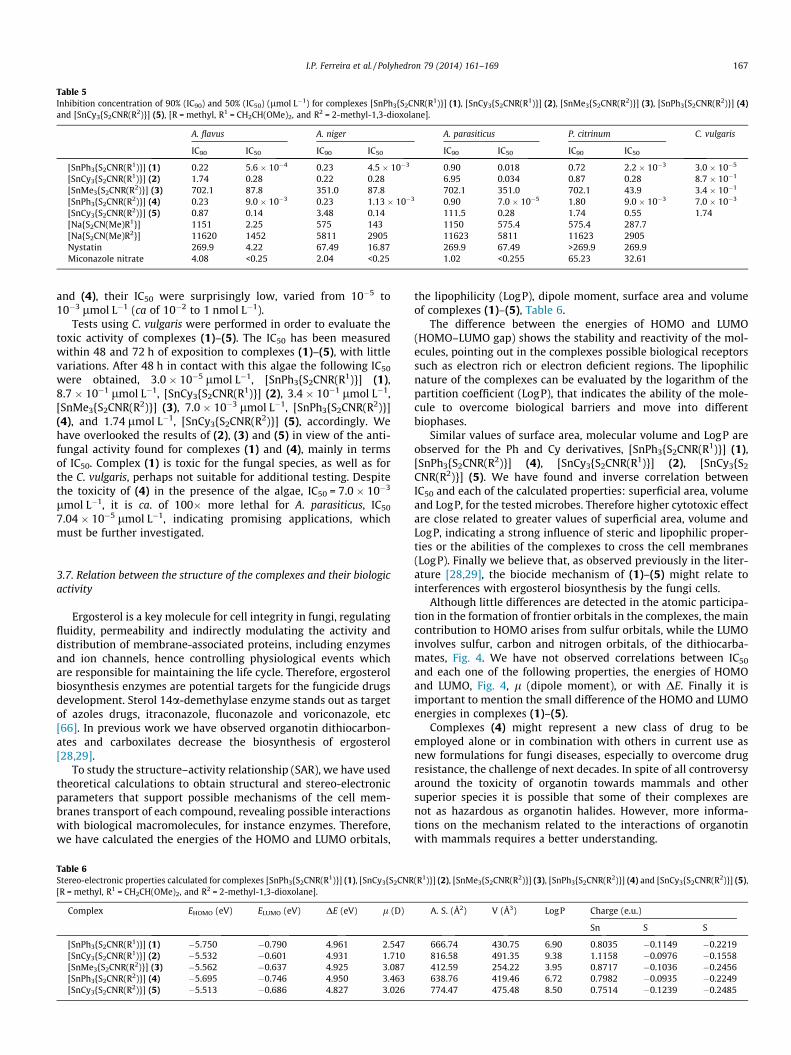
Table 5Inhibition concentration of 90% (IC90) and 50% (IC50) (lmol L�1) for complexes [SnPh3{S2CNR(R1)}] (1), [SnCy3{S2CNR(R1)}] (2), [SnMe3{S2CNR(R2)}] (3), [SnPh3{S2CNR(R2)}] (4)and [SnCy3{S2CNR(R2)}] (5), [R = methyl, R1 = CH2CH(OMe)2, and R2 = 2-methyl-1,3-dioxolane].
A. flavus A. niger A. parasiticus P. citrinum C. vulgaris
IC90 IC50 IC90 IC50 IC90 IC50 IC90 IC50
[SnPh3{S2CNR(R1)}] (1) 0.22 5.6 � 10�4 0.23 4.5 � 10�3 0.90 0.018 0.72 2.2 � 10�3 3.0 � 10�5
[SnCy3{S2CNR(R1)}] (2) 1.74 0.28 0.22 0.28 6.95 0.034 0.87 0.28 8.7 � 10�1
[SnMe3{S2CNR(R2)}] (3) 702.1 87.8 351.0 87.8 702.1 351.0 702.1 43.9 3.4 � 10�1
[SnPh3{S2CNR(R2)}] (4) 0.23 9.0 � 10�3 0.23 1.13 � 10�3 0.90 7.0 � 10�5 1.80 9.0 � 10�3 7.0 � 10�3
[SnCy3{S2CNR(R2)}] (5) 0.87 0.14 3.48 0.14 111.5 0.28 1.74 0.55 1.74[Na{S2CN(Me)R1}] 1151 2.25 575 143 1150 575.4 575.4 287.7[Na{S2CN(Me)R2}] 11620 1452 5811 2905 11623 5811 11623 2905Nystatin 269.9 4.22 67.49 16.87 269.9 67.49 >269.9 269.9Miconazole nitrate 4.08 <0.25 2.04 <0.25 1.02 <0.255 65.23 32.61
I.P. Ferreira et al. / Polyhedron 79 (2014) 161–169 167
and (4), their IC50 were surprisingly low, varied from 10�5 to10�3 lmol L�1 (ca of 10�2 to 1 nmol L�1).
Tests using C. vulgaris were performed in order to evaluate thetoxic activity of complexes (1)–(5). The IC50 has been measuredwithin 48 and 72 h of exposition to complexes (1)–(5), with littlevariations. After 48 h in contact with this algae the following IC50
were obtained, 3.0 � 10�5 lmol L�1, [SnPh3{S2CNR(R1)}] (1),8.7 � 10�1 lmol L�1, [SnCy3{S2CNR(R1)}] (2), 3.4 � 10�1 lmol L�1,[SnMe3{S2CNR(R2)}] (3), 7.0 � 10�3 lmol L�1, [SnPh3{S2CNR(R2)}](4), and 1.74 lmol L�1, [SnCy3{S2CNR(R2)}] (5), accordingly. Wehave overlooked the results of (2), (3) and (5) in view of the anti-fungal activity found for complexes (1) and (4), mainly in termsof IC50. Complex (1) is toxic for the fungal species, as well as forthe C. vulgaris, perhaps not suitable for additional testing. Despitethe toxicity of (4) in the presence of the algae, IC50 = 7.0 � 10�3
lmol L�1, it is ca. of 100� more lethal for A. parasiticus, IC50
7.04 � 10�5 lmol L�1, indicating promising applications, whichmust be further investigated.
3.7. Relation between the structure of the complexes and their biologicactivity
Ergosterol is a key molecule for cell integrity in fungi, regulatingfluidity, permeability and indirectly modulating the activity anddistribution of membrane-associated proteins, including enzymesand ion channels, hence controlling physiological events whichare responsible for maintaining the life cycle. Therefore, ergosterolbiosynthesis enzymes are potential targets for the fungicide drugsdevelopment. Sterol 14a-demethylase enzyme stands out as targetof azoles drugs, itraconazole, fluconazole and voriconazole, etc[66]. In previous work we have observed organotin dithiocarbon-ates and carboxilates decrease the biosynthesis of ergosterol[28,29].
To study the structure–activity relationship (SAR), we have usedtheoretical calculations to obtain structural and stereo-electronicparameters that support possible mechanisms of the cell mem-branes transport of each compound, revealing possible interactionswith biological macromolecules, for instance enzymes. Therefore,we have calculated the energies of the HOMO and LUMO orbitals,
Table 6Stereo-electronic properties calculated for complexes [SnPh3{S2CNR(R1)}] (1), [SnCy3{S2CNR[R = methyl, R1 = CH2CH(OMe)2, and R2 = 2-methyl-1,3-dioxolane].
Complex EHOMO (eV) ELUMO (eV) DE (eV) l (D)
[SnPh3{S2CNR(R1)}] (1) �5.750 �0.790 4.961 2.547[SnCy3{S2CNR(R1)}] (2) �5.532 �0.601 4.931 1.710[SnMe3{S2CNR(R2)}] (3) �5.562 �0.637 4.925 3.087[SnPh3{S2CNR(R2)}] (4) �5.695 �0.746 4.950 3.463[SnCy3{S2CNR(R2)}] (5) �5.513 �0.686 4.827 3.026
the lipophilicity (LogP), dipole moment, surface area and volumeof complexes (1)–(5), Table 6.
The difference between the energies of HOMO and LUMO(HOMO–LUMO gap) shows the stability and reactivity of the mol-ecules, pointing out in the complexes possible biological receptorssuch as electron rich or electron deficient regions. The lipophilicnature of the complexes can be evaluated by the logarithm of thepartition coefficient (LogP), that indicates the ability of the mole-cule to overcome biological barriers and move into differentbiophases.
Similar values of surface area, molecular volume and LogP areobserved for the Ph and Cy derivatives, [SnPh3{S2CNR(R1)}] (1),[SnPh3{S2CNR(R2)}] (4), [SnCy3{S2CNR(R1)}] (2), [SnCy3{S2
CNR(R2)}] (5). We have found and inverse correlation betweenIC50 and each of the calculated properties: superficial area, volumeand LogP, for the tested microbes. Therefore higher cytotoxic effectare close related to greater values of superficial area, volume andLogP, indicating a strong influence of steric and lipophilic proper-ties or the abilities of the complexes to cross the cell membranes(LogP). Finally we believe that, as observed previously in the liter-ature [28,29], the biocide mechanism of (1)–(5) might relate tointerferences with ergosterol biosynthesis by the fungi cells.
Although little differences are detected in the atomic participa-tion in the formation of frontier orbitals in the complexes, the maincontribution to HOMO arises from sulfur orbitals, while the LUMOinvolves sulfur, carbon and nitrogen orbitals, of the dithiocarba-mates, Fig. 4. We have not observed correlations between IC50
and each one of the following properties, the energies of HOMOand LUMO, Fig. 4, l (dipole moment), or with DE. Finally it isimportant to mention the small difference of the HOMO and LUMOenergies in complexes (1)–(5).
Complexes (4) might represent a new class of drug to beemployed alone or in combination with others in current use asnew formulations for fungi diseases, especially to overcome drugresistance, the challenge of next decades. In spite of all controversyaround the toxicity of organotin towards mammals and othersuperior species it is possible that some of their complexes arenot as hazardous as organotin halides. However, more informa-tions on the mechanism related to the interactions of organotinwith mammals requires a better understanding.
(R1)}] (2), [SnMe3{S2CNR(R2)}] (3), [SnPh3{S2CNR(R2)}] (4) and [SnCy3{S2CNR(R2)}] (5),
A. S. (Å2) V (Å3) LogP Charge (e.u.)
Sn S S
666.74 430.75 6.90 0.8035 �0.1149 �0.2219816.58 491.35 9.38 1.1158 �0.0976 �0.1558412.59 254.22 3.95 0.8717 �0.1036 �0.2456638.76 419.46 6.72 0.7982 �0.0935 �0.2249774.47 475.48 8.50 0.7514 �0.1239 �0.2485

Fig. 4. The HOMO (i) and LUMO (ii) plots for complexes [SnPh3{S2CNR(R1)}] (1), [SnCy3{S2CNR(R1)}] (2), [SnMe3{S2CNR(R2)}] (3), [SnPh3{S2CNR(R2)}] (4) and[SnCy3{S2CNR(R2)}] (5), [R = methyl, R1 = CH2CH(OMe)2, and R2 = 2-methyl-1,3-dioxolane].
168 I.P. Ferreira et al. / Polyhedron 79 (2014) 161–169
4. Conclusions
Genome studies have provided a better understanding of thecloser distance between the fungi kingdom and human species[67]. It is possible that the chemical and genetic similarities explainwhy some mammals fungi diseases are difficult to treat. Resistanceof microbes to conventional treatment has been reported andrelates most to (i) alterations in the target of the drug, (ii) interfer-ence with ergosterol biosynthesis and/or (iii) increase in theexpression of drug efflux pumps. These points altogether makethe search for alternative drugs mandatory and metal-based com-pounds might represent a novel group of antifungal agents, eitheralone or in combined formulation with drugs in current use toovercome resistance.
The synthesis, characterization and biological aspects of [SnPh3
{S2CNR(R1)}] (1), [SnCy3{S2CNR(R1)}] (2), [SnMe3{S2CNR(R2)}] (3),[SnPh3{S2CNR(R2)}] (4), [SnCy3{S2CNR(R2)}] (5), where R = methyl,R1 = CH2CH(OMe)2, and R2 = 2-methyl-1,3-dioxolane have been thefocus of this report. 13C and 119Sn couplings served to conclude thatthe structure of the compounds in solution and in the solid state arenot too different. Therefore solvation and dynamic processes in solu-tion might be neglected. In addition the complexes have been char-acterized by IR, 119Sn Mössbauer spectroscopy and X-ray diffraction,complexes (1), (4) and (5). Four species of fungus have been
cultivated in the presence of complexes (1)–(5). In terms of IC50 com-plexes (1) and (4) displayed remarkable activities, exhibiting verylow inhibition concentration against all fungus. However the biocideactivity of complex (4) is noteworthy, not only in view of its low IC50,7.04 � 10�5 lmol L�1, (nanomolar) in the presence of A. parasiticus,but also because it is 100 times more lethal for the fungi than toC. vulgaris, IC50 = 7.0 � 10�3 lmol L�1. However this results are toopremature to be conclusive. Therefore, complex (4) deserves moretests, for example concerning possible mechanism of biocide action.Finally, preliminary mechanistic studies by SAR experiments led usto suppose that the biocide activities might be influenced bytheir steric and lipophilic properties or the abilities of the complexesto bind the cell membranes interfering with the ergosterolbiosynthesis.
Acknowledgement
This work was supported by CNPq and FAPEMIG – Brazil.
Appendix A. Supplementary data
CCDC 952765, 952766, 952767; contains the supplementarycrystallographic data for complexes. These data can be obtained

I.P. Ferreira et al. / Polyhedron 79 (2014) 161–169 169
free of charge via http://www.ccdc.cam.ac.uk/conts/retriev-ing.html, or from the Cambridge Crystallographic Data Centre, 12Union Road, Cambridge CB2 1EZ, UK; fax: (+44) 1223 336 033; ore-mail: [email protected]. Supplementary data associatedwith this article can be found, in the online version, at http://dx.doi.org/10.1016/j.poly.2014.05.001.
References
[1] E.R.T. Tiekink, Appl. Organomet. Chem. 22 (2008) 533.[2] P.J. Heard, Main group dithiocarbamate complexes, Progress in Inorganic
Chemistry, vol. 53, John Wiley & Sons Inc, New York, 2005, p. 1.[3] G. Hogarth, Transition metal dithiocarbamates: 1978–2003, Progress in
Inorganic Chemistry, vol. 53, John Wiley & Sons Inc, New York, 2005, p. 71.[4] F.W. Sunderman, Ann. Clin. Lab. Sci. 9 (1979) 1.[5] D.L. Bodenner, P.C. Dedon, P.C. Keng, R.F. Borch, Cancer Res. 46 (1986) 2745.[6] J.J. Suh, H.M. Pettinati, K.M. Kampman, C.P. O’Brien, J. Clin. Psychopharmacol.
26 (2006) 290.[7] B. Cvek, Z. Dvorak, Curr. Pharm. Design 13 (2007) 3155.[8] P.J. Nieuwenhuizen, J. Reedijk, M. Van Duin, W.J. McGill, Rubber Chem.
Technol. 70 (1997) 368.[9] M. Cicotti, Compound class: alkylenebis(dithiocarbamates), in: P.W. Lee, H.
Aizawa, A.C. Barefoot, J.J. Murphy (Eds.), Handbook of Residue AnalyticalMethods for Agrochemical, vol. 2, Wiley, Chichester, 2003, p. 1089.
[10] D. Fan, M. Afzaal, M.A. Mallik, C.Q. Nguyen, P. O’Brien, P.J. Thomas, Coord.Chem. Rev. 251 (2007) 1878.
[11] A.G. Davies, M. Gielen, K.H. Pannell, E.R.T. Tiekink (Eds.), Tin Chemistry –Fundamentals, Frontiers and Applications, Wiley, Chichester, 2008.
[12] H. Tlahuext, R. Reyes-Martinez, G. Vargas-Pineda, M. Lopez-Cardoso, H. Hopfl,J. Organomet. Chem. 696 (2011) 693.
[13] A.F.A. Muthalib, I. Baba, H. Khaledi, H.M. Ali, E.R.T. Tiekink, Z. Kristallogr. 229(2014) 39.
[14] J.P. Fuentes-Martinez, I. Toledo-Martinez, P. Roman-Bravo, P.G.Y. Garcia, C.Godoy-Alcantar, M. Lopez-Cardoso, H. Morales-Rojas, Polyhedron 28 (2009)3953.
[15] A. Normah, K.N. Farahana, B. Ester, H. Asmah, N. Rajab, A.A. Halim, Res. J. Chem.Environ. 15 (2011) 544.
[16] N.A. Celis, R. Villamil-Ramos, H. Hoepfl, I.F. Hernandez-Ahuactzi, M. Sanchez,L.S. Zamudio-Rivera, V. Barba, Eur. J. Inorg. Chem. 16 (2013) 2912.
[17] E. Santacruz-Juarez, J. Cruz-Huerta, I.F. Hernandez-Ahuactzi, R. Reyes-Martinez, H. Tlahuext, H. Morales-Rojas, H. Hopfl, Inorg. Chem. 47 (2008)9804.
[18] S. Shahzadi, S. Ali, M. Fettouhi, J. Chem. Crystallogr. 38 (2008) 273.[19] M. Safari, M. Yousefi, H.A. Jenkins, M.B. Torbati, A. Amanzadeh, Med. Chem.
Res. 22 (2013) 5730.[20] L. Tian, X. Zheng, X. Zheng, Y. Sun, D. Yan, L. Tu, J. Organomet. Chem. 25 (2011)
785.[21] F. Shaheen, Z.U. Rehman, S. Ali, A. Meetsma, Polyhedron 31 (2012) 697.[22] R. Hernandez-Altamirano, V.Y. Mena-Cervantes, T.E. Chavez-Miyauchi, D.A.
Nieto-Alvarez, M.A. Dominguez-Aguilar, L.S. Zamudio-Rivera, V. Barba, F.J.Fernandez-Perrino, S. Perez-Miranda, H.I. Beltran, Polyhedron 52 (2013) 301.
[23] H.I. Beltran, C. Damian-Zea, S. Hernandez-Ortega, A. Nieto-Camacho, M.T.Ramirez-Apan, J. Inorg. Biochem. 101 (2007) 1070.
[24] J.S. White, J.M. Tobin, J.J. Cooney, Can. J. Microbiol. 45 (1999) 541.[25] J.J. Cooney, S. Wuertz, J. Ind. Microbiol. 4 (1989) 375.[26] D.C. Menezes, G.M. de Lima, A.O. Porto, M.P. Ferreira, J.D. Ardisson, R.A. Silva,
Main Group Met. Chem. 30 (2007) 49.[27] D.C. Menezes, G.M. de Lima, G.S. de Oliveira, A.V. Boas, A.M.A. Nascimento, F.T.
Vieira, Main Group Met. Chem. 31 (2008) 21.[28] D.C. Menezes, F.T. Vieira, G.M. de Lima, J.L. Wardell, M.E. Cortes, M.P. Ferreira,
M.A. Soares, A.V. Boas, Appl. Organomet. Chem. 22 (2008) 221.[29] D.C. Menezes, G.M. de Lima, F.A. Carvalho, M.G. Coelho, A.O. Porto, R. Augusti,
J.D. Ardisson, Appl. Organomet. Chem. 24 (2008) 650.[30] D.C. Menezes, F.T. Vieira, G.M. de Lima, A.O. Porto, M.E. Cortes, J.D. Ardisson,
T.E. Albrecht-Schmitt, Eur. J. Med. Chem. 40 (2005) 1277.[31] R.A. Howie, G.M. de Lima, D.C. Menezes, J.L. Wardell, S.M.S.V. Wardell, D.J.
Young, E.R.T. Tiekink, Cryst. Eng. Comm. 10 (2008) 1626.[32] D.C. Menezes, G.M. de Lima, A.O. Porto, C.L. Donnici, J.D. Ardisson, A.C.
Doriguetto, J. Ellena, Polyhedron 23 (2004) 2103.
[33] D.C. Menezes, G.M. de Lima, C.A. Cavalcanti, J.A.F. dos Santos, I.P. Ferreira, E.B.Paniago, J.L. Wardell, S.M.S.V. Wardell, K. Krambrock, I.C. Mendes, H. Beraldo, J.Mol. Struct. 988 (2011) 1.
[34] I.P. Ferreira, G.M. de Lima, E.B. Paniago, J.A. Takahashi, K. Krambrock, C.B.Pinheiro, J.L. Wardell, L.C. Visentin, J. Mol. Struct. 1048 (2013) 357.
[35] I.P. Ferreira, G.M. de Lima, E.B. Paniago, W.R. Rocha, J.A. Takahashi, C.B.Pinheiro, J.D. Ardisson, Eur. J. Med. Chem. 58 (2012) 493.
[36] I.P. Ferreira, G.M. de Lima, E.B. Paniago, J.A. Takahashi, C.B. Pinheiro, Inorg.Chim. Acta (2014) accepted for publication.
[37] I.P. Ferreira, G.M. de Lima, E.B. Paniago, J.A. Takahashi, C.B. Pinheiro, J. Coord.Chem. 67 (2014) 1097.
[38] G.M. Sheldrick, Acta Crystallogr., Sect. A 64 (2008) 112.[39] G.M. Sheldrick, SHELXL-97. Program for Crystal Structure Refinement, University
of Göttingen, Germany, 1997.[40] L.J. Farrugia, J. Appl. Crystallogr. 30 (1997) 565.[41] C.F. Macrae, P.R. Edgington, P. McCabe, E. Pidcock, G.P. Shields, R. Taylor, M.J.
van De Streek, J. Appl. Crystallogr. 39 (2006) 453.[42] R.G. Parr, W. Yang, Density-Functional Theory of Atoms and Molecule, Oxford
University Press, Oxford, 1989.[43] A.D. Becke, J. Chem. Phys. 98 (1993) 5648.[44] C.T. Lee, W.T. Yang, R.G. Parr, Phys. Rev. B 37 (1988) 785.[45] R. Ditchfie, W.J. Hehre, J.A. Pople, J. Chem. Phys. 54 (1971) 724.[46] W.J. Hehre, R. Ditchfie, J.A. Pople, J. Chem. Phys. 56 (1972) 2257.[47] P.J. Hay, W.R. Wadt, J. Chem. Phys. 82 (1985) 299.[48] M.J. Frisch, G.W. Trucks, H.B. Schlegel, G.E. Scuseria, M.A. Robb, J.R. Cheeseman,
J.A. Montgomery Jr., T. Vreven, K.N. Kudin, J.C. Burant, J.M. Millam, S.S. Iyengar,J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G.A.Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa,M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J.E. Knox,H.P. Hratchian, J.B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R.E.Stratmann, O. Yazyev, A.J. Austin, R. Cammi, C. Pomelli, J.W. Ochterski, P.Y.Ayala, K. Morokuma, G.A. Voth, P. Salvador, J.J. Dannenberg, V.G. Zakrzewski, S.Dapprich, A.D. Daniels, M.C. Strain, O. Farkas, D.K. Malick, A.D. Rabuck, K.Raghavachari, J.B. Foresman, J.V. Ortiz, Q. Cui, A.G. Baboul, S. Clifford, J.Cioslowski, B.B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R.L.Martin, D.J. Fox, T. Keith, M.A. Al-Laham, C.Y. Peng, A. Nanayakkara, M.Challacombe, P.M.W. Gill, B. Johnson, W. Chen, M.W. Wong, C. Gonzalez, J.A.Pople, Gaussian 03, Revision C.02, Gaussian Inc, Wallingford, CT, 2004.
[49] Calculator Plugins were used for structure property prediction and calculation,MarvinView 5.10.3, in, ChemAxon 2012, http://www.chemaxon.com [30/06/2013].
[50] L. Ronconi, L. Giovagnini, C. Marzano, F. Bettio, R. Graziani, G. Pilloni, D.Fregona, Inorg. Chem. 44 (2005) 1867.
[51] H.L.M. Vangaal, J.W. Diesveld, F.W. Pijpers, J.G.M. Vanderlinden, Inorg. Chem.18 (1979) 3251.
[52] D. Dakternieks, H.J. Zhu, D. Masi, C. Mealli, Inorg. Chem. 31 (1992) 3601.[53] J. Holecek, M. Nadvornik, K. Handlir, A. Lycka, J. Organomet. Chem. 315 (1986)
299.[54] T.P. Lockhart, W.F. Manders, J.J. Zuckerman, J. Am. Chem. Soc. 107 (1985) 4546.[55] T.P. Lockhart, W.F. Manders, J. Am. Chem. Soc. 109 (1987) 7015.[56] J.S. Tse, F.L. Lee, J. Gabe, Acta Crystallogr., Sect. C C42 (1986) 1876.[57] A. Asadi, C. Eaborn, P. Hitchcock, M.M. Meehan, J.D. Smith, Inorg. Chem. 42
(2003) 4141.[58] S. Calogero, P. Ganis, V. Peruzzo, G. Tagliavini, J. Organomet. Chem. 179 (1979)
145.[59] M.B. Hossain, J.L. Lefferts, K.C. Molloy, D.V.D. Helm, J.J. Zuckerman, Inorg. Chim.
Acta 36 (1979) L409.[60] A. Bondi, J. Phys. Chem. 68 (1964) 441.[61] H.A. Bent, Chem. Rev. 61 (1961) 275.[62] C. Garcia-Vidal, D. Viasus, J. Carratala, Curr. Opin. Infect. Dis. 26 (2013) 270.[63] T.J. Walsh, E.J. Anaissie, D.W. Denning, R. Herbrecht, D.P. Kontoyiannis, K.A.
Marr, V.A. Morrison, B.H. Segal, W.J. Steinbach, D.A. Stevens, J.A. van Burik, J.R.Wingard, T.F. Patterson, Clin. Infect. Dis. 46 (2008) 327.
[64] D.W. Denning, S. Park, C. Lass-Florl, M.G. Fraczek, M. Kirwan, R. Gore, J. Smith,A. Bueid, P. Bowyer, D.S. Perlin, Clin. Infect. Dis. 52 (2011) 1123.
[65] A.S. Zacchino, M.P. Gupta, Manual de técnicas in vitro para la detección decompuestos antifúngicos, Corpus Editorial y Distribuidora, Rosario, 2007. p. 85.
[66] H.K. Munayyer, P.A. Mann, A.S. Chau, T. Yarosh-Tomaine, J.R. Greene, R.S. Hare,L. Heimark, R.E. Palermo, D. Loebenberg, P.M. McNicholas, Antimicrob. AgentsChemother. 48 (2004) 3690.
[67] Q. Zeng, A.J. Morales, G. Cottarel, Trends Genet. 17 (2001) 682.





























