Embed Size (px)
Citation preview

ALUMNA: CISNEROS BELLIDO, RUTH NOEMIPROFESOR: JHONY ESCATE DIAZ
1 CICLO “A”

PROTEOMICA Y GENOMICA

PROTEOMICA ESTA DEDICADA AL ESTUDIO DE LA S PROTEOMICAS Y DE SUS CAMBIOS EN DEPONDENCIA DEL CONTEXTO

CONCEPTOS DE LA PROTEOMICAEs precisamente esta variabilidad del proteoma lo que lo hace tan atractivo
para la investigación biomédica. Recientemente la proteomica ha comenzado a dar una contribución importante a nuestra compresión de la biología y de la
medicina. Los avances logrados en genómica no se traducen directamente en nuestro conocimiento de los respectivos proteomas: muchos genes carecen de función conocida, a muchos otros se les adjudica una función por analogía con otros genomas estudiados previamente. Si se extrapola la situación encontrada
con la levadura, más de la mitad del genoma humano no tiene una función conocida en estos momentos.
Es el conjunto de proteínas expresadas por un organismo en un momento dado . La proteínica comprende todas las técnicas para el estudio en gran escala de la proteína expresada “proteoma” como las aplicaciones de estas técnicas al análisis de problemas biológicos. Mientras que el genoma es un organismo, es esencialmente constante a lo largo de la vida, el proteoma tiene un carácter dinámico: la expresión de proteínas cambia en diferentes etapas del ciclo
celular pero también en respuesta a acciones extremas.

La proteómica incluye diversos campos de investigación:
La identificación de las proteínas expresas por un organismo en una condición dada “proteómica descriptiva o estructural, todas las proteínas expresada en un momento y en un contexto”
La identificación de los cambios en el nivel de expresión de proteínas asociadas a cambios en la condiciones del organismo “proteómica comparativa, cuales proteínas cambian cuando un organismo se somete a condiciones diferentes”
La identificación de conjuntos funcionales de proteínas, es decir, grupos de proteínas que se localizan en un mismo sitio celular y que operan en mutua interacción “interacciones proteína-proteína, proteómica funcional”
La identificación de las proteínas que forman un organelo “este enfoque conduce a la elaboración de un mapa molecular de la célula”

¿PORQUE LA PROTEÓMICA?
El dogma central de la genética:
ADN ARN PROTEÍNA
Da lugar a uno de los paradigmas esenciales de la biología que prevaleció durante la segunda mitad del siglo pasado:
UN GEN UNA PROTEÍNA
Esta relación aparentemente simple.
Aunque es cierto que un gen codifica una secuencia de aminoácidos existen dos eventos que incrementan considerablemente el número de proteínas que pueden ser originadas por un gen y estar presentes en una célula en un momento dado y por tanto hace más complejo el proteoma de una célula.
Uno de esos eventos es el fenómeno conocido como empalme alternativo. En los mamíferos y otros organismos superiores el gen que codifica una proteína no esta constituido por una secuencia continua de nucleótidos.
Una parte de la secuencia del gen está formada por regiones no codificantes que interrumpen la secuencia codificante del gen y son llamados intrones. Las regiones codificantes (y que por tanto dan lugar al ARN y posteriormente a la proteína) son conocidas como exones. Cuando se forma el ARN mensajero, mARN, los intrones son eliminados quedando así la secuencia que codifica una proteína. El fenómeno de splicing consiste en que los exones pueden reordenarse de varias formas y dar lugar a mas de una proteína a partir de un solo gen.

Se calcula que alrededor del 10% de los genes de mamíferos codifica proteína cuya función es modificar otras proteínas, y que genera más de 200 tipos de modificaciones post tradicionales tales como:
Glicosilación.
Fosforilación.
Incorporcion de lípidos.
En adición, muchas proteínas participan en las rutas metabólicas que conllevan a la degradación y finalmente la eliminación de proteínas. Así, en el sistema de ubiquitinación participan 134 genes, mientras las fosfatasas son codificadas por unos 300 genes y las proteínas kinasas se estiman codificadas por mas de 1100 genes.
Por ello, una secuencia única de aminoácidos puede generar muchas especies químicas diferentes. De esta forma se estima que los aproximadamente 40 000 genes del genoma humano pueden dar lugar a mas de un millón de proteínas.

Las diferencias que existen entre las poblaciones de mARN y de proteínas están dadas, por una parte, por la amplia población de especies que portan modificaciones químicas.
, intervienen otras causas importantes: el tiempo de residencia de las moléculas de proteínas en el interior de una célula o de un compartimiento celular es variable debido a la degradación por proteasas, a la translocación intra y extracelular y a la modificación de las proteínas durante su intervención en procesos biológicos. Por ello la medición de la expresión de mARN no suministra valores confiables de los cuales se pudiera deducir la abundancia y la presencia de las proteínas traducidas.
la amplia población de especies que portan modificaciones químicas
Molécula Proteína
mRNA

El tiempo de vida media de las moléculas de proteínas en el interior de una célula o de un compartimiento celular es variable debido a la acción de proteasas, a la translocación intra- y extracelular y a la modificación de las proteínas durante su intervención en procesos biológicos. Por ello la medición de la expresión de mRNA no suministra valores confiables de los cuales se pudiera deducir la abundancia y la presencia de las proteínas traducidas. Solo el estudio directo de las proteínas, lo que es campo de la Proteómica, puede dar respuesta a estas cuestiones. Adicionalmente, una molécula de mRNA puede originar varias secuencias de proteínas, por el simple cambio del codón de iniciación o de terminación, lo que origina moléculas de secuencias más cortas o más extensas.
En resumen, al concluir el siglo XX la idea de considerar las proteínas como macromoléculas definibles por una secuencia de residuos de aminoácidos completamente determinadas por la secuencia de una molécula de ADN había cambiado sustancialmente: entre genes y proteínas existen diferencias de complejidad que definen la existencia de un campo de conocimiento singular, irreducible a formas relativamente menos complejas del conocimiento: al concluir el siglo las razones para el surgimiento de la proteómica estaban sólidamente establecidas.

ELECTROFORESIS BIDIMENSIONAL
Electroforesis bidimensional de proteínas de la fracción nuclear de
células de cáncer de pulmón humano (small cell lung cáncer) . Las proteínas
de la fracción nuclear se extrajeron con una solución de agentes caotrópicos y detergentes. La primera dimensión se
efectuó en un gel de poliacrilamida tubular con enfollinas portadoras de
rango 2 a 11. La segunda dimensión se efectuó en un gel vertical de 16.5 %
poliacrilamida en presencia de SDS. Las proteínas se detectaron mediante
tinción con plata (Gel confeccionado en el laboratorio de los autores).
. Estos geles permiten obtener
un arreglo o despliegue físico en dos dimensiones de mezclas complejas
de proteínas.

Se basan en la combinación de dos técnicas ortogonales (es decir, que
no comparten principios físicos comunes
1) la separación por carga eléctrica (focalización isoeléctrica, IEF) en la
que las proteínas migran en un gradiente de pH hasta alcanzar el sitio donde carecen de carga, es
decir su punto isoeléctrico.
2) la separación por tamaño molecular que se efectúa en un gel de poliacrilamida en presencia de
un detergente aniónico muy potente, el docecil sulfato de sodio
(SDS-PAGE).

ETAPAS DE LA IDENTIFICACIÓN DE LOS CAMBIOS POR
ESPECTROMETRÍA DE MASA

1.- PREPARACIÓN DE MUESTRA.
Eliminación de componentes no proteicos y obtención de subfraccionescelulares si es necesario.

2.- SEPARACIÓN POR PUNTO ISOELÉCTRICO.Dos técnicas a escoger: la separación en geles de anfolinas (separación en tubos) o separación en geles de immobilinas(separación en tiras IPG). En ambos casos, la focalización transcurre durante un periodo prolongado a alto voltaje (por ejemplo durante 10 a 20 horas y a voltajes entre 3.5 y 8 KVJ.

3.-SEPARACION POR TALLA.
El gel que contiene la primera separación se coloca perpendicularmente a la dirección de migración en un segundo gel, ahora de SDS.

4.- DETECCIÓN.Este paso permite ver las proteínas en el gel en forma de manchas (spots) y capturar la imagen de la separación utilizando un scanner. Algunas técnicas de detección son incompatibles con el siguiente paso de análisis C identificaciones.

5.- ANÁLISIS DE IMÁGENES DE GELES.
Se hace para identificar los cambios en la expresión de proteínas entre dos o varias condiciones experimentales diferentes, comparando varios geles de cada condición.

6.- SELECCIÓN DE PROTEÍNAS DE INTERÉS.
Se seleccionan proteínas cuyo cambio sea “a” significativo y “b” reproducible en un grupo de experimentos idénticos.
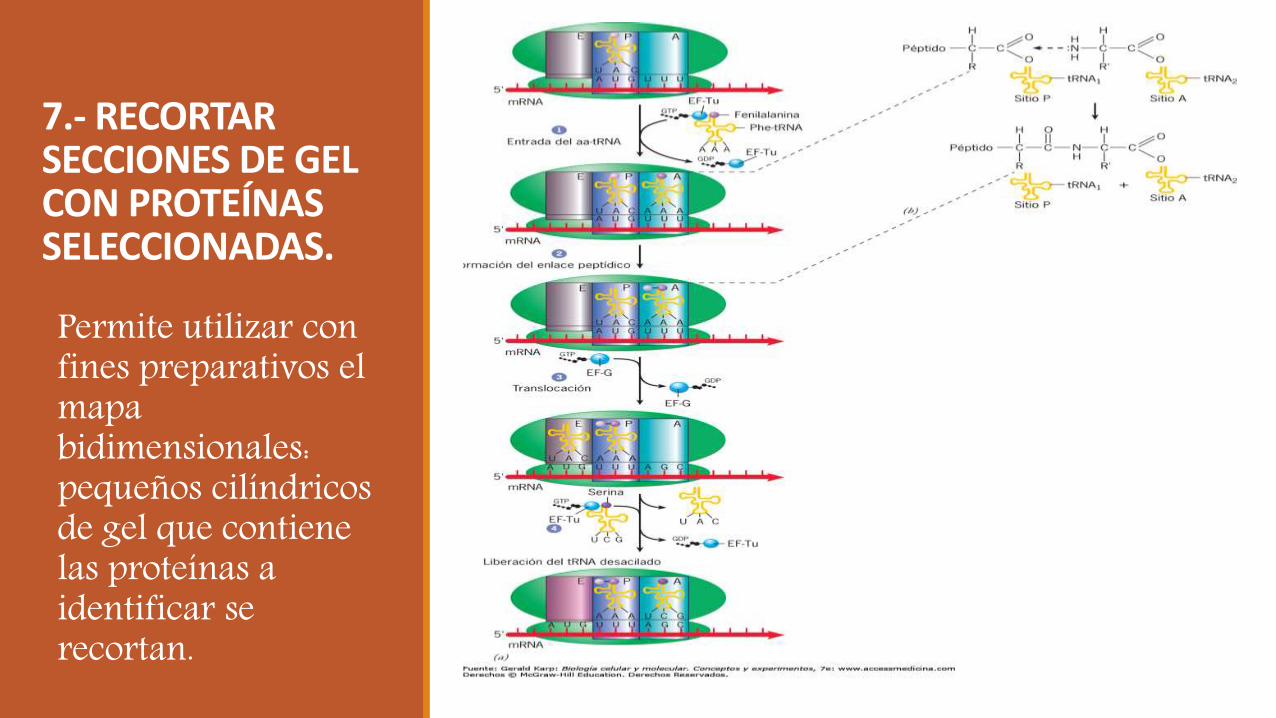
7.- RECORTAR SECCIONES DE GEL CON PROTEÍNAS SELECCIONADAS.
Permite utilizar con fines preparativos el mapa bidimensionales: pequeños cilíndricos de gel que contiene las proteínas a identificar se recortan.

8.- DIGERIR LAS PROTEÍNAS EN LOS BLOQUES DE GELES CON UNA PROTEASA ESPECIFICA (USUALMENTE TRIPSINA).Incubación de los bloques de geles con tripsina durante 3-20 horas. Se obtiene la mezcla de los péptidos trípticos, cortes en extremo C de lisina y de arginina.

9.- analizar
las mezclas
de péptidos
por
espectrometr
ía de masas.
Se obtienen las masas moleculares de los péptidos obtenidos y se puede además obtener secuencias parciales o completas de los péptidos.

10.- IDENTIFICAR POR COMPARACIÓN CON LAS BASES DE DATOS DE PROTEÍNAS.Los valores de masas y los fragmentos de secuencias, obtenidos se comparan con basas de datos de las digestiones virtuales del proteoma teórico del organismo analizado si el genoma no es conocido, se comparan los valores con las digestiones virtuales de proteomas derivados de genomas conocidos procedentes de especies cercanas.

APLICACIÓN DE LAS PROTEOMICASCaracterización del proteoma de un organismo. El propósito de este estudio es la identificación del mayor número de proteínas expresadas por un organismo en una condición biológica particular. En el caso de organismos unicelulares o de células en cultivo, muchos trabajos se han propuesto la separación y la identificación de proteínas en preparaciones de proteínas totales y ya existen mapas en bases de datos para numerosos organismos. Sin embargo, para lograr un nivel superior de información se prefiere descomponer el estudio del proteoma en el estudio de subproteomas correspondientes a organelossubcelulares. Actualmente la Organización del Proteoma Humano (HUPO) tiene en curso 3 proyectos internacionales destinados a la construcción de los primeros mapas proteómicos de órganos o tejidos. Estos son: cerebro, hígado y plasma.
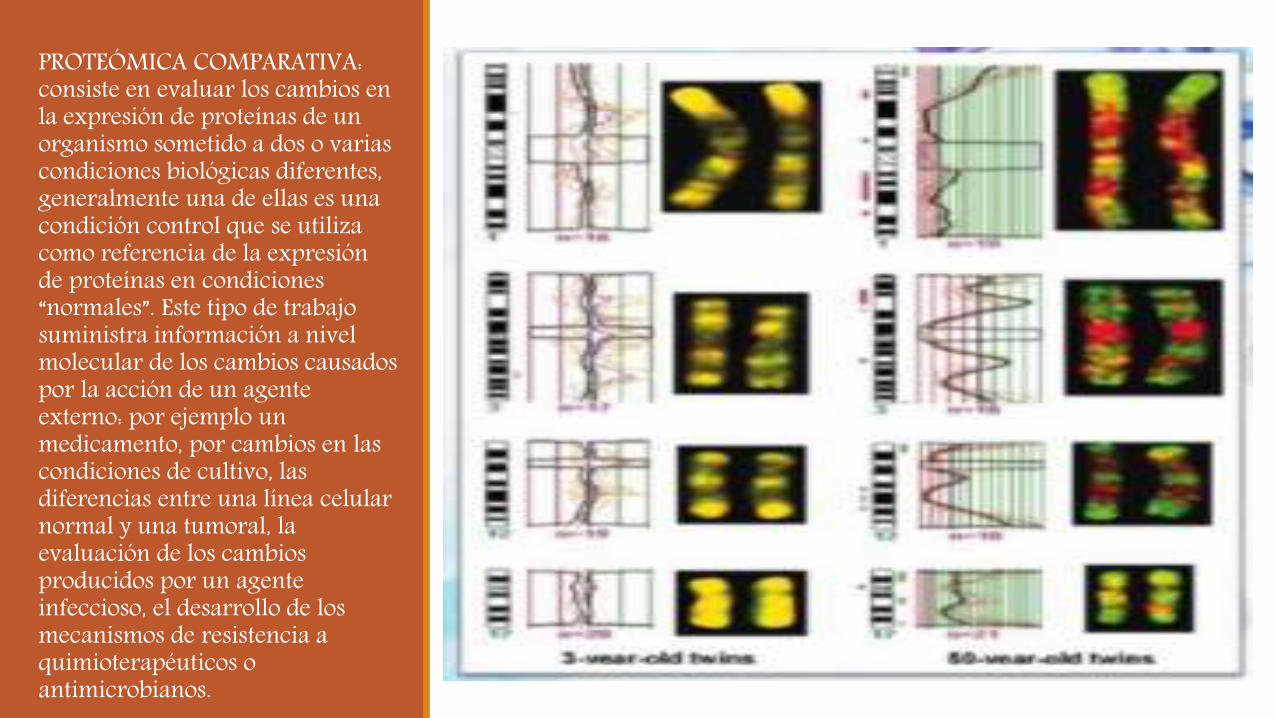
PROTEÓMICA COMPARATIVA: consiste en evaluar los cambios en la expresión de proteínas de un organismo sometido a dos o varias condiciones biológicas diferentes, generalmente una de ellas es una condición control que se utiliza como referencia de la expresión de proteínas en condiciones “normales”. Este tipo de trabajo suministra información a nivel molecular de los cambios causados por la acción de un agente externo: por ejemplo un medicamento, por cambios en las condiciones de cultivo, las diferencias entre una línea celular normal y una tumoral, la evaluación de los cambios producidos por un agente infeccioso, el desarrollo de los mecanismos de resistencia a quimioterapéuticos o antimicrobianos.

Este tipo de investigación es de alto valor en ciencias biomédicas. Para los trabajos de proteómica comparativa es necesario disponer de un diseño experimental cuidadosamente planificado. Aquí resulta de gran utilidad la existencia de una hipótesis previa que oriente la búsqueda hacia determinado tipo de proteínas que pueden ser seguidas por inmunodetección con anticuerpos específicos o que se suponen localizadas esencialmente en un orgánulo subcelular. Por ejemplo, algunos proyectos se orientan específicamente hacia la identificación de cambios en los patrones de fosforilación de proteínas.

. En este caso, la detección del subconjunto de proteínas fosforiladas puede facilitarse mediante el uso de anticuerpos específicos.Confección de n mapa bidimensional anotado. Como resultado de las identificaciones efectuadas por espectrometría de masa, el mapa bidimensional de proteínas se enriquece con abundante información que incluye la identificación de la naturaleza de las proteínas, el punto isoeléctrico y la masa experimental y teórica, y las propiedades reportadas anteriormente en la literatura.

Proteómica comparativa. El esquema resume los pasos principales que permiten la comparación de la expresión de proteínas en dos condiciones biológicas y la consecuente identificación de los cambios.
Interacción de Proteínas. En los últimos años ha quedado evidenciada la importancia de identificar las interacciones entre proteínas y de proteínas con otras moléculas. En varias publicaciones se han reportado los resultados de aislamiento de complejos de proteínas y la posterior identificación de las proteínas componentes. Estos resultados contribuyen significativamente al establecimiento de mapas de interacciones y al esclarecimiento del mecanismo de determinadas funciones biológicas.
Es posible predecir que las herramientas que se emplean en proteómica se irán perfeccionando e incluso aparecerán nuevos procedimientos y equipos que solucionarán las principales limitaciones actuales. Sin lugar a dudas la proteómicafortalecerá su posición actual en las investigaciones médicas, farmacéuticas, agrícolas y en otros campos y se multiplicarán sus aplicaciones.

HERRAMIENTAS DE LA PROTEÍNAS

Desafortunadamente, las mismas características que otorgan a las proteínas su papel fundamental como moléculas efectoras de la función celular (diversidad química y estructural y abundancia relativa) también dificultan su análisis experimental. Actualmente no existe un diagrama de flujo único para el análisis proteómico de una muestra, ya que las variables como complejidad, método de separación, concentración y estabilidad de las proteínas, además de la plataforma tecnológica disponible para su análisis, y muy especialmente el tipo de pregunta biológica que se pretende abordar, son los parámetros básicos que determinan la elección de una estrategia de estudio. Por lo tanto, no existe una metodología idónea para el estudio de proteomas en forma sistemática. En consecuencia, la investigación proteómica es el resultado de la aplicación de un conjunto de técnicas que permiten el estudio de proteínas. A continuación, se describirá en forma resumida una metodología para el análisis proteómico de una muestra.

Separación de las proteínas. Una muestra proveniente de un sistema biológico es una mezcla compleja de proteínas. Por lo tanto, las muestras provenientes de células, tejidos u otro tipo de muestras biológicas (sangre, orina, leche, líquido céfalo-raquídeo, semen, saliva, lágrimas, etc.) se separan principalmente por técnicas cromatográficas y/o electroforéticas, las cuales son tecnologías robustas, versátiles y con alta capacidad de resolución. Las más utilizadas son: electroforesis unidimensional (SDS-PAGE) y bidimensional (2-D PAGE), electroforesis capilar, cromatografía líquida de alta resolución (HPLC), cromatografía de afinidad, cromatografía de exclusión molecular y cromatografía de intercambio iónico. La electroforesis bidimensional, por ejemplo, permite la separación de hasta 2000 proteínas en un solo gel, mientras que la electroforesis unidimensional solamente 100. Estas técnicas de aislamiento pueden utilizarse separadamente, en conjunto o en sistemas de flujo continuo, como la cromatografía multidimensional, también conocida como MudPit (multidimensional protein isolationtechnology), que contiene fase reversa e intercambio iónico en forma conjugada.

La tecnología de cromatografía líquida acoplada a los espectrómetros de masas
(LC-MS, liquid chromatography-mass spectrometry) es utilizada para la separación
de péptidos provenientes de proteólisis enzimática, péptidos sintéticos o péptidos
nativos, por columnas de fase reversa micro-capilares acoplado a sistemas de
ionización tipo electrospray-MS. Esta tecnología permite la separación de mezclas
complejas de péptidos y su análisis simultaneo por espectrometría de masas. Este
sistema es compatible con columnas capilares tipo RPn y MudPit de nanoflujo, que
funcionan al mismo tiempo como agujas de producción de spray bajo la aplicación
de alto voltaje. En la actualidad, el sistema LC-MS es una de las más versátiles y
poderosas herramientas del análisis proteómico basado en la espectrometría de
masas.
Procesamiento de la muestra. El primer paso es la reducción de los puentes de
disulfuro y la alquilación de las cisteínas. Tiene como objetivo desnaturalizar las
proteínas permitiendo que se expongan los sitios específicos de corte enzimático
y, a la vez, se evita la formación de dímeros. Estas modificaciones químicas son
fundamentales, ya que para el análisis por EM se requiere que las proteínas sean
digeridas a péptidos con masas moleculares menores que 3 KDa. Eso se debe a
que las tecnologías de fragmentación de proteínas en espectrómetros de masas
(para obtención de secuencias) aún son limitadas y péptidos con mayores masas
moleculares producen informaciones estructurales no interpretables.

La muestra, reducida y alquilada, generalmente se digiere con tripsina (enzima que corta específicamente en los C-terminales
de lisina y arginina, excepto cuando éstas son seguidas de prolina); la elección de esta endoproteasa como enzima no es
casual. En su mayoría, los cortes trípticos generan péptidos con masas moleculares entre 1 y 2 KDa; permiten la diferenciación
entre los amino ácidos lisina y glutamina (128 Da), ya que todas las lisinas estarán posicionadas en el C-terminal de los
péptidos trípticos, y propicia la presencia de por lo menos dos cargas positivas en el péptido (n-terminal y C-terminal con K o
R) que posibilitarán la transferencia de carga a través de las ligaciones amídicas facilitando la fragmentación. La
descripción del proceso de reducción, alquilación y digestión enzimática de una proteína para su análisis por EM es apenas uno de los tantos procedimientos que pueden efectuarse en el
análisis proteómico. La identificación de biomarcadores, determinación de modificaciones postraduccionales,
cuantificación, identificación de mezclas complejas, entre otras aplicaciones, pueden involucrar decenas de metodologías
diferentes para el procesamiento de las muestras.

LA UNIDAD PROTEÓMICA (UPRO)DEL INSTITUTO DE BIOTECNOLOGÍA DE LA UNAM

La búsqueda de respuestas
para preguntas biológicas
complejas está directamente
relacionada con el dominio, la
aplicación y la disponibilidad de
alta tecnología. En este
sentido, la creación de
unidades de servicio
especializadas, a nivel regional
o nacional, parece ser la mejor
alternativa para la investigación
científica y tecnológica en
países en desarrollo.
Las unidades de
proteómica:
a) son entidades técnica y
operacionalmente
elaboradas.
b) por su naturaleza
requieren grandes
inversiones financieras.
c) toman un tiempo
razonable para su
establecimiento.
d) exigen recursos
humanos altamente
capacitados.

Dada la relevancia de las unidades de servicio en proteómica, tanto para la investigación básica como para la investigación biotecnológica, la comunidad de investigadores del Instituto de Biotecnología decidió la creación de la Unidad de Proteómica (UPRO-IBT) en junio de 2005. Esta unidad tiene como objetivo ofrecer servicios de análisis proteómico basados en la espectrometría de masas macromolecular.

La UPRO es la primera y única unidad de servicios en su género a nivel nacional.
Durante 2006 se analizaron más de ochocientos muestras de proteínas para
instituciones de investigación pública y privada, así como para compañías
particulares. Los servicios prestados por la UPRO incluyen determinación de masas
moleculares de proteínas y péptidos, identificación de proteínas por MALDI-TOF y
ESI-MS/MS, secuenciación parcial de proteínas desconocidas, determinación de
modificaciones postraduccionales, determinación de sitios de corte específicos de
nuevas enzimas e identificación de modificaciones sintéticas en proteínas.
Adicionalmente, la UPRO es un centro proteómico de referencia a nivel nacional
debido a la formación de recursos humanos (técnicos y de posgrado), al apoyo
brindado a otros laboratorios de proteómica y a su empeño en promover y desarrollar
la investigación proteómica en México ( http://www. smp.org.mx). El significativo
aumento de solicitudes de servicio es consecuencia del creciente interés de la
comunidad científica nacional e internacional en dilucidar a nivel cualitativo y
cuantitativo la expresión genética de los organismos, y especialmente la expresión
diferencial de proteínas debida a distintas enfermedades, estrés u otras alteraciones
físicoquímicas impuestas a los sistemas biológicos. La UPRO mantiene un esfuerzo
constante por mejorar su plataforma analítica y, de esta forma, ofrecer servicios de
primer nivel al creciente número de usuarios.



En 1974 Frederick Sanger (dos veces premio Nobel de química, en 1958 y en 1980) creó un método de secuenciación del ácido desoxirribonucleico o ADN por terminación de cadenas. Su idea consistió en obtener todas las secuencias posibles de una cadena de ADN con un único nucleótido de diferencia, para luego leerlas e inferir la secuencia completa de pares de bases de un gen. Esta técnica se aplicó manualmente en los laboratorios durante toda la década del 1980. Dada su baja resolución, afortunados eran aquellos que, repitiendo tres veces el experimento, lograban leer con pocas dudas mil pares de bases, es decir, un poco menos que el tamaño medio de un pequeño gen bacteriano. En la siguiente década, los avances técnicos automatizaron el método, redujeron los errores de lectura, permitieron la producción en paralelo, e imprimieron al costo de secuenciación una carrera descendente que aún continúa. En 1998 un secuenciador automático procesaba un millón de pares de bases por día. Ese año el consorcio público del proyecto del genoma humano se ponía como objetivo secuenciar ochenta millones de ellas. Dos años después se había duplicado el rendimiento, y en 2001 se presentó públicamente el primer esquema de nuestro genoma. Esto no constituyó más que la punta del iceberg de una nueva forma de hacer biología, cuyo objetivo inmediato fue la secuenciación de todos los genes humanos (alrededor de veintitrés mil), en especial aquellos asociados con enfermedades, que constituyen perlas para la medicina.

Siguió el estudio de su regulación y expresión, para lo cual no solo interesan los genes, sino sus secuencias regulado
Estudio de Darwin en Down House, la casa de campo en la que vivió durante cuarenta años, situada en el condado de Kent,
cerca de la localidad de Downe, unos 26km del centro de Londres en dirección sudeste.
89 Volumen 19 número 113 octubre - noviembre 2009 ARTÍCU LO
ras; los intermediarios o transcriptos de sus productos finales, las proteínas; las secuencias que separan un gen de otro, y un
conjunto de otros elementos, algunos repetidos desde unas pocas decenas hasta miles de veces, que no hace mucho
formaban parte de lo completamente desconocido del genoma de un organismo eucariota superior.
Para comprender nuestro genoma y experimentar con él debemos entender nuestras diferencias con otras especies, como
gusanos, moscas, peces, ratones y nuestros parientes primates más cercanos, lo que llevó a la genómica comparativa. Esta
revolución no solo tiene consecuencias para la medicina o la industria farmacéutica sino, también, para áreas como
agricultura, ganadería, piscicultura e ingeniería forestal. Modifica preguntas y métodos en todos los campos del conocimiento
biológico, desde la biología del desarrollo a la sistemática, desde la microbiología al comportamiento, y desde la genética de
poblaciones a la conservación de especies. Institutos como el Sanger en Inglaterra o empresas como Celera en los Estados
Unidos son en la actualidad centros robotizados dedicados a la generación masiva de datos genómicos.
Ante estas nuevas posibilidades, en un mar de A, C, T y G –como se simbolizan las cuatro bases de los ácidos nucleicos–,
¿cómo distinguir las partes que tienen alguna función de las que no la tienen? ¿Con qué herramientas manejar y guardar esa
enorme información? ¿Cómo hacer accesibles los datos a investigadores y a otros interesados? Ya habrá supuesto el lector
que de eso se ocupa la bioinformática, a la que nos referiremos después de comentar brevemente la tecnología de los chips de
expresión.

MILES DE EXPERIMENTOS EN DOS CENTÍMETROS CUADRADOS

La hibridación de ácidos nucleicos es una técnica de laboratorio usada por la biología
molecular que detecta la expresión de un gen por medio de la concentración del ARN
mensajero que da lugar a la producción de una proteína. La técnica se aplicaba
manualmente durante las décadas de 1980 y 1990. Actualmente hay empresas que venden
unas pequeñas placas de vidrio o chips que concentran en poco más de dos centímetros
cuadrados todos lo genes de un genoma, sea humano, de ratón o de mosca.
La cuantificación de ARN mensajero con estos chips de ADN, llamados microarrays, permite
medir la expresión de todos los mensajeros de los genes de un genoma sometido a una o
más condiciones experimentales y compararlo con un genoma de control. En un solo
experimento podemos obtener el transcriptoma (la totalidad de ARN mensajero expresado
en un genoma) de cualquier tipo de cáncer, en pacientes estratificados por edades, sexo y
distintos tratamientos farmacológicos. Podemos deducir cuáles genes son responsables del
crecimiento descontrolado de células, los que provocan el rendimiento de aceites en una
planta, los responsables genéticos de que una abeja reina sea diferente de una obrera o lo
que sucede durante la metamorfosis de un insecto. Esta tecnología de chips no solo se
aplica al análisis de la expresión de genes; también sirve para deducir pérdidas y ganancias
de material genético en distintas regiones cromosómicas, para la detección de mutaciones
de un solo nucleótido en poblaciones, para la interacción entre proteínas, etcétera. Si bien
las posibilidades de este tipo de tecnología son enormes, lo relevante en esta revolución
tecnológica es haber pasado del análisis individual de unos pocos genes al análisis masivo
de genes en paralelo.

FILOGENÓMICA: UNA VISIÓN GENÓMICA DEL ÁRBOL DE LA VIDA

Las relaciones de parentesco entre humanos, moscas y gusanos no segmentados o nematodos fueron definidas desde los inicios del siglo XX gracias a una característica que surgió durante la evolución de los dos primeros grupos de organismos y que no se ve en el tercero: el celoma. Este es una cavidad embrionaria característica de moluscos, gusanos segmentados o anélidos, artrópodos y vertebrados, que supuestamente habría surgido una única vez en el ancestro común de todos estos organismos.
El estudio filogenético o historia evolutiva molecular de los organismos mencionados estableció una nueva clasificación, pues postuló la existencia de los ecdisozoos (Ecdysozoa), un grupo distinto que reúne a moscas y gusanos, es decir, a artrópodos y nematodos en una sola clase. La existencia de los ecdisozoos, que forman uno de los grupos mayores del mundo animal, postula la existencia de un ancestro común diferente entre estos organismos. En la vieja clasificación los humanos, lo mismo que los cerdos y las serpientes, estábamos más cerca de las moscas y arañas que de los gusanos no segmentados. En la nueva clasificación, con el grupo de ecdisozoos quedamos a igual distancia de los insectos que de cualquier gusano parásito nematodo. Así, desde mediados de la década de 1970, la información proporcionada por la biología molecular pasó a dominar la sistemática.

La nueva clasificación fue inicialmente aceptada por algunos y discutida por otros, según qué genes utilizaran para el análisis. En 2003, cuando este autor comenzó accidentalmente a analizar el problema, se contaba ya con los genomas completos de los tres conjuntos de organismos nombrados, así como de otros muchos que tienen un lugar en el árbol de la vida, desde unicelulares como el Plasmodium (el parásito causante de la malaria) hasta los humanos. Ahora, gracias al soporte estadístico proporcionado por más de 11.000 secuencias de los genomas de diferentes organismos, el grupo de los ecdisozoos quedó firmemente establecido. La filogenómica, es decir, la historia de la vida reconstruida sobre bases genómicas o de miles de genes, aporta así una visión estadística más robusta de las relaciones genealógicas entre las especies. Requiere el manejo masivo de genes y la concatenación de miles de análisis filogenéticos por medio de programas informáticos en lenguajes como Perl, Phyton o C++.

SELECCIÓN NATURAL Y DETECCIÓN GENÓMICA DE ENFERMEDADES
En 2005 comenzamos a trabajar en la predicción de enfermedades humanas mediante el análisis evolutivo de presiones selectivas en proteínas, es decir, midiendo la fuerza con que un aminoácido es descartado de una población. El razonamiento que utilizamos se basó en el hecho de que, del mismo modo que la selección natural puede mantener el cambio adaptativo de la información genética de una proteína, su trabajo frecuente es descartar mutaciones que deterioran la información genética transmitida de padres a hijos, causa principal de que los hijos se parezcan a sus padres. Esta forma de selección se conoce como selección purificadora, y es la más frecuente en todas las especies.

En un primer estudio, trabajamos con la proteína P53, que tiene un importante cometido en el control del ciclo celular y, por lo tanto, es fundamental para evitar el crecimiento de la mayor parte de cánceres humanos. Establecimos que por debajo de cierto umbral de presiones selectivas sobre un aminoácido, podíamos distinguir residuos de proteínas asociados con diferentes frecuencias de cánceres, e identificamos los residuos de unas cuarenta proteínas con mayor probabilidad de asociación con cánceres y enfermedades inmunológicas. Después, trabajando en colaboración con un físico, extendimos esos primeros análisis a la construcción de un sistema informático que opera con los principios de inteligencia artificial y pudimos, primero, ampliar el método a todas las proteínas de la base de datos UniProt (Universal Protein Resource) y, luego, a todo el genoma humano. Finalmente, aplicamos el método en la publicación del consorcio que secuenció los genomas de todas las cepas de ratones de laboratorio. En todo esto, la combinación de conocimientos en evolución, genómica y bioinformática resultó clave para validar y predecir con éxito enfermedades.

La bioinformática utiliza algoritmos informáticos y modelos probabilísticos para dar sentido a los datos contenidos en las basesde datos biológicas. Entre las más conocidas podemos mencionar: (1) la de genes de enfermedades humanas (2) la de ontologías, que emplea un vocabulario de términos semijerárquicos organizados por procesos biológicos, componentes celulares y funciones biológicas, y (3) la de genomas completos. Entre las herramientas más populares, están las que buscan secuencias parciales o completas (exones, motivos, genes o regiones genómicas) en diferentes bases de datos. Entre los modelos probabilísticos están los ocultos de Markov, que sirven para buscar patrones definidos de secuencias. También las regresiones logísticas, los modelos basados en máxima verosimilitud y el cálculo de probabilidades bayesianas para el ajuste del modelo a los datos biológicos. El trabajo principal del bioinformático es el manejo de la información biológica. Pero no alcanza con saber buscar datos en un mar de letras y números: se debe saber qué hacer con ellos, es decir, formular hipótesis y ponerlas a prueba. El Departamentode Bioinformática y Genómica de la institución en que se desempeña el autor en Valencia ha desarrollado herramientas que integran soluciones a problemas diversos. Por ejemplo, para el análisis funcional de genes, que permite la caracterización funcional de experimentos de microarrays, y su lectura. El programa que estudia la formación de redes mínimas de proteínas, el que deduce estructuras terciarias de proteínas por comparación con secuencias con otras especies, el que se encarga del análisis de la evolución molecular, filogenia y adaptación de las secuencias moleculares, y el que busca mutaciones funcionales en el genoma humano y el de ratón. Estas herramientas y muchas otras son utilizadas en internet por una amplia comunidad de investigadores alrededor del mundo. Las herramientas bioinformáticas representan en esta época lo que fueron para la genética las técnicas de biología molecular en las décadas de 1980 y 1990. Dado que no se puede hacer un experimento si no se tiene una hipótesis de trabajo y una definición explícita de cómo manejar los números, resulta obvio que los poseedores de este conocimiento serán jugadores clave en la generación de la biología de este siglo.

PIONEROS DE LA BIOINFORMÁTICAMargaret Oakley Dayhoff (1925-1983), nacida en Filadelfia, escribió su tesis doctoral en la Universidad de Columbia sobre la medición de energías de resonancia en compuestos orgánicos. Se valió de aplicaciones informáticas que usaban tarjetas perforadas. Química de profesión, se desempeñó en las universidades Rockefeller, de Maryland y Georgetown. Inventó el código de aminoácidos de una sola letra y la matriz PAM para contabilizar el cambio evolutivo en secuencias de proteínas. Estos desarrollos la llevaron a construir el primer árbol filogenético por medio de una computadora. Su Atlas de secuencias y estructuras de proteínas, organizado por familias génicas, que le llevó trece años de trabajo desde 1965, fue su más destacada contribución al desarrollo de la disciplina. El atlas dio origen a la base de datos PIR (Protein Information Resource), hoy incluida en UniProt.
Walter Benson Goad (1925-2000), nacido en el estado norteamericano de Georgia, físico teórico del laboratorio de Los Álamos en Nuevo México, desarrolló el código informático para la extracción, almacenamiento y análisis de secuencias de ADN, que llevó a conformar en 1982 la primera base de datos de nucleótidos del mundo: Los AlamosSequence Data Bank, que se convertiría en GenBank. Junto con sus homólogas del Laboratorio Europeo de Biología Molecular y la Base de datos de ADN del Japón, actualizan su contenido diariamente y no han parado de crecer en forma exponencial desde su creación, con duplicación de su contenido cada dieciocho meses.

¿QUÉ NOS HACE HUMANOS Y NO CHIMPANCÉS?
El sueño de conocer las diferencias genéticas completas entre humanos y nuestros parientes vivientes más cercanos se hizo posible en 2005, cuando se publicó la primera versión del genoma del chimpancé. Los biólogos evolutivos de esta generación tenemos la enorme fortuna de poder responder científicamente a uno de los interrogantes que desde siempre más han interesado a la humanidad: ¿qué nos hace humanos? Con ese propósito abordamos un análisis comparativo de trece mil genes de cinco especies de mamíferos. El trabajo consistió en el alineamiento automático de esos genes, su ajuste a modelos estadísticos y la estimación de tasas o velocidades de cambio evolutivo para cada uno de ellos en cada una de las especies y en sus ancestros. Trabajamos en total con unos noventa mil genes y unas cincuenta variables a las que aplicamos diferentes pruebas para estimar si en el linaje humano había grupos de genes que cumplían funciones específicas y estaban cambiando de manera adaptativa.

Los resultados de trabajos similares venían siendo poco esclarecedores y el nuestro confirmó lo mismo: que no se han encontrado características genéticas que nos hagan distintos de un chimpancé. Sabemos algo más después de nuestro estudio: que desde la separación genealógica de nuestro ancestro común, los chimpancés cambiaron su genoma de manera adaptativa o darwiniana en mayor medida que los humanos. Asimismo, hay evidencia de que los humanos experimentaron cambios adaptativos mucho más relevantes en proteínas testiculares que cerebrales. Si eso no resulta sorprendente, sí lo es descubrir que el linaje de chimpancé tuvo muchos más cambios adaptativos en sus proteínas cerebrales que nuestro linaje. El uso de casi cinco millones de datos genéticos ofreció perspectivas desconcertantes sobre lo ocurrido en los últimos cinco millones de años de evolución de nuestra especie. Esos resultados, por fin verificados de manera completa, hicieron cambiar el rumbo de las investigaciones hacia otras regiones del genoma. En 2007 se encontraron las primeras diferencias de constitución genética entre humanos y chimpancés en materia de cambio de dieta y actividad cerebral, no en proteínas sino en las regiones del genoma que regulan la expresión de los genes.

DEL GEN A LA BIOLOGÍA DE SISTEMAS Cabe preguntarse si los avances realizados en los últimos veinte años en genómica y bioinformática aplicadas a la evolución llevaron a la biología a un estadio del conocimiento proporcionado al esfuerzo invertido. Es conveniente tener en cuenta que, en el estudio de la evolución –y posiblemente en el resto de la biología– seguimos pensando y aplicando métodos de la era pregenómica a los nuevos datos genómicos. Si antes aplicábamos una prueba de selección a una decena de genes, ahora lo hacemos a miles con la ayuda de la bioinformática. Los tres ejemplos anteriores muestran esa práctica.
Sin embargo, poco a poco va calando la idea de que este enfoque no es suficiente. Los datos genómicos muestran un comportamiento de la naturaleza que no era evidente cuando el análisis abarcaba unos pocos genes. Aquí es donde entra la biología de sistemas, una nueva rama de la biología conocida desde hace tiempo también como biología integrativa o biología holística, que tomó impulso con la revolución genómica. La disciplina, sin embargo, no resulta de la mera negación de la visión más simplista o reduccionista creada por razonar sobre pocos genes, sino de considerar las características propias del sistema complejo del que ahora tenemos datos.
Para dar un ejemplo, volvamos a la comparación de genomas entre especies que cambian con relativa rapidez y constituyen casos deadaptación molecular. En ellas, el análisis de los genes individuales con métodos anteriores a la era genómica impide conceder validez estadística al hecho de que hay, en cada especie, funciones diferentes asociadas con el cambio adaptativo. Pero reconociendo que los genes no son independientes, hemos sido capaces de encontrar esas diferentes funciones. Los genes que se modifican en forma más acelerada muestran características biológicas que no pueden ser explicadas considerándolos de manera individual, como su localización en los cromosomas, más cercanos entre ellos, o la formación de redes más apretadas, que aquellos que se modifican de manera más lenta. Es decir, existen propiedades biológicas imposibles de explicar solo por la acción de genes individuales.
![proteomica italianodef DEFSTAMPARE [modalità compatibilità] 67. italiano def/proteomica... · genomica -proteomica si concretizza nella nascita di una nuova disciplina ‘biologia](https://img.dokumen.tips/doc/110x75/5c4dbb2393f3c350ba7e2948/proteomica-italianodef-defstampare-modalita-compatibilita-67-italiano-defproteomica.jpg)














![Cap.13 Proteomica[1] REV](https://img.dokumen.tips/doc/110x75/5571f8a149795991698dcadc/cap13-proteomica1-rev.jpg)













