Embed Size (px)
DESCRIPTION
GENETICA
Citation preview

Amprentarea Amprentarea genomicãgenomicã
Sindromul Prader-WilliSindromul Prader-Willivs.vs.
Sindromul AngelmanSindromul Angelman
Popa Elena-AlinaMG V,Grupa VI

Ce intelegem prin…?
Amprentarea genomică este determinată de modificări epigenetice meiotice prin care se produce o inactivare specifică a anumitor alele, dependent de originea parentală.
Termeni sinonimi: amprentare gametică sau amprentare parentală.

Ce mecanism are?• În transmiterea mendeliană se pleacă de la premiza că
acţiunea genei este independentă de originea sa maternă sau paternă; când se formează un zigot, genele “par a uita” originea lor parentală.
• În mod obişnuit o alelă mutantă a unei gene autosomale este transmisă cu o probabilitate egală de la unul din părinţi, indiferent de sex.
• Există însă situaţii rare în care expresia unei gene mutante şi deci a bolii este diferită în funcţie de originea ei, maternă sau paternă. Această diferenţă de expresie între alela moştenită de la tată şi alela moştenită de la mamă este produsă prin amprentare parentală sau genomică.
• Amprentarea este o formă de inactivare a genei ce produce o expresie monoalelică dar nu este o mutaţie, deoarece este reversibilă.

Cand se produce?
Amprentarea se produce în gametogeneză, inainte de fecundare, marcând originea maternă sau paternă a a numitor gene.
După concepţie,expresia genei amprentate este suprimată în anumite ţesuturi şi se menţine ca atare toată viaţa.
Excepţie fac celulele germinale în care se produce o conversie a amprentării in funcţie de sex: la bărbat, alelele moştenite de la mamă, cu amprentare maternă, vor fi convertite în gametogeneză şi vor fi trecute descendenţilor cu amprentare paternă.

Unde poate fi gasita?
• In ultimii ani au fost identificate cel puţin 40 de gene amprentate .
• O listă a genelor amprentate la om poate fi gasită la adresa: www.geneimprint.com.
• Acestea sunt localizate grupat, în anumite regiuni cromosomice (de exemplu grupările de pe cromosomii 11p si 15q asa cum sunt prezentate si in tabelul de mai jos.)
• Acestea se mai pot clasifica si in functie de :1. Specie,2. Nume3. Statusul de imprimare

Preluare de pe site-ul http://www.geneimprint.com/site/genes-by-species

Genele amprentate au o serie de trăsături comune:
1. expresia monoalelică care depinde de originea parentală (expresia fie numai a alelei materne, fie numai a alelei paterne),
2. prezenta unor insule CpG în regiunea promotor sau într-un intron care suferă metilare diferenţiată (dependentă de asemeni de originea parentală),
3. prezenţa frecventă a unor molecule de ARN care sunt transcrise de pe catena ADN opusă (ARN antisens) – cu rol important în reglarea expresiei acestor gene.
Sunt asemanatoare genele amprentate?

Bialelic vs Monoalelic• Expresia monoalelică a genelor amprentate este un
fenomen extrem de complex. Numeroase gene prezintă acest model monoalelic doar în anumite etape ale dezvoltării ontogenetice sau doar în anumite ţesuturi.
• De exemplu, gena IGF2 = factorul de crestere insulin-like tip 2 este exprimată bialelic în cursul dezvoltarii embrionare precoce (şi aceasta favorizează procesele de creştere caracteristice acestei etape), pentru a dobândi apoi expresie exclusiv de pe cromosomul patern; exceptie fac unele ţesuturi (ficat etc.) în care se pastrează expresia bialelică.
• Genele amprentate sunt implicate în controlul unor procese extrem de importante în cursul dezvoltarii şi, ca urmare, alterarea expresiei normale a acestora (fenomen denumit pierderea amprentarii – de la loss of imprinting – LOI) este asociată cu numeroase boli, precum cancerele sau bolile asociate cu tulburări de comportament.

Ce importanta au genele amprentate?
• Amprentarea este un mecanism normal de reglarea genică care nu produce, prin el însăşi, o boală ereditară dar poate explica penetranţa incompletă sau expresivitatea variabilă a unei afecţiuni monogenice.
• De exemplu, tumora de glomus carotidian, transmisă AD, la care subiecţii purtători de genă mutantă sunt bolnavi sau sănătoşi după cum ei au primit gena de la tată sau de la mamă; deci gena mutantă este activă dacă o transmite tatăl sau inactivă dacă provine de la mamă.
• În ciuda numărului relativ mic de gene amprentate(aprox 40), tulburarea modului corect de expresie monoalelică (datorată UPD, deleţiei cromosomice / genice sau pierderii amprentării) s-a dovedit a avea consecinţe serioase în dezvoltarea embrionară, boli genetice şi cancer.
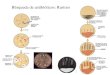
• Cel mai amplu exemplu studiat îl reprezintă sindroamele Prader-Willi (SPW) şi Angelman (SA) produse, (în mod obişnuit,dar nu exclusiv) de aceeaşi deleţie a cromosomului 15q11-13, dar pe cromosomul de origine paternă (SPW) sau maternă (SA).

SINDROMUL PRADER - WILLI SINDROMUL ANGELMAN
Fenotip Hipotonie neonatală severă, dismorfie facială caracteristică, mâini şi picioare mici, talie mică, obezitate cu debut în copilărie, retard mintal, tulburări de comportament
Dismorfie facială carcteristică, talie mică, microcefalie, retard mintal sever, spasticitate şi convulsii, lipsa vorbirii şi crize de râs.
Deleţie 15q11-13 ~ 70% cazuri cu deleţie pe cromosomul patern
~ 70% cazuri cu deleţie pe cromosomul matern
Disomie uniparentală ~ 30% cazuri (maternă) ~ 3-5 % cazuri (paternă)
Gene incriminate SNRPN şi necdina, ambele cu expresie de pe cromosomul patern.
UBE3A care este exprimată exclusiv matern, la nivel cerebral.
Mutaţie genică Neidentificată 7-9 % cazuri
Mutaţie în centrul de amprentare
1-2 % cazuri 7-9 % cazuri
Alte cauze Neidentificate 10-20 % cazuri
Tabel 5.11. Fenotipurile şi cauzele sindroamelor Prader-Willi şi Angelman

Aspecte fenotipiceBolile genetice asociate unor tulburari ale genelor
amprentate se caracterizează prin manifestări fenotipice care depind de originea parentală a defectului genetic.
În cazul sindroamelor Prader-Willi si Angelman sunt implicate cel mai adesea deleţii largi ale regiunii cromosomice 15q11-13.
1.Atunci când defectul interesează cromosomul de origine paternă manifestarile clinice corespund Sindromului Prader-Willi (OMIM 176270) caracterizat prin obezitate, hipostatură şi retard mintal moderat. Genele incriminate sunt SNRPN şi necdina, ambele cu expresie de pe cromosomul patern.
2.Defectele care interesează cromosomul de origine materna determină sindromul Angelman (OMIM 105830) caracterizat prin ataxie, hiperactivitate, retard mintal sever cu lipsa vorbirii şi crize de râs. In unele cazuri boala este determinată de defecte ale genei UBE3A care este exprimată exclusiv matern la nivel cerebral.


Ilustrare a modului în care diverse mecanisme genetice pot provoca cele două sindroame: deletii cromozomiale, defecte de amprentare și disomia uniparentala.

Copilărie Intârziere intelectuală,
Dormit excesiv,Strabism
CriptorhidiaÎntârziere in vorbire
Hiperfagie=ObezitateaPubertate întârziată
Statura scurta
Crizele convulsive se instalează la vârsta de 1–3 ani.Toate etapele de dezvoltare vor fi întârziate, astfel mersul este realizat între 2,5 şi 6 ani şi este ţeapăn, sacadat, cu membrele superioare în flexie şi pronaţie. 10% din copii nu merg niciodată. Afectarea limbajului este severă.
Prader - Willi Angelman

Aspect fenotipic Prader-Willi

Angelman
• Sunt capabili de comunicare prin arătarea obiectelor sau gesticulând
• Instalarea pubertăţii are loc în mod fiziologic şi atât bărbaţii, cât şi femeile sunt capabili de procreare. Fertilitatea este aparent normală: în 1999 s-a raportat un caz de transmitere a deleţiei la fetus de la mama afectată.
• Adulţii tineri au sănătate fizică bună, cu excepţia crizelor convulsive, a constipaţiei frecvente, iar scolioza se accentuează cu vârsta, fiind raportată la 40% din adulţi, majoritatea femei.
• Cu avansarea în vârstă, fenotipul se schimbă, fiind marcat de prognatism, macrostomie şi buză inferioară proeminentă.
• Frecvenţa crizelor convulsive scade cu înaintarea în vârstă,
-Infertilitate (bărbați și femei) -Hipogonadism-Parul pubian rar -Obezitatea -Hipotonie (tonus muscular scăzut) -Dizabilități de învățare -Predispozitie la diabet zaharatAspect fizic -Pod nazal proeminent -Mâini mici-Exces de grăsime, mai ales în porțiunea centrală a corpului -Subțierea buzei superioare -Ochii în formă de migdală -Pielea deschisa la culoare si parul în raport cu alți membri ai familiei -Lipsa de dezvoltare sexual complet
Prader Willi

Sfatul genetic• Sfatul genetic şi opţiunea testării genetice trebuie acordate tuturor
familiilor în care există un copil afectat, indiferent de mecanismul de apariţie a sindromului. Părinţii unui copil cu boala cauzata de deleţia regiunii critice sau de disomia uniparentală 15 paternă au un risc mai mic de 1% de a mai avea încă un copil afectat.
• Perioada optimă de determinare a riscului genetic şi de prezentare a disponibilităţilor diagnosticului prenatal este înaintea sarcinii. În cazul unui defect al centrului de amprentare sau a unei mutaţii în gena UBE3A, riscul de recurenţă este de 50%.
• Sfatul genetic şi opţiunea testelor genetice trebuie acordate şi rudelor mamei la care s-a identificat o mutaţie UBE3A, o deleţie a centrului de amprentare sau un rearanjament cromozomial, dar şi rudelor tatălui, în cazul disomiei uniparentale paterne. Dacă mama poartă o mutaţie a centrului de amprentare, surorile ei ar putea să aibă aceeaşi mutaţie şi acelaşi risc de 50% pentru un copil afectat cu sindrom Angelman. Fraţii ei nu au risc pentru un copil afectat, dar pot transmite defectul fiicelor lor şi pot avea nepoţi afectaţi.

Concluzii• Unele gene sunt exprimate diferit in functie de originea
materna sau paterna a alelei, fenomen denumit amprentare.• Majoritatea genelor amprentate afecteaza cresterea
fetala,proliferare celulara sau dezvoltarea sistemului nervos. • Genele cu expresie paterna promoveaza cresterea, iar cele
cu expresie materna stimuleaza proliferarea celulara. Amprenta genomica creaza un echilibru intre acestea.
• Cand un cromozom are gena amprentabila cu mutatie sau deletie, produsul genei este nefunctional deoarece gena activa este cu mutatie si gena de la celalalt parinte este inactiva.
• Fenomenul de inactivare a uneia dintre alelele unuia dintre parinti si activarea alelelor celuilalt parinte se numeste amprentare genomica
• Amprenta care identifica o alela ca fiind de origine materna sau paterna este stearsa in fiecare generatie;
• exemple de sindroame: Prader-Willi si Angelman

Amprentarea genomica pe scurt: Prader-Willi : gena incriminata : paterna
Angelman: gena incriminata: materna

Bibliografie
• www.geneimprint.com
• www.resurseacademice.org/tratat-genetica-umana-covic
• Dictionar.romedic.ro/amprentare-genomica
• www.scritube.com/biologie/Genomica

Vă mulţumesc!