Embed Size (px)
Citation preview

Dual Photoredox/HAT Chemistry in Carbohydrate Functionalization
by
Nicholas Rosano
A thesis submitted in conformity with the requirements for the degree of Master of Science
Department of Chemistry University of Toronto
© Copyright by Nicholas Rosano 2021

ii
Dual Photoredox/HAT Chemistry in Carbohydrate Functionalization
Nicholas Rosano
Master of Science
Department of Chemistry University of Toronto
2021
Abstract
Chemical synthesis of carbohydrate derivatives has attracted much attention due to their
prevalence in natural products. The methods for the derivatization of carbohydrate monomers
are, however, still mainly focused on the functionalization of hydroxyl groups rather than the
carbon-backbone. This thesis describes the exploration of a method for the regioselective C-H
alkylation of the anomeric position of 1,2-unprotected carbohydrate diols using hydrogen-atom
transfer (HAT)/photoredox chemistry and borinic acid catalysis. Initial experiments yielded
mixtures of alkylated products, culminating in the investigation of reaction kinetics and stability
of radical intermediates by computational analysis. A second research direction aimed at the
transformation of 2-O-acylated pyranosides to 3-keto-2-deoxypyranosides using another
HAT/photoredox system is also described. These products are suggested to arise by hydrogen-
atom abstraction at the C3-position of the pyranoside, followed by C2-O bond cleavage via spin-
center shift. Compatibility with variously configured mono-acylated substrates suggest wide
applicability for the preparation of rare, deoxygenated carbohydrates.

iii
Acknowledgments
Throughout the writing of this thesis, I have received a great deal of support and assistance. I
would like to thank Professor Mark S. Taylor, the Taylor group, family, and my friends for
making this work possible.

iv
Table of Contents
Abstract………………………………………………………………………….…………….....ii Acknowledgments……………………………………………………………………………….iii Table of Contents………………………………………………………………………………..iv List of Abbreviations…………………………………………………………………………….v List of Schemes………………………………………………………………………………..…vi List of Tables……………………………………………………………………………………vii 1 Introduction……………………………………..…………..……..…………………………...1 1.1.0 Properties and Applications of Carbohydrate Radicals….………………………….1 1.2.0 Dual Photoredox/HAT Catalysis……………………………………………………4
1.2.1 Cocatalysts for α-hydroxy C-H bond Weakening…………………………………..7 1.3.0 Photoredox-Mediated Functionalization of Carbohydrates…………………………8
1.4.0 Research Objectives………………………………………………………….…….10 2 Results and Discussion……………………………………………………………………….12
2.1 Regioselective C-H Alkylation of Carbohydrate Derivatives using Borinic Acid/Photoredox Catalysis……………………………………………………….12 2.1.1 Reaction Development and Optimization………………………….…….12 2.1.2 Computational Analysis………………………………………………….17
2.2 Site-Selective Redox Isomerizations of Pyranosides…………………………….19 2.2.1 Reaction Development and Optimization……………………………..…19 2.2.2 Scope Studies…………………………………………………………….22
3 Summary…………………………………………………………………………..…….26 4 Experimental……………………………………………………………………………27
4.1 Materials and Methods………………………………………………….………..27 4.1.1 General Information………………………………………..…………….27 4.1.2 Materials…………………………………………………………………27 4.1.3 Instrumentation…………………………………………………………..27
4.2 General Experimental Procedures………………..………………………………28 4.2.1 General Procedure for Diol Alkylation on 0.1 mmol Scale…………...…28 4.2.2 General Procedure for Constructing Diols………………………….……28
4.2.3 General Procedure for Pyranoside Transformation on 0.1 mmol Scale…29 4.2.4 General Procedure for Monoacylation of Carbohydrates A…………..…30 4.2.5 General Procedure for Monoacylation of Carbohydrates B…………..…30 4.2.6 General Procedure for Disaccharide Synthesis…………………..………31

v
List of Abbreviations
Ac acetyl Bn benzyl Boc tert-butyloxycarbonyl br broad Bu4N tetrabutylammonium Bu n-butyl BzO benzoyl C carbon Cu copper d doublet DART direct analysis in real time DCM dichloromethane DMAP 4-(dimethylamino)pyridine DMF N-N-dimethylformamide DBP dibutyl phosphate DPP diphenyl phosphate equiv. equivalents Et ethyl EtOAc ethyl acetate h hour(s) M molarity m multiplet Me methyl MeCN acetonitrile MeOH methanol MS molecular sieve NIS N-iodosuccinimide NMR nuclear magnetic resonance O oxygen OH hydroxyl Piv pivaloyl ppm parts per million q quartet rt room temperature t triplet TBS tert-butyldimethylsilyl TLC thin-layer chromatography

vi
List of Schemes
Scheme 1: Examples of C-glycosides in pharmaceuticals and natural products………………….1
Scheme 2: Direction of attack on glycosyl radicals……………………………………………….2
Scheme 3: Classical methods for C-glycoside linkages………………….………………….……3
Scheme 4: Carbohydrate derivatives for use synthetically and biologically……………...………4
Scheme 5: Initial photoredox works………………………………………………………………5
Scheme 6: Early dual photoredox/HAT catalysis methods……………………………………….6
Scheme 7: Various methods for α-hydroxy C(sp3)−H bond weakening………………………….7
Scheme 8: Photoredox strategies for C-glycoside synthesis………………………………………8
Scheme 9: C-H activation of carbohydrates using photoredox/HAT hybrid systems…………...10
Scheme 10. Proposed mechanism for the alkylation of the anomeric position of monomers…...11
Scheme 11. Proposed scheme for isomerization of pyranosides via spin-center shift…………..11

vii
List of Tables
Table 1. Initial results of alkylating Diol 1 with previously developed conditions……...………12
Table 2. Investigation into compatible diols…………………………………….……………….13
Table 3. Varying SOMOphile and photocatalyst components.………………………………….14
Table 4. Varying borinic acid and HAT mediator components…………………………...……..16
Table 5. 3,4,6-Tri-O-methyl glucopyranose results……………………………………..……….17
Table 6. Bond-dissociation energy (BDE) studies: 3,4,6-Tri-O-methyl glucopyranose 6………18
Table 7. Bond-dissociation energy (BDE) and transition state studies: galactose diols…………19
Table 8. Optimization of phosphate salt component………………………………….…………20
Table 9. Effect of the ratio of phosphate salt to quinuclidine ………………………...…………21
Table 10. Substrate investigation………………………………………………………...………22
Table 11. Comparison between optimized conditions……………………………………..…….24
Table 12. Substrate scope………………………………………..………………………………25

1
Introduction
1.1 Properties and Applications of Carbohydrate Radicals
Carbohydrates are ubiquitous in nature and their diverse roles in biological systems make them
interesting objects of study for the chemical and biological communities.1 Due to their
importance as building blocks, synthetic targets, and biological tools, new methodologies for
their synthesis and derivatization are of interest.2 The applications of carbon-centered radicals
derived from carbohydrates have been on the rise for some time by facilitating the preparation of
scaffolds that have not been accessible following classical synthetic routes.3 Carbohydrate-based
radicals have been used to prepare C-linked glycoconjugate compounds, enantiomerically pure
starting materials4 and tools to investigate biochemical pathways.5
Scheme 1: Examples of C-glycosides in pharmaceuticals and natural products.
C-linked glycosides are core units within several natural products and bioactive compounds
(Scheme 1). They consist of a carbohydrate unit attached to an aglycone or another carbohydrate
unit through a C-C bond linkage. Structurally similar to their O-linked analogues, they are more
inert towards enzymatic degradation, an advantage in applications as therapeutic agents.6
Various approaches have been developed for the synthesis of C-glycosides, including
intermolecular additions of glycosyl radicals to π-systems as discussed below.6
Glycosyl radicals are among the most well studied and versatile species in carbohydrate
chemistry.7 They are readily generated from the homolytic cleavage of halides, xanthates,
thioethers, selenides and nitro compounds and more recently by C-H activation methods.8
Furthermore, they react with predictable stereoselectivity. Glycosyl radicals react with
electrophilic acceptors to give preferential axial-substituted adducts (Scheme 2A), in contrast to

2
cyclohexyl radicals which favor equatorial approach.9-11 The behavior of the anomeric-radical
depends on two main stereoelectronic effects: the anomeric effect (interaction of the semi-filled
p-orbital with the ring-oxygen lone pair) and the -oxygen effect (interaction of the semi-
occupied p-orbital with the * orbital of the co-planar -C-OR bond). The combination of these
effects induce conformational changes in the glycosyl radical with respect to the original
carbohydrate in order to maximize orbital interactions.9-11 Factors, particularly the protecting
groups, that sterically hinder access to one face or restrict conformation-shifting are capable of
altering the stereoselectivity (Scheme 2B).12,13
Scheme 2: Arrows indicate preferred direction of attack on acrylonitrile. A: Stereoselectivity of
anomeric radical. B: Conformation restriction can affect the stereoselectivity.
Intermolecular additions of glycosyl radical donors to electron-deficient alkenes is the core
synthesis of C-linked glycosides. For instance, the addition of a glycosyl radical, generated by
tributyltin hydride from glucosyl bromide 1, to the exocyclic double bond of a methylene lactone
2 provides direct access to α-(1,2)-linked C-disaccharides 3 with high diastereoselectivity
(Scheme 3A).14 Approaches to the synthesis of -linked C-glycosides include using bulky
protecting groups which causes the α-face to be more sterically hindered; metal-mediated
reductive coupling reactions; and the use of silicon tethers (Scheme 3B).12,15,16 Branched-chain
sugars can also be prepared by the intermolecular addition of non-anomeric radicals to π-systems
with the stereoselectivity of the reaction being sterically controlled by the substituents adjacent
to the radical center (Scheme 3C).17

3
Scheme 3: Classical methods for C-glycoside linkages. A: Addition of glycosyl radical to an
alkene to form on α-linked C-glycoside. B. Various methods for -linked C-glycoside formation
i: Use of bulky protecting group to block α-addition. ii: Metal-mediated reductive coupling
reaction. iii: Use of silyl-tethers as a directing group. C: Formation of branched-chain
carbohydrate with C-C bond linkage.
Other interesting uses of carbohydrate-based radicals include processes involving -
fragmentation reactions. Alkoxyl radicals undergo -fragmentation transforming the anomeric
carbon into a formate group. The carbohydrate is degraded into an acyclic product, which can
allow access to functionalized chiral synthons (Scheme 4A).18 Furthermore, radical elimination
products can be used to investigate molecular processes and mechanisms. For instance,
ribonucleotide reductase catalyzes the key transformation of ribonucleotides to the corresponding
2’-deoxyribonucleotidues providing the monomeric precursors required for DNA biosynthesis.

4
The key steps involve the formation of a C-3’ nucleotide radical via hydrogen atom abstraction
by a sulfur based radical, protonation of the 2’-hydroxyl group and sequential spin-center shift to
eliminate water eventually leading to the 2’-deoxy-3’-ketonucleotide (Scheme 4B).19 In order to
model certain steps in this mechanism, Giese and co-workers synthesized a 3’-selenocarbonyl
nucleoside which allowed for the selective generation of a C-3’adenosyl radical under mild
conditions (Scheme 4C).20 Using competition kinetic methods, it was demonstrated that these
radicals rapidly eliminate the 2’-hydoxyl group providing additional support for the biosynthesis
mechanism of 2’-deoxyribonucleides in which C-3’ nucleotide radicals represent key
intermediates.20 The application of radicals in carbohydrate chemistry not only provides high
value synthetic products but also contributes to the understanding of biological processes and the
importance of stereoelectronic effects.
Scheme 4. A: -fragmentation of carbohydrates to generate chiral azirines. B: Ribonucleotide
reductase mechanism as proposed by Stubbe. C: Synthetic model for ribonucleotide reductase.
1.2 Dual Photoredox/HAT Catalysis
Over the last century, light-mediated catalysis has enabled the construction of various
unconventional bonds formations in organic chemistry.21 Recently, the field of modern
photoredox catalysis has allowed access to a variety of novel synthetic methodologies.22 As a
complement to conventional thermal methods, photocatalysis offers a unique activation mode to
generate reactive intermediates, such as radicals and radical ions. These intermediates are

5
difficult to generate using typical thermal conditions alone. A key factor in the rapid growth of
this platform has been the readily accessible organic compounds and metal complexes capable of
facilitating the conversion of visible light into chemical energy.23 This irradiation occurs at
wavelengths where common organic molecules do not absorb, allowing for the selective
excitation of the photoredox catalyst. Upon excitation, these molecules engage in single-electron
transfer (SET) events with organic substrates, providing direct access to reactive species. These
excited species generated can exist as both an oxidant and reductant simultaneously, creating
distinct organic reaction conditions.24
Scheme 5. Initial photoredox work. A: Seminal work by Kellogg. B: Work by Okada. C: Dual
Photoredox/Organocatalysis strategy by Nicewicz and MacMillan
Seminal work by Kellogg in 1978 provided the groundwork for recent developments in
photoredox catalysis. Kellogg demonstrated that the reduction of sulfonium ions to the
corresponding alkanes could be accelerated by the addition of [Ru(bpy)3]Cl2 which displayed
photocatalytic characteristics (Scheme 5A).25 Okada and co-workers would later build upon this
work by demonstrating that N-(acyloxy)phthalimides could be used as a source of alkyl radicals.
Following single electron reduction and sequential decarboxylation, the alkyl radicals formed
could be used in a variety of transformations such as chlorination, selenylation, hydrogen atom
6
abstraction and the ability to engage in conjugate additions with Michael acceptors (Scheme
5B).26 In 2008, Yoon, Nicewicz and MacMillan would demonstrate the underlying potential of
photoredox organocatalytic hybrid protocols.27,28 For instance, the direct asymmetric alkylation
of aldehydes using a catalytically generated chiral enamine intermediate proved to be a solution
to the problem of asymmetric α-carbonyl alkylation (Scheme 5C).28 These reports would
collectively trigger significant interest in the field of dual photoredox strategies.
Scheme 6. A: First dual photoredox/HAT catalysis method: hydroetherification of alkenols. B:
Alkylation of α-alcohol C-H bonds.
Hydrogen atom transfer (HAT) in a photocatalytic approach represents a unique opportunity in
organic free radical chemistry, allowing for greater versatility in the activation and
functionalization of R-H bonds. Several groups have exploited HAT catalysts for the
establishment of various dual photoredox/HAT transformations.29 Pioneering studies from the
Nicewicz group demonstrated that intermediates that do not readily interact with the
photocatalyst can be converted to products via hydrogen atom transfer with 2-phenylmalonitrile.
In their initial report, they displayed this new dual photoredox/HAT strategy by the anti-
Markovnikov hydroetherification of alkenols (Scheme 6A).30 Utilizing this catalytic strategy,
future works would entail the activation and sequential fluorination, oxygenation, alkylation and
amination of C-H bonds.31 The MacMillan group would later exploit this new approach for the
development of photoredox/HAT methodologies with the capacity to activate strong C-H
bonds.32 In 2015, MacMillan and co-workers described the use of photoredox, HAT and

7
hydrogen bonding catalysis for the selective alkylation of α-alcohol C-H bonds with Michael
acceptors in the presence of other weaker C-H bonds (Scheme 6B).33 In this protocol, a
phosphate hydrogen bonding catalyst coordinates to an alcohol group, that induces a drastic
weakening of the α-hydroxy C−H bond known as the “oxyanionic substituent effect”. The
oxidized HAT catalyst, such as a tertiary amine, phosphate, or thiol, then proceeds to abstract a
hydrogen atom at that position.34 The resultant α-hydroxy radical is then readily trapped by an
electron-deficient alkene to furnish the product. This protocol provided the fundamentals to
achieve previously elusive synthetic transformations on aliphatic and hydroxylated systems.35
1.2.1 Cocatalysts for α-Hydroxy C-H bond Weakening
Alcohols represent one of the most abundant functionalities among organic molecules. The
functionalization of α-hydroxy C(sp3)−H bonds via dual photoredox/HAT catalysis is innovative
for synthetic routes and for the late-stage derivatization of complex molecules.36 Despite the
advances by MacMillan, the methodologies for the activation of alkyl alcohols as a source of
carbon-centered radicals remain relatively underdeveloped.37 The development of novel α-C-H
bond-weakening catalysts would provide great potential for exploring unprecedented reactivity,
selectivity and substrate range in C(sp3)-H functionalizations. In recent years, various methods
have been used to alkylate the C-H bonds adjacent to alkyl alcohol groups in conjunction with
dual photoredox/HAT catalysis (Scheme 7). To name a few, MacMillan and co-workers
introduced using metal alkoxides such as zinc chloride as cooperative catalysts to accelerate the
C–H alkylation of benzyl alcohols.38 Kanai has reported the use of Martin’s spirosilane as a
bond-weakening catalyst by forming a silicate to promote the C–H alkylation of various aliphatic
alcohols.39 Recently, Taylor and co-workers have reported the use of borinic and boronic acids in
combination with cis-1,2-diol moieties to induce C-H bond weakening in carbohydrates.40,41
Scheme 7. Various methods for α-hydroxy C(sp3)−H bond weakening.

8
1.3 Photoredox-Mediated Functionalization of Carbohydrates
With the advent of light-mediated catalysis, the functionalization of complex molecules such as
carbohydrates has shown great promise.42 Currently, the majority of photoinduced strategies
have been reserved for the synthesis of oligosaccharides and glycoconjugates. Such methods
include O-glycosylations, S-glycosylations, O-arylations, dethiolations, and thiol-ene reactions.42
The use of these glycosylation protocols allow for the construction of glycosidic bonds which are
no longer confined to neighboring group participation, or the anomeric effect for directing newly
formed anomeric linkages.42 However these reaction types, while being versatile, do not
diversify the carbon-backbone of the carbohydrate itself. The application of photoredox hybrid
systems to the carbohydrate scaffold is underdeveloped.43 This represents a knowledge gap, and
only recent studies have outlined the promise of this approach.
Scheme 8. Photoredox strategies for C-glycoside synthesis. A: Conjugate addition using
glycosyl halides and electron-deficient alkenes. B: NHP-esters to activated alkenes. C: Dual
nickel/photoredox catalysis using carboxylic acid functionalized carbohydrates as coupling
partners
Synthesis of C-glycosides using photoredox catalysis was first introduced by Gagne in 2010.
Gagne and co-workers developed a reductive conjugate addition of glucosyl halides to activated

9
alkenes, leading to fully saturated C-glycosides with exclusive α-selectivity (Scheme 8A).44
Wang, Witte, and Minnaard would later build upon this approach in sequential years using
photoredox mediated additions of sugar-derived NHP-esters to activated alkenes (Scheme
8B).45,46 In 2018, Molander would introduce the synthesis of C-acyl glycosides using dual
nickel/photoredox catalysis and carboxylic acid functionalized carbohydrates as coupling
partners. Glycosyl radicals are generated via the decarboxylation of glycosyl carboxylic acids
with the organic photocatalyst, which efficiently couple with a variety of carboxylic acids under
nickel catalysis (Scheme 8C).47 Although useful, the methods to selectively functionalize
carbohydrates using photoredox catalysis are nonetheless very limited and focused mainly on the
anomeric and C-6 positions.
The currently developing photocatalytic HAT strategies for C−H bond activation has found
particular use for the modification of carbohydrates.48 As previously described, MacMillan
disclosed that HAT can occur from α-hydroxyl C-H bonds even in the presence of weaker C-H
bonds adjacent to ethers and acetals. This concept was demonstrated by the alkylation of a
protected galactose derivative in which the primary hydroxyl group remained unprotected
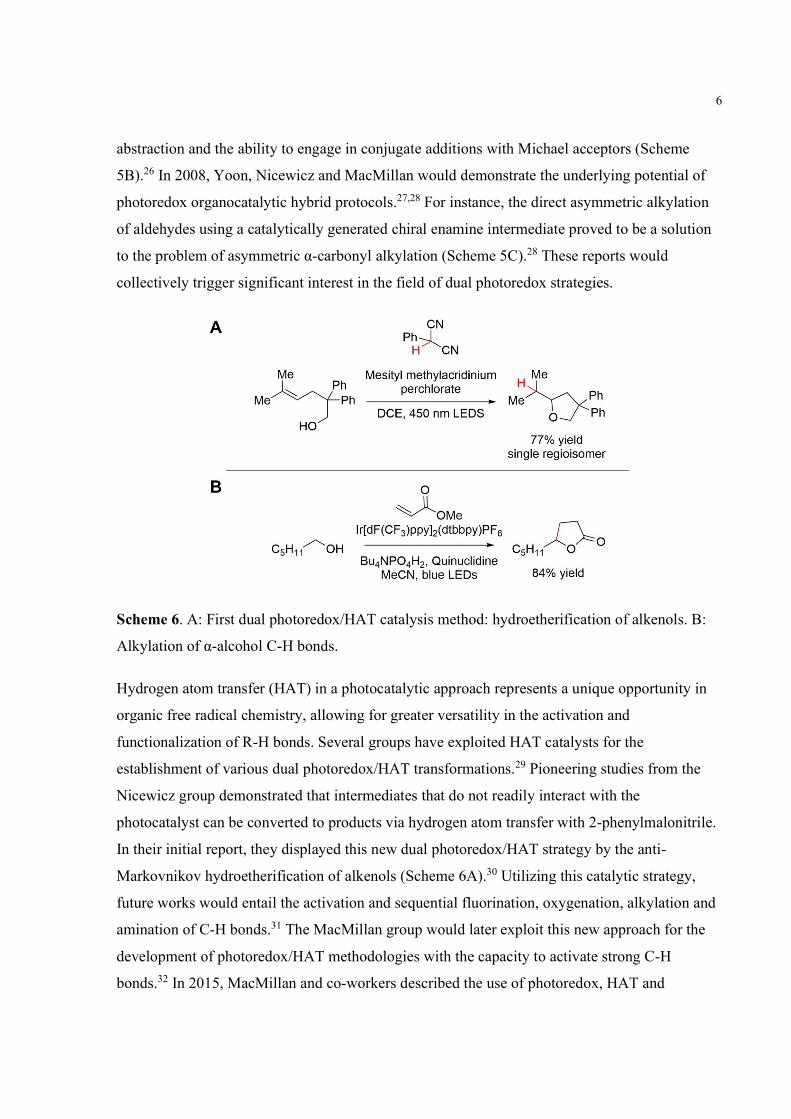
(Scheme 9A).33 The same catalytic system was then applied to the site-selective modification of
unprotected carbohydrates by Minnaard and co-workers. The transformation proceeds with C-3
alkylation on unprotected sugars being primarily favored (Scheme 9B).49 This selectively was
also used for the synthesis of rare sugar isomers through site-selective epimerization by
Wendlandt and co-workers.50 In 2019, the Taylor group achieved the stereo- and site-selective
alkylation of carbohydrates employing borinic acid and photoredox/HAT catalysis (Scheme
9C).40 These alkylated products arise from the unique interaction between cis-1,2-diols and the
organoboron cocatalyst. This interaction allows for equatorial C-H bonds to become weaker,
eventually leading to their homolytic cleavage. Due to the bicyclic nature of the borinate-
complex intermediate, the reaction proceeds with net configuration of the starting material.
Using a similar photoredox system, Taylor and co-workers also accomplished the direct
transformation of furanosides to 2’-keto-3’-deoxyfuranosides via spin-center shift (Scheme
9D).41 These products occur by boronic acid promoted hydrogen atom abstraction at the C-2’
position, followed by C-3’-O bond cleavage analogous to the previously shown mechanism of
ribonucleotide reductase enzymes.
10
Scheme 9. Functionalization of C-H bonds of carbohydrates using photoredox/HAT hybrid
systems. A: Alkylation of C6 position of galactose. B: Site-selective alkylation of unprotected
sugars. C: Site-selective alkylation of 1,2-diols using borinic acids. D: Isomerization of
furanosides to 2’-keto-3’-deoxyfuranosides via spin-center shift.
1.4 Research Objectives
Recently, colleagues in our group have developed a method to achieve site-selective C-H
alkylation of carbohydrates via diarylborinic acid and photoredox catalysis.40 However, the
scope of this reaction has yet to be applied to the anomeric position of carbohydrates. As an
ongoing effort in our laboratory, we aimed to develop a new methodology for the regioselective
C-H alkylation of the anomeric position of carbohydrate derivatives using dual hydrogen-atom
transfer (HAT)/photoredox chemistry to generate spiro-fused butyrolactones (Scheme 10). These
products would arise from the unique interaction between cis-1,2-diols and the diphenylborinic
acid cocatalyst. The resulting tetracoordinate borinic ester should only form with the α-anomer
of a carbohydrate mixture therefore selectively generating only α-products. The development of
a methodology for the formation of quaternary centers on the anomeric position would provide
access to a useful class of carbohydrate products.51 For instance, quaternary centers allow

11
carbohydrates to adopt frozen conformations which can be used to investigate molecular
recognition processes.51 Moreover, inhibitors of carbohydrate-active enzymes are often derived
from glycosides containing a quaternary anomeric position.53
Scheme 10. Proposed mechanism for the alkylation of the anomeric position of monomers.
In addition to the C-H alkylation project, another research direction we have undertaken is the
site-selective redox isomerizations of 2-O-acylated pyranosides to ketodeoxypyranosides using
another HAT/photoredox system. Previously, colleagues in our group have accomplished
applying similar chemistry for the successful isomerization of furanosides to
ketodeoxyfuranosides.41 However due to high-energy intermediates in the reaction, their protocol
gave low yields for pyranosides. By introducing a leaving group at the site of deoxygenation, we
performed the desired site-selective redox transformation via spin-center shift on pyranosides
(Scheme 11). The formation of these sugars occurs in the presence of a hydrogen-bond acceptor
catalyst and HAT/photoredox conditions. The reaction proceeds with the generation of an α-oxy
carbon radical which subsequently eliminates an adjacent leaving group to furnish the
ketodeoxypyranoside product. A potential application of this chemistry is the ability to control
the stereoselectivity in the synthesis of 2-deoxyglycosides, such as digitoxin, after a 2-O-acylated
building block is used to control the stereochemistry of the glycosidic linkage. This would be an
uncommon approach compared to other deoxygenative methods.54
Scheme 11. Proposed scheme for isomerization of pyranosides via spin-center shift.

12
Results and Discussion
2.1 Regioselective C-H Alkylation of Carbohydrate Derivatives using Borinic Acid/Photoredox Catalysis
2.1.1 Reaction Development and Optimization
Investigations into the alkylation of the anomeric position of carbohydrates first began by
identifying a class of suitable model substrates. For this reaction to take place, the hydroxyl
groups other than those in the C-1 and C-2 positions require some protection to generate the
desired borinate complex. In 2019, Takemoto demonstrated the synthesis of 1,2-cis-glycosides
by anomeric O-alkylation using borinic acid catalysis.55 The borinate complex formed in this
reaction would be analogous to the intermediate we would be interested in for weakening the
equatorial C-H bond on the anomeric position. The substrates used in that protocol involved the
protection of the O-3, O-4 and O-6 positions of carbohydrate monomers with benzylic protecting
groups. These O-alkylated sugars could be donating electron-density into the pyranose ring,
influencing the reactivity of the borinic ester intermediate towards glycosyl acceptors or C-H
bond cleavage. 3,4,6-Tri-O-benzyl glucopyranose 1 was synthesized and then subjected to the
conditions previously reported by our group for C-H activation on carbohydrates (Table 1).
Table 1. Initial results of alkylating Diol 1 with previously developed conditions.
Entry Equiv. 1 Equiv. Acrylate Yielda
1 2 1 15%
2 1 1 22%
3 1 2 24%
aYields determined by 1H NMR spectroscopic analysis of the unpurified reaction mixture using 1,3,5-trimethoxybenzene as an internal standard.
The reactions were run on a 0.1 mmol scale with Ir[dF(CF3)ppy]2(tbbpy)PF6 as the photocatalyst,
diphenylborinic acid as the cocatalyst and quinuclidine as the HAT mediator. 2 was generated in

13
poor yield. Inverting the stoichiometries of methyl acrylate to sugar did not significantly alter the
yield, neither did heating the crude mixture in acid resin for 3 hours to close any potential acyclic
alkylated product (result not displayed). The conversion of the reaction is considerable, with an
approximate 60-70% conversion in these instances. The remaining mass balance of the reaction
was complicated with numerous products being formed. Preparative TLC better enabled the
isolation and characterization of 2, whereas the identification of the other products appears to be
over-alkylation, fragmentation, degradation and seemingly dimerization. The α:β ratio of the
recovered starting material is still 1:1, suggesting that the rate of anomerization is sufficient for
the α-anomer to be replenished after capture of the borinic acid catalyst and sequential
alkylation.
Table 2. Investigation into compatible diols.
aYields determined by 1H NMR spectroscopic analysis of the unpurified reaction mixture using 1,3,5-trimethoxybenzene as an internal standard.
Due to the meager yield of 2, other diol substrates were synthesized to investigate substrate
compatibility with the reaction conditions (Table 2). The substrates were submitted to the
conditions as previously shown in Table 1., however 2 equivalents of methyl acrylate to 1
equivalent of carbohydrate were used. Once again there was little success in determining an ideal
Entry Carbohydrate Yielda Conversion
1
24% ~70%
2
26% ~60%
3
~20% >90%

14
candidate for further optimizations. The diols faced similar issues as before such as high
conversion with meager yields and complex reaction mixtures. Acylated diols proved not only
difficult to make but tended to undergo acyl migration under reaction conditions with little to no
alkylation observed (not depicted). It was determined that tri-O-benzyl galactopyranose 3 (Entry
2) would serve as the model substrate in our endeavor to optimize this reaction.
Table 3. Varying SOMOphile and photocatalyst components.
Entry SOMOphile Photocatalyst Yielda
1 R=CO2Me, R’=R’’=H Ir[dF(CF3)ppy]2(tbbpy)PF6 26%
2 R=CN, R’=R’’=H Ir[dF(CF3)ppy]2(tbbpy)PF6 11%
3 R=SO2Ph, R’=R’’=H Ir[dF(CF3)ppy]2(tbbpy)PF6 <5%
4b R=H, R’=NO2, R’’=Ph Ir[dF(CF3)ppy]2(tbbpy)PF6 0%
5c R=R’=CO2Me, R’’=Ph Ir[dF(CF3)ppy]2(tbbpy)PF6 0%
6c R=CN, R’=CO2Me, R’’=Ph Ir[dF(CF3)ppy]2(tbbpy)PF6 0%
7c R=R’=CN, R’’=Ph Ir[dF(CF3)ppy]2(tbbpy)PF6 0%
8 R=CO2Me, R’=R’’=H Ir[dF(CF3)ppy]2(bpy)PF6 28%
9 R=CO2Me, R’=R’’=H
0%
10 R=CO2Me, R’=R’’=H Ru(bpy)3Cl2 0%
aYields determined by 1H NMR spectroscopic analysis of the unpurified reaction mixture using 1,3,5-trimethoxybenzene as an internal standard. bResulted in reduction of somophile. cResulted in polymerization of somophile.

15
As part of our optimizations, the photocatalyst and SOMOphile components were addressed. It
was initially believed the radical generated at the anomeric position could not successfully be
captured by methyl acrylate and instead other more electron withdrawing SOMOphiles were
explored (Table 3). In addition, other photocatalysts were surveyed to determine the effect of
their redox potential on the reaction. As depicted in Table 3., it seems that the most ideal
SOMOphile is methyl acrylate. Popular SOMOphiles such as acrylonitrile and vinyl phenyl
sulfone saw a significant decrease in the formation of 4. These results coincide to an extent with
what was observed by our group during their optimizations of the cis-1,2-diol alkylation
project.40 It was also observed that nitrostyrenes, and various benzylidene malonates were
incompatible. These SOMOphiles would undergo reduction and polymerization, respectively. Of
the photocatalysts tested it appears that only the iridium-based photocatalysts influence the
reaction, with no significant difference between the iridium catalysts tested. Photocatalysts with
strong oxidizing and reduction potentials such as the acridinium (Entry 9) and ruthenium
catalysts (Entry 10), respectively, did not appear to form 4. These results seem to suggest that
Ir[dF(CF3)ppy]2(bpy)PF6 as the photocatalyst and methyl acrylate as the SOMOphile are the most
compatible pairings for the alkylation at the anomeric carbon.
Further optimizations were warranted to further increase the butyrolactone yield. To this end, the
borinic acid cocatalyst and HAT mediator aspect of this reaction were investigated (Table 4.). As
previously shown, standard conditions give 26% yield of 5. Concentrating the reaction to 0.50 M
has no noticeable effect on product yield. As a control to investigate the background reaction, no
borinic acid was added. As expected, no alkylation nor fragmentation of 3 was observed.
Switching the cocatalyst to a conformationally-locked borinic acid (Entry 4) halved the yield.
This may suggest that the flexibility imparted by the diphenylborinic acid may be an enhancing
factor for binding to the cis-1,2-diol moieties of carbohydrates. A couple of HAT mediators were
explored to determine if the hydrogen atom abstraction or protonation step of the mechanism
could be influencing the yield through pathways unknown. 3-Quinuclidinol, a less basic form of
quinuclidine, appears to give comparative yields as quinuclidine. The use of methyl thioglycolate
gives no observed product formation. This can be attributed to S-based HAT mediators generally
being used for hydrogen atom abstraction on weak C-H bonds such as benzylic positions.56
16
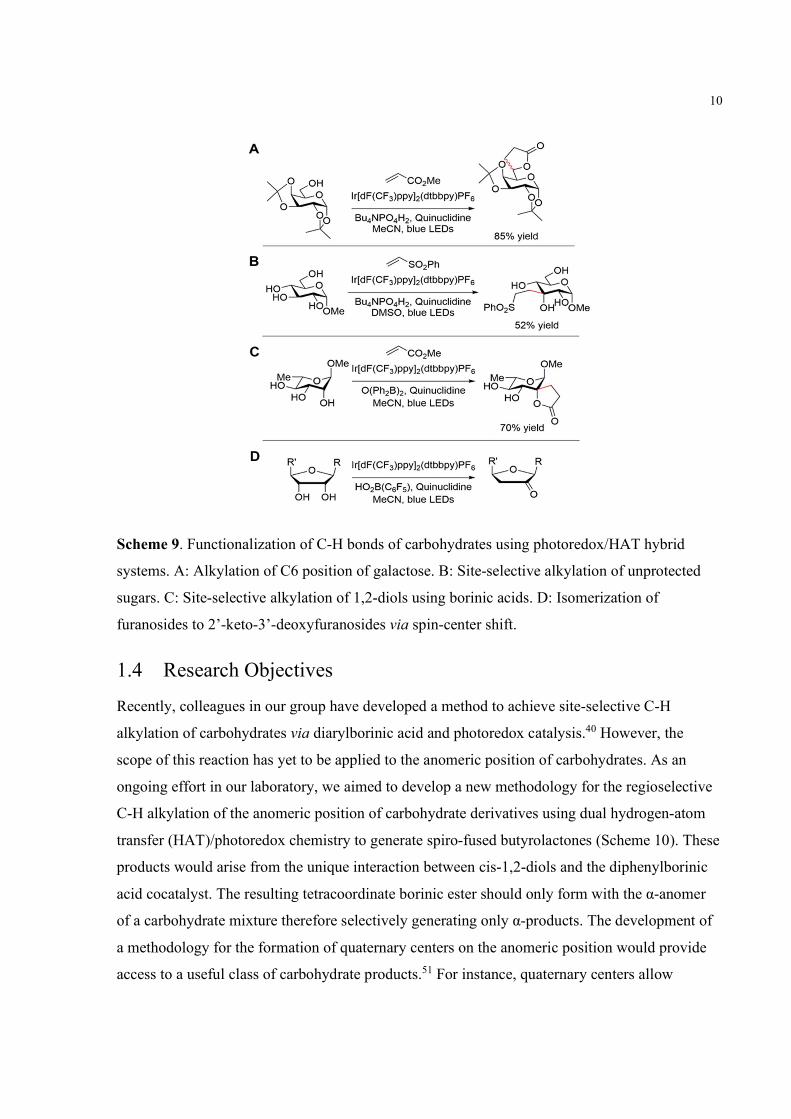
Table 4. Varying borinic acid and HAT mediator components.
Entry Borinic Acid HAT Mediator Yielda
1 (Ph2B)2O (5 mol%) Quinuclidine 26%
2b (Ph2B)2O (5 mol%) Quinuclidine 22%
3 -- Quinuclidine 0%
4
(10 mol%)
Quinuclidine 13%
5 (Ph2B)2O (5 mol%) 3-Quinuclidinol 28%
6 (Ph2B)2O (5 mol%) Methyl thioglycolate 0%
aYields determined by 1H NMR spectroscopic analysis of the unpurified reaction mixture using 1,3,5-trimethoxybenzene as an internal standard. bReaction was carried out at a concentration of 0.5 M.
Varying the components within this system does not showcase any alternatives to the already
established conditions. The conversion is high with various minor side-products unable to be
isolated or too complex to elucidate. As an effort to identify the side-products generated, 3,4,6-
tri-O-methyl glucopyranose 6 was synthesized. The rationale for the synthesis of a methylated
substrate rather than benzylated is due to two reasons. One reason is that there existed suspicion
of alkylation at the benzylic positions, leading to the various alkylated products observed.
Another reason is that the benzylic protons add another layer of complexity to the 1H NMR
spectrum. The range they occupy (5.2 – 4.2 ppm) could be overlapping upon important
carbohydrate peaks which can cause identification of side-products to be difficult. Indeed, using
6 resulted in the identification of products 7 and 8 (Table 5). The formation of butyrolactone
product 7 to 8 in a ratio of approximately 1:1 was unanticipated give given our current
understanding of the reaction mechanism. It is assumed that equatorial hydrogen atoms should
undergo hydrogen atom abstraction in preference over axial hydrogen atoms. Additionally, given
the assumed hydridicity of the anomeric position, the formation of a C-2 product should not be

17
favored kinetically or thermodynamically. Removal of the borinic acid cocatalyst did not result
in the formation of either product suggesting that the organoboron catalyst is directly responsible
for the formation of this product. The identification of isomeric product 8 motivated the
investigation of radical stabilities at the C-1 and C-2 positions as well as HAT transitions-states
studies.
Table 5. 3,4,6-Tri-O-methyl glucopyranose results
Entry Borinic Acid 7 Yielda 8 Yielda
1 Yes 24% 18%
2 No 0% 0%
aYields determined by 1H NMR spectroscopic analysis of the unpurified reaction mixture using 1,3,5-trimethoxybenzene as an internal standard.
2.1.2 Computational Analysis
Initially we sought to evaluate the bond-dissociation energies at the C-1 and C-2 positions of
3,4,6-tri-O-methyl glucopyranose 6 (Table 6). Calculations applied to the α-anomer display that
the most stable radical appears to form on the C-2 position (α-anomer) of both the free and
boron-bound sugar rather than the anomeric position, as had been assumed. However, the
magnitude of the difference was relatively low. A possible explanation for the unexpected
stability of the C-2 position in comparison to the anomeric position could be due to a variety of
factors such as the β-oxygen effect and intramolecular hydrogen bonding. Currently there are no
significant computational or experimental studies on the subject to support this rationale. In
2020, Linker described the use of a radical clock to determine the stability of carbohydrate
radicals in the C-1 and C-2 positions.57 This resulted in the preference for the C-1 radical,
however, these findings do not include hydroxyl groups at the designated positions which may
stabilize the C-2 radical to a greater extent than the C-1 radical. Efforts are underway in our
group to computationally describe the bond-dissociation energies on the carbon-backbone of
carbohydrates and their preferred HAT transition-states.

18
Table 6. Bond-dissociation energy (BDE) studies: 3,4,6-Tri-O-methyl glucopyranose 6
Position of C-H bond BDE (kcal/mol)
C-1 Position (Alpha) 82.7
C-1 Position (Beta) 81.7
C-2 Position (Alpha) 81.1
C-2 Position (Beta) 81.7
Calculations were carried out using B97-D3/Def2-TZVP (level of theory/basis set). Solvent:
acetonitrile.
For comparison, the bond-dissociation energies of the C-1 and C-2 positions on 3,4,6-tri-O-
methyl galactopyranose were also determined (Table 7). It was found that the difference in
radical stability at the C-1 and C-2 positions was insignificant as previously observed with 3,4,6-
tri-O-methyl glucopyranose 6. Changing the protecting groups on tri-protected galactose appears
to affect the radical stabilities at those two positions. Methyl protecting groups display an
insignificant difference between the radical stabilities between the two positions. Acyl protecting
groups show a preference for the C-2 position. TMS protecting groups show a preference for the
C-1 position. The stabilization of a radical at the C-2 position when using acyl protecting groups
may occur due to the adjacent ester oxygen accepting a hydrogen bond from the O-2 hydrogen.
In combination with the thermodynamics aspect, several HAT transition-states have been
modelled for the hydrogen atom abstraction at the C-1 and C-2 positions of 3,4,6-tri-O-methyl
galactopyranose. The results indicate that the HAT at the C-1 position is only favorable by 1.2
kcal/mol over the C-2 position. This is a relatively small difference for HAT at an axial versus
equatorial position on a carbohydrate. For context, Taylor and co-workers reported differences of
greater than 3.0 kcal/mol for relative HAT activation energies on methyl α-L-
rhamnopyranoside.40 Ongoing investigations into the computational analysis of the
thermodynamics and kinetics of the reaction may provide some insight into the mechanism of
HAT on the C-1 and C-2 positions of carbohydrates.

19
Table 7. Bond-dissociation energy (BDE) and transition state studies: galactose diols
Position of C-H bond BDE (kcal/mol) R = Me BDE (kcal/mol) R = Acetyl BDE (kcal/mol) R = TMS
C-1 Position (Alpha) 82.4 81.9 80.0
C-1 Position (Beta) 82.2 82.3 82.7
C-2 Position (Alpha) 82.5 81.9 82.6
C-2 Position (Beta) 82.1 79.1 82.5
Radical Position Relative Energy (kcal/mol)
C-1 Position (Alpha) 0
C-1 Position (Beta) --
C-2 Position (Alpha) 6.7
C-2 Position (Beta) 1.2
Calculations were carried out using B97-D3/Def2-TZVP (level of theory/basis set). Solvent:
acetonitrile. Transition states are in gas phase.
2.2 Site-Selective Redox Isomerizations of Pyranosides
2.2.1 Reaction Development and Optimization
As another research direction to complement the alkylation project, we have been working on the
photoredox-mediated transformation of pyranosides to ketodeoxypyranosides via spin-center
shift. As previously discussed, pyranosides were a limitation of the first-generation protocol and

20
could not isomerize pyranosides in high yields. However, this transformation may be plausible
with the assistance of a leaving group at the site of deoxygenation. Our inquiry into this subject
began when another member of this project, Julia Turner, observed that 2-O-pivaloyl-α-D-
glucopyranoside 9 undergoes isomerization to the 3-keto-2-deoxypyranoside product 10 using
tetrabutylammonium phosphate monobasic (TBAP) as a hydrogen-bond acceptor (HBA)
catalyst. Although the yield was modest, this prompted our investigation into this reaction. 9 was
chosen as our model substrate due to its ease of synthesis using methods developed by our
laboratory and its clean conversion to 10.58
Table 8. Optimization of phosphate salt component.
Salt Base Yielda
None - 27% bTBAP KHCO3 40%
Na DBP - 37%
Na DBP K2CO3 56%
K DBP K2CO3 44%
(Bu4N) DBP - 57%
(Bu4N) DBP KHCO3 46%
(Bu4N) DBP K2CO3 76%
(Bu4N) DPP - 22%
DBP K2CO3 51% aYields determined by 1H NMR spectroscopic analysis of the unpurified reaction mixture using 1,3,5-trimethoxybenzene as an internal standard. bCredit due to Julia Turner for this reaction.
Due to recent attention devoted to applications of dibutyl phosphates and electron-deficient
carboxylates as hydrogen-bond catalysts in combination with photoredox/HAT catalysis, we
began investigations into these two avenues.59,50 As part of our study, we screened a variety of
dibutyl phosphate salts in conjunction with base additives (Table 9). It appears that using K2CO3

21
as a base with the phosphates significantly increases the yield of the reaction. This is perhaps due
to the base additives neutralizing pivalic acid that presumably builds up over the reaction, which
may be detrimental to hydrogen bonding interactions between the phosphate and hydroxyl
groups. It is unknown why KHCO3 and other bicarbonates (not shown) seem to hinder the
reaction in combination with phosphates. From these findings, it appears that
tetrabutylammonium dibutyl phosphate ((Bu4N) DBP) is the most effective phosphate catalyst
for this transformation, which may be in part due to an enhanced solubility in comparison to the
other salts. The conversion of this reaction is directly correlated with the yield with no other
apparent side-products being generated when using 9 as a substrate. In addition to the
optimization of the salt and base component of the reaction, we have also been interested in
varying the salt and quinuclidine loading (Table 9). It appears that increasing and decreasing the
loading of (Bu4N) DBP has a detrimental effect on the yield of the reaction. A similar trend is
observed for varying the quinuclidine loading as well. These results suggest that the ideal salt
loading is 25 mol% with a quinuclidine loading of 20 mol%.
Table 9. Effect of the ratio of phosphate salt to quinuclidine on the yield of 10
aYields determined by 1H NMR spectroscopic analysis of the unpurified reaction mixture using 1,3,5-trimethoxybenzene as an internal standard.
Phosphate (mol%)
5% 10% 25% 50% 100%
10% - - 28% - -
Quinuclidine (mol%) 20% 43% 45% 57% 48% 38%
30% - - 40% - -
40% - - 34% - -

22
2.2.2 Scope Studies
The use of (Bu4N) DBP and K2CO3 for this reaction may be used as alternative to our current
conditions. Our conditions going forward involves the use of tetrabutylammonium 4-
chlorobenzoate (Bu4N 4-ClOBz) and KHCO3 as our HBA catalyst and base, respectively
(optimizations by Julia Turner). This system can form 3-keto-2-deoxypyranoside 10 in 79%
yield based on NMR yield using similar conditions to those shown previously. As such, my
focus has shifted from phosphate optimizations to substrate synthesis, whereas Julia Turner
would be running these reactions as well as synthesizing some substrates (where specified). With
optimized conditions in hand, we sought to evaluate the substrates and leaving groups that would
be compatible with the conditions developed (Table 10.). It appears that 2-O-benzoyl-α-D-
glucopyranoside (Entry 3) can transform into 3-keto-2-deoxypyranoside 10 although at
diminished yields in comparison to its pivaloyl counterpart. 4,6-Benzylidine groups (Entry 2)
affects the efficiency of the reaction. Currently developing computational studies may suggest
that the transition state involves a boat or half-chair conformation which may be required for
effective spin-center shift. This theory can account for the diminished yields of conformationally
locked carbohydrates such as 4,6-benzylidenes and conformationally labile carbohydrates such
as arabinose (Entry 6). Diminished yields are observed when using tert-butyloxycarbonyl (Boc)
and tosyl (Ts) groups as leaving groups. Based on the understood mechanism of the reaction,
these leaving groups should theoretically be more efficient at eliminating from the molecule
contrary to what is observed. Perhaps these leaving groups hinder the formation of the adjacent
α-oxy radical or are unable to eliminate from the molecule in the proposed concerted fashion.
Table 10. Substrate investigation.
Entry Carbohydrate Yieldc
1a,b
67%

23
2c
~15%
3a,b
47%
4b
<5%
5b
~30%
6c
~30%
aIsolated yields. Yields determined by 1H NMR spectroscopic analysis of the unpurified reaction mixture using 1,3,5-trimethoxybenzene as an internal standard. bPersonally synthesized substrates. cCredit due to Julia Turner for running these reactions and the substrates synthesized.
To broaden substrate compatibility, I compared our alternative phosphate conditions to substrates
which produced the desired keto-deoxypyranosides in poor yield using our optimized
carboxylate conditions. As shown in Table 11, there appears to be few differences between the
two conditions developed. However there exists a noticeable discrepancy in Entry 4. The
phosphate conditions can generate the keto-deoxypyranoside product from 2-O-benzoyl-α-L-
fucopyranoside in a combined yield of 75% in comparison to the carboxylate conditions
combined yield of 20%. Unfortunately, the axial O-4 undergoes epimerization to generate the
keto-deoxyrhamnoside product. Attempts at tuning the reaction conditions to favor the non-
epimerized product were unproductive (results not shown). Efforts to shorten the reaction time to
impede the epimerization of the product were unsuccessful as the rate of epimerization proceeds
quickly as the reaction progresses. However, the results gathered from this optimization did
display that the phosphate catalyzed conditions can produce the keto-deoxypyranoside product in
6 h. Going forward, products that may prove to be difficult to produce from the carboxylate
conditions may prove to be more compatible with our alternative phosphate conditions as shown
in Entry 4.
24
Table 11. Comparison between optimized conditions
Entry Carbohydrate Product Conditions A
Yielda,f
Conditions B
Yielda
1c
11% 10%
2b,f
20% (Mixture) 31% (Mixture)
3c
47% 40%
4c
15%d (5%)e 52%d (23%)e
5c
10% (Mixture) 12% (Mixture)
aYields determined by 1H NMR spectroscopic analysis of the unpurified reaction mixture using 1,3,5-trimethoxybenzene as an internal standard. bMixture of keto-deoxypyranoside isomers. cPersonally synthesized substrates. dNon-epimerized product. eEpimerized product at C-4. fCredit due to Julia Turner for running these reactions and the substrate synthesized.
Scope investigations are still underway for this reaction; however, we have found it successful
with various sugars (Table 12). As shown previously, this transformation works well with 2-O-
acylated glucose including those substituted at the 6-position with TBS groups. 2-O-benzoylated
mannose (Entry 2) produces the 3-keto-2-deoxypyranoside product in approximately 70% yield.
Monomers containing unprotected axial hydroxyl groups appear to undergo epimerization to the
more thermodynamically stable equatorial position. This is observed with substrates such as 2-O-

25
acylated galactose (Entry 3) and 4-O-acylated mannose (Entry 4) producing the epimerized
products in 42% and 57% yield, respectively. These conditions are also compatible with 6-deoxy
sugars; we can selectively acylate rhamnose at either the O-2 or O-4 position to give 2- or 4-
deoxy rhamnose in approximately 65% yield each. There are current investigations into the
disaccharide scope (general synthesis of these disaccharides described in experimental);
preliminary results suggest some compatibility as our initial attempt produced approximately
19% yield of the 3-keto-2-deoxy disaccharide (Entry 7). This substrate may not have worked due
to observed acetyl migration within the 1H NMR of the unpurified reaction mixture, effectively
shutting down the reaction. We have found that substrates containing acyl groups at the O-3
position are incompatible, giving little to no product. Previous research from the Minnaard and
Wendlandt groups suggest that hydrogen atom abstraction predominately occurs at the C-3
position of an unprotected carbohydrate.49,50 This coincides with what is observed; the ideal
radical is unable to form at the C-3 position and therefore cannot eliminate the leaving group
installed at the O-3 position via spin-center shift.
Table 12. Substrate scope for the transformation of pyranosides to 3-keto-2-deoxypyranosides.
Entry Carbohydrate Product Yieldc
1a,c
67%
2c
70%
3b
42% + 10%

26
4a,b,c
57%
5a,c
65%
6c
62%
7b
19%
8b
30%
aIsolated yields. Yields determined by 1H NMR spectroscopic analysis of the unpurified reaction mixture using 1,3,5-trimethoxybenzene as an internal standard. bPersonally synthesized substrates. cCredit due to Julia Turner for running these reactions and the substrate synthesized.
Summary
In summary, the regioselective C-H alkylation of the anomeric position of carbohydrate
derivatives is more difficult than expected. Alkylation at both the C-1 and C-2 positions in
similar ratios was unexpected given our current understanding of the reaction mechanism and
assumed radical stabilities. Additionally, the manipulation of protecting groups on the diol may
produce results different than those shown here. Future computational and mechanistic studies
will be able to shed some light on the preference for HAT on carbohydrate monomers containing
a hemiacetal. The photoredox-mediated transformation of acylated pyranosides into keto-
deoxypyranosides can proceed in 19-70% yield. These conditions appear to be compatible with a
wide variety of sugars. This method would allow for the formation of rare sugars and the ability
to control the stereochemical outcome of glycosylation with 2-deoxy sugars. There is currently
an ongoing expansion of the substrate scope in addition to computational studies on the radical
stabilities within this system.

27
Experimental
4.1.1 Materials and Methods
4.1.1.1 General Information
All reactions were stirred using teflon-coated magnetic stir bars at room temperature (23 °C)
unless otherwise stated. Stainless steel needles and gas-tight syringes were used to transfer air
and moisture-sensitive liquids. Schlenk flasks and 4 Å molecular sieves were stored at 140 ℃ for
at least 24 hours before use. Flash column chromatography was carried out using neutral silica
gel (60 Ǻ, 230-400 mesh) (Silicycle) using reagent grade solvents. Analytical TLC was carried
out using aluminium-backed silica gel 60 F254 plates (EMD Milipore) and visualized with a
UV254 lamp or with aqueous basic permanganate stain.
4.1.1.2 Materials
Where indicated, dry solvents are HPLC grade and purified using a solvent purification system
equipped with columns of activated alumina under nitrogen (Innovative Technology, Inc.).
Distilled water was obtained from an in-house supply. Nuclear magnetic resonance (NMR)
solvents were obtained from Cambridge Isotope Laboratories. Carbohydrate starting materials
were purchased from Sigma Aldrich or Carbosynth Ltd. (Berkshire, UK) or synthesized
according to literature procedures. All other reagents and solvents otherwise not indicated were
purchased from Sigma Aldrich or Caledon and used without further purification.
4.1.1.3 Instrumentation
1H, 13C and 2D NMR spectra were recorded using an Agilent DD2-600 (600MHz), Agilent DD2-
500 (13C, 126 MHz), Varian Mercury 400 (400MHz), or Bruker Avance III (400 MHz)
instrument at the Centre for Spectroscopic Investigation of Complex Organic Molecules and
Polymers (CSICOMP) at the University of Toronto. 1H NMR are reported in parts per million
(ppm) relative to tetramethylsilane and referenced to residual protium in the dominant solvent.
Spectral features are reported in the following order: chemical shift (δ, ppm); multiplicity (s-
singlet, d-doublet, t-triplet, q-quartet, m-complex multiplet, br.- broad); number of protons;
coupling constants (J, Hz); assignment. Where reported, assignments were made based on
coupling constants and 2D (gCOSY/HSQC/HMBC) NMR spectra. High-resolution mass spectra
(HRMS) were obtained on JEOL AccuTOF JMS-TL1000LC for DART+ at the Advanced
28
Instrumentation for Molecular Structure (AIMS) Mass Spectrometry Laboratory at the
University of Toronto.
4.1.2 General Experimental Procedures
4.1.2.1 General Procedure for Diol Alkylation on 0.1 mmol Scale
Carbohydrate/diol (0.1 mmol, 1 equiv.), (Ir[dF(CF3)ppy]2(dtbpy))PF6 (1 mg, 0.001 mmol, 1
mol%), quinuclidine (2.2 mg, 0.02 mmol, 20 mol%), and (Ph2B)2O borinic acid (1.7 mg, 0.005
mmol, 5 mol%) were combined in a ½ dram vial. A rubber septum was used to seal the vial,
which was then evacuated and backfilled with argon three times on a Schlenk line. Dry, degassed
acetonitrile (0.4 mL) was added under a balloon of argon. The balloon was removed, and methyl
acrylate was added to the vial using an airtight glass syringe. The rubber septum was quickly
replaced with the vial cap and sealed with Teflon tape. The vial was placed 5 inches from a blue
LED Kessil lamp and stirred at 1050 rpm at ~25 °C. After 16 hours, the crude reaction mixture
was concentrated under reduced pressure, and analyzed by 1H NMR spectroscopy.
4.1.2.2 General Procedure for Constructing Diols
A solution of pentaacetylated carbohydrate monomer A (2.00 mmol, 1 equiv.) in DCM (6 mL)
was treated with I2 (2.80 mmol, 1.4 equiv.) and Et3SiH (2.8 mmol, 1.4 equiv.). The mixture was
heated at reflux for 1 h, cooled to RT and treated with 2,6-lutidine (0.93 mL, 4 equiv.), EtOH
(0.68 mL, 6 equiv.) and TBAI (0.8 mmol, 0.4 equiv.). The mixture was heated at reflux for 3 h,

29
the volatiles were removed under reduced pressure and the residue was purified by flash
chromatography on silica gel to afford B.60
A solution of the acetylated orthoester B (1.23 mmol, 1 equiv.) in MeOH (5 mL) was treated
with NaOMe (0.12 mmol, 0.1 equiv.) in one portion and the resulting mixture was stirred at RT
until consumption of the starting material (ca. 45 min). The volatiles were removed under
reduced pressure and dried by azeotropic removal of water with toluene. The residue was
dissolved in dry DMF (5 mL). The mixture was cooled to 0 °C and NaH (5.32 mmol, 4 equiv.,
60% in mineral oil) was added in one portion. The alkyl halide (5.96 mmol, 4.5 equiv.) was
added via syringe and the resulting mixture was stirred overnight at RT. The reaction was
quenched with ice and extracted twice with EtOAc. The combined organic phases were washed
with brine, dried, concentrated and the residue was purified by flash column chromatography on
silica gel to afford C.60
Concentrated H2SO4 (0.13 mL) was added to a stirred solution of tri-O-alkylated orthoester (1.3
mmol, 1 equiv.) in 1,4-dioxane (4.80 mL) and H2O (2.55 mL), and the solution was refluxed for
7 h. Then, the reaction mixture was cooled down to room temperature, and solid NaHCO3 was
added until the mixture was neutralized. EtOAc and H2O were added and separated. The organic
layer was washed with water and brine. The organic layer was dried over Na2SO4, filtered, and
concentrated under reduced pressure. The crude product was purified by flash column
chromatography over silica gel to afford D.61,55
4.1.2.3 General Procedure for Pyranoside Transformation on 0.1 mmol Scale
O-acylated pyranoside (0.10 mmol), (Ir[dF(CF3)ppy]2(dtbpy))PF6 (1 mg, 0.001 mmol, 1 mol %),
quinuclidine (2.2 mg, 0.02 mmol, 20 mol %), salt additive (0.025 mmol, 25 mol%) and a small
magnetic stir bar were added to a ½ dram vial. A rubber septum was used to seal the vial, which
was then evacuated and backfilled with argon three times on a Schlenk line. Dry, degassed

30
acetonitrile (0.8 mL) was added to the vial under a balloon of argon. The rubber septum was
removed and quickly replaced with the vial cap which was sealed with Teflon tape and parafilm.
The vial was placed 5 inches away from a blue LED Kessil lamp and stirred at 1050 rpm for 24
hours at 25 ℃. After 24 hours the crude reaction was concentrated under reduced pressure and
analyzed by 1H NMR Spectroscopy.
4.1.2.4 General Procedure for Monoacylation of Carbohydrates
Prepared from an adapted literature procedure.58 To a round-bottomed flask containing
carbohydrate derivative (1.99 mmol, 1 equiv.) was added phenylboronic acid (1.99 mmol,1
equiv.) and toluene (10 mL). The reaction mixture stirred for 16 hours under reflux. The mixture
was then concentrated under reduced pressure and dried by azeotropic removal of water with
toluene. To the resulting colourless oil was added pyridine (4 mL), and the solution was cooled
to 0°C and acyl chloride (2.99 mmol, 1.5 equiv.) was added dropwise. The reaction was stirred at
RT until consumption of the starting material by TLC, after which it was quenched with
methanol (1 mL) and then concentrated under reduced pressure. The crude material was then
dissolved in EtOAc and transferred to a separatory funnel along with a sorbitol: sodium
carbonate solution (1M:1M) and hand shaken vigorously for 5 minutes. The aqueous layer was
extracted with EtOAc and the combined organic extracts were dried and concentrated under
reduced pressure. The resulting crude material was purified by flash chromatography on silica
gel.
4.1.2.5 General Procedure for Monoacylation of Carbohydrates
To a round-bottom flask equipped with a magnetic stir bar was added the pyranoside derivative
(1.00 mmol, 1 equiv.), copper (II) trifluoroacetic acid (1.30 mmol, 1.3 equiv.), and dissolved in

31
dry MeCN (8 mL). The anhydride (1.30 mmol, 1.3 equiv.) was added then 2,4,6-collidine (1.30
mmol, 1.3 equiv.) was added dropwise. The reaction stirred at 25 ℃ for 6 hours. The reaction was
then diluted with DCM and washed with 1 M HCl, saturated NaHCO3, then H2O. The organic
layer was dried over MgSO4 and concentrated under reduced pressure. The crude material was
then purified by flash column chromatography on silica gel.62
4.1.2.6 General Procedure for Disaccharide Synthesis
To a vigorously stirred, cooled (ice bath) biphasic solution of A63 (3.00 mmol) in DCM (12 mL),
acetone (1.2 mL) and sat. aq, NaHCO3 (20 mL), a solution of Oxone (3.59 g) in H2O (14 mL)
was added dropwise over 15 min. The mixture was vigorously stirred at 0°C for 30 min and then
at RT for an additional 2 h. The reaction was then extracted with DCM. The combined organic
phases were dried and concentrated to afford B (90%) as a white solid.64
B (1.4 mmol) was dissolved in DCM (1.2 mL), and EtSH (1 mL) was added. The mixture was
cooled to -78 °C, and trifluoroacetic acid anhydride (17 µL) was added dropwise. The reaction
mixture was stirred at -78°C for 20 min and then warmed up to RT. The volatiles were removed
under reduced pressure and the residue was purified by flash chromatography on silica gel (10%
to 50% ethyl acetate in hexane) to provide C (70%).65 C (0.5 mmol) and DMAP (5 mol%) was
dissolved in pyridine (5 mL) and cooled to 0°C. PivCl (3 equiv.) were added dropwise and the
reaction mixture was heated to 40°C for several days until TLC revealed consumption of the
starting material (ca. 4 days). The reaction was then extracted with DCM, washed with sat. aq.
NaHCO3, dried and concentrated under reduced pressure. The resulting residue was purified by
flash chromatography on silica gel (5% to 20% ethyl acetate in hexane) to give glycosyl donor
16 (85%).
A mixture containing the glycosyl donor 16 (0.10 mmol), glycosyl acceptor (0.11 mmol), and
freshly activated 4 Å molecular sieves (200 mg) in DCE (1.6 mL) was stirred under argon for 1 h

32
followed by the addition of Cu(OTf)2 (20 mol%). The reaction mixture was cooled to 0°C and
NIS (2.2 equiv.) was added in one portion. The reaction was stirred at 0°C until TLC revealed
consumption of the starting material (ca. 20 min). The reaction mixture was then diluted with
DCM and transferred to a separatory funnel. The organic layer was washed with 20% aq
NaHCO3, 10% sodium thiosulfate and water. The organic phase was separated, dried, and
concentrated under reduced pressure. The residue was purified by column chromatography on
silica gel (ethyl acetate/hexane gradient) to afford D.66
D was dissolved in EtOH, and Pd/C (10 % w/w) was added. The reaction mixture was purged
with nitrogen, then the flask was purged with hydrogen three times. The reaction mixture was
stirred at RT under hydrogen until TLC revealed consumption of the starting material (ca. 16 h).
The solution was filtered through Celite®, and the filtrate was concentrated under vacuum. The
residue was purified by column chromatography on silica gel (acetone/DCM gradient) to afford
the final disaccharide substrate.
3,4,6-Tri-O-benzyl-D-glucopyranose 1
Prepared according to general procedure (4.1.2.2). 1 was obtained as a white solid (343 mg, 69%) after flash column chromatography on silica (50% to 66% ethyl acetate in hexane). Spectral data agreed with those previously reported.55
Rf = 0.10 (EtOAc/Hexanes, 1:1)
1H NMR (400 MHz, CDCl3, 50:50 mixture of anomers): δ (ppm) = 7.377.13 (m, 15H), 5.24 (t, J = 3.5 Hz, 0.5H), 4.9079 (m, 3H), 4.6047 (m, 3.5H), 4.19 (d, J = 6.4 Hz, 0.5H), 4.04 (td, J = 6.7, 3.3 Hz, 0.5H), 3.79 (t, J = 9.0 Hz, 0.5H), 3.7046 (m, 5.5H), 2.59 (d, J = 2.3 Hz, 0.5H), 2.28 (d, J = 7.5 Hz, 0.5H)
13C NMR (126 MHz, CDCl3, 50:50 mixture of anomers): δ (ppm) = 138.5, 138.4, 138.0, 137.8, 137.6 (2C), 128.5, 128.4, 128.0 (2C), 127.9 (3C), 127.8 (4C), 96.7, 92.3, 84.3, 82.4, 77.6, 77.5, 77.2, 75.5, 75.3, 75.2, 74.9, 74.8, 73.5, 73.4, 72.7, 70.4, 68.7 (2C)
α:β = 1:1

33
(5R,7R,8R,9R,10R)-8,9-bis(benzyloxy)-7-((benzyloxy)methyl)-10-hydroxy-1,6-dioxaspiro[4.5]decan-2-one 2
Prepared from 1 according to general procedure (4.1.2.1). 2 was obtained as a white solid after Prep TLC (7% acetone in DCM). Purification of the sugar lactones was challenging.
Rf = 0.40 (Acetone/DCM, 7:93)
1H NMR (400 MHz, CDCl3): δ (ppm) = 7.40-77.27 (m, 13H), 7.20-7.16 (m, 2H), 4.92 (d, J=11.8 Hz, 1H), 4.79 (dd, J=23.8, J=11.2 Hz, 2H), 4.63-5.53 (m, 2H), 4.49 (d, J=12.2 Hz, 1H), 3.91 (d, J=8.5 Hz, 1H), 3.84-3.71 (m, 3H), 3.69-3.52 (m, 3H), 2.73-2.52 (m, 3H), 2.20-2.15 (m, 1H)
HRMS (DART+, m/z): calculated for C30H36NO7 [M+H]+: 522.24918, found: 522.24863.
Partial characterization data obtained for products whose yields could not be optimized to synthetically useful levels.
3,4,6-Tri-O-benzyl-D-galactopyranose 3
Prepared according to general procedure (4.1.2.2). 3 was obtained as a white solid (343 mg, 69%) after flash column chromatography on silica (50% to 66% ethyl acetate in hexane). Spectral data agreed with those previously reported.55
Rf = 0.15 (EtOAc/Hexanes, 2:1)
1H NMR (500 MHz, CDCl3, 70:30 mixture of anomers): δ (ppm) = 7.377.26 (m, 15H), 5.32 (d, J = 2.9 Hz, 0.7H), 4.87 (d, J = 11.5 Hz, 1H), 4.744.40 (m, 5.3H), 4.154.13 (m, 1.5H), 3.933.86 (m, 1H), 3.72 (dd, J = 10.0, 2.6 Hz, 0.7H), 3.613.44 (m, 2.6H), 3.40 (dd, J = 9.7, 2.9 Hz, 0.3H), 2.71 (br s, 0.3H), 2.37 (br s, 0.7H), 1.72 (br s, 1.0H)
13C NMR (125 MHz, CDCl3, 70:30 mixture of anomers): δ (ppm) = 138.3, 138.2, 138.0, 137.8, 137.6 (2C), 128.5 (2C), 128.4 (2C), 128.3, 128.2 (2C), 128.0 (2C), 127.9 (2C), 127.8 (2C), 127.7 (2C), 97.2, 92.7, 81.9, 79.1, 74.6, 74.5, 73.8, 73.7, 73.5 (2C), 72.9, 72.5, 72.4, 72.3, 69.6, 69.2, 68.9, 68.6

34
α:β = 7:3
(5R,7R,8S,9R,10R)-8,9-bis(benzyloxy)-7-((benzyloxy)methyl)-10-hydroxy-1,6-dioxaspiro[4.5]decan-2-one 5
Prepared from 3 according to general procedure (4.1.2.1). 5 was obtained as a white solid after Prep TLC (7% acetone in DCM). Purification of the sugar lactones was challenging.
Rf = 0.50 (Acetone/DCM, 7:93)
1H NMR (500 MHz, CDCl3): δ (ppm) = 7.36-7.27 (m, 15H), 4.86 (d, J=12.0 Hz, 1H), 4.72 (d, J=11.4 Hz, 1H), 4.60-4.54 (m, 2H), 4.48 (d, J=5.1 Hz, 2H), 4.13-4.07 (m, 4H), 3.78 (dd, J=7.4, 2.7 Hz, 1H), 3.66 (m, 1H), 3.61 (m, 1H), 3.52 (dd, J=5.6, 3.7 Hz, 1H), 2.69-2.57 (m, 3H), 2.18-2.12 (m, 1H)
13C NMR (125 MHz, CDCl3): δ (ppm) = 176.0, 138.3, 137.7, 137.6, 128.7, 128.5, 128.3, 128.1, 127.9, 127.8, 108.71, 80.0, 77.2, 74.8, 73.5, 73.0, 72.8, 72.2, 70.4, 68.0, 30.3, 28.2.
Partial characterization data obtained for products whose yields could not be optimized to synthetically useful levels.
3,4,6-Tri-O-methyl-D-glucopyranose 6
Prepared according to general procedure (4.1.2.2). 6 was obtained as a white solid (63 mg, 77%) after flash column chromatography on silica (0% to 10% methanol in ethyl acetate).
Rf = 0.30 (EtOAc)
1H NMR (500 MHz, CDCl3): δ (ppm) = 5.12 (d, J=3.6 Hz, 1H), 4.44 (d, J=7.8 Hz, 1H), 3.83 (ddd, J=10.1, 4.3, 2.7 Hz, 1H), 3.58 (d, J=2.3 Hz, 3H), 3.55 (dd, J=10.4, 2.1 Hz, 1H), 3.51-3.45 (m, 2H), 3.44 (d, J=1.6 Hz, 3H), 3.39-3.34 (m, 2H), 3.32 (d, J=2.1 Hz, 3H), 3.30-3.24 (m, 1H), 3.14-3.02 (m, 1.5H)
13C NMR (125 MHz, CDCl3): δ (ppm) = 96.6, 92.2, 86.1, 83.7, 79.6, 79.5, 74.7, 74.3, 72.2, 71.4, 69.1, 60.7 (2C), 60.2, 60.1, 59.0 (2C), 50.3

35
(5R,7R,8R,9R,10R)-10-hydroxy-8,9-dimethoxy-7-(methoxymethyl)-1,6-dioxaspiro[4.5]decan-2-one 7
Prepared according to general procedure (4.1.2.1). 7 was obtained as a clear oil after flash column chromatography on silica (5% methanol in ethyl acetate). Purification of the sugar lactones was challenging.
Rf = 0.60 (MeOH:EtOAc 5:95)
1H NMR (500 MHz, CDCl3): δ (ppm) = 3.75 (ddd, 10.0, 3.3, 2.0 Hz, 1H), 3.65 (s, 3H), 3.58-3.54 (m, 3H), 3.52 (s, 3H), 3.50-3.46 (m, 1H), 3.39 (s, 3H), 3.35-3.31 (m, 1H), 2.73-2.55 (m, 3H), 2.20-2.15 (m, 1H)
13C NMR (125 MHz, CDCl3): δ (ppm) = 175.8, 108.1, 84.3, 78.9, 73.8, 73.6, 70.4, 61.0, 60.2, 59.2, 29.9, 28.0
Partial characterization data obtained for products whose yields could not be optimized to synthetically useful levels.
(5R,8R,9R,10S)-6-hydroxy-9,10-dimethoxy-8-(methoxymethyl)-1,7-dioxaspiro[4.5]decan-2-one 8
Prepared according to general procedure (4.1.2.1). 7 was obtained as a clear oil after flash column chromatography on silica (5% methanol in ethyl acetate). Purification of the sugar lactones was challenging.
Rf = 0.65 (MeOH:EtOAc 5:95)
1H NMR (500 MHz, CDCl3): δ (ppm) = 4.78 (d, J=4.7 Hz, 1H), 4.15 (d, J=4.7 Hz, 1H), 3.62-3.60 (m, 2H), 3.58 (s, 3H), 3.54-3.51 (m, 2H), 3.51 (s, 3H), 3.39 (s, 3H), 2.98 (dd, J=9.9, 9.4 Hz, 1H), 2.68 (ddd, J=17.9, 10.9, 8.3 Hz, 1H), 2.53 (ddd, J=17.9, 11.0, 4.5 Hz, 1H), 2.35-2.16 (m, 2H)
13C NMR (125 MHz, CDCl3): δ (ppm) = 177.7, 95.7, 87.6, 85.6, 78.7, 74.8, 71.3, 61.6, 60.6, 59.3, 29.4, 19.5

36
Partial characterization data obtained for products whose yields could not be optimized to synthetically useful levels.
Methyl 2-O-pivaloyl-α-D-glucopyranoside 9
Prepared from methyl α-D-glucopyranoside (Carbosynth Ltd) according to general procedure (4.1.2.4). 9 was obtained as a white solid (343 mg, 72%) after flash column chromatography on silica (0% to 50% acetone in DCM). Spectral data agreed with those previously reported.58
Rf = 0.10 (Acetone:DCM 3:7)
1H NMR (400 MHz, CDCl3): δ (ppm) = 4.90 (d, J=3.7 Hz, 1H), 4.60 (dd, J=10.0, 3.7 Hz, 1H), 4.03–3.95 (m, 1H), 3.90–3.83 (m, 2H), 3.71–3.62 (m, 2H), 3.37 (s, 3H), 1.24 (s, 9H).
Methyl 3-keto-2-𝛂-D-deoxyglucopyranoside 10
Prepared according to general procedure (4.1.2.3). 10 was obtained as a white solid (67%) after flash column chromatography on silica (20% to 40% Acetone in DCM).
Rf = 0.33 (Acetone:DCM 4:6)
1H NMR (500 MHz, CDCl3): δ (ppm) = 5.17 (d, J = 4.4 Hz, 1H), 4.23 (dd, J = 9.8, 2.2 Hz), 3.98 (dd, J = 11.9, 2.8 Hz, 1H), 3.92 (dd, J = 12.0, 3.7 Hz, 1H), 3.76–3.72 (m, 1H), 3.35 (s, 3H), 2.82 (ddd, J = 14.0, 4.6, 1.4 Hz, 1H), 2.70 (dd, J = 14.0, 1.1 Hz, 1H).
13C NMR (125 MHz, CDCl3): δ (ppm) = 205.4, 100.1, 74.8, 73.3, 62.6, 55.1, 45.2.
IR (neat, cm-1): 3438 (w), 2930 (w), 1724 (s), 1365 (s), 1290 (s), 1194 (s), 1116(s), 1099 (s), 1034 (s), 1002 (s), 955 (s), 878 (s), 840 (s).
HRMS (DART+, m/z): calculated for C7H16NO5 [M+NH4]+: 194.10230, found: 19410223.
[𝛂]𝐃𝟐𝟎 = + 117.1 (c = 11.75 mg/mL, CHCl3)

37
Methyl 2-O-benzoyl-α-D-glucopyranoside 11
Prepared from methyl α-D-glucopyranoside (Carbosynth Ltd). Synthesized and characterized as previously reported using dibutyltin chloride.67 11 was obtained as a white solid (70%) after flash column chromatography on silica (10% Methanol in DCM).
Rf = 0.30 (MeOH;DCM 1:9)
1H NMR (400 MHz, CDCl3): δ (ppm) = 8.09 (d, J=6.9 Hz, 2H), 7.60 (t, J=7.5 Hz, 1H), 7.47 (t, J=7.5 Hz, 2H), 5.04 (d, J = 3.6 Hz, 1H), 4.91 (dd, J=3.6, 9.9 Hz, 1H), 4.16 (t, J=3.6 Hz, 1H), 3.92-3.89 (m, 2H), 3.75-3.72 (m, 2H), 3.40 (s, 3H), 2.62 (br s, 1H), 2.50 (br s, 1H), 2.00 (br s, 1H).
Methyl 2-O-tosyl-α-D-glucopyranoside 12
Prepared from methyl α-D-glucopyranoside (Carbosynth Ltd). Synthesized using a modified procedure using dibutyltin chloride.67 12 was obtained as an off white solid (14%) after flash column chromatography on silica (10% Methanol in DCM). Spectral data agreed with those previously reported.68
Rf = 0.32 (MeOH;DCM 1:9)
1H NMR (400 MHz, CDCl3): δ (ppm) = 7.84 (d, J=8.3 Hz, 1H), 7.34 (d, J=8.1 Hz, 1H), 4.65 (d, J=3.7 Hz, 1H), 4.36 (dd, J=9.7, 3.7 Hz, 1H), 4.14 (br s, 1H), 3.96-3.87 (m, 1H), 3.81 (dd, J=13.9, 2.9 Hz, 1H), 3.66-3.51 (m, 1H), 3.25 (s, 1H), 2.94 (br s, 1H), 2.43 (s, 3H)

38
Methyl 4-O-pivaloyl-6-O-(tert-butyldimethylsilyl)-𝛂-D-mannopyranoside 13
Prepared from methyl 6-O-(tert-butyldimethylsilyl)-α-D-mannopyranoside69 according to general procedure (4.1.2.4). 14 was obtained as a yellow solid (78%) after flash column chromatography on silica (30% to 50% ethyl acetate in hexanes).
Rf = 0.47 (EtOAc:Hexanes 4:6)
1H NMR (500 MHz, CDCl3): δ (ppm) = 4.93 (tt, J = 9.5, 2.0 HZ, 1H), 4.77 (d, J = 1.5 Hz, 1H), 3.92–8.85 (m, 2H), 3.7–3.67 (m, 3H), 3.39 (s, 3H), 1.22 (s, 9H), 0.88 (s, 9H), 0.06 (s, 3H), 0.05 (s, 3H).
13C NMR (125 MHz, CDCl3): δ (ppm) = 179.8, 100.4, 71.0, 70.8, 70.8, 70.6, 62.6, 55.1, 39.1, 27.2, 26.0, 18.4, -5.2, -5.2.
IR (neat, cm-1 ): 3444 (w), 2934 (w), 1731 (s), 1249 (s), 1150 (s), 1110 (s), 1038 (s), 972 (s), 836 (s), 776 (s), 569 (s).
HRMS (DART+, m/z): calculated for C18H37O7Si [M+NH4]+: 393.23031; found: 393.23089
Methyl 2-O-(tert-butyloxycarbonyl)-𝛂-D-glucopyranoside 14
Prepared from methyl α-D-glucopyranoside (Carbosynth Ltd) according to modified general procedure (4.1.2.4). Boc2O was used as the acylating agent, DMAP (10 mol%) was added. 18 was obtained as an off white solid (30%) after flash column chromatography on silica (20% to 50% acetone in DCM).
Rf = 0.22 (Acetone:DCM 1:1)
1H NMR (500 MHz, CDCl3): δ (ppm) = 4.95 (d, J=3.6 Hz, 1H), 4.47 (dd, J=10.0, 3.6 Hz, 1H), 3.96 (t, J=8.8 Hz, 1H), 3.86 (s, 2H), 3.69-3.59 (m, 2H), 3.58 (s, 1H), 3.39 (s, 3H), 3.31 (s, 1H), 1.49 (s, 9H)
13C NMR (125 MHz, CDCl3): δ (ppm) = 153.2, 97.1, 83.2, 75.7, 71.7, 70.8, 70.5, 61.9, 55.3, 27.7
IR (neat, cm-1): 3422 (w), 2989 (w), 2940 (w), 1739 (s), 1278 (s), 1154 (s), 1038 (s), 820 (s)

39
HRMS (DART+, m/z): calculated for C12H24O8 [M+NH4]+: 312.16584; found: 312.16529
[𝛂]𝐃𝟐𝟎 = +32.0 (c = 13.3 mg/mL, CHCl3)
Methyl 2-O-benzoyl-α-L-fucopyranoside 15
Prepared from methyl α-L-fucopyranoside (Carbosynth Ltd) according to general procedure (4.1.2.5). 15 was obtained as a white solid (56%) after flash column chromatography on silica (30% acetone in hexane). Spectral data agreed with those previously reported.
Rf = 0.18 (Acetone:Hexane 3:7)
1H NMR (500 MHz, CDCl3): δ (ppm) = δ 8.08–8.06 (m, 2H), 7.59–7.55 (m, 1H), 7.45–7.41 (m, 2H), 5.21 (dd, J=10.0, 4.0 Hz, 1H), 4.98 (d, J=4.0 Hz, 1H), 4.17 (dd, J=10.0, 3.2 Hz, 1H), 4.04 (q, J=6.4 Hz, 1H), 3.87 (d, J=3.2 Hz, 1H), 3.39 (s, 3H), 1.83 (br s, 2H), 1.34 (d, J=6.4 Hz, 3H)
Ethyl 3,4,6-tri-O-benzyl-2-O-pivaloyl-thio-β-D-glucopyranoside 16
Prepared according to general procedure (4.1.2.6). 16 was obtained as an off white solid (85%) after flash column chromatography on silica (5% to 20% ethyl acetate in hexane).
Rf = 0.65 (EtOAc:Hexane 2:8)
1H NMR (500 MHz, CDCl3): δ (ppm) = 7.38-7.26 (m, 13H), 7.19-7.16 (m, 2H), 5.16-5.09 (m, 1H), 4.79 (dd, J=11.0, 4.0 Hz, 2H), 4.72 (d, J=11.0 Hz, 1H), 4.63 (d, J=12.1 Hz, 1H), 4.58-4.55 (m, 2H), 4.41 (d, J=10.0 Hz, 1H), 3.78 (dd, J=11.0, 2.1 Hz, 1H), 3.75-3.71 (m, 3H), 3.54 (ddt, J=6.7, 4.7, 1.9 Hz, 1H), 2.81-2.64 (m, 2H), 1.28 (t, J=7.5, 3H), 1.22 (s, 9H)
13C NMR (125 MHz, CDCl3): δ (ppm) = 177.9, 138.2 (2C), 138.0, 128.4 (3C), 128.0, 127.8, 127.7, 127.6, 127.6, 127.4, 84.7, 83.5, 79.5, 77.8, 75.2, 75.0, 73.5, 71.5, 68.9, 38.7, 27.2, 23.6, 15.0
IR (neat, cm-1): 2976 (s), 2931 (s), 2973 (s), 1735 (s), 1503 (s), 1453 (s), 1362 (s). 1277 (s), 1134 (s), 1092 (s) 1071 (s), 749 (s) 696 (s)

40
HRMS (DART+, m/z): calculated for C34H42O6S [M+NH4]+: 596.30459; found: 596.30404
[𝛂]𝐃𝟐𝟎 = -58.0 (c = 11.0 mg/mL, CHCl3)
2-O-Pivaloyl-β-D-glucopyranoside-(1-6)-methyl 2,3,4-tri-O-acetyl-α-D-glucopyranoside 17
Prepared according to general procedure (4.1.2.6) from 16 and methyl 2,3,4-tri-O-acetyl-α-D-glucopyranoside.70 17 was obtained as a white solid (48%) after flash column chromatography on silica (0% to 10% methanol in ethyl acetate).70
Rf = 0.10 (EtOAc)
1H NMR (500 MHz, CDCl3): δ (ppm) = 5.47 (dd, J=10.2, 9.2 Hz, 1H), 4.97 (dd, J=10.1, 9.2 Hz, 1H), 4.89 (d, J=3.6, 1H), 4.83 (dd, J=10.2, 3.6 Hz, 1H), 4.73-4.69 (m, 1H), 4.51 (d, J=7.9 Hz, 1H), 3.94-3.89 (m, 2H), 3.87 (dd, J=10.5, 3.2 Hz, 1H), 3.84-3.79 (m, 1H), 3.63-3.60 (m, 2H), 3.57 (dd, J=10.6, 5.7 Hz, 1H), 3.38 (s, 3H), 2.82-2.79 (m, 2H), 2.41 (br s, 1H), 2.05 (s, 3H), 2.04 (s, 3H), 1.99 (s, 3H), 1.24 (s, 9H)
13C NMR (125 MHz, CDCl3): δ (ppm) = 178.8, 170.2, 170.0, 100.5, 96.4, 75.7, 75.3, 73.8, 71.2, 70.8, 70.0, 69.7, 68.0, 67.9, 62.1, 55.4, 39.0, 27.0, 16.3
2-O-Pivaloyl-β-D-glucopyranoside-(1-6)-1,2:3,4-di-O-isopropylidene-α-D-galactopyranoside 18
Prepared according to general procedure (4.1.2.6) from 16 and 1,2:3,4-Di-O-isopropylidene-α-D-galactopyranose (Sigma Aldrich). 18 was obtained as an off white solid (80%) after flash column chromatography on silica (20% to 50% acetone in DCM).
Rf = 0.35 (Acetone:DCM 1:1)

41
1H NMR (500 MHz, CDCl3): δ (ppm) = 5.49 (d, J=5.0 Hz, 1H), 4.69 (td, J=7.7, 2.5 Hz, 1H), 4.59 (dd, J=7.9, 2.5 Hz, 1H), 4.56 (d, J=7.8 Hz, 1H), 4.29 (dd, J=5.0, 2.4 Hz, 1H), 4.26 (dd, J=12.5, 6.0 Hz, 1H), 4.00 (dd, J=10.3, 6.1 Hz, 1H), 3.94-3.88 (m, 2H), 3.78 (dd, J=12.5, 6.0 Hz, 1H), 3.68 (dd, J=10.3, 6.1 Hz, 1H), 3.63-3.56 (m, 2H), 3.40 (ddd, J=9.0, 5.6, 3.2 Hz, 1H), 2.95 (br s, 1H), 2.90 (br s, 1H), 2.55 (br s, 1H), 1.50 (s, 3H), 1.44 (s, 3H), 1.34 (s, 3H), 1.31 (s, 3H), 1.24 (s, 9H)
13C NMR (125 MHz, CDCl3): δ (ppm) = 179.1, 109.4, 108.6, 101.0, 96.3, 75.9, 75.2, 74.3, 71.5, 70.9, 70.6, 70.5, 68.6, 66.8, 62.35, 53.8, 39.0, 29.3, 27.1, 26.1, 25.9, 24.9, 24.4
IR (neat, cm-1): 3422 (w), 2989 (s), 1739 (s), 1266 (s), 1391 (s), 1266 (s), 1182 (s), 1070 (s), 1008 (s)
HRMS (DART+, m/z): calculated for C23H38O12 [M+NH4]+: 524.27070; found: 524.27015
[𝛂]𝐃𝟐𝟎 = -37.6 (c = 5.0 mg/mL, CHCl3)
Methyl 2-O-pivaloyl-β-D-glucopyranoside 19
Prepared according to a modified general procedure (4.1.2.6). B was treated with MeOH followed by pivaloylation. 19 was obtained as an off white solid (81%) after flash column chromatography on silica (10% to 50% acetone in DCM).
Rf = 0.28 (Acetone:DCM 1:1)
1H NMR (500 MHz, CDCl3): δ (ppm) = 4.70 (dd, J=9.5, 7.9 Hz, 1H), 4.38 (d, J=7.9 Hz, 1H), 3.95-3.84 (m, 2H), 3.72-3.57 (m, 3H), 3.50 (s, 3H), 3.43-3.34 (m, 2H), 2.72 (br s, 1H), 1.22 (s, 9H)
13C NMR (125 MHz, CDCl3): δ (ppm) = 178.7, 102.1, 75.5, 75.3, 73.9, 70.7, 61.9, 57.2, 38.9, 27.0
IR (neat, cm-1): 3412 (b), 2985 (s), 1738 (s), 1405 (s), 1485 (s), 1289 (s), 1161 (s), 1060 (s)
HRMS (DART+, m/z): calculated for C12H22O7 [M+NH4]+: 296.17093; found: 296.17038
[𝛂]𝐃𝟐𝟎 = -19.4 (c = 5 mg/mL, CHCl3)

42
Sodium dibutyl phosphate 20
Prepared from dibutyl phosphoric acid (Sigma Aldrich). Synthesized as previously reported.71
1H NMR (400 MHz, CDCl3): δ (ppm) = 3.79 (q, J=6.5 Hz, 2H), 1.59 (dq, J=12.4, 7.0 Hz, 2H), 1.44-1.30 (m, 2H), 0.94 (t, J=7.3 Hz, 3H)
13C NMR (125 MHz, CDCl3): δ (ppm) = 33.0, 32.9, 19.1, 13.9
31P NMR (121 MHz, CDCl3): δ (ppm) = 1.50
Tetrabutylammonium dibutyl phosphate 21
Prepared from dibutyl phosphoric acid (Sigma Aldrich). Synthesized and characterized as previously reported.72
1H NMR (400 MHz, CDCl3): δ (ppm) = 3.85-3.80 (m, 4.2H), 3.42-3.35 (m, 8H), 1.70-1.60 (m, 12.4H), 1.49-1.35 (m, 12.4 H), 0.99 (t, J=7.4 Hz, 12H), 0.89 (t, J = 7.4 Hz, 6.5H)
Potassium dibutyl phosphate 22
Prepared from dibutyl phosphoric acid (Sigma Aldrich). Synthesized as previously reported.71
1H NMR (400 MHz, CDCl3): δ (ppm) = 3.81 (q, J=6.6 Hz, 2H), 1.59 (m, 2H), 1.45-1.33 (m, 2H), 0.94 (t, J=7.4 Hz, 3H)
13C NMR (125 MHz, CDCl3): δ (ppm) = 65.3 (2C), 32.9 (2C), 19.0, 13.8
31P NMR (121 MHz, CDCl3): δ (ppm) = -0.1

43
Tetrabutylammonium diphenyl phosphate 23
Prepared from diphenyl phosphoric acid (Sigma Aldrich). Synthesized as previously reported.72
1H NMR (400 MHz, CDCl3): δ (ppm) = 7.28=7.17 (m, 4H), 7.05 (m, 1H), 3.21-3.12 (m, 2H), 1.54 (p, J=8.1, 7.6 Hz, 2H), 1.32 (h, J=7.3 Hz, 2H), 0.90 (t, J=7.3 Hz, 3H)
13C NMR (125 MHz, CDCl3): δ (ppm) = 152.3, 152.2, 129.2, 123.6 (2C), 120.4 (2C), 58.63, 23.8, 19.6, 13.6
31P NMR (121 MHz, CDCl3): δ (ppm) = -12.9

44
References
1. de Armas, P.; Francisco, C. G.; Suarez, E. J. Am. Chem. Soc. 1993, 115(19), 8865-8866. 2. Hanessian, S.; Rancourt, G. Carbohydrate Chemistry. 1977, 1201-1214, Pergamon. 3. Pérez‐Martín, I.; Suárez, E. Radicals and carbohydrates. 2012. Wiley, Encyclopedia of
Radicals in Chemistry, Biology and Materials. 4. Suárez, E.; Rodriguez, M. S. Radicals in Organic Synthesis. 2001. Wiley, 440-454. 5. Nicolaou, K. C.; Dai, W. M. Angew. Chem. Int. Ed. 1991, 30(11), 1387-1416. 6. Koester, D. C.; Holkenbrink, A.; Werz, D. B. Synthesis. 2010, 3217−3242. 7. Xu, L. Y.; Fan, N. L.; Hu, X. G. Org. Biomol. Chem. 2020, 18, 5095-5109 8. Giese, B. Angew. Chem. Int. Ed. 1985, 24, 553-565. 9. Giese, B. Pure Appl. Chem. 1988, 60(11), 1655-1658. 10. Giese, B. Angew. Chem. Int. Ed. 1989, 28(8), 969-980. 11. Dupuis, J.; Giese, B.; Rüegge, D.; Fischer, H.; Korth, H. G.; Sustmann, R. Angew. Chem.
Int. Ed. 1984, 23(11), 896-898. 12. Roe, B. A.; Boojamra, C. G.; Griggs, J. L.; Bertozzi C. R. J. Org. Chem. 1996, 61, 6442–
6445. 13. Abe, H.; Shuto, S.; Matsuda, A. J. Am. Chem. Soc. 2001, 123, 11870–11882. 14. Giese, B.; Witzel, T. Angew. Chem. 1986, 98, 459-460. 15. Miquel, N.; Doisneau, G.; Beau, J. M. Angew. Chem. Int. Ed. 2000, 39(22), 4111-4114. 16. Rubinstenn, G.; Mallet, J. M.; Sinaÿ, P. Tetrahedron Lett., 1998, 39, 3697–3700. 17. Giese, B.; Witzel. T. Angew. Chem. Int. Ed. 1986, 25, 450-451. 18. Alonso-Cruz, C. R.; Kennedy, A. R.; Rodríguez, M. S.; Suárez, E. Org. Lett. 2003, 5(20),
3729-3732. 19. Stubbe, J. Enzymol. Relat. Areas Mol. Biol. 1990, 63, 349-417.; Stubbe, J.; van der Donk,
W. A. Chem. Biol. 1995, 2, 793-801. 20. Lenz, R.; Giese, B. J. Am. Chem. Soc. 1997, 119(12), 2784-2794.
21. Shaw, M. H.; Twilton, J.; MacMillan, D. W. J. Org. Chem. 2016, 81(16), 6898-6926.
22. Kärkäs, M. D.; Porco, J. A., Jr.; Stephenson, C. R. J. Chem. Rev. 2016, 116(17), 9683-9747
23. Romero, N. A.; Nicewicz, D. A. Chem. Rev. 2016, 116(17), 10075-10166 24. Narayanam, J. M. R.; Stephenson, C. R. J. Chem. Soc. Rev. 2011, 40, 102−113 25. Hedstrand, D. M.; Kruizinga, W. M.; Kellogg, R. M. Tetrahedron Lett. 1978, 19,
1255−1258. 26. Okada, K.; Okamoto, K.; Morita, N.; Okubo, K.; Oda, M. J. Am. Chem. Soc. 1991, 113,
9401−9402. 27. Ischay, M. A.; Anzovino, M. E.; Du, J.; Yoon, T. P. J. Am. Chem. Soc. 2008, 130,
12886−12887. 28. Nicewicz, D. A.; MacMillan, D. W. C. Science. 2008, 322, 77−80. 29. Capaldo, L.; Ravelli, D. Eur. J. Org. Chem. 2017, 15, 2056-2071.; Capaldo, L.; Quadri,
L. L.; Ravelli, D. Green Chem. 2020, 22, 3376-3396 30. Hamilton, D. S.; Nicewicz, D. A. J. Am. Chem. Soc. 2012, 134, 18577−18580. 31. Ohkubo, K.; Kobayashi, T.; Fukuzumi, S. Angew. Chem., Int. Ed. 2011, 50, 8652−8655.;
Ohkubo, K.; Fujimoto, A.; Fukuzumi, S. J. Phys. Chem. A. 2013, 117, 10719−10725.; Romero, N. A.; Margrey, K. A.; Tay, N. E.; Nicewicz, D. A. Science 2015, 349, 1326−1330.

45
32. Qvortrup, K.; Rankic, D. A.; MacMillan, D. W. C. J. Am. Chem. Soc. 2014, 136, 626−629.
33. Jeffrey, J. L.; Terrett, J. A.; MacMillan, D. W. C. Science, 2015, 349, 1532−1536. 34. Skubi, K. L.; Blum, T. R.; Yoon, T. P. Chem. Rev. 2016, 116(17), 10035-10074. 35. Twilton, J.; Christensen, M.; DiRocco, D. A.; Ruck, R. T.; Davies, I. W.; MacMillan, D.
W. Angew. Chem. 2018, 130(19), 5467-5471. 36. Cernak, T.; Dykstra, K. D.; Tyagarajan, S.; Vachal, P.; Krska, S. W. Chem. Soc. Rev.
2016, 45, 546–576. 37. Kim, S. W.; Zhang, W.; Krische, M. J. Acc. Chem. Res. 2017, 50, 2371; Alam, R.;
Molander, G. A. J. Org. Chem. 2017, 82(24), 13728-13734. 38. Twilton, J.; Christensen, M.; DiRocco, D. A.; Ruck, R. T.; Davies, I. W.; MacMillan, D.
W. Ange. Chem. 2018, 130(19), 5467-5471. 39. Sakai, K.; Oisaki, K.; Kanai, M. Adv. Synth. Catal. 2020, 362(2), 337-343. 40. Dimakos, V.; Su, H. Y.; Garrett, G. E.; Taylor, M. S. J. Am. Chem. Soc. 2019, 141(13),
5149-5153. 41. Dimakos, V.; Gorelik, D.; Su, H. Y.; Garrett, G. E.; Hughes, G.; Shibayama, H.; Taylor,
M. S. Chem. Sci. 2020, 11(6), 1531-1537.
42. Zhao, G.; Wang, T. Angew. Chem. Int. Ed. 2018, 57(21), 6120-6124. Spell, M. L.; Deveaux, K.; Bresnahan, C. G.; Bernard, B. L.; Sheffield, W.; Kumar, R.; Ragains, J. R. Angew. Chem. Int. Ed. 2016, 55(22), 6515-6519.; Ye, H.; Xiao, C.; Zhou, Q. Q.; Wang, P. G.; Xiao, W. J. J. Org. Chem. 2018, 83(21), 13325-13334.; Sangwan, R.; Mandal, P. K. RSC Adv. 2017, 7(42), 26256-26321.; Zhao, G.; Kaur, S.; Wang, T. Org. Lett. 2017, 19(12), 3291-3294.
43. Zhu, F.; Zhang, S. Q.; Chen, Z.; Rui, J.; Hong, X.; Walczak, M. A. J. Am. Chem. Soc. 2020, 142, 25, 11102–11113
44. Andrews, R. S.; Becker, J. J.; Gagné, M. R. Angew. Chem. 2010, 122(40), 7432-7434. 45. Ji, P.; Zhang, Y.; Wei, Y.; Huang, H.; Hu, W.; Mariano, P. A.; Wang, W. Org. Lett.
2019, 21(9), 3086-3092. 46. Wan, I. C. S.; Witte, M. D.; Minnaard, A. J. Org. Lett. 2019, 21, 7669–7673. 47. Badir, S. O.; Dumoulin, A.; Matsui, J. K.; Molander, G. A. Angew. Chem. Int. Ed. 2018,
57(22), 6610-6613. 48. Shaw, M. H.; Twilton, J.; MacMillan, D. W. C. J. Org. Chem. 2016, 81, 6898–6926. 49. Wan, I. C. S.; Witte, M. D.; Minnaard, A. J. Chem. Comm. 2017, 53(36), 4926-4929. 50. Wang, Y.; Carder, H. M.; Wendlandt, A. E. Nature. 2020, 578(7795), 403-408. 51. Awan, S. I.; Werz, D. B. Bioorg. Med. Chem. 2012, 20, 1846. 52. Lei, P. S.; Duchaussoy, P.; Sizun, P.; Mallet, J. M.; Petitou, M.; Sinaÿ, P. Bioorg. Med.
Chem. 1998, 6(8), 1337-1346. 53. Noort, D.; Van Straten, N. C. R.; Boons, G. J. P. H.; Van der Marel, G. A.; Bossuyt, X.;
Blanckaert, N.; ... Van Boom, J. H. Bioorg. Med. Chem. Lett. 1992, 2(6), 583-588. 54. Bennett, C. S.; Galan, M. C. Chem. Rev. 2018, 118(17), 7931-7985. 55. Izumi, S.; Kobayashi, Y.; Takemoto, Y. Org. Lett. 2019, 21(3), 665-670. 56. Hager, D.; MacMillan, D. W. J. Am. Chem. Soc. 2014, 136(49), 16986-16989. 57. Mai-Linde, Y.; Linker, T. Org. Lett. 2020, 22(4), 1525-1529. 58. Mancini, R. S.; Lee, J. B.; Taylor, M. S. Org. Biomol. Chem. 2017, 15(1), 132-143. 59. Choi, G. J.; Zhu, Q.; Miller, D. C.; Gu, C. J.; Knowles, R. R. Nature. 2016, 539(7628),
268-271.

46
60. Loscher, S.; Schobert, R. Eur. J. Chem. 2013, 19(32), 10619-10624. 61. Asai, N.; Fusetani, N.; Matsunaga, S. J. Nat. Prod. 2001, 64, 1210 62. Evtushenko, E.V. Carbohydr. Res. 2012, 359, 111-119. 63. Singh, A. K.; Kandasamy. J. Org. Biomol. Chem. 2018, 16(28), 5107-5112. 64. Cheshev, P.; Marra, A.; Dondoni, A. Carbohydr. Res. 2006, 341(16), 2714-2716. 65. Seeberger, P. H.; Eckhardt, M.; Gutteridge, C. E.; Danishefsky, S. J. J. Am. Chem. Soc.
1997, 119(42), 10064-10072. 66. Premathilake, H. D.; Mydock, L. K.; Demchenko, A. V. J. Org. Chem. 2010, 75(4),
1095-1100 67. Demizu, Y.; Kubo, Y.; Miyoshi, H.; Maki, T.; Matsumura, Y.; Moriyama, N.; Onomura,
O. Org. Lett. 2008, 10(21), 5075-5077. 68. Muramatsu, W. J. Org. Chem. 2012, 77(18), 8083-8091. 69. Doris, L.; Taylor, M. S. J. Am. Chem. 2011, 133(11), 3724 - 3727 70. Wahlstrom, J. L., & Ronald, R. C. J. Org. Chem. 1998, 63(17), 6021-6022. 71. Chen, K.; Schwarz, J.; Karl, T. A.; Chatterjee, A.; König, B. Chem. Comm. 2019, 55(87),
13144-13147. 72. Zhu, Q.; Graff, D. E.; Knowles, R. R. J. Am. Chem. Soc. 2018, 140(2), 741-747.