Embed Size (px)
Citation preview

ORIGINAL PAPER
Pamela Hill Æ Debbie Burford Æ David M.A. Martin
Andrew J. Flavell
Retrotransposon populations of Vicia species with varying genome size
Received: 10 November 2004 / Accepted: 9 March 2005 / Published online: 13 May 2005� Springer-Verlag 2005
Abstract The (non-LTR) LINE and Ty3-gypsy-typeLTR retrotransposon populations of three Vicia speciesthat differ in genome size (Vicia faba, Vicia melanops andVicia sativa) have been characterised. In each species theLINE retrotransposons comprise a complex, very het-erogeneous set of sequences, while the Ty3-gypsy ele-ments are much more homogeneous. Copy numbers ofall three retrotransposon groups (Ty1-copia, Ty3-gypsyand LINE) in these species have been estimated byrandom genomic sequencing and Southern hybridisationanalysis. The Ty3-gypsy elements are extremely numer-ous in all species, accounting for 18–35% of their ge-nomes. The Ty1-copia group elements are somewhat lessabundant and LINE elements are present in still loweramounts. Collectively, 20–45% of the genomes of thesethree Vicia species are comprised of retrotransposons.These data show that the three retrotransposon groupshave proliferated to different extents in members of theVicia genus and high proliferation has been associatedwith homogenisation of the retrotransposon population.
Key words Retrotransposon, Vicia evolution Æ CopiaGypsy Æ LINE
Introduction
Retrotransposons and the related endogenous retrovi-ruses transpose replicatively through RNA intermedi-ates. Retrotransposons are divided into the Ty1-copiagroup, the Ty3-gypsy group and the LINE elements, ornon-LTR retrotransposons. All three retrotransposongroups are found in fungi, plants and animals. Thesegenetic elements have accumulated in many eukaryotegenomes to become major components of the dispersedrepetitious DNA fraction (Marracci et al. 1996; Pearceet al. 1996; SanMiguel et al. 1996; Smit 1999; Hattoriet al. 2000).
The Ty1-copia elements are the best understood ret-rotransposon group in plants, largely due to the easewith which the complex populations of these retro-transposons can be amplified by PCR, using well con-served regions of their genomes as primers (Koniecznyet al. 1991; Flavell et al. 1992a). Several studies haveshown that the retrotransposons of the Ty1-copia groupare present as large, highly heterogeneous populationswithin all genera of higher plants studied (Koniecznyet al. 1991; Flavell et al. 1992a, b; Hirochika andHirochika 1993; Vanderwiel et al. 1993; Pearce et al.1996). These retrotransposon populations tend to spanspecies boundaries, suggesting that the Ty1-copia retro-transposons existed early in plant evolution, and wereamplified and diverged into heterogeneous subgroupsbefore modern plant species arose.
The populations of Ty3-gypsy retrotransposons andLINE retrotransposons in plants have been less studiedthan their Ty1-copia counterparts. A variety of individ-ual Ty3-gypsy and LINE retrotransposons from plantshave been described (reviewed by Kumar and Bennetzen1999) and both groups have been shown to be wide-spread in plants (Kubis et al. 1998; Suoniemi et al. 1998;Kumekawa et al. 1999; Noma et al. 1999), but com-prehensive surveys of the multiple heterogeneous LINEor Ty3-gypsy retrotransposons in a species have onlybeen carried out for Arabidopsis thaliana (Wright et al.
Electronic Supplementary Material Supplementary material isavailable for this article at http://dx.doi.org/10.1007/s00438-005-1141-x.
Communicated by M.-A. Grandbastien
P. Hill Æ D. Burford Æ A. J. Flavell (&)Plant Research Unit, University of Dundee at SCRI,Invergowrie, Dundee, DD2 5DA, UKE-mail: [email protected].: +44-13-82562731Fax: +44-13-82568587
D. M. MartinPost-Genomics and Molecular Interactions Centre,Wellcome Biocentre, University of Dundee,Dundee, DD1 5EH, UK
Mol Gen Genomics (2005) 273: 371–381DOI 10.1007/s00438-005-1141-x

1996; Wright and Voytas 1998) and rice (McCarthy et al.2002).
The contribution of retrotransposons to plant genomesize has been studied in several species. Analysis ofgenomic sequence data for A. thaliana and rice (Oryzasativa) suggest that roughly 7 and 17%, respectively, ofthese genomes are comprised of retrotransposons (KlausMayer, personal communication; McCarthy et al. 2002).In contrast, their contribution to the Zea mays (maize)genome has been estimated at over 70%, based upondetailed analysis of a 200-kb genomic clone containing 20individual retrotransposon insertions (SanMiguel et al.1996). Arabidopsis thaliana has a particularly small gen-ome (2 · 108 bp, or 30-fold smaller than that of Z. mays).For sorghum, a relative of maize with a (3.5-fold) smallergenome, the equivalent region to that studied for maizecontains no retrotransposons (SanMiguel et al. 1998),though other genomic regions of sorghum do appear tocontain large numbers of retrotransposon insertions(reviewed by Bennetzen et al. 1998).
The fact that retrotransposons can account for suchlarge proportions of their host genomes raises theinteresting question of whether they facilitate changes ingenome size. In the genus Hordeum (barley) the copynumber of the BARE-1 Ty1-copia group retrotranspo-son varies proportionally with genome size (Vicient et al.1999), suggesting a degree of cross talk between retro-transposon amplification and genome size in this genus,but there is no evidence that retrotransposons are anymore successful than other types of repetitious DNA inincreasing genome size. A second question is whetherdifferent retrotransposon groups compete with eachother for genomic space. To date little or no work hasbeen reported in this area.
Retrotransposons have also been studied in variouslegume species (Lee et al. 1990; Chavanne et al. 1998;Laten et al. 1998, Nouzova et al. 2000; Sant et al. 2000;Neuman et al. 2003), The genusVicia is an interesting onein which to address the relationship between retrotrans-posons and genome size, because Vicia species appear tobe exclusively diploid and show large differences in gen-ome size, ranging from roughly 2 · 109 (e.g. Vicia sativa)to 13 · 109 bp (Vicia faba; fava bean). Repetitious DNAshave been isolated fromVicia narbonensis,Vicia melanopsandV. sativa, and their distribution in 13Vicia species hasbeen studied by microarray and Southern analysis(Nouzova et al. 2000) but their quantitative contributionsto the genomes examined was not addressed. Anotherstudy explored the diversity of Ty1-copia group retro-transposons in Vicia species and concluded that thenumbers of Ty1-copia group retrotransposons show greatvariation between V. faba, V. melanops and V. sativa, butdo not correlate with genome size (Pearce et al. 1996). Thepurpose of the work described here was to complete thedetailed characterisation of the retrotransposons of theseVicia species by analysing the other two-retrotransposonpopulations, namely Ty3-gypsy and LINE, and todetermine their contributions to genome size variationwithin this plant genus.
Materials and methods
Isolation, subcloning and sequence determination ofTy1-copia group retrotransposons from Vicia species
Vicia DNAs were isolated from fresh leaves usingPhytopure kits (Nucleon). Reverse transcriptase (rt)gene fragments were amplified from LINE retrotrans-posons in Vicia DNAs by degenerate PCR usingprimers DVO144 (5¢-GGGATCCNGGNCCNGAYGGNWT-3¢) and 10712 (5¢-SWNARNGGRTCNC-CYTG-3¢), which were derived from those described byWright et al. (1996). The Ty3-gypsy-group retrotrans-poson sequences were amplified using primer8717 (5¢-TAYCCNHT NCCNCGNATHGA-3¢) or8718 (5¢-TAYCCNHTNC CNAGRATHGA-3¢), bothencoding YPLPRID, together with 5¢-ARCA-TRTCRTC NACRTA-3¢, encoding YVDDML (Flavellet al. 1992a). The up-stream Ty3-gypsy primers werebased on an alignment of the plant Ty3-gypsy elementsCIN-full, Huck, Leviathan, Reina, Tekay (Jeff Ben-netzen, personal communication), Del 1 (Accession No.X13886) and Ifg7 (AJ004945). The Ty1-copia groupretrotransposon primers were as described by Pearceet al. (1996). The PCR products were subclonedinto the HincII-digested M13mp19 vector. The DNAsequence analysis was performed using either theSequenase protocol (USB-Amersham) or BigDyeautomated sequencing (PE Biosystems).
Cloning and sequence determination of Vicia randomgenomic DNAs
Aliquots (10 lg) of each of the genomic DNAs weresonicated in 100 ml of Tris–HCl (pH 7.5), using anMSE probe sonicator. The sheared DNAs were iso-lated using QIAquick columns (Qiagen, Crawley,UK) and the DNA fragments prepared for cloningby treatment with Klenow polymerase (New EnglandBiolabs) following the manufacturer’s instructions.The DNAs were purified by phenol extraction andpassage through a second QIAquick column, thenligated into the cloning vector pKS+ (Stratagene, LaJolla, CA) digested with HincII. Ligations weretransformed into Escherichia coli XL10-Gold Ultra-competent cells (Stratagene), which accept methylatedand unmethylated DNAs with equal efficiency, thusavoiding the loss of retrotransposons containedwithin genomic areas of high DNA methylation.Colonies were picked and insert sizes tested by PCRusing flanking vector primers. Sublones with insertsizes between 1.5 and 2.5 kb in length were sequencedby automated sequencing (PE Biosystems). TheEMBL/GenBank Accession Nos. for the randomlycloned sequences are AJ852612–AJ853297 (Supple-mentary Table 1).
372

Sequence comparisons and construction of phylogenetictrees for Vicia retrotransposon sequences
Computer analyses were carried out at the HumanGenome Mapping Project (HGMP) Facility, Hinxton,Cambridge, UK (http://www.hgmp.mrc.ac.uk/). Retro-transposon sequences were conceptually translated in allthree reading frames, and the results were comparedvisually with a reference set of plant retrotransposonpeptide sequences to identify bona fide retrotransposonsand locate frameshifts. The retrotransposon sequenceswere named Tvf, Tvm and Tvs (for transposon of Viciafaba, etc.), with L or G added to indicate a LINE or Ty3-gypsy group sequence. The deduced peptide sequenceswere aligned (Supplementary Figs. 1, 2) and phyloge-netic trees were generated (Fig. 1) using CLUSTALW(Thompson et al. 1994). The EMBL/GenBank Acces-sion Nos. for Ty3-gypsy sequences and LINE sequencesare AJ851018–AJ851085 and AJ850210–AJ850285,respectively, and the aligned Ty3-gypsy and LINEretrotransposon sequences are EMBL AccessionsALIGN_000845 and ALIGN_000846, respectively.
Bioinformatic analysis of random Vicia sequence data
Randomly cloned Vicia genomic DNA sequences wereused to perform TBLASTX searches of the EMBL plantdatabase using standard parameters. The search resultswere parsed using the bioperl toolkit (http://bio.perl.org;script available from DMAM) to extract details of allhigh scoring pairs (HSPs), then converted to lower caseand searched for the keywords chloroplast, copia, ele-ment, env, gag, gene, gypsy, integrase, line, mite, pol,repeat, repet, retroelement, retroposon, retrotransposon,retrovirus, transcriptase, transposable and transposonusing standard Unix tools. This eliminated manyunannotated O. sativa and A. thaliana sequences, whichshowed homology to the query but provided little or noinformation on the type of DNA cloned.
Cloned sequences were classified based on the pre-dominant type of DNA to which they showed homologyby creating a relative homology indicator (RHIMS)graph for each sequence (Martin et al. 2003), using a set ofkeyword searches. This work was performed in an auto-mated manner, after placing all HSP records in a Post-
greSQL relational database (http://www.postgresql.org).Each keyword gave rise to a separate line in the plot (seebelow), allowing a rapid visual assessment of the se-quence. This was followed by rigorous examination ofeach individual sequence. Sequences were only classifiedas belonging to a particular retrotransposon group whena consensus was found; otherwise they were classified asrepetitive DNA. The complete output of this analysis,including all sequences and RHIMS plots, are availableat www.dundee.ac.uk/lifesciences/papers/ajf/ph-2002.
Southern analysis
For Southern hybridisation analysis, DNAs (1 lg each)were digested with HincII, using the buffer and reactionconditions recommended by the manufacturer, thenelectrophoresed on 0.7% agarose-TBE gels, togetherwith 10 ng each of pure retrotransposon subclone PCRproducts (three control Ty3-gypsy clones or four controlnon-LTR retrotransposon clones), prepared by PCRfrom M13 subclones and quantified on gels by com-parison with known amounts of ethidium bromide-stained DNA. The DNAs were blotted to Biodyne Anylon membranes (Pall) as follows. Gels were soaked in0.2 M HCl for 20 min, briefly rinsed with water, thensoaked in 0.5 M NaOH/1 M NaCl for 30 min. The gelwas rinsed again before soaking in 1 M Tris–HCl (pH7.5), 3 M NaCl for 1 h. The gel was blotted onto themembrane with 20 times SSC transfer buffer, thenDNAs were fixed by exposure to ultraviolet light on a300 nm transilluminator (UV Products) for 2 min.
For slot blots the method of Pearce et al. (1996) wasused. Radioactively labelled retrotransposon probeswere prepared to specific activities of approximately108 dpm/lg by random oligo-primed labelling, usingeither 5 lg of uncloned retrotransposon PCR product(obtained by degenerate PCR of genomic DNA, seeabove) or 0.1 lg each of selected subclones (38 for theTy3-gypsy group and 47 for LINE retrotransposons),chosen to represent the total respective retrotransposonpool. The subclones chosen were the same as were usedfor slot blot analysis (see below and SupplementaryFig. 4) and are indicated on the phylogenetic treesshown in Fig. 1. Blots were hybridised by the method ofSambrook et al. (1989). A final wash stringency of twotimes SSC at 65�C was used to allow the probes to hy-bridise to all members of the subgroup containing thembut not to distantly related subgroups. Blots werestripped by the method recommended by the membranemanufacturer and re-exposed to check stripping effi-ciency before re-hybridisation. The hybridised blots wereexposed to a phosphorimager screen and signals weredeveloped on a Fuji phosphorimager.
Estimation of retrotransposon copy numbers
Southern hybridisation signals were quantified usingFuji MacBAS software. The percentage (Ps) of the
Table 1 Percentages of DNA sequence types identified from ran-dom sequencing of Vicia genomic DNAs
Sequence type Vicia species
V. faba V. melanops V. sativa
Unclassified(no close homologues found)
143 (51%) 137 (61%) 106 (58%)
Ty1-copia retrotransposons 13 (5%) 10 (4.5%) 14 (8%)Ty3-gypsy retrotransposon 39 (14%) 16 (7%) 14 (8%)LINE retrotransposons 0 (0%) 0 (0%) 1 (0.5%)Other repetitive DNA 80 (28%) 58 (26%) 26 (14%)Nuclear or organellar gene 7 (2.5%) 3 (1%) 22 (12%)Total 282 224 183
373
respective genomes occupied by the retrotransposongroups was deduced from the Southern data using theformula PS ¼ Sv/SC � RS � 100, where Sv is the totalSouthern signal for 1 lg of the species DNA, SC is theaveraged signal for the control (10 ng) cloned bandsfrom the same species, and RS is the ratio of the averagesize of a plant retrotransposon (estimated as 7 kb forTy1-copia and 10 kb for Ty3-gypsy retrotransposons,respectively) to the size of the probe used (approximately230 bp for the Ty3-gypsy group and 650 bp for LINE
retrotransposons). Genomic percentages of LTR retro-transposons deduced from random sequence informa-tion (PR) were obtained using the formulaPR ¼ n/N � RR � 100, where n is the number of clonesidentified as belonging to the appropriate retrotranspo-son group, N is the total number of clones sequenced forthat genome and RR is the size of the correspondingplant retrotransposon (again, 7 kb for Ty1-copiaand 10 kb for Ty3-gypsy) divided by the sizes of the rtand int genes combined (estimated at 3 kb for both
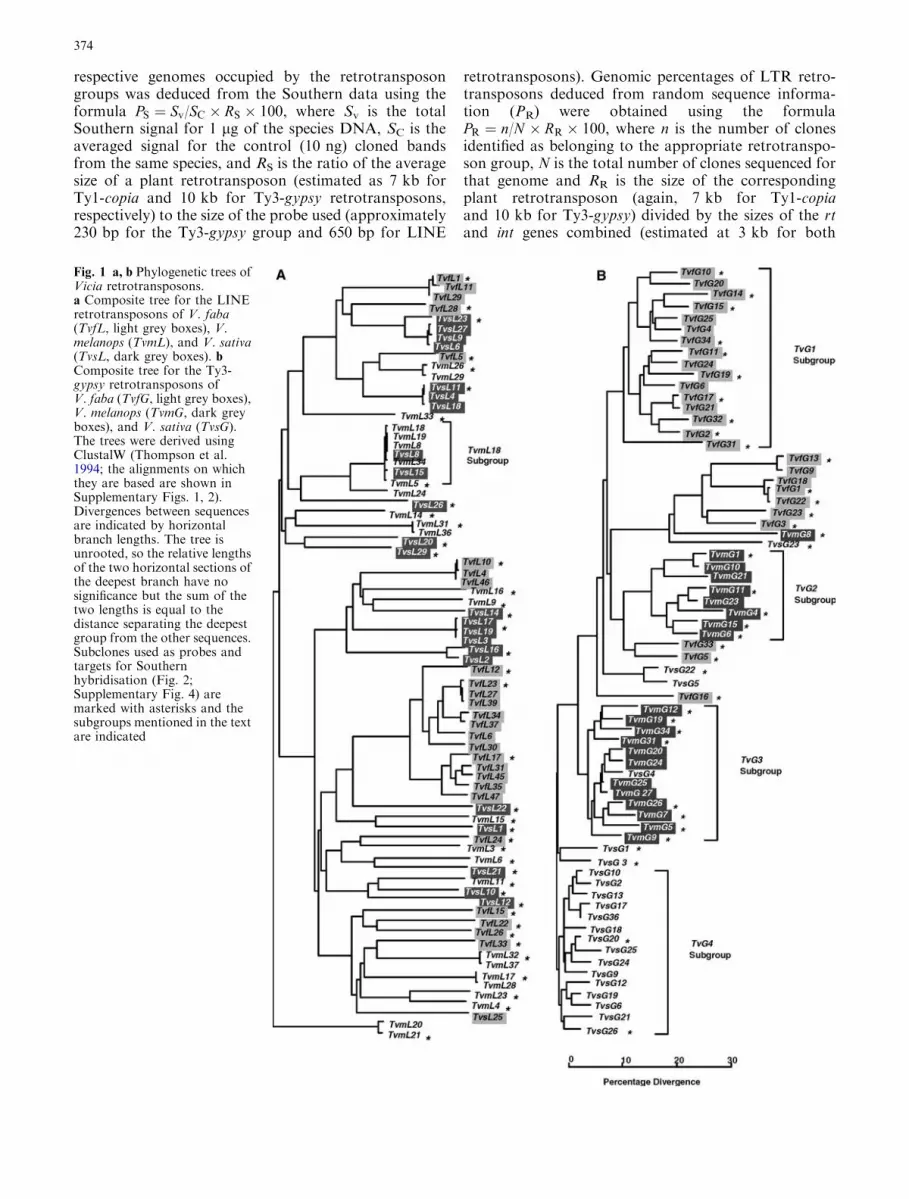
Fig. 1 a, b Phylogenetic trees ofVicia retrotransposons.a Composite tree for the LINEretrotransposons of V. faba(TvfL, light grey boxes), V.melanops (TvmL), and V. sativa(TvsL, dark grey boxes). bComposite tree for the Ty3-gypsy retrotransposons ofV. faba (TvfG, light grey boxes),V. melanops (TvmG, dark greyboxes), and V. sativa (TvsG).The trees were derived usingClustalW (Thompson et al.1994; the alignments on whichthey are based are shown inSupplementary Figs. 1, 2).Divergences between sequencesare indicated by horizontalbranch lengths. The tree isunrooted, so the relative lengthsof the two horizontal sections ofthe deepest branch have nosignificance but the sum of thetwo lengths is equal to thedistance separating the deepestgroup from the other sequences.Subclones used as probes andtargets for Southernhybridisation (Fig. 2;Supplementary Fig. 4) aremarked with asterisks and thesubgroups mentioned in the textare indicated
374

retrotransposon groups). For estimation of genomicpercentages of LTR retrotransposons from slot analysis,the relative signal strengths for rt PCR productand genomic DNAs were first compared (see below) toestimate the percentage content of the 0.28-kb rt frag-ment in a particular quantity of genomic DNA. Thispercentage was then multiplied by 7/0.28 = 25 to correctfor the size difference between the rt fragment and thetotal size of a complete Ty1-copia retrotransposon(estimated at 7 kb; Kumar and Bennetzen 1999).
Results
Characterisation of Ty3-gypsy group and LINEretrotransposons of three Vicia species
In plants, retrotransposons are usually found as complexfamilies of divergent sequences. To characterise thiscomplexity for the Ty3-gypsy and LINE retrotranspo-sons of Vicia, retrotransposon sequences were amplifiedfrom the DNAs of V. faba, V. melanops and V. sativa by
degenerate PCR of fragments of their rt genes (seeMaterials and methods). The PCR products were sub-cloned and multiple subclones were sequenced (76 LINEand 68 Ty3-gypsy group sequences).
Phylogenetic trees derived for these sequences areshown in Fig. 1. A highly heterogeneous collection ofLINE retrotransposons is revealed (Fig. 1a), character-ised by long-branch lengths, with sequences from all threespecies distributed across the tree. This suggests that anancient, diverse population of elements has survived inthe three Vicia species. Only a few LINE retrotranspo-sons of one species cluster tightly with those of another,notably TvsL8 and TvsL15 in the TvmL18 subgroup,suggesting that close LINE homologues are rare betweenVicia species. This situation differs from that seen for theTy1-copia elements in many angiosperms, includingVicia, where close homologues of many of the retro-transposons amplified from one species are found in thePCR products obtained from a related species (Flavellet al. 1992a; Pearce et al. 1996; Gribbon et al. 1998).
The corresponding phylogenetic tree for the Ty3-gypsy group retrotransposons of the three Vicia species
Fig. 2 a–d Southern analysis ofVicia retrotransposons.a, b Ty3-gypsy groupretrotransposons. c, d LINEretrotransposons. Aliquots(1 lg) of HincII-digestedgenomic DNAs (a, c) wereloaded in each lane and probedwith an equimolar mixture ofpooled Vicia retrotransposons(see Fig. 1, text and Materialsand methods for details). Bandsshared between at least twospecies are indicated by thearrows. Marker fragment sizesare given (in kb). Samples(10 ng) of each of the clonedTy3-gypsy groupretrotransposon fragments(b) or LINE retrotransposonfragments indicated (d) wereelectrophoresed on the samegel, blotted onto the samemembrane filter and probed asabove
375

(Fig. 1b) shows much lower levels of heterogeneity thanis seen either for LINEs (Fig. 1a) or Ty1-copia groupretrotransposons (Pearce et al. 1996). For example, 17out of the 21 Ty3-gypsy sequences characterised fromV. sativa (the TvG4 subgroup) are similar to each other(<17% nucleotide divergence). The tree also showsspecies-specific clustering for Ty3-gypsy group retro-transposons, with the TvG1, TvG2 and TvG4 subgroupseach derived from a different species.
Southern analysis of retrotransposons in the threeVicia species
To obtain more information on the population struc-tures of the LINE and Ty3-gypsy retrotransposons ofthe three Vicia genomes, Southern analysis was used(Fig. 2). Restriction digests of the three Vicia genomeswere probed with pooled sets of individual subclones(marked by asterisks in Fig. 1), chosen from the diver-sity tree to represent the full diversity of the retro-transposon sequences obtained. This approach reducesthe incidence of incomplete hybridisation associatedwith low copy number retrotransposons by avoiding lowprobe concentrations. A complementary experimentusing the uncloned PCR product as a probe gave vir-tually identical results, suggesting that no major retro-
transposons were missed by either method(Supplementary Fig. 3).
For the Ty3-gypsy probes a series of Southern bandssuperimposed on a background smear is visible in thegenomic digests from each of the three species (Fig. 2a).The bands represent the high copy number Ty3-gypsygroup retrotransposons in the respective species, and thesmears presumably represent a multitude of retrotrans-posons present in lower copy numbers. Several of thesebands (indicated by the arrows in Fig. 2a) are appar-ently shared between at least two of the Vicia species. ATy3-gypsy PCR probe isolated from one species tendedto hybridise with roughly similar efficiency to thegenomic DNA of another (Supplementary Fig. 3), con-firming the considerable overlap between the Ty3-gypsyretrotransposon populations in these species.
Southern data allow the estimation of copy number ifcontrol cloned sequences are included. The Southernsignal for a known amount of genomic DNA wascompared with corresponding signals for knownamounts of retrotransposon clones (see Fig. 2b andMaterials and methods; Manninen and Schulman 1993;Pearce et al. 1996; Nouzova et al. 2000). This analysissuggests that the haploid V. faba genome contains veryapproximately 240,000 Ty3-gypsy group retrotranspo-son insertions, the V. melanops genome contains veryroughly 360,000 gypsies and the V. sativa genome con-tains around 37,000 gypsies. Assuming an average sizefor a Ty3-gypsy LTR retrotransposon of 10 kb (seeDiscussion), these copy numbers imply that veryapproximately 18% of the V. faba genome is comprisedof Ty3-gypsy retrotransposons; the corresponding valuesfor V. melanops and V. sativa are 31 and 16%, respec-tively.
The results of a similar Southern-blot experimentusing pooled LINE retrotransposon clone probes areshown in Fig. 2c and d. The overall hybridisation signalis much lower with the LINE probe, indicating thatLINE retrotransposons are less common in these threeVicia species than are the Ty3-gypsy group retrotrans-posons. Furthermore, bands corresponding to promi-nent individual retrotransposon types are less apparentagainst the background smear for the LINE elementsthan is the case for Ty3-gypsy group elements, inagreement with the greater heterogeneity of the ViciaLINE elements seen in the sequence diversity trees(Fig. 1). Using a similar approach to that describedabove, we estimate that the genomic percentages forV. faba, V. melanops and V. sativa as very approximately6, 1 and 1%, although the accuracy of these estimationsis very low (standard deviations of 2, 0.5 and 1%,respectively).
Analysis of the DNA sequence content of the threeVicia species by random genomic sequencing
The Southern blot method used here and previously(Pearce et al. 1996) for retrotransposon copy number
Fig. 3 a, b Representative RHIMS graphs for LTR retrotranspo-son sequences. RHIMS plots (Martin et al. 2003) are shown for thekeywords copia, gene, gypsy, LINE, repet and retrotransposon.a Clone 3tf2 l, b Clone t3f7a
376

estimation has several major potential sources of inac-curacy. First, the method assumes that the PCR-basedprobes used in hybridisation reactions are accuraterepresentations of the corresponding genomic retro-transposon populations. This may not be the case, assequence polymorphism in the PCR primer regionscan greatly affect amplification efficiency and in aworst-case scenario, entire retrotransposon sub-groupsmay be missed in the analysis. Second, different degreesof hybridisation intensity caused by internal sequencevariation can be misinterpreted as different copynumbers.
To address these problems we used an independentapproach for estimating the retrotransposon contents ofthe three Vicia genomes, namely sequence analysis ofrandomly cloned genomic DNA. The V. faba, V. mel-anops and V. sativa DNAs were sonicated into frag-ments, cloned and sequenced. Sequences were obtainedfrom 282 V. faba, 224 V. melanops and 183 V. sativasubclones, corresponding to 143, 101 and 78 kb,respectively. To classify these DNAs, extensive databasesearches were carried out and the output data wereconverted into graphical representations of sequencetype, using the Relative Homology Index Using Math-ematical Summation method (RHIMS; Martin et al.2003). RHIMS plots accelerate the preliminary analysisand target subsequent individual inspection. Figure 3ashows an example of a RHIMS plot for a sequence(3tf2 l) that scored highly for the keywords gene, copiaand retrotransposon. This turned out to derive from aTy1-copia retrotransposon. Figure 3b shows a RHIMSplot for sequence t3f7a, which scores highly for thekeywords repet, retrotransposon, gene, Ty3-gypsy andcopia. Further analysis showed this sequence to be ofTy3-gypsy origin. This misuse of the term ‘copia’ in se-quence annotation to describe any LTR retrotransposonsequence was seen frequently.
A summary of these bioinformatics results is shownin Table 1 and the complete data, including all se-quences and RHIMS plots, are available at www.dun-dee.ac.uk/lifesciences/papers/ajf/ph-2002. Slightly morethan half of the sequences from each of the three speciescould not be classified from database searching. Between4 and 8% of the sequences obtained from each specieswere classified unambiguously as Ty1-copia group ret-rotransposons. Similar percentages to these were seenfor Ty3-gypsy elements of V. melanops and V. sativa, buttwice as many Ty3-gypsy elements (14%) were found inV. faba sequences. Surprisingly, only a single LINE se-quence was identified in the three species. The totalretrotransposon contents identified in the Vicia se-quences were quite similar: 19% for V. faba, 11.5% forV. melanops and 16% for V. sativa.
In addition to confirmed retrotransposon sequences,many repetitive DNAs were found. Repetitive DNAsinclude sequences that are possibly of retrotransposonorigin but could not be classified unambiguously, as wellas other repetitive elements, such as inverted repeat re-gions, tandem repeats and DNA transposons. Quite
similar percentages (14–28%) of the three species’ se-quences were identified as Other Repetitive DNA.Overall, the combined percentages for all identifiedrepetitious sequences (retrotransposons plus OtherRepetitive DNA) were: 47% for V. faba, 38% forV. melanops and 30% for V. sativa. Only a few percentof V. faba and V. melanops sequences were assigned tonuclear or organellar genes, but 12% of V. sativa se-quences (22 clones out of 183) were derived from nuclearor organellar genes. This is consistent with the propo-sition that V. sativa, with its smaller genome, contains ahigher proportion of genes to repetitive DNA than theother Vicia species studied here.
These percentages can be used to deduce the pro-portion of the respective genomes that are retrotrans-poson-specific, provided that allowance is made forregions of the retrotransposons that are unrecognisableby database searching. Large regions of retrotranspo-sons, particularly those, which are not protein coding,are very poorly conserved and such sequences might nothave properly annotated homologues in the genomicsequence databases. When the LTR retrotransposonsequences identified in Table 1 were mapped onto acanonical retrotransposon structure, it became clear thatlarge areas of retrotransposon are missing or under-represented (Fig. 4). More than 80% of identifiedTy1-copia and Ty3-gypsy sequences map to the two well-conserved rt and integrase (int) genes, which togetheroccupy roughly half of these retroelements (Kumar andBennetzen 1999). In particular, no sequences from theLTRs were identified at all.
To avoid this potential source of inaccuracy in cal-culating the retrotransposon content of these Vicia ge-nomes, the genomic content calculations were basedsolely upon the rt and int data (see Materials andmethods). Very many of these genes have been se-quenced in plants and other organisms, so databasesearches should pick up the large majority of them in therandom genomic sequences. Genomic percentages de-duced for the LTR retrotransposons in the three Viciagenomes from random sequencing are shown in Table 2.The Ty1-copia group retrotransposons account for 7–14% of each genome. The Ty3-gypsy group consistentlyaccounts for a larger percentage of the three genomes(18–35%). In total, roughly 25–44% of the Vicia ge-nomes are estimated to be comprised of LTR retro-transposons, based on random sequence analysis.
Reassessment of Ty1-copia group retrotransposoncopy number in Vicia
The genomic percentages of Ty1-copia obtained fromrandom sequencing are all strikingly different from thoseobtained by Southern slot blot analysis previously (9, 7and 14% for V. faba, V. melanops and V. sativa,respectively, compared to 40, 0.02 and 0.6%; Pearceet al. 1996). To resolve this discrepancy a slot blotexperiment was performed comparing the signals ob-
377

tained from each of the three genomic DNAs with thoseobtained for the corresponding mixed PCR rt fragmentsfrom the same genomes, essentially repeating theexperiment from Pearce et al. (1996) (Fig. 5). For V.faba, 50 ng of genomic DNA yields a similar hybridi-sation signal to 0.1 ng of Ty1-copia PCR product(Fig. 5a). This corresponds to a genomic percentage of5% (see Materials and methods). For V. melanops(Fig. 5b) 200 ng of genomic DNA yields the same hy-bridisation signal as 1 ng of Ty1-copia PCR product,corresponding to a genomic percentage of 13%. Finally,200 ng of V. sativa genomic DNA yields a hybridisationsignal equivalent to roughly 0.25 ng of Ty1-copia PCRproduct (Fig. 5c), corresponding to a genomic percent-age of 3%. These new values are much closer to the data
obtained from genomic sequencing (Table 2) and weconclude that the Ty1-copia content for all three Viciaspecies is within the range of 3–14%.
The nature of the prominent Ty3-gypsy groupretrotransposons in Vicia
The Southern data in Fig. 2 suggest that a relativelysmall number of Ty3-gypsy retrotransposon subgroupsare prominently represented in the three Vicia genomesstudied here. The identity of these major subgroups wasdetermined by hybridising the probes used in Fig. 2 toslot blots carrying an array of retrotransposon subclonesrepresenting the full spectrum of these sequences (Sup-plementary Fig. 4). The most prominent Ty3-gypsy ret-rotransposons identified from V. faba by this approachbelong to the TvG1 subgroup (Supplementary Fig. 4;Fig. 1b). Subsequent phylogenetic analysis showed thatthis subgroup corresponds to the BamHI repeat familiesof Vicia (Kato et al. 1985). The V. melanops andV. sativa probes also hybridise strongly to the TvG1subgroup. Similar analysis for V. melanops showed thatboth the TvG2 and TvG3 subgroups (Fig. 1a) areprominent in the genomic PCR product and the TvG4subgroup is well represented in the V. sativa genomicPCR (Supplementary Fig. 4). For LINE elements no
Table 2 Estimated percentagesof three Vicia genomes, whichare occupied by LTRretrotransposons
aThe actual numbers of se-quences obtained are shown inparentheses
Retrotransposontype
Vicia faba Vicia melanops Vicia sativa
Southernanalysis
Randomsequencea
Southernanalysis
Randomsequencea
Southernanalysis
Randomsequencea
Ty1-copia 5% 9% (11) 13% 7% (7) 3% 14% (12)Ty3-gypsy 18% 35% (32) 31% 18% (13) 16% 21% (12)LINE 6% 0% (0) 1% 0% (0) 1% 1% (1)Total 29% 44% (43) 45% 25% (20) 20% 36% (25)
Fig. 4 Regions of homology between random clones of ViciaDNA, identified as Ty1-copia or Ty3-gypsy sequences, and theirparent retrotransposons. Regions are defined based on thehomology of each sequence to one or more known Ty3-gypsyretrotransposons. The gag-pol region of the representative Ty1-copia retrotransposon is based on the SIRE-1 Ty1-copia retro-transposon (Laten et al. 1998) and the Ty3-gypsy retrotransposonis based on the RIRE2 Ty3-gypsy retrotransposon (Ohtsubo et al.1999). Clone ID numbers are shown (for Accession Nos. seeSupplementary Table 1). Regions of homology between the 900-bp, 990-bp, 1500-bp and 1750-bp BamHI repeat families (Katoet al. 1985) and the Ty3-gypsy retrotransposon are indicated
378

single subgroup was expected to dominate the LINEretrotransposons of the Vicia species (Fig. 2). The re-sults of the slot blot analysis were consistent with this,with the most strongly hybridising subclones for both V.faba and V. melanops being spread across the tree(Supplementary Fig. 5; Fig. 1b).
Discussion
This study describes the diversity of the Ty3-gypsy groupand LINE retrotransposons in three related Vicia specieswith varying genome sizes, completing and revising thedescription of the retrotransposon populations in thesespecies begun by Pearce et al. (1996). Rough estimatesfor the percentage contributions of each of the three-retrotransposon groups to their three host genomes havebeen obtained.
Diversities of the three-retrotransposon groupsin the three genomes
The three-retrotransposon groups show markedly dif-fering levels of heterogeneity (Fig. 1; Pearce et al. 1996).The Ty3-gypsy elements show low sequence diversity,with a dominant retrotransposon group present in allthree species, which corresponds to the BamHI repeatfamily (Supplementary Fig. 4; Kato et al. 1985). Thesedata are consistent with a model of evolutionarily recent,
explosive amplification of a relatively small subset ofancestral Ty3-gypsy elements in the Vicia genus. Incontrast, the LINE retrotransposons of all three speciesare highly varied (Fig. 1a), and close homologues sharedbetween Vicia species appear to be rare (Figs. 1a and 2).This is consistent with the suggestion that the LINEpopulations are dominated by ancient insertions, whichhave been lost in particular Vicia species lineages. TheTy1-copia elements of Vicia (Pearce et al. 1996) showvarying heterogeneities in the three species, and sharedelements between species are common, suggesting abehaviour, which is intermediate between those of theTy3-gypsy and LINE elements. It thus seems that thedifferent retrotransposon groups have shown differingactivities within the Vicia genus.
Accuracy of estimations of the genomic contributionsof retrotransposons
Two different methods, namely Southern and randomsequence analyses, have been used to estimate the per-centage contributions of the three-retrotransposongroups to the genomes of the three Vicia species. Bothmethods have serious potential sources of inaccuracy.The Southern-based estimations rely on the faithfulPCR amplification of the corresponding genomic ret-rotransposon populations, and some elements wereprobably missed in this study because of primer sitepolymorphism. This problem is likely to have been moreserious for the more heterogeneous LINE and Ty1-copiagroup retrotransposon populations.
A major potential source of inaccuracy with therandom sequence approach for estimating retrotrans-poson contribution derives from the incompleteness ofsequence databases for Vicia species. Sequences cannotbe recognised by database searching if they lack homo-logues in the databases. Our analysis has shown that (1)only the rt and int genes of LTR retrotransposons areconserved sufficiently well to be recognised in databasesearches (Fig. 4) and (2) Vicia LINE retrotransposonsare virtually invisible by this approach (Table 2). Thefirst problem forced us to consider only these two genesand assume that equal amounts of the other regions ofthe retrotransposons are present in these genomes. Thesecond problem is insoluble by this approach and wehave relied upon Southern data for estimating thegenomic contributions of LINEs in Vicia, bearing inmind the caveat mentioned above.
Both the Southern and the random sequence ap-proaches measure genomic contributions for a fragmentof the complete retrotransposon and deduce the valuesfor complete elements by multiplication. This can lead toerrors if the sequences amplified or sequenced are notassociated with complete retrotransposons. The LINEretrotransposons are frequently truncated at their 5¢ends, and this might have led to overestimation of thegenomic contributions made by this retrotransposongroup. Conversely, LTR numbers are known to exceed
Fig. 5 a–c Southern slot–blot quantification of Ty1-copia retro-transposons in the three Vicia species. Dilution series of genomicDNAs (200–10 ng) or uncloned 280-bp Ty1-copia retrotransposonPCR products (2–0.1 ng) were loaded. a V. faba. b V. melanops.c V. sativa
379

the numbers of genic regions for retrotransposons, atleast in barley (SanMiguel et al. 1996; Vicient et al. 1999;Kalendar et al. 2000). However, to the best of ourknowledge, there is no evidence that the internal regionsof LTR retrotransposons are differentially representedin plant genomes, so the genomic content values we haveobtained can be relied upon as reasonable lower limits.Errors in the estimations of genomic content can alsoarise if the average sizes assumed for the three-retro-transposon groups are inaccurate. There is a large rangeof size variation between different LTR retrotranspo-sons in the Viceae (4–22 kb; Lee et al. 1990; Chavanneet al. 1998; Neuman et al. 2003). We have taken theaverage of seven known Ty1-copia elements, nine Ty3-gypsy group and four LINE plant retrotransposons (Hill2003), but these experiments can only give a veryapproximate picture of the diversity and quantity ofretrotransposons in these Vicia genomes.
Genomic contributions for retrotransposonsin three Vicia species
When the estimated retrotransposon genomic percent-ages obtained by the two approaches used here arecompared with each other, significant disagreements areapparent (Table 2). Most of the estimates for a givenretrotransposon group in a given species differ byapproximately twofold between the two methods used.Nevertheless, it is clear that the contribution made byeach retrotransposon group to the genomes of all theseVicia species is considerable. The most numerous ret-rotransposons are those of the Ty3-gypsy group(25±10% for V. faba and V. melanops genomes and18±3% for V. sativa). The Ty1-copia group is less wellrepresented, accounting for approximately 8±5% for allthree genomes, and LINE retrotransposons are the leastfrequent group found. It is interesting to note thatgreater proliferation of Ty3-gypsy retrotransposons inVicia has been associated with homogenisation of thecorresponding retrotransposon population, whereas theless successful Ty1-copia and LINE retrotransposonpopulations are more diverse and presumably moreancient. The relative genomic contributions of retro-transposons for these species appear rather similar be-tween V. faba and V. melanops (35±10%), withV. sativa having perhaps less (28±8%). Retrotranspo-sons have clearly played a major role in the make-up ofat least these three Vicia genomes.
Acknowledgements PH was funded by a BBSRC Earmark Stu-dentship. This work was supported in part by Grants 960508(TEBIODIV), 31502 (TEGERM) and FP6-2002-FOOD-1-506223(GRAIN LEGUMES) from the European Commission under theFrameworks IV, V and VI. We thank Jeff Bennetzen for providingTy3-gypsy group retrotransposon sequences prior to databasesubmission, Steve Pearce for much helpful advice with the work,Gavin Ramsay for interesting discussions on Vicia phylogeny andfor Vicia seeds, and Noel Ellis, Alan Schulman, Amar Kumar,Robbie Waugh and David Marshall for many interesting discus-sions.
References
Bennetzen JL, SanMiguel P, Chen M, Tikhonov A, Francki M,Avramova Z (1998) Grass genomes. Proc Natl Acad Sci USA95:1975–1978
Chavanne F, Zhang DX, Liaud MF, Cerff R (1998) Structure andevolution of Cyclops: a novel giant retrotransposon of the Ty3/Gypsy family highly amplified in pea and other legume species.Plant Mol Biol 37:363–375
Flavell AJ, Smith DB, Kumar A (1992a) Extreme heterogeneity ofTy-copia group retrotransposons in plants. Mol Gen Genet231:233–242
Flavell AJ, Dunbar E, Anderson R, Pearce SR, Hartley R, Kumar A(1992b). Ty1-copia group retrotransposons are ubiquitous andheterogeneous in higher plants. Nucleic Acids Res 20:3639–3644
Gribbon BM, Pearce SR, Kalendar R, Schulman AH, Paulin L,Jack P, Kumar A, Flavell AJ (1999) Phylogeny and transposi-tional activity of Ty1-copia group retrotransposons in cerealgenomes. Mol Gen Genet 261:883–891
Hattori M, et al (2000) The DNA sequence of human chromosome21. Nature 405:311–319
Hill P (2003) Retrotransposons as determinants of genome size inthree Vicia species. PhD Thesis, University of Dundee
Hirochika H, Hirochika R (1993) Ty1-copia group retrotranspo-sons as ubiquitous components of plant genomes. Jpn J Genet68:35–46
Kalendar R, Tanskanen J, Immolen S, Nevo E, Schulman AH(2000) Genome evolution of wild barley (Hordeum spontaneum)by BARE-1 retrotransposon dynamics in response to sharpmicroclimate divergence. Proc Natl Acad Sci USA 97:6603–6607
Kato A, Iida Y, Yakura K, Tanifuji S (1985) Sequence analysis ofVicia faba highly repeated DNA: the BamHI repeated sequencefamilies. Plant Mol Biol 5:41–53
Konieczny A, Voytas DF, Cummings MP, Ausubel FM (1991)A superfamily of Arabidopsis thaliana retrotransposons.Genetics 127:801–809
Kubis S, Heslop-Harrison JS, Desel C, Schmidt T (1998) Thegenomic organisation of non-LTR retrotransposons from threeBeta species and five other Angiosperms. Plant Mol Biol 33:11–21
Kumar A. Bennetzen JL (1999) Plant retrotransposons. Annu RevGenet 33:479–532
Kumekawa N, Ohtsubo E, Ohtsubo H (1999) Identification andphylogenetic analysis of gypsy-type retrotransposons in theplant kingdom. Genes Genet Syst 74:299–307
Laten HM, Majumdar A, Gaucher EA (1998) SIRE-1, a copia/Ty1-like retroelement form soybean, encodes a retroviralenvelope-like protein. Proc Natl Acad Sci USA 95:6897–6902
Lee D, Ellis THN, Turner L, Hellen RP, Cleary WG (1990)A Copia-like element in Pisum demonstrates the use of dis-persed repeat sequences in genetic analysis. Plant Mol Biol15:707–722
Manninen I, Schulman AH (1993) BARE-1 a copia-like retroele-ment in barley (Hordeum vulgare L.). Plant Mol Biol 22:829–846
Marracci S, Batistoni R, Pesole G, Citt L, Nardi I (1996) gypsy/Ty3-like elements in the genome of the terrestrial salamanderHydromantes (Amphibia, Urodela). J Mol Evol 43:584–593
Martin DMA., Hill P, Barton GJ, Flavell A.J (2003) Visual rep-resentation of database search results: the RHIMS plot. Bio-informatics 19:1–2
McCarthy EM, Liu J, Lizhi G, McDonald JF (2002) Long terminalrepeat retrotransposons of Oryza sativa. Genome Biol Res3:0053.1–0053.11
Neuman P, Pozarkova D, Macas J (2003) Highly abundant pearetrotransposon Ogre is constitutively transcribed and partiallyspliced. Plant Mol Biol 53:399–410
Noma K, Ohtsubo E, Ohtsubo H (1999) Non-LTR retrotranspo-sons (LINEs) as ubiquitous components of plant genomes. MolGen Genet 261:71–79
380

Nouzova M, Neuman P, Navratilova A, Galbraith DW, Macas J(2000) Microarray-based survey of repetitive genomic sequencesin Vicia spp. Plant Mol Biol 45:229–244
Ohtsubo H, Kumekawa N, Ohtsubo E (1999) Rire2, a novel gypsy-type retrotransposon from rice. Genes Genet Syst 74:83–91
Pearce SR, Harrison G, Li D, Heslop-Harrison JS, Kumar A,Flavell AJ (1996) The Ty1-copia group retrotransposons inVicia species: copy number, sequence heterogeneity and chro-mosomal localisation. Mol Gen Genet 250:305–315
Sambrook J, Fritsch EF Maniatis T (1989) Molecular cloning: alaboratory manual, 2nd edn. Cold Spring Harbor LaboratoryPress, Cold Spring Harbor, NY
SanMiguel P, Tikhonov A, Jin Y-K, Motchoulskaia N, ZakharovD, Melake-Berhan A, Springer PS, Edwards KJ, Lee M, Av-ramova Z, Bennetzen JL (1996) Nested retrotransposons in theintergenic regions of the maize genome. Science 274:765–768
SanMiguel P, Gaut BS, Tikhonov A, Nakijama Y, Bennetzen JL(1998) The paleontology of intergene retrotransposons ofmaize. Nat Genet 20:43–45
Sant VJ, Sainani MN, Sami-Subbu R, Ranjekar PK, Gupt VS(2000) Ty1-copia retrotransposon-like elements in chickpeagenome: their identification, distribution and use for diversityanalysis. Gene 257:157–166
Smit FA (1999) Interspersed repeats and other mementos oftransposable elements in mammalian genomes. Curr OpinGenet Dev 657–663
Suoniemi A, Tanskanen J, Schulman AH. (1998) Gypsy retro-transposons are widespread in the plant kingdom. Plant J13:699–705
Thompson JD Higgins DG, Gibson TJ (1994) Clustal W:improving the sensitivity of progressive multiple sequencealignment through sequence weighting, position-specific gappenalties and weight matrix choice. Nucleic Acids Res 22:4673–4680
Vanderwiel PL, Voytas D, Wendel JF (1993) Copia-like retro-transposable element evolution in diploid and polyploid cotton(Gossypium L.). J Mol Evol 36:429–447
Vicient CM, Suoniemi A, Anamthawat-Jonsson K, Tanskanen J,Beharov A, Schulman AH (1999) Retrotransposon BARE-1and its role in genome evolution in the genus Hordeum. PlantCell 11:1769–1784
Wright DA, Voytas DF (1998) Potential retroviruses in plants:Tat1 is related to a group of Arabidopsis thaliana Ty3/gypsyretrotransposons that encode envelope-like proteins. Genetics149:703–715
Wright DA, Ke N, Smalle J, Hauge BM, Goodman HM, VoytasDF (1996) Multiple non-LTR retrotransposons in the genomeof Arabidopsis thaliana. Genetics 142:569–578
381





























