Embed Size (px)
Citation preview

Oregon Health & Science University OHSU Knight Cancer Institute
3181 SW Sam Jackson Park Rd, CR 145 Portland, OR 97239
Data and Safety Monitoring Plan
Contact Information
Bashi Ratterree, RN, BSN, CCRP
Clinical Research Management
OHSU Knight Cancer Institute
Oregon Health & Science University
Center for Health & Healing, Mail code CH15R
3303 SW Bond Ave.
Portland, OR 97239
503-494-1028
Initial NCI Approved: 11/20/01
Revision NCI Approved: 02/03/04
Revision NCI Approved: 2/21/07
Revision NCI Approved: 2/27/08

2/27/08
1
Table of Contents
IIntroduction 2
Responsibilities 2
Investigator 2
Clinical Research Review Committee (CRRC) 3
Clinical Research Management (CRM) 4
Data and Safety Monitoring Committee (DSMC) 4
DSMC Membership Selection Criteria 5
Data and Safety Monitoring Flow Chart 6
Quality Assurance Auditing 7
Locally Initiated Studies and NCI or NIH sponsored studies 7
Cooperative Groups (i.e., SWOG, CCG, GOG, etc.) 8
Pharmaceutical Industry sponsored studies 8
Central Coordination of Multicenter Studies 8
Audit Procedure 8
Audit Follow-up 14
Data and Safety Monitoring Boards (DSMB) 15
Board Membership 15
Responsibilities of the DSMB 16
Reportable Adverse Event Monitoring 16
OHSU IRB Reporting 17
FDA reporting 18
NIH/NCI reporting 18
Recombinant DNA/Gene Transfer Studies 18
SAE/UP Reporting Flow Chart 19
Appendix 1 2008 Data and Safety Monitoring Committee Member Roster 20
Appendix 2 Quality Assurance (QA) Audit Forms 21

2/27/08
2
Introduction
The OHSU Knight Cancer Institute (CI) places the highest priority on ensuring the safety of
patients participating in clinical trials. The Director of the OHSU Knight CI, the Associate
Director of Clinical Research and the Director of Clinical Research Management (CRM) hold
the overall responsibility for overseeing data and safety monitoring. Other groups with
responsibilities for data and safety monitoring include the Clinical Research Review Committee
(CRRC), the Data and Safety Monitoring Committee (DSMC), the internal audit team, individual
data safety and monitoring boards, the principal investigators of NIH grants and contracts
supporting clinical trials, and, most importantly, the principal investigator of each clinical trial.
The purpose of the OHSU Knight CI Data and Safety Monitoring Plan (DSMP) is to insure the
safety of study participants, the validity of research data and the appropriate termination of
studies for which significant benefits or risks have been uncovered or when it appears that the
trial cannot be concluded successfully.
The OHSU knight CI DSMP follows the policy of the National Cancer Institute for Data and
Safety Monitoring of Clinical Trials http://deainfo.nci.nih.gov/grantspolicies/datasafety.htm.
For purposes of this plan, a clinical trial is operationally defined as a prospective study involving
human subjects designed to answer specific questions about the effects or impact of particular
biomedical or behavioral interventions. Diagnostic studies are included only if information from
the diagnostic test somehow affects medical decision-making for the study subject.
Observational studies and those that do not test interventions are not clinical trials.
For phase I, II and III clinical trials the method and degree of monitoring will be commensurate
with the degree of risk involved in participation and the size and complexity of the clinical trial.
The protocol will define the level of monitoring. A formal study-specific Data and Safety
Monitoring Board (DSMB) will be constituted for Phase III randomized clinical trials depending
chiefly on the anticipated level of risk. DSMBs may be constituted for other trials involving
particular risk, complexity, likely decisions about early stopping or the need to obviate conflict
of interest.
For most investigator-initiated NIH grant applications, the investigator may supply this approved
institutional DSMP in the human subjects section of the grant application and describe how it
applies to the specific trial. The investigator may need to tailor the plan for their specific trial
according to the degree of risk to which participants in the trial are exposed.
Below is detailed the responsibilities for all entities involved with data and safety monitoring.
Investigator
The principal investigator of each study is ultimately responsible for every aspect of the design,
conduct, and final analysis of their protocol. The principal investigator is responsible to ensure
that:

2/27/08
3
All protocols include a data and safety monitoring plan and procedures for its
implementation
A study-specific data safety and monitoring board (DSMB) is established if required
All studies have a structured adverse event determination, monitoring and reporting
system. Serious adverse events (SAEs) are reported as required to appropriate agencies
and Unanticipated Problems (UP) are reported to the OHSU IRB.
All blinded studies describe a randomization scheme, and specific criteria and procedures
for unblinding
If the proposed protocol has additional clinical sites besides OHSU Knight CI, the
protocol describes a central coordination plan
Studies with stopping rules or interim analysis must forward results to DSMC prior to
enrolling subsequent patients
All protocols and amendments are submitted to CRM and IRB for review and approval
Clinical Research Review Committee (CRRC)
The Clinical Research Review Committee is a multidisciplinary committee charged with
providing peer review of the scientific merit of all cancer related research studies to be
conducted at OHSU. The goal of the CRRC is to ensure that all clinical trials and cancer-related
research conducted at the OHSU Knight Cancer Institute are 1) scientifically meritorious; 2)
appropriately designed; 3) feasible for completion within the specified time frame; 4) in
compliance with FDA and NIH guidelines for clinical trials; and 5) compatible with the
priorities, goals and interests of the OHSU Knight Cancer Institute. The CRRC is not intended to
duplicate or overlap the responsibilities of the IRB nor is it intended to perform an auditing or
data and safety monitoring function, but does work collaboratively with both of these other
committees to ensure human subjects protection and scientific integrity.
At the time of initial CRRC review, all aspects of the study are scrutinized. CRRC review
determines the level of risk and confirms that the protocol-specific DSMP is adequate. CRRC
may conclude that a study-specific DSMB is required. The CRRC reviewer will determine the
study level of risk as one of two levels:
Low risk: studies involving no therapeutic intervention, i.e. studies involving patient
questionnaires, blood draws, or other low-risk tissue sampling (i.e., hair, urine, sputum), voice or
video recordings, moderate exercise, existing data, documents, pathologic or diagnostic
specimens, behavioral, cognition or perception; studies involving intervention with standard
doses of nutritional agents available over the counter (i.e., vitamins, minerals). Low-risk studies
may be audited at anytime randomly or for cause.
High risk: studies involving cancer-directed chemotherapy, biologic therapy, radiation therapy,
or surgical intervention; studies involving higher-risk tissue sampling (i.e., bone marrow, or
sampling requiring any type of anesthesia); studies involving non-standard doses of agents
available over the counter; High-risk studies that are not monitored by another source will be
audited by the DSMC internal audit team. High-risk studies that are monitored by other sources
may be audited at anytime randomly or for cause.
As part of continuing scientific review, the CRRC is responsible for reviewing a monthly
summary of protocol revisions, amendments, continuing reviews and UPs for all cancer studies.

2/27/08
4
Clinical Research Management (CRM)
The OHSU Knight CI CRM provides support for all clinical trials at the OHSU Knight CI. CRM
activities specifically related to data and safety monitoring are as follows:
Assists investigators in the preparation and submission of local, investigator-initiated
studies for review by the CRRC and OHSU IRB
Distributes information on active cancer clinical trials
Provides centralized patient registration for all investigator-initiated studies
Collects, maintains and updates data on patients enrolled including data on accrual and
serious adverse events and unanticipated problems
Reviews protocol revisions, amendments and continuing reviews for all cancer studies in
real-time before forwarding to IRB
Prepares a monthly report of active trials with upcoming Stopping Rules or Interim
Analysis for review by the DSMC
Provides administrative support to the CRRC and DSMC
Ensures that cancer clinical trials are conducted in accordance with federal, state and
institutional regulations
Assists in the training of investigators and clinical trial staff in the development and
conduct of clinical trials
Data and Safety Monitoring Committee (DSMC)
The Data and Safety Monitoring Committee (DSMC) is responsible for overall coordination of
all aspects of the DSMP. The internal audit team conducts quality assurance audits on all open
clinical trials that are not monitored by another source. The DSMC meets once each month to
review the audit team’s progress and findings and to review Coordinating Center SAE and/or UP
reports, IND reports, Interim Analysis reports and outside agencies’ audit reports submitted from
investigators. The DSMC also reviews a full report of study activity for all local, active clinical
trials at the time of continuing review submission including:
protocol amendments, revisions, consent form revisions
interim analysis results
protocol violations
total number of patients enrolled on-study as compared to expected numbers
dates of patient enrollments
vital and study (on or off-study) status of each patient
all Unanticipated Problems submitted (including dates, description and relationship)
Members receive this information approximately one week prior to Committee meetings, to
allow for preliminary study and review. The Committee will vote to approve, conditionally
approve (enrollment may continue after satisfactory response by the principal investigator to
DSMC is received), suspend or close each protocol reviewed. The Committee decision will be
documented in monthly meeting minutes. Principal investigators may appeal the decision to the
Director of the CI.

2/27/08
5
The DSMC oversees the process of serious adverse event reporting to assure that reporting
requirements are met. The DSMC may require amendments, suspend or terminate any clinical
trial that falls within its jurisdiction. The DSMC has the authority to report directly to the OHSU
IRB. The DSMC communicates with and provides semi-annual summary reports to the CRRC.
The committee is made up of the following representatives: physician members of OHSU Knight
Cancer Institute, administrators of Clinical Research Management (CRM), biostatistician,
research pharmacist, research nurses, and study coordinators. A term of membership is 5 years.
Appendix 1 lists DSMC members. In the event that a Committee member is key personnel
(principal investigator, co-investigator, biostatistician, study coordinator, study investigational
pharmacist, study nurse) for a study under review, or has any other conflict of interest (including
substantial financial interest in the study sponsor agency), that member must abstain from
Committee review and discussion and must leave the room prior to final decisions on the study.
In the event that the Chair is the principal investigator for the study, the Co-Chair of the
Committee will oversee the Committee deliberations and final decisions. Our DSM Committee
includes multiple MD representatives as well as an alternate biostatistician for cases in which our
primary biostatistician has a conflict of interest.
DSMC Membership selection criteria
1. The OHSU Knight CI DSMC membership is multidisciplinary. The committee includes
the following representatives: physician members of the OHSU Knight Cancer Institute,
administrators of Clinical Research Management (CRM), biostatistician, research
pharmacist, research nurses, study coordinators.
2. Any member of the committee may nominate new members to the DSMC and after
qualifications are reviewed and approved by the committee, the Director of the OHSU
Knight CI will appoint the new member.
3. Application includes submission of a CV and a statement of interest regarding
membership to the DSMC.
4. The term of membership is five years and is renewable.
5. DSMC members should have at minimum of 2 years experience in clinical research and
have familiarity with IRB policies and procedures including, but not limited to, reporting
policies of the IRB, OHRP, FDA and NIH for adverse events, serious adverse events,
unanticipated problems, protocol deviations, and other clinical research related reporting
obligations.
6. DSMC members must be able to attend monthly DSMC meetings to assure membership
quorum.
7. The DSMC abides by all policies set forth by the University and is an equal opportunity
program.

2/27/08
6
The following flow chart illustrates the reporting structure within OHSU Knight CI and the
relationship with OHSU Research Integrity Office.
OHSU Knight Cancer Institute (CI)
Director
Associate Director Clinical Research
Clinical Research Review Committee Scientific Review
Data and Safety Monitoring Committee
Clinical Research Management
Shared Resource
Cancer Related Protocol
OHSU
Principal Investigator
OHSU Research Integrity Office
&
OHSU IRB

2/27/08
7
Quality Assurance Auditing
The DSMC internal audit team is responsible for conducting Quality Assurance (QA) audits on
active, CI-approved clinical trials that are not monitored by another source. The audit team
reports directly to the DSMC. The purpose of the QA audit is to ensure that the OHSU Knight
CI and IRB approved protocol is being followed, data is accurately recorded, and regulatory
documents are properly maintained. More specifically, the audit team members verify that
informed consent is obtained; patients are eligible for participation, properly enrolled, treated
according to protocol; and toxicities, and adverse events, serious adverse events, unanticipated
problems, and protocol deviations are properly recorded and reported. The audit also examines
patient accrual and inclusion of women, minorities, and children.
The audit team is made up of members of the DSMC. Auditors may also be selected from the
OHSU Knight CI membership at large. In addition, DSMC audit teams may include qualified
independent contractors or clinical research organizations. Audit team members will be
knowledgeable of the protocol to be reviewed, audit procedures, clinical trials methodology and
OHSU Knight CI policies. Audit team members will not be directly involved in the conduct of
the protocol to be reviewed.
The audit team will have access to consultation from clinical trial experts, biostatisticians,
bioethicists and clinicians knowledgeable about the disease and treatment under study.
Representatives from the NCI auditing group may attend audits.
Below is a listing of types of clinical trials taking place at the OHSU Knight Cancer Institute and
their levels of monitoring. In general, locally initiated studies will require internal auditing by
the DSMC audit team. Pharmaceutical studies are monitored by the sponsor or CRO.
Cooperative group trials are audited according to well-established cooperative group DSMPs.
Locally Initiated Studies and NCI or NIH sponsored studies Audits of locally initiated studies and NCI or NIH sponsored studies will be conducted by the
OHSU Knight CI DSMC audit team. If a study is audited by NCI, the principal investigator or
study coordinator will forward a copy of the summary letter and follow-up correspondence to the
DSMC for review and inclusion in the CRM study document binder.
Ideally, each high-risk locally initiated clinical trial will be audited annually, but no less than
every 3 years. Studies with potentially higher risks, special populations or high accruals will be
audited more frequently as specified in the Data and Safety Monitoring section of the protocol.
Recently initiated studies may be audited anytime after enrollment has begun.
High or low-risk locally initiated clinical trials may be audited at any time randomly or for just
cause if determined necessary by the principal investigator, DSMC, OHSU Knight CI or the
IRB.
For clinical trials for which an OHSU Knight CI Investigator holds the IND or IDE, the primary
responsibility for monitoring may be assigned to an outside contract research organization

2/27/08
8
(CRO). It would be the Investigator’s responsibility to obtain this outside CRO monitoring. The
level of direct oversight and intensity of monitoring would be assumed by the CRO according to
the specifics of the DSMP incorporated in the protocol document.
Cooperative Groups (i.e., SWOG, CCG, GOG, etc.) Each Cooperative Group will perform its own program of on-site audits, to be conducted by its
staff and/or members. DSMC members may attend as observers. The study coordinator will
forward a copy of the summary letter for each on-site audit to the DSMC for review and
inclusion in the CRM study document binder. The DSMC may conduct audits in addition to
those conducted by the Cooperative Group.
Pharmaceutical Industry sponsored studies Monitoring visits will be conducted by the sponsoring pharmaceutical company or clinical
research organization. In addition the pharmaceutical company may conduct a quality assurance
audit. DSMC members may attend as observers. The study coordinator will forward a copy of
the summary letter for each monitoring visit or audit to the DSMC for review and inclusion in
the CRM study document binder.
The DSMC may conduct audits in addition to those conducted by the pharmaceutical company
or clinical research organization. In addition, the FDA may audit the study. If a study is to be
audited by the FDA, the principal investigator or study coordinator will notify CRM of the audit.
At the completion of the audit the study team will forward a copy of the summary letter and
follow-up correspondence to the DSMC for review and inclusion in the CRM study document
binder.
Central Coordination of Multicenter Studies
OHSU central coordination of multi-center studies is the responsibility of the principal
investigator. Coordinating Center (CC) activities are to include central registration recorded in
the CRM database and central reporting of UPs (Unanticipated Problems)/SAEs (Serious
Adverse Events),. The CC will prepare Quarterly Summary Reports of UPs& SAEs from all
centers, to include the enrollment numbers. The Quarterly Summary Report will be submitted to
CRM, OHSU IRB and each individual site’s IRB. In addition, UPs and SAEs will be reported to
other sites in real-time if indicated for patient safety.
The DSMC will conduct QA audits for the OHSU clinical site only. Monitoring of other clinical
sites is the responsibility of the principal investigator and CC team according to protocol or CC
written procedures. Off-site audits will be conducted according to an approved DSMP audit
plan. Outside sites will be asked to submit their own data and safety monitoring plan. A copy of
the off-site audit summary letter will be forwarded to the DSMC.
Audit Procedure
The OHSU Knight CI’s quality assurance audit procedure is modeled after the NCI CTEP
Guidelines for Conducting the Quality Assurance Audit
http://ctep.cancer.gov/monitoring/section5.html.

2/27/08
9
Arranging the Audit
The Quality Assurance Coordinator will arrange an audit date with the principal investigator and
study coordinator at a mutually satisfactory time and place. At least 30 days notice will be given
prior to the audit. This will allow the study coordinator sufficient time to assemble and label the
study documents.
Selection of Cases
A minimum number of cases equivalent to 10 percent of the patients accrued since the last audit
or at least two will be reviewed. While most cases will be randomly selected from patients
accrued since the previous audit, any patient case may be selected for review. The study
coordinator will be notified of the cases selected for audit at least two weeks prior to the audit.
In addition, there may be an unannounced review of all or part of the total cases for appropriate
informed consent and eligibility.
Preparation for the Audit
Prior to the audit, the audit team members will review the protocol, IRB regulatory documents
on file at CRM, data in the Surveyor Clinical Trial Database and any previous audit documents.
The team members will meet to discuss the audit plan and prepare the audit forms (Appendix 3).
The study coordinator is responsible for ensuring that all relevant materials are available for
review at the time of the audit. Relevant materials include: original or copies of the patient
source documents, signed consent forms, research notes, IRB documents, drug accountability
forms, case report forms and other protocol specific documents. Diagnostic studies may be
requested.
It is recommended that the staff member most familiar with the patient cases be present at the
audit. To facilitate the review of the records, it is recommended that the staff label all documents
beforehand.
Conducting the Audit During the audit, the monitors review specific data related to the protocol and regulatory
requirements. Source documents are used to independently verify study data. Source documents
may include, but are not limited to, the following:
1. Inpatient and outpatient medical records
Progress notes
Diagnostic reports (x-rays, scans, ECGs, etc.)
Laboratory reports
Admission forms
2. Study flow sheets and other research records that are signed and dated, if this is the
source on which information is first documented (i.e. vitals, performance status)
3. Protocol or Study road maps
4. Appointment books
5. Enrollment tracking sheets
6. Subject diaries/calendars
7. Drug orders and administration records
8. NCI Drug Accountability Record Forms (DARFs) or a similar form that contains the same

2/27/08
10
information. In the remainder of this procedure document DARF will be used to mean
either of the above forms.
An audit consists of reviewing and evaluating (1) conformance to IRB and informed consent
requirements, (2) individual patient records and (3) the pharmacy and use of DARFs. A
modified version of the NCI’s Clinical Trials Monitoring Branch (CTMB) Audit Information
System audit report format will be used (see Appendix 3). This provides a common system for
assessing each component of an audit and uses a common set of terms or examples of MAJOR
and LESSER deficiencies.
Review of conformance to IRB requirements
For each protocol selected for an audit, the following items will be reviewed:
1 Documentation of full initial IRB approval of the protocol
2. Documentation of full IRB continuing re-approval
3. Documentation of IRB approval for all protocol amendments
4. Documentation of IRB approval or re-approval prior to patient registration.
The following are examples of major and minor deficiencies to be considered in assessing IRB
compliance.
Major deficiencies may include but are not limited to:
1. Protocol never approved by the IRB
2. Initial IRB approval documentation missing
3. Registration and/or treatment of patient prior to full IRB approval
4. Registration of patient on protocol during a period of delayed re-approval
5. Missing re-approval documentation
6. Expired re-approval
7. Unanticipated problems not reported to IRB
8. Reportable adverse events not reported to appropriate entity
9. Lack of documentation of full board IRB approval of a protocol amendment that
involves more than minimal risk.
Lesser deficiencies may include but are not limited to:
1. Protocol re-approval delayed less than thirty days
2. Delayed re-approval for protocol closed to accrual for which all patients have
completed therapy.
Review of conformance to Protocol requirements
For each protocol selected for an audit, the following items will be reviewed:
1. Enrollment information entered into Surveyor database
2. Drug accountability kept
3. Dose escalation/ de-escalation criteria followed
4. Interim Analysis/ Stopping Rules followed

2/27/08
11
Each audited study will be assessed for Stopping Rules or Interim Analysis requirements. If
Stopping Rules or Interim Analysis is written into the protocol, auditors will review and confirm
that the protocol was followed and that a summary report of the analysis is available.
If the audited study is a multi-center study and OHSU is the coordinating center, audit finding
letters from the DSMBs/DSMCs of all other sites will be reviewed.
1. Subjects have not been centrally registered in CRM database
2. Central reporting of UPs not documented
3. DSMP not provided/available for other sites
4. If audits conducted at other sites, audit result letter not available/not forwarded to
DSMC
If study involves IND/IDE held by the PI, the following will be reviewed:
1. IND Safety reports not submitted to FDA
2. Protocol amendments (including addition of investigators) not submitted to FDA
3. Information Amendments not submitted to FDA
4. Annual reports not submitted to FDA
Review of Patient Case Records
Selected patient records will be reviewed for major and lesser deficiencies in each of the
following categories:
1. Properly signed and dated informed consent
2. Eligibility
3. Correct treatment and treatment sequence
4. Evaluation of disease outcome/tumor response
5. Toxicities related to treatment
6. General quality of the data collected.
A major deficiency is defined as a variance from protocol-specified procedures that makes the
resulting data questionable. Following are examples of major deficiencies. This does not
represent an all-inclusive list of major deficiencies that may be found in the audit.
Failure to document properly obtained informed consent such as:
1. Consent form missing
2. Consent form not signed and dated by the patient
3. Consent form signed after the patient started treatment
4. Consent form does not contain all required signatures
5. Consent form used was not the current IRB-approved version at the time of patient
registration
6. Consent form does not contain updated information specified by the IRB.
Failure to confirm eligibility such as:
1. Review of documentation available at the time of the audit confirms patient did not
meet all eligibility criteria as specified by the protocol

2/27/08
12
2. Documentation missing. Unable to confirm eligibility.
Failure to follow protocol treatment plan such as:
1. Incorrect agent/treatment used
2. Additional agent/treatment used which is not permitted by protocol
3. Dose deviations (error greater than +/- 10%)
4. Dose modifications unjustified
5. Treatment doses incorrectly administered, calculated or documented
6. Unjustified delays in treatment.
Failure to evaluate disease outcome response according to the protocol such as:
1. Inaccurate documentation of initial sites of involvement
2. Tumor measurements/evaluation of status or disease not performed according to
protocol
3. Protocol-directed response criteria not being followed
4. Claimed response (PR, CR, etc.) cannot be verified
5. Failure to detect cancer (as in a prevention study) or failure to identify cancer
progression.
Failure to assess and report toxicities and adverse events according to the protocol such as:
1. Grades, types, or dates/duration of serious toxicities inaccurately recorded
2. Toxicities cannot be substantiated
3. Follow-up studies necessary to assess toxicities not performed
4. Failure to report a toxicity that would require filing a Serious Adverse Event Report
(SAE report) or Unanticipated Problem report
5. Recurrent under- or over-reporting of toxicities.
Failure to maintain general data quality
1. Recurrent missing documentation, e.g., charts
2. Protocol-specified laboratory tests not documented
3. Protocol-specified diagnostic studies not documented
4. Frequent data inaccuracies
5. Errors in submitted data
6. Delinquent data submission.
A lesser deficiency is a deficiency that is judged to not have a significant impact on the outcome
or interpretation of the study and is not described above as a major deficiency. An unacceptable
number of lesser deficiencies will be treated as a major deficiency in determining the final
assessment of a component.
Review of Accountability of Investigational Agents and Pharmacy Operations
Drug accountability and storage procedures described in this section are required under Federal
Regulations, CTEP and NCI policy. These drug accountability procedures refer to the guidelines
for NCI sponsored studies and CTEP supplied agents. However, these procedures should be
followed for all CI protocols involving an investigational drug or device. This would not include
approved drugs used for usual care during the study.

2/27/08
13
The auditors will review compliance with the NCI/CTEP procedures to ensure proper drug
storage and usage. Specifically, the auditors will check that the drug accountability system is
being maintained, and will spot-check the drug accountability records by comparing them with
the patients’ medical records to verify that the investigational drugs were administered per
protocol.
The NCI guidelines for Accountability of Investigational Agents and Pharmacy Operations can
be found at http://ctep.info.nih.gov/CTMB/Section5.htm. In brief, the following procedures for
drug accountability and storage will be monitored by Quality Assurance audit:
1. Investigational drugs are used only for patients entered in a CI and IRB approved
protocol.
2. The DARF is used to maintain accurate records of the disposition of all investigational
drugs. A copy of the NCI DARF can be found at
http://ctep.cancer.gov/forms/index.html.
3. Each investigational drug should be stored separately by protocol. If a drug is used for
more than one protocol, there should be separate physical storage for each protocol.
4. There should be a separate DARF for each protocol.
5. There should be a separate DARF for each drug in a multi-drug study.
6. Separate DARFs should be maintained for each different strength or dosage form of a
particular drug (e.g., a drug with a 1-mg. vial and a 5-mg. vial would require a separate
DARF).
7. The DARF will be used at each location that the investigational drug is stored, e.g., main
pharmacy, physician’s office, or other dispensing areas.
8. The DARF is used for both dispensing records and other drug transaction documentation
(e.g., receipt of drug, returns, broken vials, etc.)
9. Investigational drugs that are out-dated, damaged, or part of a study that is completed or
discontinued must be returned to the sponsor.
Audit Findings
At the conclusion of the audit, the audit team will conduct an exit interview with the investigator
and/or research staff. During the interview, the preliminary findings and any recommendations
from the audit team will be discussed. This interview provides opportunity for education,
immediate dialogue, feedback, and clarification.
After the audit is conducted, the audit team prepares a follow-up letter according to the QA
Audit SOP. Each component of the audit will be rated as Acceptable, Acceptable, needs
Follow-up or Unacceptable.
2/27/08
14
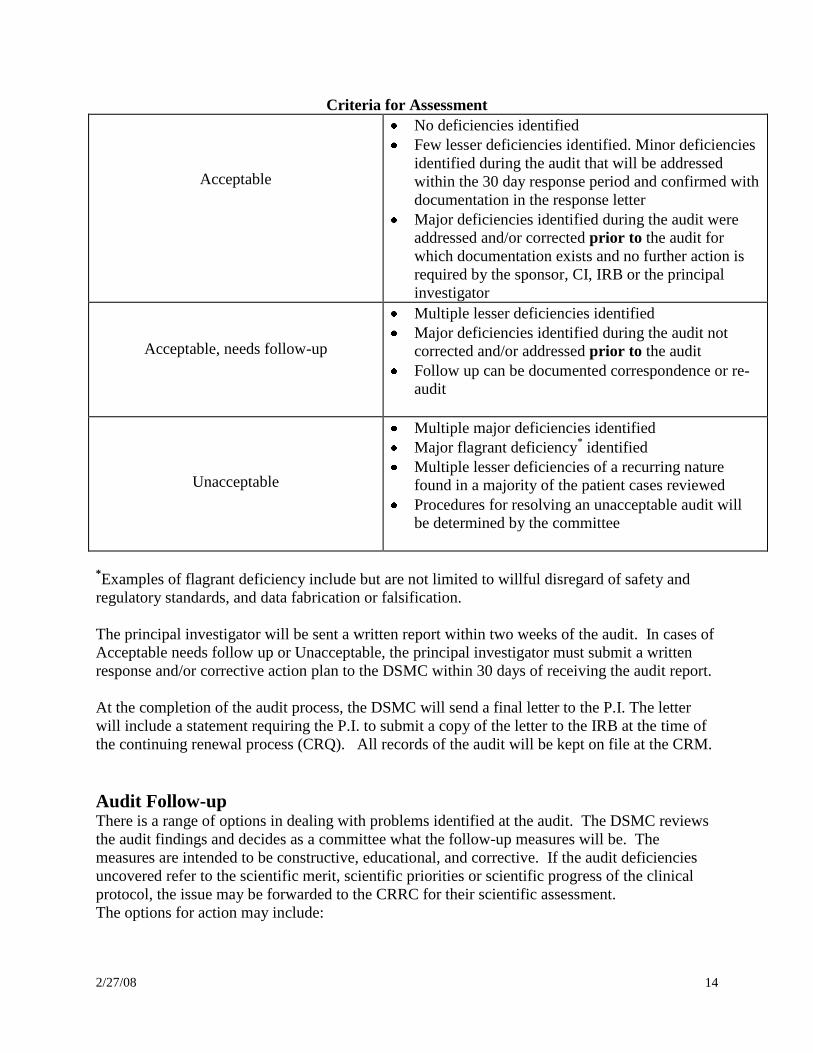
Criteria for Assessment
Acceptable
No deficiencies identified
Few lesser deficiencies identified. Minor deficiencies
identified during the audit that will be addressed
within the 30 day response period and confirmed with
documentation in the response letter
Major deficiencies identified during the audit were
addressed and/or corrected prior to the audit for
which documentation exists and no further action is
required by the sponsor, CI, IRB or the principal
investigator
Acceptable, needs follow-up
Multiple lesser deficiencies identified
Major deficiencies identified during the audit not
corrected and/or addressed prior to the audit
Follow up can be documented correspondence or re-
audit
Unacceptable
Multiple major deficiencies identified
Major flagrant deficiency* identified
Multiple lesser deficiencies of a recurring nature
found in a majority of the patient cases reviewed
Procedures for resolving an unacceptable audit will
be determined by the committee
*Examples of flagrant deficiency include but are not limited to willful disregard of safety and
regulatory standards, and data fabrication or falsification.
The principal investigator will be sent a written report within two weeks of the audit. In cases of
Acceptable needs follow up or Unacceptable, the principal investigator must submit a written
response and/or corrective action plan to the DSMC within 30 days of receiving the audit report.
At the completion of the audit process, the DSMC will send a final letter to the P.I. The letter
will include a statement requiring the P.I. to submit a copy of the letter to the IRB at the time of
the continuing renewal process (CRQ). All records of the audit will be kept on file at the CRM.
Audit Follow-up There is a range of options in dealing with problems identified at the audit. The DSMC reviews
the audit findings and decides as a committee what the follow-up measures will be. The
measures are intended to be constructive, educational, and corrective. If the audit deficiencies
uncovered refer to the scientific merit, scientific priorities or scientific progress of the clinical
protocol, the issue may be forwarded to the CRRC for their scientific assessment.
The options for action may include:

2/27/08
15
1. Letter of warning
2. Probationary status
3. Suspension of patient entry privileges
4. Immediate repeat audit
5. Removal of access to investigational drugs
6. Notification of the FDA if investigational drugs are involved
7. Notification of the Office of Research Integrity, NIH, if scientific misconduct is a
possibility
8. Notification of OHSU Scientific Integrity Committee if scientific misconduct is a
possibility (OHSU policy 04-15-005 Misconduct in Research)
9. Notification of the IRB if issues of patient rights, informed consent, protocol
violations or IRB review are involved.
The following actions may be taken by the DSMC in instances of suspected data fabrication or
falsification or other possible scientific misconduct:
1. Replacement of principal investigator
2. Termination of study
3. Reanalysis or retraction of published results
4. Debarment of investigator from future participation in OHSU Knight CI research
It is the responsibility of the DSMC to see that any action resulting in a temporary or permanent
suspension of an NCI-funded clinical trial is reported to the NCI grant program director
responsible for the grant. See http://grants.nih.gov/grants/guide/notice-files/NOT-OD-00-
053.html.
Data and Safety Monitoring Boards (DSMB)
A formal study-specific Data and Safety Monitoring Board (DSMB) will be constituted for
Phase III randomized clinical trials depending chiefly on the anticipated level of risk. DSMBs
may be constituted for other trials involving particular risk, complexity, likely decisions about
early stopping or the need to obviate conflict of interest. The CRRC may also require during the
scientific review process that a DSMB be established for high-risk Phase I or Phase II
investigator-initiated clinical trials if it is deemed necessary.
Board Membership The DSMB shall be established prior to study activation by the clinical trial’s lead principal
investigator. The DSMB members should be appointed for a fixed term of service. Members may
be within or outside the OHSU Knight CI, but a majority of the DSMB should not be affiliated
with OHSU. None of the members should be directly involved in the study design,
implementation, or outcome evaluation of the clinical trial under their review. All members of
the DSMB would be required to sign a conflict of interest and confidentiality statement to ensure
their objectivity during their service as a board member.

2/27/08
16
A DSMB shall include a physician, oncology nurse, biostatistician, at least one non-health care
professional, and other health-care professionals with expertise required by this specific board.
While these DSMB members should not be involved in any way with this specific clinical trial,
the health-care professionals chosen should be selected with their expertise in the field of
oncology under study in mind.
The study specific DSMB shall work in cooperation with the CRRC and OHSU Institutional
Review Board (IRB), but will in no way replace either of those committees in the oversight of
the clinical trial. The DSMB shall report to the Lead NCI designated OHSU Investigator, the
CRRC and OHSU IRB.
Responsibilities of the DSMB
1. Establish a meeting and reporting schedule. The NCI requires at least annual reports. The
PI, CRRC, or the DSMB may choose a more frequent meeting schedule if deemed
necessary.
2. Review the research protocol and study specific DSMP and provide a pre-activation
report to the PI and CRRC.
3. Review interim analyses of outcome data and cumulative toxicity data summaries to
determine whether the trial should continue as originally designed, should be changed, or
should be terminated based on these data. The DSMB reviews trial performance
information such as accrual information. The DSMB also determines whether and to
whom outcome results should be released prior to the reporting of study results.
4. Review reports of related studies to determine whether the monitored study needs to be
changed or terminated.
5. Review major proposed modifications to the study prior to their implementation (e.g.,
termination, dropping an arm based on toxicity results or other reported trial outcomes,
increasing target sample size).
6. Following each DSMB meeting, provide the study leadership with written information
concerning findings for the trial as a whole related to cumulative toxicity observed and
any relevant recommendations related to continuing, changing, or terminating the trial. A
copy of this information will be provided to the NCI Division Director or designee. The
study leadership will provide information on cumulative toxicity and relevant
recommendations to the local principal investigators to be shared with their IRBs.
Reportable Adverse Event and Unanticipated Problem Monitoring
OHSU Knight Cancer Institute is responsible for ensuring that all reportable adverse events and
unanticipated problems are reported in compliance with local IRB standards, FDA regulations
and NIH/NCI policies. The Data and Safety Monitoring Committee (DSMC) with

2/27/08
17
administrative support from Clinical Research Management (CRM) is responsible for review and
approval of all serious and reportable adverse events/unanticipated problems.
The purpose of reportable adverse event (serious adverse event and unanticipated problems)
monitoring is to ensure that all reportable events identified in patients who are participating in
OHSU Knight CI clinical research studies are reported to the appropriate regulatory entity and
appropriately classified by causality. Additionally, at the time of IRB continuing review
summary reports are reviewed to identify toxicity trends in study patients. The Principal
Investigator is responsible for generating the summary report of all adverse events and toxicities
occurring on study. It is also the responsibility of the PI to inform the DSMC and the IRB using
the UP reporting system if an increased frequency of an expected toxicity has been discovered.
OHSU Knight CI member investigators and affiliate investigators will follow local IRB
standards, federal regulations and NCI/NIH guidelines in the reporting of adverse events (AE)
for all OHSU Knight CI approved studies. AE reporting procedures are detailed in each protocol
and depend upon the type of study, the type and severity of AE, the trial sponsor and IND status.
The DSMC will provide real time monitoring of each unanticipated problem prior to IRB
submission. To evaluate toxicity trends, a yearly summary of reportable events (unanticipated
problems and serious adverse events) is reviewed by the DSMC at the time of continuing (annual
at minimum) IRB review. If necessary, both the DSMC and IRB are empowered to immediately
suspend accrual until concerns related to the UP are addressed. They also have the authority to
suspend or terminate a study immediately based on patient safety concerns.
OHSU IRB Reporting Deaths and potentially life-threatening events must be reported within seven (7) calendar days
after the PI learns of the event. If any of these SAEs requires a change (as determined by the PI
or the IRB) to the protocol or consent form, the PI must make those changes promptly and
submit the revised documents to the OHSU IRB.
All other UPs must be reported within fifteen (15) calendar days. If the event requires changes
(as determined by the PI or the IRB) to the protocol or consent form, the PI must make those
changes promptly and submit the revised documents to the IRB.
The OHSU IRB UP reporting requirements and definitions of reportable AEs are outlined in the
OHSU IRB Policy and Procedure Manual at
http://www.ohsu.edu/research/rda/irb/docs/regulatory/Unanticipated%20Problems%20Re
gulatory%20Sheet.pdf
The OHSU UP Report Form will be utilized by all OHSU Knight CI staff to report an
unanticipated problem to the OHSU IRB. The form and instructions may be found at
http://www.ohsu.edu/research/rda/irb/ and https://eirb.ohsu.edu/irb (for electronic submission
on our electronic IRB system). All UP reports related to a patient on an OHSU Knight CI
protocol are sent directly to CRM for review and approval by a clinician member of the DMSC.
The reports are reviewed for completeness, appropriate classification of causality and possible

2/27/08
18
need for additional reporting to FDA, NIH/NCI or sponsor. If no further information or
clarification is required, the report is forwarded to the IRB.
In order to assess reportable event trends of UPs, the DSMC generates a monthly summary report
of all reportable events including local events, multicenter events and MedWatch reports that
qualify as UPs for review by the Clinical Research Review Committee (CRRC). The summary
report lists events by study, principal investigator and causality.
The summary report is forwarded to members of the CRRC prior to the monthly meeting. The
report is presented for discussion at the meeting. If the report is considered consistent with
expectations, it is approved by the CRRC. If further action is required it is the responsibility of
the DSMC to act on the recommendation of the CRRC. The proceedings are documented in the
CRRC minutes and the summary report is attached.
FDA reporting For local studies using approved drugs, all serious unexpected possibly related events are
reported on MedWatch FDA form 3500. The form and instructions are at
http://www.fda.gov/medwatch/report/consumer/instruct.htm. A copy of the MedWatch report is
sent to the CRM and forwarded to the IRB.
For studies using investigational drugs, the SAE report will be sent to the sponsor/IND holder
according to the individual protocol. A copy of the report is sent to the CRM and forwarded to
the IRB.
NIH/NCI reporting For NIH/NCI sponsored studies, the NCI Guidelines: Expedited Adverse Event Reporting
Requirements of NCI Investigation Agents will be followed. The guidelines can be found at
CTEP Home Page http://ctep.info.nih.gov. A copy of the report is sent to the CRM. If the SAE
qualifies as a UP, a copy of the report will be forwarded to the IRB.
Recombinant DNA/Gene Transfer Studies If a trial involves recombinant DNA Molecules (gene transfer) in addition to following reporting
requirements for investigational agents as above, NIH Guidelines for Research Involving
Recombinant DNA Molecules will be followed. These guidelines are located at:
http://www4.od.nih.gov/oba/rac/guidelines/guidelines.html
The flows chart on the next page illustrates the OHSU Knight CI procedures for real-time and
cumulative Serious Adverse Event monitoring.

2/27/08 19
Real Time Unanticipated Event Monitoring
UP Report signed by Principal Investigator and submitted
to CRM for review and approval by DSMC
Submit to OHSU IRB
Deaths must be reported within 7 days of notification. All
other UPs must be reported within 15 working days.
NIH/NCI Sponsored Studies
Submit report according to
NCI Guidelines: Expedited
Adverse Event Reporting
Requirements
http://ctep.info.nih.gov
Investigational Drug
Submit SAE report to
IND holder/ sponsor for
reporting to FDA
according to protocol
Approved Drug, IND
Exempt
Report possibly related,
unexpected SAE on
MedWatch 3500 either
directly or through CC
according to protocol
CRRC reviews monthly UP
summary for toxicity trends of all
OHSU Knight CI studies
DSMC reviews annual SAE/AE
summary for local, active treatment
studies at the time of IRB
Continuing Review
Central Coordination of
Multicenter Studies
CC forwards a Quarterly Summary
of all UPs & SAEs (or more
frequent) to affiliate sites for
reporting to each site’s IRB
Cumulative Adverse Event Monitoring
Real Time Unanticipated Event and SAE
Monitoring
UP Report signed by Principal Investigator and
submitted to CRM for review and approval by
DSMC.
SAE report prepared and submitted to all required
entities.
If UP reporting required If SAE reporting required

2/27/08 20
Appendix 1:
Members of the OHSU Knight Cancer Institute
Data and Safety Monitoring Committee
January 2008
Michael Mauro, MD
Committee Chair
Principal Investigator, Center for Hematologic Malignancies
Tibor Kovasovics, MD
Committee Co-Chair
Principal Investigator, Center for Hematologic Malignancies
Bashi Ratterree, RN, BSN, CCRP
Committee Co-Chair
Director, CRM, Compliance Manager, QA Auditor
Margaret McMahon, ANP
Safety Monitor
Motomi Mori, PhD
Director, Biostatistics Shared Resource, OHSU Knight Cancer Institute
Professor and Head, Division of Biostatistics, Department of Public Health &
Preventive Medicine
Byong Park, PhD
Senior Biostatistics Associate, Biostatistics Shared Resource, OHSU Knight Cancer Institute
Kristin Hackney, MPHA, CCRP
Assistant Director, CRM, QA Auditor
Kendra Todd, BS, CCRP
Industry Trials/HR Manager, CRM, QA Auditor
Susan Aust, MSPH, RD, CCRP
Investigator Initiated Projects Coordinator, QA Auditor, CRM
Kristin Pattee, BS, CCRP
Senior Research Assistant, QA Auditor, CRM
Representative from Research Pharmacy Services – to be determined

2/27/08 21
Appendix 2:
Quality Assurance (QA) Audit Forms
A: Review of Conformance to IRB Requirements
B: Review of Conformance to Protocol Requirements
C: Review of Patient Case Records

2/27/08 22
OHSU Knight Cancer Institute
Quality Assurance Audit Review of Conformance to IRB Requirements
Protocol:
IRB Number: _____________
Date: ____________________ Auditor:
Yes
No Deficiency Identified
Comments
Protocol never approved by IRB
Initial IRB approval documentation missing
Registration and/or treatment of patient prior to IRB approval
Reapproval delayed> 30 days but < 1 year
Registration of patient on protocol during a period of delayed
reapproval
Missing reapproval
Expired reapproval
Reportable adverse events/Unanticipated Problems not
reported to IRB/appropriate entity
Lack of documentation of full IRB approval of a protocol
amendment that affects more than minimal risk
Other (Specify)

2/27/08 23
OHSU Knight Cancer Institute
Quality Assurance Audit Review of Conformance to Protocol Requirements
Protocol:
IRB Number: _____________
Date: ____________________ Auditor:
Yes
No Deficiency Identified
Comments
Enrollment information not in Surveyor database
Drug accountability records not kept
Dose escalation/ de-escalation criteria not followed
Interim Analysis/ Stopping Rules not followed
Yes
No
Coordinating Center Responsibilities
(if applicable)
Comments
Subjects. have not been centrally registered in CRM database
Central reporting of UPs not documented
DSMP not provided/available for other sites
If audits conducted at other sites, audit results letters not
available/not forwarded to DSMC
Other (Specify)
Yes
No
IND/IDE Holders
(if applicable)
Comments
IND Safety reports not submitted to FDA
Protocol Amendments (including addition of investigators)
not submitted to FDA
Information Amendments not submitted to FDA
Annual reports not submitted to FDA
Other (Specify)

2/27/08 24
OHSU Knight Cancer Institute
Quality Assurance Audit Review of Patient Case Records
Protocol:
Patient Initials: Date:
Patient Number: Auditor:
Yes
No Informed Consent
Deficiency Identified
Comments
Consent form missing
Consent form not signed and dated by patient
Consent form signed after patient started on treatment
Consent form does not contain all required signatures
Consent form used was not current IRB approved version at
time of patient registration
Consent form does not include updates or information
required by IRB
Other (Specify)
Yes
No Eligibility
Deficiency Identified
Comments
Review of documentation confirms patient did not meet all
eligibility criteria as specified by the protocol
Documentation missing; unable to confirm eligibility
Other (Specify)

2/27/08 25
OHSU Knight Cancer Institute
Quality Assurance Audit Review of Patient Case Records
Protocol:
Patient Initials: Date:
Patient Number: Auditor:
Yes
No Treatment
Deficiency Identified
Comments
Incorrect agent/treatment used
Additional agent/treatment used which is not permitted by the
protocol
Dose deviations incorrect (error greater than +/ 10%)
Dose modifications unjustified
Treatment doses incorrectly administered, calculated or
documented (dose escalation/de-escalation as per protocol)
Unjustified delays in treatment
Other (Specify)
Yes
No Disease Outcome/Response
Deficiency Identified
Comments
Inaccurate documentation of initial sites of involvement
Tumor measurements/evaluation of status or disease not
performed according to protocol
Protocol-directed response criteria not followed
Claimed response (PR, CR, etc) cannot be verified
Failure to detect cancer (as in prevention study) or failure to
identify cancer progression
Other (Specify)

2/27/08 26
OHSU Knight Cancer Institute
Quality Assurance Audit Review of Patient Case Records
Protocol:
Patient Initials: Date:
Patient Number: Auditor:
Yes
No Toxicity
Deficiency Identified
Comments
Grades, types or dates/duration of serious toxicities
inaccurately recorded
Toxicities cannot be substained
Follow-up studies necessary to access toxicities not
performed
Failure to report a toxicity that would require filing an
Adverse Event Report (AER) or Unanticipated Problem
report
Recurrent under- or over-reporting of toxicities
Other (Specify, _______________________)
Yes
No
General Data Quality
Deficiency Identified
Comments
Recurrent missing documentation e.g., charts
Protocol-specified lab tests not documented
Protocol-specified diagnostic studies not documented
Frequent data inaccuracies
Errors in submitted data
Delinquent data submission
Other (Specify)





























