Embed Size (px)
Citation preview

Metodologie di base della Chimica
Computazionale
Cenni di algebra lineare, sistemi risolvibili analiticamente,
calcolo di strutture elettroniche
Antonino Polimeno
Dipartimento di Scienze Chimiche
Universita degli Studi di Padova

1
CHIMICA COMPUTAZIONALE
Titolare: Antonino Polimeno - Dipartimento di Scienze Chimiche
Orario di ricevimento: Mercoledı dalle 15.30 alle 17.30 o per appuntamento
Tipologie didattiche: 32A + 16L; 5 crediti.
Propedeuticita: nessuna
Pre-requisiti : conoscenza dei principi della chimica quantistica
Obiettivo: apprendere le nozioni di base della chimica computazionale, e saper applicare i metodi del
calcolo a problematiche specifiche di chimica fisica, organica ed inorganica
Modalita d’esame: esame orale, con la possibilita di concordare la discussione di un problema specifico
con il docente e di discutere un breve elaborato.
Programma
Metodi per il calcolo di strutture elettroniche (20 ore + 4 ore es.)
• Richiamo dei concetti fondamentali della meccanica quantistica: hamiltoniani atomici e molecolari,
equazione di Schrodinger, principio variazionale, momenti angolari, stati di spin
• Metodi di algebra lineare per la meccanica quantistica: rappresentazione di Dirac, spazi funzionali,
basi di funzioni
• Metodo di Hartree-Fock: determinanti di Slater, principio di autoconsistenza; implementazione di
Roothaan: scelta e classificazione delle basi di orbitali atomici
• Teoria del funzionale densita: teorema KS, densita locale, gradiente, metodi ibridi
Calcolo di proprieta molecolari (8 ore + 8 ore es.)
• Metodi semiempirici: descrizione e parametrizzazione; meccanica molecolare: campi di forze e loro
parametrizzazione; implementazione, vantaggi e limitazioni dei metodi MM
• Caratterizzazione ed impiego di software computazionale: e.g. Gamess, Gaussian, ADF, Gromacs,
CHARMM; calcolo di strutture e elettroniche ed ottimizzazione di geometrie molecolari.
Metodologie computazionali per fasi condensate (4 ore + 4 ore es.)
• Determinazione di proprieta di equilibrio di fasi condensate: Dinamica Molecolare; cenni ai metodi
Monte Carlo e Dinamica Browniana
Testo adottato: dispense di lezione.
Testi consigliati : C.J. Cramer, ”Essential of Computational Chemistry” II Ed., Wiley-Interscience; D.
Young, ”Computational Chemistry: A Practical Guide for Applying Techniques to Real World Problems”,
Wiley-Interscience; M.P. Allen e D.J. Tildesley, ”Computer Simulation of Liquids”, Clarendon Press.

2

Indice generale
I Richiami di algebra lineare ed altre nozioni matematiche. 11
1 Cenni di algebra lineare 13
1.1 Spazi vettoriali . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 14
1.1.1 Definizione di spazio vettoriale . . . . . . . . . . . . . . . . . . . . . . . . . . . 14
1.1.2 Prodotti scalari . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 16
1.2 Vettori e matrici . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 18
1.2.1 Vettori tridimensionali . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 18
1.2.2 Matrici . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 22
1.3 Applicazioni . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 29
1.3.1 Sistemi lineari . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 29
1.3.2 Trasformazioni . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 31
1.3.3 Problemi agli autovalori . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 32
1.3.4 Funzioni di matrici . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 34
1.4 Esercizi . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 35
2 Meccanica quantistica 37
2.1 Spazi di Hilbert . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 37
2.1.1 Operatori lineari . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 38
2.2 Formulazione della quantomeccanica . . . . . . . . . . . . . . . . . . . . . . . . . . . 39
2.2.1 Principio Variazionale . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 41
2.2.2 Cenno alla teoria delle perturbazioni . . . . . . . . . . . . . . . . . . . . . . . . 43
3 Funzioni speciali 45
3.1 Alcune funzioni speciali . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 45
3.1.1 Integrali ellittici . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 45
3.1.2 Funzione gamma . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 46
3.1.3 Funzione dell’errore . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 46
3.1.4 Funzioni di Bessel . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 47
3.2 Polinomi ortogonali . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 47
3

4 INDICE GENERALE
3.2.1 Polinomi di Legendre . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 48
3.2.2 Polinomi di Hermite . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 48
3.2.3 Polinomi di Laguerre . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 48
II Sistemi quantomeccanici risolvibili analiticamente. 49
4 Sistemi risolvibili analiticamente 51
4.1 Sistemi monodimensionali . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 51
4.2 Fuga dal pozzo . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 55
4.3 Una scatola con una barriera interna . . . . . . . . . . . . . . . . . . . . . . . . . . . 56
4.4 Scatola tridimensionale . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 58
4.5 Particella in e su un anello . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 59
4.6 Particella in e su una sfera . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 62
4.7 Oscillatore armonico . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 65
4.8 Potenziale di Morse . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 66
4.9 Atomo idrogenoide . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 68
III Calcolo di strutture elettroniche atomiche e molecolari. 71
5 L’equazione di Schrodinger 73
5.1 L’equazione di Schrodinger . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 74
5.1.1 Insieme di punti materiali . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 74
5.1.2 Hamiltoniani atomici e molecolari . . . . . . . . . . . . . . . . . . . . . . . . . 76
5.2 Approssimazione di Born-Oppenheimer . . . . . . . . . . . . . . . . . . . . . . . . . . 79
5.2.1 Separazione delle coordinate elettroniche . . . . . . . . . . . . . . . . . . . . . 80
5.3 La molecola di idrogeno ionizzata . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 81
5.3.1 Hamiltoniano elettronico . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 81
5.3.2 Hamiltoniano nucleare . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 84
5.3.3 Soluzione approssimata della struttura elettronica . . . . . . . . . . . . . . . . 85
6 Funzioni d’onda 89
6.1 La funzione d’onda elettronica . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 89
6.1.1 Stati di spin . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 89
6.1.2 Principio di indistinguibilita . . . . . . . . . . . . . . . . . . . . . . . . . . . . 90
6.1.3 Determinanti di Slater . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 91

INDICE GENERALE 5
7 Metodi ab-initio 95
7.1 Metodo di Hartree-Fock . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 95
7.1.1 Ottimizzazione degli spinorbitali . . . . . . . . . . . . . . . . . . . . . . . . . . 96
7.1.2 Proprieta dell’operatore di Fock . . . . . . . . . . . . . . . . . . . . . . . . . . 98
7.1.3 Sistemi a guscio chiuso . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 100
7.1.4 Soluzione numerica delle equazioni HF . . . . . . . . . . . . . . . . . . . . . . 101
7.2 Interazione di configurazione . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 104
7.3 Alcuni esempi di calcolo ab initio . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 107
7.3.1 La molecola di idrogeno . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 107
7.3.2 Altri esempi . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 109
8 Teoria dei gruppi di simmetria ed orbitali molecolari 117
8.1 Gruppi di simmetria puntuale . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 117
8.1.1 Elementi ed operazioni di simmetria . . . . . . . . . . . . . . . . . . . . . . . . 119
8.1.2 Gruppi puntuali . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 120
8.2 Rappresentazioni . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 121
8.2.1 Operatori di proiezione . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 124
8.3 Applicazioni al calcolo di orbitali molecolari . . . . . . . . . . . . . . . . . . . . . . . . 124
8.3.1 Atomi . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 125
8.3.2 Molecole lineari omonucleari . . . . . . . . . . . . . . . . . . . . . . . . . . . . 126
8.3.3 L’acqua . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 126
8.3.4 Un esempio non triviale: il benzene . . . . . . . . . . . . . . . . . . . . . . . . 126
9 Metodi semiempirici 129
9.1 Metodi semiempirici . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 129
9.1.1 Metodo di Huckel . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 129
9.1.2 Neglect-of-differential-overlap (NDO) . . . . . . . . . . . . . . . . . . . . . . . 132
A L’atomo idrogenoide 135
B Separazione del baricentro 137
C Problema generalizzato agli autovalori 139
D Valore di attesa per un guscio chiuso 141
E Separabilita σ − π (Likos-Parr) 143

6 INDICE GENERALE

Indice delle Figure
1.1 Vettori nello spazio 3D . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 19
1.2 Una spirale destrorsa (Esempio 14). . . . . . . . . . . . . . . . . . . . . . . . . . . . 21
1.3 Angoli di Eulero α, β, γ per specificare la rotazione di un sistema di riferimento molecolare
rispetto ad un sistema di riferimento di laboratorio. . . . . . . . . . . . . . . . . . . . . 27
1.4 Schema del momento dipolare indotto in una molecola di CO2 da un campo elettrico
esterno. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 28
1.5 Calcolo del momento di inerzia del complesso Cu(NH3)2+6 (vedi Esempio 21). . . . . . 30
4.1 Potenziale per una particella in una scatola 1D. . . . . . . . . . . . . . . . . . . . . . . 53
4.2 Un potenziale generico in una dimensione. . . . . . . . . . . . . . . . . . . . . . . . . 54
4.3 Potenziali per una particella in una scatola 1D con una barriera interna. . . . . . . . . . 56
4.4 Varie scatole 1D. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 58
4.5 Scatola tridimensionale. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 59
4.6 Un pozzo cilindrico ed un anello. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 60
4.7 Coordinate polari sferiche. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 63
4.8 Potenziale di Morse. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 67
5.1 Sistema di riferimento e coordinate He . . . . . . . . . . . . . . . . . . . . . . . . . . 77
5.2 Sistema di riferimento e coordinate H+2 . . . . . . . . . . . . . . . . . . . . . . . . . . 82
5.3 Coordinate ellissoidali H+2 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 83
5.4 I primi due livelli energetici della molecola di H+2 [E. Teller, Z. Physik, 61, 474 (1930)] 84
5.5 Superficie che rappresenta l’orbitale 1sσg in un piano molecolare . . . . . . . . . . . . . 85
5.6 Superficie che rappresenta l’orbitale 2pσu in un piano molecolare . . . . . . . . . . . . . 86
7.1 Schema degli elementi di matrice H nel metodo CI. Sono indicati in nero i blocchi diagonali
non nulli, in grigio i blocchi non-diagonali non nulli, in bianco i blocchi non-diagonali nulli 106
7.2 Orbitali HOMO e LUMO di HCl, rappresentati come superficie . . . . . . . . . . . . . . 110
7.3 Orbitale HOMO di HCl, rappresentato come una superficie ad uguale densita di carica . 115
7.4 Orbitale LUMO di HCl, rappresentato come una superficie ad uguale densita di carica . 115
7.5 Orbitale HOMO di H2O, rappresentato come una superficie ad uguale densita di carica . 116
7

8 INDICE DELLE FIGURE
7.6 Orbitale LUMO di H2O, rappresentato come una superficie ad uguale densita di carica . 116
8.1 Set di orbitali 2p nel piano per il benzene . . . . . . . . . . . . . . . . . . . . . . . . . 128
9.1 Radicale CH3· . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 130
9.2 Esempio: 1,3-butadiene. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 131

Indice delle Tabelle
1.1 Relazioni tensoriali nella forma generica ~y = A~x . . . . . . . . . . . . . . . . . . . . . . 29
5.1 Unita atomiche . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 78
8.1 Rappresentazioni irriducibili e AO di base del benzene . . . . . . . . . . . . . . . . . . 127
9.1 Tabella dei caratteri del gruppo C2h . . . . . . . . . . . . . . . . . . . . . . . . . . . . 131
9.2 Funzioni φi in C2h . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 132
9

10 INDICE DELLE TABELLE

Parte I
Richiami di algebra lineare ed altre
nozioni matematiche.
11


Capitolo 1
Cenni di algebra lineare
Questa prima dispensa e organizzata in tre Capitoli: nel Capitolo 1 sono incluse nozioni basilari di algebra
lineare (spazi lineari, matrici, sistemi lineari e problemi agli autovalori); nel Capitolo 2 e presentata una
rapida formulazione degli assiomi della meccanica quantistica e delle due tecniche fondamentali per lo
studio di sistemi quantomeccanici: il principio variazionale ed il metodo delle perturbazioni; il Capitolo
3 e dedicato ad un riassunto delle proprieta di alcune funzioni di uso comune in vari problemi della
meccanica quantistica.
Esempi risolti sono inseriti appropriatamente nel corso di ciascun Capitolo. Esercizi non risolti (con
suggerimenti e schemi di soluzione in alcuni casi) sono inclusi alla fine del Capitolo I.
Il congresso Solvay nel 1927 a Bruxelles.
13

14 CAPITOLO 1. CENNI DI ALGEBRA LINEARE
1.1 Spazi vettoriali
Iniziamo con una breve presentazione degli spazi vettoriali. Le definizioni che seguono sono del tutto
generali: vale a dire le proprieta definite per uno spazio vettoriale non dipendono dalla natura dei suoi
componenti, o vettori, che possono essere i vettori di uno spazio tridimensionale noti dai corsi di analisi
matematica, o un insieme di funzioni e cosı via.
1.1.1 Definizione di spazio vettoriale
La definizione di uno spazio vettoriale V e fondata sulla precedente definizione di un insieme K di quantita
scalari, cioe un insieme di numeri. Il corpo K gode delle proprieta:
• Se a e b appartengono a K, allora a + b e ab appartengono a K
• 0 e 1 appartengono a K
• Se a appartiene a K, −a appartiene a K; se a non e 0 allora a−1 appartiene a K
Esempio 1. Sia N l’insieme dei numeri naturali; si vede subito che non e un corpo, poiche se n
appartiene a N , −n non e un numero naturale; analogamente per l’insieme dei numeri interi Z, per un
cui elemento generico z si ha che 1/z non appartiene a Z.
In effetti si parla di spazio vettoriale V sul corpo K di scalari. Il corpo K e in genere dato
dall’insieme dei numeri razionali Q, o reali R o complessi C, o loro sottoinsiemi. Uno spazio vettoriale
V sul corpo K e dunque un insieme di oggetti che possono essere addizionati fra loro e moltiplicati per
elementi di K (nel seguito semplicemente numeri), in modo che il risultato appartenga ancora a V , con
le seguenti proprieta aggiuntive:
Associativa. Se u, v, w sono elementi di V , allora:
(u + v) + w = u + (v + w) (1.1)
Neutro. Esiste un elemento 0 di V , tale che
0 + u = u + 0 = u (1.2)
per ogni u di V
Opposto. Per ogni u di V esiste −u di V tale che
u + (−u) = (−u) + u = 0 (1.3)

1.1. SPAZI VETTORIALI 15
Commutativa. Per ogni coppia u, v vale che
u + v = v + u (1.4)
Proprieta in K. Se a, b, c sono numeri
• c(u + v) = cu + cv
• (a + b)u = au + bu
• (ab)u = a(bu)
• 1u = u
Esempio 2. Alcuni esempi di spazi vettoriali: lo spazio PN(x) di tutti i polinomi a coefficienti reali o
complessi di grado N nella coordinata x; lo spazio F di tutte le funzioni di variabile complessa definite
in un intervallo.
Esempio 3. Dato l’insieme Rn di tutte le n-uple di numeri reali, dove la somma di due elementi e
definita come la somma ordinata delle componenti, (x1, . . . , xn) + (y1, . . . , yn) = (x1 + y1, . . . , xn + yn)
e il prodotto per uno scalare come a(x1, . . . , xn) = (ax1, . . . , axn), allora Rn e uno spazio vettoriale.
Lo studente provi a dimostrarlo verificando i vari punti della definizione di spazio vettoriale.
Esempio 4. Dato l’insieme di numeri reali positivi R+, definiamo le seguenti operazioni: ’addizione’,
x⊗y = xy (cioe l’ordinaria moltiplicazione di due reali), ’moltiplicazione per un numero reale a’, a·x = xa
(potenza); allora si dimostra che R+ e uno spazio vettoriale rispetto alle due operazioni appena definite.
Lo studente provi a dimostrarlo verificando i vari punti della definizione di spazio vettoriale.
Una somma generica di elementi di uno spazio vettoriale V ,
n∑
i=1
aixi = a1x1 + a2x2 + . . . + anxn (1.5)
e detta combinazione lineare del set di vettori xi; gli n vettori sono linearmente dipendenti se esistono
n scalari ai non tutti nulli tali che la combinazione lineare sia zero, o linearmente indipendenti se non
esistono. Se e possibile esprimere tutti gli elementi di V come combinazioni lineari del set xi, allora si
dice che il set genera (in inglese, span) lo spazio vettoriale. Una base dello spazio vettoriale e definita
come un’insieme di vettori ei che sia linearmente indipendente e generi lo spazio. Si puo dimostrare
che due basi qualunque di uno spazio, se esistono, hanno lo stesso numero di elementi. La dimensione

16 CAPITOLO 1. CENNI DI ALGEBRA LINEARE
n dello spazio V e il numero di elementi di una base, e puo anche essere infinita. Per un vettore generico
possiamo scrivere percio:
x =n∑
i=1
xiei (1.6)
il coefficiente xi e la proiezione o componente di x sull’elemento i-esimo della base. L’insieme dei
coefficienti e anche detto coordinate.
Esempio 5. Quali sono le coordinate della funzione f(θ) = 3 sin θ + 5 cos θ in un opportuno spazio di
dimensione 2?
Esempio 6. Quali sono le coordinate della funzione df/dθ dove f e la base sono definite nel precedente
esempio?
Esempio 7. Consideriamo lo spazio vettoriale delle funzioni f(θ) a valori reali e periodiche nell’intervallo
[−π, π]; l’insieme degli elementi sin θ, sin 2θ, sin 3θ etc. costituisce una base ?
1.1.2 Prodotti scalari
Se V e uno spazio vettoriale definito su un corpo K, definiamo con un prodotto scalare un modo per
associare ad ogni coppia di elementi u e v di V uno scalare di K, che indichiamo con u · v. Valgono le
seguenti proprieta
Commutativa. Per ogni coppia u, v vale che u · v = v · u
Distributiva. Per ogni terna u, v, w vale che u · (v + w) = u · v + uw
Scalare. Se a appartiene a K allora (au) · v = a(u · v) e u · (av) = a(u · v).
Due vettori u, v si dicono ortogonali o perpendicolari se u · v = 0. Data una base e1, . . . , en, questa si
dice ortogonale, se per ogni coppia di indici ei · ej = 0.
Esempio 8. Consideriamo lo spazio vettoriale delle coppie di numeri reali R2. Si noti che tale spazio e
in corrispondenza biunivoca con lo spazio dei vettori (in senso tradizionale, quantita con una intensita,
direzione e verso) nel piano. Definiamo il prodotto scalare di due vettori u = (u1, u2) e v = (v1, v2)
come:
u · v = u1v1 + u2v2 (1.7)

1.1. SPAZI VETTORIALI 17
Esempio 9. Consideriamo l’insieme di tutte le funzioni periodiche a valori reali in [−π, π]. Definiamo
il prodotto
f(θ) · g(θ) =∫ π
−πdθf(θ)g(θ) (1.8)
Lo studente dimostri che si tratta di un prodotto scalare.
Consideriamo ora nel seguito di questo paragrafo uno spazio vettoriale sul corpo R dei numeri reali (vedi
per es. sia l’Esempio 8 che l’Esempio 9). Definiamo un prodotto scalare. Si dice che il prodotto e
definito positivo se per ogni u di V si ha che u · u ≥ 0, e per ogni u non nullo, u · u > 0. Per una
coppia di vettori u e v, se v non e nullo definiamo la proiezione di u lungo v come:
a =u · vv · v (1.9)
detta anche coefficiente di Fourier. Data un base ortogonale, si puo verificare che le coordinate di un
elemento generico di V rispetto alla base stessa sono i coefficienti di Fourier rispetto agli elementi della
base
u =∑
i
uiei, ui =u · ei
ei · ei
(1.10)
Dato un vettore u, Si dice norma la grandezza scalare u =√
u · u. Se la norma vale 1, il vettore u si
dice normalizzato, o vettore unita, o versore 1.
Esempio 10. Consideriamo l’insieme dei polinomi P2(x) di grado 2 in [0, 1] a valori reali. E uno spazio
vettoriale (definendo l’addizione come somma dei coefficienti)? Di che dimensione? Possiamo definire
una base? Ed un prodotto scalare? Ed una base ortonormale? Suggerimenti: la dimensione coincide
con il numero massimo di coefficienti; una base e data semplicemente da 1, x e . . .; il prodotto scalare
puo essere definito come l’integrale da 0 ad 1 del prodotto di . . .
Consideriamo nel seguito la generalizzazione del concetto di prodotto scalare che e valido per spazi
vettoriali sul corpo dei numeri reali, al caso di spazi vettoriali sul corpo dei complessi. Parleremo in
questo caso di prodotto hermitiano tra due vettori u e v, con le seguenti proprieta
Commutativa. Per ogni coppia u, v vale che u · v = (v · u)∗
Distributiva. Per ogni terna u, v, w vale che u · (v + w) = u · v + uw
Scalare. Se a e un numero complesso allora (au) · v = a(u · v) e u · (av) = a∗(u · v).
1Supponiamo che V abbia dimensione finita n. Una sua base e1, . . . , en si dice ortonormale se essa e ortogonalee se ogni ogni suo elemento e un vettore unita; da una base ortogonale si puo sempre ricavare una base ortonormale,dividendo ogni elemento della base per la sua norma.

18 CAPITOLO 1. CENNI DI ALGEBRA LINEARE
dove con a∗ indichiamo il complesso coniugato di a. Tutte le definizioni precedenti relative al prodotto
scalare (definito positivo, ortogonalita, basi ortogonali, etc.) sono analoghe.
Esempio 11. Consideriamo lo spazio delle n-uple complesse, Cn; un elemento generico e u = (u1, . . . , un)
dove ui appartiene a C. La dimensione dello spazio e evidentemente n; definiamo il prodotto hermitiano
u · v =n∑
i=1
u∗i vi (1.11)
Il prodotto e definito positivo poiche se u 6= 0, allora almeno uno dei numeri ui e diverso da zero
(complesso) e u∗i ui e sempre positivo.
Esempio 12. Consideriamo lo spazio delle funzioni f(θ) a valori complessi definite in [−π, π]. E uno
spazio vettoriale, di dimensione infinita, e possiamo introdurre il prodotto hermitiano definito positivo:
f · g =∫ π
−πdθf(θ)∗g(θ) (1.12)
Non e difficile verificare che l’insieme di funzioni 1, e±iθ, e±2iθ, . . . costituisce una base ortogonale del
problema; se indichiamo con fn = einθ dove n e un numero intero, la norma di ciascuna funzione di base
e
fn · fn =∫ π
−πdθe−inθeinθ = 2π (1.13)
Ogni elemento generico dello spazio puo essere espresso come combinazione lineare delle funzioni di base;
il coefficiente di Fourier n-esimo e il coefficiente che misura il contributo di ciascuna funzione di base:
f(θ) =∑n
cnfn =∑
cneinθ (1.14)
cn =f · fn
fn · fn
=1
2π
∫ π
−πdθf(θ)∗einθ (1.15)
1.2 Vettori e matrici
Nel corso di questa Sezione riassumeremo alcune proprieta gia note allo studente dei vettori tridimensionali
reali. Quindi procederemo alla definizione e allo studio delle grandezze matriciali.
1.2.1 Vettori tridimensionali
Un vettore nello spazio tridimensionale ordinario e definito come una coppia ordinata di punti dello spazio.
In Figura 1.1 e riportata la rappresentazione di due vettori ~u e ~v, definiti rispetto ad un punto comune
di origine. Si deve notare che lo spazio vettoriale delle triple di numeri reali R3 e in corrispondenza

1.2. VETTORI E MATRICI 19
biunivoca con lo spazio dei vettori ordinari, che possono percio essere identificati da una terna di numeri
reali (le componenti rispetto ad un sistema di riferimento, vedi Figura 1.1). Comunque la definizione
dello spazio vettoriale dei vettori ordinari puo essere data prescindendo dalla rappresentazione: il vettore
si puo identificare come una freccia orientata, con una direzione, un verso ed un’intensita. Nel seguito
useremo sempre caratteri in grassetto per indicare n-ple di numeri o matrici (es. la tripla di numeri
v1, v2, v3 sara v); e caratteri tipografici per indicare il vettore o la matrice indipendentemente dalla sua
rappresentazione (es. v). Dove non sia necessario specificare la differenza useremo caratteri in grassetto.
La somma di due vettori e descritta come un vettore ottenibile con la regola del parallelogramma
e3
e 1
x
e2
u
u1
u2
u3
v
u+v
u v
Figura 1.1: Vettori nello spazio 3D
(vedi Figura 1.1). Il prodotto scalare e ottenuto in accordo con la definizione nota di prodotto scalare,
~u · ~v = uv cos θ, dove u e v sono il modulo dei due vettori e θ e l’angolo compreso. Alternativamente,
se si considera l’equivalente spazio R3, introducendo una base di tre versori ~e1, ~e2, ~e3, ed ogni vettore
viene rappresentato dalle sue componenti sulla base, la somma e il prodotto scalare sono definiti
u + v =3∑
i=1
(ui + vi)ei (1.16)
u · v =3∑
i=1
uivi (1.17)
Altri prodotti possono essere definiti con un chiaro significato geometrico. Il prodotto vettoriale di ~u e
~v e un vettore che ha per modulo la quantita uv sin θ, per direzione la perpendicolare al piano definito

20 CAPITOLO 1. CENNI DI ALGEBRA LINEARE
da ~u e ~v e per verso quello ottenuto avvitando una vite destrorsa lungo la direzione definita, in modo da
portare ~u su ~v (regola della vite o della mano destra). Il prodotto vettoriale e un esempio di applicazione
definita in uno spazio vettoriale: ovvero un’operazione che associa elementi dello spazio ad altri elementi
dello spazio.
Esempio 13. Alcune utili identita:
~a×~b = −~b× ~a (1.18)
~a · (~b× ~c) = ~b · (~c× ~a) = ~c · (~a×~b) (1.19)
~a× (~b× ~c) = ~b(~a · ~c)− ~c(~a ·~b) (1.20)
(~a×~b) · (~c× ~d) = (~a · ~c)(~b · ~d)− (~a · ~d)(~b · ~c) (1.21)
La notazione vettoriale e di grande utilita perche permette di riassumere in modo compatto equivalenze
ed equazioni in tre dimensioni, o in n dimensioni, la generalizzazione a Rn essendo molto semplice. In
R3 cosı l’equazione che definisce un piano nello spazio passante per un punto x0 e
(x− x0) · n = 0 (1.22)
dove n e un versore normale al piano. L’angolo diedro tra due piani e invece dato come
cos Θ = |n1 · n2| (1.23)
Molte altre identita e definizioni della geometria analitica nello spazio tridimensionale sono note allo
studente dai corsi di matematica. L’uso della notazione vettoriale e utile anche per definire funzioni
vettoriali. Possiamo distinguere funzioni scalari, o campi scalari, che associano elementi di R3 a R o
funzioni vettoriali che associano elementi di R a R3. Un campo vettoriale e infine una funzione che
associa elementi di R3 a R3.
Esempio 14. Alcuni esempi di campi scalari: la temperatura di un laboratorio, intesa come funzione
delle coordinate spaziali; la funzione f(x) = |x|; la funzione f(x) = x21 − x2x3; la concentrazione di
cloruro di terbutile in un reattore contenente alcol terbutilico, cloro e cloruro di terbutile.
Esempio 15. Alcuni esempi di funzioni vettoriali: la retta x(t) = x0+nt; la funzione x(t) = r cos ωte1+
r sin ωte2 + vte3, che rappresenta la traiettoria di una particella che si muova lungo un’elica destrorsa,
con asse parallelo a e3 (vedi Figura 1.2).
Esempio 16. Alcuni campi di vettori: la forza di gravita tra due corpi; la funzione f(x) = x × n;
l’orientazione delle molecole di cristallo liquido che formano il display di un orologio digitale.

1.2. VETTORI E MATRICI 21
z
0
5
10
y
−1
0
1 x
−1
0
1
Figura 1.2: Una spirale destrorsa (Esempio 14).

22 CAPITOLO 1. CENNI DI ALGEBRA LINEARE
1.2.2 Matrici
Una matrice di mn elementi dove m ed n sono rispettivamente il numero di righe ed il numero di colonne,
e definita come una tabella rettangolare di numeri, il cui elemento generico e aij (dove 1 ≤ i ≤ m e
1 ≤ j ≤ n, e si indica nel modo seguente
A =
a11 a12 a13 . . . a1n
a21 a22 a23 . . . a2n
. . .
am1 am2 am3 . . . amn
(1.24)
o con notazioni equivalenti: p.es. doppie linee invece di parentesi. Consideriamo l’insieme delle matrici
m×n. Possiamo definire la moltiplicazione per uno scalare α di una matrice A come la matrice ottenuta
moltiplicando ciascun elemento di A per α
(αA)ij = αaij (1.25)
La somma di due matrici si ottiene sommando gli elementi delle matrici addende
(A + B)ij = aij + bij (1.26)
La matrice nulla e formata solo da zeri
0ij = 0 (1.27)
ed infine la matrice opposta di una matrice generica e
(−A)ij = −aij (1.28)
Si verifica facilmente che l’insieme cosı definito e uno spazio vettoriale di dimensione mn. Una base e
costituita dalle mn matrici con elementi ovunque nulli, tranne che in una posizione
e11 =
(1 . . . 0
0 . . . 0
), . . . , emn =
(0 . . . 0
0 . . . 1
)(1.29)
Esempio 17. Definiamo un prodotto tra due matrici m× n come la matrice ottenuta moltiplicando gli
elementi nella stessa posizione
(A ∗B)ij = aijbij (1.30)
L’operazione ∗ e un prodotto scalare?
Il prodotto tra matrici e un’applicazione che associa a due matrici generiche A m × n e B n × p una
matrice AB m× p (il numero di colonne della prima matrice deve essere uguale al numero di righe della
seconda):
(AB)ij =n∑
k=1
aikbkj (1.31)

1.2. VETTORI E MATRICI 23
L’insieme delle n-uple di numeri, cioe gli elementi di Rn o Cn si possono mettere in relazione biunivoca
con le matrici 1×n (matrici riga) oppure n×1 (matrici colonna). Cosı la moltiplicazione di una matrice
m×n per un vettore n dimensionale significa in pratica la moltiplicazione per una matrice colonna n×1
ed il risultato e una matrice colonna m× 1:
a11 a12 a13 . . . a1n
a21 a22 a23 . . . a2n
. . .
am1 am2 am3 . . . amn
u1
u2
. . .
un
=
v1
v2
. . .
vm
≡ Au = v (1.32)
dove vi =∑n
j=1 aijuj.
Esempio 18. Il prodotto tra matrici non e commutativo, ovvero
AB 6= BA (1.33)
Si definisce il commutatore come la matrice:
[A,B] = AB−BA (1.34)
evidentemente perche il commutatore esista deve essere m = p; si dice che due matrici commutano se
[A,B] = 0.
Consideriamo nel seguito solo le matrici quadrate n×n (nel seguito, di dimensione n), e vettori (matrici
riga o colonna). Alcune matrici quadrate speciali di dimensione n sono le seguenti:
• la matrice identita
1 =
1 0 0 . . . 0
0 1 0 . . . 0
. . .
0 0 0 . . . 1
(1.35)
• le matrici scalari, A= a 1
• le matrici diagonali
D =
d1 0 0 . . . 0
0 d2 0 . . . 0
. . .
0 0 0 . . . dn
(1.36)

24 CAPITOLO 1. CENNI DI ALGEBRA LINEARE
• le matrici tridiagonali
Td =
d1 e1 0 . . . 0
e1 d2 0 . . . 0
. . .
0 0 0 . . . dn
(1.37)
• le matrici triangolari (superiori od inferiori)
T =
t11 t12 t13 . . . t1n
0 t22 t23 . . . t2n
. . .
0 0 0 . . . tnn
(1.38)
• la potenza di una matrice: Ap = A · . . .A (p volte)
• le matrici idempotenti: P2 = P
In associazione con una generica matrice A con elementi aij si possono definire alcune matrici stretta-
mente collegate
• la matrice trasposta
AT =
a11 a21 a31 . . . an1
a12 a22 a32 . . . an2
. . .
a1n a2n a3n . . . ann
(1.39)
• la matrice complessa coniugata
A∗ =
a∗11 a∗12 a∗13 . . . a∗1n
a∗21 a∗22 a∗23 . . . a∗2n
. . .
a∗1n a∗2n a∗3n . . . a∗nn
(1.40)
• la matrice aggiunta
A† =
a∗11 a∗21 a∗31 . . . a∗n1
a∗12 a∗22 a∗32 . . . a∗n2
. . .
a∗1n a∗2n a∗3n . . . a∗nn
(1.41)

1.2. VETTORI E MATRICI 25
evidentemente (AT )∗ = (A∗)T = A†. Prima di procedere oltre consideriamo il concetto di funzione
scalare di una matrice, ovvero una corrispondenza tra una matrice e l’insieme dei reali o complessi. Le
funzioni scalari piu importanti sono la traccia
Tr(A) =n∑
i=1
aii (1.42)
che e la somma di tutti gli elementi diagonali di una matrice; ed il determinante
det(A) =∑
P(−1)P Πn
j=1ajP(j) (1.43)
di definizione un poco piu complicata: si tratta della somma di n! addendi, ciascun dei quali e ottenuto
moltiplicando n quantita che coincidono con un elemento per ciascuna riga; vi sono appunto n! possibili
scelte, che coincidono con le permutazioni tra gli n indici 1, 2, . . . , n; ogni permutazione e caratterizzata
dal simbolo P ed e pari, (−1)P = 1 o dispari (−1)P = −1 a seconda che sia ottenuto con un numero
pari o dispari di trasposizioni degli indici. In pratica il calcolo dei determinanti e effettuato applicando il
seguente teorema
det(A) = (−1)1+ja1j det(A1j) + . . . + (−1)n+janj det(Anj) (1.44)
cioe scegliendo una colonna della matrice (indice j) e sommando i determinanti delle sottomatrici di
dimensione n− 1 ottenute cancellando la colonna e la riga corrispondente a ciascun elemento (e.g. A12
e la matrice ottenuta cancellando la prima riga e la seconda colonna). Un’analoga relazione sussiste per
uno sviluppo lungo una riga. Varie proprieta elementari dei determinanti sono facilmente dimostrabili:
• det(1) = 1
• det(D) = Πni=1di
• det(T) = Πni=1tii
• det(AT ) = det(A)
• det(A∗) = det(A)∗
• det(A†) = det(A)∗
• det(AB) = det(A) det(B)
• det(αB) = αn det(A)
Possiamo ora definire esplicitamente la matrice inversa di una matrice generica come la matrice A−1
tale che
AA−1 = A−1A = 1 (1.45)

26 CAPITOLO 1. CENNI DI ALGEBRA LINEARE
Gli elementi della matrice inversa sono calcolabili infatti come:
(A−1)ij = (−1)i+j det(Aji)
det(A)(1.46)
quindi una matrice ammette inversa solo se il determinante non e nullo (altrimenti si dice singolare).
Le matrici associate ad applicazioni fisiche od utilizzate nei calcoli quantomeccanici godono spesso di
alcune proprieta speciali, tra le quali
• matrice simmetrica: AT = A
• matrice hermitiana: A† = A
• matrice ortogonale: AT = A−1
• matrice unitaria: A† = A−1
Una matrice reale simmetrica o ortogonale e anche hermitiana o unitaria.
Esempio 19. Nel trattamento di Huckel dei polieni lineari e necessario calcolare il determinante della
matrice tridiagonale
AT =
x 1 0 . . . 0
1 x 1 . . . 0
. . .
0 0 0 . . . x
(1.47)
Come si puo procedere per ridurlo ad un polinomio?
In generale la moltiplicazione di un vettore per una matrice denota una trasformazione del vettore. Nello
spazio tridimensionale possiamo usare analoghi geometrici per indicare una trasformazione (rotazione,
traslazione etc.) del vettore stesso. In uno spazio n-dimensionale questo non e ovviamente possibile.
Consideriamo per esempio il problema di descrivere la rotazione di un vettore nello spazio 3D: in generale
una rotazione si puo identificare mediante tre angoli, detti angoli di Eulero. Consideriamo un vettore
generico ~u che abbia le coordinate (X, Y, Z) in un dato sistema di riferimento fisso (per esempio solidale
con il laboratorio, e definito dai versori ~X, ~Y , ~Z); vogliamo sapere quali sono le sue coordinate in un
sistema di riferimento ruotato (per esempio solidale con una molecola, e definito dai versori ~x, ~y, ~z). Il
sistema di riferimento ruotato e definito in modo complicato ma rigoroso, tramite tre rotazioni elementari
successive (vedi Figura 1.3):
• una rotazione antioraria di un angolo α intorno a ~Z; questa rotazione porta ~Y nella linea dei nodi~N ;
• una rotazione antioraria di un angolo β intorno a ~N ; questa rotazione porta ~Z in ~z;

1.2. VETTORI E MATRICI 27
β
Z
X
Y Nx
O
y
z
α
γ
Figura 1.3: Angoli di Eulero α, β, γ per specificare la rotazione di un sistema di riferimento moleco-
lare rispetto ad un sistema di riferimento di laboratorio.
• una rotazione antioraria di un angolo γ intorno a ~z; questa rotazione porta ~N in ~y;
Dopo la prima rotazione, le coordinate del vettore siano (x′, y′, z′); dopo la seconda rotazione (x′′, y′′, z′′);
dopo la terza, nel sistema di riferimento molecolare (x, y, z). Applicando la trigonometria, si vede che:
x′
y′
z′
=
cos α sin α 0
− sin α cos α 0
0 0 1
X
Y
Z
(1.48)
x′′
y′′
z′′
=
cos β 0 − sin β
0 1
sin β cos β 0
x′
y′
z′
(1.49)
x
y
z
=
cos γ sin γ 0
− sin γ cos γ 0
0 0 1
x′′
y′′
z′′
(1.50)
Moltiplicando le tre matrici di rotazione elementari si ottiene:
x
y
z
=
cos α cos β cos γ − sin α sin γ sin α cos β cos γ + cos α sin γ − sin β cos γ
− cos α cos β sin γ − sin α cos γ − sin α cos β sin γ + cos α cos γ sin β sin γ
cos α sin β sin α sin β cos β
X
Y
Z
(1.51)
La matrice di trasformazione e detta matrice di Eulero, E.

28 CAPITOLO 1. CENNI DI ALGEBRA LINEARE
Esempio 20. Qual e la trasformazione lineare che moltiplica le componenti di un vettore n-dimensionale
per uno scalare a? Scrivere la matrice che rappresenta la trasformazione. E qual e la trasformazione che
scambia la componente i-esima con la componente j-esima?
Matrici e vettori sono molto utili per la rappresentazione matematica di proprieta fisiche che dipendano
dalla direzione di misura. Consideriamo per esempio il momento di dipolo indotto µ in una molecola da
un campo elettrico E: in generale si dovra scrivere
~m = a ~E (1.52)
dove ~m e ~E sono vettori tridimensionali, mentre a e un tensore cartesiano del secondo ordine, cioe una
proprieta rappresentabile da una matrice 3 × 3 (Figura 1.4). Se la nostra molecola ha una simmetria
cilindrica, e si esprimono tutte le grandezze vettoriali in un sistema di riferimento solidale con la molecola
ed avente l’asse z diretto lungo l’asse principale della molecola, allora la matrice di polarizzabilita assume
una forma particolarmente semplice:
a =
a⊥ 0 0
0 a⊥ 0
0 0 a‖
(1.53)
a
z
E
II
I
O
C
O
a
m
Figura 1.4: Schema del momento dipolare indotto in una molecola di CO2 da un campo elettrico
esterno.
In generale, le proprieta fisiche rappresentabili da matrici 3 × 3 sono dette proprieta tensoriali, e si
puo sempre identificare un sistema di riferimento (di solito solidale con la molecola), tale che siano
rappresentate da matrici diagonali (i valori diagonali si chiamano allora valori principali). Le grandezze

1.3. APPLICAZIONI 29
Relazione tensoriale y A x
Momento angolare = tensore di inerzia × velocita angolare ~L I ~ω
Momento di dipolo = polarizzabilita × campo elettrico ~m a ~E
Induzione elettrica = tensore dielettrico × campo elettrico ~D ε ~E
Magnetizzazione = suscettibilita magnetica × campo magnetico ~M χ ~H
Velocita della luce = indice di rifrazione × velocita nel vuoto ~v n ~v0
Tabella 1.1: Relazioni tensoriali nella forma generica ~y = A~x
tensoriali in Tabella (1.1) sono sempre rappresentabili come matrici reali simmetriche. Se i tre valori
principali sono uguali, la rappresentazione della proprieta tensoriale e la stessa in qualunque sistema di
riferimento, e si parla di proprieta scalare.
Esempio 21. Consideriamo il seguente problema; e data una molecola del complesso Cu(NH3)2+6 .
Calcoliamo il tensore di inerzia nel sistema di riferimento indicato in Figura 1.5 dai versori ~x, ~y, ~z. Dal
corso di Fisica I e noto che per un sistema di punti materiali con masse mi e coordinate ri = (xi, yi, zi),
il momento di inerzia ha componenti I11 =∑
i mi(r2i − x2
i ), I12 = −∑i mixiyi e cosı via. Nel sistema
di riferimento indicato in Figura 1.5 si ha percio:
I = 4mNH3l2
1 0 0
0 1 0
0 0 1
(1.54)
Quindi il sistema di riferimento ~x, ~y, ~z e tale che il tensore di inerzia e gia in forma diagonale. Lo studente
puo verificare facilmente che anche il sistema ruotato ~x′, ~y′, ~z′ gode di questa proprieta).
1.3 Applicazioni
Dopo aver discusso le proprieta principali delle matrici e dei vettori, riassumiamo brevemente alcune
applicazioni a problemi matematici che ricorrono sistematicamente nello studio della chimica (e non solo
della chimica quantistica).
1.3.1 Sistemi lineari
La soluzione dei sistemi lineari di equazioni si presta molto bene ad una implementazione in termini di
matrici e vettori. Consideriamo solo il caso di n equazioni lineari in n incognite. Possiamo scrivere un
sistema lineare in forma estesa
a11x1 + a12x2 + . . . + a1nxn = b1
a21x1 + a22x2 + . . . + a2nxn = b2

30 CAPITOLO 1. CENNI DI ALGEBRA LINEARE
z
x y
NH
l
3
NH
NH
NH
NH
NH
3 3
3
3
3
Cu2+
x
y
’ ’
’
Figura 1.5: Calcolo del momento di inerzia del complesso Cu(NH3)2+6 (vedi Esempio 21).
... (1.55)
an1x1 + an2x2 + . . . + annxn = bn
o in forma compatta:
Ax = b (1.56)
e la soluzione si ottiene semplicemente nella forma:
x = A−1b (1.57)
Naturalmente la soluzione esiste solo se la matrice A e invertibile, cioe se il suo determinante non e
nullo.
Esempio 22. 100 ml di una soluzione di iodio I− e Cl− titola 100 ml 0.1 N di AgNO3. 100 ml della stessa
soluzione, trattati con un eccesso di AgNO3, danno 1.5 g di precipitato. Calcolare la concentrazione di
iodio e cloro nella soluzione.
Di interesse sono anche i sistemi omogenei:
Ax = 0 (1.58)
in cui b = 0; se A non e singolare, l’unica soluzione possibile e A−10 = 0, cioe l’n-upla nulla. Se A e
singolare, det(A) =, l’equazione ammette infinite soluzioni, multiple della soluzione generica:
xi = (−1)i+j det(Aji) (1.59)
dove j e un indice di riga qualunque.

1.3. APPLICAZIONI 31
1.3.2 Trasformazioni
Come un vettore tridimensionale ha diverse componenti in un diverso sistema di riferimento (o, nel
linguaggio degli spazi vettoriali, in una diversa base), cosı una matrice ha diverse componenti in un
diverso sistema di riferimento.
Nella Sezione precedente era stato discusso brevemente il concetto di rotazione di un sistema di
riferimento; inoltre nell’esempio riferito alla polarizzabilita, il momento di dipolo, il campo elettrico
e la polarizzabilita erano stati espressi implicitamente in un sistema di riferimento ‘conveniente’, che
permetteva di esprimere la polarizzabilita in una forma semplice. In generale, qual e la relazione tra la
trasformazione di un sistema di riferimento e le componenti di un vettore o di una matrice? Ancora una
volta si tratta di un problema facilmente descrivibile nel linguaggio degli spazi vettoriali. di n-uple di
numeri. Consideriamo in generale una base ei (per esempio, nel caso della rotazione tridimensionale, la
base di versori che identifica gli assi del laboratorio). Un vettore generico v ha componenti v1, . . . , vn
nella base, cioe l’n-upla v. Introduciamo un secondo sistema di riferimento, cioe una seconda base e′i,
in cui il vettore v ha componenti v′1, . . . , v′n, o v′. La relazione tra le componenti del vettore nei due
sistemi di riferimento e in generale esprimibile come una trasformazione lineare
v = Tv′ (1.60)
Nell’esempio della rotazione del sistema di riferimento T era l’inversa della matrice di Eulero, che coincide
con la trasposta della matrice di Eulero (che e ortogonale), T = ET . Consideriamo ora una relazione
tra due vettori generici, u e v, nella forma
u = Av (1.61)
Nelle due basi ei e e′i (o nei due sistemi di riferimento, se si preferisce), si possono scrivere due distinte
relazioni matriciali:
u = Av (1.62)
u′ = A′v′ (1.63)
Sostituendo nella prima equazione le espressioni u = Tu′ e v = Tv′ si ottiene
Tu′ = ATv′ → u′ = T−1ATv′ (1.64)
da cui si vede che la matrice trasformata e
A′ = T−1AT (1.65)
La precedente equazione e detta anche trasformazione di similitudine; se T e unitaria o ortogonale,
parleremo di trasformazione unitaria o ortogonale.

32 CAPITOLO 1. CENNI DI ALGEBRA LINEARE
1.3.3 Problemi agli autovalori
Un problema molto comune che ricorre nelle applicazioni fisiche e il calcolo degli autovettori e degli
autovettori di una matrice, o della risoluzione del problema agli autovalori relativo ad una matrice.
Un’autovettore x di una matrice A e un vettore tale che:
Ax = λx (1.66)
cioe tale che la moltiplicazione per A non ne modifica altro che la lunghezza, di un coefficiente scalare
λ, detto autovalore. L’insieme degli autovalori e detto spettro della matrice. La n-upla nulla, 0 e sem-
pre una soluzione, triviale; dato un autovettore x, ogni vettore ax e pure un autovettore. E‘ possibile
avere piu di un autovettore linearmente indipendente corrispondente ad uno stesso autovalore. Il nu-
mero di autovettori linearmente indipendenti che condividono lo stesso autovalore e detto molteplicita
dell’autovalore; se la molteplicita e diversa da uno, l’autovalore e degenere. La soluzione del problema
agli autovalori e impostabile come il calcolo degli zeri di un polinomio di grado n. Infatti l’equazione agli
autovalori si puo scrivere come un sistema omogeneo
(A− λ1)x = 0 (1.67)
che ammette soluzioni non triviali solo se la matrice e singolare:
det(A− λ1) = 0 (1.68)
L’equazione precedente e detta equazione secolare. Se si e in grado di trovare gli autovalori e i cor-
rispondenti autovettori, i quali possono essere eventualmente normalizzati per evitare l’arbitrarieta di un
fattore scalare, possiamo costruire un matrice diagonale Λ con gli autovalori in diagonale ed una matrice
X la cui colonna i-esima e l’autovettore i-esimo. L’insieme delle n equazioni lineari diviene dunque
AX = XΛ (1.69)
od anche
Λ = X−1AX (1.70)
La matrice degli autovettori e dunque una matrice che trasforma A secondo una trasformazione di
similitudine che la rende diagonale: se due matrici generiche A, B commutano, [A,B] = 0, allora
si possono scegliere combinazioni lineari di autovettori di A che sono anche anche autovettori di B o
viceversa. In altre parole, due matrici con commutatore nullo si possono diagonalizzare simultaneamente
con la stessa trasformazione; questa affermazione si generalizza anche ad un numero generico di matrici:
se A1, A2, . . . , An commutano fra loro, possono essere diagonalizzate simultaneamente.

1.3. APPLICAZIONI 33
Esempio 23. Nella teoria MO, basata sull’approssimazione di Huckel, per calcolare gli orbitali molecolari
π dell’alchene 1,4-butadiene e necessario diagonalizzare la seguente matrice
A =
0 1 0 0
1 0 1 0
0 1 0 1
0 0 1 0
(1.71)
Il determinante secolare e dato da
λ4 − 3λ2 + 1 = 0 (1.72)
Lo studente verifichi che e un’equazione biquadratica con le seguenti soluzioni
λ1 = −√
3 +√
5
2λ2 = −
√3−√5
2λ3 =
√3−√5
2λ4 =
√3 +
√5
2(1.73)
Nelle applicazioni quantomeccaniche, i problemi agli autovalori coinvolgono sempre matrici hermitiane
(non a caso; vedi il Capitolo 2). Le matrici hermitiane H godono delle seguenti importanti proprieta
• gli autovalori di H sono reali
• gli autovettori corrispondenti ad autovalori distinti sono ortogonali
Poiche gli autovettori corrispondenti ad un autovalore degenere costituiscono comunque un set linear-
mente indipendente, si puo trovare una trasformazione lineare che li trasforma in un set ortogonale. Di
conseguenza, il set complessivo di autovettori normalizzati di una matrice hermitiana costituisce una
base ortonormale, tale che per una coppia qualunque
x†ixj = δij (1.74)
dove δij e il simbolo di Kronecker (0 se i 6= j, 1 se i = j). In definitiva, se X e la matrice degli
autovettori di H, e Λ e la matrice diagonale degli autovalori, si ha che:
X†HX = Λ (1.75)
dove X e unitaria e Λ e reale.

34 CAPITOLO 1. CENNI DI ALGEBRA LINEARE
1.3.4 Funzioni di matrici
Concludiamo questo capitolo introducendo il concetto di funzione di matrice. Data una funzione continua
f(x) nella variabile x definita dalla serie convergente
f(x) =∞∑
n=0
cnxn (1.76)
si definisce la funzione della matrice A come la matrice ottenuta dalla serie
f(A) =∞∑
n=0
cnAn (1.77)
Esempio 24. Polinomio in A: f(A) =∑N
n=0 cnAn
Esempio 25. Esponenziale in A: f(A) = exp(A) =∑∞
n=0 An/n!
Esempio 26. Coseno in A: f(A) = cos(A) =∑∞
n=0(−1)nA2n/(2n)!
Esempio 27. Seno in A: f(A) = sin(A) =∑∞
n=0(−1)nA2n+1/(2n + 1)!
Esempio 28. Inversa di 1+A: f(A) = (1+A)−1 =∑∞
n=0(−1)nAn; con la condizione limn→∞An = 0
Esempio 29. Inversa della radice di 1 + A: f(A) = (1 + A)−1/2 =∑∞
n=0(−1)n(2n)!An/22nn!2; con
la condizione limn→∞(2n)!An/22nn!2 = 0
Dato lo sviluppo in serie di una funzione, la valutazione della funzione di matrice si persegue utilizzando
il concetto di diagonalizzazione. Sia data la funzione generica (1.77) in A. Siano note la matrice degli
autovalori Λ e la matrice degli autovettori X di A, allora
f(A) =∞∑
n=0
cnAn =
∞∑
n=0
cn(XΛX−1)n (1.78)
e lo studente puo verificare per ispezione che (XΛX−1)n = XΛnX−1 da cui segue
f(A) = X
( ∞∑
n=0
cnΛn
)X−1 = Xf(Λ)X−1 (1.79)
e f(Λ) e immediatamente valutabile come una matrice diagonale il cui i-esimo elemento diagonale e
f(λi).
Esempio 30. E data la matrice 2× 2:
A =
(√2 1
1√
2
)(1.80)

1.4. ESERCIZI 35
Valutare exp(A). Le matrici Λ e X (gli autovettori non sono normalizzati) sono:
λ =
(√2 + 1 0
0√
2− 1
)X =
(1 1
1 −1
)(1.81)
Quindi exp(A) e data da:
exp(A) =
(1 1
1 −1
) (exp(
√2 + 1) 0
0 exp(√
2− 1)
) (1/2 1/2
1/2 −1/2
)=
=
(exp(
√2 + 1) + exp(
√2− 1) exp(
√2 + 1)− exp(
√2− 1)
exp(√
2 + 1)− exp(√
2− 1) exp(√
2 + 1) + exp(√
2− 1)
)/2 (1.82)
Esempio 31. La soluzione di un sistema di equazioni lineari differenziali ordinarie per n specie chimiche
aventi concentrazioni al tempo t c1(t), c2(t), . . . , cn(t) si presta molto bene ad evidenziare l’utilita delle
funzioni di matrici. Il sistema si puo scrivere in forma compatta come:
dc(t)
dt= −Kc(t) (1.83)
dove K e una matrice di coefficienti costanti. Le condizioni iniziali, che specificano la concentrazione al
tempo iniziale, sono c(0) = c0. La soluzione del sistema e semplicemente
c(t) = exp(−Kt)c0 (1.84)
che si puo scrivere anche come:
c(t) = X exp(−Λt)X−1c0 (1.85)
Quindi il problema e equivalente alla diagonalizzazione della matrice K.
1.4 Esercizi
1. Siano x ⊗1 y e x ⊗2 y due prodotti scalari definiti nello stesso spazio vettoriale. Dimostrare che
anche le seguenti operazioni
x¯1 y = x⊗1 y + x⊗2 y
x¯2 y = λx⊗1 y + µx⊗2 y
sono prodotti scalari dove λ e µ sono numeri arbitrari non negativi e non simultaneamente nulli.
2. Definire il prodotto scalare dello spazio dei polinomi di grado ≤ n in modo che la base
1, t, t2/2!, . . . , tn/n!
sia ortonormale.

36 CAPITOLO 1. CENNI DI ALGEBRA LINEARE
3. Valutare il determinante di una matrice triangolare superiore di dimensione n.
4. Valutare il determinante di una matrice triangolare inferiore di dimensione n.
5. Porre in forma di polinomio di t il determinante della matrice
A =
−t 0 0 . . . 0 a1
a2 −t 0 . . . 0 0
0 a3 −t . . . 0 0
0 0 0 . . . −t 0
0 0 0 . . . an −t
6. Per quali valori di λ il seguente sistema ammette soluzione?
x1 + x2 + λx3 = 1
x1 + λx2 + x3 = 1
λx1 + x2 + x3 = 1
7. Trovare l’inversa della matrice
A =
0 0 . . . 0 a1
0 0 . . . a2 0
. . .
an 0 . . . 0 0
8. Trovare l’inversa della matrice
A =
1 0 . . . 0 0
a 1 . . . 0 0
0 a . . . 0 0
. . .
0 0 . . . a 1
9. Esprimere l’equazione secolare della seguente matrice in forma polinomiale:
A =
0 0 . . . 0 b1
0 0 . . . 0 b2
. . .
0 0 . . . 0 bn
c1 c2 . . . cn a
10. Trovare gli autovalori di una matrice triangolare superiore di dimensione n.

Capitolo 2
Meccanica quantistica
L’estensione dei concetti introdotti nelle Sezioni del precedente Capitolo per spazi vettoriali di dimensioni
finite e abbastanza ovvia. Un concetto completamente nuovo che deve pero essere introdotto e quello di
completezza, essenziale per il calcolo di autovalori ed autovettori e per il trattamento computazionale di
matrici di dimensioni formalmente infinite. Gli spazi vettoriali di dimensioni infinite per i quali si possa
definire la proprieta addizionale di completezza sono anche detti spazi di Hilbert e sono di importanza
fondamentale per lo studio della meccanica quantistica. Per questo motivo cambieremo la simbologia
generica del Capitolo precedente utilizzando la notazione di Dirac o notazione bracket tipica delle
applicazioni quanto-meccaniche. Un vettore di uno spazio Hilbertiano e rappresentato da un ket |φ〉.
2.1 Spazi di Hilbert
Consideriamo uno spazio vettoriale di dimensione infinita, con un prodotto hermitiano. Per due vettori
generici, |φ〉 e |ψ〉, il prodotto e 〈φ|ψ〉 (dove il primo vettore appare in forma bra). L’opposto di |φ〉 e
−|φ〉, l’elemento nullo e |0〉 e cosı via. Il modulo di un vettore e 〈φ|φ〉. Sia ora data una successione
di vettori |φ1〉, . . . , |φn〉, . . . ed un vettore limite |φ〉. La successione converge1 al vettore limite se per
ogni numero ε positivo piccolo a piacere esiste un intero N(ε) tale che per n > N(ε) vale che
〈δφn|δφn〉 < ε (2.1)
dove |δφn〉 = |φn〉−|φ〉. Si dice che lo spazio vettoriale e uno spazio di Hilbert, di cui la successione φn e
una base, se la successione converge a |φ〉 per ogni |φ〉. Nel seguito considereremo solo spazi hilbertiani.
Esempio 32. Lo spazio delle funzioni periodiche a valori complessi e uno spazio Hilbertiano. Qual e
una sua base completa?
1In senso forte: esiste anche una definizione di convergenza debole, piu generale.
37

38 CAPITOLO 2. MECCANICA QUANTISTICA
Una volta definita una base, possiamo rappresentare ogni vettore nella base definendone le componenti.
Se pensiamo agli elementi dello spazio hilbertiano come a delle funzioni generiche, queste possono essere
rappresentate da vettori colonna di dimensioni infinite. Un’applicazione ovvero una corrispondenza che
mette in relazione un vettore con un altro vettore puo essere espressa tramite il concetto, fondamentale,
di operatore
O|φ〉 = |ψ〉 (2.2)
Nell’equazione precedente l’operatore O agisce sul vettore |φ〉 trasformandolo nel vettore |ψ〉. Sia ora
data una base ortonormale |i〉; stando alle definizioni del Capitolo 4, si ha che 〈i|i〉 = δij. Un vettore
generico e
|φ〉 =∑
j
φj|j〉 (2.3)
sostituendo nell’equazione (2.2) e premoltiplicando per un generico |i〉:∑
j
〈i|O|j〉φj = ψj (2.4)
che si puo scrivere in termini matriciali come
Oφ = ψ (2.5)
La rappresentazione di un operatore e una matrice di dimensioni infinite.
2.1.1 Operatori lineari
Nella meccanica quantistica sono impiegati solo gli operatori lineari che godono della proprieta
O(a|φ〉+ b|ψ〉) = aO|φ〉+ bO|ψ〉 (2.6)
dove O e un operatore lineare, a e b due scalari generici. Tutti i concetti definiti in precedenza per le
matrici possono essere generalizzati per gli operatori
• operatore unita: 1 =∑
i |i〉〈i|
• operatore aggiunto di un operatore O: 〈ψ|O†|φ〉∗ = 〈φ|O|ψ〉
• commutatore di due operatori:[O1, O2
]= O1O2 − O2O1
e cosı via: per esempio, un’operatore O si dice hermitiano se O† = O.

2.2. FORMULAZIONE DELLA QUANTOMECCANICA 39
Esempio 33. Si dimostri, usando le proprieta degli operatori lineari, dei prodotti hermitiani e le
definizioni precedenti, che
(a1O1 + a2O2)† = a∗1O
†1 + a∗2O
†2 (2.7)
(O1O2)† = O†
2O†1 (2.8)
Il problema agli autovalori per un operatore si pone analogamente a quello di una matrice di dimensioni
finite:
O|φ〉 = λ|φ〉 (2.9)
anzi, coincide con un problema matriciale a dimensioni infinite una volta che si sia introdotto una base:
Oφ = λφ (2.10)
Valgono per gli operatori hermitiani le medesime proprieta introdotte per le matrici hermitiane: gli
autovalori sono reali, autovettori (o autofunzioni) corrispondenti ad autovalori distinti sono ortogonali;
l’operatore di trasformazione e unitario. Nella notazione di Dirac un autovettore con autovalore λ si
indica con |λ〉, ovvero O|λ〉 = λ|λ〉.
2.2 Formulazione della quantomeccanica
Formuliamo i principi generali della meccanica quantistica 2 utilizzando la simbologia matematica appena
introdotta.
Principio I: lo stato fisico di un sistema isolato e descritto da un vettore |φ〉 di un appropriato spazio
hilbertiano.
Principio II: una grandezza fisica osservabile e rappresentato da un operatore lineare hermitiano
F .
Principio III: i risultati possibili della misura di un’osservabile F sono gli autovalori fi di F :
F|fi〉 = fi|fi〉 (2.11)
se |φ〉 = |fi〉, il sistema e in un autostato di F e la misura da con certezza fi.
2Non si tratta di postulati rigorosi che costituiscano assiomi matematici da cui dedurre una struttura matematicarigorosa; piuttosto sono principi fisici, esposti in modo semplificato per comprendere le applicazioni della meccanicaquantistica nella sua formulazione non-relativistica.

40 CAPITOLO 2. MECCANICA QUANTISTICA
Principio IV: il valore d’attesa di un’osservabile F e definito come
F =〈φ|F|φ〉〈φ|φ〉 (2.12)
dove F e il risultato medio di una serie di misure di F
Principio V: l’operatore relativo ad un’osservabile si puo esprimere in funzione di operatori sposta-
mento x e momento p (in una dimensione), che obbediscono al principio
[x, p] = ih (2.13)
dove h = h/2π = 1.054× 10−34Js.
Principio VI: lo stato al tempo t di un sistema e ottenibile come la soluzione dell’equazione di
Schrodinger
ih∂
∂t|φ(t)〉 = H|φ(t)〉 (2.14)
dove H e l’operatore hamiltoniano del sistema isolato. Gli autovalori dell’hamiltoniano sono le
autoenergie del sistema:
H|E〉 = E|E〉 (2.15)
|E〉 e un autostato del sistema con energia E.
Cosa significa ‘spazio appropriato’? Dipende dalla natura fisica del sistema. Consideriamo il caso di
una particella che si muova lungo una retta: la coordinata (nel senso della meccanica classica) e x, il
momento associato e p. Gli operatori quantomeccanici corrispondenti sono x e p. Definiamo la base
continua |x〉 come gli autostati di x con autovalori x:
x|x〉 = x|x〉 (2.16)
Nella base continua |x〉, un vettore |φ〉 e rappresentato come una funzione φ(x), l’operatore spostamento
x e rappresentato dalla moltiplicazione per la funzione x, cioe x|φ〉 = xφ(x) l’operatore p e rappresentato
da h/i
∂
∂x. L’hamiltoniano e invece dato dall’espressione
H = − h2
2m
∂2
∂x2+ U(x) (2.17)
dove il primo termine rappresenta l’energia cinetica (m e la massa), il secondo l’energia potenziale.

2.2. FORMULAZIONE DELLA QUANTOMECCANICA 41
2.2.1 Principio Variazionale
Dato l’operatore hamiltoniano del sistema, uno dei problemi fondamentali in meccanica quantistica e il
calcolo degli autovalori, ovvero delle autoenergie del sistema, e dei corrispondenti autostati. Per esempio,
un chimico che conosca, con vari gradi di approssimazione, l’espressione per l’energia totale classica di
una molecola, e dunque sia in grado di costruire l’hamiltoniano quantomeccanico (applicando il principio
di corrispondenza e sostituendo alle funzioni classiche posizione e momento le corrispondenti rappresen-
tazioni nella base continua) vuole conoscere prima di ogni altra cosa l’energia dello stato fondamentale
della molecola cioe il piu basso autovalore dell’hamiltoniano. Ha cosı informazioni sull’energia ‘di riposo’
della molecola, nel suo stato fondamentale, e puo confrontare questo dato con misure di tipo calorimet-
rico e termodinamico. Il calcolo degli autovalori di un sistema richiede 1) la conoscenza dell’hamiltoniano
del sistema; 2) una rappresentazione matriciale dell’hamiltoniano. In generale comunque si tratta di un
problema non-triviale. I sistemi (gli hamiltoniani) di cui sia possibile calcolare in modo esatto (analitico)
lo spettro di autoenergie sono pochi, e saranno discussi nella II Dispensa. Tecniche di risoluzione ap-
prossimata sono disponibili e molto diffuse. Rimandiamo lo studente alla III Dispensa per una discussione
sistematica della loro applicazione al calcolo di strutture elettroniche atomiche e molecolari.
Consideriamo in questa Sezione il calcolo approssimato dell’autostato fondamentale di un sistema.
In generale si potra esprimere lo stato (il vettore) come una combinazione (non necessariamente lineare)
di vettori di base noti; i coefficienti dell’espansione possono essere determinati sulla base del principio
variazionale, che sostanzialmente sfrutta le proprieta di hermiticita dell’operatore hamiltoniano.
Definiamo innanzitutto il rapporto di Rayleigh per un generica vettore d’onda |Ψ〉
ε[|Ψ〉] =〈Ψ|H|Ψ〉〈Ψ|Ψ〉 (2.18)
Il Principio variazionale afferma che se E0 e il piu basso autovalore del sistema, il rapporto di Rayleigh
e sempre maggiore o uguale a E0
ε[|Ψ〉] ≥ E0 (2.19)
La dimostrazione e la seguente: espandiamo |Ψ〉 sulla base degli autostati esatti dell’hamiltoniano H
H|Φn〉 = En|Φn〉 E0 ≤ E1 ≤ . . . (2.20)
〈Φn|Φ′n〉 = δnn′ (2.21)
|Ψ〉 =∑n
cn|Φn〉 (2.22)
consideriamo la quantita 〈Ψ|H − E0|Ψ〉:
〈Ψ|H − E0|Ψ〉 =∑
nn′c∗ncn′〈Φn|H − E0|Φn′〉 =
∑
nn′c∗ncn′(En − E0)〈Φn|Φn′〉 =
∑n
|cn|2(En − E0) ≥ 0 (2.23)

42 CAPITOLO 2. MECCANICA QUANTISTICA
infatti |cn|2 ≥ 0, En ≥ E0; segue che:
〈Ψ|H|Ψ〉 − E0〈Ψ|Ψ〉 ≥ 0 ⇒ ε[|Ψ〉] ≥ E0 QED (2.24)
Nella sua versione piu comune, il teorema variazionale e implementato sotto forma di metodo vari-
azionale lineare. Il vettore d’onda di prova |Ψ〉 e cioe dato come una combinazione lineare di vettori
noti:
|Ψ〉 =N∑
n′=1
cn′ |Ψn′〉 (2.25)
sostituendo in H|Ψ〉 = E|Ψ〉:∑
n′cn′H|Ψn′〉 = E
∑
n′cn′|Ψn′〉 (2.26)
premoltiplicando per 〈Ψn|∑
n′cnHnn′ = E
∑
n′cnSnn′ (2.27)
dove Hnn′ = 〈Ψn|H|Ψn′〉, Snn′ = 〈Ψn|Ψn′〉; in forma matriciale:
Hc = ESc (2.28)
Il problema agli autovalori diviene piu semplice se si assumono delle funzioni di base ortonormali S = 1:
Hc = Ec (2.29)
che e ora un problema agli autovalori in forma standard, con matrici e vettori di dimensioni finite, poiche
la rappresentazione (2.25) e di solito arrestata ad un numero finito di termini.
Esempio 34. E dato l’hamiltoniano
H = − h2
2m
∂2
∂x2+ V (x) (2.30)
per un sistema monodimensionale. il potenziale V (x) e
V (x) =
V0 0 < x < L/2
0 L/2 < x < L
∞ altrove
(2.31)
Consideriamo le due funzioni di base
|1〉 =
√2
Lsin
(πx
L
)(2.32)
|2〉 =
√2
Lsin
(2πx
L
)(2.33)
Calcolare un valore approssimante all’energia dello stato fondamentale mediante il metodo variazionale.
Si deve procedere 1) calcolando gli elementi di matrice 〈i|H|j〉 con i, j pari a 1,2 (il prodotto hermitiano
e l’integrale su x); diagonalizzando la matrice H 2 × 2 e scegliendo l’autovalore piu piccolo.

2.2. FORMULAZIONE DELLA QUANTOMECCANICA 43
2.2.2 Cenno alla teoria delle perturbazioni
Di solito e possibile conoscere in modo esatto, o quantomeno accurato, la soluzione esatta di un problema
agli autovalori relativo ad un sistema fisico semplificato rispetto al sistema di cui si vogliono effettivamente
calcolare le proprieta. Se il sistema semplificato differisce dal sistema reale solo per un termine piccolo
dell’hamiltoniano (perturbazione) e possibile cercare sistematicamente la correzione alle autoenergie ed
autostati del sistema semplificato mediante la teoria delle perturbazioni indipendenti dal tempo.
Supponiamo percio che l’hamiltoniano del sistema reale sia
H = H0 + εV (2.34)
doveH0 e l’hamiltoniano del sistema semplificato (imperturbato) e V e la perturbazione; ε e un parametro
che definisce l’ordine di grandezza della perturbazione. Sia nota la soluzione al problema imperturbato
(PI)
H0|n〉 = E(0)n |n〉 (2.35)
Gli autovettori |n〉 costituiscono una base ortonormale e completa dello spazio hilbertiano. Cerchiamo
la soluzione al problema perturbato (PP)
H|Φ〉 = E|Φ〉 (2.36)
Nel seguito, per semplicita considereremo solo sistemi con spettri di autovalori discreti e non degeneri.
Possiamo rappresentare le autofunzioni del PP nella base costituita dalle autofunzioni del PI:
|Φ〉 =∑n
cn|n〉 (2.37)
Sostituendo la precedente eguaglianza nell’espressione del PP si ottiene
∑n
cn
[E(0)
n + εV]|n〉 =
∑n
cn|n〉 (2.38)
ed integrando per un generico 〈Φ(0)m |
[E − E(0)
m
]cm = ε
∑n
Vmncn (2.39)
dove Vmn = 〈m|V|n〉. Si vuole calcolare la correzione all’ m-esimo autostato, cioe si vuole che per
ε → 0 (perturbazione nulla) il sistema perturbato tenda all’autostato m-esimo del sistema perturbato.
Espandiamo ora i coefficienti cn e l’energia E in serie di ε:
E = E(0)m + εE(1) + ε2E(2) + . . . (2.40)
cn = δmn + εc(1)n + ε2c(2)
n + . . . (2.41)
come si vede, per ε → 0 l’energia diventa E(0)m , mentre i coefficienti cn diventano δmn, e quindi 1 se
m = n, e zero in caso contrario. Per calcolare le correzioni alle energie e ai coefficienti si procede

44 CAPITOLO 2. MECCANICA QUANTISTICA
sostituendo le espansioni in serie nelle equazioni (2.39), raccogliendo i termini nella stessa potenza di ε a
primo e secondo membro, ed eguagliando. Cosı per esempio la correzione al primo ordine per le energie
si ottiene conservando solo i termini di primo ordine nell’equazione (2.39) con n = m:
E(1) = Vmm (2.42)
mentre le correzioni al primo ordine per i coefficienti si ottengono con n 6= m:
c(1)n =
Vmn
E(0)m − E
(0)n
n 6= m (2.43)
il coefficiente c(1)m resta indeterminato. Si puo specificare imponendo che l’autostato del PP al primo
ordine sia normalizzato, a meno di termini al primo ordine: si puo verificare che cio avviene per c(1)m = 0.
Le approssimazioni successive si calcolano in modo analogo.

Capitolo 3
Funzioni speciali
Questo capitolo e dedicato al riassunto delle proprieta di alcune classi di funzioni che ritornano spesso
nelle soluzioni di problemi della meccanica quantistica. Si tratta di un compendio molto breve, e senza
pretese di completezza, che ha il solo scopo di fornire un ‘formulario’ allo studente per poter meglio
comprendere alcuni degli esercizi che trovera nella II Dispensa.
3.1 Alcune funzioni speciali
3.1.1 Integrali ellittici
Consideriamo gli integrali indefiniti1
I =∫
dxR[x,
√P (x)
](3.1)
dove R e una funzione razionale e P (x) e un polinomio di terzo o quarto grado. Si dimostra che I e
sempre riducibile ad una funzione elementare di∫
dx1√
(1− x2)(1− k2x2)(3.2)
∫dx
√1− k2x2
√1− x2
(3.3)
∫dx
1
(1 + n2x2)√
(1− x2)(1− k2x2)(3.4)
che sono rispettivamente integrali ellittici di prima, seconda e terza specie nella forma di Legendre.
La quantita k e detta modulo degli integrali. Altre espressioni sono le seguenti
F (ψ, k) =∫ ψ
0dα
1√(1− k2 sin2 α)
(3.5)
1Nel seguito, se non altrimenti specificato si usa x per una variabile reale e z per una variabile complessa.
45

46 CAPITOLO 3. FUNZIONI SPECIALI
E(ψ, k) =∫ ψ
0dα
√(1− k2 sin2 α) (3.6)
D(ψ, k) =F (ψ, k)− E(ψ, k)
k2(3.7)
Π(ψ, n, k) =∫ ψ
0dα
1
(1 + n2 sin2 α)√
(1− k2 sin2 α)(3.8)
Gli integrali per ψ = π/2 si dicono integrali ellittici completi.
3.1.2 Funzione gamma
Definiamo la funzione gamma sotto forma di integrale di Eulero:
Γ(z) =∫ ∞
0dttz−1e−t (3.9)
Per z = n + 1 (n intero) la funzione gamma coincide con il fattoriale di n:
Γ(n + 1) = n! (3.10)
Vale la seguente espansione:
1
Γ(z)= zeγzΠ∞
n=1
[(1 +
z
n
)e−z/n
](3.11)
dove γ e la costante di Eulero:
γ = limm→∞(1 +
1
2+
1
3+
1
4+ . . . +
1
m− ln m
)= 0.5772156649 . . . (3.12)
Di interesse e l’espansione per |z| → ∞ (formula di Stirling):
Γ(z) ∼ e−zzz−1/2√
2π(1 +
1
12z+
1
288z2− 139
51840z3− 571
2488320z4+ . . .
)(3.13)
3.1.3 Funzione dell’errore
La funzione dell’errore e definita come:
erf(z) =2√π
∫ z
0dte−t2 (3.14)
La funzione complementare erfc(z) e 1− erf(z). Alcune relazioni utili sono le seguenti:
dn+1
dzn+1erf(z) = (−1)n 2√
zHn(z)e−z2
(3.15)
erf(z) =2√z
∑
n=0
(−1)nz2n+1
n!(2n + 1)(3.16)
√πzez2
erfc(z) ∼ 1 +∑
n=1
(−1)n 1 · 3 · . . . (2n− 1)
(2z2)n|z| → ∞ (3.17)
∫dxe−(ax2+2bx+c) =
1
2
√π
ae
b2−aca erf
(√ax +
b√a
)+ costante (3.18)

3.2. POLINOMI ORTOGONALI 47
3.1.4 Funzioni di Bessel
Sia ν un numero reale non-negativo. Le funzioni di Bessel del primo tipo J±ν(z) e del secondo tipo
Y±ν(z) verificano la seguente equazione differenziale:
z2d2w
dz2+ z
dw
dz+ (z2 − ν2)w = 0 (3.19)
Sono funzioni regolari, finite a z = 0; J±ν(z) ha un estremo superiore finito per <z → ∞. Le funzioni
di Bessel modificate I±ν(z) e Kν(z) sono soluzioni dell’equazione differenziale
z2d2w
dz2+ z
dw
dz− (z2 + ν2)w = 0 (3.20)
II±ν(z) tende ad un valore finito per z → 0. Le funzioni di Bessel sono utili p.es. per la soluzione di
equazioni differenziali in problemi con una simmetria cilindrica (vedi II Dispensa). Valgono le seguenti
relazioni:
Jn(z) =1
inπ
∫ π
0dθeiz cos θ cos(nθ) (3.21)
In(z) =1
π
∫ π
0dθez cos θ cos(nθ) (3.22)
3.2 Polinomi ortogonali
E data una funzione reale non negativa w(x) ed un intervallo [a, b]. Supponiamo che∫ b
adxxnw(x) esista
per n naturale (0,1,2, . . .), e che sia positivo per n = 0. Allora esiste una successione di polinomi p0(x),
p1(x), p2(x) etc. determinata univocamente dalle condizioni:
1. pn(x) e un polinomio di grado n; il coefficiente qn di xn in pn(x) e positivo
2. i polinomi pn(x) sono ortogonali rispetto al prodotto scalare 〈f |g〉 =∫ b
adxw(x)f(x)g(x):
〈pm|pn〉 = δmnkm (3.23)
dove km e la norma km = 〈pm|pm〉. La successione costituisce un set di polinomi ortogonali su [a, b]
rispetto alla funzione peso w(x). Valgono le seguenti relazioni generali
1. Relazione di Christoffel-Darboux
∑
k=0
npk(x)pk(y) =qn
qn+1
pn+1(x)pn(y)− px(x)pn+1(y)
x− y(3.24)
2. Relazione ricorsiva
pn+1(x) = (anx + bn)pn(x)− cnpn−1(x) n ≥ 1 (3.25)

48 CAPITOLO 3. FUNZIONI SPECIALI
3.2.1 Polinomi di Legendre
I polinomi di Legendre Pn(x) sono definiti in [−1, 1]. Verificano l’equazione differenziale:
(1− x2)d2Pn(x)
dx2− 2x
dPn(x)
dx+ n(n + 1)Pn(x) = 0 (3.26)
La funzione peso e w = 1, la norma e kn = 2/2n + 1. La relazione ricorsiva e:
(n + 1)Pn+1(x) = (2n + 1)xPn(x)− nPn−1(x) (3.27)
dove P0(x) = 1, P1(x) = x.
3.2.2 Polinomi di Hermite
I polinomi di Hermite Hn(x) sono definiti in [−∞,∞]. Verificano l’equazione differenziale:
d2Hn(x)
dx2− 2x
dHn(x)
dx+ 2nHn(x) = 0 (3.28)
La funzione peso e w = e−x2, la norma e kn =
√π2nn!. La relazione ricorsiva e:
Hn+1(x) = 2nxHn(x)− 2nHn−1(x) (3.29)
dove H0(x) = 1, H1(x) = 2x.
3.2.3 Polinomi di Laguerre
I polinomi di Laguerre Ln(x) sono definiti in [0,∞]. Verificano l’equazione differenziale:
xd2Ln(x)
dx2+ (1− x)
dLn(x)
dx+ nLn(x) = 0 (3.30)
La funzione peso e w = e−x, la norma e 1. La relazione ricorsiva e:
(n + 1)Ln+1(x) = (2n + 1− x)Ln(x)− nLn−1(x) (3.31)
dove L0(x) = 1, L1(x) = 1− x.

Parte II
Sistemi quantomeccanici risolvibili
analiticamente.
49


Capitolo 4
Sistemi risolvibili analiticamente
Questa seconda dispensa e organizzata in unico Capitolo, ed e una raccolta di problemi risolti relativi
a sistemi quantomeccanici risolvibili analiticamente. Tra i vari casi trattati sono compresi gli esempi
che si trovano in tutti i libri di testo: particella nella scatola monodimensionale e bidimensionale, ro-
tatore monodimensionale, oscillatore armonico, particella su una sfera, atomo idrogenoide. In piu sono
presentate “variazioni sul tema”, come particelle in scatole monodimensionali di forme svariate, scatole
tridimensionali, scatole cilindriche e set di oscillatori interagenti.
Esempi/esercizi non risolti (con suggerimenti e schemi di soluzione in alcuni casi) sono distribuiti nel
corso del testo.
4.1 Sistemi monodimensionali
Consideriamo lo schema in Fig. (4.1), che rappresenta il potenziale confinante per una particella (in una
dimensione) imprigionata in una scatola dalle pareti infinite. Qual e l’andamento nel tempo della funzione
d’onda della particella, data una determinata configurazione iniziale? Vedremo che per rispondere a
questo quesito sara necessario definire le autofunzioni e gli autovalori dell’operatore hamiltoniano. Anzi,
poiche il problema e di natura generale, consideriamo per cominciare un sistema generico definito nello
spazio delle funzioni f(x) dove x e l’insieme delle coordinate del sistema. Al tempo t il sistema e definito
dalla funzione d’onda |ψ(x, t)〉 la cui evoluzione temporale e dettata dall’equazione di Schrodinger
ih∂
∂t|ψ(x, t)〉 = H|ψ(x, t)〉 (4.1)
al tempo t = 0 la funzione e definita da una configurazione generica (che ci dice come il sistema e stato
preparato inizialmente):
|ψ(x, 0)〉 = |ψ0(x)〉 (4.2)
51

52 CAPITOLO 4. SISTEMI RISOLVIBILI ANALITICAMENTE
Sappiamo che l’hamiltoniano ammette autostati |E〉 con autoenergie E:
H|E〉 = E|E〉 (4.3)
che sono ortonormali 〈E|E ′〉 = δE,E′ e costituiscono una base completa dello spazio. Quindi possiamo
espandere |ψ(x, t)〉|ψ(x, t)〉 =
∑
E
cE(t)|E〉 (4.4)
cE(t) = 〈ψ(x, t)|E〉 (4.5)
e al tempo t = 0 vale che
cE(0) = 〈ψ0(x)|E〉 (4.6)
Sostituendo nell’Eq. (4.1) si ottiene, moltiplicando per un generico 〈E| ed integrando
ihcE(t) = EcE(t) (4.7)
Infine, con le condizioni iniziali (4.6) si trova
cE(t) = 〈ψ0(x)|E〉 exp(Et/ih) (4.8)
e la funzione d’onda si puo scrivere al tempo t come:
|ψ(x, t)〉 =∑
E
|E〉〈ψ0(x)|E〉 exp(Et/ih) (4.9)
Dunque la funzione d’onda e una sovrapposizione degli autostati del sistema con ‘pesi’ che dipendono
dal tempo in modo esponenziale, dove le ‘costanti di decadimento’ di ciascun esponenziale sono
proporzionali agli autovalori, e dalle condizioni iniziali.
In linea di principio possiamo distinguere lo spettro di autoenergie di un sistema in due parti: lo
spettro discreto, ovvero l’insieme delle autoenergie separate da intervalli non accessibili, e lo spettro
continuo. Consideriamo un sistema monodimensionale con un potenziale U(x) che tenda a U0 per
x → +∞ e a U1 > U0 per x → −∞, vedi Fig. (4.2). Lo spettro discreto si ha per autostati con energie
E < U0: la particella e chiusa nella scatola. Se E > U0 lo spettro diviene continuo, e la partcella e
libera. Il numero di autostati discreti puo esser finito (se U0 e U1 sono quantita finite) od infinito. Nel
seguito consideremo solo stati con autovalori discreti, cioe sistemi in stati confinati.
Torniamo ora al caso della particella in una scatola monodimensionale con pareti infinite. Assumiamo
che l’energia potenziale sia nulla all’interno della scatola ed infinita all’esterna. La funzione d’onda e
dunque definita solo per 0 ≤ x ≤ L, vedi Fig. (4.1). L’equazione di Schrodinger indipendente dal tempo
assume la forma 1:
− h2
2m
d2
dx2ψE(x) = EψE(x) (4.10)
1Ci poniamo nella rappresentazione continua, in modo da lavorare direttamente con funzioni della variabile x

4.1. SISTEMI MONODIMENSIONALI 53
L x
Figura 4.1: Potenziale per una particella in una scatola 1D.
dove m e la massa della particella. Valgono le condizioni al contorno
ψE(0) = 0, ψE(L) = 0 (4.11)
L’Eq. (4.10) e un’equazione differenziale ordinaria a coefficienti costanti del secondo ordine. La sua
soluzione generica ha la forma
ψE(x, t) = A1 exp(α1x) + A2 exp(α2x) (4.12)
dove α1,2 sono le radici dell’equazione
− h2
2mα2 = E (4.13)
vale a dire
α1,2 = ±i
√2mE
h2 (4.14)
Sostituendo nelle condizioni al contorno si ottiene
A1 + A2 = 0 (4.15)
A1 exp
iL
√2mE
h2
+ A2 exp
−iL
√2mE
h2
= 0 (4.16)
che e un sistema lineare omogeneo in A1 e A2, risolvibile solo se il determinante dei coefficienti e nullo.
Si trova cosı la condizione in E:
exp
2iL
√2mE
h2
= 1 (4.17)

54 CAPITOLO 4. SISTEMI RISOLVIBILI ANALITICAMENTE
U(x)
U
U1
0
x
Figura 4.2: Un potenziale generico in una dimensione.
le cui soluzioni sono
E =h2π2k2
2mL2, k ∈ N (4.18)
Gli autostati di una particella chiusa in un pozzo infinitamente profondo sono quantizzati come i quadrati
dei numeri interi; sono anche inversamente proporzionali alla massa della particella e alla larghezza del
pozzo. Vale a dire: per una massa molto grande, od un pozzo molto largo, gli autovalori sono molto
vicini tra loro (lo spettro diviene quasi continuo), e il comportamento della particella si avvicina al
comportamento previsto dalla meccanica classica. Le autofunzioni si trovano subito tenendo conto
A1 = −A2; imponendo la condizione che siano normalizzate in [0, L] si trova:
ψk(x) =
√2
Lsin
( |k|πL
x
)(4.19)
Supponiamo ora che la particella sia inizialmente al centro del pozzo:
ψ(x, 0) = δ(x− L/2) =
√2
L
∑n
(−1)n sin
( |2n + 1|πL
x
)(4.20)

4.2. FUGA DAL POZZO 55
Da cui segue che al tempo t la funzione d’onda e
ψ(x, t) =
√2
L
∑n
(−1)n exp
[(2n + 1)2hπ2
2mL2t
]sin
( |2n + 1|πL
x
)(4.21)
Naturalmente per una scatola con pareti infinite tutte le autoenergie sono discrete (non esistono stati
“liberi” della particella).
Esempio 1. Calcolare e rappresentare in un grafico ai tempi t: 1 ps, 10 ps, 1 ns e 1 µs la probabilita
|ψ(x, t)| per un neutrone in un pozzo largo 1 A, che sia posto al centro del pozzo a t = 0.
4.2 Fuga dal pozzo
E se una le pareti del pozzo avessero un’altezza finita? Il potenziale e rappresentato in Fig. (4.3):
V (x) =
V x ≤ 0
0 0 < x < L
V L ≤ x
(4.22)
Consideriamo solo lo spettro discreto, vale a dire gli stati confinati della particella, E < V . La soluzione
generica per l’autofunzione ψE(x) con autoenergia E e definita nei singoli intervalli come
ψ(x, t) =
B1 exp(bx) x ≤ 0
A1 exp(iax) + A2 exp(−iax) 0 ≤ x < L
B2 exp(−bx) L ≤ x
(4.23)
dove a =√
2mE/h2 e b =√
2m(V − E)/h2; si noti che che per x < 0 si pone a zero il coefficiente
dell’esponenziale exp(−bx) che altrimenti divergerebbe per x → −∞ (ed analogamente per x ≥ L).
Valgono poi quattro condizioni relative alla continuita e derivabilita dell’autofunzione in x = 0 e
x = L:
A1 + A2 = B1
iaA1 − iaA2 = bB1
A1 exp(iaL) + A2 exp(−iaL) = B2 exp(−bL) (4.24)
iaA1 exp(iaL)− iaA2 exp(−iaL) = −bB2 exp(−bL)
Risolvendo il determinante 4 × 4 (scrivetelo), si ottiene una condizione in E. Saltando vari passaggi
algebrici il risultato e:
arcsinha√2mV
=nπ − kL
2(4.25)
con n ∈ N , che si risolve in a. Si verifica subito che il numero degli autostati discreti e finito.

56 CAPITOLO 4. SISTEMI RISOLVIBILI ANALITICAMENTE
4.3 Una scatola con una barriera interna
L’esempio successivo e relativo al caso di un pozzo con un barriera interna finita, Fig. (4.3). Lo schema
di soluzione e analogo ai casi precedenti: i) definizione delle forme generali delle autofunzioni; ii) impo-
sizione di condizioni al contorno; iii) determinazione degli autostati dalla soluzione di un sistema lineare
omogeneo. L’algebra e un piu complicata, ma per geometrie elementari si semplifica considerevolmente.
Il potenziale che descrive il nostro pozzo e dato in [0, L3]:
L L
L
1 2
3
V 2
V
(b)
3
x
V
xL
(a)
Figura 4.3: Potenziali per una particella in una scatola 1D con una barriera interna.
V (x) =
V1 0 ≤ x < L1
V2 L1 ≤ x < L2
V3 L2 < x ≤ L3
(4.26)
La soluzione generica per l’autofunzione ψE(x) con autoenergia E e definita nei singoli intervalli come
ψ(x, t) =
A(1)1 exp(α
(1)1 x) + A
(1)2 exp(α
(1)2 x) 0 ≤ x < L1
A(2)1 exp(α
(2)1 x) + A
(2)2 exp(α
(2)2 x) L1 ≤ x < L2
A(3)1 exp(α
(3)1 x) + A
(3)2 exp(α
(3)2 x) L2 < x ≤ L3
(4.27)
dove
α(i)1,2 = ±i
√2m(E − Vi)
h2 (4.28)

4.3. UNA SCATOLA CON UNA BARRIERA INTERNA 57
Le condizioni al contorno sono analoghe al primo caso: ai confini del pozzo l’autofunzione deve annullarsi,
poiche la particella non puo penetrare una barriera infinita. I coefficienti ignoti dell’autofunzione sono
sei, e disponiamo di due condizioni al contorno piu altre quattro condizioni: per ciascun dei punti interni,
in L1 ed L2 la funzione deve essere continua e derivabile. In definitiva, valgono le sei relazioni lineari:
A(1)1 + A
(1)2 = 0
A(1)1 exp(α
(1)1 L1) + A
(1)2 exp(α
(1)2 L1) = A
(2)1 exp(α
(2)1 L1) + A
(2)2 exp(α
(2)2 L1)
α(1)1 A
(1)1 exp(α
(1)1 L1) + α
(1)2 A
(1)2 exp(α
(1)2 L1) = α
(2)1 A
(2)1 exp(α
(2)1 L1) + α
(2)2 A
(2)2 exp(α
(2)2 L1)
A(2)1 exp(α
(2)1 L2) + A
(2)2 exp(α
(2)2 L2) = A
(3)1 exp(α
(3)1 L2) + A
(3)2 exp(α
(3)2 L2) (4.29)
α(2)1 A
(2)1 exp(α
(2)1 L2) + α
(2)2 A
(2)2 exp(α
(2)2 L2) = α
(3)1 A
(3)1 exp(α
(3)1 L2) + α
(3)2 A
(3)2 exp(α
(3)2 L2)
A(3)1 exp(α
(3)1 L3) + A
(3)2 exp(α
(3)2 L3) = 0
da cui si ricava il determinante 6 × 6 che permette di determinare E.
Esempio 2. Scrivete il determinante relativo al sistema precedente.
A questo punto lo schema di soluzione per un potenziale generico monodimensionale che sia rap-
resentabile da una serie di segmenti orizzontali e verticali, con pareti infinite, vale a dire un pozzo
monodimensionale infinitamente profonda con un “fondo” qualunque, dovrebbe risultare chiaro: 1) si
imposta la soluzione generica in ciascun intervallo; 2) si impongono condizioni di continuita e derivabilita
in ogni punto interno; 3) si annulla la soluzione alle due pareti. Il risultato e un sistema omogeneo di
equazioni per i coefficienti dell’autofunzione in ciascun intervallo, il cui determinante si deve annullare e
fornisce un’equazione per calcolare le energie.
Esempio 3. Impostare e risolvere numericamente il problema agli autostati relativo al pozzo (a) di Fig.
(4.4). Cosa succede se V → 0, o l → 0?
Esempio 4. Impostare e risolvere numericamente il problema agli autostati relativo al pozzo (b) di Fig.
(4.4). Cosa succede se il numero di gradini diviene infinito, mentre la loro larghezza tende a zero (ovvero
N →∞ con NL → cost.)?
Esempio 5. Impostare e risolvere numericamente il problema agli autostati relativo al pozzo (c) di Fig.
(4.4). Cosa succede se il numero di gradini diviene infinito, mentre la loro larghezza tende a zero (ovvero
N →∞ con NL → cost.)?
Esempio 6. Impostare e risolvere numericamente il problema agli autostati relativo ad un pozzo gener-
ico, come in (d) di Fig. (4.4).

58 CAPITOLO 4. SISTEMI RISOLVIBILI ANALITICAMENTE
l L
V
L
V
V
V
1
2
(d)
3
L
VV
V
V 43
21
V
V
VV
V 1
2
3
n-1n
(a)(b)
(c)
Figura 4.4: Varie scatole 1D.
4.4 Scatola tridimensionale
Una particella di massa m in tre dimensioni e descritto dall’hamiltoniano generico
H = − h2
2m∇2
+ U(x) (4.30)
dove ∇ e l’operatore laplaciano, che in coordinate cartesiane ha la forma
∇2=
∂2
∂x12
+∂2
∂x22
+∂2
∂x32
(4.31)
Nel caso di una particella racchiusa in una scatola in tre dimensioni, con pareti di lato a1, a2 e a3, il
potenziale e nullo all’interno della scatola, ed infinito all’esterno. Il problema e ridotto allo studio di tre
problemi separati lungo i tre assi; cioe l’autofunzione del sistema si puo scrivere come:
ψE(x) = ψ(1)E (x1)ψ
(2)E (x2)ψ
(3)E (x3) (4.32)
dove ψ(1)E (x1) e definita per 0 < x < a1 e cosı via. Le autoenergie sono date dall’espressione:
E =h2π2
2m
(k2
1
a21
+k2
2
a22
+k2
3
a23
)(4.33)

4.5. PARTICELLA IN E SU UN ANELLO 59
1
a1
a 2
a3
e
e
e3
2
Figura 4.5: Scatola tridimensionale.
dove k1, k2 e k3 sono interi. I corrispondenti autostati sono dati dall’equazione inserendo le forme
esplicite di ψ(1)E (x1) etc. nella (4.32):
ψE(x) =
√8
a1a2a3
sin
(πk1
a1
)sin
(πk2
a2
)sin
(πk3
a3
)(4.34)
E importante notare che nel caso tridimensionale le autoenergie possono essere degeneri: per una scatola
cubica (a1 = a2 = a3) una coppia di autostati identificate da terne k1, k2 e k3 che differiscano solo per
una permutazione hanno lo stesso energia.
Esempio 7. Consideriamo il caso di una scatola bidimensionale quadrata, con parametri analoghi
all’Esempio 1: i) scrivere le espressioni generali per le autoenergie e gli autostati; ii) porre in grafico le
autofunzioni corrispondenti ai primi cinque autovalori distinti.
4.5 Particella in e su un anello
Considereremo in questa Sezione e nelle Sezioni successive problemi agli autovalori relativi a sistemi che
si possono risolvere analiticamente introducendo coordinate curvilinee, non cartesiane, che sono dettate

60 CAPITOLO 4. SISTEMI RISOLVIBILI ANALITICAMENTE
naturalmente dalla geometria del potenziale. Come primo caso prendiamo in analisi una particella che
sia racchiusa in una zona dello spazio bidimensionale in una circonferenza: V = 0 se |x| < a, V = ∞ se
|x| > a, vedi Fig. (4.6) (a); a e il raggio della circonferenza; oppure che sia costretta a muoversi sulla
circonferenza stessa (rotatore planare). L’hamiltoniano per il sistema nel piano e:
P(x , x )1 2
(b)
e 1
e2
R
e
e
2
1
P
θ
r
(a)
Figura 4.6: Un pozzo cilindrico ed un anello.
H = − h2
2m
∂2
∂x12
+∂2
∂x22
(4.35)
Introduciamo le coordinate polari: r, φ, dove r e il modulo del vettore x = (x1, x2) e φ e l’angolo formato
con l’asse delle ascisse. E facile dimostrare che l’hamiltoniano, proporzionale all’operatore laplaciano in
due dimensioni, assume la forma:
H = − h2
2m
∂2
∂r2+
1
r
∂
∂r+
1
r2
∂2
∂φ2
(4.36)
Consideriamo prima il caso del rotatore planare. La particella si muove su una circonferenza a r = a,
di consequenza le derivate in r si annullano. L’hamiltoniano si riduce al solo termine dipendente da φ.
L’equazione di Schrodinger assume la forma:
− h2
2I
d2
dφ2ΨE(θ) = EΨE(φ) (4.37)
dove I = ma2 e il momento di inerzia. Risolvendo, si trova come nel caso della particella nella scatola
monodimensionale:
ΨE(θ) = A1 exp(imlφ) + A2 exp(−imlφ) (4.38)

4.5. PARTICELLA IN E SU UN ANELLO 61
dove ml =√
2IE/h2. Le condizioni al contorno sono da ricercarsi nelle proprieta fondamentali delle
funzioni d’onda di un sistema quantomeccanico, che devono sempre dar luogo ad una densita di probabilita
univoca: cioe dato x, un punto dello spazio delle coordinate del sistema, la funzione |φ(x)| deve essere
univocamente determinata. Nel caso in questione questo implica che la funzione calcolata in φ o in
φ + 2π (quindi nello stesso punto), deve essere uguale:
ΨE(φ + 2π) = ΨE(φ) (4.39)
quindi ml deve essere un numero intero. Gli autovalori sono dunque:
E =m2
l h2
2I, ml ∈ N (4.40)
Lo stato di una particella con autoenergia E e dunque descritto da una somma di due esponenziali
complessi, che rappresentano in realta due moti sovrapposti, in un senso (ml) e nel senso opposto
(−ml). Supponiamo di aver preparato la particella con un unico senso di rotazione, A2 = 0; la costante
di normalizzazione dell’autofunzione rimanente si ottiene integrando |ΨE(φ)|. Si ottiene infine:
Ψml(φ) =
√1
2πexp(imlφ) (4.41)
Ed ora consideriamo il caso di una particella racchiusa in un anello, che cioe sia costretta a muoversi
all’interno della circonferenza. L’hamiltoniano e dato dalla (4.36). L’autofunzione generica si puo scrivere
in una forma fattorizzata, poiche le coordinate r e φ sono separabili:
ΨE(r, φ) = R(r)Ψ(φ) (4.42)
Sostituendo nell’equazione di Schrodinger, HΨE = EΨE, dividendo per ΨE e moltiplicando per r2, si
ottiene:
− h2
2mR
r2d2R
dr2+ r
dR
dr
− Er2 =
h2
2mΨ
d2Ψ
dφ2(4.43)
che e un’equazione separabile; il primo e il secondo membro devono sempre essere uguali, indipendente-
mente dal valore di r e φ; scriviamo percio
d2Ψ
dφ2= −m2
l Ψ (4.44)
dove m2l e una costante; poiche la funzione deve essere univocamente definita e dunque Ψ periodica, ml
e un numero intero. Resta pero da risolvere un’equazione in R:
r2d2R
dr2+ r
dR
dr+
(2mE
h2 r2 −ml2)
R = 0 (4.45)

62 CAPITOLO 4. SISTEMI RISOLVIBILI ANALITICAMENTE
le condizioni al contorno sono: i) la funzione d’onda deve essere continua ovunque, anche all’origine
r = 0; e ii) deve annullarsi sul bordo r = a. Le funzioni che verificano l’equazione radiale con queste
condizioni sono dette funzioni di Bessel di ordine intero:
Rml(r) = Jml
√2mE
h2 r
(4.46)
con il vincolo:
Jml
√2mEml,n
h2 a
= 0 (4.47)
risolvendo l’equazione precedente si trova un set di valori di Eml,n per ogni valore di ml, con n = 1, 2, . . ..
In definitiva la soluzione generica e:
Ψml,n(r, φ) = exp(imlφ)Jml
√2mEml,n
h2 r
(4.48)
con la condizione (4.47) sulle autoenergie.
Esempio 8. Fate una ricerca bibliografica sulle proprieta delle funzioni di Bessel e valutate con l’ausilio
di tabelle i valori di E0,l con l =≤ 3, se la massa della particella e quella di un protone e il raggio della
circonferenza e pari a 2 A.
4.6 Particella in e su una sfera
Da due a tre dimensioni il passo e breve, almeno in questo caso. Generalizziamo le precedenti consider-
azioni al caso di una sfera: studiamo dunque il problema del moto di una particella di massa m su una
sfera di raggio a e poi dentro la medesima sfera. L’hamiltoniano del sistema e in coordinate cartesiane,
in analogia con il caso dell’anello:
H = − h2
2m∇2
(4.49)
dove si e fatto uso dell’operatore laplaciano. Introduciamo ora le coordinate polari sferiche r, φ, θ, vedi
Fig. (4.7):
x1 = r sin θ cos φ
x2 = r sin θ sin φ (4.50)
x3 = r cos θ (4.51)

4.6. PARTICELLA IN E SU UNA SFERA 63
φr
θ
e
e
e
1
2
3
Figura 4.7: Coordinate polari sferiche.
Il laplaciano assume una forma particolarmente semplice nelle coordinate sferiche:
∇2=
1
r2
∂
∂rr2 ∂
∂r+
1
r2M2 (4.52)
M2 =1
sin2 θ
∂2
∂φ2+
1
sin θ
∂
∂θsin θ
∂
∂θ(4.53)
dove M2 e anche detto operatore legendriano. Procediamo ora come abbiamo fatto per il caso del
rotatore planare. Se la particella e costretta a muoversi sulla superficie della sfera, trascuriamo le
derivate in r, e poniamo r = R. Dall’equazione di di Schrodinger si ottiene un’equazione in ΨE(φ, θ):
− h2
2IM2ΨE = EΨE (4.54)
dove I = ma2. La funzione ΨE dovra essere periodica in φ e in θ. Inoltre deve essere finita a θ = π. La
soluzione generale si puo scrivere in termini di funzioni speciali, note come funzioni armoniche sferiche,
che altro non sono, da un certo punto di vista, che l’equivalente tridimensionale degli esponenziali
complessi che sono stati usati nel caso bidimensionale. Varie loro proprieta sono note (e si rimanda
lo studente ad opportuni testi specializzati). Tra le altre proprieta, fondamentale e il fatto che sono
autofunzioni del legendriano:
M2Ylml(θ, φ) = −l(l + 1)Ylml
(θ, φ) (4.55)

64 CAPITOLO 4. SISTEMI RISOLVIBILI ANALITICAMENTE
dove l = 0, 1, . . . e ml = −l,−l + 1, . . . , l− 1, l. Segue dunque che le autoenergie di una particella che
si muova su una sfera sono caratterizzate da:
Elml=
h2
2Il(l + 1) (4.56)
Si noti che per ogni autostato, caratterizzato dai numeri quantici l e ml, esistono 2(l + 1) autovalori
degeneri, dato che Elmlnon dipende da ml.
Procediamo ora con il caso della particella confinata nella sfera. Ora dobbiamo considerare il lapla-
ciano nella sua forma completa. Analogamente al caso precedente, si puo assumere che la parte angolare
debba essere quella determinata dal moto sulla sfera:
ΨE(r, θ, φ) = R(r)Ylml(θ, φ) (4.57)
Sostituendo nell’Eq. di Schrodinger si trova l’equazione per la parte radiale:
1
r2
d
drr2dR
dr− l(l + 1)
r2R +
2m
h2 ER = 0 (4.58)
con le condizioni al contorno che R = 0 per r = a ed R finita a r = 0. In generale avremo dunque delle
funzioni Rnl(r) che si ottengono risolvendo la precedente equazione differenziale ordinaria. Consideriamo
nel seguito il solo caso l = 0, cioe le soluzioni del problema che siano indipendenti dagli angoli polari
(Y0,0 e una costante). L’equazione in R si puo porre nella forma ridotta
1
r
d2
dr2rR− κ2R = 0 (4.59)
dove κ =√
2mE/h2. La soluzione finita a r = 0 e
R(r) = Asin(κr)
r(4.60)
e la condizione al contorno impone che ka = 0, vale a dire ancora una volta
E =h2n2π2
2I(4.61)
Esempio 8. Normalizzate l’autofunzione radiale. Ricordate che nel passaggio da coordinate cartesiane
a coordinate sferiche si ha che:
∫dx1
∫dx2
∫dx2f(x1, x2, x3) =
∫drr2
∫dθ sin θ
∫dφf(r, θ, φ) (4.62)

4.7. OSCILLATORE ARMONICO 65
4.7 Oscillatore armonico
L’oscillatore lineare o armonico e un altro esempio di sistema risolvibile analiticamente, ovvero tale che
le sue autofunzioni sono esprimibili in termini di funzioni note. Il potenziale e definito come una parabola,
con una curvatura mω2:
V (x) =mω2
2x2 (4.63)
ω e la frequenza dell’oscillatore. Consideriamo l’hamiltoniano del sistema:− h2
2m
d2
dx2+
mω2
2x2
ψE(x) = EψE(x) (4.64)
Introduciamo per comodita di calcolo la coordinata adimensionale
ξ =
√mω
hx (4.65)
L’equazione in ψ(ξ) diviene:
d2
dξ2ψE +
(2E
hω− ξ2
)ψE = 0 (4.66)
La funzione ψ deve essere finita per ξ →∞; per x molto grandi si puo trascurare il termine in E rispetto
a ξ2, quindi la funzione si deve comportare asintoticamente come exp(−ξ2/2):
ψE(ξ) = exp(−ξ2/2)χ(ξ) (4.67)
dove χ(ξ) puo anche divergere con ξ → ∞, ma non piu velocemente di exp(−ξ2/2); possiamo percio
ottenere l’equazione in χ(ξ):
d2χ
dξ2− 2ξ
dχ
dξ+
(2E
hω− 1
)χ = 0 (4.68)
La soluzione generica all’equazione differenziale precedente, con la condizione al contorno che per ξ →∞la funzione χ non cresca piu rapidamente di una potenza finita di ξ (in modo che comunque ψE vada a
zero per ξ →∞, e data dai polinomi di Hermite, con il vincolo che
2E
hω− 1 = 2n (4.69)
ed n deve essere naturale. Le autofunzioni di un oscillatore armonico monodimensionale sono dunque pro-
porzionali ai polinomi di Hermite, con autovalori quantizzati dall’indice n. La costante di proporzionalita
e determinabile, come sempre, dalla condizione di normalizzazione della funzione d’onda. Sostituendo di
nuovo x al posto di ξ infine si trova:
ψn(x) =(
mω
πh
)1/4 1
(2nn!)1/2exp
(−mω
2hx2
)Hn
[(mω
h
)1/2
x
](4.70)
En =(n +
1
2
)hω (4.71)

66 CAPITOLO 4. SISTEMI RISOLVIBILI ANALITICAMENTE
Esempio 9. Porre in grafico le prime tre autofunzioni di un oscillatore lineare con la massa di un
elettrone e ω = 10−14 s−1.
La generalizzazione ad un oscillatore armonico in tre dimensioni e semplice. L’equazione di Schrodinger
e data da:[− h2
2m∇2
+1
2
(ω2
1x21 + ω2
2x22 + ω2
3x23
)]ΨE(x1, x2, x3) = EΨE(x1, x2, x3) (4.72)
analogamente al caso della particella in tre dimensioni (e in generale in tutti i casi in cui il potenziale sia
scritto come una somma di potenziali separati agenti ciascuno su una coordinata), possiamo scrivere gli
autostati come il prodotto degli autostati lungo ciascuna coordinata, e le autoenergie come somme:
ψn1,n2,n3(x) = Π3i=1
(mωi
πh
)1/4 1
(2nini!)1/2exp
(−mωi
2hx2
i
)Hni
[(mωi
h
)1/2
xi
](4.73)
E =∑
i
(ni +
1
2
)hωi (4.74)
Esempio 10. Qual e la degenerazione in funzione di n dell’autostato E = (3n + 3/2)hω per un
oscillatore armonico tridimensionale con ω1 = ω2 = ω3 = ω?
4.8 Potenziale di Morse
Consideriamo il potenziale
V (x) = D(1− e−αx)2 (4.75)
detto potenziale di Morse ed e mostrato in Fig. (4.8). Per x →∞ il potenziale va a D; per x → −∞ il
potenziale va a +∞. Evidentemente esisteranno autovalori discreti e positivi per E < D, ed uno spettro
continuo per E > D. L’equazione di Schrodinger e data come al solito da
− h2
2m
∂2
∂x2ψE(x) + D(1− e−αx)2ψE(x) = EψE(x) (4.76)
Introduciamo la variabile ausiliaria ξ che va da 0 a ∞
ξ =2√
2mD
αhexp(−αx) (4.77)
L’equazione di Schrodinger assume la forma ridotta:
∂2ψE
∂ξ2+
1
ξ
∂ψE
∂ξ+
(−1
4+
n + s + 1/2
ξ− s2
ξ2
)ψE = 0 (4.78)

4.8. POTENZIALE DI MORSE 67
x
D
V(x)
Figura 4.8: Potenziale di Morse.
dove
s =
√2m(D − E)
αh(4.79)
n =
√2mD
αh− s− 1
2(4.80)
Per ξ → ∞ la funzione deve evolvere come exp(−ξ/2) e per ξ → 0 come ξs. La soluzione generica si
puo scrivere percio come
ψE(ξ) = exp(−ξ/2)ξsχ(ξ) (4.81)
Sostituendo, si trova un’equazione differenziale in χ(ξ) (come si vede la procedura e sostanzialmente
analoga al caso dell’oscillatore armonico):
ξ∂2χ
∂ξ2+ (2s + 1− ξ)
∂χ
∂ξ+ nχ = 0 (4.82)
La soluzione generica e detta funzione ipergeometrica confluente e si scrive in generale χ = F (−n, 2s+
1, ξ); sappiamo che χ deve essere finita per ξ → 0; per ξ → ∞ χ non puo tendere all’infinito piu
rapidamente di una potenza finita di ξ. Queste condizioni si ottengono dallo studio delle proprieta delle

68 CAPITOLO 4. SISTEMI RISOLVIBILI ANALITICAMENTE
funzioni ipergeometriche confluenti, solo nel caso n intero non negativo. Da cio segue che le autoenergie
discrete sono:
En = D
1−
[1− αh√
2mD
(n +
1
2
)]2 (4.83)
Si noti che se αh/√
2mD > 2 non esistono autovalori discreti. Il potenziale di interazione tra gli atomi
di una molecola biatomica puo essere rappresentato in modo qualitativo da una curva di Morse. In realta
possiamo interpretare un sistema rappresentato dalla curva di Morse come un esempio di oscillatore
anarmonico.
4.9 Atomo idrogenoide
Lentamente ma inesorabilmente i sistemi considerati si vanno complicando, dal punto di vista geometrico
ed algebrico. I sistemi fisici di interesse in chimica fisica (strutture atomiche e molecolari) sono sistemi a
piu dimensioni, di impossibile risoluzione analitica. Metodi numerici approssimati saranno descritti nella
III Dispensa. Concludiamo questa Sezione con un sistema ‘chimico’ semplice, risolvibile analiticamente:
il calcolo dello spettro di autoenergie e delle relative autofunzioni per l’elettrone di un atomo di idrogeno
con un nucleo a riposo. In generale, se il nucleo non e a riposo dobbiamo semplicemente sostituire alla
massa dell’elettrone la massa ridotta (vedi III Dispensa) del sistema protone/elettrone. L’hamiltoniano
del sistema e percio:
H = − h2
2µ∇2 − e2
4πε0
1
r(4.84)
dove µ e la massa ridotta e il secondo termine e il potenziale coulombiano attrattivo tra il nucleo con
carica +e e l’elettrone con carica −e a distanza r. Il potenziale e a simmetria radiale. Possiamo percio
procedere senza perdere tempo come nel caso della particella racchiusa in una sfera: i) introduzione di
coordinate sferiche; ii) fattorizzazione della funzione d’onda in una parte angolare dipendente da θ e
φ e in una parte radiale dipendente da r; iii) imposizione di condizioni al contorno per il calcolo delle
autoenergie. La parte angolare dell’autofunzione non cambia, ed e data ancora dalle funzioni armoniche
sferiche: in realta cio e vero per qualunque potenziale a simmetria sferica, cioe dipendente solo dal
modulo r:
ΨE(r, θ, φ) = R(r)Ylml(θ, φ) (4.85)
Sostituendo nell’Eq. di Schrodinger si trova l’equazione per la parte radiale:
1
r2
d
drr2dR
dr− l(l + 1)
r2R +
2m
h2
(E +
e2
4πε0
1
r
)R = 0 (4.86)
Lo spettro degli autovalori sara discreto per E < 0, e continuo per E > 0 (il potenziale coulombiano
tende a zero per r →∞ e a −∞ per r → 0). A questo punto possiamo procedere come in tutti i casi

4.9. ATOMO IDROGENOIDE 69
precedenti: l’equazione differenziale in R, con le condizioni al contorno R finita a r → 0 e R → ∞ e
riconoscibile com l’equazione che ha per soluzione delle funzioni note: le funzioni associate di Laguerre
(moltiplicate per l’esponenziale di −r). In definitiva gli autostati e le autoenergie dell’atomo di idrogeno
a riposo sono:
Ψn,l,ml(r, θ, φ) = Rnl(r)Ylml
(θ, φ) (4.87)
Rnl = − 2
na
(n− l − 1)!
2n [(n + l)!]3ρlL2l+1
n+l (ρ) exp(−ρ/2) (4.88)
En,l,ml= − µe4
32π2ε20h
2
1
n2(4.89)
(4.90)
dove ρ = 2r/na ed a = 4πε0h2/µe2. Gli autostati discreti sono negativi e dipendono solo da 1/n2.
Esempio 11. Qual e la degenerazione di ciascun livello energetico dell’atomo di idrogeno a riposo?
Esempio 12. Valutare il valore di attesa di r rispetto per l’autofunzione con n = l = ml = 0.

70 CAPITOLO 4. SISTEMI RISOLVIBILI ANALITICAMENTE

Parte III
Calcolo di strutture elettroniche
atomiche e molecolari.
71


Capitolo 5
L’equazione di Schrodinger
In questa dispensa sara data un’introduzione schematica alle metodiche di calcolo piu comuni per la
determinazione di funzioni d’onda elettroniche. Le tecniche di calcolo quanto-meccanico permettono
la previsione ed ottimizzazione di geometrie molecolari e lo studio di proprieta molecolari (momento di
dipolo, polarizzabilita, densita elettronica etc.) a partire dalla definizione dell’operatore hamiltoniano di
una data specie chimica, e dalla soluzione numerica dell’equazione di Schrodinger corrispondente.
Tradizionalmente, le tecniche di calcolo vengono classificate in
• metodi ab initio: calcoli autoconsistenti di Hartree-Fock (HF), interazione di configurazione (CI);
l’equazione di Schrodinger viene risolta in modo ‘esatto’ (mediante un’espansione della funzione
d’onda su una base di funzioni note, o mediante l’applicazione di tecniche perturbative);
• metodi semiempirici: metodi di Huckel, Huckel esteso, PPP, i vari metodi NDO (neglect of
differential overlap); varie approssimazioni e parametrizzazioni sono introdotte per semplificare gli
integrale numerici a uno, due o piu elettroni che sono definiti nell’approccio esatto.
I metodi ab initio consentono una buona definizione del problema quantomeccanico, essendo basati
fondamentalmente sull’hamiltoniano esatto del sistema molecolare, e dunque in principio definito solo
da informazioni sulla struttura della molecola e di costanti fondamentali. La dimensione del problema di
algebra lineare (diagonalizzazione di matrice) correlato ad un calcolo ab initio per un sistema molecolare
di N elettroni cresce almeno come N2: molecole di media complessita possono gia costituire un serio
problema computazionale. D’altra parte, i metodi semiempirici sono in grado di trattare in modo veloce
ed efficiente anche il calcolo delle strutture di equilibrio di molecole organiche di notevole complessita.
Non sempre pero l’hamiltoniano approssimato e definito in modo chiaro, ne la parametrizzazione degli
integrali elettronici scelta per un determinato metodo semiempirico, in modo da ottimizzarne i risultati
per determinate classi di molecole, consente di ottenere buoni risultati in diverse condizioni o per altre
classi di sistemi molecolari.
Questa nota si articola in cinque brevi Capitoli ed alcune Appendici. Nel Capitolo 5 sono richiamate
alcune nozioni basilari sugli hamiltoniani molecolari, l’equazione di Schrodinger elettronica e la separazione
73

74 CAPITOLO 5. L’EQUAZIONE DI SCHRODINGER
tra gradi di liberta nucleari ed elettronici. Nel Capitolo 6 sono descritte le proprieta di spin della funzione
d’onda elettronica e la definizione dei determinanti di Slater. Il Capitolo 7 e dedicato al metodo di
Hartree-Fock, e a cenni relativi ad altri metodi ab initio. Nel Capitolo 8 sono riassunte le nozioni
fondamentali relative alla teoria dei gruppi applicata al calcolo di orbitali molecolari. Nel Capitolo 9
vengono richiamati i metodi semiempirici, con un particolare accento sull’approssimazione di Huckel.
HOMO del pirrolo calcolato con il metodo Extended Huckel (isosuperficie a 0.06 unita atomiche).
5.1 L’equazione di Schrodinger
5.1.1 Insieme di punti materiali
Consideriamo un insieme di N punti materiali ~ri nello spazio ordinario cui siano associate le masse
inerziali mi (i = 1, . . . , N). La descrizione del comportamento nello spazio e nel tempo dell’insieme di
punti e data in meccanica quantistica tramite la funzione d’onda Ψ(~ri, t), definita mediante l’equazione

5.1. L’EQUAZIONE DI SCHRODINGER 75
di Schrodinger dipendente dal tempo:
− h
i
∂
∂tΨ(~ri, t) = HΨ(~ri, t) (5.1)
dove H e l’operatore di Hamilton o hamiltoniano, definito come la somma dell’operatore di energia
cinetica T e dell’energia potenziale:
H = T + V (~ri) (5.2)
T =N∑
i=1
~p2i
2mi
(5.3)
L’operatore momento lineare ~pi per la particella i-esima e definito come
~pi =h
i
∂
∂~ri
(5.4)
e dunque l’operatore energia cinetica puo essere scritto anche nella forma
T = − h2
2
N∑
i=1
1
mi
∇2
i (5.5)
dove ∇2
i e l’operatore di Laplace relativo alla particella i-esima. La funzione d’onda e definita quando
siano stabilite delle condizioni iniziali, ovvero lo stato del sistema al tempo t = 0 e le condizioni al
contorno, ovvero l’integrabilita della funzione d’onda. La soluzione formale dell’equazione di Schrodinger
e data nella forma:
Ψ(~ri, t) = exp(−iHt
h
)Ψ(~ri, 0) (5.6)
con la condizione di integrabilita
〈Ψ|Ψ〉 =∫
d~rΨ(~ri, t)∗Ψ(~ri, t) = 1 (5.7)
dove d~r = d~r1d~r2 . . . d~rN . La funzione d’onda generica e ottenibile come un’espansione rispetto alle
autofunzioni dell’hamiltoniano
Ψ(~ri, t) =∑n
exp(−iEnt/h)Ψn(~ri)〈Ψn(~ri)|Ψ(~ri, , 0)〉 (5.8)
HΨn(~ri) = EnΨn(~ri) (5.9)
L’equazione (5.9) e l’equazione di Schrodinger indipendente dal tempo.
Esercizio 1. Consideriamo, per fissare le idee, il caso di un sistema di 2 particelle ~r1 e ~r2, accoppiate
da un potenziale V (|~r1 − ~r2|) L’hamiltoniano e dato nella forma:
H = − h2
2m1
∇2
1 −h2
2m2
∇2
2 + V (|~r1 − ~r2|) (5.10)

76 CAPITOLO 5. L’EQUAZIONE DI SCHRODINGER
L’hamiltoniano riportato in Eq. (5.10) e separabile, ovvero e riducibile alla somma di due hamiltoniani
non interagenti, introducendo un nuovo set di variabili, date dalla distanza relativa tra le due particelle
e dalla posizione del baricentro del sistema
~r = ~r1 − ~r2 (5.11)
~rc =m1~r1 + m2~r2
m1 + m2
(5.12)
1
m1
∇2
1 +1
m2
∇2
2 =1
µ∇2
+1
M∇2
c (5.13)
dove µ e la massa ridotta, M e la massa totale:
µ =m1m2
m1 + m2
(5.14)
M = m1 + m2 (5.15)
e ∇2, ∇2
c sono i laplaciani rispetto a ~r e ~rc. L’hamiltoniano (5.10) risulta separabile poiche si puo scrivere
come:
H = − h2
2µ∇2
+ V (r)− h2
2M∇2
c (5.16)
5.1.2 Hamiltoniani atomici e molecolari
La definizione dell’hamiltoniano completo di un sistema atomico o molecolare non e affatto un problema
semplice. Trascurando termini di interazione con campi elettrici e magnetici, che sono peraltro di
grande importanza per l’interpretazione di osservabili spettroscopici, ed ignorando correzioni di natura
relativistica, restano da considerare in dettaglio i vari termini di interazione elettrostatiche e magnetiche
dei nuclei e degli elettroni.
Esercizio 2. Consideriamo prima di tutto alcuni hamiltoniani relativi a sistemi atomici e molecolari
semplici: l’atomo H, l’atomo He e la molecola H+2 , limitandoci ad includere nell’energia potenziale i soli
termini di interazione elettrostatica.
L’atomo di idrogeno, H, e un sistema descritto da due particelle tridimensionali: il nucleo ~R di massa
mH e l’elettrone ~re di massa me L’hamiltoniano dell’atomo di idrogeno e dato come:
H = − h2
2mH
∇2R −
h2
2me
∇2e −
2e2
4πε0
1
|~R− ~re|(5.17)
dove con ∇2e e ∇2
R indichiamo il laplaciano rispetto ai vettori ~re e ~R. I gradi di liberta complessivi del
sistema sono dunque 6. L’atomo di elio, He, e un sistema descritto da tre particelle tridimensionali (il

5.1. L’EQUAZIONE DI SCHRODINGER 77
e2
e1
R
He
|R - r |
|R - r |
r
r
1
1
2
2
|r - r |1 2
Figura 5.1: Sistema di riferimento e coordinate He
nucleo e due elettroni). L’hamiltoniano dell’atomo di elio e dato come:
H = − h2
2mHe
∇2R −
h2
2me
(∇21 + ∇2
2)−2e2
4πε0
(1
|~R− ~r1|+
1
|~R− ~r2|
)+
e2
4πε0
1
|~r1 − ~r2| (5.18)
dove con ∇21,2 indichiamo i laplaciani rispetto a ~r1 e ~r2. I gradi di liberta complessivi del sistema sono
dunque 9. In Fig. 1 viene riportato uno schema delle coordinate nell’ambito di un sistema di riferimento
inerziale (di laboratorio).
La molecola di idrogeno ionizzata, H+2 , e ancora un sistema descritto da tre particelle tridimensionali
(due nuclei e un elettrone). L’hamiltoniano e dato come:
H = − h2
2mH
(∇2
A + ∇2B
)− h2
2me
∇2e −
e2
4πε0
(1
|~RA − ~re|+
1
|~RB − ~re|
)+
e2
4πε0
1
|~RB − ~RA|(5.19)
dove con ~RA ed ~RB indichiamo la posizione dei nuclei, con ~re quella dell’elettrone. I gradi di liberta
complessivi del sistema sono sempre 9.
Definiamo ora con un certo grado di generalita l’hamiltoniano, limitato ai soli termini di interazione elet-
trostatica, per una molecola di M nuclei ed N elettroni. Siano ~Rp (p = 1, . . . ,M) i vettori posizione dei

78 CAPITOLO 5. L’EQUAZIONE DI SCHRODINGER
nuclei e ~ri (i = 1, . . . , N) i vettori posizione degli elettroni. L’hamiltoniano molecolare non-relativistico
con i soli termini elettrostatici e allora scritto come:
H = − h2
2
M∑
r=1
1
mp
∇2p−
h2
2me
N∑
i=1
∇2i +
e2
4πε0
M,M∑p<q
ZpZq
|~Rp − ~Rq|+
e2
4πε0
N,N∑
i<j
1
|~ri − ~rj|−e2
4πε0
M,N∑
p,i
Zp
|~Rp − ~ri|(5.20)
E conveniente nel seguito introdurre un insieme di unita di misura ad hoc, dette unita atomiche, che
permettano di definire grandezze fisiche riscalate in modo semplice. Nella Tabella 1 sono definite le
principali unita atomiche: di particolare importanza sono l’unita di lunghezza, detta Bohr, e l’unita di
energia, chiamata Hartree. Nel seguito si useranno sempre unita atomiche, se non verra espressamente
massa mu me = 9.1091× 10−31 Kg
lunghezza lu a0 = 4πε0h2
mee2 = 0.52917× 10−10 m
momento angolare Lu h = 4.16336× 10−33 J s−1
energia Eue2
4πε0a0= 4.359× 10−18 J
carica e e = 1.602× 10−19 C
Tabella 5.1: Unita atomiche
indicato altrimenti. L’hamiltoniano molecolare elettrostatico in unita atomiche diventa:
H = −M∑
p=1
1
2mp
∇2p −
1
2
N∑
i=1
∇2i −
M,N∑
p,i
Zr
|~Rp − ~ri|+
N∑
i<j
1
|~ri − ~rj| +M∑
p<q
ZpZq
|~Rp − ~Rq|. (5.21)
Indichiamo per semplicita con gli indici i, j coordinate elettroniche, con gli indici p, q coordinate nucleari.
Esercizio 3. Vogliamo scrivere l’hamiltoniano molecolare per un sistema con nuclei a spin nullo, non-
relativistico ed in assenza di interazioni con campi esterni. L’hamiltoniano molecolare si puo descrivere
in prima approssimazione come dato da tre termini principali
H = H0 +HSO +HSS (5.22)
Il primo contributo e dato dall’operatore relativo all’energia cinetica + elettrostatica (5.21). Il secondo
contributo corrisponde al termine di interazione spin-orbitale, vale a dire tra il momento di spin (vedi
Capitolo 6) di ciascun elettrone e il moto spaziale dell’elettrone stesso
HSO = −gβ2e
∑
i6=j
1
r3ij
(2si · rij × ∇j + sj · rij × ∇j) (5.23)
Il terzo termine e riferito all’energia di interazione spin-spin
HSS = −1
2gβ2
e
∑
i,j
1
r5ij
[3(si · rij)(sj · rij)− r2ijsi · sj] (5.24)
Parecchi termini di interazione al secondo ordine sono stati trascurati nella definizione (5.22). I nuclei
sono considerati come aventi momento di spin nullo. Nei calcoli di strutture elettroniche si considera di
solito solo il primo termine.

5.2. APPROSSIMAZIONE DI BORN-OPPENHEIMER 79
5.2 Approssimazione di Born-Oppenheimer
Il primo problema che ci poniamo e la possibilita di separazione tra le coordinate nucleari ed elettroniche.
Fisicamente, la massa dei nuclei, molto maggiore rispetto a quella degli elettroni, ci consente di affermare
che il moto dei primi avviene su una scala dei tempi molto piu lenta rispetto al moto dei secondi. Cio
equivale al fatto che, con buona approssimazione, le proprieta degli elettroni possono essere definite
assumendo che lo ‘scheletro’ della molecola, formato dai nuclei, sia immobile. Fondamentalmente,
questa e l’approssimazione di Born-Oppenheimer (BO).
Consideriamo prima di tutto il problema della separazione del moto del centro di massa, che e
conseguibile in modo esatto. E dato un sistema di N elettroni ed M nuclei. L’hamiltoniano molecolare
e dato in (5.21). Definiamo il vettore baricentro
~Rc =1
M
N∑
i=1
me~ri +M∑
p=1
mp~Rp
(5.25)
dove M e la massa totale del sistema; definiamo inoltre N coordinate vettoriali ~xi che specificano la
posizione degli elettroni rispetto al baricentro dei soli nuclei ed infine 3M − 3 coordinate ’interne’ QI
(con I = 1, . . . , 3M − 3) per i nuclei, che descrivono il moto rotazionale e vibrazionale della molecola e
che si possono esprimere, per esempio [cfr. Appendice (B)] come combinazioni lineari delle coordinate
vettoriali dei nuclei. In pratica siamo esattamente nelle condizioni descritte in Appendice (B) dove gli
elettroni sono le particelle leggere e i nuclei le particelle pesanti. Usiamo le conclusioni che sono tratte
in Appendice (B) per scrivere l’hamiltoniano nelle forma
H = Hc +Hel +Hnu (5.26)
Hc = − 1
2M∇2
c (5.27)
Hel = −1
2
N∑
i
∇2
i −1
2M
∑
ij
∇i · ∇j +N∑
i<j
1
|~ri − ~rj| −M,N∑
p,i
Zp
|~Rp − ~ri|+
M∑p<q
ZpZq
|~Rp − ~Rq|(5.28)
Hnu = T nu (5.29)
dove le coordinate vettoriali elettroniche sono definite relativamente al baricentro; la forma esatta
dell’energia cinetica nucleare T nu dipende dalla scelta delle coordinate interne QI nucleari; le com-
ponenti dei vettori ~Rp − ~ri etc. sono funzioni di dette coordinate interne e dei vettori ~xi. Indichiamo
con ∇i il gradiente relativo a ~xi e cosı via. Il primo contributo Hc e relativo al moto traslazionale
del baricentro. Il secondo termine e dato dalla somma dell’energia cinetica elettronica, corretta con
un termine dipendente dall’inverso della massa totale del sistema detto di polarizzazione di massa, e
dell’energia potenziale elettroni-elettroni ed elettroni-nuclei. Il terzo termine descrive il moto nucleare
roto-vibrazionale. Si noti che l’energia potenziale nucleare e stata inclusa nell’hamiltoniano elettronico,
per convenzione.

80 CAPITOLO 5. L’EQUAZIONE DI SCHRODINGER
5.2.1 Separazione delle coordinate elettroniche
Nel seguito indichiamo con Q l’insieme di coordinate nucleari interne e con x l’insieme delle coordinate
elettroniche. L’equazione di Schrodinger indipendente dal tempo per l’hamiltoniano elettronico-nucleare,
in cui d’ora in poi non includiamo il termine, separato, relativo al moto del baricentro, e scritta nella
forma:
HΞ(Q,x) = EΞ(Q,x) (5.30)
dove Ξ(Q,x) e l’autofunzione roto-vibro-elettronica della molecola con energia E. Definiamo l’insieme
di autofunzioni del solo hamiltoniano elettronico
HelΨ(Q|x) = εn(Q)Ψ(Q|x) (5.31)
che dipendono parametricamente dalle coordinate nucleari Q, con autovalori εn(Q). Le autofunzioni Ψn
costituiscono una base ortonormale per lo spazio di Hilbert in x. Il seguente sviluppo e percio del tutto
generale:
Ξ(Q,x) =∑n
Φn(Q)Ψn(Q|x) (5.32)
Sostituendo le equazioni (5.31) e (5.32) nella (5.30), si ottiene:
∑n
[Eeln (Q) + T nu − E]Φn(Q)Ψn(Q|x) = 0 (5.33)
pre-moltiplicando per il bra 〈Ψm(Q|r)| si trova l’espressione
[Eelm(Q)− E]Φm(Q) +
∑n
〈Ψm|T nu|Ψn〉Φn(Q) = 0 (5.34)
che fornisce un sistema di equazioni per determinare le funzioni incognite Φn(Q). Si noti che l’integrazione
〈ldots〉 e rispetto a x. Sinora non e stata fatta alcuna approssimazione, e l’equazione (5.34) e equiva-
lente all’equazione di Schrodinger di partenza. Si danno ora varie possibilita. Dal punto di vista fisico e
lecito attendersi che ad ogni significativo cambiamento della configurazione nucleare gli elettroni vadano
incontro ad un rapido riaggiustamento, in una scala dei tempi molto piu veloce di quella tipica dei
moti nucleari a causa della grande differenza di massa. E quindi ragionevole attendersi che la dipen-
denza parametrica delle funzioni d’onda elettroniche Ψn(Q|x) sia ’debole’, ed addirittura trascurabile in
prima approssimazione. L’approssimazione di Born-Oppenheimer consiste nel trascurare completamente
questa dipendenza, cosicche il valor medio (in generale un’operatore agente sulle coordinate nucleari)
che compare nel secondo termine della (5.34) diventa:
〈Ψm|T nu|Ψn〉 B.O.= 〈Ψm|Ψn〉T nu = T nuδm,n (5.35)
La funzione d’onda per la parte nucleare nell’approssimazione BO e data quindi come:
[Eelm(Q) + T nu]Φm(Q) = EΦm(Q) (5.36)

5.3. LA MOLECOLA DI IDROGENO IONIZZATA 81
• L’equazione (5.31) e alla base dell’interpretazione delle osservazioni sperimentali basate sulla spet-
troscopia elettronica (UV, visibile);
• l’equazione (5.36) e utilizzata per la comprensione delle osservazioni sperimentali basate sulla
spettroscopia vibrazionale (IR);
• l’approssimazione di Born-Oppenheimer non puo essere impiegata per interpretare quei fenomeni
chimico-fisici in cui vi siano interazioni forti fra elettroni e nuclei (es. scattering elettronico),
o quando in generale non sia lecito considerare la funzione d’onda elettronica come debolmente
dipendente dalle coordinate nucleari.
5.3 La molecola di idrogeno ionizzata
Considereremo in questa sezione il sistema molecolare H+2 . Seguiamo le considerazioni generali svolte
nella sezione precedente, separando il baricentro, applicando l’approssimazione di Born-Oppenheimer,
trascurando i termini di polarizzazione di massa nell’hamiltoniano elettronico, definendo appropriate
coordinate interne nucleari - che possiamo scegliere in modo conveniente come R, θ and φ dove R e
la distanza internucleare |~RA − ~RB|, θ e φ le coordinate polari del vettore ~RA − ~RB. L’hamiltoniano
elettronico e percio
Hel = −1
2∇2 − 1
rA
− 1
rB
+1
R(5.37)
dove rA =√
r2 + (z + R/2)2 e rB =√
r2 + (z −R/2)2 [cfr. Fig. (5.2)].
5.3.1 Hamiltoniano elettronico
Gli autostati e i livelli energetici della molecola di idrogeno ionizzata saranno in generale funzioni para-
metriche di R. Per R → 0 la molecola diviene l’atomo di elio ionizzato, ed i suoi orbitali e stati energetici
sono quelli di un atomo idrogenoide con Z = 2. Per R →∞ la molecola diviene equivalente alla coppia
H+ + H. Scriviamo l’equazione di Schrodinger nella forma(−1
2∇2 − 1
rA
− 1
rB
)Ψ = EΨ (5.38)
L’energia di un autostato elettronico e data in funzione dell’autovalore E come
Eel = E +1
R(5.39)
Introduciamo le coordinate ellissoidali µ, ν e l’angolo azimutale φ relativo alla rotazione intorno all’asse
internucleare; valgono le relazioni dirette ed inverse:
µ =rA + rB
R(5.40)

82 CAPITOLO 5. L’EQUAZIONE DI SCHRODINGER
x
yz
+R/2
rA
Br
A
B
e
e
2
1
3
e
-R/2
Figura 5.2: Sistema di riferimento e coordinate H+2
ν =rA − rB
R(5.41)
x =1
R cos φ
√(µ2 − 1)(1− ν2) (5.42)
y =1
R sin φ
√(µ2 − 1)(1− ν2) (5.43)
z =µνR
2(5.44)
con µ ≥ 1, −1 ≤ ν ≤ 1; µ descrive gli ellissoidi di rivoluzione aventi come fuochi i nuclei, mentre
ν descrive gli iperboloidi confocali di rivoluzione [cfr. Fig. (5.3)]. Il laplaciano puo essere espresso in
queste coordinate nella forma:
∇2=
4
R2(µ2 − ν2)
∂
∂µ(µ2 − 1)
∂
∂µ+
∂
∂ν(1− ν2)
∂
∂ν+
∂
∂φ
(µ2 − ν2)
(µ2 − 1)(1− ν2)
∂
∂φ
(5.45)
Sostituendo nell’equazione di Schrodinger, si ottiene un’equazione differenziale separabile, cioe ponendo
Ψ(µ, ν, φ) = M(µ)N(ν)F (φ) (5.46)
si trovano le tre equazioni separate: d
dµ(µ2 − 1)
d
dµ− εµ2 + 2Rµ− λ2
ν2 − 1+ κ
M(µ) = 0 (5.47)

5.3. LA MOLECOLA DI IDROGENO IONIZZATA 83
µ A
B
ν
Figura 5.3: Coordinate ellissoidali H+2
d
dν(1− ν2)
d
dν+ εν2 +
λ2
1− ν2− κ
N(ν) = 0 (5.48)
d2
dφ2+ λ2
F (φ) = 0 (5.49)
λ e κ sono costanti di separazione, mentre ε = −R2E/2. In particolare, λ e il numero quantico
associato con la componente del momento angolare elettronico lungo l’asse internucleare, ed assume
i valori 0,±1,±2, . . .. L’autovalore E dipende da λ2, ovvero e presente una doppia degenerazione
dovuta all’equivalenza delle due direzioni di rotazione attorno all’asse. I valori permessi di κ e ε sono
determinabili numericamente. In generale i vari stati, doppiamente degeneri, di H+2 sono simboleggiati
da una notazione del tipo nlαu,g dove: n ed l sono riferiti ai numeri quantici dell’orbitale atomico
idrogenoide, cfr. Appendice (A), cui l’orbitale molecolare tende per R → 0; α e un una lettera greca
σ, π, δ, φ, . . . corrispondente a |λ| = 0, 1, 2, 3, . . .; u o g corrispondono alla simmetria dell’orbitale
molecolare rispetto all’operazione di inversione rispetto al punto centrale tra i nuclei - si puo dimostrare
che l’orbitale molecolare cambia di segno (u) o resta invariato (g) rispetto questa operazione di simmetria.

84 CAPITOLO 5. L’EQUAZIONE DI SCHRODINGER
Figura 5.4: I primi due livelli energetici della molecola di H+2 [E. Teller, Z. Physik, 61, 474 (1930)]
5.3.2 Hamiltoniano nucleare
Possiamo scrivere l’hamiltoniano risultante dall’approssimazione di Born-Oppenheimer nella seguente
forma, in cui il laplaciano e espresso in coordinate rotazionali:
− 1
mHR2
∂
∂RR2 ∂
∂R− L2
+ Eel(R)
Φ(R, θ, φ) = EΦ(R, θ, φ) (5.50)
dove L e l’operatore di momento angolare del sistema nucleare. Le coordinate angolari sono separabili e
dunque la funzione d’onda e fattorizzata in una parte radiale (vibrazionale), ed in una parte rotazionale:
Φ(R, θ, φ) = S(R)YJK(θ, φ) (5.51)
dove YJK e la generica armonica sferica
L2YJK(θ, φ) = J(J + 1)YJK(θ, φ) (5.52)
dove L2 = −M2 in unita atomiche e la parte radiale e data dall’equazione
d
dRR2 d
dR+ mR2[E − Eel(R)]− J(J + 1)
S(R) = 0 (5.53)
che puo essere risolta per via numerica od anche analitica, se si assume una particolare forma funzionale
per Eel(R) che ne approssimi l’andamento. Consideriamo per esempio lo stato elettronico 1sσg ed
approssimiamolo con un potenziale di Morse
Eel(R) = D1− exp[−β(R−Re)] (5.54)
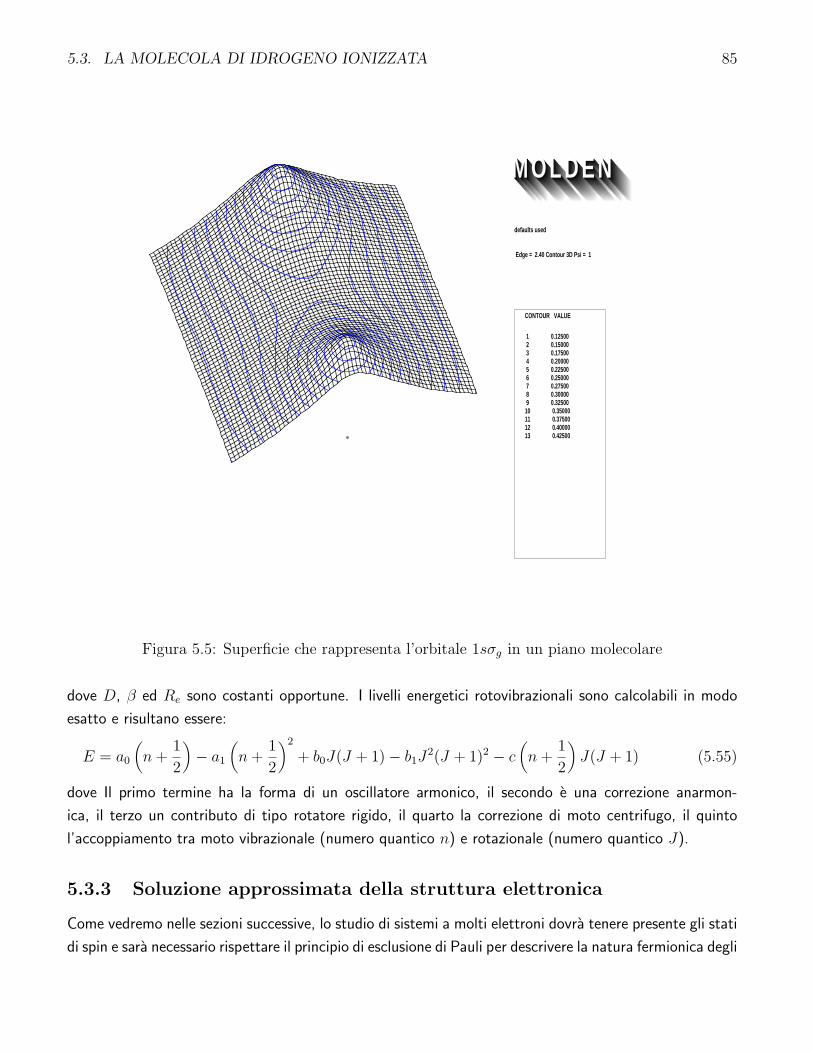
5.3. LA MOLECOLA DI IDROGENO IONIZZATA 85
CONTOUR VALUE
1 0.12500 2 0.15000 3 0.17500 4 0.20000 5 0.22500 6 0.25000 7 0.27500 8 0.30000 9 0.32500 10 0.35000 11 0.37500 12 0.40000 13 0.42500
defaults used
Edge = 2.40 Contour 3D Psi = 1
M O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E N
Figura 5.5: Superficie che rappresenta l’orbitale 1sσg in un piano molecolare
dove D, β ed Re sono costanti opportune. I livelli energetici rotovibrazionali sono calcolabili in modo
esatto e risultano essere:
E = a0
(n +
1
2
)− a1
(n +
1
2
)2
+ b0J(J + 1)− b1J2(J + 1)2 − c
(n +
1
2
)J(J + 1) (5.55)
dove Il primo termine ha la forma di un oscillatore armonico, il secondo e una correzione anarmon-
ica, il terzo un contributo di tipo rotatore rigido, il quarto la correzione di moto centrifugo, il quinto
l’accoppiamento tra moto vibrazionale (numero quantico n) e rotazionale (numero quantico J).
5.3.3 Soluzione approssimata della struttura elettronica
Come vedremo nelle sezioni successive, lo studio di sistemi a molti elettroni dovra tenere presente gli stati
di spin e sara necessario rispettare il principio di esclusione di Pauli per descrivere la natura fermionica degli

86 CAPITOLO 5. L’EQUAZIONE DI SCHRODINGER
CONTOUR VALUE
1 0.00000 2 0.02500 3 0.05000 4 0.07500 5 0.10000 6 0.12500 7 0.15000 8 0.17500 9 0.20000 10 0.22500 11 0.25000 12 0.27500 13 0.30000 14 0.32500 15 0.35000 16 -0.02500 17 -0.05000 18 -0.07500 19 -0.10000 20 -0.12500 21 -0.15000 22 -0.17500 23 -0.20000 24 -0.22500 25 -0.25000 ............
defaults used
Edge = 2.40 Contour 3D Psi = 2
M O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E N
Figura 5.6: Superficie che rappresenta l’orbitale 2pσu in un piano molecolare
elettroni. Nel caso della molecola di idrogeno ionizzata possiamo pero adottare un approccio semplice
dato che un solo elettrone e presente nel sistema. Seguiamo la metodogia illustrata nell’Appendice
(C) ed introduciamo una base di sole due funzioni, 1sA(~r) e 1sB(~r), rispettivamente il primo orbitale
idrogenoide centrato sul nucleo A ed il primo orbitale idrogenoide centrato sul nucleo B. Il calcolo
della matrice rappresentativa dell’hamiltoniano (5.37) e della matrice di overlap portano alla semplice
equazione secolare
∣∣∣∣∣∣a + 1/R− E b + s/R− Es
b + s/R− ES a + 1/R− E
∣∣∣∣∣∣= 0 (5.56)
dove a e l’elemento di matrice dell’hamiltoniano elettronico a meno del termine 1/R rispetto a 1sA(~r),
b e l’elemento di matrice dell’hamiltoniano elettronico a meno del termine 1/R rispetto a 1sB(~r), s e

5.3. LA MOLECOLA DI IDROGENO IONIZZATA 87
l’elemento di matrice di overlap tra le due funzioni, che sono normalizzate ma non ortogonali:
a = −1
2+ e−2R +
e−2R − 1
R< 0 (5.57)
b = −s
2+ e−R(1 + R) < 0 (5.58)
s = e−R
(1 + R +
R2
3
)> 0 (5.59)
Risolvendo si trovano le seguenti espressioni for i due autovalori E1 < E2 e relativi autostati
Stato fondamentale
E1 =a + b
1 + s+
1
R(5.60)
Ψ1 =1√
2(1 + s)(1sA + 1sB) (5.61)
Stato eccitato
E2 =a− b
1− s+
1
R(5.62)
Ψ1 =1√
2(1 + s)(1sA − 1sB) (5.63)
La scelta della base utilizzata per il calcolo approssimato dei primi due stati elettronici della molecola
di idrogeno ionizzata e stata effettuata in base al comportamento al limite di R → ∞ della molecola,
cioe assumendo che sostanzialmente gli orbitali molecolari siano ottenibili come combinazioni lineari degli
orbitali atomici idrogenoidi centrati sui nuclei (metodo LCAO, Linear Combination of Atomic Orbitals).

88 CAPITOLO 5. L’EQUAZIONE DI SCHRODINGER

Capitolo 6
Funzioni d’onda
6.1 La funzione d’onda elettronica
6.1.1 Stati di spin
Ad ogni particella e associato un momento di spin. La definizione completa dello coordinate di una
particella deve includere dunque anche la descrizione del suo stato di spin. La giustificazione formale
delle proprieta di spin richiede una formulazione della meccanica quantistica in modo che sia consistente
con la teoria della relativita ristretta. Il problema, affrontato da P.A.M. Dirac, conduce alla definizione in
modo non arbitrario di una proprieta assimilabile ad un momento angolare, che e descritta dall’operatore
(vettoriale) di spin ~S. Le componenti dell’operatore di spin (Sx,Sy,Sz) obbediscono alle consuete regole
di commutazione delle componenti di un operatore momento angolare generico:
[Sx,Sy] = iSz
[Sz,Sx] = iSy
[Sy,Sz] = iSx
[S2,Sx] = [S2,Sy] = [S2,Sz] = 0 (6.1)
Le autofunzioni dell’operatore di momento di spin sono ottenibili come:
S2|S,MS〉 = S(S + 1)|S, MS〉 (6.2)
Sz|S,MS〉 = MS|S, MS〉 − S ≤ MS ≤ S (6.3)
Le autofunzioni dell’operatore di spin sono ortonormali.
Es. Per S = 1/2 sono permessi i valori MS = ±1/2 :
MS = +1/2 |α〉 = |1/2, +1/2〉MS = −1/2 |β〉 = |1/2,−1/2〉 (6.4)
89

90 CAPITOLO 6. FUNZIONI D’ONDA
Le pseudo-coordinate di una particella avente spin S = 1/2 saranno quindi completamente definite solo
dopo aver specificato le coordinate spaziali (x, y, z) e il numero quantico di spin MS che puo essere 1/2
(stato α) o −1/2 (stato β).
6.1.2 Principio di indistinguibilita
Se una proprieta misurabile dipende dalle coordinate di un’insieme di particelle indistinguibili, ogni
misura di detta proprieta deve essere indifferente al tipo di classificazione delle particelle. Consideriamo
per semplicita il caso di due sole particelle indistinguibili il cui stato sia descritto da una determinata
funzione d’onda Ψ. Se definiamo:
• la probabilita di trovare 1 in q1 e 2 in q2:
|Ψ[q1(1), q2(2)]|2dq1dq2;
• la probabilita di trovare 1 in q2 e 2 in q1:
|Ψ[q2(1), q1(2)]|2dq1dq2;
ne consegue che le due probabilita devono essere uguali:
|Ψ[q1(1), q2(2)]|2 = |Ψ[q2(1), q1(2)]|2 (6.5)
Quindi, poiche’ si puo’ sempre scegliere una funzione d’onda in modo che sia reale (almeno in assenza
di campi vettoriali, come un campo magnetico esterno), la funzione d’onda puo solo cambiare di segno
quando le coordinate delle due particelle siano scambiate fra loro:
Ψ[q1(1), q2(2)] = ±Ψ[q2(1), q1(2)] (6.6)
In generale, vale la seguente classificazione:
• Tutte le particelle con momenti di spin multipli dispari di 1/2 sono descritti da funzioni d’onda
antisimmetriche; p. es.: elettrone, protone, neutrone, 32He2+ (e i nuclei a numero di massa dispari).
Tali particelle sono dette FERMIONI e seguono la statistica di Fermi-Dirac.
• Tutte le particelle con momenti di spin multipli pari di 1/2 sono descritti da funzioni d’onda
simmetriche; p. es.: fotone (S = 1), deuterone (S = 1), 42He2+ (e i nuclei a numero di massa
pari). Tali particelle sono dette BOSONI e seguono la statistica di Bose-Einstein.
Gli elettroni sono fermioni con S = 1/2 e di conseguenza la funzione d’onda elettronica di una molecola
deve essere una funzione antisimmetrica rispetto allo scambio di coordinate tra due elettroni qualunque
(principio di Pauli). In generale data una collezione di bosoni indistinguibili rappresentata dalla funzione
d’onda Ψbosoni, se si definisce con P un generico operatore di permutazione (che opera un certo numero
di scambi tra le coordinate delle particelle, argomenti della funzione d’onda), vale che
PΨbosoni = Ψbosoni (6.7)

6.1. LA FUNZIONE D’ONDA ELETTRONICA 91
cioe la funzione d’onda non cambia. Se e data invece una collezione di fermioni indistinguibili, con
funzione d’onda Ψfermioni, gli scambi di coordinate portano ad un possibile cambio di segno della funzione
d’onda:
PΨfermioni = (−)P Ψfermioni (6.8)
dove (−1)P indica la parita della permutazione P .
6.1.3 Determinanti di Slater
E data una base completa ortonormale di funzioni delle coordinate spaziali di un elettrone φj(~r), detti
anche orbitali. Le proprieta di spin dell’elettrone sono descritte dalla base |m〉, con m = ±1/2. Il
prodotto diretto della base di orbitali e di stati spin costituisce una base completa per l’elettrone, che
definiamo spin-orbitali. Un generico spin-orbitale e Sk(q) = φj(~r)|m〉 dove k indica la coppia j, m e q
indica le pseudo-coordinate (spaziali e di spin) dell’elettrone. La base di spinorbitali e ortonormale:
[Sk|Sk′ ] =∫
dqSk(q)∗Sk′(q) = 〈φj|φj′〉〈m|m′〉 = δj,j′δm,m′ = δk,k′ (6.9)
dove indichiamo con [. . .] l’integrazione in q. Una funzione d’onda elettronica e percio sempre rappre-
sentabile come una combinazione lineare di spin-orbitali:
Ψ =∑
k
ckSk(q) (6.10)
In un sistema multi-elettronico, sia Sk(n) un generico spin orbitale relativo all’elettrone n, cioe avente
come argomento le pseudo-coordinate qn. Una funzione di N elettroni puo essere scritta come il prodotto
delle N funzioni spin-orbitaliche:
ΨHartree =N∏
k=1
Sk(k) (6.11)
La funzione precedente e chiamata prodotto di Hartree e non puo rappresentare un insieme di fermioni,
poiche non verifica la (6.8). La costruzione di una funzione d’onda a N elettroni, simmetrizzata, si puo
ottenere sotto forma di determinante di una matrice di funzioni N ×N :
Ψ =1√N !
∣∣∣∣∣∣∣∣∣∣∣
S1(1) S2(1) . . . SN(1)
S1(2) S2(2) . . . SN(2)
. . . . . . . . . . . .
S1(N) S2(N) . . . SN(N)
∣∣∣∣∣∣∣∣∣∣∣
(6.12)
La funzione d’onda e detta determinante di Slater. La proprieta (6.8) e infatti verificata osservando
che dalla definizione di determinante risulta:
Ψ =1
N !
∑
P(−1)PPΨHartree (6.13)

92 CAPITOLO 6. FUNZIONI D’ONDA
Esercizio 4. Verifichiamo che la funzione d’onda soddisfi la (6.8). Le permutazioni costituiscono
un gruppo, quindi date due permutazioni P0 e P , P0P e un’altra permutazione e la sua parita e
(−1)P0+P = (−1)P0(−1)P . Quindi:
P0Ψ =1
N !
∑
P(−1)PP0PΨHartree = (−1)P0
∑
P ′(−1)P ′P ′ΨHartree = (−1)P0Ψ (6.14)
Il principio di esclusione di Pauli e implicito nella definizione della funzione d’onda multielettronica det Ψ,
poiche questa vale zero se 2 elettroni sono rappresentati dallo stesso spin-orbitale (infatti un determinante
con due colonne uguali e zero) e cambia di segno se due elettroni scambiano la loro posizione (infatti un
determinante cambia di segno se si scambiano due righe).
In letteratura sono utilizzate di frequente le notazioni alternative:
• Ψ ≡ |S1S2 . . . SN |
• φi(j) ≡ φi(j)α(j)
• φi(j) ≡ φi(j)β(j).
Una configurazione in cui gli elettroni hanno tutti gli spin accoppiati deve essere necessariamente una
autofunzione di S2 con spin 0 (singoletto)
S2Ψ = 0 (6.15)
e puo essere scritta a partire da N funzioni d’onda monoelettronica sotto forma del seguente determinante
di Slater:
Ψ = |φ1φ1φ2φ2 . . . φN−1φN−1φNφN | (6.16)
Il determinante di Slater e ottenuto dal prodotto di Hartree applicando l’operatore di anti-simmetria:
Ψ = AΨHartree (6.17)
dove
A =1√N !
∑
P(−1)PP (6.18)
Nel seguito di questa sezione, consideriamo alcune proprieta degli operatori permutazione, dell’operatore
A e dei determinanti di Slater, che ci saranno utili nel seguito.

6.1. LA FUNZIONE D’ONDA ELETTRONICA 93
P e unitario. Ovvero P† = P−1. Rappresentiamo con q l’insieme delle coordinate elettroniche.
Applicando P ad una generica funzione multielettronica Ψ(q), sostituiamo a q un set di coordinate
scambiate:
PΨ(q) = Ψ(q′) (6.19)
Vale anche, per definizione, che
Ψ(q) = P−1Ψ(q′) (6.20)
Se consideriamo ora la definizione di aggiunto di P , si ha che
〈PΨq(q)|Ψ2(q)〉 = 〈Ψ1(q)|P†|Ψ2(q)〉 (6.21)
ed applicando (6.19) al ket e (6.20) al bra del primo membro si ha
〈PΨq(q)|Ψ2(q)〉 = 〈Ψq(q)|P−1|Ψ2(q)〉 (6.22)
quindi P† = P−1.
A e hermitiano. Infatti:
A† =1√N !
∑
P(−1)PP† =
1√N !
∑
P(−1)PP−1 =
1√N !
∑
−P(−1)−PP−1 = A (6.23)
A2 e proporzionale a A. Sviluppando il quadrato si ottiene:
A2 =1
N !
∑
Pi
(−1)PiPi
∑
Pj
(−1)PjPj
=
1
N !
∑
Pi
∑
Pj
(−1)Pi+PjPiPj
=1
N !
∑
Pk
(−1)pkPk =√
N !A (6.24)
Commutazione. Dato un’operatore O simmetrico rispetto ad uno scambio di coordinate, vale natu-
ralmente che
[O,P ] = 0 → [O,A] = 0 (6.25)
Ortonormalita. I determinanti di Slater sono ortonormali. infatti:
〈Ψ|Ψ′〉 = 〈AΨHartree|AΨ′Hartree〉 = 〈ΨHartree|A2|Ψ′
Hartree〉 =√
N !〈ΨHartree|A|Ψ′Hartree〉
= 〈ΨHartree|∑P(−1)PP|Ψ′Hartree〉 = 〈ΨHartree|Ψ′
Hartree〉 = δΨ,Ψ′ (6.26)

94 CAPITOLO 6. FUNZIONI D’ONDA
Invarianza. I determinanti di Slater sono invarianti rispetto ad una trasformazione unitaria degli
spinorbitali. Sia U una matrice N×N unitaria, tale cioe che U† = U−1, il che implica | detU|2 =
1. Una trasformazione unitaria degli spinorbitali conserva l’ortonormalita
S ′i =∑
j
SjUji → [S ′i|S ′i′ ] =∑
j,j′U∗
jiUj′i′ [Sj|Sj′ ] = (U†U)ii′ = δi,i′ (6.27)
Se indichiamo con Ψ′ il determinante di Slater costruito con gli spinorbitali S ′i, abbiamo che
Ψ′ =1√N !
∣∣∣∣∣∣∣∣∣∣∣
S ′1(1) S ′2(1) . . . S ′N(1)
S ′1(2) S ′2(2) . . . S ′N(2)
. . . . . . . . . . . .
S ′1(N) S ′2(N) . . . S ′N(N)
∣∣∣∣∣∣∣∣∣∣∣
=1√N !
∣∣∣∣∣∣∣∣∣∣∣
S1(1) S2(1) . . . SN(1)
S1(2) S2(2) . . . SN(2)
. . . . . . . . . . . .
S1(N) S2(N) . . . SN(N)
∣∣∣∣∣∣∣∣∣∣∣
detU(6.28)
vale a dire Ψ′ = Ψ detU = Ψ exp(iφ), cioe il nuovo determinante di Slater differisce dal vecchio
solo per un fattore di fase, che e ininfluente nel calcolo dei valori di attesa.

Capitolo 7
Metodi ab-initio
7.1 Metodo di Hartree-Fock
L’approssimazione di Hartree-Fock e alla base del piu usato metodo di calcolo ab initio per strutture
elettroniche molecolari. Deve percio essere chiaro che si tratta di una approssimazione appunto, e
non di un metodo esatto di risoluzione dell’equazione di Schrodinger elettronica, ed ha per fine la
determinazione dello stato fondamentale di un sistema di elettroni. Considereremo dunque una molecola
di N elettroni. L’hamiltoniano elettronico in unita atomiche e:
H =N∑
n=1
h(n) +∑
n 6=n′
1
|~rn − ~rn′| (7.1)
h(n) = −1
2∇2
n −∑
k
Zk
|~rn − ~Rk|(7.2)
dove l’hamiltoniano monoelettronico di “core” di ciascun elettrone e relativo all’energia cinetica e di
interazione con i nuclei.
L’approssimazione di Hartree consiste nell’identificare la funzione d’onda multielettronica (autofun-
zione dell’hamiltoniano elettronico) come il prodotto di Hartree di N funzioni d’onda monoelettroniche.
Evidentemente, l’approssimazione di Hartree agisce a due livelli
1. si assume una forma di funzione d’onda compatibile solo con un hamiltoniano separabile dei N
elettroni;
2. si trascura il principio di indistinguibilita e dunque non si non si applica il principio di Pauli.
L’approssimazione di Hartree-Fock (HF) consiste nell’obbligare la funzione d’onda ad obbedire al principio
di Pauli, assumendo che la sua forma non sia un prodotto di Hartree, bensı un determinante di Slater:
cioe che sia data dall’equazione (6.1.3). Resta l’approssimazione fondamentale: una funzione d’onda
multielettronica e approssimata da una somma finita di prodotti di funzioni monoelettroniche. Il problema
e a questo punto formulabile nel modo seguente: nell’ambito dell’approssimazione HF, quali sono le
95

96 CAPITOLO 7. METODI AB-INITIO
funzioni monoelettroniche spaziali e di spin (spinorbitali) S1, . . . , SN che assicurino che l’energia totale
della molecola sia piu vicina a quella esatta?
Allo scopo di determinare i migliori1 spinorbitali Si si impiega il principio variazionale: il valore di
attesa dell’hamiltoniano rispetto ad una funzione d’onda arbitraria e sempre maggiore del primo autovalore
del sistema. Vale a dire che se si calcola il valore di attesa (la media) dell’hamiltoniano elettronico rispetto
al determinante di Slater, si ottiene una valore numerico che e sempre maggiore dell’energia esatta del
sistema multi-elettronico. E chiaro percio che i migliori orbitali si possono ottenere cercandone la forma
che rende minimo, e percio piu vicino al valore esatto, il valore di attesa.
7.1.1 Ottimizzazione degli spinorbitali
2 Definiamo un algoritmo di calcolo degli spinorbitali che descrivono lo stato fondamentale Ψ0 = Ψ, dove
Ψ e il determinante di Slater (6.1.3) per una molecola di N elettroni secondo l’approssimazione HF. E
dato il valore di attesa dell’hamiltoniano rispetto al determinante di Slater:
E = 〈Ψ|H|Ψ〉 (7.3)
Assumiamo che gli N spinorbitali S1, . . . , SN siano ortonormali; diciamo VHF il sottospazio generato
dalla base S1, . . . , SN ; per una variazione infinitesimale di ciascun spinorbitale
S ′i = Si + δSi (7.4)
possiamo costruire lo spinorbitale
Ψ′ = |S ′1S ′2 . . . S ′N | (7.5)
e limitando lo sviluppo ai soli termini al primo ordine si ottiene
Ψ′ = Ψ +∑
i
δΨi (7.6)
δΨi = |S1 . . . δSiSN | (7.7)
se le variazioni δSi sono ortogonali allo spazio VHF risulta che la variazione al primo ordine del valore di
attesa dell’hamiltoniano rispetto al determinante di Slater e data da
δE =∑
i
〈δΨi|H|Ψ〉+ 〈Ψ|H|δΨi〉 (7.8)
Possiamo percio affermare che la variazione infinitesimale δE e nulla al primo ordine (cioe E e minimo)
se sono indipendentemente nulli i seguenti integrali
〈δΨi|H|Ψ〉 = 0 (7.9)
1Nel senso appena definito: tali che ottimizzano il calcolo dell’energia elettronica dello stato fondamentale dellamolecola nell’approssimazione di Hartree-Fock, cioe rappresentando lo stato fondamentale stesso mediante un unicodeterminante di Slater
2Questa sezione e la successiva sono in parte basate su appunti di lezione del prof. Giorgio J. Moro

7.1. METODO DI HARTREE-FOCK 97
cioe se una variazione infinitesimale di un generico spinorbitale, ortogonale a VHF, da un contributo nullo
alla variazione del valore di attesa.
La condizione (7.9) puo essere ridotta al calcolo di un integrale monoelettronico:
〈δΨi|H|Ψ〉 =1
N !
∑
P ′,P ′′
∫dq(−1)P ′(−1)P ′′δΨi
∗HartreeP ′−1HP ′′ΨHartree (7.10)
dove si ricordi che q rappresenta l’insieme delle pseudocoordinate del sistema e ΨHartree e il prodotto di
Hartree costruito con gli N spinorbitali; nell’equazione precedente sie fatto uso della proprieta P† = P−1.
La sommatoria su P ′′ puo essere sostituita con la sommatoria su P = P ′−1P ′′, tenendo conto del fatto
che H commuta con una generica permutazione, [H,P ] = 0. Si ottiene percio che
〈δΨi|H|Ψ〉 =∑
P(−1)P
∫dqδΨi
∗HartreeHPΨHartree
=∑
P(−1)P
∫dqδΨi
∗Hartree
h(i) +
∑
n 6=i
1
rni
PΨHartree
= [δSi|h|Si] +∑
n 6=i
([δSiSi|SnSn]− [δSiSn|SnSi]) (7.11)
dove sono definiti i seguenti integrali mono e bi-elettronici:
[Sa|h|Sb] =∫
dq1Sa(1)∗h(1)Sb(1) (7.12)
[SaSb|ScSd] =∫
dq1dq2Sa(1)∗Sb(1)1
r12
Sc(2)∗Sd(2) = [SbSa|SdSc]∗ = [ScSd|SaSb] (7.13)
Possiamo definire l’operatore di Coulomb associato all’n-esimo spinorbitale
Jn(1)Sm(1) =[∫
dq2Sn(2)∗1
r12
Sn(2)]Sm(1) (7.14)
e l’operatore di scambio associato all’n-esimo spinorbitale
Kn(1)Sm(1) =[∫
dq2Sn(2)∗1
r12
Sm(2)Sn(1)]
(7.15)
In questo modo gli integrali bi-elettronici si possono scrivere come elementi di matrice monoelettronici
degli operatori di Coulomb e di scambio:
[SaSb|SnSn] = [Sa|Jn|Sb] (7.16)
[SaSn|SnSb] = [Sa|Kn|Sb] (7.17)
ed in definitiva possiamo scrivere la variazione 〈δΨi|H|Ψ〉 nella forma:
〈δΨi|H|Ψ〉 = [δSi|F|Si] (7.18)
dove F e l’operatore (monoelettronico) di Fock:
F(1) = h(1) +∑n
[J n(1)−Kn(1)] (7.19)

98 CAPITOLO 7. METODI AB-INITIO
L’operatore di Fock dipende, tramite gli operatori di Coulonb e di scambio, dal set di spinorbitali che
definisce VHF. La condizione di stazionarieta variazionale e espressa dall’equazione
[δSi|F|Si] = 0 (7.20)
per tutti gli spinorbitali Si e qualsiasi variazione δSi ortogonale a VHF, che deve percio essere uno spazio
chiuso rispetto a F :
FSi ∈ VHF. (7.21)
7.1.2 Proprieta dell’operatore di Fock
L’operatore di Fock F e definito in relazione agli spinorbitali che generano lo spazio hilbertiano VHF, ma
non dipende da una scelta specifica di questi spinorbitali. In altre parole, vale il seguente teorema
Teorema l’operatore F e invariante rispetto ad una trasformazione unitaria della base che genera VHF
Se infatti trasformiamo gli N spinorbitali secondo la trasformazione unitaria
S ′i =∑
j
SjUji (7.22)
dove U−1 = U†, si trova che sia la somma degli operatori di Coulomb che di quelli di scambio risulta
invariata
∑n
J ′(n)∑
jj′(UU∗)jj′
∫dq2Sj(2)∗
1
r12
Sj′(2) =∑n
J (n) (7.23)
∑n
K′(n)Sa(1)∑
jj′(UU∗)jj′
∫dq2Sj(2)∗
1
r12
Sa(2)Sj′(2) =∑n
K(n)Sa(1) (7.24)
e dunque l’operatore di Fock stesso risulta invariato. Un’altra importante proprieta dell’operatore di Fock
e la seguente
Teorema l’operatore F e autoaggiunto F † = F .
Infatti h(1) e autoaggiunto per definizione; per gli operatori coulombiani e di scambio vale che:
[Sa|J †
n|Sb
]= [Sb|J n|Sa]
∗ =∫
dq1Sa(1)∗[∫
dq2Sn(2)∗1
r12
Sn(2)]Sb(1) = [Sa|J n|Sb] (7.25)
[Sa|K†n|Sb
]= [Sb|Kn|Sa]
∗ =∫
dq2Sa(2)∗[∫
dq1Sn(1)∗1
r12
Sb(1)]Sn(2) = [Sa|Kn|Sb] (7.26)
Poiche F non genera spinorbitali che appartengono allo spazio complementare a VHF, una volta nota una
base ortonormale completa di spinorbitali in cui i primi N elementi siano base per lo spazio VHF stesso,
l’operatore di Fock e rappresentabile come una matrice diagonale a blocchi, ovvero (F)ij = [Si|F|Sj]
vale 0 se 1 ≤ i ≤ N e j ≥ N + 1 o i ≥ N + 1 e 1 ≤ j ≤ N . Per mezzo di una trasformazione unitaria

7.1. METODO DI HARTREE-FOCK 99
della base ortonormale iniziale possiamo sempre rendere diagonale la matrice F, vale a dire possiamo
sceglere come base ortonormale l’insieme di autofunzioni di F :
FSi = εiSi i = 1, . . . , N, N + 1, . . . (7.27)
La procedura per il calcolo di un insieme completo di spinorbitali ortonormali che verifichino l’approssimazione
HF e dunque
1. scelta di una base di spinorbitali e selezione di una sottobase, cioe dello spazio VHF
2. calcolo di F per il dato VHF
3. diagonalizzazione di F
4. confronto delle prime N autofunzioni di F e della base; iterazione della procedura dal punto 2.
fino al raggiungimento della condizione di autoconsistenza, ovvero della indistinguibilita (a meno
di qualche opportuno criterio di precisione numerica) tra le prime N autofunzioni di F e della base.
L’algoritmo cosı definito permette la determinazione di un insieme di spinorbitali che e completo, cioe
include anche gli spinorbitali SN+1, SN+2, . . ., detti orbitali virtuali, che non compaiono nella definizione
dello stato fondamentale e possono essere usati per rapresentare gli stati eccitati della molecola.
Una volta noti gli spinorbitali che verifichino le condizioni precedenti, l’energia dello stato fondamen-
tale della molecola si puo calcolare direttamente del valore di attesa dell’hamiltoniano elettronico rispetto
al determinante di Slater:
E = 〈Ψ|H|Ψ〉 =∑
i
[Si|h|Si] +
1
2
∑
n6=i
([SiSi|SnSn]− [SiSn|SnSi])
=∑
i
[Si|h +1
2
∑n
(J n −Kn)|Si] (7.28)
L’energia totale non e la somma dei primi N autovalori di F , poiche la somma∑
n(J n −Kn) compare
premoltiplicata per il fattore 1/2; se si scrive l’operatore di Fock come
F = h + G (7.29)
allora l’energia totale e
E =∑
i
[Si|F − G/2|Si] =∑
i
[Si|h + G/2|Si] (7.30)
dove G descrive l’interazione tra un elettrone e tutti gli altri elettroni (da cui segue il fattore 1/2 per non
contare l’interazione due volte).
Il significato fisico degli autovalori εi dell’operatore di Fock puo essere ricavato dal teorema di Koop-
mans

100 CAPITOLO 7. METODI AB-INITIO
Teorema a meno di correzioni di ordine superiore al primo, gli spinorbitali ottenuti secondo la procedura
HF per un sistema di N elettroni e equlli relativi ai sistemi con un’elettrone in piu o in meno sono
gli stessi
Di conseguenza l’energia Ek del sistema dopo aver estratto un elettrone si puo ottenere, secondo il
teorema di Koopmans, come
E − Ek = [Sk|h + G|Sk] = [Sk|F|Sk] = εk (7.31)
Se quindi Sk e l’autostato dell’operatore di Fock con autovalore εk piu elevato occupato (Highest Oc-
cupied Molecular Orbital, HOMO, a cui segue il Lowest Unoccupied Molecular Orbital, LUMO), −εk e
l’energia di prima ionizzazione del sistema, cioe la minima energia richiesta per la reazione
X(g) → X+(g) + e− (7.32)
dove X rappresenta il sistema molecolare di N elettroni.
Per He e Li i potenziali di ionizzazione misurati sono rispettivamente -24.5 eV e -5.39 eV contro dei
valori calcolati secondo HF pari a -24.98 eV e -5.34 eV.
7.1.3 Sistemi a guscio chiuso
La procedura descritta nella sezione precedente e riferita ad uns sistema elettronico generico, ed e percio
detta UHF, cioe “unrestricted” HF. Se ~S e l’operatore di spin elettronico totale
~S =∑n
~S(n) (7.33)
deve valere che [H,S2] = [H,Sz] = 0, poiche come abbiamo visto non includiamo termini espliciti di
spin nell’hamiltoniano elettronico. Di conseguenza la funzione multielettronica deve essere autofunzione
di S2 e di Sz: il metodo UHF non assicura questa proprieta. In pratica buona parte delle applicazioni
sono per molecole stabili in stato di singoletto di spin, con un numero N pari di elettroni. Si puo quindi
definire fin dal principio un determinante di Slater scelto in maniera tale da verificare la condizione di
essere uno stato di singoletto, cfr. Eq. (6.15). Come abbiamo visto nel capitolo precedente, se si
definiscono N/2 orbitali φ1, φ2, . . . , φN/2, il determinante di Slater si puo costruire alternando stati di
spin α e β
Ψ = |φ1φ1φ2φ2 . . . φN/2φN/2| (7.34)
In questo caso di parla di metodo RHF, cioe “restricted” HF. La procedura RHF si puo facilmente ricavare
dal caso piu generale UHF, verificando che l’operatore di Fock conserva la componente di spin
F(1)φi(1)|m(1)〉 = |m(1)〉F φi(1) (7.35)

7.1. METODO DI HARTREE-FOCK 101
dove F e un’operatore di Fock che agisce sulla sola parte orbitalica
F (1) = h(1) +N/2∑
i=n
[2Jn(1)− Kn(1)
](7.36)
dove si impiega l’operatore di Coulomb associato all’n-esimo orbitale
Jn(1)φm(1) =[∫
dr2φn(2)∗1
r12
φn(2)]φm(1) (7.37)
e l’operatore di scambio associato all’n-esimo orbitale
Kn(1)φm(1) =[∫
dr2φn(2)∗1
r12
φm(2)φn(1)]
(7.38)
L’energia totale di un sistema a guscio chiuso si puo scrivere nella forma
E = 2N/2∑
i
hi +N/2∑
i,j
(2Jij −Kij) (7.39)
dove sono definire i seguenti integrali mono e bi-elettronici
hi = 〈φi(1)|hi(1)|φi(1)〉 =∫
d~r1φi(1)∗hi(1)φi(1) (7.40)
Jij =∫
d~r1d~r2|φi(1)|2 1
r12
|φj(2)|2 (7.41)
Kij =∫
d~r1d~r2φi(1)∗φj(1)1
r12
φi(2)∗φj(2) (7.42)
dove Jij e Kij sono detti gli integrali coulombiani e di scambio. In Appendice (D) il valore di attesa
dell’hamiltoniano monoelettronico viene riderivato in modo esplicito e semplificato per il solo caso di un
sistema a guscio chiuso.
7.1.4 Soluzione numerica delle equazioni HF
La natura delle equazioni di Hartree-Fock e intrinsecamente non-lineare: gli operatori agenti sugli orbitali
φi dipendono a loro volta dagli orbitali φi stessi. Di conseguenza uno schema di soluzione numerica
naturale e di tipo auto-consistente, come e stato gia evidenziato nella sezione precedente
1. si definiscono delle funzioni in ordine 0, φ(0)1 , . . . , φ
(0)N/2;
2. si calcolano le forme esplicite degli operatori J(0)
j , K(0)
j in funzione delle funzioni φ(0)i ;
3. si calcolano nuove funzioni φ(1)1 , . . . , φ
(1)N/2, risolvendo il problema agli autovalori lineare con gli
operatori J(0)
j , K(0)
j ;
4. si calcolano nuovi operatori J(1)
j , K(1)
j in funzione delle funzioni φ(1)i ;

102 CAPITOLO 7. METODI AB-INITIO
5. il ciclo si ripete finche le funzioni φ(n+1)1 , . . . , φ
(n+1)N/2 differiscono dalle φ
(n)1 , . . . , φ
(n)N/2 a meno di un
errore prefissato.
Il modo migliore di procedere e, oggigiorno, quello di espandere linearmente le funzioni incognite φi su
un set di funzioni di base note. I coefficienti numerici dell’espansione divengono poi le vere incognite
del problema: in questo modo si riduce un sistema di equazioni differenziali ad un sistema di equazioni
ordinarie, che deve poi comunque essere risolto in modo auto-consistente.
Un set di funzioni di base permette di esprimere qualunque funzione come combinazione lineare delle
funzioni componenti:
φ = Ctrχ (7.43)
dove χm = χm e C e una matrice il cui vettore colonna m sara indicato con cm
φ1
φ2
. . .
φM
=
c11 c12 . . . c1M
c21 c22 . . . c2M
. . . . . . . . . . . .
cM1 cM2 . . . cMM
tr
χ1
χ2
. . .
χM
= (c1, c2, . . . , cM)trχ (7.44)
Si possono definire varie grandezze matriciali e vettoriali in termini dei coefficienti cm e di altri integrali
che coinvolgono gli orbitali (noti) χm:
∆pq = 〈χp|χq〉 (7.45)
hi = 〈φi|h(1)|φi〉 = c†iχ†h(1)χci = c†ihci (7.46)
Jipq = 〈χp|Ji|χq〉 (7.47)
Jij = c†iJjci = c†jJicj (7.48)
Kipq = 〈χp|Ki|χq〉 (7.49)
Kij = c†iKjci = c†jKicj (7.50)
L’energia HF assume la forma:
〈H〉av = 2∑
i
c†ihci +∑
ij
c†i (2Jj −Kj)ci (7.51)
Le equazioni di Hartree-Fock diventano le equazioni di Roothaan:
FC = ∆Cε (7.52)
dove Fpq = 〈χp|F |χq〉.La scelta delle funzioni orbitaliche di base e di importanza fondamentale per l’acccuratezza del
calcolo HF. Sostanzialmente e necessario bilanciare due fattori i) all’aumentare del numero di funzioni
di base il calcolo sara piu accurato, ma il costo in termini computazionali (CPU, spazio di memoria per

7.1. METODO DI HARTREE-FOCK 103
immagazzinare gli integrali mono e bielettronici) crescera in modo superlineare con il numero di funzioni
di base stesso; ii) funzioni di base accurate, costituite per esempio dagli orbitali dell’atomo idrogenoide,
per quanto siano piu efficienti in termini di numero di funzioni di base, possono comportare un aumento
nel costo computazionale dovuto all’aumentata complessita nel calcolo degli integrali. Per un sistema
molecolare si puo in generale scegliere un set di funzioni di base tale da riprodurre lo stato fondamentale
degli atomi componenti: e questa la cosiddetta base minima. Per esempio, nel caso della molecola CO
si possono usare come base minima gli orbitali atomici (AO) 1s, 2s, 2px,y,z per il carbonio e gli stessi
orbitali per l’ossigeno; per la molecola di ammoniaca NH3 si possono usare ancora i medesimi orbitali
centrati sull’azoto gli orbitali 1s per i tre atomi di idrogeno e cosı via. Si parla di basi estese quando si
includono AO relativi a stati non occupati dei singoli atomi.
Tra le funzioni piu utilizzate per definire gli AO di partenza, ricordiamo
orbitali idrogenoidi ottenuti dalla soluzione del problema agli autostati per un atomo idorgeonoide,
della forma generica
Ψn,l,ml(r, θ, φ) = Rnl(r)Ylml
(θ, φ) (7.53)
gia discussi in precedenza, costituiti dal prodotto delle armoniche sferiche per le funzioni di Laguerre:
Rnl =
√√√√(n− l − 1)!Z
n2[(n + l)!]3ρl+1L2l+1
n+l (ρ) exp(−ρ/2) (7.54)
Queste funzioni sono ortogonali, non-complete ed in generale computazionalmente ‘costose’, data
la loro forma particolarmente complicata.
Slater-type orbitals, STO aventi la forma generale
uα,n,l,ml(r, θ, φ) =
1√(2n)!
(2α)n+1/2rn−1 exp(−αr)Ylml(θ, φ) (7.55)
dove n ed α sono parametri variazionali; costituiscono un set completo relativamente poco costoso
computazionalmente benche non siano ortogonali.
Gaussian-type orbitals, GTO espressi come
gα,n,l,ml(r, θ, φ) = Nrn−1 exp(−αr2)Ylml
(θ, φ) (7.56)
Solitamente si impiegano combinazioni lineari di GTO per rappresentare in modo approssimato
gli orbitali di tipo STO: si parla cosı di basi STO-2G, STO-3G, . . . , STO-6G per descrivere basi
costituite da funzioni STO approssimate come combinazioni lineari di 2, 3, . . . , 6 GTO.

104 CAPITOLO 7. METODI AB-INITIO
Riassumendo: un calcolo RHF secondo il metodo di Roothaan della struttura elettronica dell’acqua H2O
con una base minima STO-4G prevede l’applicazione del metodo RHF con la base di 7 AO non-ortogonali
1sO, 2sO, 2px,y,zOe 1sH1 , 1sH2 descritti come STO approssimati da 4 GTO ciascuno. Altre basi molto
usate sono caaratterizzate da un trattamento piu accurato di alcuni elettroni rispetto ad altri, come le
funzioni 3-21G, funzioni STO approssimate da 3 GTO per gli elettroni interni e 2 per ogni elettrone di
valenza e le funzioni 6-21 o 6-31G, con 6 gaussiane per gli elettroni interni; ricordiamo anche le funzioni
6-31G∗ considerano anche AO di tipo d e 6-31G∗∗ che includono AO di tipo p centrati sugli idrogeni.
7.2 Interazione di configurazione
A differenza del metodo di Hartree-Fock, il metodo dell’interazione di configurazione (CI), e almeno in
principio in grado di fornire un risultato esatto (o arbitrariamente vicino al risultato esatto) del calcolo della
funzione d’onda elettronica. L’idea alla base del metodo CI e molto semplice: la funzione d’onda e data da
una combinazione (non necessariamente lineare) di funzioni di base note multielettroniche; i coefficienti
dell’espansione sono determinati sulla base del principio variazionale. Riassumiamo brevemente il principio
variazionale per il calcolo di una generica funzione d’onda multielettronica partendo dal rapporto di
Rayleigh per una generica funzione d’onda Ψ
ε[Ψ] =〈Ψ|H|Ψ〉〈Ψ|Ψ〉 (7.57)
Il principio variazionale afferma che se E0 e il piu basso autovalore del sistema, il rapporto di Rayleigh
e sempre maggiore o uguale di E0
ε[Ψ] ≥ E0 (7.58)
La dimostrazione e la seguente: espandiamo Ψ sulla base delle autofunzioni esatte dell’hamiltoniano H
HΦ = EnΦ E0 ≤ E1 ≤ . . . (7.59)
〈Φn|Φ′n〉 = δnn′ (7.60)
Ψ =∑n
cnΦn (7.61)
consideriamo la quantita 〈Ψ|H − E0|Ψ〉:
〈Ψ|H − E0|Ψ〉 =∑
nn′c∗ncn′〈Φn|H − E0|Φn′〉 =
∑
nn′c∗ncn′(En − E0)〈Φn|Φn′〉 =
∑n
|cn|2(En − E0) ≥ 0 (7.62)
infatti |cn|2 ≥ 0, En ≥ E0; segue che:
〈Ψ|H|Ψ〉 − E0〈Ψ|Ψ〉 ≥ 0 ⇒ ε[Ψ] ≥ E0 QED (7.63)

7.2. INTERAZIONE DI CONFIGURAZIONE 105
Nella sua versione piu comune, il metodo CI e implementato sotto forma di metodo variazionale
lineare. La funzione d’onda di prova Ψ e cioe data come una combinazione lineare di funzioni note:
Ψ =N∑
n′=1
cn′Ψn′ (7.64)
sostituendo in HΨ = EΨ:
∑
n′cn′HΨn′ = E
∑
n′cn′Ψn′ (7.65)
premoltiplicando per Ψn ed integrando:
∑
n′cnHnn′ = E
∑
n′cnSnn′ (7.66)
dove Hnn′ = 〈Ψn|H|Ψn′〉, Snn′ = 〈Ψn|Ψn′〉; in forma matriciale:
Hc = ESc (7.67)
Il problema agli autovalori diviene piu semplice se si assumono delle funzioni di base ortonormali S = 1:
Hc = Ec (7.68)
In linea di principio, un calcolo CI dovrebbe includere un numero infinito di funzioni multielettroniche.
Se ci limitiamo a rappresentare ogni possibile configurazione multielettronica come un determinante di
Slater costruito a partire dagli spinorbitali (reali e virtuali) definiti nel corso di un calcolo UHF o RHF,
dobbiamo comunque considerare un numero molto elevato di possibili configurazioni accoppiate con lo
stato fondamentale rappresentato dalla funzione Ψ0. Possiamo pero considerare in ordine di importanza
le correzioni apportate dalle varie configurazioni. Sia M lo spazio degli stati del sistema multielettronico,
che si puo ripartire come
M = M0 ⊗M1 ⊗M2 ⊗M3 ⊗ . . . (7.69)
dove M0 e lo spazio monodimensionale costituito da Ψ0; M1 e lo spazio costituito dai determinanti di
Slater ottenuti sostituendo in Ψ0 uno spinorbitale virtuale ad uno reale (configurazioni eccitate singo-
larmente); M1 e lo spazio costituito dai determinanti di Slater ottenuti sostituendo in Ψ0 due spinorbitali
virtuali a due reali (configurazioni eccitate doppiamente); e cosı via. Vale ora il seguente teorema
Teorema di Brillouin. Gli elementi di matrice tra spinorbitali appartenenti a M1 e Ψ0 sono nulli.
dal cui si puo concludere che le configurazioni singolarmente eccitate non hanno un effetto diretto
sulla correzione CI. In molti casi il metodo Ci viene applicato includendo solo le configurazioni di stato
fondamentale e quelle doppiamente eccitate (M2). Si noti che un’effetto indiretto delle configurazioni
singolarmente eccitate e naturalmente presente, dato che sono comunque accoppiate con gli elementi di
M2, M3 etc.

106 CAPITOLO 7. METODI AB-INITIO
M
M
0
M
M M M
0
1
2
1 2
Figura 7.1: Schema degli elementi di matrice H nel metodo CI. Sono indicati in nero i blocchi
diagonali non nulli, in grigio i blocchi non-diagonali non nulli, in bianco i blocchi non-diagonali nulli
La differenza tra l’energia esatta dello stato fondamentale e quella calcolata nell’approssimazione HF
e detta energia di correlazione elettronica
Ecorr = Eesatta − EHF (7.70)
Una metologia sostanzialmente equivalente per il calcolo di Ecorr e basata su un trattamento perturbativo
e va sotto il nome di correzione di Mœller-Plesset (o metodo MPPT, Mœller-Plesset perturbation
theory). E basato su una separazione dell’hamiltoniano elettronico in due contributi: uno considerato di
ordine zero secondo la teoria delle perturbazioni indipendenti dal tempo e coincidente con l’operatore di
Fock, l’altro equivalente alla differenza tra F e H:
H = H(0) +H(1) ≡ F + (H−F) (7.71)
L’energia dello stato fondamentale nel limite HF e data dai contributi perturbativi di ordine zero e di
ordine uno:
EHF = 〈Ψ0|H(0) +H(1)|Ψ0〉 (7.72)
assumendo come funzione d’onda dello stato fondamentale in ordine zero la solita funzione Ψ0 risultante
dal calcolo HF. Correzioni non banali di ordine successivo si calcolano a partire dal secondo ordine
in poi, che coinvolgono elementi di matrice tra Ψ0 e configurazioni doppiamente eccitate (metodo
MPPT2). Ordini successivi di correzioni perturbative coinvolgono configurazioni una, due e tre volte
eccitate (metodo MPPT3) e cosı via.

7.3. ALCUNI ESEMPI DI CALCOLO AB INITIO 107
7.3 Alcuni esempi di calcolo ab initio
In questa sezione sono illustrati molto brevemente alcuni esempi di calcolo ab initio RHF. I calcoli sono
stati effettuati impiegando il software Gaussian 94, un pacchetto per determinare strutture elettron-
iche, ottimizzazione di geometrie etc. La descrizione dei risultati e stata fatta impiegando Molden, un
pacchetto per la visualizzazione di vari programmi di calcolo (oltre a Gaussian 94 sono disponibili come
freeware, shareware od in commercio molti altri pacchetti software: ricordiamo tra gli altri Gamess,
Spartan, Mopac).
7.3.1 La molecola di idrogeno
L’hamiltoniano elettronico della molecola di idrogeno e
H = h(1) + h(2) +1
r12
(7.73)
La molecola e descritta nello stesso sistema di riferimento gia utilizzato per la molecola di idrogeno
ionizzata, H+2 , salvo la presenza di due elettroni definiti dalle coordinate ~r1 e ~r2. Lo stato fondamentale
e descritto dal determinante di Slater
Ψ = |φφ| (7.74)
e l’operatore di Fock (RHF, guscio chiuso, spin e parte orbitalica fattorizzate) puo essere scritto come:
F = −∇2 − 1
rA
− 1
rB
+ 2J − K (7.75)
dove J e K sono definiti come
J(~r)φ(~r) = K(~r)φ(vr) =
(∫d~r′
|φ(~r)|2|~r − ~r′|
)φ(~r) (7.76)
Adottando il metodo di Roothaan con una base minima assumiamo che un generico orbitale sia
φi = c1i1sA + c2i1sB (7.77)
poiche usiamo una base di 2 funzioni per un sistema a guscio chiuso con 2 elettroni, otteremo due
orbitali, uno reale (HOMO) e l’altro virtuale (LUMO). La diagonalizzazione dell’operatore di Fock sulla
base scelta porta alla seguente equazione secolare∣∣∣∣∣∣
a + J ′ − ε b + J ′′ − εs
b + J ′′ − εs a + J ′ − ε
∣∣∣∣∣∣= 0 (7.78)
dove a, b ed s sono state definte in precedenza per lo studio approssimato della molecola di idrogeno
ionizzata, mentre gli integrali J ′ e J ′′ sono definiti come
J ′ = 〈1sA|2J − K|1sA〉 = 〈1sB|2J − K|1sB〉 (7.79)
J ′′ = 〈1sA|2J − K|1sB〉 = 〈1sB|2J − K|1sA〉 (7.80)

108 CAPITOLO 7. METODI AB-INITIO
Gli autovalori εi e i coefficienti di espansione cij si possono trovare immediatamente, senza cioe applicare
alcun metodo iterativo, poiche il sistema e definito completamente dalle sue proprieta di simmetria. Gli
autovalori sono ottenuti come
ε1,2 =a±+J ′ ∓ J ′′
1± s(7.81)
mentre gli orbitali sono dati da
φ1,2 =1√
2(1± s)(1sA ± 1sB) (7.82)
Considerazioni di simmetria conducono al risultato corretto se si tiene conto che e possibile costruire una
base orbitalica ortonormale simmetrica rispetto all’inversione delle coordinate ~r → −~r. Data la geometria
della molecola si vede subito che un orbitale simmetrico e φg = φ1, mentre un orbitale antisimmetrico e
φu = φ2. Costruendo l’operatore di Fock direttamente rispetto a φg si ottiene che F (~r) ≡ F (−~r) da cui
segue che 〈φu|F |φg〉 = 0 cioe F e diagonale sulla base costruita per simmetria con autovalori εg = ε1 e
εu = ε2.
Una volta nota la configurazione dello stato fondamentale, avendo a disposizione un’orbitale virtuale,
possiamo costruire vari stati eccitati, come i tre stati degeneri di tripletto eccitato
ΨT1 = |φgφu| = φ1(1)φ2(2)− φ1(2)φ2(1)√2
α(1)α(2) (7.83)
ΨT0 =|φgφu|+ |φgφu|√
2=
φ1(1)φ2(2)− φ1(2)φ2(1)√2
α(1)β(2) + α(2)β(2)√2
(7.84)
ΨT−1 = |φgφu| = φ1(1)φ2(2)− φ1(2)φ2(1)√2
β(1)β(2) (7.85)
che ha un’energia calcolata come valore di attesa pari a
ET = 〈ΨTi|H|ΨTi
〉 = hg + hu + Jg,u −Kg,u (7.86)
e lo stato di singoletto eccitato
ΨS1 =|φgφu| − |φgφu|√
2=
φ1(1)φ2(2) + φ1(2)φ2(1)√2
α(1)β(2)− α(2)β(2)√2
(7.87)
avente l’energia
ES1 = 〈ΨS1|H|ΨS1〉 = hg + hu + Jg,u + Kg,u. (7.88)
Calcoli RHF effettuati su H2 con una base estesa 6-31G permettono di determinare una distanza di
equilibrio R tra i due nuclei (ottenuta ripetendo il calcolo RHF a diverse distanze R fino ad individuare
un minimo dell’energia dello stato fondamentale - un esempio di ottimizzazione di geometria molecolare)
pari a 1.38 A, contro 1.40 A determinato sperimentalmente; il potenziale di ionizzazione calcolato e invece
3.49 eV contro 4.75 eV sperimentale.

7.3. ALCUNI ESEMPI DI CALCOLO AB INITIO 109
La funzione calcolata secondo la procedura RHF per la molecola di idrogeno nel suo stato fonda-
mentale non rappresenta in modo adeguato il sistema, soprattutto per R →∞. Se infatti calcoliamo la
forma limite per R →∞ si ottiene
limR→∞
|φgφg| = [1sA(1)1sB(2) + 1sB(1)1sA(2)] + [1sA(1)1sA(2) + 1sB(1)1sB(2)] (7.89)
il primo termine indica due atomi H·, mentre il secondo indica la coppia ionica H+, H−. Cio implica
il 50 % di probabilita che una molecola di idrogeno dissoci sotto forma di coppia biradicalica o ionica,
che e del tutto irrealistico. Un calcolo realistico terra conto di configurazioni aggiuntive che vanno oltre
l’approssimazione HF. Si tratta percio sostanzialmente di uan correzione CI al risultato RHF, ottenuta
includendo configurazioni di tipo covalente e ionico scritte in modo opportuno per tener conto della
indistinguilibilita degli elettroni:
Ψcov =1
2√
1 + s2
(|1sA1sB| − |1sA1sB|
)(7.90)
Ψion =1
2√
1 + s2
(|1sA1sA|+ |1sB1sB|
)(7.91)
La generalizzazione del metodo CI per la molecola di idrogeno, basata sull’impiego di un set di funzioni
bi-elettroniche completo, permette di ottenere un accordo con i dati sperimentali decisamente buono:
il potenziale di ionizzazione calcolato con 50 configurazioni di base e 4.74 eV, mentre la distanza di
equilibrio e 1.4 A.
7.3.2 Altri esempi
Nel caso di molecule lineari l’applicazione di vincoli di simmetria alla scelta delle funzioni di base semplifica
grandemente il calcolo ab initio. L’uso di basi adattate per simmetria sara descritto nel capitolo
successivo. Qui ci limitiamo a commentare brevemente il risultato di altri due calcoli RHF effettuati per
una molecola lineare, HCl, e per l’acqua, H2O. Nel seguito, mostriamo l’esempio di un tabulato di calcolo
risultante da una ottimizzazione di geometria per la molecola di aqcua eseguita con Gaussian 94:
**********************************************************************
Population analysis using the SCF density.
**********************************************************************
Orbital Symmetries:
Occupied (A’) (A’) (A’) (A’) (A")
Virtual (A’) (A’) (A’) (A’) (A") (A’) (A’) (A’) (A’) (A’)
(A") (A’) (A’) (A’)
The electronic state is 1-A’.

110 CAPITOLO 7. METODI AB-INITIO
CONTOUR VALUE
1 0.00000 2 0.05000 3 0.10000 4 0.15000 5 0.20000 6 0.25000 7 0.30000 8 0.35000 9 0.40000 10 0.45000 11 0.50000 12 0.55000 13 0.60000 14 0.65000 15 0.70000 16 0.75000 17 -0.05000 18 -0.10000 19 -0.15000 20 -0.20000 21 -0.25000 22 -0.30000 23 -0.35000 24 -0.40000 25 -0.45000 ............
defaults used
Edge = 5.60 Contour 3D Psi = 9
M O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E N
CONTOUR VALUE
1 0.00000 2 0.05000 3 0.10000 4 0.15000 5 0.20000 6 0.25000 7 0.30000 8 0.35000 9 0.40000 10 0.45000 11 0.50000 12 0.55000 13 0.60000 14 -0.05000 15 -0.10000 16 -0.15000 17 -0.20000 18 -0.25000 19 -0.30000 20 -0.35000 21 -0.40000 22 -0.45000 23 -0.50000 24 -0.55000 25 -0.60000
defaults used
Edge = 5.60 Contour 3D Psi = 10
M O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E N
Figura 7.2: Orbitali HOMO e LUMO di HCl, rappresentati come superficie
Alpha occ. eigenvalues -- -20.54532 -1.35692 -0.73115 -0.55177 -0.50132
Alpha virt. eigenvalues -- 0.14513 0.21389 0.58263 0.58295 1.00216
Alpha virt. eigenvalues -- 1.02050 1.15363 1.43225 2.45181 2.51888
Alpha virt. eigenvalues -- 5.30956 5.41111 5.58159 51.43742
Molecular Orbital Coefficients
1 2 3 4 5
(A’)--O (A’)--O (A’)--O (A’)--O (A")--O
EIGENVALUES -- -20.54532 -1.35692 -0.73115 -0.55177 -0.50132
1 1 O 1S 0.55143 -0.11348 0.00000 -0.03853 0.00000
2 2S 0.47167 -0.19044 -0.00001 -0.06523 0.00000
3 2PX 0.00000 -0.00001 0.22078 0.00001 0.00000
4 2PY -0.00246 -0.04002 0.00000 0.25682 0.00000

7.3. ALCUNI ESEMPI DI CALCOLO AB INITIO 111
5 2PZ 0.00000 0.00000 0.00000 0.00000 0.29245
6 3S 0.00558 0.54438 0.00001 0.19936 0.00000
7 3PX 0.00000 -0.00001 0.36854 0.00001 0.00000
8 3PY 0.00090 -0.08843 0.00000 0.38806 0.00000
9 3PZ 0.00000 0.00000 0.00000 0.00000 0.43983
10 4S -0.00020 0.43887 0.00003 0.26427 0.00000
11 4PX 0.00000 0.00000 0.21489 0.00001 0.00000
12 4PY -0.00033 -0.05127 0.00000 0.40111 0.00000
13 4PZ 0.00000 0.00000 0.00000 0.00000 0.48851
14 2 H 1S 0.00020 0.09982 0.16213 -0.08409 0.00000
15 2S -0.00011 0.03908 0.19580 -0.09301 0.00000
16 3S 0.00011 -0.00396 0.04616 -0.03139 0.00000
17 3 H 1S 0.00020 0.09983 -0.16214 -0.08409 0.00000
18 2S -0.00011 0.03908 -0.19580 -0.09299 0.00000
19 3S 0.00011 -0.00396 -0.04616 -0.03138 0.00000
6 7 8 9 10
(A’)--V (A’)--V (A’)--V (A’)--V (A")--V
EIGENVALUES -- 0.14513 0.21389 0.58263 0.58295 1.00216
1 1 O 1S -0.03453 0.00000 0.00154 0.03296 0.00000
2 2S -0.05645 0.00000 0.00269 0.05733 0.00000
3 2PX 0.00001 0.12046 -0.13119 0.00615 0.00000
4 2PY -0.07330 0.00000 0.00447 0.09530 0.00000
5 2PZ 0.00000 0.00000 0.00000 0.00000 -0.24082
6 3S 0.10790 0.00000 -0.00669 -0.14264 0.00000
7 3PX 0.00001 0.13921 -0.19665 0.00922 0.00000
8 3PY -0.12323 0.00000 0.00722 0.15371 0.00000
9 3PZ 0.00000 0.00000 0.00000 0.00000 -0.83803
10 4S 0.88554 -0.00006 -0.05599 -1.19721 0.00000
11 4PX 0.00003 0.51385 -1.21714 0.05718 0.00000
12 4PY -0.20430 -0.00003 0.04192 0.89445 0.00000
13 4PZ 0.00000 0.00000 0.00000 0.00000 1.13618
14 2 H 1S -0.03829 -0.02042 0.08034 0.07574 0.00000
15 2S 0.03062 0.10816 1.59978 1.59260 0.00000
16 3S -0.83695 -1.52605 -0.76977 -0.57995 0.00000
17 3 H 1S -0.03828 0.02041 -0.07289 0.08292 0.00000
18 2S 0.03062 -0.10819 -1.44387 1.73540 0.00000
19 3S -0.83685 1.52615 0.71209 -0.64934 0.00000
11 12 13 14 15
(A’)--V (A’)--V (A’)--V (A’)--V (A’)--V
EIGENVALUES -- 1.02050 1.15363 1.43225 2.45181 2.51888

112 CAPITOLO 7. METODI AB-INITIO
1 1 O 1S 0.01558 0.00000 0.11902 -0.01185 -0.00001
2 2S 0.02682 0.00000 0.25119 -0.01343 -0.00001
3 2PX -0.00002 -0.23607 0.00000 0.00000 -0.19470
4 2PY -0.23105 0.00002 0.03803 -0.14705 -0.00001
5 2PZ 0.00000 0.00000 0.00000 0.00000 0.00000
6 3S -0.08512 0.00001 -1.81879 -0.08442 0.00004
7 3PX -0.00006 -0.90999 0.00000 -0.00004 0.01704
8 3PY -0.84364 0.00007 0.10786 -0.03304 -0.00003
9 3PZ 0.00000 0.00000 0.00000 0.00000 0.00000
10 4S -0.35054 -0.00001 2.82235 -0.48794 -0.00013
11 4PX 0.00011 1.76898 -0.00004 0.00004 0.33600
12 4PY 1.34290 -0.00012 -0.63696 0.36023 0.00005
13 4PZ 0.00000 0.00000 0.00000 0.00000 0.00000
14 2 H 1S 0.04545 -0.03403 -0.00505 -1.09309 1.10715
15 2S 0.24804 -0.21412 -0.59360 1.59577 -1.63769
16 3S 0.09448 -0.74736 -0.36175 -0.48824 0.76446
17 3 H 1S 0.04546 0.03405 -0.00508 -1.09291 -1.10733
18 2S 0.24802 0.21403 -0.59358 1.59555 1.63799
19 3S 0.09456 0.74740 -0.36184 -0.48808 -0.76451
16 17 18 19
(A")--V (A’)--V (A’)--V (A’)--V
EIGENVALUES -- 5.30956 5.41111 5.58159 51.43742
1 1 O 1S 0.00000 0.00427 0.00000 -2.24644
2 2S 0.00000 0.01050 0.00000 2.32902
3 2PX 0.00000 0.00006 -1.28379 0.00000
4 2PY 0.00000 -1.27272 -0.00006 -0.00424
5 2PZ -1.27030 0.00000 0.00000 0.00000
6 3S 0.00000 -0.07359 -0.00001 -0.24329
7 3PX 0.00000 -0.00007 1.49245 0.00000
8 3PY 0.00000 1.40721 0.00007 0.01308
9 3PZ 1.36970 0.00000 0.00000 0.00000
10 4S 0.00000 0.30033 0.00005 0.24052
11 4PX 0.00000 0.00004 -0.83840 -0.00001
12 4PY 0.00000 -0.65361 -0.00003 -0.04421
13 4PZ -0.52370 0.00000 0.00000 0.00000
14 2 H 1S 0.00000 0.21235 -0.28655 0.00635
15 2S 0.00000 -0.31639 0.35306 -0.03858
16 3S 0.00000 0.01468 0.15005 -0.03243
17 3 H 1S 0.00000 0.21233 0.28663 0.00635
18 2S 0.00000 -0.31637 -0.35316 -0.03859

7.3. ALCUNI ESEMPI DI CALCOLO AB INITIO 113
19 3S 0.00000 0.01471 -0.15005 -0.03244
Condensed to atoms (all electrons):
1 2 3
1 O 8.292901 0.261664 0.261662
2 H 0.261664 0.364534 -0.034311
3 H 0.261662 -0.034311 0.364535
Total atomic charges:
1
1 O -0.816227
2 H 0.408113
3 H 0.408114
Sum of Mulliken charges= 0.00000
Atomic charges with hydrogens summed into heavy atoms:
1
1 O 0.000000
2 H 0.000000
3 H 0.000000
Sum of Mulliken charges= 0.00000
Electronic spatial extent (au): <R**2>= 19.0015
Charge= 0.0000 electrons
Dipole moment (Debye):
X= -0.0001 Y= -2.4886 Z= 0.0000 Tot= 2.4886
Quadrupole moment (Debye-Ang):
XX= -3.8663 YY= -6.3645 ZZ= -7.2759
XY= -0.0002 XZ= 0.0000 YZ= 0.0000
Octapole moment (Debye-Ang**2):
XXX= -0.0001 YYY= -1.4797 ZZZ= 0.0000 XYY= 0.0001
XXY= -1.3015 XXZ= 0.0000 XZZ= 0.0000 YZZ= -0.4291
YYZ= 0.0000 XYZ= 0.0000
Hexadecapole moment (Debye-Ang**3):
XXXX= -5.3157 YYYY= -6.1828 ZZZZ= -5.3837 XXXY= 0.0000
XXXZ= 0.0000 YYYX= 0.0000 YYYZ= 0.0000 ZZZX= 0.0000
ZZZY= 0.0000 XXYY= -1.7032 XXZZ= -2.0640 YYZZ= -1.9605
XXYZ= 0.0000 YYXZ= 0.0000 ZZXY= 0.0000
N-N= 9.291362481815D+00 E-N=-1.993505611185D+02 KE= 7.611888714322D+01
Symmetry A’ KE= 7.153312626549D+01
Symmetry A" KE= 4.585760877728D+00
Leave Link 601 at Mon Mar 15 08:37:32 1999, MaxMem= 4000000 cpu: 0.3
(Enter /usr/local/g94/l9999.exe)
Final structure in terms of initial Z-matrix:

114 CAPITOLO 7. METODI AB-INITIO
O
H,1,r2
H,2,r3,1,a3
Variables:
r2=0.94565985
r3=1.56665299
a3=34.06847266
Test job not archived.
1\1\GINC-CFGAMMA\FOpt\RHF\6-311G\H2O1\NINOP\15-Mar-1999\0\\#P TEST OPT
HF/6-311G GFINPUT IOP(6/7=3)\\This Gaussian input file generated by B
abel 1.06e\\0,1\O,-0.0199352024,0.,-0.1040561682\H,-0.6895863854,0.,0.
5636562348\H,0.8490680049,0.,0.2687931105\\Version=SGI-G94RevE.2\State
=1-A’\HF=-76.0109546\RMSD=7.476e-09\RMSF=1.284e-04\Dipole=0.184244,0.,
0.9615714\PG=CS [SG(H2O1)]\\@
AND ALL THIS SCIENCE I DON’T UNDERSTAND
IT’S JUST MY JOB FIVE DAYS A WEEK
-- ELTON JOHN, "ROCKET MAN"
Job cpu time: 0 days 0 hours 1 minutes 18.3 seconds.
File lengths (MBytes): RWF= 5 Int= 0 D2E= 0 Chk= 2 Scr= 1
Normal termination of Gaussian 94
In entrambi i casi e stata impiegata una base 6-31G; nel caso dell’acido cloridrico il problema e
dunque a 3 (H) + 21 (Cl) funzioni di base (1s, 2s, 3s per l’idrogeno e tutto gli AO di tipo s con n
da 1 a 6 e tutti gli AO di tipo p da 1 a 5 per il Cl). In Fig. (7.2) sono mostrati gli orbitali HOMO e
LUMO per l’acido cloridrico, in corrispondenza della geometria di equilibrio. Le Figg. (7.3) e (7.4)
mostrano gli stessi orbitali, rappresentati sotto forma di superficie aventi uguale valore di densita
di carica. Infine in Figg. (7.5) e (7.6) sono raffigurati gli orbitali HOMO e LUMO ottenuti da un
calcolo RHF per l’acqua, ancora usando una base 6-31G (19 funzioni di base).

7.3. ALCUNI ESEMPI DI CALCOLO AB INITIO 115
defaults used
Edge = 5.60 Space = 0.1000 Psi = 9
M O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E N
Figura 7.3: Orbitale HOMO di HCl, rappresentato come una superficie ad uguale densita di carica
defaults used
Edge = 5.60 Space = 0.1000 Psi = 10
M O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E N
Figura 7.4: Orbitale LUMO di HCl, rappresentato come una superficie ad uguale densita di carica

116 CAPITOLO 7. METODI AB-INITIO
defaults used
Edge = 4.04 Space = 0.3000 Psi = 5
M O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E N
Figura 7.5: Orbitale HOMO di H2O, rappresentato come una superficie ad uguale densita di carica
defaults used
Edge = 4.04 Space = 0.1000 Psi = 6
M O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E NM O L D E N
Figura 7.6: Orbitale LUMO di H2O, rappresentato come una superficie ad uguale densita di carica

Capitolo 8
Teoria dei gruppi di simmetria ed
orbitali molecolari
8.1 Gruppi di simmetria puntuale
Un gruppo G e un insieme di elementi in cui e assegnata una operazione - detta legge di composizione,
e che denotiamo per ora con - che permette di associare ad ogni coppia di elementi x, y ∈ G un
elemento x y ∈ G, e che gode delle seguenti proprieta:
Associativa Per ogni terna x, y, z ∈ G vale che
(x y) z = x (y z) (8.1)
Elemento neutro Esiste un elemento e ∈ G tale che
e x = x e = x (8.2)
Inverso Per ogni x ∈ G esiste un elemento y ∈ G tale che
x y = y x = e (8.3)
indichiamo l’inverso di x con x−1.
Se il gruppo G gode della proprieta aggiuntiva
Commutativa Per ogni coppia x, y ∈ G vale che
x y = y x (8.4)
117

118 CAPITOLO 8. TEORIA DEI GRUPPI DI SIMMETRIA ED ORBITALI MOLECOLARI
allora si dice abeliano o commutativo.
Esercizio 5. Alcuni esempi di gruppi: l’insieme delle permutazioni dei nuclei di una molecola
P rispetto all’operazione di composizione (moltiplicazione delle permutazioni); l’insieme di tutte
le matrici m × n sul corpo di numeri K rispetto all’addizione; l’insieme dei numeri pari rispetto
all’operazione di addizione.
Alcune definizioni utili relative all’algebra dei gruppi saranno impiegate in seguito. I gruppi
sono infiniti o finiti, se il numero dei loro elementi e finito o infinito. L’ordine h di un gruppo
finito e il numero di elementi costituenti il gruppo. Definiamo tavola di moltiplicazione o tabella
di un gruppo finito la matrice che riassume il risultato dell’operazione del gruppo tra ogni coppia
di elementi del gruppo. Definiamo un sottogruppo come un sottoinsieme del gruppo che gode delle
proprieta di gruppo. Diciamo gruppo ciclico quel gruppo costituito da e, un elemento a e da tutte
le potenze con esponente 0 ≤ n ≤ h dell’elementoa, vale a dire a a . . . a (n volte). Definiamo
infine il concetto di classe:
• due elementi a e b di un gruppo G si dicono coniugati quando esiste un terzo elemento del
gruppo x tale che b = x−1 a x; si noti che ogni elemento e coniugato di se stesso e che se a
e coniugato a b e b e coniugato a c, allora a e coniugato a c.
• il sottoinsieme di G che contiene tutti gli elementi coniugati fra loro e una classe di G.
Nel seguito ci occuperemo dei gruppi formati dalle operazioni di simmetria di un corpo rigido:
gruppo di simmetria: l’insieme di operazioni di simmetria relative ad un corpo rigido, consider-
ato come un insieme di punti materiali verifica le condizioni di gruppo, rispetto all’operazione
di composizione (applicazione successiva) di due operazioni di simmetria.
In generale si definisce operazione di simmetria una trasformazione delle coordinate dell’insieme di
punti materiali che porta ogni punto in un punto equivalente. Nel caso di un sistema molecolare
l’insieme dei punti materiali coincide con i nuclei. Un’operazione di simmetria e definita rispetto
ad elemento di simmetria, che e un ente geometrico (punto, retta o piano) rispetto al quale possono
essere eseguite una o piu operazioni di simmetria. La composizione, cioe l’applicazione successiva
di due operazioni A1 e A2 (prima A1 e dopo A2) si indica con A2A1.
Nel seguito considereremo i soli gruppi di simmetria puntuali, vale a dire quei gruppi di operazioni
di simmetria che conservano la posizione di un punto, comune a tutte le operazioni del gruppo, e
non tratteremo dei gruppi spaziali, che includono operazioni di traslazione e non conservano percio
alcun punto fermo. I gruppi spaziali sono necessari per la comprensione delle simmetrie presenti
nei reticoli cristallini.

8.1. GRUPPI DI SIMMETRIA PUNTUALE 119
8.1.1 Elementi ed operazioni di simmetria
Nella trattazione della simmetria molecolare e necessario considerare solo quattro tipi di elementi
di simmetria e le relative operazioni di simmetria. Nel seguito useremo per chiarezza il carattere
tipografico per indicare un elemento di simmetria, distinguendolo dalle operazioni di simmetria da
esso generate. Assumiamo per semplicita che un sistema di riferimento xyz sia definito in modo
solidale con la molecola (sistema di riferimento molecolare).
identita: e l’operazione E che lascia invariata la configurazione del sistema molecolare
rotazione propria: un asse proprio di simmetria Cn e associato ad un’operazione di rotazione
propria, ovvero ad una rotazione in senso orario di un angolo 2π/n attorno all’asse dove n
e il maggior intero che genera una configurazione equivalente. Un asse di simmetria C3 per-
mettera di definire le operazioni C3 (rotazione di 2π/3), C23 (rotazione di 4π/3) e C3
3 = E
(rotazione di 2π, cioe l’identita). Facendo coincidere l’asse di simmetria con l’asse z moleco-
lare, le componenti di un generico vettore ~R saranno ruotate dall’operazione Cn secondo la
trasformazione
x′
y′
z′
=
cos 2π/n − sin 2π/n 0
sin 2π/n cos 2π/n 0
0 0 1
x
y
z
(8.5)
riflessione: un piano di simmetria molecolare σ permette di definire una operazione di riflessione
σ. Si noti che σ2 = E. Parliamo di operazioni di simmetria σv se sono associate a piani di
simmetria che contengono l’asse principale di rotazione del sistema molecolare. Se l’asse del
piano di simmetria forma un angolo φ con l’asse x del sistema molecolare le componenti di un
vettore generico sono trasformate come
x′
y′
z′
=
cos 2φ sin 2φ 0
sin 2φ − cos 2φ 0
0 0 1
x
y
z
(8.6)
Parliamo di operazioni di simmetria σh se sono associate al piano di simmetria xy perpen-
dicolare all’asse principale di rotazione del sistema molecolare; le componenti di un vettore
generico sono trasformate come
x′
y′
z′
=
1 0 0
0 1 0
0 0 −1
x
y
z
(8.7)

120 CAPITOLO 8. TEORIA DEI GRUPPI DI SIMMETRIA ED ORBITALI MOLECOLARI
inversione: un centro di inversione i permette di definire l’operazione di inversione i. Iterando i
per n volte con n pari si ottiene l’identita, con n dispari si riottiene i. Se poniamo i nell’origine
del sistema del sistema molecolare le componenti di un vettore generico sono trasformate come
x′
y′
z′
=
−1 0 0
0 −1 0
0 0 −1
x
y
z
(8.8)
rotazione impropria una rotazione impropria Sn di ordine n e costituita da una rotazione propria
e da una riflessione rispetto ad un piano perpendicolare all’asse di rotazione (indipendente-
mente dall’ordine di esecuzione). L’asse di rotazione impropria si indica con Sn. Se esistono
un asse di rotazione propria e un piano di riflessione perpendicolare esiste anche l’asse di ro-
tazione impropria, ma puo anche darsi l’esistenza di Sn senza Cn e σh. Per un vettore generico,
la rotazione impropria si puo scrivere come
x′
y′
z′
=
cos 2π/n − sin 2π/n 0
sin 2π/n cos 2π/n 0
0 0 −1
x
y
z
(8.9)
Casi particolari sono dati dalla rotazione impropria di ordine 1, che coincide con σh, S2 = σh e
dalla rotazione impropria di ordine 2, che coincide con l’inversione, S2 = i. Un asse improprio
di ordine n genera le operazioni Sn, S2n, . . . , Sn
n ; per n pari Smn = Cm
n se m e pari, quindi
per esempio per n pari Snn = Cn
n = E. Viceversa, per n dispari Snn = σh. Si noti che per n
dispari l’esistenza di Sn implica l’esistenza di σh e Cn. Un sistema molecolare che non possiede
nessun asse di simmetria improprio e dissimetrica, cioe non puo essere sovrapposta alla sua
immagine speculare (vale a dire, o ha solo assi di rotazione propria o non ha alcun elemento
di simmetria).
8.1.2 Gruppi puntuali
Possiamo classificare i vari gruppi puntuali di simmetria considerando i vari possibili insiemi completi
di operazioni di simmetria. I gruppi di simmetria sono denominati secondo due nomenclature
fondamentali, basate sulla notazione di Schœnflies ed internazionale. Nel seguito adotteremo sempre
la notazione di Schœnflies1.
gruppo C1 E costituito dall’identita E. La molecola non ha elementi di simmetria.
1La notazione di Schœnflies usa nomi simili alle operazioni di simmetria principali del gruppi considerati, cheiniziano per C, da “cyclic”, D, da “dyhedral”, S, da “spiegel” con l’aggiunta di apici quali h, v e d rispettivamenteda “horizontal”, “vertical” e “diagonal”.

8.2. RAPPRESENTAZIONI 121
gruppi Cs e Ci E presente solo un piano di riflessione σ o solo un centro di inversione i. Sono
entrambi gruppi ciclici di ordine 2.
gruppi Cn E presente un asse proprio Cn, che genera le operazioni Cn, C2n, . . . , Cn
n = E. Sono
gruppi ciclici di ordine n.
gruppi Sn E presente un asse improprio di ordine n pari. Sono gruppi ciclici di ordine n. S2 e
equivalente a i.
gruppi Dn E presente un asse proprio Cn ed n assi di rotazione binaria perpendicolari. L’ordine
e 2n.
gruppi Cnh Sono presenti un asse proprio Cn ed il piano di riflessione σh. L’ordine e 2n. Cor-
rispondono ai gruppi Sn con n dispari.
gruppi Cnv Sono presenti un asse proprio Cn ed n piani verticali di riflessione σv. L’ordine e 2n.
gruppi Dnh Sono presenti un asse proprio Cn, n assi di rotazione binaria perpendicolari ed il piano
di riflessione σh. L’ordine e 4n.
gruppi Dnd Sono presenti un asse proprio Cn, n assi di rotazione binaria perpendicolari ed n piani
diedri σd, ovvero piani di riflessione che bisecano gli angoli tra coppie adiacenti di assi binari.
L’ordine e 4n.
gruppi C∞v e D∞h Sono gruppi infiniti relativi a molecole lineari eteronucleari, C∞v, con un asse
C∞ ed infiniti assi binari perpendicolari; e molecole lineari omonucleari, D∞h, con un asse C∞,
infiniti assi binari perpendicolari ed un piano di riflessione orizzontale.
gruppi dei solidi platonici Sono i gruppi ad elevata simmetria dei cinque solidi platonici: tetrae-
dro (Td, ordine 24), cubo ed ottaedro (Oh, ordine 48), dodecaedro e icosaedro (Ih, ordine 120).
8.2 Rappresentazioni
Consideriamo un gruppo di simmetria puntuale G ed un insieme di funzioni fi tali che applicando
una qualunque delle operazioni di simmetria R ad una di esse si ottiene una combinazione lineare
delle funzioni stesse:
Rfi =g∑
k=1
Rkifk (8.10)
Se non e possibile trovare un insieme di combinazioni lineari di m tra le funzioni fi, con m < g,
diciamo che le funzioni fi formano una rappresentazione irriducibile di dimensione g del gruppo
G. Altrimenti si parla di rappresentazione riducibile. Data una rappresentazione di dimensione g,

122 CAPITOLO 8. TEORIA DEI GRUPPI DI SIMMETRIA ED ORBITALI MOLECOLARI
ad ogni operazione R e associabile la matrice R con elementi Rki. Il carattere χ(R) di una oper-
azione R rispetto ad una data rappresentazione e la traccia della matrice associata. Nel caso di una
rappresentazione (irriducibile, naturalmente) monodimensionale i caratteri sono tutti pari a +1 o
−1. In generale una rappresentazione e descritta da una tabella dei caratteri che elenca i caratteri
associati ad ogni operazione di simmetria. Le rappresentazioni irriducibili monodimensionali, bidi-
mensionali, tridimensionali sono denominate con lettere e numeri in considerazione del carattere di
alcune operazioni di simmetria secondo la seguente nomenclatura
• A e B indicano rappresentazioni monodimensionali); E bidimensionali; T tridimensionali
• gli indici 1 e 2 si aggiungono ad A o B per indicare che il carattere di ua rotazione relativa ad
un asse C2 perpendicolare all’asse principale e +1 o −1.
• gli indici ′ e ′′ si aggiungono ad tutti simboli per indicare che il carattere di una riflessione
relativa ad un piano σh e +1 o −1.
• gli indici g (da “gerade”) e u da (“ungerade”) si aggiungono a tutti i simboli per indicare che
il carattere di un’inversione relativa ad un centro σh e +1 o −1.
Classificazioni piu complesse con indici numerici, sono definite per rappresentazioni E, T e cosı
via. Il numero ed il tipo di rappresentazioni irriducibili distinte di un dato gruppo puntuale e una
proprieta univocamente definita del gruppo stesso. Data una qualunque rappresentazione, o e ir-
riducibile, o e possibile ridurla ad una somma di rappresentazioni irriducibili, tra quelle ammesse
dal gruppo. La rappresentazione monodimensionale con caratteri tutti uguali a 1 e detta rappre-
sentazione totalsimmetrica del gruppo.
Consideriamo nel seguito due rappresentazioni, di dimensione gi e gj. Vale il seguente teorema
generale:
Teorema di ortogonalita Se h e l’ordine del gruppo, e gi, gj le dimensioni di due rappresentazioni,
allora
∑
R
R(i)mn
∗R
(j)m′n′ =
h√gigj
δijδmm′δnn′ (8.11)
dove con R(i)mn indichiamo l’elemento mn della matrice rappresentativa dell’operazione R rel-
ativo alla rappresentazione i-esima.
Da questo teorema generale discendono varie conseguenze, di importanza applicativa, che si possono
riassumere nei seguenti tre vincoli
1. La somma dei quadrati delle dimensioni delle rappresentazioni irriducibili di un gruppo e
uguale all’ordine del gruppo

8.2. RAPPRESENTAZIONI 123
2. In una data rappresentazione, i caratteri di tutte le operazioni appartenenti alla stessa classe
hanno lo stesso carattere.
3. Il numero delle rappresentazioni irriducibili di un gruppo e uguale al numero di classi del
gruppo.
Date due rappresentazioni irriducibili, vale il seguente teorema di ortogonalita derivato direttamente
dal precedente teorema generale sommando i termini diagonali delle matrici rappresentativi delle
operazioni:
Teorema di ortogonalita delle rappresentazioni Se gi, gj sono le dimensioni di due rappre-
sentazioni irriducibili, allora
∑
Rχ(i)(R)
∗χ(j)(R) = gigjδij (8.12)
per i = j ritroviamo il primo vincolo. Data una rappresentazione riducibile, possiamo scrivere per
ogni operazione
χ(r)(R) =∑
i
aiχ(i)(R) (8.13)
ed utilizzando il precedente teorema di ortogonalita possiamo ricavare i coefficienti ai che per-
mettono di sviluppare la r-esima rappresentazione riducibile come una combinazione lineare delle
rappresentazioni irriducibili; moltiplicando per χ(j)(R)∗
entrambi i membri e sommando su tutte le
operazioni R si ottiene
aj =1
h
∑
Rχ(j)(R)
∗χ(r)(R) (8.14)
Noti i caratteri di una rappresentazione riducibile, e la tabella dei caratteri del gruppo di sim-
metria puntuale, e dunque possibile calcolare il coefficiente della rappresentazione irriducibile in
analogia al calcolo della componente di Fourier di un elemento generico di uno spazio vettoriale
(la rappresentazione riducibile) rispetto ad un elemento della base ortogonale (la rappresentazione
irriducibile).
Un utile concetto per la costruzione di rappresentazioni di gruppi di simmetria e dato dal prodotto
diretto di due rappresentazioni. Siano αi le funzioni che definiscono una data rappresentazione e βi
quelle che definiscono un’altra rappresentazione. Allora si puo costruire la rappresentazione prodotto
diretto come l’insieme di tutti i prodotti αiβj. Se R(α)mn e R
(β)m′n′ sono le matrici rappresentative di una
generica operazione R del gruppo, allora la matrice rappresentativa nella rappresentazione prodotto
diretto e data come
R(αβ)mm′,nn′ = R(α)
mnR(β)m′n′ (8.15)
che e il prodotto di Kronecker delle due matrici Rα e Rβ; formalmente si scrive anche R(αβ) =
Rα ⊗ Rβ. Si noti che χ(αβ)(R) = χ(α)(R)χ(β)(R), e che in generale la rappresentazione prodotto
diretto, anche di due rappresentazioni irriducibili, e riducibile.

124 CAPITOLO 8. TEORIA DEI GRUPPI DI SIMMETRIA ED ORBITALI MOLECOLARI
8.2.1 Operatori di proiezione
Data un determinato insieme di funzioni che costituisca una rappresentazione di un gruppo, e utile
disporre di un metodo sistematico per costruire le rappresentazioni irriducibili contenute. Uno
strumento formale molto utile e l’operatore di proiezione P(i) che proietta la base per la i-esima
rappresentazione irriducibile del gruppo, e che ha la forma
P(i) =gi
h
∑
Rχ(i)(R)
∗R (8.16)
L’utilita dell’operatore di proiezione e costituita dal fatto che si possono costruire basi per rap-
presentazioni irriducibili in modo semplice una volta nota basi per rappresentazioni irriducibili.
Consideriamo per esempio il caso del gruppo C3v, che ammette le rappresentazioni irriducibili A1,
A2 ed E. Costruiamo i tre operatori di proiezione
PA1 =1
6
(E + C3 + C2
3 + σ1 + σ2 + σ3
)(8.17)
PA2 =1
6
(E + C3 + C2
3 − σ1 − σ2 − σ3
)(8.18)
PE =2
3
(2E − C3 − C2
3
)(8.19)
Prendiamo in esame la funzione x2. Applicando gli operatori precedenti otteniamo:
PA1x2 ∝ x2 + y2 (8.20)
PA2x2 ∝ 0 (8.21)
PEx2 ∝ x2 − y2 (8.22)
dalla tabella del gruppo C3v troviamo infatti che x2 + y2 e base per la rappresentazione irriducibile
A1, mentre x2 − y2 e una delle funzioni di base per la rappresentazione irriducibile bidimensionale
E. La funzione x2 non ha componenti appartenenti alla rappresentazione irriducibile A2.
8.3 Applicazioni al calcolo di orbitali molecolari
L’applicabilita della teoria dei gruppi puntuali al calcolo di orbitali molecolari e evidente nel caso
del calcolo di elementi di matrice di operatori che condividono la simmetria del gruppo di punti di
un sistema molecolare.
Consideriamo un operatore F che sia invariante rispetto alle operazioni di simmetria di un
gruppo puntuale e calcoliamo l’elemento di matrice
〈φ1|F|φ2〉 =∫
dqφ∗1Fφ2 (8.23)
Il valore dell’integrale e zero a meno che la funzione integranda sia invariante rispetto a tutte le
operazioni di simmetria del gruppo, o se almeno una sua parte sia invariante, cioe se costituisce

8.3. APPLICAZIONI AL CALCOLO DI ORBITALI MOLECOLARI 125
o contiene una base per la rappresentazione totalsimmetrica del gruppo. Poiche l’operatore F e
invariante rispetto a tutte le operazioni del gruppo, l’integrale e diverso da zero se la funzione
φ∗1φ2 appartiene alla rappresentazione totalsimmetrica. Possiamo considerare la funzione prodotto
come un elemento della rappresentazione prodotto diretto α di due rappresentazioni α1 e α2, cui
appartengono rispettivamente φ∗1 e φ2. Dalle proprieta delle rappresentazioni prodotto diretto,
discende che α contiene la rappresentazione totalsimmetrica solo α1 = α2. Infatti la componente
aA della rappresentazione totalsimmetrica contenuta in α e
aA =1
h
∑
Rχ(α)(R) =
1
h
∑
Rχ(α1)(R)
∗χ(α2)(R) = δα1α2 (8.24)
Il calcolo degli elementi di matrice dell’operatore hamiltoniano o dell’operatore di Fock puo percio
essere notevolmente semplificati se si scelgono funzioni di base raggruppate secondo la loro apparte-
nenza alle rappresentazioni irriducibili del gruppo di simmetria molecolare. Combinazioni lineari di
orbitali determinate secondo questo criterio si dicono orbitali adattati per simmetria.
La simmetria e infine conservata nella definizione degli autostati degeneri di un hamiltoniano.
Nel caso infatti un hamiltoniano ammetta autostati degeneri e possibile dimostrare che
Teorema Ogni autofunzione di un hamiltoniano di un sistema quantomeccanico corrisponde ad
una rappresentazione irriducibile del gruppo di simmetria cui appartiene il sistema, vale a
dire ogni insieme di autofunzioni relative ad un autovalore degenere g volte e base per una
rappresentazione irriducibile di dimensione g.
8.3.1 Atomi
Consideriamo un sistema costituito da un solo nucleo. Il gruppo di simmetria di appartenenza e il
gruppo infinito delle rotazioni nello spazio R3, in cui la rotazione di un angolo qualunque attorno
ad un asse passante per il nucleo lascia il sistema invariato. Consideriamo ora le funzioni armoniche
sferiche Ylml(θ, φ) per un dato valore di l: poiche per definizione una rotazione applicata ad una
di queste 2l + 1 funzioni genera una loro combinazione lineare, costituiscono una base - che si puo
verificare essere irriducibile - del gruppo R3. Gli autostati dell’hamiltoniano atomico saranno percio
almeno 2l + 1 degeneri e le autofunzioni avranno la forma generica
φ(r, θ, φ) = R(r)Ylml(θ, φ) (8.25)
dove R(r) e invariante per simmetria rotazione, quindi funzione del modulo r. I vari livelli energetici
s, p, d sono dunque coincidenti con rappresentazioni irriducibili monodimensionali, tridimensionali,
pentadimensionali del gruppo R3.

126 CAPITOLO 8. TEORIA DEI GRUPPI DI SIMMETRIA ED ORBITALI MOLECOLARI
8.3.2 Molecole lineari omonucleari
Nel caso di molecole lineari omonucleari il gruppo di simmetria di appartenenza e, come abbiamo
visto, D∞h. Lo stato fondamentale di una molecola lineare omonucleare apparterra alla rappre-
sentazione irriducibile monodimensionale totalsimmetrica di D∞h, detta Σ+g (l’apice + proviene dal
fatto che χ(σv) = 1, mentre il pedice g e dovuto aχ(i) = 1). Lo stato successivo, primo singoletto
eccitato, appartiene ancora ad un rappresentazione irriducibile monodimensionale, Σ+u , antisim-
metrica all’inversione. Continuando in questo modo possiamo classificare gli autostati degeneri
appartenenti ad altre rappresentazioni, e.g. Πu, bidimensionale. Nella costruzione degli orbitali di
simmetria adattati per il calcolo HF della molecola di idrogeno la scelta intuitiva di φg ∝ 1sA +1sB
e φu ∝ 1sA − 1sB si puo quindi razionalizzare come la definizione di due funzioni di base rispetti-
vamente per Σ+g e Σ+
u .
8.3.3 L’acqua
Consideriamo la base minimale per una calcolo HF relativo alla molecola di acqua, che appartiene
al gruppo C2v: 1s per ciascun H e 1s,2s,2px,y,z per O. Gli AO si possono classificare facilmente
secondo le rappresentazioni irriducibili di C2v
A1: 1s, 2s, 2pz, (1sH1 + 1sH2)/√
2
B1: 2px
B2: 2py, (1sH1 − 1sH2)/√
2
Il calcolo HF e dunque fattorizzato in un blocco 4 × 4, in uno monodimensionale ed in uno 2 × 2.
Lo stato fondamentale della molecola, che appartiene alla rappresentazione totalsimmetrica A1 di
C2v ha la seguente configurazione: (1a1)2(2a1)
2(1b2)2(3a1)
2)(1b1)2 dove 1a1, 2a1 etc. sono orbitali
atomici calcolati appartenenti ad A1 e cosı via: la procedura HF genera percio tre orbitali reali ed
uno virtuale appartenenti alla A1, un orbitale reale appartenente alla B1, un orbitale reale ed uno
virtuale appartenente alla B2.
8.3.4 Un esempio non triviale: il benzene
Discutiamo la scelta di un insieme di orbitali atomici SALC (Symmetry Adapted Linear Combina-
tion) per il benzene C6H6. La base minima per una calcolo HF e data da 36 orbitali atomici: sei di
tipo 1s per ciascun idrogeno e sei insiemi 1s, 2s, 2p per ciascun carbonio. Nel caso degli orbitali 2p
giacenti nel piano dell’anello, definiamo per ogni carbonio un sistema di riferimento locale, orientati
in maniera tale da essere equivalenti per rotazione intorno all’asse di simmetria C6 del benzene,
che appartiene al gruppo di simmetria D6h: per ogni carbonio scegliamo poi due opportune com-
binazioni lineari degli orbitali 2px e 2py che sono equivalenti per rotazione. Gli AO cosı definiti

8.3. APPLICAZIONI AL CALCOLO DI ORBITALI MOLECOLARI 127
si possono raggruppare secondo la tabella 8.1. Cosı per esempio si possono costruire quattro or-
rappresentazione irriducibile tipo di funzioni di base numero di MO
A1g 1sH , 1s, 2s, 2px,y 4
B1u 1sH , 1s, 2s, 2px,y 4
E1u 1sH , 1s, 2s, 2px,y 10
E2g 1sH , 1s, 2s, 2px,y 10
A2u 2pz 1
B2g 2pz 1
E1g 2pz 2
E2u 2pz 2
B2u 2px,y 2
Tabella 8.1: Rappresentazioni irriducibili e AO di base del benzene
bitali molecolari appartenenti alla rappresentazione irriducibile A1g che coinvolgono gli orbitali degli
idrogeni, gli orbitali 1s, 2s e 2p nel piano dei carboni, quattro orbitali molecolari appartenenti alla
rappresentazione irriducibile B1u che coinvolgono gli orbitali degli idrogeni, gli orbitali 1s, 2s e 2p
nel piano dei carboni etc. La matrice rappresentativa dell’operatore di Fock e percio fattorizzabile
in due blocchi 4 × 4, due 10 × 10 ed uno 2× che si riferiscono ad orbitali molecolari coinvolgenti
legami nel piano dell’anello (A1g, B1u, E1u, E2u e B2u) e due blocchi 1× 1 e due blocchi 2× 2 che si
riferiscono ad orbitali molecolari coinvolgenti legami perpendicolari al piano dell’anello (A2u, B2g,
E1g, E2u).

128 CAPITOLO 8. TEORIA DEI GRUPPI DI SIMMETRIA ED ORBITALI MOLECOLARI
6
z
y
x
1
2
3
4
5
Figura 8.1: Set di orbitali 2p nel piano per il benzene

Capitolo 9
Metodi semiempirici
9.1 Metodi semiempirici
Come e stato gia accennato nell’introduzione, i vari metodi semiempirici comportano l’introduzione
di approssimazioni e semplificazioni nella forma degli hamiltoniani elettronici e nella definizione degli
integrali mono e multi-centro che devono essere valutati nel corso di un calcolo esatto. E abbastanza
difficile dare delle classificazioni generali dei vari metodi semiempirici. Di conseguenza discuteremo
il piu semplice (e forse, malgrado tutto, ancora piu usato) schema semiempirico, l’approssimazione di
Huckel, come un esempio particolare degli schemi generali di approssimazione tipici di molti metodi
semiempirici. Questa Sezione si concludera poi con un richiamo alla classe di metodi semiempirici
del tipo NDO.
9.1.1 Metodo di Huckel
Il metodo di Huckel e applicabile a molecole organiche, nelle quali sia possibile separare una classe
di elettroni relativamente localizzati ed inattivi, tipicamente impegnati in legami σ, e un’insieme di
elettroni delocalizzati e reattivi, impegnati in legami π. La natura di questa identificazione puo non
essere affatto chiara, anche se nei casi piu comuni (p. es. l’insieme di legami coniugati nei sistemi
aromatici) risulta essere abbastanza ovvia. Le classi piu comuni di molecole coinvolte sono:
• Polieni lineari: es. 1,3-butadiene;
• idrocarburi aromatici: benzene, naftalene;
• etero-molecole aromatiche: piridina, furano, acroleina;
Il sistema piu semplice e il radicale planare CH3·
129

130 CAPITOLO 9. METODI SEMIEMPIRICI
C
2pz
H
H
H
Figura 9.1: Radicale CH3·
(1s)2(Ω1)2(Ω2)
2(Ω3)2(2pz)
Ωi = 1sH + λsp2
(1s)2 : inner shell
(Ωi)2 : elettroni σ
(2pz) : elettrone π
Un tentativo di sistematizzazione dell’approssimazione di Huckel e stato dato da Likos e Parr
nell’ambito di un formalismo di partenza di tipo Hartree-Fock (vedi Appendice E). Sotto le con-
dizioni di separabilita σ − π si puo separare il calcolo della parte della funzione d’onda riferita ai
soli orbitali π. Le equazioni di Roothaan per l’hamiltoniano π−HF:
FπC = ∆Cε (9.1)
generano il seguente problema agli autovalori per la determinazione dei livelli energetici:
|Fπ − ε∆| = 0 (9.2)
Come e tipico di quasi tutte le metodologie di calcolo semiempiriche, il passo successivo e dato dalla
parametrizzazione degli elementi di matrice:
∆rs = δrs (9.3)
Frr = αr (9.4)
Frs =
βr atomi primi vicini
0 altrimenti(9.5)
αr e l’integrale di Coulomb, βr e l’integrale di risonanza. In assenza di eteroatomi:
αr = α (9.6)
βr = β (9.7)

9.1. METODI SEMIEMPIRICI 131
Ponendo x = (α− ε)/β, il calcolo e ricondotto alla diagonalizzazione della matrice topologica A:
|A− x1| = 0 (9.8)
dove Ars e 1 per atomi primi vicini, 0 altrimenti.
4
2p 2p
2p 2p
z z
z z
C
C
CC
1
2
3
Figura 9.2: Esempio: 1,3-butadiene.
Consideriamo p. es. il caso dell’1,3-butadiene. La matrice topologica e data da:
A =
0 1 0 0
1 0 1 0
0 1 0 1
0 0 1 0
(9.9)
Il gruppo di simmetria a cui appartiene il trans-1,3-butadiene e il C2h.
C2h E C2 i σh
Ag 1 1 1 1 Rz , x2, y2, z2, xy
Au 1 1 -1 -1 z
Bg 1 -1 1 -1 Rx, Ry, xz, yz
Bu 1 -1 -1 1 x, y
Tabella 9.1: Tabella dei caratteri del gruppo C2h
Possiamo percio utilizzare la simmetria delle funzioni di base nel gruppo C2h per semplificare il
calcolo. La rappresentazione riducibile e 2Au ⊕ 2Bg. I livelli energetici calcolati corrispondono a

132 CAPITOLO 9. METODI SEMIEMPIRICI
R Rφ1 Rφ2 Rφ3 Rφ4
E φ1 φ2 φ3 φ4
C2 φ4 φ3 φ2 φ1
i −φ4 −φ3 −φ2 −φ1
σh −φ1 −φ2 −φ3 −φ4
Tabella 9.2: Funzioni φi in C2h
due funzioni d’onda au e 2 funzioni d’onda bg:
ε(2bg) = α− 1.618β
ε(2au) = α− 0.618β
ε(1bg) = α + 0.618β
ε(1au) = α + 0.618β
la configurazione dello stato fondamentale e (1au)2(1bg)
2 =1 Ag Se si fosse partiti dall’isomero cis i
risultati finali non sarebbero stati diversi (poiche la matrice topologica dipende solo dalla struttura
topologica, cioe dalle connessioni tra atomi, e non dalla loro disposizione nello spazio), anche se si
sarebbe dovuto impiegare, per sfruttare proprieta di simmetria, il gruppo C2v.
9.1.2 Neglect-of-differential-overlap (NDO)
Concludiamo questa Sezione con un cenno alla classe di metodi semiempirici del tipo NDO. Il
differential overlap tra due funzioni atomiche φk e φl e definito come la probabilita di trovare
l’elettrone i-esimo in un elemento di volume comune:
dkl = φk(i)φl(i) (9.10)
L’approssimazione zero-differential-overlap (ZDO) consiste nel porre:
dkl = dkkδkl (9.11)
L’approssimazione ZDO comporta varie conseguenze nella valutazione dei gli integrali di overlap,
di Coulomb etc.:
Skl = 〈φk|φl〉 ∝ δkl overlap (9.12)
Nn = Zn〈φk| 1rni|φl〉 ∝ δkl interazione elettrone-nucleo (9.13)
J,K = 〈φk(i)φm(j)| 1rij|φl(i)φn(j)〉 ∝ δklδmn scambio e repulsione (9.14)
Lascia pero inalterati gli integrali che coinvolgono operatori differenziali:
Ii = 〈φk|∇2i |φl〉 energia cinetica (9.15)

9.1. METODI SEMIEMPIRICI 133
I vari metodi semiempirici implicano sia regole di eliminazione o semplificazione degli integrali
multicentro che di parametrizzazione di alcuni o tutti gli integrali rimanenti. I parametri ottimiz-
zabili del calcolo sono di solito gli integrali a 1 elettrone e parti degli integrali a 2 elettroni (nel
caso precedente dell’approssimazione di Huckel i numeri α e β). I vari schemi NDO corrispondono
a diverse parametrizzazioni, fatta salva la comune applicazione a diversi livelli di sofisticazione,
dell’approssimazione NDO.
• Complete NDO; Pople et al.; adotta l’approssimazione ZDO
– CNDO/1
– CNDO/2
• Intermediate NDO; adotta l’approssimazione ZDO solo per gli integrali a 2 centri
• Modified NDO; Pople et al.; INDO con una parametrizzazione piu complessa
– MNDO/1
– MNDO/2
– MNDO/3
• Partial Neglect of Diatomic DO; MNDO solo per orbitali su centri diversi.
Il metodo MNDO e stato utlizzato per calcolare calori di formazione, ottimizzare geometrie moleco-
lari, calcolare potenziali di ionizzazione e momenti di dipolo, rivelandosi utile soprattutto per idro-
carburi ciclici, ma dimostrando per esempio di non essere in grado di descrivere quantitativamente
interazioni dovute a legami idrogeno. Metodi semiempirici piu sofisticati sono ormai disponibili da
molti anni in letteratura: ricordiamo tra gli altri i metodi PM3 e AM1.

134 CAPITOLO 9. METODI SEMIEMPIRICI

Appendice A
L’atomo idrogenoide
L’hamiltoniano in unita atomiche e:
H = − 1
2mZ
∇2
R −1
2∇2
e −Z
|~R− ~re|(A.1)
dove indichiamo con ~R e ~re la posizione del nucleo e dell’elettrone, e con ∇2
R e ∇e i relativi gradi-
enti. Possiamo procedere come nell’Esercizio 1 ed introdurre le variabili di spostamento relativo
dell’elettrone e di centro di massa:
~r = ~re − ~R (A.2)
~rc =~re + mZ
~R
M(A.3)
dove M = 1 + mZ e la massa totale del sistema. Rispetto alle nuove coordinate l’hamiltoniano
risulta definito come la somma dell’hamiltoniano relativo al moto libero del baricentro con massa
M e dell’hamiltoniano relativo al moto dell’elettrone rispetto ad un nucleo a riposo, ed avente massa
ridotta µ = mZ/M in unita atomiche. In unita atomiche l’hamiltoniano separato risulta essere:
H = − 1
2µ∇2 − 1
2M∇2
c −Z
r(A.4)
dove ora r e la distanza tra il baricentro e l’elettrone. La parte relativa al baricentro determina il
moto traslazionale libero dell’atomo, e non sara considerata oltre. La parte elettronica puo essere
scritta come l’hamiltoniano di una particella di carica -1 che si muova nel campo di potenziale
elettrostatico generato da un nucleo immobile di carica Z, e avente massa µ:
H = −Z
r− 1
2µ∇2
(A.5)
Nel seguito considereremo µ ≈ 1: per Z = 1, assumendo per mZ la massa di un protone avremmo
µ = 0.9994. Il potenziale e a simmetria radiale. Possiamo percio procedere risolvendo il sistema nel
seguente modo i) introduzione di coordinate sferiche (r, θ, φ); ii) fattorizzazione della funzione d’onda
135

136 APPENDICE A. L’ATOMO IDROGENOIDE
in una parte angolare dipendente da θ e φ e in una parte radiale dipendente da r; iii) imposizione
di condizioni al contorno per il calcolo delle autoenergie. La parte angolare dell’autofunzione e data
dalle funzioni armoniche sferiche (cio e vero per qualunque potenziale a simmetria sferica)
ΨE(r, θ, φ) = R(r)Ylml(θ, φ) (A.6)
Sostituendo nell’Eq. di Schrodinger si trova l’equazione per la parte radiale:
1
r2
d
drr2dR
dr+ 2
[E +
Z
r− l(l + 1)
2r2
]R = 0 (A.7)
Lo spettro degli autovalori sara discreto per E < 0, e continuo per E > 0 (il potenziale coulombiano
tende a zero per r → ∞ e a −∞ per r → 0). L’equazione differenziale in R, con le condizioni al
contorno R finita per r → 0 e r → ∞ e riconoscibile come l’equazione che ha per soluzione delle
funzioni note: le funzioni associate di Laguerre (moltiplicate per l’esponenziale di −r). In definitiva
gli autostati e le autoenergie (in Hartree) dell’atomo di idrogeno sono:
Ψn,l,ml(r, θ, φ) = Rnl(r)Ylml
(θ, φ) (A.8)
Rnl =
√√√√(n− l − 1)!Z
n2[(n + l)!]3ρl+1L2l+1
n+l (ρ) exp(−ρ/2) (A.9)
En,l,ml= − Z2
2n2(A.10)
dove ρ = 2Zr/na; Lαn(ρ) e un polinomio di Laguerre di grado n ed ordine α. Gli autostati discreti
sono negativi e dipendono solo da 1/n2.

Appendice B
Separazione del baricentro
E data una collezione di punti materiali, che per convenienza dividiamo in due sottoinsiemi ~ri,
i = 1, . . . , N , con masse mi (nel seguito particelle ’leggere’) e ~Ri, p = 1, . . . , M , con masse Mi (nel
seguito particelle ’pesanti’). Definiamo l’hamiltoniano del sistema in unita atomiche
H = T + V (B.1)
T = −N∑
i=1
1
2mi
∇2
i −M∑
p=1
1
2Mp
∇2
p (B.2)
V =∑
i<j
V (~ri − ~rj) +∑
i,p
V (~ri − ~Rp) +∑p<q
V (~Rp − ~Rq) (B.3)
dove l’unica assunzione e che l’energia potenziale sia data da sole interazioni a coppie dipendenti
dalle distanze vettoriali relative. Vogliamo dimostrare che il baricentro del sistema e una coordinata
separabile. A questo scopo, introduciamo il nuovo set di coordinate:
~Rc =1
MT
N∑
i=1
mi~ri +M∑
p=1
Mp~Rp
(B.4)
~xi = ~ri − 1
M
M∑
p=1
Mp~Rp i = 1, . . . , N (B.5)
~Qp = ~Rp − ~Rp+1 p = 1, . . . , M (B.6)
dove M e la massa totale delle particelle pesanti, MT e la massa totale del sistema; abbiamo definito
cioe il vettore del centro di massa del sistema, le N coordinate delle particelle leggere relative al
baricentro delle particelle pesanti, ed M − 1 coordinate vettoriali ottenute come le prime m − 1
differenze tra i vettori ~Rp: complessivamente i gradi di liberta del sistema sono 3+3N +3(M−1) =
3(N +M). Dimostriamo che l’energia potenziale non dipende da ~Rc. Possiamo facilmente esprimere
le vecchie coordinate ~R2, . . . , ~RM e ~r1, . . . , ~rN in funzione delle nuove coordinate e di ~R1:
~Rp = ~R1 −p−1∑
q=1
~Qq p > 1 (B.7)
137

138 APPENDICE B. SEPARAZIONE DEL BARICENTRO
~ri = ~R1 + ~xi − 1
M
M∑
p=2
p−1∑
q=1
~Qq (B.8)
~R1 e funzione di tutte le nuove coordinate, compreso ~Rc; ma nell’energia potenziale compaiono
solo differenze tra le posizioni delle particelle, e quindi V e indipendente da ~Rc. Calcoliamo ora
l’operatore di energia cinetica nelle nuove coordinate vettoriali. Il gradiente relativo alla i-esima
particella leggera e
∇i =mi
MT
∇c + ∇xi(B.9)
dove con ∇xiindichiamo il gradiente rispetto ~xi, con ∇c rispetto a ~Rc. Segue che
1
mi
∇2
i =mi
M2T
∇2
c +1
mi
∇2
xi+
2
MT
∇c · ∇xi(B.10)
Il gradiente relativo alla p-esima particella pesante e
∇p =MP
MT
∇c − MP
M
n∑
i=1
∇xi+ ∇Qp − ∇Qp−1 (B.11)
dove con ∇Qp indichiamo il gradiente rispetto ~Qp. Segue che
1
MP
∇2
p =MP
M2T
∇2
c +MP
M
∑
i,j
∇xi· ∇xj
+1
MP
(∇Qp − ∇Qp−1
)2 − 2MP
MT M
N∑
i=1
∇c · ∇xi
+2
MT
∇c ·(∇Qp − ∇Qp−1
)− 2
M
(∇Qp − ∇Qp−1
)·
n∑
i=1
∇xi(B.12)
Si puo verificare che sostituendo le precedenti equazioni nell’espressione per l’energia cinetica otte-
niamo:
T = − 1
2MT
∇2
c −N∑
i=1
1
2mi
∇2
xi− 1
2M
∑
i,j
∇xi· ∇xj
−M∑
p=1
1
2MP
(∇Qp − ∇Qp−1
)2(B.13)
il primo termine e relativo al moto traslazionale libero del baricentro; il secondo all’energia cinetica
delle particelle leggere corretto dal terzo termine detto di polarizzazione di massa; il quarto e
l’energia cinetica delle particelle pesanti espressa nelle coordinate interne ~Qp.

Appendice C
Problema generalizzato agli autovalori
Consideriamo l’equazione di Schrodinger indipendente dal tempo per le autofunzioni dell’operatore
di Hamilton relativo ad un sistema definito rispetto alle coordinate q. Una (auto)-funzione Ψ(q) e
un elemento dello spazio di Hilbert delle funzioni in q, e sara indicata nel seguito con il ket |Ψ〉
H|Ψ〉 = E|Ψ〉 (C.1)
Introduciamo ora un insieme di funzioni linearmente indipendenti che costituiscano una base, non
necessariamente ortonormale, nello spazio di Hilbert |v1〉, |v2〉, . . .; possiamo percio esprimere |Ψ〉come una combinazione lineare degli elementi della base:
|Ψ〉 =∑
j
cj|vj〉 (C.2)
Sostituendo nella (C.1) e premoltiplicando per il generico bra 〈vi|:∑
j
〈vi|H|vj〉cj = E∑
j
〈vi|vj〉cj (C.3)
che in notazione matriciale si puo scrivere come
Hc = ESc (C.4)
dove Hij = 〈vi|H|vj〉, Sij = 〈vi|vj〉. Se la base e ortonormale, la matrice di overlap S coincide con
la matrice unita, Sij = δij, e dunque
Hc = Ec (C.5)
In pratica, se la dimensione dello spazio hilbertiano considerato e infinita, il set di funzioni di base
utilizzato viene troncato ad un numero finito N , introducendo un’approssimazione che dipende da
N . Soluzioni non-triviali del sistema N -dimensionale (C.4) si ottengono dalle radici dell’equazione
secolare:
det(H− ES) = 0 (C.6)
139

140 APPENDICE C. PROBLEMA GENERALIZZATO AGLI AUTOVALORI
Si ottengono N autovalori Ei, i = 1, . . . , N cui corrispondono N autovettori ci. Se si definisce
la matrice C le cui colonne sono autovettori, Cij = (cj)i la matrice diagonale i cui elementi sono
diagonali sono gli autovalori, Eij = δijEj, risulta che:
HC = SCE (C.7)

Appendice D
Valore di attesa per un guscio chiuso
Assumiamo che le funzioni orbitali (spaziali) siano ortonormali:
∫dqµφ
∗i (µ)φj(µ) = 〈φi|φj〉 = δi,j (D.1)
Il valore di attesa dell’hamiltoniano H si puo dividere in un contributo monoelettronico e in un
termine bi-elettronico. Indicando con Si il generico spinorbitale
E =∑
P
(−)P
〈S1(1)S2(2) . . . SN(N)|∑Nn=1 h(n) +
∑n<n′
1rn,n′
|PS1(1)S2(2) . . . SN(N)〉 (D.2)
Il contributo monoelettronico si puo calcolare distinguendo i vari casi:
Permutazione identica. Somma di N/2 termini in ordine zero:
N∑n
〈Sn(n)|h(n)|Sn(n)〉 = 2N/2∑
i=1
〈φi(n)|h(n)|φi(n)〉 = 2N/2∑
i=1
hi (D.3)
Altre permutazioni. Contributo nullo, a causa dell’ortogonalita di spin.
Si procede in modo analogo per il contributo bi-elettronico:
Permutazione identica, n dispari, n′ = n + 1. Somma di integrali a due centri:
∑
n dispari,n′=n+1
〈Sn(n)Sn(n)| 1rnn′
|Sn(n)Sn(n)〉 =N∑
i=1
〈φi(n)φi(n′)| 1
rnn′|φi(n)φi(n
′)〉
=N/2∑
i=1
Jii (D.4)
141

142 APPENDICE D. VALORE DI ATTESA PER UN GUSCIO CHIUSO
Permutazione identica, altri valori di n e n′. Somma di integrali a due centri:
∑
n,n′〈Sn(n)Sn′(n
′)| 1rnn′
|Sn(n)Sn′(n′)〉 = 4
N/2∑
i<j
〈φi(n)φj(n′)| 1
rnn′|φi(n)φj(n
′)〉
= 4N/2∑
i<j
Jij (D.5)
Permutazioni di due elettroni. Somma di integrali a due centri:
− ∑
n<n′〈Sn(n)Sn′(n
′)| 1rnn′
|Sn′(n)Sn(n′)〉 = −2N/2∑
i<j
〈φi(n)φj(n′)| 1
rnn′|φj(n)φi(n
′)〉
= −2N/2∑
i<j
Kij (D.6)
Altre permutazioni. Contributo nullo, a causa dell’ortogonalita della parte spaziale.
Poiche Jij = Jji, Kij = Kji, Jii = Kii, resta l’espressione:
E = 2N/2∑
i
hi +N/2∑
i,j
(2Jij −Kij) (D.7)
dove Jij e Kij sono gli integrali di Coulomb e di scambio.

Appendice E
Separabilita σ − π (Likos-Parr)
Le condizioni di separabilita σ−π per una funzione d’onda multielettronica si possono scrivere nella
forma:
1. e dato un sistema di N elettroni, nσ di tipo σ, nπ di tipo π:
nσ + nπ = N (E.1)
2. la funzione d’onda e approssimata da un prodotto (simmetrizzato) di una funzione σ ed una
funzione π:
Ψ(1, 2, . . . , N) = RΨσ(1, 2, . . . , nσ)Ψπ(nσ + 1, nσ+1, . . . , nπ) (E.2)
R e l’operatore somma di tutte i possibili scambi tra elettroni σ e π;
3. le funzioni σ e π sono combinazioni lineari di determinanti di Slater, costruiti a partire da
opportuni spin-orbitali
Ψσ =∑
k akDk Dk ← SσIΨπ =
∑l blDl Dl ← SπJ
(E.3)
4. le due funzioni d’onda sono normalizzate:
〈Ψσ|Ψσ〉 = 〈Ψπ|Ψπ〉 = 〈Ψ|Ψ〉 (E.4)
Le condizioni 1.-4. costituiscono l’approssimazione di separabilita σ − π.
143

144 APPENDICE E. SEPARABILITA σ − π (LIKOS-PARR)
La funzione d’onda complessiva (nel caso Ψσ e Ψπ siano date ciascuna da un solo determinante
di Slater) e data da:
Ψ(1, 2, . . . , N) =
(1
N !
)1/2
∣∣∣∣∣∣∣∣∣∣∣∣∣∣∣∣∣∣∣∣∣
Sσ1(1) . . . Sσnσ(1) Sπnσ+1(1) . . . SπN(1)...
......
...
Sσ1(nσ) . . . Sσnσ(nσ) Sπnσ+1(nσ) . . . SπN(nσ)
Sσ1(nσ + 1) . . . Sσnσ(nσ + 1) Sπnσ+1(nσ + 1) . . . SπN(nσ + 1)...
......
...
Sσ1(N) . . . Sσnσ(N) Sπnσ+1(N) . . . SπnN(N)
∣∣∣∣∣∣∣∣∣∣∣∣∣∣∣∣∣∣∣∣∣
(E.5)
L’hamiltoniano molecolare elettronico si puo scrivere nel modo consueto, evidenziando i due gruppi
di elettroni:
H = H(0)σ +H(0)
π +Hσπ (E.6)
H(0)i = −1
2
ni∑µ
∇2µ −
∑
µ,k
Zk
rµk
+ni∑µ,ν
1
rµν
i = σ, π (E.7)
Hσπ =nσ∑µ
nπ∑ν
1
rµν
(E.8)
Hπ = H(0)π +Hσπ =
nπ∑µ
hcore
µ +nπ∑
µ<ν
1
rµν
(E.9)
hcore
µ = −1
2∇2
µ −∑
k
Zk
rµk
+nσ∑ν
1
rµν
(E.10)
Se si trascura lo scambio σ − π, il valore di attesa e dato dalla somma di valori di attesa:
〈H〉av = 〈Hπ +H(0)σ 〉av = 〈Ψπ|Hπ|Ψπ〉+ 〈Ψσ|H(0)
σ |Ψσ〉 = Eπ + E(0)σ (E.11)
Si puo considerare E(0)σ costante; resta definito l’operatore π−HF, rispetto ai soli elettroni π:
Fπ = hcore
+∑
j
(2Jj − Kj) (E.12)

Bibliografia
[1] C.L. Perrin, Mathematics for Chemists, (Wiley, New York, 1970).
[2] M. Abramowitz e I. Stegun, Handbook of Mathematical Functions, (Dover, New York, 1972).
[3] H.D. Ikramov, Linear Algebra: Problems Book, (Mir Publishers, Mosca, 1983).
[4] R.J. Jacob, in Physical Chemistry: an Advanced Treatise, Vol. XI A, Cap. 1.
[5] S. Lang, Algebra Lineare, (Boringhieri, Torino, 1984).
[6] D. Henderson, in Physical Chemistry: an Advanced Treatise, Vol. XI A, Cap. 4.
[7] I.S. Gradshtein e I.M. Ryzhik, Table of Integrals, Series and Products (Academic Press, New
York, 1972).
[8] P.W. Atkins, Molecular Quantum Chemistry, (Oxford, 1983).
[9] L.D. Landau e E.M. Lifsits, Meccanica Quantistica, (Editori Riuniti, 1982).
[10] AA. VV., Modern Theoretical Chemistry voll. 3,4, ed. G.A. Segal (Plenum Press, N.Y. 1977).
[11] AA. VV., Modern Theoretical Chemistry voll. 7,8, ed. G.A. Segal (Plenum Press, N.Y. 1977).
[12] F.L. Pilar, Elementary Quantum Chemistry 2a edizione, (McGraw-Hill, N.Y. 1990).
[13] W.J. Hehre, R.F. Stewart and J.A. Pople, J. Chem. Phys 51, 2657 (1969).
[14] M.J.S. Dewar, E.G. Zoebisch, E.F. Healy and J.J.P. Stewart, J. Am. Chem. Soc. 107
145



![Cenni di anatomia e biomeccanica - oasiclubpalestra.itoasiclubpalestra.it/.../uploads/2014/12/cenniAnatomiaBiomeccanica.pdf · [CENNI DI ANATOMIA E BIOMECCANICA] Organizzazione del](https://img.dokumen.tips/doc/110x75/5c6b8d5d09d3f29a768bc868/cenni-di-anatomia-e-biomeccanica-cenni-di-anatomia-e-biomeccanica-organizzazione.jpg)

























