Embed Size (px)
Citation preview

Carbohydrate Research 346 (2011) 2244–2254
Contents lists available at ScienceDirect
Carbohydrate Research
journal homepage: www.elsevier .com/locate /carres
The efficient structure elucidation of minor components in heparin digestsusing microcoil NMR
John F. K. Limtiaco a,�, Szabolcs Beni a,b,�, Christopher J. Jones a, Derek J. Langeslay a, Cynthia K. Larive a,⇑a University of California, Department of Chemistry, Riverside, CA 92521, USAb Semmelweis University, Department of Pharmaceutical Chemistry, H}ogyes Endre u. 9, H-1092 Budapest, Hungary
a r t i c l e i n f o a b s t r a c t
Article history:Received 25 May 2011Accepted 9 July 2011Available online 20 July 2011
Keywords:Heparin diastereomersHexuronic acid epimersMicrocoil NMRNuclear magnetic resonanceUnsaturated tetrasaccharideUPLC-MS
0008-6215/$ - see front matter � 2011 Elsevier Ltd. Adoi:10.1016/j.carres.2011.07.007
⇑ Corresponding author. Tel.: +1 951 827 2990; faxE-mail address: [email protected] (C.K. Larive).
� Both authors made equal contributions to this wor
The structural complexity and microheterogeneity of the glycosaminoglycans heparin and heparan sul-fate make their characterization a daunting task. The methodology described herein utilizes a combina-tion of enzymatic digestion, size-exclusion chromatography, strong anion-exchange HPLC, reverse-phaseion-pair ultrahigh performance liquid chromatography–mass spectrometry, and microcoil NMR for theefficient sequencing of heparin-derived tetrasaccharides. The high mass sensitivity of microcoil NMRmakes this technique well suited for the characterization of mass-limited samples removing a bottleneckin the analysis workflow and permitting structural characterization of minor components isolated from aheparin enzymatic digestion. Complete characterization of one tetrasulfonated, five pentasulfonatedisomers and two hexasulfonated tetrasaccharide sequences is described. To our knowledge, two of theidentified minor tetrasaccharides are unique, and have not been previously reported: IdoA(2S)-GlcNS(6S)-IdoA(2S)-GlcNS(6S) and DUA(2S)-GlcNS(6S)-IdoA-GlcNS(6S).
� 2011 Elsevier Ltd. All rights reserved.
1. Introduction
Glycosaminoglycans (GAGs) are unbranched polysaccharidesthat are heterogeneous in molecular mass, charge density andphysico-chemical properties. The structure of GAG oligosaccharidesequences are determined through a bottom-up approach that re-lies on depolymerization reactions, size- and charge-based separa-tions and characterization by mass spectrometry (MS) and NMRexperiments to determine the structural identity of componentoligosaccharides.1–3 Because NMR is crucial for determining theconfiguration of the uronic acid residues and sulfonation positions,GAG sequencing relies heavily on 2D NMR techniques.4 However,the low inherent sensitivity of NMR often requires greater samplequantities than may be practically isolated. Microcoil NMR hasbeen shown to be a powerful technique for structural characteriza-tion of mass-limited samples,5–7 and is used in this work tostreamline the structure elucidation process.
Among the GAGs, heparin is one of the most studied com-pounds due to its anticoagulant activity and pharmaceuticalimportance. Although heparin is an important and widely pre-scribed pharmaceutical, its high degree of sequence microhetero-geneity and size polydispersity makes molecular-levelcharacterization challenging.1,3,8 Comprised of variously sulfo-nated hexuronic acid (1?4)- D-glucosamine repeating disaccharide
ll rights reserved.
: +1 951 827 4713.
k.
building blocks, heparin is the most acidic biopolymer in nature.The uronic acid residue of heparin may be either a-L-iduronic acid(IdoA) or b-D-glucuronic acid (GlcA) and can be unsubstituted orsulfonated at the 2-O position. The glucosamine residue may beeither unmodified (GlcN), N-sulfonated (GlcNS) or N-acetylated(GlcNAc) with variable patterns of O-sulfonation at the 6-O, andless frequently, the 3-O positions.9,10 Unlike nucleic acids and pro-teins that are biosynthesized through a template-driven assemblyprocess, heparin and the related GAG, heparan sulfate (HS), are ac-tively remodeled during biosynthesis through a series of enzymaticreactions that lead to variable levels of O- and N-sulfonation anduronic acid epimerization.11,12 Incomplete modification results inthe production of heparin oligosaccharides with varying sequencesof N- and O-sulfonation and IdoA content.13
Heparin and HS interact with a myriad of proteins that mediatetheir biological functions.13 To better understand the role of hepa-rin/HS in numerous physiological and pathophysiological pro-cesses such as blood coagulation,14 viral infection,15 angio- andtumorigenesis,16,17 cell differentiation and development andinflammation,18 and also to gain molecular level information abouttheir physico-chemical properties, the isolation and structuralcharacterization of pure oligosaccharide sequences is essential.
In this work, size- and charge-based chromatographic fraction-ation with MS and microcoil NMR characterization are used todetermine the structures of eight heparin-derived tetrasaccha-rides: one tetrasulfonated, five pentasulfonated isomers and twohexasulfonated sequences. Among the five pentasulfonated posi-tional isomers characterized, two uronic acid diastereomer pairs

J. F. K. Limtiaco et al. / Carbohydrate Research 346 (2011) 2244–2254 2245
were identified and the structure of a unique saturated hexasulfo-nated tetrasaccharide is also discussed. The results presentedherein demonstrate the power and versatility of microcoil NMRas a non-destructive analytical tool for the efficient sequencing ofheparin-derived oligosaccharides.
2. Results and discussion
As a result of the structural diversity of heparin and HS, mostmethods used for their structural characterization utilize a bot-tom-up approach, whereby the intact polysaccharide is chemicallyor enzymatically depolymerized to smaller oligosaccharides priorto analysis.19,20 Heparinase enzymes are highly specific with respectto the uronic acid configuration and degree of sulfonation at thecleavage site.21,22 The heparinase I enzyme used in this studycleaves the [1?4] glycosidic bond between a glucosamine residuethat is N-sulfonated (GlcNS) and an iduronic acid sulfonated at the2-O position (IdoA2S),22,23 the most common substitution motifobserved in unfractionated heparin from porcine intestinal mucosa.The enzymatic depolymerization of heparin occurs through a b-elimination reaction yielding oligosaccharide fragments that areunsaturated at the C-4,5 position of the non-reducing end of the ter-minated hexuronic acid residues, as shown in Figure S-1A.24 Theunsaturated bond at the C-4,5 position produces a characteristicabsorption maximum at 232 nm, thus allowing UV detection.24
In studies where determination of the disaccharide compositionis the goal, intact heparin is exhaustively digested with a cocktail ofheparinase enzymes I, II, and III.20,25–28 This exhaustive digestionyields almost exclusively heparin disaccharides and, with theavailability of authentic standards, compositional analysis can beachieved using HPLC20,29–32 or CE28,33–35 coupled to UV absorptionor fluorescence spectroscopy for detection and quantification. Inother cases the experimental goal of the digestion is to isolateand characterize larger heparin oligosaccharide fragments, oftenas part of a study to explore their protein binding properties or bio-logical activity. In studies where larger heparin oligosaccharidefractions are desired, partial digestion of the intact heparin isperformed producing a complex mixture of variably-sizedoligosaccharides consisting of a diverse set of positional andconfigurational isomers.36 For these samples, a single mode ofseparation is ineffective in resolving the mixture componentstherefore multiple orthogonal separation approaches are required.
2.1. Separation and isolation of heparin-derivedtetrasaccharides
Following the enzymatic digestion of heparin, the resultingsolution of oligosaccharides was separated into size-uniform frac-tions using SEC, which were isolated for further purification andanalysis using SAX-HPLC and RPIP-UPLC/MS. The SEC columnwas effective in baseline separating the components into peaksranging from disaccharides (dp2) to hexadecasaccharides (dp16)as shown in Figure S-1B. Because of the site of the enzymatic reac-tion, primarily even numbered oligomers were produced, althoughsmall quantities of trisaccharides have also been reported.37
The SEC fractions corresponding to tetrasaccharides werepooled and subjected to further separation using SAX-HPLC on aCarboPac PA1 semi-preparative scale column as described by Chu-ang and co-workers, with the resulting chromatogram shown inFigure 1A.38,39 The numbered peaks indicate the tetrasaccharidesisolated for structural characterization; SAX retention times aresummarized in Table 1. Based on their retention times we hypoth-esized that most of the SAX peaks correspond to pentasulfonatedtetrasaccharides while the largest peak at 80 min is due to thehexasulfonated tetrasaccharide DUA(2S)-GlcNS(6S)-IdoA(2S)-
GlcNS(6S). The high level of sulfonation observed in the isolatedpeaks is expected, as about 70% of the heparin isolated from por-cine intestinal mucosa is comprised of the disaccharide IdoA(2S)-GlcNS(6S).9
For comparison, the RPIP-UPLC/MS chromatogram of the tetra-saccharide SEC fraction is shown in Figure 1B. The chromatogramsshown in Figure 1 separate the different tetrasaccharides accordingto their charge. Significantly better resolution is achieved for theSAX separation (Fig. 1A) providing improved confidence in peakpurity, albeit at a cost of a much longer separation time. Also, forsamples isolated in minute amounts for NMR measurements, theion-pairing reagent (IPR) in fractions collected from RPIP-UPLC isdifficult to remove completely and this contaminant can limit theNMR receiver gain values that can be used. In contrast, SAX sepa-rations require large concentrations of salt for elution and desalt-ing of the samples is required to allow the NMR probe to beproperly tuned and matched.
2.2. RPIP-UPLC/MS analysis of the SEC tetrasaccharide fraction
RPIP-UPLC/MS was used in the preliminary analysis of tetrasac-charides present in the SEC dp4 fraction. The mass spectrum con-firmed the identity of the most abundant peak as the a-anomerof DUA(2S)-GlcNS(6S)-IdoA(2S)-GlcNS(6S) which produces an m/zof 575.96 Da for the molecular ion [M�2H]2�. The partially-resolved peak 8 at 8.0 min is the b-anomer of this tetrasaccharide.The smaller peaks eluting between 6.6 and 7.4 min in Figure 1Beach have m/z values of 535.98 Da corresponding to tetrasaccha-rides containing five sulfonate groups. The partial resolution ofthe reducing end a- and b-anomers in the RPIP-UPLC chromato-gram further complicates the separation. Because of the poor reso-lution in the RPIP-UPLC chromatogram for the SEC tetrasaccharidemixture, a small amount of each of the desalted SAX fractions wassubjected to RPIP-UPLC/MS with the retention times and m/z val-ues for the molecular ions ([M�2H]2�) reported in Table 1.
Mass spectrometry has been used for the complete structuralcharacterization of heparin hexasaccharides by Schenauer andco-workers.40 In this approach the isolated individual hexasaccha-rides were subjected to a secondary enzymatic digestion to theircorresponding tetra- and disaccharide subunits. Sequence informa-tion was derived using the differential cleavage specificities of hep-arinase enzymes to reconstruct the sequence of the originalhexasaccharide. This method may be efficient for the characteriza-tion of a few unique heparin-derived oligosaccharides especially instudies in which heparin is digested in the presence of its bindingpartner,39 but would be expensive for the large-scale characteriza-tion heparin-derived oligosaccharides. More importantly, informa-tion about the configuration of the biologically important internaluronic acid residues is lost as a result of the second enzymaticdigestion.41–43
Recent work by Amster and co-workers report the potential ofelectron detachment dissociation (EDD) for the characterizationof heparin-derived tetrasaccharides.44,45 Ion activation methods,such as EDD and electron-induced dissociation (EID), have recentlyfound more widespread application in the characterization of bio-molecules.46–49 In EDD, electron activation of precursor ionscleaves the glycosidic bonds and produces cross-ring cleavages,greatly improving the capability of MS to determine sites of sulfo-nation and acetylation.44 In addition, characteristic fragmentationpatterns and product ions can permit discrimination between GlcAand IdoA containing heparan sulfate oligosaccharides.45 In ourstudies of heparin-derived tetrasaccharides using ESI-TOF-MS,product ions from cross-ring and glycosidic bond cleavages wereinsufficient for complete structural characterization and 2D NMRspectra were required for the determination of sulfonation posi-tions and uronic acid configurations in the isolated compounds.
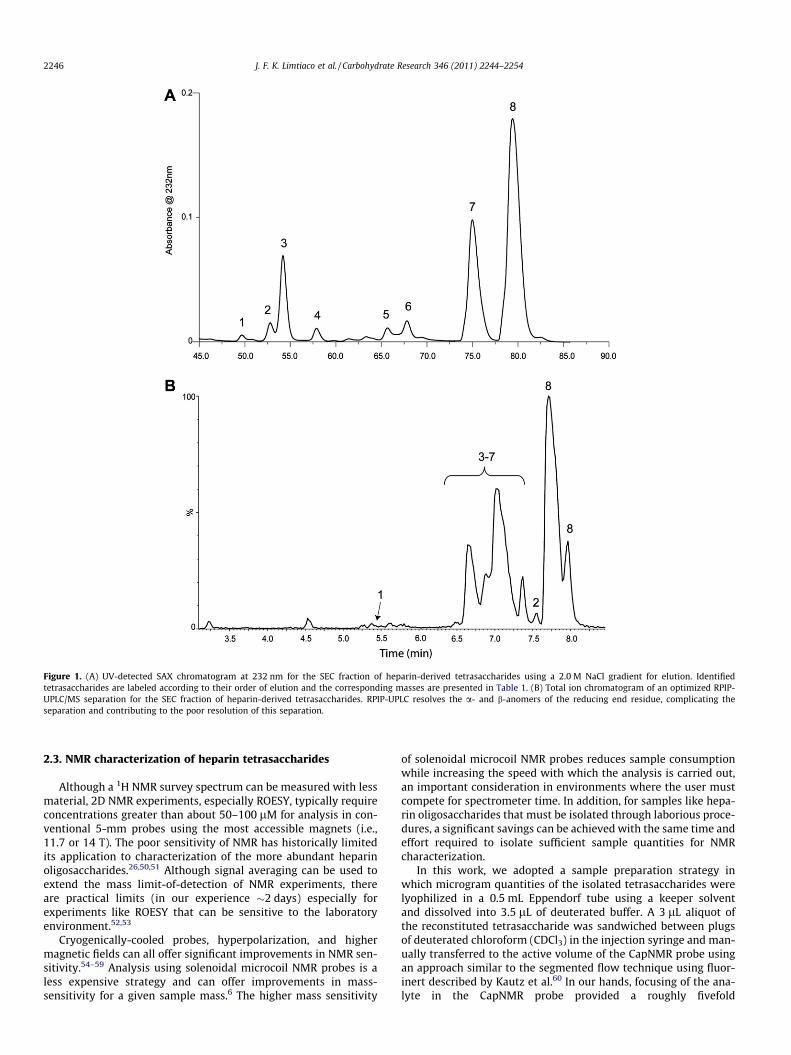
Figure 1. (A) UV-detected SAX chromatogram at 232 nm for the SEC fraction of heparin-derived tetrasaccharides using a 2.0 M NaCl gradient for elution. Identifiedtetrasaccharides are labeled according to their order of elution and the corresponding masses are presented in Table 1. (B) Total ion chromatogram of an optimized RPIP-UPLC/MS separation for the SEC fraction of heparin-derived tetrasaccharides. RPIP-UPLC resolves the a- and b-anomers of the reducing end residue, complicating theseparation and contributing to the poor resolution of this separation.
2246 J. F. K. Limtiaco et al. / Carbohydrate Research 346 (2011) 2244–2254
2.3. NMR characterization of heparin tetrasaccharides
Although a 1H NMR survey spectrum can be measured with lessmaterial, 2D NMR experiments, especially ROESY, typically requireconcentrations greater than about 50–100 lM for analysis in con-ventional 5-mm probes using the most accessible magnets (i.e.,11.7 or 14 T). The poor sensitivity of NMR has historically limitedits application to characterization of the more abundant heparinoligosaccharides.26,50,51 Although signal averaging can be used toextend the mass limit-of-detection of NMR experiments, thereare practical limits (in our experience �2 days) especially forexperiments like ROESY that can be sensitive to the laboratoryenvironment.52,53
Cryogenically-cooled probes, hyperpolarization, and highermagnetic fields can all offer significant improvements in NMR sen-sitivity.54–59 Analysis using solenoidal microcoil NMR probes is aless expensive strategy and can offer improvements in mass-sensitivity for a given sample mass.6 The higher mass sensitivity
of solenoidal microcoil NMR probes reduces sample consumptionwhile increasing the speed with which the analysis is carried out,an important consideration in environments where the user mustcompete for spectrometer time. In addition, for samples like hepa-rin oligosaccharides that must be isolated through laborious proce-dures, a significant savings can be achieved with the same time andeffort required to isolate sufficient sample quantities for NMRcharacterization.
In this work, we adopted a sample preparation strategy inwhich microgram quantities of the isolated tetrasaccharides werelyophilized in a 0.5 mL Eppendorf tube using a keeper solventand dissolved into 3.5 lL of deuterated buffer. A 3 lL aliquot ofthe reconstituted tetrasaccharide was sandwiched between plugsof deuterated chloroform (CDCl3) in the injection syringe and man-ually transferred to the active volume of the CapNMR probe usingan approach similar to the segmented flow technique using fluor-inert described by Kautz et al.60 In our hands, focusing of the ana-lyte in the CapNMR probe provided a roughly fivefold

Table 1SAX retention times, RPIP-UPLC retention times, m/z of the molecular ion [M�2H]2�, the mass used in the microcoil NMR experiments and the identities of the heparin-derivedtetrasaccharides determined, and references previously reporting this compound
SAX peaknumber
SAX retentiontime
RPIP-UPLCretention time
Molecular ionm/z
Mass for CapNMRexperiments
Tetrasaccharideidentity
References
1 49 min 5.58 min 496.02 Da 34 lg DUA(2S)-GlcNS-GlcA-GlcNS(6S) 24,482 53 7.51 584.96 49 IdoA(2S)-GlcNS(6S)-IdoA(2S)-GlcNS(6S) NA3 54 6.61 535.98 149 DUA(2S)-GlcNS(6S)-IdoA(2S)-GlcNS 24,25,644 58 6.78 535.98 21 DUA(2S)-GlcNS-IdoA(2S)-GlcNS(6S) 24,635 66 6.90 535.98 40 DUA(2S)-GlcNS(6S)-IdoA-GlcNS(6S) NA6 68 6.99 535.98 81 DUA(2S)-GlcNS(6S)-GlcA(2S)-GlcNS 247 75 7.08 535.98 192 DUA(2S)-GlcNS(6S)-GlcA-GlcNS(6S) 24,25,48,63,648 80 7.76 575.96 413 DUA(2S)-GlcNS(6S)-IdoA(2S)-GlcNS(6S) 24,25,48,63,64
J. F. K. Limtiaco et al. / Carbohydrate Research 346 (2011) 2244–2254 2247
improvement in concentration sensitivity compared to the use of astandard 5 mm NMR tube, greatly enhancing the efficiency of theNMR experiments. Although the smallest volume that could bereproducibly injected into the microcoil NMR probe without intro-ducing air bubbles was 3 lL, the residual sample is not necessarilywasted and could be dried under an N2 stream, diluted into10–20 lL of aqueous solvent and used for RPIP-UPLC/MScharacterization.
Following the introduction of the sample into the microcoilprobe, 1H NMR survey spectra were acquired to evaluate samplepurity and ensure a sufficient S/N for resonance assignment usingthe TOCSY, COSY, and ROESY experiments. The 1H NMR spectrumshown in Figure 2A was measured with 21 lg of a heparin-derivedtetrasaccharide (SAX peak 4) injected into the microcoil probe. Fig-ure 2B–D shows the COSY, TOCSY, and ROESY spectra of this sam-ple acquired over 42 h. The resonance of the unsaturated uronicacid residue is the most downfield (DUAH4, 6.02 ppm) in the COSYspectrum (Fig. 2B) and provides a good entry point into the reso-nance assignments. Using the COSY spectrum, the scalar connectiv-ity to DUAH3 (4.24 ppm), DUAH2 (4.61 ppm), and subsequently,DUAH1 (5.53 ppm), can be traced establishing the connectivitywithin the nonreducing end residue. This process was repeatedfor the other monosaccharide residues. The TOCSY spectrum wasnecessary for the assignment of the resonances for the glucosa-mine residues and was also used to confirm the COSY assignments.Figure 2C shows a portion of the TOCSY spectrum showing crosspeaks to the well resolved anomeric resonances. Chemical shiftsare summarized in Table 2. Comparison of chemical shift data withliterature values is used to determine the structural identities ofthe individual monosaccharide residues.61
ROESY spectra were used to establish the order of the monosac-charides within the intact tetrasaccharide using cross peaks be-tween resonances of the H-1 protons and the H-4 protons of thenext residue, connected via a glycosidic bond.61 Figure 2D providesa portion of the ROESY spectrum of the compound in SAX peak 4showing the important inter-residue cross peaks. From the mea-sured chemical shifts and inter-residue ROESY cross peaks theidentity of the tetrasaccharide was determined to be DUA(2S)-GlcNS-IdoA(2S)-GlcNS(6S). This structure is consistent with thespecificity of the heparinase I enzyme which requires a 2-O-sulfo-nated residue at the non-reducing end of the digested heparinfragment.
Although most of the isolated SAX fractions were fairly pure,containing a single component readily characterized using NMR,the 1H NMR spectrum obtained for SAX fraction 2 (Fig. 3A) sug-gested that it was composed of two different tetrasaccharides atroughly equal concentrations. Comparison of Figure 3A with thespectrum measured for the closely eluting SAX peak 3 (Fig. S-2A)confirms that that one of the components in fraction 2 isDUA(2S)-GlcNS(6S)-IdoA(2S)-GlcNS, (structure determined usingthe TOCSY and ROESY spectra Figures S-2B and C, respectively).Expansions of the TOCSY (Fig. 3B) and ROESY (Fig. 3C) spectra for
SAX fraction 2 were used for structural characterization of the un-known compound. Cross peaks to the anomeric resonance of theterminal uronic acid residue (IdoAH1, 5.18 ppm) were key in estab-lishing that the terminal uronate residue lacked the characteristicdouble bond introduced by enzymatic depolymerization.
To confirm the identities of the tetrasaccharides characterizedby NMR, the compounds isolated by SAX were also analyzed usingRPIP-UPLC/MS. The total ion chromatogram (TIC) for the SAX peak2 is shown in Figure 4A with the mass spectra of the componenttetrasaccharides given in Figure 4B and C. As can be seen in Fig-ure 4A, RPIP-UPLC resolves the reducing end a- and b-anomers.The identity of the contaminant in Figure 4B as DUA(2S)-GlcNS(6S)-IdoA(2S)-GlcNS is confirmed by the agreement of itsRPIP-UPLC retention time (6.41) and m/z for the doubly-chargedmolecular ion [M�2H]2� with the values observed for SAX peak3. As shown in Figure 4, IdoA(2S)-GlcNS(6S)-IdoA(2S)-GlcNS(6S)elutes at 7.30 min and has a doubly charged molecular ion[M�2H]2� of 584.96 Da, consistent with a saturated hexasulfonatedtetrasaccharide lacking the characteristic double bond at the nonre-ducing end uronic acid residue. The experiments that produced thechromatogram shown in Figure 4 and the results reported in Fig-ure 1B and Table 1 were separated by several weeks, which contrib-uted to small differences in retention times, on the order of a fewpercent.
It is interesting to note that although IdoA(2S)-GlcNS(6S)-IdoA(2S)-GlcNS(6S) is a hexasulfonated tetrasaccharide, it elutesat 53 min in the SAX chromatogram, much earlier than the corre-sponding double-bond containing hexasulfonated tetrasaccharide,DUA(2S)-GlcNS(6S)-IdoA(2S)-GlcNS(6S) (80 min), and also priorto all of the pentasulfated species we identified. This suggests acritical role of the double bond and the concomitant change inring-conformation on retention of oligosaccharides in thecharged-based SAX separation. In the RPIP-UPLC separation bothhexasulfonated tetrasaccharides, IdoA(2S)-GlcNS(6S)-IdoA(2S)-GlcNS(6S) (7.30 min) and DUA(2S)-GlcNS(6S)-IdoA(2S)-GlcNS(6S)(7.76 min), are observed to elute after the pentasulfonated tetra-saccharides as would be expected with charge-based separations.
Most of the tetrasaccharides characterized in this work and de-scribed by Tables 1 and 2 are commonly observed in a heparinasedigested mixture as the majority of the individual fragments arehighly sulfonated. The most abundant tetrasaccharide is the hexa-sulfonated tetrasaccharide, DUA(2S)-GlcNS(6S)-IdoA(2S)-GlcNS(6S), SAX peak 8, which was originally characterized by 1Hand 13C NMR in the mid 80’s.50,62 As the heparin biopolymer is richin this highly sulfonated sequence, literature corresponding to thecharacterization of this tetrasaccharide is extensive and diverse.This includes studies on the conformation and flexibility of themonosaccharide residues within the chain as well as the influencethat these residues has on its overall binding.41,63,64 Earlier investi-gations of the highly sulfonated tetrasaccharides necessitatedmilligram amounts of each compound for unambiguousidentifications. Merchant and co-workers used a 360 MHz NMR
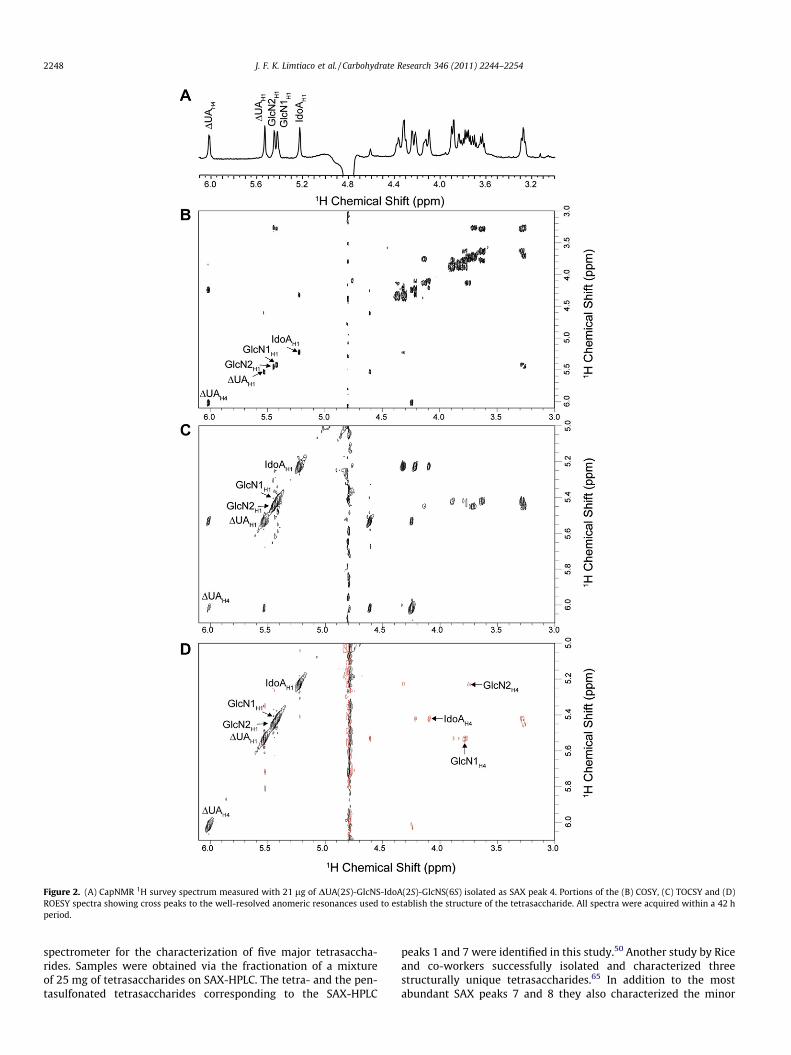
Figure 2. (A) CapNMR 1H survey spectrum measured with 21 lg of DUA(2S)-GlcNS-IdoA(2S)-GlcNS(6S) isolated as SAX peak 4. Portions of the (B) COSY, (C) TOCSY and (D)ROESY spectra showing cross peaks to the well-resolved anomeric resonances used to establish the structure of the tetrasaccharide. All spectra were acquired within a 42 hperiod.
2248 J. F. K. Limtiaco et al. / Carbohydrate Research 346 (2011) 2244–2254
spectrometer for the characterization of five major tetrasaccha-rides. Samples were obtained via the fractionation of a mixtureof 25 mg of tetrasaccharides on SAX-HPLC. The tetra- and the pen-tasulfonated tetrasaccharides corresponding to the SAX-HPLC
peaks 1 and 7 were identified in this study.50 Another study by Riceand co-workers successfully isolated and characterized threestructurally unique tetrasaccharides.65 In addition to the mostabundant SAX peaks 7 and 8 they also characterized the minor
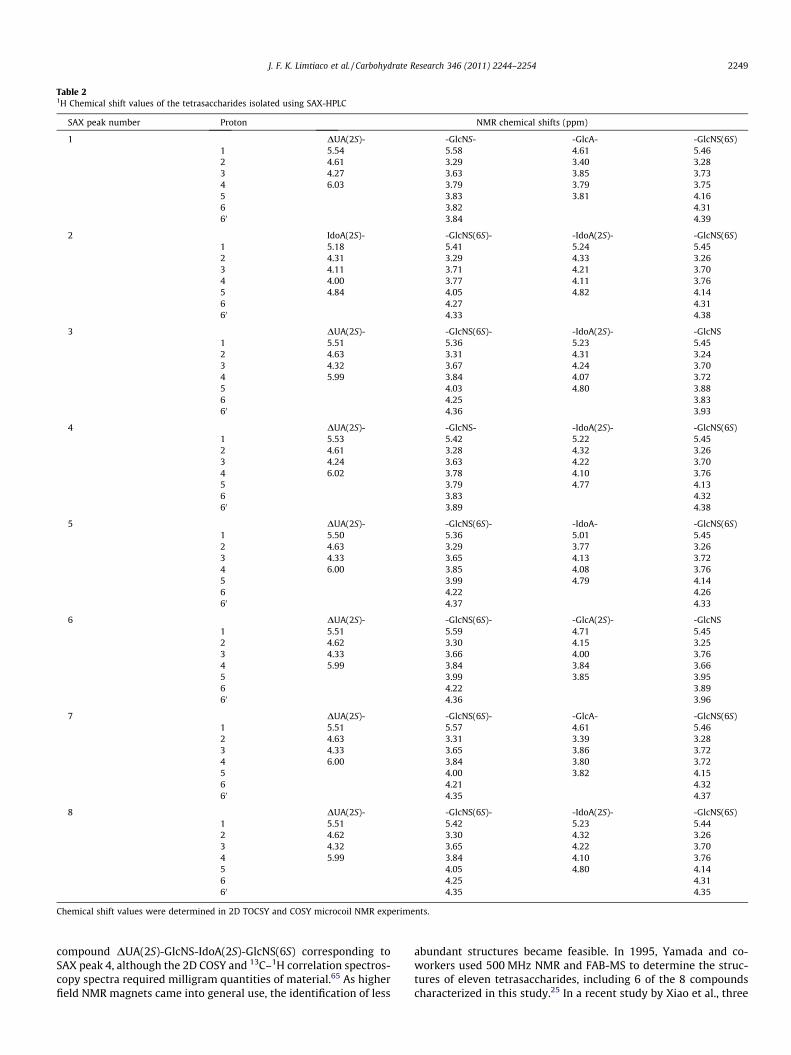
Table 21H Chemical shift values of the tetrasaccharides isolated using SAX-HPLC
SAX peak number Proton NMR chemical shifts (ppm)
1 DUA(2S)- -GlcNS- -GlcA- -GlcNS(6S)1 5.54 5.58 4.61 5.462 4.61 3.29 3.40 3.283 4.27 3.63 3.85 3.734 6.03 3.79 3.79 3.755 3.83 3.81 4.166 3.82 4.3160 3.84 4.39
2 IdoA(2S)- -GlcNS(6S)- -IdoA(2S)- -GlcNS(6S)1 5.18 5.41 5.24 5.452 4.31 3.29 4.33 3.263 4.11 3.71 4.21 3.704 4.00 3.77 4.11 3.765 4.84 4.05 4.82 4.146 4.27 4.3160 4.33 4.38
3 DUA(2S)- -GlcNS(6S)- -IdoA(2S)- -GlcNS1 5.51 5.36 5.23 5.452 4.63 3.31 4.31 3.243 4.32 3.67 4.24 3.704 5.99 3.84 4.07 3.725 4.03 4.80 3.886 4.25 3.8360 4.36 3.93
4 DUA(2S)- -GlcNS- -IdoA(2S)- -GlcNS(6S)1 5.53 5.42 5.22 5.452 4.61 3.28 4.32 3.263 4.24 3.63 4.22 3.704 6.02 3.78 4.10 3.765 3.79 4.77 4.136 3.83 4.3260 3.89 4.38
5 DUA(2S)- -GlcNS(6S)- -IdoA- -GlcNS(6S)1 5.50 5.36 5.01 5.452 4.63 3.29 3.77 3.263 4.33 3.65 4.13 3.724 6.00 3.85 4.08 3.765 3.99 4.79 4.146 4.22 4.2660 4.37 4.33
6 DUA(2S)- -GlcNS(6S)- -GlcA(2S)- -GlcNS1 5.51 5.59 4.71 5.452 4.62 3.30 4.15 3.253 4.33 3.66 4.00 3.764 5.99 3.84 3.84 3.665 3.99 3.85 3.956 4.22 3.8960 4.36 3.96
7 DUA(2S)- -GlcNS(6S)- -GlcA- -GlcNS(6S)1 5.51 5.57 4.61 5.462 4.63 3.31 3.39 3.283 4.33 3.65 3.86 3.724 6.00 3.84 3.80 3.725 4.00 3.82 4.156 4.21 4.3260 4.35 4.37
8 DUA(2S)- -GlcNS(6S)- -IdoA(2S)- -GlcNS(6S)1 5.51 5.42 5.23 5.442 4.62 3.30 4.32 3.263 4.32 3.65 4.22 3.704 5.99 3.84 4.10 3.765 4.05 4.80 4.146 4.25 4.3160 4.35 4.35
Chemical shift values were determined in 2D TOCSY and COSY microcoil NMR experiments.
J. F. K. Limtiaco et al. / Carbohydrate Research 346 (2011) 2244–2254 2249
compound DUA(2S)-GlcNS-IdoA(2S)-GlcNS(6S) corresponding toSAX peak 4, although the 2D COSY and 13C–1H correlation spectros-copy spectra required milligram quantities of material.65 As higherfield NMR magnets came into general use, the identification of less
abundant structures became feasible. In 1995, Yamada and co-workers used 500 MHz NMR and FAB-MS to determine the struc-tures of eleven tetrasaccharides, including 6 of the 8 compoundscharacterized in this study.25 In a recent study by Xiao et al., three
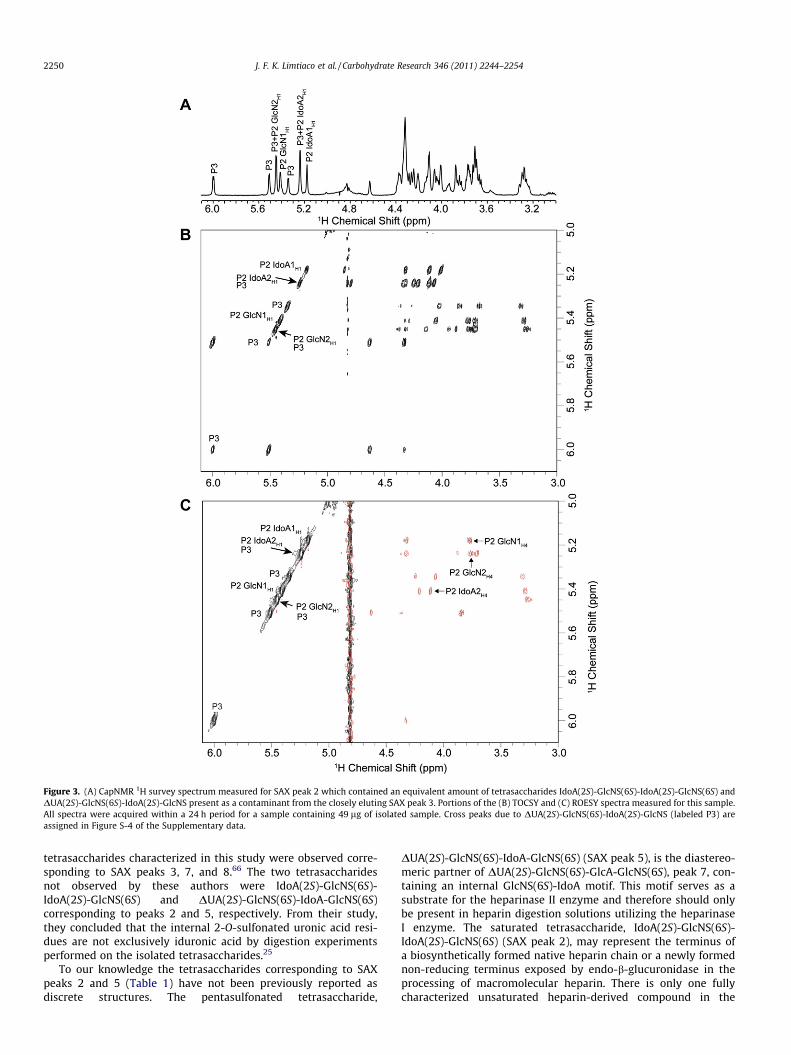
Figure 3. (A) CapNMR 1H survey spectrum measured for SAX peak 2 which contained an equivalent amount of tetrasaccharides IdoA(2S)-GlcNS(6S)-IdoA(2S)-GlcNS(6S) andDUA(2S)-GlcNS(6S)-IdoA(2S)-GlcNS present as a contaminant from the closely eluting SAX peak 3. Portions of the (B) TOCSY and (C) ROESY spectra measured for this sample.All spectra were acquired within a 24 h period for a sample containing 49 lg of isolated sample. Cross peaks due to DUA(2S)-GlcNS(6S)-IdoA(2S)-GlcNS (labeled P3) areassigned in Figure S-4 of the Supplementary data.
2250 J. F. K. Limtiaco et al. / Carbohydrate Research 346 (2011) 2244–2254
tetrasaccharides characterized in this study were observed corre-sponding to SAX peaks 3, 7, and 8.66 The two tetrasaccharidesnot observed by these authors were IdoA(2S)-GlcNS(6S)-IdoA(2S)-GlcNS(6S) and DUA(2S)-GlcNS(6S)-IdoA-GlcNS(6S)corresponding to peaks 2 and 5, respectively. From their study,they concluded that the internal 2-O-sulfonated uronic acid resi-dues are not exclusively iduronic acid by digestion experimentsperformed on the isolated tetrasaccharides.25
To our knowledge the tetrasaccharides corresponding to SAXpeaks 2 and 5 (Table 1) have not been previously reported asdiscrete structures. The pentasulfonated tetrasaccharide,
DUA(2S)-GlcNS(6S)-IdoA-GlcNS(6S) (SAX peak 5), is the diastereo-meric partner of DUA(2S)-GlcNS(6S)-GlcA-GlcNS(6S), peak 7, con-taining an internal GlcNS(6S)-IdoA motif. This motif serves as asubstrate for the heparinase II enzyme and therefore should onlybe present in heparin digestion solutions utilizing the heparinaseI enzyme. The saturated tetrasaccharide, IdoA(2S)-GlcNS(6S)-IdoA(2S)-GlcNS(6S) (SAX peak 2), may represent the terminus ofa biosynthetically formed native heparin chain or a newly formednon-reducing terminus exposed by endo-b-glucuronidase in theprocessing of macromolecular heparin. There is only one fullycharacterized unsaturated heparin-derived compound in the
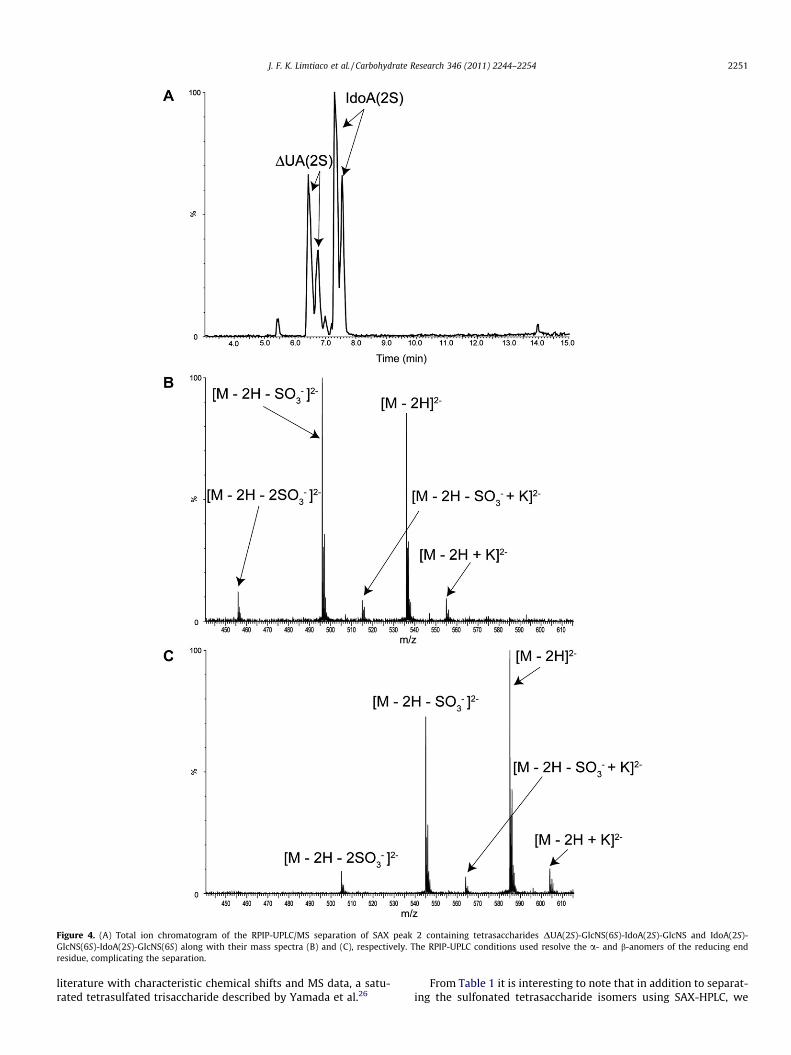
Figure 4. (A) Total ion chromatogram of the RPIP-UPLC/MS separation of SAX peak 2 containing tetrasaccharides DUA(2S)-GlcNS(6S)-IdoA(2S)-GlcNS and IdoA(2S)-GlcNS(6S)-IdoA(2S)-GlcNS(6S) along with their mass spectra (B) and (C), respectively. The RPIP-UPLC conditions used resolve the a- and b-anomers of the reducing endresidue, complicating the separation.
J. F. K. Limtiaco et al. / Carbohydrate Research 346 (2011) 2244–2254 2251
literature with characteristic chemical shifts and MS data, a satu-rated tetrasulfated trisaccharide described by Yamada et al.26
From Table 1 it is interesting to note that in addition to separat-ing the sulfonated tetrasaccharide isomers using SAX-HPLC, we

2252 J. F. K. Limtiaco et al. / Carbohydrate Research 346 (2011) 2244–2254
were also able to separate the different IdoA and GlcA epimers. Fol-lowing the identification of the isolated tetrasaccharides, we ob-served that compounds containing an internal GlcA residue havelonger SAX retention times than their IdoA-containing counterpartshaving the same sulfonation positions and net negative charge. Thesame elution order was similarly observed by Schenauer andco-workers in the case of two less charged HS-derived hexasaccha-rides, DUA-GlcNS-IdoA2S-GlcNS-IdoA-GlcNAc and DUA-GlcNS-IdoA2S-GlcNS-GlcA-GlcNAc, which differ only by C-5 epimerizationof the uronic acid at the reducing end disaccharide.40 These obser-vations confirm that the configuration of the internal uronic acidresidue plays a crucial role in the interaction between the oligosac-charides and the stationary phase of the SAX column.
The identification of tetrasaccharides is relatively straightfor-ward considering the mass of individual structures one can isolateand purify from a heparinase digested mixture, however the mainadvantage of microcoil-based structural characterization ofindividual structures resides in its extension to much larger hepa-rin- or heparan sulfate oligosaccharides where the accessibility ofunique structures with the purity desired for NMR and MS exper-iments are mass-limited.
3. Conclusions
The results presented demonstrate the potential of microcoilNMR for the structure determination of heparin oligosaccharides.The complete structural elucidation of two diastereomer pairsand various penta- and hexasulfonated heparin tetrasaccharideswas carried out using minute amounts (as low as 21 lg) of isolatedsamples. A unique, hexasulfated tetrasaccharide IdoA(2S)-GlcNS(6S)-IdoA(2S)-GlcNS(6S) was identified in which the non-reducing end lacks the double bond introduced by heparinase I.In addition, among the five pentasulfonated tetrasaccharides char-acterized, the compound DUA(2S)-GlcNS(6S)-IdoA-GlcNS(6S) hadnot been previously reported. The high aqueous solubility of theanalytes and an efficient sample handling strategy was importantin enabling complete 2D NMR characterization of small quantitiesof the isolated tetrasaccharides in 3 lL samples using a commer-cially available CapNMR probe.
4. Experimental
4.1. Materials
Porcine intestinal mucosa heparin sodium salt, grade 1-A, cal-cium acetate hydrate, tris(hydroxymethyl)aminomethane (Tris),tributyl amine (TrBA), and acetonitrile (optima grade) were ob-tained from Sigma Chemical Company (St. Louis, MO). Hydrochlo-ric acid (HCl), tert-butanol, sodium chloride, sodium hydroxide,dibasic sodium phosphate, phosphoric acid, ammonium acetate,ammonium bicarbonate, ammonium formate, formic acid, and ace-tic acid were purchased from Fisher Scientific Co. (Fair Lawn, NJ).Sephadex G-10 (superfine) was obtained from GE Healthcare(Pittsburgh, PA). Heparinase I enzyme (EC 4.2.2.7) from recombi-nant Flavobacterium heparinum was purchased from IBEX Technol-ogies Inc. (Montreal, Quebec). HPLC grade water was obtained fromBurdick and Jackson (Muskegon, MI). Low paramagnetic deuteriumoxide (D, 99.9%), chloroform-d (D, 99.8%), DMSO-d6 (D, 99.9%),EDTA-d16 (D, 98%) and TMSP-2,2,3,3-d4 (D, 98%) were purchasedfrom Cambridge Isotope Laboratories (Andover, MA).
4.2. Enzymatic depolymerization of heparin
Digestion of 1 g of heparin was carried out in 50 mL of 100 mMpH 6.8 Tris buffer, containing 2.5 mM calcium acetate. The enzyme
heparinase I (0.5 IU) was added to the mixture and incubated at28 �C in a water bath for 66 hr. The water bath temperature wascontrolled using a Fisher Scientific Isotemp 1013S scientific tem-perature regulator. To monitor the progress of the enzymatic reac-tion, UV measurements were performed at 232 nm using a ThermoScientific NanoDrop 2000 spectrophotometer (Wilmington, DE). At232 nm, the molar extinction coefficient for the monounsaturateddisaccharide is 5500 M�1 cm�1.65 At the conclusion of the enzy-matic reaction, the digest solution was quenched by placing thereaction vessel into boiling water for 5 min. The depolymerizationsolution was then lyophilized and reconstituted into 15 mL of theseparation buffer (0.5 M NH4HCO3) prior to preparative scalesize-exclusion (SEC) separation.
4.3. Size-exclusion separation (SEC)
The heparin-derived oligosaccharides were size fractionated ona 3.0 � 200 cm column packed with Bio-Rad Bio-Gel P-10 resin fine(Bio-Rad Laboratories Hercules, CA) and eluted with 0.5 MNH4HCO3 at a flow rate of 0.08 mL/min. The volume of the col-lected fractions was 4.5 mL. The progress of the separation wasmonitored offline by UV absorption measurements at 232 nmusing the NanoDrop spectrophotometer as described above. Fol-lowing SEC, similar sized fractions were pooled and stored as alyophilized powder at �20 �C before separation by SAX-HPLC.
4.4. Strong anion-exchange HPLC
Strong anion-exchange (SAX) HPLC separations of tetrasaccha-rides were performed using a Dionex 500 ion chromatography sys-tem, equipped with a GP40 gradient pump and an AD20 UV–visdetector using a CarboPac PA1 semi-preparative scale column(9 mm � 25 cm) purchased from Dionex (Sunnyvale, CA). Thelyophilized SEC fractions containing the various tetrasaccharideswere re-dissolved in HPLC grade water and 500 lL injected intothe HPLC system. Tetrasaccharides were separated with a 50 mMpH 7.0 phosphate buffer (eluent A) and 2.0 M NaCl in 50 mM pH7.0 phosphate buffer (eluent B) at a flow-rate of 2.5 mL/min for90 minutes. The gradient profile applied is as follows: 0 min:100% A, 0% B; 10 min: 71% A, 29% B; 80 min: 39% A, 61% B. Aftereach run, the column was washed with solvent B for 10 min toelute impurities, followed by solvent A for 20 min to equilibratethe column. Separated tetrasaccharide peaks were collected intovials and after the desired number of injections each fraction wasfurther purified by re-injection to concentrate the analyte and re-duce the amount of NaCl. Prior to reinjection, the collected peakswere diluted with eluent A to reduce the NaCl concentration tobelow 0.3 M, and the entire solution was pumped through thesemi-preparative column as the mobile phase. This step allowsthe tetrasaccharides to be retained on the column while the excessNaCl is eluted. The analyte was then eluted from the SAX columnwith the NaCl gradient used in its initial isolation. The purified tet-rasaccharides were lyophilized to dryness and redissolved in theminimum amount of HPLC grade water to obtain a small volumebetter suited for desalting.
4.5. Desalting procedure
Although much of the NaCl was removed by reinjection of theisolated peaks onto the SAX column, further desalting was neces-sary. The tetrasaccharide samples were desalted on a 1.6 � 70 cmSephadex G-10 (superfine) column using HPLC grade water asthe eluent at a flow-rate of 0.15 mL/min. The SAX fractions werelyophilized and reconstituted in the minimum volume of waterto obtain a saturated solution. The retention of the column for NaClwas determined by conductivity measurements, and conductivity

J. F. K. Limtiaco et al. / Carbohydrate Research 346 (2011) 2244–2254 2253
was monitored throughout the desalting step. Fractions (1.5 mL)were collected from the Sephadex G10 column and lyophilized.These samples were reconstituted into 100 lL of water for UVabsorbance measurements at 232 nm. Fractions containing the de-salted tetrasaccharide were aliquoted for analysis by NMR and MS,lyophilized, and stored at �20 �C for subsequent use.
4.6. Microcoil NMR analysis
Isolated tetrasaccharides were reconstituted in 50 lL low para-magnetic D2O and lyophilized to replace exchangeable protonswith deuterium. As a final step immediately prior to NMR analysis,samples were reconstituted in 20 lL of low paramagnetic D2O and5 lL DMSO-d6, as a keeper solvent, and lyophilized to concentratethe sample at the bottom of a 0.5 mL Eppendorf tube.67 The lyoph-ilized samples were reconstituted in 3.5 lL of pD 7.4 phosphatebuffer containing 10 mM EDTA-d16 and TMSP-d4 as the chemicalshift reference (0 ppm). The solution pD of the phosphate bufferwas calculated using the pH meter reading (pH⁄) and the equationpD = pH⁄ + 0.4.68 EDTA-d16 was added as a chelating agent to scav-enge trace paramagnetic impurities, a practice that has beenshown to improve resonance line widths of heparin.69 A 3.0 lL ali-quot of the sample was sandwiched between deuterated chloro-form plugs in a 25 lL Hamilton syringe and injected into thedouble resonance (1H, 13C) solenoidal microcoil NMR probe (Prot-asis/MRM, Marlboro, MA). This probe has a flow-cell volume of5 lL and an active volume of 1.5 lL. The sample was positionedwithin the active volume of the microcoil by monitoring the D2Olock signal. Following analysis, NMR samples were collected fromthe microcoil probe and reduced to dryness by speed vacuumand stored at �20 �C.
NMR spectra were recorded using a Bruker Avance spectrome-ter operating at 599.84 MHz for 1H. For each 1H NMR spectrum,64 transients were collected into 24,576 data points using a6613 Hz spectral window and a 3 s relaxation delay. 1H NMR spec-tra were acquired using either presaturation or the WET solventsuppression scheme included with the standard Topspin releaseversion 1.3. For WET solvent suppression, the automated Brukershape tool was used to create the sinc pulse for the selective exci-tation of the HOD resonance.
Two-dimensional correlation spectroscopy (COSY), total corre-lation spectroscopy (TOCSY), and rotating-frame Overhauser spec-troscopy (ROESY) experiments were carried out on the isolatedsamples delivered to the CapNMR flow cell. In each experiment,a spectral width of 3 kHz was used in both dimensions and 32scans per increment were acquired. Presaturation of the solventresonance was used to reduce the intensity of the residual HODsignal. Double-quantum filtered COSY spectra were obtained byacquisition of a 2048 � 256 data matrix using a relaxation delayof 1.5 s. The COSY data set was apodized using a sinebell squaredfunction with zero-filling to 4096 points in F2 and 2048 points inF1. TOCSY spectra were acquired into a 2048 � 320 data matrixusing a relaxation delay of 1.6 s and a mixing time of 200 ms.The TOCSY spectra were apodized by multiplication with a cosinefunction and zero-filled to 4096 points in F2. Both the TOCSY andROESY data were linear predicted to 2048 points in F1. ROESYspectra were acquired into a 2048 � 320 data matrix using a relax-ation delay of 1.5 s and mixing time of 350 ms. The ROESY spin lockwas applied as a continuous low power pulse with a field strengthof 2.5 kHz. The ROESY spectra were apodized by multiplicationwith a cosine squared function and zero-filled to 4096 points.
Following the completion of the NMR measurements, sampleswere collected using 50 lL CDCl3 followed by 50 lL D2O to washthem out of the probe. The collected samples were lyophilizedand reconstituted into 100 lL of water for UV absorbance
measurements at 232 nm to determine the amount of materialanalyzed using NMR.
4.7. RPIP-UPLC separation
Reversed-phase ion-pair (RPIP) chromatographic separationswere performed using a 2.1 � 100 mm Acquity UPLC BEH C18 col-umn with 1.7 lm particles purchased from Waters Corporation(Milford, MA). A guard column using the same material was alsoemployed. The column temperature was maintained at 25 �Cthroughout the separation, and a flow rate of 0.5 mL/min was used.A sample volume of 10 lL of the tetrasaccharide mixture or iso-lated tetrasaccharide was injected. Samples for MS analysis werereconstituted in water to prepare a tetrasaccharide solution withan approximate concentration of 0.2 mM for the mixture and0.1 mM for individual tetrasaccharides. A binary solvent systemwas used for gradient elution. Solvent A consisted of 5% acetonitrilein water while solvent B consisted of 80% acetonitrile in water.Both eluents contained 20 mM TrBA and 2.5 mM ammonium for-mate buffer. The pH was adjusted to 6.0 prior to the addition ofthe required volume of acetonitrile. The separation was started at30% B and the organic composition increased linearly to 47% B over4 min. The organic composition was held at 47% B for 2 min andthen increased linearly from 47% to 90% B over the next 6 min.The eluent composition was finally increased to 100% B over1 min and held for 2 min to remove any highly retained compo-nents before returning the column to its starting condition overthe next 2 min for a total run time of 17 min. A 5 min equilibrationwas utilized prior to the next injection. Day-to-day reproducibilityof UPLC retention times was 2–3%.
4.8. Mass spectrometry
Total ion chromatograms were obtained using a Waters ESIquadrupole time-of-flight mass spectrometer. Data acquisitionwas performed using MassLynx 4.1 software. All spectra were ob-tained in negative ion mode using the following instrumentparameters: capillary voltage 3 kV; cone voltage 12 V; source tem-perature 120 �C; desolvation temperature 200 �C; extraction conevoltage 1 V; radio frequency lens 0.5 V; interscan delay 0.1 s; m/zrange 230–1500. Solvent delays from 0 to 3 min were used at thebeginning of each run to reduce the amount of salt introduced intothe mass spectrometer and at the end of the run from 15 to 17 minduring the 100% B column rinse.
Acknowledgments
Support for this research is provided by National Science Foun-dation grant CHE-0848976. Sz. B. gratefully acknowledges supportfrom OTKA MB08A/80066. J.F.K.L. acknowledges support by a2008–2010 US Pharmacopeia graduate fellowship. The authorswould also like to thank Professor Dallas Rabenstein for use ofthe HPLC system for SAX separations. The authors are especiallygrateful to Orsolya Molnár for her assistance with this project.
Supplementary data
Supplementary data associated with this article can be found, inthe online version, at doi:10.1016/j.carres.2011.07.007.
References
1. Beni, S.; Limtiaco, J.; Larive, C. Anal. Bioanal. Chem. 2011, 399, 527–539.2. Jones, C. J.; Beni, S.; Limtiaco, J. F. K.; Langeslay, D. J.; Larive, C. K. Annu. Rev.
Anal. Chem. 2011, 4, 439–465.3. Minamisawa, T.; Suzuki, K.; Kajimoto, N.; Iida, M.; Maeda, H.; Hirabayashi, J.
Carbohydr. Res. 2006, 341, 230–237.

2254 J. F. K. Limtiaco et al. / Carbohydrate Research 346 (2011) 2244–2254
4. Mascellani, G.; Guerrini, M.; Torri, G.; Liverani, L.; Spelta, F.; Bianchini, P.Carbohydr. Res. 2007, 342, 835–842.
5. Wolters, A. M.; Jayawickrama, D. A.; Sweedler, J. V. J. Nat. Prod. 2005, 68, 162–167.
6. Olson, D. L.; Lacey, M. E.; Sweedler, J. V. Anal. Chem. 1998, 70, 645–650.7. Korir, A. K.; Almeida, V. K.; Malkin, D. S.; Larive, C. K. Anal. Chem. 2005, 77,
5998–6003.8. Korir, A.; Larive, C. Anal. Bioanal. Chem. 2009, 393, 155–169.9. Rabenstein, D. L. Nat. Prod. Rep. 2002, 19, 312–331.
10. Linhardt, R. J. J. Med. Chem. 2003, 46, 255–2564.11. Sugahara, K.; Kitagawa, H. IUBMB Life 2002, 54, 163–175.12. Carlsson, P.; Presto, J.; Spillmann, D.; Lindahl, U.; KjellÃ�n, L. J. Biol. Chem.
2008, 283, 20008–20014.13. Capila, I.; Linhardt, R. J. Angew. Chem., Int. Ed. 2002, 41, 391–412.14. De Mattos, D. A.; Stelling, M. P.; Tovar, A. M. F.; Mourao, P. A. S. Thromb.
Haemost. 2008, 6, 1987–1990.15. Bartlett, A. H.; Park, P. W. Expert Rev. Mol. Med. 2010, 12, 1–25.16. Muramatsu, T.; Muramatsu, H. Proteomics 2008, 8, 3350–3359.17. Iozzo, R. V.; San Antonio, J. D. J. Clin. Invest. 2001, 108, 349–355.18. Li, J.-p.; Vlodavsky, I. Thromb. Haemost. 2009, 102, 799–1006.19. Rice, K. G.; Kim, Y. S.; Grant, A. C.; Merchant, Z. M.; Linhardt, R. J. Anal. Biochem.
1985, 150, 325–331.20. Korir, A. K.; Limtiaco, J. F. K.; Gutierrez, S. M.; Larive, C. K. Anal. Chem. 2008, 80,
1297–1306.21. Linhardt, R. J.; Turnbull, J. E.; Wang, H. M.; Loganathan, D.; Gallagher, J. T.
Biochemistry 1990, 29, 2611–2617.22. Desai, U. R.; Wang, H. M.; Linhardt, R. J. Biochemistry 1993, 32, 8140–8145.23. Galliher, P. M.; Cooney, C. L.; Langer, R.; Linhardt, R. J. Appl. Environ. Microbiol.
1981, 41, 360–365.24. Lohse, D. L.; Linhardt, R. J. J. Biol. Chem. 1992, 267, 24347–24355.25. Yamada, S.; Murakami, T.; Tsuda, H.; Yoshida, K.; Sugahara, K. J. Biol. Chem.
1995, 270, 8696–8705.26. Yamada, S.; Sakamoto, K.; Tsuda, H.; Yoshida, K.; Sugahara, K.; Khoo, K.-H.;
Morris, H. R.; Dell, A. Glycobiology 1994, 4, 69–78.27. Ampofo, S. A.; Wang, H. M.; Linhardt, R. J. Anal. Biochem. 1991, 199, 249–255.28. Eldridge, S. L.; Korir, A. K.; Gutierrez, S. M.; Campos, F.; Limtiaco, J. F. K.; Larive,
C. K. Carbohydr. Res. 2008, 343, 2963–2970.29. Kerns, R. J.; Linhardt, R. J. J. Chromatogr., A 1995, 705, 369–373.30. Volpi, N.; Cusmano, M.; Venturelli, T. Biochim. Biophys. Acta, Gen. Subj. 1995,
1243, 49–58.31. Kinoshita, A.; Sugahara, K. Anal. Biochem. 1999, 269, 367–378.32. Vives, R. R.; Pye, D. A.; Salmivirta, M.; Hopwood, J. J.; Lindahl, U.; Gallagher, J. T.
Biochem. J. 1999, 339, 767–773.33. Desai, U. R.; Wang, H. M.; Ampofo, S. A.; Linhardt, R. J. Anal. Biochem. 1993, 213,
120–127.34. Ruiz-Calero, V.; Puignou, L.; Galceran, M. T. J. Chromatogr., A 1998, 828, 497–
508.35. Militsopoulou, M.; Lamari, F. N.; Hjerpe, A.; Karamanos, N. K. Electrophoresis
2002, 23, 1104–1109.36. Limtiaco, J.; Beni, S.; Jones, C.; Langeslay, D.; Larive, C. Anal. Bioanal. Chem.
2010, 399, 593–603.37. Ziegler, A.; Zaia, J. J. Chromatogr., B: Anal. Technol. Biomed. Life Sci. 2006, 837, 76–
86.
38. Chuang, W.-L.; McAllister, H.; Rabenstein, D. L. J. Chromatogr., A 2001, 932, 65–74.
39. Chuang, W.-L.; McAllister, H.; Rabenstein, D. L. Carbohydr. Res. 2002, 337, 935–945.
40. Schenauer, M. R.; Meissen, J. K.; Seo, Y.; Ames, J. B.; Leary, J. A. Anal. Chem. 2009,81, 10179–10185.
41. Mikhailov, D.; Mayo, K. H.; Vlahov, I. R.; Toida, T.; Pervin, A.; Linhardt, R. J.Biochem. J. 1996, 318, 93–102.
42. Mikhailov, D.; Linhardt, R. J.; Mayo, K. H. Biochem. J. 1997, 328, 51–61.43. Guerrini, M.; Elli, S.; Gaudesi, D.; Torri, G.; Casu, B.; Mourier, P.; Herman, F.;
Boudier, C.; Lorenz, M.; Viskov, C. J. Med. Chem. 2010, 53, 8030–8040.44. Wolff, J. J.; Amster, I. J.; Chi, L.; Linhardt, R. J. J. Am. Soc. Mass Spectrom. 2007, 18,
234–244.45. Wolff, J. J.; Chi, L.; Linhardt, R. J.; Amster, I. J. Anal. Chem. 2007, 79, 2015–2022.46. Anusiewicz, I.; Jasionowski, M.; Skurski, P.; Simons, J. J. Phys. Chem. A 2005, 109,
11332–11337.47. Yang, J.; Mo, J.; Adamson, J. T.; Hakansson, K. Anal. Chem. 2005, 77, 1876–1882.48. Wolff, J. J.; Laremore, T. N.; Busch, A. M.; Linhardt, R. J.; Amster, I. J. J. Am. Soc.
Mass Spectrom. 2008, 19, 294–304.49. Wolff, J. J.; Laremore, T. N.; Aslam, H.; Linhardt, R. J.; Amster, I. J. J. Am. Soc. Mass
Spectrom. 2008, 19, 1449–1458.50. Merchant, Z. M.; Kim, Y. S.; Rice, K. G.; Linhardt, R. J. Biochem. J. 1985, 229, 369–
377.51. Pervin, A.; Gallo, C.; Jandik, K. A.; Han, X.-J.; Linhardt, R. J. Glycobiology 1995, 5,
83–95.52. Cooper, J. W. Comput. Chem. 1976, 1, 55–60.53. Bax, A.; Grzesiek, S. ROESY; John Wiley & Sons, 2007.54. Styles, P.; Soffe, N. F.; Scott, C. A.; Crag, D. A.; Row, F.; White, D. J.; White, P. C. J.
J. Magn. Reson. 1984, 60, 397–404.55. Robosky, L.; Reily, M.; Avizonis, D. Anal. Bioanal. Chem. 2007, 387, 529–532.56. Szántay, J. C.; Béni, Z.; Balogh, G.; Gáti, T. TrAC Trends Anal. Chem. 2006, 25, 806–
820.57. Ardenkjaer, J. H.; Fridlund, B.; Gram, A.; Hansson, G.; Hansson, L.; Lerche, M. H.;
Servin, R.; Thaning, M.; Golman, K. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 10158–10163.
58. Bowen, S.; Zeng, H.; Hilty, C. Anal. Chem. 2008, 80, 5794–5798.59. Frydman, L.; Blazina, D. Nat. Phys. 2007, 3, 415–419.60. Kautz, R. A.; Goetzinger, W. K.; Karger, B. L. J. Comb. Chem. 2004, 7, 14–20.61. Chuang, W.-L.; Christ, M. D.; Rabenstein, D. L. Anal. Chem. 2001, 73, 2310–2316.62. Linker, A.; Hovingh, P. Carbohydr. Res. 1984, 127, 75–94.63. Jin, L.; Hricovíni, M.; Deakin, J. A.; Lyon, M.; Uhrín, D. Glycobiology 2009, 19,
1185–1196.64. Tjong, S.-C.; Chen, T.-S.; Huang, W.-N.; Wu, W.-g. Biochemistry 2007, 46, 9941–
9952.65. Rice, K. G.; Linhardt, R. J. Carbohydr. Res. 1989, 190, 219–233.66. Xiao, Z.; Zhao, W.; Yang, B.; Zhang, Z.; Guan, H.; Linhardt, R. J. Glycobiology
2011, 21, 13–22.67. Lin, Y.; Schiavo, S.; Orjala, J.; Vouros, P.; Kautz, R. Anal. Chem. 2008, 80, 8045–
8054.68. Glasoe, P. K.; Long, F. A. J. Phys. Chem. 1960, 64, 188–190.69. McEwen, I. J. Pharm. Biomed. Anal. 2010, 51, 733–735.





























