Embed Size (px)
Citation preview

SINDROMES MIELOPROLIFERATIVOS MARCO ADOLFO TOBAR MARCILLORESIDENTE 1ER AÑO MEDICINA INTERNA

NEOPLASIAS MIELOPROLIFERATIVAS
Son alteraciones clonales de la célula madre hematopoyética, caracterizados por la proliferación de células sanguíneas maduras, no reactivas, de las líneas celulares mieloides (granulocitos, eritrocitos,
megacariocitos y mastocitos).
Peter J. Campbell, Mechanisms of Disease, The Myeloproliferative Disorders, NEJM 2006

HISTORIA1892 VAQUEZ reconocimiento de policitemia vera.
1930 Reconocimiento de trombocitemia esencial
1951 Dameshek relación de patologías
Desordenes mieloproliferativos
1973 identificación del cromosoma Filadelfia
1976 mutaciones en células madre

HISTORIA 2002 Descripción de la recombinación mitótica que compromete el cromosoma9p como la citogenética más común lesión en la policitemia vera.
2003 Reconocimiento de las señalizaciones JAK STAT y PI3K crecimiento eritrocitario en ausencia de eritropoyetina
2005 descripción de la mutación V617F JAK2

CASIFICACION DE LA OMS 2008
Peter J. Campbell, Mechanisms of Disease, The Myeloproliferative Disorders, NEJM 2006

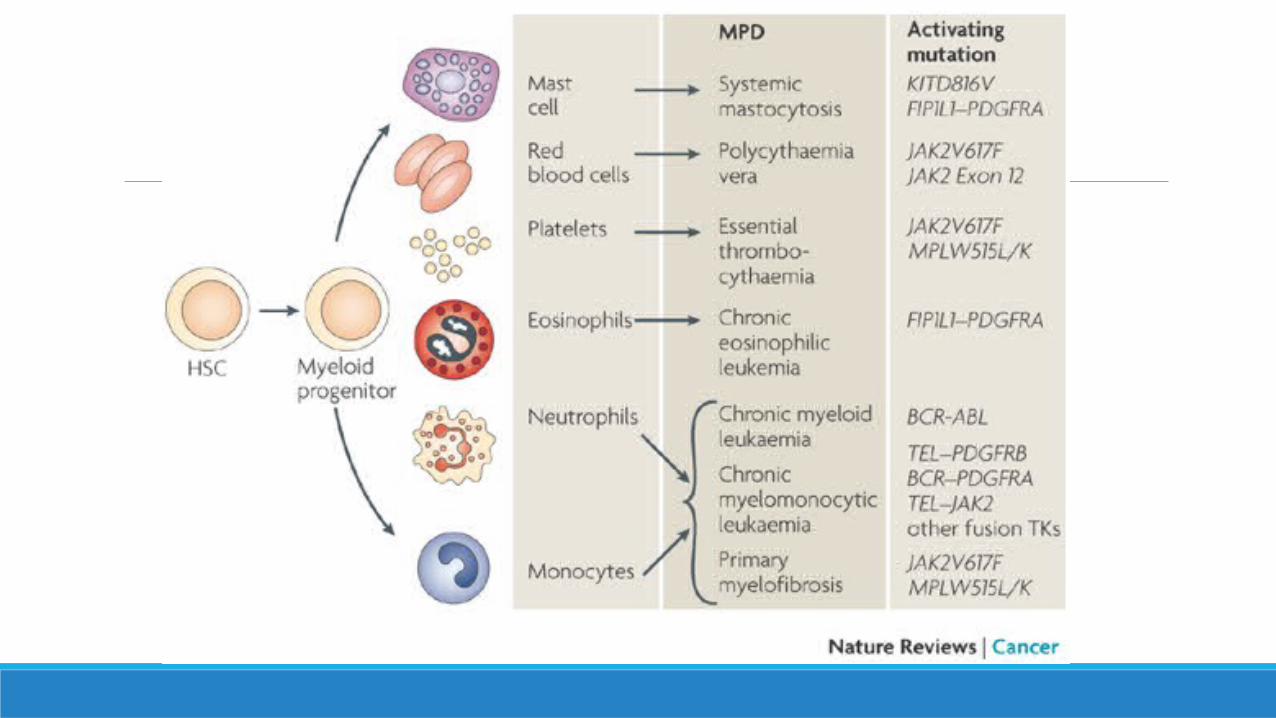
CLASIFICACION NEOPLASIAS MIELOPROLIFERATIVAS
Peter J. Campbell, Mechanisms of Disease, The Myeloproliferative Disorders, NEJM 2006

POLICITEMIA VERA

DEFINICION OMS 2008 Neoplasia mieloproliferativa crónica Incremento en la producción de células eritroides en forma independiente de los mecanismos normales de regulación de la eritropoyesis
Mutación JAK2 V617F u otra mutación similar Proliferación eritroide, de megacariocitos y de serie granular
Up To Date 2015

EPIDEMIOLOGIA La edad media al diagnóstico es de aproximadamente 60 años. La incidencia de la policitemia vera es ligeramente mayor en hombres que en mujeres (2,8 frente a 1,3 casos / 100.000 por año). Con un promedio de 1.9 casos por 100.000 habitantes por año.
La exposición a la radiación ionizante se ha sugerido como un factor de riesgo, aunque la gran mayoría de los pacientes con PV no tienen evidencia de exposición al riesgo.
Up To Date 2015

FISIOPATOLOGIAUNION DE CITOCINA AL RECEPTOR
FOSFORILACION DEL JAK
FOSFORILACION DE LAS CITOCINAS DEL RECEPTOR
DIMERIZACION DEL STAT
TRASLOCACION AL NUCLEO

FISIOPATOLOGIA
Peter J. Campbell, Mechanisms of Disease, The Myeloproliferative Disorders, NEJM 2006

FISIOPATOLOGIA
Peter J. Campbell, Mechanisms of Disease, The Myeloproliferative Disorders, NEJM 2006

MANIFESTACIONES CLINICAS La mayoría de los pacientes con policitemia vera se descubre por casualidad cuando se observa una hemoglobina o hematocrito elevado, en un conteo sanguíneo completo obtenido por alguna otra razón.
Hipertensión 46 por ciento
Prurito 36 por ciento
Síntomas vasomotores 29 por ciento
Trombosis arterial 16 por ciento
Trombosis venosa 7 por ciento
Hemorragia mayor 4 por ciento
Perturbación visual transitoria

EXPLORACION FISICA
Plétora facial (cianosis rubicunda)
La hepatomegalia en una minoría de casos
Excoriación de la piel, que puede ser extensa, lo que sugiere la presencia de prurito severo.
Estigmas de un evento trombotico venoso o arterial (por ejemplo, derrame cerebral, trombosis venosa profunda y tromboflebitis superficial)
Artritis gotosa y tofos

CARACTERISTICAS DE LABORATORIO
● Hemoglobina> 18,5 g / dl 73 por ciento
● Leucocitos totales (WBC)> 10.500 49 por ciento
● Plaquetas> 450.000 53 por ciento
recuento de plaquetas > 1.000.000 4 por ciento
● Reacción leucoeritroblastica 6 por ciento
● Elevado lactato deshidrogenasa en suero 50 por ciento
● Mutación JAK2 positividad 98 por ciento
● Nivel bajo de eritropoyetina en suero 81 por ciento
Up To Date 2015

Medula ósea y frotis periférico Se caracteriza por un aumento del hematocrito en la sangre periférica; medula ose hipercelular con un mayor número de precuersores eritroides y una variable aumento en el número de fibras de reticulina.
La hipercelularidad varia del 36 al 100% con una media de 81%.
Peter J. Campbell, Mechanisms of Disease, The Myeloproliferative Disorders, NEJM 2006

Marcadores clonales
Mutaciones JAK2 - 95 a 100 por ciento de los pacientes con policitemia vera tienen una mutación en JAK2, que implica ya sea el exón 14 o 12.
Mutación en el exón 14 95 a 97% Mutación en el exón 12 1%
Peter J. Campbell, Mechanisms of Disease, The Myeloproliferative Disorders, NEJM 2006

Otras características de laboratorio
Los pacientes con policitemia vera usualmente tienen bajas concentraciones de eritropoyetina sérica.
Los bajos niveles de EPO son altamente específicos para PV, niveles superiores a lo normal son inusuales y sugieren eritrocitosis secundaria, con una especificidad del 98 por ciento.
La formación de colonias eritroides endógenas en ausencia de eritropoyetina es característica de la policitemia vera con una sensibilidad y especificidad del 100% en ausencia de quimioterapia citotoxica previa.
Up To Date 2015

EVALUACION DIAGNOSTICA

CRITERIOS DIAGNOSTICOS OMS 2008
CRITERIOS MAYORES
1- Hb > 18,5 g/dl H, > 16,5 g/dl M, u otra evidencia de aumento de masa de células rojas
2- JAK 2 V617F u otra similar como JAK2 exon 12
CRITERIOS MENORES
1- Biopsia de medula osea con panmielosis
2- EPO sérica descendida
3- Formación de colonias eritroides in vivo
Up To Date 2015

ESTRATIFICACION DE RIESGO La mediana de supervivencia de los pacientes sintomáticos no tratados con policitemia vera se estimó inicialmente en 6 a 18 meses, mientras que la supervivencia actual de los pacientes tratados es de 13 años o más.
Incluso con el tratamiento, la mortalidad general es mayor que la de una población normal por edad y sexo emparejado.
La mortalidad cardiovascular represento el 45% de las causa, los tumores sólidos el 20% y la transformación a otras neoplasia hematológicas representaron el 13%.
Up To Date 2015

ESTRATIFICACION DE RIESGO RIESGO MAYOR DE 60 AÑOS O HISTORIA
DE TROMBOSISFACTORES DE RISGO CARDIOVASCULAR
BAJO NO NO
INTERMEDIO NO SI
ALTO SI -----
TROMBOCITOSIS: MAYOR DE 1.000.000
Polycythemia vera and essential thrombocythemia: 2013 update on diagnosis, risk-stratification, and management. AMERICAN JOURNAL OF HEMATOLOGY

TRATAMIENTO
Polycythemia vera and essential thrombocythemia: 2013 update on diagnosis, risk-stratification, and management. AMERICAN JOURNAL OF HEMATOLOGY

How I treat polycythemia vera, Francesco Passamonti, 2012 by The American Society of Hematology, BLOOD
Punto primario eventos cardiovasculares
365 pacientes en tratamiento con hidroxiurea, flebotomía o ambos

TRATAMIENTO FLEBOTOMIA
Indicadas en pacientes con bajo riesgo de complicaciones tromboticas
500 ml disminuyen 3% aproximadamente el valor del hematocrito.
CONTROL OPTIMO<45% en hombres
< 42% en mujeres
How I treat polycythemia vera, Francesco Passamonti, 2012 by The American Society of Hematology, BLOOD

TRATAMIENTO HIDROXIUREA
Inhibe la ribonucleótido-reductasa, enzima que transforma los ribonucleótidos en desoxirribonucleótidos.
Su acción es máxima en la fase S del ciclo celular.
Aumenta el riesgo de transformación a leucemia mieloide aguda.
Dosis de inicio 15 a 20 mg por kilo en 24 horas, mantenimiento 10 mg por kilo cada 24 horas
INTERFERON ALFA
Dosis de 3 milones de unidades 3 veces a la semana
35% de los pacientes abandonan el tratamiento por efectos adversos
How I treat polycythemia vera, Francesco Passamonti, 2012 by The American Society of Hematology, BLOOD

TRATAMIENTO BUSULFAN
El busulfán es un miembro de los agentes alquilantes con grupos sulfonato.
Se hidroliza para liberar los grupos metanosulfate, produciendo iones carbono reactivos que pueden alquilar el ADN y otras proteínas ocasionando citotoxicidad.
Dosis de 4mg al día por 2 semanas.
RUXOLITINIB
Inhibidor del gen JAK 2.
How I treat polycythemia vera, Francesco Passamonti, 2012 by The American Society of Hematology, BLOOD

Puntos primarios, reducción del tamaño del bazo a las 32 semanas de tratamiento y control de hematocrito a
las 35 semanas de tratamiento.
Terapia estándar 110 pacientes, Ruxolitinib 112 pacientes

TRATAMIENTO RESPUESTA
CLINICO HEMATOLOGICA Disminución de los requerimientos de flebotomías, hematocrito <45%, plaquetas <450000, leucocitos < 10000, tamaño de baso normal.
MOLECULAR Disminución de anormalidades moleculares a niveles indetectables, LDH en nivel normal
MEDULA OSEA Normocelularidad y ausencia de fibrosis reticulinica
How I treat polycythemia vera, Francesco Passamonti, 2012 by The American Society of Hematology, BLOOD

PRONOSTICO Complicaciones hematológicas 15 al 21% de los pacientes desarrollan mielofibrosis. 7% de los pacientes desarrollan leucemia mieloide aguda Supervivencia en pacientes no tratados es de 18 meses Supervivencia en pacientes tratados es de 13 años
Up To Date 2015

TROMBOCITOSIS ESCENCIAL

DEFINICION También conocida como: trombocitosis idiopática, trombosis primaria y trombocitemia hemorrágica
Es un trastorno clonal de etiología desconocida que involucra a una célula hematopoyética multipotencial y que se manifiesta por sobreproducción de plaquetas sin causa identificable.
Aumento mantenido de plaquetas mayor de 450,000, aumento de eritrocitos y granulocitos.
Incidencia 2/100,000 con predominio en mujeres 2:1, sin afectar la esperanza de vida.
Up To Date 2015

ETIOLOGIA La megacariocitopoyesis y la producción de plaquetas dependen de la trombopoyetina, interleucina 3 y de su receptor MPL
Su desarrollo subsecuente también se favorecen presencia de la quimiocina factor 1 derivado de células del estroma (SDF-1).
En la trombocitosis esencial existe una mutación de trombopoyetina y MPL que genera aumento de trombopoyetina y aumento de células progenitoras
Up To Date 2015

ETIOLOGIA TROMBOCITEMIA HEREDITARIA
Enfermedad rara 0.9 casos por millón de habitantes
Relacionado en 15% con MPL W515L/K.
TROMBOCITOSIS ADQUIRIDARelacionado en 50 a 60% a mutación JAK2 V617F
Up To Date 2015

MANIFESTACIONES CLINICASLa mitad de los pacientes son asintomáticos.
En 40% se presentan síntomas vasomotores que incluyen: dolor de cabeza, mareo, síncope, dolor torácico atípico, parestesias en mandibula, livedo reticularis, trombocitosis con eritromelalgia, amaurosis fugaz.
Prurito se presenta en 5% de los pacientes.
Aborto Temprano en 43% de pacientes
Trombosis y hemorragia son complicaciones comunes, probablemente debido a alteraciones cualitativas y cuantitativas de plaquetas se presenta en el 18% y 26% respectivamente.
Up To Date 2015

EXAMEN FISICO Entre el 25 al 40% de los pacientes tienen esplenomegalia palpable.
La hepatomegalia y adenopatías son infrecuentes
Plétora facial y cianosis distal
En una minoría de pacientes se presenta petequias
La severidad de las manifestaciones no se relacionan con el nivel de plaquetas.

EVALUACION DIAGNOSTICA La única patología dentro de los síndromes mielo proliferativos que se diagnostica por exclusión es la trombocitopenia esencial.
Frotis de sangre periférica: plaquetas hipogranulares grandes, lucocitosis
neutrófila leve.
Bx de Médula ósea: Hiperplasia e hipertrofia megacariocítica, ausencia de
fibrosis.
Up To Date 2015

EVALUACION DIAGNOSTICA
HARRISON, manual de oncología, 2012

CRITERIOS DIAGNOSTICOS CRITERIOS DEL GRUPO DE ESTUDIO DE POLICITEMIA VERA
Recuento de plaquetas> 600.000 / microlitroHiperplasia megacariocítica en biopsia de medula oseaAusencia del cromosoma FiladelfiaAusencia de infección, inflamación, y otras causas de trombocitosis reactivaGlobulos rojos normales o una concentración de hemoglobina <13 g / dlPresencia de hierro en una aspiración de médula ósea o aumento ≤1 / dl g en
la concentración de hemoglobina después de una prueba de un mes de tratamiento con hierro oral.
Up To Date 2015

ESTRATIFICACION DE RIESGO RIESGO MAYOR DE 60 AÑOS O HISTORIA
DE TROMBOSISFACTORES DE RISGO CARDIOVASCULAR
BAJO NO NO
INTERMEDIO NO SI
ALTO SI -----
TROMBOCITOSIS: MAYOR DE 1.000.000
Polycythemia vera and essential thrombocythemia: 2013 update on diagnosis, risk-stratification, and management. AMERICAN JOURNAL OF HEMATOLOGY

TRATAMIENTO
Polycythemia vera and essential thrombocythemia: 2013 update on diagnosis, risk-stratification, and management. AMERICAN JOURNAL OF HEMATOLOGY

Puntos primarios, control del número de plaquetas, hemoglobina y leucocitos a 36 meses de seguimiento.

PRONOSTICO Riesgo de trasformación a leucemia aguda 2-3%
Supervivencia igual al grupo control.
Up To Date 2015

MIELOFIBROSIS PRIMARIA

DEFINICION Mieloproliferación clonal con fibrosis medular reactiva y hematopoyesis extramedular Es un trastorno por clonación de una célula progenitora hematopoyética multipotencial, de etiología desconocida y se caracteriza por fibrosis de la médula ósea, hematopoyesis extramedular y esplenomegalia.
Antes llamada: metaplasia mieloide agnógena con mielofibrosis También llamada: mielofibrosis idiopática o mielofibrosis con metaplasia mieloide
Up To Date 2015

EPIDEMIOLOGIA La mielofibrosis primaria es el menos frecuente entre las enfermedades mieloproliferativas crónicas, incidencia estimada de 1,5 por 100.000 por año.
Se presenta principalmente en pacientes especialmente hombre de mediana edad y ancianos, la edad media de presentación es de 67 años, aproximadamente el 17 por ciento de los pacientes son diagnosticados antes de los 50 años.
Up To Date 2015

ETIOLOGIA
HARRISON, manual de oncología, 2012

FISIOPATOLOGIA Citoquinas liberadas por megacariocitos, histiocitos y monocitos clonales, provocan un depósito reactivo
de tejido fibroblástico policlonal.
Fibrosis, neoangiogénesis y osteosclerosis
Jean-Jacques Lataillade, Does primary myelofibrosis involve a defective stem cell niche? From concept to evidence, BLOOD 2008.

FISIOPATOLOGIA
Jean-Jacques Lataillade, Does primary myelofibrosis involve a defective stem cell niche? From concept to evidence, BLOOD 2008.

MANIFESTACIONES CLINICASFatiga 80 por cientoDolor abdominal 68 por cientoSudoración nocturna 55 por ciento Dolor oseo 51 por ciento Prurito 45 por cientoPerdida de peso 38 por cientoFiebre 25 por ciento
Up To Date 2015

EXPLORACION FISICA ESPLENOMEGALIA Presente en el 96% de los pacientes, de los cuales 10% tienen esplenomegalia masiva.
HEPATOMEGALIA Presente en el 50 al 60% de los paciente Palidez mucocutanea
Up To Date 2015

HALLAZGOS DE LABORATORIO ANEMIA
Hemoglobina < 10 gr/dl se encuntra presente en el 50% de los pacientes y en 20% se encuentra por debajo de 8 gr/dl.
PLAQUETAS Y LEUCOCITOS
Son variables entre el 10 y 15% de los paciente se pueden encontrar elevadas al momento del diagnostico.
La trombocitopenia se vuelve común a medida que la enfermedad avanza relacioandose con complicaciones hemorrágicas.
Células progenitoras endoteliales circulantes con CD34, CD133
Up To Date 2015

EXTENDIDO DE SANGRE PERIFERICA
La medula osea presenta arquitectura celular desordenada, megacariocitos displásicas, nueva formación de hueso en la médula ósea y la formación de fibras de colágeno. En el extendido periférico reacción leucoeritroblastica con dismorfismos en línea eritroide Anisocitosis, poiquilocitosis, dacriocitosis.
Up To Date 2015

MUTACIONES ADQUIRIDAS

CRITERIOS DIAGNOSTICOS CRITERIOS MAYORES (debe cumplir todos)
1. Hiperplasia megacariocítica atípica con fibrosis reticulínica y/o colágena
2. Ausencia de criterios diagnósticos para otra neoplasia mieloide o mielodisplasia
3. Presencia de la mutación V617F u otro marcador clonal o ausencia de fibrosis medular reactiva
CRITERIOS MENORES (debe cumplir dos)
1. Leucoeritroblastosis
2. Elevación de la LDH sérica
3. Anemia
4. Esplenomegalia palpable
Up To Date 2015

ESTRATIFICACION DE RIESGO
Edad > 65 añosSíntomas constitucionalesAnemia <10 gr/dlLeucocitosis >25.000Blastos >1% en sangre periférica
Up To Date 2015

TRATAMIENTO En ausencia de factores pronósticos adversos (anemia o síntomas) NO es necesario tratamiento.
Trasplante de células alogénico es el único tx curativo. A considerarse en pacientes jóvenes con MAL pronóstico.
Tratamiento de soporte◦ Trasfusiones. ◦ Esplenectomía en casos refractarios o esplenomegalia dolorosa.◦ Ruxolitinib. (inhibidor de JAK 1/JAK 2), disminuye síntomas, disminuye esplenomegalia,
aumenta supervivencia◦ Talidomida y Lenalidomida (mejoran el recuento eritrocítico)◦ Alopurinol en casos de hiperuricemia◦ Hidroxiurea◦ En combinación con Glucocorticoides para controlar síntomas generales.
Up To Date 2015

PRONOSTICO Mediana de supervivencia= 5 años.
Evolución a leucemia mieloide aguda del 8%/año
Los pacientes con positividad al receptor MPL, tienden a producir anemia más intensa
Factores de mal pronóstico de Internacional Working Group:
Edad > 65 añosSíntomas constitucionalesAnemia <10 gr/dlLeucocitosis >25.000Blastos >1% en sangre periférica

LEUCEMIA NEUTROFILICA CRONICA
Es un raro trastorno caracterizado por la proliferación granulocítica madura en sangre, médula ósea y la infiltración en los órganos que resultan en hepatoesplenomegalia.
Existen granulaciones tóxicas en los neutrófilos, hipersegmentación nuclear y aumento de la fosfatasa alcalina.
Un subconjunto de pacientes demuestra mutaciones puntuales en el gen CSF3R, que codifica el receptor para CSF3, regulación de la producción de neutrófilos.
Up To Date 2015

LEUCEMIA EOSINOFILICA CRONICA
Se caracteriza por hipereosinofilia en médula ósea y sangre periférica mayor de 1500/ml. En su forma pura, es secundaria a alteraciones cromosómicas.
La más frecuente, es la ocasionada por el reordenamiento 4q12 que origina el producto de fusión FIP1L1/PDGFRA.
Otras menos frecuentes son los reordenamientos en los genes PDGFRB y FGFR1, asociándose también este último a neoplasias linfoides.
Responden a tratamiento con inhibidores de tirocincinasa.
Up To Date 2015

MASTOCITOSIS Afecta preferentemente a la piel, su forma mas frecuente es la urticaria pigmentosa.
La leucemia mastocitaria corresponde al 1% de las mastocitosis, relacionada con mutaciones en el gen KIT D816V.
Se diagnostica por análisis de medula osea con mastocitos medulares > 20%
Diagnostico con biopsia, triptasa sérica
Pronostico malo con sobrevida de meses, tratamiento paleativo con hidroxiurea.
Up To Date 2015





























