Embed Size (px)
Citation preview

Kaposi’s sarcoma: a computational approach through protein-protein interaction and gene regulatory networks analysis
Aubhishek Zaman1*, Md. Habibur Rahman1, Samsad Razzaque2
1Department of Genetic Engineering and Biotechnology, University of Dhaka, Dhaka-1000, Bangladesh.
2Biotechnology program, Department of Mathematics and Natural Science, BRAC university, Mohakhali, Dhaka-1212, Bangladesh
* Corresponding author; email: [email protected]

Kaposi’s sarcoma: a computational approach through protein-protein interaction and gene regulatory networks analysis

Abstract
Interactomic data for Kaposi’s Sarcoma Associated Herpes virus (KSHV)- the causative agent of vascular origin tumor called Kaposi’s sarcoma- is relatively modest to date. The objective of this study was to assign functions to the previously uncharacterized ORFs in the virus using computational approaches and subsequently fit them to the host interactome landscape on protein, gene and cellular level. On the basis of expression data, predicted RNA interference data, reported experimental data and sequence based functional annotation we also tried to hypothesize the ORFs role in lytic and latent cycle during viral infection. We studied 17 previously uncharacterized ORFs in KSHV and the host virus interplay seems to work in three major functional pathways- cell division, transport, metabolic and enzymatic in general. Studying the host virus crosstalk for lytic phase predicts ORF 10 and ORF 11 as a predicted virus hub whereas PCNA is predicted as a host hub. On the other hand, ORF31 has been predicted as a latent phase inducible protein. KSHV invests a lion’s share of its coding potential to suppress host immune response; various inflammatory mediators such as IFN-γ, TNF, IL-6 and IL-8 are negatively regulated by the ORFs while Il-10 secretion is stimulated in contrast. Although, like any other computational prediction, the study requires further validation, keeping into account the reproducibility and vast sample size of the systems biology approach the study allows us to propose an integrated network for host-virus interaction with good confidence. We hope that the study, in the long run, would help us identify effective dug against potential molecular targets.
Key words: Kaposi sarcoma, Herpes Virus 8, Protein-Protein Interaction, Gene regulatory network, Host virus crosstalk, Immunosuppression

1. Introduction
Kaposi’s sarcoma (KS), caused by Kapsosi’s Sarcoma Associated Herpes Virus (KSHV) is a tumor of vascular origin [1, 2]. One of the major symptoms of this disease primarily involves lesions in the skin with red and purple patches [3]. The virus infection spreads to visceral organs including gastrointestinal tract and respiratory tract [4, 5]. It has been shown to co-occur with AIDS (Acquired Immune-Deficiency Syndrome) [6]; though the relationship between HIV-1 and KSHV is not fully clear [7, 8]. KSHV, alternatively known as human herpes virus 8 (HHV8) infects Peripheral Blood Mononuclear Cells (PBMC) [9, 10]. Like all other viruses in Herpes virus group, KSHV too has two distinct phase of growth- lytic and latent. Often an acute infection of KSHV turns latent in immune-compromised patients [11]. In immunocompetent individuals, KSHV establishes latent infection following an acute infection [12]. The virus contains a double stranded DNA as its genome and the genome is maintained as an extra-nuclear episome in the host while in latent phase [13]. The latent phase is characterized by a literal shut off of most of the genes except a few on the latency locus. The consequence is that in latency period the virus produces no virion structure and thus remains asymptomatic with prolonged persistence. The lytic phase, on the contrary, involves extensive metabolic activity [14]. Upon exposure to favorable conditions the virus comes out of its latency period and causes chronic infection which can take up a tumorigenic course with time [15]. Latency is not only responsible for an escape from lytic phase, but also is required as an essential phase to establish tumor in the host [16, 17]. Up until now, however, there have been very few attempts to associate the unknown ORFs of KSHV to known proteins in other virus [18-24]. For example ORF 50 has been characterized as a major lytic inducer in KSHV. ORF 54, another protein with dUTPase function, has been characterized as a major signal transducer; it dampen down Natural Killer cell mediated immune response against the virus infected cells [25]. dUTPase function in virus has been of much interest in recent years [26]. Many perplexing activity of this motifs are being explored day by day; but what is even more interesting is the redundancy of this motif. However, insight on proteins with dUTPase function is surprisingly poor [27]. A systems level understanding of these proteins can really be of much help to understand the mechanism of the action in detail. Using systems biology to understand the underlying mechanism of virus-host crosstalk and designing drugs against viral proliferation have been a popular approach in the post genomic era [28-30]. Systems biology considers genes and proteins to be integrated to each other and hence takes up an integrated approach [31]. Protein-protein interaction (PPI) and gene regulatory networks (GRN) are one of the most recurrent themes of systems biology in this regard [32]. PPI exposes novel information about whole proteome interaction status for both host and the virus [33-35]. A common strategy of the systems biology investigators has been looking for most densely interconnected protein, known as hub proteins, in the systems map; hub proteins often make a good target for drug designing against the virus [30, 36]. Similarly DNA-protein interaction and GRN has been important to understand host pathogen interaction and host cellular mechanism [37]. Systems biology has been applied for assaying drugs against virus with complex life cycle [38]. Detecting a universal drug against these pathogens has been difficult as they drug target proteins are often poorly understood [39]. For example, a proteome-wide interaction map of KSHV proteins has not yet been provided [40]. Although systems biology studies often involve large scale data in this study a comparatively smaller scale, more specific interaction maps were generated. in silico analyses have been performed, collecting information on host interactions from hypothesis-driven studies [41] or from studies that identified interactions on the basis of conserved orthologs [42]- often referred to as interologs [43].

KSHV infection is followed by interplay of a range of different proteins of both host and of the virus itself [44, 45]. Although there have been emphatic proof of such crosstalks in closely linked viruses, for example Epstein-Barr virus (EBV), Human Papilloma virus (HPV), Cytomegalovirus and Herpes virus family, not much is known about such crosstalks in KSHV [46-48]. Furthermore not all ORFs in the viral genome have been characterized yet. Therefore the objective of the study was to at first characterize the ORFs using various computational tools and establish a host virus crosstalk network in proteomic, genomic and cellular level. In total 96 ORFs have been reported in KSHV genome. Among these, characterization for 24 ORFs is yet to be carried out. In this study 17 ORFs- ORF 10, 11, 18, 23, 24, 27, 28, 30, 31, 33, 34, 35, 42, 49, 52, 53 and 44 were taken and characterized through motif based sequence analysis, structural analysis and microarray and SAGE based functional analysis. The data were also validated by text mining and searching functions of the homologous proteins in other viruses.

2. Method 2.1. Selection of the host genes Through extensive text mining and literature search the initial disease related gene pool for the host genes was generated. For searching literatures associated to KSHV, the most important collection of scientific publications used was PubMed (http://www.ncbi.nlm.nih.gov/pubmed). Other data-mining tools used for literature search were HighWire Topicmap and AliBaba (http://alibaba.informatik.huberlin.de) (Supplementary figure 1). Apart from the text mined data, the gene pool was also enriched by analyzing microarray data associated to KSHV from Gene Expression Omnibus (GEO) database. In this regard, from KS related GSE1880 dataset the genes that were differentially expressed during KSHV lytic and latent phase were taken into consideration. See Supplementary method for details. 2.2. Characterization of the viral ORF 18 previously uncharacterized viral ORFs were characterized using several computational tools. Gene Ontology (GO), InterPro, Motif Finder, I-Tasser, SWISS-Model, DNA BindR, Kyte Dolittle were some of the tools used for the process. SAGE data was collected from Gene Expression Omnibus (GEO) database from GSM3241 dataset. SAGE data was analyzed using The SAGE Digital Gene Expression Displayer tool keeping Q value at 0.1. See Supplementary method for details. 2.3. Establishing interaction network for the selected host protein The selected host proteins were looked for their probable interaction partner by using STRING 9.0 [51] and BIND database [52]. From these databases the interaction data were pooled on the basis of physical binding and its experimental proof as well as co-expression and co-appearance in literatures. The interaction network for each protein sequence was merged using Advanced Network Merge plug-in in Cytoscape platform. 2.4. Establishing interaction network for the viral ORFs Because interaction data for uncharacterized viral ORFs were lacking in the databases, homologous proteins of them, for which interaction data was available, were first searched. The uncharacterized viral protein sequences were PSIBLASTed in the NCBI database. The ORF homologues found this way were searched for partners in STRING9.0 database. The interaction network for the homologues were further integrated between them by using Advanced Network Merge plug-in integrated in Cytoscape platform. The integration here was based on functional resembles between the nodes. See Supplementary method for details. 2.5. Integrating host and viral networks and predicting potential hubs As mentioned earlier, to integrate the host and virus networks the viral proteins were searched at STRING9.0 database for their human homologues. When no human homologues for the viral proteins were found, the ORF homologues were selected from other organisms. Hence, different ORF homologues

belonged to different organisms and they were finally integrated within the same organism on the basis of functional and sequence homology between them. For example ORF 10 was homologous to Acel_1402 protein belonging to Acidothermus cellulolyticus. On the other hand ORF 11 was homologous to SDFABSDF1895 of Acinetobacter baumannii. The two ORF homologues were made to integrate in the same organism that is by searching for Acel_1402’s homologue in the Acinetobacter baumannii. 2.6. Identifying interfering RNA regulated host genes in the network It has been reported that the KSHV interfering RNAs play an important role in host gene regulation. To identify this interfering RNA within the viral genome the virus genome sequence was downloaded from NCBI (National Center for Biotechnology Information) (http://www.ncbi.nlm.nih.gov) database. To detect potential non-coding RNA in the genome EMBOSS E-inverter and Repeat around repeat finding tool was used. The interfering RNA stability was predicted by RNAfold and MiPred web servers. Also MirCheck stand-alone tool was used to predict probable microRNA like interfering RNA comparing the sequences with mirDB and mirBASE database on the basis of homology. Minimum free energy (MFE) and partition function was chosen as the parameter for antisense RNA prediction. RNAhybrid tool was used to predict the target of the antisense RNA. From the predicted structures RNA structure with scores below -30.00 kcal/mol p value were sorted and predicted as significant hits. 2.7. Analysis of the data The robustness of the network was analyzed by checking several different parameters of the network. According to the power law, the probability that a new vertex k will be connected to vertex i depends on the connectivity of that vertex in the following way:
Clustering coefficient of a network represents how tightly a node in the network is linked. It can be thought to be the ability of drawing triangles through a given node. The equation of clustering co-efficient is as follows:
N (v) is set of the direct neighbors of node v and d (v) is the number of the direct neighbors of node v. Betweeness Centrality is another measure of centraliy of a node. It refers to the number of shortest paths from all vertices to all others that pass through that node. It was calculated using the following equation.
( ) ii
jj
kkk
∏ =∑
( )
( )1)()(
,2)( )(,
−= ∈
vdvd
jivC vNji

stρ is the number of shortest paths from node s to t and (v) the number of shortest paths from s to t that pass through the node v. See Supplementary method for details
Furthermore, analysis of the network was done in functional and structural level. The data from Microarray, SAGE and interfering RNA were made to correlate to each other and by the virtue of the expression data a binary and dynamic status of the genes in the network was established.
3. Result 3.1. Selection of host gene reveals a wide range of genes involving immune signaling, cell division and cell adhesion The heat map showed that the 7 different clusters for the host gene pool could have been generated on the basis of functional information. Genes expressed could have been classified on the basis of function in the following way- Cluster 7: Matrix proteins and cell division proteins, Cluster 6: translational regulation, tumor suppressors and receptor genes, Cluster 5: Hormone and cell signaling genes, Cluster 4: Transcription factors, DNA repair and enzymes, Cluster 3: Complex proteins with various functions; Cluster 2: Transcription factor and enzymatic activity and Cluster 1: Heat shock proteins and growth signaling proteins. Microarray data reveals information not only about the association of the genes with the disease but also about the relative expression of the genes with respect to one another. The expression pattern for the proteins was used furthermore to design the regulatory network (Supplementary Table 1 and File: Host_gene_list.xlsx).
From the microarray data, SAGE data (File: SAGE_data.docx) and literature information 42 host genes were selected and their interaction partners were taken and merged to produce 4 different network modules accommodating 680 numbers of nodes. The PPI network topology reveals a similar behavior with respect to general topology of PPI following a power law behavior and therefore scale-free properties. These types of networks have the particular feature that some nodes are highly connected compared with others on the same network. These highly connected nodes (hubs) in general, represent important proteins/genes in biological terms and therefore are treated with special attention.
The network was modified not to have any self loops. The global clustering coefficient of the network turned out to be of 0.657 whereas the local clustering coefficient- a single node’s clustering ability- was found to be ~11 neighboring nodes. The shortest path of the network covered 73% of the nodes. The high clustering coefficient of the network fits well with the characteristic biological networks. A clustering coefficient for a biological network usually range between 0.4 to 0.7 [31] and hence the network, albeit highly edge abundant, fitted in the range suitable for biological network. This means the false positives for the results were well within the permitted level (Supplementary figure 3). 3.2. Characterization of the viral ORF predicted the uncharacterized ORFs to be multifunctional
∑∈≠≠
=Vvts st
stB
vvCρ
ρ )()(

The principle for characterizing the viral ORFs were based on a principle of redundancy- the sequence or motif based results had to match well with the structure based results. The probable characterization of the ORFs is enlisted in File: ORF_characterization.docx.
It showed that the proteins had a wide array of functions. ORF 10 and ORF11 were functionally related to each other as both of them had dUTPase function. ORF 23, 28, 33 and 34 were predicted to be a set of structural proteins- either Capsid or Tegument protein. ORF 35, 49, 53 and 55 were predicted to be Transcription factors. Unlike other ORFs for which not very prominent indication of cell level regulation was found, ORF 27, 30, 31, 34 were predicted to take part in the immune regulation of the cells. However, one key aspect of the whole study was that many of the proteins, mostly the catalytic ones, were multifunctional in themselves. This overlapping in function is often abundant in virus genes as the remarkably small living system has to multitask to hijack the host system.
3.3. The network for the selected viral ORFs clusters the ORFs in few recurrent functional groups
One key limitation of the viral ORFs was that they were poorly characterized and hence their interactions too were so. Therefore, the sequences were needed to be brought in and merged in an organism for which sufficient interactome data has already been deposited. The network for the viral ORFs was made to merge together in different hierarchies. Firstly, their sequence was searched for homologues using a PSIPRED based techniques. In this technique the homologues were selected from an organism other than the virus itself. Most of the homologues found by this technique belonged to bacterial proteins. For the technique a control experiment was also run where distantly related but associated to a similar broad function viral and bacterial protein sequences were compared using PSIBLAST (Supplementary Table 2). The data for this comparison were termed as negative control. As a positive control protein sequence from virus that are known to have good homology with the bacterial protein sequences such as Mimivirus were used to compare with proteins from their bacterial counterpart. Surprisingly enough, even though the match was poor except two ORFs all the viral ORFs of KSHV showed better identity and length of match than that of the negative control. They are listed in Supplementary Table 3
The PSIBLAST generated homologues were searched further for human homologues; but when this approach failed they were merged into the organism for which a human homologue has been found. No human homologues were found for ORF 27, 30, 33, 34, 35, 39 49. The integration of the ORF homologues was done into 7 different networks belonging to different bacterial organisms. One network among these 7 was further integrated in to a network of integrating 3 ORFs (Supplementary Figure 4).
ORF 10, 11, 49, 55 and 35 were predicted to work as catalytic proteins responsible for nucleotide processing and DNA replication regulation. ORF 27, 53, 30, 54 were included in the genes responsible for transport, sorting and migratory processes within the host cell. ORF 33 and 52 interacted with host chaperone like proteins. ORF 31 and 35 were functional in the extracellular matrix related function (Supplementary Table 2).
ORF24, 28, 42 and 53 were not taken further into consideration as their scores were lower than that of the scores of the controls.
3.4. Virus and host network integration predicts the rout of virus host crosstalk

The viral ORFs network generated earlier were made to integrate within the host network (Supplementary File: Final network 1.pdf and Final network 2.pdf). The results are summarized in Figure 1. The function of the host protein are mentioned in Supplementary Table 5. The crosstalk reveals dUTPase activity as a potential viral hub for host interaction. Dut protein interacted with as many as 14 different other host proteins reflected by its dense edge connections (Figure 1). ORF 10, 49, 55 and 35 directly associated with it. Whereas PCNA was found to be the corresponding host hub. Apart from ORF 10, 11, 24, 27, 30, 31, 33, 34, 35, 42, 49, 52, 53 and 55 the other ORFs failed to fit into the host network.
The virus ORFs interacted with the host proteins belonging to functions similar to Cluster 1 (heat shock proteins), Cluster 4 (DNA repair, transcription factors), Cluster 6 (translational regulation) and Cluster 7 (Cell division proteins) of the microarray data.

Figure 1: Truncated image of the virus ORF integrated into the host ORF. The viral ORFs once integrated into the host network shows association to many. The pink nodes stand for nodes exclusive for hosts. The purple and blue nodes are shared by host and virus both- however the blue node is a predicted one on the basis of broadly related function whereas the purple one stands for direct link on the basis of homologue search. The red nodes search for viral ORF. The cyan nodes stand for linker proteins- they arise from the viral ORF homologues network and are of bacterial origin. Edges are represented by blue straight line. ORF 10 and ORF 11 are represented as a single node ‘DUT.ORF10’. It is to be noted that both ORF 10 and ORF 11 had dUTPase function. See text for details abd Supplementary file: Final netork 1 and 2 for detailed image.
The merged network shows that ORF 10/11 is located center of cell cycle, DNA replication, transcription and cytokine regulation pathways. It was predicted that this ORFs, by the virtue of accelerating cell cycle activities hijack the cellular machinery and switch it towards a lytic cycle.
The function of ORF 31 could not have been proposed here. However, a text-mining based regulatory circuit along with microarray data and sequence analysis predicted it to be a potent latent cycle inducer.
Clustering coefficient of the network is 0.62. Network diameter was 14 units. Network centralization valued 0.094. Shortest path covered 86% of the proteins. Average number of neighbors for the nodes was 10.35. Network heterogeneity was 0.868. All these values fit well with the standard biological network parameters.
3.5. Interfering RNA prediction showed possibility of a wide array of genes as a potential target The interfering RNA in the whole genome of the recently deposited Herpes Virus 8 genome were predicted. 5 different precursor interfering RNA could have been predicted (File: Interfering RNA.xlsx). Both sense and antisense RNAs from the precursor RNA could have been matured into functioning RNAi effector.
RNA interference is another important mechanism how KSHV regulates host gene expression. To predict underexpression of the host genes due to non coding RNA SAGE data available for KSHV was checked and the results showed that Angioprotein like, Claudin, Glutathione peroxidase, Heat shock protein, Ribonuclease, Solute carrier family member 6 are predicted to be downregulated in SAGE count and they were also predicted to have significant homology with interfering RNA sequence. This means they are probable targets for interfering RNA interference mediated silencing in KSHV (Supplementary File: Interfering RNA). The functions of the genes were collected from GeneCard database (Supplementary figure 16).
CCL3L3, CRADD, DDX5, HSP90B1, IGFBP5, RAB5C, TSKS, and RANB2-AS1 were also found to be possible hits as they were shown to be downregulated in microarray data. Gene targets that were only found through antisense RNA but was consistent with the predicted PPI, GRN and cellular network were NFKBIA, MAF, GEMIN8, RBL2 were found to be potential target molecules. The functions of the genes were collected from GeneCard database.

3.6. Analyzing the established network along with expression data and interfering RNA results predicts a dynamic gene e in lytic and latent phases
SAGE, microarray and RNAi data gives out dynamic data about the cellular protein concentration. It also provides information on status of the host and viral proteins in lytic and latent phases. It has been reported that the latency is mediated by LANA, vCyclin, vFLIP, v-miRNAs, and Kaposin proteins. They act to maintain viral episome and inactivation of the lytic genes.
Overexpressed and underexpressed genes are enlisted in Supplementary file- SAGE_Table.docx. The genes that were overexpressed and also reinforced by Microarray data and subsequently by RNA interference regulation data are- CCL3L3, CRADD, DDX5, HSP90B1, IGFBP5, RAB5C, TSKS, RANB2-AS1. On the contrary, the overexpressed genes are ANGPTL4, CLDN5, CXCL14, DARC, GPX3, HSPB1, MT2A, RNASE1, RPS17 and SLC2A6.
From Microarray data the genes could have been placed on 7 functional clusters (Supplementary Table 1, Figure 2). The microarray data shows that Cluster 1, 2, 3 and 4 (heat shock proteins, mostly transcription factor and related enzymes) are underexpressed in Lana expressed samples. LANA (Latency Associated Nuclear Antigen) is a negative modulator of host genes and hence a potent latency triggering protein. Cluster 5, 6 and 7 are overexpressed during LANA expression (mostly cell signaling proteins and tumor related proteins).
Cluster 2, 3 and 6 are mostly underexpressed in the initial phase of the Lytic cycle (Column 16, Figure 2). However, in the later period of the infection in lytic they are upregulated. Other clusters are more or less upregulated in the course of 3 months to 10 months.
On the other hand the generated heat map for the ORFs show that ORF 10 and ORF 11- previously held to be a potential hub- are highly expressed during lytic phase. ORF 27, 34, 49 52, 53 and 57 are also upregulated during lytic cycle.
1
2
3
4
5
6
7
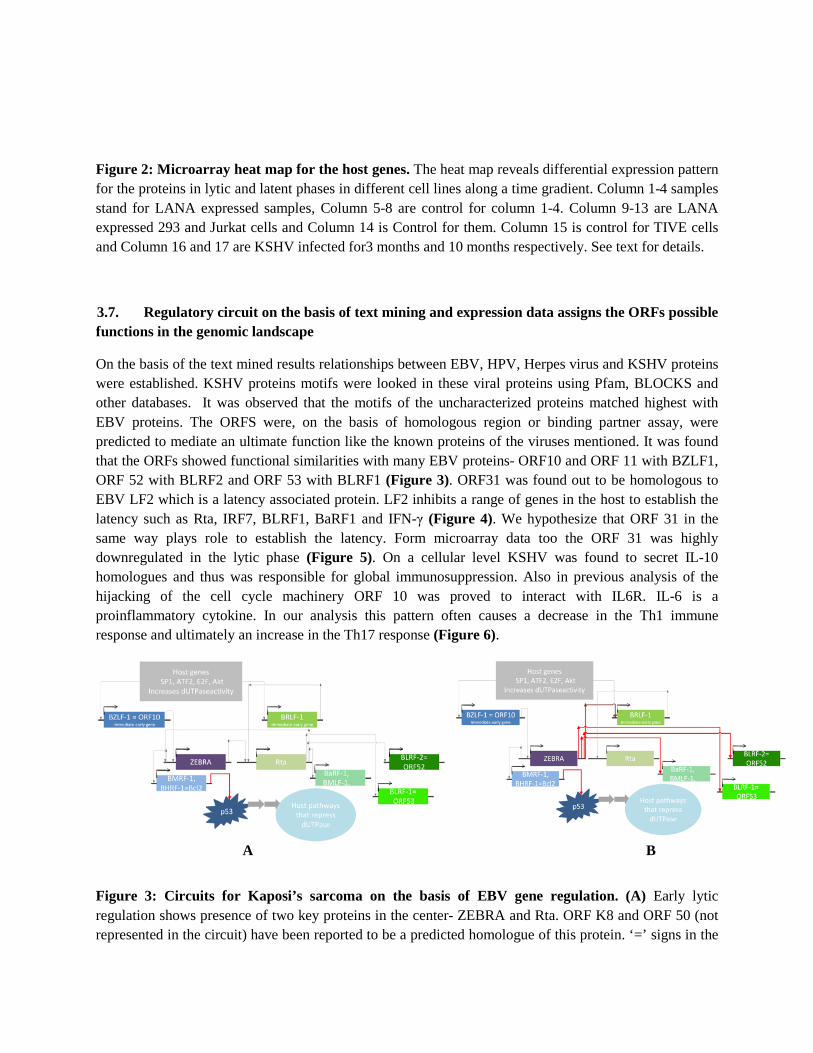
Figure 2: Microarray heat map for the host genes. The heat map reveals differential expression pattern for the proteins in lytic and latent phases in different cell lines along a time gradient. Column 1-4 samples stand for LANA expressed samples, Column 5-8 are control for column 1-4. Column 9-13 are LANA expressed 293 and Jurkat cells and Column 14 is Control for them. Column 15 is control for TIVE cells and Column 16 and 17 are KSHV infected for3 months and 10 months respectively. See text for details.
3.7. Regulatory circuit on the basis of text mining and expression data assigns the ORFs possible functions in the genomic landscape
On the basis of the text mined results relationships between EBV, HPV, Herpes virus and KSHV proteins were established. KSHV proteins motifs were looked in these viral proteins using Pfam, BLOCKS and other databases. It was observed that the motifs of the uncharacterized proteins matched highest with EBV proteins. The ORFS were, on the basis of homologous region or binding partner assay, were predicted to mediate an ultimate function like the known proteins of the viruses mentioned. It was found that the ORFs showed functional similarities with many EBV proteins- ORF10 and ORF 11 with BZLF1, ORF 52 with BLRF2 and ORF 53 with BLRF1 (Figure 3). ORF31 was found out to be homologous to EBV LF2 which is a latency associated protein. LF2 inhibits a range of genes in the host to establish the latency such as Rta, IRF7, BLRF1, BaRF1 and IFN-γ (Figure 4). We hypothesize that ORF 31 in the same way plays role to establish the latency. Form microarray data too the ORF 31 was highly downregulated in the lytic phase (Figure 5). On a cellular level KSHV was found to secret IL-10 homologues and thus was responsible for global immunosuppression. Also in previous analysis of the hijacking of the cell cycle machinery ORF 10 was proved to interact with IL6R. IL-6 is a proinflammatory cytokine. In our analysis this pattern often causes a decrease in the Th1 immune response and ultimately an increase in the Th17 response (Figure 6).
Figure 3: Circuits for Kaposi’s sarcoma on the basis of EBV gene regulation. (A) Early lytic regulation shows presence of two key proteins in the center- ZEBRA and Rta. ORF K8 and ORF 50 (not represented in the circuit) have been reported to be a predicted homologue of this protein. ‘=’ signs in the
A B

diagram equates Kapsosi’s Sarcoma virus genes with that of the EBV ones on the basis of function as a whole. It is not to be taken as a sign for indicating homologs. (B) Latent lytic gene circuity for the virus shows that the gene expression of Rta is negatively regulated. However, ZEBRA expression remains unaltered. See text for detail. Red lines stand for inhibition.
Figure 4: Expression of the viral ORFs. Column 1-8: Latent cycle (0 hour, 2 hour, 4 hour, 10 hour, 24 hour, 34 hour, 48 hour, 72 hour); Column 9-21 (0 hours+TPA, 2 hours+TPA, 4 hours+TPA, 10 hours+TPA, 24 hours+TPA, 24 hours+TPA (2), 34 hours+TPA, 34 hours+TPA (2), 48 hours+TPA, 48 hours+TPA (2), 72 hours+TPA, 72 hours+TPA(2)): tetradecanoyl phorbol acetate (TPA) induced Lytic cycle. From this image it can be viewed that ORF10 and ORF11 is highly expressed in lytic induction whereas ORF 31 is lowly induced in lytic phase.
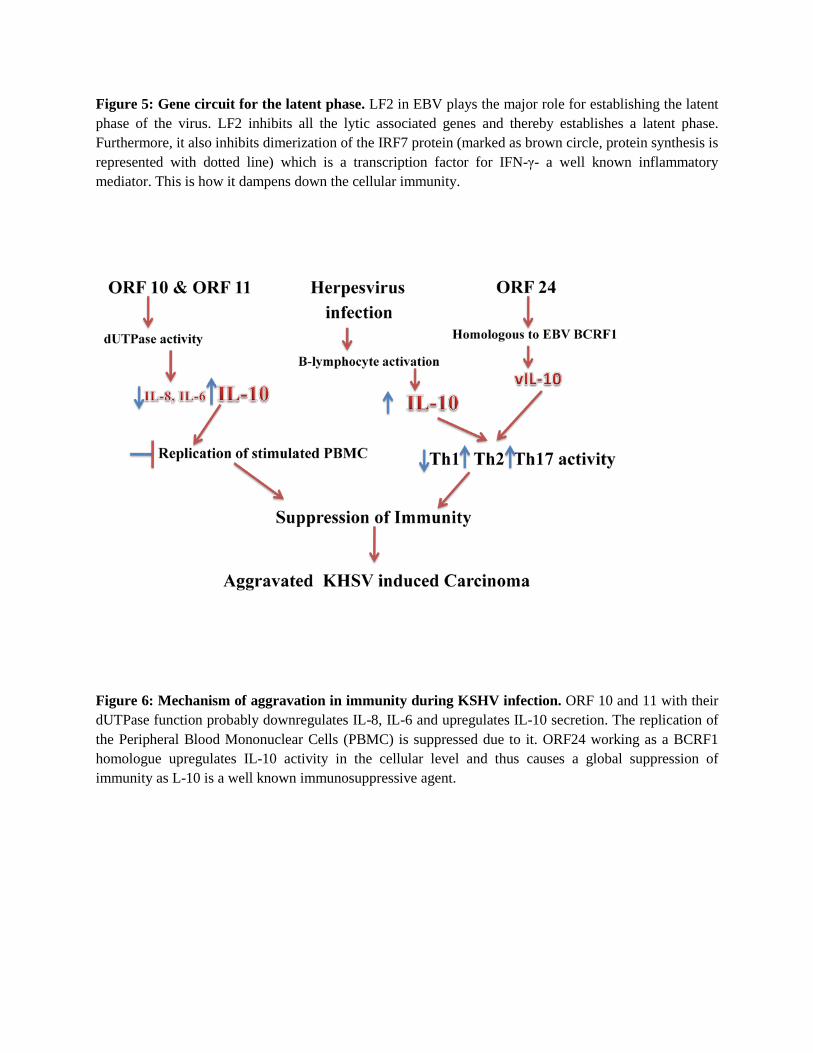
Figure 5: Gene circuit for the latent phase. LF2 in EBV plays the major role for establishing the latent phase of the virus. LF2 inhibits all the lytic associated genes and thereby establishes a latent phase. Furthermore, it also inhibits dimerization of the IRF7 protein (marked as brown circle, protein synthesis is represented with dotted line) which is a transcription factor for IFN-γ- a well known inflammatory mediator. This is how it dampens down the cellular immunity.
Figure 6: Mechanism of aggravation in immunity during KSHV infection. ORF 10 and 11 with their dUTPase function probably downregulates IL-8, IL-6 and upregulates IL-10 secretion. The replication of the Peripheral Blood Mononuclear Cells (PBMC) is suppressed due to it. ORF24 working as a BCRF1 homologue upregulates IL-10 activity in the cellular level and thus causes a global suppression of immunity as L-10 is a well known immunosuppressive agent.

4. Discussion
The experiment was designed to identify a protein protein interaction (PPI) network for differentially expressed host genes and uncharacterized viral ORFs in KS. Subsequently the two networks were merged to produce an integrated network. A gene regulatory and cellular network was also produced using information that came out from the PPI study as well as using ORF homology with EBV sequences. But before all this could have been performed the ORFs of interest needed to be characterized. The characterization revealed novel information regarding them. It was found that the uncharacterized ORFs belong to a wide array of types; types as varying as structural proteins, membrane bound proteins, cytosolic enzymes, transcription factors etc.
ORF 24 was predicted to be a membrane bound protein with ATP and carbohydrate binding potential and ORF 30 had transcription factor activity which probably leads to an apoptosis dysregulation in the host cell (Supplementary file: ORF_characterization). From STRING results ORF 30 also appeared to have oxidoreductase activity. Keeping both the results in mind, we predict that the protein probably plays dual role- it catalyzes a redox reaction and also takes part in changing host transcription in favor of the virus. ORF 30 possesses a CARD (Caspase Recruitment Domain) like domain. CARD proteins have been shown to participate in recognition of intracellular double-stranded RNA, a common constituent of a number of viral genomes. RIG-I (retionic acid-inducible gene) proteins encoded from DDX gene has CARD motif as its integral part [74, 75]. Differential expression of DDX and RIG-I is apparent from the SAGE and microaaray data mentioned in this manuscript. From our results we assume that ORF 30 might work as a viral form of RIG-I that acts as a molecular competitor for cellular RIG-I causing apoptosis dysregulation. The interferring RNA prediction results also correlated well with the findings; a predicted antisense RNA was found against DDX.
The results also indicate that ORF10 and 11, with its dUTPase function, triggers viral lytic cycle. According to published literatures dUTPases are known to downregulate a ligand for the NK activating receptor [25]. Earlier it has been reported that Matrix Metalloproteinase-9 expression is redox sensitive [76, 77] and MMP-9 downregulation dampens down Nuclear Factor B Signaling Pathway in carcinoma cells. The viral ORF, by the virtue of its oxidoreductase activity, may be able to shift the cellular balance towards activation of MMP-9. The microarray data suggests that MMP 2 is significantly upregulated during KSHV infection; from this information it was assumed that the virus infection also alters MMP 9 as MMP 2 and 9 both are functionally closely related to each other as they both have collegenase activity. There were also reports that vanillin inhibits MMP 9 [78]. The fact that the protein sequence, although marginally, had homology with vanillin dehydrogenase seems to be reinforcing our assumption (Supplementary file: ORF_characterization).
ORF24 was predicted to interact with ADAMTS5 (Figure 1). ADAMTS5 cleaves aggrecan- a cartilage proteoglycan, and could be involved in its turnover. ADAMTS5 plays an important role in the destruction of aggrecan which may lead to arthritic diseases [79-81]. ORF24-ADAMTS5 interaction reveals how arthritis is often associated with Kaposi’s sarcoma. Apoptosis dysregulation is another key molecular indicator for arthritis. The study predicts ORF30 to be responsible for this. It is to be noted that apoptosis dysregulation is one basic signaling slip-up that pushes the cell towards a tumorigenic course [82, 83].
ORF27 was predicted to be an extracellular catalytic enzyme (Supplementary file: ORF_characterization). On the other hand ORF53 was found to be an envelope glycoprotein with

transcription factor activity (Supplementary file: ORF_characterization.docx). Additionally, according to STRING results ORF 27 is required for vacuolar transport. The results indicate that they are associated with host CLTC or clathrin heavy chain protein (Figure 1). CTLC is required for TNF mediated inflammatory response and from the findings ORF27 and host interaction may explain how inflammatory response in KS is downregulated [84].
In our analysis ORF 42 was predicted to be a transcription factor with lytic transglycosylase activity (Supplementary file: ORF_characterization.docx). Lytic transglycosylase are reported to be needed for degrading murein and are structurally rich in alpha helices. The viral ORF was also rich in alpha helices [85, 86].
ORF 34 was predicted to be a DNA binding tegument protein; they aid the virus in DNA replication. ORF 34 and 42 are closely linked to response regulators transcription factors, recO DNA repair system and methylating enzymes (Supplementary file: ORF_characterization.docx). Through interaction with these two predicted interactors they interact with human protein RAD51 and thereby participate in a common DNA damage response pathway associated with activation of homologous recombination and double-strand break repair (Figure 1). RAD51 binds to single and double stranded DNA and exhibits DNA-dependent ATPase activity and unwinds duplex DNA to forms helical nucleoprotein filaments. It has been reported that RAD51 plays important role in viral clearance [87].
ORF 33 was also predicted to be a tegument protein with enzymatic activity. ORF 52 is probably another tegument protein that initiates the lytic cycle by triggering transcription of the viral genes (Supplementary file: ORF_characterization.docx). They associate with the hosts oxidative stress recognizing system that includes proteins RSAD1 (radical S-adenosyl methionine domain containing 1), HSPs (Heat Shock Proteins), Succinyl transferase etc.
ORF49, ORF10 and ORF11 plays role during lytic switch on through their dUTPase (Figure 1). Their host interactor, PCNA protein, is an auxiliary protein of DNA polymerase delta and is involved in the control of eukaryotic DNA replication by increasing the polymerase's processibility during elongation of the leading strand. PCNA has been earlier reported to interact with other viral proteins [88-92].
In EBV the lytic cycle is governed by two important hub proteins- ZEBRA and Rta [93-98]. In KSHV ORF K8 and ORF 50 plays the similar role. Here we report, previously uncharacterized ORF10 of KSHV as a potent lytic inducer. ORF31 on the other hand probably is an inducer of latent phase (Figure 1). The analysis also reveals that ORF 55 and 35 probably work in more or less in the same pathway as ORF 10 and 11 did. Their host interactors include Werner helicase interacting protein 1 which functions as a modulator for initiation or reinitiating events during DNA polymerase delta-mediated DNA synthesis [99, 100]. They ultimately interact with PCNA the same way ORF 10 and 11 does.
ORF 31 and ORF35 interacts with host protein Cadherin- a tumor suppressor gene. It was therefore assumed that it causes a repression in its tumor suppressing ability and consequently increases the metastatic potential of the cells during KSHV infection [101, 102] (Figure 1).
A methodology was developed for characterization of the capsid proteins. Generally membrane binding proteins are located outside the cell and do not enter the cell and hence do not show any DNA binding ability. However, in case of few ORFs (e.g. ORF 23) the protein showed both DNA binding potential as

well as membrane localization (Supplementary Figure 9). After further studies they were predicted to be capsid protein. Because the genome has dsDNA the capsid proteins showed a membrane binding as well as a DNA binding potential. This assumption was validated by screening known capsid proteins (HPV GenBank: AAX54678.1) in the same way- the results show both membrane binding and DNA binding potential.
RNA interference is another important mechanism how KSHV regulates host gene expression. To predict underexpression of the host genes due to non coding RNA SAGE and microarray data available for KSHV was checked. The results showed that Angioprotein like, Claudin, Glutathione peroxidase, Heat shock protein, Ribonuclease, Solute carrier family member 6, CCL3L3, CRADD, DDX5, HSP90B1, IGFBP5, RAB5C, TSKS, RANB2-AS1, NFKBIA, MAF, GEMIN8 and RBL2 were also found to be possible hits as they were shown to be downregulated in microarray data The functions of the genes were collected from GeneCard database (Supplementary Table 7).
The association of Angioprotein like, IGFBP3 explains the molecular basis why diabetes is a common risk factor for KS [103, 104]. On the other hand Caludin, Glutathione peroxidase, Heat shock protein, Ribonuclease and Solute carrier family member 6 downregulation indicates a metabolic shift that favors viral infection and global cellular stress causing an increase in cellular oncogenic potential [105-108]. CCL3, TSKS, RAB5C, NFKB1, MAF, GEMIN8, RBL2 downregulation, on the other hand, are known to be associated to tumor progression [109-116].
Apart from these, the KSHV genome has several hijacked gene of host origin within its large genome of 165 kb. For example it possesses IL-6, BCL-2, cyclin-D, IRF, vMIP, FLIP etc proteins. In earlier studies knocking down vIRF-3 expression resulted in increased activity of caspase-3 and/or caspase-7 and hence caused viral efficiency of infection to drop significantly [117]. Our gene regulatory network based on homology information also showed that IRF-7 dimerization is hampered by viral IRF (Figure 4). In another earlier study it was suggested that human IL-6 plays an important role in the pathogenesis of HHV-8-associated multicentric Castleman disease (MCD). However, it was also reported that vIL-6 is not absolutely necessary for lymphatic reprogramming during viral infection [118]. In our study we have showed that cellular effector role of IL6 is decreased and the virus uses its encoded Il-6 to cause so (Figure 6). KSHV also encodes a number of different chemokine homologues such as viral macrophage inflammatory protein or alternatively known as vMIP [119]. vMIP-I has been earlier reported as a specific agonist for the CC chemokine receptor (CCR)8 [119]. This receptor accelerates a Th2 immune response. In our cellular network this relationship has been incorporated (Figure 6). In another article it was reported that viral FADD-like interleukin-1-beta-converting enzyme or FLICE/caspase 8-inhibitory protein (FLIP) activates NF-kappaB more potently than cellular FLIP [116]. Besides, KSHV encodes K-cyclin. The level of K-cyclin, a homologue of cellular D cyclin, is expressed constitutively throughout the cell cycle and thus breaches the boundaries among cell cycle phases and [120].
In biological networks small world property applies and thus checking robustness of the network is of great importance. Several standard parameters of the network had to be checked in a bid to ensure that the network can withstand destabilizing forces. For example, a weak host network would mean that due to false prediction of one ORF host interaction the other interactions may also get affected. The network was found to be robust with high clustering coefficient. The large sample size and reproducible results

obtained through multiple bioinformatic approaches gives us good confidence to claim the propriety of the results as a whole.

References
1. Kempf, W. and V. Adams, Viruses in the pathogenesis of Kaposi's sarcoma--a review. Biochem Mol Med, 1996. 58(1): p. 1-12.
2. Armes, J., A review of Kaposi's sarcoma. Adv Cancer Res, 1989. 53: p. 73-87. 3. Zurrida, S., et al., Classic Kaposi's sarcoma: a review of 90 cases. J Dermatol, 1992. 19(9): p. 548-
52. 4. Safai, B., Kaposi's sarcoma: a review of the classical and epidemic forms. Ann N Y Acad Sci, 1984.
437: p. 373-82. 5. Akasbi, Y., et al., Non-HIV Kaposi's sarcoma: a review and therapeutic perspectives. Bull Cancer. 6. Mitsuyasu, R.T., AIDS-related Kaposi's sarcoma: a review of its pathogenesis and treatment.
Blood Rev, 1988. 2(4): p. 222-31. 7. Stein, M., et al., AIDS-related Kaposi's sarcoma: a review. Isr J Med Sci, 1994. 30(4): p. 298-305. 8. Febrer Bosch, M.I., et al., [Kaposi's sarcoma associated with human immunodeficiency virus
infection: a clinical, histological, immunohistochemical, serological and development review]. Med Clin (Barc), 1991. 96(16): p. 601-6.
9. Ramirez-Amador, V., G. Anaya-Saavedra, and G. Martinez-Mata, Kaposi's sarcoma of the head and neck: a review. Oral Oncol. 46(3): p. 135-45.
10. Bahia, F. and C. Brites, Human Herpes Virus 8 and Kaposi's Sarcoma: A Review. Braz J Infect Dis, 1999. 3(5): p. 166-175.
11. Hunt, R.D. and L.V. Melendez, Herpes virus infections of non-human primates: a review. Lab Anim Care, 1969. 19(2): p. 221-34.
12. Finelli, P.F., Herpes simplex virus and the human nervous system: current concepts and review. Mil Med, 1975. 140(11): p. 765-71.
13. Sarid, R., et al., Characterization and cell cycle regulation of the major Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) latent genes and their promoter. J Virol, 1999. 73(2): p. 1438-46.
14. Sadagopan, S., et al., Kaposi's sarcoma-associated herpesvirus-induced angiogenin plays roles in latency via the phospholipase C gamma pathway: blocking angiogenin inhibits latent gene expression and induces the lytic cycle. J Virol. 85(6): p. 2666-85.
15. Belopasova, L.A. and Z.V. Markina, [Kaposi's sarcoma associated with acute leukemia]. Klin Med (Mosk), 1975. 53(6): p. 117-8.
16. Babini, G., [A case of Kaposi's disease associated with chronic lymphatic leukemia]. Arch Ital Dermatol Venereol Sessuol, 1966. 34(6): p. 450-62.
17. Dotz, W.I. and B. Berman, Kaposi's sarcoma, chronic ulcerative herpes simplex, and acquired immunodeficiency. Arch Dermatol, 1983. 119(1): p. 93-4.
18. Wang, L., et al., Identification and functional characterization of a spliced rhesus rhadinovirus gene with homology to the K15 gene of Kaposi's sarcoma-associated herpesvirus. J Gen Virol, 2009. 90(Pt 5): p. 1190-201.
19. Glenn, M., et al., Identification of a spliced gene from Kaposi's sarcoma-associated herpesvirus encoding a protein with similarities to latent membrane proteins 1 and 2A of Epstein-Barr virus. J Virol, 1999. 73(8): p. 6953-63.
20. Rossetto, C.C. and G. Pari, KSHV PAN RNA associates with demethylases UTX and JMJD3 to activate lytic replication through a physical interaction with the virus genome. PLoS Pathog. 8(5): p. e1002680.

21. Jackson, B.R., et al., An interaction between KSHV ORF57 and UIF provides mRNA-adaptor redundancy in herpesvirus intronless mRNA export. PLoS Pathog. 7(7): p. e1002138.
22. Seifi, A., et al., The lytic activation of KSHV during keratinocyte differentiation is dependent upon a suprabasal position, the loss of integrin engagement, and calcium, but not the interaction of cadherins. Virology. 410(1): p. 17-29.
23. Papugani, A., et al., The interaction between KSHV RTA and cellular RBP-Jkappa and their subsequent DNA binding are not sufficient for activation of RBP-Jkappa. Virus Res, 2008. 131(1): p. 1-7.
24. Liang, Y., et al., The lytic switch protein of KSHV activates gene expression via functional interaction with RBP-Jkappa (CSL), the target of the Notch signaling pathway. Genes Dev, 2002. 16(15): p. 1977-89.
25. Madrid, A.S. and D. Ganem, Kaposi's sarcoma-associated herpesvirus ORF54/dUTPase downregulates a ligand for the NK activating receptor NKp44. J Virol. 86(16): p. 8693-704.
26. Chen, R., H. Wang, and L.M. Mansky, Roles of uracil-DNA glycosylase and dUTPase in virus replication. J Gen Virol, 2002. 83(Pt 10): p. 2339-45.
27. Fleischmann, J., et al., Expression of viral and human dUTPase in Epstein-Barr virus-associated diseases. J Med Virol, 2002. 68(4): p. 568-73.
28. Tategu, M., et al., Systems biology-based identification of crosstalk between E2F transcription factors and the Fanconi anemia pathway. Gene Regul Syst Bio, 2007. 1: p. 1-8.
29. Westerhoff, H.V., Systems biology: new paradigms for cell biology and drug design. Ernst Schering Res Found Workshop, 2007(61): p. 45-67.
30. Stumpf, M.P., et al., Systems biology and its impact on anti-infective drug development. Prog Drug Res, 2007. 64: p. 1, 3-20.
31. Kohn, K.W., et al., Molecular interaction maps of bioregulatory networks: a general rubric for systems biology. Mol Biol Cell, 2006. 17(1): p. 1-13.
32. Bader, S., S. Kuhner, and A.C. Gavin, Interaction networks for systems biology. FEBS Lett, 2008. 582(8): p. 1220-4.
33. Damm, E.M. and L. Pelkmans, Systems biology of virus entry in mammalian cells. Cell Microbiol, 2006. 8(8): p. 1219-27.
34. Pelkmans, L., Systems biology of virus infection in mammalian cells. Curr Opin Microbiol, 2009. 12(4): p. 429-31.
35. Peng, X., et al., Virus-host interactions: from systems biology to translational research. Curr Opin Microbiol, 2009. 12(4): p. 432-8.
36. Silver, P.A. and J.C. Way, Molecular systems biology in drug development. Clin Pharmacol Ther, 2007. 82(5): p. 586-90.
37. Chen, B.S., et al., A systems biology approach to construct the gene regulatory network of systemic inflammation via microarray and databases mining. BMC Med Genomics, 2008. 1: p. 46.
38. Green, M., Adenoviruses-model systems of virus replication, human cell molecular biology, and neoplastic transformation. Perspect Biol Med, 1978. 21(3): p. 373-97.
39. Xue, Q. and K. Miller-Jensen, Systems biology of virus-host signaling network interactions. BMB Rep. 45(4): p. 213-20.
40. Si, H., S.C. Verma, and E.S. Robertson, Proteomic analysis of the Kaposi's sarcoma-associated herpesvirus terminal repeat element binding proteins. J Virol, 2006. 80(18): p. 9017-30.
41. Peri, S., et al., Development of human protein reference database as an initial platform for approaching systems biology in humans. Genome Res, 2003. 13(10): p. 2363-71.
42. Lehner, B. and A.G. Fraser, A first-draft human protein-interaction map. Genome Biol, 2004. 5(9): p. R63.

43. Matthews, L.R., et al., Identification of potential interaction networks using sequence-based searches for conserved protein-protein interactions or interologs . Genome Res, 2001. 11(12): p. 2120-6.
44. Lan, K., D.A. Kuppers, and E.S. Robertson, Kaposi's sarcoma-associated herpesvirus reactivation is regulated by interaction of latency-associated nuclear antigen with recombination signal sequence-binding protein Jkappa, the major downstream effector of the Notch signaling pathway. J Virol, 2005. 79(6): p. 3468-78.
45. Valiya Veettil, M., et al., Interaction of c-Cbl with myosin IIA regulates Bleb associated macropinocytosis of Kaposi's sarcoma-associated herpesvirus. PLoS Pathog. 6(12): p. e1001238.
46. Villa, M.L. and E. Bombardieri, LYDMA-antigens and immunity against EBV-infected cells Epstein-Barr virus as a model for the study of host-infection interaction. Int J Biol Markers, 1987. 2(2): p. 125-32.
47. Rundell, B.B. and R.F. Betts, Interaction of cytomegalovirus immune complexes with host cells. Infect Immun, 1981. 33(3): p. 658-65.
48. Vanover, J., et al., Interaction of herpes simplex virus type 2 (HSV-2) glycoprotein D with the host cell surface is sufficient to induce Chlamydia trachomatis persistence. Microbiology. 156(Pt 5): p. 1294-302.
49. Ferreira, J.A., The Benjamini-Hochberg method in the case of discrete test statistics. Int J Biostat, 2007. 3(1): p. Article 11.
50. Diehn, M., et al., Large-scale identification of secreted and membrane-associated gene products using DNA microarrays. Nat Genet, 2000. 25(1): p. 58-62.
51. Szklarczyk, D., et al., The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 39(Database issue): p. D561-8.
52. Bader, G.D., D. Betel, and C.W. Hogue, BIND: the Biomolecular Interaction Network Database. Nucleic Acids Res, 2003. 31(1): p. 248-50.
53. Bhagwat, M. and L. Aravind, PSI-BLAST tutorial. Methods Mol Biol, 2007. 395: p. 177-86. 54. Jeudy, S., et al., Dissecting the unique nucleotide specificity of mimivirus nucleoside diphosphate
kinase. J Virol, 2009. 83(14): p. 7142-50. 55. Lambeth, D.O., et al., Characterization and cloning of a nucleoside-diphosphate kinase targeted
to matrix of mitochondria in pigeon. J Biol Chem, 1997. 272(39): p. 24604-11. 56. Morales, M., et al., Genome comparison of a nonpathogenic myxoma virus field strain with its
ancestor, the virulent Lausanne strain. J Virol, 2009. 83(5): p. 2397-403. 57. Zhang, Y., et al., Chlorella virus-encoded deoxyuridine triphosphatases exhibit different
temperature optima. J Virol, 2005. 79(15): p. 9945-53. 58. Upton, C., et al., Myxoma virus expresses a secreted protein with homology to the tumor
necrosis factor receptor gene family that contributes to viral virulence. Virology, 1991. 184(1): p. 370-82.
59. Schneiker, S., et al., Complete genome sequence of the myxobacterium Sorangium cellulosum. Nat Biotechnol, 2007. 25(11): p. 1281-9.
60. Geisbrecht, B.V., et al., The crystal structures of EAP domains from Staphylococcus aureus reveal an unexpected homology to bacterial superantigens. J Biol Chem, 2005. 280(17): p. 17243-50.
61. Crouse, C.A. and R.J. Pauley, Molecular cloning and sequencing of the MTV-1 LTR: evidence for a LTR sequence alteration. Virus Res, 1989. 12(2): p. 123-37.
62. Pullen, A.M., et al., The open reading frames in the 3' long terminal repeats of several mouse mammary tumor virus integrants encode V beta 3-specific superantigens. J Exp Med, 1992. 175(1): p. 41-7.
63. Guan, K.L. and J.E. Dixon, Bacterial and viral protein tyrosine phosphatases. Semin Cell Biol, 1993. 4(6): p. 389-96.

64. Woellmer, A., J.M. Arteaga-Salas, and W. Hammerschmidt, BZLF1 Governs CpG-Methylated Chromatin of Epstein-Barr Virus Reversing Epigenetic Repression. PLoS Pathog. 8(9): p. e1002902.
65. Ma, S.D., et al., An Epstein-Barr Virus (EBV) mutant with enhanced BZLF1 expression causes lymphomas with abortive lytic EBV infection in a humanized mouse model. J Virol. 86(15): p. 7976-87.
66. Kalla, M., et al., AP-1 homolog BZLF1 of Epstein-Barr virus has two essential functions dependent on the epigenetic state of the viral genome. Proc Natl Acad Sci U S A. 107(2): p. 850-5.
67. Adamson, A.L., N. Wright, and D.R. LaJeunesse, Modeling early Epstein-Barr virus infection in Drosophila melanogaster: the BZLF1 protein. Genetics, 2005. 171(3): p. 1125-35.
68. Lee, B.S., et al., Suppression of tetradecanoyl phorbol acetate-induced lytic reactivation of Kaposi's sarcoma-associated herpesvirus by K1 signal transduction. J Virol, 2002. 76(23): p. 12185-99.
69. Chang, P.J., Y.S. Chang, and S.T. Liu, Role of Rta in the translation of bicistronic BZLF1 of Epstein-Barr virus. J Virol, 1998. 72(6): p. 5128-36.
70. Sun, R., et al., Kinetics of Kaposi's sarcoma-associated herpesvirus gene expression. J Virol, 1999. 73(3): p. 2232-42.
71. Bechtel, J.T., R.C. Winant, and D. Ganem, Host and viral proteins in the virion of Kaposi's sarcoma-associated herpesvirus. J Virol, 2005. 79(8): p. 4952-64.
72. Zhu, F.X., et al., Virion proteins of Kaposi's sarcoma-associated herpesvirus. J Virol, 2005. 79(2): p. 800-11.
73. Calderwood, M.A., A.M. Holthaus, and E. Johannsen, The Epstein-Barr virus LF2 protein inhibits viral replication. J Virol, 2008. 82(17): p. 8509-19.
74. Palacios-Rodriguez, Y., et al., Polypeptide modulators of caspase recruitment domain (CARD)-CARD-mediated protein-protein interactions. J Biol Chem. 286(52): p. 44457-66.
75. Hofmann, K., P. Bucher, and J. Tschopp, The CARD domain: a new apoptotic signalling motif. Trends Biochem Sci, 1997. 22(5): p. 155-6.
76. Gargiulo, S., et al., Plaque oxysterols induce unbalanced up-regulation of matrix metalloproteinase-9 in macrophagic cells through redox-sensitive signaling pathways: Implications regarding the vulnerability of atherosclerotic lesions. Free Radic Biol Med. 51(4): p. 844-55.
77. Buhimschi, I.A., et al., Reduction-oxidation (redox) state regulation of matrix metalloproteinase activity in human fetal membranes. Am J Obstet Gynecol, 2000. 182(2): p. 458-64.
78. Liang, J.A., et al., Vanillin inhibits matrix metalloproteinase-9 expression through down-regulation of nuclear factor-kappaB signaling pathway in human hepatocellular carcinoma cells. Mol Pharmacol, 2009. 75(1): p. 151-7.
79. Durigova, M., et al., MMPs are less efficient than ADAMTS5 in cleaving aggrecan core protein. Matrix Biol. 30(2): p. 145-53.
80. Casoli, P. and B. Tumiati, Kaposi's sarcoma, rheumatoid arthritis and immunosuppressive and/or corticosteroid therapy. J Rheumatol, 1992. 19(8): p. 1316-7.
81. Schottstaedt, M.W., E.R. Hurd, and M.J. Stone, Kaposi's sarcoma in rheumatoid arthritis. Am J Med, 1987. 82(5): p. 1021-6.
82. Stanton, H., et al., ADAMTS5 is the major aggrecanase in mouse cartilage in vivo and in vitro. Nature, 2005. 434(7033): p. 648-52.
83. McCulloch, D.R., et al., Adamts5, the gene encoding a proteoglycan-degrading metalloprotease, is expressed by specific cell lineages during mouse embryonic development and in adult tissues. Gene Expr Patterns, 2009. 9(5): p. 314-23.

84. Escobar, G.A., et al., Clathrin heavy chain is required for TNF-induced inflammatory signaling. Surgery, 2006. 140(2): p. 268-72.
85. Yu, Y.C., et al., A putative lytic transglycosylase tightly regulated and critical for the EHEC type three secretion. J Biomed Sci. 17: p. 52.
86. Dijkstra, B.W. and A.M. Thunnissen, 'Holy' proteins. II: The soluble lytic transglycosylase. Curr Opin Struct Biol, 1994. 4(6): p. 810-3.
87. Pasaje, C.F., et al., Lack of association of RAD51 genetic variations with hepatitis B virus clearance and occurrence of hepatocellular carcinoma in a Korean population. J Med Virol. 83(11): p. 1892-9.
88. Bagewadi, B., et al., PCNA interacts with Indian mung bean yellow mosaic virus rep and downregulates Rep activity. J Virol, 2004. 78(21): p. 11890-903.
89. Ehrmann, J., Jr., et al., Apoptosis-related proteins, BCL-2, BAX, FAS, FAS-L and PCNA in liver biopsies of patients with chronic hepatitis B virus infection. Pathol Oncol Res, 2000. 6(2): p. 130-5.
90. Jin, Z.Y., G.R. Qi, and C.D. Lu, Expression of Anti-PCNA mRNA Ribozyme by T7 Vaccinia Virus System in HeLa Cells. Sheng Wu Hua Xue Yu Sheng Wu Wu Li Xue Bao (Shanghai), 1997. 29(1): p. 107-109.
91. Kanavaros, P., et al., Mycosis fungoides: expression of C-myc p62 p53, bcl-2 and PCNA proteins and absence of association with Epstein-Barr virus. Pathol Res Pract, 1994. 190(8): p. 767-74.
92. Luzzatto, R., M.M. Bosch, and M.E. Boon, Proliferating cell nuclear antigen (PCNA) in herpes virus infected cells. Diagn Cytopathol, 1994. 10(3): p. 304.
93. Dreyfus, D.H., et al., Analysis of an ankyrin-like region in Epstein Barr Virus encoded (EBV) BZLF-1 (ZEBRA) protein: implications for interactions with NF-kappaB and p53. Virol J. 8: p. 422.
94. Chen, L., et al., Induction of Epstein-Barr virus lytic replication by recombinant adenoviruses expressing the zebra gene with EBV specific promoters. Acta Biochim Biophys Sin (Shanghai), 2005. 37(4): p. 215-20.
95. Dreyfus, D.H., et al., Inactivation of NF-kappaB by EBV BZLF-1-encoded ZEBRA protein in human T cells. J Immunol, 1999. 163(11): p. 6261-8.
96. Drouet, E., et al., High Epstein-Barr virus serum load and elevated titers of anti-ZEBRA antibodies in patients with EBV-harboring tumor cells of Hodgkin's disease. J Med Virol, 1999. 57(4): p. 383-9.
97. Chen, Y.J., et al., Epstein-Barr virus (EBV) Rta-mediated EBV and Kaposi's sarcoma-associated herpesvirus lytic reactivations in 293 cells. PLoS One. 6(3): p. e17809.
98. Pepperl, S., et al., Immediate-early transactivator Rta of Epstein-Barr virus (EBV) shows multiple epitopes recognized by EBV-specific cytotoxic T lymphocytes. J Virol, 1998. 72(11): p. 8644-9.
99. Bish, R.A. and M.P. Myers, Werner helicase-interacting protein 1 binds polyubiquitin via its zinc finger domain. J Biol Chem, 2007. 282(32): p. 23184-93.
100. Tsurimoto, T., et al., Human Werner helicase interacting protein 1 (WRNIP1) functions as a novel modulator for DNA polymerase delta. Genes Cells, 2005. 10(1): p. 13-22.
101. Nijkamp, M.M., et al., Expression of E-cadherin and vimentin correlates with metastasis formation in head and neck squamous cell carcinoma patients. Radiother Oncol. 99(3): p. 344-8.
102. Nguyen, P.T., et al., N-cadherin expression is correlated with metastasis of spindle cell carcinoma of head and neck region. J Oral Pathol Med. 40(1): p. 77-82.
103. Gori, A. Reversible Diabetes in Patient with AIDS-related Kaposi’s Sarcoma Treated with Interferon $alpha;-2a. The Lancet, 1995, 345 (8962): 1438–1439.
104. Ronchese F, Kern AB. 1953. KAposi’s Sarcoma and Diabetes Mellitus. Archives of Dermatology 67 (1): 95–96.

105. Shang, Xiying, et al. Tight Junction Proteins Claudin-3 and Claudin-4 Control Tumor Growth and Metastases. Neoplasia, 2012 14 (10): 974–985.
106. Schneider, et al. Absence of Glutathione Peroxidase 4 Affects Tumor Angiogenesis Through Increased 12/15-lipoxygenase Activity. Neoplasia, 2010 12 (3): 254–263.
107. Glaunsinger, Britt, and Don Ganem. Highly Selective Escape from KSHV-mediated Host mRNA Shutoff and Its Implications for Viral Pathogenesis. The Journal of Experimental Medicine, 2004 200 (3): 391–398.
108. Glaunsinger, Britt, and Don Ganem. Lytic KSHV Infection Inhibits Host Gene Expression by Accelerating Global mRNA Turnover. Molecular Cell, 2004 13 (5): 713–723.
109. Barcelos, Lucíola S. et al., Role of the Chemokines CCL3/MIP-1α and CCL5/RANTES in Sponge-induced Inflammatory Angiogenesis in Mice. Microvascular Research, 2009 78 (2): 148–154.
110. Scorilas, Andreas, et al. Identification and Characterization of a Novel Human Testis-Specific Kinase Substrate Gene Which Is Downregulated in Testicular Tumors. Biochemical and Biophysical Research Communications, 2001 285 (2): 400–408.
111. Onodera, Yasuhito, et al. Rab5c Promotes AMAP1–PRKD2 Complex Formation to Enhance Β1 Integrin Recycling in EGF-induced Cancer Invasion. The Journal of Cell Biology, 2012 197 (7): 983–996.
112. Sharma, H W, et al. Transcription Factor Decoy Approach to Decipher the Role of NF-kappa B in Oncogenesis. Anticancer Research, 1996 16 (1): 61–69.
113. Landi, Stefano, et al. Association of Common Polymorphisms in Inflammatory Genes Interleukin (IL)6, IL8, Tumor Necrosis Factor Α, NFKB1, and Peroxisome Proliferator-activated Receptor γ with Colorectal Cancer. Cancer Research, 2003 63 (13): 3560–3566.
114. Hurt, Elaine M, et al. Overexpression of C-maf Is a Frequent Oncogenic Event in Multiple Myeloma That Promotes Proliferation and Pathological Interactions with Bone Marrow Stroma. Cancer Cell, 2004 5 (2) 191–199.
115. Suzuki, Takeshi, et al. Tumor Suppressor Gene Identification Using Retroviral Insertional Mutagenesis in Blm-deficient Mice. The EMBO Journal, 2006 25 (14): 3422–3431.
116. Zaman A., Characterization of Coilin protein nucleotide binding region in Homo sapiens, Online J Bioinform, 2012 13(2):314-330, 2012.
117. Morris, Valerie A, et al. The KSHV Viral IL-6 Homolog Is Sufficient to Induce Blood to Lymphatic Endothelial Cell Differentiation. Virology, 2012 428 (2): 112–120.
118. Endres, M J, et al. The Kaposi’s Sarcoma-related Herpesvirus (KSHV)-encoded Chemokine vMIP-I Is a Specific Agonist for the CC Chemokine Receptor (CCR)8. The Journal of Experimental Medicine 1999 189 (12): 1993–1998.
119. Guasparri, Ilaria, et al. KSHV vFLIP Is Essential for the Survival of Infected Lymphoma Cells. The Journal of Experimental Medicine 2004 199 (7): 993–1003.
120. Van Drosset, et al. Constitutively Active K-cyclin/cdk6 Kinase in Kaposi Sarcoma-associated Herpesvirus-infected Cells Journal of the National Cancer Institute 2005 97 (9): 656–666.





























