Embed Size (px)
Citation preview

MASARYK UNIVERSITY
Faculty of Medicine
DOCTORAL THESIS
Brno 2016 Mgr. Zdenka HAŠANOVÁ

MASARYK UNIVERSITY
Faculty of Medicine
Department of Biology
Structure-specific endonucleases in
DNA repair
A thesis submitted for the degree of Doctor Philosophy
Program: Medical Biology
Supervisor: Author:
Doc. Mgr. Lumír Krejčí, Ph.D. Mgr. Zdenka Hašanová
Brno, 2016

ABSTRACT
Background. Genome integrity is continuously challenged by exogenous or endogenous factors.
To prevent global DNA damage potentially leading to tumorigenesis, DNA lesions need to be
permanently recognized and repaired. A large set of proteins contribute to DNA repair. In this
thesis we focus on structure-specific endonucleases functioning in resolution of various
intermediates arising during DNA replication and the repair of double-strand breaks (DSBs) by
homologous recombination (HR). In particular, we describe cooperation of human structure-
specific endonuclease MUS81-EME1 with RecQ helicase RECQL5β. In addition, DNA repair
enzymes also need to be tightly regulated to ensure their proper function. One such mechanism
involves SUMOylation where a small peptide (SUMO) is post-translationally attached to an
enzyme thus potentially modulating its activity and function. In the other part of this thesis we
have identified and characterized the effect of SUMOylation on yeast structure-specific
endonuclease Rad1-Rad10 operating during a HR subpathway and nucleotide excision repair.
Methods. Our goals were achieved by using recombinant purified proteins in in vitro assays.
Results. MUS81 and RECQL5β physically interact and RECQL5β enhances MUS81 activity by
stimulating its cleavage on branched DNA substrates specific for MUS81. Further, RECQL5β
enables MUS81 to access DNA by dissociating RAD51 from single-stranded DNA occurring at
common fragile sites (CFSs). We designated the Rad1 SUMO conjugation-site and provide
evidence that SUMOylated Rad1-Rad10 complex had modified DNA binding activities.
Additionally, we found that SUMO-Rad1 enhances SUMOylation of its interaction partner Saw1.
Conclusions. MUS81 and RECQL5β together process late replication intermediates and promote
common fragile site expression. SUMO modification of Rad1 during DNA repair is responsible
for protein turnover. SUMOylation of Saw1 modulates the preference of Saw1 for an alternative
DNA repair pathway independent of Rad1-Rad10. Our studies shed more light into the
complicated process and regulation of structure specific endonucleases during DNA repair.
Key words: homologous recombination, structure-specific endonuclease, RecQ helicase,
MUS81, RECQL5β, common fragile sites, Rad1-Rad10, SUMOylation

ABSTRAKT
Úvod. Genómová stabilita je neustále ohrozovaná exogénnymi a endogénnymi faktormi. Ochranu
genetického materiálu zabezpečujú rôzne DNA opravné mechanizmy, ktoré neustále rozoznávajú
a opravujú DNA poškodenia, ktorého môžu potenciálne viesť ku karcinogenéze. DNA oprava je
vykonávaná rôznymi typmi enzýmov. V tejto dizertačnej práci sa zameriavame na štruktúrne
špecifické endonukleázy, ktoré štiepia rôzne intermediáty, ktoré vznikajú počas DNA replikácie a
homologickej rekombinácie (HR) (oprave dvojvláknových zlomov). Konkrétne, popisujeme
kooperáciu medzi ľudskou štruktúrne špecifickou endonukleázou MUS81-EME1 a RecQ
helikázou RECQL5β. Ďalej DNA opravné enzými musia byť striktne regulované, aby spĺňali
svoju požadovanú funkciu. Jeden typ takejto regulácie je SUMOylácia, kde malý peptid (SUMO)
je posttranslačne naviazaný na enzým a tým modifikuje aktivitu a funkciu daného proteínu.
Charakterizovali sme úlohu SUMOylácie pri kvasinkovej štruktúrne špecifickej endonukleáze
Rad1-Rad10, ktorá opravuje poškodenia počas podtypu HR a nukleotidovej excíznej opravy.
Metódy. Využívali sme rekombinantné purifikované proteíny v in vitro esejach.
Výsledky. MUS81 a RECQL5β fyzicky interagujú a RECQL5β stimuluje štiepenie MUS81 na
rozvetvených DNA substrátoch, ktoré sú špecifické pre MUS81. RECQL5β umožňuje naviazanie
MUS81 na DNA disociáciou RAD51 z jednovláknovej DNA, ktorá sa vyskytuje na neskorých
replikačných intermediátoch. Ďalej sme určili pozíciu SUMOylácie Rad1 proteínu a ukázali sme,
že SUMOylácia Rad1-Rad10 komplexu vedie k modifikácii DNA afinity. Okrem toho sme
ukázali, že SUMO-Rad1 stimuluje SUMOyláciu jeho interakčného partnera Saw1.
Záver. MUS81 a RECQL5β spolu štiepia neskoré replikačné intermediáty, čo vedie k vzniku
dvojvlákonvých zlomov. SUMO modifikácia Rad1 počas DNA opravy je zodpovedná za
disociáciu proteínu z DNA po rozštiepení DNA. SUMOylácia Saw1 moduluje jeho preferencie
pre alternatívne DNA opravné mechanizmy nezávislé od Rad1-Rad10. Dokopy naše výsledky
viacej objasňujú komplexnú funkciu a regulaciu štruktúrne špecifických endonukleáz v DNA
oprave.
Kľúčové slová: homologická rekombinácia, štruktúrne špecifické endonukleázy, RecQ helikázy,
MUS81, RECQL5β, Rad1-Rad10, SUMOylácia

DECLARATION
I hereby declare, that I have worked on this thesis independently using only primary and
secondary sources listed in the bibliography, under the supervision of doc. Mgr. Lumír Krejčí,
Ph.D.
..........................................
Author signature

ACKNOWLEDGEMENT
First of all, I would like to thank my supervisor Lumír for giving me the opportunity to work in his
dynamic lab, allowing me to grow professionaly and learn not only new biochemical methods but also to
get in to the topic of homologous recombination, which, during my masters study, I found kind of scary.
Also for great motivation to work on myself knowing that it can always be better. Besides his hard
professional side, also giving me alot of valuble lessons – mostly patience, I would like to thank him for
showing his social-prone side during my maternity leave, allowing me to raise maybe a new generation of
scientists (who knows :-)).
Uncountable thanks goes to my dear collegue Verchika for major lab help and for wrapping my
mind about that pure altruism really exists. But mostly I thank her for being a great friend (even as time
passes) and for making a great work atmosphere.
Special thanks goes to Marek who somehow convinced me that recombination is cool and letting
me know that there is this Laboratory of Recombination and DNA Repair in Brno and I should definitely
apply for PhD. Sometimes I was not sure I would get to an end – but it seems its here and that it was worth
it.
A special place in my heart has Sashika and Bara for being great friends, for our Pekanda sessions
and for making the lab a great and fun place to work at.
Of course I thank all the LORDs who I came across with and especially those who had
contributions to my projects: Melita, Pištík, Hanka, Vicky, Mário, Petra and Lenka for outstanding
technical support. I thank Pavel Janščák for helpful comments to my thesis and cooperation in the MUS81-
RECQ5 project.
Special thanks goes to my home supporting team – my husband Palik, for patience and support
during this crazy PhD finishing. I thank him for turning aside all gender stereotypes that mothers need to
be housewives and for a change he tried it instead of me, while I was experimenting in the lab. Mostly, I
want to thank my dearest children Emilko and Adamko for every minute of sleep, so I could write this
thesis, but more importantly for being the greatest motivation to do so.

Contents
1. THEORETICAL BACKGROUND .................................................................................................... 9
1.1. DNA repair .................................................................................................................................. 9
1.2. Repair of double-strand breaks ................................................................................................. 11
1.2.1. Double-strand break formation ......................................................................................... 11
1.2.2. Non-homologous end joining ........................................................................................... 11
1.2.3. Choice between NHEJ and HR ......................................................................................... 12
1.2.4. Homologous recombination .............................................................................................. 12
1.2.4.1. Double-strand break repair ....................................................................................... 14
1.2.4.2. Synthesis-dependent strand-annealing ...................................................................... 14
1.2.4.3. Break-induced replication ......................................................................................... 14
1.2.4.4. Single-strand annealing............................................................................................. 16
1.3. Structure-specific endonucleases .............................................................................................. 17
1.3.1. MUS81-EME1/EME2 complex ........................................................................................ 17
1.3.2. XPF-ERCC1 (Rad1-Rad10) ............................................................................................. 24
1.4. RecQ helicases .......................................................................................................................... 26
1.4.1. Structure of RecQ helicases .............................................................................................. 27
1.4.2. RecQ helicases in yeast .................................................................................................... 29
1.4.3. RecQ helicases in humans ................................................................................................ 29
1.4.3.1. RECQL1 ................................................................................................................... 29
1.4.3.2. WRN ......................................................................................................................... 30
1.4.3.3. BLM .......................................................................................................................... 31
1.4.3.4. RECQL4 ................................................................................................................... 32
1.4.3.5. RECQL5 ................................................................................................................... 33
1.4.4. Interactions between RecQ helicases ................................................................................ 35
1.5. Post-translational modifications ............................................................................................... 36
1.5.1. SUMOylation .................................................................................................................... 36
1.5.2. SUMOylation in HR ......................................................................................................... 38
2. AIMS OF THESIS ............................................................................................................................ 40
3. MATERIAL AND METHODS ........................................................................................................ 41
3.1. Material ..................................................................................................................................... 41
3.1.1. Chemicals ......................................................................................................................... 41

3.1.2. Buffers and stock solutions ............................................................................................... 42
3.1.3. Growth media ................................................................................................................... 43
3.1.4. Bacterial strains ................................................................................................................ 43
3.1.5. Molecular weight standards .............................................................................................. 43
3.1.6. Plasmids ............................................................................................................................ 44
3.1.7. DNA primers .................................................................................................................... 44
3.1.8. DNA substrates ................................................................................................................. 45
3.1.9. Proteins ............................................................................................................................. 45
3.2. Methods .................................................................................................................................... 46
3.2.1. DNA techniques................................................................................................................ 46
3.2.1.1. Visualization of DNA ............................................................................................... 46
3.2.1.2. DNA purification from E. coli .................................................................................. 47
3.2.1.3. Site-directed mutagenesis ......................................................................................... 47
3.2.1.4. Restriction analysis ................................................................................................... 48
3.2.1.5. DNA annealing ......................................................................................................... 48
3.2.2. Cell techniques.................................................................................................................. 49
3.2.2.1. Bacterial transformation by heat-shock .................................................................... 49
3.2.2.2. Preparation of chemocompetent bacterial cells ........................................................ 49
3.2.3. Protein techniques ............................................................................................................. 50
3.2.3.1. Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) ............. 50
3.2.3.2. Silver staining ........................................................................................................... 50
3.2.3.3. Protein expression ..................................................................................................... 51
3.2.3.4. Protein purification ................................................................................................... 51
3.2.3.5. Immunoblotting (Western blot) ................................................................................ 57
3.2.4. Functional assays .............................................................................................................. 57
3.2.4.1. Pull-down assay ........................................................................................................ 57
3.2.4.2. Nuclease assay .......................................................................................................... 58
3.2.4.3. Electromobility shift assay (EMSA) ......................................................................... 58
3.2.4.4. In vitro SUMOylation assay ..................................................................................... 59
3.2.4.5. Gel filtration chromatography .................................................................................. 59
3.2.4.6. RAD51 removal assay .............................................................................................. 59
4. RESULTS ......................................................................................................................................... 61

4.1. MUS81-EME1 nuclease and RECQ5 helicase cooperatively promote stability of CFSs ........ 61
4.1.1. RECQ5 physically interacts with MUS81-EME1 in vitro ................................................ 62
4.1.2. RECQ5 stimulates MUS81-EME1 endonuclease activity in vitro ................................... 63
4.1.3. Full-length RECQ5 protein is needed for the interaction with MUS81 ........................... 64
4.1.4. RECQ5 stimulates the nuclease activity of yeast Mus81-Mms4 ...................................... 65
4.1.5. RECQ5 can dissociate RAD51 from DNA to enable MUS81 cleavage........................... 66
4.1.6. Discussion ......................................................................................................................... 68
4.2. Characterization of SUMOylation of Rad1-Rad10 complex and its interacting partner Saw1 72
4.2.1. Characterization of Rad1 SUMOylation in vitro .............................................................. 72
4.2.1.1. E3 ligase dependence ................................................................................................ 73
4.2.1.2. The effect of DNA on Rad1 SUMOylation .............................................................. 74
4.2.1.3. The effect of Rad1-Rad10 interaction partners on its SUMOylation ....................... 75
4.2.2. Identification of SUMO-binding site in Rad1 .................................................................. 77
4.2.2.1. Characterization of Rad1-K32R-Rad10 enzymatic activity ..................................... 78
4.2.3. Function of Rad1 SUMOylation ....................................................................................... 80
4.2.3.1. Effect on nuclease activity ........................................................................................ 80
4.2.3.2. Effect on DNA binding ............................................................................................. 81
4.2.3.3. Effect on oligomeric status ....................................................................................... 82
4.2.3.4. Effect on interaction with Saw1 ................................................................................ 82
4.2.3.5. Interplay between Rad1 and Saw1 SUMOylation .................................................... 84
4.2.3.6. Effect of SUMOylation on DNA binding of Saw1 ................................................... 85
4.2.3.7. SUMOylated complex Rad1-Rad10-Saw1 has unchanged nuclease activity on Y-
form 86
4.2.4. Discussion ......................................................................................................................... 87
5. CONCLUSIONS .............................................................................................................................. 90
6. BIBLIOGRAPHY............................................................................................................................. 91
7. LIST OF ABBREVIATIONS ......................................................................................................... 112
8. LIST OF FIGURES ........................................................................................................................ 114
9. LIST OF TABLES .......................................................................................................................... 116
10. LIST OF PUBLICATIONS ........................................................................................................ 118
11. SUMMARY ................................................................................................................................ 119
12. SUPPLEMENTS ........................................................................................................................ 120

9
1. THEORETICAL BACKGROUND
1.1. DNA repair
DNA is a matrix for the genetic code programming all life. To ensure its homogeneity
throughout evolution it needs to be protected from deleterious mutations and this is achieved by
a robust cooperation of proteins – DNA repair enzymes and DNA damage checkpoint enzymes.
Since each cell encounters damages to its DNA roughly 10,000 times a day, several DNA
repair pathways evolved to combat this1. In eukaryotes DNA repair mechanisms include
nucleotide excision repair (NER), base excision repair (BER), mis-match repair (MMR),
translesion synthesis (TLS), non-homologous end-joining (NHEJ) and homologous
recombination (HR) (summarized in Table 1.1).
Table 1.1. Summary of DNA repair pathways.
DNA repair pathway DNA damage
NER helical distortions of nucleotides caused by UV
BER base modifications leading to mispairing, ssDNA gaps
MMR mismatches, small insertions, deletions
TLS tolerance/synthesis through DNA damage
NHEJ DSBs
HR DSBs
DNA damage can arise spontaneously by endogenous metabolic processes and biological
elements or can be induced by exogenous sources. Cell’s own metabolism has a wide range of
mechanisms for altering the genetic information. For example, during replication, tautomeric
changes, base methylation or deamination or mutator genes leading to substitutions.
Replication hot spots, such as repetitions or incomplete palindromes, are prone for
insertions/deletions. Inefficient repair enzymes likely produce mutations, often associated with
some disease or syndrome. By-products of cell’s metabolism, e.g. mitochondrial oxidative
phosphorylation, are sources of free radicals attacking bases leading to deletions, cross-links

10
and finally DNA breaks. Biological elements such as retroviruses or retrotransposons can also
cause gene and chromosomal mutations or affect gene expression therefore influencing cell
homeostasis.
Exogenous sources include physical agents, such as ionizing radiation (IR) and ultraviolet light
(UV), or chemical compounds. These agents function in various mechanisms (summarized in
Table 1.2.). While they can affect gene transcription, they are mostly toxic during DNA
replication, where their final effect is replication fork stalling. This leads to checkpoint
activation and replication arrest, when the cell has time to cope with the replication obstacle. If
this is unsuccessful the replication fork is very likely to collapse, thus generating a double-
strand break (DSB), initiating DSB repair.
Table 1.2: Summary of DNA damaging agents. RF – replication fork.
DNA damaging agent DNA damage mechanism Type of DNA damage
ionizing radiation (IR) reactive oxygen species attacking bases and
sugar- phosphate backbone
SSB and DSB
ultraviolet light (UV) forms pyrimidine dimers RF stalling/RF collapse
hydroxyurea (HU) depletion of dNTP pool RF stalling/RF collapse
aphidicolin (Aph) DNA polymerase inhibitor RF stalling/RF collapse
methyl methansulfonate (MMS) methylates DNA RF stalling/RF collapse
mitomycin C (MMC) inter-strand DNA cross-links RF stalling/RF collapse
cisplatin intra-strand DNA cross-links RF stalling/RF collapse
camptothecin (CPT) Topoisomerase I inhibitor SSB, RF stalling/RF collapse
etoposide Topoisomerase II inhibitor DSB
Repair pathways do not act only alone but can also cooperate to ensure repair of complicated
DNA lesions, e.g. inter-strand crosslinks (ICLs). ICL repair combines NER, TLS, MMR and
HR to ensure DNA lesion unhooking, bypass and subsequent restoration of the collapsed
replication fork, reviewed in2.

11
1.2. Repair of double-strand breaks
DNA double strand breaks represent the most toxic kind of lesion for cells. Unrepaired DSBs
can be lethal for the cell or result in chromosome rearrangements leading to genome instability,
where tumorigenesis is a likely outcome.
1.2.1. Double-strand break formation
As previously mentioned, DSBs can be formed either as a direct result of DNA damaging
agents or can be introduced during meiosis by a topoisomerase-like protein Spo11 to initiate
HR thus ensuring the diversity of the genetic information during sexual reproduction3.
Depending on the phase of the cell cycle DSBs are repaired by two different pathways: non-
homologous end-joining (NHEJ) or homologous recombination (HR). NHEJ is a preferred
pathway of DSB repair in humans unlike in budding yeast where HR is a major pathway4.
1.2.2. Non-homologous end joining
Classical NHEJ is an error-prone repair pathway of DSBs and it is conserved throughout the
evolution from prokaryotes to eukaryotes5,6
. It is referred to as non-homologous because it
involves a direct ligation of broken DNA ends7,8
. The advantages of NHEJ are cell cycle
independence and the unnecessary presence of dNTP pool9. On the other hand, NHEJ might
potentially result in loss of genetic information thus leading to genome instability and cancer.
KU70/80 heterodimer binds and protects the ends from nucleolytic degradation, mainly the
MRN complex10
. KU-complex with DNA-dependent protein kinase catalytic subunit (DNA-
PKs) brings together the non-homologous regions and recruits DNA ligase IV and its accessory
factor XRCC4 to ligate the overhangs11–13
.

12
Similar to classical NHEJ mechanism, a backup system called alternative NHEJ (altNHEJ) or
microhomology-mediated end-joining (MMEJ) (also known as micro-SSA) has been recently
discovered, reviewed in14
. This KU-independent pathway requires end resection and annealing
occurs at regions containing microhomology (<10 nt). It is more related to further discussed
single-strand annealing (SSA) pathway than to classical NHEJ7,15–17
. Molecular details of
altNHEJ pathway are not yet clear and seem to differ within interspecies14
. Figure 1.1 shows a
schematic view of NHEJ and altNHEJ pathways.
1.2.3. Choice between NHEJ and HR
The choice between NHEJ and HR is tightly regulated. The critical step in pathway
decision between NHEJ and HR is DNA end resection initiated by MRN complex. In G1
phase, 53BP1 and RIF1 proteins bind DSBs promoting NHEJ through tethering of KU70/KU80
complex, which inhibits further exonucleolytic cleavage of DNA ends. In G2 phase, BRCA1
with CtIP suppress 53BP1-RIF1 DNA binding promoting resection and thus switching to HR18–
22.
1.2.4. Homologous recombination
HR is a process where nucleotide sequences are exchanged between two homologous
sequences. The exchange may include homologous sequences between sister chromatids
(mitotic recombination) or between two homologous chromosomes (meiotic recombination).
Mitotic recombination ensures precise repair of DSBs during S/G2 phase without loosing any
genetic information. Meiotic recombination takes place in M phase and is responsible for the
genetic diversity of organisms, which is vital for adaptations throughout evolution.

13
The process of HR is evolutionary conserved and the human HR proteins as well as their yeast
homologs or orthologues are listed in Table 1.3. The recombination process can be divided into
three phases: pre-synapsis, synapsis and post-synapsis.
In humans during pre-synaptic phase DSBs are first recognized by the MRN complex with
CtIP23
. MRN binds the DSBs and activates the DSB signaling ATM protein kinase24,25
leading
to cell cycle arrest. Two redundant mechanisms cooperate with MRN to ensure long resection
of 5’-ends to generate a 3’-single-stranded overhang at both sides of the DSB. One mechanism
depends on EXO1 exonuclease, meanwhile the second pathway involves DNA2 nuclease with
helicase-topoisomerase complex BLM-TOPO3α-RMI1/2 (BTR complex)26–33
. Phosphorylation
of CtIP and DNA2 by CDK1 is essential for efficient resection34–36
. Generated ssDNA is
immediately covered by ssDNA-binding protein RPA, which is also a signal for cell cycle
arrest and DNA damage response by the ATM and ATR kinase signaling37–39
. RPA is a
heterotrimer, which protects the ssDNA from degradation and formation of secondary
structures40
. Recombination is performed mainly by the proteins of the RAD52 epistasis
group41
. RPA is removed from ssDNA-overhangs by BRCA2 and RAD52 with other accessory
proteins (RAD51B/D/C) promotes the formation/stabilization of a RAD51 presynaptic
filament42–47
. RAD51 forms right-handed nucleoprotein filament on ssDNA48
. This step, which
initiates HR, may be counteracted by the action of helicases, making it an important regulation
point. In humans it is not yet clear, which helicase among –RECQL5β, PARI or FBH1 has a
major role in disrupting the RAD51 presynaptic filament, but this antirecombinase activity was
first discovered in budding yeast Srs2 helicase49–54
.
During synapsis RAD51-ssDNA filament invades the homologous DNA duplex and forms a
so-called D-loop (displacement loop). Recombination factors RAD54/RAD54B greatly
stimulate RAD51-dependent strand-exchange41,55
.
In post-synaptic phase RAD51 is dissociated from the D-loop and DNA polymerase delta
(Polδ) starts the DNA synthesis on the new template utilizing the free 3’-overhang as a primer.
The further outcome of the recombination process can be separated into three different
subpathways - double-strand break repair (DSBR), synthesis-dependent strand annealing
(SDSA), and break induced replication (BIR). Schematic view of HR subpathways is
represented in Figure 1.1.

14
1.2.4.1. Double-strand break repair
In DSBR pathway, the D-loop stabilized by capturing by the second DNA end (termed
second-end capture) and a dHJ is formed41,56
. This recombination intermediate can be resolved
either by structure-specific endonucleases (MUS81-EME1, XPF-ERCC1, SLX1-SLX4 or
GEN1) or by dissolution with BTR helicase-topoisomerase complex28,57–61
. Depending on the
symmetry of the cut the outcome can be either a non-crossover or a crossover product. This
type of resolution is not favored during mitotic recombination since a crossover can lead to loss
of heterozygosity (LOH) or genome rearrangements, which may lead to cancer development62–
64. Thus preferred mechanism is dissolution of the dHJ by the BTR complex, which utilizes the
branch migration activity of BLM and decatenation activity of TOPO3α65,66
. This reaction
generates a non-crossover product (gene conversion).
1.2.4.2. Synthesis-dependent strand-annealing
In SDSA pathway the extended nascent DNA strand is displaced from the D-loop by helicase
activity of FANCM, BLM, RTEL1 and the DNA strand anneals to the 3’-overhang of the
original molecule67,68,69,70
. This prevents the formation of a dHJ, thus ensuring an exclusively
NCO product of HR. Moreover, RECQL5β helicase promotes SDSA by preventing RAD51
filament formation on the extended DNA strand to avoid second-end capture leading to
possibly undesired crossovers71
.
1.2.4.3. Break-induced replication
BIR events occur on one-ended DSBs, collapsed replication forks or DSBs near telomeres,
where second-end capture cannot proceed. After the formation of a D-loop, the synthesis of the
lost strand continues to the end of the chromatid. This can lead to large loss of heterozygosity
since a whole chromatid can be lost72
. Genetic characterization revealed that this mechanism is

15
RAD52-dependent, but some fraction is RAD51-independent. RAD51-independent BIR
requires other recombination factors including MRN and RAD54B73,74
.
Collapsed replication forks can be re-assembled by BIR pathway in G2 phase64,75
. This type of
repair is especially important in the maintenance of telomeric regions in telomerase negative
cells (ALT cells) where it facilitates a survivor76,77
. The difference between SDSA and BIR is
that BIR initiation requires a non-essential subunit of Polδ - Pol32 and the replication
machinery of both lagging and leading strand, but Polε may be dispensable. Interestingly, this
replication is highly mutagenic likely due to repeated initiation of replication by strand
invasion, decreased fidelity of Polδ due to increased dNTP pool and reduced repair at
replication forks78–80
.
In the case that BIR occurs at non-homologous chromatids due to microhomology segmental
duplications, deletions, nonreciprocal translocations and complex rearrangements can occur. In
humans this may lead to a number of human diseases (hemophilia A, Pelizaeus-Merzbacher
disease, some neurogical diseases) and cancer81,82
.
Figure 1.1: Scheme of HR pathways with human proteins. Repair of DSBs by NHEJ, MMEJ, SSA, BIR,
SDSA and DSBR. * - DNA damaging agents summarized in Table 1.1.

16
Table 1.3: Summary of budding yeast and human recombination factors. Reviewed in41,83,84
.
Yeast protein Human homolog/ortholog Function
Mre11/Rad50/Xrs2 MRE11/RAD50/NBS1 end resection
Ku70/80 KU70/80 overhang binding and protection
Sae2 CtIP end resection
Dna2 DNA2 end resection
Exo1 EXO1 end resection
Tel1 ATM DSB DNA-damage signaling
Mec1 ATR DNA-damage signaling
RPA RPA ssDNA binding
Rad51 RAD51 presynaptic filament formation
Rad52 RAD52
BRCA2/DSS1
homology search
RPA removal
Rad55-Rad57 RAD51B-RAD51C
RAD51D-XRCC2
RAD51C- XRCC3
accessory proteins in presynaptic filament
formation
- BRCA1 loading of BRCA2-RAD51 (together with
PALB2)
Rad54 RAD54 translocase
Rdh54/Tid1 RAD54B dsDNA ATPase
Srs2 RECQL5β/PARI/FBH1 (?) antirecombinase
Mph1 FANCM, BLM, RTEL1 disruption of D-loop
Mus81-Mms4 MUS81-EME1 dHJ cleavage
Sgs1-TOPO3-Rrm1 BLM-TOPO3α-RRM1-
RRM2
dHJ dissolution
Rad59 RAD52 annealing of complementary ssDNA
Rad1-Rad10 XPF-ERCC1 flap cleavage, dHJ cleavage
Slx1-Slx4 SLX1-SLX4 cleavage of HJs, scaffold for other nucleases
Msh2-Msh3 MSH2-MSH3 mis-match recognition
Saw1 - recruitment of Rad1-Rad10 to DNA lesions
Polδ Polδ DNA synthesis
Yen1 GEN1 dHJ cleavage
DNA PKs DNA PKs sensor for DNA damage
DNA lig IV LIG4 ligation of ssDNA
XRCC4 XRCC4 stimulation of ligation step in NHEJ
Cdk1 CDK1 cell cycle regulation kinase, G2-M transition
Rad9 53BP1 promotes NHEJ pathway
Rif1 RIF1 promotes NHEJ pathway
1.2.4.4. Single-strand annealing
SSA is a subpathway of homologous recombination where DSBs are repaired by direct
annealing of two DNA strands in repeat sequences84–86
. This results in deletion of sequences

17
between the repeats and this pathway is mutagenic. After the resection of the DSB ends by
MRN, the revealed complementary strands are annealed together in a RAD51-independent
manner87–89
. The annealing generates 3’-single-stranded non-homologous tails that need to be
cleaved by the structure-specific endonuclease XPF-ERCC1 and mismatch recognition
complex MSH2-MSH390–94
. MSH2-MSH3 complex has no effect on SSA between longer
repeats (1 kb) as, its function is to stabilize the annealing intermediates by binding to the
ssDNA-dsDNA junction prior to 3’-tail removal94–96
.
In humans XPF-ERCC1 directly interacts with RAD52, while the yeast homologue Rad1-
Rad10 is recruited to overhangs by structure-specific binding protein Saw195,97,98
.
Phosphorylated Slx4 also has a role in SSA and even though it interacts with Rad1-Rad10 and
not other SSA proteins (Rad52, Saw1, Msh2, Msh3), it does not recruit Rad1-Rad10 to the
damaged DNA95,99
.
1.3. Structure-specific endonucleases
Structure-specific endonucleases (SSEs) are enzymes, which cut various intermediates based
on their structure, therefore individual SSEs recognize and cleave specific DNA substrates.
Several SSE families have been identified including the XPF family (MUS81-EME1/EME2,
XPF-ERCC1), SLX1 family (SLX1-SLX4), and the XPG/Rad2 family (FEN1, GEN1, EXO1).
We will focus on the XPF family but for further interest in SSEs and their implications for
cancer therapy see review100
.
1.3.1. MUS81-EME1/EME2 complex
MUS81-EME1 is an endonuclease belonging to the XPF family. MUS81 is evolutionary
conserved in eukaryotes from yeast to humans and the complex differs in the non-catalytic
subunit - EME1 (Mms4 in budding yeast; Eme1 in fission yeast). EME1/Mms4/Eme1 are

18
probably responsible for the DNA binding of MUS81/Mus81 even though they have distant
protein sequences101–104
.
MUS81 interaction with EME1 is essential for its activity and forms a dimer of
heterodimers101,105,106
, even though it has been reported that Mus81-Mms4 forms a single
heterodimer107
. In fission yeast Mus81-Eme1 forms a dimer of heterodimers in low magnesium
concentrations making the nuclease proficient in processing recombination intermediates108
.
Human MUS81 also forms a complex with EME2102,109
. MUS81-EME2 is dispensable for
processing of recombination intermediates opposed to MUS81-EME1 and also contributes to
the maintenance of telomeres in ALT cells110,111
.
Substrate specificity
Purified MUS81-EME1/Mms4 complex has the highest affinity for DNA structures with an
exposed 5’-end near the DNA junction including 3’-flap, nicked-Holliday junction (nHJ), D-
loop and fork (cut on leading strand). On the other hand it cleaves poorly intact HJs, Y-form
and 5’-flaps102,105,112–115
. This fact has been supported by determining the crystal structure of
MUS81 bound to 3’-flap DNA116
. MUS81-EME1 complex makes an asymmetrical incision
with preference to the 5’-side (3-7 nt) of the homologous core unlike other typical resolvases
(e.g. Resolvase A). Resulting nicked duplexes are not directly ligatable59,117
. MUS81-EME1
purified from HeLa cells cleaves intact HJs better than from insect cells suggesting a specific
post-translational modification or a cofactor is needed for its action106
. Indeed, it has been later
reported that interaction with SLX4 promotes MUS81 cleavage of intact HJs through a nick-
counternick mechanism118
.
MUS81-EME2 complex exhibits broader substrate specificity than MUS81-EME1. In addition
to EME1 substrates it cleaves also intact HJs, 5’-flaps, nicked and gapped duplexes, and D-
loops by cleaving the 3’-invading strand119,120
(Summarized in Table 1.4, Figure 1.2). These
distinct substrate requirements suggest a non-overlapping role for these complexes in vivo.

19
Table 1.4: Substrate specificity of MUS81-EME1/EME1. HJ – Holliday junction, nHJ – nicked HJ, iHJ – intact
HJ.
Complex DNA substrate
MUS81-EME1 3’-flap, fork, nHJ, D-loop
MUS81-EME2 3’-flap, 5’-flap, fork, Y-form, iHJ, nHJ, mHJ, D-loop, nicked/gapped duplex
Figure 1.2: Schematic view of MUS81-EME1/EME2 specific DNA substrates. Arrows indicate the position of
incision.
Protein structure
The endonuclease activity of MUS81 is encoded by ERCC4 (VERK) nuclease domain (typical
for the XPF family) (Figure 1.3). It is dependent on magnesium ions but independent on
ATP59,101
. MUS81 and EME1 also contain two helix-hairpin-helix (HhH) domains at the C-
terminus, which are responsible for DNA binding at ss/dsDNA junctions121,122
. MUS81 has an
additional N-terminal HhH domain implicated in protein-protein interactions123
. MUS81-DNA
binding through WH domain (DNA recognition motif consisting of wings, α-helices and β-
sheets) also stimulates the nuclease activity of both MUS81-EME1/EME2 complexes and
influences the position of incision at synthetic DNA structures in the MUS81-EME2
complex123
. The WH domain in MUS1-EME2 complex also modifies its substrate specificity
by stimulating the cleavage of Y-form DNA.

20
Figure 1.3: Schematic view of human and yeast MUS81 complex domains. aa – aminoacids, WH – winged
helix, HhH – helix-hairpin-helix.
MUS81 in S-phase
Several pieces of evidence support the role of MUS81 during S phase. First, it has a higher
abundance in S-phase and more severe nuclear localization after inducing DNA damage by
HU, thymidine and UV59
. Recently, it has been established that in human cells MUS81
interacts with EME2 prior to replication start and functions in S-phase110
(Figure 1.4). CHK1
regulated MUS81-EME2-dependent cleavage of stalled replication forks also affects replication
dynamics such as the dNTP pool and RF speed110,124
. MUS81 stabilizes PCNA binding to
chromatin and it is responsible for the replication fork collapse after replication arrest induced
by DNA damaging agents such as HU, aphidicolin, MMC, cisplatin, CPT, MMS125–129
and
CHK1 inhibition130
. The generated DSB (collapsed fork) is a substrate for Rad54-mediated
repair by HR enabling the replication fork to restart128
.
SSEs (MUS81, SLX4, GEN1) also impact S phase progression in human cells.
MUS81/SLX4/GEN1-depleted cells show significant RF impediments and CHK2 activation
leading to chromosome segmentation and micronucleation131
. This suggests a role for MUS81
in stabilizing the DNA replication process.
Fission yeast Mus81-Eme1 is activated in a DNA damage-dependent manner that is regulated
by Cdc2 (CDK1)- and Rad3 (ATR)-dependent phosphorylation of Eme1 subunit132
. Mus81
subunit is also phosphorylated in a Cds1-dependent manner. Cds1 replication checkpoint kinase
promotes replication fork stability after HU treatment by dissociating Mus81 from chromatin to
avoid unwanted cleavage133
.

21
MUS81 in G2-M
In G2-M MUS81 interacts with EME1 and this subunit is activated by CDK1 and PLK1
kinases at this stage (Figure 1.4). EME1 phosphorylation is essential for association with SLX4
scaffold endonuclease110,118
. Formation of SLX-MUS stable holoenzyme solves MUS81
cleavage problem with intact HJs and is crucial for HJ resolution in the absence of GEN1.
A proportion of cellular MUS81 (through W24, L25 aa) interacts with SLX4 to ensure ICL
repair occurring during G2 in human cells60,118,134–137
. MUS81 probably, cooperatively with
other enzymes, functions in early and late steps of ICL repair - introducing an incision adjacent
to the crosslink to ensure its unhooking, DSB formation and HJ resolution26,27,59,61,112,125,127,138–
140.
MUS81-EME1 endonuclease was shown to resolve late replication intermediates at hard to
replicate regions - common fragile sites (CFSs), leading to the appearance of gaps or breaks in
metaphase chromosomes141,142
. Such cleavage during prometaphase ensures proper segregation
of chromosomes even at the expense of a generated. Unprocessed joint molecules lead to
accumulation of ultra-fine bridges (UFBs) in anaphase and 53BP1 bodies in the daughter cell in
the ensuing G1 phase141,143,144
.
In yeast, Mms4 is also regulated by phosphorylation during late S-phase and G2-M in a Cdc28-
/Cdc5-dependent manner resulting in the hyperactivation of Mus81-Mms4 complex107,145–148
.
Mus81 is required for segregation of chromosomes during meiosis I in a crossover
manner101,114
. Failure to process recombination intermediates leads to decreased spore viability
and aberrant asci114
.
MUS81 interactions
Besides SLX4, MUS81 endonuclease activity is stimulated by several translocases: RAD54,
FBH1, BLM, Srs2, Rqh1126,149–153
(summarized in Figure 1.5). These translocases cooperate
with MUS81 usually by its targeting to DNA, but through different mechanisms.

22
Figure 1.4: Schematic view of cell-cycle dependent MUS81 function. Adapted from110,142
.
In yeast, Rad54 targets Mus81-Mms4 to DNA substrates independent of its ATPase activity
and Rad54 is epistatic to Mus81151
. Similarly, human RAD54 targets MUS81 to DNA
substrates but paradoxly in an ATP-dependent manner152
. Both yeast and human RAD54
greatly stimulate MUS81 endonuclease activity. In mouse cells Mus81 and Rad54
cooperatively mediate DSB repair and function in chromosome segregation154
. Human FBH1
helicase uses its ATPase/helicase activity to cooperate with MUS81 to promote DSB formation
after prolonged replication fork stalling caused by HU126
. BLM, a RecQ helicase, colocalizes
with MUS81 in human cells after HU exposure and enhances its DNA binding in an ATP-
independent manner153
. Budding yeast Srs2 helicase stimulates Mus81-Mms4 cleavage
independently of its helicase activity through a direct interaction and enables Mus81 to reach
DNA for cleavage by utilizing its antirecombinase activity to disrupt Rad51-ssDNA
filament149
. In fission yeast stalled replication forks are processed by Mus81-Eme1 in
cooperation wit Rqh1, but independently of its ATPase/helicase activity150
.
Besides translocases MUS81 can also be targeted and stimulated also by a protein belonging to
the Fanconi anemia complementation group - FANCA to make a 5’-end incision of a psoralen-
induced damage residing the leading strand of a replication fork. FANCA regulates MUS81

23
nuclease activity in a damage-dependent manner, protecting DNA in the absence of damage
and enhancing MUS81 activity in the presence of ICLs138
.
Figure 1.5: Schematic view of some MUS81 interactions.
On the other hand, MUS81-EME1/EME2 stimulates another structure-specific endonuclease
FEN1 through the MUS81 catalytic subunit. Similarly in budding yeast, Mus81 and Rad27
mutually stimulate each other155,156
. This suggests that both nucleases function together during
replication to process flap structures, including Okazaki fragments.
MUS81 in cancer
Cells with depleted MUS81 exhibit proliferation defects and accumulate various chromosomal
aberrations157,158
. Haploinsufficiency of MUS81 was shown to result in chromosomal
abnormalities and increased sensitivity to crosslinking agents140,159
, thus indicating that already
a single copy of MUS81 can cause genomic instability. Accordingly, decreased levels of
MUS81 expression have been found in hepatic metastasis and correlated with poor cancer
prognosis160
. A role for MUS81 in tumorigenesis is further supported by evidence of a
synergistic effect with inactivation of p53 and, on the other hand, suppression by inactivating
CHK2161
. This all together suggests that MUS81 could be a potential target for cancer therapy
in combination with other synthetically lethal proteins.

24
1.3.2. XPF-ERCC1 (Rad1-Rad10)
Another member of the XPF family is XPF-ERCC1 complex (Rad1-Rad10 in budding yeast),
which participates in multiple repair pathways such as NER, BER, ICL repair, SSA and MMEJ,
reviewed in162
. In this thesis we focused in more detail on yeast Rad1-Rad10 nuclease.
Rad1 substrate specificity and protein structure
Rad1 (XPF) protein forms a stable heterodimer with a non-catalytic subunit Rad10 (ERCC1),
which is essential for its endonuclease activity by ensuring DNA binding and promoting other
protein-protein interactions163,164
. XPF, similarly to MUS81 possesses an ERCC4 nuclease
domain, but it also contains a N-terminal inactive helicase (DEAH) domain (Figure 1.6). Rad1-
Rad10 is a 3’-flap endonuclease, which cleaves dsDNA at a 5’-end of a DNA junction,
preferably with the 5’-end further away opposed to Mus81-Mms4. It also cleaves bubble
substrates, D-loops and G-rich telomeric overhangs105,134,135,165–170
(Figure 1.6).
Figure 1.6: Rad1/XPF protein structure and DNA specific substrates. (A) Schematic view of Rad1-Rad10 and
XPF-ERCC1 protein domains. ERCC4 – nuclease domain, HhH- helix-hairpin-helix (DNA binding) domain,
DEAH – helicase domain. (B) Schematic view of XPF-ERCC1/Rad1-Rad10 specific DNA substrates. Arrows
indicate the position of incision.
B) A) B)

25
Rad1-Rad10 in DNA repair
Rad1-Rad10 complex is essential for the repair of UV-induced damage by NER pathway171
.
Recruitment of Rad1-Rad10 to UV-induced adducts is mediated by Rad14 (XPA in humans), a
damage recognition protein172,173
. Rad1-Rad10 (XPF-ERCC1) is responsible for a 5’ incision of
a bubble DNA lession, a consequence of pyrimidine dimers. The second incision is made by
Rad2 (XPG in humans) and a ssDNA gap is formed, which is filled by a DNA
polymerase167,174,175
.
Human XPF mutants are hypersensitive to ICLs176
. After ICL promoted replication fork
collapse, XPF-ERCC1 is tethered to ICL-sites by SLX4 scaffold nuclease and is capable of
DNA incision on both sides of the DNA lesion134,135,137,177–180
. XPF complex is later utilized in
the NER and HR step of ICL repair but it is dispensable for DSB formation181
.
Role of Rad1 was also shown to include processing of 3’-nonhomologous tails occurring after
strand annealing of complementary regions during SSA57,182–187
. Slx4 mediates efficient Rad1-
Rad10 cleavage of 3’-tails188
. In yeast, Rad1-Rad10 is targeted to overhangs by a mediator
Saw1, but not in the case of short flaps during G1 phase95,97,98,189
. Rad1-Rad10 forms a stable
complex with Saw1, which stimulates its endonuclease activity95
.
Rad1-Rad10 is also involved in other DNA repair pathways. For example it was shown to be
required during BER, where it has a redundant role with Apn1 and Apn2 nucleases in
processing 3’-blocked termini at DNA breaks caused by oxidative agents190–192
. Rad1-Rad10
also participates in processing of overhang substrates in altNHEJ193,194
. Moreover, during DSB
repair via HR, Rad1-Rad10 was shown to colocalize with SDSA sites through interaction with
Saw1 in S and G2 phase of the cell cycle195
.
XPF and diseases
Mutations in the XPF protein cause a serious syndrome of skin malignancies and melanoma
called Xeroderma pigmentosum (XP) and progeroid syndrome (XFE). XP is an autosomal
recessive genetic disorder, where cells fail to repair DNA damage caused by UV and XFE
patients display premature aging syndromes196,197
, probably caused by telomeric instability.
Consistent with this hypothesis, XPF-ERCC1 was found in a complex with TRF2, which

26
protects the telomeres from degradation. Moreover, ERCC1-defective cells show telomere
deletions as a result of telomeric fusions by NHEJ as a consequence of the presence of an
unprocessed telomeric overhang198
. A number of various cancers show a link between
increased expression of ERCC1 and poor response to platinum-based therapy 199–202
. This
makes XPF-ERCC1 a suitable potential target for sensitization of cancer cells.
1.4. RecQ helicases
Structure-specific endonucleases were shown to cooperate with several RecQ enzymes during
DNA replication and repair125,153
. This makes it interesting to study both types of enzymatic
families to provide molecular understanding of the intricate network ensuring chromosome
stability. The RecQ-like helicase family belongs to the SF2 superfamily of helicases. It is a
highly conserved group with one representative in prokaryotes and two in lower eukaryotes:
Escherichia coli - RecQ (Nakayama, 1984), Saccharomyces cerevisiae - Sgs1, Hrq1,
Schizosaccharomyces pombe - Rqh1, Hrq1. In humans five RecQ homologs were identified –
RECQL1, WRN, BLM, RECQL4, RECQL5. RecQ helicases function in multiple cell
processes such as DNA replication, transcription, DNA repair, telomere maintenance, and
DNA-damage signaling203–207
.
They are also referred to as the guardians of genome stability as their depletion or mutations
may lead to chromosome aberrations and cancer. In humans three RecQ helicases are
associated with syndromes featuring genome instability, premature aging and cancer
predisposition, namely Bloom (mutations in the BLM gene), Werner (mutations in the WRN
gene), and Rothmund-Thomson, RAPADILINO, Baller-Gerold syndromes (all mutations in the
RECQL4 gene)208–212
. The phenotypes are a result of increased sister-chromatid exchange
(SCE) and telomere shortening in BLM- and WRN-patients, respectively. Recently, also
RECQL1 was implicated in cancer, associated with tongue squamous cell carcinoma and it also
serves as a prognosis factor in epithelial ovarian cancer213–215

27
RecQ helicases unwind DNA in a 3’-5’ polarity and they are, to certain extent, capable of
unwinding a various types of structures such as dsDNA, forks, D-loops, three-way and four-
way junctions, triple helices, and G-quadruplex DNA210,216–221
. On the contrary to their helicase
activity, some RecQ helicases possess also a DNA strand-annealing activity219,222–226
. Together
this enables some enzymes to perform branch migration of DNA junctions or fork regression at
stalled replication forks utilizing their translocase activity on both ssDNA and dsDNA through
hydrolysis of ATP218,227,228
.
1.4.1. Structure of RecQ helicases
RecQ helicases contain three conserved domains – the core helicase domain (DEAH box)
(consisting of 7 conserved motifs), the RecQ C-terminal (RQC) domain, and the helicase-and-
RNaseD-like-C-terminal (HRDC) domain (Figure 1.7). The RQC domain contains a zinc-
binding, HhH, WH and β-hairpin motifs, and it mediates DNA binding229–232
. While all RecQ
enzymes posses the helicase domain, some of them may be missing the RQC and HRDC
domain. However, only BLM and WRN contain the HRDC domain, which is essential for their
localization to specific DNA lesions233–236
. In BLM it has been demonstrated how RQC and
HDRC domains cooperatively mediate DNA binding and unfolding of G-quadruplexes237
.
Within the helicase domain, RecQ helicases contain a highly conserved sequence – an aromatic
loop adjacent to the ATP hydrolysis motif - Walker B implicated in dimerization and coupling
of ATPase activity to DNA binding238–240
.

28
Figure 1.7: Schematic view of protein structure of RecQ helicases in bacteria, yeast and humans. HRDC –
helicase and RNaseD-like C-terminal, RQC – RecQ C-terminal, NLS – nuclear localization signal, Zn – zinc
finger, SRI – RNA polymerase II interaction.
RecQ enzymes may function in various oligomeric states. For all DNA helicases it is essential
to possess at least two DNA binding domains, so they can translocate along DNA and unwind
duplex DNA241,242
. If these two DNA binding sites are not distributed along one molecule,
oligomerization is required. The oligomeric state is also affected by the binding to ssDNA and
ATP. While nucleotide binding favors smaller oligomeric molecules, ssDNA stabilizes higher
oligomeric forms. Interestingly, RECQL1 in form of a pentamer or hexamer exhibits strand
annealing activity, but to utilize its unwinding activity it has to be in a monomeric or dimeric
state221,238,243
. Recently, RECQL1 crystal structure was solved and it suggests a tetrameric
conformation for this enzyme to mediate HJ recognition244
. WRN also exists in two oligomeric
states depending on DNA binding. In solutions WRN exists in a dimeric state but if bound to
HJs or forks it forms a tetramer245,246
. BLM can form hexameric ring-like structures in the
absence of DNA and ATP 247
. RECQL4 exists primarily as a dimer248
and its flap structure and
RNA binding is mediated through a N-terminally situated Zinc knuckle249
. Bacterial RecQ and
human RECQL5 function like monomers in a free and also ssDNA-bound form223,229,250,251
.

29
Oligomerization is probably needed for its strand-annealing activity which is inhibited by NTP
binding222,224,226,243
.
1.4.2. RecQ helicases in yeast
Saccharomyces cerevisiae Sgs1 (BLM ortholog) in complex with TOPO3-Rmi1 (STR
complex) plays a role in processing DSBR intermediates occurring during perturbed DNA
replication. Furthermore, Sgs1 affects replication fork stability, DNA damage checkpoint
response, telomere maintenance, meiotic recombination and processing of DNA ends252
.
During HR and replication template switch the STR complex utilizes its branch migration
activity to transform a dHJ into a single HJ and dissolve it in a non-crossover fashion65,253–256
.
Similarly to Sgs1, its Schizosaccharomyces pombe ortholog Rqh1 functions in HR, DNA
replication and protection of telomeres252,257–260
.
Hrq1 (a RECQL4 ortholog) was described in both budding and fission yeast. Hrq1 deficiency
shows similar hyperrecombination phenotypes as RECQL4 dysfunction in humans. It is critical
for ICL repair, NER and genome stability261–264
.
1.4.3. RecQ helicases in humans
1.4.3.1. RECQL1
RECQL1 is the most abundant of the RecQ helicases in human cells. RECQL1 is
overexpressed in many tumors making it a promising prognosis factor in cancer
treatment213,214,265
.
During replication RECQL1 has a unique role in replication fork restart after CPT treatment.
Using its helicase activity RECQL1 remodels chicken foot structures into replication forks but

30
it is incapable of fork regression and this process is inhibited by PARP1266,267
. RECQL1 (like
WRN) unwinds the leading strand of replication forks, which is stimulated by presence of RPA.
RECQL1 coordinates replication origin firing and governs RPA's availability in order to
preserve normal replication dynamics and suppress DNA damage268
. After replication stress
RECQL1 is enriched at CFSs to promote checkpoint activation and chromosome stability269
.
Besides the role during replication, RECQL1 facilitates the unwinding of HR intermediates
such as immobile and mobile HJs in a RPA-independent manner221
. Its role in HR is further
supported by its interaction with RAD51. Moreover, RECQL1 depletion leads to elevated levels
of SCEs, accumulation of spontaneous DSBs and sensitivity to DNA damage inducing agents
(IR, CPT)270
.
RECQL1 also functions in telomere maintenance in ALT cells by actively dissolving telomeric
D-loops and HJs271
. The importance of RECQL1 function in protection of telomeres is
supported by the fact that deletion of RECQL1 results in telomere loss and/or shortening,
elevated telomeric SCE and telomere fragility due to treatment with aphidicolin271
. In addition
telomere binding proteins, TRF2 and POT1, regulate RECQL1 helicase activity on telomeres.
1.4.3.2. WRN
Mutations in the WRN (RECQL2) gene cause an autosomal recessive disorder known as
Werner syndrome (WS), which is characterized by premature aging and early onset of cancer
especially osteosarcoma and mesenchymal tumors. Additional features include early onset of
osteoporosis, atherosclerosis, arteriosclerosis, type II diabetes and cataracts272,273
. WS cells
accumulate chromosome aberrations like deletions, translocations and other rearrangements,
and are hypersensitive to DNA crosslinking agents, topoisomerase inhibitors and IR274–281
.
Among the RecQ helicases only WRN possesses a unique 3’-5’ exonuclease activity for DNA
degradation at resected ends and ssDNA gaps32,282,283
. WRN is implicated in resolving
recombination intermediates in a gene conversion manner during mitotic recombination and
participates in choice of DSB repair pathway preferring suppression of recombination284–286
. In

31
the absence of WRN DSBs are repaired by a highly mutagenic pathway - alternative NHEJ287
.
WRN is an accessory protein in classical NHEJ, it interacts with KU70-KU86 and XRCC4-
DNA ligase IV complex, which stimulate its exonuclease and helicase activity, respectively288–
290.
WRN stabilizes stalled replication forks preventing their collapse in an ATM/ATR-dependent
manner283,291
. Together by interactions with FEN1 and RAD52 it promotes fork regression,
accurate processing and replication fork restart292–294
. At CFSs, Polδ processivity is stimulated
by WRN to ensure replication progression295,296
.
WRN also participates in telomere maintenance in cooperation with shelterin proteins by
promoting efficient replication at G-rich telomeric DNA, preventing telomeric SCEs and
disrupting G-quadruplexes297–302
. WRN favors acting on strand invasion intermediates
especially if a G-rich ssDNA is present and promotes strand invasion and strand exchange303
.
The significant role of WRN in preventing telomere erosion may explain the progeroid
phenotype of WS patients.
WRN has been also implicated in BER, NER, TLS and ICL repair304–308
underlining its
importance in genome stability.
1.4.3.3. BLM
Mutations in the BLM (RECQL3) gene cause a rare autosomal recessive disorder the Bloom
syndrome (BS), which is characterized by premature aging, mental retardation, sunlight
sensitivity and cancer predisposition with normal distribution of type and tissue208,309,310
. BS
cells exhibit high rates of SCEs and loss of heterozygosity (LOH) indicating a role for BLM in
the regulation of HR. Correspondingly expression of BLM is cell cycle regulated with the peak
in S and G2-M phases309,311,312
. BLM has a sensory mechanism for DNA unwinding where it
unwinds DNA to a critical length, then reanneals the separated strands and reinitiates further
unwinding313
.
BLM together with WRN is important in the initiation of HR by stimulating DNA2 in the
resection of DSBs and generating a 3’-ssDNA overhang30,32
. BLM predominantly negatively
regulates HR by dismantling the RAD51 presynaptic filament in D-loops314
, but it has also a

32
pro-recombination role by stimulating primer extension by Polη in a D-loop promoting
SDSA314
. In later stages of HR, BLM suppresses crossovers in cooperation with TOPOIIIα and
RMI1/RMI2 (BTR complex) by dissolving dHJs in DSBR and also sister chromatid junctions
in anaphase315–320
.
At stalled replication forks BLM interacts with MUS81-EME1 and it also utilizes its strand-
annealing activity for branch migration to promote fork regression in an ATP-dependent
manner but strand-annealing itself is inhibited by ATP153,222,321,322
. BLM is likely to function in
Okazaki fragment maturation through its interaction with FEN1323
. Furthermore BLM has a
role in telomere maintenance facilitating DNA replication at telomeres at G-rich strands324
.
1.4.3.4. RECQL4
Mutations in RECQL4 gene cause three rare autosomal recessive disorders: Rothmund-
Thomson (RTS), Baller-Gerold (BGS) and RAPADILINO syndromes. Main manifestations of
these syndromes are poikiloderma, growth retardation, juvenile cataracts and early onset of
cancer, especially osteosarcoma. The RECQL4 mutations lead to chromosome aberrations such
as trisomy and isochromosomes. The mutation spectrum of these diseases usually include
generation of premature STOP codons leading to truncated versions of the RECQL4 protein325
.
RECQL4 is proposed to be required for replication initiation as it interacts with replisome
factors MCM10, MCM2-7, CDC45, GINS, and CTF4 under the control by CDK (cyclin-
dependent kinase) and DDK (Dbf4-dependent kinase)326–330
. RECQL4 is the only protein
among the RecQ helicases that possesses two NLS domains at its N-terminus suggesting a
crucial role of this domain in DNA metabolism. Mutations in RTS patients generating truncated
RECQL4 fragments mostly contain this domain325
. The importance of the “replication” domain
was further confirmed in Xenopus xRTS (RECQL4 homolog) where the N-terminal domain is
essential for loading Polα onto replication origins329,331
. Among RecQ helicases RECQL4 has
an unique role in mitochondrial replication and transport of p53 to mitochondria in the absence
of DNA stress. Mutations in RECQL4 gene associated with RTS cause deregulation (higher
levels) of mtDNA synthesis332–335
.

33
RECQL4 functions in multiple repair pathways such as NER and BER, and cooperates in
maintaining telomere stability during S-phase336–339
. RECQL4 probably also functions in HR as
RECQL4 depleted cells are sensitive to induced DSBs340,341
. RECQL4 was found to colocalize
and coimmunoprecipitate with RAD51342
, but the role of this interaction is not clear. RECQL4
also participates in NHEJ by a direct interaction with KU70/KU80 heterodimer, part of the
DNA-PK complex, to stimulate its DNA binding thus promoting efficient NHEJ343
.
1.4.3.5. RECQL5
Dysfunctional RECQL5 has not yet been associated with any syndromes, but its depletion
leads to genomic instability, e.g. in osteosarcoma344
.
RECQL5 has three isoforms α, β, γ from which only the β-form localizes to the nucleus since it
contains a NLS345–347
. These isoforms are a result of alternative splicing. The RECQL5β form
does not contain the HRDC and WH domains, and its expression is cell-cycle independent348
.
In this study we will further refer to RECQL5β as RECQL5 for simplicity.
RECQL5 interacts throughout the cell cycle with the MRN complex and this interaction is
independent on DNA damage or DNA itself. By this interaction MRN recruits RECQL5 to
sites of DNA damage and RECQL5 inhibits its exonuclease activity to avoid HR initiation349
.
RECQL5 plays an essential role in DNA recombination as well as in DNA replication (see
Figure 1.8.). In HR, it dismantles the RAD51 presynaptic filament in an ATP-dependent
manner thus it negatively regulates homologous recombination similarly to Saccharomyces
cerevisiae Srs249,350,351
. RECQL5 interaction with RAD51 is critical for RAD51 removal from
ssDNA52
. During SDSA RECQL5 dismantles aberrant RAD51 filaments, potentially forming
during post-synapsis, thus protecting the cell from cross-over pathway71
.

34
Figure 1.8: Scheme of RECQL5 involvement in recombination pathway and replication. Orange triangle
depicts a DNA lession, blue arrow indicates the leading strand synthesis and green arrows depict lagging strand
synthesis by Okazaki fragments.
RECQL5 plays also a role in replication, where it interacts with PCNA in early and late S-
phase and accumulates at stalled replication forks caused by UV, HU and cisplatin352
. RECQL5
promotes ATP-dependent branch migration of Holliday junctions and strand-exchange activity
on the lagging strand223,352
. It possesses an intrinsic strand-annealing activity inhibited by
RPA223
. In vitro RECQL5 is also capable of annealing DNA and RNA in an ATP-independent
manner353
. It also prevents DNA breakage by its role in Okazaki fragment removal at stalled
replication forks exploiting its helicase activity and stimulating the endonuclease activity of
FEN1354
. The role of RECQL5 in stabilizing stalled replication forks is supported by in vivo
data from mouse embryonic fibroblasts where Recql5 prevents RF collapse after treatment with

35
CPT351
. During replication it also inhibits RNA-polymerase II by dissociating it from active
replication forks ensuring unperturbed replication fork progression355–357
.
Depletion of RECQL5 leads to metaphase chromosome defects like uncondensed and
entangled chromosomes358
. RECQL5 stimulates the decatenation activity of TOPOIIα, which
ensures proper segregation of chromatids358,359
. It also interacts with TOPOIIIα and TOPOIIIβ
but it can not dissolve dHJ as the BTR complex due to the lack of HRDC domain in
RECQL5347
.
1.4.4. Interactions between RecQ helicases
Since lower organisms (bacteria, yeast) possess only one or two RecQ helicases it is important
to map the functional divergence and redundancy of RecQ properties in humans. RecQ
helicases cooperate with each other in DNA repair and replication, while also having their
unique functions (Table 1.5). This is demonstrated by the ability of RECQL4 to stimulate BLM
helicase activity on fork substrates and BLM to enhances RECQL4 retention at DSBs in
vivo360
. In addition, RECQL5 stimulates the helicase activity of WRN, but not BLM, on fork
substrates and it partially compensates for the loss of WRN in WS cells. On the other hand,
BLM inhibits WRN exonuclease activity361
. The separate roles of RecQ helicases is
demonstrated by synthetic lethality observed in cells co-depleted for WRN and RECQL5362
.
Table 1.5: Summary of some interaction partners of RecQ helicases. Sgs1 interactions cited in65,363–368
. Human
RecQ helicase interactions reviewed in205
.
RecQ helicase Interaction partners
Sgs1 Dna2, Cdc28, Mlh1, Mre11, Rad51, TOPO3, Rmi1, Srs2, Smt3, Ubc9
RECQL1 RPA, RAD51, PARP1, EXO1, MLH1, MSH2, KU
BLM MUS81-EME1, WRN, TOPO3α, TOP1, TRF1, TRF2, POT1, RAD51, RPA, MRE11, NBS1,
RAD52, FEN1, Polη, Polδ, PARP1, BRCA1, FANCJ, FANCM, RECQL4, RMI1, RMI2,
MLH1, EXO1
WRN MRN (NBS1), BRCA1, RAD52, RAD51, BLM, RPA, ATM, p53, EXO1, PARP1, FEN1, DNA
Polβ, Polδ, Polη, Polε, Polλ, POT1, TOP1, TRF2, KU70-KU80, BLM, PCNA, RECQL5,
RECQL4, RAD54
RECQL4 RPA, FEN1, PARP1, APE1, RAD51, Polβ, Polϒ, p53, MCM10, TRF1, TRF2, BLM, WRN
RECQL5 WRN, RPA, FEN1, PARP1, RAD51, RNA PolII, TOPIIβ, MRN

36
1.5. Post-translational modifications
Not only protein-protein interactions, but also post-translational modifications are involved in
regulation of protein biological functions, including e.g. phosphorylation, acetylation,
methylation, ubiquitination and SUMOylation. These modifications provide a dynamic system
to modulate protein activity depending on specific requirements of the cell environment. Such
requirements include protein activation/inactivation, interaction modifications, subcellular
localization and proteosomal degradation. During the course of this study we focused on post-
translational modification SUMOylation.
1.5.1. SUMOylation
SUMOylation is a similar process to ubiquitination, but instead of mostly tagging proteins for
proteosomal degradation, it rather alters the conformation, activity, stability, localization or
interaction of the target protein. The importance of SUMOylation is further signified as
numerous proteins participating in many biological processes including signal transduction,
senescence, DNA repair, ion transport, intracellular transport, or gene transcription are
modified by SUMO369–371
. All the biological outcomes of SUMOylation are not yet well
understood, but it seems that dysfunction of this process may lead to cancer, developmental
abnormalities and some neurodegenerative diseases372
.
SUMO stands for small ubiquitin-like modifier and its molecular mass is 11 kDa. It shares 18
% homology with ubiquitin373–376
. SUMOylation is a three-step process where a SUMO peptide
is attached to a target protein. SUMO is usually attached to a lysine in a consensus sequence
ψKx(D/E), where ψ corresponds to a large aliphatic branched hydrophobic amino acid, K is the
target lysine, x corresponds to any amino acid, D is aspartic acid and E is glutamic acid377
.
First, the SUMO peptide needs to be activated by proteolytic enzymes that cleave the carboxyl-
terminus to expose a di-glycine motif (Figure 1.9A). The SUMOylation pathway starts by the
activation of the SUMO peptide by a SUMO-activating enzyme (E1) and this process is ATP-
dependent374
. SUMO is then transferred to a SUMO conjugating enzyme (E2), which facilitates
37
its conjugation to the target protein378
. This process is usually in vivo enhanced by SUMO E3
ligases370,379–383
. SUMO itself may be SUMOylated and form SUMO chains384
, but the
relevance of this process is yet to be understood. SUMOylation is a dynamic reversible process
and the SUMO protein can be also cleaved off by SUMO proteases385
. The individual proteins
of the SUMOylation pathway for budding yeast and mammals are listed in Table 1.6.
In addition, proteins can noncovalently interact also with SUMO through a SUMO-specific
interacting site called SIM (SUMO-interacting motif). SIMs consist of a hydrophobic core of 3-
4 amino acids (usually valine and isoleucine) that are flanked by acidic amino acids (aspartic
and glutamic acids)386
. SUMO-SIM interaction may affect the interactions with other proteins
or DNA. Moreover, SUMOylation of protein which also contains SIM motif, can lead to
conformational changes resulting in altered interaction properties or activity of the protein
(Figure 1.9B)387–389
.
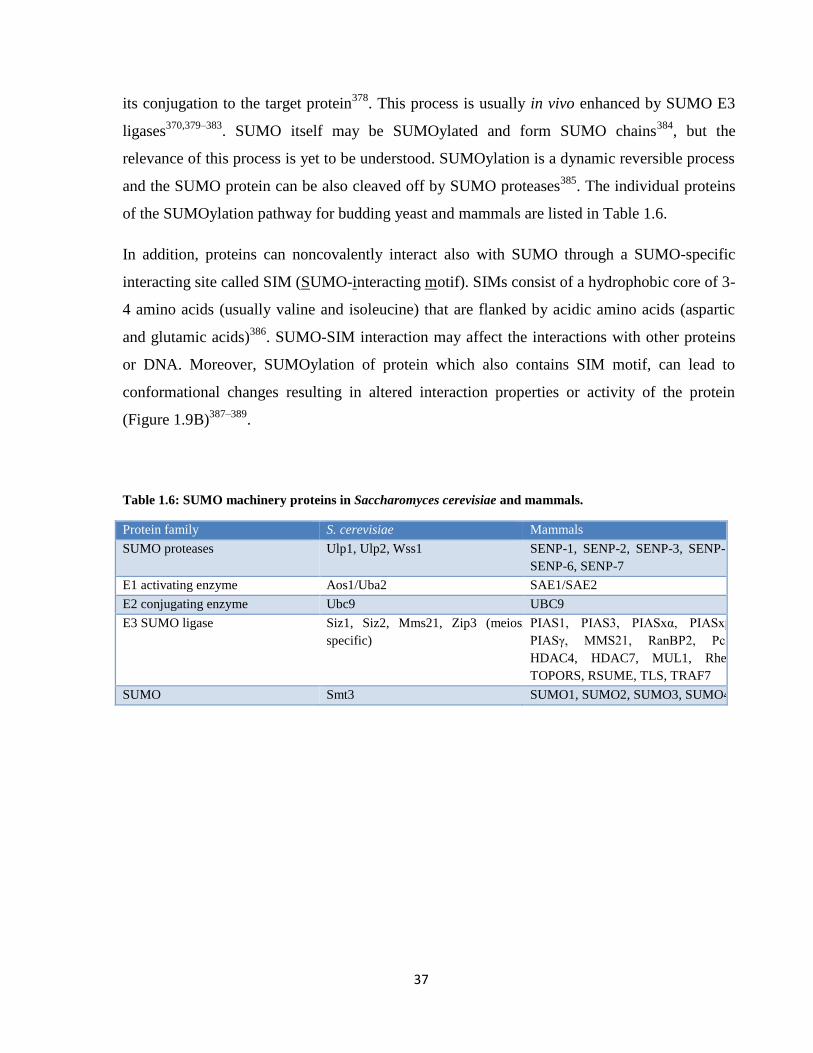
Table 1.6: SUMO machinery proteins in Saccharomyces cerevisiae and mammals.
Protein family S. cerevisiae Mammals
SUMO proteases Ulp1, Ulp2, Wss1 SENP-1, SENP-2, SENP-3, SENP-5,
SENP-6, SENP-7
E1 activating enzyme Aos1/Uba2 SAE1/SAE2
E2 conjugating enzyme Ubc9 UBC9
E3 SUMO ligase Siz1, Siz2, Mms21, Zip3 (meiosis
specific)
PIAS1, PIAS3, PIASxα, PIASxβ,
PIASγ, MMS21, RanBP2, Pc2,
HDAC4, HDAC7, MUL1, Rhes,
TOPORS, RSUME, TLS, TRAF7
SUMO Smt3 SUMO1, SUMO2, SUMO3, SUMO4

38
Figure 1.9: SUMOylation mechanism. (A) Schematic view of SUMOylation pathway. G-glycine, A-alanine, C-
cysteine, K-lysine. (B) Protein-protein interaction mediated by SUMO-SIM. i) Protein A containing a SIM
noncovalently interacts with SUMOylated Protein B. ii) SUMO peptide of SUMOylated Protein A interacts with
its own SIM leading to conformational changes.
1.5.2. SUMOylation in HR
Many proteins participating in HR are SUMOylated and their SUMOylation leads to both
negative and positive regulation of HR83,390,391
. SUMOylation usually affects DNA-protein,
protein-protein interactions, solubility and localization of the target protein thus modulating its
function in maintaining genome stability. Protein SUMOylation is elevated after DNA damage
supporting the model where SUMOylation is a crucial regulator of DNA repair392,393
. Few
examples of effects of SUMO attachment during HR are listed below.
SUMOylation/ubiquitination of PCNA is a solid example of the importance of post-
translational modifications in regulation/choice of DNA repair pathway. Crosstalk between
post-translational modifications of a highly conserved lysine (K164) of PCNA determines the
repair pathway in favor of TLS or HR after encountering a DNA lession during

39
replication394,395
. Monoubiquitination of PCNA is DNA-damage dependent and recruits error-
prone DNA polymerases for TLS396–398
. Polyubiquitination of PCNA facilitates error-free
repair by utilizing an undamaged sister chromatid. PCNA is also SUMOylated, which results in
recruitment of Srs2 through its SIM in S phase in a DNA-damage independent manner to
inhibit unscheduled DNA recombination395,399
. The Srs2 SIM can bind SUMO-PCNA or
SUMO peptide attached to Srs2 itself forming an intramolecule interaction probably changing
its conformation388,400
. SUMOylation of Srs2 thus serves as a regulatory mechanism for
promoting HR by SUMO-Srs2 or its inhibition by SUMO-PCNA-Srs2.
Table 1.7. Effect of SUMOylation on HR proteins. Citations not mentioned in text 401–405
.
HR protein Function of SUMOylated form
PCNA inhibits HR by recruitment of Srs2
Srs2 promotes HR
Sae2 decreases solubility
EXO1 inhibits HR by impeding end resection of DSB by promoting protein degradation
Lif1 inhibits NHEJ by impeding ligation step
Sgs1 promotes HR at telomeres
BLM promotes HR by increased binding of RAD51
WRN colocalization with RAD51
BRCA1 promotes HR
Rad52 inhibits HR by compromising strand-annealing
yRPA inhibits SSA and BIR
SUMOylation has been shown to affect biochemical activities of several HR proteins. It
inhibits DNA binding and annealing activities of recombination mediator Rad52 protein thus
shifting DNA repair from SSA to gene-conversion products406,407
. Moreover, SUMO-Lif1
exhibits decreased self-association and ssDNA binding impeding its activity during the ligation
step in NHEJ408
. A novel mechanism of SUMOylation function was described in case of end
resection protein Sae2 where SUMOylation together with phosphorylation increases solubility
of Sae2409
. In humans, SUMOylation negatively influences EXO1 by promoting its
ubiquitination and protein degradation410
. Summary and other examples of HR regulation by
SUMOylation are shown in Table 1.7.

40
2. AIMS OF THESIS
1. Characterize interaction of MUS81-EME1 endonuclease with RECQL5 helicase and
determine biological importance of this interaction.
2. Biochemically characterize effect of SUMOylation on Rad1-Rad10 complex.

41
3. MATERIAL AND METHODS
3.1. Material
3.1.1. Chemicals
Table 3.1: List of chemicals and companies of their purchase.
AppliChem β-mercaptoethanol (β-ME), 2-propanol, aceton, acetic acid, acrylamide 4K,
adenosintriphosphate (ATP), ammonium persulfate (APS), aprotinine, benzamidine
hydrochloride, bis-acrylamide, boric acid, bromphenol blue, Coomassie Brilliant
Blue, disodium hydrogenphosphate (NaH2PO4), dithiothreitol (DTT), ethanol,
ethylenediaminetetraacetic acid (EDTA), formic acid, glucose, gluthatione, glycerol,
glycine, imidazole, isopropyl-beta-D-thiogalactopyranoside (IPTG), leupeptine,
magnesium chloride hexahydrate (MgCl2.6H2O), methanol, Nonidet P40 (NP40),
OrangeG, pepstatin, phenylmethanesulfonylfluoride (PMSF), Ponceau S, potassium
dihydrogenphosphate (KH2PO4), potassium chloride (KCl), Silver Nitrate (AgNO3),
sodium chloride (NaCl), Sodium Thiosulfate Na2S2O3, sucrose,
tetramethylethylenediamine (TEMED), Trichloroacetic acid (TCA), Tris, Tris-HCl,
Tris-ultrapure, tryptone enyzymatic digest, Tween 20, yeast extract
Bio-Rad hydroxyapatite
Fermentas GeneRulerTM 1kb DNA Ladder, PageRulerTM Prestained Protein Ladder, dATP,
dCTP, dGTP, dTTP
Fluka agar
GE Healthcare MonoS, MonoQ, SP-Sepharose, Q-Sepharose, Glutathione Sepharose
New England BioLabs Chitin affinity beads, 10x NEB3 buffer, 10x bovine serum albumin (BSA),
restriction endonucleases: AseI, DpnI
Nippon Genetics Midori Green DNA stain
Serva agarose, sodium dodecylsulfate (SDS), ethanol (for molecular biology)
Sigma-Aldrich ampicillin, bovine serum albumin (BSA), His-Select nickel affinity gel, chymostatin
Stratagene PFU Turbo DNA polymerase, 10 x Cloned PFU DNA polymerase reaction buffer

42
3.1.2. Buffers and stock solutions
Table 3.2: List of used buffers, stock solutions and their composition.
30 % acrylamide 30 % (w/v) acrylamide, 0.8 % (w/v) bis-acrylamide
Blocking buffer 5 % (w/v) non-fat milk, 1 x PBS , 0.05 % (v/v) Tween 20
2 x CBB (Cell breakage
buffer)
100 mM Tris–HCl, 20 % (w/v) sucrose, 20 mM EDTA, pH 7.5
Coomassie Blue 40 % (v/v) methanol, 20 % (v/v) acetic acid, 0.32 % (w/v) Coomassie Brilliant Blue
5 x D (EMSA buffer) 200 mM Tris-HCl, pH 7.5, 250 mM KCl, 5 mM DTT, 500 μg/ml BSA
Destain 40 % (v/v) methanol, 20 % (v/v) acetic acid
Hybridization buffer 50 mM NaCl, 50 mM MgCl2.6H2O
2 x K 40 mM K2HPO4, 20 % (v/v) glycerol, 1 mM EDTA, pH 7.5
6 x DNA loading buffer 10 mM Tris-HCl, 0.12 % (w/v) Orange G, 60 % (v/v) glycerol, 60 mM EDTA, pH 7.6
5 x ME 250 mM Tris-HCl, pH 7.5, 5 mM DTT, 25 mM MgCl2, 500 µg/ml BSA
5 x MM 100 mM Tris-HCl, 1 mM DTT, 50 mM MgCl2, 500 µg/ml BSA, 25 % (v/v) glycerol,
pH 8
6x native DNA loading buffer 10 mM Tris–HCl, 60 % (v/v) glycerol, 60 mM EDTA, pH 7.4
10 % native gel in 0.5x TBE 0.5 x TBE, 10 % (w/v) acrylamide, 0.15 % (w/v) APS, 0.015 % (v/v) TEMED
10 % native gel in 1x TBE 1 x TBE, 10 % (w/v) acrylamide, 0.15 % (w/v) APS, 0.015 % (v/v) TEMED
10x PBS 1.37 M NaCl, 27 mM KCl, 100 mM Na2HPO4, 18 mM KH2PO4, pH 7.4
10x PBST 1.37 M NaCl, 27 mM KCl, 100 mM Na2HPO4, 18 mM KH2PO4, 0.05 % (v/v) Tween,
pH 7.4
PI cocktail (Protease
inhibitors)
aprotinine, benzamidine, chymostatin, leupeptin, pepstatine, each 5 μg/ml
Ponceau S 0.2 % (w/v) Ponceau S, 5 % (v/v) acetic acid
5 x R 250 mM Tris-HCl, pH 7.5, 5 mM DTT, 5 mM MgCl2, 500 µg/ml BSA
Resolving buffer 3 M Tris-HCl, pH 8.8
RF1 buffer 100 mM RbCl, 50 mM MnCl2.4H2O, 30 mM KOAc, 10 mM CaCl2.2H2O, 15% glycerol,
pH 5.8
RF2 buffer 10 mM MOPS, 10 mM RbCl, 75 mM CaCl2.2H2O, 15% glycerol, pH 6.8
7.5 % Running gel solution resolving buffer, 7.5 % (w/v) acrylamide, 0.1 % (w/v) SDS, 0.09 % (w/v) APS, 0.015 %
(v/v) TEMED
8 % Running gel solution resolving buffer, 8 % (w/v) acrylamide, 0.1 % (w/v) SDS, 0.09 % (w/v) APS, 0.015 %
(v/v) TEMED
10 % Running gel solution resolving buffer, 10 % (w/v) acrylamide, 0.1 % (w/v) SDS, 0.09 % (w/v) APS, 0.015 %
(v/v) TEMED
12 % Running gel solution resolving buffer, 12 % (w/v) acrylamide, 0.1 % (w/v) SDS, 0.09 % (w/v) APS, 0.015 %
(v/v) TEMED
5 x S 500 mM Tris-HCl, pH 7.5, 50 mM MgCl2
10x SDS buffer 0.25 M Tris-HCl, 0.192 M glycine, 2 % SDS (w/v), pH 8.3
2x SDS-Laemmli buffer 125 mM Tris–HCl, 4 % (w/v) SDS, 10 % (v/v) β-ME, 20 % (v/v) glycerol, 0.004 %
(w/v) bromophenol blue, pH 6.8
Stacking buffer 0.5 M Tris-HCl, pH 6.8
3.75 % Stacking gel solution stacking buffer, 3.75 % (w/v) acrylamide, 0.08 % (w/v) SDS, 0.071 % (w/v) APS,
0.036 % (v/v) TEMED
2 x T 25 mM Tris-HCl, 10 % (v/v) glycerol, 0.5 mM EDTA, pH 7.5
10 x TBE 900 mM Tris–HCl, pH 7, 900 mM boric acid, 20 mM EDTA, pH 7.5
Transfer buffer 25 mM Tris-HCl, 190 mM glycine, 10 % methanol, 0.1 % (w/v) SDS

43
3.1.3. Growth media
All media were sterilized at 121°C, 120 kPa for 15 minutes. The media (see Table 3.3) were
supplemented with appropriate antibiotics based on the plasmid resistance (ampicillin 100
μg/ml).
Table 3.3: Growth media information.
2 x TY 2 % tryptone (w/v), 1 % yeast extract (w/v), 0.5 % NaCl (w/v)
LB 1 % tryptone (w/v), 0.5 % yeast extract (w/v), 1 % NaCl (w/v), 2 % agar (w/v) (for solid LB plates)
SOC 2 % tryptone (w/v), 0.5 % yeast extract (w/v), 10 mM NaCl, 20 mM glucose, 2.5 mM KCl, 10 mM
MgCl2.6H2O, pH 7.2
3.1.4. Bacterial strains
The bacterial strains used in our studies are listed in Table 3.4.
Table 3.4: Bacterial strain information.
Strain Genotype
E. coli DH5α F- endA1 glnV44 thi-1 recA1 relA1 gyrA96 deoR nupG Φ80dlacZΔM15 Δ(lacZYA-
argF)U169, hsdR17(rK- mK+), λ–
E. coli Rosetta (DE3) pLysS F- ompT hsdSB(RB- mB-) gal dcm λ(DE3 [lacI lacUV5-T7 gene 1 ind1 sam7 nin5])
pLysSRARE (CamR)
E. coli BL21 (DE3) pLysS F–, ompT, lon, hsdSB (rB–, mB–), dcm, gal, λ(DE3), pLysS (Cam
r)
E. coli Arc RIL F– ompT hsdS (rB–mB–) dcm
+ Tet
r gal endA Hte [cpn10 cpn60 Gent
r] [argUileY leuW
Strr]
3.1.5. Molecular weight standards
The molecular weight standards used in our experiments are listed in Table 3.5.
Table 3.5: Molecular weight standards information.
Name Sizes Company
GeneRulerTM
1 kb DNA ladder 250, 500, 750, 1000, 1500, 2000, 2500, 3000,
3500, 4000, 5000, 6000, 8000, 10000 bp
Fermentas
HMW 29, 45, 66, 97, 116 kDa Sigma Aldrich
PageRulerTM Prestained Protein Ladder 10, 25, 34, 43, 55, 72, 95, 130, 170 kDa Fermentas

44
3.1.6. Plasmids
Detail information about the plasmids used in our work are listed in Table 3.6.
Table 3.6: Plasmid information.
Plasmid Resistance Size Source
pET21d-MUS81/EME16xHis AMP 8821 bp Stephen West
pGEX-GSTMUS81/EME16xHis AMP 8281 bp Stephen West
pET14b-Mus81/Mms46xHis AMP 8166 bp Matthew Whitby
pTXB1-RECQL5β AMP 9679 bp Pavel Janščák
pET11a-6xHisRad1/Rad10 AMP 9783 bp Steven Brill
pGEX-4T-Saw1 AMP 5683 bp Eun Li
pET21b-Siz11-465 AMP 6852 bp Yoshimitsu Takahashi
pSUPER-retro.puro AMP 6343 bp R. Venkitaraman
3.1.7. DNA primers
DNA primers were purchased from Sigma-Aldrich or VBC Biotech (fluorescently labelled).
Table 3.7: DNA primers information. SM – SUMO-mutant, NDM – nuclease-dead mutant, seq. – sequencing, F
– forward, R – reverse, * - FITC (fluorescent label)
Primer # Name Sequence 5’-3’ Application
pR595 Rad1-K32R F caagcatctcaaagttctaaaattagaaatgaagatgaacccgacgactccaatc Rad1 SM
pR596 Rad1-K32R R gattggagtcgtcgggttcatcttcatttctaattttagaactttgagatgcttg Rad1 SM
pR1112 Rad1-D869A F gttattctgtaatgacccaattaaTGcagaaatcgattttctttcgagaca Rad1 NDM
pR1113 Rad1-D869A R tgtctcgaaagaaaatcgatttctgCAttaattgggtcattacagaataac Rad1 NDM
pR599 Rad1 seq1 acccatttgctgtccaccag Rad1 seq.
pR600 Rad1 seq2 ctaaagaaaacgactgtcct Rad1 seq.
pR601 Rad1 seq3 tgttctgcccataaatcctt Rad1 seq.
pR602 Rad1 seq4 ttgtgcctcatcgaccaatag Rad1 seq.
pR603 Rad1 seq5 gctcaagattccgccagaaa Rad1 seq.
pR706 Rad1 seq6 ttacacaggtgcttcaggaac Rad1 seq.
pR604 Rad10 seq1 cagcaaaaaacccctcaaga Rad10 seq.
pR25 D27 agctatgaccatgattacgaattgctt 5’-flap, 5’-overhang
pR26 D22 aattcgtgcaggcatggtagct fork, 3’-flap
pR27 49N* agctaccatgcctgcacgaattaagcaattcgtaatcatggtcatagct fork, flap, dsDNA
pR28 49R agctatgaccatgattacgaattgcttaattcgtgcaggcatggtagct dsDNA
pR29 F3-d47 agctatgaccatgattacgaattgcttggaatcctgacgaactgtag fork, 3’-flap, Y
pR30 F5-d42 gatgtcaagcagtcctaaggaattcgtgcaggcatggtagct 5’-flap
pR31 D20 ctacagttcgtcaggattcc fork
pR45 X-0 oligo 2 tgggtcaacgtgggcaaagatgtcctagcaatgtaatcgtctatgacgtt HJ, nHJ
pR46 X-0 oligo 5 tgccgaattctaccagtgccagtgatggacatctttgcccacgttgaccc HJ
pR47 X-0 oligo 6 gtcggatcctctagacagctccatgatcactggcactggtagaattcggc HJ
pR48 X-0 oligo 7 * caacgtcatagacgattacattgctacatggagctgtctagaggatccga HJ, nHJ
pR49 X-0 nick oligo10 ggacatctttgcccacgttgaccc nHJ
pR50 X-0 nick oligo 15 tgccgaattctaccagtgccagtgat nHJ

45
3.1.8. DNA substrates
Various fluorescently labelled DNA substrates were used for EMSAs and nuclease assays, and
unlabeled DNA for SUMOylation assays. DNA substrates were prepared by Victoria Marini as
described in 151
. Indicated oligonucleotides were annealed and cleaned by HPLC. Unlabeled Y-
form was prepared by simple hybridization (see Chapter 3.2.1.5.).
Figure 3.1: Schematic view of DNA substrates. * - FITC label
3.1.9. Proteins
SUMO machinery proteins (Aos1/Uba2, Ubc9, Smt3-KR, Siz11-465, Siz2) were purified by
Veronika Altmannová and Peter Kolesár as described in388,406
. Human RAD51-K133R was
purified by Mário Špírek as in411
. RECQL5β truncation mutants were purified by Marek
Šebesta and RECQL5β point mutants (K58R and F666A) were purified by Hana Sedláčková,
Mário Špírek and Veronika Altmannová as described in52
.

46
3.2. Methods
3.2.1. DNA techniques
3.2.1.1. Visualization of DNA
A) Agarose gel electrophoresis
Agarose gels were prepared by melting agarose (1 % (w/v)) in 1 x TBE buffer. While cooling,
0.02 μl/ml Midori Green DNA stain was added into the agarose solution. The solution was
poured into horizontal electrophoresis unit. DNA samples were prepared by adding 1/6 volume
of 6 x DNA loading buffer. 1 kb ladder was used as a molecular weight marker. DNA species
were separated at voltage gradient of approximately 5-7 V/cm. DNA was visualized in the
GelDoc system.
B) Native gel electrophoresis
Native gels were used for separation of small fluorescently labelled oligonucleotide DNA
species with or without bound proteins. Native DNA gel solutions were poured into vertical
electrophoresis units. 10 % polyacrylamide native gels in 1 x TBE were used for the nuclease
assays, meanwhile EMSAs were done using 10 % polyacrylamide native gels in 0.5 x TBE.
Gels were run in 1 x TBE or 0.5 x TBE running buffer depending on the gel composition at a
constant voltage 80-110 V for 45 minutes (nuclease assay) or 90 minutes (EMSA). The gels
were visualized by Image Reader FLA-9000 (FujiFilm) and the results were quantified using
MultiGauge V3.2 software.

47
3.2.1.2. DNA purification from E. coli
Plasmids were typically purified from 5 ml of overnight bacterial culture grown in LB medium
with appropriate antibiotics at 37°C. DNA isolation was performed from cell pellet using
commercial NucleoSpin Plasmid kit (Macherey–Nagel) or QIAprep Spin Miniprep kit (Qiagen)
according to the user’s manual.
3.2.1.3. Site-directed mutagenesis
Site-directed mutagenesis was used to produce point mutations leading to substitutions
generating Rad1-K32R-Rad10, Rad1-D869A-Rad10 and Rad1-K32R, D869A-Rad10 mutants.
The point mutations were introduced into original plasmids (pET11a-Rad10-Rad1) (in the case
of Rad1-K32R, D869A mutant pET11a-Rad10-Rad1-K32R was used) by PCR using mutagenic
primers summarized in Table 3.8. The PCR mixture contained 1 x TurboPFU cloning buffer,
0.5 mM dNTP mix, 0.5 μM forward and reverse primer, ~ 10 ng of plasmid DNA, and 5 U of
TurboPFU DNA polymerase in a final volume 50 μl. PCR program for the thermocycler is
summarized in Table 3.9. The presence of desired mutations in plasmid DNA was verified by
digestion with restriction enzyme (new AseI site) or by sequencing (see Table 3.8.). Entire
Rad1-Rad10 ORFs were sequenced to exclude random mutations. Generated expression
plasmids include pET11a-Rad10-Rad1-K32R, pET11a-Rad10-Rad1-D869A, and pET11a-
Rad10-Rad1-K32R, D869A.
Table 3.8: Mutagenic primers.
Original plasmid Protein mutation DNA mutation Primers Verification
pET11a-Rad10-Rad1 K32R aaa→aga pR595, pR596 sequencing
pET11a-Rad10-Rad1 D869A gtc→tgc pR1112, pR1113 sequencing, AseI digestion
pET11a-Rad10-Rad1-K32R D869A gtc→tgc pR1112, pR1113 sequencing

48
Table 3.9: Mutagenic PCR thermocycler program.
92°C 2 min.
92°C 30 sec.
16 x 55°C 30 sec.
72°C 23 min.
72°C 10 min.
4°C ∞
To disrupt the original plasmid from the newly synthetized one containing the desired mutation
DpnI digestion was performed. DpnI enzyme specifically cleaves the methylated parental
plasmid but not the PCR product. 1 μl of DpnI was added to the PCR mixture and incubated at
37°C for 2 hours. The enzyme was inactivated at 80°C for 20 min.
4 μl of the mixture were transformed into E. coli DH5α by heat-shock. Cells were plated on LB
plates with the appropriate antibiotics and incubated O/N at 37°C. After purification of plasmid
DNA the presence of the mutation was verified by restriction analysis and/or sequencing.
3.2.1.4. Restriction analysis
To verify plasmid identity and mutation presence after site-directed mutagenesis DNA was
digested by restriction endonucleases. Restriction mixtures contained 1 x corresponding buffer,
1 x BSA if required, 100 ng of DNA and 0.2 U of restriction enzyme in a final volume 20 μl.
The restriction reaction was incubated under conditions suggested by the supplier usually at
37°C for 2 hours. DNA species were separated and visualized on the agarose gel.
3.2.1.5. DNA annealing
To generate an unlabelled Y-form substrate 1 μl of pR29 and pR39 oligonucleotides
(both 100 μM) were mixed in 98 μl of hybridization buffer and incubated 3 minutes at 78°C.
Then the mixture was slowly cooled to RT overnight. The resulting concentration of Y-form
substrate was 1 μM.

49
3.2.2. Cell techniques
3.2.2.1. Bacterial transformation by heat-shock
10 ng of plasmid DNA or 4 μl of PCR mixture was added to 50 μl of chemocompetent E. coli
cells and the mixture was incubated on ice for 30 min. The heat-shock was induced at 42°C for
1 minute in water bath. After incubation of the mixture on ice for 2 minutes, 500 μl of
prewarmed SOC media was added to the mixture for cell regeneration (only for transformation
of PCR mixtures). The culture was incubated at 37°C for 30 min at 250 rpm and then it was
plated on solid LB media containing the appropriate antibiotics and grown at 37°C overnight.
3.2.2.2. Preparation of chemocompetent bacterial cells
Hannah competent cell protocol
Competent cells from a frozen stock were plated on LB plate (with appropriate antibiotics if
necessary) and grown overnight at 37°C. A single colony was inoculated into 5 ml of SOB
medium (with antibiotics if necessary) and grown in a shaker overnight at 37°C. 0.5 ml of
overnight culture was diluted into fresh 50 ml of SOB media (with antibiotics if necessary) and
the cells were grown at 37°C till OD600 reached 0.5. Bacteria were pelleted by spinning in
centrifuge (3000 rpm, 4°C, 10 minutes). The pellet was gently resuspended in 13.4 ml of RF1
buffer. Cells were incubated on wet ice in cold room for 15 minutes and pelleted as previously
mentioned. Pellet was gently resuspended in 2 ml of RF2 buffer and incubate on wet ice in cold
room for 15 min. Cells were aliquoted (50 μl), frozen in liquid nitrogen and stored at -80°C.

50
3.2.3. Protein techniques
3.2.3.1. Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE)
Proteins were separated on SDS gels (7,5 % - 12 %) depending on their molecular weight. Gels
consist of two layers - separating resolving gel and stacking gel for sample loading. Gels were
run in vertical electrophoretic SDS-PAGE units in 1 x SDS buffer. Protein samples were
diluted 1:1 with 2 x SDS-Laemmli buffer, boiled (1 min) and loaded into the wells of the gel.
Protein weight markers were used to determine the molecular weight of analyzed protein
samples. The electrophoresis was run under constant voltage 215 V for 45 min. Gels were
stained with Coomassie Brilliant Blue dye for 20 minutes and then transferred into destain
solution for 20 minutes, under continuous shaking. Alternatively proteins were visualized by
silver staining, which is a more sensitive visualization method.
3.2.3.2. Silver staining
Silver staining is a very sensitive method for protein and DNA visualization. In our
case, it was mainly used for detection of SUMO-modified proteins. SDS gels were fixed in 50
% (v/v) acetone + 15 % (v/v) trichloroacetic acid (TCA) + 0.0055 % (v/v) formaldehyde
(HCHO) for 5 minutes, then rinsed with distilled water and washed with 50 % (v/v) acetone.
Gels were sensitized for 1 minute in 0.02 % Sodium Thiosulfate (Na2S2O3) and two times
rinsed in water. For the silver reaction gels were incubated 8 minutes in 0.1 % (w/v) Silver
Nitrate (AgNO3) + 0.08 % (v/v) formaldehyde, then 2 times rinsed for 5 seconds with H2O.
Developing reaction was done by incubation for 10-20 seconds in 2 % (w/v) sodium carbonate
(Na2CO3) with 0.0055 % (v/v) formaldehyde and 0.0015 % (w/v) Na2S2O3. Developing
reaction was stopped by washing with 1 % (v/v) acetic acid for 10 minutes. Finally, gels were
washed in H2O for 10 minutes.
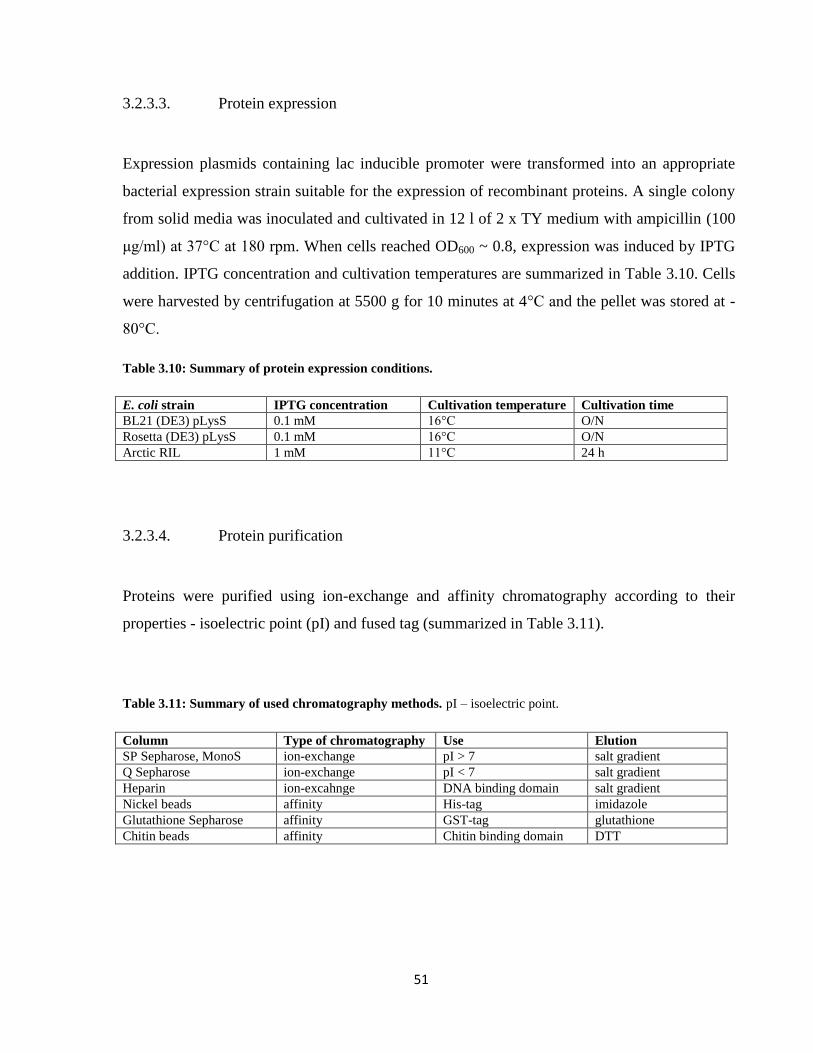
51
3.2.3.3. Protein expression
Expression plasmids containing lac inducible promoter were transformed into an appropriate
bacterial expression strain suitable for the expression of recombinant proteins. A single colony
from solid media was inoculated and cultivated in 12 l of 2 x TY medium with ampicillin (100
μg/ml) at 37°C at 180 rpm. When cells reached OD600 ~ 0.8, expression was induced by IPTG
addition. IPTG concentration and cultivation temperatures are summarized in Table 3.10. Cells
were harvested by centrifugation at 5500 g for 10 minutes at 4°C and the pellet was stored at -
80°C.
Table 3.10: Summary of protein expression conditions.
E. coli strain IPTG concentration Cultivation temperature Cultivation time
BL21 (DE3) pLysS 0.1 mM 16°C O/N
Rosetta (DE3) pLysS 0.1 mM 16°C O/N
Arctic RIL 1 mM 11°C 24 h
3.2.3.4. Protein purification
Proteins were purified using ion-exchange and affinity chromatography according to their
properties - isoelectric point (pI) and fused tag (summarized in Table 3.11).
Table 3.11: Summary of used chromatography methods. pI – isoelectric point.
Column Type of chromatography Use Elution
SP Sepharose, MonoS ion-exchange pI > 7 salt gradient
Q Sepharose ion-exchange pI < 7 salt gradient
Heparin ion-excahnge DNA binding domain salt gradient
Nickel beads affinity His-tag imidazole
Glutathione Sepharose affinity GST-tag glutathione
Chitin beads affinity Chitin binding domain DTT

52
Expression and purification of MUS81-EME1 and Mus81-Mms4
MUS81-EME16xHis was expressed from pET21d-MUS81-EME1, GST-tagged MUS81-
EME16xHis from pGEX-MUS81-EME1 and the yeast ortholog Mus81-Mms46xHis from pET14b-
Mus81-Mms4 expression plasmids. Overexpression of all constructs was induced in E. coli
Rosetta (DE3) pLysS bacterial strains by addition of 0.1 mM IPTG and incubation at 16°C
overnight. 20 g of harvested cells was resuspended in 80 ml of CBB buffer containing 200 mM
KCl, 0.01 % NP40, PI cocktail (aprotinine, benzamidine, pepstatine, chymostatin, leupeptin -
final concentration 5 μg/ml), 1 mM PMSF with 1 mM β-ME (His-tagged proteins) or 1 mM
DTT (GST-tagged proteins). For further lysis and DNA disruption the lysate was sonicated 3
times for 10 s at 80 % amplitude in ice bath (heilscher Ultrasound technology sonicator). The
lysate was clarified by ultracentrifugation at 100,000 g for 1 h at 4°C.
Purification of MUS81-EME16xHis and Mus81-Mms46xHis
Supernatant was sequentially loaded on a 20 ml SP-Sepharose column. Proteins were eluted
from the SP-Sepharose column with a 200-ml linear gradient of 150-800 mM KCl in buffer K
supplemented with 0.01 % NP40 and 1 mM β-ME. The peak fractions were pooled and mixed
with 1 ml of His-Select nickel affinity gel for 2 h at 4 °C. The beads were washed with 10 ml of
buffer K containing 150 mM KCl (NP40, β-ME) and 10 mM imidazole. The bound proteins
were eluted with buffer K containing 150 mM KCl (NP40, β-ME) and 150, 300, 500 and 1000
mM imidazole, respectively. Peak fractions were loaded onto a 1 ml heparin column. Proteins
were eluted with 10 ml of buffer K (NP40, β-ME) in a gradient of 150-500 mM KCl. The peak
fractions were concentrated on Vivaspin 2, aliquoted and stored at -80°C.
Purification of GSTMUS81-6xHisEME1
The supernatant was incubated overnight at 4°C with 1 ml of GTH-Sepharose preequilibrated
in buffer K containing 350 mM KCl (NP40, DTT). Bound proteins were eluted with 7 x 1 ml
of buffer K containing 350 mM KCl (NP40, DTT) and 150 mM glutathione. Peak fractions
were loaded on a 1 ml heparin column and the proteins were eluted with 10 ml buffer K (NP40,

53
DTT) using a 150 to 1000 mM KCl gradient. Peak fractions were concentrated on Vivaspin 2,
aliquoted and stored at -80°C.
Expression and purification of RECQ5
For the expression of RECQ5 we used the vector pTXB1-RECQ5. This produced RECQ5 in a
C-terminal fusion with an intein cleavable chitin binding domain (CBD) 223
. Plasmids were
transformed into E. coli Arctic RIL strain and expression was induced with 1 mM IPTG
followed by the incubation at 11°C for 24 hours. Cells were pelleted and stored at -80°C. Cell
pellet (10 g) was resuspended in 40 ml of CBB containing 200 mM KCl, 0.01 % NP40, PI
cocktail, PMSF and sonicated. The lysate was clarified by ultracentrifugation at 100,000 g for 1
hour at 4°C. The supernatant was incubated for 2 hours with 2 ml of chitin affinity beads (New
England BioLabs), preequilibrated in buffer T containing 500 mM KCl (NP40). Beads were
then washed with 40 ml of buffer T containing 500 mM KCl (NP40). Intein cleavage was
induced with a quick wash with 2 ml of buffer T containing 500 mM KCl and 100 mM DTT
and 1 ml was left for overnight incubation. Proteins released from chitin beads were eluted by 5
ml of buffer T containing 500 mM KCl (NP40) and 1 mM DTT. Peak fractions were pooled
and loaded on a 1 ml heparin column. Proteins were eluted with 10 ml of buffer T (NP40, DTT)
in a gradient of 150 – 1000 mM KCl. Peak fractions were pooled and loaded on a 0.5 ml
MonoS column and eluted with 5 ml of buffer T (NP40, DTT) in a gradient of 150 – 1000 mM
KCl. Fractions containing RECQ5 were pooled and concentrated on Vivaspin 2, aliquoted and
stored at -80°C.
Purified proteins and their properties are summarized in Table 3.12 and shown in Figure 3.2.

54
Table 3.12: Summary of used proteins MUS81, RECQ5 and RAD51. MW – molecular weight, pI – isoelectric
point.
Protein Tag MW protein [kDa] MW protein + tag [kDa] pI (+tag)
MUS81-EME16xHis 6xHis on EME1 61 + 63 61 + 64 9.78 + 6.7
GST-MUS81-EME16xHis GST, 6xHis 61 + 63 87 + 64 9.21 + 6.7
Mus81-Mms46xHis 6x His on Mms4 72 + 79 72 + 80 8.68 + 7.6
RECQ5 - 109 109 8.86
RECQ5-K58R - 109 109 8.86
RECQ5-F666A - 109 109 8.86
RECQ51-475 - 52.8 52.8 9.01
RECQ5410-991 - 63.2 63.2 8.66
hRAD51-K133R - 37 37 5.42
GST - 26 26 6.09
Figure 3.2: Purified proteins MUS81, RECQ5 and RAD51. Proteins were resolved on 12 % SDS-PAGE
followed by Coomassie staining.
Expression and purification of Rad1-Rad10 complex and its mutants
E. coli Rosetta(DE3) pLysS was transformed with expression plasmid pET11a-Rad10-
6xHisRad1 or its mutated versions (K32R, D869A and K32R-D869A). Expression was induced
with 0.1 mM IPTG and cells were incubated at 16°C overnight. Cell pellet (10 g) was
resuspended in 40 ml of CBB containing 200 mM KCl, 0.01 % NP40, 1 mM β-ME, PI cocktail
and 1 mM PMSF followed by sonication. The lysate was clarified by ultracentrifugation. The
supernatant was incubated overnight with 1 ml of HIS-Select nickel affinity gel. The beads
were preequilibrated in buffer K containing 150 mM KCl (NP40, β-ME). After incubation the
beads were washed with 20 ml of buffer K containing 150 mM KCl (NP40, β-ME) and 10 mM

55
imidazole, and the bound proteins were eluted using 150 mM, 300 mM, 500 mM and 1000 mM
imidazole in buffer K containing 150 mM KCl (NP40, β-ME). Eluted fractions containing
Rad1-Rad10 or its mutants were pooled and loaded on a 1 ml heparin column and eluted with
10 ml of buffer K in a 150 to 1000 mM KCl gradient. Fractions eluted from 320 to 570 mM
KCl were pooled and loaded on a 0.5 ml MonoQ column. The column was washed and bound
proteins eluted with 5 ml of buffer K in a 150 to 1000 mM KCl gradient. Peak fractions were
pooled, concentrated in Vivaspin 2, aliquoted and stored at -80°C.
Expression and purification of Saw1
The expression plasmid pGEX-4T-Saw1 97
was transformed into E. coli BL21 (DE3) pLysS
cells. Expression was induced with 0.1 mM IPTG and cells were incubated at 16°C overnight.
Cell pellet (10 g) was resuspended in 50 ml of CBB containing 200 mM KCl, 0.01 % NP40, 1
mM DTT, PI cocktail, 1 mM PMSF and sonicated. The lysate was clarified by
ultracentrifugation and the supernatant was loaded on a 20 ml SP-sepharose column. The
column was preequilibrated with buffer K containing 150 mM KCl (NP40, DTT). Bound
proteins were eluted in a 150 to 1000 mM KCl gradient. Fractions eluted from 255 to 570 mM
KCl were pooled and incubated with 1 ml of GTH-sepharose preequilibrated in buffer K
containing 150 mM KCl (NP40, DTT) overnight at 4°C. Bound proteins were eluted with 7 ml
of buffer K containing 150 mM KCl (NP40, DTT) and 150 mM glutathione as a competitor.
The elution fractions containing Saw1 were loaded on a 1 ml MonoS column followed by
washing, and bound proteins were eluted in a 150 to 1000 mM KCl gradient in buffer K. Peak
fractions were concentrated on Vivaspin 2, aliquoted and stored at -80°C.
Expression and purification of Siz1
E.coli BL21(DE3) were transformed with expression plasmid pET21b-Siz11-465 412
generating a
Siz1 truncation proficient as a E3 ligase. Expression was induced with 0.1 mM IPTG and cells
were incubated at 16°C overnight. Cell pellet (10 g) was resuspended in 40 ml of CBB
containing 200 mM KCl, 0.01 % NP40, 1 mM β-ME, PI cocktail and 1 mM PMSF and

56
sonicated. The lysate was clarified by ultracentrifugation. The supernatant was incubated
overnight with 1 ml of HIS-Select nickel affinity gel. The beads were preequilibrated in buffer
K containing 150 mM KCl (NP40, β-ME). After incubation the beads were washed with 20 ml
of buffer K containing 150 mM KCl (NP40, β-ME) and 10 mM imidazole and the bound
proteins were eluted using 150 mM, 300 mM, 500 mM and 1000 mM imidazole in buffer K
containing 150 mM KCl (NP40, β-ME). Eluted fractions containing Siz1 were pooled and
concentrated on Vivaspin 2, aliquoted and stored at -80°C.
Purified proteins and their properties are summarized in Table 3.13 and shown in Figure 3.3.
Table 3.13: Summary of used proteins Rad1-Rad10, Saw1, SUMO proteins. MW – molecular weight, pI –
isoelectric point.
Protein Tag MW protein [kDa] MW protein + tag [kDa] pI (+tag)
Rad1-Rad10 6xHis on Rad1 126 + 24 127 + 24 5.12, 9.18
Rad1-K32R-Rad10 6xHis on Rad1 126 + 24 127 + 24 5.12, 9.18
Rad1-D869A-Rad10 6xHis on Rad1 126 + 24 127 + 24 5.12, 9.18
Rad1-K32R, D869A-Rad10 6xHis on Rad1 126 + 24 127 + 24 5.12, 9.18
Saw1 GST 29.7 55.7 8.79
Aos1-Uba2 GST on Aos1 39 + 71 65 + 71 5.37 + 4.74
Ubc9 6xHis 17.9 18.9 9.65
Smt3-K11, 15, 19R 6xHis-Flag 11.6 13.6 4.66
Siz1 (1-465 aa) 6xHis 52.7 53.7 7
Siz2 6xHis 82 83 7.28
GST - 26 26 6
Figure 3.3: Purified Rad1-Rad10 variants and Saw1. Each purified protein (1 ug) was run on 10% SDS-PAGE
followed Coomassie staining. Lane 1 – HMW marker.

57
3.2.3.5. Immunoblotting (Western blot)
Proteins from SDS gels were blotted on a nitrocellulose membrane (AppliChem) in Trans Blot
Turbo Transfer System (Bio-Rad) according to the user’s manual at 1 A for 14 minutes using
transfer buffer. Transfer of proteins onto membrane was verified by short incubation with
Ponceau S solution. After rinsing Ponceau S with distilled water the membrane was blocked for
1 hour in blocking buffer. Primary antibodies were diluted in 5% non-fat milk in 1 x PBS and
added to the membrane for 2 h at RT (Table 3.14). The membrane was washed three times in
PBST and secondary antibodies with conjugated horseradish peroxidase were applied for 30
minutes at RT. Membrane was washed three times with PBST. Followed by the incubation for
1 min with LumiGLO (R) solutions (Cell Signaling Technology), the membrane was exposed
to Luminescent Image Analyzer LAS-4000 (Genetica) to visualize the signal.
Table 3.14: Summary of used antibodies for Western blots.
Antibodies Type Animal Dilution Source
α-Rad1 polyclonal IgG primary goat 1:1000 Santa Cruz
α-Smt3 primary rabbit 1:2000 Helle Ulrich lab
α-GST primary rabbit 1:2000 Sigma-Aldrich
α-rabbit IgG-HRP secondary goat 1:1000 Sigma-Aldrich
α-goat IgG-HRP secondary donkey 1:2000 Santa Cruz
3.2.4. Functional assays
3.2.4.1. Pull-down assay
Affinity beads (usually 30 μl) were preequilibrated with 10 volumes of appropriate buffer (K or
T buffer) containing 100 mM KCl, 0.01 % NP40 and 1 mM β-ME or DTT. β-ME is used for
nickel affinity beads and DTT for GTH-Sepharose. Reaction mixtures contained 5 μg of
purified proteins. The final salt concentration (100 mM KCl) includes salt concentrations of
purified proteins. The mixtures were incubated for 30 minutes at 4°C with gentle shaking.
Samples were centrifuged (6000 g, 30 seconds) and the supernatants were collected. The beads

58
were washed with 10 volumes of corresponding buffer, centrifuged and the supernatant was
collected as the wash fraction. Proteins bound to the beads were eluted by the addition of 25 μl
of SDS-Laemmli buffer. Protein interactions were analyzed by SDS-PAGE.
3.2.4.2. Nuclease assay
Nuclease assays determine the cleavage activity of purified enzymes on various DNA
substrates. Increasing concentrations of the tested protein were added to the reaction mixture
containing fluorescently labelled DNA substrate (6 nM), 1 x appropriate buffer (ME (for
MUS81-EME1), MM (for Mus81-Mms4), R (for Rad1-Rad10)), 100 mM KCl and 0.1 mg/ml
BSA in a final volume 10 μl. Reaction mixtures were incubated at 37°C (human proteins) or
30°C (yeast proteins) for usually 20 minutes. The nuclease reaction was stopped by protein
degradation by addition of 0.1 % (w/v) SDS and 500 µg/ml of Proteinase K followed by further
incubation for 10 minutes. After adding 2 μl of 6 x native gel loading buffer the reaction
mixtures were separated on 10 % native polyacrylamide gel in 1 x TBE buffer at a constant
voltage 110 V for 1 hour at 4°C. Fluorescent DNA species were visualized by Image Reader
FLA-9000 and quantified using MultiGauge V3.2 software (Fujifilm).
3.2.4.3. Electromobility shift assay (EMSA)
EMSA determines DNA binding properties of a tested protein. Increasing amounts of protein
was added to the reaction mixture containing fluorescently labelled DNA substrate (4 nM), 1 x
D buffer, 100 mM KCl and 0.1 mg/ml BSA in a final volume 10 μl. The reaction mixtures were
incubated at 30°C for 30 min. After adding 2 μl of 6 x native gel loading buffer the reaction
mixtures were separated on 10 % native polyacrylamide gel in 0.5 x TBE buffer at a constant
voltage 80 V for 90 minutes at 4°C. Fluorescent DNA species were visualized by Image Reader
FLA-9000 and quantified using MultiGauge V3.2 software (Fujifilm).

59
3.2.4.4. In vitro SUMOylation assay
The SUMOylation of a target protein can be performed in vitro by adding all the essential
proteins of the SUMOylation pathway as described in406
. Usually 20 μl reaction mixtures
contained 1 x S buffer, 150 mM KCl, 150 nM Aos1/Uba2, 0.5 μM Ubc9, 4.3 μM Smt3-KR, 10-
100 nM Siz11-465 (or Siz2), 2 μg of target protein and the reaction was started by the addition of
2.5 mM ATP. Reaction mixtures were incubated at 30°C for at least 30 minutes and SUMO
conjugation was stopped by protein denaturation by the addition of SDS-Laemmli buffer. The
reaction mixtures were analyzed by SDS-PAGE followed by the silver staining.
3.2.4.5. Gel filtration chromatography
Oligomeric status of proteins can be determined by gel filtration chromatography where the
protein complexes are eluted from the matrix according to their molecular weight (size). In our
study, a 23 ml Sephacryl S400 column (molecular weight range 20-8000 kDa) was used to
analyze the oligomerization of our protein of interest (Rad1-Rad10). 100 μg of protein was
filtered through the column at a flow rate 0.11 ml/min in buffer K containing 300 mM KCl and
0.35 ml fractions were collected. Indicated fractions were separated on SDS-PAGE and
subjected to immunoblotting.
3.2.4.6. RAD51 removal assay
In the RAD51 removal assay we first blocked the endonucleolytic cleavage of MUS81-EME1
(1 nM) on 3’-flap DNA substrate. Then a helicase was added to remove RAD51 bound to DNA
and renew nuclease cleavage. Reaction mixtures contained fluorescently labelled DNA
substrate (6 nM), 1 x ME buffer, 100 mM KCl, ATP regeneration system creatine
kinase/creatine phosphatase, ATP (2 mM) and 0.1 mg/ml BSA in a final volume 10 μl. First
RAD51 was preincubated with DNA for 10 minutes at 37°C, then RECQL5 was added and
incubated with RAD51 for 10 minutes followed by addition of MUS81-EME1 for additional 20

60
minutes. The nuclease reaction was stopped by protein degradation by addition of 0.1 % (w/v)
SDS and 500 µg/ml of Proteinase K followed by further incubation for 10 minutes. After
adding 2 μl of 6 x native gel loading buffer the reaction mixtures were separated on 10 %
native polyacrylamide gel in 1 x TBE buffer at a constant voltage 110 V for 1 hour at 4°C.
Fluorescent DNA species were visualized by Image Reader FLA-9000 and quantified using
MultiGauge V3.2 software (Fujifilm).

61
4. RESULTS
According to our aims the first part of this thesis is focused on the functional interaction
between human MUS81-EME1 endonuclease and RECQL5β helicase. The second part
describes the characterization of consequences of SUMOylation on the function of budding
yeast Rad1-Rad10 nuclease.
4.1. MUS81-EME1 nuclease and RECQ5 helicase cooperatively promote
stability of CFSs
Common fragile sites (CFSs) are genomic regions where the progression of replication forks is
compromised under conditions of replication stress, leading to the appearance of gaps or breaks
in metaphase chromosomes413
. MUS81-EME1 endonuclease was shown to resolve aberrant
structures at CFSs in order to promote correct sister chromatid disjunction during
anaphase141,142
. Here we describe a novel functional interaction between human MUS81-EME1
endonuclease and RECQL5β (used abbreviation RECQ5) helicase in the resolution of late
replication intermediates in early mitosis.
My part in this project was the biochemical analysis of the MUS81-EME1 and RECQ5
interaction. In vivo experiments were carried out by Stefano Di Marco, Naga Raja Chappidi,
Radhakrishnan Kanagaraj from Pavel Janščák’s group at the Institute of Molecular Cancer
Research (Zurich, Switzerland).
In this section are shown only my own results and data from our collaborators are attached in
Supplement in attached publications. MUS81-RECQ5 data are not yet published.

62
4.1.1. RECQ5 physically interacts with MUS81-EME1 in vitro
Repair enzymes present a robust cooperation network to ensure genome stability. Investigation
of these interactions sheds light on the complex process of DNA repair and helps us understand
the consequences of its abnormalities. There are indications that MUS81 has a functional
relationship with RecQ helicases (BLM, Rqh1), therefore it made sense to investigate another
protein belonging to this helicase family - RECQ5150,153,321,322
. Both MUS81 and RECQ5
function in promoting correct sister chromatid disjunction, indicating that both proteins could
act together141,142,358
.
Figure 4.1: MUS81-EME1 interacts with RECQ5. RECQ5 was incubated with GST-Mus81-EME1 or GST on
GTH-Sepharose for 30 min. Fraction of unbound (F – flow) and bound (B – beads) proteins were resolved on 10
% SDS-PAGE followed by silver staining.
To test this hypothesis, we first tested their direct physical interaction. We used recombinant
purified GST-MUS81-EME1 and untagged RECQ5 in a pull-down assay. Proteins were mixed
together with glutathione-Sepharose (GTH-Sepharose) and GST mixed with RECQ5 was used
as a negative control to rule out unspecific binding. Reaction mixtures consisted of 5 μg of
purified proteins in 1 x T buffer (NP40, DTT) with a final salt concentration 100 mM KCl.
Proteins and affinity beads were incubated for 30 minutes at 4°C under constant shaking. Flow
fraction was stored separately (with 1 x SDS-Laemmli buffer) and beads were washed 2 times
with 1 x T buffer containing 100 mM KCl (NP40, DTT). Proteins bound to beads were eluted
with 1 x SDS-Laemmli buffer. Flow and bead fractions were analyzed on 12 % SDS gel. As

63
shown in lane 7 (Figure 4.1), RECQ5 specifically interacts with GST-MUS81-EME1 complex,
as no binding was observed with GST alone (lanes 4-5, Figure 4.1). This interaction is
mediated through the MUS81 subunit, confirmed by pull-down with only GST-MUS81 (data
Hana Sedláčková (Krejčí group)).
4.1.2. RECQ5 stimulates MUS81-EME1 endonuclease activity in vitro
The physical interaction between MUS81 and RECQ5 prompted us to test whether RECQ5 is
able to influence the endonuclease activity of the MUS81 complex. We used DNA substrates
specific for MUS81 such as 3´-flap, nicked Holliday junction (nHJ) and fork and as a negative
control Y-form, intact Holliday junction (HJ) and 5’-flap61,102,106,117
. These substrates mimic
various kinds of replication or recombination intermediates. As shown in Figure 4.2, RECQ5
dramatically stimulated the MUS81-EME1 nuclease activity on 3’-flap, fork and nHJ in a
concentration-dependent manner. This stimulation is not due to unspecific nuclease activity of
RECQ5 protein, as its addition alone has no effect on any of the DNA substrates tested (Figure
4.2 (A), lane 6). Since Y-form, HJ and 5’-flap structures are not cleaved by MUS81, no further
cleavage was observed after RECQ5 addition, indicating that RECQ5 does not modify MUS81
substrate specificity. Preincubation of RECQ5 with DNA did not lead to increased MUS81-
mediated DNA cleavage (data not shown), suggesting that the stimulation effect occurs by a
mechanism other than tethering of MUS81-EME1 to DNA.
To further corroborate on the stimulation of MUS81-EME1 activity by RECQ5 we measured
the rates of MUS81-EME1 cleavage of DNA in the presence and absence of RECQ5. To this
end, 3’-flap DNA substrate was incubated with MUS81-EME1 (0.2 nM) in the presence or
absence of RECQ5 (5 nM) for indicated time-points (5, 10, 15 and 20 minutes). While the
cleavage of the MUS81 complex alone reached maximum of 8 % after 20 minutes, in the
presence of RECQ5 we observed significant increase of DNA cleavage to about 44 % (Figure
4.3 C, D).
The enhanced nucleolytic cleavage is not a result of RECQ5 ATPase activity since these assays
were performed without ATP. Addition of ATP to the nuclease assay did not lead to further
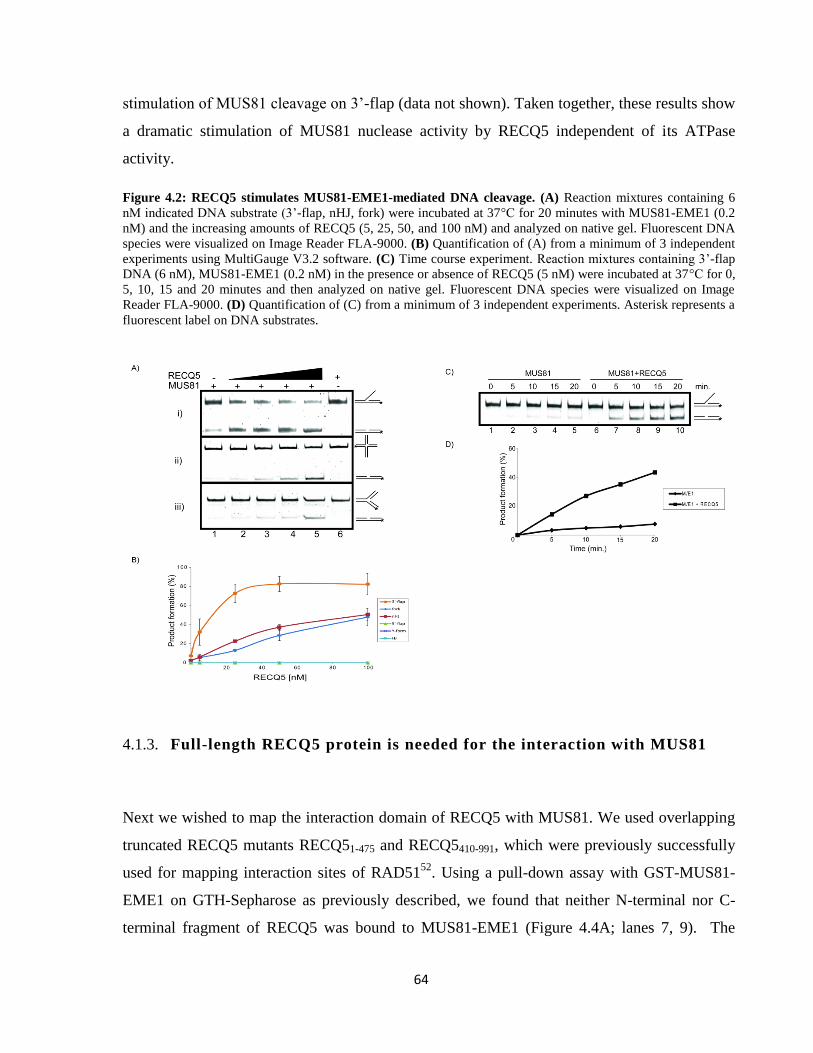
64
stimulation of MUS81 cleavage on 3’-flap (data not shown). Taken together, these results show
a dramatic stimulation of MUS81 nuclease activity by RECQ5 independent of its ATPase
activity.
Figure 4.2: RECQ5 stimulates MUS81-EME1-mediated DNA cleavage. (A) Reaction mixtures containing 6
nM indicated DNA substrate (3’-flap, nHJ, fork) were incubated at 37°C for 20 minutes with MUS81-EME1 (0.2
nM) and the increasing amounts of RECQ5 (5, 25, 50, and 100 nM) and analyzed on native gel. Fluorescent DNA
species were visualized on Image Reader FLA-9000. (B) Quantification of (A) from a minimum of 3 independent
experiments using MultiGauge V3.2 software. (C) Time course experiment. Reaction mixtures containing 3’-flap
DNA (6 nM), MUS81-EME1 (0.2 nM) in the presence or absence of RECQ5 (5 nM) were incubated at 37°C for 0,
5, 10, 15 and 20 minutes and then analyzed on native gel. Fluorescent DNA species were visualized on Image
Reader FLA-9000. (D) Quantification of (C) from a minimum of 3 independent experiments. Asterisk represents a
fluorescent label on DNA substrates.
4.1.3. Full-length RECQ5 protein is needed for the interaction with MUS81
Next we wished to map the interaction domain of RECQ5 with MUS81. We used overlapping
truncated RECQ5 mutants RECQ51-475 and RECQ5410-991, which were previously successfully
used for mapping interaction sites of RAD5152
. Using a pull-down assay with GST-MUS81-
EME1 on GTH-Sepharose as previously described, we found that neither N-terminal nor C-
terminal fragment of RECQ5 was bound to MUS81-EME1 (Figure 4.4A; lanes 7, 9). The

65
overlapping region 410-475 aa excludes the possibility that the truncations are directly in the
interaction site.
Figure 4.3: Truncated RECQ5 does not bind or stimulate MUS81-EME1. (A) Pull-down assay: proteins
mixed on GTH-Sepharose, incubated for 30 min, resolved on 7.5 % SDS gel and silver-stained. F – flow, B –
beads. Lane 1 – HMW (molecular weight marker), lanes 2-3 GST+RECQ51-475, lanes 4-5 GST+RECQ5410-991,
lanes 6-7 MUS81-EME1+RECQ51-475, lanes 8-9 MUS81-EME1+RECQ5410-991. (B) Reaction mixtures containing
3’-flap DNA (6 nM) were incubated at 37°C for 20 minutes with MUS81-EME1 (0.2 nM) and the increasing
concentration of indicated RECQ5 fragments (5, 25, 50, and 100 nM) and analyzed on 10 % native gel in 1 x TBE.
The fluorescent species were visualized on Image Reader FLA-9000. (C) Quantification of (B) from a minimum
of three independent experiments using MultiGauge V3.2 software.
Next, we analyzed if the N- or C-terminal fragments are sufficient to stimulate the nuclease
activity of RECQ5. As shown in Figure 4.3.B neither of these fragments was able to stimulate
MUS81 cleavage activity. We also tested other RECQ5 truncations (1-410, 1-725, 1-651, 675-
991 aa), however no interaction nor stimulation of MUS81 was observed (data not shown).
4.1.4. RECQ5 stimulates the nuclease activity of yeast Mus81-Mms4
To investigate if RECQ5 stimulates specifically human MUS81 complex we tested its effect on
the budding yeast homologue using the in vitro nuclease assay as described previously.
Similarly to human MUS81-EME1, increasing concentrations of RECQ5 stimulated Mus81-

66
Mms4-mediated cleavage of 3’-flap DNA to an even greater extent (90 %) as observed for
MUS81-EME1 (80 %), indicating that the interaction between RECQ5 and MUS81 is
evolutionary conserved (Figure 4.4).
Figure 4.4: RECQ5 stimulates Mus81-Mms4-mediated DNA cleavage. (A) Reaction mixtures containing 3’-
flap DNA (6 nM) were incubated at 37°C for 20 minutes with Mus81-Mms4 (0.2 nM) and the increasing
concentrations of RECQ5 (5, 25, 50, 100 nM) and analyzed on 10 % native gel in 1 x TBE. Fluorescent species
were visualized on Image Reader FLA-9000. (B) Quantification of (A) from a minimum of three independent
experiments using MultiGauge V3.2 software.
4.1.5. RECQ5 can dissociate RAD51 from DNA to enable MUS81 cleavage
Results from Janscak group suggest that both MUS81 and RECQ5 accumulate at CFSs
(FRA3B, FRA16D and FRA7H) and are required for DSB formation at the loci after
replication stress induced by aphidicolin. As shown in yeast, late replication intermediates
contain ssDNA, which may be covered by Rad51 and promote illegitimate recombination
resulting in the formation of anaphase bridges414
. Correspondingly, depletion of RECQ5 in
humans led to RAD51 accumulation at CFSs and elevated levels of anaphase bridges (Janscak
group). Also the presence of micronuclei and 53BP1 bodies was detected, indicating a
chromosome segregation defect observed in MUS81-depleted cells141,142
.
The above described data prompted us to test whether RAD51 can physically block the
overhang cleavage by MUS81-EME1. We titrated MUS81-EME1 to cleave about 90 % of 3’-
flap. Then we preincubated DNA with increasing amounts (100 – 800 nM) of RAD51 in almost

67
blocked cleavage (500 nM) (data not shown). ATPase dead RAD51 mutant, which can form a
stable DNA-protein complex and not dissociate from DNA was used415
.
Since RECQ5 acts as an antirecombinase and disrupts RAD51 filaments in an ATP-dependent
manner through a direct interaction with RAD5152,71,350
, we tested the effect of RECQ5 on
MUS81 cleavage in the presence of RAD51. First, we pre-incubated RAD51-K133R (500 nM)
with DNA to block its cleavage by MUS81-EME1 (to about 15 %) (Fig 4.5A, lanes 2,5). Next,
the increasing amounts of RECQ5 were added to the reactions followed by the addition of
MUS81-EME1. As shown in Figure 4.5, wild-type RECQ5 was able to restore MUS81-
dependent 3’-flap cleavage to about 30 % in a concentration dependent manner suggesting that
RECQ5 is capable of removing RAD51 from ssDNA thus allowing MUS81-EME1 cleavage.
To determine if the ATPase activity of RECQ5 or its interaction with RAD51 are essential for
this activity, we also tested the effect of RECQ5-K58R and RECQ5-F666A, respectively52
.
Indeed, RECQ5-F666A and were not able to restore 3’-flap cleavage by MUS81-EME1. In the
case of RECQ5-K58R, we can even see a further inhibition of MUS81 activity, probably due to
the fact, that RECQ5-K58R binds RAD51 but is unable to remove it, hence resulting in further
blockage of MUS81 access. A weak third band (Figure 4.5, lanes 4, 6-10) in the RECQ5 wild-
type reaction corresponds to a DNA product generated by its helicase activity in the presence of
ATP, confirmed by addition of RPA to the assay, which stimulates RECQ5 helicase activity
(data not shown)352
.
Collectively, RECQ5 is able to suppress the RAD51-mediated inhibition of cleavage of 3’-flap
DNA by MUS81-EME1 in a concentration dependent manner.

68
Figure 4.5: RECQ5 rescues the 3’-flap cleavage mediated by MUS81-EME1 after RAD51 inhibition. (A) RAD51-K133R (500 nM) was preincubated with 3’-flap DNA (6 nM) and 2 mM ATP, followed by addition of
indicated concentrations of RECQ5 (or F666A, K58R mutants) (10 minutes at 37°C). Finally MUS81-EME1 (8
nM) was added, reaction mixture incubated at 37°C for 20 minutes and analyzed on 10 % native gel in 1 x TBE
and fluorescent species were visualized on Image Reader FLA-9000. (B) Quantification of (A) from a minimum of
three independent experiments.
4.1.6. Discussion
In this study we show a novel functional relationship between MUS81-EME1 and RECQ5.
These proteins physically interact both in vitro and in vivo (Co-IP, Janscak group). Moreover,
Co-IP data have shown that the interaction is independent of DNA damage and DNA binding.
This is similar to co-IP of MUS81-BLM showing DNA-damage independence, but
colocalization experiments suggested a physical interaction after replication stress induced by
HU153
. Similarly, ChIP analysis place both MUS81 and RECQ5 at CFSs after aphidicolin
treatment, proposing a cooperative function in CFS expression (Janscak group). This together
suggests that despite a physical interaction MUS81-BLM, MUS81-RECQ5, in vivo MUS81
function with these helicases is a response to DNA damage.
Biochemically we show that RECQ5 stimulates MUS81-mediated DNA cleavage on
replication and recombination intermediates, but not through targeting of MUS81. This is in
A) B)

69
contrast with yeast, where DNA targeting of Mus81-Mms4 was observed with Srs2 and Rad54
helicases149,151
. The stimulation effect is independent of RECQ5 helicase activity, in agreement
with the stimulation mechanism of other helicases/translocases on Mus81 like Rqh1 and
Rad54128,150,151
, but in contrast to BLM, which utilizes its ATPase activity to affect
MUS81153,321,322
.
N-terminal and C-terminal RECQ5 fragments, with overlapping regions, were not able to
interact or stimulate MUS81. Thus, we conclude that the entire RECQ5 protein is needed for
the interaction with MUS81, suggesting a dispersed interaction domain or a domain depending
on the 3D structure. This is not an unique case where full-length protein is needed for
functional interaction, similarly only full-length FANCA stimulates MUS81 during ICL
repair138
.
RECQ5 was able to stimulate yeast Mus81-Mms4 suggesting an evolutionary conserved
mechanism. On the contrary, yeast Srs2 did not stimulate human MUS81-EME1 endonuclease
activity149
, suggesting unique properties of RECQ5 compared to Srs2.
Under-replicated DNA regions caused by partial inhibition of DNA polymerases contain
ssDNA. These stretches of DNA can occur in front of the replication fork and during Okazaki
fragment maturation. Normally, ssDNA is coated with RPA leading to ATR-dependent
activation of G2-M checkpoint causing cell-cycle arrest and replication restart. Obviously, cells
with unreplicated DNA at CFSs can escape cell-cycle checkpoint control and continue into
mitosis413,416,417
.
It is possible that ATR activation leads to the replacement of RPA with RAD51 recombinase at
ssDNA through BRCA1-BRCA2-PALB2418–420
. It is not clear, if this can lead to G2-M
checkpoint escape, but indeed, increased levels of RAD51 were implicated in chromosome
missegregation and aneuploidy421
. Accordingly, RAD51 accumulates at CFSs in the absence of
fully functional RECQ5 and prevents CFSs expression leading to the formation of anaphase
bridges (Janscak group). In avian DT40 and yeast cells TopBP1/Dpb11 binds DNA at anaphase
bridges to stimulate Mec1/ATR checkpoint and prevent HR by RAD51414
. It is very likely that
lack of CFS expression, accompanied with increased number of UFBs associated with RAD51
accumulation at CFSs, is caused by inhibition of MUS81 cleavage. In vitro we were able to

70
reconstitute such a situation where RAD51 preincubation with flap DNA inhibited MUS81
nuclease activity. RECQ5 via its antirecombinase activity was capable to disrupt RAD51
filament and stimulate MUS81 cleavage. Both ATPase/translocase activity and RAD51 binding
of RECQ552,350
were required for RAD51 removal in nuclease assay. In agreement RECQ5-KR
and RECQ5-FA mutants were defective in CFS expression and accumulated RAD51 at late
replication intermediates after aphidicolin treatment in vivo (Janscak group).
RECQ5 may have a broader role in ensuring proper chromosome segregation. It is known to
stimulate TOPOIIα, which dissolves catenates impeding proper sister chromatid
disjunction358,359,414
. On the other hand, it cannot dissolve HJs. This is the role of BLM in
complex with TOPOIIIα-RMI1/RMI2, which serves as a backup mechanism in anaphase to
free entangled DNA143,144
. Surprisingly, WRN, which is synthetically lethal with RECQ5, was
shown to prevent MUS81-mediated cleavage at CFSs and prevent ssDNA gap accumulation by
stimulating polymerase δ296,362,422
. It would be interesting to see if RECQ5 protects CFSs also
by aiding replication by dissociating RNA polymerase II355,357
.
Together, we provide insights into a mechanism for ensuring chromosome stability at CFSs.
First, we show that RECQ5 and MUS81 function together in the same pathway to generate a
DSB at under-replicated regions at CFSs. The role RECQ5 is probably to disrupt bound
RAD51 and allow MUS81 to access DNA for cleavage. It is yet to be addressed, if this is the
final function of RECQ5 in promoting proper chromosome segregation. After DSB formation
by MUS81 these ends may be again protected by RECQ5 through inhibition of MRN-mediated
DNA end resection avoiding HR. For a cell, a DSB during prometaphase is a more suitable
damage than mitotic catastrophe due to entangled DNA.

71
Figure 4.6: Hypothetical model for the roles of RECQ5 and MUS81 in processing of persistent replication
intermediates in early mitosis. (A) Schematic view of involved proteins. (B) ssDNA generated at CFSs due to
stalling of DNA polymerases on both leading and lagging strand, while DNA helicase unwinds parental DNA
strands, is coated with RPA, which is a signal for ATR-mediated G2-M checkpoint activation. Due to the
involvement of BRCA1 it is possible that RAD51 is loaded on ssDNA through BRCA2-PALB2. How such a
structure escapes the G2-M checkpoint is questionable. (C) Phosphorylated RECQ5 (by CDK1) utilizes its
antirecombinase activity to remove RAD51 to facilitate MUS81-EME1 (phosphorylated by CDK1) access to
DNA. Green rectangles depict explicit data from this study. (D) Through protein-protein interactions RECQ5
enables MUS81 DNA cleavage generating a DSB. Green rectangles depict explicit data from this study. (E) The
DSB ends might be protected from MRN exonucleolytic cleavage by RECQ5 to prevent HR and RF restoration.
(F) Untangled, but broken, chromatids ready for proper disjunction during anaphase.

72
4.2. Characterization of SUMOylation of Rad1-Rad10 complex and its
interacting partner Saw1
Recent studies have shown that a number of DNA repair enzymes are SUMOylated in response
to DNA damage392,423–425
. Among these enzymes are present also structure-specific
endonucleases like XPF-ERCC1. We focused our effort on establishing the effect of
SUMOylation on properties of its yeast homolog Rad1-Rad10 complex. My part on this project
was the in vitro biochemical characterization of Rad1-Rad10 SUMOylation, while in vivo
implications were investigated by Prabha Sarangi from Xiaolan Zhao’s lab (Memorial Sloan-
Kettering Cancer Center, New York). During our work on Rad1 SUMOylation project, we also
discovered that the Rad1-interacting protein Saw1, which is crucial for Rad1 recruitment to
branched DNA during single-strand annealing, can be modified by SUMO97
. This prompted us
to further investigate the effect of SUMOylation on Saw1 function and its role in DNA repair.
Our data were published in two papers Sarangi et al., Nucleic Acids Research, 2013 and
Sarangi et al., Cell Reports, 2013 viewed in Supplement.
4.2.1. Characterization of Rad1 SUMOylation in vitro
Rad1 was found as a SUMOylation target in response to DNA damage in proteomic and
biochemical screens392,424,425
. Some basal level of Rad1 SUMOylation is present under normal
growth conditions, but this is rapidly enhanced upon treatment with DNA damaging agents
such as UV, MMS, or CPT (Zhao group). This is in agreement with a role of Rad1-Rad10-
dependent pathways (NER or SSA) in repair of lesions generated by UV and MMS171,182–186
.
To further corroborate that Rad1 SUMOylation is DNA damage dependent and possibly Rad1
needs to be recruited to damaged sites, other NER and SSA upstream factors were assessed. It
has been previously reported that Rad14 and Saw1 target Rad1-Rad10 to sites of damage in
NER and SSA respectively97,172,173
, hence Δrad14 and Δsaw1 mutants exhibit no Rad1
SUMOylation upon genotoxic stress. Similarly, also Δrad4, Δrad52, Δslx4 had abolished Rad1

73
SUMOylation. Taken together this suggests that Rad1 is SUMOylated after its recruitment to
damaged DNA (Zhao group).
4.2.1.1. E3 ligase dependence
To determine the effect of SUMOylation on Rad1 activity we performed in vitro SUMOylation
assay with purified Rad1-Rad10 protein complex. The SUMOylation assay was carried out
with 0.4 μM recombinant Rad1-Rad10 and mixed with other SUMO machinery proteins –
Aos1/Uba2, Ubc9, Smt3 (see Material and Methods) including the increasing amount (10 nM
and 100 nM) of various E3 ligases (Siz1, Siz2, Mms21). The reaction was started by addition
of 2.5 mM ATP. Control samples did not contain any ATP. Mixtures were incubated at 30°C
for 14 minutes. SUMOylation of Rad1 was analyzed by SDS-PAGE and western blot using
both α-Rad1 and α-Smt3 antibodies. As shown in Figure 4.7, we observed a higher migrating
band corresponding to mono-SUMOylated Rad1 protein only in the reactions containing Siz1
or Siz2. We do not exclude the requirement of Mms21 for Rad1 SUMOylation, because we
were unable to verify the ligase activity of the purified protein (data not shown). In vivo data
confirmed that Rad1 is SUMOylated in Siz-dependent manner but they also showed a minor
effect of Mms21, which is in accordance with our data (Zhao group).

74
Figure 4.7: SUMOylation of Rad1 is dependent on E3 ligases. (A) SUMOylation assay. Reaction mixtures
containing 0.4 μM Rad1-Rad10 without E3 ligases (lane 2, 3), with 10 nM and 100 nM Siz1 (lane 4-6), Siz2 (lane
7-9), Mms21 (lane 10-12) in the presence or absence of ATP as indicated were incubated at 30°C for 14 minutes
and the reaction mixtures were resolved on 8 % SDS-PAGE and silver-stained. The control reaction containing
only SUMO machinery proteins and ATP are shown in lanes 13-15. Position of individual proteins is indicated
according to lane 1 – HMW marker. (B) Western blot. SDS gel from (A) was transferred on nitrocellulose
membrane and treated with α-Rad1 and α-Smt3 antibodies. Horseradish-peroxidase signal was visualized using
ChemiDoc system.
4.2.1.2. The effect of DNA on Rad1 SUMOylation
As previously mentioned Rad14 and Saw1 proteins, recruit Rad1-Rad10 to damaged
DNA, and their deletion abolishes Rad1 SUMOylation (Zhao group). This suggests that Rad1
is SUMOylated upon binding to DNA. To test this hypothesis we performed in vitro

75
SUMOylation assay in the presence of various DNA substrates such as 49-mer single-stranded
DNA, plasmid dsDNA and Y-form structure, which best mimic Rad1-Rad10 in vivo substrates.
We used a molar ratio 1:20 (DNA:protein) where all DNA should be bound by Rad1-Rad10
according to EMSA (data not shown) and also a 2-fold excess of DNA (ratio 1:10). Rad1
SUMOylation was titrated to a suboptimal level (with 10 nM Siz1) to better visualize the
possible enhancement of SUMOylation. As shown in Figure 4.8., the presence of various DNA
substrates did not have any effect on the level of Rad1 SUMOylation suggesting that DNA
binding per se does not trigger SUMO conjugation to Rad1.
Figure 4.8: SUMOylation of Rad1 is not dependent on DNA binding. SUMOylation reaction mixtures
containing 0.4 μM Rad1-Rad10 and 10 nM Siz1 without (lane 2) and with 2.5 mM ATP (lane 3). Indicated DNA
substrates were added to the SUMOylation reaction with ATP (lane 4-9). Lane 10 represents the control reaction
without Rad1-Rad10. All mixtures were incubated at 30°C for 14 minutes and then proteins were separated on 8 %
SDS-PAGE and silver stained. Lane 1 – HMW marker.
4.2.1.3. The effect of Rad1-Rad10 interaction partners on its SUMOylation
During DNA repair Rad1-Rad10 complex interacts with several proteins, which can
affect its function and potentially also its SUMOylation level. In collaboration with Zhao group
we observed that Rad1 SUMOylation is dependent on Saw1, Rad14, Rad52, Rad4 and Slx4.
Therefore we hypothesized that among others Saw1 could trigger Rad1 SUMOylation. To test
this, Rad1 SUMOylation was titrated to a suboptimal level, and increasing concentrations of
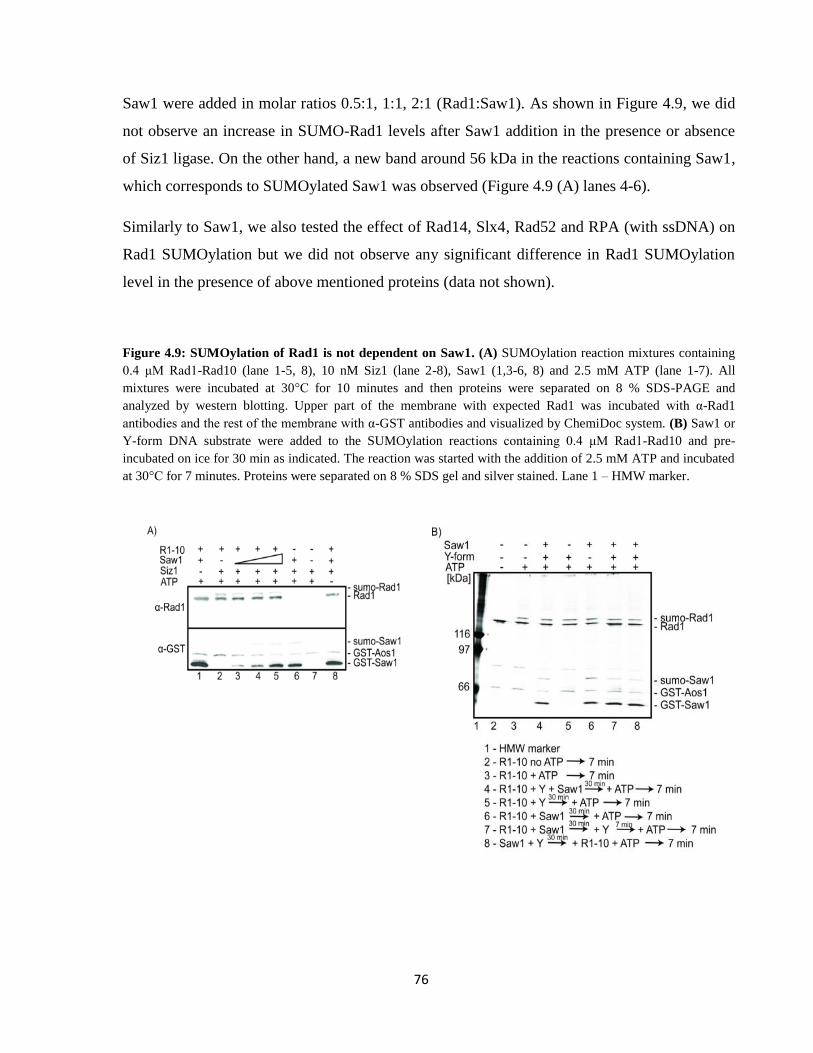
76
Saw1 were added in molar ratios 0.5:1, 1:1, 2:1 (Rad1:Saw1). As shown in Figure 4.9, we did
not observe an increase in SUMO-Rad1 levels after Saw1 addition in the presence or absence
of Siz1 ligase. On the other hand, a new band around 56 kDa in the reactions containing Saw1,
which corresponds to SUMOylated Saw1 was observed (Figure 4.9 (A) lanes 4-6).
Similarly to Saw1, we also tested the effect of Rad14, Slx4, Rad52 and RPA (with ssDNA) on
Rad1 SUMOylation but we did not observe any significant difference in Rad1 SUMOylation
level in the presence of above mentioned proteins (data not shown).
Figure 4.9: SUMOylation of Rad1 is not dependent on Saw1. (A) SUMOylation reaction mixtures containing
0.4 μM Rad1-Rad10 (lane 1-5, 8), 10 nM Siz1 (lane 2-8), Saw1 (1,3-6, 8) and 2.5 mM ATP (lane 1-7). All
mixtures were incubated at 30°C for 10 minutes and then proteins were separated on 8 % SDS-PAGE and
analyzed by western blotting. Upper part of the membrane with expected Rad1 was incubated with α-Rad1
antibodies and the rest of the membrane with α-GST antibodies and visualized by ChemiDoc system. (B) Saw1 or
Y-form DNA substrate were added to the SUMOylation reactions containing 0.4 μM Rad1-Rad10 and pre-
incubated on ice for 30 min as indicated. The reaction was started with the addition of 2.5 mM ATP and incubated
at 30°C for 7 minutes. Proteins were separated on 8 % SDS gel and silver stained. Lane 1 – HMW marker.
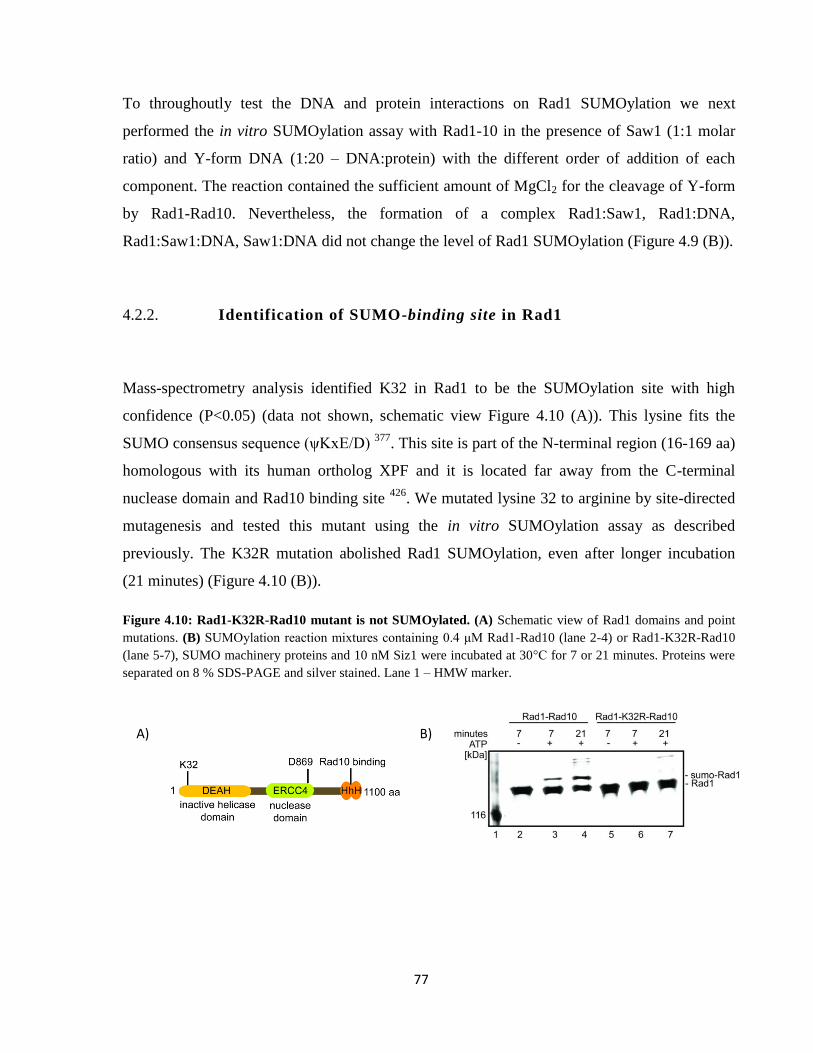
77
To throughoutly test the DNA and protein interactions on Rad1 SUMOylation we next
performed the in vitro SUMOylation assay with Rad1-10 in the presence of Saw1 (1:1 molar
ratio) and Y-form DNA (1:20 – DNA:protein) with the different order of addition of each
component. The reaction contained the sufficient amount of MgCl2 for the cleavage of Y-form
by Rad1-Rad10. Nevertheless, the formation of a complex Rad1:Saw1, Rad1:DNA,
Rad1:Saw1:DNA, Saw1:DNA did not change the level of Rad1 SUMOylation (Figure 4.9 (B)).
4.2.2. Identification of SUMO-binding site in Rad1
Mass-spectrometry analysis identified K32 in Rad1 to be the SUMOylation site with high
confidence (P<0.05) (data not shown, schematic view Figure 4.10 (A)). This lysine fits the
SUMO consensus sequence (ψKxE/D) 377
. This site is part of the N-terminal region (16-169 aa)
homologous with its human ortholog XPF and it is located far away from the C-terminal
nuclease domain and Rad10 binding site 426
. We mutated lysine 32 to arginine by site-directed
mutagenesis and tested this mutant using the in vitro SUMOylation assay as described
previously. The K32R mutation abolished Rad1 SUMOylation, even after longer incubation
(21 minutes) (Figure 4.10 (B)).
Figure 4.10: Rad1-K32R-Rad10 mutant is not SUMOylated. (A) Schematic view of Rad1 domains and point
mutations. (B) SUMOylation reaction mixtures containing 0.4 μM Rad1-Rad10 (lane 2-4) or Rad1-K32R-Rad10
(lane 5-7), SUMO machinery proteins and 10 nM Siz1 were incubated at 30°C for 7 or 21 minutes. Proteins were
separated on 8 % SDS-PAGE and silver stained. Lane 1 – HMW marker.
A) B)

78
This mutation was also introduced in to the Rad1 locus in yeast genome and under genotoxic
stress (UV, MMS) no Rad1 SUMOylation was observed (Zhao group), which was consistent
with our in vitro data.
4.2.2.1. Characterization of Rad1-K32R-Rad10 enzymatic activity
To further characterize the molecular function of Rad1 SUMOylation, we analyzed
Rad1-K32R properties in several ways. In vivo the K32R mutation does not alter Rad1 binding
with Rad10 as well as the protein level (Zhao group) so likely DNA involving activities should
stay unaffected. To test this hypothesis we compared endonuclease activity of Rad1-Rad10
with Rad1-K32R-Rad10 on a Rad1 specific DNA substrate. Increasing concentrations (10-80
nM) of wild-type and mutant Rad1-Rad10 were mixed with 6 nM FITC labeled Y-form and
incubated at 30°C for 30 minutes. As shown in Figure 4.11 (A, B), there was no difference in
the cleavage efficiency between wild-type and SUMO-deficient mutant of Rad1.
Next, we tested DNA binding properties of Rad1-K32R-Rad10 compared to wild-type
protein on ssDNA and dsDNA using EMSA. Increasing concentrations (10 - 80 nM) of wild-
type or SUMO-deficient mutant of Rad1-Rad10 were mixed with 4 nM FITC labeled ssDNA
(49-mer) or dsDNA (49 bp) and incubated at 30°C for 30 minutes. Similarly to the results from
nuclease assay, DNA binding activity of Rad1-K32R/Rad10 mutant was comparable to wild-
type (Figure 4.11 (C, D)), indicating that K32R mutation to Rad1 does not alter its DNA
binding properties.
Finally, we also tested if SUMOylation deficiency alters the interaction of Rad1 with
other proteins. While in vivo data confirmed that it does not affect the interaction with Rad10
(Zhao group), using in vitro pull-down assay we analyzed the potential effect on Saw1 binding.
GST-tagged Saw1 (or GST alone as a negative control) were mixed with Rad1 or Rad1-K32R
and bound to GTH-Sepharose. Mixtures were incubated on ice for 30 minutes and then
separated on SDS-PAGE. As shown in Figure 4.11(E), Rad1-K32R-Rad10 interacts with Saw1
with the same efficiency as wild-type.
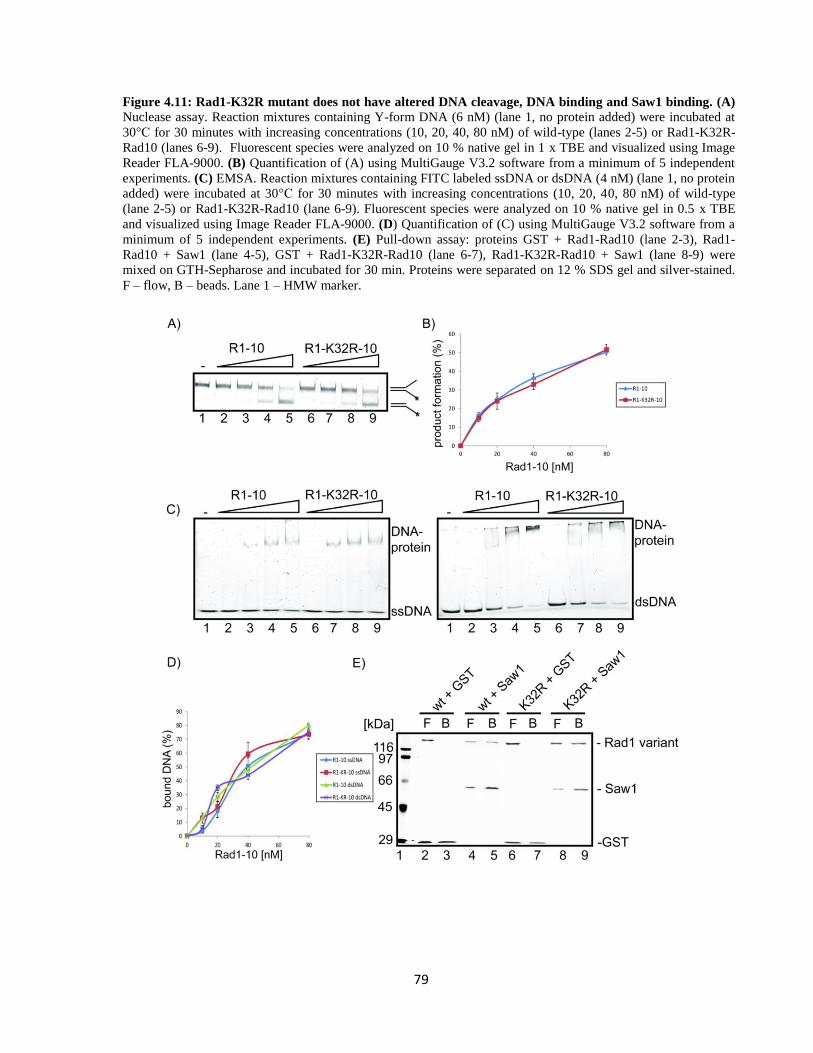
79
Figure 4.11: Rad1-K32R mutant does not have altered DNA cleavage, DNA binding and Saw1 binding. (A) Nuclease assay. Reaction mixtures containing Y-form DNA (6 nM) (lane 1, no protein added) were incubated at
30°C for 30 minutes with increasing concentrations (10, 20, 40, 80 nM) of wild-type (lanes 2-5) or Rad1-K32R-
Rad10 (lanes 6-9). Fluorescent species were analyzed on 10 % native gel in 1 x TBE and visualized using Image
Reader FLA-9000. (B) Quantification of (A) using MultiGauge V3.2 software from a minimum of 5 independent
experiments. (C) EMSA. Reaction mixtures containing FITC labeled ssDNA or dsDNA (4 nM) (lane 1, no protein
added) were incubated at 30°C for 30 minutes with increasing concentrations (10, 20, 40, 80 nM) of wild-type
(lane 2-5) or Rad1-K32R-Rad10 (lane 6-9). Fluorescent species were analyzed on 10 % native gel in 0.5 x TBE
and visualized using Image Reader FLA-9000. (D) Quantification of (C) using MultiGauge V3.2 software from a
minimum of 5 independent experiments. (E) Pull-down assay: proteins GST + Rad1-Rad10 (lane 2-3), Rad1-
Rad10 + Saw1 (lane 4-5), GST + Rad1-K32R-Rad10 (lane 6-7), Rad1-K32R-Rad10 + Saw1 (lane 8-9) were
mixed on GTH-Sepharose and incubated for 30 min. Proteins were separated on 12 % SDS gel and silver-stained.
F – flow, B – beads. Lane 1 – HMW marker.

80
Taken together our data suggest that while the substitution of lysine 32 to arginine abolished
Rad1 SUMOylation both in vitro and in vivo, it does not alter any functional properties of
Rad1-Rad10.
4.2.3. Function of Rad1 SUMOylation
4.2.3.1. Effect on nuclease activity
Rad1-Rad10 complex is a structure-specific endonuclease with a preference for
cleaving Y-form DNA substrate91
. We therefore tested if the SUMOylation of Rad1 has an
effect on its nuclease activity. As previously described, we performed in vitro SUMOylation
assay to obtain SUMOylated Rad1 (with efficiency about 10, 30, or 80%) and its nuclease
activity with non-SUMOylated Rad1-Rad10 (Figure 4.12 (B) – 30 % SUMOylation, data not
shown – 10 % and 80 % SUMOylation).
Figure 4.12: SUMOylated Rad1 exhibits the same nuclease activity as wild-type. (A) Nuclease assay.
Reaction mixtures containing FITC labeled Y-form DNA (6 nM) (lane 1, no protein added) were incubated at
30°C for 30 minutes with increasing concentrations (10, 20, 40, 80 nM) of non-SUMOylated (lane 2-5) or
SUMOylated Rad1 (lane 6-9). Fluorescent species were analyzed on 10 % native gel in 1 x TBE and visualized
using Image Reader FLA-9000. (B) In vitro SUMOylation assay on 10 % SDS gel - silver stained. Non-
SUMOylated Rad1 (lane 1, no Ubc9), SUMOylated Rad1 (lane 2), SUMO machinery proteins (lane 3). (C)
Quantification of (A) using MultiGauge V3.2 software from a minimum of 5 independent experiments.
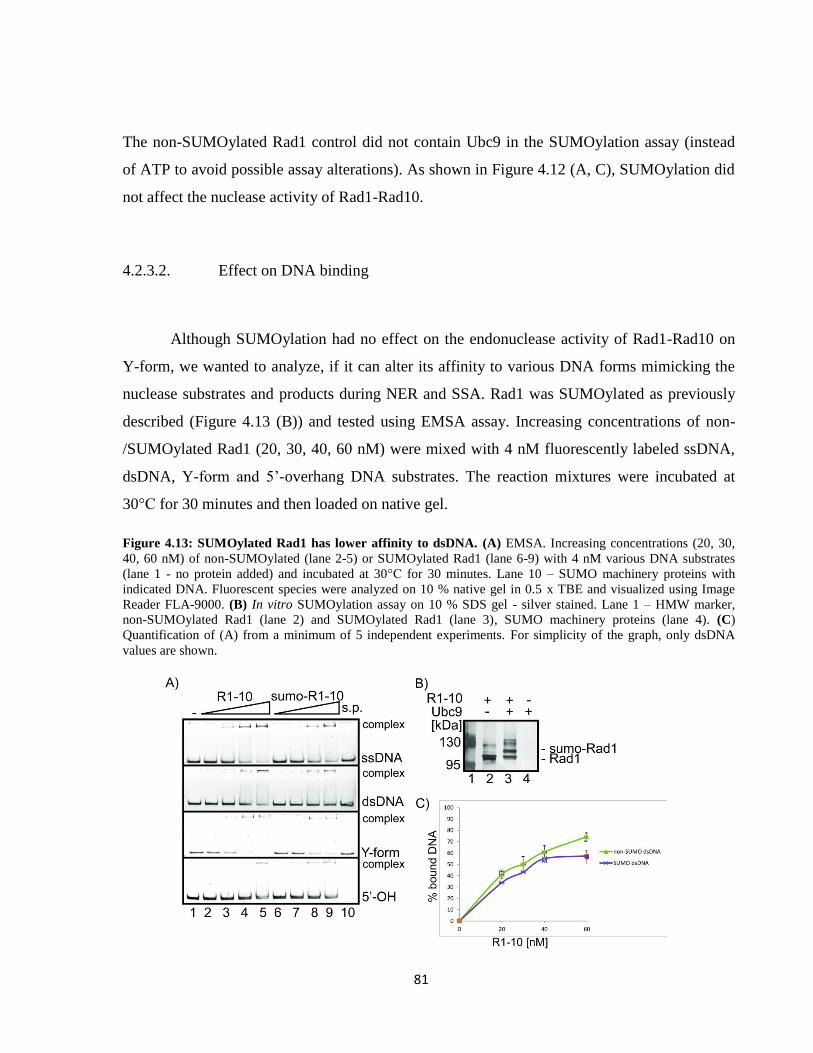
81
The non-SUMOylated Rad1 control did not contain Ubc9 in the SUMOylation assay (instead
of ATP to avoid possible assay alterations). As shown in Figure 4.12 (A, C), SUMOylation did
not affect the nuclease activity of Rad1-Rad10.
4.2.3.2. Effect on DNA binding
Although SUMOylation had no effect on the endonuclease activity of Rad1-Rad10 on
Y-form, we wanted to analyze, if it can alter its affinity to various DNA forms mimicking the
nuclease substrates and products during NER and SSA. Rad1 was SUMOylated as previously
described (Figure 4.13 (B)) and tested using EMSA assay. Increasing concentrations of non-
/SUMOylated Rad1 (20, 30, 40, 60 nM) were mixed with 4 nM fluorescently labeled ssDNA,
dsDNA, Y-form and 5’-overhang DNA substrates. The reaction mixtures were incubated at
30°C for 30 minutes and then loaded on native gel.
Figure 4.13: SUMOylated Rad1 has lower affinity to dsDNA. (A) EMSA. Increasing concentrations (20, 30,
40, 60 nM) of non-SUMOylated (lane 2-5) or SUMOylated Rad1 (lane 6-9) with 4 nM various DNA substrates
(lane 1 - no protein added) and incubated at 30°C for 30 minutes. Lane 10 – SUMO machinery proteins with
indicated DNA. Fluorescent species were analyzed on 10 % native gel in 0.5 x TBE and visualized using Image
Reader FLA-9000. (B) In vitro SUMOylation assay on 10 % SDS gel - silver stained. Lane 1 – HMW marker,
non-SUMOylated Rad1 (lane 2) and SUMOylated Rad1 (lane 3), SUMO machinery proteins (lane 4). (C)
Quantification of (A) from a minimum of 5 independent experiments. For simplicity of the graph, only dsDNA
values are shown.

82
Interestingly, we observed a significant difference in binding to dsDNA, where the non-
SUMOylated Rad1 [60 nM] bound about 73 % of dsDNA and SUMO-Rad1 [60 nM] only
about 59 % (Figure 4.13 (A, C)).
4.2.3.3. Effect on oligomeric status
SUMOylation can cause conformational changes of proteins or modify their
oligomerization389,427
. Since oligomerization of structure-specific nucleases is essential for
cleavage105,106,112
, we tested if the SUMOylation of Rad1 has an effect on the oligomeric status
of Rad1-Rad10 heterodimer by gel filtration. Purified Rad1-Rad10 (100 μg) was SUMOylated
in vitro as previously described and applied to a S400 Sephacryl column. Thyroglobulin and
catalase were used as markers with known molecular weight (669, 232 kDa, respectively). The
fractions containing Rad1 were analyzed using a Western blot with α-Rad1 antibodies.
Modified and unmodified Rad1 were eluted in the same fractions around 300 kDa, suggesting
that Rad1-Rad10 forms a dimer of heterodimers (Figure 4.14).
Figure 4.14: Modified and unmodified Rad1-Rad10 form a dimer of heterodimers. SUMOylated and non-
SUMOylated Rad1-Rad10 were separated on S400 Sephacryl column. Indicated fractions were loaded on 8 %
SDS gel and blotted on nitrocellulose membrane treated with α-Rad1 antibodies and visualized by ChemiDoc
system. Below are indicated thyroglobulin and catalase markers.
4.2.3.4. Effect on interaction with Saw1
SUMOylation has been shown to mediate protein-protein interaction via SUMO-interacting
motif within proteins400
. A reliable consensus sequence for SIMs is still of a major scientific

83
interest and is yet to be established428
. In many reported cases it corresponds to 3-4
hydrophobic aminoacids flanked by few acidic aminoacids386
. By protein sequence analysis we
identified a putative SUMO-interacting motif (SIM) (EDLLLIV) in Saw1 (Figure 4.15 (D)).
Figure 4.15: SUMOylated Rad1 has enhanced Saw1 binding. (A) Indicated proteins (5 μg) were mixed on
GTH-Sepharose in two salt concentrations (150 mM (lane 2-5, 10-11) and 400 mM (lane 6-9, 12-13) KCl). F –
flow and B – bead fractions were separated on 8 % gel to better distinguish Rad1 SUMOylation and silver stained.
Lane 1 – HMW marker. (B) Quantification of (A) by MultiGauge V3.2 software from 3 independent experiments
showing the percentage of bound Rad1 (+/- SUMO) to Saw1 in 150 mM KCl. (C) Indicated proteins (5 μg) were
mixed on GTH-Sepharose and separated on 12 % SDS gel and silver stained. Lane 1 – HMW marker. F – flow, B
– bead fractions. (D) Saw1 protein sequence (yeastgenome.org) – underlined is a putative SIM motif.
This prompted us to analyze if Saw1 can directly bind to SUMO alone and if SUMOylation of
Rad1 exhibits changed binding affinity towards Saw1. We performed a pull-down assay with
SUMOylated (to about 30 %) and non-SUMOylated Rad1 with GST-Saw1 on GTH-Sepharose
in two different salt concentrations – 150 mM and 400 mM KCl, to see a potentially more

84
profound effect. Samples from flow and bead fractions were separated on SDS-PAGE. As
shown in Figure 4.15 (A, B), SUMOylated Rad1 interacts slightly better with Saw1. On the
other hand, we did not observe a direct interaction between Saw1 and SUMO in pull-down
assay (Figure 4.15 (C), suggesting that Saw1 does not posses a SIM. Further analysis would be
necessary to confirm the presence of active SIM motif within Saw1, which could potentially
mediate the higher affinity of SUMOylated Rad1 to Saw1. The other possibility is that
SUMOylation can cause conformational changes within Rad1 thus improving the interaction
with Saw1.
4.2.3.5. Interplay between Rad1 and Saw1 SUMOylation
As mentioned earlier, we have discovered that Saw1 can also be modified by SUMO. Since
Saw1 closely cooperates with Rad1-Rad10 during DNA repair, we further investigated if Rad1-
Rad10 could influence SUMOylation of Saw1. We performed in vitro SUMOylation assay with
increasing concentrations of wild-type Rad1 and Rad1-K32R and Rad1-D869A mutants in
molar ratios (0.5:1, 1:1, 2:1) with Saw1. Rad1-D869A is a nuclease-dead mutant, which also
exhibits significantly lower binding affinity to DNA (Vance and Wilson 2002, Figure 4.16 (C)).
Interestingly, only Rad1 wild-type was able to stimulate Saw1 SUMOylation in contrast to both
mutants of Rad1 (Figure 4.16 (A)). The increased level of SUMOylated Saw1 could be caused
either by direct interaction with Rad1 or by SUMOylated Rad1. To further analyze this
hypothesis, we performed in vitro pull-down assay with Saw1 and Rad1 mutants as well as in
vitro SUMOylation assay with Rad1-D869A. As shown in Figure 4.18 B and C, SUMO-
deficient Rad1-K32R mutant effectively binds Saw1 similarly to wild-type Rad1, meanwhile
Saw1 interaction with Rad1-D869A mutant is abolished. On the other hand, SUMOylation
level of Rad1-D869A mutant is comparable to that of the wild-type Rad1. Taken together these
results suggest that stimulation of Saw1 SUMOylation by Rad1-Rad10 is dependent on their
direct interaction as well as on modification of Rad1 by SUMO.
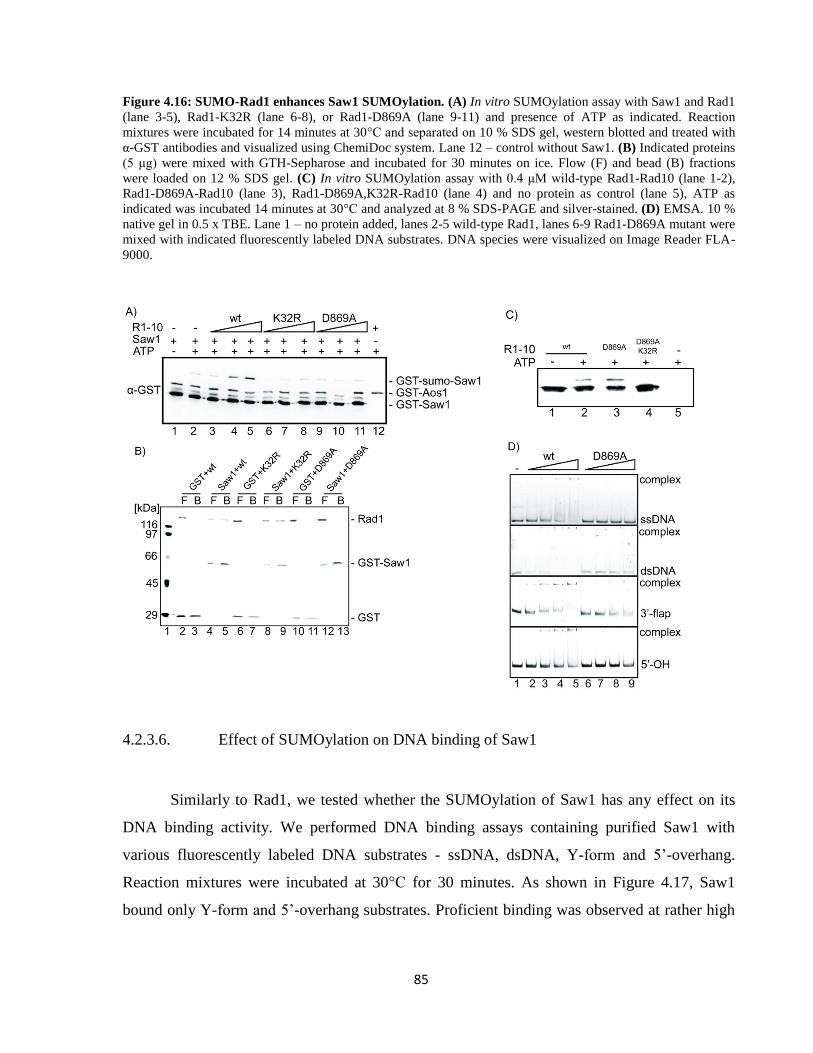
85
Figure 4.16: SUMO-Rad1 enhances Saw1 SUMOylation. (A) In vitro SUMOylation assay with Saw1 and Rad1
(lane 3-5), Rad1-K32R (lane 6-8), or Rad1-D869A (lane 9-11) and presence of ATP as indicated. Reaction
mixtures were incubated for 14 minutes at 30°C and separated on 10 % SDS gel, western blotted and treated with
α-GST antibodies and visualized using ChemiDoc system. Lane 12 – control without Saw1. (B) Indicated proteins
(5 μg) were mixed with GTH-Sepharose and incubated for 30 minutes on ice. Flow (F) and bead (B) fractions
were loaded on 12 % SDS gel. (C) In vitro SUMOylation assay with 0.4 μM wild-type Rad1-Rad10 (lane 1-2),
Rad1-D869A-Rad10 (lane 3), Rad1-D869A,K32R-Rad10 (lane 4) and no protein as control (lane 5), ATP as
indicated was incubated 14 minutes at 30°C and analyzed at 8 % SDS-PAGE and silver-stained. (D) EMSA. 10 %
native gel in 0.5 x TBE. Lane 1 – no protein added, lanes 2-5 wild-type Rad1, lanes 6-9 Rad1-D869A mutant were
mixed with indicated fluorescently labeled DNA substrates. DNA species were visualized on Image Reader FLA-
9000.
4.2.3.6. Effect of SUMOylation on DNA binding of Saw1
Similarly to Rad1, we tested whether the SUMOylation of Saw1 has any effect on its
DNA binding activity. We performed DNA binding assays containing purified Saw1 with
various fluorescently labeled DNA substrates - ssDNA, dsDNA, Y-form and 5’-overhang.
Reaction mixtures were incubated at 30°C for 30 minutes. As shown in Figure 4.17, Saw1
bound only Y-form and 5’-overhang substrates. Proficient binding was observed at rather high

86
concentrations 100 - 400 nM, which is in accordance with 95
where 200 nM Saw1 was used.
Low concentrations (10 - 60 nM Saw1) showed very poor DNA binding (data not shown).
Figure 4.17: Saw1 binds Y-form and 5’-overhang DNA substrates. (A) EMSA. Increasing concentrations of
Saw1 (60, 120, 240, 400 nM) were incubated with 4 nM FITC labeled DNA substrates (ssDNA (lane 2-5), dsDNA
(lane 7-10), Y-form (lane 12-15), 5’-OH (lane 17-20)) at 30°C for 30 minutes. Lanes 1, 5, 11, 16 represent free
ssDNA, dsDNA, Y-form and 5’-overhang, respectively. Samples were separated on 10 % native gel in 0.5 x TBE
and visualized by Image Reader FLA-9000. (B) Quantification of (A) with MultiGauge V3.2 software from 3
independent experiments.
4.2.3.7. SUMOylated complex Rad1-Rad10-Saw1 has unchanged nuclease activity on
Y-form
Saw1 was previously shown to stimulate Rad1-Rad10 nuclease activity95
. Since Rad1-Rad10
forms a stable complex with Saw1 and Saw1 SUMOylation is stimulated by Rad1-Rad10
interaction, we aimed to test if SUMOylation of the whole complex affects its nuclease activity,
that is SUMO-Rad1-Rad10-SUMO-Saw1. Rad1-Rad10 and Saw1 were SUMOylated as
previously described. Increasing concentrations (20, 30, 40, 60 nM) of unmodified and
modified complex were mixed with fluorescently labeled Y-form DNA and incubated as in
standard nuclease assay. SUMOylated complex cleaved the Y-form with the same efficiency
as unmodified complex suggesting that similarly to Rad1-Rad10 alone, SUMOylation does not
affect nuclease activity of Rad1-Rad10-Saw1 complex (Figure 4.18).

87
Figure 4.18: SUMO-Saw1 does not affect SUMO-Rad1 nuclease activity. (A) Nuclease assay. Increasing
concentrations (20, 30, 40, 60 nM) of unmodified (lane 2-5) and SUMOylated Rad1-Rad10-Saw1 (lane 6-9) were
incubated with 4 nM FITC labeled Y-form DNA substrate (lane 1 – free DNA) at 30°C for 30 minutes. Samples
were separated on 10 % native gel in 0.5 x TBE and visualized by Image Reader FLA-9000. (B) In vitro
SUMOylation assay of Rad1-Rad10-Saw1 used in (A). Reaction mixtures contained SUMO machinery proteins,
Rad1-Rad10-Saw1 and were incubated for 14 minutes at 30°C and analyzed on 10 % SDS-PAGE and silver
stained. Lane 1 – unmodified Rad1-Rad10-Saw1 (without ATP). Lane 2 – modified Rad1-Rad10-Saw1 (with
ATP). Lane 3 – control containing SUMO machinery proteins with ATP. (C) Quantification of (A) with
MultiGauge software from 3 independent experiments.
4.2.4. Discussion
Rad1-Rad10 is a structure-specific endonuclease with preference for Y-form DNA structures,
which are processed during NER, SSA and ICL repair91
. In this work we discovered that Rad1
is SUMOylated at a single lysine K32 and the SUMOylation process is dependent on E3 ligases
(mainly Siz1 and Siz2). Perhaps another yet unknown factor can also contribute to Rad1
SUMOylation, but we were unable to identify it in vitro (DNA binding, interaction partners).
Maybe because Rad1 SUMOylation has a specific biological purpose in living cells regarding
localization, or as a biological clock for enzymatic activity alteration perhapes through timely

88
targeting of SUMO machinery proteins especially Siz1 (Siz2) during DNA damage induced by
UV and MMS agents (Zhao group).
Using in vitro biochemical assays we investigated the impact of SUMOylation on the
properties and biochemical activities of Rad1-Rad10. We have shown that SUMOylated Rad1-
Rad10 exhibits a lower affinity to dsDNA, meanwhile the other enzyme properties (nuclease
activity, protein interactions, oligomerization state) stayed unaffected. Reduced affinity of
SUMOylated Rad1-Rad10 specifically for dsDNA may suggest that SUMOylation might
regulate the dissociation of Rad1-Rad10 from dsDNA after the cleavage promoting protein
turnover. Double-stranded DNA generated after Rad1 cleavage is still a suitable substrate for
Rad1-Rad10 binding. Enhanced dissociation of Rad1 from resected DNA lesions could allow
other repair enzymes to finish repair of the damaged site and/or free Rad1 from DNA to ensure
its use at other sites of damage or in other repair pathways. This is supported by in vivo data,
showing that Rad1 is not very abundant in cells and robust Rad1 SUMOylation is observed at
high doses of DNA damage (Zhao group). This was similarly seen in case of thymine DNA
glycosylase where SUMO conjugation dramatically reduced AP-site affinity389,429,430
.
Saw1 is a structure-specific binding protein with preference for branched DNA substrates95
. It
recruits Rad1-Rad10 nuclease to DNA lesions during SSA95,97
and also cooperates with Rad1-
Rad10 during BER and ICL repair (Zhao group). SUMO-Rad1 stimulates Saw1 SUMOylation,
which could hypothetically point to a turnover mechanism from DNA for the whole complex
Rad1-Rad10-Saw1. Free Saw1 can then engage with other repair proteins to fulfill its role in
other pathways.
SUMOylated Saw1 did not have an altered DNA binding activity compared to unmodified
Saw1 (Veronika Altmannová, Krejčí group), suggesting that SUMOylation might not directly
change the protein properties but rather affect its function. Indeed, in vivo data have shown that
Saw1 has a role not only during SSA by recruitment of Rad1-Rad10, but it also participates in
the repair of base lesions, UV damage and protein-DNA adducts. Interestingly, SUMOylation
stimulates the interaction of Saw1 with another DNA nuclease Slx1-Slx4 in a specific manner
where SUMO binds preferentially to Slx1, while Saw1 interacts with Slx4. Moreover this
interaction is preferred on the expense of Saw1-Rad1 interaction (Zhao group). This suggests a

89
control mechanism between SUMOylation of Saw1 and its interacting partners depending on
the type of damage.
Collectively, we provide new evidence about the molecular details and mechanisms how
SUMOylation of Rad1 affects protein turnover after cleavage at damaged sites. Moreover, we
show that SUMO-Rad1 enhances Saw1 SUMOylation targeting it towards another repair
pathway through a specific interaction with Slx1, highlighting the importance of SUMOylation
in the regulation of DNA repair.
Figure 4.19: Model of SUMO-Rad1-Rad10 and SUMO-Saw1 role in DNA repair. Rad1-Rad10 complex is
tethered to flap substrate during SSA in a Saw1-dependent manner. After Slx4-dependent Rad1-Rad10 flap
removal, Rad1 is SUMOylated at K32 and dissociates from dsDNA. SUMO-Rad1 enhances SUMOylation of
Saw1 shifting its preference toward binding with Slx1 on the Slx4 scaffold mediating a Rad1-independent
repair.

90
5. CONCLUSIONS
1. We characterized protein-protein interactions of structure-specific endonuclease
MUS81-EME1 with RECQ5 helicase and their cooperative role in DNA repair. MUS81-EME1
physically interacts with RECQ5 helicase. The whole RECQ5 protein is needed for this
interaction. RECQ5 stimulates MUS81 endonuclease activity on its specific DNA substrates.
RECQ5 is capable of stimulating also yeast Mus81-Mms4 nuclease. To investigate a more in
vivo situation we carried out RAD51 removal assays, where we show that RECQ5 is capable of
suppressing RAD51-mediated inhibition of MUS81 cleavage. Both RAD51 binding and
translocase activities of RECQ5 are required for efficient disruption of RAD51 filament. Our
biochemical data confirm genetic data, implicating MUS81-RECQ5 cooperation in resolution
of late replication intermediates at CFSs during early mitosis. Depletion of RECQ5 inhibited
CFSs expression and exhibited RAD51 accumulation at CFSs thus impairing MUS81-
dependent cleavage of entangled DNA strands.
2. We investigated the effect of a specific post-translational modification (SUMOylation)
on a yeast structure-specific endonuclease Rad1-Rad10, which has a role mainly in NER and
HR subpathway. SUMOylation of Rad1 subunit of the Rad1-Rad10 complex occurs at K32 and
is dependent on E3 ligases (mainly Siz1 and Siz2). Genetic data show an increase in Rad1
SUMOylation after DNA damage. SUMO-Rad1-Rad10 has a decreased DNA binding activity
towards dsDNA, pointing to its function in protein turnover from DNA after flap cleavage
supported by in vivo data. SUMO-Rad1 stimulates SUMOylation of Saw1, probably leading to
complex turnover from DNA and complex dissociation. SUMO-Saw1 can then engage with
Slx1 and function in other repair processes.

91
6. BIBLIOGRAPHY
1. Lindahl, T. Instability and decay of the primary structure of DNA. Nature 362, 709–715 (1993). 2. Haynes, B., Saadat, N., Myung, B. & Shekhar, M. P. V. Crosstalk between translesion synthesis,
Fanconi anemia network, and homologous recombination repair pathways in interstrand DNA crosslink repair and development of chemoresistance. Mutat. Res. Rev. Mutat. Res. 763, 258–266 (2015).
3. Keeney, S., Giroux, C. N. & Kleckner, N. Meiosis-specific DNA double-strand breaks are catalyzed by Spo11, a member of a widely conserved protein family. Cell 88, 375–384 (1997).
4. Guirouilh-Barbat, J. et al. Impact of the KU80 pathway on NHEJ-induced genome rearrangements in mammalian cells. Mol. Cell 14, 611–623 (2004).
5. Gong, C. et al. Mechanism of nonhomologous end-joining in mycobacteria: a low-fidelity repair system driven by Ku, ligase D and ligase C. Nat. Struct. Mol. Biol. 12, 304–312 (2005).
6. Weller, G. R. et al. Identification of a DNA nonhomologous end-joining complex in bacteria. Science 297, 1686–1689 (2002).
7. Deriano, L. & Roth, D. B. Modernizing the nonhomologous end-joining repertoire: alternative and classical NHEJ share the stage. Annu. Rev. Genet. 47, 433–455 (2013).
8. Moore, J. K. & Haber, J. E. Cell cycle and genetic requirements of two pathways of nonhomologous end-joining repair of double-strand breaks in Saccharomyces cerevisiae. Mol. Cell. Biol. 16, 2164–2173 (1996).
9. Burkhalter, M. D., Roberts, S. A., Havener, J. M. & Ramsden, D. A. Activity of ribonucleotide reductase helps determine how cells repair DNA double strand breaks. DNA Repair 8, 1258–1263 (2009).
10. Stracker, T. H. & Petrini, J. H. J. The MRE11 complex: starting from the ends. Nat. Rev. Mol. Cell Biol. 12, 90–103 (2011).
11. Grundy, G. J., Moulding, H. A., Caldecott, K. W. & Rulten, S. L. One ring to bring them all--the role of Ku in mammalian non-homologous end joining. DNA Repair 17, 30–38 (2014).
12. Tomkinson, A. E. & Sallmyr, A. Structure and function of the DNA ligases encoded by the mammalian LIG3 gene. Gene 531, 150–157 (2013).
13. Wu, D., Topper, L. M. & Wilson, T. E. Recruitment and dissociation of nonhomologous end joining proteins at a DNA double-strand break in Saccharomyces cerevisiae. Genetics 178, 1237–1249 (2008).
14. Decottignies, A. Alternative end-joining mechanisms: a historical perspective. Front. Genet. 4, 48 (2013).
15. Deng, S. K., Gibb, B., de Almeida, M. J., Greene, E. C. & Symington, L. S. RPA antagonizes microhomology-mediated repair of DNA double-strand breaks. Nat. Struct. Mol. Biol. 21, 405–412 (2014).
16. Ma, J.-L., Kim, E. M., Haber, J. E. & Lee, S. E. Yeast Mre11 and Rad1 proteins define a Ku-independent mechanism to repair double-strand breaks lacking overlapping end sequences. Mol. Cell. Biol. 23, 8820–8828 (2003).
17. Simsek, D. et al. DNA ligase III promotes alternative nonhomologous end-joining during chromosomal translocation formation. PLoS Genet. 7, e1002080 (2011).
18. Chapman, J. R. et al. RIF1 is essential for 53BP1-dependent nonhomologous end joining and suppression of DNA double-strand break resection. Mol. Cell 49, 858–871 (2013).
19. Di Virgilio, M. et al. Rif1 prevents resection of DNA breaks and promotes immunoglobulin class switching. Science 339, 711–715 (2013).

92
20. Escribano-Díaz, C. et al. A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Mol. Cell 49, 872–883 (2013).
21. Ferrari, M. et al. Functional interplay between the 53BP1-ortholog Rad9 and the Mre11 complex regulates resection, end-tethering and repair of a double-strand break. PLoS Genet. 11, e1004928 (2015).
22. Zimmermann, M., Lottersberger, F., Buonomo, S. B., Sfeir, A. & de Lange, T. 53BP1 regulates DSB repair using Rif1 to control 5’ end resection. Science 339, 700–704 (2013).
23. Sartori, A. A. et al. Human CtIP promotes DNA end resection. Nature 450, 509–514 (2007). 24. Lee, J.-H. & Paull, T. T. Direct activation of the ATM protein kinase by the Mre11/Rad50/Nbs1
complex. Science 304, 93–96 (2004). 25. Lee, J.-H. & Paull, T. T. ATM activation by DNA double-strand breaks through the Mre11-
Rad50-Nbs1 complex. Science 308, 551–554 (2005). 26. Daley, J. M., Chiba, T., Xue, X., Niu, H. & Sung, P. Multifaceted role of the Topo IIIα-RMI1-RMI2
complex and DNA2 in the BLM-dependent pathway of DNA break end resection. Nucleic Acids Res. 42, 11083–11091 (2014).
27. Mimitou, E. P. & Symington, L. S. Sae2, Exo1 and Sgs1 collaborate in DNA double-strand break processing. Nature 455, 770–774 (2008).
28. Mimitou, E. P. & Symington, L. S. Nucleases and helicases take center stage in homologous recombination. Trends Biochem. Sci. 34, 264–272 (2009).
29. Mimitou, E. P. & Symington, L. S. DNA end resection--unraveling the tail. DNA Repair 10, 344–348 (2011).
30. Nimonkar, A. V. et al. BLM-DNA2-RPA-MRN and EXO1-BLM-RPA-MRN constitute two DNA end resection machineries for human DNA break repair. Genes Dev. 25, 350–362 (2011).
31. Paull, T. T. Making the best of the loose ends: Mre11/Rad50 complexes and Sae2 promote DNA double-strand break resection. DNA Repair 9, 1283–1291 (2010).
32. Sturzenegger, A. et al. DNA2 cooperates with the WRN and BLM RecQ helicases to mediate long-range DNA end resection in human cells. J. Biol. Chem. 289, 27314–27326 (2014).
33. Zhu, Z., Chung, W.-H., Shim, E. Y., Lee, S. E. & Ira, G. Sgs1 helicase and two nucleases Dna2 and Exo1 resect DNA double-strand break ends. Cell 134, 981–994 (2008).
34. Chen, X. et al. Cell cycle regulation of DNA double-strand break end resection by Cdk1-dependent Dna2 phosphorylation. Nat. Struct. Mol. Biol. 18, 1015–1019 (2011).
35. Huertas, P., Cortés-Ledesma, F., Sartori, A. A., Aguilera, A. & Jackson, S. P. CDK targets Sae2 to control DNA-end resection and homologous recombination. Nature 455, 689–692 (2008).
36. Peterson, S. E. et al. Cdk1 uncouples CtIP-dependent resection and Rad51 filament formation during M-phase double-strand break repair. J. Cell Biol. 194, 705–720 (2011).
37. Ball, H. L. et al. Function of a conserved checkpoint recruitment domain in ATRIP proteins. Mol. Cell. Biol. 27, 3367–3377 (2007).
38. Choi, J.-H. et al. Reconstitution of RPA-covered single-stranded DNA-activated ATR-Chk1 signaling. Proc. Natl. Acad. Sci. U. S. A. 107, 13660–13665 (2010).
39. Zou, L. & Elledge, S. J. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 300, 1542–1548 (2003).
40. Alani, E., Thresher, R., Griffith, J. D. & Kolodner, R. D. Characterization of DNA-binding and strand-exchange stimulation properties of y-RPA, a yeast single-strand-DNA-binding protein. J. Mol. Biol. 227, 54–71 (1992).
41. Krogh, B. O. & Symington, L. S. Recombination proteins in yeast. Annu. Rev. Genet. 38, 233–271 (2004).
42. Bhattacharyya, A., Ear, U. S., Koller, B. H., Weichselbaum, R. R. & Bishop, D. K. The breast cancer susceptibility gene BRCA1 is required for subnuclear assembly of Rad51 and survival

93
following treatment with the DNA cross-linking agent cisplatin. J. Biol. Chem. 275, 23899–23903 (2000).
43. Bi, B., Rybalchenko, N., Golub, E. I. & Radding, C. M. Human and yeast Rad52 proteins promote DNA strand exchange. Proc. Natl. Acad. Sci. U. S. A. 101, 9568–9572 (2004).
44. Krejci, L. et al. Interaction with Rad51 is indispensable for recombination mediator function of Rad52. J. Biol. Chem. 277, 40132–40141 (2002).
45. Shinohara, A. & Ogawa, T. Stimulation by Rad52 of yeast Rad51-mediated recombination. Nature 391, 404–407 (1998).
46. Thorslund, T. & West, S. C. BRCA2: a universal recombinase regulator. Oncogene 26, 7720–7730 (2007).
47. Wooster, R. et al. Identification of the breast cancer susceptibility gene BRCA2. Nature 378, 789–792 (1995).
48. Shinohara, A., Ogawa, H. & Ogawa, T. Rad51 protein involved in repair and recombination in S. cerevisiae is a RecA-like protein. Cell 69, 457–470 (1992).
49. Krejci, L. et al. DNA helicase Srs2 disrupts the Rad51 presynaptic filament. Nature 423, 305–309 (2003).
50. Mankouri, H. W., Chu, W. K. & Hickson, I. D. A novel antirecombinase gains PARIty. Mol. Cell 45, 6–7 (2012).
51. Moldovan, G.-L. et al. Inhibition of homologous recombination by the PCNA-interacting protein PARI. Mol. Cell 45, 75–86 (2012).
52. Schwendener, S. et al. Physical interaction of RECQ5 helicase with RAD51 facilitates its anti-recombinase activity. J. Biol. Chem. 285, 15739–15745 (2010).
53. Simandlova, J. et al. FBH1 helicase disrupts RAD51 filaments in vitro and modulates homologous recombination in mammalian cells. J. Biol. Chem. 288, 34168–34180 (2013).
54. Veaute, X. et al. The Srs2 helicase prevents recombination by disrupting Rad51 nucleoprotein filaments. Nature 423, 309–312 (2003).
55. Petukhova, G., Stratton, S. & Sung, P. Catalysis of homologous DNA pairing by yeast Rad51 and Rad54 proteins. Nature 393, 91–94 (1998).
56. Szostak, J. W., Orr-Weaver, T. L., Rothstein, R. J. & Stahl, F. W. The double-strand-break repair model for recombination. Cell 33, 25–35 (1983).
57. Mazón, G., Lam, A. F., Ho, C. K., Kupiec, M. & Symington, L. S. The Rad1-Rad10 nuclease promotes chromosome translocations between dispersed repeats. Nat. Struct. Mol. Biol. 19, 964–971 (2012).
58. Svendsen, J. M. & Harper, J. W. GEN1/Yen1 and the SLX4 complex: Solutions to the problem of Holliday junction resolution. Genes Dev. 24, 521–536 (2010).
59. Chen, X. B. et al. Human Mus81-associated endonuclease cleaves Holliday junctions in vitro. Mol. Cell 8, 1117–1127 (2001).
60. Castor, D. et al. Cooperative control of holliday junction resolution and DNA repair by the SLX1 and MUS81-EME1 nucleases. Mol. Cell 52, 221–233 (2013).
61. Blais, V. et al. RNA interference inhibition of Mus81 reduces mitotic recombination in human cells. Mol. Biol. Cell 15, 552–562 (2004).
62. Allers, T. & Lichten, M. Differential timing and control of noncrossover and crossover recombination during meiosis. Cell 106, 47–57 (2001).
63. Hunter, N. & Kleckner, N. The single-end invasion: an asymmetric intermediate at the double-strand break to double-holliday junction transition of meiotic recombination. Cell 106, 59–70 (2001).

94
64. Malkova, A., Ivanov, E. L. & Haber, J. E. Double-strand break repair in the absence of RAD51 in yeast: a possible role for break-induced DNA replication. Proc. Natl. Acad. Sci. U. S. A. 93, 7131–7136 (1996).
65. Cejka, P., Plank, J. L., Bachrati, C. Z., Hickson, I. D. & Kowalczykowski, S. C. Rmi1 stimulates decatenation of double Holliday junctions during dissolution by Sgs1-TOPO3. Nat. Struct. Mol. Biol. 17, 1377–1382 (2010).
66. Mankouri, H. W., Ashton, T. M. & Hickson, I. D. Holliday junction-containing DNA structures persist in cells lacking Sgs1 or TOPO3 following exposure to DNA damage. Proc. Natl. Acad. Sci. U. S. A. 108, 4944–4949 (2011).
67. Mitchel, K., Zhang, H., Welz-Voegele, C. & Jinks-Robertson, S. Molecular structures of crossover and noncrossover intermediates during gap repair in yeast: implications for recombination. Mol. Cell 38, 211–222 (2010).
68. Nassif, N., Penney, J., Pal, S., Engels, W. R. & Gloor, G. B. Efficient copying of nonhomologous sequences from ectopic sites via P-element-induced gap repair. Mol. Cell. Biol. 14, 1613–1625 (1994).
69. Prakash, R. et al. Yeast Mph1 helicase dissociates Rad51-made D-loops: implications for crossover control in mitotic recombination. Genes Dev. 23, 67–79 (2009).
70. Zheng, X.-F. et al. Processing of DNA structures via DNA unwinding and branch migration by the S. cerevisiae Mph1 protein. DNA Repair 10, 1034–1043 (2011).
71. Paliwal, S., Kanagaraj, R., Sturzenegger, A., Burdova, K. & Janscak, P. Human RECQ5 helicase promotes repair of DNA double-strand breaks by synthesis-dependent strand annealing. Nucleic Acids Res. 42, 2380–2390 (2014).
72. Bosco, G. & Haber, J. E. Chromosome break-induced DNA replication leads to nonreciprocal translocations and telomere capture. Genetics 150, 1037–1047 (1998).
73. Ruiz, J. F., Gómez-González, B. & Aguilera, A. Chromosomal translocations caused by either pol32-dependent or pol32-independent triparental break-induced replication. Mol. Cell. Biol. 29, 5441–5454 (2009).
74. Signon, L., Malkova, A., Naylor, M. L., Klein, H. & Haber, J. E. Genetic requirements for RAD51- and RAD54-independent break-induced replication repair of a chromosomal double-strand break. Mol. Cell. Biol. 21, 2048–2056 (2001).
75. Nickoloff, J. A., Sweetser, D. B., Clikeman, J. A., Khalsa, G. J. & Wheeler, S. L. Multiple heterologies increase mitotic double-strand break-induced allelic gene conversion tract lengths in yeast. Genetics 153, 665–679 (1999).
76. Le, S., Moore, J. K., Haber, J. E. & Greider, C. W. RAD50 and RAD51 define two pathways that collaborate to maintain telomeres in the absence of telomerase. Genetics 152, 143–152 (1999).
77. Lundblad, V. & Blackburn, E. H. An alternative pathway for yeast telomere maintenance rescues est1- senescence. Cell 73, 347–360 (1993).
78. Deem, A. et al. Break-induced replication is highly inaccurate. PLoS Biol. 9, e1000594 (2011). 79. Lydeard, J. R., Jain, S., Yamaguchi, M. & Haber, J. E. Break-induced replication and telomerase-
independent telomere maintenance require Pol32. Nature 448, 820–823 (2007). 80. Lydeard, J. R. et al. Break-induced replication requires all essential DNA replication factors
except those specific for pre-RC assembly. Genes Dev. 24, 1133–1144 (2010). 81. Hastings, P. J., Ira, G. & Lupski, J. R. A microhomology-mediated break-induced replication
model for the origin of human copy number variation. PLoS Genet. 5, e1000327 (2009). 82. Payen, C., Koszul, R., Dujon, B. & Fischer, G. Segmental duplications arise from Pol32-
dependent repair of broken forks through two alternative replication-based mechanisms. PLoS Genet. 4, e1000175 (2008).

95
83. Krejci, L., Altmannova, V., Spirek, M. & Zhao, X. Homologous recombination and its regulation. Nucleic Acids Res. 40, 5795–5818 (2012).
84. Pâques, F. & Haber, J. E. Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. MMBR 63, 349–404 (1999).
85. Elliott, B., Richardson, C. & Jasin, M. Chromosomal translocation mechanisms at intronic alu elements in mammalian cells. Mol. Cell 17, 885–894 (2005).
86. Liang, F., Han, M., Romanienko, P. J. & Jasin, M. Homology-directed repair is a major double-strand break repair pathway in mammalian cells. Proc. Natl. Acad. Sci. U. S. A. 95, 5172–5177 (1998).
87. Ivanov, E. L., Sugawara, N., Fishman-Lobell, J. & Haber, J. E. Genetic requirements for the single-strand annealing pathway of double-strand break repair in Saccharomyces cerevisiae. Genetics 142, 693–704 (1996).
88. Sugawara, N., Ira, G. & Haber, J. E. DNA length dependence of the single-strand annealing pathway and the role of Saccharomyces cerevisiae RAD59 in double-strand break repair. Mol. Cell. Biol. 20, 5300–5309 (2000).
89. Symington, L. S. Role of RAD52 epistasis group genes in homologous recombination and double-strand break repair. Microbiol. Mol. Biol. Rev. MMBR 66, 630–670, table of contents (2002).
90. Bardwell, A. J., Bardwell, L., Tomkinson, A. E. & Friedberg, E. C. Specific cleavage of model recombination and repair intermediates by the yeast Rad1-Rad10 DNA endonuclease. Science 265, 2082–2085 (1994).
91. Ciccia, A., McDonald, N. & West, S. C. Structural and functional relationships of the XPF/MUS81 family of proteins. Annu. Rev. Biochem. 77, 259–287 (2008).
92. Fishman-Lobell, J. & Haber, J. E. Removal of nonhomologous DNA ends in double-strand break recombination: the role of the yeast ultraviolet repair gene RAD1. Science 258, 480–484 (1992).
93. Ivanov, E. L. & Haber, J. E. RAD1 and RAD10, but not other excision repair genes, are required for double-strand break-induced recombination in Saccharomyces cerevisiae. Mol. Cell. Biol. 15, 2245–2251 (1995).
94. Sugawara, N., Pâques, F., Colaiácovo, M. & Haber, J. E. Role of Saccharomyces cerevisiae Msh2 and Msh3 repair proteins in double-strand break-induced recombination. Proc. Natl. Acad. Sci. U. S. A. 94, 9214–9219 (1997).
95. Li, F. et al. Role of Saw1 in Rad1/Rad10 complex assembly at recombination intermediates in budding yeast. EMBO J. 32, 461–472 (2013).
96. Surtees, J. A. & Alani, E. Mismatch repair factor MSH2-MSH3 binds and alters the conformation of branched DNA structures predicted to form during genetic recombination. J. Mol. Biol. 360, 523–536 (2006).
97. Li, F. et al. Microarray-based genetic screen defines SAW1, a gene required for Rad1/Rad10-dependent processing of recombination intermediates. Mol. Cell 30, 325–335 (2008).
98. Motycka, T. A., Bessho, T., Post, S. M., Sung, P. & Tomkinson, A. E. Physical and functional interaction between the XPF/ERCC1 endonuclease and hRad52. J. Biol. Chem. 279, 13634–13639 (2004).
99. Flott, S. et al. Phosphorylation of Slx4 by Mec1 and Tel1 regulates the single-strand annealing mode of DNA repair in budding yeast. Mol. Cell. Biol. 27, 6433–6445 (2007).
100. Bartosova, Z. & Krejci, L. Nucleases in homologous recombination as targets for cancer therapy. FEBS Lett. 588, 2446–2456 (2014).
101. Boddy, M. N. et al. Mus81-Eme1 are essential components of a Holliday junction resolvase. Cell 107, 537–548 (2001).

96
102. Ciccia, A., Constantinou, A. & West, S. C. Identification and characterization of the human mus81-eme1 endonuclease. J. Biol. Chem. 278, 25172–25178 (2003).
103. Fu, Y. & Xiao, W. Functional domains required for the Saccharomyces cerevisiae Mus81-Mms4 endonuclease complex formation and nuclear localization. DNA Repair 2, 1435–1447 (2003).
104. Mullen, J. R., Kaliraman, V., Ibrahim, S. S. & Brill, S. J. Requirement for three novel protein complexes in the absence of the Sgs1 DNA helicase in Saccharomyces cerevisiae. Genetics 157, 103–118 (2001).
105. Fricke, W. M., Bastin-Shanower, S. A. & Brill, S. J. Substrate specificity of the Saccharomyces cerevisiae Mus81-Mms4 endonuclease. DNA Repair 4, 243–251 (2005).
106. Taylor, E. R. & McGowan, C. H. Cleavage mechanism of human Mus81-Eme1 acting on Holliday-junction structures. Proc. Natl. Acad. Sci. U. S. A. 105, 3757–3762 (2008).
107. Schwartz, E. K. et al. Mus81-Mms4 functions as a single heterodimer to cleave nicked intermediates in recombinational DNA repair. Mol. Cell. Biol. 32, 3065–3080 (2012).
108. Gaskell, L. J., Osman, F., Gilbert, R. J. C. & Whitby, M. C. Mus81 cleavage of Holliday junctions: a failsafe for processing meiotic recombination intermediates? EMBO J. 26, 1891–1901 (2007).
109. Ciccia, A. et al. Identification of FAAP24, a Fanconi Anemia Core Complex Protein that Interacts with FANCM. Mol. Cell 25, 331–343 (2007).
110. Pepe, A. & West, S. C. MUS81-EME2 promotes replication fork restart. Cell Rep. 7, 1048–1055 (2014).
111. Zeng, S. et al. Telomere recombination requires the MUS81 endonuclease. Nat. Cell Biol. 11, 616–623 (2009).
112. Bastin-Shanower, S. A., Fricke, W. M., Mullen, J. R. & Brill, S. J. The mechanism of Mus81-Mms4 cleavage site selection distinguishes it from the homologous endonuclease Rad1-Rad10. Mol. Cell. Biol. 23, 3487–3496 (2003).
113. Ehmsen, K. T. & Heyer, W.-D. A junction branch point adjacent to a DNA backbone nick directs substrate cleavage by Saccharomyces cerevisiae Mus81-Mms4. Nucleic Acids Res. 37, 2026–2036 (2009).
114. Osman, F., Dixon, J., Doe, C. L. & Whitby, M. C. Generating crossovers by resolution of nicked Holliday junctions: a role for Mus81-Eme1 in meiosis. Mol. Cell 12, 761–774 (2003).
115. Whitby, M. C., Osman, F. & Dixon, J. Cleavage of model replication forks by fission yeast Mus81-Eme1 and budding yeast Mus81-Mms4. J. Biol. Chem. 278, 6928–6935 (2003).
116. Gwon, G. H. et al. Crystal structures of the structure-selective nuclease Mus81-Eme1 bound to flap DNA substrates. EMBO J. 33, 1061–1072 (2014).
117. Constantinou, A., Chen, X.-B., McGowan, C. H. & West, S. C. Holliday junction resolution in human cells: two junction endonucleases with distinct substrate specificities. EMBO J. 21, 5577–5585 (2002).
118. Wyatt, H. D. M., Sarbajna, S., Matos, J. & West, S. C. Coordinated actions of SLX1-SLX4 and MUS81-EME1 for Holliday junction resolution in human cells. Mol. Cell 52, 234–247 (2013).
119. Amangyeld, T., Shin, Y.-K., Lee, M., Kwon, B. & Seo, Y.-S. Human MUS81-EME2 can cleave a variety of DNA structures including intact Holliday junction and nicked duplex. Nucleic Acids Res. (2014). doi:10.1093/nar/gku237
120. Pepe, A. & West, S. C. Substrate specificity of the MUS81-EME2 structure selective endonuclease. Nucleic Acids Res. 42, 3833–3845 (2014).
121. Chang, J. H., Kim, J. J., Choi, J. M., Lee, J. H. & Cho, Y. Crystal structure of the Mus81-Eme1 complex. Genes Dev. 22, 1093–1106 (2008).
122. Newman, M. et al. Structure of an XPF endonuclease with and without DNA suggests a model for substrate recognition. EMBO J. 24, 895–905 (2005).

97
123. Fadden, A. J. et al. A winged helix domain in human MUS81 binds DNA and modulates the endonuclease activity of MUS81 complexes. Nucleic Acids Res. 41, 9741–9752 (2013).
124. Técher, H. et al. Signaling from Mus81-Eme2-Dependent DNA Damage Elicited by Chk1 Deficiency Modulates Replication Fork Speed and Origin Usage. Cell Rep. 14, 1114–1127 (2016).
125. Doe, C. L., Ahn, J. S., Dixon, J. & Whitby, M. C. Mus81-Eme1 and Rqh1 involvement in processing stalled and collapsed replication forks. J. Biol. Chem. 277, 32753–32759 (2002).
126. Fugger, K. et al. FBH1 co-operates with MUS81 in inducing DNA double-strand breaks and cell death following replication stress. Nat. Commun. 4, 1423 (2013).
127. Hanada, K. et al. The structure-specific endonuclease Mus81-Eme1 promotes conversion of interstrand DNA crosslinks into double-strands breaks. EMBO J. 25, 4921–4932 (2006).
128. Hanada, K. et al. The structure-specific endonuclease Mus81 contributes to replication restart by generating double-strand DNA breaks. Nat. Struct. Mol. Biol. 14, 1096–1104 (2007).
129. Regairaz, M. et al. Mus81-mediated DNA cleavage resolves replication forks stalled by topoisomerase I-DNA complexes. J. Cell Biol. 195, 739–749 (2011).
130. Forment, J. V., Blasius, M., Guerini, I. & Jackson, S. P. Structure-specific DNA endonuclease Mus81/Eme1 generates DNA damage caused by Chk1 inactivation. PloS One 6, (2011).
131. Sarbajna, S., Davies, D. & West, S. C. Roles of SLX1-SLX4, MUS81-EME1, and GEN1 in avoiding genome instability and mitotic catastrophe. Genes Dev. 28, 1124–1136 (2014).
132. Dehé, P.-M. et al. Regulation of Mus81-Eme1 Holliday junction resolvase in response to DNA damage. Nat. Struct. Mol. Biol. 20, 598–603 (2013).
133. Kai, M., Boddy, M. N., Russell, P. & Wang, T. S.-F. Replication checkpoint kinase Cds1 regulates Mus81 to preserve genome integrity during replication stress. Genes Dev. 19, 919–932 (2005).
134. Fekairi, S. et al. Human SLX4 is a Holliday junction resolvase subunit that binds multiple DNA repair/recombination endonucleases. Cell 138, 78–89 (2009).
135. Muñoz, I. M. et al. Coordination of structure-specific nucleases by human SLX4/BTBD12 is required for DNA repair. Mol. Cell 35, 116–127 (2009).
136. Nair, N., Castor, D., Macartney, T. & Rouse, J. Identification and characterization of MUS81 point mutations that abolish interaction with the SLX4 scaffold protein. DNA Repair 24, 131–137 (2014).
137. Svendsen, J. M. et al. Mammalian BTBD12/SLX4 assembles a Holliday junction resolvase and is required for DNA repair. Cell 138, 63–77 (2009).
138. Benitez, A. et al. Damage-dependent regulation of MUS81-EME1 by Fanconi anemia complementation group A protein. Nucleic Acids Res. 42, 1671–1683 (2014).
139. Fabre, F., Chan, A., Heyer, W.-D. & Gangloff, S. Alternate pathways involving Sgs1/TOPO3, Mus81/ Mms4, and Srs2 prevent formation of toxic recombination intermediates from single-stranded gaps created by DNA replication. Proc. Natl. Acad. Sci. U. S. A. 99, 16887–16892 (2002).
140. McPherson, J. P. et al. Involvement of mammalian Mus81 in genome integrity and tumor suppression. Science 304, 1822–1826 (2004).
141. Naim, V., Wilhelm, T., Debatisse, M. & Rosselli, F. ERCC1 and MUS81-EME1 promote sister chromatid separation by processing late replication intermediates at common fragile sites during mitosis. Nat. Cell Biol. 15, 1008–1015 (2013).
142. Ying, S. et al. MUS81 promotes common fragile site expression. Nat. Cell Biol. 15, 1001–1007 (2013).
143. Harrigan, J. A. et al. Replication stress induces 53BP1-containing OPT domains in G1 cells. J. Cell Biol. 193, 97–108 (2011).

98
144. Lukas, C. et al. 53BP1 nuclear bodies form around DNA lesions generated by mitotic transmission of chromosomes under replication stress. Nat. Cell Biol. 13, 243–253 (2011).
145. Gallo-Fernández, M., Saugar, I., Ortiz-Bazán, M. Á., Vázquez, M. V. & Tercero, J. A. Cell cycle-dependent regulation of the nuclease activity of Mus81-Eme1/Mms4. Nucleic Acids Res. (2012). doi:10.1093/nar/gks599
146. Matos, J., Blanco, M. G., Maslen, S., Skehel, J. M. & West, S. C. Regulatory control of the resolution of DNA recombination intermediates during meiosis and mitosis. Cell 147, 158–172 (2011).
147. Saugar, I. et al. Temporal regulation of the Mus81-Mms4 endonuclease ensures cell survival under conditions of DNA damage. Nucleic Acids Res. 41, 8943–8958 (2013).
148. Szakal, B. & Branzei, D. Premature Cdk1/Cdc5/Mus81 pathway activation induces aberrant replication and deleterious crossover. EMBO J. 32, 1155–1167 (2013).
149. Chavdarova, M. et al. Srs2 promotes Mus81-Mms4-mediated resolution of recombination intermediates. Nucleic Acids Res. 43, 3626–3642 (2015).
150. Froget, B., Blaisonneau, J., Lambert, S. & Baldacci, G. Cleavage of stalled forks by fission yeast Mus81/Eme1 in absence of DNA replication checkpoint. Mol. Biol. Cell 19, 445–456 (2008).
151. Matulova, P. et al. Cooperativity of Mus81.Mms4 with Rad54 in the resolution of recombination and replication intermediates. J. Biol. Chem. 284, 7733–7745 (2009).
152. Mazina, O. M. & Mazin, A. V. Human Rad54 protein stimulates human Mus81-Eme1 endonuclease. Proc. Natl. Acad. Sci. U. S. A. 105, 18249–18254 (2008).
153. Zhang, R. et al. BLM helicase facilitates Mus81 endonuclease activity in human cells. Cancer Res. 65, 2526–2531 (2005).
154. Ghamrasni, S. E. et al. Rad54 and Mus81 cooperation promotes DNA damage repair and restrains chromosome missegregation. Oncogene (2016). doi:10.1038/onc.2016.16
155. Kang, M.-J. et al. Genetic and functional interactions between Mus81-Mms4 and Rad27. Nucleic Acids Res. 38, 7611–7625 (2010).
156. Shin, Y.-K., Amangyeld, T., Nguyen, T. A., Munashingha, P. R. & Seo, Y.-S. Human MUS81 complexes stimulate flap endonuclease 1. FEBS J. 279, 2412–2430 (2012).
157. Abraham, J. et al. Eme1 is involved in DNA damage processing and maintenance of genomic stability in mammalian cells. EMBO J. 22, 6137–6147 (2003).
158. Dendouga, N. et al. Disruption of murine Mus81 increases genomic instability and DNA damage sensitivity but does not promote tumorigenesis. Mol. Cell. Biol. 25, 7569–7579 (2005).
159. Hiyama, T. et al. Haploinsufficiency of the Mus81-Eme1 endonuclease activates the intra-S-phase and G2/M checkpoints and promotes rereplication in human cells. Nucleic Acids Res. 34, 880–892 (2006).
160. Wu, F. et al. Decreased expression of methyl methansulfonate and ultraviolet-sensitive gene clone 81 (Mus81) is correlated with a poor prognosis in patients with hepatocellular carcinoma. Cancer 112, 2002–2010 (2008).
161. El Ghamrasni, S. et al. Inactivation of chk2 and mus81 leads to impaired lymphocytes development, reduced genomic instability, and suppression of cancer. PLoS Genet. 7, e1001385 (2011).
162. Ciccia, A., McDonald, N. & West, S. C. Structural and functional relationships of the XPF/MUS81 family of proteins. Annu. Rev. Biochem. 77, 259–287 (2008).
163. Sancar, A. DNA excision repair. Annu. Rev. Biochem. 65, 43–81 (1996). 164. Wood, R. D. Nucleotide excision repair in mammalian cells. J. Biol. Chem. 272, 23465–23468
(1997). 165. de Laat, W. L., Appeldoorn, E., Jaspers, N. G. & Hoeijmakers, J. H. DNA structural elements
required for ERCC1-XPF endonuclease activity. J. Biol. Chem. 273, 7835–7842 (1998).

99
166. Niedernhofer, L. J. et al. The structure-specific endonuclease Ercc1-Xpf is required for targeted gene replacement in embryonic stem cells. EMBO J. 20, 6540–6549 (2001).
167. Sijbers, A. M. et al. Xeroderma pigmentosum group F caused by a defect in a structure-specific DNA repair endonuclease. Cell 86, 811–822 (1996).
168. Svendsen, J. M. et al. Mammalian BTBD12/SLX4 assembles a Holliday junction resolvase and is required for DNA repair. Cell 138, 63–77 (2009).
169. Tripsianes, K. et al. The structure of the human ERCC1/XPF interaction domains reveals a complementary role for the two proteins in nucleotide excision repair. Struct. Lond. Engl. 1993 13, 1849–1858 (2005).
170. Zhu, X.-D. et al. ERCC1/XPF removes the 3’ overhang from uncapped telomeres and represses formation of telomeric DNA-containing double minute chromosomes. Mol. Cell 12, 1489–1498 (2003).
171. Reynolds, R. J. & Friedberg, E. C. Molecular mechanisms of pyrimidine dimer excision in Saccharomyces cerevisiae: incision of ultraviolet-irradiated deoxyribonucleic acid in vivo. J. Bacteriol. 146, 692–704 (1981).
172. Guzder, S. N., Sommers, C. H., Prakash, L. & Prakash, S. Complex formation with damage recognition protein Rad14 is essential for Saccharomyces cerevisiae Rad1-Rad10 nuclease to perform its function in nucleotide excision repair in vivo. Mol. Cell. Biol. 26, 1135–1141 (2006).
173. Li, L., Peterson, C. A., Lu, X. & Legerski, R. J. Mutations in XPA that prevent association with ERCC1 are defective in nucleotide excision repair. Mol. Cell. Biol. 15, 1993–1998 (1995).
174. de Boer, J. & Hoeijmakers, J. H. Nucleotide excision repair and human syndromes. Carcinogenesis 21, 453–460 (2000).
175. Prakash, S. & Prakash, L. Nucleotide excision repair in yeast. Mutat. Res. 451, 13–24 (2000). 176. Wood, R. D. Mammalian nucleotide excision repair proteins and interstrand crosslink repair.
Environ. Mol. Mutagen. 51, 520–526 (2010). 177. Andersen, S. L. et al. Drosophila MUS312 and the vertebrate ortholog BTBD12 interact with
DNA structure-specific endonucleases in DNA repair and recombination. Mol. Cell 35, 128–135 (2009).
178. Kim, Y. et al. Mutations of the SLX4 gene in Fanconi anemia. Nat. Genet. 43, 142–146 (2011). 179. Kuraoka, I. et al. Repair of an interstrand DNA cross-link initiated by ERCC1-XPF
repair/recombination nuclease. J. Biol. Chem. 275, 26632–26636 (2000). 180. Stoepker, C. et al. SLX4, a coordinator of structure-specific endonucleases, is mutated in a new
Fanconi anemia subtype. Nat. Genet. 43, 138–141 (2011). 181. Niedernhofer, L. J. et al. The structure-specific endonuclease Ercc1-Xpf is required to resolve
DNA interstrand cross-link-induced double-strand breaks. Mol. Cell. Biol. 24, 5776–5787 (2004).
182. Adair, G. M. et al. Role of ERCC1 in removal of long non-homologous tails during targeted homologous recombination. EMBO J. 19, 5552–5561 (2000).
183. Al-Minawi, A. Z., Saleh-Gohari, N. & Helleday, T. The ERCC1/XPF endonuclease is required for efficient single-strand annealing and gene conversion in mammalian cells. Nucleic Acids Res. 36, 1–9 (2008).
184. Fishman-Lobell, J. & Haber, J. E. Removal of nonhomologous DNA ends in double-strand break recombination: the role of the yeast ultraviolet repair gene RAD1. Science 258, 480–484 (1992).
185. Schiestl, R. H. & Prakash, S. RAD10, an excision repair gene of Saccharomyces cerevisiae, is involved in the RAD1 pathway of mitotic recombination. Mol. Cell. Biol. 10, 2485–2491 (1990).

100
186. Tomkinson, A. E., Bardwell, A. J., Bardwell, L., Tappe, N. J. & Friedberg, E. C. Yeast DNA repair and recombination proteins Rad1 and Rad10 constitute a single-stranded-DNA endonuclease. Nature 362, 860–862 (1993).
187. Welz-Voegele, C. & Jinks-Robertson, S. Sequence divergence impedes crossover more than noncrossover events during mitotic gap repair in yeast. Genetics 179, 1251–1262 (2008).
188. Toh, G. W.-L. et al. Mec1/Tel1-dependent phosphorylation of Slx4 stimulates Rad1-Rad10-dependent cleavage of non-homologous DNA tails. DNA Repair 9, 718–726 (2010).
189. Mardirosian, M. et al. Saw1 localizes to repair sites but is not required for recruitment of Rad10 to repair intermediates bearing short non-homologous 3’ flaps during single-strand annealing in S. cerevisiae. Mol. Cell. Biochem. 412, 131–139 (2016).
190. Boiteux, S. & Guillet, M. Abasic sites in DNA: repair and biological consequences in Saccharomyces cerevisiae. DNA Repair 3, 1–12 (2004).
191. Fisher, L. A., Samson, L. & Bessho, T. Removal of reactive oxygen species-induced 3’-blocked ends by XPF-ERCC1. Chem. Res. Toxicol. 24, 1876–1881 (2011).
192. Guzder, S. N. et al. Requirement of yeast Rad1-Rad10 nuclease for the removal of 3’-blocked termini from DNA strand breaks induced by reactive oxygen species. Genes Dev. 18, 2283–2291 (2004).
193. Ma, J.-L., Kim, E. M., Haber, J. E. & Lee, S. E. Yeast Mre11 and Rad1 proteins define a Ku-independent mechanism to repair double-strand breaks lacking overlapping end sequences. Mol. Cell. Biol. 23, 8820–8828 (2003).
194. Yan, C. T. et al. IgH class switching and translocations use a robust non-classical end-joining pathway. Nature 449, 478–482 (2007).
195. Diamante, G. et al. SAW1 is required for SDSA double-strand break repair in S. cerevisiae. Biochem. Biophys. Res. Commun. 445, 602–607 (2014).
196. Matsumura, Y., Nishigori, C., Yagi, T., Imamura, S. & Takebe, H. Characterization of molecular defects in xeroderma pigmentosum group F in relation to its clinically mild symptoms. Hum. Mol. Genet. 7, 969–974 (1998).
197. Niedernhofer, L. J. et al. A new progeroid syndrome reveals that genotoxic stress suppresses the somatotroph axis. Nature 444, 1038–1043 (2006).
198. Zhu, X.-D. et al. ERCC1/XPF removes the 3’ overhang from uncapped telomeres and represses formation of telomeric DNA-containing double minute chromosomes. Mol. Cell 12, 1489–1498 (2003).
199. Olaussen, K. A. et al. DNA repair by ERCC1 in non-small-cell lung cancer and cisplatin-based adjuvant chemotherapy. N. Engl. J. Med. 355, 983–991 (2006).
200. Simon, G. R., Sharma, S., Cantor, A., Smith, P. & Bepler, G. ERCC1 expression is a predictor of survival in resected patients with non-small cell lung cancer. Chest 127, 978–983 (2005).
201. Song, L., Ritchie, A.-M., McNeil, E. M., Li, W. & Melton, D. W. Identification of DNA repair gene Ercc1 as a novel target in melanoma. Pigment Cell Melanoma Res. 24, 966–971 (2011).
202. Takenaka, T. et al. Combined evaluation of Rad51 and ERCC1 expressions for sensitivity to platinum agents in non-small cell lung cancer. Int. J. Cancer J. Int. Cancer 121, 895–900 (2007).
203. Bachrati, C. Z. & Hickson, I. D. RecQ helicases: guardian angels of the DNA replication fork. Chromosoma 117, 219–233 (2008).
204. Bohr, V. A. Rising from the RecQ-age: the role of human RecQ helicases in genome maintenance. Trends Biochem. Sci. 33, 609–620 (2008).
205. Croteau, D. L., Popuri, V., Opresko, P. L. & Bohr, V. A. Human RecQ helicases in DNA repair, recombination, and replication. Annu. Rev. Biochem. 83, 519–552 (2014).

101
206. Ouyang, K. J., Woo, L. L. & Ellis, N. A. Homologous recombination and maintenance of genome integrity: cancer and aging through the prism of human RecQ helicases. Mech. Ageing Dev. 129, 425–440 (2008).
207. Sharma, S., Doherty, K. M. & Brosh, R. M., Jr. Mechanisms of RecQ helicases in pathways of DNA metabolism and maintenance of genomic stability. Biochem. J. 398, 319–337 (2006).
208. Ellis, N. A. et al. The Bloom’s syndrome gene product is homologous to RecQ helicases. Cell 83, 655–666 (1995).
209. Kitao, S., Lindor, N. M., Shiratori, M., Furuichi, Y. & Shimamoto, A. Rothmund-thomson syndrome responsible gene, RECQL4: genomic structure and products. Genomics 61, 268–276 (1999).
210. Van Maldergem, L. et al. Revisiting the craniosynostosis-radial ray hypoplasia association: Baller-Gerold syndrome caused by mutations in the RECQL4 gene. J. Med. Genet. 43, 148–152 (2006).
211. Siitonen, H. A. et al. Molecular defect of RAPADILINO syndrome expands the phenotype spectrum of RECQL diseases. Hum. Mol. Genet. 12, 2837–2844 (2003).
212. Yu, C. E. et al. Positional cloning of the Werner’s syndrome gene. Science 272, 258–262 (1996). 213. Matsushita, Y. et al. The level of RECQL1 expression is a prognostic factor for epithelial ovarian
cancer. J. Ovarian Res. 7, 107 (2014). 214. Sanada, S. et al. RECQL1 DNA repair helicase: a potential therapeutic target and a proliferative
marker against ovarian cancer. PloS One 8, e72820 (2013). 215. Tao, J. et al. RECQL1 plays an important role in the development of tongue squamous cell
carcinoma. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 33, 1579–1590 (2014).
216. Constantinou, A. et al. Werner’s syndrome protein (WRN) migrates Holliday junctions and co-localizes with RPA upon replication arrest. EMBO Rep. 1, 80–84 (2000).
217. Fry, M. & Loeb, L. A. Human werner syndrome DNA helicase unwinds tetrahelical structures of the fragile X syndrome repeat sequence d(CGG)n. J. Biol. Chem. 274, 12797–12802 (1999).
218. Karow, J. K., Constantinou, A., Li, J. L., West, S. C. & Hickson, I. D. The Bloom’s syndrome gene product promotes branch migration of holliday junctions. Proc. Natl. Acad. Sci. U. S. A. 97, 6504–6508 (2000).
219. Keller, H. et al. The intrinsically disordered amino-terminal region of human RecQL4: multiple DNA-binding domains confer annealing, strand exchange and G4 DNA binding. Nucleic Acids Res. 42, 12614–12627 (2014).
220. Mohaghegh, P., Karow, J. K., Brosh, R. M., Jr, Bohr, V. A. & Hickson, I. D. The Bloom’s and Werner’s syndrome proteins are DNA structure-specific helicases. Nucleic Acids Res. 29, 2843–2849 (2001).
221. Popuri, V. et al. The Human RecQ helicases, BLM and RECQ1, display distinct DNA substrate specificities. J. Biol. Chem. 283, 17766–17776 (2008).
222. Cheok, C. F., Wu, L., Garcia, P. L., Janscak, P. & Hickson, I. D. The Bloom’s syndrome helicase promotes the annealing of complementary single-stranded DNA. Nucleic Acids Res. 33, 3932–3941 (2005).
223. Garcia, P. L., Liu, Y., Jiricny, J., West, S. C. & Janscak, P. Human RECQ5beta, a protein with DNA helicase and strand-annealing activities in a single polypeptide. EMBO J. 23, 2882–2891 (2004).
224. Machwe, A., Xiao, L., Groden, J., Matson, S. W. & Orren, D. K. RecQ family members combine strand pairing and unwinding activities to catalyze strand exchange. J. Biol. Chem. 280, 23397–23407 (2005).
225. Macris, M. A., Krejci, L., Bussen, W., Shimamoto, A. & Sung, P. Biochemical characterization of the RECQ4 protein, mutated in Rothmund-Thomson syndrome. DNA Repair 5, 172–180 (2006).

102
226. Sharma, S. et al. Biochemical analysis of the DNA unwinding and strand annealing activities catalyzed by human RECQ1. J. Biol. Chem. 280, 28072–28084 (2005).
227. Bugreev, D. V., Brosh, R. M. & Mazin, A. V. RECQ1 possesses DNA branch migration activity. J. Biol. Chem. 283, 20231–20242 (2008).
228. Opresko, P. L., Sowd, G. & Wang, H. The Werner syndrome helicase/exonuclease processes mobile D-loops through branch migration and degradation. PloS One 4, e4825 (2009).
229. Bernstein, D. A., Zittel, M. C. & Keck, J. L. High-resolution structure of the E.coli RecQ helicase catalytic core. EMBO J. 22, 4910–4921 (2003).
230. Huber, M. D., Duquette, M. L., Shiels, J. C. & Maizels, N. A conserved G4 DNA binding domain in RecQ family helicases. J. Mol. Biol. 358, 1071–1080 (2006).
231. Kitano, K., Kim, S.-Y. & Hakoshima, T. Structural basis for DNA strand separation by the unconventional winged-helix domain of RecQ helicase WRN. Struct. Lond. Engl. 1993 18, 177–187 (2010).
232. Pike, A. C. W. et al. Structure of the human RECQ1 helicase reveals a putative strand-separation pin. Proc. Natl. Acad. Sci. U. S. A. 106, 1039–1044 (2009).
233. Karmakar, P. et al. BLM is an early responder to DNA double-strand breaks. Biochem. Biophys. Res. Commun. 348, 62–69 (2006).
234. Lan, L. et al. Accumulation of Werner protein at DNA double-strand breaks in human cells. J. Cell Sci. 118, 4153–4162 (2005).
235. Samanta, S. & Karmakar, P. Recruitment of HRDC domain of WRN and BLM to the sites of DNA damage induced by mitomycin C and methyl methanesulfonate. Cell Biol. Int. 36, 873–881 (2012).
236. Wu, L. et al. The HRDC domain of BLM is required for the dissolution of double Holliday junctions. EMBO J. 24, 2679–2687 (2005).
237. Chatterjee, S. et al. Mechanistic insight into the interaction of BLM helicase with intra-strand G-quadruplex structures. Nat. Commun. 5, 5556 (2014).
238. Lucic, B. et al. A prominent β-hairpin structure in the winged-helix domain of RECQ1 is required for DNA unwinding and oligomer formation. Nucleic Acids Res. 39, 1703–1717 (2011).
239. Vindigni, A., Marino, F. & Gileadi, O. Probing the structural basis of RecQ helicase function. Biophys. Chem. 149, 67–77 (2010).
240. Zittel, M. C. & Keck, J. L. Coupling DNA-binding and ATP hydrolysis in Escherichia coli RecQ: role of a highly conserved aromatic-rich sequence. Nucleic Acids Res. 33, 6982–6991 (2005).
241. von Hippel, P. H. Helicases become mechanistically simpler and functionally more complex. Nat. Struct. Mol. Biol. 11, 494–496 (2004).
242. Lohman, T. M. & Bjornson, K. P. Mechanisms of helicase-catalyzed DNA unwinding. Annu. Rev. Biochem. 65, 169–214 (1996).
243. Muzzolini, L. et al. Different quaternary structures of human RECQ1 are associated with its dual enzymatic activity. PLoS Biol. 5, e20 (2007).
244. Pike, A. C. W. et al. Human RECQ1 helicase-driven DNA unwinding, annealing, and branch migration: insights from DNA complex structures. Proc. Natl. Acad. Sci. U. S. A. 112, 4286–4291 (2015).
245. Compton, S. A., Tolun, G., Kamath-Loeb, A. S., Loeb, L. A. & Griffith, J. D. The Werner syndrome protein binds replication fork and holliday junction DNAs as an oligomer. J. Biol. Chem. 283, 24478–24483 (2008).
246. Perry, J. J. P. et al. WRN exonuclease structure and molecular mechanism imply an editing role in DNA end processing. Nat. Struct. Mol. Biol. 13, 414–422 (2006).
247. Karow, J. K., Newman, R. H., Freemont, P. S. & Hickson, I. D. Oligomeric ring structure of the Bloom’s syndrome helicase. Curr. Biol. CB 9, 597–600 (1999).

103
248. Suzuki, T., Kohno, T. & Ishimi, Y. DNA helicase activity in purified human RECQL4 protein. J. Biochem. (Tokyo) 146, 327–335 (2009).
249. Marino, F. et al. Structural and biochemical characterization of an RNA/DNA binding motif in the N-terminal domain of RecQ4 helicases. Sci. Rep. 6, 21501 (2016).
250. Xu, H. Q. et al. The Escherichia coli RecQ helicase functions as a monomer. J. Biol. Chem. 278, 34925–34933 (2003).
251. Zhang, X.-D. et al. Escherichia coli RecQ is a rapid, efficient, and monomeric helicase. J. Biol. Chem. 281, 12655–12663 (2006).
252. Ashton, T. M. & Hickson, I. D. Yeast as a model system to study RecQ helicase function. DNA Repair 9, 303–314 (2010).
253. Bzymek, M., Thayer, N. H., Oh, S. D., Kleckner, N. & Hunter, N. Double Holliday junctions are intermediates of DNA break repair. Nature 464, 937–941 (2010).
254. Ira, G., Malkova, A., Liberi, G., Foiani, M. & Haber, J. E. Srs2 and Sgs1-TOPO3 suppress crossovers during double-strand break repair in yeast. Cell 115, 401–411 (2003).
255. Liberi, G. et al. Rad51-dependent DNA structures accumulate at damaged replication forks in sgs1 mutants defective in the yeast ortholog of BLM RecQ helicase. Genes Dev. 19, 339–350 (2005).
256. Oh, S. D. et al. BLM ortholog, Sgs1, prevents aberrant crossing-over by suppressing formation of multichromatid joint molecules. Cell 130, 259–272 (2007).
257. Lambert, S. et al. Homologous recombination restarts blocked replication forks at the expense of genome rearrangements by template exchange. Mol. Cell 39, 346–359 (2010).
258. Langerak, P., Mejia-Ramirez, E., Limbo, O. & Russell, P. Release of Ku and MRN from DNA ends by Mre11 nuclease activity and Ctp1 is required for homologous recombination repair of double-strand breaks. PLoS Genet. 7, e1002271 (2011).
259. Nanbu, T. et al. Fission Yeast Exo1 and Rqh1-Dna2 Redundantly Contribute to Resection of Uncapped Telomeres. PloS One 10, e0140456 (2015).
260. Zhang, J.-M. et al. Fission yeast Pxd1 promotes proper DNA repair by activating Rad16XPF and inhibiting Dna2. PLoS Biol. 12, e1001946 (2014).
261. Bochman, M. L., Paeschke, K., Chan, A. & Zakian, V. A. Hrq1, a homolog of the human RecQ4 helicase, acts catalytically and structurally to promote genome integrity. Cell Rep. 6, 346–356 (2014).
262. Choi, D.-H., Lee, R., Kwon, S.-H. & Bae, S.-H. Hrq1 functions independently of Sgs1 to preserve genome integrity in Saccharomyces cerevisiae. J. Microbiol. Seoul Korea 51, 105–112 (2013).
263. Groocock, L. M., Prudden, J., Perry, J. J. P. & Boddy, M. N. The RecQ4 orthologue Hrq1 is critical for DNA interstrand cross-link repair and genome stability in fission yeast. Mol. Cell. Biol. 32, 276–287 (2012).
264. Kwon, S.-H., Choi, D.-H., Lee, R. & Bae, S.-H. Saccharomyces cerevisiae Hrq1 requires a long 3’-tailed DNA substrate for helicase activity. Biochem. Biophys. Res. Commun. 427, 623–628 (2012).
265. Mendoza-Maldonado, R. et al. The human RECQ1 helicase is highly expressed in glioblastoma and plays an important role in tumor cell proliferation. Mol. Cancer 10, 83 (2011).
266. Berti, M. et al. Human RECQ1 promotes restart of replication forks reversed by DNA topoisomerase I inhibition. Nat. Struct. Mol. Biol. 20, 347–354 (2013).
267. Popuri, V., Croteau, D. L., Brosh, R. M. & Bohr, V. A. RECQ1 is required for cellular resistance to replication stress and catalyzes strand exchange on stalled replication fork structures. Cell Cycle Georget. Tex 11, 4252–4265 (2012).

104
268. Banerjee, T., Sommers, J. A., Huang, J., Seidman, M. M. & Brosh, R. M. Catalytic strand separation by RECQ1 is required for RPA-mediated response to replication stress. Curr. Biol. CB 25, 2830–2838 (2015).
269. Lu, X., Parvathaneni, S., Hara, T., Lal, A. & Sharma, S. Replication stress induces specific enrichment of RECQ1 at common fragile sites FRA3B and FRA16D. Mol. Cancer 12, 29 (2013).
270. Sharma, S. & Brosh, R. M., Jr. Human RECQ1 is a DNA damage responsive protein required for genotoxic stress resistance and suppression of sister chromatid exchanges. PloS One 2, e1297 (2007).
271. Popuri, V. et al. Human RECQL1 participates in telomere maintenance. Nucleic Acids Res. 42, 5671–5688 (2014).
272. Goto, M., Ishikawa, Y., Sugimoto, M. & Furuichi, Y. Werner syndrome: a changing pattern of clinical manifestations in Japan (1917~2008). Biosci. Trends 7, 13–22 (2013).
273. Lauper, J. M., Krause, A., Vaughan, T. L. & Monnat, R. J. Spectrum and risk of neoplasia in Werner syndrome: a systematic review. PloS One 8, e59709 (2013).
274. Fukuchi, K., Martin, G. M. & Monnat, R. J., Jr. Mutator phenotype of Werner syndrome is characterized by extensive deletions. Proc. Natl. Acad. Sci. U. S. A. 86, 5893–5897 (1989).
275. Martin, G. M. Genetic syndromes in man with potential relevance to the pathobiology of aging. Birth Defects Orig. Artic. Ser. 14, 5–39 (1978).
276. Ogburn, C. E. et al. An apoptosis-inducing genotoxin differentiates heterozygotic carriers for Werner helicase mutations from wild-type and homozygous mutants. Hum. Genet. 101, 121–125 (1997).
277. Okada, M., Goto, M., Furuichi, Y. & Sugimoto, M. Differential effects of cytotoxic drugs on mortal and immortalized B-lymphoblastoid cell lines from normal and Werner’s syndrome patients. Biol. Pharm. Bull. 21, 235–239 (1998).
278. Pichierri, P., Franchitto, A., Mosesso, P. & Palitti, F. Werner’s syndrome cell lines are hypersensitive to camptothecin-induced chromosomal damage. Mutat. Res. 456, 45–57 (2000).
279. Poot, M., Gollahon, K. A. & Rabinovitch, P. S. Werner syndrome lymphoblastoid cells are sensitive to camptothecin-induced apoptosis in S-phase. Hum. Genet. 104, 10–14 (1999).
280. Poot, M., Gollahon, K. A., Emond, M. J., Silber, J. R. & Rabinovitch, P. S. Werner syndrome diploid fibroblasts are sensitive to 4-nitroquinoline-N-oxide and 8-methoxypsoralen: implications for the disease phenotype. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 16, 757–758 (2002).
281. Salk, D., Au, K., Hoehn, H. & Martin, G. M. Cytogenetic aspects of Werner syndrome. Adv. Exp. Med. Biol. 190, 541–546 (1985).
282. Huang, S. et al. The premature ageing syndrome protein, WRN, is a 3’-->5’ exonuclease. Nat. Genet. 20, 114–116 (1998).
283. Machwe, A., Xiao, L., Lloyd, R. G., Bolt, E. & Orren, D. K. Replication fork regression in vitro by the Werner syndrome protein (WRN): holliday junction formation, the effect of leading arm structure and a potential role for WRN exonuclease activity. Nucleic Acids Res. 35, 5729–5747 (2007).
284. Franchitto, A. et al. Replication fork stalling in WRN-deficient cells is overcome by prompt activation of a MUS81-dependent pathway. J. Cell Biol. 183, 241–252 (2008).
285. Prince, P. R., Emond, M. J. & Monnat, R. J., Jr. Loss of Werner syndrome protein function promotes aberrant mitotic recombination. Genes Dev. 15, 933–938 (2001).
286. Saintigny, Y., Makienko, K., Swanson, C., Emond, M. J. & Monnat, R. J., Jr. Homologous recombination resolution defect in werner syndrome. Mol. Cell. Biol. 22, 6971–6978 (2002).

105
287. Sallmyr, A., Tomkinson, A. E. & Rassool, F. V. Up-regulation of WRN and DNA ligase IIIalpha in chronic myeloid leukemia: consequences for the repair of DNA double-strand breaks. Blood 112, 1413–1423 (2008).
288. Cooper, M. P. et al. Ku complex interacts with and stimulates the Werner protein. Genes Dev. 14, 907–912 (2000).
289. Kusumoto, R. et al. Werner protein cooperates with the XRCC4-DNA ligase IV complex in end-processing. Biochemistry (Mosc.) 47, 7548–7556 (2008).
290. Oshima, J., Huang, S., Pae, C., Campisi, J. & Schiestl, R. H. Lack of WRN results in extensive deletion at nonhomologous joining ends. Cancer Res. 62, 547–551 (2002).
291. Cheng, W.-H. et al. WRN is required for ATM activation and the S-phase checkpoint in response to interstrand cross-link-induced DNA double-strand breaks. Mol. Biol. Cell 19, 3923–3933 (2008).
292. Baynton, K. et al. WRN interacts physically and functionally with the recombination mediator protein RAD52. J. Biol. Chem. 278, 36476–36486 (2003).
293. Sharma, S. et al. WRN helicase and FEN-1 form a complex upon replication arrest and together process branchmigrating DNA structures associated with the replication fork. Mol. Biol. Cell 15, 734–750 (2004).
294. Sharma, S. et al. The interaction site of Flap Endonuclease-1 with WRN helicase suggests a coordination of WRN and PCNA. Nucleic Acids Res. 33, 6769–6781 (2005).
295. von Kobbe, C. et al. Central role for the Werner syndrome protein/poly(ADP-ribose) polymerase 1 complex in the poly(ADP-ribosyl)ation pathway after DNA damage. Mol. Cell. Biol. 23, 8601–8613 (2003).
296. Shah, S. N., Opresko, P. L., Meng, X., Lee, M. Y. W. T. & Eckert, K. A. DNA structure and the Werner protein modulate human DNA polymerase delta-dependent replication dynamics within the common fragile site FRA16D. Nucleic Acids Res. 38, 1149–1162 (2010).
297. Crabbe, L., Verdun, R. E., Haggblom, C. I. & Karlseder, J. Defective telomere lagging strand synthesis in cells lacking WRN helicase activity. Science 306, 1951–1953 (2004).
298. Laud, P. R. et al. Elevated telomere-telomere recombination in WRN-deficient, telomere dysfunctional cells promotes escape from senescence and engagement of the ALT pathway. Genes Dev. 19, 2560–2570 (2005).
299. Li, B., Jog, S. P., Reddy, S. & Comai, L. WRN controls formation of extrachromosomal telomeric circles and is required for TRF2DeltaB-mediated telomere shortening. Mol. Cell. Biol. 28, 1892–1904 (2008).
300. Machwe, A., Xiao, L. & Orren, D. K. TRF2 recruits the Werner syndrome (WRN) exonuclease for processing of telomeric DNA. Oncogene 23, 149–156 (2004).
301. Opresko, P. L. et al. Telomere-binding protein TRF2 binds to and stimulates the Werner and Bloom syndrome helicases. J. Biol. Chem. 277, 41110–41119 (2002).
302. Opresko, P. L. et al. POT1 stimulates RecQ helicases WRN and BLM to unwind telomeric DNA substrates. J. Biol. Chem. 280, 32069–32080 (2005).
303. Edwards, D. N., Machwe, A., Chen, L., Bohr, V. A. & Orren, D. K. The DNA structure and sequence preferences of WRN underlie its function in telomeric recombination events. Nat. Commun. 6, 8331 (2015).
304. Brosh, R. M., Jr et al. Werner syndrome protein interacts with human flap endonuclease 1 and stimulates its cleavage activity. EMBO J. 20, 5791–5801 (2001).
305. Otterlei, M. et al. Werner syndrome protein participates in a complex with RAD51, RAD54, RAD54B and ATR in response to ICL-induced replication arrest. J. Cell Sci. 119, 5137–5146 (2006).

106
306. Sale, J. E., Lehmann, A. R. & Woodgate, R. Y-family DNA polymerases and their role in tolerance of cellular DNA damage. Nat. Rev. Mol. Cell Biol. 13, 141–152 (2012).
307. Trego, K. S. et al. The DNA repair endonuclease XPG interacts directly and functionally with the WRN helicase defective in Werner syndrome. Cell Cycle Georget. Tex 10, 1998–2007 (2011).
308. Zhang, N. et al. The Pso4 mRNA splicing and DNA repair complex interacts with WRN for processing of DNA interstrand cross-links. J. Biol. Chem. 280, 40559–40567 (2005).
309. Luo, G. et al. Cancer predisposition caused by elevated mitotic recombination in Bloom mice. Nat. Genet. 26, 424–429 (2000).
310. Manthei, K. A. & Keck, J. L. The BLM dissolvasome in DNA replication and repair. Cell. Mol. Life Sci. CMLS 70, 4067–4084 (2013).
311. Chaganti, R. S., Schonberg, S. & German, J. A manyfold increase in sister chromatid exchanges in Bloom’s syndrome lymphocytes. Proc. Natl. Acad. Sci. U. S. A. 71, 4508–4512 (1974).
312. Kitao, S. et al. Cloning of two new human helicase genes of the RecQ family: biological significance of multiple species in higher eukaryotes. Genomics 54, 443–452 (1998).
313. Yodh, J. G., Stevens, B. C., Kanagaraj, R., Janscak, P. & Ha, T. BLM helicase measures DNA unwound before switching strands and hRPA promotes unwinding reinitiation. EMBO J. 28, 405–416 (2009).
314. Bugreev, D. V., Yu, X., Egelman, E. H. & Mazin, A. V. Novel pro- and anti-recombination activities of the Bloom’s syndrome helicase. Genes Dev. 21, 3085–3094 (2007).
315. Chan, K. L. & Hickson, I. D. New insights into the formation and resolution of ultra-fine anaphase bridges. Semin. Cell Dev. Biol. 22, 906–912 (2011).
316. Chan, K.-L., North, P. S. & Hickson, I. D. BLM is required for faithful chromosome segregation and its localization defines a class of ultrafine anaphase bridges. EMBO J. 26, 3397–3409 (2007).
317. Hu, P. et al. Evidence for BLM and Topoisomerase IIIalpha interaction in genomic stability. Hum. Mol. Genet. 10, 1287–1298 (2001).
318. Raynard, S., Bussen, W. & Sung, P. A double Holliday junction dissolvasome comprising BLM, topoisomerase IIIalpha, and BLAP75. J. Biol. Chem. 281, 13861–13864 (2006).
319. Wu, L. & Hickson, I. D. The Bloom’s syndrome helicase suppresses crossing over during homologous recombination. Nature 426, 870–874 (2003).
320. Wu, L. et al. BLAP75/RMI1 promotes the BLM-dependent dissolution of homologous recombination intermediates. Proc. Natl. Acad. Sci. U. S. A. 103, 4068–4073 (2006).
321. Ralf, C., Hickson, I. D. & Wu, L. The Bloom’s syndrome helicase can promote the regression of a model replication fork. J. Biol. Chem. 281, 22839–22846 (2006).
322. Shimura, T. et al. Bloom’s syndrome helicase and Mus81 are required to induce transient double-strand DNA breaks in response to DNA replication stress. J. Mol. Biol. 375, 1152–1164 (2008).
323. Sharma, S. et al. Stimulation of flap endonuclease-1 by the Bloom’s syndrome protein. J. Biol. Chem. 279, 9847–9856 (2004).
324. Lillard-Wetherell, K. et al. Association and regulation of the BLM helicase by the telomere proteins TRF1 and TRF2. Hum. Mol. Genet. 13, 1919–1932 (2004).
325. Siitonen, H. A. et al. The mutation spectrum in RECQL4 diseases. Eur. J. Hum. Genet. EJHG 17, 151–158 (2009).
326. Im, J.-S. et al. Assembly of the Cdc45-Mcm2-7-GINS complex in human cells requires the Ctf4/And-1, RecQL4, and Mcm10 proteins. Proc. Natl. Acad. Sci. U. S. A. 106, 15628–15632 (2009).
327. Im, J.-S. et al. RecQL4 is required for the association of Mcm10 and Ctf4 with replication origins in human cells. Cell Cycle Georget. Tex 14, 1001–1009 (2015).

107
328. Kliszczak, M. et al. Interaction of RECQ4 and MCM10 is important for efficient DNA replication origin firing in human cells. Oncotarget 6, 40464–40479 (2015).
329. Sangrithi, M. N. et al. Initiation of DNA replication requires the RECQL4 protein mutated in Rothmund-Thomson syndrome. Cell 121, 887–898 (2005).
330. Xu, X., Rochette, P. J., Feyissa, E. A., Su, T. V. & Liu, Y. MCM10 mediates RECQ4 association with MCM2-7 helicase complex during DNA replication. EMBO J. 28, 3005–3014 (2009).
331. Matsuno, K., Kumano, M., Kubota, Y., Hashimoto, Y. & Takisawa, H. The N-terminal noncatalytic region of Xenopus RecQ4 is required for chromatin binding of DNA polymerase alpha in the initiation of DNA replication. Mol. Cell. Biol. 26, 4843–4852 (2006).
332. Croteau, D. L. et al. RECQL4 localizes to mitochondria and preserves mitochondrial DNA integrity. Aging Cell 11, 456–466 (2012).
333. De, S. et al. RECQL4 is essential for the transport of p53 to mitochondria in normal human cells in the absence of exogenous stress. J. Cell Sci. 125, 2509–2522 (2012).
334. Gupta, S. et al. RECQL4 and p53 potentiate the activity of polymerase γ and maintain the integrity of the human mitochondrial genome. Carcinogenesis 35, 34–45 (2014).
335. Wang, J.-T., Xu, X., Alontaga, A. Y., Chen, Y. & Liu, Y. Impaired p32 regulation caused by the lymphoma-prone RECQ4 mutation drives mitochondrial dysfunction. Cell Rep. 7, 848–858 (2014).
336. Fan, W. & Luo, J. RecQ4 facilitates UV light-induced DNA damage repair through interaction with nucleotide excision repair factor xeroderma pigmentosum group A (XPA). J. Biol. Chem. 283, 29037–29044 (2008).
337. Ghosh, A. K. et al. RECQL4, the protein mutated in Rothmund-Thomson syndrome, functions in telomere maintenance. J. Biol. Chem. 287, 196–209 (2012).
338. Schurman, S. H. et al. Direct and indirect roles of RECQL4 in modulating base excision repair capacity. Hum. Mol. Genet. 18, 3470–3483 (2009).
339. Woo, L. L., Futami, K., Shimamoto, A., Furuichi, Y. & Frank, K. M. The Rothmund-Thomson gene product RECQL4 localizes to the nucleolus in response to oxidative stress. Exp. Cell Res. 312, 3443–3457 (2006).
340. Kumata, Y. et al. Possible involvement of RecQL4 in the repair of double-strand DNA breaks in Xenopus egg extracts. Biochim. Biophys. Acta 1773, 556–564 (2007).
341. Singh, D. K. et al. The involvement of human RECQL4 in DNA double-strand break repair. Aging Cell 9, 358–371 (2010).
342. Petkovic, M., Dietschy, T., Freire, R., Jiao, R. & Stagljar, I. The human Rothmund-Thomson syndrome gene product, RECQL4, localizes to distinct nuclear foci that coincide with proteins involved in the maintenance of genome stability. J. Cell Sci. 118, 4261–4269 (2005).
343. Shamanna, R. A. et al. RECQ helicase RECQL4 participates in non-homologous end joining and interacts with the Ku complex. Carcinogenesis 35, 2415–2424 (2014).
344. Wu, J., Zhi, L., Dai, X., Cai, Q. & Ma, W. Decreased RECQL5 correlated with disease progression of osteosarcoma. Biochem. Biophys. Res. Commun. 467, 617–622 (2015).
345. Sakurai, H. et al. RecQ5 protein translocation into the nucleus by a nuclear localization signal. Biol. Pharm. Bull. 36, 1159–1166 (2013).
346. Sekelsky, J. J., Brodsky, M. H., Rubin, G. M. & Hawley, R. S. Drosophila and human RecQ5 exist in different isoforms generated by alternative splicing. Nucleic Acids Res. 27, 3762–3769 (1999).
347. Shimamoto, A., Nishikawa, K., Kitao, S. & Furuichi, Y. Human RecQ5beta, a large isomer of RecQ5 DNA helicase, localizes in the nucleoplasm and interacts with topoisomerases 3alpha and 3beta. Nucleic Acids Res. 28, 1647–1655 (2000).

108
348. Kawabe, T. et al. Differential regulation of human RecQ family helicases in cell transformation and cell cycle. Oncogene 19, 4764–4772 (2000).
349. Zheng, L. et al. MRE11 complex links RECQ5 helicase to sites of DNA damage. Nucleic Acids Res. 37, 2645–2657 (2009).
350. Hu, Y. et al. RECQL5/Recql5 helicase regulates homologous recombination and suppresses tumor formation via disruption of Rad51 presynaptic filaments. Genes Dev. 21, 3073–3084 (2007).
351. Hu, Y., Lu, X., Zhou, G., Barnes, E. L. & Luo, G. Recql5 plays an important role in DNA replication and cell survival after camptothecin treatment. Mol. Biol. Cell 20, 114–123 (2009).
352. Kanagaraj, R., Saydam, N., Garcia, P. L., Zheng, L. & Janscak, P. Human RECQ5beta helicase promotes strand exchange on synthetic DNA structures resembling a stalled replication fork. Nucleic Acids Res. 34, 5217–5231 (2006).
353. Khadka, P., Croteau, D. L. & Bohr, V. A. RECQL5 has unique strand annealing properties relative to the other human RecQ helicase proteins. DNA Repair 37, 53–66 (2016).
354. Speina, E. et al. Human RECQL5beta stimulates flap endonuclease 1. Nucleic Acids Res. 38, 2904–2916 (2010).
355. Aygün, O., Svejstrup, J. & Liu, Y. A RECQ5-RNA polymerase II association identified by targeted proteomic analysis of human chromatin. Proc. Natl. Acad. Sci. U. S. A. 105, 8580–8584 (2008).
356. Aygün, O. et al. Direct inhibition of RNA polymerase II transcription by RECQL5. J. Biol. Chem. 284, 23197–23203 (2009).
357. Kanagaraj, R. et al. RECQ5 helicase associates with the C-terminal repeat domain of RNA polymerase II during productive elongation phase of transcription. Nucleic Acids Res. 38, 8131–8140 (2010).
358. Ramamoorthy, M. et al. RECQL5 cooperates with Topoisomerase II alpha in DNA decatenation and cell cycle progression. Nucleic Acids Res. 40, 1621–1635 (2012).
359. Sakurai, H., Okado, M., Ito, F. & Kawasaki, K. Anaphase DNA bridges induced by lack of RecQ5 in Drosophila syncytial embryos. FEBS Lett. 585, 1923–1928 (2011).
360. Singh, D. K. et al. The human RecQ helicases BLM and RECQL4 cooperate to preserve genome stability. Nucleic Acids Res. 40, 6632–6648 (2012).
361. von Kobbe, C. et al. Colocalization, physical, and functional interaction between Werner and Bloom syndrome proteins. J. Biol. Chem. 277, 22035–22044 (2002).
362. Popuri, V. et al. RECQL5 plays co-operative and complementary roles with WRN syndrome helicase. Nucleic Acids Res. 41, 881–899 (2013).
363. Chiolo, I. et al. Srs2 and Sgs1 DNA helicases associate with Mre11 in different subcomplexes following checkpoint activation and CDK1-mediated Srs2 phosphorylation. Mol. Cell. Biol. 25, 5738–5751 (2005).
364. Mullen, J. R., Nallaseth, F. S., Lan, Y. Q., Slagle, C. E. & Brill, S. J. Yeast Rmi1/Nce4 controls genome stability as a subunit of the Sgs1-TOPO3 complex. Mol. Cell. Biol. 25, 4476–4487 (2005).
365. Ptacek, J. et al. Global analysis of protein phosphorylation in yeast. Nature 438, 679–684 (2005).
366. Sollier, J. et al. The Saccharomyces cerevisiae Esc2 and Smc5-6 proteins promote sister chromatid junction-mediated intra-S repair. Mol. Biol. Cell 20, 1671–1682 (2009).
367. Wang, T.-F. & Kung, W.-M. Supercomplex formation between Mlh1-Mlh3 and Sgs1-TOPO3 heterocomplexes in meiotic yeast cells. Biochem. Biophys. Res. Commun. 296, 949–953 (2002).
368. Wu, L., Davies, S. L., Levitt, N. C. & Hickson, I. D. Potential role for the BLM helicase in recombinational repair via a conserved interaction with RAD51. J. Biol. Chem. 276, 19375–19381 (2001).

109
369. Cheng, C.-H. et al. SUMO modifications control assembly of synaptonemal complex and polycomplex in meiosis of Saccharomyces cerevisiae. Genes Dev. 20, 2067–2081 (2006).
370. Takahashi, Y., Kahyo, T., Toh-E, A., Yasuda, H. & Kikuchi, Y. Yeast Ull1/Siz1 is a novel SUMO1/Smt3 ligase for septin components and functions as an adaptor between conjugating enzyme and substrates. J. Biol. Chem. 276, 48973–48977 (2001).
371. Ulrich, H. D. The SUMO system: an overview. Methods Mol. Biol. Clifton NJ 497, 3–16 (2009). 372. Zhao, J. Sumoylation regulates diverse biological processes. Cell. Mol. Life Sci. CMLS 64, 3017–
3033 (2007). 373. Bayer, P. et al. Structure determination of the small ubiquitin-related modifier SUMO-1. J. Mol.
Biol. 280, 275–286 (1998). 374. Johnson, E. S., Schwienhorst, I., Dohmen, R. J. & Blobel, G. The ubiquitin-like protein Smt3p is
activated for conjugation to other proteins by an Aos1p/Uba2p heterodimer. EMBO J. 16, 5509–5519 (1997).
375. Mahajan, R., Delphin, C., Guan, T., Gerace, L. & Melchior, F. A small ubiquitin-related polypeptide involved in targeting RanGAP1 to nuclear pore complex protein RanBP2. Cell 88, 97–107 (1997).
376. Müller, S., Hoege, C., Pyrowolakis, G. & Jentsch, S. SUMO, ubiquitin’s mysterious cousin. Nat. Rev. Mol. Cell Biol. 2, 202–210 (2001).
377. Xu, J. et al. A novel method for high accuracy sumoylation site prediction from protein sequences. BMC Bioinformatics 9, 8 (2008).
378. Johnson, E. S. & Blobel, G. Ubc9p is the conjugating enzyme for the ubiquitin-like protein Smt3p. J. Biol. Chem. 272, 26799–26802 (1997).
379. Gareau, J. R. & Lima, C. D. The SUMO pathway: emerging mechanisms that shape specificity, conjugation and recognition. Nat. Rev. Mol. Cell Biol. 11, 861–871 (2010).
380. Hickey, C. M., Wilson, N. R. & Hochstrasser, M. Function and regulation of SUMO proteases. Nat. Rev. Mol. Cell Biol. 13, 755–766 (2012).
381. Li, S. J. & Hochstrasser, M. A new protease required for cell-cycle progression in yeast. Nature 398, 246–251 (1999).
382. Li, S. J. & Hochstrasser, M. The yeast ULP2 (SMT4) gene encodes a novel protease specific for the ubiquitin-like Smt3 protein. Mol. Cell. Biol. 20, 2367–2377 (2000).
383. Takahashi, Y., Iwase, M., Strunnikov, A. V. & Kikuchi, Y. Cytoplasmic sumoylation by PIAS-type Siz1-SUMO ligase. Cell Cycle Georget. Tex 7, 1738–1744 (2008).
384. Ulrich, H. D. The fast-growing business of SUMO chains. Mol. Cell 32, 301–305 (2008). 385. Hay, R. T. SUMO-specific proteases: a twist in the tail. Trends Cell Biol. 17, 370–376 (2007). 386. Kerscher, O. SUMO junction-what’s your function? New insights through SUMO-interacting
motifs. EMBO Rep. 8, 550–555 (2007). 387. Baba, D. et al. Crystal structure of thymine DNA glycosylase conjugated to SUMO-1. Nature
435, 979–982 (2005). 388. Kolesar, P., Sarangi, P., Altmannova, V., Zhao, X. & Krejci, L. Dual roles of the SUMO-interacting
motif in the regulation of Srs2 sumoylation. Nucleic Acids Res. 40, 7831–7843 (2012). 389. Steinacher, R. & Schär, P. Functionality of human thymine DNA glycosylase requires SUMO-
regulated changes in protein conformation. Curr. Biol. CB 15, 616–623 (2005). 390. Altmannová, V., Kolesár, P. & Krejčí, L. SUMO Wrestles with Recombination. Biomolecules 2,
350–375 (2012). 391. Sarangi, P. & Zhao, X. SUMO-mediated regulation of DNA damage repair and responses.
Trends Biochem. Sci. 40, 233–242 (2015). 392. Cremona, C. A. et al. Extensive DNA damage-induced sumoylation contributes to replication
and repair and acts in addition to the mec1 checkpoint. Mol. Cell 45, 422–432 (2012).

110
393. Cremona, C. A., Sarangi, P. & Zhao, X. Sumoylation and the DNA damage response. Biomolecules 2, 376–388 (2012).
394. Hoege, C., Pfander, B., Moldovan, G.-L., Pyrowolakis, G. & Jentsch, S. RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature 419, 135–141 (2002).
395. Papouli, E. et al. Crosstalk between SUMO and ubiquitin on PCNA is mediated by recruitment of the helicase Srs2p. Mol. Cell 19, 123–133 (2005).
396. Kannouche, P. L., Wing, J. & Lehmann, A. R. Interaction of human DNA polymerase eta with monoubiquitinated PCNA: a possible mechanism for the polymerase switch in response to DNA damage. Mol. Cell 14, 491–500 (2004).
397. Stelter, P. & Ulrich, H. D. Control of spontaneous and damage-induced mutagenesis by SUMO and ubiquitin conjugation. Nature 425, 188–191 (2003).
398. Watanabe, K. et al. Rad18 guides poleta to replication stalling sites through physical interaction and PCNA monoubiquitination. EMBO J. 23, 3886–3896 (2004).
399. Burkovics, P. et al. Srs2 mediates PCNA-SUMO-dependent inhibition of DNA repair synthesis. EMBO J. 32, 742–755 (2013).
400. Kolesar, P., Altmannova, V., Silva, S., Lisby, M. & Krejci, L. Pro-recombination role of Srs2 requires SUMO but is independent of PCNA interaction. J. Biol. Chem. (2016). doi:10.1074/jbc.M115.685891
401. Hass, C. S., Chen, R. & Wold, M. S. Detection of posttranslational modifications of replication protein A. Methods Mol. Biol. Clifton NJ 922, 193–204 (2012).
402. Lu, C.-Y., Tsai, C.-H., Brill, S. J. & Teng, S.-C. Sumoylation of the BLM ortholog, Sgs1, promotes telomere-telomere recombination in budding yeast. Nucleic Acids Res. 38, 488–498 (2010).
403. Morris, J. R. et al. The SUMO modification pathway is involved in the BRCA1 response to genotoxic stress. Nature 462, 886–890 (2009).
404. Ouyang, K. J. et al. SUMO modification regulates BLM and RAD51 interaction at damaged replication forks. PLoS Biol. 7, e1000252 (2009).
405. Woods, Y. L. et al. p14 Arf promotes small ubiquitin-like modifier conjugation of Werners helicase. J. Biol. Chem. 279, 50157–50166 (2004).
406. Altmannova, V. et al. Rad52 SUMOylation affects the efficiency of the DNA repair. Nucleic Acids Res. 38, 4708–4721 (2010).
407. Sacher, M., Pfander, B., Hoege, C. & Jentsch, S. Control of Rad52 recombination activity by double-strand break-induced SUMO modification. Nat. Cell Biol. 8, 1284–1290 (2006).
408. Vigasova, D. et al. Lif1 SUMOylation and its role in non-homologous end-joining. Nucleic Acids Res. 41, 5341–5353 (2013).
409. Sarangi, P. et al. Sumoylation influences DNA break repair partly by increasing the solubility of a conserved end resection protein. PLoS Genet. 11, e1004899 (2015).
410. Bologna, S. et al. Sumoylation regulates EXO1 stability and processing of DNA damage. Cell Cycle Georget. Tex 14, 2439–2450 (2015).
411. Burkovics, P. et al. The PCNA-associated protein PARI negatively regulates homologous recombination via the inhibition of DNA repair synthesis. Nucleic Acids Res. (2016). doi:10.1093/nar/gkw024
412. Takahashi, Y., Toh-E, A. & Kikuchi, Y. Comparative analysis of yeast PIAS-type SUMO ligases in vivo and in vitro. J. Biochem. (Tokyo) 133, 415–422 (2003).
413. Durkin, S. G. & Glover, T. W. Chromosome fragile sites. Annu. Rev. Genet. 41, 169–192 (2007). 414. Germann, S. M. et al. TopBP1/Dpb11 binds DNA anaphase bridges to prevent genome
instability. J. Cell Biol. 204, 45–59 (2014). 415. Chi, P., Van Komen, S., Sehorn, M. G., Sigurdsson, S. & Sung, P. Roles of ATP binding and ATP
hydrolysis in human Rad51 recombinase function. DNA Repair 5, 381–391 (2006).

111
416. Arlt, M. F. et al. BRCA1 is required for common-fragile-site stability via its G2/M checkpoint function. Mol. Cell. Biol. 24, 6701–6709 (2004).
417. Casper, A. M., Nghiem, P., Arlt, M. F. & Glover, T. W. ATR regulates fragile site stability. Cell 111, 779–789 (2002).
418. Roy, R., Chun, J. & Powell, S. N. BRCA1 and BRCA2: different roles in a common pathway of genome protection. Nat. Rev. Cancer 12, 68–78 (2012).
419. Xia, B. et al. Control of BRCA2 cellular and clinical functions by a nuclear partner, PALB2. Mol. Cell 22, 719–729 (2006).
420. Yuan, S. S. et al. BRCA2 is required for ionizing radiation-induced assembly of Rad51 complex in vivo. Cancer Res. 59, 3547–3551 (1999).
421. Richardson, C., Stark, J. M., Ommundsen, M. & Jasin, M. Rad51 overexpression promotes alternative double-strand break repair pathways and genome instability. Oncogene 23, 546–553 (2004).
422. Murfuni, I. et al. Perturbed replication induced genome wide or at common fragile sites is differently managed in the absence of WRN. Carcinogenesis 33, 1655–1663 (2012).
423. Blomster, H. A. et al. Novel proteomics strategy brings insight into the prevalence of SUMO-2 target sites. Mol. Cell. Proteomics MCP 8, 1382–1390 (2009).
424. Psakhye, I. & Jentsch, S. Protein group modification and synergy in the SUMO pathway as exemplified in DNA repair. Cell 151, 807–820 (2012).
425. Silver, H. R., Nissley, J. A., Reed, S. H., Hou, Y.-M. & Johnson, E. S. A role for SUMO in nucleotide excision repair. DNA Repair 10, 1243–1251 (2011).
426. Bardwell, A. J., Bardwell, L., Johnson, D. K. & Friedberg, E. C. Yeast DNA recombination and repair proteins Rad1 and Rad10 constitute a complex in vivo mediated by localized hydrophobic domains. Mol. Microbiol. 8, 1177–1188 (1993).
427. Smet-Nocca, C., Wieruszeski, J.-M., Léger, H., Eilebrecht, S. & Benecke, A. SUMO-1 regulates the conformational dynamics of thymine-DNA Glycosylase regulatory domain and competes with its DNA binding activity. BMC Biochem. 12, 4 (2011).
428. Zhao, Q. et al. GPS-SUMO: a tool for the prediction of sumoylation sites and SUMO-interaction motifs. Nucleic Acids Res. 42, W325-330 (2014).
429. Anckar, J. et al. Inhibition of DNA binding by differential sumoylation of heat shock factors. Mol. Cell. Biol. 26, 955–964 (2006).
430. Hardeland, U., Steinacher, R., Jiricny, J. & Schär, P. Modification of the human thymine-DNA glycosylase by ubiquitin-like proteins facilitates enzymatic turnover. EMBO J. 21, 1456–1464 (2002).

112
7. LIST OF ABBREVIATIONS
A - alanine
ALT – alternative lengthening of telomeres
Aph - aphidicolin
ATM – Ataxia Telangiectasia Mutated
ATP - adenosine triphosphate
ATR - Ataxia Telangiectasia - related
BER – base excision repair
BIR – break-induced repair
BGS – Barel-Garold syndrome
β-ME - betamercaptoethanol
BTR complex – BLM-TOPIIIα-RMI1/RMI2
complex
BS – Bloom syndrome
CDK – cyclin-dependent kinase
CFS – common fragile site
CO – cross-over
CPT – camptothecin
dHJ – double Holliday junctions
D – aspartic acid
DDK – Dbf4-dependent kinase
DNA – deoxyribonucleic acid
D-loop – displacement loop
dNTP – deoxyribonucleotide triphosphate
DTT - dithiothreitol
DSB – double-strand break
DSBR – double-strand break repair
dsDNA – double-strand DNA
E – glutamic acid
EMSA – electromobility shift assay
FA – Fanconi anemia
G – glycine
GTH - glutathione
HRDC – helicase and RNaseD-like C-
terminal
HhH – helix-hairpin-helix
HJ – Holliday junction
HR – homologous recombination
HU – hydroxyurea
ICL – interstrand crosslink
ICLR – interstrand crosslink repair
IR – ionizing radiation
K - lysine
LOH – loss of heterozygosity
LP-BER – long patch base excision repair
MMC – mitomycin C
MMEJ – microhomology-mediated end
joining
MMR – mismatch repair
MMS – methyl methansulfonate

113
MRN – MRE11/RAD50/NBS1
MRX – Mre11/Rad50/Xrs2
Mw – molecular weight
NCO – non-crossover
NER – nucleotide excision repair
NHEJ – non-homologous end joining
NLS – nuclear localization signal
NTP – nucleoside triphosphate
PCNA – proliferating cell nuclear antigen
pI – isoelectric point
Polβ – polymerase beta
Polδ – polymerase delta
Polη – polymerase eta
TBE – Tris/Borate/EDTA
TDG - thymine DNA glycosylase
TLS – translesion repair
RF – replication fork
RQC – RecQ C-terminal
RTS – Rothmund-Thomson syndrome
SCE – sister chromatid-exchange
ssDNA – single-strand DNA
SDS-PAGE – sodium dodecyl sulfate
polyacrylamide gel electrophoresis
SDSA – synthesis-dependent strand
annealing
SIM – SUMO interacting motif
SSE – structure-specific endonuclease
SRI domain – RNA polymerase II
interacting domain
SSA – single-strand annealing
STR complex – Sgs1-TOPO3-Rmi1 complex
SUMO – small ubiquitin-like modifier
UFB – ultrafine bridges
UV – ultraviolet light
WH – winged helix
WS – Werner syndrome
XFE – progeroid syndrome
XP – Xeroderma pigmentosum

114
8. LIST OF FIGURES
Figure 1.1: Scheme of HR pathways with human proteins.....................................................15
Figure 1.2: Schematic view of MUS81-EME1/EME2 specific DNA substrates....................19
Figure 1.3: Schematic view of human and yeast MUS81 complex domains..........................20
Figure 1.4: Schematic view of cell-cycle dependent MUS81 function..................................22
Figure 1.5: Schematic view of some MUS81 interactions.....................................................23
Figure 1.6: Rad1/XPF protein structure and DNA specific substrates....................................25
Figure 1.7: Schematic view of protein structure of RecQ helicases in bacteria, yeast and
humans. ...................................................................................................................................28
Figure 1.8: Scheme of RECQL5 involvement in recombination pathway and replication.....34
Figure 1.9: SUMOylation mechanism.....................................................................................38
Figure 3.1: Schematic view of DNA substrates......................................................................46
Figure 3.2: Purified proteins MUS81, RECQ5 and RAD51...................................................55
Figure 3.3: Purified Rad1-Rad10 variants and Saw1..............................................................57
Figure 4.1: MUS81-EME1 interacts with RECQ5..................................................................63
Figure 4.2: RECQ5 stimulates MUS81-EME1-mediated DNA cleavage...............................65
Figure 4.3: Truncated RECQ5 does not bind or stimulate MUS81-EME1.............................66
Figure 4.4: RECQ5 stimulates Mus81-Mms4-mediated DNA cleavage................................67
Figure 4.5: RECQ5 rescues the 3’-flap cleavage mediated by MUS81-EME1 after RAD51
inhibition..................................................................................................................................69

115
Figure 4.6: Hypothetical model for the roles of RECQ5 and MUS81 in processing of
persistent replication intermediates in early mitosis................................................................72
Figure 4.7: SUMOylation of Rad1 is dependent on E3 ligases...............................................75
Figure 4.8: SUMOylation of Rad1 is not dependent on DNA binding...................................76
Figure 4.9: SUMOylation of Rad1 is not dependent on Saw1................................................77
Figure 4.10: Rad1-K32R-Rad10 mutant is not SUMOylated.................................................78
Figure 4.11: Rad1-K32R mutant does not have altered DNA cleavage, DNA binding and
Saw1 binding...........................................................................................................................80
Figure 4.12: SUMOylated Rad1 exhibits the same nuclease activity as wild-type.................81
Figure 4.13: SUMOylated Rad1 has lower affinity to dsDNA...............................................82
Figure 4.14: Modified and unmodified Rad1-Rad10 form a dimer of heterodimers..............83
Figure 4.15: SUMOylated Rad1 has enhanced Saw1 binding................................................84
Figure 4.16: SUMO-Rad1 enhances Saw1 SUMOylation......................................................86
Figure 4.17: Saw1 binds Y-form and 5’-overhang DNA substrates.......................................87
Figure 4.18: SUMO-Saw1 does not affect SUMO-Rad1 nuclease activity............................88
Figure 4.19: Model of SUMO-Rad1-Rad10 and SUMO-Saw1 role in DNA repair...............90

116
9. LIST OF TABLES
Table 1.1. Summary of DNA repair pathways..........................................................................9
Table 1.2: Summary of DNA damaging agents......................................................................10
Table 1.3: Summary of budding yeast and human recombination factors..............................16
Table 1.4: Substrate specificity of MUS81-EME1/EME1......................................................19
Table 1.5: Summary of some interaction partners of RecQ helicases.....................................36
Table 1.6: SUMO machinery proteins in Saccharomyces cerevisiae and mammals..............37
Table 1.7. Effect of SUMOylation on HR proteins.................................................................39
Table 3.1: List of chemicals and companies of their purchase................................................42
Table 3.2: List of used buffers, stock solutions and their composition...................................43
Table 3.3: Growth media information.....................................................................................44
Table 3.4: Bacterial strain information....................................................................................44
Table 3.5: Molecular weight standards information................................................................44
Table 3.6: Plasmid information...............................................................................................45
Table 3.7: DNA primers information......................................................................................45
Table 3.8: Mutagenic primers..................................................................................................48
Table 3.9: Mutagenic PCR thermocycler program..................................................................49
Table 3.10: Summary of protein expression conditions..........................................................52
Table 3.11: Summary of used chromatography methods........................................................52
Table 3.12: Summary of used proteins MUS81, RECQ5 and RAD51...................................55

117
Table 3.13: Summary of used proteins Rad1-Rad10, Saw1, SUMO proteins........................57
Table 3.14: Summary of used antibodies for Western blots...................................................58

118
10. LIST OF PUBLICATIONS
Bartosova, Z., and Krejci, L. (2014). Nucleases in homologous recombination as targets for
cancer therapy. FEBS Lett. 588, 2446–2456.
Sarangi, P., Bartosova, Z., Altmannova, V., Holland, C., Chavdarova, M., Lee, S.E., Krejci,
L., and Zhao, X. (2014). SUMOylation of the Rad1 nuclease promotes DNA repair and
regulates its DNA association. Nucleic Acids Res. 42, 6393–6404.
Sarangi, P., Altmannova, V., Holland, C., Bartosova, Z., Hao, F., Anrather, D., Ammerer, G.,
Lee, S.E., Krejci, L., and Zhao, X. (2014). A versatile scaffold contributes to damage survival
via sumoylation and nuclease interactions. Cell Rep. 9, 143–152.

119
11. SUMMARY
Our effort was to investigate structure-specific endonucleases involved in homologous
recombination, their protein-protein interactions and post-translational modifications. We
discovered a cooperative role in DNA repair of human structure-specific endonuclease
MUS81-EME1 with RECQ5 helicase. MUS81-EME1 physically interacts with full-length
RECQ5 helicase. MUS81 endonuclease activity is stimulated by RECQ5 on its specific DNA
substrates. This process is evolutionary conserved as RECQ5 is capable of stimulating also
yeast Mus81-Mms4 nuclease. We show that RECQ5 is capable of suppressing RAD51-
mediated inhibition of MUS81 cleavage, together with genetic data implicating MUS81-
RECQ5 in the resolution of late replication intermediates at CFSs during early mitosis. Both
RAD51 binding and translocase activities of RECQ5 are required for efficient disruption of
RAD51 filament.
Further, we investigated the effect of a specific post-translational modification
(SUMOylation) on an yeast structure-specific endonuclease Rad1-Rad10 (belonging to the
XPF nuclease family as MUS81), which has a role mainly in NER and HR subpathway.
SUMOylation of Rad1 subunit of the Rad1-Rad10 complex occurs at K32 and is dependent
on E3 ligases (mainly Siz1 and Siz2). SUMO-Rad1-Rad10 has a decreased DNA affinity
towards dsDNA, pointing to its function in protein turnover. SUMO-Rad1 stimulates
SUMOylation of Saw1 interaction partner. SUMO-Saw1 has a higher affinity for Slx1
interaction rather than Rad1 thus probably engages in another repair process.
Mgr. Zdenka Hašanová Doc. Mgr. Lumír Krejčí, Ph.D.

120
12. SUPPLEMENTS

FEBS Letters 588 (2014) 2446–2456
journal homepage: www.FEBSLetters .org
Review
Nucleases in homologous recombination as targets for cancer therapy
http://dx.doi.org/10.1016/j.febslet.2014.06.0100014-5793/� 2014 Federation of European Biochemical Societies. Published by Elsevier B.V. All rights reserved.
⇑ Corresponding author at: Department of Biology and National Centre forBiomolecular Research, Masaryk University, Kamenice 5/A7, Brno 625 00, CzechRepublic.
E-mail address: [email protected] (L. Krejci).
Zdenka Bartosova a, Lumir Krejci a,b,c,⇑a Department of Biology, Masaryk University, Kamenice 5/A7, Brno 625 00, Czech Republicb National Centre for Biomolecular Research, Masaryk University, Kamenice 5/A7, Brno 625 00, Czech Republicc International Clinical Research Center, Center for Biomolecular and Cellular Engineering, St. Anne’s University Hospital Brno, Brno, Czech Republic
a r t i c l e i n f o a b s t r a c t
Article history:Received 7 May 2014Revised 2 June 2014Accepted 2 June 2014Available online 10 June 2014
Edited by Wilhelm Just
Keywords:Genomic integrityHomologous recombinationNucleaseInhibitorCancer therapy
Genomic DNA is constantly challenged from endogenous as well as exogenous sources. The DNAdamage response (DDR) mechanism has evolved to combat these challenges and ensure genomicintegrity. In this review, we will focus on repair of DNA double-strand breaks (DSB) by homologousrecombination and the role of several nucleases and other recombination factors as suitable targetsfor cancer therapy. Their inactivation as well as overexpression have been shown to sensitize cancercells by increasing toxicity to DNA-damaging agents and radiation or to be responsible for resistanceof cancer cells. These factors can also be used in targeted cancer therapy by taking advantage ofspecific genetic abnormalities of cancer cells that are not present in normal cells and that resultin cancer cell lethality.� 2014 Federation of European Biochemical Societies. Published by Elsevier B.V. All rights reserved.
1. Introduction
Both endogenous as well as exogenous insults are constantlychallenging our DNA, the integrity of which is essential for genomestability as well as carcinogenesis. Endogenous sources includereactive oxygen species produced during cell metabolism orreplication of damaged DNA template. Exogenous factors includechemotherapeutic agents and ionizing radiation. Among the mosttoxic types of damage are dsDNA breaks. Two major pathwayshave evolved for their repair: non-homologous end joining (NHEJ)and homologous recombination (HR) (Fig. 1). During NHEJ, theends of DNA are directly joined with the help of KU70/KU80 com-plex, which protects them from degradation and recruits DNAligase IV and its accessory factor XRCC4 for their ligation. Recently,a third alternative, microhomology-mediated end joining (MMEJ),was described. This requires a greater extent of ends resection inas much as microhomology (<10 nt) demands that the ends beannealed. That is in contrast to classical NHEJ, where up to 3 ntcan be used [1–3]. For more detailed description, see the moreextensive reviews regarding NHEJ [4,5]. These mechanisms areerror-prone, however, as they frequently result in loss of geneticinformation.
On the other hand, HR constitutes an error-free pathway thatrequires undamaged homologous sequence on a sister chromatidfrom which the information is copied (Fig. 1). The HR mechanismcan be separated into three steps: pre-synapsis, synapsis andpost-synapsis. In the first step, the MRE11/RAD50/NBS1 complextogether with CtIP is required for recognition of the ends, damagesignaling, and to initiate end resection at both sides of the double-strand break (DSB) [6]. Two redundant pathways are required forlong resection to produce a 30-overhang. One depends on EXO1nuclease and the other involves the activity of DNA2 together withthe BLM/TOP3/RMI1 complex [7,8]. The resulting 30-overhang isprotected by RPA and serves also as a sensor for DNA damagecheckpoint activation [9]. Nevertheless, the RAD51 recombinaseneeds to obtain access to DNA in order to form nucleoproteinfilament capable of homology search. Therefore, recombinationmediators, including BRCA2 and RAD51 paralogues, are requiredfor loading RAD51 onto RPA-coated ssDNA and stabilizing thefilament. During synapsis, the RAD51 filament searches for ahomologous sequence, resulting in formation of a stable intermedi-ate known as a D-loop with the 30-end of the invading strandserving as a primer for DNA repair synthesis (Fig. 1). Withinpost-synapsis, the intact chromosome is restored. This constitutesthe most complex step, however, as several mechanistically differ-ent scenarios can arise, namely: synthesis-dependent strandannealing (SDSA), classical DSB repair (DSBR), and break-inducedreplication (BIR) (Fig. 1). During SDSA, the extended D-loop is dis-placed by the action of helicase. There then follows annealing with

D-loop
NHEJ / MMEJ
SSA
DSB
dHJ
gene conversion gene conversion crossoverhalf crossover gene conversion
DSBRSDSABIR
MRE11/RAD50/NBS1EXO1, DNA2, BLM
RAD51
BLM
RAD52
MRE11XPF/ERCC1
XPF/ERCC1
dissolution
MUS81/EME1, SLX1/SLX4, GEN1, MLH1, EXO1
Fig. 1. Model of pathways involved in repair of double-strand breaks. Possible targets for therapeutic intervention are highlighted in green
Z. Bartosova, L. Krejci / FEBS Letters 588 (2014) 2446–2456 2447
the second strand of DSB and a consequent second round of DNAsynthesis and ligation. Here, Mus81/EME1 is believed to cleavepossible 30-flaps generated as a consequence of annealing over-synthesized invading strand. In DSBR, the extended D-loop is notdisplaced (which is in contrast to SDSA), but rather it is stabilizedby BRCA2- and RAD52-mediated capturing of the second end of thebreak. After a second round of DNA synthesis, a double Hollidayjunction (HJ) is formed. From here, either dissolution by BLM/TOP3/RMI1 or resolution by the coordinated action of several nuc-leases (including SLX1-SLX4, GEN1, XPF/ERCC1 and MUS81/EME1)restores the integrity of the genome. Finally, BIR operates via apathway where one end of the DSB is lost and only the other endis used for repair. Here, the D-loop is converted to a replicationfork-like structure that ensuring synthesis along the chromosomalarm. Again, most of the nucleases have been shown to be involvedin BIR. Not only are frequent gross chromosomal rearrangementsand loss of heterozygosity associated with this pathway, however,but the synthesis also is more mutagenic compared to normal rep-lication. Finally, when extension resection of DSB reveals regions ofhomology, single strand annealing (SSA) pathway comes into play.In contrast to HR, the ends are healed using the same DNA mole-cule containing the break. RAD52 anneals the complementarystrands of the homologous region, thereby generating 30-tails thatare processed by XPF-ERCC1 nuclease, and this is followed by DNAsynthesis and ligation. Several recent reviews provide moredetailed description [10–14].
We will describe below some of the promising targets of HR orDDR pathways for chemical biology or therapeutic interventionwhile specifically focusing on nucleases. As already noted above,various nucleases participating in HR are responsible for theprocessing end of DSBs and resolution of DNA replication or
recombination intermediates (Fig. 1). However, they often playredundant and overlapping roles in other DNA repair pathways.Mutations in some of them are directly associated with diseasesor they show defects in DNA repair, accumulation of damage,and greater risk of genome instability (thereby creating a predispo-sition to cancer). On the other hand, DNA repair enzymes, includ-ing nucleases, are also commonly upregulated in some cancertypes to ensure efficient repair of DNA damage caused by chemo-or radiotherapy, and this can lead to drug resistance. Down-regulation or identification of suitable nuclease inhibitors canpotentiate the effects of current anti-cancer therapies and providenew tools in targeted cancer therapy, especially in the light ofsynthetic lethal approaches. To achieve this, detailed understand-ing of various repair mechanisms, their inhibitors, interactionpartners, associated diseases, localization, expression levels andpost-translational modifications is required.
1.1. MUS81
MUS81 is a member of the XFP family of endonucleases. It isevolutionarily conserved and forms a complex with a non-catalyticsubunit – either EME1 or EME2 (which for simplicity’s sake we willrefer to as MUS81 complex) – that differs among species (Mms4 inbudding yeast; Eme1 in fission yeast). Despite having little homol-ogy, these subunits have been shown to be involved in DNA bind-ing and required for enzymatic activity of the complex [15–19].
Purified MUS81 complex has the highest affinity for DNA struc-tures. It has an exposed 50-end near the DNA junction, including a30-flap, nicked Holliday junction (nHJ) and forks (Table 1). It intro-duces a nick at the duplex 3–7 nt away from the junction point[16,20–23]. MUS81’s activity, together with increased expression

Table 1Summary of substrate specificities of selected nucleases and their role in various pathways.
Nuclease Substrates for endo-activity Substrates for exo-activity Pathways
Mus81-EME1 30-flap, nHJ, mHJ, fork, D-loop RF restart, HR, ICLR, TMXPF-ERCC1 30-flap, bubble, D-loop, G-tail NER, HR, ICLR, NHEJ, MMEJ, SSA, TMSLX1-SLX4 50-flap, HJ HR, ICLR, SSA, RF restart, BER, NER, TMGEN1 50-flap, HJ HRFEN1 50-flap, double flap 50–30 on nicked dsDNA BER, OFM, TMMRE11 50-flap 30–50 on dsDNA HR, NHEJ, MMEJ, TMEXO1 50-flap 50–30 , 30–50 on dsDNA HR, SSA, MMR, NER, OFM, TMDNA2 50-flap OFM, HR, SSA, TM
RF restart – replication fork restart, HR – homologous recombination, ICLR – interstrand DNA crosslink repair, TM – telomere maintenance, NER – nucleotide excision repair,NHEJ – non-homologous end joining, SSA – single strand annealing, BER – base excision repair, MMEJ – microhomology mediated end joining, OFM – Okazaki fragmentmaturation.
Table 2Expression levels of selected HR factors identified in primary cancers.
Nuclease Increased expression Decreased expression
MUS81 Gastric, colorectal,hepatic cancer
ERCC1 Ovarian, colorectal, gastric, head andneck, non-small-cell lung cancer
Gastric, colorectal,non-small-cell lungcancer
FEN1 Testes, lung, brain, breast, kidney,pancreatic, colon cancer
Lung, prostate,gastrointestinalcancer
MRE11 Ovarian, breast,colorectal cancer
RAD50 breast cancerNBS1 Neck and head cancer, melanoma Breast, colorectal
cancer, melanomaEXO1 Atypical HNPCCDNA2 Breast, pancreatic, ovarian cancer,
pancreatic ductal adenocarcinomaRAD51 Breast, pancreatic, head and neck,
non-small-cell lung cancerColorectal, breastcancer
RAD52 Colorectal cancer
Expression confirmed by mRNA/protein level measurements from cancer patients.
2448 Z. Bartosova, L. Krejci / FEBS Letters 588 (2014) 2446–2456
during S phase and the peak in G2 [24], suggests its role in process-ing replication and recombination intermediates. During S phase, itis responsible for cleavage of collapsed replication fork induced byDNA damaging agents [25–28]. Interestingly, EME1/Mms4 istightly regulated during S phase in a cell cycle dependent mannerto prevent untimely cleavage of replication forks [29–31].
DSBs produced as a consequence of cleaved and/or collapsedreplication forks are substrates for homologous recombination toallow replication fork restart [27]. MUS81 is not involved only ingenerating a substrate for HR, however, but is also required forprocessing of recombination intermediates. MUS81 complex is tar-geted by RAD54 to the downstream DNA intermediates, which dra-matically stimulate its nuclease activity, and it has been shown tobe involved in the SDSA sub-pathway of HR to provide an alterna-tive mechanism for BLM-dependent dissolution of HJ [25,32–34].Similarly, deletion of MUS81 results in severe meiotic defects con-sistent with a failure to process recombination intermediates[15,22]. Because its phenotypes in yeast could be rescued byexpression of RusA, its function as a Holliday junction resolvasehas been suggested by [15]. Nevertheless, nicked Holliday junc-tions and D-loops are preferred substrates in vitro. In addition,MUS81 cleaves the substrates asymmetrically, which is in contrastto typical resolvases, and this results in nicks that are not directlyligatable [35,36]. Recently, SLX1-SLX4 and MUS81-EME1 havebeen shown to cooperate in HJ resolution, with the SLX complexgenerating the initial nick and thus creating a more suitable sub-strate for MUS81-EME1 counter cleavage [37,38].
MUS81-deficient cells exhibit proliferation defects and accumu-late various chromosomal aberrations [39,40]. Haploinsufficiencyof MUS81 has been shown to result in chromosomal abnormalitiesand increased sensitivity to crosslinking agents [41,42], thus indi-cating that already a single copy of MUS81 can result in genomicinstability. Accordingly, decreased levels of MUS81 expressionhave been found in hepatic metastasis and correlated with poorcancer prognosis [43] (Table 2). A role for MUS81 in tumorigenesisis further supported by evidence of a synergistic effect with inacti-vation of p53 and, on the other hand, suppression by inactivatingCHK2 [44]. This all together suggests MUS81 to be a potential tar-get for cancer therapy.
1.2. XPF-ERCC1
XPF-ERCC1 (yeast homolog Rad1-Rad10) is a structure-specificendonuclease participating in multiple repair pathways, includingnucleotide excision repair (NER), interstrand crosslink repair(ICLR), HR and NHEJ. XPF protein forms a stable heterodimer witha non-catalytic subunit, ERCC1, which is essential for its endonu-clease activity and also ensures binding to DNA and other proteins.Preferred DNA structures are splayed-arm, bubble, stem-loop andD-loop structures (Table 1). Cuts are introduced in duplex DNA at
a 50-side of a ssDNA/dsDNA junction, preferably with the 50-endfurther away [45–51].
The XPF complex plays an essential role in NER, as correspond-ing mutants show sensitivity to UV lesions [52] and it is responsi-ble for a 50 incision of a bubble DNA lesion generated during NER.XPG is responsible for a corresponding 30 cleavage that results in arelease of ssDNA oligonucleotide containing the lesion. The newlyformed gap is subsequently filled by a DNA polymerase and ligated[49,53–55]. In contrast to other NER factors, the XPF is hypersensi-tive to ICL. This suggests its more specific role in crosslink repair[56]. Although the precise mechanism of ICL repair is not clear, itemploys XPF in the incision step due to its ability to cleave theDNA on either side of the ICL [57]. In addition to ICL, XPF complexis also involved in HR and NHEJ, as mutations in XPF-ERCC1 causesensitivity to DSBs [58–61]. XPF-ERCC1 processes 30-non-homolo-gous single-strand tails during RAD52-mediated SSA and in MMEJ[59,58,62,60,63]. XPF activity is also required during meioticrecombination to process recombination intermediates [64,65].While in Drosophila this might constitute the major pathway inHJ processing [66], in other organisms its activity is more redun-dant and linked with other nucleases. Indeed, such coordinationhas been reported for human MUS81 and XPF complexes [67,68],thus indicating a high level of complexity to ensure resolution ofvarious intermediates.
Mutations in XPF protein have been identified in patients withXeroderma pigmentosum (XP) and a progeroid syndrome (XFE).While XP patients fail to repair DNA damage caused by UV, theXFE patients display premature aging syndromes [69,70], thus

Z. Bartosova, L. Krejci / FEBS Letters 588 (2014) 2446–2456 2449
implying a role of XPF in telomere maintenance. Indeed, XPF-ERCC1 was found in a complex with telomeric protein TRF2, whichprotects the chromosome ends from degradation. Moreover,ERCC1-defective cells show telomere deletions and formation ofT-loop structures as a consequence of processing telomeric recom-bination intermediates [71]. A number of various cancers show alink between increased expression of ERCC1 and poor responseto platinum-based therapy [72–74] (Table 2). Accordingly, whileERCC1-proficent xenografts were shown to be resistant to cisplatin,ERCC1-deficient ones were completely cured [75]. Together withits defensive role in cancer cells by means of repairing the damagecaused to those cells by chemotherapeutics, this makes XPF a valu-able target for sensitization and chemoresistance of cancer cells. Italso should be noted that not only inhibition of nuclease activitybut also helicase-like domain as well as interaction with RPA andXPA proteins may present attractive targets for drug discoveryprograms.
1.3. SLX4 (FANCP)
SLX4 (FANCP) is a structure-specific subunit that serves as ascaffold for such other structure-specific endonucleases asMUS81-EME1, XPF-ERCC1 and SLX1 [45,48,50]. Together theseform a multiprotein complex responsible for repair of damagedDNA and are essential for replication fork restart and ICL repair.The SLX1-SLX4 complex has a preference for branched structureslike 50-flaps, but, in contrast to the fungal protein, the humancomplex can also cleave static and mobile HJs and thus has beensuggested to function as a HJ resolvase [50,48,76,45,77] (Table 1).It introduces symmetrical cuts in duplex DNA near junctions withssDNA, resulting in nicked duplex products which are readily ligat-able [45,48,50].
SLX4 enhances endonuclease activity of associated nucleasesXPF-ERCC1, MUS81-EME1 and SLX1, and it plays a crucial role inICL repair [48,50,45]. An interaction with XPF-ERCC1 promotescleavage of bubble structures during processing of UV-damage inNER and is also required during the SSA pathway of DSB repair[78,79]. Similarly, the interaction with MUS81-EME1 leads tocooperation in HJ resolution [38,37]. SLX4 is involved in telomeremaintenance, as it associates with TRF2 and localizes to thetelomeric regions; moreover, the RTEL1-deficient cells showSLX4-mediated replication stalling at telomeres and their cleavagegenerating telomeric circles [50,80,81].
The importance of SLX4 in ICL repair and genomic stability isunderscored by the identification of biallelic mutations in SLX4in patients with Fanconi anemia complementation group P andhereditary breast cancer [82–84]. While inhibition of SLX4 shouldsensitize the cells to DNA crosslinking agents, as reported forSLX4-deficient cells [48,85,84], the inhibition of its mediated inter-actions should have specific and/or broad effect on various repairpathways.
1.4. GEN1
GEN1 (yeast Yen1) belongs to the Rad2/XPG family of structure-specific endonucleases [86]. GEN1 is unique for its activity inresolving HJs, which occurs through its ability to dimerize on HJsand cleave these symmetrically to produce two nicked duplexesthat can be easily ligated [87] (Table 1). An absence of humanGEN1 leads to defects in HR and accumulation of DNA damage[88]. Its co-depletion with MUS81 or SLX4 in BLM-deficient cellsresults in severe chromosomal instability [89], thus indicating arole of GEN1 in HJ resolution in vivo. The functional overlap withMus81, Slx4 and Rad1 nucleases also has been suggested to playan important role in processing recombination intermediates inyeast [86,90–92]. Cytological and cell cycle analysis supports the
idea that GEN1 could be the last resort in resolving unprocessedHJ to ensure genetic stability, as GEN1 gains access to DNA at theinitiation of mitosis [88,93].
The synthetic lethality of cells lacking GEN1 and Fanconi ane-mia-linked SLX4 [37] further indicate its requirement for process-ing HJ in vivo and make this a good candidate for therapeuticintervention. Finally, GEN1 mutations predicted to affect nucleaseactivity were identified in a cohort of breast cancer patients, thusindicating possible association with carcinogenesis [86]. Morestudies are required, however, in order to confirm this observation.
1.5. FEN1
FEN1 (yeast Rad27) has several essential roles during DNA rep-lication and repair [94]. It is a member of the Rad2/XPG family andhas 50–30exonuclease, gap endonuclease and RNaseH activities[95,96]. These different modes of hydrolysis share a predominantincision 1 nt into the downstream dsDNA with minor cleavage at50 or 30 of the site [97]. While, double-flap DNA is the most pre-ferred substrate for FEN1 in vivo, a wide range of substrates ishydrolyzed by FEN1 using the same active center (Table 1). Thissuggests differences in the manner of FEN1’s binding to variousDNA [98–100].
Removal of 50-flap containing RNA primer is crucial for Okazakifragment maturation during lagging strand DNA synthesis [97].This has been validated in vivo, as FEN1-deficent mice demonstratedefects in DNA replication, cell proliferation and embryonic lethal-ity [101–103]. The FEN1 activity is also required during long-patchBER (LP-BER), where a long flap is formed and serves as a substratefor FEN1 [104]. Concerted action of FEN1 activities is also requiredfor apoptotic fragmentation and resolution of structures derivedfrom trinucleotide repeats (TNRs). The role of FEN1 in apoptoticDNA fragmentation was suggested based on a cell death phenotypesimilar to endonuclease G-mediated apoptosis that is observed inthe case of FEN1 deficiency [103]. FEN1 also certainly contributesto TNR stability, regardless of whether it is mediated directly oras part of redundant pathways [104–107]. Since TNR instabilityis observed in many neurodegenerative and neuromuscular dis-eases, including Huntington disease [108], FEN1 has been sug-gested to play a role in their development [109]. These findingsare contradictory, however, and this needs to be further character-ized. FEN1 is also implicated in other DNA metabolic pathways,including rescue of stalled replication forks, as well as stabilityof minisatellites, telomeres and other repetitive sequences [110–114]. Accordingly, depletion of FEN1 results in telomere dysfunc-tion and end-to-end fusions in ALT-positive cancer cells and MEFs[113,115,116].
The multifaceted activities of FEN1 suggest its essential role ingenomic stability and cancer predisposition. Accordingly, inknockout mice it is embryonic lethal and its combination withAPC results in cancer development [101,117]. Furthermore, nucle-ase-deficient mice show an increased mutator phenotype, defectsin apoptosis and susceptibility to cancers [102,103]. Two germlinemutations resulting in lowered FEN1 expression are associ-ated with increased lung and gastrointestinal cancers [118,119](Table 2). In Fanconi anemia, FEN1 haploinsufficiency supportscancer cell proliferation and tumor progression [101]. On the otherhand, once malignant transformation has occurred, FEN1 overex-pression confers a growth advantage in many types of cancer[120,121]. Despite risks to cancer-prone patients, inhibition ofFEN1 offers a potent target for use in chemotherapies, and espe-cially in ones to which resistance is developing. Furthermore,FEN1 inhibitors can be used in targeting HR-deficient cancers, asFEN1 mutations accumulate damage that is processed by HR[122]. Since the multifaceted role of FEN1 requires several regula-tory modes, including specific protein interactions, localization,

2450 Z. Bartosova, L. Krejci / FEBS Letters 588 (2014) 2446–2456
and post-translational modifications and localization, these alsocan be targeted therapeutically.
1.6. MRE11
MRE11 functions in a complex with RAD50 and NBS1, referredto collectively as the MRN complex. This complex has multipleroles in repair of DSBs via HR, NHEJ and MMEJ, as it is responsiblefor sensing the DSBs, activating the DNA damage checkpoint,tethering the ends, evicting nucleosomes in the vicinity of thebreak, and initiating end resection (for a review see [123]). Theessentiality of any of the components of the MRN complex forthe cell is underlined by their embryonic lethality in mice[124–126]. MRE11 possesses 30–50 exonuclease activity and ssDNAendonuclease activity with preference for blunt ends and branchedsubstrates such as 50-overhangs [127]. Interestingly, structuralintegrity of the complex is more important for the end resectionthan for its nuclease activity. MRE11’s nuclease activity togetherwith NBS1 is nevertheless required for MMEJ, and it also uses itsendonuclease activity to cleave a covalently bound SPO11 at the50-ends of the DNA after DSB formation during meiosis to initiatetheir resection [128–131]. In addition, the MRN complex localizesto the telomeres and regulates telomeric length either by recruit-ment of the telomerase RNA subunit or as a sensor of damagedtelomeres promoting ATM activation and alternative lengtheningof telomeres [132–134]. The MRN-mediated end resection and uti-lization of both HR and NHEJ pathways is promoted by CtIP protein[135–137]. CtIP has recently been shown to possess nuclease activ-ity required for processing DNA adducts or secondary structures atthe sites of breaks [138,139], making it also possible target forchemical intervention.
Germline mutations of MRE11, NBS1 and RAD50 cause ataxia-telangiectasia-like disease (ATLD), Nijmegen breakage syndrome(NBS) and NBS-like disorder (NBSLD), respectively. ATLD andNBSLD have similar features as does ataxia-telangiectasia (AT),caused by mutations in the ATM gene, which include hypersensi-tivity to DSB-inducing agents, chromosome fragility, DNA dam-age-dependent cell-cycle arrest and high predisposition to cancer[140–144]. Mutations in CtIP have been identified as causal inthe Seckel and Jawad syndromes and have demonstrated defectsin processing DNA damage and ATR activation [145]. Moreover,impaired expression of MRE11 is associated with some cancersand MRE11 depletion sensitizes these to poly(ADP-ribose) poly-merase (PARP) inhibition [146–149] (Table 2). Together with theMRN complex’s central role in HR and genome stability, these datahighlight MRE11 and its partners as constituting a valuable phar-macological target. Indeed, the first described MRE11 inhibitors(mirin and telomelysin) have been confirmed in preclinical studiesto have potential as sensitizers of cancer cells [150–152]. Inaddition, MRE11-deficient cells are also sensitive to topoisomerasepoisons [153], thus suggesting a role for MRN in removal of TOP1/TOP2-DNA intermediates and in stimulating an effect of topoinhibitors. Indeed, triapine (RNR inhibitor) was recently shown toblock MRN/CtIP-mediated HR and sensitize ovarian cancer cellsto PARP and topo inhibitors [154,155]. Further work is required,however, to potentiate MRN inhibitors for clinical trials.
1.7. EXO1
EXO1 belongs to the RAD2/XPG nuclease family possessing 50–30
exonuclease, 50-overhang endonuclease and RNase H activities(Table 1). It participates in several genome stability pathways,including MMR, DSB repair, and telomere maintenance. In MMR,EXO1 is involved in the 50 and 30 directed excision of DNAmismatches [156,157]. A nuclease-independent and more struc-tural scaffold-like role of EXO1 in MMR has also been suggested
in yeast [158], but this needs to be further analyzed. During DSBrepair, EXO1 is involved in long resection of DNA ends generating30-overhangs required for HR [7,159]. During NER, EXO1 insuresgap enlargement, which is a signal for an ATR-mediated DNAdamage response [160,161]. At telomeres, EXO1 promotes telo-meric recombination by extensive end resection of G-rich strandsand genetic instability in telomerase-deficient cells [162–164].Such instability, together with involvement in DNA repairpathways, makes EXO1 a logical target for mutation duringtumorigenesis.
It has been suggested that EXO1 contributes to non-polyposiscolorectal cancer (HNPCC) and sporadic colorectal cancers (CRC)(Table 2). While missense mutations in EXO1 have been describedin patients with atypical HNPCC [165], the role in suppressing CRCremains unclear. Nevertheless, EXO1-deficient mice show reducedsurvival, higher mutation rates and increased susceptibility to gen-erate lymphomas [166]. Several studies in yeast point to a possiblemulti-mutation hypothesis, where a weaker mutation phenotypeof EXO1 might be combined with other mutator alleles to resultin pathogenesis [158,167]. On the other hand, overexpression ofEXO1 may also increase genetic instability and provide an advan-tage for cancer’s progression, as suggested by loss of mutant allelein 12 of 13 atypical HNPCC patients [168]. For more details, seealso [169,170].
1.8. DNA2
DNA2 is a conserved helicase/nuclease contributing to DNA rep-lication and DSB repair. It is an ssDNA-dependent ATPase possess-ing weak 50–30 helicase activity. The helicase activity is attenuatedby its own endonuclease activity [171]. During lagging strand syn-thesis, DNA2 interacts with FEN1, where it cleaves long 50-flaps toprovide a suitable substrate for FEN1 [172]. Within DSB repair,DNA2 is one of the nucleases involved in long resection by process-ing DNA ends into 30-overhangs together with BLM/TOP3/RMI1complex [173,8].
In several cancer cells, DNA2 expression is significantlyincreased and facilitates massive DNA repair by HR [174], thusproviding a possible advantage toward survival in cases of replica-tion-associated stress (Table 1). Accordingly, depletion of DNA2sensitizes normal cells but rescues the sensitivity of FANCD2 cellsto cisplatin [175] and thus has possible implications in therapy ofFA patients. A high-throughput screen in Saccharomyces cerevisiaehas revealed that most replication and chromatin dynamics pro-teins are synthetically lethal with DNA2 [176], making this,together with its role in genomic stability, a suitable biomarkeras well as a target for chemical intervention.
2. Other HR-related targets for pharmacological inhibition
2.1. Checkpoint kinases
Checkpoint kinases in DNA damage response present, in parallelto individual DNA repair pathways, a main mechanism that sensesDNA damage and orchestrates appropriate responses. DNA damageresults in an induction of cell-cycle arrest allowing sufficient timefor efficient DNA repair. ATM and ATR play central roles in the DDRpathway by controlling the checkpoint via other downstreameffectors (CHK1, CHK2, p53, BRCA1, etc.). Both activation anddeactivation of the DNA damage response have been observed incancers, and both present important condition factors in currentand future clinical trials. The roles of these kinases in tumorigene-sis and their pharmacological inhibition has been describedextensively elsewhere and will not be included in this review[177–179].

Z. Bartosova, L. Krejci / FEBS Letters 588 (2014) 2446–2456 2451
2.2. RAD51
RAD51 is one of the key factors in HR and genomic stability. Itsdeficiency results in early embryonic lethality, and mutations inRAD51 have been associated with increased risk of breast and colo-rectal cancers [180–182]. Increased expression, on the other hand,also has been identified in a wide range of cancers, thus suggestinga possible mechanism for resistance [183–186] (Table 2). In addi-tion, RAD51 paralogues may also constitute potent targets, espe-cially as RAD51C was recently linked with Fanconi anemia andidentified as a novel breast cancer susceptibility gene [187,188].Recently developed RAD51 inhibitors as well as inhibitors of onco-gene tyrosine kinases [189–191] are expected to sensitize cancercells to DNA damage.
2.3. RAD52
RAD52 forms an oligomeric ring, binding both ss- as well asdsDNA and catalyzing the annealing of complementary strandsduring SSA, an alternative pathway to BRCA2-mediated HR. Recentdiscovery of synthetic lethality of RAD52- and BRCA2-deficentcells [192] indicates that RAD52 represents an essential backuppathway for HR and thus is a very interesting target for BRCA-associated tumors.
2.4. RecQ
RecQ helicases also represent very promising targets for anti-cancer therapy through DNA repair inhibition. They function inmultiple cell processes such as DNA replication, transcription,DNA repair, telomere maintenance and DNA damage signaling[193–196]. In humans, five RecQ homologs (RECQL1, WRN, BLM,RECQL4, RECQL5) have been identified. Three of these are associ-ated with syndromes featuring genome instability, prematureaging and cancer predisposition, namely Bloom (mutations in theBLM gene), Werner (mutations in the WRN gene), Rothmund–Thomson, RAPADILINO, and Baller–Gerold syndromes (mutationsin the RECQL4 gene) [197–201]. For more details on targeting heli-case in cancer therapy, see [202].
3. Inhibitors
Inhibition of a gene of interest can be achieved by its downreg-ulation or alternatively through small chemical compounds. Use ofinhibitors may be a great advantage since proteins function in mul-tiple pathways and their complete deletion could negatively affectthe entire cell’s metabolism. Medicinal chemistry programs couldprovide inhibitors of specific enzymatic activity, interaction orlocalization to block a certain repair pathway while leaving otherfunctions intact.
However, identification of effective small molecule inhibitors ishindered by the specificities of cancer cells. Cancer cells have a sig-nificantly different metabolism than do normal cells and can utilizealternative pathways to cope with treatment, thereby resulting indrug resistances. This can be achieved by mutating a specific targetof chemotherapeutic intervention, thus making it insensitive to thedrug [203]. Similarly, epigenetics have been suggested to play aleading role in creating resistance to cisplatin [204]. Another pos-sibility is re-activation of a blocked signal pathway by a gain-of-function mutation in downstream effectors, although upstreamhyperactivation also has been observed [205]. In addition, multi-drug resistance-associated proteins greatly contradict cancertreatment simply by excluding foreign compounds from the cellthrough increased efflux achieved via overexpression of ABC trans-porters [206]. Alternatively, enzymatic deactivation of the drug by
glutation-S-transferase or decreased membrane permeabilitycan result in multidrug resistance [207]. Cancer cells are also capa-ble of inhibiting apoptosis, thus making treatment with DNA-damaging agents and DNA-repair inhibitors ineffective [208,209],unless an excessive amount of damage leads to mitotic catastrophedue to a loss of essential genetic material. Another factor that canimpede success of DNA repair inhibitors is selectivity to tumors.This could mean that unless these inhibitors are selectively deliv-ered to cancer cells they may cause adverse defects also to normalcells. Nevertheless, recent developments in drug delivery, includ-ing nanomedicine and targeted nano-sized carriers, could haveenormous effects on therapy [210].
Another complication is the treatment of patients with germ-line syndromes, as all the cells will be sensitive to damagingagents. This is the case mainly in homozygous mutations, but alsoheterozygous cells show haploinsufficiency. For example, patientswith Fanconi anemia are hypersensitive to crosslinking agentsand these drugs must be omitted in therapy [211]. Moreover,tumors are comprised of a population of both dividing and dor-mant cells, with highly proliferating cells oriented on the surfaceof the tumor. Hence, drugs causing replication blocks are not effec-tive for eliminating the whole tumor and non-replicating cancercells can also show different capacities for repair [212]. Finally,numerous difficulties arise in individuals and highly specific‘‘personalized’’ cancer therapy may be required that combines amechanistic understanding of cancer’s progression and its patho-genesis with the development of targeted drugs that are stratifiedby genetic characteristics and individualized drug administration.Despite all these complications, there are positive results thatpromise further success and better understanding in the treatmentof cancer and of biological mechanisms generally.
4. Synthetic lethality (SL)
The concept of synthetic lethality or sickness (SLS) representsone of the most promising perspectives for cancer treatment inthe future. The SLS phenotype requires two mutations. Each ofthese alone has no effect on cell viability, but when they are com-bined this leads to slow growth or cell death. If either of the twoSLS partners is cancer-specific, then inactivation of the second geneby a drug-mediated inhibition will provide highly effective andselective killing of cancer cells without toxic effect to the normalcells. Since several cancers are defective in DDR and DNA repairpathways, use of specific repair inhibitors can take full advantageof this concept. Regarding HR, this approach was pioneered byAshworth and Helleday [213,214]. They combined chemicalinhibition of the base excision repair factor Poly(ADP-ribose) poly-merase (PARP) in the context of breast cancer where patients aredefective in recombination mediator BRCA2. The great sensitiza-tion observed in these cells is a consequence of blocked repair ofssDNA breaks by PARP inhibition. These are converted to dsDNAbreaks as a result of stalled replication forks that cannot berepaired in these recombination-deficient cells lines. Accordingly,other HR-deficient cells are also sensitive to PARP inhibition[215,216], thus suggesting their broader use.
Numerous other examples of SLS are now being reported,including sensitization of FA cells to ATM inhibitors [217]. Regard-ing the nucleases involved in HR, ERCC1-deficient cancer cells aresynthetically lethal with ATR [218]. FEN1 depletion or inhibitionhas been shown to cause synthetic lethality with CDC4 andMRE11 mutations in cancer cell lines [219]. FEN1-depletion alsoresults in SLS of RAD54B-deficient colorectal cells [220]. Further-more, SLX1 and MUS81 complexes are synthetic lethal in theabsence of BLM, thus confirming evolutionary conservation of theprocessing of replication-induced intermediates [19,37].

2452 Z. Bartosova, L. Krejci / FEBS Letters 588 (2014) 2446–2456
While whole-genome synthetic lethality screens in yeast havehelped to characterize the genetic interaction network, to conductsimilar studies in humans is technically more demanding. Never-theless, identification of SLS genes will not only help to understandredundancy and complexity of repair pathways as potential targetsand consequently to select more powerful patient-specific treat-ments, it also will allow to circumvent resistance mechanisms. Inany case, detailed biochemical characterization of potential targets,design of throughput screening methods, as well as identificationof reliable biomarkers are essential for future clinical trials andapplications.
Acknowledgments
This work was supported by the Czech Science Foundation[GACR 13-26629S and 207/12/2323] and the European RegionalDevelopment Fund (Project FNUSA-ICRC) [No. CZ.1.05/1.1.00/02.0123].
References
[1] Deng, S.K., Gibb, B., de Almeida, M.J., Greene, E.C. and Symington, L.S. (2014)RPA antagonizes microhomology-mediated repair of DNA double-strandbreaks. Nat. Struct. Mol. Biol. 21, 405–412.
[2] Guirouilh-Barbat, J., Huck, S., Bertrand, P., Pirzio, L., Desmaze, C., Sabatier, L.and Lopez, B.S. (2004) Impact of the KU80 pathway on NHEJ-induced genomerearrangements in mammalian cells. Mol. Cell 14, 611–623.
[3] Simsek, D., Brunet, E., Wong, S.Y.-W., Katyal, S., Gao, Y., McKinnon, P.J., Lou, J.,Zhang, L., Li, J., Rebar, E.J., Gregory, P.D., Holmes, M.C. and Jasin, M. (2011)DNA ligase III promotes alternative non-homologous end-joining duringchromosomal translocation formation. PLoS Genet. 7, e1002080.
[4] Boboila, C., Alt, F.W. and Schwer, B. (2012) Classical and alternative end-joining pathways for repair of lymphocyte-specific and general DNA double-strand breaks. Adv. Immunol. 116, 1–49.
[5] Deriano, L. and Roth, D.B. (2013) Modernizing the non-homologous end-joining repertoire: alternative and classical NHEJ share the stage. Annu. Rev.Genet. 47, 433–455.
[6] Paull, T.T. (2010) Making the best of the loose ends: Mre11/Rad50 complexesand Sae2 promote DNA double-strand break resection. DNA Repair 9, 1283–1291.
[7] Mimitou, E.P. and Symington, L.S. (2008) Sae2, Exo1 and Sgs1 collaborate inDNA double-strand break processing. Nature 455, 770–774.
[8] Nimonkar, A.V., Genschel, J., Kinoshita, E., Polaczek, P., Campbell, J.L., Wyman,C., Modrich, P. and Kowalczykowski, S.C. (2011) BLM-DNA2-RPA-MRN andEXO1-BLM-RPA-MRN constitute two DNA end resection machineries forhuman DNA break repair. Genes Dev. 25, 350–362.
[9] Zou, L. and Elledgez, S.J. (2003) Sensing DNA damage through ATRIPrecognition of RPA-ssDNA complexes. Science 300, 1542–1548.
[10] Krejci, L., Altmannova, V., Spirek, M. and Zhao, X. (2012) Homologousrecombination and its regulation. Nucleic Acids Res. 40, 5795–5818.
[11] Li, X. and Heyer, W.-D. (2008) Homologous recombination in DNA repair andDNA damage tolerance. Cell Res. 18, 99–113.
[12] San Filippo, J., Sung, P. and Klein, H. (2008) Mechanism of eukaryotichomologous recombination. Annu. Rev. Biochem. 77, 229–257.
[13] Sung, P. and Klein, H. (2006) Mechanism of homologous recombination:mediators and helicases take on regulatory functions. Nat. Rev. Mol. Cell Biol.7, 739–750.
[14] Symington, L.S. (2002) Role of RAD52 epistasis group genes in homologousrecombination and double-strand break repair. Microbiol. Mol. Biol. Rev. 66,630–670. table of contents.
[15] Boddy, M.N., Gaillard, P.H., McDonald, W.H., Shanahan, P., Yates 3rd, J.R. andRussell, P. (2001) Mus81-Eme1 are essential components of a Hollidayjunction resolvase. Cell 107, 537–548.
[16] Ciccia, A., Constantinou, A. and West, S.C. (2003) Identification andcharacterization of the human mus81-eme1 endonuclease. J. Biol. Chem.278, 25172–25178.
[17] Fu, Y. and Xiao, W. (2003) Functional domains required for the Saccharomycescerevisiae Mus81-Mms4 endonuclease complex formation and nuclearlocalization. DNA Repair 2, 1435–1447.
[18] Gaskell, L.J., Osman, F., Gilbert, R.J.C. and Whitby, M.C. (2007) Mus81 cleavageof Holliday junctions: a failsafe for processing meiotic recombinationintermediates? EMBO J. 26, 1891–1901.
[19] Mullen, J.R., Kaliraman, V., Ibrahim, S.S. and Brill, S.J. (2001) Requirement forthree novel protein complexes in the absence of the Sgs1 DNA helicase inSaccharomyces cerevisiae. Genetics 157, 103–118.
[20] Bastin-Shanower, S.A., Fricke, W.M., Mullen, J.R. and Brill, S.J. (2003) Themechanism of Mus81-Mms4 cleavage site selection distinguishes itfrom the homologous endonuclease Rad1-Rad10. Mol. Cell. Biol. 23,3487–3496.
[21] Gaillard, P.-H.L., Noguchi, E., Shanahan, P. and Russell, P. (2003) Theendogenous Mus81-Eme1 complex resolves Holliday junctions by a nickand counternick mechanism. Mol. Cell 12, 747–759.
[22] Osman, F., Dixon, J., Doe, C.L. and Whitby, M.C. (2003) Generating crossoversby resolution of nicked Holliday junctions: a role for Mus81-Eme1 in meiosis.Mol. Cell 12, 761–774.
[23] Whitby, M.C., Osman, F. and Dixon, J. (2003) Cleavage of model replicationforks by fission yeast Mus81-Eme1 and budding yeast Mus81-Mms4. J. Biol.Chem. 278, 6928–6935.
[24] Gao, H., Chen, X.-B. and McGowan, C.H. (2003) Mus81 endonuclease localizesto nucleoli and to regions of DNA damage in human S-phase cells. Mol. Biol.Cell 14, 4826–4834.
[25] Doe, C.L., Ahn, J.S., Dixon, J. and Whitby, M.C. (2002) Mus81-Eme1 and Rqh1involvement in processing stalled and collapsed replication forks. J. Biol.Chem. 277, 32753–32759.
[26] Hanada, K., Budzowska, M., Modesti, M., Maas, A., Wyman, C., Essers, J. andKanaar, R. (2006) The structure-specific endonuclease Mus81-Eme1promotes conversion of interstrand DNA crosslinks into double-strandsbreaks. EMBO J. 25, 4921–4932.
[27] Hanada, K., Budzowska, M., Davies, S.L., van Drunen, E., Onizawa, H., Beverloo,H.B., Maas, A., Essers, J., Hickson, I.D. and Kanaar, R. (2007) The structure-specific endonuclease Mus81 contributes to replication restart by generatingdouble-strand DNA breaks. Nat. Struct. Mol. Biol. 14, 1096–1104.
[28] Regairaz, M., Zhang, Y.-W., Fu, H., Agama, K.K., Tata, N., Agrawal, S., Aladjem,M.I. and Pommier, Y. (2011) Mus81-mediated DNA cleavage resolvesreplication forks stalled by topoisomerase I-DNA complexes. J. Cell Biol.195, 739–749.
[29] Gallo-Fernández, M., Saugar, I., Ortiz-Bazán, M.Á., Vázquez, M.V. and Tercero,J.A. (2012) Cell cycle-dependent regulation of the nuclease activity of Mus81-Eme1/Mms4. Nucleic Acids Res. 40, 8325–8335.
[30] Matos, J., Blanco, M.G., Maslen, S., Skehel, J.M. and West, S.C. (2011)Regulatory control of the resolution of DNA recombination intermediatesduring meiosis and mitosis. Cell 147, 158–172.
[31] Szakal, B. and Branzei, D. (2013) Premature Cdk1/Cdc5/Mus81 pathwayactivation induces aberrant replication and deleterious crossover. EMBO J. 32,1155–1167.
[32] Fabre, F., Chan, A., Heyer, W.-D. and Gangloff, S. (2002) Alternate pathwaysinvolving Sgs1/Top3, Mus81/Mms4, and Srs2 prevent formation of toxicrecombination intermediates from single-stranded gaps created by DNAreplication. Proc. Natl. Acad. Sci. USA 99, 16887–16892.
[33] Matulova, P., Marini, V., Burgess, R.C., Sisakova, A., Kwon, Y., Rothstein, R.,Sung, P. and Krejci, L. (2009) Cooperativity of Mus81.Mms4 with Rad54 in theresolution of recombination and replication intermediates. J. Biol. Chem. 284,7733–7745.
[34] Mazina, O.M. and Mazin, A.V. (2008) Human Rad54 protein stimulateshuman Mus81-Eme1 endonuclease. Proc. Natl. Acad. Sci. USA 105, 18249–18254.
[35] Constantinou, A., Chen, X.-B., McGowan, C.H. and West, S.C. (2002) Hollidayjunction resolution in human cells: two junction endonucleases with distinctsubstrate specificities. EMBO J. 21, 5577–5585.
[36] Chen, X.B., Melchionna, R., Denis, C.M., Gaillard, P.H., Blasina, A., Van deWeyer, I., Boddy, M.N., Russell, P., Vialard, J. and McGowan, C.H. (2001)Human Mus81-associated endonuclease cleaves Holliday junctions in vitro.Mol. Cell 8, 1117–1127.
[37] Garner, E., Kim, Y., Lach, F.P., Kottemann, M.C. and Smogorzewska, A. (2013)Human GEN1 and the SLX4-associated nucleases MUS81 and SLX1 areessential for the resolution of replication-induced Holliday junctions. CellRep. 5, 207–215.
[38] Wyatt, H.D.M., Sarbajna, S., Matos, J. and West, S.C. (2013) Coordinatedactions of SLX1-SLX4 and MUS81-EME1 for Holliday junction resolution inhuman cells. Mol. Cell 52, 234–247.
[39] Abraham, J., Lemmers, B., Hande, M.P., Moynahan, M.E., Chahwan, C., Ciccia,A., Essers, J., Hanada, K., Chahwan, R., Khaw, A.K., McPherson, P., Shehabeldin,A., Laister, R., Arrowsmith, C., Kanaar, R., West, S.C., Jasin, M. and Hakem, R.(2003) Eme1 is involved in DNA damage processing and maintenance ofgenomic stability in mammalian cells. EMBO J. 22, 6137–6147.
[40] Dendouga, N., Gao, H., Moechars, D., Janicot, M., Vialard, J. and McGowan, C.H.(2005) Disruption of murine Mus81 increases genomic instability and DNAdamage sensitivity but does not promote tumorigenesis. Mol. Cell. Biol. 25,7569–7579.
[41] Hiyama, T., Katsura, M., Yoshihara, T., Ishida, M., Kinomura, A., Tonda, T.,Asahara, T. and Miyagawa, K. (2006) Haploinsufficiency of the Mus81-Eme1endonuclease activates the intra-S-phase and G2/M checkpoints andpromotes rereplication in human cells. Nucleic Acids Res. 34, 880–892.
[42] McPherson, J.P., Lemmers, B., Chahwan, R., Pamidi, A., Migon, E., Matysiak-Zablocki, E., Moynahan, M.E., Essers, J., Hanada, K., Poonepalli, A., Sanchez-Sweatman, O., Khokha, R., Kanaar, R., Jasin, M., Hande, M.P. and Hakem, R.(2004) Involvement of mammalian Mus81 in genome integrity and tumorsuppression. Science 304, 1822–1826.
[43] Wu, F., Liu, S.-Y., Tao, Y.-M., Ou, D.-P., Fang, F. and Yang, L.-Y. (2008)Decreased expression of methyl methansulfonate and ultraviolet-sensitivegene clone 81 (Mus81) is correlated with a poor prognosis in patients withhepatocellular carcinoma. Cancer 112, 2002–2010.
[44] El Ghamrasni, S., Pamidi, A., Halaby, M.J., Bohgaki, M., Cardoso, R., Li, L.,Venkatesan, S., Sethu, S., Hirao, A., Mak, T.W., Hande, M.P., Hakem, A. andHakem, R. (2011) Inactivation of chk2 and mus81 leads to impaired

Z. Bartosova, L. Krejci / FEBS Letters 588 (2014) 2446–2456 2453
lymphocytes development, reduced genomic instability, and suppression ofcancer. PLoS Genet. 7, e1001385.
[45] Fekairi, S., Scaglione, S., Chahwan, C., Taylor, E.R., Tissier, A., Coulon, S., Dong,M.-Q., Ruse, C., Yates 3rd, J.R., Russell, P., Fuchs, R.P., McGowan, C.H. andGaillard, P.-H.L. (2009) Human SLX4 is a Holliday junction resolvase subunitthat binds multiple DNA repair/recombination endonucleases. Cell 138, 78–89.
[46] De Laat, W.L., Sijbers, A.M., Odijk, H., Jaspers, N.G. and Hoeijmakers, J.H.(1998) Mapping of interaction domains between human repair proteinsERCC1 and XPF. Nucleic Acids Res. 26, 4146–4152.
[47] Li, L., Peterson, C.A., Lu, X. and Legerski, R.J. (1995) Mutations in XPA thatprevent association with ERCC1 are defective in nucleotide excision repair.Mol. Cell. Biol. 15, 1993–1998.
[48] Muñoz, I.M., Hain, K., Déclais, A.-C., Gardiner, M., Toh, G.W., Sanchez-Pulido,L., Heuckmann, J.M., Toth, R., Macartney, T., Eppink, B., Kanaar, R., Ponting,C.P., Lilley, D.M.J. and Rouse, J. (2009) Coordination of structure-specificnucleases by human SLX4/BTBD12 is required for DNA repair. Mol. Cell 35,116–127.
[49] Sijbers, A.M., de Laat, W.L., Ariza, R.R., Biggerstaff, M., Wei, Y.F., Moggs, J.G.,Carter, K.C., Shell, B.K., Evans, E., de Jong, M.C., Rademakers, S., de Rooij, J.,Jaspers, N.G., Hoeijmakers, J.H. and Wood, R.D. (1996) Xerodermapigmentosum group F caused by a defect in a structure-specific DNA repairendonuclease. Cell 86, 811–822.
[50] Svendsen, J.M., Smogorzewska, A., Sowa, M.E., O’Connell, B.C., Gygi, S.P.,Elledge, S.J. and Harper, J.W. (2009) Mammalian BTBD12/SLX4 assembles aHolliday junction resolvase and is required for DNA repair. Cell 138, 63–77.
[51] Tripsianes, K., Folkers, G., Ab, E., Das, D., Odijk, H., Jaspers, N.G.J., Hoeijmakers,J.H.J., Kaptein, R. and Boelens, R. (2005) The structure of the human ERCC1/XPF interaction domains reveals a complementary role for the two proteinsin nucleotide excision repair. Structure 1993 (13), 1849–1858.
[52] Reynolds, R.J. and Friedberg, E.C. (1981) Molecular mechanisms of pyrimidinedimer excision in Saccharomyces cerevisiae: incision of ultraviolet-irradiateddeoxyribonucleic acid in vivo. J. Bacteriol. 146, 692–704.
[53] Fagbemi, A.F., Orelli, B. and Schärer, O.D. (2011) Regulation of endonucleaseactivity in human nucleotide excision repair. DNA Repair 10, 722–729.
[54] O’Donovan, A., Davies, A.A., Moggs, J.G., West, S.C. and Wood, R.D. (1994) XPGendonuclease makes the 30 incision in human DNA nucleotide excisionrepair. Nature 371, 432–435.
[55] Prakash, S. and Prakash, L. (2000) Nucleotide excision repair in yeast. Mutat.Res. 451, 13–24.
[56] Wood, R.D. (2010) Mammalian nucleotide excision repair proteins andinterstrand crosslink repair. Environ. Mol. Mutagen. 51, 520–526.
[57] Kuraoka, I., Kobertz, W.R., Ariza, R.R., Biggerstaff, M., Essigmann, J.M. andWood, R.D. (2000) Repair of an interstrand DNA cross-link initiated byERCC1-XPF repair/recombination nuclease. J. Biol. Chem. 275, 26632–26636.
[58] Ahmad, A., Robinson, A.R., Duensing, A., van Drunen, E., Beverloo, H.B.,Weisberg, D.B., Hasty, P., Hoeijmakers, J.H.J. and Niedernhofer, L.J. (2008)ERCC1-XPF endonuclease facilitates DNA double-strand break repair. Mol.Cell. Biol. 28, 5082–5092.
[59] Sargent, R.G., Rolig, R.L., Kilburn, A.E., Adair, G.M., Wilson, J.H. and Nairn, R.S.(1997) Recombination-dependent deletion formation in mammalian cellsdeficient in the nucleotide excision repair gene ERCC1. Proc. Natl. Acad. Sci.USA 94, 13122–13127.
[60] Al-Minawi, A.Z., Saleh-Gohari, N. and Helleday, T. (2008) The ERCC1/XPFendonuclease is required for efficient single-strand annealing and geneconversion in mammalian cells. Nucleic Acids Res. 36, 1–9.
[61] Niedernhofer, L.J., Essers, J., Weeda, G., Beverloo, B., de Wit, J., Muijtjens, M.,Odijk, H., Hoeijmakers, J.H. and Kanaar, R. (2001) The structure-specificendonuclease Ercc1-Xpf is required for targeted gene replacement inembryonic stem cells. EMBO J. 20, 6540–6549.
[62] Fishman-Lobell, J. and Haber, J.E. (1992) Removal of non-homologous DNAends in double-strand break recombination: the role of the yeast ultravioletrepair gene RAD1. Science 258, 480–484.
[63] Niedernhofer, L.J., Odijk, H., Budzowska, M., van Drunen, E., Maas, A., Theil,A.F., de Wit, J., Jaspers, N.G.J., Beverloo, H.B., Hoeijmakers, J.H.J. and Kanaar, R.(2004) The structure-specific endonuclease Ercc1-Xpf is required to resolveDNA interstrand cross-link-induced double-strand breaks. Mol. Cell. Biol. 24,5776–5787.
[64] Hsia, K.-T., Millar, M.R., King, S., Selfridge, J., Redhead, N.J., Melton, D.W. andSaunders, P.T.K. (2003) DNA repair gene Ercc1 is essential for normalspermatogenesis and oogenesis and for functional integrity of germ cell DNAin the mouse. Development 130, 369–378.
[65] Yildiz, O., Kearney, H., Kramer, B.C. and Sekelsky, J.J. (2004) Mutationalanalysis of the Drosophila DNA repair and recombination gene mei-9.Genetics 167, 263–273.
[66] Yildiz, O., Majumder, S., Kramer, B. and Sekelsky, J.J. (2002) DrosophilaMUS312 interacts with the nucleotide excision repair endonuclease MEI-9 togenerate meiotic crossovers. Mol. Cell 10, 1503–1509.
[67] Naim, V., Wilhelm, T., Debatisse, M. and Rosselli, F. (2013) ERCC1 andMUS81-EME1 promote sister chromatid separation by processing latereplication intermediates at common fragile sites during mitosis. Nat. CellBiol. 15, 1008–1015.
[68] Ying, S., Minocherhomji, S., Chan, K.L., Palmai-Pallag, T., Chu, W.K., Wass, T.,Mankouri, H.W., Liu, Y. and Hickson, I.D. (2013) MUS81 promotes commonfragile site expression. Nat. Cell Biol. 15, 1001–1007.
[69] Matsumura, Y., Nishigori, C., Yagi, T., Imamura, S. and Takebe, H. (1998)Characterization of molecular defects in xeroderma pigmentosum group F inrelation to its clinically mild symptoms. Hum. Mol. Genet. 7, 969–974.
[70] Niedernhofer, L.J., Garinis, G.A., Raams, A., Lalai, A.S., Robinson, A.R.,Appeldoorn, E., Odijk, H., Oostendorp, R., Ahmad, A., van Leeuwen, W.,Theil, A.F., Vermeulen, W., van der Horst, G.T.J., Meinecke, P., Kleijer, W.J.,Vijg, J., Jaspers, N.G.J. and Hoeijmakers, J.H.J. (2006) A new progeroidsyndrome reveals that genotoxic stress suppresses the somatotroph axis.Nature 444, 1038–1043.
[71] Zhu, X.-D., Niedernhofer, L., Kuster, B., Mann, M., Hoeijmakers, J.H.J. and deLange, T. (2003) ERCC1/XPF removes the 30 overhang from uncappedtelomeres and represses formation of telomeric DNA-containing doubleminute chromosomes. Mol. Cell 12, 1489–1498.
[72] Olaussen, K.A., Dunant, A., Fouret, P., Brambilla, E., André, F., Haddad, V.,Taranchon, E., Filipits, M., Pirker, R., Popper, H.H., Stahel, R., Sabatier, L.,Pignon, J.-P., Tursz, T., Le Chevalier, T., Soria, J.-C. and Bio Investigators, I.A.L.T.(2006) DNA repair by ERCC1 in non-small-cell lung cancer and cisplatin-based adjuvant chemotherapy. N. Engl. J. Med. 355, 983–991.
[73] Simon, G.R., Sharma, S., Cantor, A., Smith, P. and Bepler, G. (2005) ERCC1expression is a predictor of survival in resected patients with non-small celllung cancer. Chest 127, 978–983.
[74] Takenaka, T., Yoshino, I., Kouso, H., Ohba, T., Yohena, T., Osoegawa, A., Shoji, F.and Maehara, Y. (2007) Combined evaluation of Rad51 and ERCC1expressions for sensitivity to platinum agents in non-small cell lungcancer. Int. J. Cancer J. Int. Cancer 121, 895–900.
[75] Song, L., Ritchie, A.-M., McNeil, E.M., Li, W. and Melton, D.W. (2011)Identification of DNA repair gene Ercc1 as a novel target in melanoma.Pigment Cell Melanoma Res. 24, 966–971.
[76] Coulon, S., Gaillard, P.-H.L., Chahwan, C., McDonald, W.H., Yates 3rd, J.R. andRussell, P. (2004) Slx1-Slx4 are subunits of a structure-specific endonucleasethat maintains ribosomal DNA in fission yeast. Mol. Biol. Cell 15, 71–80.
[77] Fricke, W.M. and Brill, S.J. (2003) Slx1-Slx4 is a second structure-specificendonuclease functionally redundant with Sgs1-Top3. Genes Dev. 17, 1768–1778.
[78] Flott, S., Alabert, C., Toh, G.W., Toth, R., Sugawara, N., Campbell, D.G., Haber,J.E., Pasero, P. and Rouse, J. (2007) Phosphorylation of Slx4 by Mec1 and Tel1regulates the single-strand annealing mode of DNA repair in budding yeast.Mol. Cell. Biol. 27, 6433–6445.
[79] Li, F., Dong, J., Pan, X., Oum, J.-H., Boeke, J.D. and Lee, S.E. (2008) Microarray-based genetic screen defines SAW1, a gene required for Rad1/Rad10-dependent processing of recombination intermediates. Mol. Cell 30, 325–335.
[80] Vannier, J.-B., Pavicic-Kaltenbrunner, V., Petalcorin, M.I.R., Ding, H. andBoulton, S.J. (2012) RTEL1 dismantles T loops and counteracts telomeric G4-DNA to maintain telomere integrity. Cell 149, 795–806.
[81] Wilson, J.S.J., Tejera, A.M., Castor, D., Toth, R., Blasco, M.A. and Rouse, J. (2013)Localization-dependent and -independent roles of SLX4 in regulatingtelomeres. Cell Rep. 4, 853–860.
[82] Kim, Y., Spitz, G.S., Veturi, U., Lach, F.P., Auerbach, A.D. and Smogorzewska, A.(2013) Regulation of multiple DNA repair pathways by the Fanconi anemiaprotein SLX4. Blood 121, 54–63.
[83] Schuster, B., Knies, K., Stoepker, C., Velleuer, E., Friedl, R., Gottwald-Mühlhauser, B., de Winter, J.P. and Schindler, D. (2013) Whole exomesequencing reveals uncommon mutations in the recently identified Fanconianemia gene SLX4/FANCP. Hum. Mutat. 34, 93–96.
[84] Stoepker, C., Hain, K., Schuster, B., Hilhorst-Hofstee, Y., Rooimans, M.A.,Steltenpool, J., Oostra, A.B., Eirich, K., Korthof, E.T., Nieuwint, A.W.M., Jaspers,N.G.J., Bettecken, T., Joenje, H., Schindler, D., Rouse, J. and de Winter, J.P.(2011) SLX4, a coordinator of structure-specific endonucleases, is mutated ina new Fanconi anemia subtype. Nat. Genet. 43, 138–141.
[85] Crossan, G.P., van der Weyden, L., Rosado, I.V., Langevin, F., Gaillard, P.-H.L.,Sanger Mouse Genetics Project, McIntyre, R.E., Gallagher, F., Kettunen, M.I.,Lewis, D.Y., Brindle, K., Arends, M.J., Adams, D.J. and Patel, K.J. (2011)Disruption of mouse Slx4, a regulator of structure-specific nucleases,phenocopies Fanconi anemia. Nat. Genet. 43, 147–152.
[86] Ip, S.C.Y., Rass, U., Blanco, M.G., Flynn, H.R., Skehel, J.M. and West, S.C. (2008)Identification of Holliday junction resolvases from humans and yeast. Nature456, 357–361.
[87] Rass, U., Compton, S.A., Matos, J., Singleton, M.R., Ip, S.C.Y., Blanco, M.G.,Griffith, J.D. and West, S.C. (2010) Mechanism of Holliday junction resolutionby the human GEN1 protein. Genes Dev. 24, 1559–1569.
[88] Gao, M., Rendtlew Danielsen, J., Wei, L.-Z., Zhou, D.-P., Xu, Q., Li, M.-M., Wang,Z.-Q., Tong, W.-M. and Yang, Y.-G. (2012) A novel role of human hollidayjunction resolvase GEN1 in the maintenance of centrosome integrity. PLoSOne 7, e49687.
[89] Wechsler, T., Newman, S. and West, S.C. (2011) Aberrant chromosomemorphology in human cells defective for Holliday junction resolution. Nature471, 642–646.
[90] Blanco, M.G., Matos, J., Rass, U., Ip, S.C.Y. and West, S.C. (2010) Functionaloverlap between the structure-specific nucleases Yen1 and Mus81-Mms4 forDNA-damage repair in S. cerevisiae. DNA Repair 9, 394–402.
[91] Muñoz-Galván, S., Tous, C., Blanco, M.G., Schwartz, E.K., Ehmsen, K.T., West,S.C., Heyer, W.-D. and Aguilera, A. (2012) Distinct roles of Mus81, Yen1, Slx1-Slx4, and Rad1 nucleases in the repair of replication-born double-strandbreaks by sister chromatid exchange. Mol. Cell. Biol. 32, 1592–1603.

2454 Z. Bartosova, L. Krejci / FEBS Letters 588 (2014) 2446–2456
[92] Pardo, B. and Aguilera, A. (2012) Complex chromosomal rearrangementsmediated by break-induced replication involve structure-selectiveendonucleases. PLoS Genet. 8, e1002979.
[93] García-Luis, J., Clemente-Blanco, A., Aragón, L. and Machín, F. (2014) Cdc14targets the Holliday junction resolvase Yen1 to the nucleus in early anaphase.Cell Cycle 13, 1392–1399.
[94] Balakrishnan, L. and Bambara, R.A. (2013) Flap endonuclease 1. Annu. Rev.Biochem. 82, 119–138.
[95] Harrington, J.J. and Lieber, M.R. (1994) The characterization of a mammalianDNA structure-specific endonuclease. EMBO J. 13, 1235–1246.
[96] Zheng, L., Zhou, M., Chai, Q., Parrish, J., Xue, D., Patrick, S.M., Turchi, J.J.,Yannone, S.M., Chen, D. and Shen, B. (2005) Novel function of the flapendonuclease 1 complex in processing stalled DNA replication forks. EMBORep. 6, 83–89.
[97] Zheng, L., Jia, J., Finger, L.D., Guo, Z., Zer, C. and Shen, B. (2011) Functionalregulation of FEN1 nuclease and its link to cancer. Nucleic Acids Res. 39, 781–794.
[98] Bornarth, C.J., Ranalli, T.A., Henricksen, L.A., Wahl, A.F. and Bambara, R.A.(1999) Effect of flap modifications on human FEN1 cleavage. Biochemistry(Mosc.) 38, 13347–13354.
[99] Kaiser, M.W., Lyamicheva, N., Ma, W., Miller, C., Neri, B., Fors, L. andLyamichev, V.I. (1999) A comparison of eubacterial and archaeal structure-specific 50-exonucleases. J. Biol. Chem. 274, 21387–21394.
[100] Liu, R., Qiu, J., Finger, L.D., Zheng, L. and Shen, B. (2006) The DNA-proteininteraction modes of FEN-1 with gap substrates and their implication inpreventing duplication mutations. Nucleic Acids Res. 34, 1772–1784.
[101] Kucherlapati, M., Yang, K., Kuraguchi, M., Zhao, J., Lia, M., Heyer, J., Kane, M.F.,Fan, K., Russell, R., Brown, A.M.C., Kneitz, B., Edelmann, W., Kolodner, R.D.,Lipkin, M. and Kucherlapati, R. (2002) Haploinsufficiency of Flapendonuclease (Fen1) leads to rapid tumor progression. Proc. Natl. Acad. Sci.USA 99, 9924–9929.
[102] Larsen, E., Gran, C., Saether, B.E., Seeberg, E. and Klungland, A. (2003)Proliferation failure and gamma radiation sensitivity of Fen1 null mutantmice at the blastocyst stage. Mol. Cell. Biol. 23, 5346–5353.
[103] Zheng, L., Dai, H., Zhou, M., Li, M., Singh, P., Qiu, J., Tsark, W., Huang, Q.,Kernstine, K., Zhang, X., Lin, D. and Shen, B. (2007) Fen1 mutations result inautoimmunity, chronic inflammation and cancers. Nat. Med. 13, 812–819.
[104] Klungland, A. and Lindahl, T. (1997) Second pathway for completion ofhuman DNA base excision-repair: reconstitution with purified proteins andrequirement for DNase IV (FEN1). EMBO J. 16, 3341–3348.
[105] Goula, A.-V., Berquist, B.R., Wilson 3rd, D.M., Wheeler, V.C., Trottier, Y. andMerienne, K. (2009) Stoichiometry of base excision repair proteins correlateswith increased somatic CAG instability in striatum over cerebellum inHuntington’s disease transgenic mice. PLoS Genet. 5, e1000749.
[106] Liu, Y., Zhang, H., Veeraraghavan, J., Bambara, R.A. and Freudenreich, C.H.(2004) Saccharomyces cerevisiae flap endonuclease 1 uses flap equilibrationto maintain triplet repeat stability. Mol. Cell. Biol. 24, 4049–4064.
[107] Singh, P., Zheng, L., Chavez, V., Qiu, J. and Shen, B. (2007) Concerted action ofexonuclease and Gap-dependent endonuclease activities of FEN-1contributes to the resolution of triplet repeat sequences (CTG)n- and(GAA)n-derived secondary structures formed during maturation of Okazakifragments. J. Biol. Chem. 282, 3465–3477.
[108] McMurray, C.T. (2010) Mechanisms of trinucleotide repeat instability duringhuman development. Nat. Rev. Genet. 11, 786–799.
[109] Freudenreich, C.H., Kantrow, S.M. and Zakian, V.A. (1998) Expansion andlength-dependent fragility of CTG repeats in yeast. Science 279, 853–856.
[110] Lopes, J., Debrauwère, H., Buard, J. and Nicolas, A. (2002) Instability of thehuman minisatellite CEB1 in rad27Delta and dna2-1 replication-deficientyeast cells. EMBO J. 21, 3201–3211.
[111] Muftuoglu, M., Wong, H.K., Imam, S.Z., Wilson 3rd, D.M., Bohr, V.A. andOpresko, P.L. (2006) Telomere repeat binding factor 2 interacts with baseexcision repair proteins and stimulates DNA synthesis by DNA polymerasebeta. Cancer Res. 66, 113–124.
[112] Parenteau, J. and Wellinger, R.J. (1999) Accumulation of single-stranded DNAand destabilization of telomeric repeats in yeast mutant strains carrying adeletion of RAD27. Mol. Cell. Biol. 19, 4143–4152.
[113] Saharia, A., Guittat, L., Crocker, S., Lim, A., Steffen, M., Kulkarni, S. andStewart, S.A. (2008) Flap endonuclease 1 contributes to telomere stability.Curr. Biol. 18, 496–500.
[114] Sharma, S., Sommers, J.A., Gary, R.K., Friedrich-Heineken, E., Hübscher, U. andBrosh Jr., R.M. (2005) The interaction site of Flap Endonuclease-1 with WRNhelicase suggests a coordination of WRN and PCNA. Nucleic Acids Res. 33,6769–6781.
[115] Saharia, A. and Stewart, S.A. (2009) FEN1 contributes to telomere stability inALT-positive tumor cells. Oncogene 28, 1162–1167.
[116] Sampathi, S., Bhusari, A., Shen, B. and Chai, W. (2009) Human flapendonuclease I is in complex with telomerase and is required fortelomerase-mediated telomere maintenance. J. Biol. Chem. 284, 3682–3690.
[117] Kucherlapati, M., Nguyen, A., Kuraguchi, M., Yang, K., Fan, K., Bronson, R.,Wei, K., Lipkin, M., Edelmann, W. and Kucherlapati, R. (2007) Tumorprogression in Apc(1638N) mice with Exo1 and Fen1 deficiencies.Oncogene 26, 6297–6306.
[118] Liu, L., Zhou, C., Zhou, L., Peng, L., Li, D., Zhang, X., Zhou, M., Kuang, P., Yuan,Q., Song, X. and Yang, M. (2012) Functional FEN1 genetic variants contributeto risk of hepatocellular carcinoma, esophageal cancer, gastric cancer andcolorectal cancer. Carcinogenesis 33, 119–123.
[119] Yang, M., Guo, H., Wu, C., He, Y., Yu, D., Zhou, L., Wang, F., Xu, J., Tan, W.,Wang, G., Shen, B., Yuan, J., Wu, T. and Lin, D. (2009) Functional FEN1polymorphisms are associated with DNA damage levels and lung cancer risk.Hum. Mutat. 30, 1320–1328.
[120] Nikolova, T., Christmann, M. and Kaina, B. (2009) FEN1 is overexpressed intestis, lung and brain tumors. Anticancer Res. 29, 2453–2459.
[121] Singh, P., Yang, M., Dai, H., Yu, D., Huang, Q., Tan, W., Kernstine, K.H., Lin, D.and Shen, B. (2008) Overexpression and hypomethylation of flapendonuclease 1 gene in breast and other cancers. Mol. Cancer Res. 6,1710–1717.
[122] Tishkoff, D.X., Filosi, N., Gaida, G.M. and Kolodner, R.D. (1997) A novelmutation avoidance mechanism dependent on S. cerevisiae RAD27 is distinctfrom DNA mismatch repair. Cell 88, 253–263.
[123] Stracker, T.H. and Petrini, J.H.J. (2011) The MRE11 complex: starting from theends. Nat. Rev. Mol. Cell Biol. 12, 90–103.
[124] Buis, J., Wu, Y., Deng, Y., Leddon, J., Westfield, G., Eckersdorff, M., Sekiguchi,J.M., Chang, S. and Ferguson, D.O. (2008) Mre11 nuclease activity hasessential roles in DNA repair and genomic stability distinct from ATMactivation. Cell 135, 85–96.
[125] Luo, G., Yao, M.S., Bender, C.F., Mills, M., Bladl, A.R., Bradley, A. and Petrini,J.H. (1999) Disruption of mRad50 causes embryonic stem cell lethality,abnormal embryonic development, and sensitivity to ionizing radiation.Proc. Natl. Acad. Sci. USA 96, 7376–7381.
[126] Zhu, J., Petersen, S., Tessarollo, L. and Nussenzweig, A. (2001) Targeteddisruption of the Nijmegen breakage syndrome gene NBS1 leads to earlyembryonic lethality in mice. Curr. Biol. 11, 105–109.
[127] De Jager, M., Wyman, C., van Gent, D.C. and Kanaar, R. (2002) DNA end-binding specificity of human Rad50/Mre11 is influenced by ATP. NucleicAcids Res. 30, 4425–4431.
[128] Deriano, L., Stracker, T.H., Baker, A., Petrini, J.H.J. and Roth, D.B. (2009) Rolesfor NBS1 in alternative non-homologous end-joining of V(D)J recombinationintermediates. Mol. Cell 34, 13–25.
[129] Keeney, S., Giroux, C.N. and Kleckner, N. (1997) Meiosis-specific DNA double-strand breaks are catalyzed by Spo11, a member of a widely conservedprotein family. Cell 88, 375–384.
[130] Rass, E., Grabarz, A., Plo, I., Gautier, J., Bertrand, P. and Lopez, B.S. (2009) Roleof Mre11 in chromosomal non-homologous end joining in mammalian cells.Nat. Struct. Mol. Biol. 16, 819–824.
[131] Xie, A., Kwok, A. and Scully, R. (2009) Role of mammalian Mre11 in classicaland alternative non-homologous end joining. Nat. Struct. Mol. Biol. 16, 814–818.
[132] Jiang, W.-Q., Zhong, Z.-H., Henson, J.D., Neumann, A.A., Chang, A.C.-M. andReddel, R.R. (2005) Suppression of alternative lengthening of telomeres bySp100-mediated sequestration of the MRE11/RAD50/NBS1 complex. Mol.Cell. Biol. 25, 2708–2721.
[133] Porro, A., Feuerhahn, S. and Lingner, J. (2014) TERRA-reinforced association ofLSD1 with MRE11 promotes processing of uncapped telomeres. Cell Rep. 6,765–776.
[134] Zhu, X.D., Küster, B., Mann, M., Petrini, J.H. and de Lange, T. (2000) Cell-cycle-regulated association of RAD50/MRE11/NBS1 with TRF2 and humantelomeres. Nat. Genet. 25, 347–352.
[135] Sartori, A.A., Lukas, C., Coates, J., Mistrik, M., Fu, S., Bartek, J., Baer, R., Lukas, J.and Jackson, S.P. (2007) Human CtIP promotes DNA end resection. Nature450, 509–514.
[136] Jazayeri, A., Falck, J., Lukas, C., Bartek, J., Smith, G.C.M., Lukas, J. and Jackson,S.P. (2006) ATM- and cell cycle-dependent regulation of ATR in response toDNA double-strand breaks. Nat. Cell Biol. 8, 37–45.
[137] Quennet, V., Beucher, A., Barton, O., Takeda, S. and Löbrich, M. (2011) CtIPand MRN promote non-homologous end-joining of etoposide-induced DNAdouble-strand breaks in G1. Nucleic Acids Res. 39, 2144–2152.
[138] Makharashvili, N., Tubbs, A.T., Yang, S.-H., Wang, H., Barton, O., Zhou, Y.,Deshpande, R.A., Lee, J.-H., Lobrich, M., Sleckman, B.P., Wu, X. and Paull, T.T.(2014) Catalytic and non-catalytic roles of the CtIP endonuclease in double-strand break end resection. Mol. Cell.. in press.
[139] Wang, H., Li, Y., Truong, L.N., Shi, L.Z., Hwang, P.Y.-H., He, J., Do, J., Cho, M.J.,Li, H., Negrete, A., Shiloach, J., Berns, M.W., Shen, B., Chen, L. and Wu, X.(2014) CtIP maintains stability at common fragile sites and invertedrepeats by end resection-independent endonuclease activity. Mol. Cell. inpress.
[140] Carney, J.P., Maser, R.S., Olivares, H., Davis, E.M., Le Beau, M., Yates 3rd, J.R.,Hays, L., Morgan, W.F. and Petrini, J.H. (1998) The hMre11/hRad50 proteincomplex and Nijmegen breakage syndrome: linkage of double-strand breakrepair to the cellular DNA damage response. Cell 93, 477–486.
[141] Lavin, M.F. (2007) ATM and the Mre11 complex combine to recognize andsignal DNA double-strand breaks. Oncogene 26, 7749–7758.
[142] Stewart, G.S., Maser, R.S., Stankovic, T., Bressan, D.A., Kaplan, M.I., Jaspers,N.G., Raams, A., Byrd, P.J., Petrini, J.H. and Taylor, A.M. (1999) The DNAdouble-strand break repair gene hMRE11 is mutated in individuals with anataxia-telangiectasia-like disorder. Cell 99, 577–587.
[143] Taylor, A.M.R., Groom, A. and Byrd, P.J. (2004) Ataxia-telangiectasia-likedisorder (ATLD)-its clinical presentation and molecular basis. DNA Repair 3,1219–1225.
[144] Waltes, R., Kalb, R., Gatei, M., Kijas, A.W., Stumm, M., Sobeck, A., Wieland, B.,Varon, R., Lerenthal, Y., Lavin, M.F., Schindler, D. and Dörk, T. (2009) HumanRAD50 deficiency in a Nijmegen breakage syndrome-like disorder. Am. J.Hum. Genet. 84, 605–616.

Z. Bartosova, L. Krejci / FEBS Letters 588 (2014) 2446–2456 2455
[145] Qvist, P., Huertas, P., Jimeno, S., Nyegaard, M., Hassan, M.J., Jackson, S.P. andBørglum, A.D. (2011) CtIP mutations cause Seckel and Jawad syndromes.PLoS Genet. 7, e1002310.
[146] Bartkova, J., Tommiska, J., Oplustilova, L., Aaltonen, K., Tamminen, A.,Heikkinen, T., Mistrik, M., Aittomäki, K., Blomqvist, C., Heikkilä, P., Lukas, J.,Nevanlinna, H. and Bartek, J. (2008) Aberrations of the MRE11-RAD50-NBS1DNA damage sensor complex in human breast cancer: MRE11 as a candidatefamilial cancer-predisposing gene. Mol. Oncol. 2, 296–316.
[147] Desai-Mehta, A., Cerosaletti, K.M. and Concannon, P. (2001) Distinctfunctional domains of nibrin mediate Mre11 binding, focus formation, andnuclear localization. Mol. Cell. Biol. 21, 2184–2191.
[148] Giannini, G., Rinaldi, C., Ristori, E., Ambrosini, M.I., Cerignoli, F., Viel, A.,Bidoli, E., Berni, S., D’Amati, G., Scambia, G., Frati, L., Screpanti, I. and Gulino,A. (2004) Mutations of an intronic repeat induce impaired MRE11 expressionin primary human cancer with microsatellite instability. Oncogene 23, 2640–2647.
[149] Vilar, E., Bartnik, C.M., Stenzel, S.L., Raskin, L., Ahn, J., Moreno, V., Mukherjee,B., Iniesta, M.D., Morgan, M.A., Rennert, G. and Gruber, S.B. (2011) MRE11deficiency increases sensitivity to poly(ADP-ribose) polymerase inhibition inmicrosatellite unstable colorectal cancers. Cancer Res. 71, 2632–2642.
[150] Dupré, A., Boyer-Chatenet, L., Sattler, R.M., Modi, A.P., Lee, J.-H., Nicolette,M.L., Kopelovich, L., Jasin, M., Baer, R., Paull, T.T. and Gautier, J. (2008) Aforward chemical genetic screen reveals an inhibitor of the Mre11-Rad50-Nbs1 complex. Nat. Chem. Biol. 4, 119–125.
[151] Kuroda, S., Fujiwara, T., Shirakawa, Y., Yamasaki, Y., Yano, S., Uno, F., Tazawa,H., Hashimoto, Y., Watanabe, Y., Noma, K., Urata, Y., Kagawa, S. and Fujiwara,T. (2010) Telomerase-dependent oncolytic adenovirus sensitizes humancancer cells to ionizing radiation via inhibition of DNA repair machinery.Cancer Res. 70, 9339–9348.
[152] Shibata, A., Moiani, D., Arvai, A.S., Perry, J., Harding, S.M., Genois, M.-M.,Maity, R., van Rossum-Fikkert, S., Kertokalio, A., Romoli, F., Ismail, A., Ismalaj,E., Petricci, E., Neale, M.J., Bristow, R.G., Masson, J.-Y., Wyman, C., Jeggo, P.A.and Tainer, J.A. (2014) DNA double-strand break repair pathway choice isdirected by distinct MRE11 nuclease activities. Mol. Cell 53, 7–18.
[153] Llorente, B. and Symington, L.S. (2004) The Mre11 nuclease is not requiredfor 50–30 resection at multiple HO-induced double-strand breaks. Mol. Cell.Biol. 24, 9682–9694.
[154] Lin, Z.P., Ratner, E.S., Whicker, M.E., Lee, Y. and Sartorelli, A.C. (2014) Triapinedisrupts CtIP-mediated homologous recombination repair and sensitizesovarian cancer cells to PARP and topoisomerase inhibitors. Mol. Cancer Res.12, 381–393.
[155] Ying, S., Hamdy, F.C. and Helleday, T. (2012) Mre11-dependent degradationof stalled DNA replication forks is prevented by BRCA2 and PARP1. CancerRes. 72, 2814–2821.
[156] Genschel, J., Bazemore, L.R. and Modrich, P. (2002) Human exonuclease I isrequired for 50 and 30 mismatch repair. J. Biol. Chem. 277, 13302–13311.
[157] Tishkoff, D.X., Boerger, A.L., Bertrand, P., Filosi, N., Gaida, G.M., Kane, M.F. andKolodner, R.D. (1997) Identification and characterization of Saccharomycescerevisiae EXO1, a gene encoding an exonuclease that interacts with MSH2.Proc. Natl. Acad. Sci. USA 94, 7487–7492.
[158] Amin, N.S., Nguyen, M.N., Oh, S. and Kolodner, R.D. (2001) Exo1-dependentmutator mutations: model system for studying functional interactions inmismatch repair. Mol. Cell. Biol. 21, 5142–5155.
[159] Zhu, Z., Chung, W.-H., Shim, E.Y., Lee, S.E. and Ira, G. (2008) Sgs1 helicase andtwo nucleases Dna2 and Exo1 resect DNA double-strand break ends. Cell 134,981–994.
[160] Lindsey-Boltz, L.A., Kemp, M.G., Reardon, J.T., DeRocco, V., Iyer, R.R., Modrich,P. and Sancar, A. (2014) Coupling of human DNA excision repair and the DNAdamage checkpoint in a defined in vitro system. J. Biol. Chem. 289, 5074–5082.
[161] Sertic, S., Pizzi, S., Cloney, R., Lehmann, A.R., Marini, F., Plevani, P. and Muzi-Falconi, M. (2011) Human exonuclease 1 connects nucleotide excision repair(NER) processing with checkpoint activation in response to UV irradiation.Proc. Natl. Acad. Sci. USA 108, 13647–13652.
[162] Hackett, J.A. and Greider, C.W. (2003) End resection initiates genomicinstability in the absence of telomerase. Mol. Cell. Biol. 23, 8450–8461.
[163] Maringele, L. and Lydall, D. (2002) EXO1-dependent single-stranded DNA attelomeres activates subsets of DNA damage and spindle checkpointpathways in budding yeast yku70Delta mutants. Genes Dev. 16, 1919–1933.
[164] Wu, P., Takai, H. and de Lange, T. (2012) Telomeric 30 overhangs derive fromresection by Exo1 and Apollo and fill-in by POT1b-associated CST. Cell 150,39–52.
[165] Sun, X., Zheng, L. and Shen, B. (2002) Functional alterations of humanexonuclease 1 mutants identified in atypical hereditary non-polyposiscolorectal cancer syndrome. Cancer Res. 62, 6026–6030.
[166] Wei, K., Clark, A.B., Wong, E., Kane, M.F., Mazur, D.J., Parris, T., Kolas, N.K.,Russell, R., Hou Jr., H., Kneitz, B., Yang, G., Kunkel, T.A., Kolodner, R.D., Cohen,P.E. and Edelmann, W. (2003) Inactivation of exonuclease 1 in mice results inDNA mismatch repair defects, increased cancer susceptibility, and male andfemale sterility. Genes Dev. 17, 603–614.
[167] Tran, H.T., Gordenin, D.A. and Resnick, M.A. (1999) The 30 ? 50 exonucleasesof DNA polymerases delta and epsilon and the 50 ? 30 exonuclease Exo1 havemajor roles in postreplication mutation avoidance in Saccharomycescerevisiae. Mol. Cell. Biol. 19, 2000–2007.
[168] Wu, Y., Berends, M.J., Post, J.G., Mensink, R.G., Verlind, E., Van Der Sluis, T.,Kempinga, C., Sijmons, R.H., van der Zee, A.G., Hollema, H., Kleibeuker, J.H.,
Buys, R.M. and Hofstra, R.M. (2001) Germline mutations of EXO1 gene inpatients with hereditary non-polyposis colorectal cancer (HNPCC) andatypical HNPCC forms. Gastroenterology 120, 1580–1587.
[169] Liberti, S.E. and Rasmussen, L.J. (2004) Is hEXO1 a cancer predisposing gene?Mol. Cancer Res. 2, 427–432.
[170] Tran, P.T., Erdeniz, N., Symington, L.S. and Liskay, R.M. (2004) EXO1-A multi-tasking eukaryotic nuclease. DNA Repair 3, 1549–1559.
[171] Levikova, M., Klaue, D., Seidel, R. and Cejka, P. (2013) Nuclease activity ofSaccharomyces cerevisiae Dna2 inhibits its potent DNA helicase activity.Proc. Natl. Acad. Sci. USA 110, E1992–2001.
[172] Bae, S.H., Bae, K.H., Kim, J.A. and Seo, Y.S. (2001) RPA governs endonucleaseswitching during processing of Okazaki fragments in eukaryotes. Nature 412,456–461.
[173] Cejka, P., Cannavo, E., Polaczek, P., Masuda-Sasa, T., Pokharel, S., Campbell, J.L.and Kowalczykowski, S.C. (2010) DNA end resection by Dna2-Sgs1-RPA andits stimulation by Top3-Rmi1 and Mre11-Rad50-Xrs2. Nature 467, 112–116.
[174] Peng, G., Dai, H., Zhang, W., Hsieh, H.-J., Pan, M.-R., Park, Y.-Y., Tsai, R.Y.-L.,Bedrosian, I., Lee, J.-S., Ira, G. and Lin, S.-Y. (2012) Human nuclease/helicaseDNA2 alleviates replication stress by promoting DNA end resection. CancerRes. 72, 2802–2813.
[175] Karanja, K.K., Lee, E.H., Hendrickson, E.A. and Campbell, J.L. (2014) Preventingover-resection by DNA2 helicase/nuclease suppresses repair defects inFanconi anemia cells. Cell Cycle 13, 1540–1550.
[176] Budd, M.E., Tong, A.H.Y., Polaczek, P., Peng, X., Boone, C. and Campbell, J.L.(2005) A network of multi-tasking proteins at the DNA replication forkpreserves genome stability. PLoS Genet. 1, e61.
[177] Cimprich, K.A. and Cortez, D. (2008) ATR: an essential regulator of genomeintegrity. Nat. Rev. Mol. Cell Biol. 9, 616–627.
[178] Hosoya, N. and Miyagawa, K. (2014) Targeting DNA damage response incancer therapy. Cancer Sci. 105, 370–388.
[179] Smith, J., Tho, L.M., Xu, N. and Gillespie, D.A. (2010) The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv. Cancer Res. 108,73–112.
[180] Galamb, O., Sipos, F., Dinya, E., Spisak, S., Tulassay, Z. and Molnar, B. (2006)MRNA expression, functional profiling and multivariate classification ofcolon biopsy specimen by cDNA overall glass microarray. World J.Gastroenterol. 12, 6998–7006.
[181] Lim, D.S. and Hasty, P. (1996) A mutation in mouse rad51 results in an earlyembryonic lethal that is suppressed by a mutation in p53. Mol. Cell. Biol. 16,7133–7143.
[182] Tsuzuki, T., Fujii, Y., Sakumi, K., Tominaga, Y., Nakao, K., Sekiguchi, M.,Matsushiro, A., Yoshimura, Y. and Morita, T. (1996) Targeted disruption ofthe Rad51 gene leads to lethality in embryonic mice. Proc. Natl. Acad. Sci.USA 93, 6236–6240.
[183] Connell, P.P., Jayathilaka, K., Haraf, D.J., Weichselbaum, R.R., Vokes, E.E. andLingen, M.W. (2006) Pilot study examining tumor expression of RAD51 andclinical outcomes in human head cancers. Int. J. Oncol. 28, 1113–1119.
[184] Hannay, J.A.F., Liu, J., Zhu, Q.-S., Bolshakov, S.V., Li, L., Pisters, P.W.T., Lazar,A.J.F., Yu, D., Pollock, R.E. and Lev, D. (2007) Rad51 overexpression contributesto chemoresistance in human soft tissue sarcoma cells: a role for p53/activatorprotein 2 transcriptional regulation. Mol. Cancer Ther. 6, 1650–1660.
[185] Maacke, H., Jost, K., Opitz, S., Miska, S., Yuan, Y., Hasselbach, L., Lüttges, J.,Kalthoff, H. and Stürzbecher, H.W. (2000) DNA repair and recombinationfactor Rad51 is over-expressed in human pancreatic adenocarcinoma.Oncogene 19, 2791–2795.
[186] Yoshikawa, K., Ogawa, T., Baer, R., Hemmi, H., Honda, K., Yamauchi, A.,Inamoto, T., Ko, K., Yazumi, S., Motoda, H., Kodama, H., Noguchi, S., Gazdar,A.F., Yamaoka, Y. and Takahashi, R. (2000) Abnormal expression of BRCA1and BRCA1-interactive DNA-repair proteins in breast carcinomas. Int. J.Cancer J. Int. Cancer 88, 28–36.
[187] Meindl, A., Hellebrand, H., Wiek, C., Erven, V., Wappenschmidt, B.,Niederacher, D., Freund, M., Lichtner, P., Hartmann, L., Schaal, H., Ramser,J., Honisch, E., Kubisch, C., Wichmann, H.E., Kast, K., Deissler, H., Engel, C.,Müller-Myhsok, B., Neveling, K., Kiechle, M., Mathew, C.G., Schindler, D.,Schmutzler, R.K. and Hanenberg, H. (2010) Germline mutations in breast andovarian cancer pedigrees establish RAD51C as a human cancer susceptibilitygene. Nat. Genet. 42, 410–414.
[188] Vaz, F., Hanenberg, H., Schuster, B., Barker, K., Wiek, C., Erven, V., Neveling, K.,Endt, D., Kesterton, I., Autore, F., Fraternali, F., Freund, M., Hartmann, L.,Grimwade, D., Roberts, R.G., Schaal, H., Mohammed, S., Rahman, N.,Schindler, D. and Mathew, C.G. (2010) Mutation of the RAD51C gene in aFanconi anemia-like disorder. Nat. Genet. 42, 406–409.
[189] Budke, B., Kalin, J.H., Pawlowski, M., Zelivianskaia, A.S., Wu, M., Kozikowski,A.P. and Connell, P.P. (2013) An optimized RAD51 inhibitor that disruptshomologous recombination without requiring Michael acceptor reactivity. J.Med. Chem. 56, 254–263.
[190] Huang, F., Motlekar, N.A., Burgwin, C.M., Napper, A.D., Diamond, S.L. andMazin, A.V. (2011) Identification of specific inhibitors of human RAD51recombinase using high-throughput screening. ACS Chem. Biol. 6, 628–635.
[191] Slupianek, A., Schmutte, C., Tombline, G., Nieborowska-Skorska, M., Hoser, G.,Nowicki, M.O., Pierce, A.J., Fishel, R. and Skorski, T. (2001) BCR/ABL regulatesmammalian RecA homologs, resulting in drug resistance. Mol. Cell 8, 795–806.
[192] Lok, B.H. and Powell, S.N. (2012) Molecular pathways: understanding therole of Rad52 in homologous recombination for therapeutic advancement.Clin. Cancer Res. 18, 6400–6406.

2456 Z. Bartosova, L. Krejci / FEBS Letters 588 (2014) 2446–2456
[193] Bachrati, C.Z. and Hickson, I.D. (2008) RecQ helicases: guardian angels of theDNA replication fork. Chromosoma 117, 219–233.
[194] Bohr, V.A. (2008) Rising from the RecQ-age: the role of human RecQ helicasesin genome maintenance. Trends Biochem. Sci. 33, 609–620.
[195] Ouyang, K.J., Woo, L.L. and Ellis, N.A. (2008) Homologous recombination andmaintenance of genome integrity: cancer and aging through the prism ofhuman RecQ helicases. Mech. Ageing Dev. 129, 425–440.
[196] Sharma, S., Doherty, K.M. and Brosh Jr., R.M. (2006) Mechanisms of RecQhelicases in pathways of DNA metabolism and maintenance of genomicstability. Biochem. J. 398, 319–337.
[197] Ellis, N.A., Groden, J., Ye, T.Z., Straughen, J., Lennon, D.J., Ciocci, S., Proytcheva,M. and German, J. (1995) The Bloom’s syndrome gene product is homologousto RecQ helicases. Cell 83, 655–666.
[198] Kitao, S., Lindor, N.M., Shiratori, M., Furuichi, Y. and Shimamoto, A. (1999)Rothmund–thomson syndrome responsible gene, RECQL4: genomicstructure and products. Genomics 61, 268–276.
[199] Van Maldergem, L., Siitonen, H.A., Jalkh, N., Chouery, E., De Roy, M., Delague,V., Muenke, M., Jabs, E.W., Cai, J., Wang, L.L., Plon, S.E., Fourneau, C., Kestilä,M., Gillerot, Y., Mégarbané, A. and Verloes, A. (2006) Revisiting thecraniosynostosis-radial ray hypoplasia association: Baller–Gerold syndromecaused by mutations in the RECQL4 gene. J. Med. Genet. 43, 148–152.
[200] Siitonen, H.A., Kopra, O., Kääriäinen, H., Haravuori, H., Winter, R.M.,Säämänen, A.-M., Peltonen, L. and Kestilä, M. (2003) Molecular defect ofRAPADILINO syndrome expands the phenotype spectrum of RECQL diseases.Hum. Mol. Genet. 12, 2837–2844.
[201] Yu, C.E., Oshima, J., Fu, Y.H., Wijsman, E.M., Hisama, F., Alisch, R., Matthews,S., Nakura, J., Miki, T., Ouais, S., Martin, G.M., Mulligan, J. and Schellenberg,G.D. (1996) Positional cloning of the Werner’s syndrome gene. Science 272,258–262.
[202] Gupta, R. and Brosh Jr., R.M. (2008) Helicases as prospective targets for anti-cancer therapy. Anticancer Agents Med. Chem. 8, 390–401.
[203] Jänne, P.A. (2008) Challenges of detecting EGFR T790M in gefitinib/erlotinib-resistant tumours. Lung Cancer 60 (Suppl. 2), S3–9.
[204] Taniguchi, T., Tischkowitz, M., Ameziane, N., Hodgson, S.V., Mathew, C.G.,Joenje, H., Mok, S.C. and D’Andrea, A.D. (2003) Disruption of the Fanconianemia-BRCA pathway in cisplatin-sensitive ovarian tumors. Nat. Med. 9,568–574.
[205] Bernards, R. (2013) Finding effective cancer therapies through loss offunction genetic screens. Curr. Opin. Genet. Dev. 24C, 23–29.
[206] Choi, Y.H. and Yu, A.-M. (2014) ABC transporters in multidrug resistance andpharmacokinetics, and strategies for drug development. Curr. Pharm. Des. 20,793–807.
[207] Townsend, D.M. and Tew, K.D. (2003) The role of glutathione-S-transferase inanti-cancer drug resistance. Oncogene 22, 7369–7375.
[208] Ventura, A., Kirsch, D.G., McLaughlin, M.E., Tuveson, D.A., Grimm, J., Lintault,L., Newman, J., Reczek, E.E., Weissleder, R. and Jacks, T. (2007) Restoration ofp53 function leads to tumour regression in vivo. Nature 445, 661–665.
[209] Xue, W., Zender, L., Miething, C., Dickins, R.A., Hernando, E., Krizhanovsky, V.,Cordon-Cardo, C. and Lowe, S.W. (2007) Senescence and tumour clearance istriggered by p53 restoration in murine liver carcinomas. Nature 445, 656–660.
[210] Peer, D., Karp, J.M., Hong, S., Farokhzad, O.C., Margalit, R. and Langer, R.(2007) Nanocarriers as an emerging platform for cancer therapy. Nat.Nanotechnol. 2, 751–760.
[211] Kee, Y. and D’Andrea, A.D. (2010) Expanded roles of the Fanconi anemiapathway in preserving genomic stability. Genes Dev. 24, 1680–1694.
[212] Lin, W.-C., Rajbhandari, N. and Wagner, K.-U. (2014) Cancer cell dormancy innovel mousemodels for reversible pancreatic cancer: a lingering challenge inthe development of targeted therapies. Cancer Res. 74, 2138–2143.
[213] Bryant, H.E., Schultz, N., Thomas, H.D., Parker, K.M., Flower, D., Lopez, E., Kyle,S., Meuth, M., Curtin, N.J. and Helleday, T. (2005) Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature434, 913–917.
[214] Farmer, H., McCabe, N., Lord, C.J., Tutt, A.N.J., Johnson, D.A., Richardson, T.B.,Santarosa, M., Dillon, K.J., Hickson, I., Knights, C., Martin, N.M.B., Jackson, S.P.,Smith, G.C.M. and Ashworth, A. (2005) Targeting the DNA repair defect inBRCA mutant cells as a therapeutic strategy. Nature 434, 917–921.
[215] Bryant, H.E. and Helleday, T. (2006) Inhibition of poly (ADP-ribose)polymerase activates ATM which is required for subsequent homologousrecombination repair. Nucleic Acids Res. 34, 1685–1691.
[216] McCabe, N., Turner, N.C., Lord, C.J., Kluzek, K., Bialkowska, A., Swift, S.,Giavara, S., O’Connor, M.J., Tutt, A.N., Zdzienicka, M.Z., Smith, G.C.M. andAshworth, A. (2006) Deficiency in the repair of DNA damage by homologousrecombination and sensitivity to poly(ADP-ribose) polymerase inhibition.Cancer Res. 66, 8109–8115.
[217] Kennedy, D.R. and Beerman, T.A. (2006) The radiomimetic enediyne C-1027induces unusual DNA damage responses to double-strand breaks.Biochemistry (Mosc.) 45, 3747–3754.
[218] Mohni, K.N., Kavanaugh, G.M. and Cortez, D. (2014) ATR pathway inhibitionis synthetically lethal in cancer cells with ERCC1 deficiency. Cancer Res. 74,2835–2845.
[219] Van Pel, D.M., Barrett, I.J., Shimizu, Y., Sajesh, B.V., Guppy, B.J., Pfeifer, T.,McManus, K.J. and Hieter, P. (2013) An evolutionarily conserved syntheticlethal interaction network identifies FEN1 as a broad-spectrum target foranticancer therapeutic development. PLoS Genet. 9, e1003254.
[220] McManus, K.J., Barrett, I.J., Nouhi, Y. and Hieter, P. (2009) Specific syntheticlethal killing of RAD54B-deficient human colorectal cancer cells by FEN1silencing. Proc. Natl. Acad. Sci. USA 106, 3276–3281.

Published online 20 April 2014 Nucleic Acids Research, 2014, Vol. 42, No. 10 6393–6404doi: 10.1093/nar/gku300
Sumoylation of the Rad1 nuclease promotes DNArepair and regulates its DNA associationPrabha Sarangi1,2, Zdenka Bartosova3, Veronika Altmannova3, Cory Holland4,Melita Chavdarova5, Sang Eun Lee4,6, Lumir Krejci3,5,7 and Xiaolan Zhao1,*
1Molecular Biology Program, Memorial Sloan-Kettering Cancer Center, New York, NY 10065, USA, 2Programs inBiochemistry, Cell, and Molecular Biology, Weill Cornell Graduate School of Medical Sciences, New York, NY 10065,USA, 3Department of Biology, Masaryk University, Brno 62500, Czech Republic, 4Department of Molecular Medicine,Institute of Biotechnology, University of Texas Health Science Center at San Antonio, San Antonio, TX 78229, USA,5National Centre for Biomolecular Research, Masaryk University, Brno 62500, Czech Republic, 6Division of RadiationBiology, Department of Radiation Oncology, University of Texas Health Science Center at San Antonio, San Antonio,TX 78229, USA and 7International Clinical Research Center, St. Anne’s University Hospital in Brno, Brno 60200,Czech Republic
Received December 16, 2013; Revised March 26, 2014; Accepted March 29, 2014
ABSTRACT
The Saccharomyces cerevisiae Rad1-Rad10 complexis a conserved, structure-specific endonuclease im-portant for repairing multiple types of DNA lesions.Upon recruitment to lesion sites, Rad1-Rad10 re-moves damaged sequences, enabling subsequentgap filling and ligation. Acting at mid-steps of repair,the association and dissociation of Rad1-Rad10 withDNA can influence repair efficiency. We show thatgenotoxin-enhanced Rad1 sumoylation occurs afterthe nuclease is recruited to lesion sites. A single ly-sine outside Rad1’s nuclease and Rad10-binding do-mains is sumoylated in vivo and in vitro. Mutation ofthis site to arginine abolishes Rad1 sumoylation andimpairs Rad1-mediated repair at high doses of DNAdamage, but sustains the repair of a single double-stranded break. The timing of Rad1 sumoylation andthe phenotype bias toward high lesion loads point toa post-incision role for sumoylation, possibly affect-ing Rad1 dissociation from DNA. Indeed, biochemi-cal examination shows that sumoylation of Rad1 de-creases the complex’s affinity for DNA without affect-ing other protein properties. These findings suggesta model whereby sumoylation of Rad1 promotes itsdisengagement from DNA after nuclease cleavage,allowing it to efficiently attend to large numbers ofDNA lesions.
INTRODUCTION
Structure-specific nucleases occupy a central position inDNA repair due to their ability to remove a wide rangeof damaged sequences and resolve joint DNA structures inthe genome. Consequently, they greatly influence genomestability and cell survival upon exposure to environmentalmutagens and cancer therapeutic drugs. Optimized nucle-ase function in vivo is achieved by multiple layers of regu-lation. Many structure-specific nucleases interact with andare regulated by other repair factors that help recruit the nu-cleases to specific DNA lesion sites or stimulate their activ-ities (1–4). In addition, many nucleases are modified post-translationally (4–19). A few detailed studies show that dy-namic and reversible protein modifications can alter nucle-ase activities or protein levels in order to meet specific cellcycle needs for DNA cleavage (11–16). However, the effectsof many of these protein modifications on nucleases are cur-rently unknown.
Recent studies have revealed that a large number of DNArepair proteins, including several nucleases, are sumoylatedin response to DNA damage in yeast and humans (7–9,20).Although sumoylation as a whole can increase DNA repaircapacity (7,20–25), it is unclear how this is achieved at thelevel of each substrate and what principles underlie SUMO-mediated regulation of DNA repair. A comprehensive un-derstanding of these questions requires detailed studies ofsumoylation’s effects on each target. Here we look into therole of sumoylation in regulating the Rad1 nuclease in bud-ding yeast. Rad1 forms a heterodimer with Rad10, whichis required for Rad1 catalytic activity on branched DNAsubstrates (26–28). Rad1-Rad10 and their human homologsXPF-ERCC1 can remove several types of DNA lesions,such as those generated by UV radiation, topoisomerase
*To whom correspondence should be addressed. Tel: +1 212 639 5579; Fax: +1 646 422 2062; Email: [email protected]
C© The Author(s) 2014. Published by Oxford University Press on behalf of Nucleic Acids Research.This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/3.0/), whichpermits unrestricted reuse, distribution, and reproduction in any medium, provided the original work is properly cited.

6394 Nucleic Acids Research, 2014, Vol. 42, No. 10
inhibitors and DNA break-inducing agents (1,29). Theirimportant physiological roles are highlighted by the asso-ciation of XPF-ERCC1 mutations with cancer-prone dis-eases, including xeroderma pigmentosum, Cockayne syn-drome and Fanconi anemia (30–32).
In yeast, Rad1-Rad10 acts in nucleotide excision repair(NER) to remove bulky DNA lesions, such as those in-duced by UV (29). DNA distortion generated by these le-sions is recognized by the NER factors Rad4 and Rad23(33–35). A pre-incision complex is subsequently formed atlesion sites to unwind the DNA surrounding the lesion, gen-erating a bubble structure (29,36,37). The Rad14 proteinof this pre-incision complex recruits Rad1-Rad10 to DNAbubbles via direct physical interaction (38,39). Dual inci-sions by Rad1-Rad10 and another nuclease, Rad2, at the5′ and 3′ ends of the bubble, respectively, remove lesion-containing fragments (40,41). This allows subsequent re-pair synthesis and ligation. Besides involvement in NER,Rad1-Rad10 also acts as a back-up nuclease to removeprotein-DNA adducts generated by the Top1 inhibitorcamptothecin (CPT) (42,43). Moreover, Rad1-Rad10 func-tions in single-strand annealing (SSA) repair of double-stranded breaks, where its cleavage of 3′ flaps enables subse-quent ligation (44,45). Recruitment and nucleolytic activityof Rad1-Rad10 in SSA are regulated by the lesion-bindingfactor Saw1 and the scaffolding protein Slx4, respectively(46–48).
Here, we determined that Rad1 is sumoylated on a sin-gle lysine and generated an unsumoylatable rad1 allele. Ex-amining the phenotype of this mutant and the timing ofmodification in vivo, in conjunction with in vitro analysisof the sumoylated Rad1 protein, suggests that sumoylationof Rad1 promotes repair efficiency, most likely by enhanc-ing the dissociation of Rad1-Rad10 from DNA after nucle-olytic cleavage.
MATERIALS AND METHODS
Yeast strains and genetic manipulations
Strains used are listed in Table 1. Standard yeast protocolswere used for strain generation, growth, medium prepara-tion and DNA damage sensitivity assays. As siz1Δ siz2Δresults in amplification of 2-micron plasmids (49), strainswith siz1Δ siz2Δ mutations were cured of the plasmid asdescribed (50). Spot assays were performed as describedpreviously (7). Briefly, log phase cells were diluted 10-foldor 3-fold and spotted onto YPD (Yeast extract-Peptone-Dextrose) media with or without CPT, or irradiated withUV. Plates were incubated at 30◦C and photographed after24–72 h.
Detection of sumoylated proteins and immunoprecipitation
These were performed as described previously (7). In brief,cells were lysed by bead beating under denaturing condi-tions and TAP-tagged proteins were immunoprecipitatedusing immunoglobulin G (IgG)-Sepharose. These werethen washed and eluted with loading dye, followed bysodium dodecylsulphate-polyacrylamide gel electrophore-sis (SDS-PAGE) and western blotting with antibodiesagainst SUMO (22) and the protein A portion of the TAP
tag (Sigma-Aldrich). Damage-induced sumoylation was as-sayed by subjecting log-phase cells to 0.03–0.3% methyl-methane sulfonate (MMS, Sigma-Aldrich), 50 ug/ml CPT(Sigma-Aldrich), or 0.2 M hydroxyurea (HU, US Biologi-cals) for 2 h, or UV irradiation using UV Stratalinker 1800(Stratagene). For Figure 1A, UV and MMS doses were cho-sen based on comparable survival posttreatment as shownin Supplementary Figure S1A–B. Co-immunoprecipitationexperiments were performed as described previously (51).
SSA assays
Chromosomal and plasmid-based SSA assays were per-formed as described (46). In brief, for chromosomal assay,log-phase cells grown in YP-Glycerol were plated on ei-ther YPD or YP-Galactose media. Only the latter inducesHO expression, and thus double -strand break formationand SSA repair. Colonies were counted after incubation at30◦C for 3–4 days. SSA survival quantification was doneby dividing the number of viable colonies on YP-galactoseplates by that on YPD and multiplying this factor by 100.For plasmid-based assay, cells were transformed with eitherBsu36I- or mock-digested pNSU208 plasmid, plated on me-dia lacking leucine and incubated at 30◦C for 3–4 days. Per-cent survival estimating SSA efficiency was calculated bydividing number of viable colonies of the Bsu36I-digestedtransformants by those of mock-digested ones.
Rad1-Rad10 protein expression, purification and gel filtra-tion
The Escherichia coli strain Rosetta(DE3)pLysS was trans-formed with a bicistronic plasmid (gift from Dr. SteveBrill (52)) expressing (His)6-Rad1 and Rad10, or (His)6-Rad1-K32R and Rad10. The Rad1-K32R mutant wasgenerated using site-directed mutagenesis. Protein expres-sion was induced by 0.1 mM IPTG (Isopropyl �-D-1-thiogalactopyranoside) at 16◦C overnight. A total of 9 g ofcell paste was sonicated in 40 ml breakage buffer (50 mMTris-HCl, pH 7.5, 10% sucrose, 2 mM ethylenediaminete-traacetic acid (EDTA), 200 mM KCl, 0.01% NP40, 1 mM�-mercaptoethanol and protease inhibitor cocktail contain-ing pepstatin, aprotinin, benzamidine, chymostatin and le-upeptin). The lysate was clarified by centrifugation (100kg, 4◦C, 1 h), and the supernatant was incubated with 1 mlof HIS-Select nickel affinity gel (Sigma) for 1 h at 4◦C. Thebeads were washed with 12 ml of buffer K (20 mM K2HPO4,10% glycerol, 0.5 mM EDTA) containing 150 mM KCl. Thebound proteins were eluted with buffer containing 50 mMKCl and imidazole (from 50 to 1000 mM). Pooled fractionscontaining Rad1-Rad10 (150–500 mM imidazole) were ap-plied to a 1-ml Heparin column (GE Healthcare) followedby elution using 8 ml gradient of 275–1000 mM KCl inbuffer K. The peak fractions of Rad1-Rad10 at ∼500 mMKCl elution were pooled, loaded onto a 0.5-ml MonoQcolumn (GE Healthcare) and eluted using a 5-ml gradientof 275–1000 mM KCl in buffer K. The Rad1-Rad10 frac-tions were concentrated to 400 �l in a VivaSpin-2 concen-trator, and fractionated on a 23-ml Sephacryl S400 column(GE Healthcare) in K buffer containing 300 mM KCl. PeakRad1-Rad10 fractions were concentrated to 2 �g/�l. To

Nucleic Acids Research, 2014, Vol. 42, No. 10 6395
Table 1. Yeast strains used in this study
Strain Genotype Source
W1588-4A Mat alpha leu2-3,112 ade2-1 can1-100his3-11,15 ura3-1 trp1-1 RAD5
R. Rothstein
T581 Mat a RAD1-TAP::HIS This studyX3456-2C Mat a RAD1-TAP::HIS rad14Δ::KAN This studyX3711-1B RAD1-TAP::HIS rad4Δ::KAN This studyX3527-9A RAD1-TAP::HIS saw1Δ::KAN This studyX3539-8B Mat alpha RAD1-TAP::HIS slx4Δ::KAN This studyX3527-1A Mat a RAD1-TAP::HIS rad14Δ::KAN
saw1Δ::KANThis study
X3458-1A Mat alpha RAD1-TAP::HIS rad52Δ::KAN This studyX3526-13B Mat alpha RAD1-TAP::HIS rad14Δ::KAN
rad52Δ::KANThis study
T1302-2 Mat a rad1-D825A-TAP::HIS This studyT908-2 Mat alpha rad1-D869A-TAP::HIS This studyX3563-1B Mat alpha rad1-D869A-TAP::HIS
saw1Δ::KANThis study
X3541-10D Mat alpha rad1-D869A-TAP::HISrad14Δ::KAN
This study
X3541-10A Mat alpha rad1-D869A-TAP::HISrad52Δ::KAN
This study
X3629-20A RAD1-TAP::HIS mec1Δ::TRP sml1Δ::HIS This studyX3580-12A Mat alpha RAD1-TAP::HIS siz1Δ::KAN cir0 This studyX3580-4C Mat a RAD1-TAP::HIS siz2Δ::URA cir0 This studyX3201-6D RAD1-TAP::HIS mms21-11::HIS cir0 This studyX3580-5C Mat alpha RAD1-TAP::HIS siz1Δ::KAN
siz2Δ::URA cir0This study
X3401-1C Mat alpha rad1Δ::LEU This studyX3840-6B rad1-K32R-TAP::HIS This studyX5692-7A RAD1-TAP::HIS apn2Δ::HIS This studyX3919-2D rad1-K32R-TAP::HIS apn2Δ::HIS This studyX6004-1B rad1Δ::LEU apn2Δ::HIS This studyX4373-17A RAD1-TAP::HIS tdp1Δ::KAN This studyX3916-1C rad1-K32R-TAP::HIS tdp1Δ::KAN This studyX3404-2D rad1Δ::LEU tdp1Δ::HIS This studyX3727-1A RAD1-TAP::HIS RAD10-13Myc::KAN This studyX3840-5C rad1-K32R-TAP::HIS RAD10-13Myc::KAN This studyX3729-1A RAD1-TAP::HIS RAD14-13Myc::KAN This studyX4491-5A rad1-K32R-TAP::HIS RAD14-13Myc::KAN This studyX4965-2C RAD1-TAP::HIS SAW1-FLAG::KAN This studyX4966-9A rad1-K32R-TAP::HIS SAW1-FLAG::KAN This studyX4489-1C RAD1-TAP::HIS SLX4-3HA::KAN This studyX4490-1B rad1-K32R-TAP::HIS SLX4-3HA::KAN This studyX3728-1A RAD10-13Myc::KAN This studyX4491-3B RAD14-13Myc::KAN This studyX4965-2B SAW1-FLAG::KAN This studyX4489-1B SLX4-3HA::KAN This studySLY5151 EAY1141 ho HML mat::leu2::hisG hmrΔ3
leu2-3,112 ura3-52 trp1THR4-ura3-A(205bp)-HOcs-URA3-Aade3::GAL10-HO::NAT
This study
SLY5136 SLY5151 rad1-K32R-3HA::HYG This study
Yeast strains are listed in Table 1. Strains in this study except SSA strains are derivatives of W1588–4C, a RAD5 derivative of W303.Thomas, B.J. and Rothstein, R. (1989) Elevated recombination rates in transcriptionally active DNA. Cell, 56, 619–630.Sugawara, N., Goldfarb, T., Studamire, B., Alani, E. and Haber, J.E. (2004) Heteroduplex rejection during SSA requires Sgs1 helicase and mismatch repairproteins Msh2 and Msh6 but not Pms1. Proc. Natl. Acad. Sci. U. S. A., 101, 9315–9320.
determine the oligomeric status of Rad1-Rad10, a 23 mlSephacryl S400 column was eluted with buffer K containing300 mM KCl (0.11 ml/min flow rate), and 0.35 ml fractionswere collected. The indicated fractions were separated bySDS-PAGE and detected by western blotting using �-Rad1antibody (Santa Cruz).
In vitro sumoylation assay
Purification of GST-Aos1/Uba2, His-Ubc9, His-Flag-Smt3, His-Flag-Smt3-KR, His-Siz1 (1–465) and Siz2 and
the sumoylation assay were performed as previously (53).In brief, the 10 �l reaction contained 150 nM Aos1/Uba2,0.5 �M Ubc9, 4.3 �M Smt3 or Smt3-KR, 0.4 �M Rad1-Rad10, 1 mM adenosine triphosphate (ATP) and buffer S1(100 mM Tris-HCl pH 7.5, 10 mM MgCl2) (53). In the indi-cated cases, 10–100 nM Siz1 or 10–100 nM Siz2 was addedto the reaction.

6396 Nucleic Acids Research, 2014, Vol. 42, No. 10
Figure 1. DNA damage-induced sumoylation of Rad1 occurs upon recruitment to lesion sites. (A) Rad1 sumoylation is induced by UV, MMS and CPT,but not HU treatment. TAP-tagged Rad1 was immunoprecipitated from yeast strains untreated or treated with 100 J/m2 UV, 50 ug/ml CPT, 0.2 M HU or0.2% MMS and was western blotted for SUMO (top) and Protein A (bottom). Note that in the SUMO blot, the lower band representing the unmodifiedform arises from interaction of the nonspecific region of the antibody with the Protein A (ProA) portion of TAP (7). (B–D) Rad1 sumoylation inductionrequires upstream NER and SSA factors. Cells containing Rad1-TAP and indicated mutations were treated with 200 J/m2 UV (B) or 0.3% MMS (C–D)and examined as in (A). (E–G) Hypersumoylation of nuclease dead Rad1-D825A (−nd1) and Rad1-D869A (−nd2) depends on upstream NER and SSAfactors. Cells containing indicated mutations were examined as in (B–C). In (E) and (F), the relative ratios of modified to unmodified forms were quantifiedusing the band intensities from the SUMO blot and are shown below. Different intensities of unmodified and sumoylated Rad1 bands on different blotsare due to variable antibody quality and exposure time.
DNA substrates
All substrates were prepared as described (54). Syntheticoligonucleotides were purchased from VBC Biotech; se-quences are available upon request.
Electrophoretic mobility shift assay (EMSA)
Rad1-Rad10 was incubated with fluorescently labeled DNAsubstrates (4 nM) in 10 �l buffer D (40 mM Tris-HCl, pH7.5, 50 mM KCl, 1 mM DTT and 100 �g/ml bovine serumalbumin) at 37◦C for 10 min. The reactions were stoppedby adding loading buffer (60% glycerol, 10 mM Tris, pH 7.4and 60 mM EDTA). Samples were separated on native poly-acrylamide gel (10%) in 0.5×TBE buffer (40 mM Tris-HCl,20 mM boric acid, 2 mM EDTA, pH 7.5). DNA was visu-alized by a scanner FLA9000 Starion (Fujifilm) and quan-tified using MultiGauge software (Fujifilm).
Nuclease assay
Rad1-Rad10 was incubated with fluorescently labeled DNAsubstrate (4 nM) in 10 �l buffer R (50 mM Tris-HCl, pH 7.5,
1 mM MgCl2, 1 mM DTT and 100 �g/ml bovine serumalbumin). The reaction mixtures were incubated at 30◦Cfor 30 min and deproteinized by adding 0.1% SDS and 500�g/ml of proteinase K for 5 min at 37◦C. Samples were re-solved on a 10% native polyacrylamide gel in TBE buffer.The fluorescent DNA species were visualized as in EMSA.
RESULTS
Rad1 sumoylation is enhanced by DNA damaging agents andrequires Rad1 recruitment to DNA lesions
Recent proteomic and biochemical screens have identifiedRad1 as one of the sumoylation targets during the DNAdamage response (7,8,20). We confirmed Rad1 sumoyla-tion in our W303 strain background using endogenouslyexpressed and functional TAP-tagged Rad1 (Figure 1A,and data not shown). The sumoylated form of Rad1 wasdetected as a single band above the unmodified proteinband on western blots when immunoprecipitated Rad1 wasexamined with anti-SUMO antibody (Figure 1A). Rad1sumoylation was observed under normal growth conditionsand an increase was seen upon exposure to UV, the alkyla-

Nucleic Acids Research, 2014, Vol. 42, No. 10 6397
tion agent MMS and the Top1 poison CPT (Figure 1A andSupplementary Figure S1C). Treatment with the ribonu-cleotide reductase inhibitor HU that is not known to re-quire Rad1-mediated repair had no effect (Figure 1A). Wenote that even upon UV, MMS and CPT exposure, only asmall proportion of Rad1 was sumoylated, as the modifiedRad1 band was barely detectable on blots probed with anti-tag antibody after short exposures. We conclude that a smallfraction of Rad1 is mono-sumoylated and this modificationis enhanced by UV, MMS and CPT treatment.
The low level of Rad1 sumoylation and its DNA dam-age inducibility raised the possibility that Rad1 is sumoy-lated only in specific situations, such as when the proteinis actively engaged in DNA repair. We addressed this pos-sibility by examining whether Rad1 sumoylation dependedon its recruitment to lesions. Under UV treatment, Rad14recruits Rad1-Rad10 to DNA lesions by physical interac-tion (38). We found that rad14Δ greatly reduced the level ofRad1 sumoylation (Figure 1B). We also examined whetherSaw1, a Rad1 recruitment factor in SSA (46,47), influencesits sumoylation. Since SSA mutants are most sensitive toMMS treatment, we conducted the test in MMS and foundthat saw1Δ also reduced Rad1 sumoylation (Figure 1C).Residual sumoylation in rad14Δ and saw1Δ cells likely re-flects Rad1 recruitment to DNA lesions by the remainingfactor. Indeed, rad14Δ saw1Δ double mutants exhibitedfurther reduction of Rad1 sumoylation than either singlemutant (Figure 1D).
As Rad14 and Saw1 both recruit and form complexeswith Rad1-Rad10, the observed Rad1 sumoylation decreasecould be due to either impaired recruitment or impairedcomplex formation in their absence. To discern if Rad1-Rad10 recruitment per se is important for Rad1 sumoyla-tion, we examined more upstream NER and SSA proteinsthat are necessary for Rad1-Rad10 recruitment but do notphysically interact with the nuclease. In NER, Rad1 recruit-ment to UV lesions requires the lesion recognition factorRad4, which does not bind to Rad1-Rad10 (55). We foundthat rad4Δ, like rad14Δ, reduced Rad1 sumoylation (Fig-ure 1B), suggesting that it is the presence of Rad1 at DNAlesions that is essential for its sumoylation. Similarly, in mu-tants of the SSA factor Rad52, which is required for forma-tion of 3′ flaps and Rad1 recruitment but does not phys-ically interact with Rad1 (44), we also detected reducedRad1 sumoylation, as in saw1Δ cells (Figure 1D). Also as insaw1Δ cells, the residual sumoylation in rad52Δ was largelydependent on Rad14, as the rad52Δ rad14Δ double mu-tant showed a further decrease in Rad1 sumoylation (Fig-ure 1D), consistent with both NER and SSA contributingto the repair of MMS lesions. Taken together, the reductionin Rad1 sumoylation in mutants lacking two NER proteins,Rad4 and Rad14, and two SSA proteins, Saw1 and Rad52,suggests that the initiation of repair and subsequent recruit-ment of Rad1-Rad10 to DNA lesions are required for Rad1sumoylation.
Mutations blocking Rad1-mediated cleavage affect Rad1sumoylation
The above results suggest that Rad1 sumoylation occursafter it is recruited to DNA lesions. Next we addressed
whether DNA lesion recruitment is sufficient to induceRad1 sumoylation, using the slx4Δ mutant. Slx4 is requiredfor Rad1 nucleolytic cleavage but not its recruitment to3′ flaps during SSA (46,47,56). In slx4Δ cells, Rad1 is re-cruited to SSA lesions but cannot initiate cleavage (46,47).We found that slx4Δ reduced Rad1 sumoylation similarly tosaw1Δ (Figure 1C). This effect suggests that Rad1 sumoy-lation requires an Slx4-dependent step after DNA lesion as-sociation.
To test if nucleolytic cleavage is required for sumoyla-tion, we used two nuclease dead Rad1 mutants. The D825Amutant (Rad1-nd1) lacks a conserved catalytic residue andis trapped on DNA (46), and the D869A mutant (Rad1-nd2) corresponds to D720A in XPF that shows no nu-clease activity in vitro (42,57). Interestingly, both catalyticsite mutations increased Rad1 sumoylation, as relative lev-els of SUMO-Rad1 over unmodified forms were higher inrad1-nd1 and –nd2 cells than in wild-type (Figure 1E andF). Moreover, both mutants showed additional sumoylatedRad1 species (Figure 1E and F). The increased sumoyla-tion still depended on Rad1 recruitment to DNA lesionsas it was reduced in mutants of upstream NER and SSAfactors, including Rad14, Saw1 and Rad52 (Figure 1G).This result, in conjunction with our other data, suggeststhat Rad1 sumoylation occurs at lesion sites and requires anSlx4-dependent step but not Rad1-Rad10 nucleolytic activ-ity.
Sumoylation of Rad1 after DNA damage is Mec1-independent and requires the Siz ligases
Several aspects of DNA repair are regulated by the DNAdamage checkpoint (58,59). However, we have previouslyshown that the DNA damage-induced sumoylation of nu-merous proteins is checkpoint-independent, indicating thatthis sumoylation response is largely separable from thephosphorylation-based checkpoint pathway (7,60). To un-derstand if Rad1 sumoylation follows this rule or is an ex-ception, we analyzed its sumoylation in the absence of themain checkpoint kinase Mec1. As shown in Figure 2A, thelevel of Rad1 sumoylation was the same in mec1Δ cells asin wild-type after treatment with UV or MMS. Thus, weconclude that damage-induced sumoylation of Rad1, as formany other repair proteins, is not dependent on Mec1.
Yeast, like most organisms, contains only one SUMOE2, but multiple mitotic SUMO E3 ligases. Yeast E3 lig-ases include the homologous Siz1 and Siz2 proteins, andthe more distant Mms21. The Siz enzymes, and sometimesMms21, show redundancy in sumoylation of various sub-strates (61,62). To understand whether Rad1 sumoylationis dependent on a particular E3 or can be carried out by re-dundant E3s, we examined single and double E3 mutants invivo. We found that none of the single E3 mutants affectedRad1 sumoylation level (Figure 2B). For combinatorial E3mutants, only the siz1Δ siz2Δ double mutant significantlydecreased Rad1 sumoylation, while combining the siz singlemutations with mms21–11, which lacks the Mms21 ligasedomain, resulted in smaller decreases (Figure 2C and D).The redundancy of the two Siz proteins is consistent withthe observation that the siz1Δ siz2Δ double mutant, butneither single mutant, is sensitive to UV and MMS ((7,20)

6398 Nucleic Acids Research, 2014, Vol. 42, No. 10
Figure 2. Sumoylation induction of Rad1 is dependent on Siz1 and Siz2 but not Mec1. (A) Rad1 sumoylation upon MMS and UV treatment doesnot require Mec1. Rad1-TAP from wild-type or mec1Δ strains was assayed as in Figure 1B and C. (B–D) Siz1 and Siz2 primarily contribute to Rad1sumoylation in vivo. Cells containing Rad1-TAP and indicated mutations were assayed as in Figure 1B and C. (E) Coomassie stain showing purified His-tagged recombinant Rad1-Rad10 and Rad1-K32R-Rad10 complexes. (F) Rad1 sumoylation is stimulated by the Siz1 and Siz2 ligases in vitro. Reactionswere incubated with increasing concentrations of Siz enzymes (10–100 nM) for 1 h at 30◦C and analyzed by 10% SDS–PAGE, followed by western blottingwith antibodies against Rad1 (top) and SUMO (bottom).
and data not shown). Thus, the three E3s are redundant insumoylating Rad1 in vivo, with Siz1 and Siz2 playing promi-nent roles.
To test Siz-dependent sumoylation in vitro, we purifiedrecombinant Rad1-Rad10, as well as SUMO machineryproteins, including SUMO (Smt3), E1 (Aos1/Uba2), E2(Ubc9) and the Siz E3s, as previously described ((53), Fig-ure 2E and data not shown). As shown in Figure 2F, Siz1or Siz2 can efficiently sumoylate Rad1 in the presence ofATP. In both cases, a single Rad1 sumoylation band wasdetected by antibodies against Rad1 or SUMO on west-ern blots. These observations are consistent with our in vivofindings, indicating that Rad1 can be sumoylated by bothSiz ligases. We note that in vitro sumoylation requirementsare more relaxed than in vivo, likely due to the high con-centration of sumoylation machinery components (63–65).Consistent with this, the presence of DNA and/or Saw1 didnot appear to affect Rad1 sumoylation (data not shown).
Rad1 sumoylation occurs on a single lysine in vivo and in vitro
Despite the less stringent requirements for Rad1 sumoyla-tion in vitro, the similar sumoylation patterns and the sameSiz E3 dependence in vitro and in vivo suggested that invitro sumoylated Rad1 can be informative for determin-ing the sumoylation site using mass spectrometry. This ex-periment identified lysine 32 as the sumoylation site withhigh confidence (P < 0.05, Supplementary Figure S2). K32lies within a sumoylation consensus motif (� KxE/D, where� is a hydrophobic amino acid, (66,67)). This residue inthe N-terminal region of the protein is far from Rad1’snuclease domain and the helix–hairpin–helix (HhH) do-main involved in interaction with Rad10 and DNA (Fig-ure 3A). The Rad1 N-terminal domain has not been stud-ied in detail, though the same region in the human ho-molog, XPF, contributes to DNA interaction (68). Thus, theRad1 sumoylation site localizes to a region likely involvedin DNA binding.
To verify that K32 is indeed the bona fide sumoylated ly-sine, we first purified a recombinant K32R mutant of theRad1 protein and subjected it to in vitro sumoylation. Asshown in Figure 3B, wild-type Rad1, but not the K32R mu-tant, can be sumoylated in the presence of ATP. We thentested if K32R also abolishes sumoylation in vivo, by in-troducing the K32R mutation at the endogenous RAD1 lo-cus and assaying sumoylation. rad1-K32R eliminated Rad1sumoylation under both UV and MMS conditions (Figure3C), confirming that K32 is the Rad1 sumoylation site invivo.
The rad1-K32R mutant shows sensitivity to high doses of UVand CPT, but is proficient for repair of a single DNA break
Next, we examined how lack of Rad1 sumoylation affectscell survival in the presence of genotoxins. We first queriedthe UV sensitivity of cells expressing the nonsumoylatablerad1-K32R allele using both spotting and plate-out assays.In both assays, blocking Rad1 sumoylation decreased re-sistance to high doses of UV (Figure 3D and E), with anestimated reduction of ∼20% in the survival of rad1-K32Rcells compared to wild-type cells (P < 0.05).
We did not detect increased sensitivity of rad1-K32Rto other types of DNA damaging agents, including CPT,MMS and HU (data not shown). As Rad1 acts in a back-up pathway for CPT repair and its sumoylation is inducedby CPT treatment ((42); Figure 1A), we examined if rad1-K32R affects resistance to this drug when other proteins in-volved in this repair are absent. Like rad1Δ but to a less de-gree, rad1-K32R sensitized cells lacking Tdp1, a phosphodi-esterase that processes the majority of CPT lesions ((42,69);Figure 3F). As reported previously, rad1Δ did not sensitizecells lacking Apn2, an endonuclease that functions in baseexcision and CPT repair, as well as in processing blockedDNA ends with Rad1 (43,70,71); unexpectedly, rad1-K32Rshowed sensitization (Figure 3F). It is possible that un-sumoylated Rad1 prevents downstream or other repair in

Nucleic Acids Research, 2014, Vol. 42, No. 10 6399
Figure 3. Rad1 is monosumoylated at K32 and its sumoylation contributes to UV and CPT repair. (A) Schematic depicting Rad1 domains and sumoylationsite. HhH denotes helix–hairpin–helix. (B) Rad1-K32R is not sumoylated in vitro. Proteins purified as in Figure 2E were assayed for sumoylation as inFigure 2F. (C)rad1-K32R abolishes Rad1 sumoylation in vivo. Rad1-TAP from wild-type or rad1-K32R cells was assayed as in Figure 1B and C. (D andE)rad1-K32R is sensitive to UV at high doses. Spot assay showing 3-fold serial dilutions of yeast strains either untreated or treated with indicated UV doses(D). Survival curves after exposure to the indicated UV doses is presented based on 5 independent trials; averages and standard deviations are shown (E).Asterisks denote statistically significant differences. (F)rad1-K32R sensitizes tdp1Δ and apn2Δ to CPT. Spot assay showing 10-fold serial dilutions of yeaststrains on indicated media. (G and H)rad1-K32R is not defective in chromosomal (G) and plasmid-based (H) SSA assays. Assays measuring SSA between205 bp ura3 repeats on yeast chromosome V (G) or plasmid pNSU208 carrying 240 bp of directly repeated LacZ sequences (H). Survival of wild-type andrad1-K32R derivative upon HO induction is shown.
the absence of Apn2 (see Discussion). In both tdp1Δ andapn2Δ backgrounds, rad1-K32R sensitization was observedonly at high CPT doses (Figure 3F).
The manifestation of rad1-K32R’s effects at high drugdoses suggests that sumoylation influences aspects of Rad1function that become more critical when the lesion burden islarge. To test this idea, we examined how rad1-K32R affectsthe repair of a single double-strand break via SSA. Rad1-Rad10 cleaves at the ds-ssDNA junction in this process asin NER and CPT repair. If the above idea were correct,one would expect that rad1-K32R is proficient for repair-ing a single break. We used two well-established SSA assayswhere a double-strand break is generated either on a plas-mid or at a chromosomal locus by the HO endonuclease(46). As the HO cut site is flanked by two complementarysequences, repair of the break is mediated by SSA. We foundthat rad1-K32R was proficient for SSA repair of both plas-mid and chromosomal breaks, based on survival rates afterHO induction (Figure 3G-H). Taken together, the pheno-type of rad1-K32R supports the notion that Rad1 sumoyla-tion becomes more important when large numbers of DNAlesions need to be repaired.
Lack of Rad1 sumoylation affects neither its protein interac-tions nor its nuclease activity
To gain a molecular understanding of the effect of Rad1sumoylation and of the reason underlying the rad1-K32R
phenotype, we examined Rad1 protein properties in severalways. First, rad1-K32R did not affect Rad1 protein levelsunder normal and genotoxin conditions (Figure 4A; datanot shown). Second, Rad1-K32R behaved similarly to wild-type Rad1 in DNA binding assays (Supplementary FigureS3A and B). Third, rad1-K32R did not affect interactionwith Rad10, as wild-type and mutant Rad1 proteins exhib-ited similar levels of Rad10 association in vitro and in vivo(Figures 2E and 4B). Additionally, the gel filtration pro-files of unmodified and sumoylated Rad1 in complex withRad10 were similar, with sumoylated Rad1 being eluted inthe same fractions as the unmodified protein (Figure 4C).Thus, we conclude that the sumoylation status of Rad1 doesnot affect Rad10 interaction. We note that the gel filtrationprofiles of Rad1-Rad10 proteins are consistent with a dimerof heterodimers with an apparent molecular mass of 300kDa (Figure 4C), though further work is necessary to gaina detailed understanding of this potential oligomerization.
Next, we examined Rad1 protein interactions importantfor its recruitment and/or nuclease activity. First, we testedRad1 interaction with Rad14 after UV treatment, as NER iscritical for UV repair. No difference was seen between Rad1and Rad1-K32R in their ability to pull down Rad14 pro-tein after UV treatment (Figure 4D). Similar results wereobtained when wild-type Rad1 and the K32R mutant wereassessed for Saw1 or Slx4 interactions (Figure 4E and F).Note that the Slx4 band shift after MMS treatment was seenas previously reported and is due to phosphorylation of the

6400 Nucleic Acids Research, 2014, Vol. 42, No. 10
Figure 4. Rad1 sumoylation affects neither Rad1 protein levels nor its protein–protein interactions. (A)rad1-K32R does not affect Rad1 or Rad10 proteinlevels. Rad1-TAP and Rad10-Myc protein levels in crude extracts from wild-type and rad1-K32R mutant cells were assayed by western blotting withantibodies against corresponding tags. (B, D–F) Rad1-K32R is proficient for interaction with Rad10, Rad14, Saw1 and Slx4 in vivo. Extracts from cellscontaining indicated tagged proteins treated with MMS or UV were immunoprecipitated with IgG-Sepharose to pull down Rad1-TAP, and probed bywestern blot using antibodies against either the corresponding tag or Saw1. Note that in (D), the faint Rad14 band present in the untagged Rad1 sample(first lane) is due to nonspecific binding of Rad14-Myc to the resin. The Rad14-Rad1 interaction is manifested in the increased amount of Rad14 pulleddown from Rad1-TAP compared with untagged Rad1. (C) Sumoylated Rad1 has the same gel filtration profile as unmodified protein. Recombinant Rad1-Rad10 was assayed by gel filtration before and after being subjected to sumoylation reaction. Molecular markers are indicated below the gel filtrationprofiles.
protein (72). The reason for lower levels of Slx4 pulled downafter MMS treatment is unclear, though it could be due toeither reduced Slx4-Rad1 interaction or less efficient detec-tion. Nevertheless, the Rad1-K32R mutant behaved simi-larly to the wild-type protein in its ability to interact withSlx4 in this assay (Figure 4F). Taken together, we concludethat Rad1-K32R is proficient for interactions with Rad14,Saw1 and Slx4, suggesting that sumoylation of Rad1 doesnot cause major changes in protein–protein interactions.
To test the effect of sumoylation on Rad1 nuclease ac-tivity, we compared the activity of equal amounts of wild-type Rad1-Rad10 with the K32R variant complex on Y-form substrates. The fluorescently labeled DNA cleavageproduct migrated faster than the reactant on gels and themutant complex exhibited activity similar to its wild-typecounterpart (Supplementary Figure S3C). We also opti-mized our in vitro sumoylation system such that ∼80% ofRad1 was sumoylated (Figure 5A). When we comparedequal amounts of sumoylated and unmodified Rad1 in thesecleavage assays, again, no difference was detected (Figure5B). As control reactions that contain only the sumoylationreaction proteins did not show any nuclease activity (Sup-plementary Figure S4A), the cleaved product was producedby sumoylated Rad1, rather than other components of thesumoylation reaction. We note that the presence of 20% un-modified Rad1 may exclude the detection of small effects ofsumoylation. Together, these results show that neither thelack of Rad1 sumoylation nor using a protein prep contain-ing predominantly sumoylated Rad1 affects nuclease activ-ity.
Sumoylation of Rad1 decreases its affinity for dsDNA andY-forms
Our in vivo data suggest that Rad1 sumoylation occurs af-ter DNA lesion recruitment and that the modification be-comes more important when dealing with large numbers ofDNA lesions. Both observations suggest that sumoylationmay aid the release of Rad1 from the product after nucleasecleavage. Thus, we examined whether sumoylation of Rad1favors DNA dissociation.
We tested the interaction of unmodified and sumoylatedRad1-Rad10 with both Y-form DNA and dsDNA, whichmimic the substrate and product of the nucleolysis reac-tion, respectively. We found that unmodified Rad1-Rad10binds to both dsDNA and Y-forms (Figure 5C and D). Al-though Rad1-Rad10 is thought to be released from DNAafter cleavage simply by intrinsic differences in the enzyme’saffinity for different forms of DNA, we found that Rad1-Rad10 interaction with dsDNA is only slightly weaker thanthat with Y-forms. This result suggests that additional regu-lation exists to enable efficient dissociation of Rad1-Rad10from dsDNA. Consistent with this, the excised oligomer inNER is released at a faster rate in vivo than in vitro (73), im-plying the existence of regulatory mechanisms specificallyfor achieving shorter turnaround times in NER in vivo.
When sumoylated Rad1 obtained as above was assayed,we detected a reproducible decrease in affinity for both Y-forms and dsDNA, compared with unmodified Rad1 (Fig-ure 5C–E). This is not due to the sumoylation reaction com-ponents, as this mixture did not show DNA association(Supplementary Figure S4B and C). In addition, the effectof sumoylated Rad1 is not mimicked by Rad1-K32R-Rad10in combination with free SUMO (Supplementary FigureS4D and E), suggesting that only the Rad1-conjugated form

Nucleic Acids Research, 2014, Vol. 42, No. 10 6401
Figure 5. Rad1 sumoylation leads to decreased affinity for DNA but does not affect nuclease activity. (A) Rad1 sumoylation reaction in vitro. s.p. denotessumoylation reaction mixture without Rad1-Rad10. (B) Sumoylated Rad1 complexed with Rad10 has the same nuclease activity on Y-forms as the unmodi-fied complex. Increasing concentrations of sumoylated (lanes 6–9) and nonsumoylated (lanes 2–5) Rad1-Rad10 proteins (0.06–1.2 nM) were incubated withY-form DNA (4 nM) and assayed as described in Methods. (C–E) Sumoylated Rad1 complexed with Rad10 exhibit decreased affinity for Y-forms (C) anddsDNA (D). Increasing concentrations of sumoylated (lanes 6–9) and nonsumoylated (lanes 2–5) Rad1–10 proteins (5–50 nM) were tested. Quantificationof several binding trials is shown in (E); Rad1–10 denotes Rad1-Rad10. Asterisks indicate statistically significant differences. (F) Model: Sumoylation ofRad1 promotes dissociation of the Rad1-Rad10 complex from the cleavage product. This effect likely occurs in NER (depicted), CPT and SSA repair (notshown). Details in Discussion.Discussion.
of SUMO exerts the observed effect on DNA association.We also found that sumoylated Rad1 exhibited less bind-ing to 5′ overhang, but not ssDNA, compared to unmodi-fied Rad1 (Supplementary Figure S4F–H). It is conceivablethat the observed sumoylation-mediated reductions in affin-ity for the product of the cleavage reaction enable efficientdisengagement of Rad1-Rad10 from DNA postcleavage.
DISCUSSION
The Rad1-Rad10 nuclease and homologs are involved inmultiple DNA repair pathways and are tightly regulatedat the levels of recruitment and activation. Despite the ad-vances in understanding the regulation of this importanttype of nucleases, the full picture of how they can efficientlyattend to large numbers of DNA lesions via multiple path-ways has been unclear. Here, we show that budding yeastRad1 is modified by SUMO after recruitment to lesionsites. Lack of Rad1 sumoylation leads to cellular sensitivityto high doses of DNA damage. These results suggest thatsumoylation may affect Rad1 function by enabling efficientrecycling of the enzyme. Our biochemical studies provide di-rect evidence that sumoylation of Rad1 reduces its associa-tion with DNA but does not affect other protein properties.Together, these findings suggest a new regulatory mode forRad1-Rad10 whereby sumoylation promotes Rad1’s DNAdissociation.
Several lines of evidence support the idea that Rad1sumoylation occurs at DNA lesion sites. First, only a smallfraction of Rad1 is sumoylated and only DNA damaging
agents that elicit Rad1-mediated repair enhance its sumoy-lation (Figure 1A). These results hint that the timing ofRad1 sumoylation is strictly regulated. Second, and impor-tantly, sumoylation of Rad1 largely depends on proteinsthat recruit it to DNA lesion sites (Figure 1B–D). Thesedata strongly suggest that Rad1 needs to be present at sitesof damage in order to be sumoylated. Third, Rad1 sumoy-lation exhibited a strong dependence on Slx4 (Figure 1C),which affects the nucleolysis step after Rad1 recruitment.This further delineates the timing of Rad1 sumoylation toan Slx4-dependent step after lesion recruitment. Finally,nuclease-dead Rad1 that is trapped at lesion sites is hyper-sumoylated (Figure 1E and F), suggesting that sumoylationdoes not require catalysis per se but rather increases as theinactive protein associates longer with DNA.
Rad1 sumoylation requires either of the two homologousSiz SUMO ligases in vivo or in vitro (Figure 2). The lack ofspecific ligase could mean that the SUMO E2 plays impor-tant roles in determining the modification site. Mass spec-trometry identification of the sumoylation site, as well as invitro and in vivo confirmation (Figure 3B and C and Sup-plementary Figure S2), show that Rad1 is sumoylated atK32, which is within the E2 substrate-recognition site (67).The K32 residue is located outside the Rad1 nuclease andRad10-binding domains, but is within a domain implicatedin DNA association in XPF (68). This location fits with theproposed effect of sumoylation (see below).
The nonsumoylatable rad1-K32R mutant exhibited mod-erate UV sensitivity only at high doses (Figure 3D and E),suggesting that sumoylation of Rad1 may be particularly

6402 Nucleic Acids Research, 2014, Vol. 42, No. 10
useful when large numbers of lesions are present. Consistentwith this notion, rad1-K32R sensitized tdp1Δ and apn2Δ athigh doses of CPT (Figure 3F). This phenotype bias towardlarge lesion loads is in line with normal repair of a singleDNA break in rad1-K32R cells (Figure 3G and H). A sim-ple explanation of the phenotype is that high lesion loadsdemand the efficient recycling of the enzyme, a feature notso important for the repair of a single lesion.
Our biochemical results show that while sumoylationdoes not alter Rad1 protein levels, protein interactions ornuclease activity, it causes a decrease in DNA binding affin-ity (Figures 4 and 5), consistent with the location of themodified lysine in a putative DNA association domain. In-tegrating these biochemical findings with our other results,a plausible model is that sumoylation facilitates the disen-gagement of Rad1 from DNA after nuclease cleavage (Fig-ure 5F). Although sumoylation of Rad1 reduced bindingto both dsDNA (product) and Y-forms (substrate) in vitro,only the former likely has functional consequences in vivo,as Rad14 and Saw1 may dictate Rad1-Rad10 binding toDNA substrates. We envision that alterations occur in thefollowing steps mediated by Slx4, such that Rad1-Rad10can better access the substrate DNA and become amenablefor sumoylation. Subsequently, sumoylation could impactRad1-Rad10’s affinity for the dsDNA product of the reac-tion, facilitating its release, thus contributing to the timelyrepair of large numbers of lesions. Our data do not excludeother possible effects of Rad1 sumoylation in DNA repair.For example, sumoylation could reduce the interaction ofRad1-Rad10 with dsDNA at other regions, thus providinga ‘sweeping’ mechanism to disfavor unproductive DNA as-sociation or impedance of other repair pathways, as sug-gested by the genetic interaction with apn2Δ. As Rad1 isnot an abundant protein but is required for multiple repairpathways, ensuring high turnover may well be importantfor Rad1 function in the cell. Further biochemical work isneeded to elucidate how sumoylation of Rad1 promotes itsdissociation from DNA; for example, sumoylation may in-duce a conformational change in the N-terminal domain ofRad1 leading to decreased DNA binding ability, similar tothe case of sumoylation of TDG (thymine-DNA glycosy-lase), a human base excision repair enzyme (74,75).
The mild effect of sumoylation on Rad1 function seenhere is not an exception, rather it adds to a growing listof cases where sumoylation of DNA repair substrates hasmoderate but functional effects (53,76–81). These accumu-lating findings point to the possibility that sumoylationwields a strong influence on DNA repair by collectively ex-erting small changes on the functions of many proteins. Wethus propose that at least part of the SUMO-dependentDNA damage response is mediated by a ‘group effort’ whereSUMO moderately alters many protein functions all atonce, rather than by a ‘star effect’ hinging on only a few keytargets. These two strategies have different biological impli-cations: the former offers a well-buffered and, therefore, ro-bust system for coping with large numbers of DNA lesions,whereas the latter is more sensitive to perturbations and alsoimplies a waste of cellular resources as more substrates aremodified than necessary. Importantly, we highlight that the‘group effort’ strategy entails diverse functional alterationsof protein-DNA or protein–protein interactions or enzy-
matic activities, and not solely by providing molecular glue.Further studies of additional sumoylation substrates will beable to thoroughly test the above notions.
In summary, our results reveal a new contribution ofsumoylation to achieve efficient DNA repair by targetingRad1, and suggest a new way to regulate nucleases bysumoylation. Considering that XPF, the human homologof Rad1, is sumoylated upon stress (10), and plays multipleimportant roles in DNA repair, it will be interesting to de-termine whether sumoylation makes similar functional con-tributions in human cells.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGMENTS
We thank Isabel Lam for her initial work in identifying thesumoylation site on Rad1. We thank current and former labmembers, especially Inn Chung and Catherine Cremona,for discussions and useful suggestions.
FUNDING
National Institutes of Health (NIH) grant [GM071011to S.E.L.]; Czech Science Foundation [GACR 13–26629S,207/12/2323]; European Regional Development Fund(Project FNUSA-ICRC) [CZ.1.05/1.1.00/02.0123 to L.K.];‘Employment of Newly Graduated Doctors of Sciencefor Scientific Excellence’ (CZ.1.07/2.3.00/30.0009) co-financed from European Social Fund [to V.A.]; NIH grant[GM080670]; American Cancer Society grant [RSG-12-013-01-CCG]; Leukemia and Lymphoma Society ScholarAward [to X.Z.]. Funding for open access charge: NIH.Conflict of interest statement. None declared.
REFERENCES1. Ciccia,A., McDonald,N. and West,S.C. (2008) Structural and
functional relationships of the XPF/MUS81 family of proteins.Annu. Rev. Biochem., 77, 259–287.
2. Schwartz,E.K. and Heyer,W.D. (2011) Processing of joint moleculeintermediates by structure-selective endonucleases duringhomologous recombination in eukaryotes. Chromosoma, 120,109–127.
3. Rouse,J. (2009) Control of genome stability by SLX proteincomplexes. Biochem. Soc. Trans., 37, 495–510.
4. Zheng,L., Jia,J., Finger,L.D., Guo,Z., Zer,C. and Shen,B. (2011)Functional regulation of FEN1 nuclease and its link to cancer.Nucleic Acids Res., 39, 781–794.
5. Chen,S.H., Albuquerque,C.P., Liang,J., Suhandynata,R.T. andZhou,H. (2010) A proteome-wide analysis of kinase-substratenetwork in the DNA damage response. J. Biol. Chem., 285,12803–12812.
6. Emanuele,M.J., Elia,A.E., Xu,Q., Thoma,C.R., Izhar,L., Leng,Y.,Guo,A., Chen,Y.N., Rush,J., Hsu,P.W. et al. (2011) Globalidentification of modular cullin-RING ligase substrates. Cell, 147,459–474.
7. Cremona,C.A., Sarangi,P., Yang,Y., Hang,L.E., Rahman,S. andZhao,X. (2012) Extensive DNA damage-induced sumoylationcontributes to replication and repair and acts in addition to the Mec1checkpoint. Mol. Cell, 45, 422–432.
8. Psakhye,I. and Jentsch,S. (2012) Protein group modification andsynergy in the SUMO pathway as exemplified in DNA repair. Cell,151, 807–820.

Nucleic Acids Research, 2014, Vol. 42, No. 10 6403
9. Blomster,H.A., Hietakangas,V., Wu,J., Kouvonen,P., Hautaniemi,S.and Sistonen,L. (2009) Novel proteomics strategy brings insight intothe prevalence of SUMO-2 target sites. Mol. Cell Proteomics, 8,1382–1390.
10. Golebiowski,F., Matic,I., Tatham,M.H., Cole,C., Yin,Y.,Nakamura,A., Cox,J., Barton,G.J., Mann,M. and Hay,R.T. (2009)System-wide changes to SUMO modifications in response to heatshock. Sci. Signal, 2, ra24.
11. Guo,Z., Kanjanapangka,J., Liu,N., Liu,S., Liu,C., Wu,Z., Wang,Y.,Loh,T., Kowolik,C., Jamsen,J. et al. (2012) Sequentialposttranslational modifications program FEN1 degradation duringcell-cycle progression. Mol. Cell, 47, 444–456.
12. Chen,X., Niu,H., Chung,W.H., Zhu,Z., Papusha,A., Shim,E.Y.,Lee,S.E., Sung,P. and Ira,G. (2011) Cell cycle regulation of DNAdouble-strand break end resection by Cdk1-dependent Dna2phosphorylation. Nat. Struct. Mol. Biol., 18, 1015–1019.
13. Gallo-Fernandez,M., Saugar,I., Ortiz-Bazan,M.A., Vazquez,M.V.and Tercero,J.A. (2012) Cell cycle-dependent regulation of thenuclease activity of Mus81-Eme1/Mms4. Nucleic Acids Res., 40,8325–8335.
14. Matos,J., Blanco,M.G., Maslen,S., Skehel,J.M. and West,S.C. (2011)Regulatory control of the resolution of DNA recombinationintermediates during meiosis and mitosis. Cell, 147, 158–172.
15. Szakal,B. and Branzei,D. (2013) Premature Cdk1/Cdc5/Mus81pathway activation induces aberrant replication and deleteriouscrossover. EMBO J., 32, 1155–1167.
16. Fu,Q., Chow,J., Bernstein,K.A., Makharashvili,N., Arora,S.,Lee,C.F., Person,M.D., Rothstein,R. and Paull,T.T. (2014)Phosphorylation-regulated transitions in an oligomeric state controlthe activity of the Sae2 DNA repair enzyme. Mol. Cell Biol., 34,778–793.
17. Baroni,E., Viscardi,V., Cartagena-Lirola,H., Lucchini,G. andLonghese,M.P. (2004) The functions of budding yeast Sae2 in theDNA damage response require Mec1- and Tel1-dependentphosphorylation. Mol. Cell Biol., 24, 4151–4165.
18. Huertas,P., Cortes-Ledesma,F., Sartori,A.A., Aguilera,A. andJackson,S.P. (2008) CDK targets Sae2 to control DNA-end resectionand homologous recombination. Nature, 455, 689–692.
19. D’Amours,D. and Jackson,S.P. (2001) The yeast Xrs2 complexfunctions in S phase checkpoint regulation. Genes Dev., 15,2238–2249.
20. Silver,H.R., Nissley,J.A., Reed,S.H., Hou,Y.M. and Johnson,E.S.(2011) A role for SUMO in nucleotide excision repair. DNA Repair10, 1243–1251.
21. Morris,J.R., Boutell,C., Keppler,M., Densham,R., Weekes,D.,Alamshah,A., Butler,L., Galanty,Y., Pangon,L., Kiuchi,T. et al.(2009) The SUMO modification pathway is involved in the BRCA1response to genotoxic stress. Nature, 462, 886–890.
22. Zhao,X. and Blobel,G. (2005) A SUMO ligase is part of a nuclearmultiprotein complex that affects DNA repair and chromosomalorganization. Proc. Natl. Acad. Sci. U.S.A., 102, 4777–4782.
23. Galanty,Y., Belotserkovskaya,R., Coates,J., Polo,S., Miller,K.M. andJackson,S.P. (2009) Mammalian SUMO E3-ligases PIAS1 and PIAS4promote responses to DNA double-strand breaks. Nature, 462,935–939.
24. Branzei,D., Sollier,J., Liberi,G., Zhao,X., Maeda,D., Seki,M.,Enomoto,T., Ohta,K. and Foiani,M. (2006) Ubc9- andMms21-mediated sumoylation counteracts recombinogenic events atdamaged replication forks. Cell, 127, 509–522.
25. Maeda,D., Seki,M., Onoda,F., Branzei,D., Kawabe,Y. andEnomoto,T. (2004) Ubc9 is required for damage-tolerance anddamage-induced interchromosomal homologous recombination in S.cerevisiae. DNA Repair 3, 335–341.
26. Bardwell,A.J., Bardwell,L., Tomkinson,A.E. and Friedberg,E.C.(1994) Specific cleavage of model recombination and repairintermediates by the yeast Rad1-Rad10 DNA endonuclease. Science,265, 2082–2085.
27. Davies,A.A., Friedberg,E.C., Tomkinson,A.E., Wood,R.D. andWest,S.C. (1995) Role of the Rad1 and Rad10 proteins in nucleotideexcision repair and recombination. J. Biol. Chem., 270, 24638–24641.
28. Sung,P., Reynolds,P., Prakash,L. and Prakash,S. (1993) Purificationand characterization of the Saccharomyces cerevisiae Rad1/Rad10endonuclease. J. Biol. Chem., 268, 26391–26399.
29. Scharer,O.D. (2013) Nucleotide excision repair in eukaryotes. ColdSpring Harb. Perspect. Biol., 5, a012609.
30. Kirschner,K. and Melton,D.W. (2010) Multiple roles of theERCC1-XPF endonuclease in DNA repair and resistance toanticancer drugs. Anticancer Res., 30, 3223–3232.
31. Gregg,S.Q., Robinson,A.R. and Niedernhofer,L.J. (2011)Physiological consequences of defects in ERCC1-XPF DNA repairendonuclease. DNA Repair 10, 781–791.
32. Sijbers,A.M., de Laat,W.L., Ariza,R.R., Biggerstaff,M., Wei,Y.F.,Moggs,J.G., Carter,K.C., Shell,B.K., Evans,E., de Jong,M.C. et al.(1996) Xeroderma pigmentosum group F caused by a defect in astructure-specific DNA repair endonuclease. Cell, 86, 811–822.
33. Batty,D.P. and Wood,R.D. (2000) Damage recognition in nucleotideexcision repair of DNA. Gene, 241, 193–204.
34. Guzder,S.N., Sung,P., Prakash,L. and Prakash,S. (1998) Affinity ofyeast nucleotide excision repair factor 2, consisting of the Rad4 andRad23 proteins, for ultraviolet damaged DNA. J. Biol. Chem., 273,31541–31546.
35. Jansen,L.E., Verhage,R.A. and Brouwer,J. (1998) Preferential bindingof yeast Rad4.Rad23 complex to damaged DNA. J. Biol. Chem., 273,33111–33114.
36. Sung,P., Guzder,S.N., Prakash,L. and Prakash,S. (1996)Reconstitution of TFIIH and requirement of its DNA helicasesubunits, Rad3 and Rad25, in the incision step of nucleotide excisionrepair. J. Biol. Chem., 271, 10821–10826.
37. Evans,E., Moggs,J.G., Hwang,J.R., Egly,J.M. and Wood,R.D. (1997)Mechanism of open complex and dual incision formation by humannucleotide excision repair factors. EMBO J., 16, 6559–6573.
38. Guzder,S.N., Sommers,C.H., Prakash,L. and Prakash,S. (2006)Complex formation with damage recognition protein Rad14 isessential for Saccharomyces cerevisiae Rad1-Rad10 nuclease toperform its function in nucleotide excision repair in vivo. Mol. CellBiol., 26, 1135–1141.
39. Mardiros,A., Benoun,J.M., Haughton,R., Baxter,K., Kelson,E.P. andFischhaber,P.L. (2011) Rad10-YFP focus induction in response toUV depends on Rad14 in yeast. Acta Histochem., 113, 409–415.
40. Guzder,S.N., Habraken,Y., Sung,P., Prakash,L. and Prakash,S.(1995) Reconstitution of yeast nucleotide excision repair with purifiedRad proteins, replication protein A, and transcription factor TFIIH.J. Biol. Chem., 270, 12973–12976.
41. Habraken,Y., Sung,P., Prakash,S. and Prakash,L. (1996)Transcription factor TFIIH and DNA endonuclease Rad2 constituteyeast nucleotide excision repair factor 3: implications for nucleotideexcision repair and Cockayne syndrome. Proc. Natl. Acad. Sci.U.S.A., 93, 10718–10722.
42. Vance,J.R. and Wilson,T.E. (2002) Yeast Tdp1 and Rad1-Rad10function as redundant pathways for repairing Top1 replicativedamage. Proc. Natl. Acad. Sci. U.S.A., 99, 13669–13674.
43. Guillet,M. and Boiteux,S. (2002) Endogenous DNA abasic sitescause cell death in the absence of Apn1, Apn2 and Rad1/Rad10 inSaccharomyces cerevisiae. EMBO J., 21, 2833–2841.
44. Krogh,B.O. and Symington,L.S. (2004) Recombination proteins inyeast. Annu. Rev. Genet., 38, 233–271.
45. San Filippo,J., Sung,P. and Klein,H. (2008) Mechanism of eukaryotichomologous recombination. Annu. Rev. Biochem., 77, 229–257.
46. Li,F., Dong,J., Pan,X., Oum,J.H., Boeke,J.D. and Lee,S.E. (2008)Microarray-based genetic screen defines SAW1, a gene required forRad1/Rad10-dependent processing of recombination intermediates.Mol. Cell, 30, 325–335.
47. Li,F., Dong,J., Eichmiller,R., Holland,C., Minca,E., Prakash,R.,Sung,P., Yong Shim,E., Surtees,J.A. and Eun Lee,S. (2013) Role ofSaw1 in Rad1/Rad10 complex assembly at recombinationintermediates in budding yeast. EMBO J., 32, 461–472.
48. Toh,G.W., Sugawara,N., Dong,J., Toth,R., Lee,S.E., Haber,J.E. andRouse,J. (2010) Mec1/Tel1-dependent phosphorylation of Slx4stimulates Rad1-Rad10-dependent cleavage of non-homologousDNA tails. DNA Repair 9, 718–726.
49. Burgess,R.C., Rahman,S., Lisby,M., Rothstein,R. and Zhao,X.(2007) The Slx5-Slx8 complex affects sumoylation of DNA repairproteins and negatively regulates recombination. Mol. Cell Biol., 27,6153–6162.
50. Tsalik,E.L. and Gartenberg,M.R. (1998) Curing Saccharomycescerevisiae of the 2 micron plasmid by targeted DNA damage. Yeast,14, 847–852.

6404 Nucleic Acids Research, 2014, Vol. 42, No. 10
51. Hang,L.E., Liu,X., Cheung,I., Yang,Y. and Zhao,X. (2011)SUMOylation regulates telomere length homeostasis by targetingCdc13. Nat. Struct. Mol. Biol., 18, 920–926.
52. Bastin-Shanower,S.A., Fricke,W.M., Mullen,J.R. and Brill,S.J. (2003)The mechanism of Mus81-Mms4 cleavage site selection distinguishesit from the homologous endonuclease Rad1-Rad10. Mol. Cell Biol.,23, 3487–3496.
53. Altmannova,V., Eckert-Boulet,N., Arneric,M., Kolesar,P.,Chaloupkova,R., Damborsky,J., Sung,P., Zhao,X., Lisby,M. andKrejci,L. (2010) Rad52 SUMOylation affects the efficiency of theDNA repair. Nucleic Acids Res., 38, 4708–4721.
54. Matulova,P., Marini,V., Burgess,R.C., Sisakova,A., Kwon,Y.,Rothstein,R., Sung,P. and Krejci,L. (2009) Cooperativity ofMus81.Mms4 with Rad54 in the resolution of recombination andreplication intermediates. J. Biol. Chem., 284, 7733–7745.
55. Prakash,S. and Prakash,L. (2000) Nucleotide excision repair in yeast.Mutat. Res., 451, 13–24.
56. Lyndaker,A.M. and Alani,E. (2009) A tale of tails: insights into thecoordination of 3’ end processing during homologous recombination.Bioessays, 31, 315–321.
57. Enzlin,J.H. and Scharer,O.D. (2002) The active site of the DNArepair endonuclease XPF-ERCC1 forms a highly conserved nucleasemotif. EMBO J., 21, 2045–2053.
58. Friedel,A.M., Pike,B.L. and Gasser,S.M. (2009) ATR/Mec1:coordinating fork stability and repair. Curr. Opin. Cell Biol., 21,237–244.
59. Flott,S., Alabert,C., Toh,G.W., Toth,R., Sugawara,N.,Campbell,D.G., Haber,J.E., Pasero,P. and Rouse,J. (2007)Phosphorylation of Slx4 by Mec1 and Tel1 regulates the single-strandannealing mode of DNA repair in budding yeast. Mol. Cell Biol., 27,6433–6445.
60. Cremona,C.A., Sarangi,P. and Zhao,X. (2012) Sumoylation and theDNA Damage Response. Biomolecules, 2, 376–388.
61. Johnson,E.S. (2004) Protein modification by SUMO. Annu. Rev.Biochem., 73, 355–382.
62. Ulrich,H.D. (2009) The SUMO system: an overview. Methods Mol.Biol., 497, 3–16.
63. Sarge,K.D. and Park-Sarge,O.K. (2009) Detection of proteinssumoylated in vivo and in vitro. Methods Mol. Biol., 590, 265–277.
64. Windecker,H. and Ulrich,H.D. (2008) Architecture and assembly ofpoly-SUMO chains on PCNA in Saccharomyces cerevisiae. J. Mol.Biol., 376, 221–231.
65. Takahashi,Y., Toh,E.A. and Kikuchi,Y. (2003) Comparative analysisof yeast PIAS-type SUMO ligases in vivo and in vitro. J. Biochem.,133, 415–422.
66. Rodriguez,M.S., Dargemont,C. and Hay,R.T. (2001) SUMO-1conjugation in vivo requires both a consensus modification motif andnuclear targeting. J. Biol. Chem., 276, 12654–12659.
67. Sampson,D.A., Wang,M. and Matunis,M.J. (2001) The smallubiquitin-like modifier-1 (SUMO-1) consensus sequence mediatesUbc9 binding and is essential for SUMO-1 modification. J. Biol.Chem., 276, 21664–21669.
68. Bowles,M., Lally,J., Fadden,A.J., Mouilleron,S., Hammonds,T. andMcDonald,N.Q. (2012) Fluorescence-based incision assay for humanXPF-ERCC1 activity identifies important elements of DNA junctionrecognition. Nucleic Acids Res., 40, e101.
69. Nitiss,K.C., Malik,M., He,X., White,S.W. and Nitiss,J.L. (2006)Tyrosyl-DNA phosphodiesterase (Tdp1) participates in the repair ofTop2-mediated DNA damage. Proc. Natl. Acad. Sci. U. S. A., 103,8953–8958.
70. Boiteux,S. and Guillet,M. (2004) Abasic sites in DNA: repair andbiological consequences in Saccharomyces cerevisiae. DNA Repair 3,1–12.
71. Guzder,S.N., Torres-Ramos,C., Johnson,R.E., Haracska,L.,Prakash,L. and Prakash,S. (2004) Requirement of yeast Rad1-Rad10nuclease for the removal of 3’-blocked termini from DNA strandbreaks induced by reactive oxygen species. Genes Dev., 18, 2283–2291.
72. Flott,S. and Rouse,J. (2005) Slx4 becomes phosphorylated after DNAdamage in a Mec1/Tel1-dependent manner and is required for repairof DNA alkylation damage. Biochem J., 391, 325–333.
73. Hu,J., Choi,J.H., Gaddameedhi,S., Kemp,M.G., Reardon,J.T. andSancar,A. (2013) Nucleotide excision repair in human cells: fate ofthe excised oligonucleotide carrying DNA damage in vivo. J. Biol.Chem., 288, 20918–20926.
74. Steinacher,R. and Schar,P. (2005) Functionality of human thymineDNA glycosylase requires SUMO-regulated changes in proteinconformation. Curr. Biol., 15, 616–623.
75. Hardeland,U., Steinacher,R., Jiricny,J. and Schar,P. (2002)Modification of the human thymine-DNA glycosylase byubiquitin-like proteins facilitates enzymatic turnover. EMBO J., 21,1456–1464.
76. Vigasova,D., Sarangi,P., Kolesar,P., Vlasakova,D., Slezakova,Z.,Altmannova,V., Nikulenkov,F., Anrather,D., Gith,R., Zhao,X. et al.(2013) Lif1 SUMOylation and its role in non-homologousend-joining. Nucleic Acids Res., 41, 5341–5353.
77. Chen,X., Ding,B., LeJeune,D., Ruggiero,C. and Li,S. (2009) Rpb1sumoylation in response to UV radiation or transcriptionalimpairment in yeast. PLoS One, 4, e5267.
78. Hoege,C., Pfander,B., Moldovan,G.L., Pyrowolakis,G. and Jentsch,S.(2002) RAD6-dependent DNA repair is linked to modification ofPCNA by ubiquitin and SUMO. Nature, 419, 135–141.
79. Papouli,E., Chen,S., Davies,A.A., Huttner,D., Krejci,L., Sung,P. andUlrich,H.D. (2005) Crosstalk between SUMO and ubiquitin onPCNA is mediated by recruitment of the helicase Srs2p. Mol. Cell,19, 123–133.
80. Hang,L.E., Lopez,C.R., Liu,X., Williams,J.M., Chung,I., Wei,L.,Bertuch,A.A. and Zhao,X. (2014) Regulation of Ku-DNAassociation by Yku70 C-terminal tail and SUMOmodification. J. Biol. Chem., doi:10.1074/jbc.M113.526178; epubahead of print February 24, 2014.
81. Sacher,M., Pfander,B., Hoege,C. and Jentsch,S. (2006) Control ofRad52 recombination activity by double-strand break-inducedSUMO modification. Nat. Cell Biol., 8, 1284–1290.

Article
A Versatile Scaffold Contribu
tes to Damage Survival viaSumoylation and Nuclease InteractionsGraphical Abstract
Highlights
The Saw1 scaffold has multiple roles and copes with diverse
types of DNA lesions
Saw1 assists the Rad1-Rad10 nuclease in a range of DNA dam-
age conditions
Sumoylation of Saw1 facilitates its interaction with another
nuclease Slx1-Slx4
Saw1 sumoylation promotes UV resistance independently of
two repair pathways
Sarangi et al., 2014, Cell Reports 9, 143–152October 9, 2014 ª2014 The Authorshttp://dx.doi.org/10.1016/j.celrep.2014.08.054
Authors
Prabha Sarangi, Veronika Altmannova, ...,
Lumir Krejci, Xiaolan Zhao
[email protected] (L.K.),[email protected] (X.Z.)
In Brief
Scaffold proteins are not DNA repair en-
zymes themselves but make important
contributions to DNA repair by regulating
and coordinating various enzymes with
their DNA substrates. Sarangi et al. reveal
the versatility of the Saw1 scaffold by
identifying how it copes with several
types of DNA damage that depend on its
nuclease interactions and sumoylation.
These findings highlight the diverse
ways in which multifunctional scaffolds
can operate under genotoxic stress and
how this is directed by protein
modification.

Cell Reports
Article
A Versatile Scaffold Contributesto Damage Survival via Sumoylationand Nuclease InteractionsPrabha Sarangi,1,2,9 Veronika Altmannova,3,9 Cory Holland,4,11 Zdenka Bartosova,3 Fanfan Hao,1 Dorothea Anrather,5
Gustav Ammerer,5 Sang Eun Lee,4,6 Lumir Krejci,3,7,8,10,* and Xiaolan Zhao1,2,10,*1Molecular Biology Program, Memorial Sloan-Kettering Cancer Center, New York, NY 10065, USA2Programs in Biochemistry, Cell, and Molecular Biology, Weill Cornell Graduate School of Medical Sciences, New York, NY 10065, USA3Department of Biology, Masaryk University, Brno 62500, Czech Republic4Department of Molecular Medicine, Institute of Biotechnology, University of Texas Health Science Center at San Antonio, San Antonio,TX 78229, USA5Department of Biochemistry and Cell Biology, Max F. Perutz Laboratories, University of Vienna, Vienna 1030, Austria6Division of Radiation Biology, Department of Radiation Oncology, University of Texas Health Science Center at San Antonio, San Antonio,TX 78229, USA7National Centre for Biomolecular Research, Masaryk University, Brno 62500, Czech Republic8International Clinical Research Center, St. Anne’s University Hospital in Brno, Brno 60200, Czech Republic9Co-first author10Co-senior author11Present address: Department of Biochemistry, Vanderbilt University School of Medicine, Nashville, TN 37232, USA
*Correspondence: [email protected] (L.K.), [email protected] (X.Z.)
http://dx.doi.org/10.1016/j.celrep.2014.08.054This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/3.0/).
SUMMARY
DNA repair scaffolds mediate specific DNA and pro-tein interactions in order to assist repair enzymesin recognizing and removing damaged sequences.Many scaffold proteins are dedicated to repairinga particular type of lesion. Here, we show that thebudding yeast Saw1 scaffold is more versatile. Ithelps cells cope with base lesions and protein-DNAadducts through its known function of recruiting theRad1-Rad10 nuclease to DNA. In addition, it pro-motes UV survival via a mechanism mediated by itssumoylation. Saw1 sumoylation favors its interactionwith another nuclease Slx1-Slx4, and this SUMO-mediated role is genetically separable from twomain UV lesion repair processes. These effectsof Saw1 and its sumoylation suggest that Saw1 is amultifunctional scaffold that can facilitate diversetypes of DNA repair through its modification andnuclease interactions.
INTRODUCTION
Timely repair of the large number of DNA lesions occurring in the
genome is critical to prevent mutations and other alterations of
the genetic information. This task requires collaborations be-
tween individual DNA repair enzymes, as well as with scaffold
proteins that aid some of these enzymes. In particular, DNA nu-
cleases that remove damaged sequences from the genome
often carry out their functions in conjunction with scaffold pro-
teins (e.g., Guzder et al., 2006; Hammel et al., 2011; Prolla
et al., 1994; Vidal et al., 2001).
Most repair scaffolds are thought to assist a particular repair
process (Guzder et al., 2006; Hammel et al., 2011; Prolla et al.,
1994; Vidal et al., 2001). The budding yeast scaffold protein
Saw1 was recently shown to support single-strand annealing
(SSA) repair of double-strand breaks (DSBs) (Li et al., 2008,
2013). SSA entails the annealing of resected DNA at repeat
sequences adjacent to the break, the subsequent removal of
nonhomologous flaps, and final ligation (Fishman-Lobell et al.,
1992; reviewed in Heyer et al., 2010; Krogh and Symington,
2004). In SSA, Saw1 recruits the Rad1-Rad10 nuclease to the
break sites for flap removal (Li et al., 2008, 2013). This recruit-
ment requires the coordinated interactions of Saw1 with the
nuclease, the flap DNA, and upstream SSA factors (Li et al.,
2008, 2013). SSA is considered error-prone repair as it leads to
deletions or translocations (Fishman-Lobell et al., 1992; Heyer
et al., 2010; Krogh and Symington, 2004).
Although Saw1 is thought to be an SSA-specific scaffold,
Rad1-Rad10 is involved in processes that repair other types of
DNA lesions (Figure 1A). These include the repair of UV lesions
via the nucleotide excision repair (NER) pathway (reviewed in
Scharer, 2013), as well as backup repair of base lesions and pro-
tein-DNA adducts (Guillet and Boiteux, 2002; Vance and Wilson,
2002). Compared with error-prone SSA repair, these processes
contribute to cellular survival in specific genotoxic environments.
It has not been explored whether Saw1 can aid Rad1-Rad10 in
these repair contexts, nor is it known if Saw1 has Rad1-indepen-
dent roles in DNA repair.
Here, we show that Saw1 promotes survival in different geno-
toxic environments that generate base lesions, protein-DNA ad-
ducts, and UV lesions. Saw1 interactions with Rad1 and DNA
Cell Reports 9, 143–152, October 9, 2014 ª2014 The Authors 143

C
Untreated UV 70 J/m2
WT
saw1
rad1
B
Untreated CPT 15 ug/ml
saw1 tdp1
saw1
tdp1
D
E
rad59
saw1 rad59
saw1Untreated UV 75 J/m2
saw1 apn1 apn2
rad59 apn1 apn2
A Rad1-Rad10-mediated DNA repair processes examined
Double strand breaks (flanked by repeats)
annealing (Rad59)
UV lesions
(Rad7, Rad16 and Rad26)
Bold: the only known Saw1-mediated repair process thus far
Base lesions (enhanced by MMS)
Backup repair in apn cells
Protein-DNA adducts (induced by CPT)
Backup repair in tdp1 cells
saw1 /+ apn1 /+ apn2 /+
rad59 /+ apn1 /+ apn2 /+
Tetrad 1
Tetrad 2
Tetrad 1
Figure 1. Saw1 Promotes Resistance to Multiple Types of DNA Lesions
(A) Summary of Rad1-Rad10-mediated DNA repair processes examined in this study.
(B) saw1D and rad1D cells are sensitive to UV radiation.
(C) saw1D sensitizes rad59D to UV.
(D) apn1D apn2D is synthetically lethal with saw1D but not rad59D. Representative tetrads dissected from diploids with indicated genotypes are shown. Triple
mutants are labeled and spore clones of other genotypes grow similarly.
(E) saw1D enhances the CPT sensitivity of tdp1D cells.
In (B) and (E), 10-fold serial dilutions of cell cultures were spotted and either untreated or treated with the indicated UV dose (B) or on media containing CPT (E). In
(C), 3-fold dilutions were used.
flaps are required in the first two situations, suggesting that
Saw1 assists Rad1-Rad10 in a broader range of DNA damage
contexts than previously appreciated. In contrast, these known
functions of Saw1 are not critical under UV condition, indicating
that Saw1 also has Rad1-independent roles in specific lesion
contexts. To elucidate this previously unknown aspect of
Saw1’s roles, we examined whether it is enabled by alteration
of Saw1 function through protein modification. The only known
modification of Saw1 is sumoylation, as reported by two recent
proteomic screens (Cremona et al., 2012; Psakhye and Jentsch,
2012). We found that this modification is critical for Saw1-medi-
ated UV resistance partly due to collaboration with another DNA
nuclease, Slx1-Slx4. Our findings highlight the versatility of the
Saw1 nuclease scaffold in multiple damage contexts via collab-
orations with different repair factors and also provide an example
whereby sumoylation of a repair scaffold differently regulates its
functions.
RESULTS
Saw1-Mediated UV Resistance Is Separable from ItsSSA FunctionTo understand if Saw1 has broader effects in repairing different
types of DNA lesions beyond its known SSA function, we exam-
ined how cells lacking Saw1 cope with several DNA damaging
144 Cell Reports 9, 143–152, October 9, 2014 ª2014 The Authors
agents. We first examined UV treatment, as the Saw1 binding
partner, the Rad1-Rad10 nuclease, is critical for UV repair via
the NER pathway (reviewed in Scharer, 2013; Figure 1A). We
found that saw1D cells exhibited increased UV sensitivity
compared to wild-type cells (Figure 1B; see Table 1 for strain
list). Because this sensitivity was less severe than that of
rad1D cells (Figure 1B), Saw1 is not the main Rad1 recruitment
factor during UV repair, a notion consistent with the NER protein
Rad14 being mainly responsible for Rad1 recruitment to UV
lesions (Guzder et al., 2006).
Next, we asked whether the newly found UV sensitivity of
saw1D is attributable to defective SSA. To this end, we per-
formed epistasis analysis with mutants lacking Rad59, a protein
essential for SSA (Bai and Symington, 1996) (Figure 1A). We
found that saw1D rad59D cells were more UV sensitive than
rad59D cells (Figure 1C), indicating that the Saw1 contribution
to UV resistance extends beyond SSA.
Saw1 Promotes Survival in Other Damage ConditionsIndependently of SSANext, we examined if saw1D cells exhibit a phenotype indicative
of defects in the repair of other types of DNA damage in which
Rad1-Rad10 plays backup roles (Figure 1A). In the absence of
base excision repair that requires the endonucleases Apn1 and
Apn2, Rad1 becomes essential for cell growth (Boiteux and

Table 1. Yeast Strains Used in This Study
Strain Genotype
W1588-4A MATalpha ade2-1 his3-11,15 leu2-3,112 trp1-1 ura3-1 can1-100 RAD5
X3401-1C MATalpha rad1D::LEU2
T956-1 MATalpha saw1D::KAN
X5318-9B rad59D::LEU2
X5318-11B saw1D::KAN rad59D::LEU2
X5316-1A saw1D::KAN tdp1D::KAN
T958-3 SAW1-TAP::HIS3
X4505-3A SAW1-TAP::HIS3 siz1D::KAN
X4505-5A SAW1-TAP::HIS3 siz2D::URA3
X4506-9A SAW1-TAP::HIS3 mms21-11::HIS3
X4506-9D SAW1-TAP::HIS3 siz1D::KAN mms21-11::HIS3
X4507-1A SAW1-TAP::HIS3 siz2D::URA3 mms21-11::HIS3
X4505-2D SAW1-TAP::HIS3 siz1D::KAN siz2D::URA3
T1490-2 saw1-K221R-TAP::HIS3
X5314-1A saw1-DRBD-TAP::HIS3
X5313-1A saw1-DFBD-TAP::HIS3
X5519-1C SAW1-TAP::HIS3 rad59D::LEU2
X5624-1A saw1-K221R-TAP::HIS3 rad59D::LEU2
SLY5151 ho HML mat::leu2::hisG hmrD3 leu2-3,112 ura3-52 trp1 THR4-ura3-A(205bp)-HOcs-URA3-A ade3::GAL10-HO::NAT
X5638-11B SAW1-TAP::HIS3 apn1D::KAN apn2D::HIS3
X5639-7C saw1-K221R-TAP::HIS3 apn1D::KAN apn2D::HIS3
X5359-9A SAW1-TAP::HIS3 tdp1D::KAN
X5360-5A saw1-K221R-TAP::HIS3 tdp1D::KAN
X5643-2C saw1-DRBD-TAP::HIS3 tdp1D::KAN
X5644-8A saw1-DFBD-TAP::HIS3 tdp1D::KAN
X4965-2D RAD1-TAP::HIS3
X4965-2B SAW1-3FLAG::KAN
X4965-2C RAD1-TAP::HIS3 SAW1-3FLAG::KAN
X4967-6B RAD1-TAP::HIS3 saw1-K221R-3FLAG::KAN
X5536-6A rad55D::KAN
X5535-5A saw1D::KAN rad55D::KAN
X5530-2D rad26D::KAN
X5529-1B saw1D::KAN rad26D::KAN
X5532-11A rad16D::KAN
X5531-10C saw1D::KAN rad16D::KAN
X5559-3B SAW1-TAP::HIS3 rad55D::KAN
X5536-8D saw1-K221R-TAP::HIS3 rad55D::KAN
X5561-1D SAW1-TAP::HIS3 rad26D::KAN
X5530-2A saw1-K221R-TAP::HIS3 rad26D::KAN
X5557-1C SAW1-TAP::HIS3 rad16D::KAN
X5532-10B saw1-K221R-TAP::HIS3 rad16D::KAN
X5900-3C slx1D::KAN
X5899-1B saw1D::KAN slx1D::KAN
X5881-3A slx4D::KAN
X5881-3C saw1D::KAN slx4D::KAN
All strains, except those for assaying SSA, are in the W303 background that has wild-type RAD5, and the full genotype is listed only for W1588-4A
(Chen et al., 2013). Experiments were performed with at least two different spore clones; only one is listed in the table.
Cell Reports 9, 143–152, October 9, 2014 ª2014 The Authors 145

A
B D
Saw1
SUMO-Saw1
Saw1
SUMO
TAP
WT K22
1R
C
+MM
S
+UV
SUMO
TAP
SUMO-Saw1
Saw1
Saw1
SUMO-Saw1
+CP
T
mm
s21
SUMO-Saw1
WT siz1
siz2
siz1
siz
2
siz1
mm
s21
siz2
mm
s21
TAP
SUMO SUMO-Saw1
Saw1
Saw1
55
75 kDa
+ :ATP
SUMO-Saw1
Saw1
+ + + + +
+ + + :Siz2 + + + :Siz1
1 2 3 4 5 6 Lane: 7
Saw1
All reactions contain SUMO, SUMO E1 and E2
Figure 2. Saw1 Monosumoylation In-
creases after DNA Damage Treatment
(A) Saw1 sumoylation level in SUMO ligase mu-
tants. Indicated strains were treated with UV and
immunoprecipitated Saw1-TAP was examined by
western blotting using antibody recognizing TAP
(bottom) and SUMO (top). Note that the modified
form of Saw1 runs �20 kDa higher than the un-
modified form on SDS-PAGE gels, a signature shift
caused by sumoylation.
(B) In vitro sumoylation of Saw1 is stimulated by
the SUMO E3s Siz1 and Siz2. Recombinant Saw1
was subjected to standard in vitro sumoylation
reactions; all lanes have SUMO (Smt3), E1 (Aos1/
Uba2), and E2 (Ubc9). Saw1 sumoylation in the
absence of E3 and presence of ATP is due to
Ubc9-mediated direct conjugation and is further
stimulated by the Siz1 and Siz2 E3s.
(C) Saw1 sumoylation is induced byMMS, UV, and
CPT treatment. Saw1 sumoylation in cells treated
with different DNA damaging agents was exam-
ined as in (A). Note that the increased sumoylation
of Saw1 after DNA damage treatment can be seen
on both blots.
(D) Saw1-K221R is not sumoylated in vivo. Indi-
cated strains were examined for Saw1 sumoyla-
tion after exposure to 100 J/m2 UV.
See also Figure S1.
Guillet, 2004; Guillet and Boiteux, 2002). We found that saw1D
also showed the same genetic interaction with apn1D apn2D
as does rad1D. This finding is consistent with idea that Saw1 is
required for the backup repair of base lesions (Figure 1D). This
function of Saw1 is separable from SSA, because rad59D did
not show similar synthetic lethality (Figure 1D).
The Rad1-Rad10 nuclease also acts in the backup repair of
DNA linked to the topoisomerase Top1 (Figure 1A) (Vance and
Wilson, 2002). Top1-DNA adducts are stabilized by camptothe-
cin (CPT) and are primarily removed by the phosphodiesterase
Tdp1 (Pouliot et al., 1999). In the absence of Tdp1, repair of
Top1-DNA adducts by Rad1-Rad10 becomes critical, because
tdp1D rad1D cells are inviable on CPT-containing media (Vance
and Wilson, 2002). We found that tdp1D saw1D cells were also
inviable when treated with CPT (Figure 1E), suggesting that
Saw1 also contributes to Top1-DNA adduct situations. Again,
this function of Saw1 is unrelated to SSA, because rad59D
does not sensitize tdp1D cells (Vance and Wilson, 2002).
Taken together, the genetic evidence supports SSA-indepen-
dent roles for Saw1 in survival under different DNA damage con-
ditions. Next, we aimed to understand how a scaffold protein
performs these multiple tasks by examining whether posttrans-
lational modification contributes to its diverse functions.
Saw1 Sumoylation Increases upon DNA DamageTreatmentSaw1 was found to be sumoylated in recent proteomic screens
(Cremona et al., 2012; Psakhye and Jentsch, 2012). Consistent
with these reports, a single sumoylated form of Saw1 from immu-
nopurified samples was detected by western blotting using
antibodies against SUMO or the TAP tag fused to the protein
(Figure 2A). We note that as the Fc region of the SUMO antibody
146 Cell Reports 9, 143–152, October 9, 2014 ª2014 The Authors
interacts with the Protein A part of TAP tag, it detects the unmod-
ified protein, but more strongly so for the sumoylated form due to
additional high affinity for SUMO (Cremona et al., 2012). Saw1
sumoylation was also detected in vitro in the presence of
SUMO, sumoylation E1 and E2 enzymes, and ATP (Figure 2B,
lane 2) (Altmannova et al., 2010).
To determine the SUMO E3s responsible for Saw1 sumoyla-
tion, we examined its modification levels in cells lacking function
of the threemitotic E3s, namely, Siz1, Siz2, andMms21 (Johnson
andGupta, 2001; Takahashi et al., 2001; Zhao and Blobel, 2005).
Saw1 sumoylation was reduced in siz1D siz2D and siz1Dmms21
double mutants, but not in siz2D mms21 or single E3 mutants
in vivo (Figure 2A). In vitro, both Siz1 and Siz2 stimulated Saw1
sumoylation (Figure 2B, lanes 3 and 4). Thus, more than one
SUMO ligase contributes to Saw1 sumoylation, making Saw1
yet another redundant E3 substrate (reviewed in Ulrich, 2009).
Because our findings suggest that Saw1 contributes to sur-
vival in the presence of multiple types of lesions, we examined
Saw1 sumoylation under these DNA damage conditions. Saw1
sumoylation was greatly enhanced by treatment with UV, meth-
ylmethane sulfonate (MMS) that generates base lesions, and to a
smaller extent by CPT (Figure 2C). This is in line with a role for
Saw1 sumoylation in the repair of these lesions.
Saw1 Sumoylation Occurs at a Lysine outside Its Rad1and Flap Binding MotifsTo examine whether and how sumoylation affects the different
functions of Saw1, we first mapped its sumoylation site. To
this end, the sumoylated form of recombinant Saw1 was sub-
jected to mass spectrometry analysis. This analysis identified
lysine K221 as a candidate sumoylation site (Figure S1). Replac-
ing this lysine with arginine at the endogenous locus eliminated

A B
C
D
EG
FH
Figure 3. Differential Effects of Saw1 Attributes under Several Damage Situations
(A) Schematic of Saw1 depicting three main features. Motifs required for binding to Rad1 (RBD) and flap DNA (FBD) and sumoylation site (K221) are shown.
(B) saw1-K221R is proficient for SSA repair. Schematic of SSA assay is on the right. saw1-K221R is denoted as saw1-KR here and in other panels. Data from three
trials are represented as mean ± SD.
(C) saw1-K221R behaves like saw1D and is more sensitive to UV than saw1-DRBD and saw1-DFBD. As in Figure 1B, 3-fold serial dilutions were spotted.
(D) saw1-K221R is additive with rad59D for UV sensitivity. As in Figure 1A, 10-fold serial dilutions were spotted.
(E) saw1-DRBD and saw1-DFBD, but not saw1-K221R, are synthetically lethal with apn1D apn2D. Diploids heterozygotic for the indicated mutations were
dissected, and a representative tetrad is shown for each diploid. Triple mutants are labeled.
(F) saw1-DRBD and saw1-DFBD cells exhibit stronger sensitization of tdp1D than saw1-K221R on CPT. As in Figure 1E, 3-fold serial dilutions were spotted. Note
that none of the saw1 mutants shows sensitivity to CPT at this concentration.
(G) saw1-K221R slows apn1D apn2D cell growth and exacerbates its MMS sensitivity.
(H) Schematic depicting the different contributions of the three Saw1 attributes to its functions under diverse DNA damage conditions. Newly found contributions
are in blue. Thicker lines indicate greater contributions.
Saw1 sumoylation in vivo (Figure 2D), confirming that K221 is the
SUMO acceptor site in vivo.
Saw1 is a small proteinwith only twomotifs identified thus far: a
sixaminoacidRad1-bindingmotif at theN terminus (referred toas
RBD), and another six amino acid motif at the C terminus that is
required for 30 flap binding in vitro (referred to as FBD) (Figure 3A)
(Li et al., 2008, 2013). Bothmotifs are absolutely required forRad1
recruitment to 30 flaps in SSA, and thus SSA repair (Li et al., 2008,
2013). Lysine 221 lies outside both motifs and is conserved
among homologs in yeast species (Figure 3A; SGD database).
Saw1-Mediated UV Resistance, but Not SSA, Relies onIts SumoylationWe examined the phenotype of saw1-K221R and compared it
with those of saw1 null or mutants lacking either the Rad1 bind-
ing (saw1-DRBD) or the flap binding (saw1-DFBD) motifs. First,
SSA efficiency was examined using an assay where the HO
endonuclease-induced DSB is flanked by direct repeats (Li
et al., 2008). Repair of this DSB is primarily mediated by SSA
and can be scored by counting the colonies that survive DSB
induction. saw1D, -DRBD, and -DFBD mutants that cannot
recruit Rad1 to 30 flaps show very poor survival and hence low
SSA repair levels (Li et al., 2013). However, colony number for
saw1-K221R cells was similar to that of wild-type (Figure 3B),
suggesting that sumoylation of Saw1 is not required for SSA.
Next, we tested UV resistance. Figure 3C shows that
saw1-K221R exhibited UV sensitivity similarly to saw1D. This is
in striking contrast to the SSA results and suggests that Saw1
sumoylation is required for its role in UV condition. As in
the case of saw1D, saw1-K221R sensitized rad59D to UV
Cell Reports 9, 143–152, October 9, 2014 ª2014 The Authors 147

A C
B
D
IP: Rad1-TAP IB: Saw1-FLAG
TAP
FLAG
RAD1-TAP: SAW1-FLAG:
Saw1
WT WT K221R + + +
Rad1
FLAG
K22
1R
WT
Stain
Saw1 SAW1:
Complex
Y-form
WT K221RSaw1:
1 2 3 4 5 6 7
Complex
Y-form
Saw1 Saw1-SUMO
1 2 3 4 5 6 7 8 9
0 300100 200
25
50
75
100
0
Concentration of Saw1 (nM)
Per
cent
age
boun
d
Per
cent
age
boun
d
Concentration of Saw1 (nM)100 150 200
0
25
50
75
100
500
WTK221R
Saw1-SUMOSaw1
Figure 4. Saw1 Sumoylation Affects Neither Its
Protein Level nor Its Interactions with Rad1
and Y-Form DNA
(A) saw1-K221R does not affect Saw1 protein level
after UV treatment. Extracts from cells with Saw1 and
Saw1-K221R tagged with FLAG at its own chromo-
somal locus were examined by western blotting (top).
Loading is shown on the bottom.
(B) Saw1-K221R is indistinguishable from wild-type
protein for binding to Y-form DNA. Increasing con-
centrations of recombinant wild-type (lanes 2–4) and
mutant Saw1 (lanes 5–7) (30–280 nM) were tested by
EMSA for binding to Y-form DNA (6 nM). Protein-DNA
binding is manifested by the upshift of the fluo-
rescently labeled DNA (complex). Percentages of
Y-form DNA shifted from three trials were quantified
as mean ± SD (bottom).
(C) Sumoylation ofSaw1doesnot alter interactionwith
Y-form DNA in vitro. Recombinant GST-Saw1 was
subjected to in vitro sumoylation as in Figure 2B to
yield about 40%sumoylated Saw1. Themixture of the
products (40–200nM)was tested for binding toY-form
DNA (6 nM) (lanes 6–9) and compared with similar
amounts of Saw1 that underwent the same procedure
in the absence of SUMO E1 (lanes 2–5). Percentages
of Y-form shifted from three trials were quantified as
mean ± SD (bottom). Note that the different DNA shift
pattern here compared with that in (B) is likely due
to changes caused by incubation for sumoylation
reactions or other proteins in the reactions.
(D) Saw1-K221R is proficient for Rad1 interaction in vivo after UV treatment. Rad1-TAP was pulled down, and coimmunoprecipitated Saw1-FLAG was detected
by antibody against FLAG. The ratio of copurified Saw1 to Rad1 is similar between wild-type and saw1-K221R cells.
See also Figure S2.
(Figure 3D), further supporting the notion that saw1-K221R’s UV
sensitivity is not due to an SSA defect.
Different from saw1-K221R, saw1-DFBD showed only slight
UV sensitivity, suggesting that flap binding is largely dispensable
for UV resistance (Figure 3C). The UV sensitivity of saw1-DRBD
was in between that of saw1-DFBD and saw1-K221R or null (Fig-
ure 3C), suggesting that the Saw1 contribution in theUV situation
is only partly via assistance of Rad1-Rad10.
Saw1-Mediated Survival inOther LesionContexts Relieson Its Rad1 and DNA Flap BindingWe examined saw1 mutants for phenotype indicative of defects
in base lesion and CPT repair. Like saw1D, saw1-DRBD, and
-DFBD were synthetically lethal with apn1D apn2D, and strongly
sensitized tdp1D to CPT (Figures 1D, 1E, 3E, and 3F). Thus,
Saw1 interactions with Rad1 and 30 flap DNA are important for
survival in the presence of base lesions and Top1-DNA adducts
in these genetic backgrounds. Different from saw1-DRBD and
-DFBD, saw1-K221R apn1D apn2D cells were viable but ex-
hibited slower growth and stronger MMS sensitivity than
apn1D apn2D (Figures 3E and 3G), and only moderate sensitiza-
tion of tdp1D cells to CPT (Figure 3F). These results suggest that
Saw1 sumoylation only moderately promotes survival in the
presence of base lesions and protein-DNA adducts.
Taken together, our genetic analyses suggest that the three at-
tributes of Saw1, namely, Rad1 interaction, flap binding, and su-
moylation, contribute to different extents in coping with different
lesions (Figure 3H). In the UV case, Saw1 sumoylation is critical,
148 Cell Reports 9, 143–152, October 9, 2014 ª2014 The Authors
whereas Rad1 and flap binding are less important. The reverse is
true for MMS and CPT situations, as in SSA repair. Our data sug-
gest that whereas Saw1 contributes to the latter three situations
via the known mechanism of Rad1-Rad10 recruitment, its su-
moylation affects the UV situation largely independently of this
mechanism. Next, we focused our efforts on understanding
how Saw1 sumoylation promotes UV survival.
Saw1 Sumoylation Does Not Affect Protein Level, DNABinding, or Rad1 InteractionWe first assessed protein levels of Saw1 in untreated and UV-
treated cells and detected no difference between wild-type and
saw1-K221R cells (Figures 4A and S2A), indicating that sumoyla-
tion of Saw1 does not affect bulk protein levels. Next, we exam-
ined how Saw1 sumoylation affects its DNA binding. Saw1 is a
structure-specific DNA binding protein with affinity for branched
DNA structures such as Y-forms (Li et al., 2013). We found that
recombinant Saw1-K221R protein exhibited similar binding to
Y-form DNA as its wild-type counterpart (Figure 4B). In addition,
SUMO-Saw1 obtained by subjecting the protein to in vitro
sumoylation that yielded �40% modified protein as shown in
Figure 2B (lane 3) showed no difference in binding affinity for
Y-form DNA when compared with equal amounts of unmodified
protein (Figure 4C). We also found that the Saw1-K221R mutant
was proficient for Rad1 interaction in vivo, in both UV- andMMS-
treated conditions (Figures 4D and S2B). These results suggest
that sumoylation unlikely influences Saw1 protein stability or its
known interactions with Rad1 and Y-form DNA.

saw1-KR rad55
rad55Untreated UV 40 J/m2 A
B
C
saw1 rad55
rad55
UV 45 J/m2 Untreated
D
rad26
saw1-KR rad26
Untreated UV 40 J/m2
Untreated UV 25 J/m2
rad7
saw1-KR rad7
rad26
saw1 rad26
Untreated UV 40 J/m2
rad7
saw1 rad7
Untreated UV 15 J/m2
Figure 5. Saw1 and Its Sumoylation Contribute to UV Resistance Independently of Rad51-Dependent HR and NER
(A–D) saw1D and saw1-K221R exacerbate theUV sensitivities of rad55D, rad26D, and rad7D cells. In (A) and (B), cells were spotted in 10-fold serial dilutions; in (C)
and (D), 3-fold serial dilutions were used. Note that neither saw1D nor saw1-KR exhibit noticeable sensitivity at the UV doses shown. See also Figure S3.
Saw1 Contributes to UV Resistance Independently ofNER and Homologous RecombinationThe observation that sumoylation of Saw1 does not affect the
above properties raised the possibility that its effect could be
throughmechanisms not hitherto associated with Saw1. We first
assessed whether Saw1’s effect in the UV situation is related to
two main UV lesion removal pathways, Rad51-mediated homol-
ogous recombination (HR) and NER (Krogh and Symington,
2004; Scharer, 2013). In each case, we examined the combina-
torial mutant between saw1-K221R or saw1D with the null
of representative proteins of the pathway. saw1-K221R and
saw1D sensitized mutants that either lack Rad55 and Rad57 in
the Rad51-mediated recombination pathway (Figures 5A, 5C,
and S3) or lack Rad26 and Rad7-Rad16 in the two branches of
NER (Figures 5B, 5D, and S3). These results suggest that UV
resistance mediated by Saw1 and its sumoylation is separable
from Rad51-dependent HR or NER.
SUMO Favors Saw1 Interaction with Slx1-Slx4, and theTwo Are Epistatic in the UV SituationBecause Saw1 is a scaffold for the Rad1-Rad10 nuclease, we
queried whether Saw1 interacts with other structure-specific nu-
cleases. An interaction with Slx4was detected in both yeast two-
hybrid and in vitro pull-down assays (Figures 6A–6B). Slx4 binds
to Slx1 to form a nuclease that cleaves 50 flaps with opposite
polarity as Rad1-Rad10 (Fricke and Brill, 2003). Although no
Saw1-Slx1 interaction was detected in 2H assay, Slx1 showed
interaction with SUMO (Figure 6A). The Slx1-SUMO and Slx4-
Saw1 interactions suggest a dual interaction mode between
SUMO-Saw1 and Slx1-Slx4. In support of this idea, fusing
SUMO to Saw1 enhanced Slx4 interaction in two-hybrid assay,
compared with Saw1 (Figure 6C). Interestingly, this fusion
reduced interaction with Rad1 (Figure 6C). These results suggest
competition between Slx4 and Rad1 for Saw1 binding, and that
SUMO favors the former at the expense of the latter. Consistent
with this notion, the Saw1-DRBD mutant that cannot interact
with Rad1 showed stronger interaction with Slx4 than its wild-
type counterpart (Figures 6 and S4). Taken together, our results
suggest that SUMO could act as a switch to favor Saw1 inter-
action with Slx4 over Rad1.
We next examined whether the SUMO-enhanced Saw1-Slx4
interaction pertains to the UV situation using epistasis analysis.
Figure 6D shows that slx4D cells reproducibly showed slightly
more sensitivity than wild-type cells in the higher UV dose range,
and that the saw1D slx4D doublemutant behaved like the saw1D
single mutant. This genetic relationship supports a functional
relationship between Saw1 and Slx4 in the UV condition.
DISCUSSION
Saw1 is a recently identified DNA repair scaffold protein that re-
cruits the Rad1-Rad10 nuclease to flap DNA during SSA repair of
DNA breaks (Li et al., 2008, 2013). Here, we show that Saw1 also
contributes to survival in the presence of other types of DNA le-
sions. Its roles in situations that require the repair of base lesions
and Top1-DNA adducts depend on Rad1 and flap binding, as in
the case of SSA. We thus propose that Saw1 recruits Rad1-
Rad10 to flap DNA in multiple repair contexts, both as those
tested here and possibly others that require Rad1-Rad10 flap
cleavage, such as recombination between dispersed repeats
or synthesis-dependent strand annealing (Diamante et al.,
2014; Mazon et al., 2012) (Figure 6E).
Distinct from these processes, Saw1’s role in the UV situation
only partially depends on Rad1 binding, and not on flap binding.
These results suggest that Saw1 uses a distinct mechanism in
this situation, likely involving interaction with different DNA
structures and nucleases. As Saw1 binds to DNA bubbles (Li
et al., 2013), this interaction may contribute to UV repair when
the region of local distortion caused by bulky photoproducts
is unwound. One candidate nuclease that Saw1 collaborates
with is Slx1-Slx4. The observed Saw1-Slx4 and Slx1-SUMO in-
teractions suggest a two-pronged interaction mode to confer
Cell Reports 9, 143–152, October 9, 2014 ª2014 The Authors 149

TT TTTTTTTTTTTTTTTTTTTTTTTTTTTTTT
GST
GST-Saw1
Slx4
D B
C
Rad1/10 S Slx1 Slx1
Saw1 Slx4
A
Saw1
Em
pty
Slx
4
Empty
Saw1-SUMO
-L-T -L-T-H -L-T-H+3AT
Rad
1
Em
pty
Slx
4
Rad
1
Em
pty
Slx
4
Rad
1
Bai
t
Prey
Saw1
Empty E
mpt
y
Slx
4
-L-T -L-T-H E
mpt
y
Slx
4
Slx1
Empty
Em
pty
SU
MO
-L-T -L-T-H
Em
pty
SU
MO
Bai
t Prey
Per
cent
age
surv
ival
600
10
20
30
40
70 80 90UV dose (J/m2)
WT
saw1
saw1 slx4
slx4
*
*
E
S E S E
116 97
66
45
29
GST-Saw1 + Slx4
GST + Slx4
Saw1 SSA
SUMO-Saw1: Bubble and Slx1/4 binding
Saw1: flap and Rad1/10 binding
Base lesion repair
Protein-DNA adduct repair
S Saw1
UV repair kDa
Rad1/10
Model
Figure 6. Saw1 Physical and Genetic Interactions with Slx4
(A) Saw1 interacts with Slx4 and SUMO interacts with Slx1 in yeast two-hybrid assay. Cells transformed with the indicated plasmids were patched onto selection
plates. Growth on SC–L-T plates indicates presence of plasmids, and growth on SC–L-T-H plates indicates interaction.
(B) GST-Saw1, but not GST, pulls down Slx4 in vitro. Supernatant (S) and eluate (E) of each of the GST pull-down reactions are shown.
(C) Fusing SUMO to the C terminus of Saw1 enhances Slx4 interaction and reduces Rad1 interaction. Similar to (A), except that growth on SC–L-T-H+3AT
indicates stronger interaction.
(D) saw1D is epistatic to slx4D for UV sensitivity. Data from at least three trials are represented as mean ± SD. Asterisks denote statistically significant differences
between survival of wild-type and slx4D cells (p < 0.05).
(E) Top: possible model for Saw1 and its sumoylation in promoting UV repair. This could involve Saw1 binding to bubble DNA structures and SUMO-enhanced
binding of Saw1 to the Slx1-Slx4 nuclease. Saw1 interaction with Rad1-Rad10 plays partial roles here, and the two nucleases may be coordinated for dual
cleavage of the DNA lesion. Bottom: Saw1 and its ability to bind Rad1 and 30 flap DNA are required for SSA and likely the repair of base lesions and protein-DNA
adducts. Note that for simplicity, partial contributions of Saw1 sumoylation to the latter two repair processes are not drawn.
See also Figure S4.
binding specificity to the sumoylated form of Saw1 for the
nuclease. In addition, we found that SUMO favors the Saw1-
Slx4 interaction at the expense of the Saw1-Rad1 interaction.
These results suggest a SUMO-based switch of Saw1 binding
partner toward Slx4. As saw1D is epistatic with slx4D for UV
sensitivity, Saw1 can partly collaborate with Slx1-Slx4 in UV
repair. However, as slx4D is not as sensitive to UV as saw1D,
Saw1 may have other nuclease partners or other roles. Though
these roles are currently unclear, our data suggest that they are
genetically separable from SSA, Rad51-dependent HR, and
NER. Although lesion tolerance mechanisms are candidates,
an interesting possibility is that Saw1 may be part of an alterna-
tive excision repair pathway, which mimics a minimal UV exci-
sion repair pathway found in fission yeast and N. crassa
(Bowman et al., 1994; McCready et al., 2000; Takao et al.,
1996; Yajima et al., 1995; Yasui, 2013; Yonemasu et al.,
1997). In this scenario, Saw1 and its sumoylation may coordi-
nate Slx1-Slx4 and Rad1-Rad10 for the cleavage reaction in
this repair (Figure 6E).
It is noteworthy that mammalian SLX4 contains a large N-ter-
minal extension that is absent in the yeast Slx4 protein. This
150 Cell Reports 9, 143–152, October 9, 2014 ª2014 The Authors
extended region interacts with the Rad1-Rad10 homolog
(ERCC4-ERCC1), whereas the conserved region interacts with
SLX1 (Figure S5) (Fekairi et al., 2009; Munoz et al., 2009; Svend-
sen et al., 2009). Dual nuclease interaction in this case may be
functionally similar to the Saw1 interactions with Rad1-Rad10
and Slx1-Slx4 in yeast. This raises the possibility that Saw1
serves the function of the N-terminal region of mammalian
SLX4. In both cases, the scaffolds assist their associated nucle-
ases in multiple molecular settings. Further testing of this notion
will shed light on the evolutionarily important mechanisms in
scaffold-mediated nuclease coordination.
Our findings expand the roles of SUMO in coping with UV
lesions beyond the previously reported effects on Rad1 and
XRCC1 (Sarangi et al., 2014; Wang et al., 2005). Unique to this
case, sumoylation dictates a specific function for Saw1, rather
than affecting general protein attributes. This is an example of
SUMO specifying a DNA repair factor to a particular function.
Our findings suggest one possible mechanism involving Slx1-
Slx4 and rule out several others. As our understanding of Saw1
function in UV repair grows, this and additional mechanisms
can be tested thoroughly. In conclusion, our findings highlight

the versatility of Saw1 as a nuclease scaffold in promoting cell
survival in different genotoxic stress conditions and reveal an
additional role for sumoylation in promoting UV resistance.
These findings open up avenues to explore the roles of this
nuclease scaffold in DNA repair.
EXPERIMENTAL PROCEDURES
Yeast Strains and Genetic Manipulations
Strains used are listed in Table 1. Standard yeast protocols were used
for strain generation, growth, medium preparation, and DNA damage sensi-
tivity assays. For DNA damage sensitivity tests, log phase cells were diluted
10- or 3-fold and spotted onto YPD media with or without MMS or CPT, or
irradiated with UV. For UV treatment, cells were irradiated on plates, and all
subsequent steps were done in conditions that prevent light exposure. For
survival curves, colonies were counted after incubation for 48 hr. For spot as-
says, plates were incubated at 30�C and photographed after 24–72 hr. Yeast
two hybrid assays were performed as described (Hang et al., 2011). Note that
3AT was added to SC–L-T-H media to detect only the stronger two hybrid
interactions (Joung et al., 2000).
Detection of Sumoylated Proteins and Immunoprecipitation
These were performed as described previously (Cremona et al., 2012). In brief,
cells were lysed by bead beating in denaturing conditions and TAP-tagged
proteins were immunoprecipitated using immunoglobulin (Ig) G-Sepharose.
These were washed and eluted with loading dye, followed by SDS-PAGE
and western blotting with antibodies against SUMO and the protein A part of
the TAP tag (Sigma-Aldrich). Damage-induced sumoylation was assessed
by exposing log-phase cells to 100 or 200 J/m2 UV using UV Stratalinker
1800 (Stratagene), 0.3% methylmethane sulfonate (MMS, Sigma-Aldrich), or
50 mg/ml camptothecin (CPT, Sigma-Aldrich) for 2 hr. We note that, unlike
most sumoylated proteins characterized thus far whose sumoylation levels
are very low (Ulrich, 2009), sumoylation of Saw1 can be readily detected by
the antibody against the tag (Figure 2C). Quantification of the bands showed
that approximately 7% of Saw1 is sumoylated under normal growth conditions
and around 26% after damage treatment. This makes Saw1 one of the rare
substrates with high levels of sumoylation. Coimmunoprecipitation was
done as described previously (Hang et al., 2011).
His6- and GST-Saw1 Protein Purification
The plasmid expressing Saw1 protein with (His)6-affinity tag was introduced
into E. coli strain Rosetta(DE3)pLysS. Protein expression was induced by
1 mM isopropyl-beta-D-thiogalactopyranoside (IPTG) at 37�C for 4 hr. Extract
from 13 g of cell paste was prepared by sonication in 50ml of buffer containing
50 mM Tris-HCl (pH 8.0), 150 mM NaCl, 10% glycerol, 0.5% Triton X-100,
1 mM b-mercaptoethanol, and protease inhibitor cocktail. The lysate was clar-
ified by ultracentrifugation, and the resulting supernatant was incubated with
1 ml Ni-NTA agarose (QIAGEN) for 2 hr at 4�C. The beads were washed with
12 ml of buffer T (25 mM Tris-HCl, 10% glycerol, 0.5 mM EDTA [pH 7.5]) con-
taining 100 mM KCl. The bound proteins were eluted with buffer T containing
50 mM KCl and imidazole (from 50 to 1,000 mM). Fractions containing Saw1
(500–1,000 mM imidazole) were applied onto a 0.5 ml MonoS column (GE
Healthcare), and eluted using 200–1,000 mM KCl in buffer T. The peak Saw1
fractions (550–1,000 mM KCl) were concentrated to 3 mg/ml in a Vivaspin-2
concentrator. For GST-Saw1, the E. coli strain BL21(DE3)pLysS was trans-
formed with a plasmid expressing GST-tagged Saw1 protein. Protein expres-
sion was induced by addition of 0.1 mM IPTG at 16�C overnight. Ten grams of
cell paste was sonicated in 50 ml of buffer CBB (50 mM Tris-HCl [pH 7.5], 10%
sucrose, 2 mM EDTA, 150 mM KCl, 0.01% NP40, 1 mM DTT, and protease
inhibitor cocktail). The lysate was clarified by ultracentrifugation and the super-
natant was loaded on a 7-ml Sp-Sepharose column (GE Healthcare). The col-
umn was eluted using 150–1,000 mM KCl in buffer K (20 mM K2HPO4, 10%
glycerol, 0.5 mM EDTA [pH 7.5]). Peak Saw1 fractions eluting around 400–
600 mM KCl were incubated with 700 ml glutathione-Sepharose (GE Health-
care) for 1 hr at 4�C. The beads were washed with 10 ml of buffer K containing
100 mM KCl and eluted in steps with 50–200 mM glutathione in buffer K con-
taining 100 mM KCl. The fractions containing Saw1 (100–200 mM glutathione)
were applied onto a 1 ml MonoS column (GE Healthcare) and eluted using
200–1,000 mM KCl in buffer K. The peak fractions (500–800 mM KCl) were
concentrated to 10 mg/ml in a Vivaspin-2 concentrator. The saw1-K221R
mutant was generated by site-directed mutagenesis.
Pull-Down Assay
PurifiedGST-Saw1 (3 mM) and Slx4 (0.2 mM) proteins were incubated with 25 ml
of glutathione-Sepharose 4 Fast Flow (GE Healthcare) in 25 ml of buffer T
(20 mM Tris-HCl [pH 7.5], 80 mM KCl, 1 mM DTT, 0.5 mM EDTA, and 0.01%
NP40) for 30 min at 4�C, with gentle shaking. Following incubation, the super-
natants were collected and mixed with 20 ml of SDS Laemmli buffer. After
washing the beads with 100 ml of buffer T, the bound proteins were eluted
with 30 ml of SDS Laemmli buffer. The supernatant and SDS eluate fractions
were subjected to SDS-PAGE analysis.
Other Assays
In vitro sumoylation assay, mass spectrometry detection of sumoylated
lysines, and electrophoretic mobility shift assays (EMSAs) were performed
as described previously, except that the EMSA used a 5% polyacrylamide
gel in 0.5 3 Tris-borate-EDTA and 6 nM DNA substrate (Sarangi et al.,
2014). His-taggedSlx4was purified as described (Fricke andBrill, 2003). Chro-
mosomal SSA assay was performed as described earlier (Li et al., 2008). In
brief, log phase cells were grown in YP-glycerol and then plated on YP-glucose
or YP-galactose plates, and colonies were counted after 3–4 days. Percentage
survival was calculated as number of colonies on YP-galactose plates divided
by that on YP-glucose plates.
SUPPLEMENTAL INFORMATION
Supplemental Information includes five figures and can be found with this
article online at http://dx.doi.org/10.1016/j.celrep.2014.08.054.
AUTHOR CONTRIBUTIONS
P.S., V.A., L.K., and X.Z. conceived and designed the experiments, P.S., V.A.,
Z.B., and C.H. performed the experiments, F.H. generated some reagents,
D.A. and G.A. performed the mass spectrometry experiments, P.S. and X.Z.
analyzed the in vivo data, C.H. and S.E.L. analyzed the SSA results, V.A.
and L.K. analyzed the in vitro data, P.S. and X.Z. wrote the paper, and V.A.,
C.H., S.E.L., and L.K. commented on the manuscript.
ACKNOWLEDGMENTS
We thank Steve Brill for the Slx4 expression plasmid.We are grateful to X.Z. lab
members for discussions and useful suggestions. This work is supported by
NIH grant GM071011 to S.E.L.; Czech Science Foundation (GACR 13-
26629S and 207/12/2323), European Regional Development Fund (Project
FNUSA-ICRC; no. CZ.1.05/1.1.00/02.0123) and CZ.1.07/2.3.00/20.0011 cofi-
nanced by European Social Fund and the state budget of the Czech Republic
to L.K.; ‘‘Employment of Newly Graduated Doctors of Science for Scientific
Excellence’’ (CZ.1.07/2.3.00/30.0009) cofinanced by European Social Fund
to V.A.; NIH grant GM080670, American Cancer Society grant RSG-12-013-
01-CCG, and Leukemia and Lymphoma Society Scholar Award to X.Z. Fund-
ing for open access charge was provided by the NIH.
Received: June 16, 2014
Revised: July 23, 2014
Accepted: August 22, 2014
Published: September 25, 2014
REFERENCES
Altmannova, V., Eckert-Boulet, N., Arneric, M., Kolesar, P., Chaloupkova, R.,
Damborsky, J., Sung, P., Zhao, X., Lisby, M., and Krejci, L. (2010). Rad52
SUMOylation affects the efficiency of the DNA repair. Nucleic Acids Res. 38,
4708–4721.
Cell Reports 9, 143–152, October 9, 2014 ª2014 The Authors 151

Bai, Y., and Symington, L.S. (1996). A Rad52 homolog is required for RAD51-
independent mitotic recombination in Saccharomyces cerevisiae. Genes Dev.
10, 2025–2037.
Boiteux, S., and Guillet, M. (2004). Abasic sites in DNA: repair and biological
consequences in Saccharomyces cerevisiae. DNA Repair (Amst.) 3, 1–12.
Bowman, K.K., Sidik, K., Smith, C.A., Taylor, J.S., Doetsch, P.W., and Freyer,
G.A. (1994). A new ATP-independent DNA endonuclease from Schizosacchar-
omyces pombe that recognizes cyclobutane pyrimidine dimers and 6-4 photo-
products. Nucleic Acids Res. 22, 3026–3032.
Chen, Y.H., Szakal, B., Castellucci, F., Branzei, D., and Zhao, X. (2013). DNA
damage checkpoint and recombinational repair differentially affect the replica-
tion stress tolerance of Smc6 mutants. Mol. Biol. Cell 24, 2431–2441.
Cremona, C.A., Sarangi, P., Yang, Y., Hang, L.E., Rahman, S., and Zhao, X.
(2012). Extensive DNAdamage-induced sumoylation contributes to replication
and repair and acts in addition to the mec1 checkpoint. Mol. Cell 45, 422–432.
Diamante, G., Phan, C., Celis, A.S., Krueger, J., Kelson, E.P., and Fischhaber,
P.L. (2014). SAW1 is required for SDSA double-strand break repair in
S. cerevisiae. Biochem. Biophys. Res. Commun. 445, 602–607.
Fekairi, S., Scaglione, S., Chahwan, C., Taylor, E.R., Tissier, A., Coulon, S.,
Dong, M.Q., Ruse, C., Yates, J.R., 3rd, Russell, P., et al. (2009). Human
SLX4 is a Holliday junction resolvase subunit that binds multiple DNA repair/
recombination endonucleases. Cell 138, 78–89.
Fishman-Lobell, J., Rudin, N., and Haber, J.E. (1992). Two alternative
pathways of double-strand break repair that are kinetically separable and
independently modulated. Mol. Cell. Biol. 12, 1292–1303.
Fricke, W.M., and Brill, S.J. (2003). Slx1-Slx4 is a second structure-specific
endonuclease functionally redundant with Sgs1-Top3. Genes Dev. 17, 1768–
1778.
Guillet, M., and Boiteux, S. (2002). Endogenous DNA abasic sites cause cell
death in the absence of Apn1, Apn2 and Rad1/Rad10 in Saccharomyces
cerevisiae. EMBO J. 21, 2833–2841.
Guzder, S.N., Sommers, C.H., Prakash, L., and Prakash, S. (2006). Complex
formation with damage recognition protein Rad14 is essential for Saccharo-
myces cerevisiae Rad1-Rad10 nuclease to perform its function in nucleotide
excision repair in vivo. Mol. Cell. Biol. 26, 1135–1141.
Hammel, M., Rey, M., Yu, Y., Mani, R.S., Classen, S., Liu, M., Pique, M.E.,
Fang, S., Mahaney, B.L., Weinfeld, M., et al. (2011). XRCC4 protein interac-
tions with XRCC4-like factor (XLF) create an extended grooved scaffold for
DNA ligation and double strand break repair. J. Biol. Chem. 286, 32638–32650.
Hang, L.E., Liu, X., Cheung, I., Yang, Y., and Zhao, X. (2011). SUMOylation
regulates telomere length homeostasis by targeting Cdc13. Nat. Struct. Mol.
Biol. 18, 920–926.
Heyer, W.D., Ehmsen, K.T., and Liu, J. (2010). Regulation of homologous
recombination in eukaryotes. Annu. Rev. Genet. 44, 113–139.
Johnson, E.S., and Gupta, A.A. (2001). An E3-like factor that promotes SUMO
conjugation to the yeast septins. Cell 106, 735–744.
Joung, J.K., Ramm, E.I., and Pabo, C.O. (2000). A bacterial two-hybrid selec-
tion system for studying protein-DNA and protein-protein interactions. Proc.
Natl. Acad. Sci. USA 97, 7382–7387.
Krogh, B.O., and Symington, L.S. (2004). Recombination proteins in yeast.
Annu. Rev. Genet. 38, 233–271.
Li, F., Dong, J., Pan, X., Oum, J.H., Boeke, J.D., and Lee, S.E. (2008). Micro-
array-based genetic screen defines SAW1, a gene required for Rad1/Rad10-
dependent processing of recombination intermediates. Mol. Cell 30, 325–335.
Li, F., Dong, J., Eichmiller, R., Holland, C., Minca, E., Prakash, R., Sung, P.,
Yong Shim, E., Surtees, J.A., and Eun Lee, S. (2013). Role of Saw1 in Rad1/
152 Cell Reports 9, 143–152, October 9, 2014 ª2014 The Authors
Rad10 complex assembly at recombination intermediates in budding yeast.
EMBO J. 32, 461–472.
Mazon, G., Lam, A.F., Ho, C.K., Kupiec, M., and Symington, L.S. (2012). The
Rad1-Rad10 nuclease promotes chromosome translocations between
dispersed repeats. Nat. Struct. Mol. Biol. 19, 964–971.
McCready, S.J., Osman, F., 1, and Yasui, A. (2000). Repair of UV damage in the
fission yeast Schizosaccharomyces pombe. Mutat. Res. 451, 197–210.
Munoz, I.M., Hain, K., Declais, A.C., Gardiner, M., Toh, G.W., Sanchez-Pulido,
L., Heuckmann, J.M., Toth, R., Macartney, T., Eppink, B., et al. (2009). Coor-
dination of structure-specific nucleases by human SLX4/BTBD12 is required
for DNA repair. Mol. Cell 35, 116–127.
Pouliot, J.J., Yao, K.C., Robertson, C.A., and Nash, H.A. (1999). Yeast gene
for a Tyr-DNA phosphodiesterase that repairs topoisomerase I complexes.
Science 286, 552–555.
Prolla, T.A., Pang, Q., Alani, E., Kolodner, R.D., and Liskay, R.M. (1994). MLH1,
PMS1, and MSH2 interactions during the initiation of DNA mismatch repair
in yeast. Science 265, 1091–1093.
Psakhye, I., and Jentsch, S. (2012). Protein group modification and synergy
in the SUMO pathway as exemplified in DNA repair. Cell 151, 807–820.
Sarangi, P., Bartosova, Z., Altmannova, V., Holland, C., Chavdarova, M., Lee,
S.E., Krejci, L., and Zhao, X. (2014). Sumoylation of the Rad1 nuclease pro-
motes DNA repair and regulates its DNA association. Nucleic Acids Res. 42,
6393–6404.
Scharer, O.D. (2013). Nucleotide excision repair in eukaryotes. Cold Spring
Harb. Perspect. Biol. 5, a012609.
Svendsen, J.M., Smogorzewska, A., Sowa, M.E., O’Connell, B.C., Gygi, S.P.,
Elledge, S.J., and Harper, J.W. (2009). Mammalian BTBD12/SLX4 assembles
a Holliday junction resolvase and is required for DNA repair. Cell 138, 63–77.
Takahashi, Y., Toh-e, A., and Kikuchi, Y. (2001). A novel factor required for the
SUMO1/Smt3 conjugation of yeast septins. Gene 275, 223–231.
Takao, M., Yonemasu, R., Yamamoto, K., and Yasui, A. (1996). Characteriza-
tion of a UV endonuclease gene from the fission yeast Schizosaccharomyces
pombe and its bacterial homolog. Nucleic Acids Res. 24, 1267–1271.
Ulrich, H.D. (2009). The SUMO system: an overview. Methods Mol. Biol. 497,
3–16.
Vance, J.R., and Wilson, T.E. (2002). Yeast Tdp1 and Rad1-Rad10 function
as redundant pathways for repairing Top1 replicative damage. Proc. Natl.
Acad. Sci. USA 99, 13669–13674.
Vidal, A.E., Boiteux, S., Hickson, I.D., and Radicella, J.P. (2001). XRCC1
coordinates the initial and late stages of DNA abasic site repair through pro-
tein-protein interactions. EMBO J. 20, 6530–6539.
Wang, Q.E., Zhu, Q., Wani, G., El-Mahdy, M.A., Li, J., and Wani, A.A. (2005).
DNA repair factor XPC is modified by SUMO-1 and ubiquitin following UV irra-
diation. Nucleic Acids Res. 33, 4023–4034.
Yajima, H., Takao, M., Yasuhira, S., Zhao, J.H., Ishii, C., Inoue, H., and Yasui,
A. (1995). A eukaryotic gene encoding an endonuclease that specifically
repairs DNA damaged by ultraviolet light. EMBO J. 14, 2393–2399.
Yasui, A. (2013). Alternative excision repair pathways. Cold Spring Harb.
Perspect. Biol. 5, a012617.
Yonemasu, R., McCready, S.J., Murray, J.M., Osman, F., Takao, M., Yama-
moto, K., Lehmann, A.R., and Yasui, A. (1997). Characterization of the alterna-
tive excision repair pathway of UV-damaged DNA in Schizosaccharomyces
pombe. Nucleic Acids Res. 25, 1553–1558.
Zhao, X., and Blobel, G. (2005). A SUMO ligase is part of a nuclear multiprotein
complex that affects DNA repair and chromosomal organization. Proc. Natl.
Acad. Sci. USA 102, 4777–4782.





























