Embed Size (px)
Citation preview

A WORKSHOP ON:VIRTUAL GENETIC ENGINEERING USING BIO-SOFTWARES:PRIMER DESIGN
May 07, 2015
National Institute of Genetic Engineering and Biotechnology
Saeid Kadkhodaei, PhD
SynHiTech

Sequence Data ManipulationSequence Data Manipulation
• Data Formats (input, output)• Conversions• Alignment• Codon optimization• Restriction mapping• In silico cloning• ORF finding• Contig assembly• …
Part 1

Sequence Data ManipulationSequence Data Manipulation
• Formats Single files FASTA Gene Bank ABI SEQ ….
Multiple sequences

Sequence Data ManipulationSequence Data Manipulation
Class
Definition
CON Entry constructed from segment entry sequences; if unannotated, annotation may be drawn from segment entries
PAT Patent EST Expressed Sequence Tag GSS Genome Survey Sequence HTC High Thoughput CDNA sequencing HTG High Thoughput Genome sequencing MGA Mass Genome Annotation WGS Whole Genome Shotgun TSA Transcriptome Shotgun Assembly STS Sequence Tagged Site STD Standard (all entries not classified as above)
• Sequence Data Classes in Databases

Sequence Data ManipulationSequence Data Manipulation
• Taxonomic Division
Division CodeBacteriophage PHGEnvironmentalSample ENVFungal FUNHuman HUMInvertebrate INVOtherMammal MAMOtherVertebrate VRTMusmusculus MUSPlant PLNProkaryote PROOtherRodent RODSynthetic SYNTransgenic TGNUnclassified UNCViral VRL

Sequence Data ManipulationSequence Data Manipulation
• Structure of an Entry
ID-identification RA-referenceauthor(s)AC-accessionnumber RT-referencetitlePR-projectidentifier RL-referencelocationDT-date DR-databasecross-referenceDE-description CC-commentsornotesKW-keyword AH-assemblyheaderOS-organismspecies AS-assemblyinformationOC-organismclassification FH-featuretableheaderOG-organelle FT-featuretabledataRN-referencenumber XX-spacerlineRC-referencecomment SQ-sequenceheaderRP-referencepositions CO-contig/constructlineRX-referencecross-reference bb-(blanks)sequencedataRG-referencegroup //-terminationline

Sequence Data ManipulationSequence Data Manipulation
• Standard Base Codes (IUPAC-IUB)
Code Base Description
G Guanine G A Adenine A T Thymine T C Cytosine C R Purine (A or G) Y Pyrimidine (C or T or U) M Amino (A or C) K Ketone (G or T) S Strong interaction (C or G) W Weak interaction (A or T) H Not-G (A or C or T) H follows G in the
alphabet B Not-A (C or G or T) B follows A V Not-T (not-U) (A or C or G) V follows U D Not-C (A or G or T) D follows C N Any (A or C or G or T)

A Simple Genetic Engineering ProcedureA Simple Genetic Engineering Procedure

Plasmid Preparation
Plasmid Preparation
Insert Preparation
Insert Preparation
Vector Selection
Vector Selection
Primer Design
Primer Design
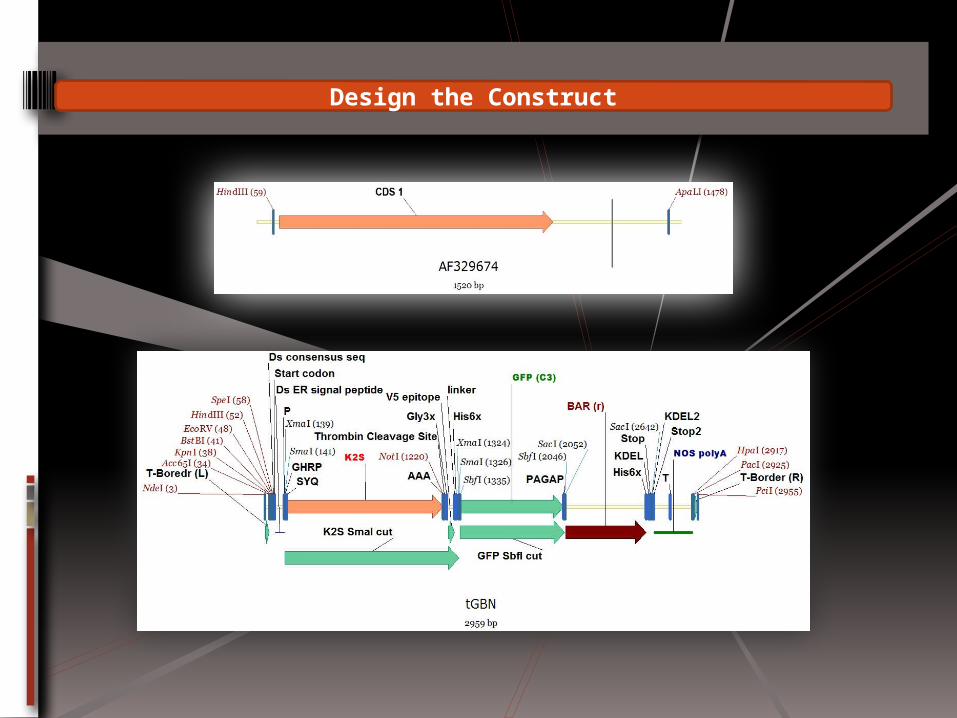
Design the Construct
Design the Construct
PCR: Insert Amplificati
on
PCR: Insert Amplificati
on
Electrophoresis
DNA Dilution
Electrophoresis
DNA DilutionDouble Digestion with
same REsDouble Digestion with
same REsGel
ExtractionGel
ExtractionLigatio
nLigatio
nCompetent CellCompetent
Cell Transformation to E.coli
Transformation to E.coli
Confirmation
Confirmation
Colony PCRColony
PCRDigestionDigestion
Sequencing
Sequencing
Transformation to the HostTransformation to
the Host
Flow Chart of a Simple Genetic Transformation Project
Design the Construct

Primer Design
• Purpose
• Principles
• Softwares
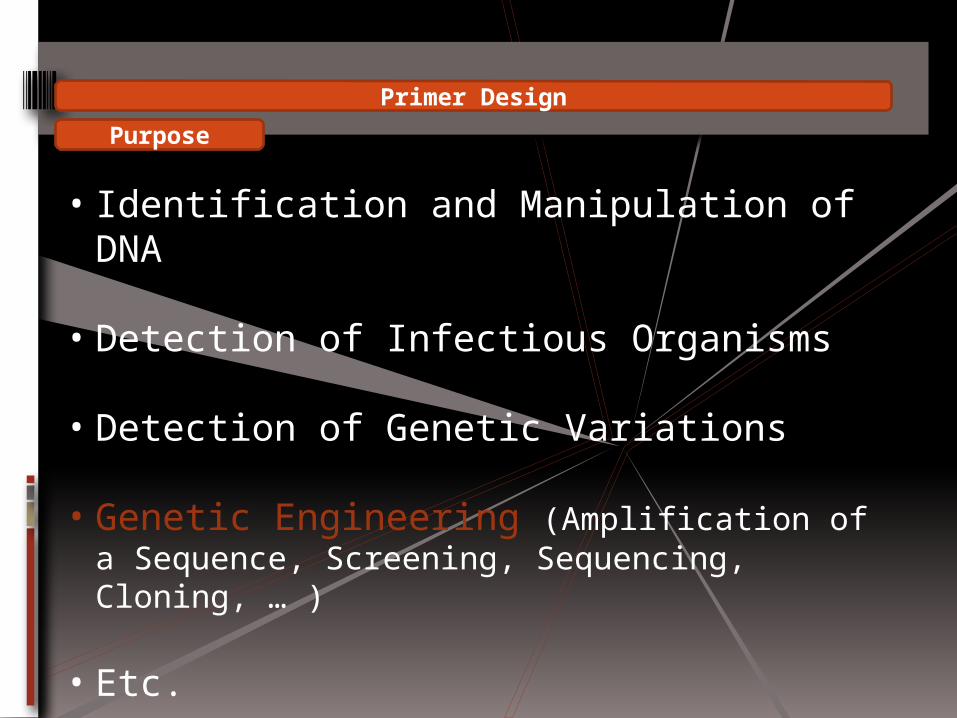
Primer Design
• Identification and Manipulation of DNA
• Detection of Infectious Organisms
• Detection of Genetic Variations
• Genetic Engineering (Amplification of a Sequence, Screening, Sequencing, Cloning, … )
• Etc.
Purpose

Primer Design
1. Primer Length: 18-22 bp • At least 15 bases to ensure uniqueness• Usually, 17-28 bases. This range varies based on if you
can find unique primers with appropriate annealing temperature within this range.
2. Primer Melting Temperature: 52-58oC• Nearest Neighbor Thermodynamic Theory: Tm(oC) = {ΔH/ ΔS + R ln(C)} - 273.15
3. Primer annealing temperature: • Ta = 0.3 x Tm(primer) + 0.7 Tm (product) – 14.9• Ta = Tm(primer) – 4C
Principles

Primer Design
4. Base Composition:• GC Content: 40-60%• Avoid long A+T and G+C rich region if possible
5. GC Clamp: • Not more than 3 G's or C's in the last 5 bases at the 3'
end of the primer. • Let the primer end in A or T. • The better the 3’ end binds the more efficient is DNA
synthesis.
Principles

Primer Design
6. Primer Secondary Structures: • Hairpins : ΔG of -2 kcal/mol for a 3' end hairpin ΔG of -3 kcal/mol for an internal hairpin Larger negative value for ΔG indicates stable, undesirable hairpins.
Principles

Primer Design
6. Primer Secondary Structures:
• Self Dimer : ΔG of -5 kcal/mol for a 3' end self dimer ΔG of -6 kcal/mol for an internal self dimer
Principles

Primer Design
6. Primer Secondary Structures: • Cross Dimer : ΔG of -5 kcal/mol for a 3' end cross dimer ΔG of -6 kcal/mol for an internal cross dimer
Principles

Primer Design
7. Repeats: A maximum number of 4 di-nucleotide
8. Runs: A maximum number of 4 single base runs
9. 3' End Stability: An unstable 3' end (less negative ΔG) will result in less false priming.
10. Avoid Template secondary structure11. Avoid Cross homology: Commonly, primers are designed and then BLASTed to test the specificity.
12. Amplicon Length13. Product position: Generally, the sequence close to the 3' end is known with greater confidence and hence preferred most frequently.
Principles

Primer Design
14. Tm of Product
15. Optimum Annealing temperature (Ta Opt):Ta Opt = 0.3 x(Tm of primer) + 0.7 x(Tm of product) - 14.9whereTm of primer is the melting temperature of the less stable primer-template pair
16. Primer Pair Tm Mismatch Calculation: The difference of 5oC or more can lead no amplification.
Principles

Primer Design
Degenerate Primers (guessmer)
In some cases, DNA sequences are either unavailable or difficult to align. Then, a single/group of related proteins can be back translated into nucleotide sequences that will be used as template to design primers/probes. Back translation is both problematic and feasible. While the genetic codes are degenerate, different organisms do show preferential biases in codon usage, which can be used to limit the possible back-translated nucleotide sequences.
Principles

Primer Design
Degenerate Primers (guessmer)Strategy:
• Back translate the protein sequence using corresponding codon usage table. Identify 5’ and 3’ regions where there is the least ambiguity.
• Design and match forward and reverse primers as before. But the primers shall be about 30 bases long in order to offset the decreased hybridization specificity caused by mismatched bases.
• Set higher annealing temperature to increase the primer annealing stringency.
Principles

Primer Design
Semi-Universal PrimersPrimers can be designed to amplify only a subset of
template sequences from a large group of similar sequences. For example, design primer to amplify HPV type 1 and type 6 gene, but not other types.
Strategy:1. Align all types of HPV genes.2. Identify a subset of genes that are more similar to each other than to other
subsets. In this case, type 1 and type 6.3. Find the 5’ and 3’ regions that are conserved between type 1 and type 6, but are
variable in other types.4. Design forward primers from the 5’ region and reverse primers from the 3’
region.5. Matching forward and reverse primers to find the best pair.6. Ensure uniqueness in all template sequences.7. Ensure uniqueness in possible contaminant sources.
Principles

Primer Design
The basic rules :• Appropriate hybridization • Specificity and • Stability
Principles
Summary

Primer Design
• Primer Premier follows all the guidelines specified for PCR primer design. Primer Premier can be used to design primers for single templates, alignments, degenerate primer design, restriction enzyme analysis. contig analysis and design of sequencing primers.
• Oligo: Life Science Software, standalone application
• GCG: Accelrys, ICBR maintains the server.
• Primer3: MIT, standalone / web application
Softwares

Primer Design
• BioTools: BioTools, Inc. ICBR distributes the license.• Primer select• Perlprimer• AlleleID • Beacon Designer• PrimerPlex • GeneFisher • xprimer • Web Primer (Stanford)• EMBL Primer Design (caution, uses Java)
• Others: Primer!, NBI oligo program, idtDNA oligo analyzer, etc.
Softwares

Primer design principles
Include the start and the stop codons in the primers, unless you are using a fusion protein (GST, His-tag etc.) in one or both ends.
Make sure the amplified DNA will be inserted into the vector in the correct reading frame and in correct orientation.
Add enough nucleotides in the 5' ends to guarantee efficient cleavage by restriction enzymes. Most enzymes are less efficient the closer the cleavage site is to the 5' end of the DNA. 5-6 nucleotides should be sufficient. NEB website
Primer Design

The enzymes you can use depends, in addition to the results of the restriction analysis, on the vector's polylinker or multiple cloning site.
If the start and stop codons are to be inserted by PCR, restriction sites carrying the codon ATG for initiation methionine or TAA/TGA/TAG stop codons become very convenient. It is also wise to plan ahead and anticipate which other vectors you might need to try and choose enzymes which are present in those polylinkers as well.
NcoI recognizes hexa-nucleotide CCATGG and is my favourite for the 5‘ end of the gene. The reasons are many:It cleaves a sequence with the ATG codon, it leaves a four-base overhang (unlike the much used and almost as often cursed NdeI which leaves only two-base overhang)
Primer design principles Primer Design

REBASE / CLC / …To check the sequences of REs or
to find specific sequences in REs
Primer Design

pay attention to the open reading frame, both in the N- and C-termini of the expressed protein, to avoid introducing frameshifts and hence ruining your construct. Many vectors are also available in three versions which differ in the reading frame of (part of) the polylinker and one should choose carefully the correct version.
Primer design principles Primer Design

Introduce different restriction sites to the 5' and 3' oligos as this forces the orientation of the fragment in the vector and reduces the possibility of the vector to self-ligate and thus lower the cloning efficiency
Include always at least 5 to 6 nucleotides (Ts and As have lower annealing temperature as Cs and Gs) before the restriction enzyme recognition site to ensure the enzymes can cleave the DNA. Some enzymes are very reluctant to act on sequences at the very end of a DNA fragment
Primer design principles Primer Design

The annealing part of the oligo should be ca. 21-24 nt and carry similar and fairly even composition of nucleotides in both primers for efficient annealing in the PCR tube
Some people recommend to end the primer in a C or G to make sure the end where polymerase acts upon is tightly bound to the template. It doesn’t seem to be absolutely necessary, but there is certainly no harm in doing that either.
Primer design principles Primer Design

Primer OrderPrimer Design
For PCR you need very small amounts and I would suggest you to order the smallest scale the service offers.
From a 40 nmol synthesis you can easily get enough oligo to run hundreds of PCRs. And if everything works OK, a single run will be enough.

which synthesis scale to orderThe synthesis scale is based on the amount of the first base attached to the solid support to start the oligo synthesis. For larger scales, the amount of solid support is increased. It is NOT the expected final yield. The yield depends on the size of the oligo, the coupling efficiency, and the base composition.
Primer OrderPrimer Design
Decreasing yield as the length of the oligo increases
All the synthesis steps involve an increasing inefficiency as the oligo length is increased. Additional purification such as PAGE or HPLC will also result in the loss of some product. Therefore, synthesis inefficiency coupled with losses from purification, the quantity of oligo ultimately received is always lower than the theoretical yield.
Primer OrderPrimer Design

To decide which scale synthesis to order, first determine the amount of oligo required (no. of PCR reactions to carry out). After that, compare required yield with the yield guaranteed for the scale. Please note that modifications (if any), and purification will reduce the final yield due to increased processing. As such, order such oligos at a higher synthesis scale.For most PCR and sequencing needs, only a minute amount of oligo is needed.
For example, the majority of sequencing protocols call for 10 picomoles of primer. For an average 25-mer oligonucleotide, 1 OD260 unit is equal to about 4 nanomoles, or enough primer to do 400 PCR or sequencing reactions. Unless a very large number of reactions are planned using a given primer, it is seldom necessary to order more than a 50 nmole scale primer.
Primer OrderPrimer Design

Scale Guaranteed Amount of Oligo
50 nmole scale, PCR Grade
2 - 3 OD
200 nmole scale, PCR Grade
10 OD
1umole scale, PCR Grade 40 ODPAGE Purified Oligos 1 ODModified Oligos + TOP purified
3 ODNote: Above mentioned final yield are applicable to Oligos up to 50mers only. For longer Oligos, please enquire.
Primer OrderPrimer Design

Scale of Synthesis Estimated Number of
Reactions 25 nmole 500 to 2,500 50 nmole 1,000 to 5,000 200 nmole 4,000 to 20,000 1 µmole 20,000 to 100,000 10 µmole 100,000 to 1,000,000
Synthesis Scale for PCR Applications When ordering custom oligos for PCR applications, the scale of synthesis determines the number of reactions provided. The table below assumes a 100 µl PCR reaction and a final oligo concentration of 0.1 to 0.5 µM.
Primer OrderPrimer Design

What purification methods are available?
Several types of purification are available :
1. Desalting: Every oligo is desalted to remove residual by-products from synthesis deprotection and cleavage.
2. RP1 (Reverse-Phase Cartridge): Separation on a reverse-phase cartridge offers the next level of purity.
3. HPLC: Efficient purification method for oligos with fluorophores and large scale synthesis.
4. Poly-Acrylamide Gel Electrophoresis (PAGE): Recommended when a highly purified product is required.
Primer OrderPrimer Design

Do I use TE buffer or DI water?
Either can be used though DI water is increasing favored now for the simple fact that EDTA from TE buffer inhibits PCR, where oligos are most widely employed. DI water on the other hand would require careful handling which is easily achieved through proper sterile technique.Under acidic conditions, DNA oligos can become depurinated. On the other hand, the phosphodiester bond of RNA oligos can be hydrolyzed under basic conditions.
Primer OrderPrimer Design

Primer OrderPrimer Design
How do I store my oligos?
Oligos are chemically stable. If stored dry (lyophilized), they will be stable for years. However upon hydration, they are susceptible to degradation by nucleases. Even so, hydrated oligos, if handled correctly, should still be stable for years. Oligos can undergo degradation from exposure to low pH (<3) or heat, leading to depurination and cleavage. Any oligo can be degraded by contaminating environmental nucleases.You can store hydrated oligos by refrigeration or freezing but refrigeration is increasingly preferred. Refrigeration avoids freeze-thaw cycles and offers convenience. Even when oligos are stored in DI water and refrigerated, they have been observed to be stable for over two years.When freezing is preferred, it is recommended to aliquot stock concentrates to several tubes.

Primer OrderPrimer Design
How to calculate the Tm of an oligo? Why does the Tm value from my own primer software differ from the Tm reading on the datasheet? The two standard approximation calculations. For sequences less than 14 nucleotides the formula is:Tm= (wA+xT) * 2 + (yG+zC) * 4
where w,x,y,z are the number of the bases A,T,G,C in the sequence, respectively.
For sequences longer than 14 nucleotides, the equation used is (Wallace Rule):Tm= 64.9 +41*(yG+zC-16.4)/(wA+xT+yG+zC)
ASSUMPTIONS:Both equations assume that the annealing occurs under the standard conditions of 50 nM primer, 50 mM Na+, and pH 7.0.
There are up to 9 different algorithms used to calculate the Tm of oligos and the different Tm values does not indicate that the primers will differ from one oligo service provider to another. Simply use the same algorithm or software to optimize the Tm of your oligos. Then perform your experiments as you designed it to be. In the process of synthesis, we don't change anything with regards to the Tm of the oligos and the difference shown in the datasheet is just a matter of different ways of calculations.

Primer OrderPrimer Design
Is PCR grade pure enough for routine PCR and sequencing?
Purification options incur additional cost to oligos. PCR Grade oligos rack up considerable saving and also facilitate speed in completion and delivery.For PCR and sequencing, oligos below 30 bases in length are acceptable with PCR Grade. PCR and sequencing can tolerate up to 50% truncated sequences. Anything above 30 bases, you are highly advised against using crude oligos for these applications. If sequencing targets are large templates like BAC, cosmids or bacterial genomes, it is highly advised to have them TOP purified.

Primer OrderPrimer Design
What is TOP purification and what can it be used for?TOP: Trityl-On Oligonucleotide Purification
TOP is a simple, high-throughput approach to oligonucleotide purification. TOP provides greater than 90% pure full-length oligo and is ideal for applications that require high-quality DNA. It efficiently removes truncated failure sequences generated during the synthesis process that do not contain a 5' dimethoxytrityl group. In addition, deprotection solution salts and by-products (i.e. benzamide protecting groups) are also removed simultaneously.TOP chemistry is based on the principle of reverse-phase (RP) chromatography. Oligonucleotides purified by TOP are synthesized with the final 5' terminus protecting group [trityl or dimethoxyltrityl (DMT)] left on the oligo. The hydrophobic nature of the trityl group permits tighter retention of the desired full-length oligo than the truncated failure sequences that do not contain a trityl group. The failure sequences are washed from the tube with a low percent acetonitrile rinse. Retained oligonucleotides are then detritylated on column with trifluoroacetic acid (TFA) to remove the acid-labile trityl group. Residual acid is washed from the tube with two rinses. The full-length oligo is recovered in its purified form with an aqueous-organic solvent.

Primer OrderPrimer Design
ALL Oligos manufactured will consist of a population with mutations. Mutations arise in some regions in Oligo sequences which, because of their folding properties, are very difficult to synthesize and therefore have an increased mutation rate at precisely these points.

Primer OrderPrimer Design
What are these mutations exactly?Ans: Assuming a worst case scenario with an average coupling efficiency of 98.5%, a typical PCR grade 30 residue long oligo synthesis would have yielded around 50% full length products. Theoretically, if a PCR is performed and the PCR products cloned and sequenced, out of 100 clones, 50 will yield full-length sequences. The rest will be made of truncated oligos of various degrees ranging from 1 to 6 residue truncations. Picking a 6 residue truncation clone is rare but 1-3 residue truncations are common. There are also insertions and deletions seen in these cases though they are encountered less frequently than truncations.
Assuming we take this same oligo and performed some sort of purification like PAGE, this will raise the number full length clones obtained from 50% to 85%. A rather comforting number but why is it not close to 99%? Other factors like cloning artefacts, PCR related secondary structures and even bacterial recombination events contribute to the loss of that 14%. As you can see, this is a numbers game. 1st BASE recommends you pick at least 3-5 clones before reporting to us any wayward oligo. Re-synthesis followed by PCR and then cloning is a time consuming process. A lot can be saved if that next clone that you could have picked is correct.

Primer OrderPrimer Design
An interesting note about these aberrations is that they are distributed evenly throughout the length of the oligo. When analyzed by MALDI-TOF or PAGE, these aberrations are not even significantly visible!
Therefore even with the highest purity purification, stringent QC using MALDI-TOF and CE, mutations should always be expected therefore temper your optimism by picking at least 3-5 clones.

Primer OrderPrimer Design
Will I eliminate mutations with purified Oligos?Additional purification is definitely recommended especially for Oligos used for cloning projects. However, the TOP-, PAGE- or HPLC-purified oligos (and the PCR products obtained with them) should be expected to give some mutant clones (clones with sequence infidelities, originating in the oligos). Purification reduces the amount of mutant Oligos but does not eliminate them.
Inclusion of restriction sites on the 5' ends of the oligos to facilitate cloning PCR products can ensure fidelity. These enzymes will only recognize their specific palindromes thus eliminating majority of the internally deleted (or addition) products. With the advent of TA cloning, most have forgotten that this can actually reduce mutant numbers by a large degree even without using purified Oligos.
Keeping Oligos below 35mers in length also keeps the mutations low because synthesis infidelities are increased as the oligo length is increased.
In general, selection and sequence analysis of 3-5 independent clones is still advised (not just one or two clones).

Primer OrderPrimer Design
Why does my DNA oligo have mutations? It is important to differentiate naturally occurring mutations linked to the chemical nature of the oligo manufacturing process from the perceived mutations that occur when desalted oligos are used in certain applications.
The naturally occurring mutations is an event inherent to the chemical synthesis of the oligos and the chances of having one single insertion or deletion in a given oligo of about 30 bases is about 2%. Invitrogen will be happy to replace any oligo that falls into this category.
With regards to the perceived mutations, following DNA synthesis, the completed DNA chain is released from the solid support by incubation in basic solutions such as ammonium hydroxide. This solution contains the required full-length oligo but also contains all of the DNA chains that were aborted during synthesis (failure sequences). If a 30-mer was synthesized, the solution would also contain 29 mer failures, 28 mer failures, 27 mer failures etc. The amount of failure sequences present is influenced by the coupling efficiency. For an oligo of this type, the percentage of full-length oligo would be between 74 and 54%, assuming a 99 or 98% coupling efficiency. This percentage is even lower when you consider oligos that are longer.

Primer OrderPrimer Design
Because the oligos are synthesized from 3' to 5' end, the primers that are desalted and not purified for length will have missing bases at the 5' end. Hence, oligos that are desalted are only recommended for diagnostic PCR, micro array or sequencing. Invitrogen recommends purification of the oligos if they will be used in certain demanding applications such as mutagenesis or cloning, especially if restriction sites are added to the 5' end of them.
Other sources of perceived mutations for both desalted and purified oligos are sequencing artifacts, point mutation introduced during PCR, unstable stem loop structures in the primers, propagation of the plasmid DNA after cloning in an E. coli strain that is muS, mutD or mutT or a silent mutation selected by the bacterial strain because of codon usage in that strain.

Primer OrderPrimer Design
How do I determine the percentage of full-length oligonucleotide? The percentage of full-length oligonucleotide depends on the coupling efficiency of the chemical synthesis. The average efficiency is close to 99%. To calculate the percentage of full-length oligonucleotide, use the formula: 0.99n-1 Therefore, 79% of the oligonucleotide molecules in the tube are 25 bases long; the rest are <25 bases. If you are concerned about starting with a preparation of oligonucleotide that is full-length you may want to consider cartridge, PAGE, or HPLC purification.

Primer OrderPrimer Design
Can the companies make the oligos have a high percentage of 'G' residues?It is known that oligos having a high percentage of 'G' residues are difficult to synthesize, especially if the sequence contains a run of 'G'. It is also reported if there is a run of four or more 'G', oligos tend to aggregate and form guanine tetraplex. (Poon and MacGregor, Biopolymers, 1998, 45, 427-434.) By substituting inosine for some of the 'G', the formation of guanine tetraplex can be disrupted.

Primer OrderPrimer Design
Why can't I clone double stranded Oligo directly?All synthetic oligos do not have the 5' phosphate group necessary for ligase to work, instead they contain the hydroxyl groups (-OH) only. To clone directly you must add a 5' phosphate using Polynucleotide Kinase(PNK) or order them phosphorylated (at an extra charge).

Primer OrderPrimer Design
What is the maximum length of oligo that can be produced? Coupling efficiency is the major factor affecting the length of DNA that can be synthesized. Base composition and synthesis scales will also be contributing factors. Table 2 shows that at 99% coupling efficiency, a crude solution of synthesized 95-mers would contain 38% full-length product and 62% (nx) failure sequences. This is before other chemical effects have been taken into account such as depurination. Depurination mainly affects the base A. The frequency of depurination is small but will increase significantly with primer length. For these reasons, we specify a maximum length of 100 bases, which we believe is the maximum length that can be synthesized routinely and economically.

Primer OrderPrimer Design
How do I make a 100 µMolar stock solution of my oligo?
A general rule states that for any oligo, the number of nmoles x 10 will give you the amount of solvent to add in microliters for a 100 µM stock solution. 100 µM is also equal to 100 pmol/microliter for those who wish to work with pmole amounts.

Primer Design
HiFiedelity Polymerases

Primer Design
Thermal Cycling Programs

Genetic Engineering SotwaresGenetic Engineering Sotwares
Hands on:
• Primer design (Primer premier 6)• Making a recombinant construct and
DNA manipulation, Virtual cloning, Alignment algorithms, Codon optimization, Back translation, Restriction mapping, ... (VectorNTI 11, Serial Cloner 2.1, CLCbio, etc.)
Part 2

GOOD LUCK!





























