Embed Size (px)
Citation preview

The involvement of nicotinic cholinergic receptors in drug abuse
UNIVKRSIT V OF
SURREY
By
Athanasios Metaxas, M.Sc.
A thesis submitted in accordance with the requirements of the University of Surrey forthe Degree of Doctor of Philosophy
Faculty of Health and Medical Sciences March 2010
University of Surrey
Guildford
Surrey
GU2 7XH
U.K.
UNIVERSITY OF SURREY LIBRARY

ProQuest Number: U516352
All rights reserved
INFORMATION TO ALL USERS The quality of this reproduction is dependent upon the quality of the copy submitted.
In the unlikely event that the author did not send a com p le te manuscript and there are missing pages, these will be noted. Also, if material had to be removed,
a note will indicate the deletion.
uestProQuest U516352
Published by ProQuest LLO (2019). Copyright of the Dissertation is held by the Author.
All rights reserved.This work is protected against unauthorized copying under Title 17, United States C ode
Microform Edition © ProQuest LLO.
ProQuest LLO.789 East Eisenhower Parkway
P.Q. Box 1346 Ann Arbor, Ml 48106- 1346

11
ACKNOWLEDGEMENTS
I would like to thank my supervisor Professor Ian Kitchen, who gave me the opportunity
to undertake this project, providing invaluable scientific advice and support throughout
my studies. More than three years down the line, I now completely agree that ‘a day
without reading a paper is a day wasted’ and that ‘research without publications is like
blinking at a woman in the dark’. Also, I hereby confirm that Ian never says no to an
experiment!
Dr. Bailey’s help has been enormous during these years, from the ‘nyxta partying’ MSc
times up to now. Many thanks Alexis for being such a ‘kick ass’ friend, and for patiently
teaching me all o f the experimental techniques I have used in this thesis (including
loKopoK)! JiHoonio I am expecting to see you and your alter ego, Jerry Korea, very soon.
Our frequent scientific gatherings outside the AY building have given birth to concepts
such as shem, prv, kokiri, y \m , y \m , to mention but a few!! A million thanks go to Dr.
Al-Hasani for helping me in so many ways inside and out of the lab; Reamy, it has been a
pleasure to implant, talk, drive, and work-out with you (in the gym).
Dr. Ying Chen your help and critical discussion of the cocaine experiments is much
appreciated, and I wish I had time to do more of the work we discussed. Professor
Susanna Hourani thanks for helping me get my US VISA and for discussing work during
the lab meetings, which I really think should be resumed, along with more cakes and
baklava! Thanks go to Shelagh Hampton, Bennita Middleton, and Penny Giorgio for
their help with radiation work, and to Rebecca Holland for subbing 1,000,000 slides for
us all, on top of genotyping. Thanks to the EBU staff: to Kim Morton for organising the
unit, to Graham Moorey for animal handling tutorials, and to Graham and Alison for
being so helpful and nice to talk to. Special thanks to Zak Ghouze, as he is the best IT
support one could have ever wished for!
I would also like to thank everyone who works or has worked for longer or shorter
periods of time in 19AY02 and 23AY02, for being great friends and colleagues, and for
‘living a little’! Thanks to Lisa Wells, Martin Hussey, Matt Cooper, Bram Bekaert, Alys
Dreux, Josh Foster, Ana Slak (ex Voncina), Sapna Thakur, Helen Keyworth (aka Pye),

Ill
Sherie Rosie Smith (aka Miss Swinton), Sophie Wehrens and her Captain, Dr. Nikos,
Simone Maentele, Kat Lederle, Lloyd Morgan, Christiana Papamichael, Kylie as Such,
Vanessa Viegas, Dannyelza Pereira, Sibah Hasan, Rianne van der Spek, Andreas
Gerondopoulos, Alba Lopez y Helena Silva, who have all been proper parteh animals
and great scientists. Thanks to members of the study room with whom we didn’t have
the opportunity of visiting the pub on such a regular basis, like Shahida Shafi, Lesley
Beeton, George Vinoj, Mary Kelly, Danni Otway, David Ribe, Vicky Revell, Tracey
Sletten, Priti Chivers, Lei Teng, James Du, Margaret Gompers, Irina Vinogradova,
Thomas Kantermann, and Jenny Tickner. You have all made Surrey a great place to
work in.
Outside the lab, I would particularly like to thank my housemates, mama Vera Neves and
Elena Heister, for putting up with me, cooking for me, and discussing LOST with me.
You are the perfect people to live with! ! Bruno and daddy John, I am sure I will be seeing
both of you real soon. Last but not least, a special thanks to Elena Paraskevopoulou for
opening up the world of sustainability research, which we all know is the precondition
to.... ;-))
I would like to dedicate my thesis to my parents and sister, for their endless love and
support.
‘KOKOS Y KOKOS PRESENT AN’:

IV
ABSTRACT
The prevalence of nicotine addiction suggests that interactions of neuronal nicotinic
cholinergic receptors (nAChRs) with illicit substances and multiple neurotransmitter
systems are involved in mediating the rewarding properties of nicotine, cocaine, and
morphine. Hence, the overall aim of this thesis was to investigate the role of nAChRs in
rendering nicotine, a weak primary reinforcer, to be so widely abused.
Interactions between nAChRs and cocaine were investigated at the behavioural and
receptor density levels, following chronic cocaine treatment and withdrawal. The effects
of blockade of different nAChR subtypes on the development of cocaine-induced
behaviours were investigated using dihydro-fi-erythroidine (DhBE; 2.0 mg/kg/inj, i.p.)
and methyllycaconitine (MLA; 5.0 mg/kg/inj, i.p.). Nicotinic receptor antagonism
produced very distinct behaviours in C57BL/6 mice, treated for 14 days with a steady
dose, ‘binge’ administration protocol of cocaine (15.0 mg/kg/inj, i.p.). a4(32* nAChR
blockade by DhBE prolonged cocaine-induced horizontal locomotor activity, reduced
vertical activity, and diminished stereotypy sensitization. On the contrary, a l nicotinic
receptor antagonism by MLA resulted in the delayed expression of cocaine-induced
rearing sensitization and induced a unique, intense grooming phenotype in cocaine-
treated animals. To further understand the mechanisms behind this behavioural
dissociation, quantitative autoradiography of heteromeric and homomeric nAChRs was
performed using [^^^Ijepibatidine and [^^^I]a-bungarotoxin, respectively. Following
‘binge’ cocaine administration, a4p2* nAChR density was increased in the ventral
tegmental area (VTA) and substantia nigra compacta o f cocaine-treated animals,
compared to saline controls. Moreover, following 14 days of abstinence from cocaine
treatment, a l nAChR density was decreased in the VTA of cocaine withdrawn animals.

V
compared to controls. These cocaine-induced, subtype- and brain region-specific
alterations in nAChR density were not observed following 8 days of treatment with
escalating doses of ‘intermittent’ morphine, nor after the acute and prolonged withdrawal
of C57BL/6 mice from morphine administration.
An investigation into the mechanism of interactions between nicotinic cholinergic and
adenosinergic systems was performed using CDl mice with a genetic deletion of the
adenosine A2A receptor. Following 14 days of nicotine hydrogen salt administration at 24
mg/kg/day via omotic minipumps, quantitative autoradiography of [^^^I]epibatidine and
[^^^I]a-bungarotoxin binding sites revealed significant genotype x treatment interaction
effects on a7 nAChR density. Although chronic nicotine equally increased heteromeric
nAChR density in wild type and mutant mice, its effects on a7 nAChRs were
predominantly observed in the brains of wild type, rather than adenosine A2A receptor
knockout animals.
The mechanisms of nicotine reinforcement were investigated using quantitative
autoradiography of cytisine-sensitive [^^^I]epibatidine binding sites, following the
acquisition of nicotine self-administration by drug naïve C57BL/6 mice. Using an
operant yoked-control protocol of nicotine treatment, increased a4p2* nAChR density
was observed in the ventral tegmental area and dorsal lateral geniculate nucleus of self-
administering animals, compared to mice that passively received nicotine.
Overall, the results of the present thesis suggest that alterations in a4p2* and/or a7
nAChR density occur following chronic treatment with nicotine and illicit substances,
which are indicative of the interactions that render a weak primary reinforcer as broadly
abused as nicotine.

VI
CONTENTS
ACKNOWLEDGEMENTS...................................................................................................üABSTRACT...............................................................................................................................LIST OF FIGURES................. .............................................................................................. ...LIST OF TABLES............................................................................................................... xiiiLIST OF PRESENTATIONS & PUBLICATIONS........................................................xvLIST OF ABBREVIATIONS........................................................................................... xvii
1 GENERAL INTRODUCTION ..............................................................................2
1.1 Nicotinic acetylcholine receptors in the CNS...........................................................41.1.1 Structure of the nicotinic acetylcholine receptor.................................................. 41.1.2 Localisation of neuronal nAChRs.......................................................................... 7
1.1.2.1 Distribution and classification o f nAChR subtypes....................................... 71.1.2.2 Presynaptic, postsynaptic and non synaptic location..................................11
1.1.3 Functional aspects of nAChRs.............................................................................. 121.1.3.1 Neuronal nAChR transition states................................................................ 121.1.3.2 Current rectification.......................................................................................141.1.3.3 Neuronal nAChR upregulation......................................................................14
1.1.4 Physiology of neuronal nAChRs..........................................................................181.1.4.1 Presynaptic, postsynaptic and non synaptic nAChRs..................................181.1.4.2 nAChR subunit knockout mice.......................................................................20
1.1.5 Pharmacology of neuronal nAChRs.................................................................... 231.1.5.1 Neuronal nAChR agonists..............................................................................251.1.5.2 Neuronal nAChR antagonists........................................................................27
1.2 nAChRs in drug addiction........................................................................................ 291.2.1 Drug addiction: a chronic disorder o f neuronal adaptation............................... 291.2.2 The central role of dopamine................................................................................ 31
1.2.2.1 The Dopaminergic system: localisation, function, receptors.....................311.2.2.2 The Dopaminergic signal............................................................................... 33
1.2.3 nAChRs and dopaminergic signalling................................................................. 351.2.3.1 Anatomical foundations..................... 351.2.3.2 Cholinergic modulation ofDAergic signalling in the VTA ........................371.2.3.3 Cholinergic modulation o f DAergic signalling in the NAc ............... 38
1.3 Interactions between nAChRs and drugs of abuse............................ 401.3.1 nAChRs and cocaine..............................................................................................40
1.3.1.1 Cocaine’s mode o f action ......................................................................4Q1.3.1.2 Neurochemical evidence fo r interactions between nAChRs and cocaine. 411.3.1.3 Behavioural evidence fo r interactions between nAChRs and cocaine 43
1.3.2 nAChRs and opioid abuse..................................................................................... 451.3.2.1 The opioid system: Localisation, function, receptors................................. 461.3.2.2 Neurochemical evidence fo r interactions between nAChRs and opioids. 481.3.2.3 Behavioural evidence fo r interactions between nAChRs and opioids 50

vil
1.3.3 nAChRs and adenosine systems...........................................................................521.3.3.1 The adenosine system: Localisation, function, receptors........................... 531.3.3.2 Neurochemical evidence fo r interactions between nAChRs and adenosine ....................................................................................................................................... 551.3.3.3 Behavioural evidence fo r interactions between nAChRs and adenosine.. 59
1.3.4 Interactions between nAChRs and other drugs of abuse....................................61
2 THESIS AIMS AND HYPOTHESIS.........................................................................63
3 BEHAVIOURAL AND NEUROCHEMICAL INTERACTIONS BETWEEN COCAINE AND THE NICOTINIC CHOLINERGIC SYSTEM................................ 67
3.1 Introduction.................................................................................................................. 673.2 Materials and Methods...............................................................................................69
3.2.1 nAChR antagonist dose selection studies............................................................693.2.1.1 Antagonism o f acute nicotine-induced hypolocomotion by DhfiE 693.2.1.2 Chronic nicotine pretreatment and MLA-precipitated withdrawal........... 69
3.2.2 Animals and ‘binge’ cocaine treatment............................................................... 703.2.3 Minipump preparation and nicotine pretreatment experiments.........................713.2.4 Motor activity measurements................................................................................ 723.2.5 Cocaine-induced stereotypy and grooming.........................................................733.2.6 Quantitative receptor autoradiography................................................................ 74
3.2.6.1 Autoradiography o f dopamine D1 and D2, nicotinic cholinergic a4p2"^ and a7 receptors, and o f dopaminergic and cholinergic transporters.................. 743.2.6.2 Film exposure and development............................................... 773.2.6.3 Quantitative analysis......................................................................................78
3.2.7 Statistical analysis.................................................................................................. 793.3 Results............................................................................................................................80
3.3.1 Preliminary studies................................................................................................. 803.3.1.1 DhfiE dose selection........................................................................................803.3.1.2 MLA dose selection.........................................................................................81
3.3.2 Effect of chronic nAChR blockade on cocaine-induced horizontal activity... 823.3.3 Effect o f chronic nAChR blockade on cocaine-induced vertical activity 853.3.4 Effect of chronic nAChR blockade on cocaine-induced grooming and stereotypy..........................................................................................................................863.3.5 Quantitative autoradiography of a4p2* and a7 nAChRs, and of the high affinity choline transporter, following chronic, steady dose, ‘binge’ cocaine treatment...........................................................................................................................893.3.6 Effects of nicotine pretreatment on cocaine-induced locomotor stimulation.. 963.3.7 Quantitative autoradiography of D1 and D2 receptors, and of the dopamine transporter, following chronic, steady dose, ‘binge’ treatment....................................993.3.8 Quantitative autoradiography of a4p2* and a7 neuronal nAChRs, following prolonged withdrawal from steady dose, ‘binge’ cocaine treatment........................ 1033.3.9 Quantitative autoradiography of D1 and D2 receptors, and of the dopamine transporter, following prolonged withdrawal from steady dose, ‘binge’ cocaine treatment.........................................................................................................................106
3.4 Discussion.................................................................................................................... 109

Vlll
4 QUANTITATIVE AUTORADIOGRAPHY OF HETEROMERIC AND HOMOMERIC NEURONAL nAChR SUBTYPES IN C57BL/6 MICE, FOLLOWING CHRONIC MORPHINE TREATMENT AND WITHDRAWAL 120
4.1 Introduction................................................................................................................1 2 04.2 Materials and Methods...................................................... 122
4.2.1 Animals and drug treatment................................................................................ 1224.2.2 Quantitative autoradiography of a4p2* and a? nicotinic cholinergic receptors............... 1234.2.3 Statistical analysis................................................................................................ 124
4.3 Results......................................................................................................................... 1254.3.1 Quantitative autoradiography of heteromeric nAChRs following chronic morphine administration and withdrawal....................................................................1254.3.2 Quantitative autoradiography of a? nAChRs following chronic morphine administration and withdrawal......................................................................................130
4.4 Discussion.................................................................................................................... 132
5 CHANGES IN THE NICOTINIC CHOLINERGIC AND DOPAMINERGIC SYSTEMS OF MICE DEFICIENT IN THE A2A GENE, EXPLORED BY QUANTITATIVE AUTORADIOGRAPHY.................................................................. 139
5.1 Introduction................................................................................................................ I3 95.2 Materials and methods.............................................................................................141
5.2.1 Generation of the A2A KG mouse and experimental conditions...................... 1415.2.2 Minipump preparation and implantation........................................................... 1425.2.3 Quantitative receptor autoradiography.............................................................. 1425.2.4 Determination of nicotine and cotinine levels ........................................ 1445.2.5 Statistical analysis................................................................................................ I44
5.3 Results..........................................................................................................................I4 55.3.1 Naïve study........................................................................................................... I45
5.3.1.1 Nicotinic receptor autoradiography in naïve WT and adenosine A2A receptor KO mice.................................................. 1455.3.1.2 Dopaminergic receptor and transporter autoradiography in naïve WT and adenosine A2A receptor KO mice ............................................................................. 757
5.3.2 Nicotine treatment................................................................................................ I 535.3.2.1 Confirmation o f genotype............................................................................ 7555.3.2.2 Levels o f plasma nicotine and cotinine following chronic nicotine administration......................................... 7555.3.2.3 Nicotinic receptor autoradiography in WT and adenosine A2A receptor KO mice, following chronic nicotine treatment............................................................ 7555.3.2.4 Dopaminergic receptor and transporter autoradiography in WT and adenosine A2A receptor KO mice, following chronic nicotine treatment 162
5.4 Discussion.................................................................................................................... 1 6 6

IX
6 QUANTITATIVE AUTORADIOGRAPHY OF HETEROMERIC NICOTINIC AND DOPAMINERGIC RECEPTORS, AND OF THE DOPAMINETRANSPORTER IN C57BL/6 MICE, FOLLOWING CONTINGENT AND NONCONTINGENT NICOTINE ADMINISTRATION 174
6.1 Introduction................................................................................................................1746.2 Materials and methods.............................................................................................176
6.2.1 Self administration paradigm.............................................................................. 1766.2.1.1 Animals..........................................................................................................1766.2.1.2 Surgery and self-administration procedure............................................... 176
6.2.2 Quantitative receptor autoradiography.............................................................. 1786.2.3 Statistical analysis................................................................................................ 179
6.3 Results......................................................................................................................... 1806.3.1 Nicotine self-administration................................................................................ 1806.3.2 Quantitative receptor autoradiography.............................................................. 181
6.3.2.1 Effects o f response contingency on nAChR density...................................1816.3.2.2 Effects o f response contingency on dopaminergic D1 and D2 receptors, and on dopamine transporter density......................................................................189
6.4 Discussion..................................................................................................................193
7 CONCLUSIONS AND FUTURE WORK..............................................................200
REFERENCES..................................................................................................................... 208

X
LIST OF FIGURES
Figure 1.1 Structure of the nicotinic acetylcholine receptor subunit................................... 5
Figure 1.2 Organisation and structure of neuronal nAChRs................................................. 6
Figure 1.3 Transition states of neuronal nAChRs................................................................ 13
Figure 1.4 Ligand binding sites on a neuronal nicotinic receptor...................................... 24
Figure 1.5 Subtypes of nAChRs involved in DAergic neurotransmission........................36
Figure 3.1 Antagonism of nicotine-induced hypolocomotion by DhBE............................ 80
Figure 3.2 MLA-precipitated nicotine withdrawal..............................................................81
Figure 3.3 Effect of nicotinic acetylcholine receptor blockade on cocaine-induced horizontal activity.................................................................................................................... 82
Figure 3.4 Effect of daily ‘binge’ treatment on total horizontal activity...........................84
Figure 3.5 Effect of nicotinic acetylcholine receptor blockade on cocaine-induced rearing ....................................................................................................................................................85
Figure 3.6 Effect of nicotinic acetylcholine receptor blockade on cocaine-induced grooming (A) and correlation between grooming and locomotor behaviour in cocaine+MLA treated animals (B)..........................................................................................87
Figure 3.7 Effect of nicotinic acetylcholine receptor blockade on cocaine-induced stereotypy................................................................................................................... 89
Figure 3.8 Representative autoradiograms of heteromeric (A), and a7 nAChR binding (B), following chronic ‘binge’ cocaine.................................................................................. 93
Figure 3.9 Representative autoradiograms of [^Hjhemicholinium binding following chronic ‘binge’ cocaine (± nAChR antagonists) administration..........................................95
Figure 3.10 Effect of nicotine pretreatment on cocaine-induced motor activity 98
Figure 3.11 Representative autoradiograms of [^HJraclopride binding following chronic ‘binge’ cocaine........................................................................................................................100
Figure 3.12 Representative autoradiograms of [^H]mazindol binding following chronic ‘binge’ cocaine........................................................................................................................102
Figure 3.13 Quantitative autoradiography of heteromeric nAChR binding following withdrawal from chronic ‘binge’ cocaine............................................................................ 103

XI
Figure 3.14 Quantitative autoradiography of cytisine-sensitive (A) and cytisine-resistant(B) [^^^IJepibatidine binding following withdrawal from chronic ‘binge’ cocaine 104
Figure 3.15 Quantitative autoradiography of a7 nAChR binding following withdrawal from chronic ‘binge’ cocaine................................................................................................ 105
Figure 3.16 Representative autoradiograms of [^^^I]a-bungarotoxin binding following withdrawal from chronic ‘binge’ cocaine............................................................................106
Figure 3.17 Quantitative autoradiography of D1 receptor binding following withdrawal from chronic ‘binge’ cocaine................................................................................................ 107
Figure 3.18 Quantitative autoradiography of D2 receptor binding following withdrawal from chronic ‘binge’ cocaine.............................................................. 108
Figure 3.19 Quantitative autoradiography of dopamine transporter binding following withdrawal from chronic ‘binge’ cocaine............................................................................ 109
Figure 4.1 Representative autoradiograms of [^^^IJepibatidine binding following A. chronic morphine treatment and acute morphine withdrawal, and B. long term withdrawal from ‘intermittent’ morphine administration.......................................................................126
Figure 4.2 Representative autoradiograms of [^^^I]a-bungarotoxin binding following morphine treatment and withdrawal.....................................................................................130
Figure 5.1 Quantitative autoradiography of heteromeric nAChR binding in naïve WT and adenosine A2A receptor KO mice..........................................................................................146
Figure 5.2 Quantitative autoradiography of cytisine-sensitive nAChR binding in naïve WT and adenosine A2A receptor KO mice........................................................................... 147
Figure 5.3 Quantitative autoradiography of cytisine-resistant nAChR binding in naïve WT and adenosine A2A receptor KO mice........................................................................... 148
Figure 5.4 Quantitative autoradiography of a7 nAChR binding in naïve WT and adenosine A2A receptor KO mice..........................................................................................149
Figure 5.5 Representative autoradiograms of [^^^I]epibatidine (A) and [^^^I]a- bungarotoxin (B) binding in naïve wild type and adenosine A2A receptor KO m ice 150
Figure 5.6 Quantitative autoradiography of D2 receptor binding in naïve WT and adenosine A2A receptor KO mice..........................................................................................151
Figure 5.7 Quantitative autoradiography of D1 receptors (A) and dopamine transporters (B) in naïve WT and adenosine A2A receptor KO mice......................................................152
Figure 5.8 Adenosine A2A receptor expression in naïve CDl wildtype and adenosine A2A receptor KO mice................................................................................................................... I53

X ll
Figure 5.9 Mean plasma nicotine and cotinine levels following chronic nicotine administration....................................................................................................................... ..
Figure 5.10 Representative autoradiograms of [^^^I]epibatidine binding in nicotine- treated wild type and adenosine A2A receptor KO mice.....................................................159
Figure 5.11 Representative autoradiograms of [^^^IJbungarotoxin binding in nicotine- treated wild type and adenosine A2A receptor KO m ice.....................................................160
Figure 5.12 Quantitative autoradiography of dopaminergic D1 receptor binding in nicotine-treated WT and adenosine A2A receptor KO mice............................................... 163
Figure 5.13 Quantitative autoradiography of dopaminergic D2 receptor binding in nicotine-treated WT and adenosine A2A receptor KO mice............................................... 164
Figure 5.14 Quantitative autoradiography of dopaminergic transporter binding in nicotine-treated WT and adenosine A2A receptor KO mice............................................... 165
Figure 6.1 Acquisition and maintenance of nicotine self-administration by C57BL/6 mice 180
Figure 6 .2 Computer-enhanced colour autoradiograms of total, cytisine-resistant, and non-specific [^^^IJepibatidine binding in coronal sections from saline-treated C57BL/6 mouse brain..............................................................................................................................
Figure 6.3 Representative autoradiograms of heteromeric nAChR binding in coronal brain sections from nicotine contingent, non contingent, and saline treated animals 188
Figure 6.4 Quantitative autoradiography of D2 receptor binding in nicotine contingent, non contingent, and saline treated animals.......................................................................... 190
Figure 6.5 Quantitative autoradiography of D1 receptors (A) and dopamine transporters (B) in nicotine contingent, non contingent, and saline treated animals............................ 191
Figure 6 .6 Computer-enhanced colour autoradiograms of dopamine D2 (A) and D1 receptors (B), and of DA transporters (C), in coronal brain sections from nicotine contingent, non contingent, and saline treated animals......................................................192
Figure 7.1 Proposed mechanism for the modulation of the effects of cocaine via nAChRs in the VTA and SNc...............................................................................................................201

X lll
LIST OF TABLES
Table 1.1 Classification of neuronal nAChRs....................................................................... 8
Table 1.2 Phenotypes of nAChR subunit gene knockout mice...........................................21
Table 1.3 Opioid receptors in the mouse brain.................................................................... 47
Table 1.4 Adenosine receptors in the brain...........................................................................54
Table 3.1 Quantitative autoradiography of heteromeric nAChRs, following ‘binge’ treatment of cocaine ± nAChR antagonists...........................................................................90
Table 3.2 Quantitative autoradiography of a4g2* nAChRs, following ‘binge’ treatment of cocaine ± nAChR antagonists.............................................................................................91
Table 3.3 Quantitative autoradiography of cytisine-resistant nAChR density, following ‘binge’ treatment of cocaine ± nAChR antagonists..............................................................92
Table 3.4 Quantitative autoradiography of a7 nAChRs following ‘binge’ treatment ofcocaine ± nAChR antagonists................................................................................................. 94Table 3.5 Quantitative autoradiography of choline transporters, following ‘binge’ treatment of cocaine ± nAChR antagonists......................................................................... 96
Table 3.6 Quantitative autoradiography of D2 receptors, following ‘binge’ treatment of cocaine ± nAChR antagonists................................................................................................. 99
Table 3.7 Quantitative autoradiography of D1 receptors, following ‘binge’ treatment of cocaine ± nAChR antagonists............................................................................................... 101
Table 3.8 Quantitative autoradiography of dopamine transporters, following ‘binge’treatment of cocaine ± nAChR antagonists....................................................................... 102
Table 4.1 Daily morphine intake........................................................................................ 123
Table 4.2 Quantitative autoradiography of total [^^^IJepibatidine binding, followingchronic morphine treatment and withdrawal.......................................................................127
Table 4.3 Quantitative autoradiography of cytisine-sensitive [^^^IJepibatidine binding, following chronic morphine treatment and withdrawal......................................................128
Table 4.4 Quantitative autoradiography of cytisine-resistant [^^^IJepibatidine binding, following morphine treatment and withdrawal....................................................................129
Table 4.5 Quantitative autoradiography of [^^^I]a-bungarotoxin binding, following morphine treatment and withdrawal.....................................................................................131

XIV
Table 5.1 Quantitative autoradiography of total [^^^I]epibatidine binding in WT and adenosine A2A receptor KO mice, following chronic nicotine administration.................156
Table 5.2 Quantitative autoradiography of cytisine-sensitive [^^^IJepibatidine binding in WT and adenosine A2A receptor KO mice, following chronic nicotine administration.. 157
Table 5.3 Quantitative autoradiography of cytisine-resistant [^^^IJepibatidine binding in WT and adenosine A2A receptor KO mice, following chronic nicotine administration .158
Table 5.4 Quantitative autoradiography of a7 nAChR binding in nicotine-treated WT and adenosine A2A receptor KO mice.......................................................................................... 161
Table 6.1 Quantitative autoradiography of total [^^^I]epibatidine binding in nicotine contingent, non contingent, and saline treated animals......................................................184
Table 6.2 Quantitative autoradiography of cytisine-sensitive [^^^I]epibatidine binding in nicotine contingent, non contingent, and saline treated anim als....................................... 186
Table 6.3 Quantitative autoradiography of cytisine-resistant [^^^IJepibatidine binding in nicotine contingent, non contingent, and saline treated animals....................................... 187

XV
LIST OF PRESENTATIONS & PUBLICATIONS
Journals
A. Bailey, A. Metaxas, JH. Yoo, T. McGee, and I. Kitchen (2008) Decrease o f D2
receptor binding but increase in D2-stimulated G-protein activation, dopamine transporter
binding and behavioural sensitization of mice treated with a chronic escalating dose
‘binge’ cocaine administration paradigm. Eur. J. Neurosci. 28, 759-70
A. Bailey, A. Metaxas, R. Al-Hasani, D. Forster, H. Keyworth, and I. Kitchen (2010)
Mouse strain differences in locomotor, sensitization and rewarding effect of heroin;
association with alterations in MOP-r activation and dopamine transporter binding. Eur. J.
Neurosci. 31, 742-53
Unpublished Conference Proceedings
A. Metaxas, F. Barbano, L. Galeote, R. Maldonado, and I. Kitchen (2007) Quantitative
autoradiography of a4p2* nicotinic, D1 and D2 receptors, and the dopamine transporter
in C57BL/6 mice, following self-administration of nicotine. International Narcotics
Research Conference', IX 20
A. Bailey, A. Metaxas, D. Forster, and I. Kitchen (2007) A comparison of locomotor,
rewarding and neurochemical effects of chronic heroin treatment on the opioid and
dopaminergic systems in C57BL/6J and DBA/2J mice. International Narcotics Research
Conference', IX 21
J. Cabello, K. Wells, A. Metaxas, A. Bailey, I. Kitchen, A. Clark, M. Prydderch, and R.
Turchetta (2007) Digital autoradiography imaging using CMOS technology: First tritium,
autoradiography with a back thinned CMOS detector and comparison of CMOS imaging
performance with autoradiography film. IEEE Nuclear Science Symposium Conference
Record', M l9-91

XVI
A. Metaxas, JH. Yoo, A. Bailey, and I. Kitchen (2008) Effect o f a4^2* or a l nicotinic
acetylcholine receptor blockade on the initiation and development of cocaine
sensitization in C57BL/6 mice. Nicotinic Acetylcholine Receptors', P53
A. Metaxas, R. Al-Hasani, J. Foster, S. Hourani, I. Kitchen, and Y.Chen (2008) Nicotinic
acetylcholine receptor expression and function in the adenosine A2A receptor knockout
mouse. Nicotinic Acetylcholine Receptors', P I7
A. Metaxas, H. Keyworth, JH. Yoo, Y. Chen, A. Bailey, and I. Kitchen (2009) Neuronal
nicotinic receptor subtypes differentially influence cocaine-induced locomotor activity
and stereotypy following a chronic ‘binge’ treatment. Society fo r Neurosciences', S9
253.30
R. Al-Hasani, A. Metaxas, A. Bailey, S. Hourani, and I. Kitchen (2009) Differences in
dopamine and D2 binding between CDl mice that respond and those that do not respond
to cocaine-induced conditioned place preference. Society fo r Neurosciences', Z21 553.8
Oral presentations
A. Metaxas, H. Keyworth, JH. Yoo, Y. Chen, A. Bailey, and I. Kitchen (2009)
Behavioural and neurochemical interactions between cocaine and the nicotinic
cholinergic system. Gennadict annual workshop, Reykjavik, Iceland

X V ll
LIST OF ABBREVIATIONS
AC adenylyl cyclase
AcbC nucleus accumbens, core
AcbSh nucleus accumbens, shellACh acetylcholine
Amy amygdala
AV anteroventral thalamic nucleus
a-BgTx alpha bungarotoxin
a-CtxMII alpha conotoxin Mil
BLA basolateral amygdaloid nucleus
CAl hippocampus, CAl
CA3 hippocampus, CAB
cAMP 3', 5'-cyclic adenosine monophosphate
Cg cingulate cortex
CHT choline transporter
Cl claustrum
CNS central nervous system
CPP conditioned place preference
CPu caudate putamen
CPuC caudate putamen, caudal part
CPuR caudate putamen, rostral part
CREB cAMP response element binding proteinDA dopamine
DAT dopamine transporter
DEn dorsal endopiriform nucleus
DG dentate gyrus
DhBE dihydro-p-erythroidine
DLG dorsal lateral geniculate nucleus
DM dorsomedial hypothalamic nucleusDOP delta opioid
ER endoplasmic reticulum
fr fasciculus retroflexus

XVlll
FrA frontal association cortex
GABA y-aminobutyric acid
5 -HT3 5-hydroxytryptamine 3Hip hippocampus
Hyp hypothalamus
InG intermediate gray layer of the superior colliculusIP interpeduncular nucleus
i.p. intraperitoneal
i.v. intravenous
KO knockout
KOP kappa opioid
LH lateral hypothalamus
LPMR lateral posterior thalamic nucleusM l primary motor cortex
M2 secondary motor cortex
MAN medial amygdaloid nucleus
mAChRs muscarinic acetylcholine receptors18-MC 18-methoxycoronaridineMCx motor cortex
MG medial geniculate nucleus
MHb medial habenula
MLA methyllycaconitine
MOP mu opioid
mRNA messenger ribonucleic acid
pM micromolar
NAc nucleus accumbens
nAChRs nicotinic acetylcholine receptorsNET norepinephrine transporter
nM nanomolar
NOP nociceptin opioid
ns nigrostriatal bundle
NSB non specific binding
PLCo posteromedial cortical amygdaloid nucleus

XIX
pM picomolar
Po posterior thalamic nuclear group
PrL prelimbic cortex
S subiculum
s.c. subcutaneous
SERT serotonin transporter
SN substantia nigra
SNc substantia nigra pars compacta
SuG superficial gray layer of the superior colliculus
THC delta9-tetrahydrocannabinol
Tu olfactory tubercle
VI visual cortex
VLG ventral lateral geniculate nucleus
VM ventromedial hypothalamic nucleus
VTA ventral tegmental area
WT wildtype
ZI zona incerta

CHAPTER
ONE

1 GENERAL INTRODUCTION
Cholinergic signalling in the central nervous system (CNS) is mediated by the
endogenous neurotransmitter acetylcholine (ACh), exerting its effects through two
distinct classes of receptors: the muscarinic and nicotinic ACh receptors (mAChRs and
nAChRs, respectively). The latter are multimeric ligand-gated ion channels that have
been demonstrated to participate in cognitive processes, such as learning, memory, and
attention, as well as in the perception of pain and the control of movement in normal
subjects (reviewed in Dani and Bertrand, 2007). Recent advantages in our understanding
of brain nAChR structure, distribution and subtype diversity, have unmasked the
contribution of nicotinic cholinergic signalling in several CNS dysftinctions, such as
epilepsy, schizophrenia, Parkinson’s and Alzheimer’s disease, autism, dementia with
Lewy bodies, and addiction (reviewed in Albuquerque et al., 2009).
In the beginning of the 1990’s, there was still a great deal of controversy as to whether
cigarette smoking constituted a form of drug-addiction, or was simply a social habit. A
large amount of published research has since established that persistent smoking
behaviour is the result of the reinforcing properties of nicotine, binding in the CNS to its
corresponding pharmacological targets, the nAChRs. In this respect, nicotine shares with
other addictive drugs the common property of activating the mesolimbic dopaminergic
pathway, which originates in the ventral tegmental area and projects towards limbic
forebrain regions, including the nucleus accumbens (Di Chiara and Imperato, 1988). This
shared property establishes a neural substrate for the examination of a striking feature of
nicotine addiction, which is its high co-morbidity with substance abuse disorders. Indeed,
not only is nicotine addictive, but it has also been implicated as a ‘gateway’ to increased
substance consumption, since it is often self-administered in combination with several

illicit and licit substances, including the psychostimulants (Weinberger and Sofuoglu,
2009), the opioids (Pomerleau, 1998), ethanol (Funk et al., 2006), and caffeine (Swanson
et al., 1994). This positive correlation between smoking and enhanced drug use has
promped the investigation of the nicotinic cholinergic mechanisms that may contribute to
the prevalence of poly-drug dependence in cigarette smokers. To this end, the regulation
of different subtypes o f nAChRs in response to chronic cocaine or morphine treatment
and withdrawal has been examined in mice by means of quantitative receptor
autoradiography. To investigate interactions between nicotine and the adenosine system,
the regulation of different nAChR subtypes following chronic nicotine treatment has been
examined in mice with a genetic deletion of the adenosine Aia receptor.
The activation of midbrain dopaminergic pathways is a neurobiological commonality that
is shared by nicotine and other drugs of abuse, potentially accounting for their concurrent
consumption. Nevertheless, it is still argued that nicotine is only a weak primary
reinforcer, and that the prevalence of nicotine addiction stems from its ability to enhance
the potency of other reinforcers, rather than from the drug’s direct rewarding effects
(reviewed in Chaudhri et al., 2006). To unravel the mechanisms of nicotine
reinforcement, the regulation of the nicotinic cholinergic and dopaminergic systems
during the acquisition of nicotine self-administration by drug naïve mice has been studied
using an operant yoked-control protocol. The latter has been employed to examine
differences produced by the active or passive nicotine administration, and to determine
the extent to which nicotine-induced neuroadaptation is the consequence of the drug’s
direct pharmacological action or the result of the contingency between a subject’s
response and the delivery of a reinforcer.

1.1 Nicotinic acetylcholine receptors in the CNS
1.1.1 Structure of the nicotinic acetylcholine receptor
Nicotinic acetylcholine receptors (nAChRs) are typical members of the ‘cys-loop’ ligand-
gated ion channel superfamily, which also includes y-aminobutyric acid (GABAa,
GAB Ac), glycine, and 5-hydroxytryptamine 3 (5-HTs) serotonin receptors (reviewed in
Karlin and Akabas, 1995). Based on their major site of expression, nAChRs can be
subdivided into muscle and neuronal types, the latter of which form hetero- or
homomeric complexes of related subunits. All members of the nicotinic cholinergic
family are characterised by a pentameric structure. They comprise five membrane-
spanning subunits, arranged symmetrically around a central ion channel to form a barrel
like structure (reviewed in Karlin, 2002). Figure 1.1 shows the topography of a single
nAChR subunit, which is composed of a long ( -2 1 0 amino acids) N-terminal region,
four hydrophobic transmembrane helical domains (Mi - M4), and a shorter C-terminal.
The N- and C- hydrophilic termini are located on the extracellular region of the
membrane, and an intracellular loop is formed between the third and fourth
transmembrane domains. The second membrane-spanning helical segment (M2) from
each subunit shapes the central channel, which has a diameter of -25 Â at the synaptic
level and it progressively becomes narrower at the transmembrane level (Unwin et al.,
1988). As the M2 helix contains far more negatively than positively charged amino acids,
the central pore in the ion channel becomes highly cation selective. Indeed, all nAChRs
identified so far are permeable to positively charged ions (reviewed in Changeux and
Edelstein, 1998).

Mt
Figure 1.1 Structure o f the nicotinic acetylcholine receptor subunit
Schematic linear representation of a single nAChR subunit: the N-terminal hydrophilic portion, the transmembrane
domain, and the small hydrophilic C-terminal domain. Taken from Karlin, (2002).
To date, genes encoding for eight a (a2, a3, a4, a5, a6 , a l, a9, and alO) and three p (p2,
P3, and p4) mammalian neuronal nicotinic subunits have been cloned (reviewed in
Nashmi and Lester, 2006). In addition, subunit a 8 has been identified in avian species
(Schoepfer et al., 1990). Subunits a2 through alO, and P2 through p4 combine to form
either heteromeric pentameric structures, made up by a or p subunits in an axPy
stoichiometry, or homomeric (a7)5 and (a9)s nAChRs. Structural data obtained from the
crystallisation of an acetylcholine-binding protein from the European snail Lymnaea
stagnalis have greatly improved our understanding of the nicotinic receptor binding sites,
which lie at the interface between two adjacent subunits at the N-terminal domain
(Corringer et al., 1995; Prince and Sine, 1996; Brejc et al., 2001). In analogy with
muscle type nAChRs, and with the exception of subunit a5, all a subunits carry the
principal agonist binding site, defined by a pair of vicinal cysteines (Wang et al., 1996).
Subunits P2 and p4 lack the N-terminal cysteine residues and provide the complementary
binding site, whereas p3 subunits do not contribute to an agonist binding site. Thus, in
the case of the homomeric nAChRs, it is the vicinity of the a subunits that defines the

acetylcholine (ACh) binding pocket, and five identical binding sites exist per receptor
molecule. In heteromeric receptors, such as the (u4)2((32)3 receptor, the binding pocket is
formed between the a4 and its neighbouring |32 subunit. Thus, two ligand binding sites
exist per receptor molecule, and both ct and (3 subunits contribute to the pharmacology of
the heteromeric receptor (Luetje and Patrick, 1991; Figure 1.2).
(A)
a l
o 7 0.7
«7 Y «7.
Homomericreceptor
ACh
(B)Ion c h a n n e l p o r e
ACh
■ E x tr a c e l ju la r
(C)
a4
In t race l lu la r
Heteromericreceptor
Figure 1.2 Organisation and structure o f neuronal nAChRs
(A) and (C); Rotational symmetry of nicotinic subunits and localisation o f the ACh binding sites in transversal sections
o f homomeric a7 and heteromeric a4p2 nAChR subtypes. Subunits are arranged in an anticlockwise manner along the
axis. Taken from Gotti and Clementi, (2004). (B): Schematic representation o f the subunits in the heteromeric
receptor, the location o f the two acetylcholine (ACh) binding sites, and the central ion conducting channel. Adapted by
Karlin, (2002).
Such wide variety of nicotinic subunit associations gives rise to multiple neuronal
nAChR subtypes, with distinct physiological, pharmacological and biochemical
properties.

1.1.2 Localisation o f neuronal nAChRs
1.1.2.1 Distribution and classification o f nAChR subtypes
In order to describe the topographical distribution of various nAChR subtypes, it is
necessary to first summarise the main cholinergic pathways in the brain. Nearly every
region of the CNS receives cholinergic input from five clusters of cholinergic nuclei
(reviewed in Woolf, 1991). These are briefly described below:
• The Magnocellular basal complex provides the majority of cortical and hippocampal
cholinergic input.
• The Peduncolopontine-laterodorsal tegmental complex originates from the
peduncolopontine tegmental nucleus and projects to the thalamic nuclei and the
midbrain dopamine neurons.
• The Striatum is densely innervated by intrinsic cholinergic neurons that do not project
beyond the borders of the striatum.
• The Habenulo-interpeduncular system originates from the medial habenula and
projects to the interpeduncular nucleus via the fasciculus retroflexus. This system
receives input from the thalamus and is part of a limbic network thought to affect
visceral and behavioural functions, as well as responses to pain.
• The cholinergic neurons of the Lower brain stem innervate the superior colliculus,
cerebral nuclei, and cortex.
The diffuse nature of cholinergic neurons throughout the CNS accounts for the broad
distribution of neuronal nAChRs, which were initially distinguished on a
pharmacological basis (Clarke et ah, 1985b). High resolution autoradiography, using
radiolabeled nicotine, acetylcholine and the neuromuscular antagonist a-bungarotoxin (a-
BgTx), revealed a separation of the a-BgTx binding sites from the nicotine and

acetylcholine binding proteins. It was therefore deduced that more than one neuronal
nicotinic receptor population exists in the CNS. With the advent of molecular cloning
techniques, this pharmacological heterogeneity has been confirmed and extended.
Structurally divergent nicotinic agonists and antagonists, as well as mice lacking the p2
subunit have been employed to map the composition of distinct nAChR subtypes in
different brain regions, using autoradiographic, in situ hybridization, and patch-clamp
techniques (Zoli et al., 1998; see also Section 1.2.3.1). Expanding on a previous
classification from Alkondon and Albuquerque, (1993), and along with data from
previous anatomical and electrophysiological studies, four main types of neuronal
nAChRs have been indentified, which are summarised in Table 1.1.
Receptortype
Subunitcomposition
Predominant localisation in CNS
Pharmacology (Equilibrium binding &
electrophysiology)
I a?Cortex, Limbic area. Hippocampus,
Hypothalamus, and other telencephalic areas
MLA and a-BgTx sensitive
very fast desensitising epibatidine>nicotine>cytisine
II
a4p2 (a5?) Throughout CNS
a-BgTx insensitive epibatidine>cytisine=nicotine
(a2?)P2 Interpeduncular nucleus
(a3?)P2Retina, Superior Colliculus, Lateral Geniculate nucleus. Hippocampus
(a6p3?)P2 Catecholaminergic nuclei
III a3p4 (a5?)Medial Habenula, Interpeduncular
nucleus, Pineal glandMLA-insensitive
epibatidine
IV(a4p4?) Lateral Habenula MLA-insensitive
epibatidine=cytisine>nicotine(a2p4?) Dorsal Interpeduncular nucleus
Table 1.1 Classification o f neuronal nAChRs
Four classes o f neuronal nAChRs have been distinguished by means o f autoradiographic, in situ hybridization, and patch-clamp
techniques. The table shows the putative subunit composition o f each class o f receptor, its predominant localisation in the CNS, and its
characteristic pharmacology. The question mark denotes the nicotinic subunit most likely to participate in the formation o f functional
nAChRs, from experiments on the anatomy and electrophysiology o f nAChRs. Abbreviations: CNS; central nervous system, a-BgTx; a
bungarotoxin, MLA; methyllycaconitine. Adapted from Zoli et al., (1998).

9
Type I receptors. Neuronal nAChRs of this class bind a-bungarotoxin (a-BgTx) and
the competitive antagonist methyllycaconitine (MLA) with high affinity. The pattern
of labelling by a-BgTx is not altered in p2 subunit knockout animals (p2 -/-), whereas
in a7 knockout mice a-BgTx binding sites completely disappear (Orr-Urtreger et al.,
1997), indicating that the P2 subunit is not a component of type I receptors. The
distribution of type I receptors represents the localisation of the a7 homomeric
nAChRs, which is higher in the hippocampal formation, the hypothalamus, and other
telencephalic areas (Marks et al., 1986).
Type II receptors. Containing the p2 subunit (p2*), this class of receptors is a-
bungarotoxin insensitive, representing the majority of neuronal nAChRs in the mouse
brain. The most ubiquitously expressed receptor in this group is composed of a4 and
p2 subunits and it binds nicotinic agonists with nanomolar or subnanomolar affinities.
This high affinity binding is lost in p2 or a4 subunit knockout animals (Picciotto et al.,
1995; Marubio et al., 1999). a4p2* receptors are highly expressed throughout the
mammalian CNS, including dopaminergic areas and the striatum. The p2 subunit is
likely to co-assemble with others, namely the a2, a3, a5 (Vemallis et al., 1993; Wang
et al., 1996; Zoli et al., 2002), and the p4 (Conroy and Berg, 1995). Despite the fact
that such combinations have a relatively restricted distribution in the brain, they may
constitute the main functional subpopulations of nAChRs in the neurons where they
are expressed. The a3|32* subtype has been demonstrated to be mostly expressed in
the visual pathway (Gotti et al., 2005), whereas the density of the a6|32* subtype is
higher in catecholaminergic nuclei, the locus coeruleus, the substantia nigra, the VTA,
and the striatum (Le Novere et al., 1996; Champtiaux et al., 2003). Overall, type II
receptors can either constitute simple subtypes, or assemble in more complex forms,
in axUy p combinations (see Gotti et al., 2006).

1 0
• Type III receptors. They do not contain the p2 subunit and they bind epibatidine with
high affinity in equilibrium binding studies. The distribution of type III receptors
represents the localisation of a3p4* nAChRs, which is predominant in the habenulo-
interpeduncular system, locus coeruleus, cerebellum, area postrema, and the pineal
gland (Flores et al., 1996; Marks et al., 2002; Hernandez et al., 2004). Type III
receptors may comprise the a5 subunit, albeit this expression pattern does not change
their binding properties.
• Type IV receptors. Containing the P4 subunit in combination with the o2 or a4
subunit, this class of receptors binds epibatidine and cytisine with equally high
affinity. They are highly expressed in the lateral habenula and the interpeduncular
nucleus. Type IV can be distinguished from type III receptors on the basis of their
faster desensitisation rates in electrophysiological experiments (Zoli et al., 1998).
At this point, it should be mentioned that the exact subunit composition of neuronal
nAChRs is an ongoing question. In view of the fact that the assembly of individual
nAChRs depends on the availability of related subunits (Conroy and Berg, 1995;
Ramirez-Latorre et al., 1996) and the common observation that multiple nicotinic
receptor channels can be expressed by a single neuron (Alkondon and Albuquerque,
1993; Connolly et al., 1995), characterising the composition and distribution of multiple
nAChR subtypes is an extremely demanding task. Nevertheless, a large body of evidence
suggests that the most common hetero- and homo- oligomeric nAChRs expressed in the
brain are assembled by a4/p2 and a7 subunits, respectively (Wada et al., 1989; Couturier
et al., 1990; Chen and Patrick, 1997).

11
1.1.2.2 Presynaptic, postsynaptic and non synaptic location
Confocal and electron microscopy studies have revealed that neuronal nAChRs can be
distributed not only pre- and post- synaptically, but also at extrasynaptic locations, such
as the somatodendritic and axonal parts of an individual neuron. In addition, nAChRs
can be localised intracellularly, where they are often associated with the endoplasmic
reticulum before translocation to the cell membrane (Jones and Wonnacott, 2004). The
relative synaptic vs. extrasynaptic distribution of nAChRs on the cell membrane differs
for distinct nicotinic subtypes. For instance, glutamatergic nerve terminals in the ventral
tegmental area (VTA) that predominantly express the homomeric a7 nAChR, have 73 %
of the nicotinic receptor located at presynaptic and preterminal sites, compared with 27 %
that is situated at extrasynaptic locations (Jones and Wonnacott, 2004). For nigrostriatal
dopaminergic axon terminals predominantly expressing the p2 subunit, however, nearly
all P2 containing nAChRs are observed at extrasynaptic sites (Jones et al., 2001).
Therefore, it seems that distinct nicotinic receptor subtypes are strategically expressed
along the length of a neuron; depending on their pre-, post-, and non synaptic distribution,
nAChRs can influence a plethora of synaptic events, generating or altering action
potentials and modifying membrane excitability (reviewed in McKay et al., 2007). The
physiological significance of the diverse localisation of nAChRs in neurons is discussed
in more detail in Section 1.1.4.

1 2
1.1.3 Functional aspects o f nAChRs
1.1.3.1 Neuronal nAChR transition states
Nicotinic receptors in the CNS can exist in three different fiinctional states: resting (R),
active (A), and desensitised (D) (reviewed in Galzi and Changeux, 1995). Upon binding
of ACh the pore-lining helices rotate and the receptor channel extensively changes its
conformation (Unwin, 2005). The central pore opens to allow conductance of small
monovalent (Na" and K^) and divalent cations (C a ^ down their electrochemical gradient,
causing the receptor to switch from the resting to an activated state. In this open pore
state the receptor displays low micromolar (pM) affinity for ACh, whose continued
presence will eventually lead to receptor desensitisation and ion pore closure. Albeit the
fact that desensitised nAChRs are refractory to activation, they exhibit 2-4 fold higher
affinity for ACh (Heidmann and Changeux, 1980). Dissociation of the agonist from its
binding site will cause the receptor to isomerise to the resting state in the millisecond
time range (Lester and Dani, 1995). Prolonged exposure to low concentrations of
agonists, however, can induce a pronounced desensitised state from which recovery
occurs with slower rates (Sakmann et al., 1980; Lester and Dani, 1995). Desensitisation
is thus a reversible phenomenon that can occur in stages, with a transient, fast-onset
desensitised state converting into a more stable, slow-onset desensitised state in the
persisting presence of an agonist (Wilson and Karlin, 2001). The cycle of activation,
desensitisation, and recovery is depicted in Figure 1.3. The kinetics for these processes is
highly dependent on the subunit composition of neuronal nicotinic receptors. Under
normal physiological conditions, where high concentrations of ACh (~lmM) diffuse in
the synaptic cleft and acetylcholinesterases hydrolyse the neurotransmitter in
milliseconds, the homomeric a7 nicotinic receptor displays fast desensitisation and
recovery kinetics (Castro and Albuquerque, 1993; Anand et al., 1998), whereas

heteromeric nAChRs, such as the a4p2, desensitise slower (Fenster et a l, 1997; Corringer
et al., 1998). During prolonged periods of sustained exposure to low (~1 pM) agonist
concentrations, however, a4p2 nAChRs will significantly desensitize, whereas a l
nAChRs will effectively remain unaffected (Dani et al., 2000; Quick and Lester, 2002;
Wooltorton et al., 2003). Therefore, desensitisation is an intrinsic property of individual
nAChR subtypes and it depends on the concentration and length of exposure to agonists.
Its physiological role becomes extremely important, specifically when considering the
low levels of nicotine (30-600 nM) that chronically bathe the brains of smokers (Russell
et al., 1980; Henningfield et al., 1993).
(R) RESTING Channel closed
nA ChR a g o n is ts b in d w ith LOW AFFINITY
&(A) ACTIVE channel open
(D) DESENSITISEDchannel closed
rx
(I) INACTIVE
nAChR a g o n is ts b in d w ith HIGH AFFINITY
Figure 1.3 Transition states o f neuronal nAChRs
Schematic representation of the functional states that nAChRs undergo. The diagram shows the transition from resting
(R) and desensitised (D) nonconducting states (closed pore), to active (A), conducting conditions (open pore).
Prolonged exposure to agonists leads the receptor to switch from a desensitised to an inactive state (I), from which the
recovery rate is very slow. Each transition state is characterised by different receptor-ligand kinetics.

14
1.1.3.2 Current rectification
Activated nicotinic receptors pass current freely when a cell is hyperpolarised, providing
strong inward voltage to drive cations into the channel. This functional feature of the
neuronal nAChR is distinct from the NMDA subtype of glutamate receptors or other
voltage-gated ion channels that are normally blocked by external Mg " at a cell’s resting
potential. As the transmembrane potential becomes more positive than -40 mV the
current is reduced, and at very active depolarised postsynaptic membranes nAChRs
progressively contribute less to signal transmission (Couturier et al., 1990; Mathie et al.,
1990). The whole process is termed current rectification, and it is considered to be a
molecular mechanism that facilitates the propagation of action potentials towards nerve
terminals (Haghighi and Cooper, 2000).
1.1.3.3 Neuronal nAChR upregulation
In general, prolonged exposure to agonists induces receptor down-regulation, as a
consequence of overstimulation (see Creese and Sibley, 1981). In the case of neuronal
nAChRs, however, the opposite is true; nicotinic receptor density has been shown to
increase following continued exposure to agonists. This has been well documented for
nicotine, the principal psychoactive ingredient in tobacco. Postmortem radioligand
binding studies reveal a dose-dependent increase in [^HJnicotine and [^HJepibatidine
binding sites in the brains of chronic smokers, compared with non-smokers (Benwell et
al., 1988; Breese et al., 1997). The observed upregulation displays marked regional
variability, with many areas of the brain participating and showing different sensitivity to
nAChR upregulation (Benwell et al., 1988; Perry et al., 1999). The results described
above have confirmed in humans the findings of numerous rodent studies, which first
showed that neuronal nAChR upregulate following long term exposure to nicotine. Since

15
the discovery that nicotinic binding sites are increased after repeated injections of
nicotine in rats and following chronic nicotine infusion in mice, the phenomenon of
nAChR upregulation has been established as the hallmark of chronic nicotine treatment
(Marks et al., 1983; Schwartz and Kellar, 1983). In both rats and mice, neuronal nAChR
upregulation is a process that requires hours to weeks to develop, depending on the
nicotine dose and the duration of treatment (Marks et al., 1985; Rowell and Li, 1997).
Moreover, subtypes of nAChRs show different sensitivity to upregulation in vivo.
Following 14 days of nicotine administration to rats, at concentrations that are compatible
with those observed in the blood of smokers, a4p2* nAChRs seem to be readily
upregulated in the range of 20-100% over controls, compared with other heteromeric
subtypes, containing a3, a6 or g4 subunits (Nguyen et al., 2003). In addition, chronic
infusion of nicotine in the mouse robustly upregulates a4^2*, rather than a l nAChRs
(Pauly et al., 1991). Therefore, it seems that the affinity of individual receptor subtypes
for nicotine greatly contributes to nAChR upregulation, with «4^2* nAChRs being
affected the most. Evidence from in vitro experiments, however, suggests that chronic
nicotine has the potential to upregulate several nAChR subtypes in a dose and time
dependent manner, and that this numerical upregulation occurs with magnitudes and rates
that depend on the individual subunit composition of each receptor (Ke et al., 1998;
Warpman et al., 1998; Fenster et al., 1999). Neuronal nAChR upregulation is a non
permanent, reversible phenomenon. As revealed in rodent and human post-mortem brain
studies, the upregulation of nAChR binding returns to control levels with a time course
that depends on the dose and duration of prior nicotine exposure (Marks et al., 1985;
Breese et al., 1997; Miao et al., 1998).

16
Neuronal nAChR upregulation seems to be associated with an increase in receptor
numbers, without an increase in the mRNA expression of associated subunits. In situ
hybridisation studies in the mouse revealed no treatment effects on the levels of a4 and
P2 subunit mRNA, following chronic nicotine infusions at a rate of 4.0mg/kg/hr (Marks
et al., 1992; Pauly et al., 1996). Following 7 or 10 days of nicotine administration,
however, an average increase of 42 % or 67 % in nicotine binding sites was observed
respectively, indicating that the numerical upregulation of nAChRs is independent of
transcriptional events. Similar results have been obtained in the rat after repeated
postnatal injections of 0.1 mg/kg of nicotine, twice for 20 days (Miao et al., 1998). In
vitro, MIO cells that stably express recombinant a4p2 nAChRs exhibit 2.5-fold increases
in the amount of a4p2 protein, but fail to show increased transcription of a4 or p2 subunit
mRNA following three days of treatment with 5pM of nicotine (Peng et al., 1994).
Therefore, it seems that post-transcriptional events, such as decreased surface receptor
internalisation (Peng et al., 1994) or increased receptor trafficking to the cell surface
(Harkness and Millar, 2002), increased assembly of subunits from existing pools in the
endoplasmic reticulum (ER) (Kuryatov et al., 2005) or blockade of nicotinic subunit
degradation in the ER (Corringer et al., 2006), as well as nicotine-induced conformational
changes of nAChRs (Vallejo et al., 2005) mediate receptor upregulation.
This unusual phenomenon of agonist-induced receptor upregulation has been the subject
of intense investigation as to its functional consequences. It has been proposed that
upregulation reflects receptor desensitisation and loss of nAChR function. Indeed,
following chronic incubation of oocytes expressing the a4(32 nAChR with nicotine, the
half maximal concentration necessary to produce desensitisation is the same as that
needed to induce upregulation of the receptor (Fenster et al., 1999). Moreover, rats

17
injected twice daily with nicotine for a period of 10 days, show increased a4p2* nAChR
density, but decreased nicotine-induced striatal dopamine release, compared with saline
controls (Jacobs et al., 2002). Accordingly, the increase in a4g2* nAChR binding sites in
brain regions of mice chronically treated with nicotine highly correlates with a decrease
in functionality of the receptor in the same brain areas, as measured by ACh-stimulated
^^Rb efflux assays (Marks et al., 2004). On the other hand, substantial evidence that
upregulated nAChRs retain function also exists. Measurement of epibatidine-stimulated
^^Rb efflux from rat brain slices, suggests that chronic nicotine administration not only
induces receptor upregulation, but also that these receptors are functional (Nguyen et al.,
2004). Similarly, functional potentiation of human neuronal nAChRs stably expressed in
cell lines has been demonstrated using patch clamp techniques (Wang et al., 1998;
Buisson and Bertrand, 2001). The complex relation between nAChR upregulation and
function can be demonstrated by the fact that, even for a particular receptor subtype, the
effects of nicotine have been found to differ, depending on brain region (Nguyen et al.,
2004) and cellular specific neural distribution (Nashmi et al., 2007). Therefore, further
studies are needed to determine the functional consequences of nAChR upregulation,
since data on the effects of chronic nicotine treatment on receptor function are still
controversial. It seems probable, however, that the relative subunit association and final
assembly of an individual nAChR subtype will define its functional response to agonist-
induced upregulation.

18
1.1.4 Physiology o f neuronal nAChRs
1.1.4.1 Presynaptic, postsynaptic and non synaptic nAChRs
In contrast to nAChRs of the neuromuscular junction, which are located post-synaptically
to mediate fast, excitatory transmission, the general consensus is that nicotinic receptors
in the CNS are predominantly found at presynaptic locations, where they play a
modulatory role in neurotransmission (see Role and Berg, 1996). Their activation by
nicotinic agonists has been implicated in the enhanced release of glutamate (McGehee et
al., 1995), GAB A (Endo et al., 2005), noradrenaline (Barik and Wonnacott, 2006),
acetylcholine (Wilkie et al., 1996), dopamine (Whiteaker et al., 1995), and 5-
hydroxytryptamine (Reuben and Clarke, 2000). Application of nicotinic antagonists, on
the other hand, decreases neurotransmitter release. The mechanism behind this
modulation seems to be calcium mediated. Nicotinic receptors allow the passage of a
small direct calcium efflux, which in turn triggers calcium-induced calcium release from
intracellular stores (Sharma and Vijayaraghavan, 2003). In addition, activation of
nAChRs may indirectly promote exocytosis through presynaptic voltage-gated calcium
channels (Tredway et al., 1999). The overall effect is an elevation of intraterminal
calcium, which is sufficient to evoke neurotransmitter release (reviewed in Wonnacott,
1997). As neuronal nAChRs exhibit different cation permeability, each receptor subtype
makes a distinct contribution to the modulation of neurotransmission. Often, the highly
calcium permeable homomeric a l receptor is involved, but in other cases different
nAChR subtypes mediate enhanced neurotransmitter release.
Fast nicotinic transmission has been rarely documented in the CNS. Structural evidence
suggests that only 7-14 % of acetylcholine release sites make synaptic contacts in the
mammalian brain (Descarries, 1998). Therefore, due to the low density of nicotinic

19
synapses, and because of the diffuse nature of cholinergic neurons in the CNS, the
detection of fast nicotinic signalling in brain slices has been experimentally difficult.
Indeed, functional nAChRs have only been recently documented at postsynaptic sites, in
brain structures that have well-established sources of cholinergic afferents, such as the
hippocampus and the neocortex. For instance, postsynaptic nicotinic transmission has
been observed in the rodent hippocampus as a small excitatory input onto GABAergic
intemeurons (Alkondon et al., 1998; Frazier et al., 1998). Moreover, direct nAChR-
mediated signalling can be evoked onto both glutamatergic pyramidal and GABAergic
neurons in the developing visual cortex (Roerig et al., 1997). Given the wide distribution
of nAChRs throughout the CNS, however, it is likely that nAChR-mediated synaptic
activity also occurs in brain regions with less well-established cholinergic inputs. This
has been shown in the supraoptic nucleus of the hypothalamus, and other subcotircal
areas, including the VTA (Pidoplichko et al., 1997; Hatton and Yang, 2002). Overall,
however, evidence for a postsynaptic role of nAChRs in mediating synaptic transmission
is scarce, suggesting that fast nicotinic signalling is not a major excitatory mechanism in
the CNS.
Beyond their specific pre- and post- synaptic roles at discrete synapses, nAChRs are also
widely expressed at non synaptic sites on an individual neuron. Pre-terminal nAChRs,
located before the presynaptic bouton, have been shown to indirectly potentiate GABA
release by activating voltage-gated ion channels (Lena et al., 1993; Alkondon et al., 1997).
Axonal, dendritic and somatic nAChRs may also influence synaptic transmission, by
altering membrane impedance and thereby influencing the spread and efficiency of an
action potential (see Dani and Bertrand, 2007). It is thus conceivable that nAChRs are

2 0
strategically located along neuronal cells, in order to modulate communication between
pre-synaptic neurons and their targets in a broader sense.
1.1.4.2 nAChR subunit knockout mice
The wide variety of nicotinic receptor subtypes expressed in the CNS, coupled with the
lack of subtype-specific ligands, have rendered knockout (KO) mice an extremely
important tool for investigating the contribution of particular nAChRs to specific
nicotine-induced behaviours (reviewed in Cordero-Erausquin et al., 2000; Marubio and
Changeux, 2000). In addition, genetic manipulations of the nicotinic cholinergic system
have revealed new information not only for the subunit composition of functional
nAChRs, but also for the role of defined receptors in mediating normal physiological
function. To date several different nicotinic subunit genes have been silenced, as listed in
Table 1.2. Of them, only the a3 subunit seems to be necessary for survival, since a3 KO
mice survive 1-8 weeks postnatal, showing severe autonomic nervous system defects (Xu
et al., 1999). Deletion of the a4 nAChR subunit diminishes nicotine-induced
antinociception in the hot-plate and tail-flick tests (Marubio et al., 1999), and produces
mutant mice that show increased basal levels of anxiety and are less sensitive to the
locomotor depressant effects of nicotine, compared with their wild type (WT) littermates
(Ross et al., 2000). The a5 subunit seems to be directly involved in the pathogenesis of
nicotine-induced seizures (Salas et al., 2003b), and its expression in presynaptic vs.
extrasynaptic locations differentially contributes to nAChR function (Fischer et al., 2005).
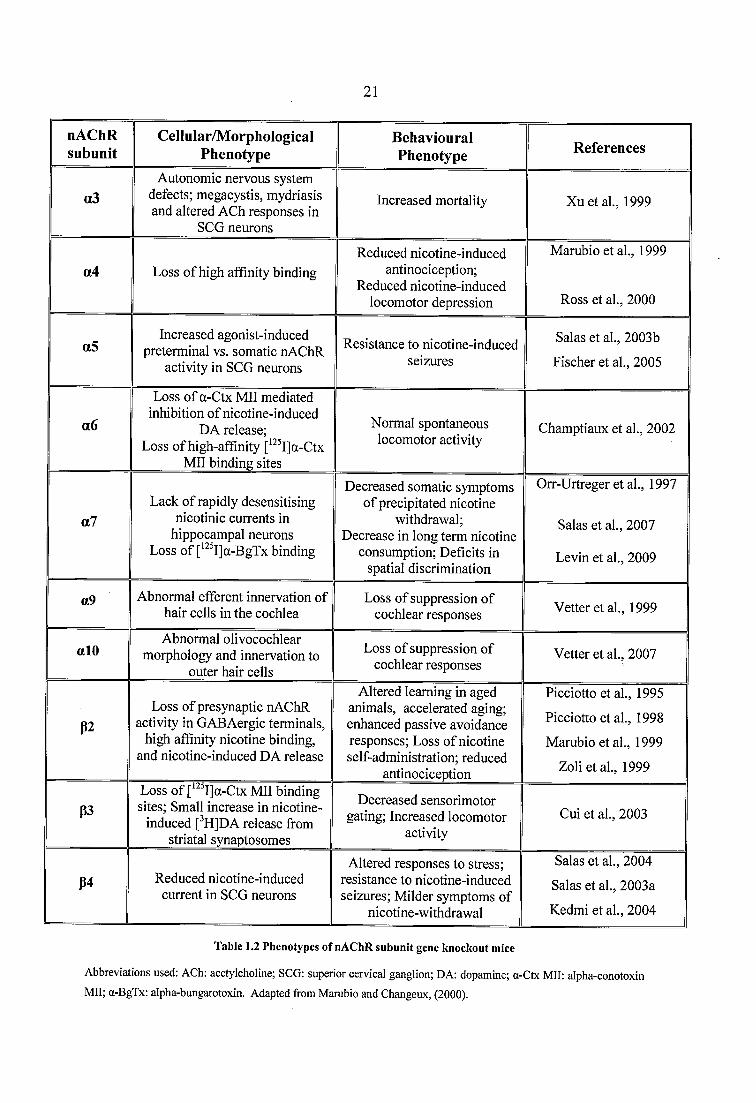
2 1
nAChRsubunit
Cellular/MorphologicalPhenotype
BehaviouralPhenotype References
o3Autonomic nervous system
defects; megacystis, mydriasis and altered A(3h responses in
SCG neurons
Increased mortality Xu et al., 1999
a4 Loss of high affinity bindingReduced nicotine-induced
antinociception; Reduced nicotine-induced
locomotor depression
Marubio et al., 1999
Ross et al., 2000
a5Increased agonist-induced
preterminal vs. somatic nAChR activity in SCG neurons
Resistance to nicotine-induced seizures
Salas et al., 2003b Fischer et al., 2005
a6
Loss of a-Ctx Mil mediated inhibition of nicotine-induced
DA release;Loss of high-affmity [*^ I]a-Ctx
Mil binding sites
Normal spontaneous locomotor activity
Champtiaux et al., 2002
a7Lack of rapidly desensitising
nicotinic currents in hippocampal neurons
Loss of [^^ I]a-BgTx binding
Decreased somatic symptoms of precipitated nicotine
withdrawal;Decrease in long term nicotine
consumption; Deficits in spatial discrimination
Orr-Urtreger et al., 1997
Salas et al., 2007
Levin et al, 2009
a9 Abnormal efferent innervation of hair cells in the cochlea
Loss of suppression of cochlear responses Vetter et al, 1999
alOAbnormal olivocochlear
morphology and innervation to outer hair cells
Loss of suppression of cochlear responses
Vetter et al, 2007
P2Loss of presynaptic nAChR
activity in GABAergic terminals, high affinity nicotine binding,
and nicotine-induced DA release
Altered learning in aged animals, accelerated aging; enhanced passive avoidance responses; Loss of nicotine self-administration; reduced
antinociception
Picciotto et al, 1995
Picciotto et al, 1998 Marubio et al, 1999
Zoli et al, 1999
p3Loss of a-Ctx Mil binding sites; Small increase in nicotine-
induced [^H]DA release from striatal synaptosomes
Decreased sensorimotor gating; Increased locomotor
activityGui et al, 2003
P4 Reduced nicotine-induced current in SCG neurons
Altered responses to stress; resistance to nicotine-induced seizures; Milder symptoms of
nicotine-withdrawal
Salas et al, 2004 Salas et al, 2003a Kedmi et al, 2004
Table 1.2 Phenotypes o f nAChR subunit gene knockout mice
Abbreviations used: ACh: acetylcholine; SCG: superior cervical ganglion; DA: dopamine; a-Ctx M il: alpha-conotoxin
M il; a-BgTx: alpha-bungarotoxin. Adapted from Marubio and Changeux, (2000).

2 2
a6 KO mice do not exhibit any gross neurological or behavioural defects; their generation,
however, has revealed that a6 instead of a3 subunits combine with the (32 to form
functional nAChRs that modulate nicotine-induced dopamine release in the mouse brain
(Champtiaux et al., 2002). Mice lacking the a l subunit are viable and anatomically
normal, despite completely lacking a-BgTx binding sites (Orr-Urtreger et al., 1997). In
the open field test, a l mutants spent more time in the centre of the open field compared to
WT animals, which is suggestive of a decreased baseline anxiety level (Paylor et al.,
1998). Subsequent studies, however, have failed to replicate this finding using a battery
of anxiety-related behavioural tests (Salas et al., 2007). Somatic symptoms of nicotine
withdrawal are decreased in mice lacking the a l nicotinic subunit (Salas et al., 2007),
which has been recently implicated in spatial learning and discrimination, as well as in
regulating long term nicotine consumption (Levin et al., 2009). The a9 subunit is
necessary for the development and function of the synaptic connections in cochlear outer
hair cells (Vetter et al., 1999). alO subunit KO mice exhibit abnormal olivocochlear
morphology and innervation to outer hair cells, indicating that expression of the alO
subunit is necessary for normal olivocochlear synaptic activity (Vetter et al., 2007).
Aged (32 subunit KO mice show degenerative alterations in the cortex and the
hippocampus, which resemble those observed during physiological aging and
Alzheimer’s disease in humans. The neurodegeneration is accompanied by significant
decreases of performance in the Morris maze task, suggesting impaired cognitive
functioning in aged animals, following deletion of the p2 subunit (Zoli et al., 1999). In
addition to their role in cognition and neuronal survival, p2 subunits have also been
implicated in learning and memory, as P2 KO mice fail to show the nicotine-induced
improvement of performance in the passive avoidance task, which tests for associative
memory (Picciotto et al., 1995). Moreover, nicotine self-administration and nicotine-

23
elicited antinociception are attenuated in the p2 subunit KO mouse, indicating that the
reinforcing and antinociceptive properties of the drug are exerted via p2* nAChRs
(Picciotto et al., 1998; Marubio et al., 1999). Deletion of the p3 nAChR subunit increases
locomotor activity in the open field and decreases prepulse inhibition of the acoustic
startle response in p3 null mutant mice, compared to WT littermates (Cui et al., 2003).
p4 nAChR mutant mice exhibit milder signs of mecamylamine-precipitated nicotine
withdrawal (Salas et al., 2004), are resistant to nicotine-induced seizures (Kedmi et al.,
2004), and differentially respond to stress, depending on the type of anxiety-provoking
experience (Salas et al., 2003a). In conclusion, the generation of KO mice has greatly
elucidated the relationship between individual nicotinic subunits, functional nAChR
subtypes and nAChR-mediated behaviour. Despite the fact that compensatory
developmental changes can occur in response to the deletion of a particular subunit gene
(reviewed in Lowenstein and Castro, 2001), a wealth of data has been generated
regarding the physiology of neuronal nAChRs using homologous recombination
technologies.
1.1.5 Pharmacology o f neuronal nAChRs
The structural features of the ligand binding site determine the pharmacological profile of
an individual nAChR subtype. As the ligand binding pocket of distinct nAChRs is
formed by the assembly of different nicotinic subunits, neuronal nicotinic receptors
display pharmacological properties that highly depend on their subunit composition. In
addition, there are ligands that bind to locations other than the agonist binding sites and
modulate nAChR function (allosteric modulators). Overall, four groups of ligands can be
distinguished, depending on their location of binding to the nicotinic ion channel (Fig.
1.4):

24
• Agonists, such as acetylcholine, nicotine and epibatidine occupy the agonist binding
site and activate the receptor.
• Antagonists, such as dihydro-P-erythroidine (DhBE) and methyllycaconitine (MLA),
compete with agonists for the nAChR binding site, but do not activate the receptor.
• Allosteric modulators bind to the interface between adjacent subunits, albeit at
locations different from the agonist binding pocket. Allosteric modulators can either
potentiate or decrease nAChR channel activity. Examples include the positive
modulator of receptor function, estradiol (Curtis et al., 2002), and the negative
allosteric modulator progesterone (Valera et al., 1992).
• Channel blockers, such as mecamylamine, bind within the ion channel pore and
block ion fluxes.
agonists and non-competitivecompetitive i inhibitors andantagonists ^ modulators
non-com petittve channel blockers ' >' cytoskeieton
Figure 1.4 Ligand binding sites on a neuronal nicotinic receptor
A representation o f the different locations that ligands can bind on a nAChR. Depending on the site o f binding, four
groups of ligands have been distinguished; agonists and antagonists, which compete for the same binding site, allosteric
modulators, which can occupy multiple binding sites, and non competitive channel blockers, which bind within the
channel pore. Adapted from Dani, (2001).
Detailed reviews on nicotinic receptor ligands can be found elsewhere (Lloyd and
Williams, 2000; Romanelli et al., 2007). In this section, the pharmacology of ligands that
were used in the present set of experiments will be addressed in more detail.

25
1.1.5.1 Neuronal nAChR agonists
• (-)-Nicotine [3-(l-methyl-2-pyrrolidinyl)pyridine] is an alkaloid isolated from the
leafs of the plant Nicotiana tabacum, and it is the prototype nAChR agonist.
Radioligand and competition binding studies have demonstrated that nicotine binds to
heteromeric nAChRs with high affinity (in the nM range), but has a much lower
affinity towards the homomeric a l nAChR (in the pM range) (Marks et al., 1986;
Pauly et al., 1989). Its pharmacokinetics has been well established (see Hukkanen et
al., 2005). In humans, approximately 70 to 80% of nicotine is primarily metabolized
in the liver to cotinine, mainly by action of the CYP2A6 enzyme. The drug has a
wide volume of distribution and it readily penetrates the blood brain barrier and the
placenta. The half life of elimination for plasma nicotine is ~2h in humans, but with
regular, chronic smoking it slows down to ~20h. Being the main psychoactive
ingredient in tobacco, nicotine is widely used in behavioural studies.
• Epibatidine [exo-2-(6-chloro-3-pyridyl)-7-azabicyclo-[2.2.1]heptane] is an alkaloid
compound originally isolated from the skin of the Ecuadorian tree frog, Epipedobates
tricolor. It binds with high affinity to both heteromeric and homomeric nAChRs, in
the pM or low nM range, respectively (Gerzanich et al., 1995; Houghtling et al.,
1995). Specifically, epibatidine binds not only to a4p2* nAChRs, which can be
recognised by nicotine and cytisine, but also to nicotinic receptors containing the a3
and the P4 subunits (Marks et al., 1998; Whiteaker et al., 2000b). Indeed,
[^H]epibatidine binding persists in several brain regions of the P2 knockout mouse
(Zoli et al., 1998). The tritiated ligand binds to a4p2* nAChRs with a Kd o f 8 pM; its
iodinated analogue [^^^Ijepibatidine, however, displays a slightly lower affinity for
the same receptor subtype (Kd =50pM) (Davila-Garcia et al., 1997; Whiteaker et al..

26
2000b). This reduction in affinity is probably due to the substitution of a for a
chlorine atom. Nevertheless, the highly specific activity of the iodinated analogue,
coupled with its high affinity and low non-specific binding, have established
[^^^IJepibatidine as one of the most potent nAChR ligands. Moreover, its outstanding
binding characteristics make it a useful tool for the identification of novel receptor
subtypes. In vivo, epibatidine has been shown to exhibit remarkable non-opioid
antinociceptive properties (Badio and Daly, 1994; Rupniak et al., 1994). Despite its
narrow therapeutic window, the compound has generated interest in nAChR as
potential analgesic targets.
Cytisine [l,2,3,4,5,6-hexahydro-l,5-methano-pyrido[l,2-a][l,5]diazocin-8-one] is an
alkaloid found in the seeds of Laburnum anagyroides and it binds to a4p2* nAChRs
with higher affinity than nicotine (Kocyt=0.15 nM, Künic=0.89 nM, Anderson and
Americ, 1994). As the compound differentially interacts with other nAChRs
subtypes, it has been used in competition binding experiments with radiolabeled
epibatidine to distinguish between subpopulations of nicotinic binding sites with high
(cytisine-sensitive) and low (cytisine-resistant) affinity for cytisine (Marks et al.,
2006). The pharmacology and distribution of [^HJcytisine binding sites highly
correlates with that of [^Hjnicotine and [^HJacetylcholine, indicating that the ligand
identifies the same population of heteromeric a4p2* nAChRs (Pabreza et al., 1991).
Its high affinity and low non-specific binding have made [^HJcytisine a very useful
ligand for neuronal nAChRs. In in vivo studies cytisine readily penetrates the blood
brain barrier, and shows a longer half life than nicotine (Sloan et al., 1988), although
it is less potent than nicotine (Coe et al., 2005).

27
• A-85350 [3-((R)-l-azetidin-2-ylmethoxy)pyridine] is a synthetic compound, derived
after introducing a methyl ether moiety into the nicotine molecule. It has been shown
to be a potent and selective ligand for the a4p2* nAChR subtype in vitro (K d=10 pM,
Mukhin et al., 2000). A-85350 has been used in competition binding experiments
with [^^^IJepibatidine to distinguish between subpopulations of nicotinic binding sites
with high and low affinity for A-85350; the latter represent the distribution of a3p4
nAChRs in the habenulointerpeduncular tract (Whiteaker et al., 2002). The iodinated
ligand readily crosses the blood brain barrier and exhibits low toxicity in mice
(Vaupel et al., 1998) and Rhesus monkeys (Chefer et al., 1998). Therefore, it has
been used as a subtype specific ligand for positron emission tomography studies.
1.1.5.2 Neuronal nAChR antagonists
• a-bungarotoxin (a-BgTx) is a long, 75-amino acid peptide that is isolated from
Bungarus multicinctus, a species of East Asian snake (Lee, 1972). Initially
recognised for its potency to block postsynaptic, muscle-type nAChRs, a-
bungarotoxin labels a unique class of neuronal nicotinic receptors, which bind [^^^I]a-
bungarotoxin with high affinity (K d=0.65-1.7 nM), but [^H]nicotine with lower,
micromolar affinity (Marks et al., 1986). This population of nAChRs is considered to
correspond to homomeric a l receptors, and [^^^I]a-BgTx has been the prototypical
ligand for studying their distribution and pharmacology (Clarke et al., 1985b).
• Methyllycaconitine (MLA), isolated from the seeds of Delphinium brownii, is a
natural alkaloid that potently displaces the binding of [^^^I]a-bungarotoxin from rat
membranes (Ki=4 nM) and exhibits selectivity for a-BgTx, compared to heteromeric
nAChR binding sites (Ward et al., 1990). MLA has also been shown to be a

28
competitive antagonist on other nAChR subtypes, containing a4/p2 (Buisson et a l,
1996), a3/a6 (Mogg et al., 2002), and a3/p4 (Free et al., 2003) subunits, albeit with
lower affinity. Unlike a-BgTx, MLA discriminates between a l and muscle nAChRs
(Ward et al., 1990). The compound displays low blood brain barrier penetration.
Following its peripheral administration, however, MLA accumulates into brain tissue
at concentrations that do not produce any overt signs of toxicity and that are
behaviourally relevant (Turek et al., 1995). [^H]methyllycaconitine has been used as
an alternative to a-bungarotoxin in equilibrium binding studies, because of its
faster association kinetics and better signal to noise ratio (Davies et al., 1999).
• Dihydro-p-erythroidine (DhBE) is a plant alkaloid isolated from the seeds of
Erythina americana. In addition to its curare-like effects, DhBE at nanomolar
concentrations is a competitive antagonist at heteromeric neuronal nicotinic subtypes
containing the P2 subunit (Harvey et al., 1996). [^H]DhBE binding sites colocalise
with high-affmity nicotine binding sites, suggesting that the ligand predominantly
labels the widely distributed a4p2* nAChRs (Williams and Robinson, 1984). The
compound penetrates the blood brain barrier and has been shown to antagonise a
variety of nicotine-induced behaviours thought to be exerted by a4p2* nAChRs,
including nicotine reward and nicotine-induced enhancement of contextual fear
conditioning (Corrigall et al., 1994; Davis and Gould, 2006; Kenny and Markou,
2006).

29
1.2 nAChRs in drug addiction
1.2.1 Drug addiction: a chronic disorder of neuronal adaptation
Drug addiction is a state that develops after prolonged exposure to certain
pharmacological agents, notably the psychostimulants, the opiates, alcohol, nicotine, and
the cannabinoids (reviewed in Pierce and Kumaresan, 2006). It is characterised by the
progressive loss of control over drug intake, which ultimately leads to compulsive drug
use, despite severe medical and social consequences. Moreover, addiction is a chronic
and recrudescent disorder, defined by the persistence of a high relapse risk, long after an
individual has stopped taking drugs. Research currently aims to understand the cellular
and neuronal processes that underlie the transition from occasional drug use to the onset
of chronic drug addiction. Indeed, while the initial neuronal targets for abused substances
have been well characterised, little is known about the downstream effects of prolonged
drug administration, and the long-term adaptations that take place in the CNS in response
to repeated drug use. These cellular and circuit adaptations manifest with phenomena
that collectively define addiction, such as dependence, tolerance, sensitisation,
withdrawal, and relapse. Two major types of neurobiological adaptation are thought to
account for the characteristic features of compulsive drug use; homeostatic physiological
adaptations, and synaptic adaptations of the type that mediate associative learning
mechanisms (reviewed in Berke and Hyman, 2000). Each of these processes has been
linked to distinct motivational aspects of drug abuse. Homeostatic alterations have been
held responsible for reinforcing drug taking behaviour by producing tolerance,
withdrawal, and dependence (reviewed in Koob, 2000). Tolerance can be defined as the
requirement for increased doses of a drug in order to maintain a stable effect during
chronic administration. Dependence is an adaptive process that manifests with the onset
of withdrawal symptoms upon drug cessation. Because of tolerance development, the

30
addict increases dosage to maintain the rewarding effects of a drug. During periods of
abstinence, dependence is expressed with symptoms of withdrawal; in an attempt to
escape from the negative affective state that accompanies them, drug-intake resumes and
a vicious circle is established with detrimental results for the health and function of the
addicted individual. Homeostatic neuroadaptation theories can therefore explain some of
the cardinal symptoms of compulsive drug use, suggesting that hedonic tolerance and
affective withdrawal may be the driving forces of addiction. Tolerance, withdrawal and
dependence are drug- and cell circuit-specific phenomena (reviewed in Nestler and
Aghajanian, 1997). Their potency and likelihood of occurrence depends highly on the
type of abused substance and its interactions with specific receptors in relevant cells. For
instance, heroin produces a powerful physical withdrawal symptom, whereas cocaine
does not. Tolerance develops to the hedonic properties of heroin, but not to the
characteristic opiate-induced papillary constriction.
Although informative, the homeostatic adaptation model cannot explain a key aspect of
compulsive drug use, which is its persistent nature. Addiction is a chronic and relapsing
disorder that can be reinstated even after prolonged periods of abstinence. Exposure to
drugs, drug-associated cues, and stress has been linked to the reinstatement of drug-
taking behaviour, long after withdrawal symptoms have subsided (reviewed in Hyman et
al., 2006). This suggests that in addition to the avoidance of withdrawal, reward-related
associative mechanisms contribute to ongoing drug-use. In rat self-administration studies
for example, drug-seeking can be more efficiently primed by small doses of drugs, rather
than by withdrawal (Stewart and Wise, 1992). Moreover, drug-seeking in rodents and
craving in humans can be reliably elicited by exposure to drug-associated cues, when no
measurable symptoms of withdrawal can be observed (O'Brien et al., 1998; Semenova

31
and Markon, 2003). Stress-induced reinstatement involves the activation of reward
pathways, further suggesting that positive reinforcement rather than withdrawal is
involved in re-establishing drug-taking behaviour (reviewed in Marinelli and Piazza,
2002). In addition, patients on morphine for cancer treatment, as well as subjects who
require benzodiazepines for anxiety disorders, exhibit dependence and withdrawal
without the compulsion to seek drugs following treatment cessation (Jage, 2005; O'Brien,
2005). Therefore, considerable evidence suggests that in addition to homeostatic
neuroadaptation, long-term, reward-related learning processes play a crucial role in
mediating drug addiction. Indeed, for addicted individuals drugs become valued over all
other goals, including natural rewards, such as food and sex.
1.2.2 The central role o f dopamine
The full pharmacological profile of different classes of abused drugs is mediated by
several neurotransmitter systems throughout the CNS. Their addictive potential, however,
seems to stem from their shared property for increasing dopamine release in striatal areas
(Di Chiara and Imperato, 1988). Therefore, midbrain DAergic pathways are considered
as the neural substrates that account for compulsive drug use, dependence and late relapse.
1.2.2.1 The Dopaminergic system: localisation, function, receptors
The dopaminergic system comprises an anatomically and functionally diverse group of
neurons, localised in the diencephalon, mesencephalon, and the olfactory bulb.
Mesodiencephalic dopaminergic neurons have been categorised into three main groups
(Fallon and Moore, 1978; reviewed in Bjorklund and Dunnett, 2007).

32
• The nigrostriatal pathway, originating in the substantia nigra pars compacta (SNc),
extends its axons into the dorsal striatum. This system plays a pivotal role in the
control of voluntary movement (reviewed in Gerfen, 1992).
• The mesolimbic system is more medial to the nigrostriatal pathway, and arises from
dopaminergic cells present in the VTA. Mesolimbic fibres mainly project to the
nucleus accumbens and olfactory bulb, but also innervate the hippocampus, amygdala,
and the septum.
• The mesocortical pathway also originates in the VTA and projects to the frontal,
cingulate and perirhinal cortex.
Due to the overlapping nature of their projections, the mesolimbic and mesocortical
pathways are collectively referred to as the mesocorticolimbic system. This is involved
in emotion-based behaviour, including motivation and reward (Mogenson et al., 1980).
To date, five subtypes of DA receptors have been indentified, all of which belong to the
superfamily of G-protein coupled receptors (reviewed in Missale et al., 1998). Dl-like
receptor subtypes (Dl and D5) are Gs coupled, and their activation stimulates adenylyl
cyclase (AC), leading to increased 3', 5’-cyclic adenosine monophosphate (cAMP)
production. On the contrary, D2-like receptors (D2, D3, and D4) are Gi coupled and their
activation inhibits AC. In addition to AC coupling, both receptor subtypes can interact
with different signalling pathways; Dl-like receptors couple to the stimulation of
phosphoinositide metabolism and mobilise intracellular calcium stores (Liu et al., 1992;
Undie et al., 1994), whereas D2-like receptor activation stimulates inwardly rectifying
potassium channels (Werner et al., 1996). Dopamine receptors exhibit different patterns
of neuroanatomical distribution. D l and D2 receptors are widespread compared to other
DA subtypes, with higher levels observed in the dorsal and ventral striatum, and the

33
olfactory tubercle, and lower levels in the hippocampus, neocortex, hypothalamus, and
thalamus (Weiner et ah, 1991). Presynaptic D2 receptors are expressed by dopaminergic
neurons in the axon terminal and somatodendritic regions, where they function as
autoreceptors to regulate dopamine release (Tepper et al., 1987; Tepper et al., 1997). D3
receptors are localised in the shell of the nucleus accumbens and the islands of Calleja
(Sokoloff et al., 1990; Bouthenet et al., 1991), whereas D4 are weakly expressed in
cortical and limbic areas (Svingos et al., 2000; Wedzony et al., 2000). D5 receptor
mRNA is low in the striatum and neocortex, but higher in the hippocampus,
hypothalamus and certain thalamic nuclei (Meador-Woodruff et al., 1992; Khan et al.,
2000). Transport of DA from the extracellular space into the presynaptic bouton is
essential for the termination of DAergic transmission and the maintenance of presynaptic
storage. This is performed by the dopamine transporter (DAT), a transmembrane, Na"
and C r dependent carrier protein (Volz and Schenk, 2005) that is localised in areas of DA
innervation, including the ventral mesencephalon and the striatum (Ciliax et al., 1995).
1.2.2.2 The Dopaminergic signal
Dopamine neurons exhibit two main patterns of discharge activity, a sustained, single
spike, irregular mode (tonic signal), and a transient, homogenous burst of action potential
firing (phasic signal). In both primates and rodents, DAergic neurons switch from tonic
activity to either a phasic increase or pause of firing rates, in response to the presentation
or omission of reward and reward-predicting stimuli, respectively (Schultz et al., 1997;
Hyland et al., 2002). In this way, mesostriatal DA neurons carry information about the
occurrence of reward-related events to brain regions that are dedicated to processing their
hedonic value, such the frontal cortex. The tonic vs. phasic patterns of activation at the
level of the cell body differentially relates to DA release into the terminal projection areas

34
of the mesolimbic pathway. Thus, the phasic stimulation of DA cells is believed to
produce a much higher efflux of extracellular DA compared to tonic firing frequencies
(Chergui et al., 1994; Garris et al., 1994), thereby effectively conveying the signal of
reward to corticostriatal areas. Several findings suggest that DA release is a highly
dynamic rather than a static, fixed-amplitude process. Indeed, the release of extracellular
DA is accompanied by short-term changes in DA release probability, in such a way that
DA release in the striatum undergoes a use-dependent depression of release probability at
rapidly successive pulses (Gragg, 2003; Montague et al., 2004). This type of plasticity in
DA release probability suggests that DA levels do not depend linearly on the frequency
of DAergic neuron activity, but that extensive neuromodulation occurs at the terminal
release sites. Drugs of abuse bypass all the aforementioned physiological processes of
reward-related signalling by pharmacologically inducing DA release in the striatum, and
therefore producing a sustained reward signal. The participation of nicotinic cholinergic
mechanisms on midbrain DAergic neurotransmission will be examined in the following
section.

35
1.2,3 nAChRs and dopaminergic signalling
1.2.3.1 Anatomical foundations
Dopaminergic and cholinergic innervation is denser in the striatum than anywhere else in
the brain. Moreover, there is a precise overlap in the distribution of the rate limiting
enzymes for ACh and DA synthesis, suggesting that cholinergic and DAergic fibres are
closely intertwined in striatal areas (Zhou et al., 2001). In addition, cholinergic
projections from the mesopontine tegmentum, namely from the pedunculopontine (PPTg)
and the laterodorsal tegmental nuclei (LDTg), innervate the substantia nigra (SN) and the
VTA, respectively, providing a source of endogenous acetylcholine that modulates the
firing of midbrain dopaminergic neurons via nAChRs that are expressed in these areas
(reviewed in Maskos, 2008). Nicotinic cholinergic mRNA for a4, a5, a6, a3, a7, p2, P3,
and P4 subunits is widely expressed on the cell bodies of midbrain DA neurons (Azam et
al., 2002). This potentially gives rise to multiple pentameric nAChR combinations with
distinct functional properties. The exact subunit composition, stoichiometry and function
of different nAChRs have been characterised using immunoprécipitation and
electrophysiology studies, specific ligands, and subunit-specific KO mice. a7 and p4
subunits are expressed in the SNc and VTA, but are not encountered on striatal DAergic
terminals (Champtiaux et al., 2003; Quik et al., 2005). All nAChRs that are expressed
presynaptically on DA terminals in the striatum contain the P2 subunit (Jones et al., 2001;
Champtiaux et al., 2003). In addition to the p2, a4 and a6 subunits are also abundant in
the rodent striatum. The a6/a3 selective antagonist a-conotoxin Mil (a-CtxMII) has been
used to distinguish between two subpopulations of p2-containing nAChRs, those that are
a-CtxMII sensitive and resistant, respectively (Kulak et al., 1997; Whiteaker et al.,
2000a; Whiteaker et al., 2002). The a-CtxMII sensitive population comprises a6 subunits,
colocalised with P3, and to a smaller proportion a4 subunits (Le Novere et al., 1996;

36
Champtiaux et al., 2003; Gui et al., 2003). The a-CtxMII resistant population is
composed of a4 subunits, which sometimes associates with the a5 subunit (Zoli et al.,
2002; Champtiaux et al., 2003; Salminen et al., 2004; Salminen et al., 2005). Altogether,
evidence supports the existence of functional a4p2, a4a5p2, a6a4p2p3, a6p2p3, a6p2,
a6a4p2 in DA axons of the rodent striatum.
Glu
DA
GABA
al al
Figure 1.5 Subtypes o f nAChRs involved in DAergic neurotransm ission
The subunit composition of functional nAChR subtypes expressed by DAergic, GABAergic and glutamatergic neurons
is shown. The exact contribution o f subunits in brackets remains remains to be elucidated. Adapted from Champtiaux
et al., (2003).
In addition, nAChRs are expressed presynaptically by GABAergic and glutamatergic
neurons in both the VTA and NAc, and are therefore capable of modulating DA release
indirectly, by facilitating inhibitory and excitatory input onto DAergic neurons.
Specifically, presynaptic a7 nAChRs are located on glutamatergic nerve terminals, and
a4p2* nAChRs on GABAergic interneurons (Mansvelder and McGehee, 2000;
Mansvelder et al., 2002). Therefore, the cholinergic modulation of striatal DA release
seems to involve sophisticated connections between distinct nAChR subtypes, localised
on several different neurons.

37
1.2.3.2 Cholinergic modulation o f DAergic signalling in the VTA
There is overwhelming evidence that all drugs of abuse activate the mesolimbic
dopaminergic pathway in order to exert their reinforcing effects (reviewed in Pierce and
Kumaresan, 2006). Nicotine obtained from tobacco smoke constitutes no exception. The
responses of DA neurons to endogenous ACh or exogenous nicotine are mediated by
nAChRs localised on three different cell types; a4p2* nAChRs on the DAergic neuron
itself, presynaptic a4p2* nAChRs on GABAergic intemeurons, and presynaptic a7
nAChRs on glutamatergic fibres. In the VTA microcircuitry these nAChR subtypes
differentially affect DAergic, GABAergic and glutamatergic responses to a concentration
of nicotine similar to that acquired by smokers, as a result of their inherently distinct
desensitisation properties (see Section 1.1.3.1). Initially, nicotine rapidly activates high
affinity, p2 subunit containing receptors on DA and GABA neurons. Indeed, the direct
depolarisation of DA neurons coincides with an increase of GABAergic synaptic
transmission (Mansvelder et al., 2002). Desensitisation of p2 subunit containing nAChRs,
however, results in the decline of inhibitory GABAergic input onto DA neurons. At the
same time, nicotine activates presynaptic a7 nAChRs that are located on glutamatergic
nerve terminals. As these receptors are resistant to desensitisation in the range of nicotine
concentrations experienced by smokers, an increase of glutamate transmission onto DA
neurons occurs (Pidoplichko et al., 1997). When this enhanced presynaptic glutamate
release coincides with a sufficient postsynaptic depolarisation of the DA neuron, long
term potentiation of the excitatory inputs onto the dopaminergic system takes place
(Mansvelder and McGehee, 2000). These mechanisms explain why a single exposure to
nicotine causes prolonged increases in striatal DA release in vivo (Imperato et al., 1986;
Di Chiara and Imperato, 1988).

38
1.2.3.3 Cholinergic modulation o f DAergic signalling in the NAc
The striatal effects of nicotine on DAergic neurotransmission were until recently
neglected. The long-held view that nAChRs in the VTA primarily mediate nicotine’s
addictive potential has been supported by several reports, which indicate that systemic
nicotine injections or nicotine infiised into the VTA, but not the striatum, sustains
increased levels of DA in the NAc (Nisell et al., 1994; Ferrari et al., 2002). Moreover,
blockade of nAChRs in the VTA and not the striatum prevents nicotine self
administration (Corrigall et al., 1994). Nevertheless, recent insight into nicotine’s mode
of action reveals that potent cholinergic mechanisms are in place in the striatum, which
provide an amplification of reward-related signalling in terminal DAergic regions.
Contrary to microdialysis studies, which show an enhancement of DA release in the
striatum by nicotine, fast-cyclic voltammetry in mice striatal brain slices reveals that
desensitising doses of nicotine decrease DA release evoked by tonic stimuli by -80%
(Zhou et al., 2001). This decrease is also observed after depletion of endogenous ACh
with vesamicol and following the administration of nicotinic antagonists. In addition, the
cholinergic modulation of DA release is absent in mutant mice lacking the p2 subunits,
suggesting that tonic cholinergic activity regulates DA release at the target, via p2-
containing nAChRs (Zhou et al., 2001). Interestingly, the inhibition of single pulse-
evoked DA release by nicotine is only observed at low tonic frequencies. Similarly to
cocaine and amphetamine, nicotine-induced DA release during the phasic activation of
DAergic neurons has been shown to increase (Rice and Cragg, 2004). This dual effect of
nicotine - amplification of phasic and suppression of tonic DA release - is explained by
the dynamic plasticity in DA release probability that occurs at the circuit-level (see
Section 1.2.2.2). At striatal synapses, short-term depression of DA release takes place,
diminishing release probability at consecutive pulses (Cragg, 2003). Nicotine, by

39
suppressing single pulse-evoked DA release contributes to the relief of short-term
depression, an effect that is equivalent per successive pulse (Rice and Cragg, 2004).
Therefore, desensitising doses of nicotine, or a reduction of ACh tone at nAChRs, can
both enhance reward-related bursts of DA neuron activity and further suppress the DA
signal that accompanies the omission of rewards. Together, somatodendritic and axonal
nAChRs serve as a dynamic and diverse cholinergic filter of DA neurotransmission.
Striatal ACh neurons cease action potential firing in response to reward-related events
(Aosaki et al., 1994; Shimo and Hikosaka, 2001), contrary to mesostriatal DA cells,
which switch from tonic to burst firing patterns of activity (Bayer and Glimcher, 2005;
Tobler et ah, 2005). Interestingly, these reward-related neuronal responses coincide
temporally when recorded in the same tasks, occurring with similar latency and duration
(Morris et al., 2004). This functional cooperation suggests that nicotinic cholinergic
mechanisms may play a broader role in mediating reward-related processes. Because
drugs of abuse reliably increase synaptic DA, it seems highly probable that cholinergic
modulation via nAChRs may provide an additional neural substrate for the therapeutic
intervention of drug addiction. In the following section, evidence for the role of nAChRs
in mediating the abuse potential of different classes of drugs will be reviewed.

40
1.3 Interactions between nAChRs and drugs of abuse
1.3.1 nAChRs and cocaine
The prevalence of poly-drug abuse is characteristically depicted in the concurrent
consumption of nicotine with cocaine. In humans, the extent of smoking has been
associated both with the probability and the frequency of cocaine abuse (Henningfield et
al., 1990; Schorling et al., 1994; Konings et al., 1995). Moreover, several studies have
suggested that the two drugs may facilitate each other’s intake (Budney et al., 1993;
Higgins et al., 1994; Roll et al., 1996), producing subjective effects that considerably
overlap (Foltin and Fischman, 1991; Jones et al., 1999). The fact that a single
administration of nicotine can enhance cue-induced cocaine craving in cocaine-abstinent
subjects, while mecamylamine can dose-dependently attenuate it, indicates that there are
direct and persistent psychopharmacological interactions between the two substances
(Reid et al., 1998; Reid et al., 1999).
1.3.1.1 Cocaine’s mode o f action
Cocaine exerts its effects on the CNS through its interaction with the dopamine (DAT),
norepinephrine (NET), and serotonin (SERT) cell membrane transporters (reviewed in
Carrera et al., 2004). By blocking the carrier proteins, cocaine prevents the reuptake of
catecholamine neurotransmitters into the presynaptic cleft, and consequently increases
their synaptic availability, thereby acutely activating DAergic, adrenergic and
serotonergic receptors. The relative importance o f DAT, NET, and SERT inhibition in
the reinforcing properties of cocaine has been the subject of intense investigation.
Evidence suggests that blockade of the DAT, rather than the NET and SERT, is critically
involved in mediating cocaine’s abuse-related potential (Ritz et al., 1987). Indeed, drugs
that inhibit noradrenergic or serotonergic reuptake do not prevent cocaine self-

41
administration in rodents and non-human primates (De Wit and Wise, 1977; Howell and
Byrd, 1995). On the contrary, direct DA agonists are self-administered by animals
trained to self-administer cocaine (Woolverton et al., 1984; Wise et al., 1990; Caine and
Koob, 1993), and lesions of DAergic nerve terminals in the NAc (Roberts et al., 1977;
Roberts et al., 1980) or cell bodies in the VTA (Roberts and Koob, 1982) lead to the
extinction of cocaine self-administration behaviour. Moreover, cocaine is not self
administered by animals with genetic deletion of the DAT gene (Thomsen et al., 2009),
or by DAT knockin mice carrying a mutant transporter that is functional, albeit
insensitive to cocaine inhibition (Chen et al., 2006). These data suggest that DAT
blockade is necessary for cocaine reward.
As there is currently no available pharmacotherapy for the treatment of cocaine addiction,
a major goal of the scientific community is to characterise the neuropharmacological
determinants of the drug’s high abuse potential. Given the role of nAChRs in DAergic
signalling, manipulation of the nicotinic cholinergic system has the potential to influence
strategies for cocaine addiction.
1.3.1.2 Neurochemical evidence fo r interactions between nAChRs and cocaine
The anatomical foundations for the nicotinic cholinergic influence on DAergic
neurotransmission have been described in Section 1.2.3.1. Through different molecular
mechanisms, cocaine and nicotine increase DA release in the NAc, indicating that the
mesolimbic DAergic pathway is the principal site for interactions between the two drugs
(Di Chiara and Imperato, 1988; Pontieri et al., 1996). In rats, co-administration of
nicotine at 0.4 mg/kg with either 10 or 20 mg/kg of cocaine, increases extracellular NAc
DA levels in an additive or synergistic manner, respectively, suggesting that the degree of

42
DA transporter occupancy contributes to the interactions between these drugs (Gerasimov
et al., 2000). Indeed, by inhibiting DA reuptake through the blockade of dopamine
transporters, cocaine’s efficacy is determined by the amount of DA that is released and
available within the synaptic cleft. In the NAc, depletion of endogenous ACh,
administration of nicotinic antagonists or the genetic deletion of the g2 subunit
dramatically decreases DA release by -80%, suggesting that endogenous cholinergic
activity tonically stimulates the mesolimbic DAergic pathway (Zhou et al., 2001).
Therefore, administration of nAChR antagonists within the NAc is anticipated to
decrease cocaine-induced DA release, by antagonising the striatal cholinergic tone.
Indeed, intra-NAc perftision of the broad spectrum nAChR antagonist mecamylamine, or
co-perfusion of the heteromeric nAChR antagonist DhBE with the homomeric nAChR
antagonist MLA, attenuates DA increase following an acute i.p. injection of 5 mg/kg
cocaine in mice (Zanetti et al., 2007). The fact that experimenter- and se lf administered
cocaine enhances acetylcholine efflux in various brain regions, including the cerebral
cortex (Day et al., 1997), hippocampus (Imperato et al., 1992), interpeduncular nucleus
(Hussain et al., 2008), nucleus accumbens (Mark et al., 1999; Crespo et al., 2006) and
striatum (Zocchi and Pert, 1994), further suggests that a targeted increase in ACh tone
may be responsible for significant adaptations of cocaine-induced DAergic
neurotransmission. In mice that have been repeatedly treated with cocaine, co
administration of the psychostimulant with nAChR antagonists prevents the development
of sensitisation to cocaine-induced increases of extracellular DA, strongly implying that
an enhanced cholinergic tone acting on nAChRs is necessary not only for the acute, but
also for the long-term effects of cocaine on mesolimbic DAergic signalling (Zanetti et al.,
2006).

43
Data on the cholinergic modulation of cocaine reinforcement, however, are as yet
equivocal. The ablation of cholinergic intemeurons with immunotoxins within the NAc
has been shown to potentiate cocaine-induced locomotion and conditioned place
preference, while acetylcholinesterase inhibition suppresses these behaviours, suggesting
that inhibition rather than activation of the striatal cholinergic neuron is involved in
cocaine’s stimulant effects (Hikida et al., 2001; Hikida et al., 2003). Similarly, cocaine
administration has been paralleled with a decrease in ACh turnover in the NAc (Smith et
al., 2004). Moreover, contrary to their effects within the NAc, the nAChR antagonists
DhBE and MLA increase cocaine-induced extracellular release of DA when infused
directly into the VTA (Zanetti et al., 2007). Differences in the experimental conditions
among these and the aforementioned studies may account for the inconclusive results
obtained so far as to the cholinergic modulation of cocaine-induced DAergic
neurotransmission. All the same, given the heterogeneity of nAChR subtypes that are
expressed by midbrain DAergic neurons, and the dynamic filtering of DAergic
neurotransmission by nAChRs, no single effect of cocaine on nicotinic cholinergic
signalling can account for these discrepancies.
1.3.1.3 Behavioural evidence fo r interactions between nAChRs and cocaine
Among the paradigms employed to model different aspects of drug reinforcement, the
self-administration procedure has been favoured as the most direct way of investigating
the behavioural and neurobiological processes that lead to addiction. In rodents, the
acquisition of cocaine self-administration can be dose-dependently suppressed by the
broad nAChR antagonist mecamylamine (Blokhina et al., 2005), and facilitated by pre
exposure to nicotine (Horger et al., 1992). In rats that have been trained to lever press for
0.32 mg/kg/infusion of intravenous (i.v.) cocaine, pretreatment with mecamylamine dose-

44
dependency reduces cocaine se lf administration (Levin et al., 2000). Moreover, in the
presence of mecamylamine, the escalation from moderate to excessive cocaine intake is
prevented in rats with extended daily access to the psychostimulant (Hansen and Mark,
2007). When the antagonist is removed, drug self-administration is increased, indicating
that nAChRs prevent the consumption of cocaine under conditions that promote an
escalation of its intake. Repeated exposure to 0.6 mg/kg of s.c. nicotine prior to the
initiation of cocaine self administration sessions progressively leads to increased
responding for 0.1 mg/kg/infusion of i.v. cocaine over the course of 14 days, indicating
that nicotine can potentiate cocaine’s rewarding effects (Bechtholt and Mark, 2002).
Following the extinction of self-administration, acute nicotine emits a burst of non
reinforced responses, indicating that the drug elicits cocaine-seeking behaviour. These
results suggest that nAChRs are involved in all aspects of cocaine se lf administration,
including the acquisition, maintenance, escalation and relapse to this behaviour. In the
conditioned place preference paradigm (GPP), pharmacological disruption of nicotinic
neurotransmission with 1 mg/kg of mecamylamine inhibits place preference to 5 mg/kg
of cocaine, which is the lowest dose that can condition this response in C57BL/6 mice.
On the contrary, nicotine at 0.2 mg/kg potentiates place preference to a subthreshold dose
of 3.0 mg/kg of cocaine. The effects of nicotine on cocaine-induced GPP are mediated by
receptors containing the (32 subunit, since (32 mutant mice fail to condition to 5 mg/kg of
cocaine (Zachariou et al., 2001). In the drug discrimination paradigm, the discriminative
stimulus effects of 0.1 mg/kg nicotine fully substitute for those of cocaine, whereas
cocaine partially substitutes for nicotine, indicating a considerable, yet assymetric overlap
in the subjective effects of the two drugs (Desai et al., 1999).

45
Cocaine has been shown to progressively enhance locomotor activation in rodents during
its repeated administration. This sensitised response reflects long-term neuroadaptation
that is considered to contribute to cocaine’s addictive properties. Systemic blockade of
nAChRs with mecamylamine has been shown to prevent the development of cocaine-
induced locomotor sensitization. (Schoffelmeer et al., 2002) The same effect can be
exerted by intra-VTA infusion of the heteromeric, (32 subunit containing nAChR
antagonist DhBE, but not o f the homomeric nAChR antagonist MLA, suggesting that
heteromeric receptors within the VTA are involved in the neuroadaptive changes
underlying cocaine sensitisation (Champtiaux et al., 2006). Repeated administration of
nicotine, on the other hand, has been shown to cause no sensitisation to the locomotor
stimulant effects of cocaine in adult rats (Horger et al., 1992) and mice (Itzhak and
Martin, 1999). Nevertheless, locomotor cross-sensitisation between nicotine and cocaine
is observed preferentially during periadolescence rather than adulthood, demonstrating
that this age may be more vulnerable to the risk of cocaine abuse after nicotine exposure
(Collins and Izenwasser, 2004). Indeed, long-term, low-level adolescent nicotine
administration produces dose-dependent changes in cocaine-induced motor effects in
adult mice (Kelley and Rowan, 2004). Altogether, a large body of evidence supports the
notion that nAChRs are a common denominator for the neurochemical and behavioural
effects of cocaine.
1.3.2 nAChRs and opioid abuse
There is a large amount of published research that supports interactions between the
nicotinic cholinergic and opioid systems in humans. Indeed, tobacco smoking can be
reduced by the MOP receptor antagonist naloxone (Karras and Kane, 1980),
administration of which dose-dependently induces opioid-like withdrawal symptoms in

46
smokers (Krishnan-Sarin et al., 1999). The longer lasting opioid antagonist naltrexone
has been found to significantly reduce the total number of cigarettes smoked, as well as
the desire to smoke (King and Meyer, 2000), and it has showed beneficial effects in
smoking cessation treatment (Covey et al., 1999). Nevertheless, other reports have not
reached similar conclusions in humans (Nemeth-Coslett and Griffiths, 1986; Sutherland
et al., 1995; Wong et al., 1999). It is therefore necessary to better identify the
mechanisms involved in the interaction between nAChRs and the endogenous opioid
system.
1.3.2.1 The opioid system: Localisation, function, receptors
In the mid-1970s, endogenous peptides with morphine-like activity were isolated:
methionine and leucine enkephalins were derived from the pig brain (Hughes et al., 1975),
whereas P-endorphin was isolated from camel pituitary glands (Li and Chung, 1976).
Another group of peptides, the dynorphins, were subsequently isolated from porcine
pituitary in the early 1980s (Goldstein et al., 1981). A fourth endogenous opioid agonist
was reported in 1995 by two groups and named nociceptin/orphanin FQ (Meunier et al.,
1995; Reinscheid et al., 1995). To date, more than 20 endogenous opioid peptides have
been identified, which are mainly derived from four precursor proteins, pro
opiomelanocortin, proenkephalin, prodynorphin, and pronociceptin/orphanin FQ
(reviewed in Vaccarino and Kastin, 2001). Endogenous peptides exert their action by
acting on specific receptors, namely the MOP, KOP, DOP, and NOP receptors.
Collectively, opioid peptides and their receptors form the endogenous opioid system. All
opioid receptors belong to the G-protein coupled family, interacting with pertussis-toxin
sensitive Gi proteins to inhibit AC, thus reducing intracellular cAMP content. In addition,
both the a and the Py subunits of the G protein interact with effector systems to inhibit the
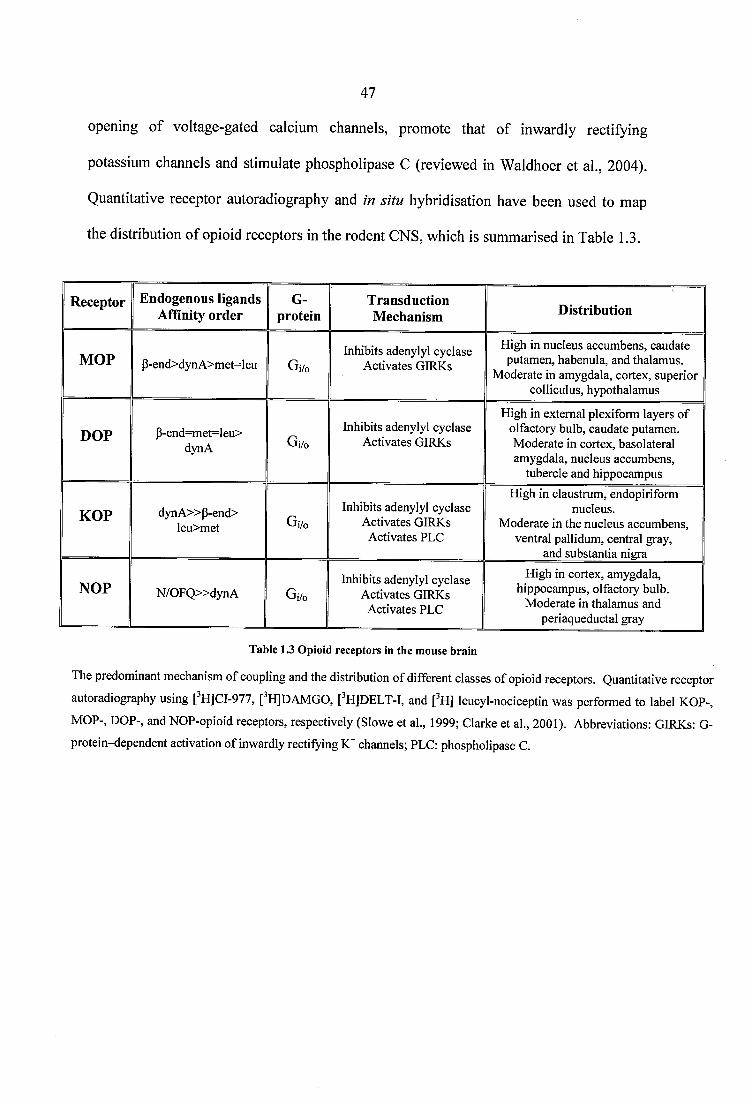
47
opening of voltage-gated calcium channels, promote that of inwardly rectifying
potassium channels and stimulate phospholipase C (reviewed in Waldhoer et al., 2004).
Quantitative receptor autoradiography and in situ hybridisation have been used to map
the distribution of opioid receptors in the rodent CNS, which is summarised in Table 1.3.
Receptor Endogenous ligands Affinity order
G-protein
TransductionMechanism Distribution
MOP P-end>dynA>met=leu Gi/oInhibits adenylyl cyclase
Activates GIRKs
High in nucleus accumbens, caudate putamen, habenula, and thalamus.
Moderate in amygdala, cortex, superior colliculus, hypothalamus
DOP P-end=met=leu>dynA Gi/o
Inhibits adenylyl cyclase Activates GIRKs
High in external plexiform layers o f olfactory bulb, caudate putamen. Moderate in cortex, basolateral amygdala, nucleus accumbens,
tubercle and hippocampus
KOP dynA »P-end>leu>met Gi/o
Inhibits adenylyl cyclase Activates GIRKs
Activates PLC
High in claustrum, endopiriform nucleus.
Moderate in the nucleus accumbens, ventral pallidum, central gray,
and substantia nigra
NOP N / OF Q » d y n A Gi/oInhibits adenylyl cyclase
Activates GIRKs Activates PLC
High in cortex, amygdala, hippocampus, olfactory bulb.
Moderate in thalamus and periaqueductal gray
Table 1.3 Opioid receptors in the m ouse brain
The predominant mechanism o f coupling and the distribution o f different classes o f opioid receptors. Quantitative receptor
autoradiography using [ H]CI-977, [^HJDAMGO, [^HJDELT-I, and [^H] leucyl-nociceptin was performed to label KOP-,
MOP-, DOP-, and NOP-opioid receptors, respectively (Slowe et al., 1999; Clarke et al., 2001). Abbreviations: GIRKs: G-
protein-dependent activation o f inwardly rectifying channels; PLC: phospholipase C.

48
1.5.2.2 Neurochemical evidence fo r interactions between nAChRs and opioids
Administration of nicotine or opioids has been shown to increase the accumbal release of
dopamine (DA), an effect that is considered to be critical for the addictive nature of many
drugs of abuse (Pontieri et a l, 1996). Indeed, the systemic or focal administration of
either class of substances into the VTA or NAc raises extracellular DA in striatal areas, as
measured by in vivo microdialysis (Devine et a l, 1993; Hemby et a l, 1995; Ferrari et a l,
2002; Hirose et a l, 2005). More importantly, chronic oral pretreatment of mice with
nicotinic solutions enhances morphine-induced DA release in the CPu and the NAc
(Vihavainen et a l, 2008b), and a single intraperitoneal (i.p.) injection of the selective
a3p4 nAChR antagonist 18-methoxycoronaridine (18-MC) abolishes the sensitized
dopamine response in rats that have been repeatedly exposed to morphine (Maisonneuve
and Click, 1999; Szumlinski et a l, 2000; Taraschenko et a l, 2007a). In addition, lesions
of striatal cholinergic intemeurons with cholinotoxins eliminate MOP receptor agonist-
mediated increases in DA release (Dourmap et a l, 1997). Therefore, a large body of
evidence points to the mesolimbic DA system as the neuronal pathway involved in
nicotinic-opioid interactions. Midbrain dopaminergic neurons switch from tonic to burst
patterns of action potential firing in response to rewarding or reward-predicting stimuli,
and this transition contributes to increased striatal DA release (Schultz, 2007). Recently,
electrochemical and electrophysiology studies have shown that MOP and DOP receptor
agonists inhibit tonic DA release indirectly, by suppressing cholinergic intemeuron
activity within the NAc (Britt and McGehee, 2008). As shown by in vivo microdialysis,
cholinergic inhibition lowers baseline levels of ACh in the NAc (Rada et a l, 1996),
which would consequently decrease nAChR activation on DA terminals. The endpoint of
this cholinergic-mediated decrease in tonic DA release by opioids is an enhanced ratio of
burst evoked DA levels relative to baseline, and thus an indirect increase in the

49
magnitude of the DAergic signal (Britt and McGehee, 2008). Interestingly, nicotine
similarly acts in the striatum to increase the contrast between tonic and burst-evoked
dopaminergic signals, by directly desensitising nAChRs on DA nerve terminals (Zhang
and Sulzer, 2004). This profound mechanistic overlap between nicotine and opioids has
strong anatomical foundations, as MOP receptors within the shell of the NAc are
expressed not on dopaminergic, but on cholinergic intemeurons (Svingos et al., 2001). In
addition, the transition of midbrain DA neurons from tonic to burst firing pattems
involves the modulation of afferent inputs to the VTA, a structure that highly expresses
both MOP receptors and nAChRs (Mansour et al., 1995; Picciotto et al., 1998).
Altogether, substantial evidence suggests that opioids partly exert their effects within the
reward circuitry indirectly, by modulating DA release through nAChRs.
Following 0.5 mg/kg of subcutaneous nicotine, extracellular DA release in the NAc is
impaired in preproenkephalin gene KOs compared to WT mice, suggesting that
endogenous opioids derived from preproenkephalin participate in the modulation of DA
neurotransmission by nicotine (Berrendero et al., 2005). Indeed, acute and chronic
nicotine administration increases the release of endogenous opioids (Dhatt et al., 1995;
Houdi et al., 1998; Isola et al., 2002). Morphine has been shown to excite DA neurons by
hyperpolarising GABAergic cells in the VTA via MOP receptors (Johnson and North,
1992). Nicotine-induced increases in the release of endogenous opioids can therefore
contribute to the disinhibition of DA neurons, and consequently to the reinforcing
properties of the drug, by activating presynaptic MOP receptors on GABAergic cells. In
support of this, nicotine-induced conditioned placed preference is not observed in
preproenkephalin or MOP receptor KO mice, indicating that opioid-dependent
mechanisms mediate the rewarding effects of nicotine (Berrendero et al., 2002;

50
Berrendero et al., 2005). Overall, DAergic transmission within the mesolimbic system
seems to be a common neurochemical mechanism, both for the cholinergic modulation of
opioid-induced effects and for the involvement of the opioid system in the reinforcing
properties of nicotine.
1.3.2.3 Behavioural evidence fo r interactions between nAChRs and opioids
Nicotine and morphine have been shown to influence each other’s effects in various
experimental settings, designed to model the addictive properties of the two drugs. Thus,
rats that have received 5 daily injections of 0.5 mg/kg of nicotine (Biala and Weglinska,
2004), and rats that have been pre-exposed to 0.4 mg/kg/day of nicotine for 5 days
(Shippenberg et al., 1996) show enhanced conditioned place preference (CPP) to 5 mg/kg
of morphine. Similarly, 7-week oral nicotine pretreatment in mice potentiates morphine-
induced CPP (Vihavainen et al., 2008a). Furthermore, pretreatment with the broad
spectrum nicotinic antagonist mecamylamine reverses morphine-induced CPP, while the
opioid antagonist naloxone diminishes nicotine-induced CPP in mice (Zarrindast et al.,
2003). The participation of different components of the opioid system in the rewarding
effect of nicotine has been demonstrated by the fact that 8 days of 0.5 mg/kg of s.c.
nicotine does not induce CPP in MOP-receptor (Berrendero et al., 2002),
preproenkephalin (Berrendero et al., 2005), and (3-endorphin (Trigo et al., 2009) gene KO
mice, compared to WT animals. Overall, the interaction between nicotinic receptors and
the opioid system has been clearly demonstrated in the CPP paradigm of drug abuse. The
application of the self-administration paradigm, however, has produced mixed results.
Corrigall and Coen, (1991), failed to show any effect of naltrexone on nicotine-self
administration in rats. In a study with a positive outcome, the antagonist markedly
suppressed nicotine-induced increases in responding to a fixed-interval schedule of food

51
presentation in mice (Corrigall et al., 1988). In an intravenous morphine self
administration report, the potent aSp4 nAChR antagonist 18-MC produced a downward
shift in the entire morphine dose-response curve, indicating that the drug reduced the
reinforcing efficacy of morphine, without altering is potency (Maisonneuve and Click,
1999). Therefore, further research is required to determine the nature of nAChR-opioid
receptor interactions in the self-administration paradigm, particularly with respect to
specific nAChR subtypes that may modulate opioid intake.
Chronic exposure to nicotine or opioids results in the development of physical
dependence, which manifests with somatic symptoms of withdrawal upon cessation of
drug-intake (spontaneous withdrawal) or upon the administration of nicotinic and opioid
antagonists (precipitated withdrawal). Interestingly, the nicotine abstinence syndrome
closely resembles opioid withdrawal, including responses such as shakes and tremors,
teeth chattering, writhes, gasps, and chews (Malin et al., 1993; Hildebrand et al., 1997).
Indeed, mecamylamine at 3.0 mg/kg and naloxone at 1.0 mg/kg precipitate qualitatively
similar symptoms of nicotine withdrawal in mice that have been exposed to intermittent
nicotine injections for 7 days (Biala et al., 2005). In rats that have been exposed to 3.2
mg/kg/day of nicotine for 7 days via osmotic minipumps, a single s.c. injection of 4.5
mg/kg naloxone precipitates withdrawal, whereas 2 mg/kg of morphine alleviate the
symptoms of spontaneous nicotine withdrawal (Malin et al., 1993). Moreover, 2 mg/kg
of naloxone and 1 mg/kg of mecamylamine precipitate a qualitatively similar withdrawal
symptom in rats that have been pretreated with nicotine via osmotic minipumps at a rate
of 3.16 mg/kg/day for 8 days (Carboni et al., 2000). The above data imply an important
role for opioid-dependent mechanisms in nicotine abstinence. Indeed, deletion of the
MOP receptor (Berrendero et al., 2002) or of the preproenkephalin gene (Berrendero et

52
a l, 2005) attenuates the mecamylamine-precipitated somatic expression of the nicotine
withdrawal syndrome, indicating that an endogenous opioid tone is crucial for the
manifestation of physical abstinence signs. In addition to the somatic signs, the affective
aspects of nicotine abstinence may also involve interactions between nAChRs and
opioids, as the administration of naloxone induces conditioned place aversion in nicotine-
treated rats (Watkins et al., 2000). Moreover, mecamylamine-induced nicotine-
withdrawal aversion can be significantly attenuated by morphine pretreatment (Ise et al.,
2000). Similarly to the participation of opioid mechanisms in nicotine-abstinence,
physical signs of opioid withdrawal can be reversed in morphine-dependent rats. A
single injection of nicotine 15 min prior to naloxone diminishes the severity of the opioid
withdrawal syndrome, in a dose-dependent and mecamylamine-reversible manner
(Zarrindast and Farzin, 1996). Moreover, the repeated, concurrent administration of
nicotine with morphine dose-dependently abolishes the incidence of withdrawal jumping
in mice (Haghparast et al., 2008). In conclusion, common neurobiological mechanisms
seem to subserve nicotine and opioid physical dependence.
1.3.3 nAChRs and adenosine systems
A significant positive correlation for the concurrent consumption of nicotine and caffeine,
a non selective adenosine Ai and A%A receptor antagonist, has been derived from six
epidemiological studies, which collectively examined the smoking and coffee drinking
habits of almost 40,000 people (reviewed in Swanson et al., 1994). The joint use of
nicotine and caffeine has been further supported by controlled human studies, which
show that caffeine consumption is larger in smokers compared with non-smokers (Istvan
and Matarazzo, 1984; Budney et al., 1993; Kozlowski et al., 1993; Rose et al., 1993), and
that nicotine-intake is enhanced while drinking coffee (Parsons and Neims, 1978;

53
Marshall et al., 1980; Emurian et al., 1982; Nellis et al., 1982; Brown and Benowitz,
1989). Therefore, a number of different reports suggest that interactions exist between
nAChRs and the adenosine system.
1.3.3.1 The adenosine system: Localisation, function, receptors
Adenosine is a neuromodulator that plays a very important role in basal ganglia function
(reviewed in Ferre et al., 1997). Its actions are mediated by the adenosine receptors,
which are part of the G-protein coupled family and classified into Ai, A2A, A2B and A3
subtypes. Their differential regional distribution in the brain and the heterogeneity of
receptor coupling reflect adenosine’s diverse neuromodulatory role. Compared to A2B
and As, Ai and A2A receptors exhibit a much higher affinity for adenosine, and are
thought to mediate the tonic adenosine tone encountered in most brain regions (see Table
1.4). Ai receptors are widely distributed throughout the CNS and their stimulation leads
to inhibition of AC activity via Gi-dependent coupling. In addition, Ai receptor
activation hyperpolarises the resting potential of the cell membrane, by inhibiting calcium
channels (MacDonald et al., 1986), and stimulating inwardly rectifying potassium
channels (Trussell and Jackson, 1985).
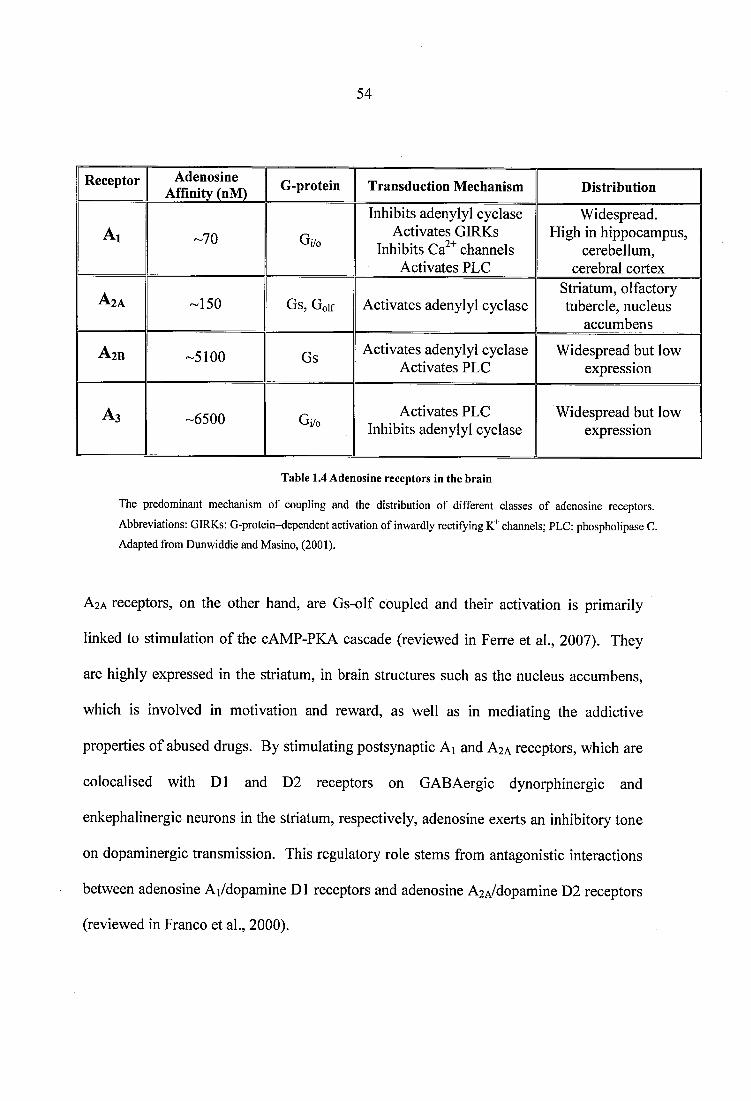
54
Receptor Adenosine Affinity (nM) G-protein Transduction Mechanism Distribution
A i -70 Gi/o
Inhibits adenylyl cyclase Activates GIRKs
Inhibits Ca " channels Activates PLC
Widespread. High in hippocampus,
cerebellum, cerebral cortex
A 2A -150 Gs, Golf Activates adenylyl cyclaseStriatum, olfactory tubercle, nucleus
accumbens
A 2B -5100 Gs Activates adenylyl cyclase Activates PLC
Widespread but low expression
A 3 -6500 Gi/o Activates PLC Inhibits adenylyl cyclase
Widespread but low expression
Table 1.4 Adenosine receptors in the brain
The predominant mechanism of coupling and the distribution of different classes of adenosine receptors.
Abbreviations: GIRKs: G-protein-dependent activation of inwardly rectifying channels; PLC: phospholipase C. Adapted from Dunwiddie and Masino, (2001).
A2A receptors, on the other hand, are Gs-olf coupled and their activation is primarily
linked to stimulation of the cAMP-PKA cascade (reviewed in Ferre et al., 2007). They
are highly expressed in the striatum, in brain structures such as the nucleus accumbens,
which is involved in motivation and reward, as well as in mediating the addictive
properties of abused drugs. By stimulating postsynaptic Ai and A2A receptors, which are
colocalised with D l and D2 receptors on GABAergic dynorphinergic and
enkephalinergic neurons in the striatum, respectively, adenosine exerts an inhibitory tone
on dopaminergic transmission. This regulatory role stems from antagonistic interactions
between adenosine Ai/dopamine Dl receptors and adenosine A2A/dopamine D2 receptors
(reviewed in Franco et al., 2000).

55
1.3.3.2 Neurochemical evidence fo r interactions between nAChRs and adenosine
The underlying neurochemical mechanism of interactions between nicotinic and
adenosine receptors is poorly understood. By stimulating neuronal nAChRs, nicotine
plays an important neuromodulatory role in the CNS and participates in a large number of
physiological processes (reviewed in Dani and Bertrand, 2007). Several lines of evidence,
however, have linked the reinforcing and addictive properties of the drug with a
facilitation of DA neurotransmission in the mesolimbic dopaminergic system (reviewed
in Picciotto, 1998). Caffeine elicits its stimulant effects through a non-selective
competitive antagonism at adenosine Ai and A%A receptors (reviewed in Ferre, 2008).
Despite the large consumption of caffeine-containing beverages worldwide, no consistent
reports have been obtained as to the drug’s potential of increasing extracellular DA levels
in the NAc, an elevation that is characteristic o f all abused substances (Di Chiara and
Imperato, 1988). Consistent with caffeine’s role as a weak primary reinforcer, reports on
the administration of behaviourally relevant doses of caffeine in rats have either shown
relatively small significant increases in extracellular DA (Solinas et a l, 2002; Quarta et
a l, 2004a), or failed to demonstrate caffeine-induced effects on DA levels in the NAc
(Tanda and Goldberg, 2000; Acquas et a l, 2002). Nevertheless, rats that have been
exposed to 3 mg/ml of caffeine in their drinking water for three weeks, show enhanced
extracellular DA levels in the shell of the NAc compared with tap-water drinking animals,
following s.c. nicotine administration at 0.2 or 0.4 mg/kg (Tanda and Goldberg, 2000).
Therefore, chronic exposure to caffeine alters the response of the DAergic system to
nicotine, implying that mesolimbic neurotransmission contributes to the interactions
between the two drugs. The facilitatory mode of caffeine’s action on nicotine-induced
DA signalling is most probably the result of antagonism at adenosine A% and A2A
receptors. Due to the antagonistic nature of interactions between adenosine and

56
dopamine receptors, inhibition of Ai and A2A receptors by caffeine would allow a more
efficient transduction of the DA signal, which may account for the nicotine-induced
increases in DA release in caffeine pre-exposed animals. In addition to postsynaptic
interactions, presynaptic adenosine-mediated mechanisms are also involved in the
regulation of DAergic transmission. Adenosine Ai receptors, for instance, are located
presynaptically on DAergic synapses, where they inhibit DA release (Borycz et al., 2007).
Moreover, Ai and A2A receptors are localised presynaptically on glutamatergic nerve
terminals in the striatum, where they inhibit or induce glutamate release, respectively,
depending on the extracellular concentration of adenosine (Ciruela et al., 2006).
Therefore, adenosine antagonists can release the pre- and postsynaptic brakes that the
neuromodulator exerts on DA signalling, thereby potentiating nicotine’s effects on
mesolimbic neurotransmission.
The role of the A2A receptor in the neurochemical effects of nicotine has been elucidated
by the generation of the A2A KO mouse (Ledent et al., 1997). As demonstrated by in vivo
microdialysis, genetic inactivation of adenosine A2A receptors abolishes nicotine-induced
increases in NAc DA levels of mutant mice compared to WT animals, suggesting that
adenosine A2A receptors contribute to the reinforcing properties of nicotine (Castane et al.,
2006). Indeed, conditioned placed preference to 0.5 mg/kg of nicotine is not observed in
the absence of the A2A receptor (Castane et al., 2006). Adenosine Ai and A2A receptors
are expressed on striatal cholinergic neurons (Dixon et al., 1996; Preston et al., 2000),
and functional evidence from synaptosome preparations and electrophysiology suggests
that their activation exerts opposing actions on ACh release; stimulation of A, decreases,
whereas activation of A2A receptors increases striatal acetylcholine (Brown et al., 1990;
Kurokawa et al., 1994; Gubitz et al., 1996; Preston et al., 2000). A2A-mediated ACh

57
release in the rat striatum has also been demonstrated in vivo (Kurokawa et ah, 1996),
suggesting that striatal cholinergic neurotransmission is tonically mediated by adenosine.
In turn, tonically active cholinergic neurons have been shown to regulate DA release by
acting at (32 nicotinic subunits on striatal DA axon terminals (Zhou et al., 2001; Rice and
Cragg, 2004). Depletion of endogenous ACh or silencing of the (32 subunit gene
decreases evoked DA release by -80%, an effect that can also be evoked by
mecamylamine, but not the muscarinic antagonist atropine (Zhou et al., 2001). Therefore,
striatal DA release seems to be under the regulation of ongoing nicotinic cholinergic
activity. Thus, in animals lacking A2A receptors a corresponding decrease of basal ACh
tone would most likely relate to decreased basal levels of DA. Indeed, deletion o f the
A2A gene leads to a hypo-dopaminergic mouse phenotype, with mutant animals showing a
45% reduction in extracellular DA concentration compared to WT littermates, as shown
by in vivo microdialysis (Dassesse et al., 2001). A lower basal level of DA following
deletion of the A2A gene could therefore explain the lack of nicotine-induced DA efflux
observed in the A2A KO mouse (Castane et al., 2006). Note, however, that contrary to the
Dassesse et al., (2001) report, lower baseline DA levels were not observed in the A2A KO
control group in the Castane et al., (2006) study. In view of this discrepancy, one cannot
exclude that mechanisms other than the direct impact of a decreased cholinergic tone on
DA signalling, mediate nicotine-induced changes in striatal DA levels of the A2A KO
mutant. For instance, nicotine has been shown to exert its effects on mesolimbic DAergic
transmission via presynaptic a l nAChRs, localized on glutamatergic nerve terminals
(Mansvelder and McGehee, 2000). Given the colocalisation between adenosine A2A and
glutamatergic mGluR5 receptors on striatal glutamatergic terminals (Ciruela et al., 2006),
which has been shown to facilitate glutamate release in a synergistic manner (Rodrigues
et al., 2005), the possibility exists that the absence of A2A receptors impairs glutamatergic

58
neurotransmission, thereby indirectly blunting DA signalling in nicotine-treated A2A KO
mice. Similarly, antagonistic A2A and D2 receptor interactions on GABAergic
enkephalinergic neurons, which also express nAChRs of the non-a7 subtype (de Rover et
al., 2002), are absent in mice lacking the A2A receptor. Therefore, altered GABAergic
signalling is potentially involved in the diminished nicotine-induced striatal DA release in
the A2A KO mouse. Altogether, it seems reasonable to suggest that at the level of
neuronal function adenosine A2A receptors play an essential role in the modulation of
nicotine-induced DA neurotransmission.
At an intracellular level, chronic nicotine administration in rats at a rate of 6 mg/kg/day
has been shown to stimulate the adenylyl cyclase (AC) pathway in medium spiny neurons
(Abreu-Villaca et al., 2003). Importantly, during either spontaneous (Abreu-Villaca et al.,
2003) or mecamylamine-induced (Tzavara et al., 2002) withdrawal, both AC activity and
cyclic AMP (cAMP) are further increased in nicotine-withdrawn animals. Enhanced AC
activity results in increased levels of cAMP, which is the precursor of extracellular
adenosine. These adaptive cellular changes imply functional interactions between
nicotine and adenosine systems, which might contribute towards the development of
physical dependence.

59
1.3.3.3 Behavioural evidence fo r interactions between nAChRs and adenosine
The effects of nicotine and caffeine on locomotion are biphasic. Small doses of caffeine
stimulate, whereas higher doses depress locomotor activity (reviewed in Daly and
Fredholm, 1998). In a familiar environment, low doses of nicotine result in locomotor
activation, whereas acute nicotine treatment in a novel environment rapidly induces
locomotor depression (reviewed in Picciotto et al., 2000). Interactions between
adenosine and nicotinic systems have been clearly documented using locomotion as the
behavioural endpoint. The subcutaneous coadministration of nicotine (0.2 mg/kg) and
caffeine (8mg/kg) abolishes the depressant locomotor effects of a single nicotine injection
in rats. Moreover, chronic nicotine at 0.4 mg/kg induces tolerance to the acute
behavioural depressive effects of the drug, which can be overcome by the
coadministration of nicotine with caffeine, but not by either drug alone in nicotine-
tolerant rats (Cohen et al., 1991). In mice, chronic caffeine pretreatment diminishes
nicotine-induced depression of locomotor activity (Nikodijevic et al., 1993), and nicotine-
pretreatment at 1.0 or 2.0 mg/kg for 5 days enhances the locomotor stimulant effects of
5.0 mg/kg caffeine (Celik et al., 2006). Therefore, synergistic interactions between
caffeine and nicotine have been extensively reported on locomotor activity. In addition,
although ablation of the A2A receptor gene does not affect acute locomotor responses to
nicotine, it produces animals with a characteristic hypolocomotor basal phenotype
(Ledent et al., 1997; Castane et al., 2006), suggesting that adenosine A2A receptors play a
key role in modulating motor activity.
In paradigms of drug abuse that more directly measure the reinforcing properties of
psychoactive substances, pre-exposure of rats to caffeine for 7 days at a dose of 150-180
mg/kg/day accelerates the acquisition of nicotine self-administration. When caffeine is

60
excluded from the drinking solution, nose-poking for 0.03 mg/kg/infusion of nicotine
returns to baseline, whereas adding caffeine to the drinking water of control animals
increases responding by over 90%, demonstrating that chronic caffeine potentiates the
reinforcing properties of nicotine (Shoaib et al., 1999). Caffeine’s rewarding effects have
not been extensively examined using CPP. The drug can induce place conditioning that
is biphasic in nature, with lower doses (3.0mg/kg) producing place-preference and higher
(30 mg/kg) place aversion (Brockwell et al., 1991). The relative contribution of Ai and
A2A receptors in the conditioning effect of caffeine has been examined using subtype-
selective compounds (Brockwell and Beninger, 1996). The A2A antagonist CGS 15943A
dose dependently produces CPP, whereas the Ai antagonist CGS 21680 fails to do so,
underlying the importance of adenosine A2A receptors in establishing place conditioning.
Importantly, genetic deletion of the A2A receptor attenuates nicotine-induced CPP, which
identifies A2A as an important factor contributing to the rewarding properties of the drug
(Castane et al., 2006). In drug-discrimination experiments, chronic caffeine does not
generalise to a nicotine cue and vice versa, suggesting that there are no similarities in the
subjective-like effects of the two substances (Modrow et al., 1981; Rosecrans, 1989).
This lack of cross-generalisation, however, does not preclude that caffeine can acutely
enhance sensitivity to the subjective effects of nicotine. Indeed, rats chronically exposed
to 0.25 mg/ml of caffeine in their drinking water discriminate 0.4 mg/kg of nicotine faster
than tap water-drinking animals (Gasior et al., 2000). This effect is also acutely observed
to sub-threshold doses of nicotine (0.05 mg/kg) and does not undergo tolerance (Gasior et
al., 2002). In addition, the discriminative-stimulus effects of nicotine can also be
potentiated by the A2A antagonist MSX-3 and the Ai antagonist cyclopentyltheophylline,
suggesting that both adenosine receptors are capable of modulating nicotine
discrimination (Justinova et al., 2009).

61
1.3.4 Interactions between nAChRs and other drugs o f abuse
Psychosocial and neurobiological reasons may account for the high co-morbidity of
cigarette smoking with substance abuse disorders. For a detailed overview of the
‘gateway’ mechanisms that link smoking to drug addiction, the reader is referred to
excellent reviews by Lindsay and Rainey, (1997), and Slotkin, (2002). Briefly, the
participation of the nicotinic cholinergic system in stimulant abuse is not only restricted
to cocaine (see Section 1.3.1). The prevalence of cigarette smoking, for instance, is even
higher in methamphetamine abusers, compared to cocaine addicts and the general
population (Baker et al., 2004; Brecht et al., 2007; Weinberger and Sofuoglu, 2009). In
animal models of drug addiction, three weeks of nicotine pretreatment increases, whereas
mecamylamine attenuates sensitisation of amphetamine-induced locomotor activity
(Schoffelmeer et al., 2002). Consistent with its behavioural effects, repeated nicotine
administration enhances DA efflux in the NAc of rats treated with amphetamine (Birrell
and Balfour, 1998). This suggests that nAChRs are involved in producing persistent
alterations in the psychomotor and reinforcing effects o f stimulants. Furthermore,
smoking is robustly correlated not only with stimulant, but also with ethanol consumption.
Indeed, more than 80% of alcoholics are smokers (DiFranza and Guerrera, 1990), while
nicotine dependence yields a 2.7 fold increased probability of leading to ethanol addiction
(Breslau, 1995). Importantly, the two disorders are associated at a genetic level, since
studies on twins have shown that the genetic factors linked to increased risk for
alcoholism are similar with those associated for nicotine dependence (Koopmans et al.,
1997). The co-abuse of nicotine and alcohol suggests that common neurobiological
mechanisms mediate the reinforcing effects of the two drugs.

62
CHAPTER
TWO

63
2 THESIS AIMS AND HYPOTHESIS
Tobacco use is the single most important preventable risk to human health globally
(Hatsukami et ah, 2008). Nicotine is only a weak primary reinforcer, but there is
evidence to suggest that the predominance of nicotine addiction stems from the drug’s
ability to enhance the potency of other reinforcers, rather than from its direct rewarding
effects (reviewed in Chaudhri et al., 2006). Indeed, a striking feature of nicotine
addiction is the prevalence of the drug’s use in combination with licit and illicit
substances. Tobacco smoking positively correlates with the probability and frequency of
psychostimulant abuse (Patkar et al., 2002), as well as with the severity of opiate
dependence (Frosch et al., 2000). In addition, the concurrent consumption of nicotine
with caffeine places the two substances among the most widely used licit drugs in
modem societies (reviewed in Fredholm et al., 1999). This high comorbidity raises
fundamental questions about the extent to which nicotinic cholinergic mechanisms
contribute to poly-drug abuse. Hence, the overall working hypothesis of the present
thesis is that alterations in a4(32* and/or a7 nAChR binding occur following chronic
treatment with nicotine or illicit substances, which are indicative of the interactions that
render a weak primary reinforcer as broadly abused as nicotine.
• To investigate the influence of distinct subtypes of nAChRs on cocaine related
behaviour, the effects of chronic a4p2* and a7 nAChR antagonism on cocaine-induced
locomotor activity, stereotypy and grooming have been studied using dihydro-g-
erythroidine (DhbE) and methyllycaconitine (MLA), respectively. To establish possible
neurochemical and neuroanatomical loci for the effects of chronic cocaine administration
on nAChR regulation, quantitative receptor autoradiography of a4p2* and a7 nAChRs
has been conducted in brain sections from cocaine-treated C57BL/6 mice.

64
Autoradiography of dopaminergic D1 and D2 receptors, as well of the dopamine
transporters has been conducted in order to identify the impact of specific nAChR
blockade on the dopaminergic system of cocaine-treated animals. To investigate the
participation of the nicotinic cholinergic and dopaminergic systems in cocaine
withdrawal, quantitative receptor autoradiography of a4p2* and a7 nAChRs, and of
dopaminergic D l, D2 receptors and transporters has been conducted in brains of mice,
chronically withdrawn from ‘binge’ cocaine treatment.
• To investigate whether morphine administration or the acute and prolonged
withdrawal from morphine treatment alter the receptor component of the nicotinic
cholinergic system, quantitative autoradiography of a4p2* and a7 nAChRs has been
performed in mice treated and withdrawn from an escalating-dose, ‘intermittent’ protocol
of morphine administration.
• An investigation into the mechanism of interactions between nicotine and
adenosine has been performed by examining whether genetic deletion of the A2A receptor
gene leads to compensatory changes in the binding or distribution characteristics of
nicotinic and dopaminergic receptors. To further characterise the effects of adenosine
A2A gene deletion on nicotine-induced neuroadaptation, quantitative autoradiography of
a4p2* and a7 nAChRs, and of dopaminergic D l, D2 receptors and transporters has been
conducted in CDl wild type and adenosine A2A receptor knockout animals, following
chronic nicotine treatment.
• The extent to which nicotine-induced neuroadaptation is the consequence of the
drug’s direct pharmacological action has been examined in C57BL/6 mice, treated with a
yoked-control paradigm of nicotine-self administration. The consequences of active vs.
passive nicotine administration on nicotinic cholinergic and dopaminergic regulation

65
have been investigated by means of quantitative autoradiography of a4p2* and a7
nAChRs, and of dopaminergic D l, D2 receptors and transporters.
The ambition of the current work has therefore been to contribute towards unraveling the
mechanisms of nicotine reinforcement, and to characterise the neurobiological substrates
that underlie the concurrent consumption of nicotine with illicit and licit drugs.

66
CHAPTER
THREE

67
3 BEHAVIOURAL AND NEURO CHEMICAL INTERACTIONS BETWEEN
COCAINE AND THE NICOTINIC CHOLINERGIC SYSTEM
3.1 Introduction
The prevalence of poly-drug abuse is characteristically depicted in the concurrent consumption
of nicotine with cocaine. Cigarette smoking in cocaine abusers is greater compared to the
general population, and nicotine intake has been associated both with the probability and the
frequency of cocaine use (Henningfield et al., 1990; Schorling et al., 1994; Breslau, 1995;
Martinez-Ortega et al., 2006). Moreover, cocaine addiction is linked to an increased incidence of
psychotic-like behaviours (Tang et al., 2007), with cocaine-dependent individuals reporting both
stimulant enhancing and calming effects of nicotine consumption (Wiseman and McMillan,
1998). A prima facie case for these apparently opposing effects of nicotine in cocaine addiction
is that distinct subtypes of nicotinic acetylcholine receptors (nAChRs) differentially mediate
cocaine-induced behaviour.
The principal mode of cocaine’s action, responsible for its reinforcing potential and high abuse
liability, involves activation of the mesolimbic dopaminergic system via blockade of the
monoamine dopamine transporter (DAT) (Anderson and Pierce, 2005). Nevertheless, it is
becoming increasingly accepted that cocaine impacts on dopaminergic neurotransmission partly
via the activation of the nicotinic cholinergic system. The development of cocaine-induced
locomotor sensitization, for instance, is mediated by nicotinic receptor activation, as pre
treatment with nicotinic antagonists reduces locomotor activity in cocaine-sensitized rats
(Schoffelmeer et al., 2002). Moreover, nicotine injections prior to cocaine self-administration
progressively enhance cocaine intake in rats (Bechtholt and Mark, 2002), whereas treatment with
nicotinic antagonists diminishes the rewarding effect of cocaine in mice (Zachariou et al., 2001).

68
Data from in vivo microdialysis studies point to the mesolimbic dopaminergic system as the site
for cocaine-nAChR interactions (Gerasimov et al., 2000; Zanetti et al., 2007), suggesting that
nicotinic cholinergic signalling within dopamine-rich brain regions mediates the effects of
cocaine.
Neuronal nAChRs are a heterogeneous family of pentameric structures, formed by the assembly
of five a and (3 subunits (see Section 1.1.1). Of the multiple receptor subtypes that exist,
heteromeric a4p2* and homomeric a7 nAChRs are abundantly expressed throughout the central
nervous system (CNS), where they have been shown to differentially mediate the effects of
nicotine on several CNS functions (Dani and Bertrand, 2007). Although the involvement of the
nicotine system in cocaine related addictive behaviour has been well documented, the exact
contribution of distinct nAChR subtypes in the behavioural effects of chronic cocaine is unclear.
To determine the role of the two major classes of nAChRs in the behavioural effects of chronic
cocaine, the impact of a4p2* or a7 receptor antagonism on cocaine-induced locomotor activity,
stereotypy and grooming was studied using a ‘binge’ drug administration protocol, which
mimics the temporal patterns of cocaine abuse in humans (Koob and Kreek, 2007). To
determine the neurochemical effects of chronic cocaine treatment and nAChR antagonism on the
nicotinic and dopaminergic systems, quantitative autoradiography of a4|32* and a l nAChRs, D l
and D2 dopaminergic receptors, and of cholinergic and dopamine transporters was carried out.
Based on the behavioural and neurochemical observations from the ‘binge’ administration
experiments, the role of a4(32* nAChR upregulation in the stimulant effects of cocaine was
further investigated following pretreatment with different doses of nicotine. To investigate the
involvement of the nicotinic cholinergic system in cocaine withdrawal, quantitative
autoradiography of nAChRs was performed in brain sections of mice, chronically withdrawn
from ‘binge’ cocaine administration.

69
3.2 Materials and Methods
3.2.1 nAChR antagonist dose selection studies
In the current set of experiments, dihydro-B-erythroidine (DhBE, Sigma-Aldrich, UK) and
methyllycaconitine (MLA, Sigma-Aldrich, UK) were used to block (32* and a l nicotinic
subunits, respectively. The doses of nicotinic antagonists were based on literature
(Damaj et al., 2003; Schoffelmeer et al., 2002), and their potency to antagonize nicotine-
induced effects was confirmed in preliminary experiments, as detailed in Sections 3.2.1.1
and 3.2.1.2. For all experiments, 6-8 week old, male C57BL/6 mice (Charles River Ltd.,
Kent, UK), weighing 20-25 g, were used. Animals were housed individually in a
temperature-controlled room, under a 12 h light/dark schedule. Food and water were
available ad libitum, except during the experimental sessions. All studies were conducted
in accordance with protocols approved by the Home Office, UK, and in accordance with
European Community Council directive 86/609/EEC.
3.2.1.1 Antagonism o f acute nicotine-induced hypolocomotion by DhfiE
C57BL/6 mice were habituated to the locomotor chambers for 4 days, prior to treatment
(n=6). On the day of the experiment, animals were pretreated with saline (0.1 ml/kg) or
DhBE (2.0 mg/kg), 5 min prior to the injection of saline (0.1 ml/kg) or nicotine (1.0
mg/kg, free-base weight). 5 min after acute nicotine, total locomotor activity was
measured for 10 minutes. All injections were delivered intraperitoneally.
3.2.1.2 Chronic nicotine pretreatment and MLA-precipitated withdrawal
Chronic nicotine hydrogen salt was delivered to C57BL/6 mice (n=6) using miniosmotic
pumps (ALZET®2002 model, Charles River, UK), at a dose of 7.8 mg/kg/day (free-base
weight), at a rate of 0.5 p 1/hour, for a period of 14 days. For minipump implantation.

70
animals were anaesthetised with a volatile isofluorane anaesthetic (3.5%-4.5%) (Isoflo,
Abbott Laboratories Ltd, Kent, UK), which was vaporised in 95% O2 / 5% CO2 gas and
delivered by a U400 anaesthetic unit (Univentor, Royem Scientific, UK), at a flow rate of
-450 ml/min. The animals were placed in the anaesthetic chamber for 3-4 min until all
righting reflex was lost, and were subsequently connected to a mask delivering
anaesthesia throughout the surgery. A single incision was made along the midline of the
back, to reveal the subcutaneous layer. Blunt ended scissors were used to open up a
subcutaneous compartment, where an osmotic minipump was placed parallel to the spine,
with the flow moderator pointing away from the incision. The incision was closed with 2
Michelle clips. Upon completion of the surgical procedure mice were allowed to recover
in a heated recovery chamber until their righting reflex returned, and were transferred to
their home cage.
After 14 days of nicotine treatment, animals received an acute injection of MLA (5.0
mg/kg, i.p.) and withdrawal signs were measured for 20 min immediately afterwards in
an observation chamber. The number o f head shakes, paw tremors, scratches,
retropulsive movements, body tremor, genital licks and jumps were counted. A global
withdrawal score was subsequently calculated for each animal, by adding individual signs
of MLA-precipitated symptoms, as described previously (Berrendero et al., 2002).
3.2.2 Animals and ‘binge* cocaine treatment
6-8 week old, male C57BL/6 mice (Charles River Ltd., Kent, UK), weighing 20-25 g,
were used in all experiments. Studies were conducted in accordance with protocols
approved by the Home Office, UK, and in accordance with European Community
Council directive 86/609/EEC, under housing conditions that have been described in

71
Section 3.2.1. Animals were habituated to the testing environment and to handling for a
period of 7 days, prior to treatment. For the chronic treatment protocol, saline or cocaine
(Sigma-Aldrich, UK) were delivered alone or were co-administered with nicotinic
antagonists for a period of 14 days, in a steady dose, ‘binge’ administration paradigm.
Animals received three daily intraperitoneal (i.p.) injections, one hour apart, with the first
injection delivered one hour after the beginning of the light phase of the light/dark cycle.
Dailv administration protocol:
Group 1 (n=6): 3 x Saline (0.1 ml/kg/inj)
Group 2 (n=7): 3 x Cocaine (15.0 mg/kg/inj)
Group 3 (n=6): 3 x Saline + DhBE (2.0 mg/kg/inj)
Group 4 (n=7): 3 x Cocaine + DhBE
Group 5 (n=6): 3 x Saline + MLA (5.0 mg/kg/inj)
Group 6 (n=7): 3 x Cocaine + MLA
For the chronic withdrawal study, two groups of animals (n=6) were treated with ‘binge’
saline or cocaine (3 x 15 mg/kg/inj for 14 days) and were allowed to withdraw from
chronic treatment for a period of 14 days. All drugs (salt weights) were dissolved in
sterile 0.9% saline.
3.2.3 Minipump preparation and nicotine pretreatment experiments
Osmotic minipumps (ALZET®2002 model, Charles River, UK) were filled with either
nicotine hydrogen salt in 0.9% sterile saline, or vehicle (0.9% sterile saline; pH=7.4), and
were implanted to C57BL/6 mice (n=4), as described in Section 3.2.1.2. Nicotine
hydrogen salt (Sigma-Aldrich, UK) was delivered at 0.2 mg/kg/hr, 0.45 mg/kg/hr, or 0.8

72
mg/kg/hr, at a rate of 0.5pl/hour, for a period of 8 days. Doses are reported as the free
base of nicotine, and were selected as to their ability to upregulate neuronal nAChRs in
C57BL/6 mice (Marks et al., 2004). A single i.p. injection of cocaine (15.0 mg/kg/inj) +
DhBE (2.0 mg/kg/inj) was administered to individual groups of mice, 8 days after
minipump implantation. This time point was chosen based on the temporal pattern with
which differences emerged among groups o f ‘binge’ treated animals.
3.2.4 Motor activity measurements
Total horizontal activity and total vertical activity (rearing) were measured in 12 motility
chambers (40cm length x 20 cm wide x 20 cm height; Linton Instrumentation, Norfolk,
UK). Each cage had two sets of 16 photocells located at right angles to each other,
projecting horizontal infrared beams 2.5 cm apart and 1 and 6 cm above the cage floor.
Each daily session began with habituating the animals in the motility chambers for 60
min (assessment of basal activity). Subsequently, mice received an i.p. injection of saline
or cocaine (± nAChR antagonists), and were returned immediately to the chambers where
activity was assessed for 60 min. Animals received a second and third injection 60 min
and 120 min respectively after the first one. Behavioural activities for each mouse after
each of the three daily injections of saline or cocaine (± nAChR antagonists) were
monitored daily for a period of 14 days. Total horizontal and vertical motor activity were
defined by the measurement of sequential infrared beam breaks, recorded every 5 min,
beginning immediately after placing the animals in the cage following an injection of
saline or cocaine (± nAChR antagonists) and continued for a 60 min period.

73
3.2.5 Cocaine-induced stereotypy and grooming
Animals were videotaped 20 minutes after each injection, and were later rated for
stereotypy and grooming by a trained observer, blind as to the animal’s treatment group.
The time point was chosen based on the half maximal brain concentrations of cocaine,
following its i.p. administration in C57BL/6 mice (Azar et al., 1998). Observations were
limited to 40 s for each mouse, to enable all animals to be observed at a similar time point
following each injection (Schlussman et al., 2005). The rating for stereotypy was based
on the scale used by Bailey et al., (2007), and consisted of a graded scale of drug-induced
behaviours: 1) asleep, inactive; 2) alert, ‘comfort’ grooming; 3) increased sniffing
(occasional light sniffing, often while exploring the cage); 4) intermittent rearing and
sniffing (two or three rears in a 20s period, with sniffing frequently at the apex of the
rear); 5) increased locomotion; 6) intense sniffing in one location (rapid sniffing, often
with head down is the predominant behaviour displayed); 7) continuous pivoting and
sniffing (no rearing, no locomotion); 8) intermittent rearing and sniffing (intermittent up
and down rearing behaviour with locomotion); 9) maintained rearing and sniffing (animal
remains up on hind legs throughout most of the observed period); 10) splayed limbs.
Each animal received a single score following each injection, which corresponded to the
specific stereotypic behaviour predominantly observed during the 40 sec observation
period. Stereotypy was scored every day throughout the duration of the experiment. The
median score for each mouse on each day was used for later analysis.
For grooming behaviour, a percentage of time spent grooming was calculated for the
entire observation period for each animal, on each day, throughout the duration of the
study. The mean percentage of time spent grooming from each group was then used in
the analysis.

74
3.2,6 Quantitative receptor autoradiography
3.2.6.1 Autoradiography o f dopamine D l and D2, nicotinic cholinergic a4p2"^ and a l
receptors, and o f dopaminergic and cholinergic transporters
Animals were killed by cervical dislocation 30 min following the last injection of saline
or cocaine (± nAChR antagonists) and brains were immediately removed, quickly frozen
in isopentane and stored at -80°C for subsequent processing. Tissue sectioning was
carried out at -21°C using a Zeiss Microm HM505E cryostat. 20 pm frozen coronal
sections were cut at 300 pm intervals, from rostral to caudal levels. Sections were stored
at -20°C for radioligand binding.
Quantitative autoradiography was performed in order to measure binding to a4p2* and
a l nAChRs, the high affinity choline transporter, dopaminergic D l and D2 receptors, and
the dopamine transporter (DAT). On the day of each experiment, sections were thawed
and processed according to established protocols (Javitch et al., 1984; Orr-Urtreger et al.,
1997; Whiteaker et al., 2000b; Marks et al., 2002; Lena et al., 2004; Besson et al., 2007),
with minor modifications. Sections for analysis were derived from six to seven brains
from each of the six treatment groups (n=6-7). Multiple, adjacent sections from all
groups were processed together in a paired binding protocol.
For a4p2* nicotinic receptor binding, sections were pre-incubated for 10 min at room
temperature in Tris-HCl buffer, containing 120 mM NaCl, 5 mM KCl, 2.5 mM CaCL,
and 1 mM MgCb, pFI 7.4, followed by incubation with 100 pM [^^^Ijepibatidine
(PerkinElmer, Boston, USA; specific activity 2,200 Ci/mmol) for two hours at room
temperature. Determination of subtype-specific binding was performed using adjacent
sections from each brain, in order to measure total [^^^Ijepibatidine binding (no

75
rl25ncompeting ligand), and [^^HJepibatidine binding in the presence of 20 nM cytisine
(Sigma-Aldrich, UK). Competition of [^^^I]epibatidine binding by unlabeled cytisine has
been used to reveal two subpopulations of nAChRs with high and low affinity for cytisine,
termed cytisine-sensitive and cytisine-resistant [^^^I]epibatidine binding sites,
respectively. The epibatidine binding sites with high affinity for cytisine correspond to
a4p2* nicotinic receptors, and were calculated following the subtraction of cytisine-
resistant from total epibatidine binding. To determine non-specific binding (NSB),
further adjacent sections were incubated with epibatidine in the presence of 300 pM
of (-) nicotine hydrogen tartrate (Sigma-Aldrich, UK). Incubations were terminated by
two, ten minute washes into ice-cold 50 mM Tris-HCL buffer (pH 7.4), and a rapid rinse
in ice-cold water.
For the homomeric a l nAChR, sections were pre-incubated for 30 minutes in 50 mM
Tris-HCl, containing 1% w/v BSA (pH 7.4, room temperature). Adjacent, non-specific
binding sections were pre-incubated in the same buffer, containing ImM of nicotine
bitartrate salt. Sections were then incubated at room temperature with 3nM of [^^^I]a-
bungarotoxin (GE Healthcare, Little Chalfont, UK; specific activity 250 Ci/mmol;
PerkinElmer, Boston, USA; specific activity 159 Ci/mmol) for 3 hours. Adjacent
sections were incubated in the same buffer, in the presence of ImM (-) nicotine hydrogen
tartrate, to calculate NSB. Incubations were terminated by 3 xIO minute washes into ice
cold 50 mM Tris buffer (pH 7.4), and a rapid rinse in ice cold water.
For the high affinity choline transporter (CHT), sections were incubated with 8 nM
[^H]Hemicholinium-3 (PerkinElmer, Boston, USA; specific activity 144.5 Ci/mmol) at
4°C for 60 min in 50 mM Tris (pH 7.4), containing 300 mM NaCl. After incubation.

76
sections were rinsed six times for 1 min each in ice-cold 50 mM Tris (pH 7.4) and briefly
in distilled water. Non-specific binding was measured in the presence of 100 pM
unlabeled hemicholinium-3.
For D l and D2 receptor binding, all sections were first pre-incubated for 20 min in 50
mM Tris-HCl buffer, pH 7.4, containing 120 mM NaCl, 5 mM KCl, 2 mM CaC^ and 1
mM MgCh, at room temperature. For D l receptors, sections were then incubated for 90
min in the same buffer, at room temperature, in the presence of 4 nM [^H]SCH23390
(PerkinElmer, Boston, USA; specific activity 70.3 Ci/mmol) and 1 pM mianserin, in
order to avoid binding of [^H]SCH23390 to 5-HT2 and 5-HTic receptors. To label D2
receptors, incubation was carried out for 60 min in the presence of 4 nM [^H]raclopride
(PerkinElmer, Boston, USA; specific activity 60.1 Ci/mmol), under identical pH and
temperature conditions. Non-specific binding was determined on adjacent sections in the
presence of 10 pM of cis-flupenthixol for D l receptors or 10 pM of sulpiride for D2
receptors. The incubations were terminated by rapid rinses (6x1 min) in ice-cold 50 mM
Tris-HCl buffer (pH 7.4) followed by a dip into ice-cold distilled water.
For the DAT binding, all slides were pre-incubated for 5 min at 4°C in 50 mM Tris-HCl
buffer, pH 7.4, containing 300 mM NaCl and 5 mM KCl. The slides were subsequently
incubated at 4°C for 45 min in the same buffer, containing 4 nM [^Hjmazindol
(PerkinElmer, Boston, USA; specific activity 20.6 Ci/mmol) and 0.3 pM desipramine, to
block the binding to norepinephrine uptake sites. Non-specific binding was determined
in the presence of 10 pM mazindol. After incubation, sections were rinsed twice for 1
min in ice-cold Tris buffer, and briefly dipped in ice-cold distilled water.

77
3.2.6.2 Film exposure and development
Following binding, sections were dried for two hours under a cold stream of air, and they
were subsequently dried overnight using anhydrous calcium sulphate (BDH Chemicals,
Poole, UK). Adjacent total and non-specific labelled sections from all animals were laid
down to Kodak BioMax MR-1 film in parallel. To allow quantification, autoradiographic
microscales of known radioactive concentration were also apposed to each film.
[^^^IJepibatidine and [^^^l]a-bungarotoxin bound sections were exposed to film for a
period of 24 h and 1 week, respectively, along with a set of " C microscale standards,
which had been cross-calibrated to iodinated standards (Miller and Zahniser, 1987;
Baskin and Wimpy, 1989). [^Hjradioligand bound sections were exposed to film along
with microscale calibration standards for a period of 5 weeks for D l receptors and the
DAT, 6 weeks for D2 receptors, and 25 days for the high affinity choline transporter.
Kodak BioMax MR-1 films, [^H], [ ^ 1], and microscale standards were purchased
from GE Healthcare Life sciences, Amersham, UK.
For development, films were placed into a tray and were covered with an aqueous
solution of 50% v/v Kodak D19 developer (Sigma-Aldrich, UK). The reaction was
stopped after 1 min by placing films in a solution o f distilled water, containing a drop of
glacial acetic acid for 1 min. Fixation of images was carried out for 3min in Kodak rapid
fix solution (Sigma-Aldrich, UK). Films were then rinsed under running water for 30
min and dried overnight in a fume cupboard.

78
3.2.6.3 Quantitative analysis
Quantitative analysis of receptor binding was performed by video-based computerised
densitometry, using an MCID image analyser (Imaging Research, Canada), as previously
described (Kitchen et al., 1997). Briefly, optical density values quantified from the [^H]
and microscale standards were entered with their corresponding radioactivity values
(fmol/mg tissue equivalents) into a calibration table, and the relationship between tissue
radioactivity and optical density was determined using the MCID software. Appropriate
adjustments were undertaken to allow for the radioactive decay of both the standards and
the radioligands. Values of specific receptor binding were derived after subtraction of
non-specific binding from total binding images.
Where applicable, measurements were taken from both brain hemispheres and thus
represent a duplicate determination for each brain region examined. Computerised tools
were used to take measurements from each area. Areas of the cortex, olfactory tubercle,
and superior colliculus were analysed by sampling with a box tool, and all other areas
were analysed by freehand drawing. All structures were identified by reference to the
mouse brain atlas of Franklin and Paxinos, 2001.

79
3.2.7 Statistical analysis
Preliminary data from the antagonism of nicotine-induced hypolocomotion by DhBE
were analysed using one-way ANOVA; Mann-Whitney’s non-parametric test was used
for the analysis of MLA-precipitated nicotine withdrawal.
For the ‘binge’ treatment protocol, all cocaine-induced behaviours were analysed by two-
way analysis of variance (ANOVA) for the factors treatment and time, with repeated
measures for time. Tukey’s Honest Significance (HSD) post hoc test for unequal N
(Spjotvoll-Stoline test) was applied when appropriate, to compare differences in
behavioural responses between groups, on individual test days. In addition, one way
repeated measures ANOVA was used to analyze changes over time within each group.
Since the expression of behavioural stereotypy was measured with a behavioural rating
scale, non parametric Mann-Whitney U tests were also used in order to confirm results
obtained by the ANOVA.
For the nicotine-pretreatment experiments, two-way ANOVA for the factors pretreatment
(saline vs. nicotine) and treatment (cocaine vs. cocaine+DhBE) was used to compare
motor activity of pretreated animals, followed by Fischer’s LSD post hoc test.
Two-way ANOVA (for the factors treatment and region) was used for the comparison of
quantitative measures of a4p2* and a l nicotinic receptors, choline transporters,
dopaminergic D l and D2 receptors, and dopamine transporters in brain regions of chronic
‘binge’ saline and cocaine treated mice. Where ANOVA yielded significant treatment
effects, and depending on the n number of the treatment groups, Duncan’s multiple
ranges or Fisher’s LSD post hoc analysis was applied to investigate differences in
radioligand binding between groups in individual regions.
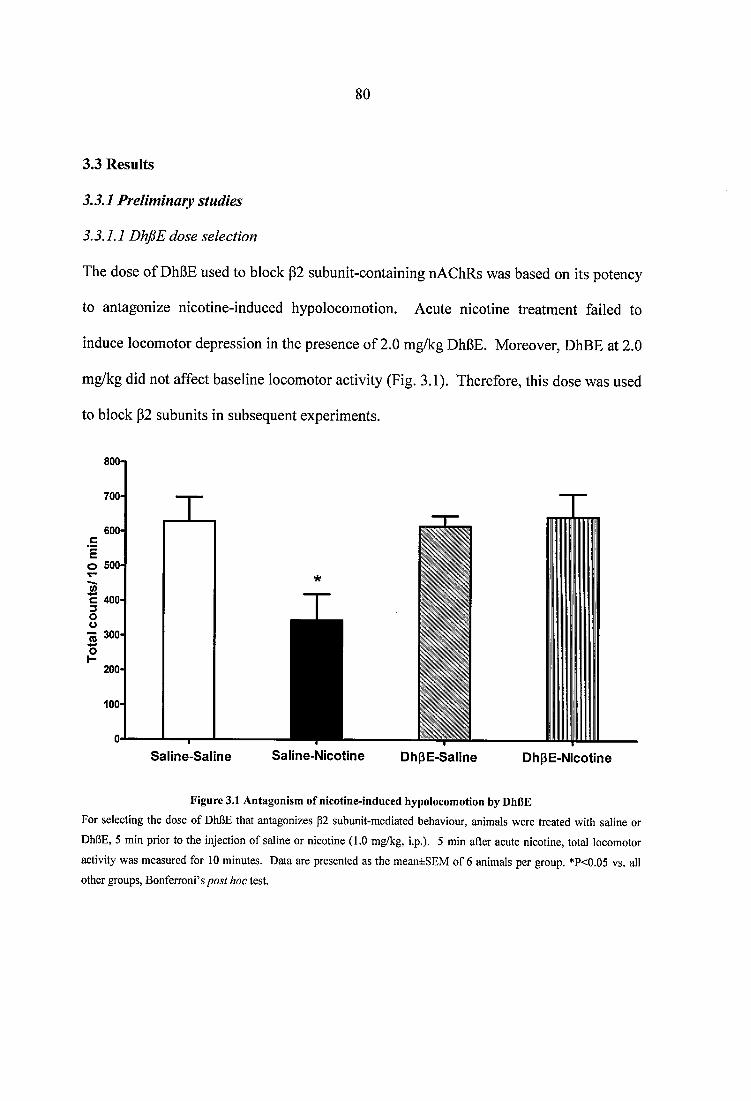
8 0
3.3 Results
3.3.1 Preliminary studies
3.3.1.1 DhfiE dose selection
The dose of DhBE used to block P2 subunit-containing nAChRs was based on its potency
to antagonize nicotine-induced hypolocomotion. Acute nicotine treatment failed to
induce locomotor depression in the presence of 2.0 mg/kg DhBE. Moreover, DhBE at 2.0
mg/kg did not affect baseline locomotor activity (Fig. 3.1). Therefore, this dose was used
to block P2 subunits in subsequent experiments.
800
O 500
S alin e -S a lin e Saline-N ico tine D hpE -S aiine D hpE -N icotine
Figure 3.1 Antagonism of nicotine-induced hypolocomotion by DhRE
For selecting the dose of DhBE that antagonizes p2 subunit-mediated behaviour, animals were treated with saline or
DhBE, 5 min prior to the injection of saline or nicotine (1.0 mg/kg, i.p.). 5 min after acute nicotine, total locomotor
activity was measured for 10 minutes. Data are presented as the mean±SEM of 6 animals per group. *P<0.05 vs. all other groups, Bonferroni’s post hoc test.
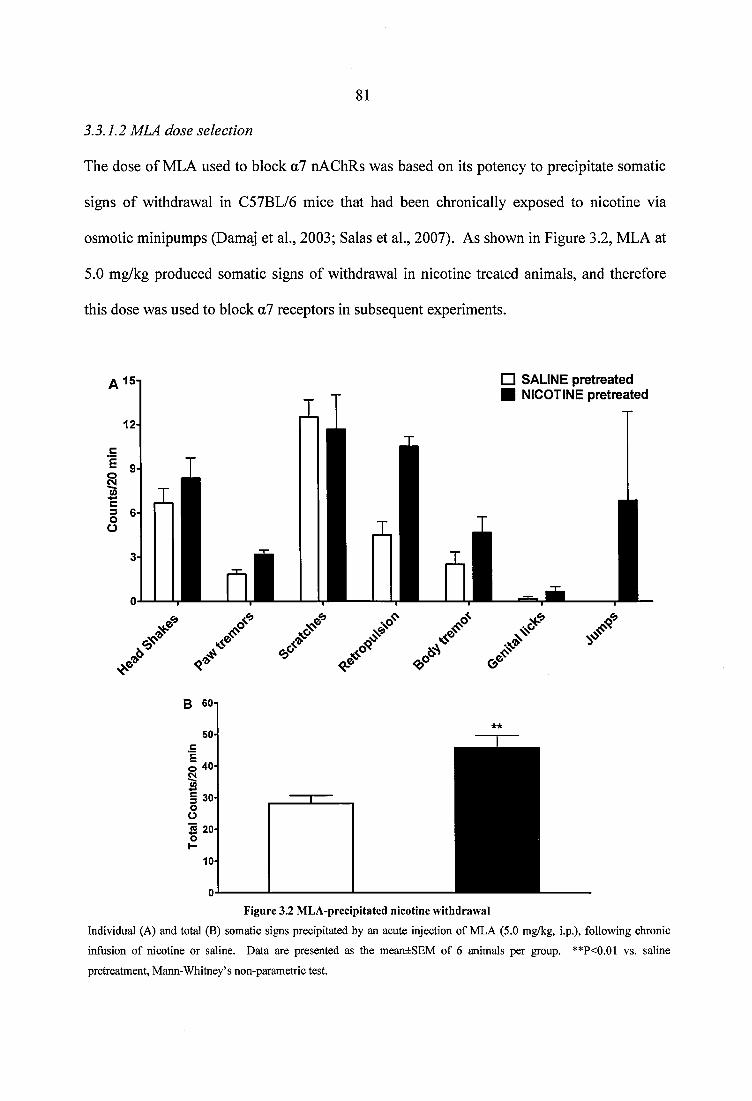
81
3.3.1.2 MLA dose selection
The dose of MLA used to block a7 nAChRs was based on its potency to precipitate somatic
signs of withdrawal in C57BL/6 mice that had been chronically exposed to nicotine via
osmotic minipumps (Damaj et ah, 2003; Salas et al., 2007). As shown in Figure 3.2, MLA at
5.0 mg/kg produced somatic signs of withdrawal in nicotine treated animals, and therefore
this dose was used to block a7 receptors in subsequent experiments.
□ SALINE p re trea ted ■ NICOTINE p re trea ted
B 60
50
i:oÜ
jS 20oH
10 -
Figure 3.2 M LA-precipitated nicotine withdraw al
Individual (A) and total (B) somatic signs precipitated by an acute injection of MLA (5.0 mg/kg, i.p.), following chronic
infusion of nicotine or saline. Data are presented as the mean±SEM of 6 animals per group. **P<0.01 vs. saline
pretreatment, Mann-Whitney’s non-parametric test.
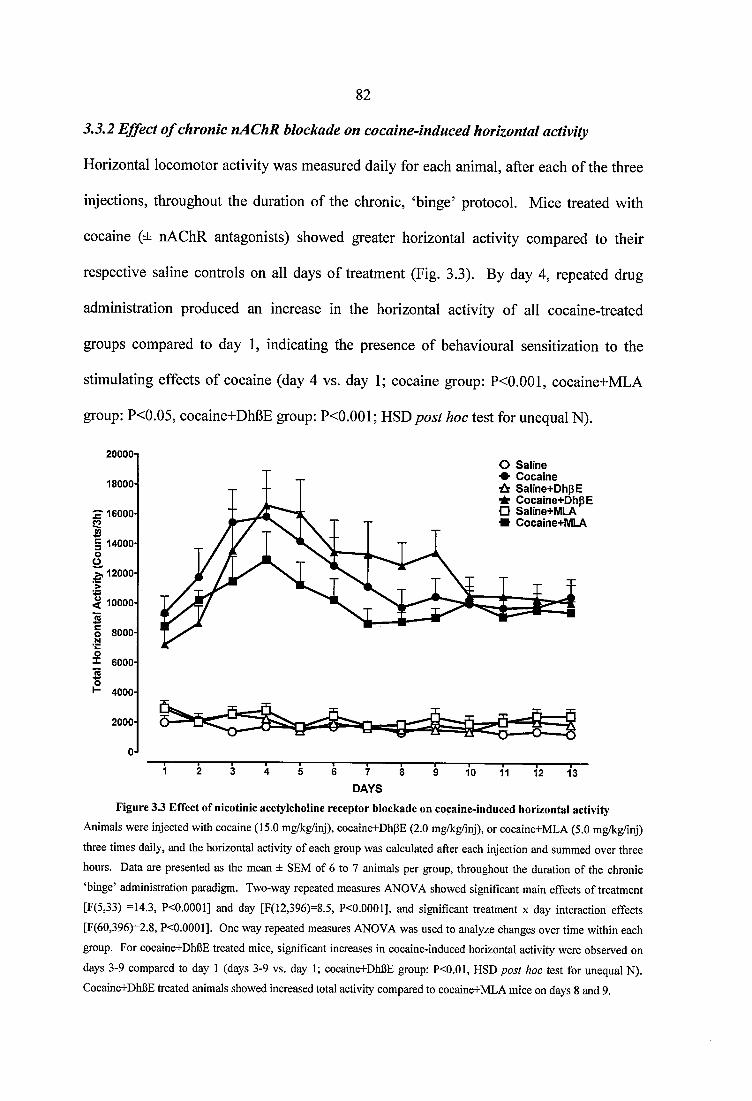
82
3.3.2 Effect o f chronic nACHR blockade on cocaine-induced horizontal activity
Horizontal locomotor activity was measured daily for each animal, after each of the three
injections, throughout the duration of the chronic, ‘binge’ protocol. Mice treated with
cocaine (± nAChR antagonists) showed greater horizontal activity compared to their
respective saline controls on all days of treatment (Fig. 3.3). By day 4, repeated drug
administration produced an increase in the horizontal activity of all cocaine-treated
groups compared to day 1, indicating the presence of behavioural sensitization to the
stimulating effects of cocaine (day 4 vs. day 1; cocaine group: P<0.001, cocaine+MLA
group: P<0.05, cocaine+DhbE group: P<0.001; HSD post hoc test for unequal N).
200001
18000*
S ' 16000
§I 14000
â 12000I< 10000-
£1
8000
6000
4000
2000
O Saline C o cain e
•ÙS S aline+ D hpE C ocaine+ D hpE
□ Saline+MLA * Cocaine+M LA
7
DAYS10 11 12 13
Figure 3.3 E ffect o f nicotinic acetylcholine receptor blockade on cocaine-induced horizontal activity
Animals were injected with cocaine (15.0 mg/kg/inj), cocaine+DhpE (2.0 mg/kg/inj), or cocaine+MLA (5.0 mg/kg/inj)
three times daily, and the horizontal activity of each group was calculated after each injection and summed over three
hours. Data are presented as the mean ± SEM of 6 to 7 animals per group, throughout the duration of the chronic
‘binge’ administration paradigm. Two-way repeated measures ANOVA showed significant main effects of treatment
[F(5,33) =14.3, P<0.0001] and day [F(12,396)=8.5, P<0.0001], and significant treatment x day interaction effects
[F(60,396)=2.8, P<0.0001]. One way repeated measures ANOVA was used to analyze changes over time within each
group. For coeaine+DhBE treated mice, significant increases in cocaine-induced horizontal activity were observed on
days 3-9 compared to day 1 (days 3-9 vs. day 1; eocaine+DhBE group: P<0.01, HSD post hoc test for unequal N).
Coeaine+DhBE treated animals showed increased total activity compared to cocaine+MLA mice on days 8 and 9.

83
The duration and magnitude of this response was modified by the addition of nicotinic
antagonists. Cocaine and cocaine+MLA treated animals quickly returned to day 1 levels
of total activity (day 5 vs. day 1, P>0.05), whereas co-administration of cocaine with the
P2 subunit antagonist DhGE prolonged the effects of the psychostimulant on total activity
up to day 9 (day 9 vs. day 1, P<0.01). Between groups comparison showed a significant
enhancement in cocaine-induced horizontal activity in cocaine+DhGE treated mice
compared to cocaine+MLA treated animals (P<0.05). Post hoc comparison revealed that
the largest differences in motor stimulation between the two groups were observed on
days 8 and 9 (P<0.05). Following day 4, cocaine-induced motor activity in DhGE-treated
animals was markedly enhanced during each hourly session, predominantly within the
first 30 minutes after drug administration (Fig. 3.4). The effects of binge treatment for all
treatment groups are shown in Figure 3.4 on days 1 ,4 ,8 and 13, throughout the daily 3h
observation period. Within each individual group, no differences in cocaine-induced
activity were observed among the three Ih observation sessions following drug treatment.
Horizontal activity after the first drug administration was similar to that observed after
the second or third treatment, on each daily session. Two-way repeated measures
ANOVA confirmed significant main effects of treatment and day on total horizontal
activity, with significant treatment x day interaction effects.
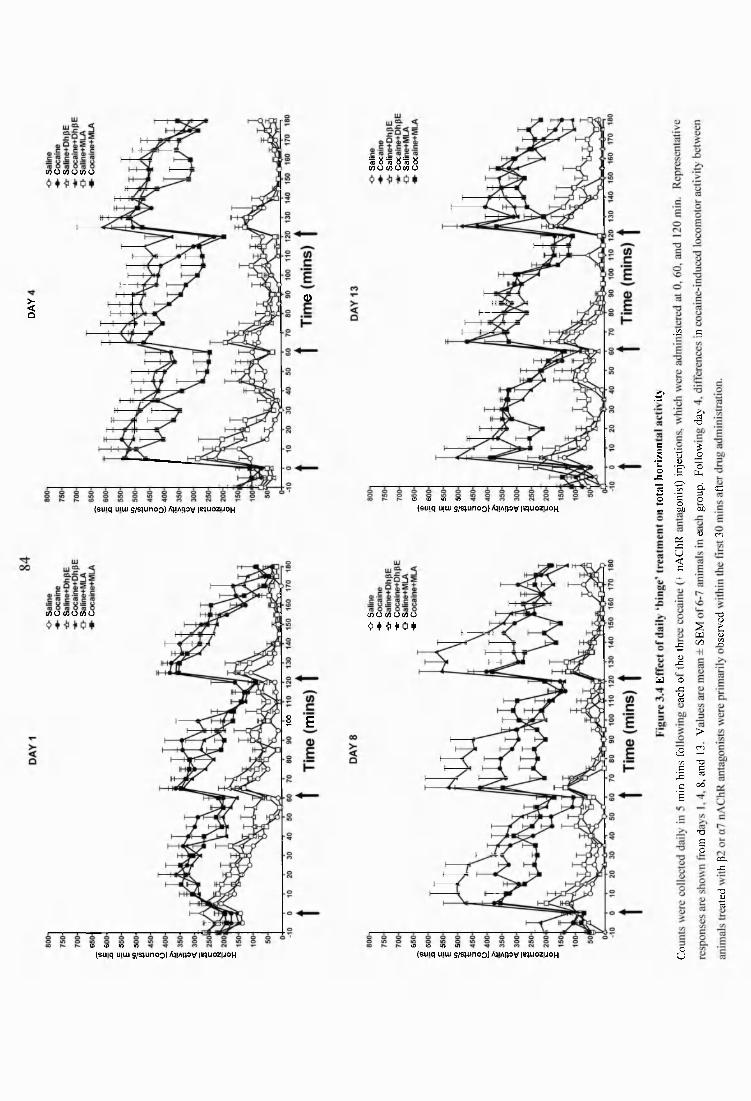
C/)OC0 O(/)O
I
(suiq uiiu 9/siunoo) A;!A!iov IBJUOZuoh
Q)
%= y = y = y (0 o (0 o (0 o (fiotnouio
£
.22 ç
t
(D O tn m(suiq ujiu s/s}unoo) Aiiarov |b»uozijoh
1 1 1
1 1 1(K
COO W ü « ü
i Ê
<o <0 to m
COO(/)OCÔO*+4+0*
ï- H
•£I01
tâ
u rn
i l
>a =
l îs
•oso
<0 <D to to(suiq uiui s/sjunoo) AjjAqov |B)uozuoh (suiq uiiu s/s}unoo) À iAqov IBJUozuoh Ô
CCLs:
1
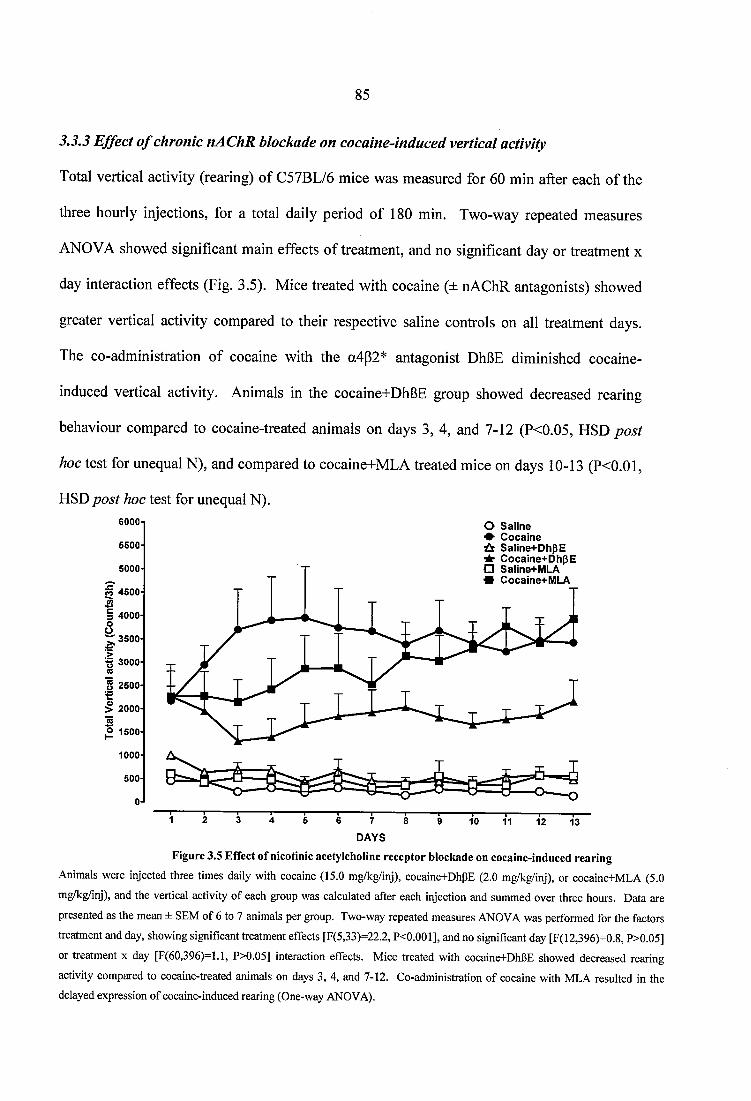
85
3.3.3 Effect o f chronic nAChR blockade on cocaine-induced vertical activity
Total vertical activity (rearing) of C57BL/6 mice was measured for 60 min after each of the
three hourly injections, for a total daily period of 180 min. Two-way repeated measures
ANOVA showed significant main effects of treatment, and no significant day or treatment x
day interaction effects (Fig. 3.5). Mice treated with cocaine (± nAChR antagonists) showed
greater vertical activity compared to their respective saline controls on all treatment days.
The co-administration of cocaine with the a4p2* antagonist DhBE diminished cocaine-
induced vertical activity. Animals in the cocaine+DhBE group showed decreased rearing
behaviour compared to cocaine-treated animals on days 3, 4, and 7-12 (P<0.05, HSD post
hoc test for unequal N), and compared to cocaine+MLA treated mice on days 10-13 (P<0.01,
HSD post hoc test for unequal N).GOOOi5500-
5000
CÔ 4500
I3 40000% 3500 ■>■g 3000
8 2500
1> 2000 SO 1500
1000
500-
0 -
O Saline# C ocaine
Saline+D hpE^ C ocaine+ D hpE D Saline+MLA* Cocaine+M LA
7
DAYS10 11 12 13
Figure 3.5 Effect o f nicotinic acetylcholine receptor blockade on cocaine-induced rearing
Animals were injeeted three times daily with coeaine (15.0 mg/kg/inj), coeaine+DhpE (2.0 mg/kg/inj), or cocaine+MLA (5.0
mg/kg/inj), and the vertical activity of each group was calculated after each injection and summed over three hours. Data are
presented as the mean ± SEM of 6 to 7 animals per group. Two-way repeated measures ANOVA was performed for the factors
treatment and day, showing significant treatment effects [F(5,33)=22.2, P<0.001], and no significant day [F(12,396)-0.8, P>0.05]
or treatment x day [F(60,396)=l.l, P>0.05] interaction effects. Mice treated with coeaine+DhPE showed decreased rearing
activity compared to cocaine-treated animals on days 3, 4, and 7-12. Co-administration of cocaine with MLA resulted in the delayed expression of coeaine-indueed rearing (One-way ANOVA).

86
One-way repeated measures ANOVA revealed that cocaine-induced rearing was not
sensitized neither in cocaine, nor in cocaine+DhBE treated animals. Co-administration of
cocaine with MLA, however, resulted in the delayed expression of cocaine-induced
rearing sensitization, with animals in this group showing enhanced rearing during the last
three days of treatment, compared to day 1.
3.3.4 Effect o f chronic nAChR blockade on cocaine-induced grooming and stereotypy
For grooming activity, a percentage of time spent engaged in this behaviour was
calculated for the entire observation period for each animal, on each day, throughout the
duration of the study. It should be noted that assessing the effects of nAChR blockade on
cocaine-induced grooming was not an original aim of the present study. Instead,
grooming behaviour was analysed because of an intense and previously uncharacterised
phenotype following the co-administration of cocaine with the a7 nAChR antagonist
MLA. Behaviour in cocaine+MLA treated mice was constantly interrupted by bursts of
grooming, with animals in this group spending as much as 50% of the entire observation
period engaged in grooming activity from day 5 onwards (Fig. 3.6A). A negative
correlation between the increased time spent grooming and the locomotor activity of
cocaine+MLA treated animals was derived, indicating the competing nature of the
measured behaviours (Fig. 3.6B).
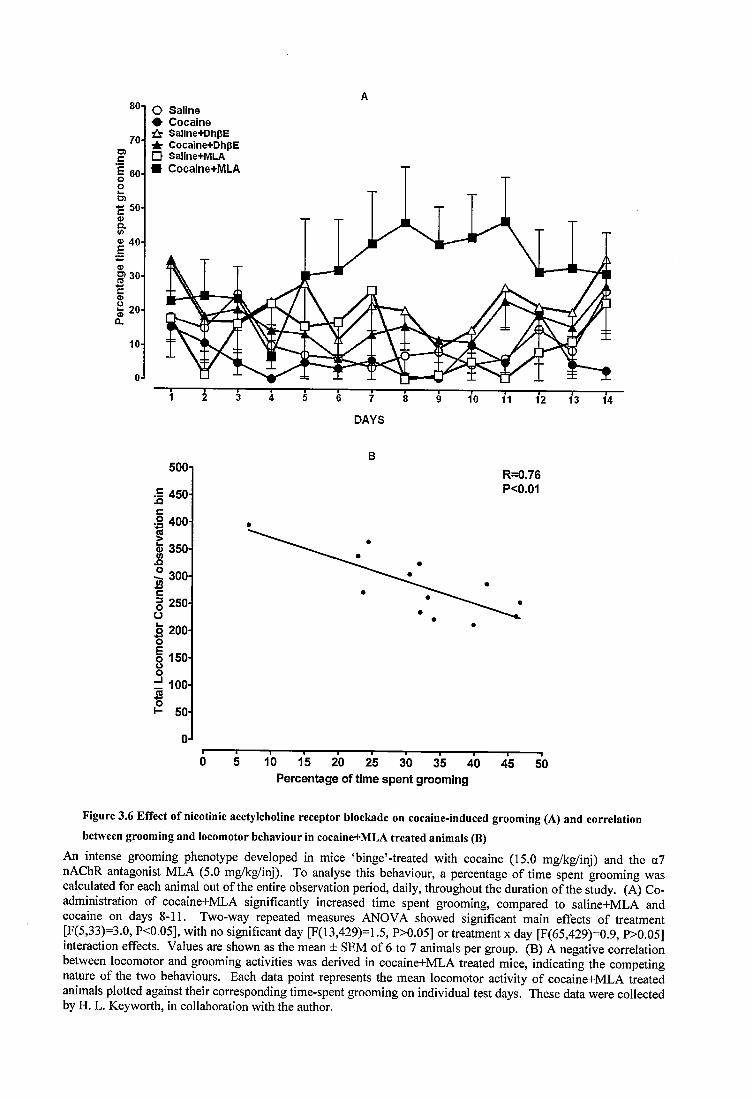
80-1
TO
GO-
D)-g 50 0)I 40
0g» 30-1
120-
10-
SalineC o ca in eSaline+DhpECocaine+DhpESaline+MLAC ocaine+M LA
DAYS
5 0 0 i
•i 450JQI 4 0 0
Ip 3 50Ê| 300-
§ 2 5 0 ü
IE8 o
200 -
150-
_ 100-
o ,I - 504
R = 0 .7 6P< 0 .01
10 15 " S ' 2 5 30 3 5 4 0P e r c e n ta g e o f tim e s p e n t g ro o m in g
— I—
4 5 50
Figure 3.6 Effect o f nicotinic acetylcholine receptor blockade on cocaine-induced groom ing (A) and correlation
between groom ing and locom otor behaviour in cocaine+M LA treated anim als (B)
An intense grooming phenotype developed in mice ‘binge’-treated with cocaine (15.0 mg/kg/inj) and the a7 nAChR antagonist MLA (5.0 mg/kg/inj). To analyse this behaviour, a percentage o f time spent grooming was calculated for each animal out o f the entire observation period, daily, throughout the duration o f the study. (A) Coadministration o f cocaine+MLA significantly increased time spent grooming, compared to saline+MLA and cocaine on days 8-11. Two-way repeated measures ANOVA showed significant main effects o f treatment [F(5,33)=3.0, P<0.05], with no significant day [F(13,429)=1.5, P>0.05] or treatment x day [F(65,429)=0.9, P>0.05] interaction effects. Values are shown as the mean ± SEM o f 6 to 7 animals per group. (B) A negative correlation between locomotor and grooming activities was derived in cocaine+MLA treated mice, indicating the competing nature o f the two behaviours. Each data point represents the mean locomotor activity o f cocaine+MLA treated animals plotted against their corresponding time-spent grooming on individual test days. These data were collected by H. L. Keyworth, in collaboration with the author.

Grooming activity was intense and interrupted, progressed randomly rather than
sequentially, and left the fur of mice substantially worse than that of animals in other
groups. As a result, the evaluation of cocaine-induced stereotypy in cocaine+MLA
mice was not included in the analysis. Two-way repeated measures ANOVA showed
significant main effects of treatment on grooming behaviour, with no significant day
or treatment x day interaction effects. Post hoc analysis confirmed that co
administration of cocaine+MLA significantly increased time spent grooming,
compared to administration of saline+MLA and cocaine on days 8-11 (P<0.05, HSD
test for unequal N).
Stereotypy was scored for 40 seconds, 20 min after each of the three hourly injections
and the median score was calculated daily for each animal. Two-way repeated
measures ANOVA showed significant main effects of treatment, day, and significant
treatment x day interaction effects (Fig. 3.7). Administration of cocaine resulted in
the expression of behavioural stereotypy. The magnitude of this effect differed
between cocaine-treated groups. Animals receiving cocaine+DhBE showed
diminished stereotypy on days 2, 3, 4 , 7 , 11 and 13 compared to cocaine-treated
animals (P<0.05, HSD post hoc test for unequal N). Stereotypy sensitization was
observed only in the cocaine group from day 3 onwards (day 3 vs. day 1, P<0.01,
HSD post hoc test for unequal N), and it was maintained for the duration of this study
(days 4-14 vs. day 1, P<0.001 HSD post hoc test for unequal N). In contrast, cocaine-
induced stereotypy sensitization was attenuated by the administration of the nicotinic
antagonist DhBE.
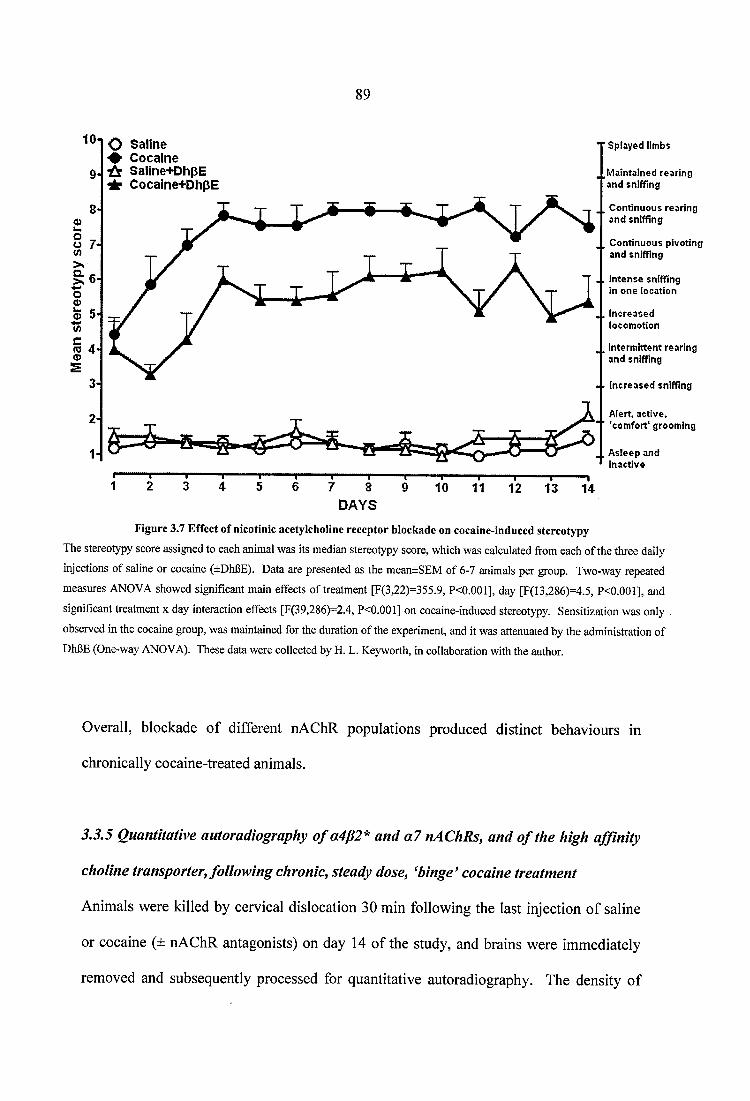
10
9
8Q)u.
I 6-o0
5toM 4'<D
2*
1-
O Saline # C ocaine ■Û Saline+D hpE - if C ocaine+D hpE
Splayed limbs
j,Maintained rearing and sniffing
J, Continuous rearing and sniffing
Continuous pivoting and sniffing
4. Intense sniffing in one location
Increased locomotion
Intermittent rearing and sniffing
Increased sniffing
Alert, active, 'comfort' grooming
X Asleep and inactive
7 8 DAYS
“ T “10
—r- 11 12
-T-13
—114
Figure 3.7 E ffect o f nicotinic acetylcholine receptor blockade on cocaine-induced stereotypy
The stereotypy score assigned to each animal was its median stereotypy score, which was calculated from each of the three daily
injections of saline or cocaine (±Dhl3E). Data are presented as the mean±SEM of 6-7 animals per group. Two-way repeated
measures ANOVA showed significant main effects of treatment [F(3,22)=355.9, P<0.001], day [F(13,286)=4.5, P<0.001], and
significant treatment x day interaction effects [F(39,286)-2.4, P<0.001] on cocaine-induced stereotypy. Sensitization was only
observed in the cocaine group, was maintained for the duration of the experiment, and it was attenuated by the administration of
DhBE (One-way ANOVA). These data were collected by H. L. Keyworth, in collaboration with the author.
Overall, blockade of different nAChR populations produced distinct behaviours in
chronically cocaine-treated animals.
3.3.5 Quantitative autoradiography o f a4p2* and a l nACHRs, and o f the high affinity
choline transporter, following chronic, steady dose, binge* cocaine treatment
Animals were killed by cervical dislocation 30 min following the last injection of saline
or cocaine (± nAChR antagonists) on day 14 of the study, and brains were immediately
removed and subsequently processed for quantitative autoradiography. The density of
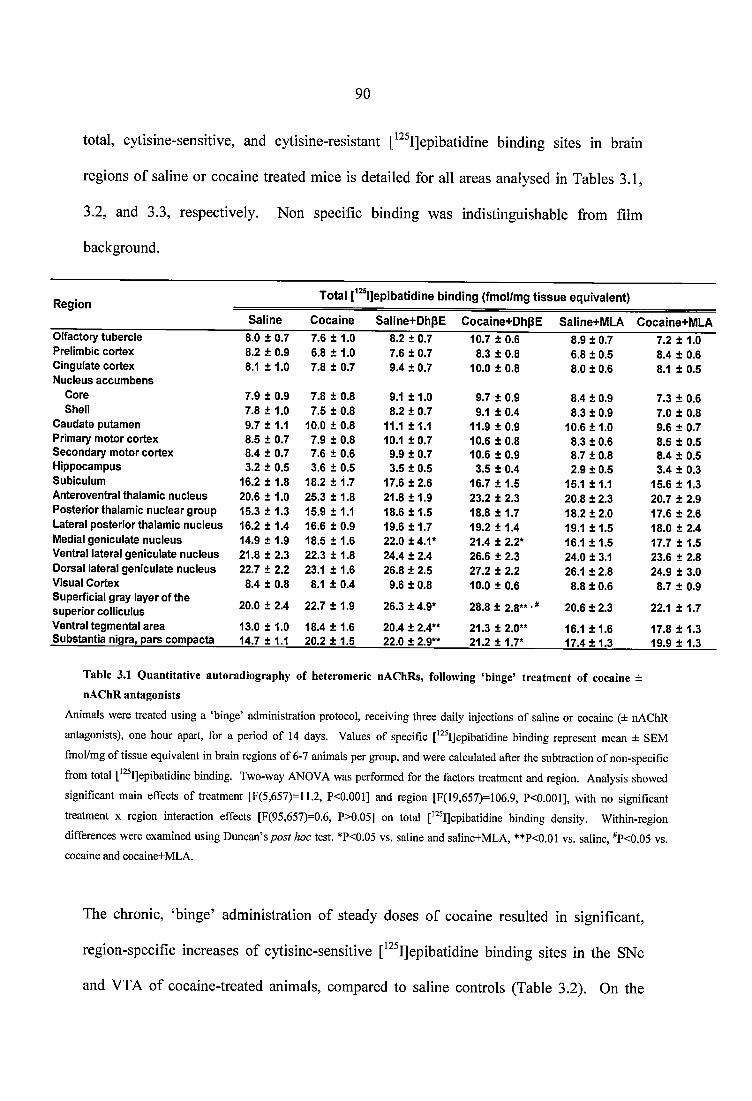
90
total, cytisine-sensitive, and cytisine-resistant [^^^I]epibatidine binding sites in brain
regions of saline or cocaine treated mice is detailed for all areas analysed in Tables 3.1,
3.2, and 3.3, respectively. Non specific binding was indistinguishable from film
background.
R e g io nT o ta l 1 ]e p ib a tid in e b in d in g (fm o l/m g t i s s u e e q u iv a le n t)
S a lin e C o c a in e S a lin e + D h p E C o c a in e + D h p E Saline+ M L A C o cain e+ M L AOlfactory tu b erc le 8.0 ± 0.7 7.6 ± 1.0 8.2 ± 0.7 10.7 ± 0.6 8.9 ± 0.7 7.2 ± 1.0Prelim bic co rtex 8.2 ± 0.9 6.8 ± 1.0 7.6 ± 0.7 8.3 ± 0.8 6.8 ± 0.5 8.4 ± 0.6C ingulate co rtex 8.1 ± 1.0 7.8 ± 0.7 9.4 ± 0.7 10.0 ± 0.8 8.0 ± 0.6 8.1 ± 0.5N ucleus a c c u m b e n s
Core 7.9 ± 0.9 7.8 ± 0.8 9.1 ± 1 .0 9.7 ± 0.9 8.4 ± 0.9 7.3 ± 0.6Shell 7.8 ± 1.0 7.5 ± 0.8 8.2 ± 0.7 9.1 ± 0.4 8.3 ± 0.9 7.0 ± 0.8
C au d ate pu tam en 9.7 ± 1.1 10.0 ± 0.8 11.1 ±1 .1 11.9 ± 0.9 10.6 ± 1 .0 9.6 ± 0.7Prim ary m oto r co rtex 8.5 ± 0.7 7.9 ± 0.8 10.1 ± 0 .7 10.6 ± 0.8 8.3 ± 0.6 8.5 ± 0.5S e co n d ary m oto r co rtex 8.4 ± 0.7 7.6 ± 0.6 9.9 ± 0.7 10.6 ± 0.9 8.7 ± 0.8 8.4 ± 0.5H ippocam pus 3.2 ± 0.5 3.6 ± 0.5 3.5 ± 0.5 3.5 ± 0.4 2.9 ± 0.5 3.4 ± 0.3Subiculum 16.2 ± 1.8 18.2 ± 1.7 17.6 ± 2.6 16.7 ± 1.5 15.1 ±1.1 15.6 ± 1.3A nteroventral thalam ic n u c leu s 20.6 ± 1.0 25.3 ± 1.8 21.8 ± 1 .9 23.2 ± 2.3 20.8 ± 2.3 20.7 ± 2.9P o s te rio r thalam ic n u c lea r g ro u p 15.3 ± 1.3 15.9 ± 1.1 18.6 ± 1 .5 18.8 ± 1.7 18.2 ± 2 .0 17.6 ± 2.6Lateral p o s te rio r thalam ic n u c leu s 16.2 ± 1.4 16.6 ± 0.9 19.6 ± 1 .7 19.2 ± 1.4 19.1 ± 1 .5 18.0 ± 2.4Medial gen icu la te n u c leu s 14.9 ± 1.9 18.5 ± 1.6 22.0 ± 4.1* 21.4 ± 2.2* 16.1 ± 1 .5 17.7 ± 1.5V entral lateral g en icu la te n u c leu s 21.8 ± 2.3 22.3 ± 1.8 24.4 ± 2.4 26.6 ± 2.3 24.0 ± 3.1 23.6 ± 2.8D orsal lateral gen icu la te n u c leu s 22.7 ± 2.2 23.1 ± 1.6 26.8 ± 2.5 27.2 ± 2.2 26.1 ± 2.8 24.9 ± 3.0V isual C ortex 8.4 ± 0.8 8.1 ± 0.4 9.6 ± 0.8 10.0 ± 0.6 8.8 ± 0.6 8.7 ± 0.9Superficial g ray layer o f th e
20.0 ± 2.4 22.7 ± 1.9 26.3 ± 4.9* 28.8 ± 2.8** ’^su p e rio r colliculus 20.6 ± 2.3 22.1 ± 1.7
V entral tegm en ta l a re a 13.0 ± 1.0 18.4 ± 1.6 20.4 ± 2.4** 21.3 ± 2.0** 16.1 ± 1 .6 17.8 ± 1.3S u b s tan tia nigra, p a rs co m p acta 14.7 ± 1.1 20.2 ± 1.5 22.0 ± 2.9** 21.2 ± 1.7* 17.4 ± 1 .3 19.9 ± 1.3
Table 3.1 Q uantitative autoradiography o f heterom eric nAChRs, following ‘binge’ treatm ent o f cocaine ±
nA ChR antagonists
Animals were treated using a ‘binge’ administration protocol, receiving three daily injeetions of saline or eoeaine (± nAChR
antagonists), one hour apart, for a period of 14 days. Values of speeifie [’ ^I]epibatidine binding represent mean ± SEM
fmol/mg of tissue equivalent in brain regions of 6-7 animals per group, and were caleulated after the subtraetion of non-specific
from total [’^^I]epibatidine binding. Two-way ANOVA was performed for the factors treatment and region. Analysis showed
significant main effects of treatment [F(5,657)=11.2, P<0.001] and region [F(19,657)=106.9, P<0.001], with no significant
treatment x region interaction effects [F(95,657)=0.6, P>0.05] on total ['^^I]epibatidine binding density. Within-region
differences were examined using Duncan’s post hoc test. *P<0.05 vs. saline and saline+MLA, **P<0.01 vs. saline, ^P<0.05 vs. coeaine and coeaine+MLA.
The chronic, ‘binge’ administration of steady doses of cocaine resulted in significant,
region-specific increases of cytisine-sensitive [^^^IJepibatidine binding sites in the SNc
and VTA of cocaine-treated animals, compared to saline controls (Table 3.2). On the
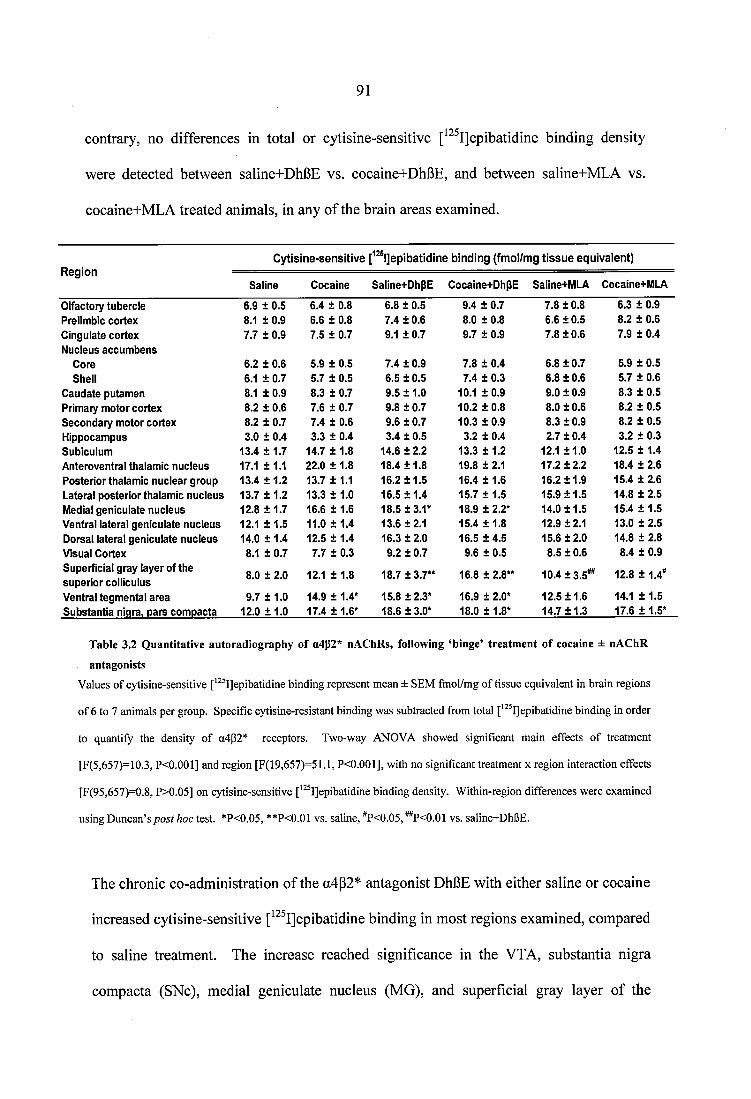
91
contrary, no differences in total or cytisine-sensitive [^^^I]epibatidine binding density
were detected between saline+DhBE vs. cocaine+DhBE, and between saline+MLA vs.
cocaine+MLA treated animals, in any of the brain areas examined.
R egionC y tis in e -se n s itiv e f ® l]epibatidine b in d in g (fm ol/m g t is s u e eq u iv a len t)
Saline Cocaine Saline+DhpE Cocaine+DhpE Saline+MLA Cocaine+MLA
Olfactory tubercle 6.9 ± 0.5 6.4 ± 0.8 6.8 ± 0.5 9.4 ± 0.7 7.8 ± 0.8 6.3 ± 0.9Prelimbic cortex 8.1 ± 0.9 6.6 ± 0.8 7.4 ± 0.6 8.0 ± 0.8 6.6 ± 0.5 8.2 ± 0.6Cingulate cortex Nucleus accum bens
7.7 ± 0.9 7.5 ± 0.7 9.1 ± 0.7 9.7 ± 0.9 7.8 ± 0.6 7.9 ± 0.4
Core 6.2 ± 0.6 5.9 ± 0.5 7.4 ± 0 .9 7.8 ± 0.4 6.8 ±0.7 5.9 ± 0.5Shell 6.1 ± 0.7 5.7 ± 0.5 6.5 ± 0.5 7.4 ± 0.3 6.8 ± 0.6 5.7 ± 0.6
C audate putam en 8.1 + 0.9 8.3 ± 0.7 9.5 ±1 .0 10.1 ± 0.9 9.0 ± 0.9 8.3 ± 0.5Primary m otor cortex 8.2 ± 0.6 7.6 ± 0.7 9.8 ± 0.7 10.2 ± 0.8 8.0 ± 0.6 8.2 ± 0.5Secondary m otor cortex 8.2 ± 0.7 7.4 ± 0.6 9.6 ± 0.7 10.3 ± 0.9 8.3 ± 0.9 8.2 ± 0.5Hippocam pus 3.0 ± 0.4 3.3 ± 0.4 3.4 ± 0.5 3.2 ± 0.4 2.7 ± 0.4 3.2 ± 0.3Subiculum 1 3 .4 + 1 .7 14.7 ± 1.8 14.6 ± 2.2 13.3 ± 1.2 12.1 ±1.0 12.5 ± 1.4Anteroventral thalamic nucleus 17.1 ± 1.1 22.0 ± 1.8 18.4 ±1 .8 19.8 ± 2.1 17.2 ±2.2 18.4 ± 2.6Posterior thalam ic nuclear group 13.4 ± 1 .2 13.7 ± 1.1 16.2 ± 1 .5 16.4 ± 1.6 16.2 ±1.9 15.4 ± 2.6Lateral posterior thalamic nucleus 13.7 ± 1.2 13.3 ± 1.0 16.5 ± 1 .4 15.7 ± 1.5 15.9 ±1 .5 14.8 ± 2.5Medial geniculate nucleus 12.8 ± 1 .7 16.6 ± 1.6 18.5 ±3.1* 18.9 ± 2.2* 14.0 ±1 .5 15.4 ± 1.5Ventral lateral geniculate nucleus 12.1 ± 1.5 11.0 ± 1.4 13.6 ±2.1 15.4 ± 1.8 12.9 ±2.1 13.0 ± 2.5Dorsal lateral geniculate nucleus 14.0 ± 1.4 12.5 ± 1.4 16.3 ±2 .0 16.5 ± 4 .5 15.6 ±2 .0 14.8 ± 2.8Visual Cortex 8.1 ± 0.7 7.7 ± 0.3 9.2 ± 0.7 9.6 ± 0.5 8.5 ± 0.6 8.4 ± 0.9Superficial gray layer of the superio r colliculus
8.0 ± 2.0 12.1 ± 1.8 18.7 ±3.7** 16.8 ± 2.8** 10.4 ±3.5** 12.8 ± 1.4*
Ventral tegm ental area 9.7 ± 1.0 14.9 ± 1.4* 15.8 ±2.3* 16.9 ± 2.0* 12.5 ±1.6 14.1 ± 1.5Substantia nigra, pars com pacta 12.0 ± 1 .0 17.4 ± 1.6* 18.6 ±3.0* 18.0 ±1.8* 14.7 ±1.3 17.6 ± 1.5*
Table 3.2 Quantitative autoradiography of u4p2* nAChRs, following ‘binge’ treatment of cocaine ± nAChR
antagonistsValues of cytisine-sensitive ['^^I]epibatidine binding represent mean ± SEM fmol/mg of tissue equivalent in brain regions
of 6 to 7 animals per group. Specific cytisine-resistant binding was subtracted from total ['^^I]epibatidine binding in order
to quantify the density of a4p2* receptors. Two-way ANOVA showed significant main effects of treatment
[F(5,657)=10.3, P<0.001] and region [F(19,657)=51.1, P<0.001], with no significant treatment x region interaction effects
[F(95,657)=0.8, P>0.05] on cytisine-sensitive ['^ IJepibatidine binding density. Within-region differences were examined
using Duncan’s post hoc XqsX. *P<0.05, **P<0.01 v s . saline, ^P<0.05,^P<0.01 vs. saline+DhBE.
The chronic co-administration of the a4p2* antagonist DhBE with either saline or cocaine
increased cytisine-sensitive [^^^I]epibatidine binding in most regions examined, compared
to saline treatment. The increase reached significance in the VTA, substantia nigra
compacta (SNc), medial geniculate nucleus (MG), and superficial gray layer of the
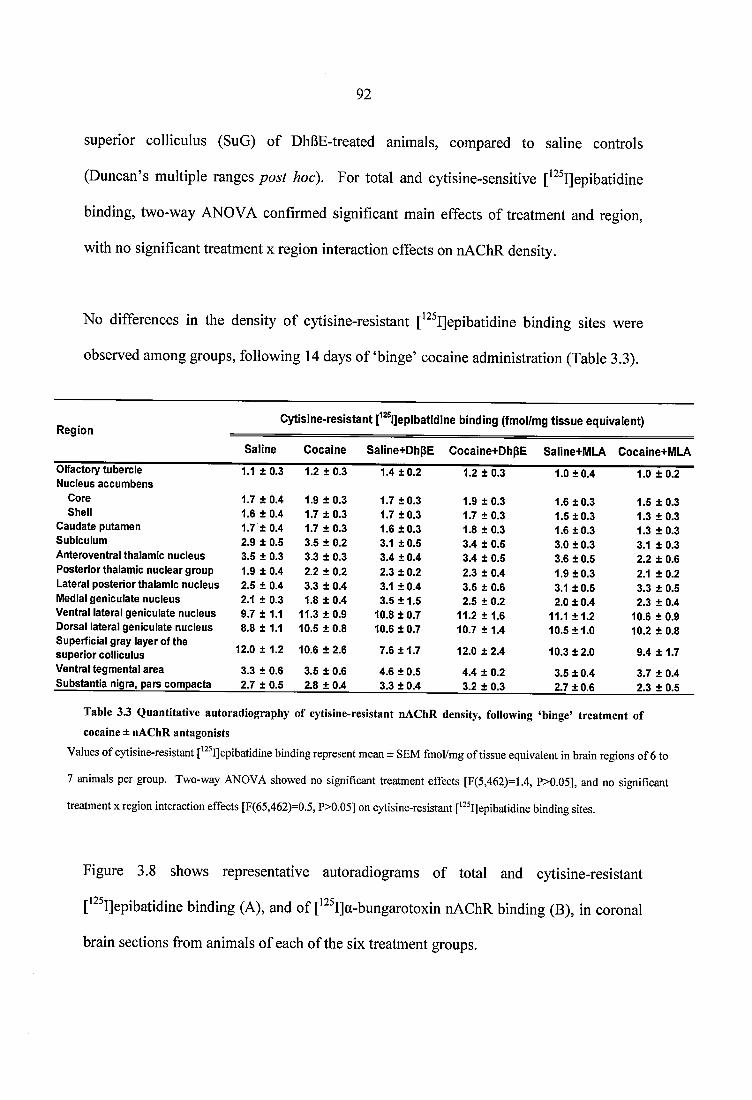
92
superior colliculus (SuG) of DhBE-treated animals, compared to saline controls
(Duncan’s multiple ranges post hoc). For total and cytisine-sensitive [^^^IJepibatidine
binding, two-way ANOVA confirmed significant main effects of treatment and region,
with no significant treatment x region interaction effects on nAChR density.
No differences in the density of cytisine-resistant [^^^IJepibatidine binding sites were
observed among groups, following 14 days o f ‘binge’ cocaine administration (Table 3.3).
R eg io nC y tis in e - re s is ta n t [^^^l]epibatidine b in d in g (fm ol/m g t is s u e eq u iv a len t)
S a lin e C o c a in e Saline+ D hpE C o cain e+ D h p E Saline+M LA C ocaine+M LA
Olfactory tubercle 1.1 ± 0.3 1.2 ± 0.3 1.4 ± 0 .2 1.2 ± 0.3 1.0 ± 0 .4 1.0 ± 0.2Nucleus accum bens
Core 1.7 ± 0.4 1.9 ± 0.3 1.7 ±0 .3 1.9 ± 0.3 1.6 ±0 .3 1.5 ± 0 .3Shell 1.6 ± 0.4 1.7 ± 0.3 1.7 ± 0 .3 1.7 ± 0.3 1.5 ±0 .3 1.3 ± 0.3
C audate putam en 1.7 ± 0 .4 1.7 ± 0.3 1.6 ± 0 .3 1.8 ± 0.3 1.6 ±0 .3 1.3 ± 0 .3Subiculum 2.9 ± 0.5 3.5 ± 0.2 3.1 ± 0 .5 3.4 ± 0.5 3.0 ± 0.3 3.1 ± 0.3A nteroventral thalam ic nucleus 3.5 ± 0.3 3.3 ± 0.3 3.4 ± 0.4 3.4 ± 0.5 3.6 ± 0.5 2.2 ± 0.6Posterio r thalam ic nuclear group 1.9 ± 0.4 2.2 ± 0.2 2.3 ± 0.2 2.3 ± 0.4 1.9 ±0 .3 2.1 ± 0.2Lateral posterio r thalam ic nucleus 2.5 ± 0.4 3.3 ± 0.4 3.1 ± 0.4 3.5 ± 0.6 3.1 ± 0.5 3.3 ± 0.5Medial geniculate nucleus 2.1 ± 0.3 1.8 ± 0.4 3.5 ± 1 .5 2.5 ± 0.2 2.0 ± 0.4 2.3 ± 0.4Ventral lateral geniculate nucleus 9.7 ± 1.1 11.3 ± 0.9 10.8 ± 0.7 11.2 ± 1.6 11.1 ±1 .2 10.6 ± 0.9Dorsal lateral geniculate nucleus 8.8 ± 1.1 10.5 ± 0.8 10.6 ± 0.7 10.7 ± 1.4 10.5 ±1 .0 10.2 ± 0.8Superficial gray layer of the su perio r colliculus 12.0 ± 1.2 10.6 ± 2.6 7.6 ± 1 .7 12.0 ± 2.4 10.3 ±2 .0 9.4 ± 1.7
Ventral tegm ental area 3.3 ± 0.6 3.5 ± 0.6 4.6 ± 0.5 4.4 ± 0.2 3.5 ± 0.4 3.7 ± 0.4S ubstan tia nigra, pars com pacta 2.7 ± 0.5 2.8 ± 0.4 3.3 ± 0.4 3.2 ± 0.3 2.7 ± 0.6 2.3 ± 0.5
Table 3 3 Quantitative autoradiography of cytisine-resistant nAChR density, following ‘binge’ treatment of cocaine ± nAChR antagonists
Values of cytisine-resistant ['^ IJepibatidine binding represent mean ± SEM fmol/mg of tissue equivalent in brain regions of 6 to
7 animals per group. Two-way ANOVA showed no significant treatment effects [F(5,462)=1.4, P>0.05], and no significant
treatment x region interaction effects [F(65,462)=0.5, P>0.05] on cytisine-resistant [*^ I]epibatidine binding sites.
Figure 3.8 shows representative autoradiograms of total and cytisine-resistant
[^^^IJepibatidine binding (A), and of [^^^I]a-bungarotoxin nAChR binding (B), in coronal
brain sections from animals of each of the six treatment groups.
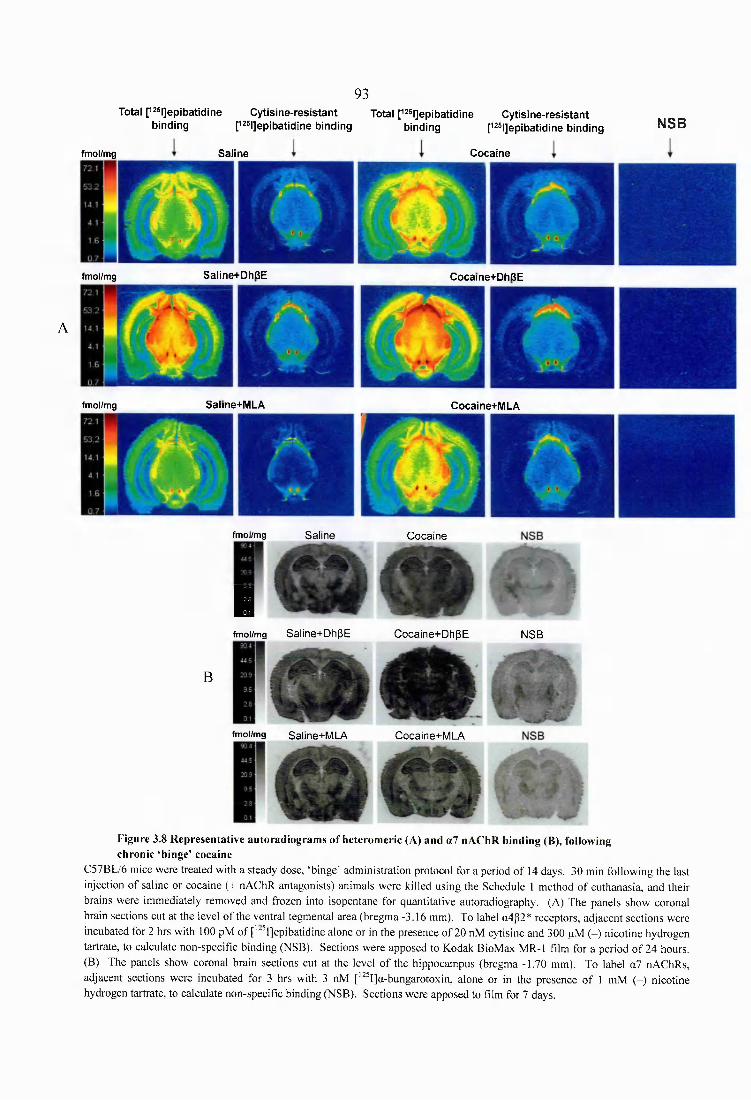
93Total p2S|]epibatidine
bindingC y tis in e -resistan t
P^^IJepibatidine bindingTotal p^®l]epibatidine
bindingC y tis in e -resistan t
r^sijep ibatid ine binding NSB
fmol/mg
A
C ocaineSaline
Saline+DhBEfmol/mg Cocaine+DhBE
Saline+MLAfmol/mg Cocaine+MLA
fmol/mg Saline Cocaine
■fmol/mg Saline+DhgE Cocaine+DhpE NSB
B
fmol/mg Saline+MLA Cocaine+MLA
Figure 3.8 Representative autoradiogram s o f heteromeric (A) and a l nAChR binding (B), following chronic ‘binge’ cocaine
C57BL/6 mice were treated with a steady dose, ‘binge’ administration protoeol for a period of 14 days. 30 min following the last injeetion of saline or cocaine (± nAChR antagonists) animals were killed using the Schedule 1 method of euthanasia, and their brains were immediately removed and frozen into isopentane for quantitative autoradiography. (A) The panels show coronal brain sections cut at the level of the ventral tegmental area (bregma -3.16 mm). To label a4p2* receptors, adjacent sections were incubated for 2 hrs with 100 pM of ['“ Ijepibatidine alone or in the presence of 20 nM cytisine and 300 pM (-) nicotine hydrogen tartrate, to calculate non-specific binding (NSB). Sections were apposed to Kodak BioMax MR-1 film for a period of 24 hours. (B) The panels show coronal brain sections cut at the level of the hippocampus (bregma -1.70 mm). To label a l nAChRs, adjacent sections were incubated for 3 hrs with 3 nM ['^^I]a-bungarotoxin, alone or in the presence of 1 mM (-) nicotine hydrogen tartrate, to calculate non-specific binding (NSB). Sections were apposed to film for 7 days.
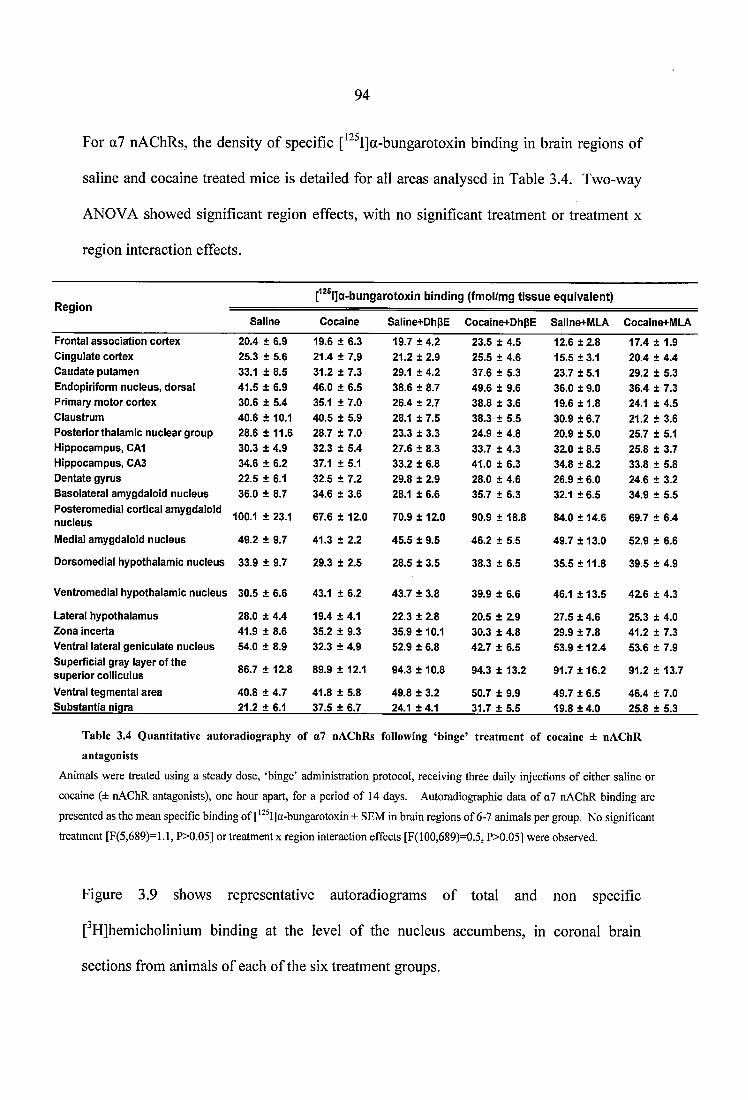
94
For a l nAChRs, the density of specific [^^^I]a-bungarotoxin binding in brain regions of
saline and cocaine treated mice is detailed for all areas analysed in Table 3.4. Two-way
ANOVA showed significant region effects, with no significant treatment or treatment x
region interaction effects.
R eg io n[^^^ l]a-b u n g arcto x in b in d in g (fm ol/m g t i s s u e e q u iv a le n t)
Saline C ocaine Saiine+DhpE Cocaine+DhpE Saline+MLA Cocaine+MLA
Frontal asso c ia tio n cortex 20.4 ± 6.9 19.6 ± 6.3 19.7 ± 4 .2 23.5 ± 4.5 12.6 ± 2 .8 17.4 ± 1.9Cinguiate cortex 25.3 ± 5.6 21.4 ± 7.9 21.2 ± 2.9 25.5 ± 4.6 15.5 ±3.1 20.4 ± 4.4C audate putam en 33.1 ± 8.5 31.2 ± 7.3 29.1 ± 4.2 37.6 ± 5.3 23.7 ± 5.1 29.2 ± 5.3Endopiriform nucleus, dorsal 41.5 ± 6.9 46.0 ± 6.5 38.6 ± 8.7 49.6 ± 9.6 36.0 ± 9.0 36.4 ± 7.3Primary m otor cortex 30.6 ± 5.4 35.1 ± 7.0 26.4 ± 2.7 38.8 ± 3.6 19.6 ± 1 .8 24.1 ± 4.5Claustrum 40.6 ± 10.1 40.5 ± 5.9 28.1 ± 7.5 38.3 ± 5.5 30.9 ± 6.7 21.2 ± 3.6Posterio r thalam ic nuclear group 28.6 ± 1 1 .6 28.7 ± 7.0 23.3 ± 3.3 24.9 ± 4.8 20.9 ± 5.0 25.7 ± 5.1H ippocam pus, CA1 30.3 ± 4.9 32.3 ± 5.4 27.6 ± 8.3 33.7 ± 4.3 32.0 ± 8.5 25.8 ± 3.7H ippocam pus, CA3 34.6 ± 6.2 37.1 ± 5.1 33.2 ± 6.8 41.0 ± 6.3 34.8 ± 8.2 33.8 ± 5.8Dentate gyrus 22.5 ± 6.1 32.5 ± 7.2 29.8 ± 2.9 28.0 ± 4.6 26.9 ± 6.0 24.6 ± 3.2Basolateral am ygdaloid nucleus 36.0 ± 8.7 34.6 ± 3.6 28.1 ± 6.6 35.7 ± 6.3 32.1 ± 6.5 34.9 ± 5.5Posterom edial cortical am ygdaloid nucleus 100.1 ± 23.1 67.6 ± 12.0 70.9 ± 12.0 90.9 ± 18.8 84.0 ± 14.6 69.7 ± 6.4
Medial am ygdaloid nucleus 49.2 ± 9.7 41.3 ± 2.2 45.5 ± 9.5 46.2 ± 5.5 49.7 ± 13 .0 52.9 ± 6.6
Dorsom edial hypothalam ic nucleus 33.9 ± 9.7 29.3 ± 2.5 28.5 ± 3.5 38.3 ± 6.5 35.5 ± 11 .8 39.5 ± 4.9
Ventrom edial hypothalam ic nucleus 30.5 ± 6.6 43.1 ± 6.2 43.7 ± 3.8 39.9 ± 6.6 46.1 ± 13 .5 42.6 ± 4.3
Lateral hypothalam us 28.0 ± 4.4 19.4 ± 4.1 22.3 ± 2.8 20.5 ± 2.9 27.5 ± 4.6 25.3 ± 4.0Z ona incerta 41.9 ± 8.6 35.2 ± 9.3 35.9 ± 10.1 30.3 ± 4.8 29.9 ± 7.8 41.2 ± 7.3Ventral lateral geniculate nucleus 54.0 ± 8.9 32.3 ± 4.9 52.9 ± 6.8 42.7 ± 6.5 53.9 ± 12 .4 53.6 ± 7.9Superficial gray layer of th e su perio r colliculus 86.7 ± 12.8 89.9 ± 12.1 94.3 ± 10.8 94.3 ± 13.2 91.7 ± 16 .2 91.2 ± 13.7
Ventral tegm ental area 40.8 ± 4.7 41.8 ± 5.8 49.8 ± 3.2 50.7 ± 9.9 49.7 ± 6.5 46.4 ± 7.0S u b stan tia nigra 21.2 ± 6.1 37.5 ± 6.7 24.1 ± 4.1 31.7 ± 5.5 19.8 ± 4 .0 25.8 ± 5.3
Table 3.4 Quantitative autoradiography of a l nAChRs following ‘binge’ treatment of coeaine ± nAChR
antagonists
Animals were treated using a steady dose, ‘binge’ administration protocol, receiving three daily injeetions of either saline or
eoeaine (± nAChR antagonists), one hour apart, for a period of 14 days. Autoradiographic data of a l nAChR binding are
presented as the mean specific binding of [^a-bungarotoxin ± SEM in brain regions of 6-7 animals per group. No significant
treatment [F(5,689)=l.l, P>0.05] or treatment x region interaetion effects [F(100,689)=0.5, P>0.05] were observed.
Figure 3.9 shows representative autoradiograms of total and non specific
[^HJhemicholinium binding at the level of the nucleus accumbens, in coronal brain
sections from animals of each of the six treatment groups.
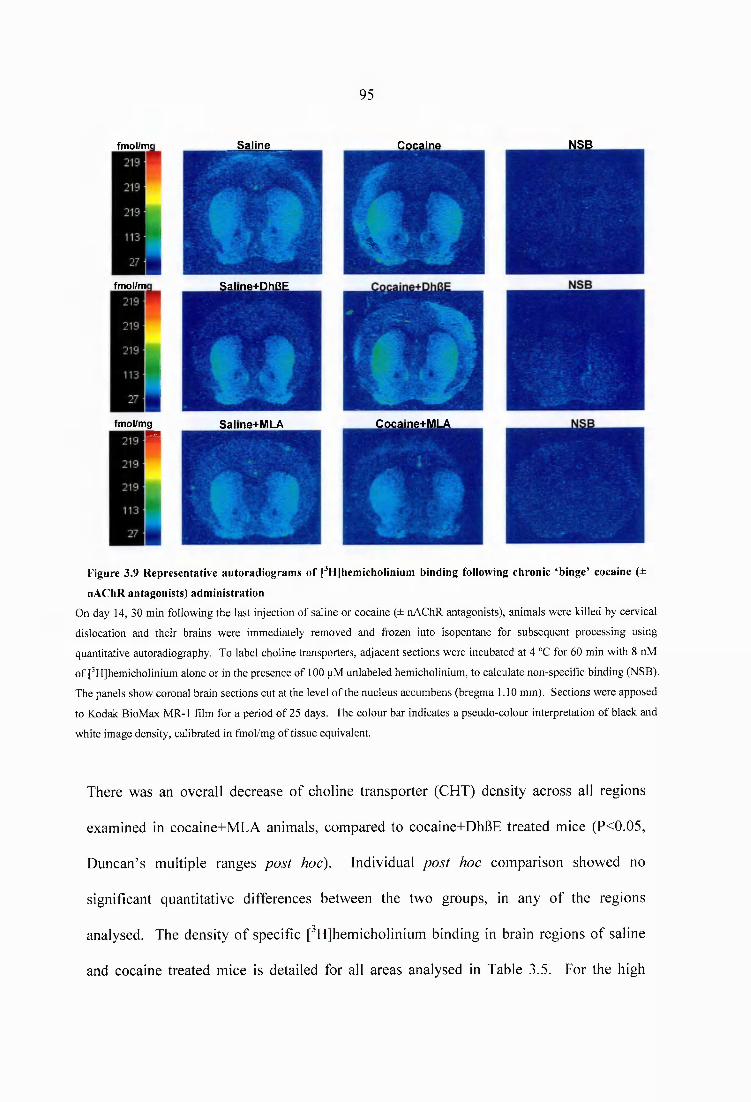
95
fmol/mç S a lin e C o c a in e N SB
%
fmol/m S aline+ D hB E
C ocaine+ M L Afmol/mg 1 ^
Saline+ M L A
Figure 3.9 Representative autoradiogram s o f fH ]hem icholinium binding following chronic ‘binge’ cocaine (±
nAChR antagonists) adm inistration
On day 14, 30 min following the last injection of saline or cocaine (± nAChR antagonists), animals were killed by cervical
dislocation and their brains were immediately removed and frozen into isopentane for subsequent processing using
quantitative autoradiography. To label choline transporters, adjacent sections were incubated at 4 °C for 60 min with 8 nM
of f Hjhemicholinium alone or in the presence of 100 pM unlabeled hemicholinium, to calculate non-specific binding (NSB).
The panels show coronal brain sections cut at the level of the nucleus accumbens (bregma 1.10 mm). Sections were apposed
to Kodak BioMax MR-1 film for a period of 25 days. The colour bar indicates a pseudo-colour interpretation of black and
white image density, calibrated in fmol/mg of tissue equivalent.
There was an overall decrease of choline transporter (CHT) density across all regions
examined in cocaine+MLA animals, compared to cocaine+DhBE treated mice (P<0.05,
Duncan’s multiple ranges post hoc). Individual post hoc comparison showed no
significant quantitative differences between the two groups, in any of the regions
analysed. The density of specific [^H]hemicholinium binding in brain regions of saline
and cocaine treated mice is detailed for all areas analysed in Table 3.5. For the high
96
affinity CHT, two-way ANOVA revealed significant main effects of treatment and region,
with no significant treatment x region interaction effects.
pH]hem icholinium binding (fmol/mg tis su e equivalent)
Saline Cocaine Saline+DhpE Cocaine+DhpE Saiine+MLA Cocaine+MLA
Olfactory tubercle Nucleus accumbens
41.5 ± 5.6 35.9 ± 4.4 42.0 ± 7.1 44.4 ± 4.0 26.9 ± 8.3 31.0 ±4.9
Core 23.4 + 3.7 17.5 ± 2.8 20.3 ± 3.4 21.0 ± 3.0 15.5 ±4.2 14.9 ± 3.4Shell
Caudate putamen27.8 ± 3.1 22.8 + 4.1 27.7 ± 3.9 28.2 ± 3.8 17.5 ±5.4 20.8 ± 4.2
Rostral part 42.4 ± 7.7 31.1 ± 4.0 35.0 ± 4.7 38.4 ± 3.7 28.9 ± 8.1 25.6 ± 4.3Caudal part 44.0 ± 7.2 41.3 ± 5.5 48.8 ± 8.8 37.7 ± 4.4 27.0 ± 5.6 31.9 ± 5.8
Basolateral amygdaloid nucleus 36.3 ± 5.8 31.5 ± 5.5 25.7 ± 3.0 35.7 + 3.4 28.7 ±4.3 25.9 ± 2.9Island of Calleja, major island 39.1 ± 5.6 44.6 + 4.8 43.1 ± 2.3 48.1 ± 6.5 36.1 ± 14.3 27.7 ± 6.5Anteroventral thalamic nucleus 46.8 ± 9.1 29.6 ± 3.9 31.1 +6..6 49.4 ± 8.1 28.2 ±7.7 28.7 ± 7.8Interpeduncular nucleus 152.1 ± 25.5 137.0 ± 13.4 112.2 ± 9.6 131.9 ± 12.2 151.2 ±38.3 128.0 ± 15.4
Table 3.5 Q uantitative autoradiography o f eholine transporters, following ‘binge’ treatm ent o f coeaine ± nAChR
antagonists
Animals were injected with cocaine (15.0 mg/kg/inj), cocaine+DhpE (2.0 mg/kg/inj), or cocaine+MLA (5.0 mg/kg/inj)
three times daily, for a period of 14 days. 30 min after the last injection, mice were killed and their brains removed and
processed for quantitative receptor autoradiography. Data for the binding of choline transporters are shown as the mean
specific binding of [^HJhemicholinium ± SEM in brain regions of 6 to7 animals per group. Two-way ANOVA was
conducted for the factors treatment and region, showing significant treatment [F(5,287)=3.2, P<0.01] and region
[F(8,287)=101.7, P<0.001] effects, and no significant treatment x region interaction [F(40,287)=0.6, P>0.5]. Analysis
revealed an overall decrease in the density of high affinity choline transporters across all regions in cocaine+MLA
animals, compared to cocaine+DhBE treated mice (P<0.05, Duncan’s multiple ranges post hoc).
3.3,6 Effects o f nicotine pretreatment on cocaine-induced locomotor stimulation
In view of the region-specific upregulation of cytisine-sensitive nAChRs by ‘binge’
cocaine, it was hypothesized that the behavioural phenotype displayed by cocaine+DhBE
treated animals was due to blockade of a4p2* nAChR upregulation in the SNc and VTA.
Figure 3.10 A&B shows acute locomotor activity of mice, pretreated with different doses
of nicotine, which have been shown to induce maximal, half-maximal, or sub-maximal
upregulation of nAChRs in the mouse brain (Marks et al., 1991; Marks et al., 2004).
Nicotine at 0.8 mg/kg/hr significantly enhanced locomotor activity in cocaine+DhBE
treated mice, compared with saline pretreatment and with nicotine at 0.45 mg/kg/hr or 0.2

97
mg/kg/hr. At the nicotine dose of 0.2 mg/kg/hr, antagonism of a4p2* nAChRs reduced
cocaine-induced locomotion (P<0.05). This effect was abolished with higher doses of
nicotine pretreatment. The disinhibitory effect of a4p2* nAChR antagonism on cocaine-
induced locomotion was clearly evident when motor activity of cocaine-treated mice was
expressed as percentage change from baseline activity of saline-pretreated animals (Fig.
3.1GB). Differences in cocaine-induced motor stimulation between groups emerged as a
fraction of nicotine dose. Pre exposure to nicotine at doses that marginally upregulate
a4p2* nAChRs similarly enhanced cocaine-induced motor activity in cocaine-treated
animals. Nicotine doses that half-maximally or maximally upregulate nAChRs increased
locomotor stimulation in cocaine+DhBE treated mice accordingly. A positive correlation
between nicotine pretreatment dose and cocaine+DhBE-induced motor stimulation was
derived. In mice treated with cocaine alone, an inverted U-shaped curve was observed
for the effects of nicotine pretreatment on acute motor activity. No locomotor cross
sensitization was observed between cocaine and nicotine, at any of the nicotine
pretreatment doses employed. Two-way ANOVA confirmed significant effects of
nicotine pretreatment and significant pretreatment x treatment interaction effects on acute
cocaine-induced motor stimulation.
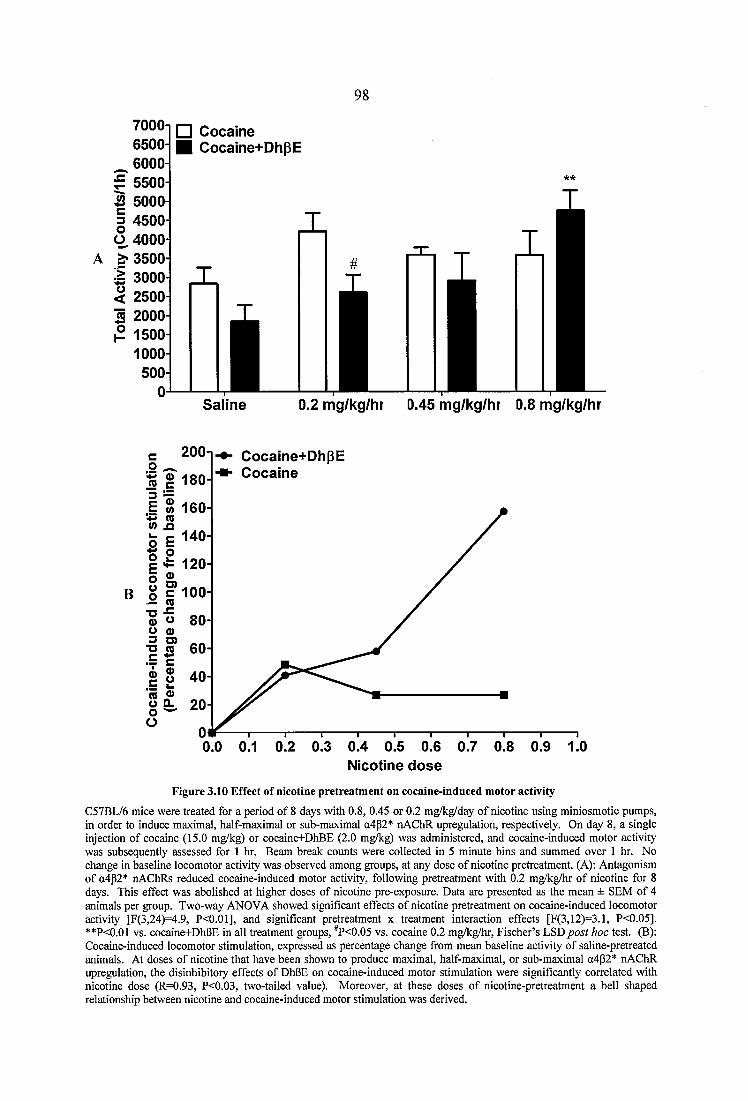
98
7000-I 6500-
^ 6000- g 5500- 2 5000 I 4500- Ü 4000-
A ^ 3500-
□ Cocaine ■ Cocaine+DhpE
3000-2500-2000-
1500-1000-
Saline mg/kg/hr 0.45 mg/kg/hr 0.8 mg/kg/hr
200-1 Cocaine+DhpECocaine
(A 160
* 120
c 100
Nicotine dose
Figure 3.10 Effect o f nicotine pretreatm ent on cocaine-induced m otor activity
C57BL/6 mice were treated for a period of 8 days with 0.8, 0.45 or 0.2 mg/kg/day of nicotine using miniosmotic pumps, in order to induce maximal, half-maximal or sub-maximal a4p2* nAChR upregulation, respectively. On day 8, a single injection of cocaine (15.0 mg/kg) or cocaine+DhBE (2.0 mg/kg) was administered, and cocaine-induced motor activity was subsequently assessed for 1 hr. Beam break counts were collected in 5 minute bins and summed over 1 hr. No change in baseline locomotor activity was observed among groups, at any dose of nicotine pretreatment. (A): Antagonism of o4p2* nAChRs reduced cocaine-induced motor activity, following pretreatment with 0.2 mg/kg/hr of nicotine for 8 days. This effect was abolished at higher doses of nicotine pre-exposure. Data are presented as the mean ± SEM of 4 animals per group. Two-way ANOVA showed significant effects of nicotine pretreatment on cocaine-induced locomotor activity [F(3,24)=4.9, P<0.01], and significant pretreatment x treatment interaction effects [F(3,12)=3.1, P<0.05]. **P<0.01 vs. cocaine+DhBE in all treatment groups, *P<0.05 vs. cocaine 0.2 mg/kg/hr, Fischer’s LSD post hoc test. (B): Cocaine-induced locomotor stimulation, expressed as percentage change from mean baseline activity of saline-pretreated animals. At doses of nicotine that have been shown to produce maximal, half-maximal, or sub-maximal o4p2* nAChR upregulation, the disinhibitory effects of DhBE on cocaine-induced motor stimulation were significantly correlated with nicotine dose (R=0.93, P<0.03, two-tailed value). Moreover, at these doses of nicotine-pretreatment a bell shaped relationship between nicotine and cocaine-induced motor stimulation was derived.
99
3.3.7 Quantitative autoradiography o f D1 and D2 receptors, and o f the dopamine
transporter, following chronic, steady dose, ^binge’ treatment
The density of specific [^HJraclopride binding in brain regions of saline and cocaine
treated mice is detailed for all areas analysed in Table 3.6. Figure 3.11 shows
representative autoradiograms of total and non specific [^HJraclopride binding at the level
of the nucleus accumbens (bregma 1.10 mm), in coronal brain sections from animals of
each of the six treatment groups. An increase in D2 receptor density was observed in the
rostral part of the caudate putamen of cocaine+DhBE mice, compared to saline treated
animals (P<0.05, Duncan’s multiple ranges post hoc). In the same region, the increase of
[^HJraclopride binding in cocaine+DhBE compared to cocaine+MLA animals was close
to significance (P=0.06).
R egion[^H ]raclopride b in d in g (fm ol/m g t is s u e e q u iv a len t)
Saline Cocaine Saline+DhpE Cocaine+DhpE Saline+MLA Cocaine+MLA
Olfactory tubercle 45.4 ± 3.0 52.3 ± 4.0 48.4 ± 6.3 57.1 ± 2.8 48.9 ± 3.5 54.6 ± 2.9Nucleus accum bens
Core 38.2 ± 3.4 42.5 ± 3.4 38.1 ± 6.2 41.1 ± 5.6 40.7 ± 6.9 41.6 ± 3.5
Shell 33.9 ± 2.3 39.5 ± 3.4 35.1 ± 5.8 36.8 ± 5.6 35.7 ± 6.4 38.4 ± 3.8Caudate putam en
Rostral part 62.6 ± 3.3 67.2 ± 4.0 67.7 ± 3.9 76 ± 2.9* 65.8 ± 5.6 66.6 ± 2.8Caudal part 60.2 ± 4.2 64.3 ± 5.0 68.2 ± 3.5 71.4 ± 5.8 68.6 ± 5.7 62.9 ± 4.1
Ventral tegm ental area 15.6 ± 2.2 15.9 ± 3.3 16.4 ± 1.7 18.8 ± 1.7 15.0 ± 1.4 15.7 ± 1.5
Substantia nigra, pars com pacta 15.5 ± 1.4 19.7 + 2.7 17.5 ± 2.0 18.5 ± 1.9 15.2 ± 1.9 14.9 ± 2.0
Table 3.6 Quantitative autoradiography of D2 receptors, following ‘binge’ treatment of cocaine ± nAChR
antagonists
On day 14 of the steady dose ‘binge’ treatment protoeol, animals were killed by cervical dislocation and their brains were
immediately removed and frozen into isopentane for subsequent processing using quantitative autoradiography. Values of D2
receptor binding represent the mean specific binding of [^HJraelopride ± SEM in brain regions of 6 to7 animals per group.
Two-way ANOVA was conducted for the factors treatment and region, showing significant treatment [F(5,229)= 2.4, P<0.01]
and region [F(6,229)=178.3, P<0.001] effects, and no significant treatment x region interaetion [F(30,229)=0.4, P>0.05].
Within-region differences were examined using Duncan’s post hoc test, *P<0.05 vs. saline.

100
Saline Cocainefmol/mg
Cocaine+DhBEfmol/m Saline+DhBE
Cocaine+MLAfmol/m
Figure 3.11 Representative autoradiograms of pHjracIopride binding following chronic ‘binge’ cocaine
To label D2 receptors, adjacent sections were incubated for 60 min with 4 nM [^Hjraclopride alone or in the presence
of 10 pM of sulpiride, to calculate non-specific binding (NSB). Sections were apposed to Kodak BioMax MR-1 film
for a period of 6 weeks. The colour bar indicates a pseudo-colour interpretation of black and white image density,
calibrated in fmol/mg of tissue equivalent.
For D1 receptors, the density of specific [^H]SCH-23390 binding in brain regions of
saline and cocaine treated mice is detailed for all areas analysed in Table 3.7. Two-way
ANOVA showed no significant main effects of treatment and no significant treatment x
region interaction effects.
101
R egion[®H]SCH-23390 b ind ing (fm ol/m g t is s u e equ iva ien t)
Saline Cocaine Saline+DhpE Cocaine+DhpE Saline+MLA Cocaine+MLA
Olfactory tubercle Nucleus accum bens
288.3 ± 14.4 347.5 ± 23.6 335.9 ±21.0 330.2 ± 21.1 342.0 ± 13.5 329.6 ± 14.9
Core 278.0 ± 17.4 278.7 ± 16.4 244.8 ±21.0 273.3 ± 39.5 281.9 ±20.6 236.7 ± 14.4
Shell 267.6 ± 18.8 269.8 ± 12.1 243.8 ± 16.3 267.8 ± 35.3 280.8 ±21.1 232.0 ± 15.4
Claustrum 64.4 ± 6.8 67.1 ± 5.5 70.0 ± 9.1 69.2 ± 5.5 61.0 ±3.3 65.9 ± 3.0
Endopiriform nucleus, dorsal 76.9 ± 3.3 83.2 ± 3.2 80.7 ± 3.2 79.6 ± 5.0 82.6 ± 3.9 81.6 ± 3.1
Nigrostriatal bundle Caudate putamen
53.4 ± 8.7 63.7 + 6.0 49.3 ± 4.0 55.9 ± 8.0 52.9 ± 8.4 52.4 ± 5.0
Rostral part 402.1 ± 15.2 404.0 ± 16.8 408.9 ± 10.4 390.9 ± 28.3 410.1 ± 9.0 394.1 ± 13.0
Caudal part 331.0 ± 19.1 343.6 ± 21.0 347.9 ± 17.1 339.5 ± 23.8 334.6 ± 9.9 308.8 ± 23.5
Amygdala 46.7 ± 3.2 48.7 ± 2.3 47.2 ± 1.0 48.4 ± 2.3 50.1 ± 2.8 45.9 ± 2.1
Hippocampus 21.5 ± 2.6 28.1 ± 1.7 21.8 ± 1.9 20.8 ± 1.1 22.7 ± 2.4 23.8 ± 2.7
Hypothalamus 20.3 ± 2.1 22.5 ± 1.4 19.5 ± 0.9 20.5 ± 1.1 20.3 ± 2.0 20.5 ± 2.2
Ventral tegmental area 36.2 ± 3.5 39.4 ± 2.3 40.2 ± 2.9 35.8 ± 3.6 34.8 ± 4.9 46.5 ± 6.1
Substantia nigra, pars com pacta 162.3 ± 4.8 187.6 ± 4.3 183.0 ± 5.1 184.3 ± 8.3 170.0 ± 2.6 176.2 ± 2.8
Table 3.7 Quantitative autoradiography of D1 receptors, following ‘binge’ treatment of cocaine ± nAChR
antagonists
C57BL/6 mice were treated with a steady dose, ‘binge’ administration protocol for a period of 14 days. 30 min following
the last injection of saline or cocaine (± nAChR antagonists), animals were killed using the Schedule 1 method of
euthanasia, and their brains were immediately removed and frozen into isopentane for quantitative autoradiography. Data
for D1 receptor binding are presented as the mean specific binding of [^HjSCH-23390 ± SEM in brain regions of 6-7
animals per group. Two-way ANOVA revealed a significant effect of region [F(12,408)=422.2, P<0.001], and no
significant treatment [F(5,408)=1.6, P>0.05] or treatment x region interaetion effect [F(60,408)=0.6, P>0.05].
The density of specific [^H]mazindol binding in brain regions of saline and cocaine (±
nAChR antagonists) treated mice is detailed for all areas analysed in Table 3.8. Chronic,
‘binge’ cocaine administration increased DA transporter binding in the rostral part of the
caudate putamen of cocaine+DhBE animals, compared to saline treated mice (P<0.05,
Duncan’s multiple ranges post hoc). Two-way ANOVA showed significant main effects
of treatment and region on [^H]mazindol binding sites, with no significant treatment x
region interaction effects. Figure 3.12 shows representative autoradiograms of total and
non specific [^H]mazindol binding at the level of the nucleus accumbens, in coronal brain
sections from animals of each of the six treatment groups.
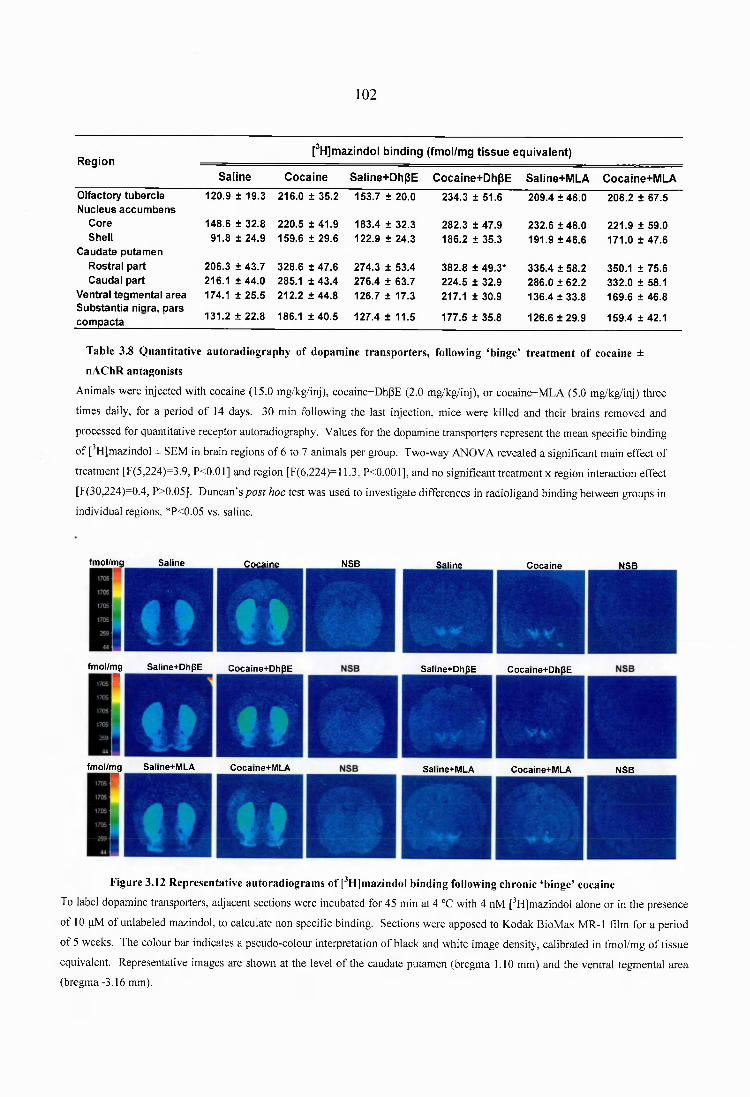
102
R eg io n[^H ]m azlndol b in d in g (fm ol/m g t i s s u e e q u iv a le n t)
S a lin e C o c a in e S a lin e+ D h p E C o c a in e+ D h p E Saline+M LA C ocaine+M L AOlfactory tubercle 120.9 ± 19.3 216.0 ± 35.2 153.7 ± 20.0 234.3 ± 51.6 209.4 ±46.0 206.2 ± 67.5Nucleus accum bens
Core 148.6 ± 32.8 220.5 ± 41.9 183.4 ± 32.3 282.3 ± 47.9 232.6 ±48.0 221.9 ± 59.0Shell 91.8 ± 24.9 159.6 ± 29.6 122.9 ± 24.3 186.2 ± 35.3 191.9 ±46.6 171.0 ± 47.6
Caudate putamenRostral part 206.3 ± 43.7 328.6 ± 47.6 274.3 ± 53.4 382.8 ± 49.3* 335.4 ± 58.2 350.1 ± 75.6Caudal part 216.1 ± 44.0 285.1 ± 43.4 276.4 ± 63.7 224.5 ± 32.9 286.0 ± 62.2 332.0 ± 58.1
Ventral tegmental area 174.1 ± 25.5 212.2 ± 44.8 126.7 ± 17.3 217.1 ± 30.9 136.4 ±33.8 169.6 ± 46.8Substantia nlqra, parscompacta 131.2 ± 22.8 186.1 ±40.5 127.4 ± 11.5 177.5 ± 35.8 126.6 ±29.9 159.4 ± 42.1
Table 3.8 Quantitative autoradiography of dopamine transporters, following ‘binge’ treatment of cocaine ± nAChR antagonists
Animals were injected with cocaine (15.0 mg/kg/inj), cocaine+DhpE (2.0 mg/kg/inj), or cocaine+MLA (5.0 mg/kg/inj) three
times daily, for a period of 14 days. 30 min following the last injection, mice were killed and their brains removed and
processed for quantitative receptor autoradiography. Values for the dopamine transporters represent the mean specific binding
of f Hjmazindol ± SEM in brain regions of 6 to 7 animals per group. Two-way ANOVA revealed a significant main effect of
treatment [F(5,224)=3.9, P<0.01] and region [F(6,224)=l 1.3, P<0.001], and no significant treatment x region interaction effect
[F(30,224)=0.4, P>0.05]. Duncan’s post hoc test was used to investigate differences in radioligand binding between groups in
individual regions, *P<0.05 vs. saline.
fmol/mg Saline Cocaine NSB Saline Cocaine NSB
fmol/mg Saline+DhpEi
Cocaine+DhpE Salme+DhPE Cocame+DhpE
fmol/mg Saline+MLA
!Cocaine+MLA Saline+MLA Cocaine+MLA NSB
Figure 3.12 Representative autoradiograms of fHjmazindoi binding following chronic ‘binge’ cocaine
To label dopamine transporters, adjacent sections were incubated for 45 min at 4 °C with 4 nM [^Hjmazindol alone or in the presence
of 10 pM of unlabeled mazindol, to calculate non specific binding. Sections were apposed to Kodak BioMax MR-1 film for a period
of 5 weeks. The colour bar indicates a pseudo-colour interpretation of black and white image density, calibrated in fmol/mg of tissue
equivalent. Representative images are shown at the level o f the caudate putamen (bregma 1.10 mm) and the ventral tegmental area
(bregma -3.16 mm).
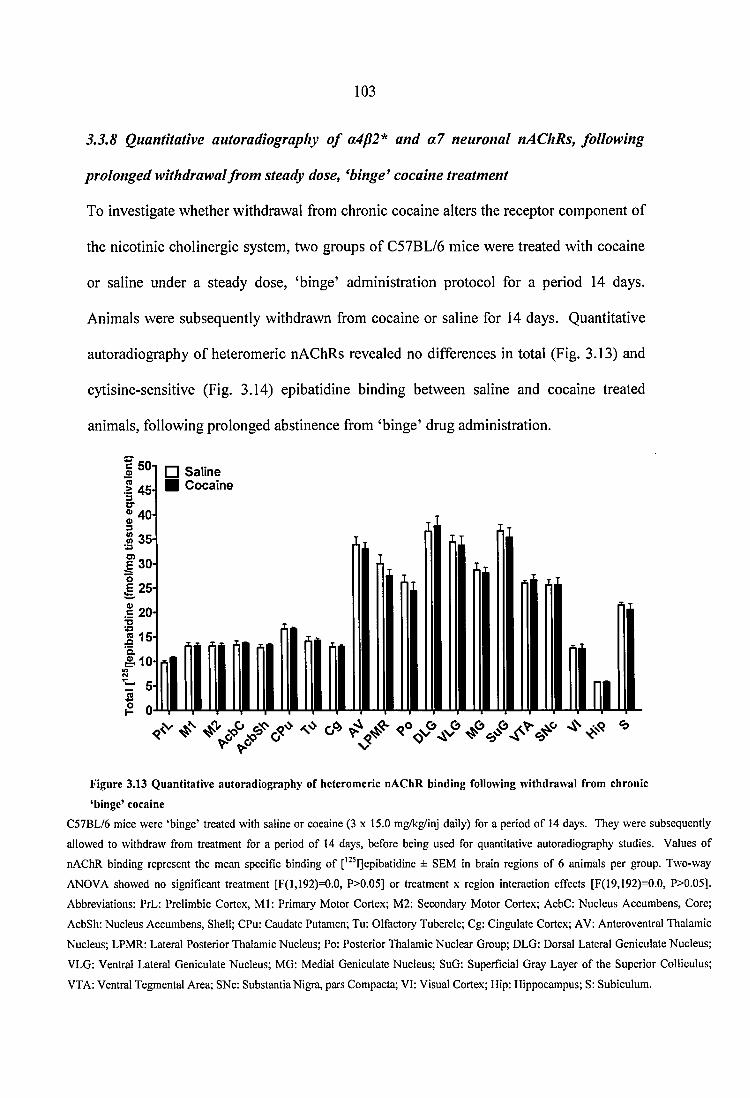
103
3.3.8 Quantitative autoradiography o f a4p2* and a l neuronal nAChRs, following
prolonged withdrawal from steady dose, ^binge’ cocaine treatment
To investigate whether withdrawal from chronic cocaine alters the receptor component of
the nicotinic cholinergic system, two groups of C57BL/6 mice were treated with cocaine
or saline under a steady dose, ‘binge’ administration protocol for a period 14 days.
Animals were subsequently withdrawn from cocaine or saline for 14 days. Quantitative
autoradiography of heteromeric nAChRs revealed no differences in total (Fig. 3.13) and
cytisine-sensitive (Fig. 3.14) epibatidine binding between saline and cocaine treated
animals, following prolonged abstinence from ‘binge’ drug administration.
| 5 0 i
I 454
1 40 i 35
:304oè 25jÏ20.ii5 1 10
□ Saline ■ C ocaine
iA
Figure 3.13 Q uantitative autoradiography o f heterom erie nA ChR binding following withdrawal from chronie
‘binge’ cocaine
C57BL/6 mice were ‘binge’ treated with saline or coeaine (3 x 15.0 mg/kg/inj daily) for a period of 14 days. They were subsequently
allowed to withdraw from treatment for a period of 14 days, before being used for quantitative autoradiography studies. Values of
nAChR binding represent the mean speeifie binding of ['^^Ijepibatidine ± SEM in brain regions of 6 animals per group. Two-way
ANOVA showed no signifieant treatment [F(l,192)=0.0, P>0.05] or treatment x region interaction effects [F(19,192)=0.0, P>0.05].
Abbreviations: PrL: Prelimbic Cortex, Ml: Primary Motor Cortex; M2: Secondary Motor Cortex; AcbC: Nucleus Accumbens, Core;
AcbSh: Nucleus Accumbens, Shell; CPu: Caudate Putamen; Tu: Olfaetory Tubercle; Cg: Cingulate Cortex; AV: Anteroventral Thalamic
Nucleus; LPMR: Lateral Posterior Thalamic Nucleus; Po: Posterior Thalamic Nuclear Group; DLG: Dorsal Lateral Geniculate Nucleus;
VLG: Ventral Lateral Geniculate Nucleus; MG: Medial Geniculate Nucleus; SuG: Superficial Gray Layer of the Superior Colliculus;
VTA: Ventral Tegmental Area; SNc: Substantia Nigra, pars Compacta; VI: Visual Cortex; Hip: Hippocampus; S: Subiculum.
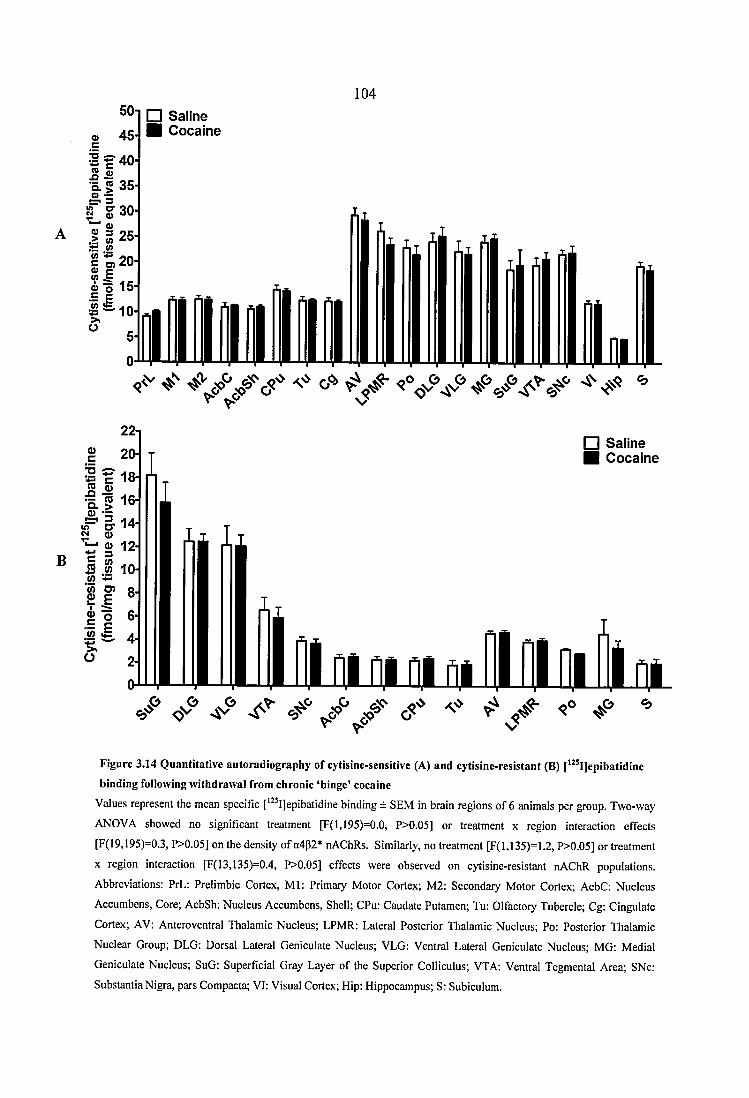
104
IIO .C— 5-: ?
il;50i45-40-3530425
20
1510
5
□ S a lin e ■ C o c a ïn e
L . I
m
□ S a lin e ■ C o c a in e
ro (u=§5 16
® I l 10-
Figure 3.14 Q uantitative autoradiography o f cytisine-sensitive (A) and cytisine-resistant (B) [*“ l]epibatid ine
binding following withdrawal from chronic ‘binge’ cocaine
Values represent the mean specific epibatidine binding ± SEM in brain regions of 6 animals per group. Two-way
ANOVA showed no signifieant treatment [F(l,195)=0.0, P>0.05] or treatment x region interaction effects
[F(19,195)=0.3, P>0.05] on the density of a4p2* nAChRs. Similarly, no treatment [F(l,135)=1.2, P>0.05] or treatment
X region interaction [F(13,135)=0.4, P>0.05] effects were observed on cytisine-resistant nAChR populations.
Abbreviations: PrL: Prelimbic Cortex, Ml: Primary Motor Cortex; M2: Secondary Motor Cortex; AcbC: Nucleus
Aceumbens, Core; AcbSh: Nucleus Accumbens, Shell; CPu: Caudate Putamen; Tu: Olfactory Tubercle; Cg: Cingulate
Cortex; AV: Anteroventral Thalamic Nucleus; LPMR: Lateral Posterior Thalamic Nucleus; Po: Posterior Thalamic
Nuclear Group; DLG: Dorsal Lateral Geniculate Nucleus; VLG: Ventral Lateral Geniculate Nueleus; MG: Medial
Geniculate Nucleus; SuG: Superfieial Gray Layer of the Superior Colliculus; VTA: Ventral Tegmental Area; SNc:
Substantia Nigra, pars Compacta; VI: Visual Cortex; Hip: Hippocampus; S: Subiculum.
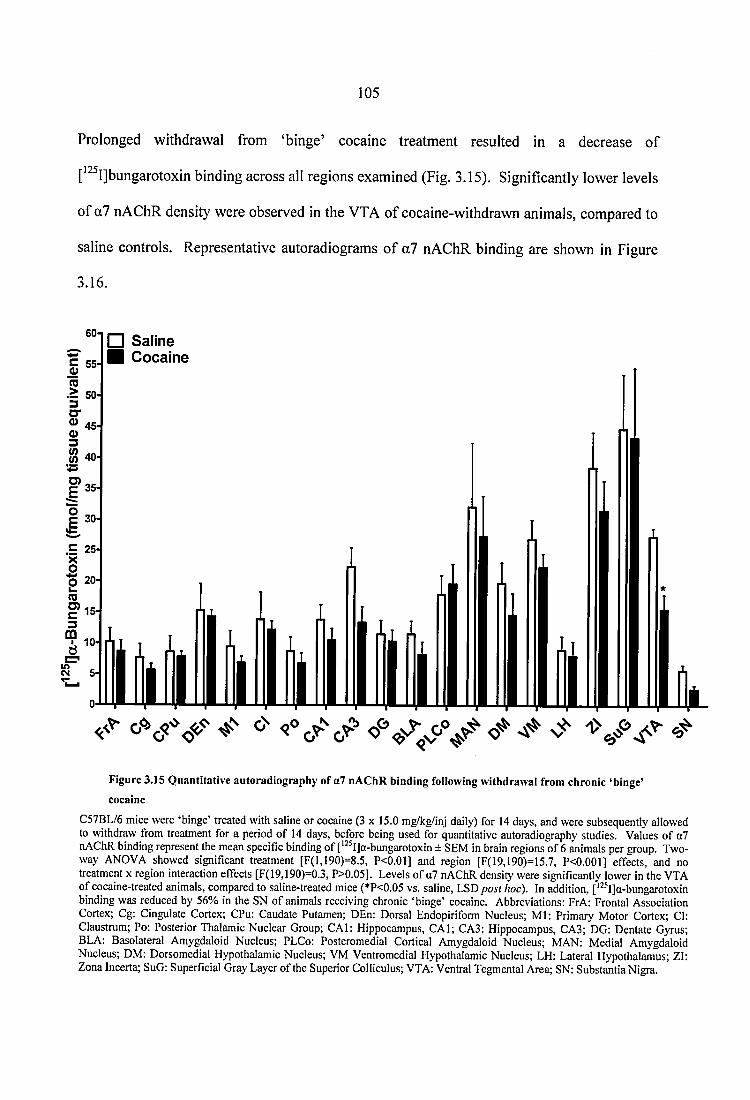
105
Prolonged withdrawal from ‘binge’ cocaine treatment resulted in a decrease of
[^^^IJbungarotoxin binding across all regions examined (Fig. 3.15). Significantly lower levels
of a l nAChR density were observed in the VTA of cocaine-withdrawn animals, compared to
saline controls. Representative autoradiograms of a l nAChR binding are shown in Figure
3.16.
□ Saline Cocaine
Figure 3.15 Quantitative autoradiography of a l nAChR binding following withdrawal from chronie ‘binge’ cocaine
C57BL/6 mice were ‘binge’ treated with saline or cocaine (3 x 15.0 mg/kg/inj daily) for 14 days, and were subsequently allowed to withdraw from treatment for a period of 14 days, before being used for quantitative autoradiography studies. Values of a l nAChR binding represent the mean specific binding of [*^ l]a-bungarotoxin ± SEM in brain regions of 6 animals per group. Two- way ANOVA showed significant treatment [F(l,190)=8.5, P<0.01] and region [F(19,190)=15.7, P<0.001] effects, and no treatment x region interaction effects [F( 19,190)^0.3, P>0.05]. Levels of a? nAChR density were significantly lower in the VTA of cocaine-treated animals, compared to saline-treated mice (*P<0.05 vs. saline, LSD post hoc). In addition, [' 1]a-bungarotoxin binding was reduced by 56% in the SN of animals receiving chronic ‘binge’ cocaine. Abbreviations: FrA: Frontal Association Cortex; Cg: Cingulate Cortex; CPu: Caudate Putamen; DEn: Dorsal Endopiriform Nucleus; Ml: Primary Motor Cortex; Cl: Claustrum; Po: Posterior Thalamic Nuclear Group; CAl: Hippocampus, CAl; CA3: Hippocampus, CA3; DG: Dentate Gyrus; BLA: Basolateral Amygdaloid Nucleus; PLCo: Posteromedial Cortical Amygdaloid Nucleus; MAN: Medial Amygdaloid Nucleus; DM: Dorsomedial Hypothalamic Nucleus; VM Ventromedial Hypothalamic Nucleus; LH: Lateral Hypothalamus; Zl: Zona Incerta; SuG: Superficial Gray Layer of the Superior Colliculus; VTA: Ventral Tegmental Area; SN: Substantia Nigra.
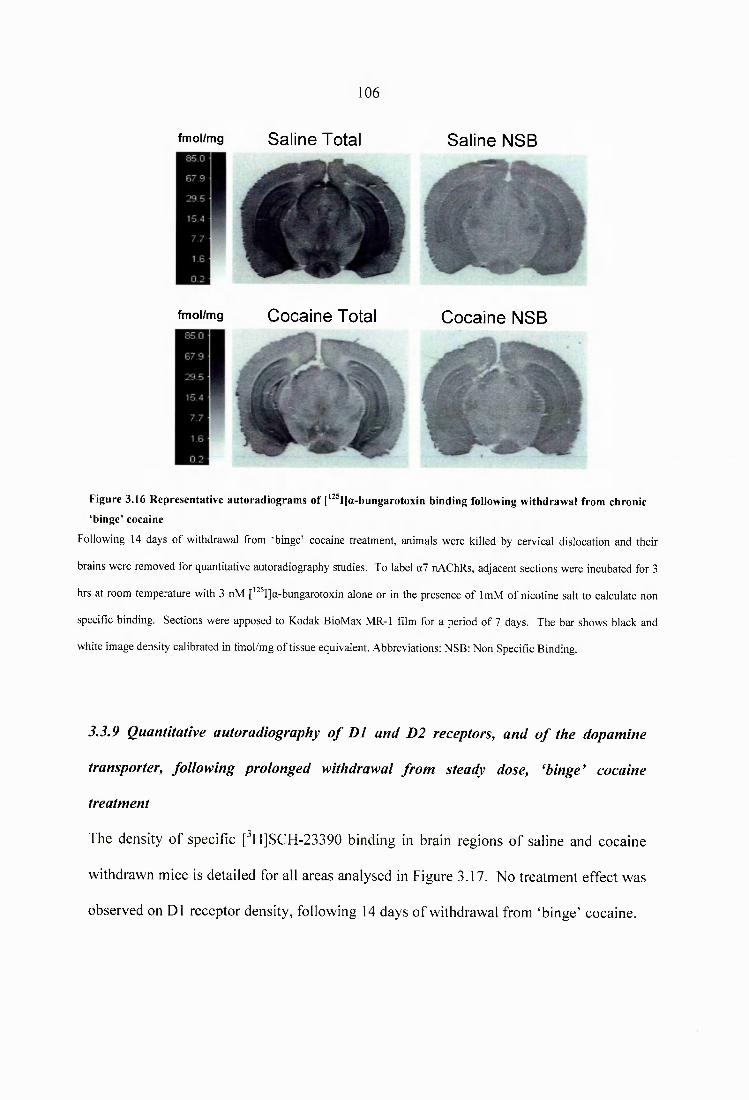
106
fm o l/m g Saline Total Saline NSB
fm o l/m g Cocaine Total Cocaine NSB
Figure 3.16 Representative autoradiogram s o f [‘^®I]a-bungarotoxin binding following withdrawal from chronic
‘binge’ cocaine
Following 14 days of withdrawal from ‘binge’ cocaine treatment, animals were killed by cervical dislocation and their
brains were removed for quantitative autoradiography studies. To label a7 nAChRs, adjacent sections were incubated for 3
hrs at room temperature with 3 nM [’^^I]a-bungarotoxin alone or in the presence of ImM of nicotine salt to calculate non
specific binding. Sections were apposed to Kodak BioMax MR-1 film for a period of 7 days. The bar shows black and
white image density calibrated in fmol/mg of tissue equivalent. Abbreviations: NSB: Non Specific Binding.
3.3.9 Quantitative autoradiography of D1 and D2 receptors, and o f the dopamine
transporter, following prolonged withdrawal from steady dose, ^binge' cocaine
treatment
The density of specific [^H]SCH-23390 binding in brain regions of saline and cocaine
withdrawn mice is detailed for all areas analysed in Figure 3.17. No treatment effect was
observed on D1 receptor density, following 14 days of withdrawal from ‘binge’ cocaine.
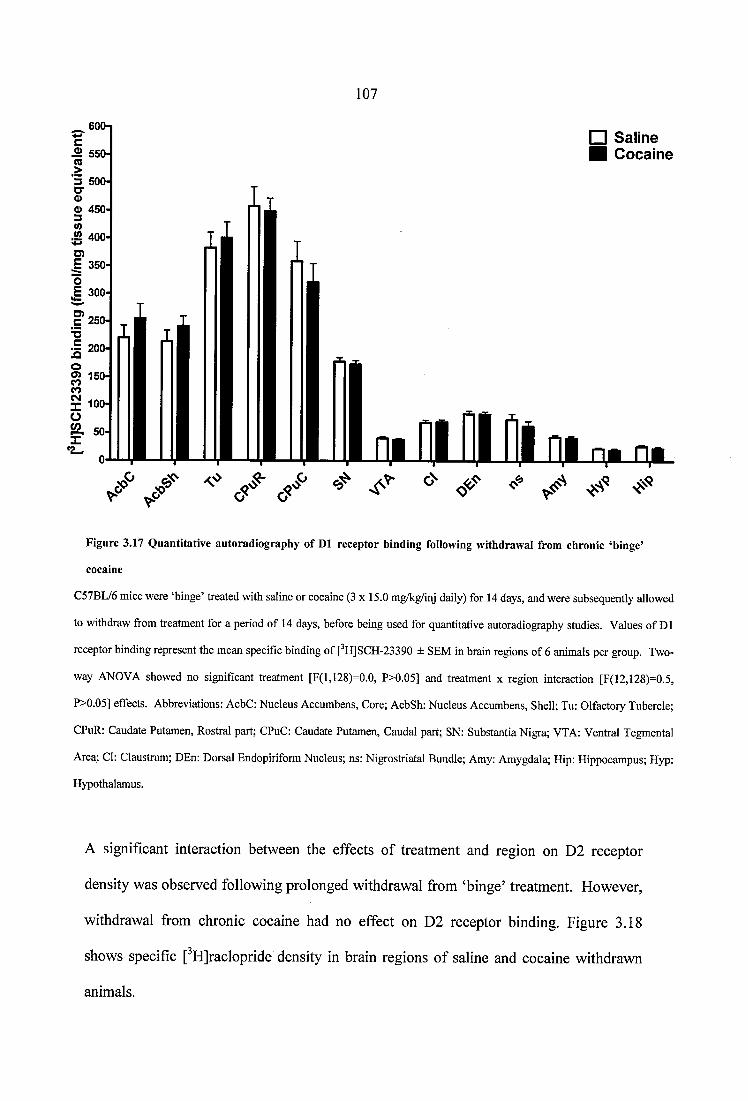
107
600□ Saline■ Cocaine^ 550
0> 450
W 400
= 200oo> 150
Figure 3.17 Q uantitative autoradiography o f D1 receptor binding following withdrawal from chronic ‘binge’
cocaine
C57BL/6 mice were ‘binge’ treated with saline or cocaine (3 x 15.0 mg/kg/inj daily) for 14 days, and were subsequently allowed
to withdraw from treatment for a period of 14 days, before being used for quantitative autoradiography studies. Values of D1
receptor binding represent the mean specific binding of [^HJSCH-23390 ± SEM in brain regions of 6 animals per group. Two-
way ANOVA showed no signifieant treatment [F(l,128)=0.0, P>0.05] and treatment x region interaction [F(12,128)=0.5,
P>0.05] effects. Abbreviations: AcbC: Nucleus Aecumbens, Core; AcbSh: Nucleus Accumbens, Shell; Tu: Olfactory Tubercle;
CPuR: Caudate Putamen, Rostral part; CPuC: Caudate Putamen, Caudal part; SN: Substantia Nigra; VTA: Ventral Tegmental
Area; Cl: Claustrum; DEn: Dorsal Endopiriform Nucleus; ns: Nigrostriatal Bundle; Amy: Amygdala; Hip: Hippocampus; Hyp:
Hypothalamus.
A significant interaction between the effects of treatment and region on D2 receptor
density was observed following prolonged withdrawal from ‘binge’ treatment. However,
withdrawal from chronic cocaine had no effect on D2 receptor binding. Figure 3.18
shows specific [^HJraclopride density in brain regions of saline and cocaine withdrawn
animals.
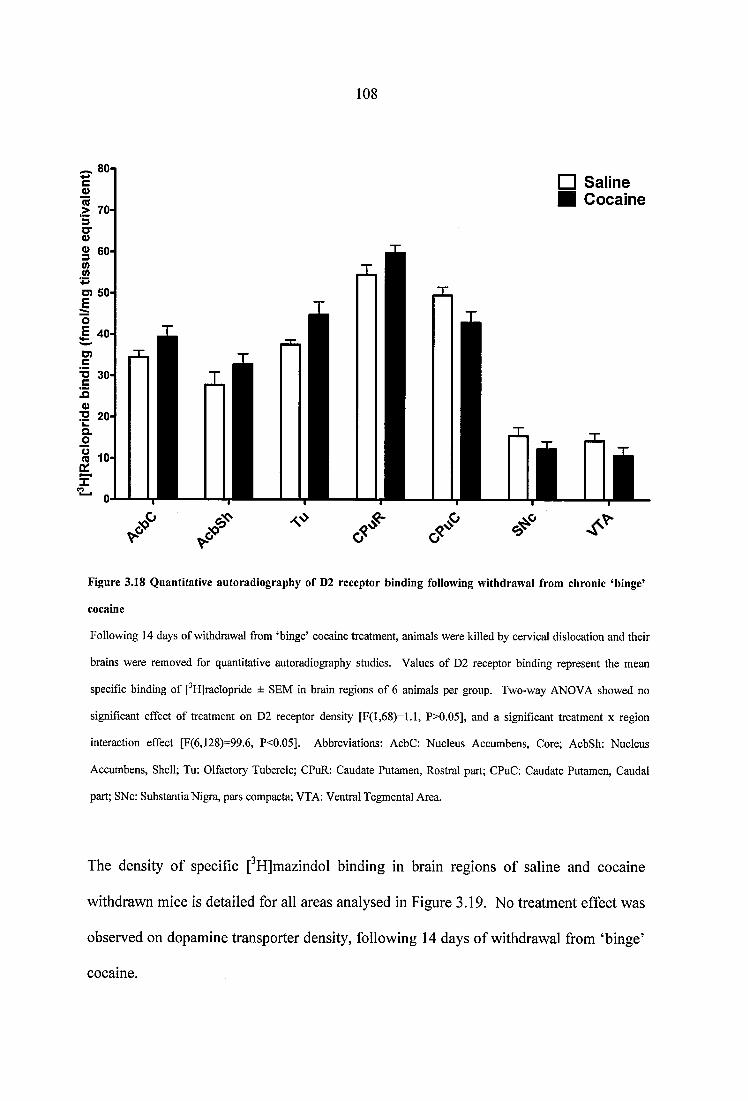
108
^ 80-1□ Saline■ Cocaine
O 50
2 20-
Figure 3.18 Quantitative autoradiography o f D2 receptor binding following withdrawal from chronic ‘binge’
cocaine
Following 14 days of withdrawal from ‘binge’ cocaine treatment, animals were killed by cervical dislocation and their
brains were removed for quantitative autoradiography studies. Values of D2 receptor binding represent the mean
specific binding of [^HJraclopride ± SEM in brain regions of 6 animals per group. Two-way ANOVA showed no
significant effect of treatment on D2 receptor density [F(l,68)=l.l, P>0.05], and a significant treatment x region
interaction effect [F(6,128)==99.6, P<0.05]. Abbreviations: AcbC: Nucleus Accumbens, Core; AcbSh: Nucleus
Accumbens, Shell; Tu: Olfactory Tubercle; CPuR: Caudate Putamen, Rostral part; CPuC: Caudate Putamen, Caudal
part; SNc: Substantia Nigra, pars compacta; VTA: Ventral Tegmental Area.
The density of specific [^H]mazindol binding in brain regions of saline and cocaine
withdrawn mice is detailed for all areas analysed in Figure 3.19. No treatment effect was
observed on dopamine transporter density, following 14 days of withdrawal from ‘binge’
cocaine.
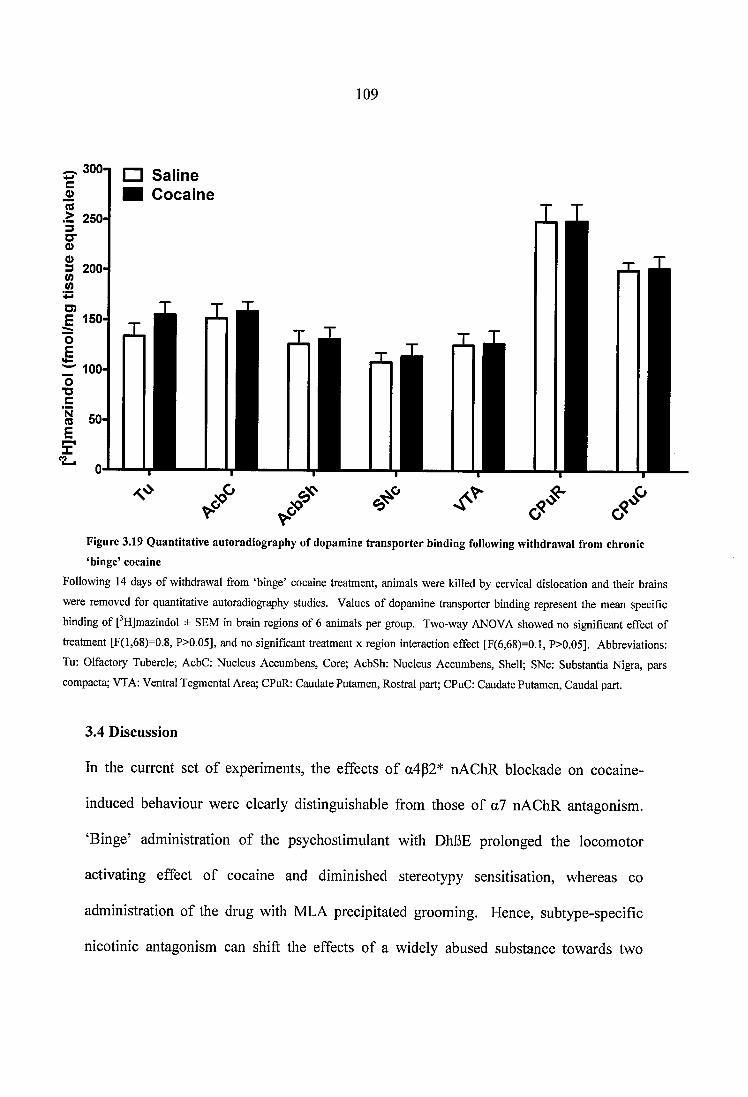
109
300-
> 250-
□ Saline■ Cocaine
3 200-
O)E 150
to 50-
Figure 3.19 Quantitative autoradiography of dopamine transporter binding following withdrawal from chronic ‘binge’ cocaine
Following 14 days of withdrawal from ‘binge’ cocaine treatment, animals were killed by cervical dislocation and their brains
were removed for quantitative autoradiography studies. Values of dopamine transporter binding represent the mean specific
binding of f HJmazindol ± SEM in brain regions of 6 animals per group. Two-way ANOVA showed no significant effect of
treatment [F(l,68)=0.8, P>0.05], and no significant treatment x region interaction effect [F(6,68)=0.1, P>0.05]. Abbreviations:
Tu: Olfactory Tubercle; AcbC: Nucleus Accumbens, Core; AcbSh: Nucleus Accumbens, Shell; SNc: Substantia Nigra, pars
compacta; VTA: Ventral Tegmental Area; CPuR: Caudate Putamen, Rostral part; CPuC: Caudate Putamen, Caudal part.
3.4 Discussion
In the current set of experiments, the effects of a4p2* nAChR blockade on cocaine-
induced behaviour were clearly distinguishable from those of a7 nAChR antagonism.
‘Binge’ administration of the psychostimulant with DhBE prolonged the locomotor
activating effect of cocaine and diminished stereotypy sensitisation, whereas co
administration of the drug with MLA precipitated grooming. Hence, subtype-specific
nicotinic antagonism can shift the effects of a widely abused substance towards two

110
markedly different directions, suggesting that each nicotinic subtype makes a distinct
contribution to cocaine-induced behaviour.
Consistent with studies that have applied a similar protocol of steady-dose, ‘binge’ drug
administration, chronic cocaine resulted in the expression of stereotypy sensitisation, and
produced sensitisation and then tolerance to its locomotor activating effects (Schlussman
et al., 2003; Schlussman et al., 2005; Bailey et al., 2008). These behavioural responses
were accompanied by increases in cytisine-sensitive [^^^I]epibatidine binding within the
VTA and SNc of cocaine-treated mice, suggesting that chronic cocaine region-
specifically regulates a4p2* nAChRs. Cholinergic projections from the mesopontine
tegmentum innervate the SN and the VTA, providing a source of endogenous
acetylcholine that tonically activates nAChRs (Maskos, 2008). Furthermore, self
administered and passively received cocaine enhances acetylcholine release in the VTA,
implicating the neurotransmitter and its cholinergic afférents to the VTA in the
reinforcing effects of the psychostimulant (You et al., 2008). Therefore, increased
acetylcholine release after chronic cocaine may account for the enhanced cytisine-
sensitive [^^^Ijepibatidine binding in ‘binge’ treated mice. Indeed, a4p2* nAChRs are
readily upregulated compared to other nAChR subtypes (Pauly et al., 1991; Nguyen et al.,
2003), and have been shown to increase after chronic exposure to several nicotinic
agonists, including the structural analogues of acetylcholine, methylcarbamylcholine and
N, N-dimethylcarbmylcholine (Warpman et al., 1998; Whiteaker et al., 1998).
Chronic a4p2* nAChR antagonism abolished cocaine-induced stereotypy sensitisation
and prolonged the locomotor activating effects of the psychostimulant. The ability of

I l l
DhBE to attenuate sensitisation is in agreement with previous reports, which show that
administration of antagonists at heteromeric nAChRs blocks cocaine-induced
sensitisation, whether behaviour is measured as locomotor activity or stereotypy
(Schoffelmeer et al., 2002; Champtiaux et al., 2006). Psychomotor stimulants have been
consistently shown to induce motor activation and stereotyped behaviour by acting
predominantly on the mesolimbic and nigrostriatal dopaminergic (DAergic) systems,
respectively (Kelly and Iversen, 1976; Ljungberg and Ungerstedt, 1985; Amalric and
Koob, 1993; Aliane et al., 2009). Moreover, heteromeric a4p2* nAChRs are
conveniently placed on excitatory and inhibitory neurons within the VTA and the SNc
(Klink et al., 2001; Zoli et al., 2002), to inhibit and stimulate DAergic neurotransmission,
respectively, upon the administration of cocaine (Zanetti et al., 2007) or nicotinic agonists
(Lichtensteiger et al., 1982; Clarke et al., 1985a; Sorenson et al., 1998; Tapper et al.,
2004; Nashmi et al., 2007). Since it is shown here that ‘binge’ cocaine region-
specifically increases a4p2* receptors after its chronic administration, the emergence of a
protracted locomotor activity response in place of stereotypy in cocaine+DhBE mice may
reflect the consequences of blocking a4p2* nAChRs at a certain level of cocaine-induced
upregulation.
To further investigate the physiological significance of a4p2* nAChR upregulation in
cocaine-related behaviour, C57BL/6 mice were pretreated with nicotine at doses that are
known to induce maximal, half-maximal, or sub-maximal upregulation of cytisine-
sensitive epibatidine binding in the mouse brain (Marks et al., 1991; Marks et al., 2004),
and a single injection of cocaine or cocaine+DhBE was subsequently administered.
Notwithstanding that the effects of nicotine on cocaine-induced locomotion are age and

112
sex dependent (Collins and Izenwasser, 2004), the results of this experiment confirm the
lack of motor cross-sensitisation between the two substances, extending the findings of
previous reports across a range of nicotine pretreatment doses (Schenk et ah, 1991;
Birrell and Balfour, 1998; Itzhak and Martin, 1999; Desai and Terry, 2003). Importantly
for the present discussion, the pharmacological blockade of a4p2* nAChRs at increasing
doses of nicotine pretreatment unmasks a crucial role for nicotinic receptor upregulation
in the motor stimulant effect of acute cocaine. The inverted U-shaped relationship
between the effects of nicotine-pretreatment and cocaine-induced motor stimulation
suggests a negative correlation between a4p2* nAChR upregulation and cocaine-induced
activity. In support of this, the antagonism of a4p2* nAChRs at increasing doses of
nicotine pretreatment was significantly and linearly linked to acute motor stimulation.
Therefore, the chronic ‘binge’ and acute cocaine administration data strongly imply that
the upregulation of a4p2* nAChRs is a behaviourally relevant neurobiological
commonality that is important for the cholinergic modulation of cocaine-induced effects.
Indeed, a progressive, region-specific upregulation of nAChRs by chronic cocaine may
explain why nAChR antagonists are only effective at preventing sensitisation or the
escalation of cocaine self-administration when repeatedly co-administered with the
psychostimulant, and not upon their acute administration following chronic cocaine
treatment (Corrigall et al., 1994; Schoffelmeer et al., 2002; Champtiaux et al., 2006;
Zanetti et al., 2006; Hansen and Mark, 2007). Moreover, it is shown here that under
conditions of diminished a4p2* receptor availability, the relative potency of cocaine to
stimulate locomotion is enhanced by increasing doses of nicotine, without a change in the
absolute maximal effect of the psychostimulant on motor activity.

113
Co-administration of cocaine with MLA resulted in a unique behavioural modification.
Excessive grooming emerged exclusively following a7 nAChR antagonism, and it was
evident in the increased time cocaine+MLA treated mice spent engaged in this behaviour.
Animals in this group were observed to groom to the extent of hair removal, indicating
the intense nature of their phenotype. The fact that grooming was displayed in a novel,
habituated context (i.e. activity chambers) might also reflect its severity, since novel
environments have been suggested to suppress grooming (Berridge et al., 2005).
Moreover, cocaine+MLA treated mice frequently disrupted their locomotor activity to
groom, confirming the persisting nature of this behaviour. This type of excessive activity
is far from what is termed normal, ‘comfort’ grooming (Berridge et al., 1987). Instead, it
is reminiscent of animal models of stress-induced or compulsive disorders, in which
paradoxical patterns of grooming are observed (Greer and Capecchi, 2002; Berridge et al.,
2005; Kalueff and Tuohimaa, 2005). Interestingly, stress and obsessive compulsion have
both been associated with the chronic abuse of cocaine in the clinical setting (Crum and
Anthony, 1993; Kranzler et al., 1994; S inha et al., 2006).
Evidence suggests that activation of the nigrostriatal system mediates the generation of
grooming patterns (Berridge and Whishaw, 1992; Aldridge and Berridge, 1998) and that
enhancement of DAergic nigrostriatal neurotransmission boosts grooming behaviour
(Campbell et al., 1999; Berridge and Aldridge, 2000). In the present study, no
differences in dopamine receptor and transporter binding were observed between
cocaine-treated groups, which is in accordance with studies that have employed the same
protocol of steady-dose, ‘binge’ administration (Unterwald et al., 1994; Bailey et al.,
2008). Although this does not preclude alterations in receptor function (Bailey et al..

114
2008), the decrease in [^H]hemicholinium binding in cocaine+MLA mice, compared to
cocaine+DhBE treated animals, suggests that the grooming phenotype relates to
perturbations of cholinergic neurotransmission following a7, but not a4p2* nAChR
antagonism. Selective labelling of the high affinity choline transporters by
[^HJhemicholinium is considered to depict in vivo cholinergic neuron activity (Ribeiro et
al., 2006). The expression and functional capacity of choline transporters are reduced in
the dopamine transporter knockdown (DAT-KD) mouse, suggesting that a
hyperdopaminergic system deregulates cholinergic neurotransmission (Parikh et al.,
2006). Of note, DAT-KD mice groom more than wild-type animals, exhibiting abnormal
patterns of grooming behaviour (Greer and Capecchi, 2002; Berridge et al., 2005). In
addition, ablation of striatal cholinergic neurons leads to compensatory upregulation of
a4 nAChR subunits in the SN (Kitabatake et al., 2003). In analogy with all of the
aforementioned studies, ‘binge’ administration of a dopamine transporter antagonist with
MLA precipitated an intense grooming phenotype, reduced [^H]hemicholinium binding
sites, and increased density of a4p2* receptors in the SNc of cocaine+MLA treated
animals, compared to saline alone. Therefore, the protective role of a7 nAChRs against
cocaine-induced behavioural disorders may arise by sustaining a balance between
cholinergic and dopaminergic neurotransmission during repeated exposure to cocaine.
Antagonistic interactions between cholinergic and dopaminergic systems are often
described in the literature (Hikida et al., 2001; Hikida et al., 2003). In support of an
important role for a7 nAChRs in mediating them, intra-accumbal or intra-VTA
administration of MLA has been shown to significantly enhance cocaine-induced
dopamine release (Zanetti et al., 2007).

115
Following 14 days of withdrawal from ‘binge’ cocaine treatment, the enhanced cytisine-
sensitive [^^^IJepibatidine binding in the VTA and SNc of cocaine-treated animals
returned to baseline levels. This observation is in agreement with findings of previous
studies in rodents and humans, which have established that the numerical upregulation of
P2 subunit-containing nAChRs is a reversible phenomenon (Marks et al., 1985; Collins et
al., 1990; Breese et al., 1997). Indeed, heteromeric nAChR binding levels have been
shown to return to control values within 7 to 10 days after the cessation of nicotine
administration in mice (Marks et al., 1985). The present results thus demonstrate that
cocaine regulates the a4p2* component of the nicotinic cholinergic system in a similar
way to nicotine, both during its chronic administration, and upon subsequent
psychostimulant withdrawal. This suggests that the neurochemical mechanisms
mediating the effects of nicotine and cocaine in the SNc and the VTA share common
features, at least with regards to the impact of the two substances on a4p2* nAChR
density.
Drug-induced decreases of nAChR binding have only occasionally been reported in the
literature, since both agonists and antagonists at nicotinic receptors have been
consistently shown to induce receptor upregulation (see Section 1.1.3.3). Moreover, as
mentioned before, nAChR upregulation does not occur permanently; it persists with a
time course that depends on the dose and duration of prior nicotine exposure. [^^^I]a-
bungarotoxin binding sites, in particular, have been shown to return to contol levels
within 2 to 4 days following termination of nicotine treatment (Miner and Collins, 1989;
Collins et al., 1990). In this respect, the observation that a7 nicotinic receptors are
significantly reduced in the VTA of animals chronically withdrawn from cocaine, albeit

116
novel and intriguing is difficult to interpret. Decreased a7 nAChR binding has been
implicated in the pathohysiology of various neuropsychiatrie disorders, including
schizophrenia (Mathew et al., 2007), Parkinson’s (Burghaus et al., 2003) and Alzheimer’s
(Wevers et al., 1999) disease. Interestingly, prolonged cocaine abuse is characterised by
severe mood disturbances, ranging from depression and anxiety into the etreme of
paranoia (Cadet and Bolla, 1996; Lejoyeux et al., 2000). In the current study, blockade
of a7 receptors during ‘binge’ cocaine treatment induced an intense grooming behaviour,
which is reminiscent of animal models of compulsive or stress-induced syndromes. One
can therefore speculate that withdrawal from chronic cocaine unmasks a permanent
neuroadaptation in the regulation of a7 nAChRs, which relates to the observed grooming
phenotype and that is solely characteristic of this drug class. Importantly, the observation
that a7 nAChRs decrease following prolonged withdrawal from cocaine confirms that the
nicotinic cholinergic system is part of the neurochemical circuitry that is affected by
chronic cocaine administration.
From a neurobiological perpective, the decrease in [^^^I]a-bungarotoxin binding sites in
the VTA of cocaine withdrawn animals may simply reflect some compensatory response
to the increased a4p2* nAChR density, which was observed in the same brain region
following chronic ‘binge’ administration of cocaine. It has been shown that chronic
nicotine cell-specifically desensitises p2* nAChRs in the mouse VTA, but also triggers
an a7 mediated mechanism that counteracts this inactivation (Besson et al., 2007). To the
extent that cocaine and nicotine share common neurobiological processes, the observed
reduction in the density of a7 nAChRs may constitute the long term expression of this
compensatory process in the VTA of cocaine withdrawn mice. Although a7 nAChRs do

117
not upregulate in response to chronic ‘binge’ cocaine, one cannot preclude that the
psychostimulant mobilises and permanently depletes a large reserve pool of nicotinic
receptors in the VTA, a neuroadaptation that is expressed after the cessation of chronic
drug administration. Indeed, while quantitative autoradiography measures nAChRs
present in the cytosol and on cell membranes, the majority of a7 nAChRs in the rodent
VTA has been shown to reside in the cytoplasm (Jones and Wonnacott, 2004). Therefore,
although the absolute numbers of a7 nAChRs within a cell may not change, perhaps the
numbers of receptors with functional, synaptic roles in the VTA is actually increased
after prolonged cocaine use, and decreases upon cocaine withdrawal. It is thus possible
that the reduction in [^^^I]a-bungarotoxin binding sites of cocaine withdrawn animals
represents drug-induced, permanent alterations in the functional balance between p2* and
a7 nAChR in the VTA.
No changes in the density of D1 and D2 dopaminergic receptors and transporters were
observed following prolonged withdrawal from ‘binge’ cocaine administration. This is
consistent with several other reports, which have applied a similar protocol of intermittent
cocaine administration to examine alterations in the regulation of the dopaminergic
system during cocaine abstinence (King et al., 1994; Maggos et al., 1998). Nevertheless,
it has been demonstrated that receptor density does not necessarily relate to receptor
function, and it would therefore be interesting to examine dopaminergic receptor
coupling in response to chronic cocaine administration and withdrawal (Bailey et al.,
2004b; Bailey et al., 2008).

1 1 8
In conclusion, nicotinic receptor subunit antagonism caused a progressive, subtype
specific dissociation of cocaine-induced behaviours, following the chronic, ‘binge’
administration of the psychostimulant. Genetic evidence has linked a risk allele for
nicotine dependence with a protective role in cocaine addiction (Grucza et al., 2008).
The present results also imply a protective role for nicotine in cocaine-induced behaviour,
and further suggest a subunit-specific involvement of the nicotinic cholinergic system in
the behavioural effects of chronic cocaine. Upregulation of a4p2* nAChRs mediates the
stimulatory and stereotypic effects of the psychostimulant, whereas a7 nAChRs are
protective against an intense phenotype that is reminiscent of cocaine-related behavioural
disorders. An important implication of the present report is that the co-abuse of nicotine
with cocaine can both enhance stimulation and protect against cocaine-induced
behavioural abnormalities, depending on the availability of distinct nAChR subtypes.
The face-validity of this assumption is supported by surveys in cocaine dependent
individuals, who report both stimulant-enhancing and calming effects of cigarette
smoking (Wiseman and McMillan, 1998). The clear-cut behavioural dissociation that
arises from the antagonism of different nAChR subunits after chronic cocaine
administration suggests that manipulation of the nicotinic cholinergic system has the
potential to influence treatment strategies for cocaine dependence, and implicates a tailor-
made pharmacological approach for the alleviation of cocaine abuse symptomatology.

119
CHAPTER
FOUR

120
4 QUANTITATIVE AUTORADIOGRAPHY OF HETEROMERIC AND
HOMOMERIC NEURONAL nAChR SUBTYPES IN C57BL/6 MICE,
FOLLOWING CHRONIC MORPHINE TREATMENT AND WITHDRAWAL
4.1 Introduction
Opiate drugs have a long medical history in the treatment of pain, but their clinical use is
limited by severe side effects, including respiratory depression, constipation, and
vomiting (reviewed in Raehal and Bohn, 2005). In addition, opiates are highly addictive,
and their abuse-related potential renders them a leading cause of death, morbidity, and
productivity loss in society (reviewed in Gruber et al., 2007). Morphine is the
prototypical opioid that is isolated from the plant extracts of opium poppy, and its
analgesic and addictive properties are mainly exerted through MOP receptors in the CNS
(reviewed in Kieffer, 1999). While the role of dopamine in mediating the reinforcing
effects of morphine is well established, the involvement of other neurotransmitter
systems has been less investigated. Recently, several studies have suggested that the
nicotinic cholinergic system influences morphine-induced behaviour and neurochemistry
(see Section 1.3.2). Thus, morphine-induced conditioned place prereference (GPP) is
enhanced following pretreatment with nicotine in both rats (Shippenberg et al., 1996;
Biala and Weglinska, 2004) and mice (Vihavainen et al., 2008a), whereas morphine’s
locomotor activity in mice is potentiated after nicotine administration (Biala and
Weglinska, 2004; Vihavainen et al., 2006). In morphine-treated animals, nicotine has
been shown to attenuate naloxone-induced conditioned place aversion (Araki et al., 2004)
and withdrawal jumping (Zarrindast and Farzin, 1996; Haghparast et al., 2008),
suggesting that nAChRs are involved not only in morphine reward, but also in the

1 2 1
negative affective and somatic states that characterise morphine abstinence. Furthermore,
infusion of the potent aSp4 nAChR antagonist 18-methoxycoronaridine (18-MC) into
brain rich cholinergic nuclei, such as the medial habenula and the interpeduncular nucleus,
reduces morphine self-administration and attenuates sensitisation of morphine-induced
dopamine release (Click et al., 2002; Click et al., 2006; Taraschenko et al., 2007a).
Therefore, an increasing body of evidence indicates that nAChRs can modulate the
effects of morphine administration.
Neuronal nAChRs constitute a heterogeneous family of pentameric ligand-gated ion
channels, with unique pharmacological properties and distinct topographical distribution
(see Section 1.1.2). Based on the evidence for interactions between morphine and the
nicotinic cholinergic system, it was hypothesized that the effects of morphine treatment
and withdrawal could be depicted at the neurochemical level, as changes in the density of
different nicotinic receptor subtypes. To this end, quantitative autoradiography of
heteromeric and homomeric nAChRs was performed in brain sections of morphine-
treated mice. To investigate the involvement of the nicotinic cholinergic system in
morphine withdrawal, quantitative autoradiography of nAChRs was performed in brain
sections of mice that were acutely and chronically withdrawn from morphine
administration. For the purposes of this study, an escalating, ‘intermittent’ protocol of
morphine treatment was applied, which resembles human patterns of opiate abuse (Kreek
et al., 2002).

1 2 2
4.2 Materials and Methods
4.2.1 Animals and drug treatment
Male C57BL/6 mice (B&K Universal Ltd, Hull, UK), weighing 20-25 g were used.
Animals were housed individually in a temperature-controlled room, under a 12 h
light/dark schedule. Food and water were available ad libitum. All studies were
conducted according to protocols approved by the Home Office, UK, and in accordance
with European Community Council directive 86/609/EEC. Animals were habituated to
handling for a period of 7 days, prior to treatment. Following acclimatization,
subcutaneous (s.c.) injections of either saline or morphine were administered to C57BL/6
mice in an ‘intermittent’ escalating dose paradigm, in order to mimic the pattern of opiate
self-administration in human abusers (Kreek et al., 2002). Animals were separated into
five groups:
Group 1 (n=6): Saline for 7 days, followed by Saline on day 8
Group 2 (n=6): Morphine for 7 days, followed by Morphine on day 8 (chronic treatment)
Group 3 (n=6): Morphine for 7 days, followed by Saline on day 8 (acute withdrawal)
Group 4 (n=6): Saline for 7 days, followed by 7 day withdrawal
Group 5 (n=6): Morphine for 7 days, followed by 7 day withdrawal (chronic withdrawal)
Two injections were given daily (5 pm and 9 am) in accordance with protocols
investigating opiate-induced addictive processes in animal models of drug abuse
(Spanagel, 1995; Muller and Unterwald, 2004). Mice in the escalating group received
morphine at 2 x 20 ml/kg/inj on days 1 and 2, 2 x 40 mg/kg/inj on days 3 and 4, 2 x 80
mg/kg/inj on days 5 and 6 , and 2 x 100 mg/kg/inj on day 7, followed by a single
morphine injection (100 mg/kg) on day 8 (chronic treatment group). A separate group of

123
animals was treated under the same protocol, with the exception of receiving an injection
of saline instead of morphine on day 8 (acute withdrawal group). Two separate groups
received ‘intermittent’ injections of morphine or saline for 7 days, and were subsequently
allowed to withdraw from treatment for a period of 7 days (chronic withdrawal). The
maximum daily morphine dose used in the study was 200 mg/kg, as illustrated in Table
4.1.
Day 1 2 3 4 5 6 7 8
Dose per injection (mg/kg)
20 20 40 40 80 80 100 100
Daily dose (mg/kg) 40 40 80 80 160 160 200 100
Table 4.1 Daily morphine intake
The dose per injection and the maximum daily morphine dose, administered to C57BL/6 mice subcutaneously, using a
chronic ‘intermittent’ escalating dose paradigm.
Animals in the chronic treatment and acute withdrawal groups were killed 2 h following
the last injection of saline or morphine on day 8 . Mice in the chronic withdrawal groups
were killed 7 days after the end of the ‘intermittent’ escalating dose morphine treatment.
All mice were killed by cervical dislocation. Brains were immediately removed, snap
frozen into isopentane at -25 °C and stored at -80 °C until further processed for
quantitative autoradiography.
4.2.2 Quantitative autoradiography o f a4p2* and a?nicotinic cholinergic receptors
Tissue sectioning was carried out at -21 °C using a Zeiss Microm HM505E cryostat. 20
pm frozen coronal sections were cut at 300 pm intervals, from rostral to caudal levels.
On the day of assay, slides for labeling a4p2* and a7 nAChRs were thawed to room

124
temperature for 20 min, prior to incubation with [^^^I]epibatidine and
bungarotoxin, respectively. Sections for analysis were derived from 6 brains from each
of the five treatment groups (n=6 per group). Multiple, adjacent sections from all groups
were processed together in a paired binding protocol. Radioligand bound sections were
apposed to Kodak BioMax MR-1 film (Sigma-Aldrich, UK), along with appropriate
microscale standards, to allow quantification. Detailed experimental protocols for
autoradiographic procedures can be found in Section 3.2.6.
For nAChRs, radioligand bound sections were apposed to film for 24h. Structures of
exceptionally high nAChR density, including the medial habenula, fasciculus retrofiexus,
and the interpeduncular nucleus were apposed for a period of 6h, to avoid film saturation.
All structures were identified by reference to the mouse brain atlas of Franklin and
Paxinos, (2001), and analysed using an MCID image analyser (Image research, Linton,
UK).
4.2.3 Statistical analysis
Two-way ANOVA for the factors treatment and region was used for the comparison of
quantitative measures of a4(32* and a7 nicotinic receptors in brain regions of chronically
treated and acutely withdrawn morphine and saline C57BL/6 mice. The effects of
prolonged withdrawal from morphine treatment on nAChR density were analysed
separately using two-way ANOVA (independent variables treatment and region).
Structures of the habenulo-interpeduncular pathway were quantified using a 6h exposure
to film, because of the high densities of heteromeric nAChRs in this cholinergic system.
Therefore, comparisons between structures of high nicotinic receptor density were

125
performed using a separate two away ANOVA (independent factors treatment and
region).
4.3 Results
4.3.1 Quantitative autoradiography o f heteromeric nAChRs following chronic
morphine administration and withdrawal
The present study was conducted to determine whether morphine or withdrawal from
morphine alters the receptor component of the nicotinic cholinergic system. Figure 4.1
shows representative autoradiograms of total and cytisine-resistant [^^^I]epibatidine
binding in coronal brain sections of morphine-treated, and of acutely and chronically
morphine-withdrawn animals. The ‘intermittent’ administration of escalating doses of
morphine, as well as the acute withdrawal from morphine treatment had no effect on the
density of total, cytisine-sensitive, and cytisine-resistant [^^^I]epibatidine binding,
compared to saline treatment. Similarly, no significant differences in heteromeric
nAChR binding density were observed between groups that were chronically withdrawn
from saline or morphine. The density of total, cytisine-sensitive, and cytisine-resistant
[^^^I]epibatidine binding is detailed for all areas analysed in Tables 4.2, 4.3, and 4.4,
respectively. Non specific [^^^I]epibatidine binding was indistinguishable from film
background.
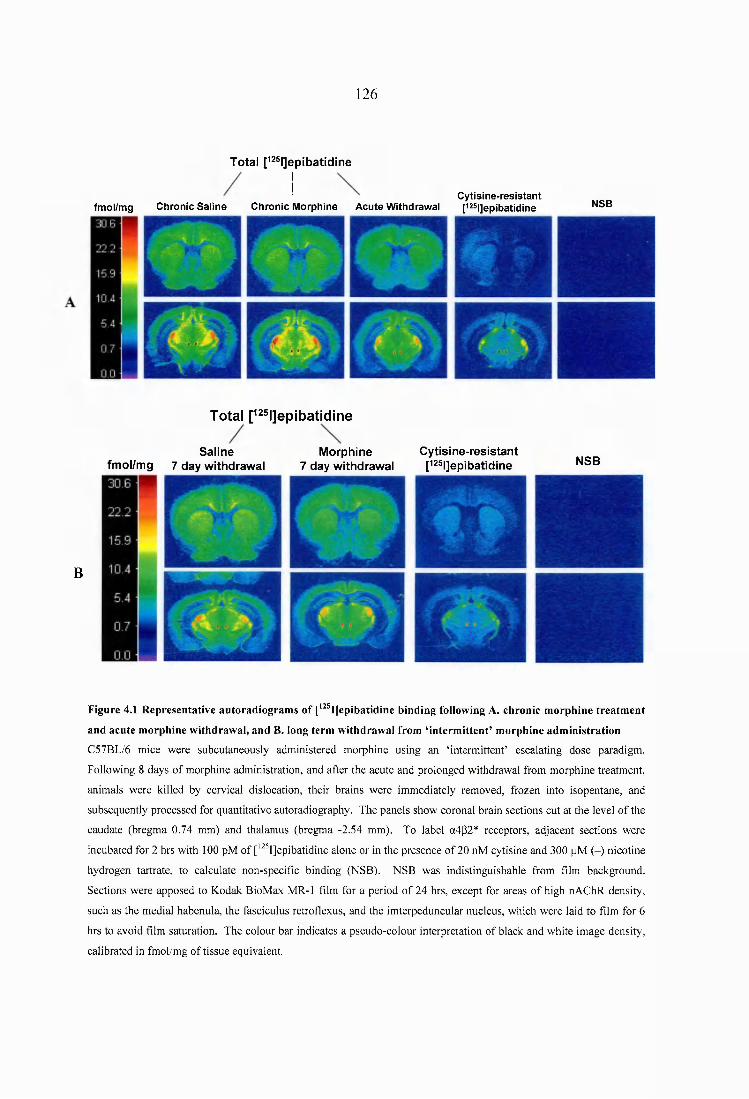
126
T o ta l p ^ ^ l] e p lb a t ld in e
Cytisine-resistantfmoi/mg Chronic Saiine Chronic Morphine Acute Withdrawai p2S|]epibatidine NSB
T o ta l p 2 ® l]ep ib a tld in e
B
S a l in e M o rp h in efm o l/m g 7 d a y w ith d ra w a l 7 d a y w ith d ra w a l
C y t is in e - r e s i s t a n tp25i]epibatidine N SB
Figure 4.1 Representative autoradiogram s o f [*^T]epibatidine binding following A. chronic m orphine treatm ent
and acute morphine withdrawal, and B. long term withdrawal from ‘interm ittent’ m orphine adm inistration
C57BL/6 mice were subcutaneously administered morphine using an ‘intermittent’ escalating dose paradigm.
Following 8 days of morphine administration, and after the acute and prolonged withdrawal from morphine treatment,
animals were killed by cervical dislocation, their brains were immediately removed, frozen into isopentane, and
subsequently processed for quantitative autoradiography. The panels show coronal brain sections cut at the level of the
caudate (bregma 0.74 mm) and thalamus (bregma -2.54 mm). To label a4|32* receptors, adjacent sections were
incubated for 2 hrs with 100 pM of ['^ IJepibatidine alone or in the presence of 20 nM cytisine and 300 pM (-) nicotine
hydrogen tartrate, to calculate non-specific binding (NSB). NSB was indistinguishable from film background.
Sections were apposed to Kodak BioMax MR-1 film for a period of 24 hrs, except for areas of high nAChR density,
such as the medial habenula, the fasciculus retrofiexus, and the imterpeduncular nucleus, which were laid to film for 6
hrs to avoid film saturation. The colour bar indicates a pseudo-colour interpretation of black and white image density,
calibrated in fmol/mg of tissue equivalent.
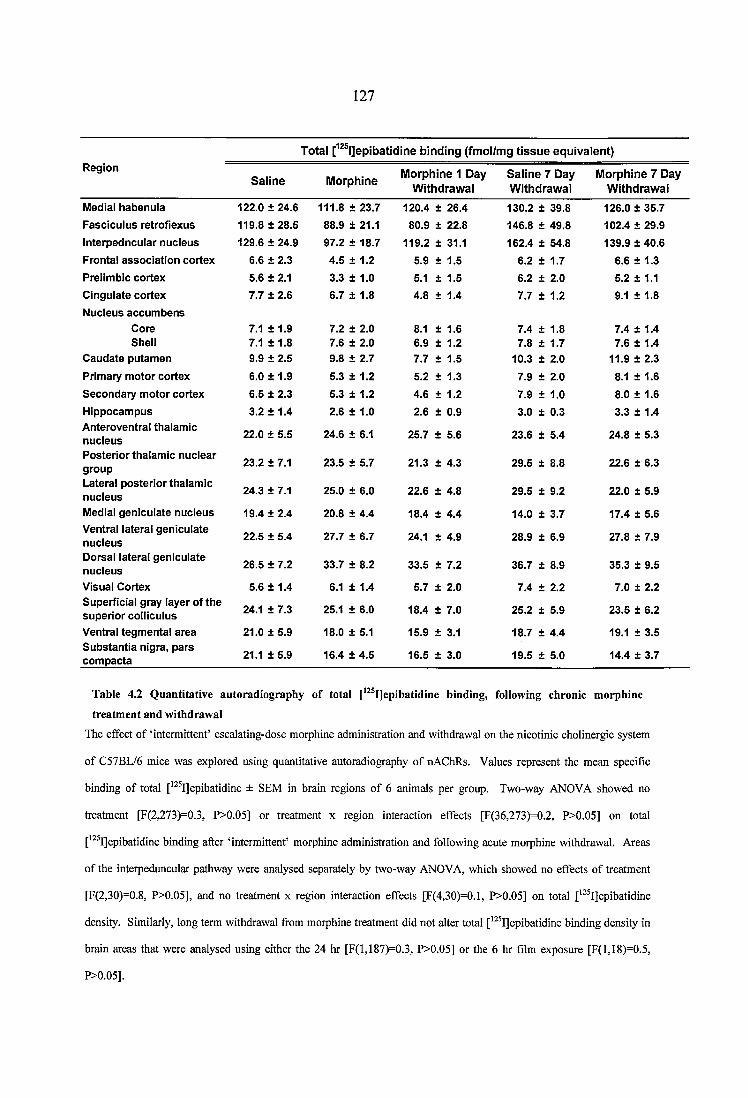
127
T o ta l [ l ] e p ib a t id ln e b in d in g (fm o l/m g t i s s u e e q u iv a le n t)Region
S a l in e M o rp h in eM o rp h in e 1 D ay
W ith d ra w a lS a l in e 7 D ay W ith d ra w a l
M o rp h in e 7 D ay W ith d ra w a l
Medial h ab en u la 122.0 ± 24.6 111.8 ± 23.7 120.4 ± 26.4 130.2 ± 39.8 126.0 ± 35.7
F a sc icu lu s re tro fiex u s 119.8 ± 28.5 88.9 ± 21.1 80.9 ± 22.8 146.8 ± 49.8 102.4 ± 29.9
In te rp ed n cu la r n u c le u s 129.6 ± 24.9 97.2 ± 18.7 119.2 ± 31.1 162.4 ± 54.8 139.9 ± 40.6
Frontal a s so c ia tio n co rtex 6.6 ± 2.3 4.5 ± 1.2 5.9 ± 1.5 6.2 ± 1.7 6.6 ± 1 .3
Preiim bic co rtex 5.6 ± 2.1 3.3 ± 1.0 5.1 ± 1.5 6.2 ± 2.0 5.2 ± 1.1
C ingu la te co rtex 7.7 ± 2.6 6.7 ± 1.8 4.8 ± 1.4 7.7 ± 1.2 9.1 ± 1.8
N uc ieus a c c u m b e n s C ore 7.1 ± 1.9 7.2 ± 2.0 8.1 ± 1.6 7.4 ± 1.8 7.4 ± 1.4Sheli 7.1 ± 1.8 7.6 ± 2.0 6.9 ± 1.2 7.8 ± 1.7 7.6 ± 1.4
C a u d a te pu tam en 9.9 ± 2.5 9.8 ± 2.7 7.7 ± 1.5 10.3 ± 2.0 11.9 ± 2.3
Prim ary m o to r co rtex 6.0 ± 1.9 5.3 ± 1.2 5.2 ± 1.3 7.9 ± 2.0 8.1 ± 1.6
S e c o n d ary m o to r co rtex 6.5 ± 2.3 5.3 ± 1.2 4.6 ± 1.2 7.9 ± 1.0 8.0 ± 1.6
H ip p o cam p u s 3.2 ± 1.4 2.6 ± 1.0 2.6 ± 0.9 3.0 ± 0.3 3.3 ± 1.4A nteroven tra l th alam ic n u c ie u s 22.0 ± 5.5 24.6 ± 6.1 25.7 ± 5.6 23.6 ± 5.4 24.8 ± 5.3
P o s te r io r th a lam ic n u c le a r g ro u p 23.2 ± 7.1 23.5 ± 5.7 21.3 + 4.3 29.5 ± 8.8 22.6 ± 6.3
L ateral p o s te rio r th a lam ic n u c ie u s 24.3 ± 7.1 25.0 ± 6.0 22.6 ± 4.8 29.5 ± 9.2 22.0 ± 5.9
Medial g en icu la te n u c ie u s 19.4 ± 2.4 20.8 ± 4.4 18.4 ± 4.4 14.0 ± 3.7 17.4 ± 5.6V entral latera l g en icu la te n u c le u s 22.5 ± 5.4 27.7 ± 6.7 24.1 ± 4.9 28.9 ± 6.9 27.8 ± 7.9
D orsal latera l g en icu la te n u c le u s 26.5 ± 7.2 33.7 ± 8.2 33.5 ± 7.2 36.7 ± 8.9 35.3 ± 9.5
Visual C ortex 5.6 ± 1.4 6.1 ± 1.4 5.7 ± 2.0 7.4 ± 2.2 7.0 ± 2.2Superficial gray layer o f th e su p e rio r co iiicu iu s 24.1 ± 7.3 25.1 ± 6.0 18.4 ± 7.0 25.2 ± 5.9 23.5 ± 6.2
V entral teg m en ta l a rea 21.0 ± 5.9 18.0 ± 5.1 15.9 ± 3.1 18.7 ± 4.4 19.1 ± 3.5S u b s ta n tia nigra, p a rs co m p a c ta 21.1 ± 5.9 16.4 ± 4.5 16.5 ± 3.0 19.5 ± 5.0 14.4 ± 3.7
Table 4.2 Q uantitative autoradiography o f total [^^®I]epibatidine binding, following chronic m orphine
treatm ent and withdrawal
The effect of ‘intermittent’ escalating-dose morphine administration and withdrawal on the nicotinic cholinergic system
of C57BL/6 mice was explored using quantitative autoradiography of nAChRs. Values represent the mean specific
binding of total [ ^ IJepibatidine ± SEM in brain regions of 6 animals per group. Two-way ANOVA showed no
treatment [F(2,273)=0.3, P>0.05] or treatment x region interaction effects [F(36,273)=0.2, P>0.05] on total
[ ^ IJepibatidine binding after ‘intermittent’ morphine administration and following acute morphine withdrawal. Areas
of the interpeduncular pathway were analysed separately by two-way ANOVA, which showed no effects of treatment
[F(2,30)=0.8, P>0.05], and no treatment x region interaction effects [F(4,30)=0.1, P>0.05] on total ['^^I]epibatidine
density. Similarly, long term withdrawal from morphine treatment did not alter total [^ IJepibatidine binding density in
brain areas that were analysed using either the 24 hr [F(l,187)=0.3, P>0.05] or the 6 hr film exposure [F(l,18)=0.5,
P>0.05].
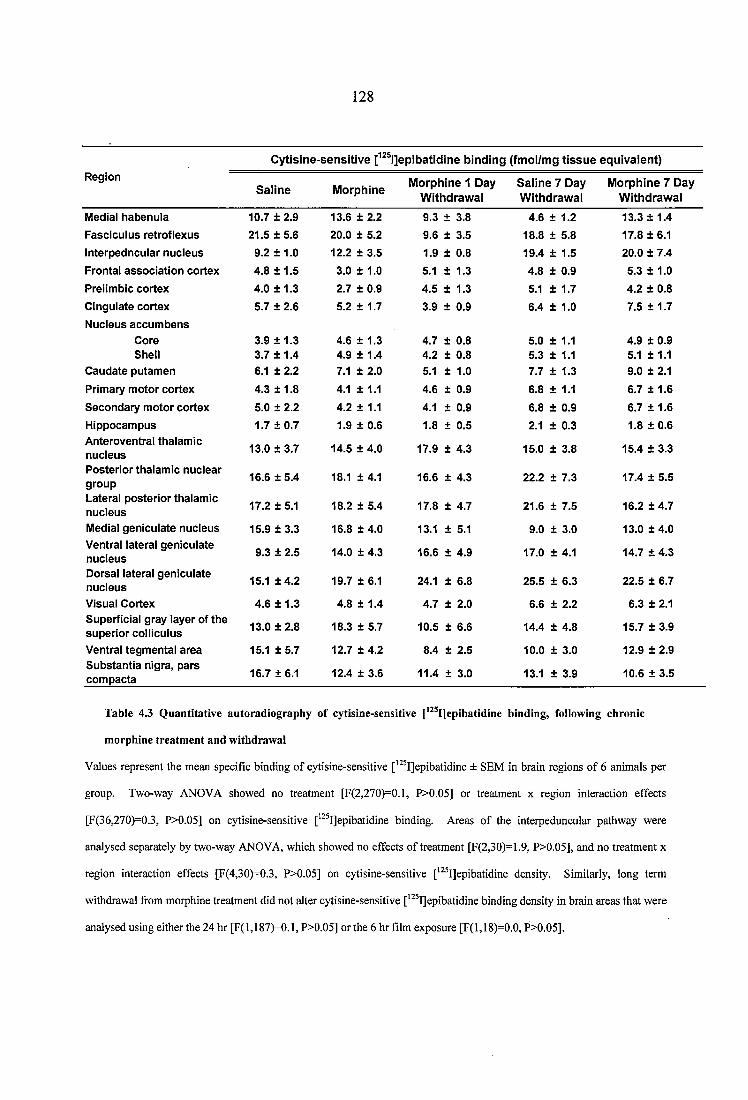
128
C y t is in e - s e n s i t iv e [ l ] e p lb a t id ln e b in d in g ( fm o l/m g t i s s u e e q u iv a le n t )Region
S a l in e M o rp h in eM o rp h in e 1 D ay
W ith d ra w a lS a l in e 7 D ay W ith d ra w a l
M o rp h in e 7 D ay W ith d ra w a l
M edial h a b en u la 10.7 ± 2.9 13.6 ± 2.2 9.3 ± 3.8 4.6 ± 1.2 13.3 ± 1.4
F a sc ic u lu s re tro fiex u s 21.5 ± 5 .6 20.0 ± 5.2 9.6 ± 3.5 18.8 ± 5.8 17.8 ± 6.1
In te rp e d n cu la r n u c ie u s 9.2 ± 1 .0 12.2 ± 3.5 1.9 ± 0.8 19.4 ± 1.5 20.0 ± 7.4
F ron ta l a s s o c ia t io n co rte x 4.8 ± 1 .5 3.0 ± 1.0 5.1 ± 1.3 4.8 ± 0.9 5.3 ± 1.0
P re iim bic c o rte x 4.0 ± 1 .3 2.7 ± 0.9 4.5 ± 1.3 5.1 ± 1.7 4.2 ± 0.8
C in g u la te c o rte x 5.7 ± 2.6 5.2 ± 1.7 3.9 ± 0.9 6.4 ± 1.0 7.5 ± 1.7
N u c leu s a c c u m b e n sC ore 3.9 ± 1.3 4.6 ± 1.3 4.7 ± 0.8 5.0 ± 1.1 4.9 ± 0.9Shell 3 .7 ± 1 .4 4.9 ± 1.4 4.2 ± 0.8 5.3 ± 1.1 5.1 ± 1.1
C a u d a te p u tam e n 6.1 ± 2.2 7.1 ± 2.0 5.1 ± 1.0 7.7 ± 1.3 9.0 ± 2.1
P rim ary m o to r c o rte x 4.3 ± 1.8 4.1 ± 1.1 4.6 ± 0.9 6.8 ± 1.1 6.7 ± 1.6
S e c o n d a ry m o to r c o rte x 5.0 ± 2.2 4.2 ± 1.1 4.1 ± 0.9 6.8 ± 0.9 6.7 ± 1.6
H ip p o cam p u s 1.7 ± 0 .7 1.9 ± 0.6 1.8 ± 0.5 2.1 ± 0.3 1.8 ± 0.6A nte ro v en tra l th a lam ic n u c le u s 13.0 ± 3.7 14.5 ± 4.0 17.9 ± 4.3 15.0 ± 3.8 15.4 ± 3.3
P o s te r io r th a la m ic n u c le a r g ro u p
16.6 ± 5 .4 18.1 ± 4.1 16.6 ± 4.3 22.2 ± 7.3 17.4 ± 5.5
L ateral p o s te r io r th a lam ic n u c ie u s 17.2 ± 5.1 18.2 ± 5.4 17.8 ± 4.7 21.6 ± 7.5 16.2 ± 4.7
M edial g e n ic u la te n u c ie u s 15.9 ± 3 .3 16.8 ± 4.0 13.1 ± 5.1 9.0 ± 3.0 13.0 ± 4.0V entral la te ra l g e n ic u la te n u c le u s 9.3 ± 2.5 14.0 ± 4.3 16.6 ± 4.9 17.0 ± 4.1 14.7 ± 4.3
D orsal latera l g e n ic u la te n u c le u s 15.1 ± 4 .2 19.7 ± 6.1 24.1 ± 6.8 25.5 ± 6.3 22.5 ± 6.7
V isual C ortex 4.6 ± 1.3 4.8 ± 1.4 4.7 ± 2.0 6.6 ± 2.2 6.3 ± 2.1S up erfic ia l g ray lay er o f th e su p e r io r c o iiicu iu s 13.0 ± 2 .8 18.3 ± 5.7 10.5 ± 6.6 14.4 ± 4.8 15.7 ± 3.9
V entral teg m e n ta l a re a 15.1 ± 5 .7 12.7 ± 4.2 8.4 ± 2.5 10.0 ± 3.0 12.9 ± 2.9S u b s ta n tia n ig ra , p a rs c o m p a c ta 16.7 ± 6 .1 12.4 ± 3.6 11.4 ± 3.0 13.1 ± 3.9 10.6 ± 3.5
Table 4.3 Quantitative autoradiography o f cytisine-sensitive epibatidine binding, follow ing chronic
morphine treatm ent and withdrawal
Values represent the mean specific binding of cytisine-sensitive ['^^I]epibatidine ± SEM in brain regions of 6 animals per
group. Two-way ANOVA showed no treatment [F(2,270)=0.1, P>0.05] or treatment x region interaction effects
[F(36,270)=0.3, P>0.05] on cytisine-sensitive [ ^ IJepibatidine binding. Areas of the interpeduncular pathway were
analysed separately by two-way ANOVA, which showed no effects of treatment [F(2,30)=1.9, P>0.05], and no treatment x
region interaction effects [F(4,30)-0.3, P>0.05] on cytisine-sensitive ['^ IJepibatidine density. Similarly, long term
withdrawal from morphine treatment did not alter cytisine-sensitive ['^ IJepibatidine binding density in brain areas that were
analysed using either the 24 hr [F(l,187)=0.1, P>0.05] or the 6 hr film exposure [F(l,18)=0.0, P>0.05].
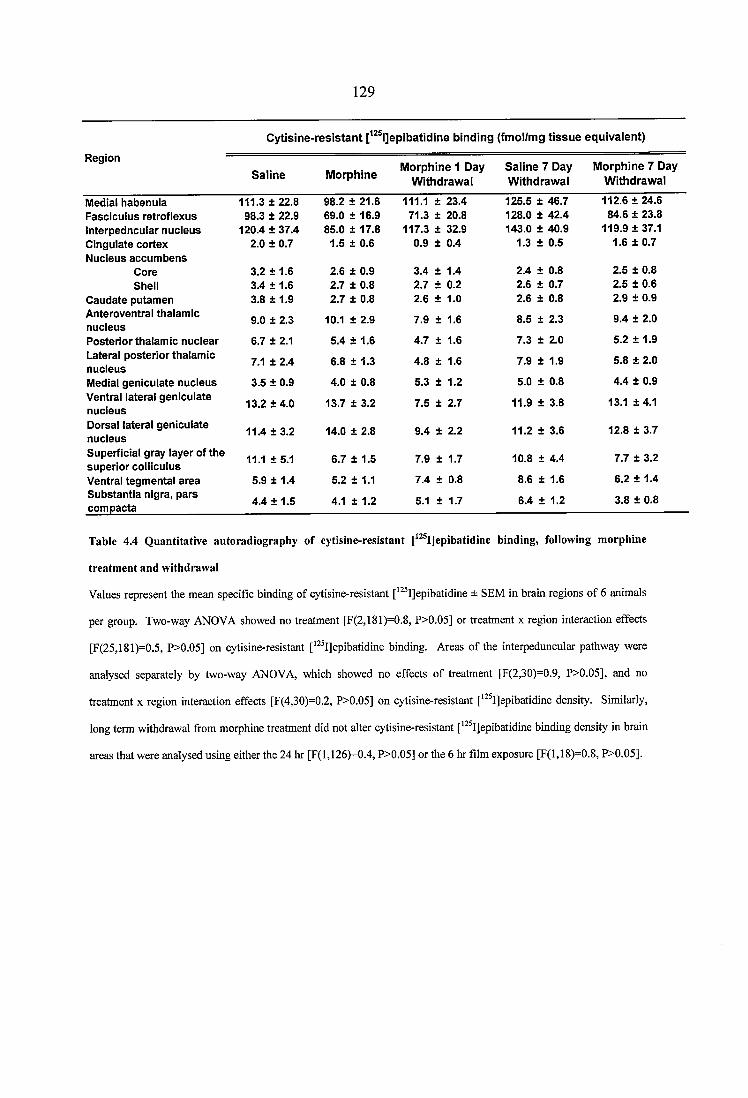
129
C y tis in e - r e s i s ta n t l]e p ib a tid in e b in d in g (fm o l/m g t i s s u e e q u iv a le n t)
R egion
S a lin e M o rp h in eM o rp h in e 1 D ay
W ith d ra w a iS a lin e 7 D ay W ith d raw a l
M o rp h in e 7 D ay W ith d ra w a l
Medial h ab en u la 111.3 ± 22.8 98.2 ± 21.6 111.1 ± 23.4 125.5 ± 46.7 112.6 ± 24.6F a sc icu lu s re tro fiex u s 98.3 ± 22.9 69.0 ± 16.9 71.3 ± 20.8 128.0 ± 42.4 84.6 ± 23.8In terp ed n cu la r n u c ie u s 120.4 ± 37.4 85.0 ± 17.8 117.3 ± 32.9 143.0 ± 40.9 119.9 ± 37.1C ingulate co rtex N ucieus a cc u m b e n s
2.0 ± 0.7 1.5 ± 0.6 0.9 ± 0.4 1.3 ± 0.5 1.6 ± 0.7
C ore 3.2 ± 1.6 2.6 ± 0.9 3.4 ± 1.4 2.4 ± 0.8 2.5 ± 0.8Shell 3.4 ± 1.6 2.7 ± 0.8 2.7 ± 0.2 2.6 ± 0.7 2.5 ± 0.6
C au d ate pu tam en 3.8 + 1.9 2.7 ± 0.8 2.6 ± 1.0 2.6 ± 0.8 2.9 ± 0.9A nteroventral th alam ic n u c ieu s
9.0 ± 2.3 10.1 ± 2.9 7.9 ± 1.6 8.5 ± 2.3 9.4 ± 2.0
P o s te rio r th a lam ic n u c lea r 6.7 ± 2.1 5.4 ± 1.6 4.7 ± 1.6 7.3 ± 2.0 5.2 ± 1.9Lateral p o ste rio r thalam ic n u c ieu s
7.1 ± 2.4 6.8 ± 1 .3 4.8 ± 1.6 7.9 ± 1.9 5.8 ± 2.0
Medial g en icu la te n u c leu s 3.5 ± 0.9 4.0 ± 0.8 5.3 ± 1.2 5.0 ± 0.8 4.4 ± 0.9Ventral lateral g en icu la te n u c ieu s
13.2 ± 4.0 13.7 ± 3.2 7.5 ± 2.7 11.9 ± 3.8 13.1 ± 4.1
D orsal lateral g en icu la te n u c leu s
11.4 ± 3 .2 14.0 ± 2.8 9.4 ± 2.2 11.2 ± 3.6 12.8 ± 3.7
Superficial gray layer o f th e su p e rio r co iiicu ius
11.1 ± 5.1 6.7 ± 1.5 7.9 ± 1.7 10.8 ± 4.4 7.7 ± 3.2
V entral teg m en ta l a rea 5.9 ± 1.4 5.2 ± 1.1 7.4 ± 0.8 8.6 ± 1.6 6.2 ± 1.4S u b s ta n tia nigra, p a rs co m p acta 4.4 ± 1.5 4.1 ± 1.2 5.1 ± 1.7 6.4 ± 1.2 3.8 ± 0.8
Table 4.4 Quantitative autoradiography o f cytisine-resistant [ “ l]epibatidine binding, following m orphine
treatm ent and w ithdraw al
Values represent the mean specific binding of cytisine-resistant epibatidine ± SEM in brain regions of 6 animals
per group. Two-way ANOVA showed no treatment [F(2,181)=0.8, P>0.05] or treatment x region interaction effects
[F(25,181)=0.5, P>0.05] on cytisine-resistant [*^ I]epibatidine binding. Areas of the interpeduncular pathway were
analysed separately by two-way ANOVA, which showed no effects of treatment [F(2,30)=0.9, P>0.05], and no
treatment x region interaction effects [F(4,30)=0.2, P>0.05] on cytisine-resistant [ ^ IJepibatidine density. Similarly,
long term withdrawal from morphine treatment did not alter cytisine-resistant ['^ IJepibatidine binding density in brain
areas that were analysed using either the 24 hr [F(l,126)-0.4, P>0.05] or the 6 hr film exposure [F(l,18)=0.8, P>0.05].
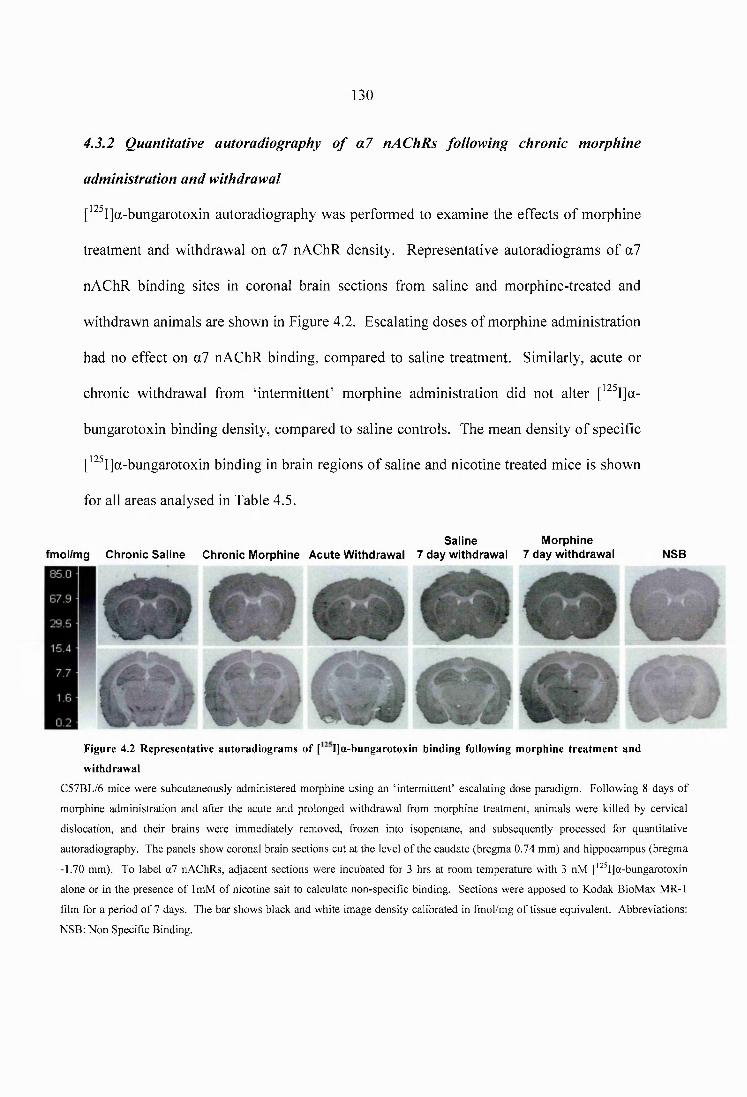
130
4.3.2 Quantitative autoradiography of a l nAChRs following chronic morphine
administration and withdrawal
[^^^I]a-bungarotoxin autoradiography was performed to examine the effects of morphine
treatment and withdrawal on a7 nAChR density. Representative autoradiograms of a7
nAChR binding sites in coronal brain sections from saline and morphine-treated and
withdrawn animals are shown in Figure 4.2. Escalating doses of morphine administration
had no effect on a7 nAChR binding, compared to saline treatment. Similarly, acute or
chronic withdrawal from ‘intermittent’ morphine administration did not alter [^^^I]a-
bungarotoxin binding density, compared to saline controls. The mean density of specific
['^^I]a-bungarotoxin binding in brain regions of saline and nicotine treated mice is shown
for all areas analysed in Table 4.5.
S a lin e M o rp h in efm o l/m g C h ro n ic S a l in e C h ro n ic M o rp h in e A c u te W ith d ra w a l 7 d a y w ith d ra w a l 7 d a y w ith d ra w a l N SB
Figure 4.2 Representative autoradiogram s o f [ I]a-bungarotoxin binding following m orphine treatm ent and
withdrawal
C57BL/6 mice were subcutaneously administered morphine using an ‘intermittent’ escalating dose paradigm. Following 8 days of
morphine administration and after the acute and prolonged withdrawal from morphine treatment, animals were killed by cervical
dislocation, and their brains were immediately removed, frozen into isopentane, and subsequently processed for quantitative
autoradiography. The panels show coronal brain sections cut at the level of the caudate (bregma 0.74 mm) and hippocampus (bregma
-1.70 mm). To label a l nAChRs, adjacent sections were incubated for 3 hrs at room temperature with 3 nM ['^^I]a-bungarotoxin
alone or in the presence of ImM of nicotine salt to calculate non-specific binding. Sections were apposed to Kodak BioMax MR-1
film for a period of 7 days. The bar shows black and white image density calibrated in fmol/mg of tissue equivalent. Abbreviations:
NSB: Non Specific Binding.
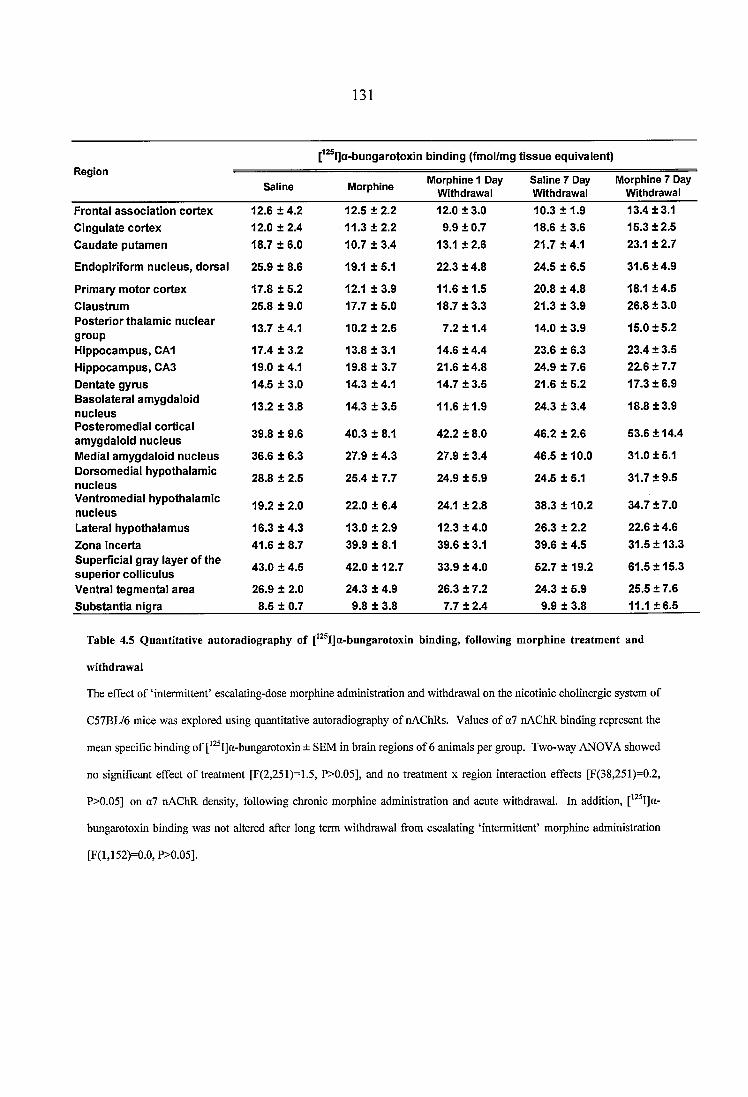
131
[^^® l]a-bungarotoxin b in d in g (fm ol/m g t i s s u e e q u iv a le n t)i\eg ion
Saline M orphine M orphine 1 Day W ithdrawal
Saline 7 Day W ithdrawal
M orphine 7 Day W ithdrawal
F ro n ta l a s s o c ia t io n c o r te x 12.6 ± 4 .2 12.5 ± 2.2 12.0 ± 3 .0 10.3 ± 1.9 13.4 ± 3 .1
C in g u la te c o r te x 12.0 ± 2 .4 11.3 ± 2 .2 9.9 ± 0.7 18.6 ± 3.6 15.3 ± 2 .5
C a u d a te p u ta m e n 18.7 ± 6 .0 10.7 ± 3 .4 13.1 ± 2 .6 21 .7 ± 4 .1 23.1 ± 2.7
E n d o p irifo rm n u c le u s , d o rsa l 25.9 ± 8 .6 19.1 ± 5.1 22 .3 ± 4.8 24 .5 ± 6.5 31.6 ± 4 .9
P rim ary m o to r c o r te x 17.8 ± 5 .2 12.1 ± 3.9 11.6 ± 1 .5 20 .8 ± 4 .8 18.1 ± 4 .5
C la u s tru m 25.8 ± 9 .0 17.7 ± 5.0 18.7 ± 3 .3 21 .3 ± 3 .9 26.8 ± 3 .0P o s te r io r th a la m ic n u c le a r g ro u p
13.7 ± 4 .1 10.2 ± 2 .5 7.2 ± 1 .4 14.0 ± 3.9 15.0 ± 5 .2
H ip p o c a m p u s , CA1 17.4 ± 3 .2 13.8 ± 3.1 14.6 ± 4 .4 23 .6 ± 6.3 23 .4 ± 3.5
H ip p o c a m p u s , CA3 19.0 ± 4 .1 19.8 ± 3.7 21.6 ± 4 .8 24 .9 ± 7.6 22 .6 ± 7 .7
D e n ta te g y ru s 14.5 ± 3 .0 14.3 ± 4.1 14.7 ± 3 .5 21 .6 ± 5 .2 17.3 ± 6 .9B a so la te ra l a m y g d a lo id n u c le u s
13.2 ± 3 .8 14.3 ± 3.5 11.6 ± 1 .9 24.3 ± 3.4 18.8 ± 3 .9
P o s te ro m e d ia l c o r tic a l a m y g d a lo id n u c le u s 39.8 ± 9 .6 4 0 .3 ± 8.1 42 .2 ± 8.0 46 .2 ± 2.6 53.6 ± 14.4
M edial a m y g d a lo id n u c le u s 38.6 ± 6 .3 27 .9 ± 4 .3 27.9 ± 3.4 46 .5 ± 10.0 3 1 .0 ± 5 .1D o rso m e d ia l h y p o th a la m ic n u c le u s
28.8 ± 2 .5 25.4 ± 7.7 24.9 ± 5.9 24 .5 ± 5.1 31 .7 ± 9 .5
V e n tro m e d ia l h y p o th a la m ic n u c le u s
19.2 ± 2 .0 22.0 ± 6.4 24.1 ± 2.8 38 .3 ± 10 .2 34 .7 ± 7 .0
L atera l h y p o th a la m u s 16.3 ± 4 .3 13.0 ± 2.9 12.3 ± 4 .0 26.3 ± 2.2 22.6 ± 4 .6
Z o n a in c e r ta 4 1 .6 ± 8 .7 39.9 ± 8.1 39.6 ± 3.1 39 .6 ± 4 .5 31 .5 ± 1 3 .3S u p e rf ic ia l g ra y la y e r o f th e s u p e r io r c o llic u lu s
43.0 ± 4 .5 42 .0 ± 12.7 33.9 ± 4 .0 52 .7 ± 19.2 61.5 ± 1 5 .3
V en tra l te g m e n ta l a r e a 26.9 ± 2 .0 24.3 ± 4.9 26.3 ± 7.2 24.3 ± 5.9 25 .5 ± 7 .6
S u b s ta n t ia n ig ra 8.5 ± 0 .7 9 .8 ± 3.8 7.7 ± 2.4 9.9 ± 3.8 11.1 ± 6 .5
Table 4.5 Q uantitative autoradiography o f [^^®I]a-bungarotoxin binding, following m orphine treatm ent and
withdrawal
The effect of ‘intermittent’ escalating-dose morphine administration and withdrawal on the nicotinic cholinergic system of
C57BL/6 mice was explored using quantitative autoradiography of nAChRs. Values of a7 nAChR binding represent the
mean specific binding of [*^^I]a-bungarotoxin ± SEM in brain regions of 6 animals per group. Two-way ANOVA showed
no significant effect of treatment [F(2,251)=1.5, P>0.05], and no treatment x region interaction effects [F(38,251)=0.2,
P>0.05] on o7 nAChR density, following chronic morphine administration and acute withdrawal. In addition, [’^ I]a-
bungarotoxin binding was not altered after long term withdrawal from escalating ‘intermittent’ morphine administration
[F(l,152)=0.0, P>0.05].

132
4.4 Discussion
Previously published reports suggest that nicotine and mophine influence each other’s
effects in various experimental settings (see Section 1.3.2). The aim of the present study
was to investigate whether the reported behavioural and neurochemical interactions
between the two substances could be depicted at the nicotinic receptor binding level,
following morphine treatment and withdrawal. The current results demonstrate no clear
effects of escalating morphine administration, and of acute and prolonged withdrawal
from ‘intermittent’ morphine treatment on [^^^Ijepibatidine and [^^^I]a-bungarotoxin
binding sites. These negative results suggest that interactions between morphine and the
nicotinic cholinergic sytem do not directly occur at the nAChR density level.
Among the nAChR subtypes that may mediate interactions between morphine and the
nicotinic cholinergic system, a3p4 receptors have been demonstrated to influence
morphine’s addictive properties at both the behavioural and neurochemical levels. Thus,
administration of the synthetic aSp4 antagonist 18-methoxycoronaridine into the medial
habenula (MHb) and the interpeduncular nucleus (IP) reduces morphine self
administration (Click et al., 1996), alleviates symptoms of morphine withdrawal (Panchal
et al., 2005), and blocks dopamine sensitization to repeated morphine treatment
(Taraschenko et al., 2007a), strongly implying that a3p4 nAChRs in these brain regions
are involved in morphine reinforcement and withdrawal. Cytisine-resistant
[^^^I]epibatidine sites constitute the predominant nicotinic receptor binding population in
the MHb and IP, and are believed to correspond to nAChRs with a pharmacology
suggestive of aSp4 receptors (Zoli et al., 1998; Whiteaker et al., 2000b; Marks et al..

133
2002). These nAChRs, however, are relatively resistant to agonist-induced upregulation
both in vivo (Nguyen et al., 2003) and in vitro (Wang et al., 1998). Therefore, the
inherent lack of sensitivity of aSp4 nAChRs to upregulation may account for the absence
of morphine-induced alterations in [^^^IJepibatidine binding sites in the present study.
Contrary to the effects of chronic cocaine treatment on nAChR density, no change in
[^^^I]epibatidine and [^^^I]a-bungarotoxin binding was observed following morphine
administration and withdrawal, suggesting that cocaine-induced nicotinic receptor
regulation does not generalize to opiates. Upregulation of binding to nAChRs is
observed after exposure to several nicotinic agonists, including the structural analogues
of acetylcholine (ACh), methylcarbamylcholine and N, N-dimethylcarbmylcholine
(Warpman et al., 1998; Whiteaker et al., 1998). Differences in the regulation of ACh
release between morphine and cocaine may therefore account for the effects of the two
drugs on nAChR density. Administration of cocaine has been consistently shown to
enhance acetylcholine release in various brain regions, including the hippocampus
(Imperato et al., 1992), cerebral cortex (Day et al., 1997), nucleus accumbens (Crespo et
al., 2006) and striatum (Zocchi and Pert, 1994). On the contrary, in vivo micodialysis of
opiate-induced ACh release reveals that the levels of neurotransmitter increase, decrease,
or remain unchanged in response to morphine, depending on its dose and dosing regimen,
as well as on the brain area under investigation (Rada et al., 1991; Imperato et al., 1996;
Rada et al., 1996; Taraschenko et al., 2007b). In the interpeduncular nucleus, the acute
administration of morphine at 5 mg/kg increases ACh release, whereas 20 mg/kg of
morphine exert the opposite effect, reducing extracellular ACh concentrations
(Taraschenko et al., 2007b). MOP receptor-mediated activation of cholinergic

134
intemeurons in the nucleus accumbens (NAc) has been shown to suppress cholinergic
activity (Britt and McGehee, 2008), diminishing ACh release following escalating doses
of moprhine for a period of 8 days (Rada et ah, 1996). Spontaneously or electrically
evoked ACh release after high doses of morphine is also reduced in slices from the
mammalian striatum (Arenas et ah, 1990), hippocampus (Jackisch et ah, 1986), thalamus
(Beani et ah, 1982) and cerebral cortex (Lapchak et ah, 1989). Hence, a possible
explanation for the distinct effects of cocaine and morphine on nAChR density lies in the
fact that the two substances differentially regulate cholinergic neuron activity. It is worth
mentioning that nicotine-induced changes in MOP receptor density have similarly been
shown to depend on the paradigm of drug administration used. Thus, chronic nicotine
increases (Wewers et ah, 1999; Walters et ah, 2005), decreases (Galeote et ah, 2006;
Marco et ah, 2007), or induces no change (Vihavainen et ah, 2008a) in MOP receptor
binding, depending on the dose and length of its administration.
Several reports have demonstrated that naloxone-precipitated withdrawal from chronic
morphine increases brain ACh levels in various regions, including the nucleus accumbens
and areas of the cortex (Bhargava and Way, 1975; Antonelli et ah, 1986; Rada et ah,
1996). In the present study, however, neither the acute, nor the prolonged withdrawal
from ‘intermittent’ morphine treatment altered [^^^IJepibatidine and [^^^I]a-bungarotoxin
binding sites in the brains of morphine withdrawn animals. To the extent that enhanced
concentrations of extracellular ACh would lead to increased nAChR density, the current
data suggest that ACh release during morphine abstinence depends on whether
withdrawal occurs spontaneously or is induced by opioid-antagonists. Indeed, contrary to
the acute increases in neurotransmitter release following naloxone-precipitated

135
withdrawal, basal levels of extracellular ACh in the rat nucleus accumbens have been
shown to increase fifteen days after the termination of escalating morphine treatment,
normalizing on day thirtyfive of abstinence (Fiserova et al., 1999). Although
concentrations of ACh were not measured in the current study, it could be that changes in
nAChR density during morphine abstinence may take longer to develop, reflecting the
long term adaptive changes that characterize cholinergic responses to spontaneous
morphine withdrawal.
The lack of morphine-induced effects on nAChR density does not preclude that
alterations in nicotinic receptor function occur following morphine treatment and
withdrawal. Using the acetylcholine binding protein as a model o f nicotinic receptor
binding domains, for instance. X-ray crystallography demonstrates that morphine binds to
non-a nicotinic subunit interfaces with pM affinity, at sites that are similar to those of
modulating benzodiazepines on GABAa receptors (Hansen and Taylor, 2007; Taly et al.,
2009). In addition, in cell lines that express a3p4 nAChRs, morphine has been shown to
inhibit nicotine-induced ^^Rb efflux by directly binding to the central lumen of the ion
channel, indicating that the opiate is a non-competitive antagonist at this nicotinic
receptor subtype (Jozwiak et al., 2006). Together, these data suggest that morphine has
the potential to interact and alter heteromeric nAChR function by acting both as as
channel blocker, and as a negative allosteric modulator. Conceivably, if a morphine-
induced reduction in the function of nAChRs has occurred in the present study, it would
not be detectable by quantitative autoradiography.

136
Several alternative explanations can be offered for the absence of changes in nAChR
binding in the current study. First, the effects of morphine treatment and withdrawal on
nAChR binding may only have occurred in certain neuronal populations. Morphine’s
effects on dopaminergic regulation are exerted via MOP receptor hyperpolarization of
GABAergic neurons (Johnson and North, 1992), and the alteration in the GABAergic
neuronal input to mesolimbic dopaminergic neurons has been suggested to mediate the
interactions between morphine and nicotine (Vihavainen et al., 2008b). Therefore,
morphine administration may have selectively altered nAChR density on GABAergic
cells. Second, morphine appears to selectively alter distribution of dentritic, and not
somatic MOP receptors (Haberstock-Debic et al., 2003). By way of analogy, similar
changes in nAChRs may have occurred only across certain neuronal subcompartments.
Third, signaling cascades downstream of nicotinic receptors, rather than changes in
nAChR abundance and function may mediate the nicotinic modulation of morphine-
induced effects. Evidence suggests that adaptations in the cAMP signaling transduction
pathway critically mediate the rewarding properties of many drugs of abuse. In particular,
chronic morphine increases levels of the cAMP response element-binding protein
(CREB), whereas morphine withdrawal symptoms are reduced in CREB“ mutant mice
(Widnell et al., 1994; Maldonado et al., 1996). Similarly, nicotine-induced conditioned
place preference requires CREB phosphorylation, and so does the expression of the
nicotinic withdrawal symptom (Brunzell et al., 2003; Brunzell et al., 2009). Importantly,
a link between nicotine, MOP receptor activation, and CREB phosphorylation has been
established (Walters et al., 2005). These findings support the concept that indirect
actions at the anatomical and/or molecular levels are involved in the interactions between
morphine and nicotine.

137
As nicotine exerts its effects by binding in the CNS to neuronal nAChRs, an obvious
hypothesis for the mechanism of interactions between nicotine and morphine is that
administration of the opioid alters DAergic neurotransmission via nicotinic cholinergic
receptors. To test this, a protocol that mimics the pattern of opiate abuse in human
addicts was employed, and escalating ‘intermittent’ morphine was administered at doses
that have been shown to produce sensitisation and dependence (reviewed in Spanagel,
1995). Notwithstanding that morphine-induced changes in ACh release depend on the
dosing regimen employed, and that changes in receptor abundance do not always
translate to nAChR function, the present report suggests that alterations in nAChR
density do not contribute to morphine-nicotine interactions.

138
CHAPTER
FIVE

139
5 CHANGES IN THE NICOTINIC CHOLINERGIC AND DOPAMINERGIC
SYSTEMS OF MICE DEFICIENT IN THE Aja GENE, EXPLORED BY
QUANTITATIVE AUTORADIOGRAPHY
5.1 Introduction
The purine neuromodulator adenosine exerts its effects through four classes of G-protein
coupled receptors, namely A%, A2A, Ais, and A3 adenosine receptors (see Section 1.3.3.1).
Among them, A2A receptors have attracted much interest as potential modulators of the
effects of abused substances, because of their restricted localization in brain pathways
that are involved in drug addiction, and due to the altered behavioural phenotype that A2A
receptor knockout (KO) mice exhibit in response to drugs of abuse (reviewed in Ferre et
al., 2007). Indeed, adenosine A2A receptors are abundantly expressed in structures of the
mesolimbic dopaminergic system, where they have been shown to mediate the properties
of several addictive substances, including opioids (Bailey et al., 2004b), cannabinoids
(Soria et al., 2004), psychostimulants (Soria et al., 2006), and nicotine (Castane et al.,
2006). Thus, A2A receptor KO mice exhibit enhanced naloxone-precipitated morphine
withdrawal (Bailey et al., 2004b), diminished delta9-tetrahydrocannabinol (THC)-
induced place conditioning (Soria et al., 2004), and reduced rate of cocaine self-
administraiton (Soria et al., 2006). The participation of adenosine A2A receptors in
nicotine addiction, in particular, is revealed by the fact that caffeine, a non selective
antagonist at Ai and A2A receptors, is commonly consumed in combination with nicotine
by tobacco smokers. Evidence suggests that caffeine intake leads to increased cigarette
smoking (Brown and Benowitz, 1989), and that smokers tend to consume more caffeine
compared to the general population (reviewed in Swanson et al., 1994). Moreover,
preclinical studies show that adding caffeine into the drinking water of rats potentiates

140
nicotine self-administration, whereas its exclusion brings nicotine intake back to baseline
levels (Shoaib et ah, 1999). A specific role for adenosine A2A receptors to enhance the
reinforcing properties of nicotine has been futher revealed in mice with a genetic ablation
of the A2A receptor gene. Using the conditioned place preference (CPP) paradigm, it has
been shown that nicotine-induced CPP is attenuated in A2A receptor KO animals, which
do not exhibit an increase in their extracellular levels of accumbal dopamine following
the acute administration of nicotine (Castane et al., 2006). Hence, a growing body of
evidence demonstrates the occurence of interactions between adenosine A2A receptors
and the nicotinic cholinergic system, and suggests that the adenosinergic modulation of
nicotine-induced effects takes place within the mesolimbic DAergic system.
The generation of the A2A KO mouse provides a powerful genetic tool to analyse the
involvement of A2A receptors in nicotine addicition in vivo. It may be a prima facie case,
however, that the reported nicotine-induced phenotypes are precipitated in animals with
permanent and global inactivation of the A2A gene, as a result of compensatory
developmental changes in the nicotinic cholinergic and dopaminergic systems. Therefore,
to investigate whether alterations in nicotinic and dopaminergic receptor binding occur in
the absence of A2A receptors, quantitative autoradiographic analysis of heteromeric and
homomeric nAChRs, and of dopaminergic D1 and D2 receptors and transporters, has
been performed in brain sections from drug naïve, wild type (WT) and homozygous (-/-)
A2A receptor KO GDI mice. In addition, to further investigate the involvement of A2A
receptors in modulating nicotine-induced neuroadaptation, the regulation of the nicotinic
and dopaminergic systems has been assessed in WT and A2A receptor KO mice,
following chronic nicotine treatment.

141
5.2 Materials and methods
5.2.1 Generation o f the A 2A KO mouse and experimental conditions
CDl adenosine Aia receptor KO mice, originally generated by Ledent et al., (1997), were
obtained from the breeding colony of the University of Surrey, as previously described
(Prentice et al., 2002). In brief, mutant mice were bred from heterozygote breeding pairs,
which originated from the individual crossing of a number of heterozygote males from
the existing colony, each with a wildtype female of the CDl strain, which was obtained
from an external supplier (Charles River, Margate, Kent). The male and female
heterozygotes from the litters of the newly created breeding pairs were paired and these
formed the main breeding colonies, which were maintained for approximately a year
before new breeding pairs were created. All mice were genotyped at weaning using a
polymerase chain reaction (PCR) based method. Tail tip samples were collected at three
weeks of age, from which DNA was extracted using the DNeasy tissue kit, according to
the manufacturer’s protocol (Qiagen, Hilden, Germany).
8-12 week old male CDl mice (n=5-7) were used for in vivo and autoradiography
experiments. The animals were single-housed in a temperature controlled environment,
under a 12 h light/dark cycle. Food and water were available ad libitum. All studies
were conducted in accordance with protocols approved by the Home Office, UK.

142
5.2.2 Minipump preparation and implantation
Prior to surgical implantation, osmotic minipumps (ALZET®2002 model, Charles River,
UK) were filled with either nicotine hydrogen salt in 0.9% sterile saline, or vehicle (0.9%
sterile saline). Nicotine hydrogen salt (Sigma-Aldrich, UK) was delivered to CDl mice
at a dose of 7.8 mg/kg/day (free-base weight), at a rate of 0.5pl/hour, for 14 days. The
procedure followed for the implantation of the miniosmotic pumps has been described in
Section 3.2.1.2.
Fourteen days after the minipumps had been implanted, mice were rapidly killed by
cervical dislocation and the intact brains immediately removed and rapidly frozen in
isopentane at -20 °C. Brains were stored at -80 °C for a maximum period of 1 month
before sectioning.
5.2.3 Quantitative receptor autoradiography
Quantitative autoradiography was performed in order to measure density of a4|32* and a7
nAChRs, dopaminergic D1 and D2 receptors, and of the dopamine transporter (DAT), in
drug naïve and nicotine-treated WT and adenosine Aia receptor KO mice. In addition,
binding to adenosine A2A receptors was performed in nicotine-treated WT and A2A KO
animals, in order to confirm genotype. For receptor binding experiments, sections were
thawed and processed according to established protocols (Javitch et al., 1984; Orr-
Urtreger et al., 1997; Whiteaker et al., 2000b; Marks et al., 2002; Bailey et al., 2004a;
Lena et al., 2004), with minor modifications. Sections for analysis were derived from
five to seven brains from each group (n=5-7). Multiple, adjacent sections were processed
together in a paired binding protocol. Radioligand bound sections were apposed to
Kodak BioMax MR-1 film (Sigma-Aldrich, UK), along with appropriate microscale

143
standards to allow quantification. All structures were identified by reference to the
mouse brain atlas of Franklin and Paxinos, (2001), and analysed using an MCID image
analyser (Image research, Linton, UK).
To confirm the genotype of nicotine-treated animals, sections for A2A receptor binding
were pre-incubated for 30 minutes in 170 mM Tris-HCl, containing ImM EDTA (pH 7.4,
room temperature). For the determination of total receptor binding, sections were
incubated at room temperature with 10 nM of [^H]CGS21680 (PerkinElmer; specific
activity 30 Ci/mmol) for 2 hours, in incubation buffer containing 170 mM Tris-HCl, 10
nM MgCh, and 2 U/ml adenosine deaminase (pH 7.4 at 25 °C). Incubations were
terminated by 3 x 5 minute washes into ice cold 50 mM Tris buffer (ph 7.4), and a rapid
rinse in ice cold water. Radioligand bound sections were apposed to film for a period of
3 weeks.
For nAChRs, radioligand bound sections were apposed to film for 24h. Structures of
exceptionally high nAChR density, including the medial habenula, fasciculus retrofiexus,
and the interpeduncular nucleus were apposed for a period of 6h, to avoid film saturation.
Detailed experimental protocols for autoradiographic procedures can be found in Section
3.2.6.

144
5.2.4 Determination o f nicotine and cotinine levels
Chronic nicotine-treated WT and adenosine A2A receptor KO mice were killed by
cervical dislocation at the end of the experiment and core blood from each animal was
collected in ice cold tubes. Plasma was obtained by centrifugation for 20 min. at 2500
rpm in a refrigerated centrifuge, and transported in dry ice to ABS Laboratories Ltd,
Welwyn Garden city, UK, for analysis of nicotine and cotinine levels by gas liquid
chromatography (Feyerabend and Russell, 1990).
5.2.5 Statistical analysis
Two-way ANOVA for the factors genotype and region was used for the comparison of
quantitative measures of a4p2* and a7 nicotinic receptors, dopaminergic D1 and D2
receptors, and of dopamine transporters in brain regions of drug naïve CDl mice. For
nAChR binding, structures of the habenulo-interpeduncular pathway were quantified
using a 6 h exposure to film, because of the high densities of heteromeric nAChRs in this
cholinergic system. Therefore, comparisons between structures of high nicotinic receptor
density were performed using a separate two away ANOVA (independent factors
genotype and region). Three-way ANOVA for the factors treatment, genotype, and
region, was used to compare differences in receptor binding between nicotine-treated WT
and adenosine A2A receptor KO animals.
Where ANOVA yielded significant treatment effects, and depending on the n number of
the treatment groups, Duncan’s multiple ranges or Fisher’s LSD post hoc analysis was
used to investigate differences in radioligand binding between groups in individual
regions.

145
5.3 Results
5.3.1 Naïve study
5.3.1.1 Nicotinic receptor autoradiography in naïve WT and adenosine A2A receptor KO
mice
To investigate whether deletion of the genes encoding for the adenosine A2A receptor
leads to any alterations in nicotinic receptor density, quantitative autoradiography of
a4p2* and a7 nAChRs was carried out in brain sections from drug naïve wild type (WT)
and A2A receptor knockout (KO) CDl mice. The pattern of distribution of all nicotinic
receptor subtypes studied in homozygous adenosine A2A receptor KO mice was identical
to wild type. For heteromeric nicotinic receptors, no change in total, cytisine-sensitive,
and cytisine-resistant [^^^I]epibatidine binding was observed between naïve WT and A2A
receptor KO mice, in any of the brain regions examined (Fig. 5.1 & 5.2 & 5.3). The
majority of CDl mouse brain [^^^Ijepibatidine binding sites were of the cytisine-sensitive,
a4p2* nAChR subtype. High levels of cytisine-resistant [^^^Ijepibatidine binding were
observed in the medial habenula, fasciculus retrofiexus, and interpeduncular nucleus.
Non specific binding (NSB) was indistinguishable from film background.
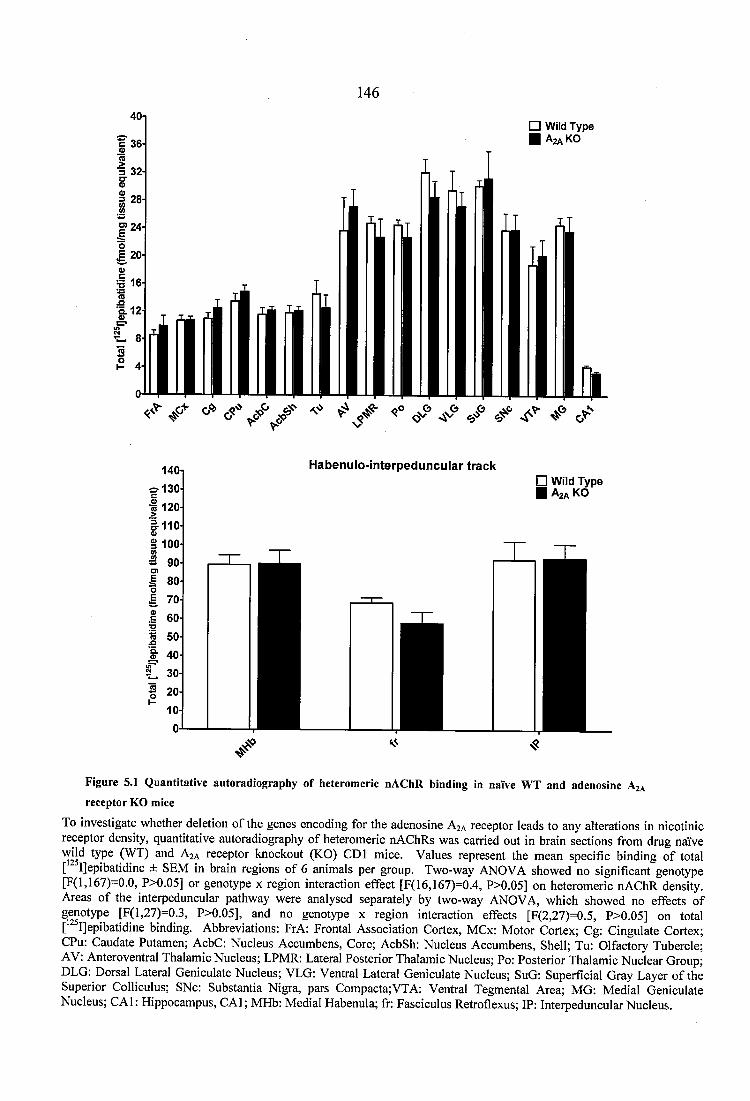
14640-1
□ Wild Type I ^ 2A KOc 36-
3 32-
3 28-
o>24-
g 20-
^ ^ ^
140 Habenulo-interpeduncular track
■5 1 20
.5 60
ra 50
g 40
□ W ild T ype ■ A2aK0
X
Figure 5.1 Q uantitative autoradiography o f heterom erie nA ChR binding in naïve W T and adenosine A ja
receptor KO mice
To investigate whether deletion o f the genes encoding for the adenosine AgA receptor leads to any alterations in nicotinic receptor density, quantitative autoradiography o f heteromeric nAChRs was carried out in brain sections from drug naïve wild type (WT) and AzA receptor knockout (KO) CDl mice. Values represent the mean specific binding o f total [ IJepibatidine ± SEM in brain regions o f 6 animals per group. Two-way ANOVA showed no significant genotype [F(l,167)=0.0, P>0.05] or genotype x region interaction effect [F(16,167)=0.4, P>0.05] on heteromeric nAChR density. Areas o f the interpeduncular pathway were analysed separately by two-way ANOVA, which showed no effects o f genotype [F(l,27)=0.3, P>0.05], and no genotype x region interaction effects [F(2,27)=0.5, P>0.05] on total [ IJepibatidine binding. Abbreviations: FrA: Frontal Association Cortex, MCx: Motor Cortex; Cg: Cingulate Cortex; CPu; Caudate Putamen; AcbC: Nucleus Accumbens, Core; AcbSh: Nucleus Accumbens, Shell; Tu: Olfactory Tubercle; AV: Anteroventral Thalamic Nucleus; LPMR: Lateral Posterior Thalamic Nucleus; Po: Posterior Thalamic Nuclear Group; DLG: Dorsal Lateral Geniculate Nucleus; VLG: Ventral Lateral Geniculate Nucleus; SuG: Superficial Gray Layer o f the Superior Colliculus; SNc: Substantia Nigra, pars Compacta;VTA: Ventral Tegmental Area; MG: Medial Geniculate Nucleus; CAl: Hippocampus, C Al; MHb: Medial Habenula; ff: Fasciculus Retrofiexus; IP: Interpeduncular Nucleus.
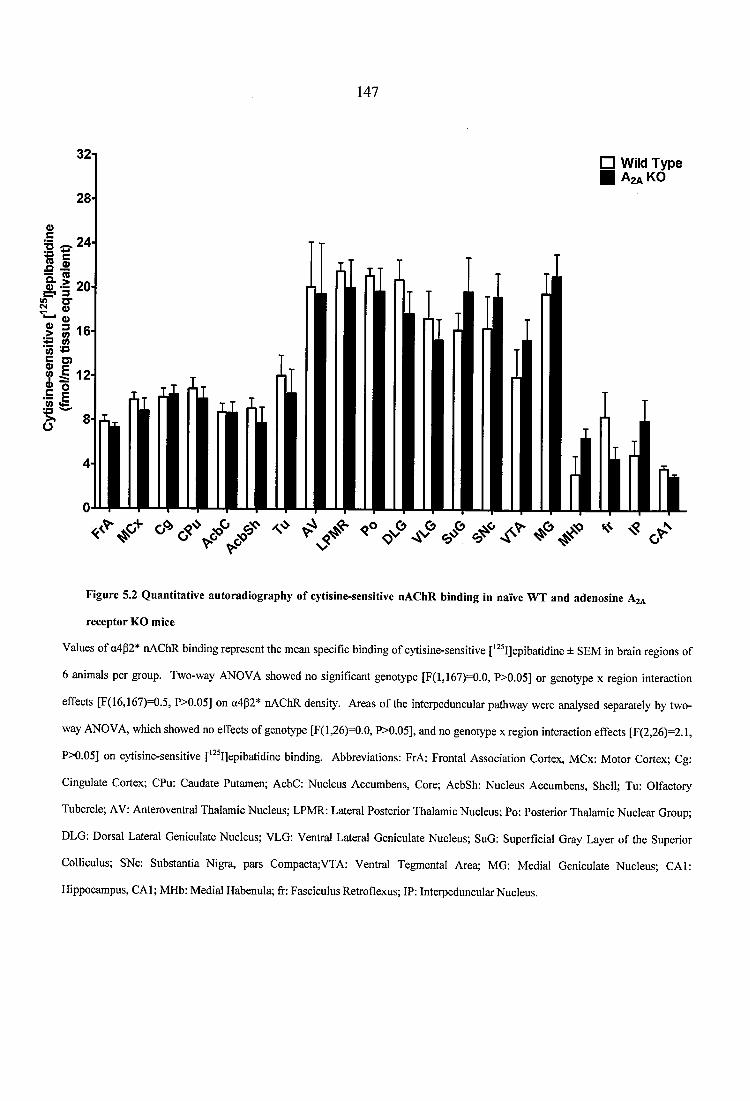
147
32n□ Wild T ype ■ A2aK 0
28-
24-
A 3 20-
Figure 5.2 Q uantitative autoradiography o f cytisine-sensitive nA ChR binding in naïve W T and adenosine A ja
receptor KO mice
Values o f a4p2* nAChR binding represent the mean specific binding o f cytisine-sensitive ['^^I]epibatidine ± SEM in brain regions o f
6 animals per group. Two-way ANOVA showed no significant genotype [F(l,167)=0.0, P>0.05] or genotype x region interaction
effects [F(16,167)=0.5, P>0.05] on o4p2* nAChR density. Areas o f the interpeduncular pathway were analysed separately by two-
way ANOVA, which showed no effects o f genotype [F(l,26)=0.0, P>0.05], and no genotype x region interaction effects [F(2,26)=2.1,
P>0.05] on cytisine-sensitive [^^^I]epibatidine binding. Abbreviations: FrA: Frontal Association Cortex, MCx: Motor Cortex; Cg:
Cingulate Cortex; CPu: Caudate Putamen; AcbC: Nucleus Accumbens, Core; AcbSh: Nucleus Accumbens, Shell; Tu: Olfactory
Tubercle; AV: Anteroventral Thalamic Nucleus; LPMR: Lateral Posterior Thalamic Nucleus; Po: Posterior Thalamic Nuclear Group;
DLG: Dorsal Lateral Geniculate Nucleus; VLG: Ventral Lateral Geniculate Nucleus; SuG: Superficial Gray Layer o f the Superior
Colliculus; SNc: Substantia Nigra, pars Compacta;VTA: Ventral Tegmental Area; MG: Medial Geniculate Nucleus; C A l:
Hippocampus, CA l; MHb: Medial Habenula; ff: Fasciculus Retrofiexus; IP: Interpeduncular Nucleus.
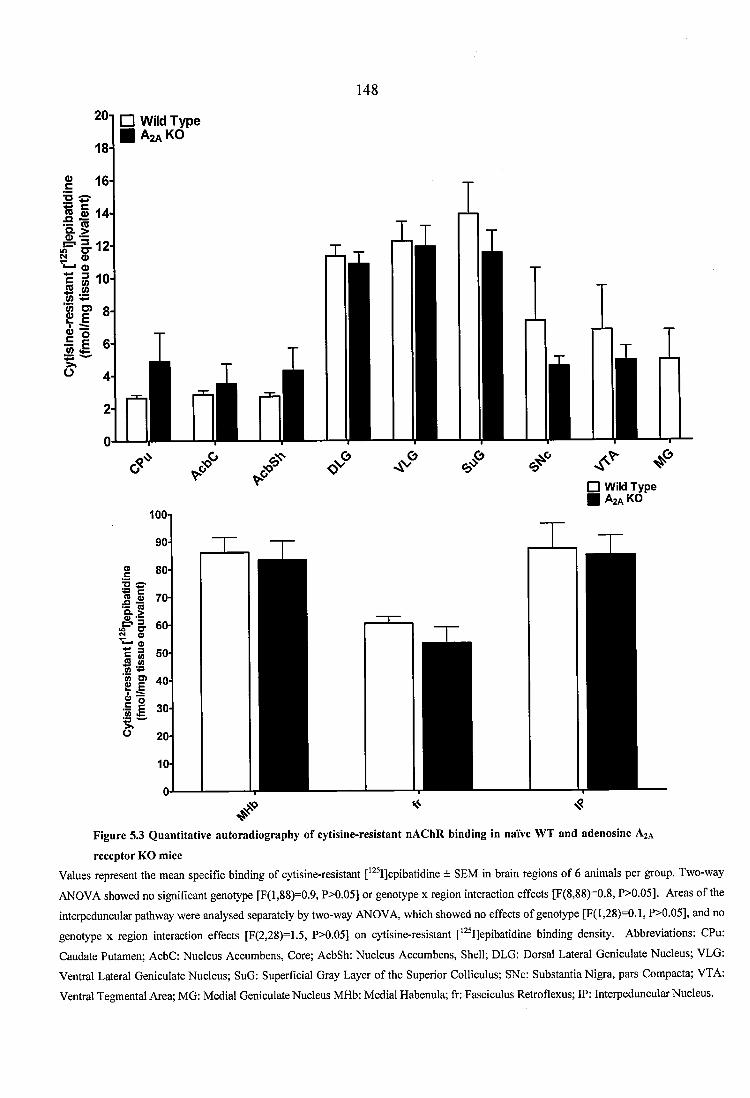
14820-1
18-
□ Wild T ype ■ A z a K O
® 164
i?« S 14
t hÏ I ”
I I
I IÜ
8-
6-
4-
lOOi
□ W ild T y p e ■ A2aK0
(A O )
Figure 5.3 Q uantitative autoradiography o f cytisine-resistant nA ChR binding in naïve W T and adenosine A%A
receptor KG mice
Values represent the mean spécifié binding o f cytisine-resistant [^^^I]epibatidine ± SEM in brain regions o f 6 animals per group. Two-way
ANOVA showed no significant genotype [F(l,88)=0.9, P>0.05] or genotype x region interaction effects [F(8,88)=0.8, P>0.05]. Areas o f the
interpeduncular pathway were analysed separately by two-way ANOVA, which showed no effects o f genotype [F(l,28)=0.1, P>0.05], and no
genotype x region interaction effects [F(2,28)=1.5, P>0.05] on cytisine-resistant [’^^I]epibatidine binding density. Abbreviations; CPu
Caudate Putamen; AcbC: Nucleus Accumbens, Core; AcbSh: Nucleus Accumbens, Shell; DLG: Dorsal Lateral Geniculate Nucleus; VLG
Ventral Lateral Geniculate Nucleus; SuG: Superficial Gray Layer o f the Superior Colliculus; SNc: Substantia Nigra, pars Compacta; VTA
Ventral Tegmental Area; MG: Medial Geniculate Nucleus MHb: Medial Habenula; ff: Fasciculus Retrofiexus; IP: Interpeduncular Nucleus.
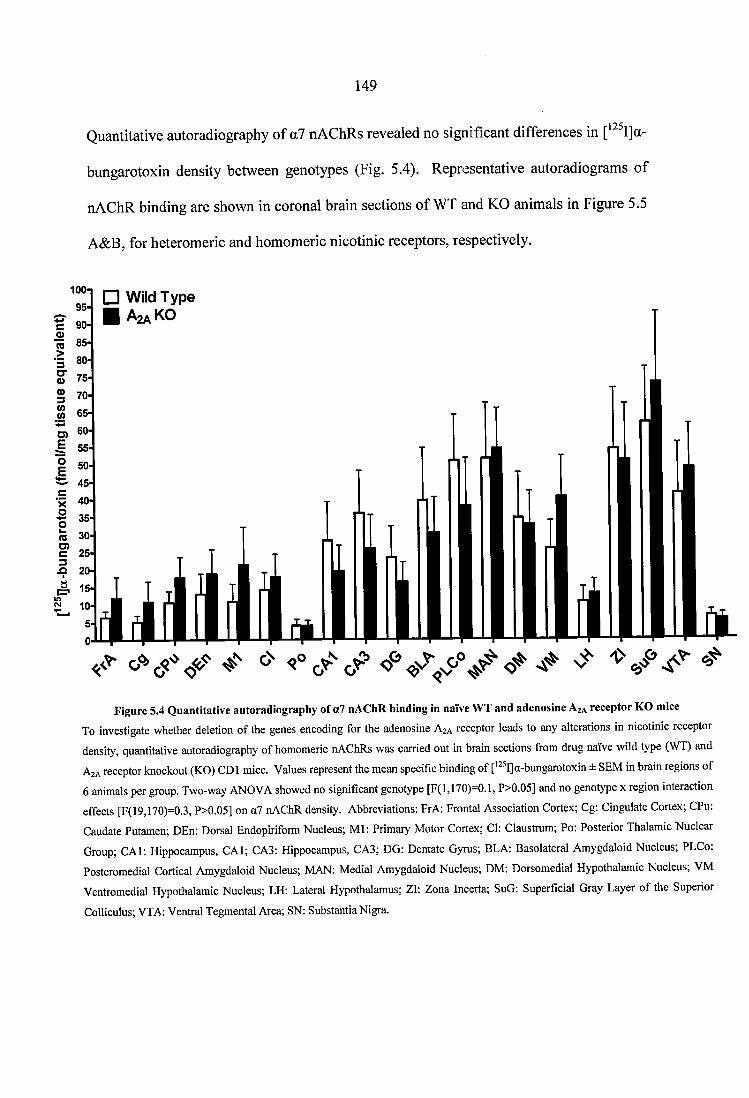
149
Quantitative autoradiography of a? nAChRs revealed no significant differences in
bungarotoxin density between genotypes (Fig. 5.4). Representative autoradiograms of
nAChR binding are shown in coronal brain sections of WT and KO animals in Figure 5.5
A&B, for heteromeric and homomeric nicotinic receptors, respectively.
Wild Type A2A K O
W 65
<0 30-
Figure 5.4 Quantitative autoradiography of a7 uAChR binding in naïve WT and adenosine Aja receptor KG mice
To investigate whether deletion of the genes encoding for the adenosine A2A receptor leads to any alterations in nicotinic receptor
density, quantitative autoradiography of homomeric nAChRs was carried out in brain sections from drug naïve wild type (WT) and
A2A receptor knockout (KG) CDl mice. Values represent the mean specific binding of ['^^I]a-bungarotoxin ± SEM in brain regions of
6 animals per group. Two-way ANOVA showed no significant genotype [F(l,170)=0.1, P>0.05] and no genotype x region interaction
effects [F(19,170)=0.3, P>0.05] on a7 nAChR density. Abbreviations; FrA: Frontal Association Cortex; Cg: Cingulate Cortex; CPu:
Caudate Putamen; DEn: Dorsal Endopiriform Nucleus; Ml: Primary Motor Cortex; Cl: Claustrum; Po: Posterior Thalamic Nuclear
Group; CAl: Hippocampus, CAl; CA3: Hippocampus, CA3; DG: Dentate Gyrus; BLA: Basolateral Amygdaloid Nucleus; PLCo:
Posteromedial Cortical Amygdaloid Nucleus; MAN: Medial Amygdaloid Nucleus; DM: Dorsomedial Hypothalamic Nucleus; VM
Ventromedial Hypothalamic Nucleus; LH: Lateral Hypothalamus; Zl: Zona Incerta; SuG: Superficial Gray Layer of the Superior
Colliculus; VTA: Ventral Tegmental Area; SN: Substantia Nigra.
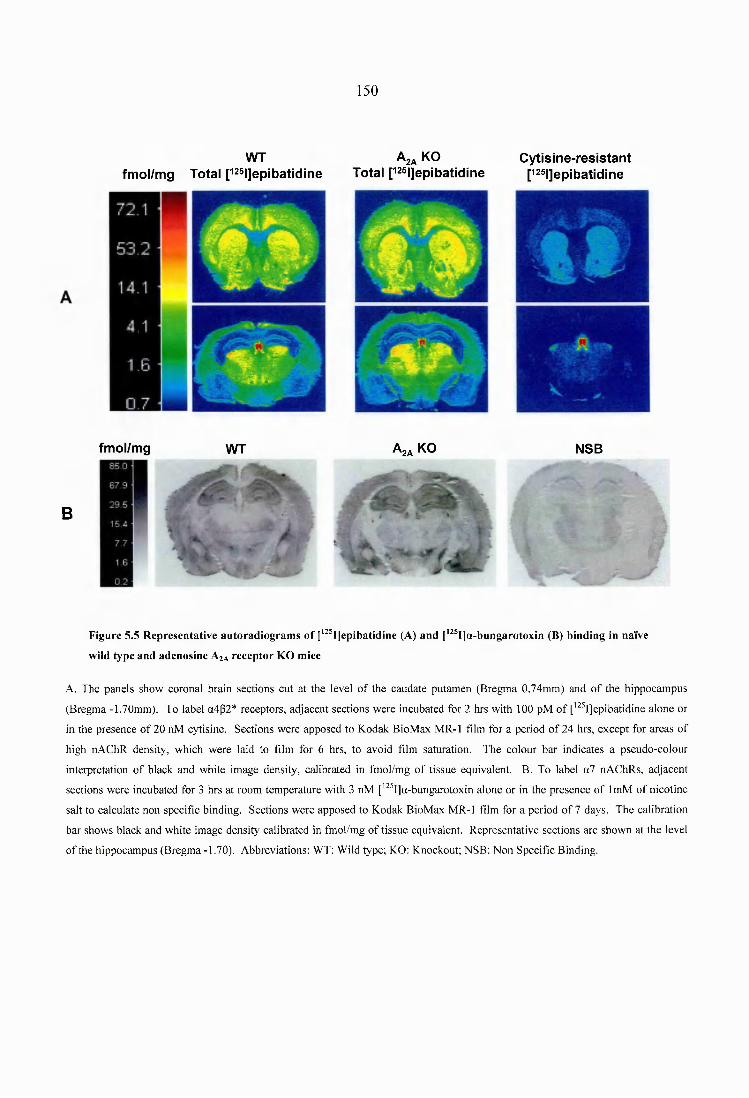
150
WT A2a KOfm o l/m g T o ta l r^ ^ l]ep ib a tid in e T o ta l [^^sijepibatidine
C y tis in e - re s is ta n tp25|]epibatidine
fm ol/m g
B
WT AgA KO NSB
Figure 5.5 Representative autoradiograms of [‘^^IJepibatidine (A) and [‘^®I]a-bungarotoxin (B) binding in naïve
wild type and adenosine A%A receptor KO mice
A. The panels show coronal brain sections cut at the level of the caudate putamen (Bregma 0.74mm) and o f the hippocampus
(Bregma -1.70mm). To label a4p2* receptors, adjacent sections were incubated for 2 hrs with 100 pM o f ['^^IJepibatidine alone or
in the presence of 20 nM cytisine. Sections were apposed to Kodak BioMax M R-1 film for a period o f 24 hrs, except for areas of
high nAChR density, which were laid to film for 6 hrs, to avoid film saturation. The colour bar indicates a pseudo-colour
interpretation o f black and white image density, calibrated in fmol/mg o f tissue equivalent. B. To label a7 nAChRs, adjacent
sections were incubated for 3 hrs at room temperature with 3 nM ['^^IJa-bungarotoxin alone or in the presence of ImM o f nicotine
salt to calculate non specific binding. Sections were apposed to Kodak BioMax MR-1 film for a period of 7 days. The calibration
bar shows black and white image density calibrated in fmol/mg of tissue equivalent. Representative sections are shown at the level
o f the hippocampus (Bregm a-1.70). Abbreviations: WT: Wild type; KO: Knockout; NSB: Non Specific Binding.
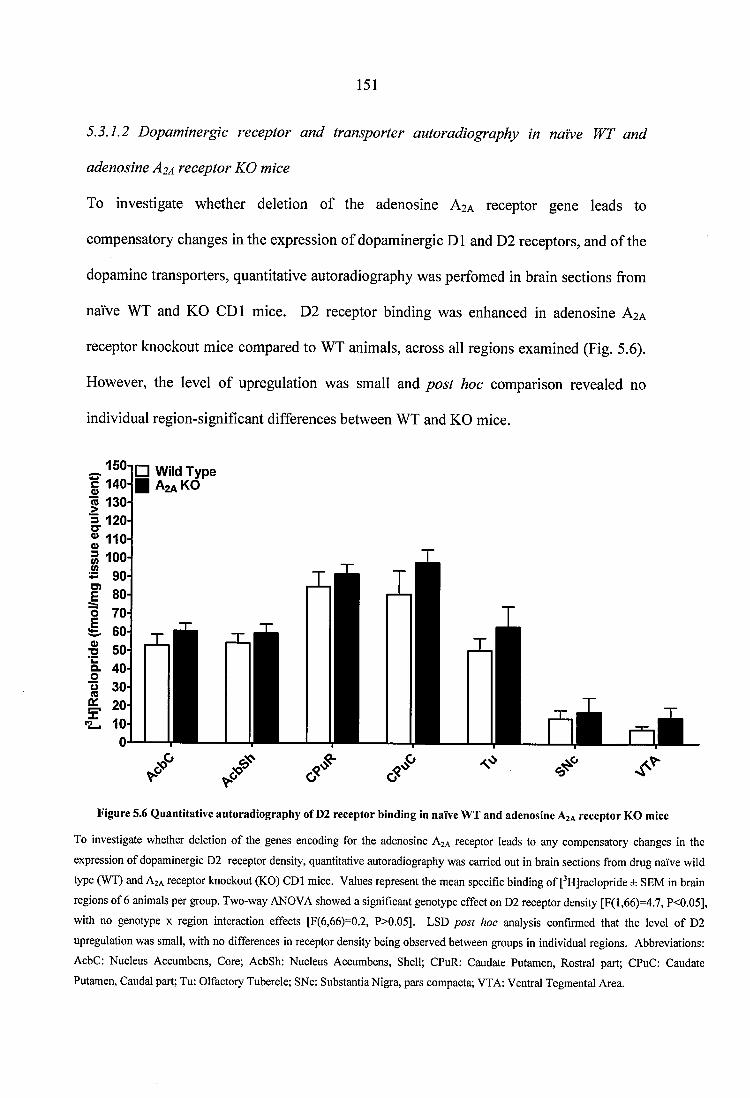
151
5.3.1.2 Dopaminergic receptor and transporter autoradiography in naïve WT and
adenosine A2A receptor KO mice
To investigate whether deletion of the adenosine A2A receptor gene leads to
compensatory changes in the expression of dopaminergic D1 and D2 receptors, and of the
dopamine transporters, quantitative autoradiography was perfomed in brain sections from
naïve WT and KO CDl mice. D2 receptor binding was enhanced in adenosine A2A
receptor knockout mice compared to WT animals, across all regions examined (Fig. 5.6).
However, the level of upregulation was small and post hoc comparison revealed no
individual region-significant differences between WT and KG mice.
□ Wild Type H A2aK0
« 130
*s 90
2010
Figure 5.6 Q uantitative autoradiography o f D2 receptor binding in naïve W T and adenosine A 2A receptor K O m ice
To investigate whether deletion of the genes encoding for the adenosine A2A receptor leads to any compensatory changes in the
expression of dopaminergic D2 receptor density, quantitative autoradiography was carried out in brain sections from drug naïve wild
type (WT) and A2A receptor knockout (KO) CDl mice. Values represent the mean specific binding of fHJraclopride ± SEM in brain
regions of 6 animals per group. Two-way ANOVA showed a significant genotype effect on D2 receptor density [F(l,66)=4.7, P<0.05],
with no genotype x region interaction effects [F(6,66)=0.2, P>0.05]. LSD post hoc analysis confirmed that the level of D2
upregulation was small, with no differences in receptor density being observed between groups in individual regions. Abbreviations;
AcbC: Nucleus Accumbens, Core; AcbSh: Nucleus Accumbens, Shell; CPuR: Caudate Putamen, Rostral part; CPuC: Caudate
Putamen, Caudal part; Tu: Olfactoiy Tubercle; SNe: Substantia Nigra, pars compacta; VTA: Ventral Tegmental Area.
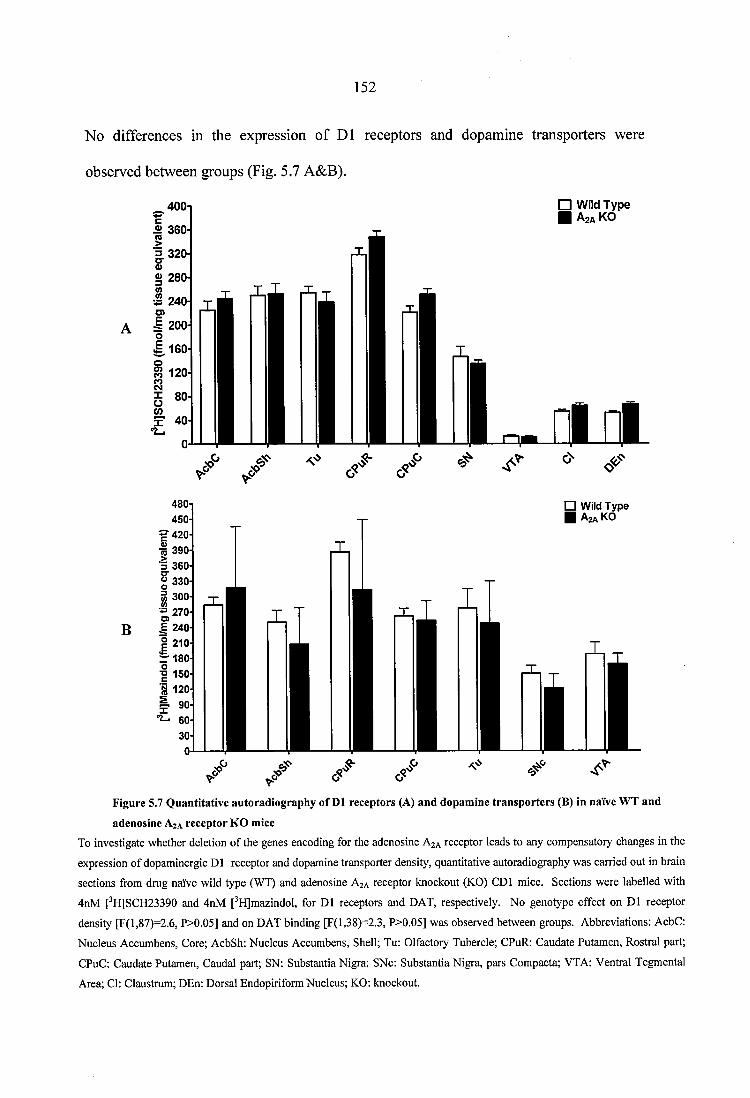
152
No differences in the expression of D1 receptors and dopamine transporters were
observed between groups (Fig. 5.7 A&B).
B
400i □ W ild T y p e ■ A2aK0
i l l
□ Wild Type A2A KO
-R 390-■5 360? 330-
» 270- E 240I 210-r i 8 0 -o■o 150
Figure 5.7 Quantitative autoradiography of D1 receptors (A) and dopamine transporters (B) in naïve WT and
adenosine Aja receptor KO miceTo investigate whether deletion of the genes encoding for the adenosine A2A receptor leads to any compensatory changes in the
expression of dopaminergic D1 receptor and dopamine transporter density, quantitative autoradiography was carried out in brain
sections from drug naïve wild type (WT) and adenosine A2A receptor knockout (KG) CDl mice. Sections were labelled with
4nM [^H]SCH23390 and 4nM [^HJmazindol, for D1 receptors and DAT, respectively. No genotype effect on D1 receptor
density [F(l,87)=2.6, P>0.05] and on DAT binding [F(l,38)=2.3, P>0.05] was observed between groups. Abbreviations: AebC:
Nucleus Accumbens, Core; AcbSh: Nucleus Accumbens, Shell; Tu: Olfactory Tubercle; CPuR: Caudate Putamen, Rostral part;
CPuC: Caudate Putamen, Caudal part; SN: Substantia Nigra; SNe: Substantia Nigra, pars Compacta; VTA: Ventral Tegmental
Area; Cl: Claustrum; DEn: Dorsal Endopiriform Nucleus; KG: knockout.

153
5.3.2 Nicotine treatment
5.3.2.1 Confirmation o f genotype
Binding of [^H]CGS21680 to adenosine Asa receptors showed a complete absence of A
binding sites in the brains of homozygous mice, confirming the genotype of the animals
used in this study (Fig. 5.8).
W T A2a KO
2A
miceFigure 5.8 Adenosine Az* receptor expression in naïve CDl wildtype and adenosine A^ receptor KO
To confirm the genotype o f the animals used in the present experiments, sections were labeled with lOnM
[ HJCGS21680 for a period of 2 hrs. From top to bottom, the horizontal panels show Ag* receptor binding at the level
o f the caudate putamen (bregma 0.98mm) and of the ventral tegmental area (bregma -3.40mm).
5.3.2.2 Levels o f plasma nicotine and cotinine following chronic nicotine administration
Plasma nicotine and cotinine levels were determined after 14 days of treatment with
nicotine at a dose of 7.8 mg/kg/day, in WT and adenosine A2A receptor KO mice. In
nicotine-treated groups, the mean plasma levels of nicotine (34.9±3.7 ng/ml) and cotinine
(72.0±5.7 ng/ml) were similar between genotypes (Fig. 5.9).
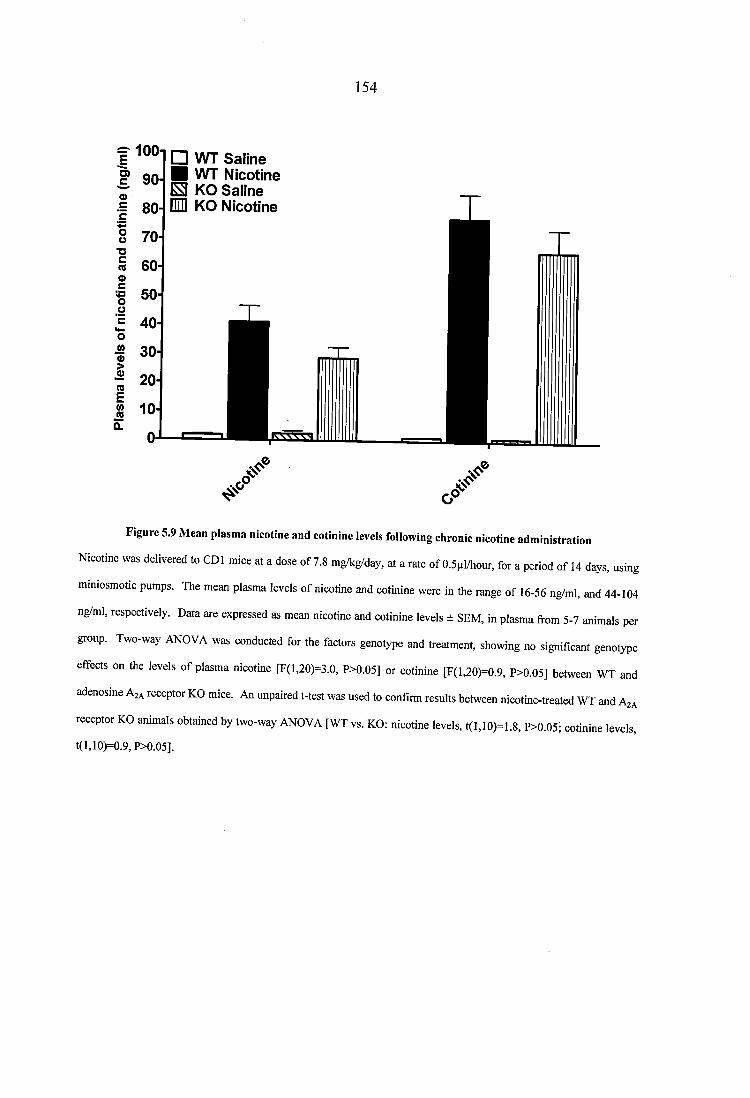
154
I* 100-O)
□ W T Saline ■ W T Nicotine E KO Saiine Em KG Nicotine
<2 10
Figure 5.9 M ean plasm a nicotine and cotinine levels follow ing chronic nicotine adm inistration
Nicotine was delivered to CDl mice at a dose of 7.8 mg/kg/day, at a rate of O.Spldiour, for a period of 14 days, using
miniosmotic pumps. The mean plasma levels of nicotine and cotinine were in the range of 16-56 ng/ml, and 44-104
ng/ml, respectively. Data are expressed as mean nicotine and cotinine levels ± SEM, in plasma from 5-7 animals per
group. Two-way ANOVA was conducted for the factors genotype and treatment, showing no significant genotype
effects on the levels of plasma nicotine [F(l,20)=3.0, P>0.05] or cotinine [F(l,20)=0.9, P>0.05] between WT and
adenosine AzA receptor KO mice. An unpaired t-tcst was used to confirm results between nicotine-treated WT and Aja
receptor KO animals obtained by two-way ANOVA [WT vs. KO: nicotine levels, t(l,10)=1.8, P>0.05; cotinine levels,
t(l,10)=0.9,P>0.05].

155
J.3.2.3 Nicotinic receptor autoradiography in WT and adenosine A2A receptor KO mice,
following chronic nicotine treatment
The density of total, cytisine-sensitive, and cytisine-resistant [^^^Ijepibatidine binding is
detailed for all brain regions analysed in Tables 5.1, 5.2, and 5.3. Administration of
nicotine for 14 days resulted in a significant increase of total and cytisine-sensitive
[^^^I]epibatidine binding sites in nicotine-treated WT and adenosine A2A receptor KO
mice, compared to saline controls (Tables 5.1 & 5.2). The degree of upregulation was
identical between genotypes, and was primarily observed in cortical areas of nicotine-
treated wildtype and knockout animals. Duncan’s post hoc analysis was performed in
order to investigate differences in radioligand binding between groups in individual brain
regions. Higher levels of total and cytisine-sensitive [^^^IJepibatidine binding were
observed in the frontal association cortex, primary motor cortex, visual cortex, and
auditory cortex of nicotine-treated mice, compared to saline controls. No changes in
cytisine-resistant [^^^IJepibatidine binding were observed among groups (Table 5.3).
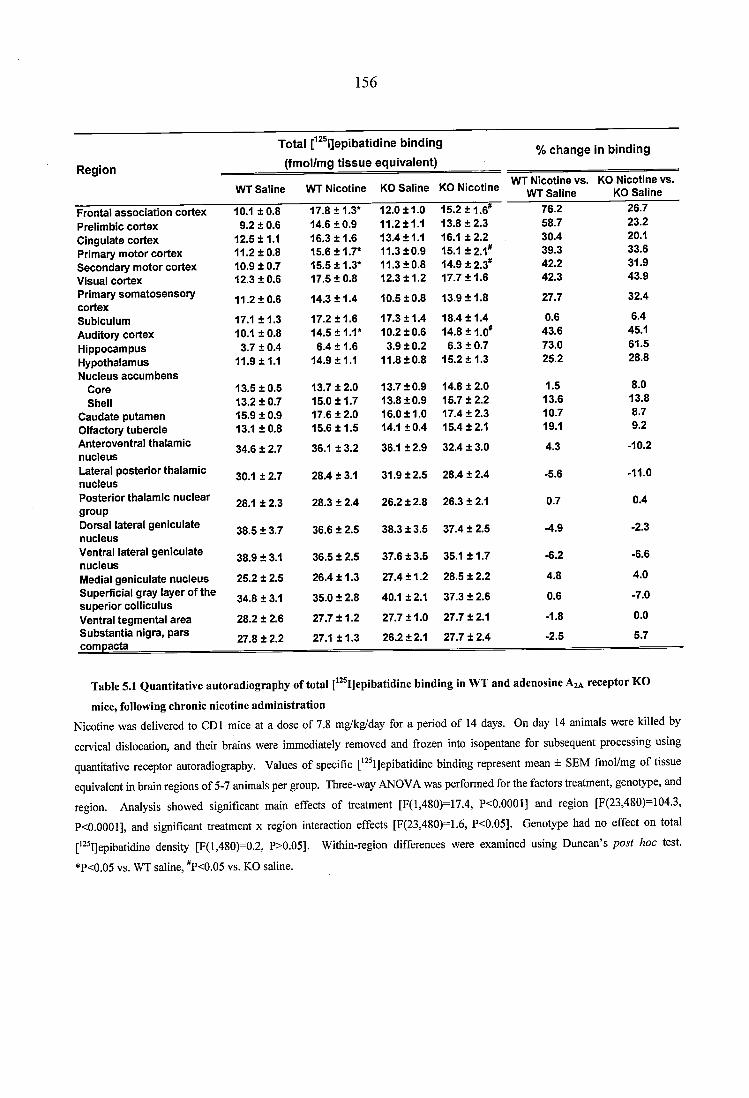
156
R e a io n
T o ta l [^^® l]epibatid ine b in d in g
( fm o l/m g t i s s u e e q u iv a le n t )% c h a n g e In b in d in g
WT Saline ’WT N icotine KO S aline KO N icotineWT N icotine vs.
WT S alineKO N icotine v s.
KO Saline
Frontal a sso c ia tio n co rtex 10.1 ± 0 .8 17.8 ±1.3* 12.0 ±1 .0 15.2 ±1.6*' 76.2 26.7
Preiim bic co rtex 9.2 ± 0.6 14.6 ± 0 .9 1 1 .2±1 .1 13.8 ± 2 .3 58.7 23.2
C ingulate co rtex 12.5 ±1.1 16.3 ± 1 .6 13.4 ±1.1 16.1 ± 2 .2 30.4 20.1
Prim ary m o to r co rtex 11.2 ± 0 .8 15.6 ±1.7* 11.3 ±0 .9 15.1 ±2.1" 39.3 33.6
S eco n d ary m o to r co rtex 10.9 ± 0.7 15.5 ±1.3* 11.3 ±0 .8 14.9 ± 2.3" 42.2 31.9
Visual co rtex 12.3 ± 0 .6 17.5 ± 0.8 12.3 ± 1 .2 17.7 ± 1 .6 42.3 43.9
Prim ary so m a to sen so ry co rtex
11.2 ± 0 .6 14.3 ± 1 .4 10.5 ±0 .8 13.9 ± 1 .8 27.7 32.4
Subiculum 17.1 ± 1 .3 17.2 ± 1 .6 17.3 ±1 .4 18.4 ± 1 .4 0.6 6.4
Auditory co rtex 10.1 ± 0 .8 14.5 ±1.1* 10.2 ±0 .6 14.8 ±1.0" 43.6 45.1
H ippocam pus 3.7 ± 0.4 6.4 ± 1 .6 3.9 ±0 .2 6.3 ± 0.7 73.0 61.5
H ypothalam us N ucleus acc u m b e n s
11.9 ±1.1 14.9 ±1.1 11.8 ±0.8 15.2 ± 1 .3 25.2 28.8
C ore 13.5 ± 0 .5 13.7 ± 2.0 13.7 ±0.9 14.8 ± 2.0 1.5 8.0
Shell 13.2 ± 0 .7 15.0 ± 1 .7 13.8 ±0.9 15.7 ± 2 .2 13.6 13.8
C audate pu tam en 15.9 ± 0 .9 17.6 ± 2 .0 16.0 ±1 .0 17.4 ± 2 .3 10.7 8.7
O lfactory tu b erc le 13.1 ± 0 .8 15.6 ± 1 .5 14.1 ±0 .4 15.4 ±2.1 19.1 9.2
A nteroventral thalam ic nu c leu s
34.6 ± 2.7 36.1 ± 3.2 36.1 ±2 .9 32.4 ± 3.0 4.3 -10.2
Lateral p o s te rio r thalam ic nu c leu s
30.1 ± 2.7 28.4 ± 3.1 31.9 ±2 .5 28.4 ± 2.4 -5.6 -11.0
P o s te rio r thalam ic n u c lear group
28.1 ± 2.3 28.3 ± 2.4 26.2 ±2 .8 26.3 ± 2.1 0.7 0.4
Dorsal lateral g en icu la te nu c leu s
38.5 ± 3.7 36.6 ± 2.5 38.3 ± 3 .5 37.4 ± 2.5 ■4.9 -2.3
V entral lateral g en icu la te nu c leu s
38.9 ± 3.1 36.5 ± 2.5 37.6 ± 3 .5 35.1 ± 1 .7 -6.2 -6.6
Medial g en icu la te n u c leu s 25.2 ± 2.5 26.4 ± 1 .3 27.4 ± 1 .2 28.5 ± 2.2 4.8 4.0
Superficial gray layer of th e su p e rio r co lllcu lus
34.8 ± 3.1 35.0 ± 2.8 40.1 ±2.1 37.3 ± 2.6 0.6 -7.0
V entral tegm en ta l a rea 28.2 ± 2.6 27.7 ± 1 .2 27.7 ±1 .0 27.7 ± 2.1 -1.8 0.0
S u b s ta n tia nigra, p a rs co m p acta
27.8 ± 2.2 27.1 ± 1 .3 26.2 ±2.1 27.7 ± 2.4 -2.5 5.7
Table 5.1 Q uantitative autoradiography o f total [‘“ ijepibatid ine binding in W T and adenosine A ;* receptor KO
mice, following chronic nicotine adm inistration
Nicotine was delivered to GDI mice at a dose of 7.8 mg/kg/day for a period of 14 days. On day 14 animals were killed by
cervical dislocation, and their brains were immediately removed and frozen into isopentane for subsequent processing using
quantitative receptor autoradiography. Values of specific [*^ I]epibatidine binding represent mean ± SEM fmol/mg of tissue
equivalent in brain regions of 5-7 animals per group. Three-way ANOVA was performed for the factors treatment, genotype, and
region. Analysis showed significant main effects of treatment [F(1,480)=17.4, P<0.0001] and region [F(23,480)—104.3,
P<0.0001], and significant treatment x region interaction effects [F(23,480)=1.6, P<0.05]. Genotype had no effect on total
[*^ I]epibatidine density [F(l,480)=0.2, P>0.05]. Within-region differences were examined using Duncan’s post hoc test.
*P<0.05 vs. WT saline, ^P<0.05 vs. KO saline.
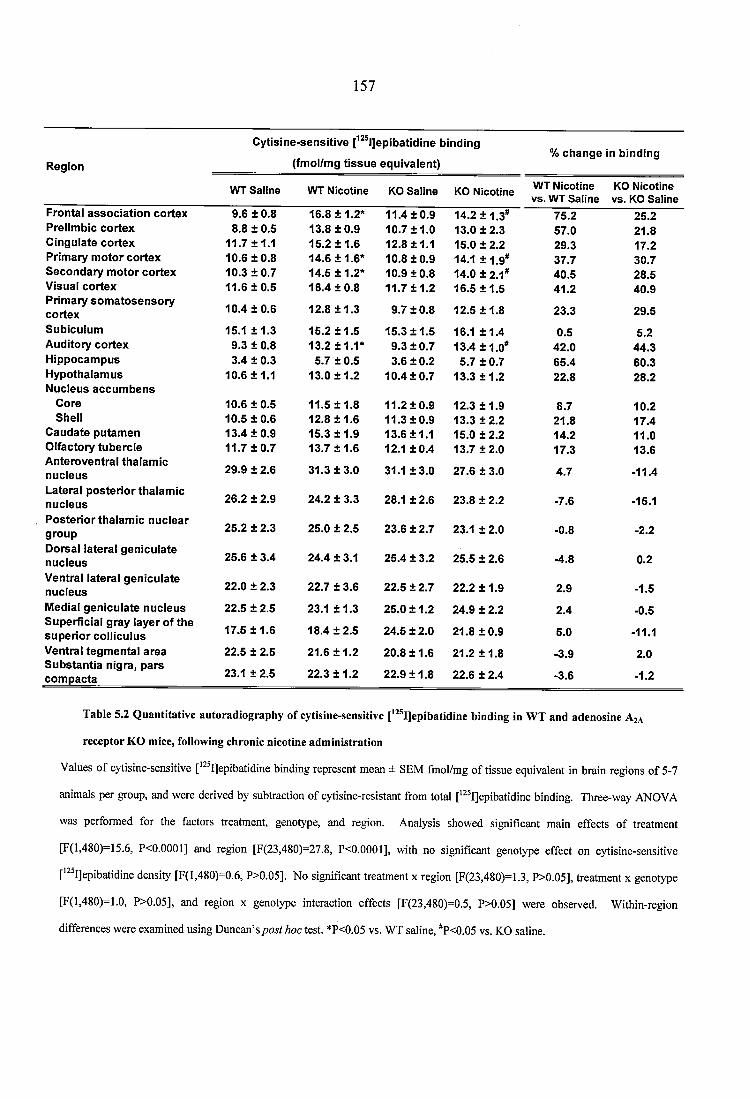
157
R e g io n
C y tis in e -s e n s i t iv e [^^^l]ep lbatid lne b in d in g
(fm o l/m g t i s s u e e q u iv a le n t)% c h a n g e In b in d in g
F ro n ta l a s s o c ia t io n c o r te x P re llm b ic c o r te x C in g u la te c o r te x P r im a ry m o to r c o r te x S e c o n d a r y m o to r c o r te x V isu a l c o r te x P rim a ry s o m a to s e n s o r y c o r te x S u b ic u lu m A u d ito ry c o r te x H ip p o c a m p u s H y p o th a la m u s N u c le u s a c c u m b e n s
C o re S h e ll
C a u d a te p u ta m e n O lfa c to ry tu b e rc le A n te ro v e n tra l th a la m ic n u c le u sL a te ra l p o s te r io r th a la m ic n u c le u sP o s te r io r th a la m ic n u c le a r g ro u pD o rsa l la te ra l g e n ic u la te n u c le u sV e n tra l la te ra l g e n ic u la te n u c le u sM edial g e n ic u la te n u c le u s S u p e rf ic ia l g ra y la y e r o f th e s u p e r io r c o ll lc u lu s V e n tra l te g m e n ta l a r e a S u b s ta n t ia n ig ra , p a r s c o m p a c ta ________
WT S aline WT Nicotine KO S aline KO Nicotine WT Nicotine vs. WT S aline
KO Nicotine vs. KO Saline
9 .6 ± 0.8 16.8 ± 1 .2 * 1 1 .4 ± 0 .9 14 .2 ± 1 .3 * 7 5 .2 25 .28 .8 ± 0.5 13.8 ± 0 .9 1 0 .7 ± 1 .0 13 .0 ± 2 .3 57 .0 21.8
11 .7 ± 1 .1 1 5 .2 ± 1 .6 12 .8 ± 1 .1 15 .0 ± 2 .2 29 .3 17.210 .6 ± 0 .8 1 4 .6 ± 1 .6 * 10 .8 ± 0 .9 14.1 ± 1 .9 * 37 .7 30 .71 0 .3 ± 0 .7 14 .5 ± 1 .2 * 10.9 ± 0 .8 1 4 .0 ± 2 .1 * 4 0 .5 28 .51 1 .6 ± 0 .5 1 6 .4 ± 0 .8 11 .7 ± 1 .2 16.5 ± 1 .5 4 1 .2 4 0 .9
1 0 .4 ± 0 .6 12 .8 ± 1 .3 9 .7 ± 0 .8 12 .5 ± 1 .8 23 .3 2 9 .5
15.1 ± 1 .3 15 .2 ± 1 .5 15 .3 ± 1 .5 16.1 ± 1 .4 0.5 5.29 .3 ± 0.8 13.2 ± 1 .1 * 9 .3 ± 0 .7 1 3 .4 ± 1 .0 * 4 2 .0 4 4 .33 .4 ± 0.3 5 .7 ± 0.5 3 .6 ± 0 .2 5 .7 ± 0.7 6 5 .4 6 0 .3
1 0 .6 ± 1 .1 13 .0 ± 1 .2 10 .4 ± 0 .7 13 .3 ± 1 .2 2 2 .8 28 .2
10 .6 ± 0 .5 11.5 ± 1 .8 11 .2 ± 0 .9 12.3 ± 1 .9 8 .7 10 .210 .5 ± 0 .6 12.8 ± 1 .6 1 1 .3 ± 0 .9 13.3 ± 2 .2 2 1 .8 17 .41 3 .4 ± 0 .9 15.3 ± 1 .9 1 3 .6 ± 1 .1 15.0 ± 2 .2 14 .2 11.011 .7 ± 0 .7 13.7 ± 1 .6 12.1 ± 0 .4 13.7 ± 2 .0 17 .3 13.6
29 .9 ± 2.6 31 .3 ± 3 .0 31.1 ± 3 .0 27 .6 ± 3.0 4 .7 -11 .4
26 .2 ± 2.9 2 4 .2 ± 3.3 28.1 ± 2 .6 23 .8 ± 2.2 -7.6 -15.1
25 .2 ± 2.3 2 5 .0 ± 2.5 2 3 .6 ± 2.7 23.1 ± 2.0 -0.8 -2.2
25 .6 ± 3.4 2 4 .4 ± 3.1 2 5 .4 ± 3.2 2 5 .5 ± 2.6 -4.8 0.2
22 .0 ± 2.3 2 2 .7 ± 3 .6 22 .5 ± 2.7 2 2 .2 ± 1 .9 2 .9 -1.5
2 2 .5 ± 2.5 23.1 ± 1 .3 25 .0 ± 1 .2 2 4 .9 ± 2.2 2 .4 -0.5
17 .5 ± 1 .6 1 8 .4 ± 2 .5 24 .5 ± 2.0 2 1 .8 ± 0 .9 5 .0 -11.1
2 2 .5 ± 2.5 21 .6 ± 1 .2 20 .8 ± 1 .6 2 1 .2 ± 1 .8 -3.9 2.0
23.1 ± 2.5 22 .3 ± 1 .2 22 .9 ± 1 .8 2 2 .6 ± 2 .4 -3.6 -1.2
Table 5.2 Q uantitative autoradiography o f cytisine-sensitive [*“ l]epibatid ine binding in W T and adenosine A za
receptor KG mice, following chronic nicotine adm inistration
Values of cytisine-sensitive ['^ IJepibatidine binding represent mean ± SEM fmol/mg of tissue equivalent in brain regions of 5-7
animals per group, and were derived by subtraction of cytisine-resistant from total [*^ I]epibatidine binding. Three-way ANOVA
was performed for the factors treatment, genotype, and region. Analysis showed significant main effects of treatment
[F(1,480)=15.6, P<0.0001] and region [F(23,480)=27.8, P<0.0001], with no significant genotype effect on cytisine-sensitive
[*^ I]epibatidine density [F(l,480)=0.6, P>0.05]. No significant treatment x region [F(23,480)=1.3, P>0.05], treatment x genotype
[F(l,480)=1.0, P>0.05], and region x genotype interaction effects [F(23,480)=0.5, P>0.05] were observed. Within-region
differences were examined using Duncan’s post hoc test. *P<0.05 vs. WT saline, "P<0.05 vs. KG saline.
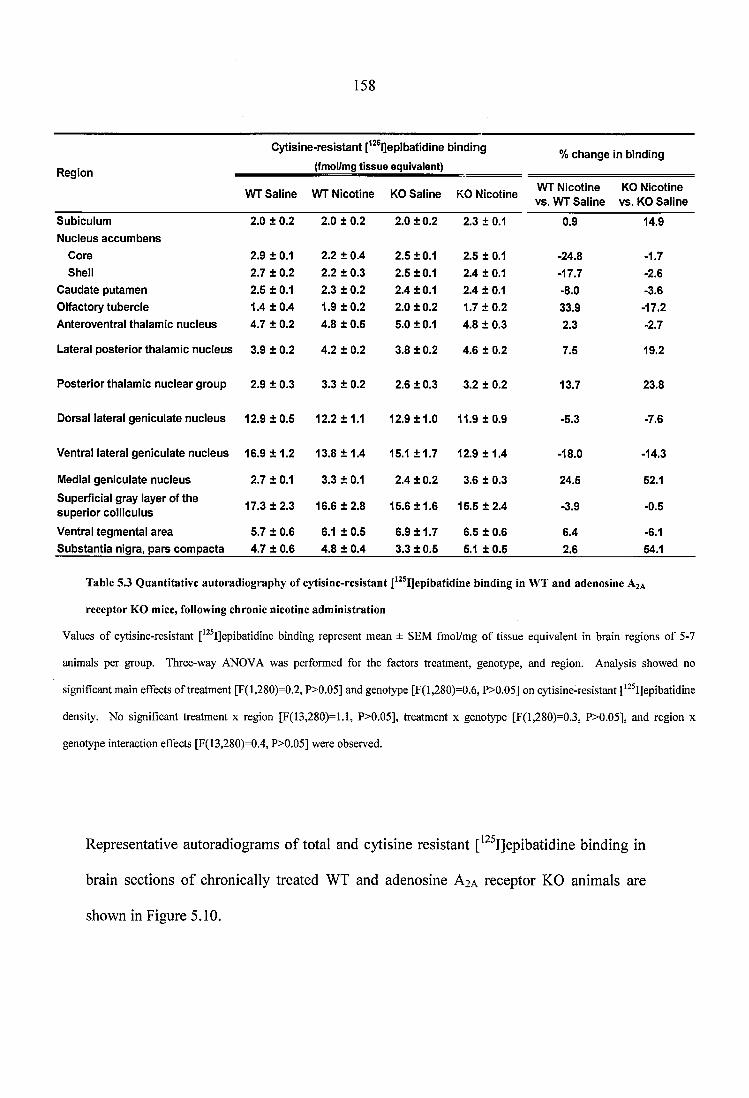
158
R sg io n
C y tis in e -re s is ta n t [^^®l]eplbatidine b in d in g
(fmol/mg tis su e equivalent)% c h a n g e In b ind ing
WT S a lin e WT N icotine KO S a lin e KG N icotineWT N ico tine v s. WT S a lin e
KO N icotine v s . KO S a lin e
S u b icu lu m
N u c leu s a c c u m b e n s
2.0 ± 0.2 2.0 ± 0.2 2.0 ± 0.2 2.3 ± 0.1 0.9 14.9
C ore 2.9 ± 0.1 2.2 ± 0.4 2.5 ±0 .1 2.5 ± 0.1 -24.8 -1.7Shell 2.7 ± 0.2 2.2 ± 0.3 2.5 ±0 .1 2.4 ± 0.1 -17.7 -2.6
C a u d a te p u tam en 2.5 ± 0.1 2.3 ± 0.2 2.4 ± 0.1 2.4 ± 0.1 -8.0 -3.6O lfacto ry tu b e rc le 1.4 ± 0.4 1.9 ± 0 .2 2.0 ± 0.2 1.7 ± 0 .2 33.9 -17.2
A n te ro v en tra l th a lam ic n u c le u s 4.7 ± 0.2 4.8 ± 0.5 5.0 ±0 .1 4.8 ± 0.3 2.3 -2.7
L ateral p o s te r io r th a la m ic n u c le u s 3.9 ± 0.2 4.2 ± 0.2 3.8 ± 0.2 4.6 ± 0.2 7.5 19.2
P o s te r io r th a la m ic n u c le a r g ro u p 2.9 ± 0.3 3.3 ± 0.2 2.6 ± 0 .3 3.2 ± 0.2 13.7 23.8
D orsal la te ra l g e n ic u la te n u c le u s 12.9 ± 0 .5 12.2 ± 1.1 12.9 ± 1 .0 11.9 ± 0 .9 -5.3 -7.6
V entral la te ra l g e n ic u la te n u c le u s 16.9 ± 1 .2 13.8 ± 1.4 15.1 ± 1 .7 12.9 ± 1 .4 -18.0 -14.3
M edial g e n ic u la te n u c le u s 2.7 ± 0.1 3.3 ± 0.1 2.4 ± 0.2 3.6 ± 0.3 24.5 52.1
S up erfic ia l g ray layer o f th e su p e r io r co lllcu lu s 17.3 ± 2.3 16.6 ± 2 .8 15.6 ± 1 .6 15.5 ± 2 .4 -3.9 -0.5
V entral teg m e n ta l a re a 5.7 ± 0.6 6.1 ± 0.5 6.9 ± 1 .7 6.5 ± 0.6 6.4 -6.1S u b s ta n tia n ig ra , p a rs c o m p a c ta 4.7 ± 0.6 4.8 ± 0.4 3.3 ± 0.5 5.1 ± 0.5 2.6 54.1
Table 5.3 Q uantitative autoradiography o f cytisine-resistant [^^®I]epibatidine binding in W T and adenosine A za
receptor K G mice, following chronic nicotine adm inistration
Values of cytisine-resistant ['^ IJepibatidine binding represent mean ± SEM fmol/mg of tissue equivalent in brain regions of 5-7
animals per group. Three-way ANOVA was performed for the factors treatment, genotype, and region. Analysis showed no
significant main effects of treatment [F(l,280)=0.2, P>0.05] and genotype [F(l,280)=0.6, P>0.05] on cytisine-resistant [^^ IJepibatidine
density. No significant treatment x region [F(13,280)=l.l, P>0.05], treatment x genotype [F(l,280)=0.3, P>0.05], and region x
genotype interaction effects [F(13,280)=0.4, P>0.05] were observed.
Representative autoradiograms of total and cytisine resistant ['^^Ijepibatidine binding in
brain sections of chronically treated WT and adenosine A2A receptor KO animals are
shown in Figure 5.10.
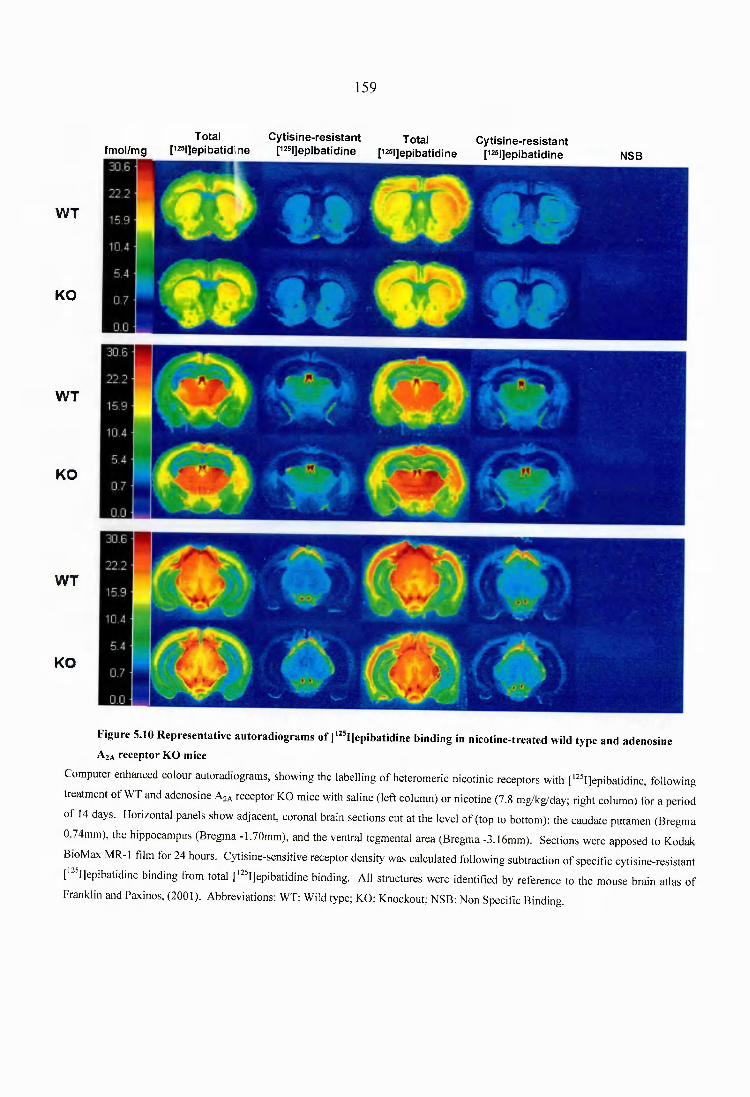
159
W T
KO
W T
KO
W T
fm ol/m gTotal
[i25|]epibatid,ineC y tis in e - re s is ta n t
p25|]epibatidineTotal
p25 |]epibatid ineC y tis in e - re s is ta n t
[i25|]epibatidine NSB
1KO
Figure 5.10 Representative autoradiogram s o f [‘^^IJepibatldine binding in nicotine-treated wild type and adenosine
A za receptor KO mice
Computer enhanced colour autoradiograms, showing the labelling of heteromeric nicotinic receptors with ['^^I]epibatidine, following
treatment of WT and adenosine Aza receptor KO mice with saline (left column) or nicotine (7.8 mg/kg/day; right column) for a period
of 14 days. Horizontal panels show adjacent, coronal brain sections cut at the level of (top to bottom): the caudate putamen (Bregma
0.74mm), the hippocampus (Bregma -1.70mm), and the ventral tegmental area (Bregma -3.16mm). Sections were apposed to Kodak
BioMax MR-1 film for 24 hours. Cytisine-sensitive receptor density was calculated following subtraction o f specific cytisine-resistant
['""Ijepibatidine binding Ifom total ['^Ijepibatidine binding. All structures were identified by reference to the mouse brain atlas o f
Franklin and Paxinos, (2001). Abbreviations: WT: Wild type; KO: Knockout; NSB: Non Specific Binding.
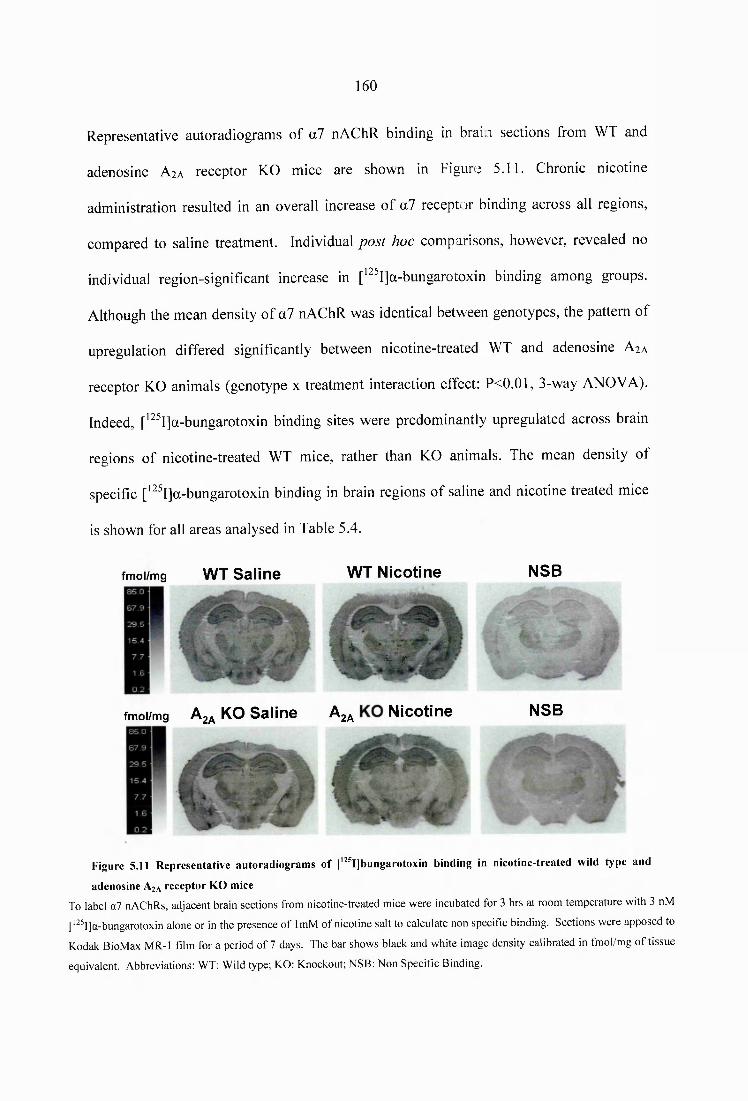
160
Representative autoradiograms of a7 nAChR binding in brai..i sections from WT and
adenosine Aia receptor KO mice are shown in Figure 5.11. Chronic nicotine
administration resulted in an overall increase of a7 receptor binding across all regions,
compared to saline treatment. Individual post hoc comparisons, however, revealed no
individual region-significant increase in [^^^I]a-bungarotoxin binding among groups.
Although the mean density of a7 nAChR was identical between genotypes, the pattern of
upregulation differed significantly between nicotine-treated WT and adenosine A2A
receptor KO animals (genotype x treatment interaction effect: P<0.01, 3-way ANOVA).
Indeed, ['^^I]a-bungarotoxin binding sites were predominantly upregulated across brain
regions of nicotine-treated WT mice, rather than KO animals. The mean density of
specific ['^^I]a-bungarotoxin binding in brain regions of saline and nicotine treated mice
is shown for all areas analysed in Table 5.4.
fm o l/m g W T Saline W T N icotine
* -* 5 %■ . " . f -
fm o l/m g ^ 2^ KO Saline ^ 2a N icotine
NSB
NSB
Figure 5.11 Representative autoradiogram s o f [*^T]bungarotoxin binding in nicotine-treated wild type and
adenosine A ja receptor KO mice
To label a7 nAChRs, adjacent brain sections from nicotine-treated mice were incubated for 3 hrs at room temperature with 3 nM
['^^I]a-bungarotoxin alone or in the presence of ImM of nicotine salt to calculate non specific binding. Sections were apposed to
Kodak BioMax MR-1 film for a period of 7 days. The bar shows black and white image density calibrated in fmol/mg of tissue
equivalent. Abbreviations: WT: Wild type; KO: Knockout; NSB: Non Specific Binding.
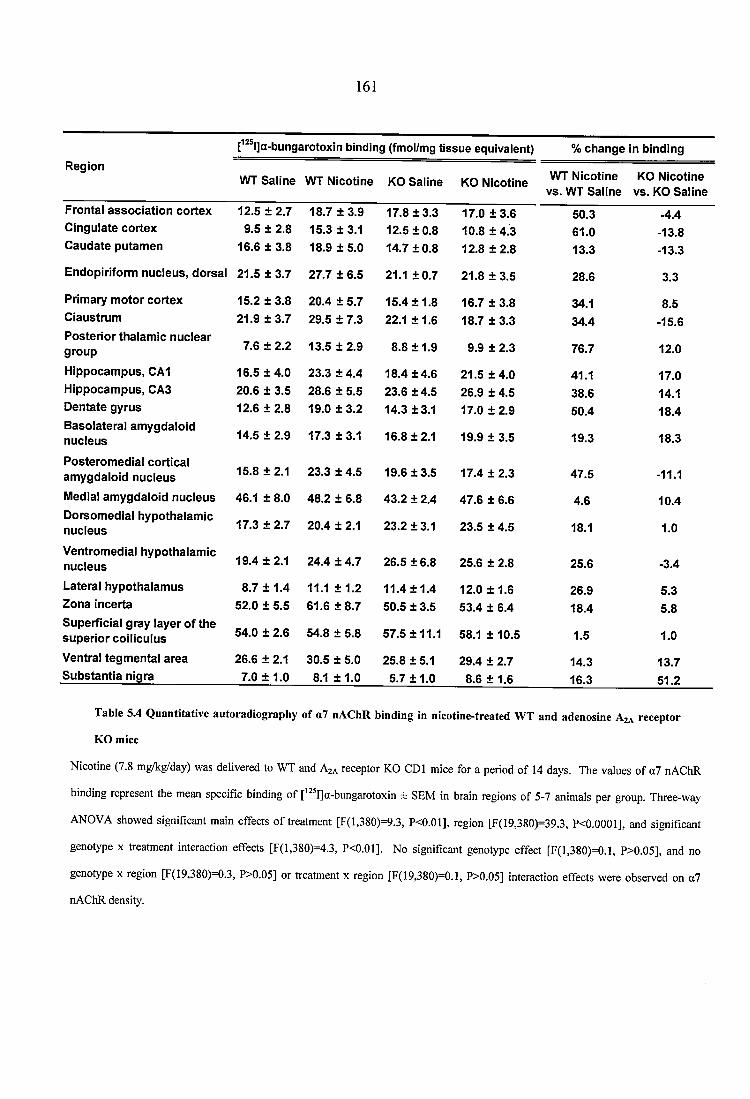
161
f ® l]a-bungaro tox in b in d in g (fm ol/m g t i s s u e e q u iv a le n t) % c h a n g e in b in d in g
R e g io nW T S a l in e W T N ic o tin e KO S a l in e KO N ic o tin e
W T N ic o tin e v s . W T S a l in e
KO N ic o tin e v s . KO S a l in e
F ro n ta l a s s o c i a t i o n c o r te x 1 2 .5 ± 2 .7 1 8 .7 ± 3 .9 1 7 .8 ± 3 .3 1 7 .0 ± 3 .6 5 0 .3 -4 .4C in g u la te c o r te x 9 .5 ± 2 .8 1 5 .3 ± 3 .1 1 2 .5 ± 0 .8 1 0 .8 ± 4 .3 6 1 .0 -1 3 .8C a u d a te p u ta m e n 1 6 .6 ± 3 .8 1 8 .9 ± 5 .0 1 4 .7 ± 0 .8 1 2 .8 ± 2 .8 1 3 .3 -1 3 .3
E n d o p ir ifo rm n u c le u s , d o r s a l 2 1 .5 ± 3 .7 2 7 .7 ± 6 .5 21.1 ± 0 .7 2 1 .8 ± 3 .5 2 8 .6 3 .3
P r im a ry m o to r c o r te x 1 5 .2 ± 3 .8 2 0 .4 ± 5 .7 1 5 .4 ± 1 .8 1 6 .7 ± 3 .8 34.1 8 .5C ia u s tru m 2 1 .9 ± 3 .7 2 9 .5 ± 7 .3 22.1 ± 1 .6 1 8 .7 ± 3 .3 3 4 .4 -1 5 .6P o s te r io r th a la m ic n u c le a r g r o u p 7 .6 ± 2 .2 1 3 .5 ± 2 .9 8 .8 ± 1 .9 9 .9 ± 2 .3 7 6 .7 12 .0
H ip p o c a m p u s , CA1 1 6 .5 ± 4 .0 2 3 .3 ± 4 .4 1 8 .4 ± 4 .6 2 1 .5 ± 4 .0 41.1 1 7 .0H ip p o c a m p u s , CA3 2 0 .6 ± 3 .5 2 8 .6 ± 5 .5 2 3 .6 ± 4 .5 2 6 .9 ± 4 .5 3 8 .6 14.1D e n ta te g y r u s 1 2 .6 ± 2 .8 1 9 .0 ± 3 .2 1 4 .3 ± 3 .1 1 7 .0 ± 2 .9 5 0 .4 1 8 .4B a s o la te ra l a m y g d a lo id n u c le u s 1 4 .5 ± 2 .9 1 7 .3 ± 3 .1 1 6 .8 ± 2 .1 1 9 .9 ± 3 .5 1 9 .3 1 8 .3
P o s te r o m e d ia l c o r t ic a l a m y g d a lo id n u c le u s 1 5 .8 ± 2.1 2 3 .3 ± 4 .5 1 9 .6 ± 3 .5 1 7 .4 ± 2 .3 4 7 .5 -11.1
M edial a m y g d a lo id n u c le u s 46 .1 ± 8 .0 4 8 .2 ± 6 .8 4 3 .2 ± 2 .4 4 7 .6 ± 6 .6 4 .6 1 0 .4D o rs o m e d ia l h y p o th a la m ic n u c le u s 1 7 .3 ± 2 .7 2 0 .4 ± 2.1 2 3 .2 ± 3.1 2 3 .5 ± 4 .5 18.1 1 .0
V e n tro m e d ia l h y p o th a la m ic n u c le u s 1 9 .4 ± 2.1 2 4 .4 ± 4 .7 2 6 .5 ± 6 .8 2 5 .6 ± 2 .8 2 5 .6 -3 .4
L a te ra l h y p o th a la m u s 8 .7 ± 1 .4 11.1 ± 1 .2 1 1 .4 ± 1 .4 1 2 .0 ± 1.6 2 6 .9 5 .3Z o n a in c e r ta 5 2 .0 ± 5 .5 6 1 .6 ± 8 .7 5 0 .5 ± 3 .5 5 3 .4 ± 6 .4 1 8 .4 5 .8S u p e r f ic ia l g ra y la y e r o f th e s u p e r i o r c o i l ic u lu s 5 4 .0 ± 2 .6 5 4 .8 ± 5 .8 5 7 .5 ± 1 1 .1 58.1 ± 10 .5 1 .5 1 .0
V e n tra l t e g m e n ta l a r e a 2 6 .6 ± 2.1 3 0 .5 ± 5 .0 2 5 .8 ± 5 .1 2 9 .4 ± 2 .7 1 4 .3 1 3 .7S u b s ta n t i a n ig ra 7 .0 ± 1 .0 8.1 ± 1 .0 5 .7 ± 1 .0 8 .6 ± 1.6 1 6 .3 5 1 .2
Table 5.4 Q uantitative autoradiography o f a7 nAChR binding in nicotine-treated W T and adenosine A za receptor
KO mice
Nicotine (7.8 mg/kg/day) was delivered to WT and Aza receptor KO GDI mice for a period of 14 days. The values of a l nAChR
binding represent the mean specific binding of [ I]a-bungarotoxin ± SEM in brain regions of 5-7 animals per group. Three-way
ANOVA showed significant main effects of treatment [F(l,380)=9.3, P<0.01], region [F(19,380)=39.3, P<0.0001], and significant
genotype x treatment interaction effects [F(l,380)=4.3, P<0.01]. No significant genotype effect [F(l,380)=0.1, P>0.05], and no
genotype x region [F( 19,380)—0.3, P>0.05] or treatment x region [F(19,380)=0.1, P>0.05] interaction effects were observed on a7
nAChR density.

162
53.2.4 Dopaminergic receptor and transporter autoradiography in WT and adenosine
A ia receptor KO mice, following chronic nicotine treatment
The impact of deletion of the adenosine A2A receptor gene on nicotine-induced
dopaminergic binding was investigated in WT and adenosine A2A receptor KO animals
by means of quantitative autoradiography of D1 and D2 receptors, and of the dopamine
transporter (DAT). Three-way ANOVA revealed that chronic administration of nicotine
increased D1 receptor density, compared to saline treatment (Fig. 5.12). [^F1]SCH23390
binding was significantly enhanced in the nucleus accumbens core of nicotine-treated
GDI mice, compared to controls. Following fourteen days of nicotine administration, D2
receptor binding was significantly decreased in nicotine-treated animals, compared to
saline controls. [^FI]raclopride binding was significantly downregulated in the rostral and
caudal caudate putamen of nicotine-treated mice, compared to saline-treated animals (Fig.
5.13). Importantly, the effect of nicotine on D1 and D2 receptor binding was similarly
observed in both WT and adenosine A2A receptor KO mice. No treatment or genotype
effects on dopamine transporter density were observed among groups (Fig. 5.14).
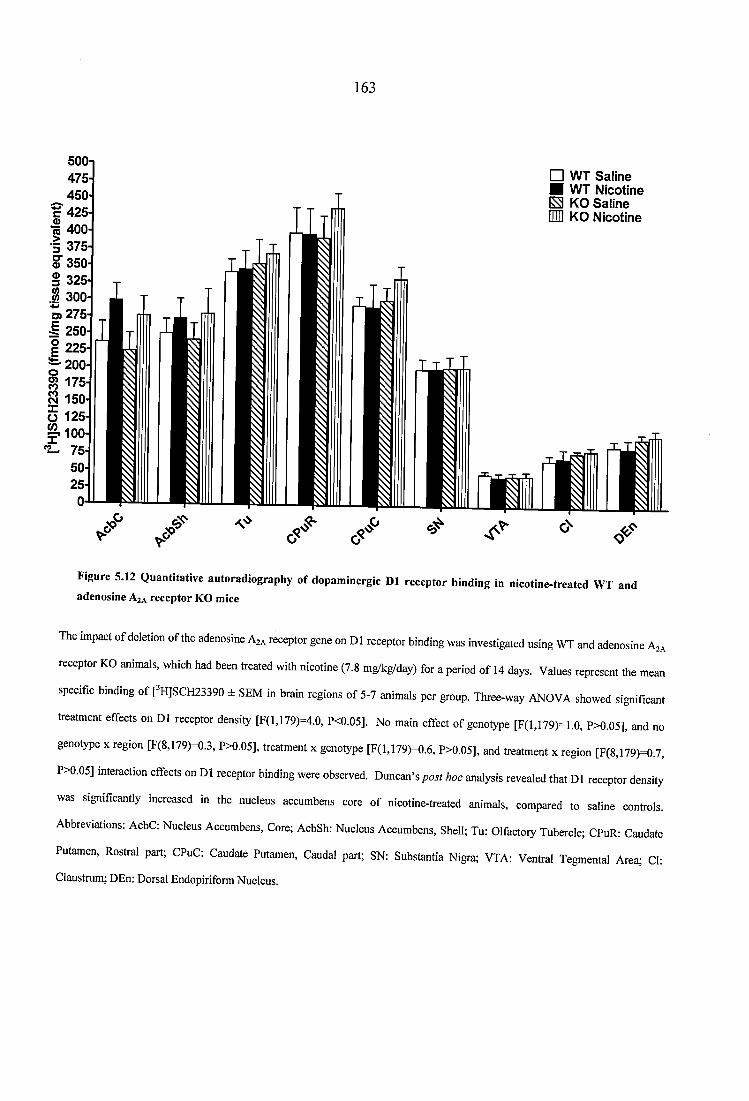
163
□ WT Saline ■ WT N icotine S KO Saline inn KO N icotine
oj 400 I 375
« 300O) 275-I 250
R 175
Ü 125-
Figure 5.12 Quantitative autoradiography of dopaminergic D1 receptor binding in nicotine-treated WT and adenosine Aza receptor KO mice
The impact of deletion of the adenosine Aza receptor gene on D1 receptor binding was investigated using WT and adenosine Aza
receptor KO animals, which had been treated with nicotine (7.8 mg/kg/day) for a period of 14 days. Values represent the mean
spedfic binding ofFH]SClT23390 i SEIW in bmin regions of 5-7 animals per group. Three-way ANC)V/L showed wgnifkant
treatment effects on D1 receptor density [F(l,179)=4.0, P<0.05]. No main effect of genotype [F(l,179)=1.0, P>0.05], and no
genotype x region [F(8,179)=0.3, P>0.05], treatment x genotype [F(l,179)=0.6, P>0.05], and treatment x region [F(8,179)=0.7,
P>0.05] interaction effects on D1 receptor binding were observed. Duncan’s hoc analysis revealed that D1 receptor density
was significantly increased in the nucleus accumbens core of nicotine-treated animals, compared to saline controls.
Abbreviations: AcbC: Nucleus Accumbens, Core; AcbSh: Nucleus Accumbens, Shell; Tu: Olfactory Tubercle; CPuR: Caudate
Putamen, Rostral part; CPuC: Caudate Putamen, Caudal part; SN: Substantia Nigra; VTA: Ventral Tegmental Area; Cl:
Ciaustrum; DEn: Dorsal Endopiriform Nucleus.
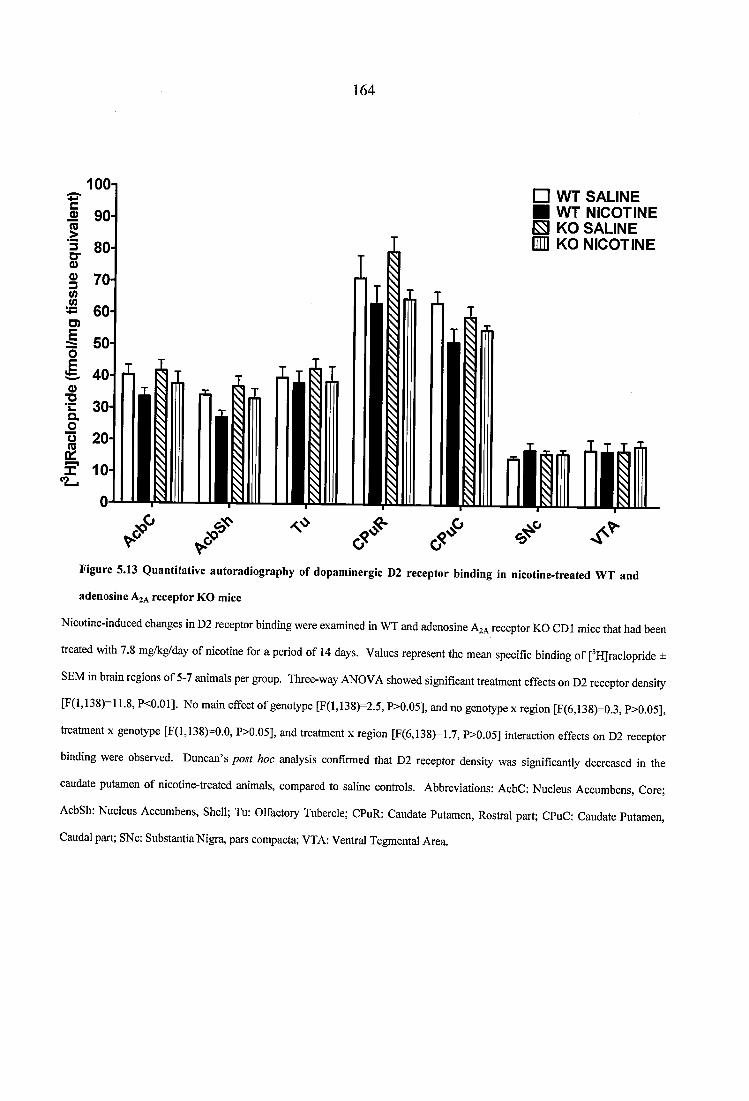
164
□ WT SALINE ■ WT NICOTINE E KO SALINE IQ KO NICOTINE
o 90-
S 70
» 60-
o 20
Figure 5.13 Q uantitative autoradiography o f dopam inergie D2 reeeptor binding in nicotine-treated W T and
adenosine A za receptor K O mice
Nicotine-induced changes in D2 receptor binding were examined in WT and adenosine Aza receptor KO GDI mice that had been
treated with 7.8 mg/kg/day of nicotine for a period of 14 days. Values represent the mean specific binding of [^H]raclopride ±
SEM in brain regions of 5-7 animals per group. Three-way ANOVA showed significant treatment effects on D2 receptor density
[F(l,138)-11.8, P<0.01]. No main effect of genotype [F(l,138)=2.5, P>0.05], and no genotype x region [F(6,138)=0.3, P>0.05],
treatment x genotype [F(l,138)=0.0, P>0.05], and treatment x region [F(6,138)=1.7, P>0.05] interaction effects on D2 receptor
binding were observed. Duncan’s post hoc analysis confirmed that D2 receptor density was significantly decreased in the
caudate putamen of nicotine-treated animals, compared to saline controls. Abbreviations: AcbC: Nucleus Accumbens, Core;
AcbSh: Nucleus Accumbens, Shell; Tu: Olfactoiy Tubercle; CPuR: Caudate Putamen, Rostral part; CPuC: Caudate Putamen,
Caudal part; SNc: Substantia Nigra, pars compacta; VTA: Ventral Tegmental Area.
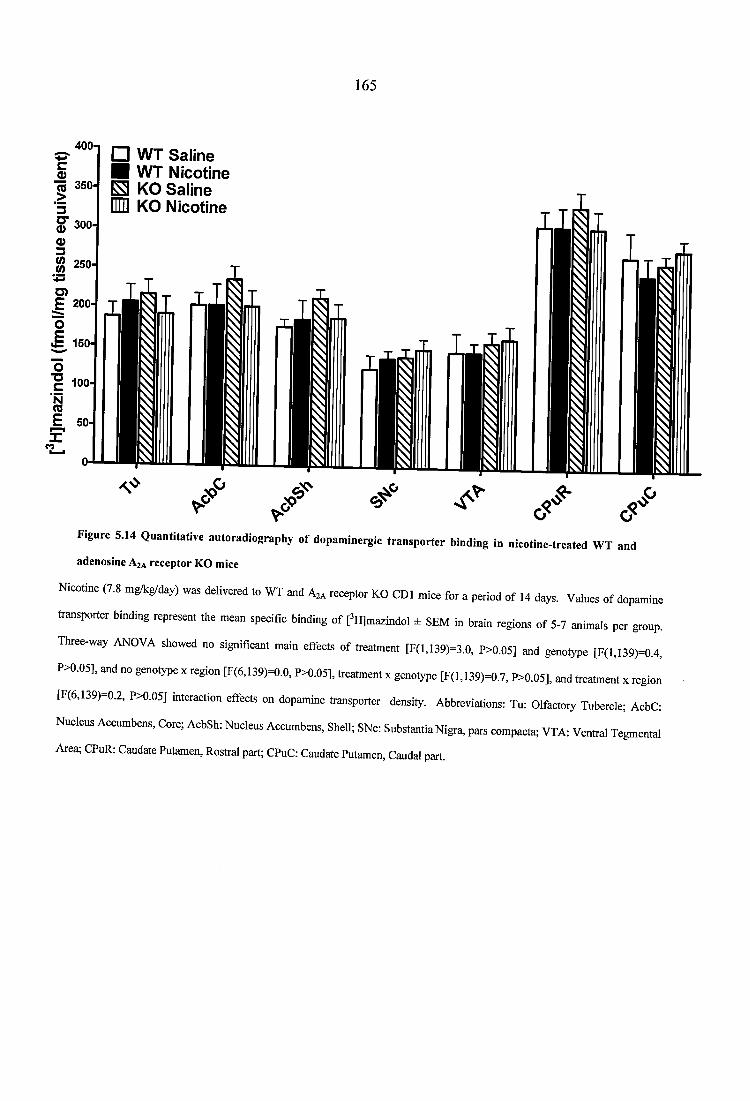
165
400-1□ WT Saline H WT Nicotine ^ KO Saline En KO Nicotine
Figure 5.14 Quantitative autoradiography of dopaminergie transporter binding in nicotine-treated WT and
adenosine Aza receptor KO mice
Nicotine (7.8 mgAg/day) was delivered to WT and A ,, receptor KO CDl mice for a period o f 14 days. Values of dopamine
transporter binding represent the mean speeific binding of [’H]mazindoi ± SEM in brain regions o f 5-7 animals per group.
Three-way ANOVA showed no significant main effects of treatment [F(l,139)=3.0, P>0.05] and genotype [F(l,139)=0.4,
P>0.05], and no genotype x region [F(6,139)=0.0. P>0.05], treatment x genotype [F(l,139)=0.7. P>0.05]. and treatment x region
[F(6,139)=0.2, P>0.05] interaction effects on dopamine transporter density. Abbreviations: Tu: Olfactoiy Tubercle; AcbC:
Nucleus Accumbens, Core; AcbSh: Nucleus Accumbens, Shell; SNc: Substantia Nigra, pars compacta; VTA: Ventral Tegmental
Area; CPuR: Caudate Putamen, Rostral part; CPuC: Caudate Putamen, Caudal part.

166
5.4 Discussion
The development of adenosine A2A receptor KO mice has generated a wealth of
information on the incidence of interactions between A2A receptors and the nicotinic
cholinergic and dopaminergic systems, both at the behavioural (Castane et ah, 2006) and
molecular levels (Svenningsson et ah, 2000). To further characterize nicotinic and
dopaminergic regulation in mice with ablation of adenosine A2A receptors, quantitative
autoradiography of a4p2* and a? nAChRs, dopaminergic D1 and D2 receptors, and of
the dopamine transporters was performed in brain sections of drug naïve and nicotine-
treated wild type (WT) and homozygous A2A receptor knockout (KO) animals.
In the current set of experiments, no changes in the distribution or density of a4p2* and
a7 nAChRs were observed between drug naïve WT and KO animals, suggesting that
deletion of the adenosine A2A receptor gene does not alter the constitutional expression of
nicotinic cholinergic receptors. This lack of interdependence between nAChRs and A2A
receptors builds on previous autoradiographic studies, which have examined the
modification of several neurotransmitter systems in response to A2A gene deletion,
drawing similar conclusions. Indeed, A2A receptor KO mice exhibit no compensatory
alterations in the distribution and expression of MOP, KOP, DOP, and ORL-1 opioid
receptors (Bailey et ah, 2002). Similarly, both the expression and the functional activity
of cannabinoid CB-1 receptors remain unchanged in the adenosine A2A KO mouse (Soria
et ah, 2004). Moreover, [^H]CGP-39653 (Dassesse et ah, 2001) and [^H]MK-801 (Chen
et ah, 1999) autoradiography reveals no change in glutamatergic NMDA receptor binding
between wild type and mutant mice, while no quantitative difference in striatal and
cortical density of AMP A receptors has been detected between WT and A2A receptor KO
animals (Snell et ah, 2000). In line with the above reports, it is shown here that the lack

167
of adenosine A2A receptors does not impact on the distribution or binding characteristics
of two major neuronal nAChR subtypes, the heteromeric a4g2* and the homomeric a7
nicotinic receptors.
Within the mesolimbic dopaminergic pathway, a wide body of literature describes
reciprocal antagonistic interactions between adenosine A2A and dopaminergic D2
receptors (reviewed in Fuxe et al., 2007). In various cell lines, activation of A2A
receptors has been shown to induce a reduction in D2 receptor affinity, Gi protein
coupling, and signal transduction (Ferre et al., 1991; Dasgupta et al., 1996; Salim et al.,
2000; Hillion et al., 2002). Moreover, co-immunoprecipitation and resonance energy
transfer studies show that A2a/D2 receptor heteromeric comlplexes are constitutively
expressed in striatal cells (Hillion et al., 2002; Canals et al., 2003). The modest increase
in D2 receptor density that was observed across all brain structures in drug naïve mutant
mice may therefore reflect a global compensatory mechanism of the dopaminergic system
in response to ablation of adenosine A2A receptors. Indeed, small increases in D2
receptor mRNA expression have been consistently reported upon disruption of the A2A
receptor gene (Dassesse et al., 2001; Short et al., 2006b). In keeping with these studies,
which show that slightly increased D2 receptor mRNA does not translate to additional D2
binding sites in individual areas of the A2A receptor KO brain, the quantification of
[^HJraclopride binding revealed no region-specific differences in D2 receptor density
between mutant and wild type animals. Nevetheless, the behavioural consequences o f a
modest overall D2 receptor upregulation may be significant, as evidenced by the
diminished haloperidol-induced catalepsy observed in A2A receptor KO mice (Chen et al.,
2001b).

168
Previous reports on the regulation of D1 receptors in adenosine A2A receptor KO mice
have produced contradictory results. In agreement with the current data, D1 receptor
density is unaltered in A2A receptor KO animals developed on a C57BL/6 background
(Chen et al., 2000; Chen et al., 2001b). On the other hand, evidence exists for increased
DI receptor expression in mutant mice with mixed C57BL/6-CD1 genetic background
(Short et al., 2006a), as well as in A2A receptor KO animals bred for fourteen generations
on a CD I background (Dassesse et al., 2001). Similarly, the observed lack of alteration
in DAT density between wild type and adenosine A2A receptor KO mice is consistent
with studies on inbred (Chen et al., 1999; Chen et al., 2001a), but not mixed background
strains (Short et al., 2006b). Therefore, the absence of a uniform genetic preparation
between this and the aforementioned studies compromises the direct comparison and
interpretation of results. In view of the significant impact that genes from parental strains
have on the mutant phenotype (Crawley et al., 1997), it is highly probable that regulation
of dopaminergic D l receptors and transporters depends on the mouse line being assessed.
Behavioural responses induced by nicotine have been shown to differ between wild type
and adenosine A2A receptor KO mice, providing direct evidence for the involvement of
A2A receptors in nicotine addiction (Castane et al., 2006). To further elucidate the
neurochemical basis of this interaction, wild type and A2A receptor mutant mice were
chronically treated with nicotine, and changes in the nicotinic cholinergic and
dopaminergic systems were subsequently assessed by means of quantitative receptor
autoradiography. The blood plasma concentrations of nicotine observed after fourteen
days of subcutaneous treatment at 7.8 mg/kg/day (base-weight) are comparable with the
nicotine levels achieved in C57BL/6 mice following chronic nicotine infusion at similar
doses (Marks et al., 2004). At this nicotine concentration, enhanced a4p2* nAChR

169
density was observed across brain areas that are particularly susceptible to upregulation,
including regions of the cerebral cortex. Moreover, an increase of 72% and 25% was
observed in the hippocampus and hypothalamus of nicotine-treated mice, respectively.
On the contrary, cytisine-sensitive [^^^I]epibatidine binding sites in thalamic structures
were resistant to nicotine-induced upregulation. These region-specific alterations in
a4p2* nAChR density are in agreement with several reports, which have established that
upregulation of high affinity binding sites in the mouse differs in individual brain areas,
as a function of treatment dose (Pauly et al., 1991; Marks et al., 2004). Importantly for
the present experiment, the observed changes in cytisine-sensitive [^^^I]epibatidine
binding sites occurred with similar intensity and regardless of genotype, suggesting that
direct interactions between A2A receptors and the nicotinic cholinergic system do not
occur at the a4p2* nAChR level. Although this does not preclude functional alterations
in a4p2* nAChR regulation, [^^^I]a-bungarotoxin autoradiography suggests that a7
nAChRs mediate the reported nicotine-induced behavioural differences between WT and
A2A mutant mice. In line with evidence that demonstrate upregulation of the low affinity
nicotinic receptor in response to behaviourally relevant nicotine doses (Pauly et al., 1991;
Rasmussen and Perry, 2006), chronic administration of the drug increased a l nAChR
density across all regions analysed, compared to saline treatment. However, enhanced
nicotine-induced [^^^I]a-bungarotoxin binding sites were predominantly observed in brain
regions of WT mice, rather than adenosine A2A receptor KO mutants, suggesting that a l
nAChRs comprise the neurochemical substrate for interactions between nicotine and A2A
receptors.
The enhancement of striatal DA release after acute nicotine administration is abolished in
A2A receptor KO mice, indicating that the receptors mediate nicotine’s reinforcing

170
properties by modulating mesolimbic DAergic neurotransmission (Castane et a l, 2006).
Nevertheless, adenosine A2A receptors are not expressed on dopamine nerve terminals
(Hettinger et al., 2001; Rosin et al., 2003). Therefore, hypodopaminergic responses in
nicotine-treated A2A receptor mutants are probably secondary to nicotine’s effects on
other neurotransmitter systems. The lack of nicotine-induced a l nAChR upregulation in
A2A receptor KO mice, implicates glutamatergic signalling in the mechanism of
interactions between adenosine A2A receptors and nicotine. Indeed, by acting on a l
nAChRs that are located presynaptically on glutamatergic nerve terminals, nicotinic
agonists have been shown to modulate glutamate release in various brain regions,
including the stiatum (Kaiser and Wonnacott, 2000), ventral tegmental area (Mansvelder
and McGehee, 2000), hippocampus (Gray et al., 1996), amygdala (Girod et al., 2000),
olfactory bulb (Alkondon et al., 1996), lateral geniculate nucleus (Guo et al., 1998), and
synapses of the interpeduncular pathway (McGehee et al., 1995). In addition, nicotine
induces adaptive changes in glutamatergic regulation postsynaptically, in brain areas that
express high levels of [^^^I]a-bungarotoxin binding sites. Thus, nicotine administration
upregulates metabotropic I glutamate receptors in the amygdala and VTA (Kane et al.,
2005), and increases NMDA receptor density and subunit expression in the hippocampus
and prefrontal cortex, respectively (Levin et al., 2005; Wang et al., 2007). Accordingly,
antagonism of mGluR5 receptors (Paterson et al., 2003) or a l nAChRs (Markou and
Paterson, 2001) reduces nicotine self-administration, and NMDA receptor blockade
attenuates nicotine-induced a l nAChR upregulation (Levin et al., 2005), suggesting that
a l nAChR can modulate glutamatergic signaling both at pre- and postsynaptic levels.
The finding that increased a l nAChR density was robustly observed in nicotine-treated
WT mice may thus reflect impaired glutamatergic neurotransmission in the absence of
the A2A gene. Functional synergistic interactions that favour glutamate signalling occur

171
between A2A/mGluR5 receptors both pre- and postsynaptically (Ferre et a l, 2002;
Rodrigues et al., 2005), as well as between A2a/NMDA receptors postsynaptically in the
striatum (Quarta et al., 2004b). Furthermore, activation of presynaptic A2A receptors on
glutamatergic terminals has been shown to potentiate glutamate release (Ciruela et al.,
2006). Therefore, the constitutional deletion of A2A receptors presumably removes
adenosine’s pre- and postsynaptic facilitatory action on glutamate neurotransmission,
further suggesting that the lack of upregulation in a-bungarotoxin binding sites in
nicotine-treated A2A receptor mutants reflects decreased nicotine-induced glutamatergic
neurotransmission.
In support of a substantial role for a l nAChRs in mediating nicotine-adenosine A2A
interactions primarily via glutamatergic, rather than dopaminergic mechanisms, similar
changes in D l and D2 receptor density were observed between WT and adenosine A2A
receptor KO mice following fourteen days of nicotine treatment. Chronic continuous
nicotine administration has been shown to decrease D2 receptor binding in the rat basal
ganglia (Janson et al., 1992), as well as D2 receptor availability in the dorsal striatum of
human smokers (Fehr et al., 2008). In keeping with these reports, nicotine treatment in
the current study resulted in a decrease of D2 receptor binding in the caudate putamen of
CD I mice, irrespective of genotype. In addition, there were no genotype effects on
dopamine transporter density, while nicotine-induced D l receptor upregulation was
equally observed in the nucleus accumbens (NAc) core of WT and A2A receptor mutants.
These results tie in with evidence for increased D l receptor mRNA expression in the
NAc of nicotine-treated rats (Bahk et al., 2002), and with data from systemically or
intracerebrally delivered nicotine, which show that acute and long-term changes in the
mesolimbic dopaminergic pathway of nicotine-treated animals do not involve the

172
dopamine transporter (Ferrari et al., 2002). Importantly, the lack of differential nicotine-
induced dopaminergic regulation between WT and adenosine A2A receptor KO mice
supports the hypothesis that a l nAChR-mediated signalling is the predominant
mechanism by which A2A receptors modulate the effects of chronic nicotine
administration.
Of note, autoradiographic studies on the regulation of dopaminergic D l and D2 receptors
following nicotine treatment have produced contradictory results. In contrast to this and
the aforementioned studies, no change in DI and D2 receptor binding has been observed
after seven weeks of oral nicotine administration in mice (Pietila et al., 1996), or
following 21 days of continuous nicotine delivery via osmotic minipumps in the rat
(Kirch et al., 1992). Moreover, dopamine receptor autoradiography in the human post
mortem brain reveals no differences in D l or D2 binding density between smokers and
the general population (Court et al., 1998). Hence, nicotine-induced dopamine receptor
regulation seems to depend on the treatment paradigm used, as well as on the duration
and dose of nicotine administration. Nevertheless, the present findings have been
obtained at a behaviourally-relevant nicotine dose (Davis et al., 2005; Castane et al.,
2006), which resulted in nicotine plasma concentration that are compatible with those
observed in human smokers (Russell et al., 1980). Notwithstanding that dopaminergic
binding levels do not always translate to receptor function (Bailey et al., 2008), the
current study suggests that the modulatory effects of adenosine A2A receptors on nicotine-
induced neuroadaptation are exerted via actions on the a l nAChR.

173
CHAPTER
SIX

174
6 QUANTITATIVE AUTORADIOGRAPHY OF HETEROMERIC NICOTINIC
AND DOPAMINERGIC RECEPTORS, AND OF THE DOPAMINE
TRANSPORTER IN C57BL/6 MICE, FOLLOWING CONTINGENT AND
NON-CONTINGENT NICOTINE ADMINISTRATION
6.1 Introduction
Nicotine exerts its reinforcing effects by binding in the brain to its corresponding
pharmacological targets, the neuronal nicotinic receptors (nAChRs). These are a
heterogeneous family of pentameric structures, formed by the assembly of five a and p
subunits. To date twelve different subunits have been identified (a2-al0, p2-p4), whose
combinations give rise to nAChRs with unique pharmacological properties and distinct
topographical distribution (reviewed in Gotti and Clementi, 2004). This prominent
heterogeneity accounts for many of nicotine’s acute reinforcing effects (Pidoplichko et al.,
1997). However, it cannot explain the transition from the activation of nicotinic receptors to
addictive behaviour, which is a complicated and multistage process, involving the acquisition,
maintenance, extinction, and reinstatement of drug-taking. The observation that nicotine
administration leads to increased brain nAChR density in both cigarette smokers (Perry et al.,
1999) and animal models of nicotine consumption (Marks et al., 1983) is considered to be
central to the course of development of nicotine addiction, and constitutes the hallmark of
chronic nicotine exposure. Of the multiple nicotinic subtypes that exist in the mammalian
brain, a4p2* receptors are abundantly expressed throughout the central nervous system, and
they readily upregulate following exposure to nicotine (Nguyen et al., 2003; Marks et al.,
2004). As shown in a conditioned place preference paradigm, a mutation that renders a4
subunits hypersensitive to agonists creates mice that are more susceptible to nicotine
reinforcement than wild type animals (Tapper et al., 2004). Moreover, deletion of the p2

175
subunit gene attenuates nicotine self-administration (Picciotto et al., 1998) and its re
expression in the VTA of p2 subunit knockout animals re-establishes the behaviour (Maskos
et al., 2005). Altogether, these data demonstrate an important role for a4p2* nAChRs in
nicotine addiction.
Among the paradigms employed to model different aspects of drug reinforcement, self
administration has been favoured as the most direct way of investigating the behavioural and
neurobiological processes that lead to addiction. Its face validity is supported by increasing
evidence, which suggests that drugs of abuse exert different effects, depending on whether
their administration is forced by the experimenter or is under the control of the experimental
animal (reviewed in Jacobs et al., 2003). The yoked-control paradigm has been particularly
designed to examine differences produced by the active or passive drug administration. In
this model, self-administering animals have control over the rate and amount of drug intake
that a yoked littermate passively receives in an identical environment. Thus, potential
differences in the consequences of active versus passive administration can be attributed to
having control over the pattern of drug exposure. To determine the extent to which nicotine-
induced neuroadaptation is the consequence of the drug’s direct pharmacological action or
the result of the contingency between a subject’s response and the delivery of a reinforcer, a
yoked-control paradigm of chronic nicotine administration in mice has been employed, using
nose-poking as the operating response. The latter is considered to more naturally mimic
smoking behaviour in mice, requiring less motor and motivational output than lever pressing
responding (Pons et al., 2008). The consequences of active versus passive nicotine exposure
on the nicotinic cholinergic and dopaminergic systems of C57BL/6 mice have been examined
by means of quantitative autoradiography of cytisine-sensitive heteromeric nicotinic
receptors, Dl and D2 dopaminergic receptors, and dopamine transporters.

176
6.2 Materials and methods
The self administration experiments were carried out at the University of Pompeu Fabra,
Barcelona, Spain, and the brains from the animals were provided for quantitative
autoradiography. In order to fully interpret the autoradiography results, the methods and
results for the self-administration protocol are presented in full.
6.2.1 Self administration paradigm
6.2.1.1 Animals
All animal procedures were conducted in accordance with the guidelines of the European
Communities Directive 86/609/EEC regulating animal research, and were approved by
the local ethical committee of the University of Pompeu Fabra, Barcelona, Spain (CEEA-
IMAS-UPF). C57BL/6 male mice (Charles River Ltd., France), weighing 24-26 g at the
beginning of the study were used. Animals were housed individually in controlled
laboratory conditions under a reversed 13h/l Ih dark/light cycle (lights off at 7.30h, lights
on at 20.30h), with the temperature maintained at 21 ± 1 °C, and humidity at 55 ± 10%.
Mice were tested during the dark phase of the dark/light cycle. Food and water were
available ad libitum, except during the experimental sessions.
6.2.1.2 Surgery and self-administration procedure
Mice were anaesthetised with a mixture of ketamine (100 mg/kg; Imalgène 1000; Rhône
Mérieux, Lyon, France) and xylazine (20 mg/kg; Sigma, Madrid, Spain), and then
implanted with indwelling i.v. catheters as previously described (Soria et al., 2005).
Following surgery, all incisions were sutured and coated with antibiotic ointment
(Bactroban, GlaxoSmithKline, Spain), and animals were allowed to recover for 3 days.
Mice were subsequently randomly assigned to contingent or yoked groups (n=l 1). Drug

177
self-administration experiments were conducted in mouse operant chambers (Model
ENV-307A-CT, Medical Associates, Georgia, VT, USA), housed in sound- and light-
attenuated boxes that were equipped with fans to provide ventilation and white noise.
The house light was on at the beginning of the session for 3 sec and off during the
remaining time. The chambers were equipped with two holes, of which one was selected
as the active and the other as the inactive hole. Responding in both holes was recorded
for all groups, throughout the experiments. The contingent group was trained to self-
administer nicotine (0.03 mg/kg/infusion delivered in a volume of 23.5 pi over 2s; Sigma,
Madrid, Spain) in single daily 1-h sessions for a period of 12 days. Each session started
with a priming injection of nicotine or saline (depending on the group). Acquisition of
nicotine self-administration was conducted under a fixed ratio 1 (FR-1) schedule of
reinforcement; one poke in the active nose hole resulted in one nicotine infusion, while
nose poking in the inactive hole had no programmed consequences. A 10 sec time-out
period was established following each nicotine infusion. During this period, responses on
the active and inactive holes were recorded but had no programmed consequences. The
session was terminated after 50 infusions had been delivered, or after one hour,
whichever occurred first. Each contingent mouse was connected to two animals, one in
the yoked nicotine and one in the yoked saline groups. Yoked mice passively received
the same number of drug infusions as compared with their self-administration partners, at
identical times during each session. Their nose-poking activity on the active and inactive
holes had no consequences. A stimulus light located above the active hole was paired
with the delivery of nicotine or saline, according to the response o f the contingent mouse.
The patency of intravenous catheters was evaluated periodically, and whenever drug self
administration behaviour appeared to deviate dramatically from that observed previously,
by the infusion of 0.1 ml of thiobarbital (5 mg/ml) through the catheter. If prominent

178
signs of anaesthesia were not apparent within 3 sec of the infusion, the mouse and its
corresponding littermates were removed from the experiment. The criteria for the
acquisition of self-administration behavior have been described previously (Orejarena et
al., 2009; Trigo et al., 2009), and were achieved when all o f the following conditions had
been met: a stable responding, with less than 20% deviation from the mean of the total
number o f reinforces earned in three consecutive sessions (80% of stability), at least 65%
of responding on the active hole, and a minimum of 4 nicotine infusions earned per
experimental session. After each session, animals were returned to their home cages. At
the end of the study mice were killed by cervical dislocation, their brains were rapidly
removed and frozen for storage at -80 °C, and subsequently sent to the University of
Surrey, UK, for quantitative autoradiography studies.
6.2.2 Quantitative receptor autoradiography
Quantitative autoradiography was performed in order to measure nAChRs with a4p2*
subtype properties, dopaminergic Dl and D2 receptors, and the dopamine transporters (DAT),
according to the procedures described in Section 3.2.6. On the day of each experiment,
sections were thawed and processed according to established protocols, with minor
modifications (Javitch et al., 1984; Marks et al., 2002; Lena et al., 2004). Sections for
analysis of nAChR bindings were derived from 11 brains from each of the three treatment
groups. Dopaminergic receptor and transporter binding was performed in 3 self-
administering mice that achieved acquisition criteria, 3 that did not, and their corresponding
littermates (n=6 per group). Multiple, adjacent sections from all groups were processed in a
paired binding protocol. Radioligand bound sections were apposed to Kodak BioMax MR-1
film (Sigma-Aldrich, UK), along with appropriate microscale standards, to allow
quantification. For nAChRs, radioligand bound sections were apposed to film for 24h. All

179
structures were identified by reference to the mouse brain atlas of Franklin and Paxinos,
(2001), and analysed using an MCID image analyser (Image research, Linton, UK).
6.2.3 Statistical analysis
Behavioural results were analysed using a mixed-design multivariate analysis of variance
(MANOVA), with group (contingent, non-contingent, and saline-treated) as the between-
subjects variable. Daily sessions and hole (active vs. inactive) represented the within-
subjects variable. Overall interactions were further analysed using the Newman-Keuls post
hoc test, when the initial P value was smaller than 0.05. All the results are expressed as
mean ± standard error of the mean (SEM).
For quantitative autoradiography, the mean±SEM of radioligand binding was calculated in
each region for all three treatment groups, as previously described (Kitchen et al., 1997).
Two-way ANOVA for the factors treatment and region was carried out in order to compare
quantitative measurements of autoradiographic binding in brain areas of contingent, non
contingent, and saline-treated animals. Moreover, quantitative differences in nicotinic and
dopaminergic binding sites between self-administering mice that achieved acquisition criteria
and those that did not were analysed using a separate two-way ANOVA. LSD post hoc
analysis was applied, when appropriate, to investigate differences in radioligand binding
between groups in individual regions. All data were analysed using the Statistica software
(StatSoft Inc., France).
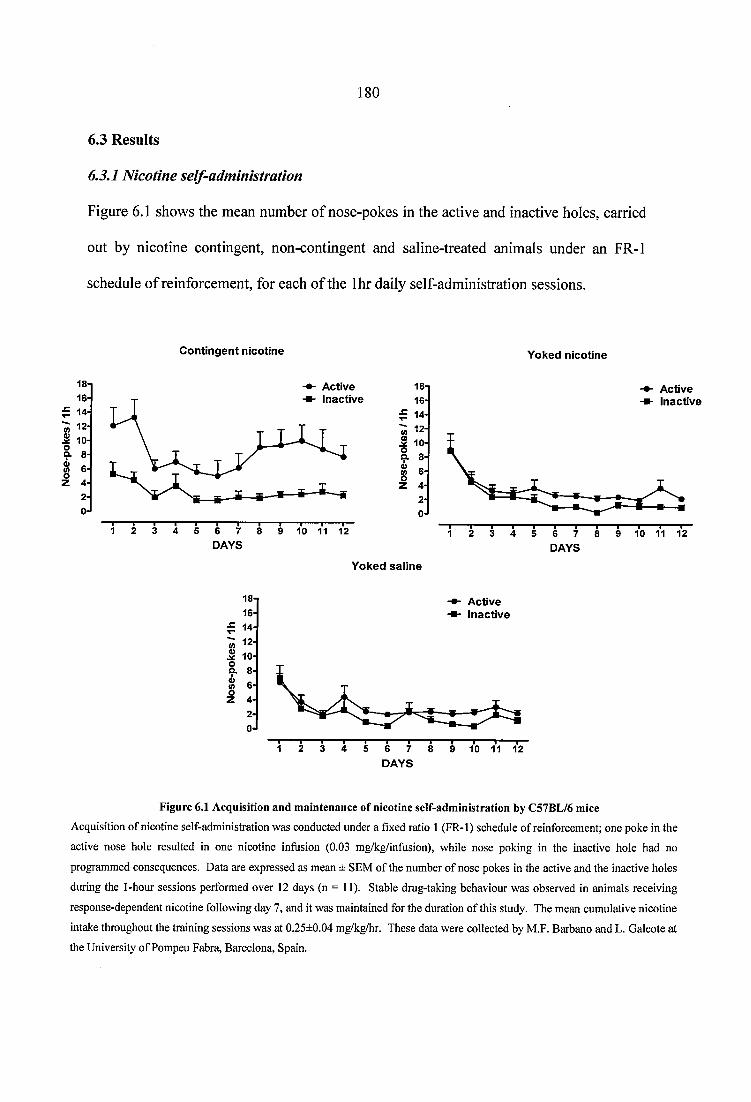
180
6.3 Results
6.3.1 Nicotine self-administration
Figure 6.1 shows the mean number of nose-pokes in the active and inactive holes, carried
out by nicotine contingent, non-contingent and saline-treated animals under an FR-1
schedule of reinforcement, for each of the Ihr daily self-administration sessions.
C o n tin g e n t n ic o tin e
18- 16-
^ 14-
? 2- jg 10-9- 8-
A ctiveIn ac tiv e
8-4-2 -
0-f6 7 DAYS
—I---- I--- I—10 11 12
Y o k ed n ic o tin e
18- 16-
^ 14-
i 10 -
A ctiveIn ac tiv e
8-
6 -
4-2 -
0
DAYS
Y o k e d s a l in e
A ctiv ein a c tiv e
8 -
4-2-
0-J
DAYS
F ig u re 6.1 A cquisition an d m ain ten an ce o f n icotine se lf-ad m in is tra tio n by C57B L/6 m ice
Acquisition of nicotine self-administration was conducted under a fixed ratio 1 (FR-1) schedule of reinforcement; one poke in the
active nose hole resulted in one nicotine infusion (0.03 mg/kg/infusion), while nose poking in the inactive hole had no
programmed consequences. Data are expressed as mean ± SEM of the number of nose pokes in the active and the inactive holes
during the 1-hour sessions performed over 12 days (n = 11). Stable drug-taking behaviour was observed in animals receiving
response-dependent nicotine following day 7, and it was maintained for the duration of this study. The mean cumulative nicotine
intake throughout the training sessions was at 0.25±0.04 mg/kg/hr. These data were collected by M.F. Barbano and L. Galeote at
the University of Pompeu Fabra, Barcelona, Spain.

181
At the nicotine dose of 0.03 mg/kg/inflision, contingent mice started to discriminate
between the active and inactive holes from the first training session. The percentage of
animals that proceeded to complete all o f the criteria for stable nicotine self
administration was 54.5% (six out of eleven), and the mean time of acquisition was
8.83±1.19 days. On the contrary, nicotine- and saline- yoked mice did not discriminate
between holes in any of the experimental sessions. On days 1 and 2, the number of nose
pokes was enhanced in all groups, compared to days 3-12. The mean nicotine intake
decreased from 0.38±0.01 mg/kg/hr on days 1 and 2, to 0.22±0.01 mg/kg/hr for days 3-12.
Contingent mice showed a mean cumulative nicotine intake of 0.25±0.04 mg/kg/hr
throughout the training sessions. In the case of the animals that achieved acquisition
criteria, the mean intake of nicotine during the last three days of self-administration was
0.35±0.09 mg/kg/hr. The overall ANOVA revealed a statistically significant effect of the
factors group [F(2,31)=10.4, P<0.001], time [F(ll,341)=11.3, P<0.001], and nose-poke
[F(l,31)=35.7, P<0.001], as well as significant group x nose-poke interaction effects
[F(2,31)=l 1.9, P<0.001]. Newman-Keulspost hoc analysis confirmed that the responses
on the active nose-poke were significantly higher only in the contingent group and not in
the yoked groups.
6.3.2 Quantitative receptor autoradiography
6.3.2.1 Effects of response contingency on nAChR density
Quantitative autoradiography was conducted for a4p2* nAChRs, using 100 pM of
[*^^I]epibatidine alone or in the presence of cytisine (20 nM). The majority o f mouse
brain [^^^Ijepibatidine binding sites were of the cytisine-sensitive, a4g2* subtype. Non
specific [^^^Ijepibatidine binding was indistinguishable from film background (Fig. 6.2).
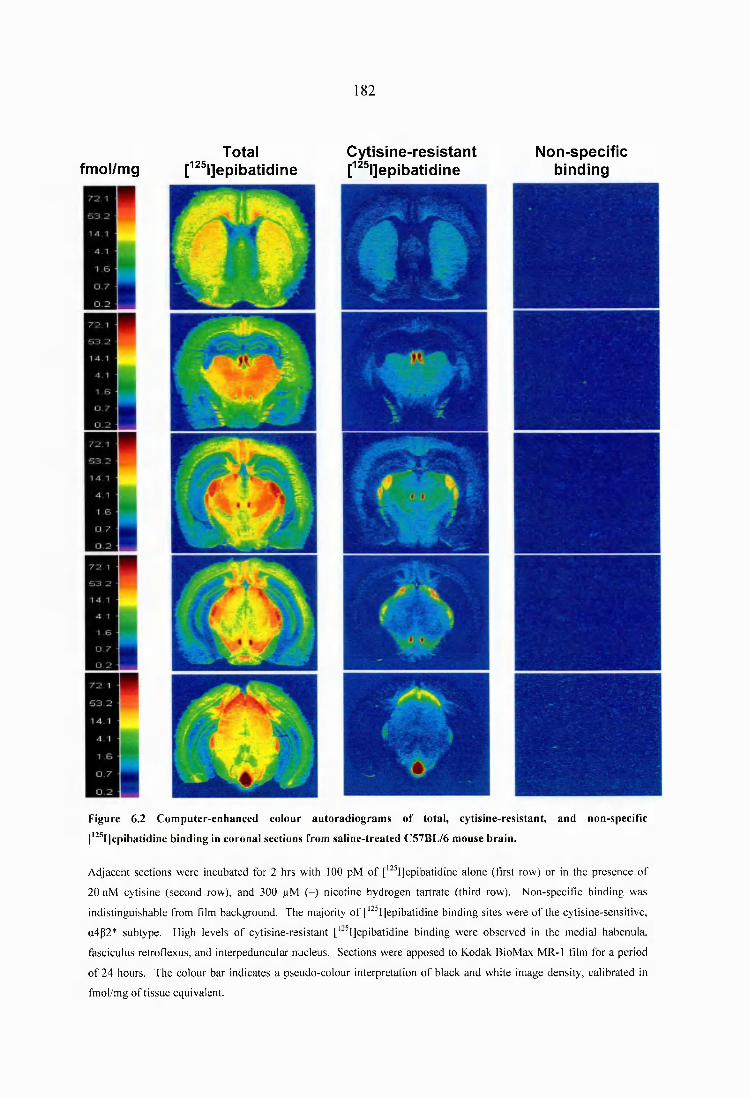
182
Totalfmol/mg [^^®l]epibatidine
Cytisine-resistantf^®[]epibatidine
Non-specificbinding
Figure 6.2 Com puter-enhanced colour autoradiogram s o f total, cytisine-resistant, and non-specific
l‘^^I|epibatidine binding in coronal sections from saline-treated C57BL/6 mouse brain.
Adjacent sections were incubated for 2 hrs with 100 pM o f ['^^Ijepibatidine alone (first row) or in the presence of
20 nM cytisine (second row), and 300 pM (-) nicotine hydrogen tartrate (third row). Non-specific binding was
indistinguishable from film background. The majority of ['^^Ijepibatidine binding sites were of the cytisine-sensitive,
a4p2* subtype. High levels of cytisine-resistant [ '“^Ijepibatidine binding were observed in the medial habenula,
fasciculus retroflexus, and interpeduncular nucleus. Sections were apposed to Kodak BioMax MR-1 film for a period
of 24 hours. The colour bar indicates a pseudo-colour interpretation of black and white image density, calibrated in
fmol/mg of tissue equivalent.

183
The density o f total, cytisine-sensitive, and cytisine-resistant [^^^I]epibatidine binding in
brain regions of saline, contingent, and non-contingent mice is detailed in Tables 6.1, 6.2,
and 6.3, respectively. For total [^^^I]epibatidine binding, significantly higher levels of
heteromeric nAChR density were observed in the superficial gray layers of the superior
colliculus (SuG), the dorsal (DLG) and ventral (VLG) lateral geniculate nucleus, and the
lateral posterior thalamic nucleus (LPMR) of contingent mice, compared to saline
controls. In non-contingent nicotine animals, higher levels of heteromeric receptor
binding were observed in the VLG compared to saline-treated mice. Contingent nicotine
increased total [^^^IJepibatidine binding in the ventral tegmental area and the SuG of self-
administering mice, compared to non-contingent animals. Two-way ANOVA (treatment
and region) showed significant main effects of treatment [F(2,462)=13.3, P<0.001] and
region [F( 17,462)= 183.0, P<0.001], with no significant treatment x region interaction
effects [F(34,462)=1.0, P>0.05].
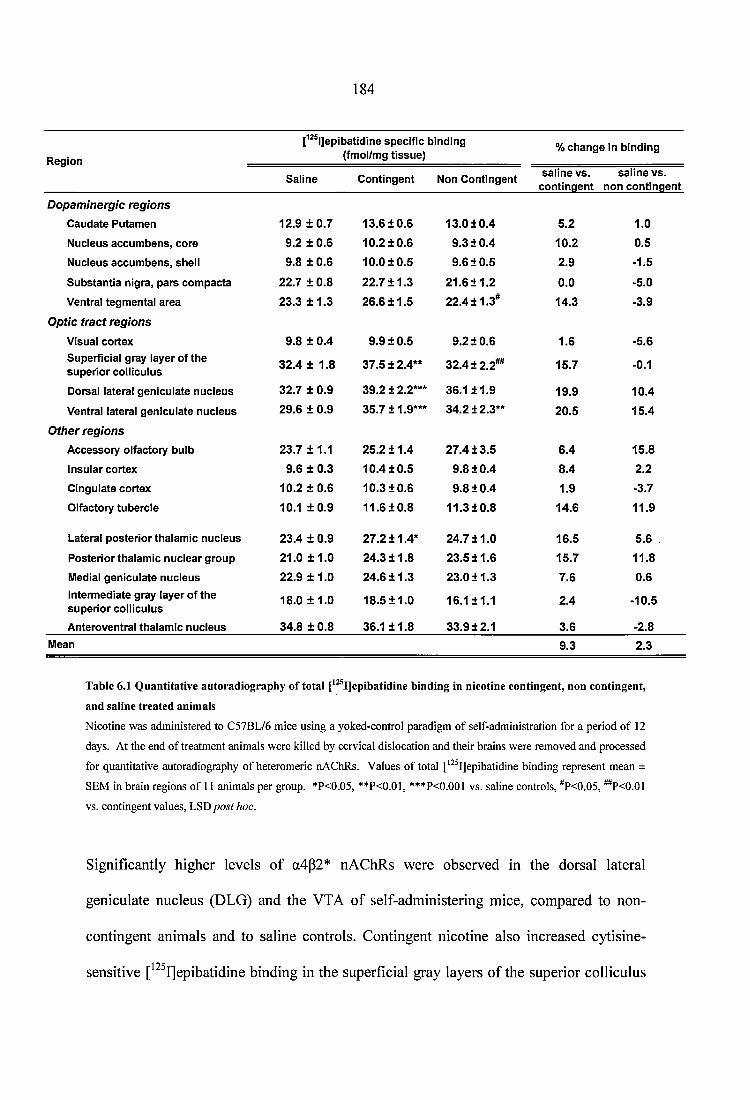
184
R egion
f ®l]epibatidine sp e c if ic b ind ing (fm ol/m g tis su e )
% c h a n g e in b in d in g
S a lin e C o n tin g en t Non C o n tin g en t sa lin e v s. sa lin e v s. c o n tin g e n t n o n c o n tin g e n tDopaminergic regions
C a u d a te P u tam en 1 2 .9 ± 0 .7 1 3 .6 ± 0 .6 1 3 .0 + 0 .4 5 .2 1.0N ucleus a c c u m b e n s , co re 9 .2 ± 0 .6 10.2 ± 0 .6 9 .3 + 0 .4 10.2 0 .5
N ucleus a c c u m b e n s , sh e ll 9 .8 ± 0 .6 1 0 .0 ± 0 .5 9 .6 + 0 .5 2 .9 -1 .5
S u b s ta n tia n igra, p a rs co m p a c ta 2 2 .7 ± 0 .8 2 2 .7 ± 1 .3 21.6 + 1 .2 0.0 -5 .0
V entral teg m en ta l a rea 2 3 .3 ± 1 .3 2 6 .6 ± 1 .5 2 2 .4 + 1.3*' 1 4 .3 -3 .9
Optic tract regionsVisual co rtex 9 .8 ± 0 .4 9 .9 ± 0 .5 9 .2 + 0 .6 1.6 -5 .6Superficial gray layer o f th e su p e r io r co llicu lu s
3 2 .4 ± 1 .8 3 7 .5 ±2.4** 3 2 .4 + 2.2** 1 5 .7 -0.1
D orsal lateral g e n ic u la te n u c le u s 3 2 .7 ± 0 .9 3 9 .2 ± 2.2*** 3 6 .1 + 1 .9 1 9 .9 1 0 .4
V entral lateral g e n ic u la te n u c le u s 2 9 .6 ± 0 .9 3 5 .7 ±1.9*** 3 4 .2 + 2.3** 2 0 .5 1 5 .4
Other regionsA ccesso ry o lfactory bu lb 2 3 .7 ± 1 .1 2 5 .2 ± 1 .4 2 7 .4 + 3 .5 6 .4 1 5 .8
In su la r co rtex 9 .6 ± 0 .3 1 0 .4 ± 0 .5 9 .8 + 0 .4 8 .4 2.2C in g u la te co rtex 10.2 ± 0 .6 1 0 .3 ± 0 .6 9 .8 + 0 .4 1 .9 -3 .7
O lfactory tu b e rc le 10.1 ± 0 .9 11.6 ± 0 .8 1 1 .3 + 0 .8 1 4 .6 1 1 .9
Lateral p o s te r io r th a lam ic n u c le u s 2 3 .4 ± 0 .9 2 7 .2 ± 1 .4 * 2 4 .7 + 1.0 1 6 .5 5 .6
P o s te r io r th a lam ic n u c le a r g ro u p 21.0 ± 1 .0 2 4 .3 ± 1 .8 2 3 .5 + 1 .6 1 5 .7 11.8Medial g en ic u la te n u c le u s 2 2 .9 ± 1 .0 2 4 .6 ± 1 .3 2 3 .0 + 1 .3 7 .6 0.6In term ed ia te g ray layer o f th e su p e r io r co llicu lu s
1 8 .0 ± 1 .0 1 8 .5 ± 1 .0 16.1 + 1.1 2 .4 -1 0 .5
A nteroven tra l th a lam ic n u c le u s 3 4 .8 ± 0 .8 36.1 ± 1 .8 3 3 .9 + 2.1 3 .6 -2 .8
Mean 9 .3 2 .3
Table 6.1 Q uantitative autoradiography o f total p^^Ijepibatidine binding in nicotine contingent, non contingent,
and saline treated animals
Nicotine was administered to C57BL/6 mice using a yoked-control paradigm of self-administration for a period of 12
days. At the end of treatment animals were killed by cervieal dislocation and their brains were removed and processed
for quantitative autoradiography of heteromeric nAChRs. Values of total [*^ I]epibatidine binding represent mean ±
SEM in brain regions of 11 animals per group. *P<0.05, **P<0.01, ***P<0.001 vs. saline controls, ^P<0.05, ^P<0.01
vs. contingent values, LSD post hoc.
Significantly higher levels of a4p2* nAChRs were observed in the dorsal lateral
geniculate nucleus (DLG) and the VTA of self-administering mice, compared to non
contingent animals and to saline controls. Contingent nicotine also increased cytisine-
sensitive [^^^IJepibatidine binding in the superficial gray layers of the superior colliculus

185
(SuG) and certain thalamic nuclei, such as the ventral lateral geniculate nucleus (VLG),
the posterior thalamic nuclear group (Po), and the lateral posterior thalamic nucleus
(LPMR), compared to saline treatment. In non-contingent animals, higher levels of
a4p2* receptor binding were observed in the VLG and SuG compared to saline controls.
For nAChRs with a4p2* nAChR properties, two-way ANOVA showed significant main
effects of treatment [F(2,463)=20.4, P<0.001] and region [F(17,463)=48.3, P<0.001],
with no treatment x region interaction effects [F(34,463)=1.2, P>0.05]. LSD post hoc
analysis confirmed significant, region specific differences in cytisine-sensitive
[^^^I]epibatidine binding between self-administering and nicotine-yoked mice in the VLG
and the VTA.
No differences in cytisine-sensitive [^^^I]epibatidine binding sites were detected between
contingent mice that achieved acquisition criteria, and animals that did not stably respond
for nicotine [F(l,140)=1.8, P>0.05].
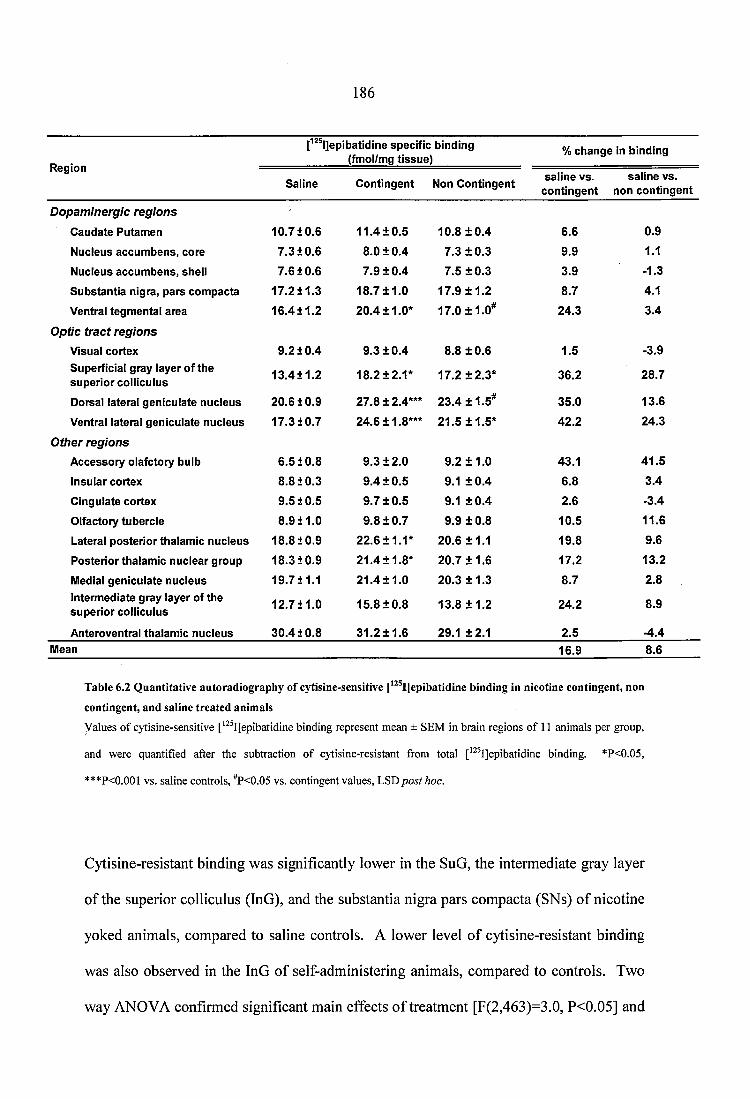
186
R eg io n
[^^^l]epibatidine sp e c if ic b in d in g (fm ol/m g t is s u e )
% c h a n g e in b in d in g
sa l in e v s . c o n tin g e n t
sa l in e v s. n o n c o n tin g e n tS a lin e C o n tin g e n t Non C o n tin g e n t
Dopaminergic regionsC a u d a te P u ta m e n 1 0 .7 1 0 .6 1 1 .4 ± 0 .5 1 0 .8 ± 0 .4 6.6 0 .9
N u c leu s a c c u m b e n s , c o re 7 .3 1 0 .6 8 .0 ± 0 .4 7 .3 ± 0 .3 9 .9 1.1N u c leu s a c c u m b e n s , sh e ll 7 .6 1 0 .6 7 .9 ± 0 .4 7 .5 ± 0 .3 3 .9 -1 .3
S u b s ta n tia n ig ra , p a rs c o m p a c ta 1 7 .2 1 1 .3 1 8 .7 ± 1 .0 1 7 .9 ± 1 .2 8 .7 4.1
V entral te g m e n ta l a re a 1 6 .4 1 1 .2 2 0 .4 ± 1 .0 * 1 7 .0 ± 1 .0 * 2 4 .3 3 .4
Optic tract regionsV isual c o r te x 9 .2 1 0 .4 9 .3 ± 0 .4 8.8 ± 0 .6 1 .5 -3 .9
S u p e rfic ia l g ra y lay e r o f th e s u p e r io r c o llic u lu s
1 3 .4 1 1 .2 1 8 .2 ± 2 .1 * 1 7 .2 ± 2 .3 * 3 6 .2 2 8 .7
D orsal la te ra l g e n ic u la te n u c le u s 2 0 .6 1 0 .9 2 7 .8 ± 2.4*** 2 3 .4 ± 1 .5 * 3 5 .0 1 3 .6
V entral la te ra l g e n ic u la te n u c le u s 1 7 .3 1 0 .7 2 4 .6 ±1.8*** 2 1 .5 ± 1 .5 * 4 2 .2 2 4 .3
Other regionsA c c e s s o ry o la fc to ry b u lb 6 .5 1 0 .8 9 .3 ± 2 .0 9 .2 ± 1 .0 4 3 .1 4 1 .5
In su la r c o rte x 8 .8 1 0 .3 9 .4 ± 0 .5 9.1 ± 0 .4 6.8 3 .4
C in g u la te c o r te x 9 .5 1 0 .5 9 .7 ± 0 .5 9.1 ± 0 .4 2.6 -3 .4
O lfac to ry tu b e rc le 8 .9 1 1 .0 9 .8 ± 0 .7 9 .9 ± 0 .8 1 0 .5 11.6L ateral p o s te r io r th a la m ic n u c le u s 1 8 .8 1 0 .9 22.6 ± 1 .1 * 20.6 ± 1 .1 1 9 .8 9 .6
P o s te r io r th a la m ic n u c le a r g ro u p 1 8 .3 1 0 .9 2 1 .4 ± 1 .8 * 2 0 .7 ± 1 .6 1 7 .2 1 3 .2
M edial g e n ic u la te n u c le u s 1 9 .7 1 1 .1 2 1 .4 ± 1 .0 2 0 .3 ± 1 .3 8 .7 2.8In te rm ed ia te g ra y lay er o f th e s u p e r io r c o llic u lu s
1 2 .7 1 1 .0 1 5 .8 ± 0 .8 1 3 .8 ± 1 .2 2 4 .2 8 .9
A n te ro v en tra l th a la m ic n u c le u s 3 0 .4 1 0 .8 3 1 .2 ± 1 .6 29 .1 ± 2 .1 2 .5 -4 .4M ean 1 6 .9 8.6
Table 6.2 Q uantitative autoradiography o f cytisine-sensitive [^^®I]epibatidine binding in nicotine contingent, non
contingent, and saline treated animals
Values of cytisine-sensitive [*^ I]epibatidine binding represent mean ± SEM in brain regions of 11 animals per group,
and were quantified after the subtraction of cytisine-resistant from total ['^ IJepibatidine binding. *P<0.05,
***P<0.001 vs. saline controls, ^P<0.05 vs. contingent values, LSD post hoc.
Cytisine-resistant binding was significantly lower in the SuG, the intermediate gray layer
of the superior colliculus (InG), and the substantia nigra pars compacta (SNs) o f nicotine
yoked animals, compared to saline controls. A lower level of cytisine-resistant binding
was also observed in the InG of se lf administering animals, compared to controls. Two
way ANOVA confirmed significant main effects of treatment [F(2,463)=3.0, P<0.05] and
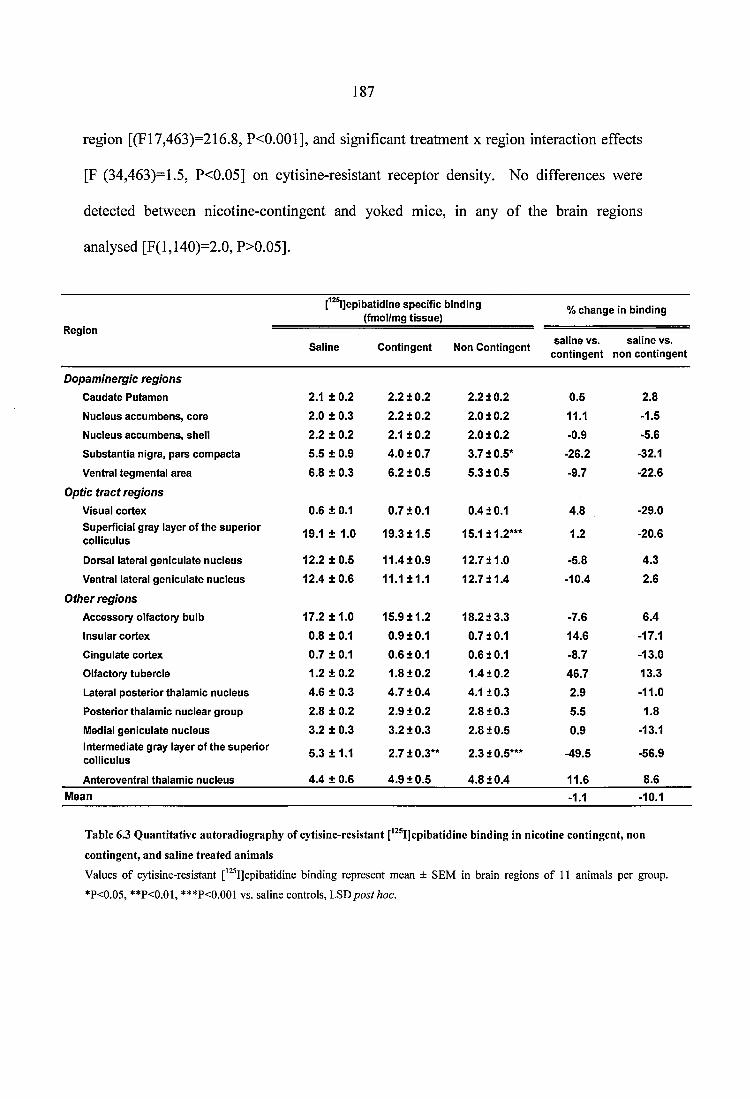
187
region [(F17,463)=216.8, P<0.001], and significant treatment x region interaction effects
[F (34,463)=!.5, P<0.05] on cytisine-resistant receptor density. No differences were
detected between nicotine-contingent and yoked mice, in any of the brain regions
analysed [F(l,140)=2.0, P>0.05].
f^^l]epibatidine spec ific binding (fmol/mg tissu e )
% ch an g e in binding
RegionSaline C ontingent Non C ontingent
sa iine v s. sa line vs. co n tin g en t non con tingen t
Dopaminergic regionsC audate Putam en 2.1 ± 0 .2 2.2 ± 0 .2 2.2 1 0 .2 0 .5 2.8N ucleus accum bens, core 2 .0 ± 0.3 2.2 ± 0 .2 2.0 1 0 .2 11.1 -1.5
N ucleus accum bens, shell 2.2 ± 0 .2 2.1 ± 0 .2 2.0 1 0 .2 -0.9 -5.6
S u b stan tia nigra, pa rs com pacta 5 .5 ± 0.9 4 .0 ± 0 .7 3 .7 1 0 .5 * -26.2 -32.1
Ventral tegm ental area 6.8 ± 0 .3 6 .2 ± 0 .5 5 .3 1 0 .5 -9.7 -22.6
Optic tract regionsVisual cortex 0.6 ± 0.1 0 .7 ±0 .1 0 .4 1 0 .1 4 .8 -29.0Superficial gray layer o f th e su p erio r colliculus 19.1 ± 1.0 19.3 ± 1 .5 15.111.2*** 1.2 -20.6
Dorsal lateral gen icu late nucleus 12.2 ± 0 .5 1 1 .4 ± 0 .9 1 2 .7 1 1 .0 -5.8 4 .3
Ventral lateral gen icu late nucleus 12.4 ± 0 .6 11.1 ±1 .1 1 2 .7 1 1 .4 -10.4 2.6Other regions
A ccessory olfactory bulb 17.2 ± 1 .0 15.9 ± 1 .2 1 8 .2 1 3 .3 -7.6 6 .4
Insular cortex 0.8 ± 0.1 0 .9 ±0 .1 0 .7 1 0 .1 14.6 -17.1
C ingulate cortex 0 .7 ± 0.1 0.6 ±0 .1 0 .6 1 0 .1 -8.7 -13.0
Olfactory tubercle 1.2 ± 0 .2 1.8 ± 0 .2 1 .4 1 0 .2 4 6 .7 13.3
Lateral p o ste rio r thalam ic n ucleus 4 .6 ± 0.3 4 .7 ± 0 .4 4 .1 1 0 .3 2.9 -11.0
Posterio r thalam ic nuc lear group 2.8 ± 0 .2 2 .9 ± 0 .2 2 .8 1 0 .3 5.5 1.8Medial gen icu late n ucleus 3 .2 ± 0.3 3 .2 ± 0 .3 2 .8 1 0 .5 0.9 -13.1Interm ediate gray layer of th e su p erio r colliculus 5 .3 ± 1 .1 2 .7 ±0.3** 2.310.5*** -49.5 -56.9
Anteroventral thalam ic nucleus 4 .4 ± 0.6 4 .9 ± 0 .5 4 .8 1 0 .4 11.6 8.6M ean -1.1 -10.1
Table 6.3 Q uantitative autoradiography o f cytisine-resistant [*^®I]epibatidine binding in nicotine contingent, non
contingent, and saline treated anim als
Values o f cytisine-resistant ['^^IJepibatidine binding represent mean ± SEM in brain regions o f 11 animals per group.
*P<0.05, **P<0.01, ***P<0.001 vs. saline controls, LSD post hoc.
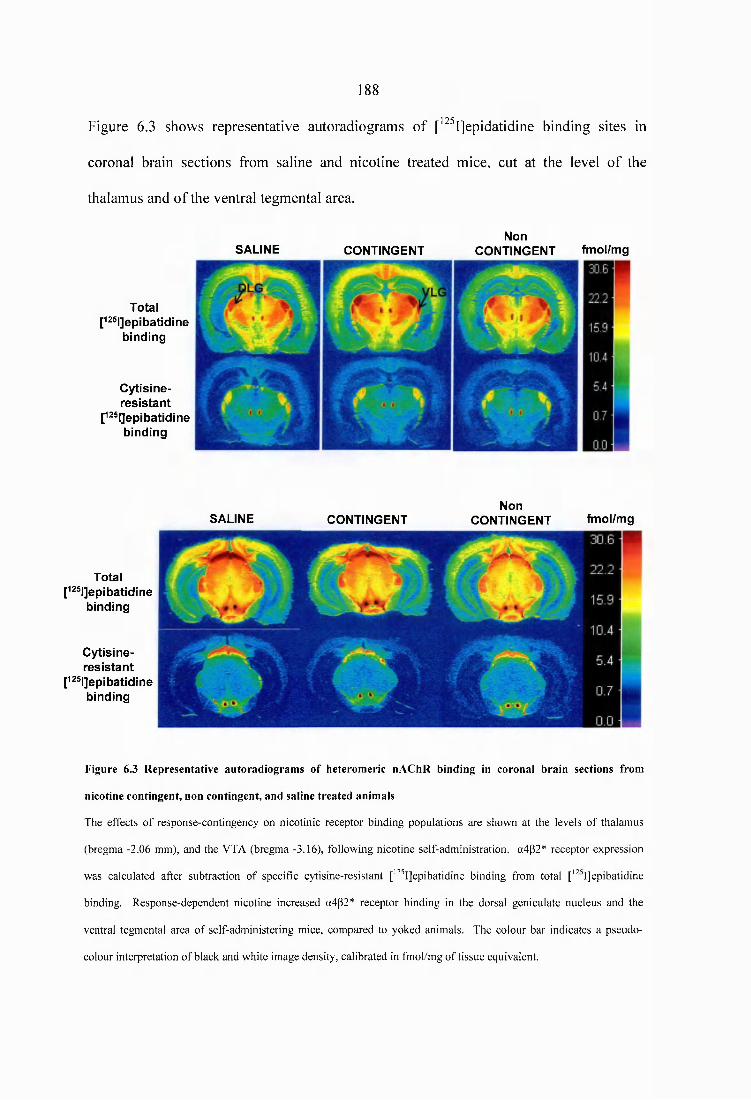
188
Figure 6.3 shows representative autoradiograms of [’ ^Ijepidatidine binding sites in
coronal brain sections from saline and nicotine treated mice, cut at the level of the
thalamus and of the ventral tegmental area.
T o ta lP ^ ^ IJ e p ib a t id ln e
b in d in g
C y t is in e -r e s i s t a n t
[izsqepibatidineb in d in g
SA L IN E C O N T IN G E N TN o n
C O N T IN G E N T fm o l/m g
T o ta lP ^ ® l]e p ib a tid in e
b in d in g
C y t is in e -r e s i s t a n t
[i2S|]epibatidineb in d in g
SA L IN E C O N T IN G E N TN o n
C O N T IN G E N T fm o l/m g
Figure 6.3 Representative autoradiogram s o f heteromeric nAChR binding in coronal brain sections from
nicotine contingent, non contingent, and saline treated animals
The effects of response-contingency on nicotinic receptor binding populations are shown at the levels of thalamus
(bregma -2.06 mm), and the VTA (bregma -3.16), following nicotine self-administration. a4p2* receptor expression
was calculated after subtraction of specific cytisine-resistant ['^ IJepibatidine binding from total [' "’IJepibatidine
binding. Response-dependent nicotine increased a4p2* receptor binding in the dorsal geniculate nucleus and the
ventral tegmental area of self-administering mice, compared to yoked animals. The colour bar indicates a pseudo
colour interpretation of black and white image density, calibrated in fmol/mg of tissue equivalent.

189
6.3.2.2 Effects o f response contingency on dopaminergic D1 and D2 receptors, and on
dopamine transporter density
Full quantification of dopaminergic D1 and D2 receptor binding, and of the dopamine
transporters (DAT) was carried out on brain sections from all treatment groups, using
£^H]SCH23390, [^H]raclopride, and [^H]mazindol, respectively. For D2 receptor binding,
two-way ANOVA revealed significant main effects of treatment [F(2,95)=6.1, P<0.01]
and region [F(6,95)=74.4, P<0.001], with no significant treatment x region interaction
effect [F(12,95)=0.3, P>0.05] (Fig. 6.4). There was an overall increase in D2 receptor
binding across all regions, in both nicotine contingent and non-contingent mice,
compared to saline controls. However, post hoc comparison revealed no individual
region significant increase among groups. The small level of increase in D2 receptor
density was observed in self-administering animals, as well as in contingent mice that did
not fulfil acquisition criteria [(Fl,24)=0.0, P>0.05].
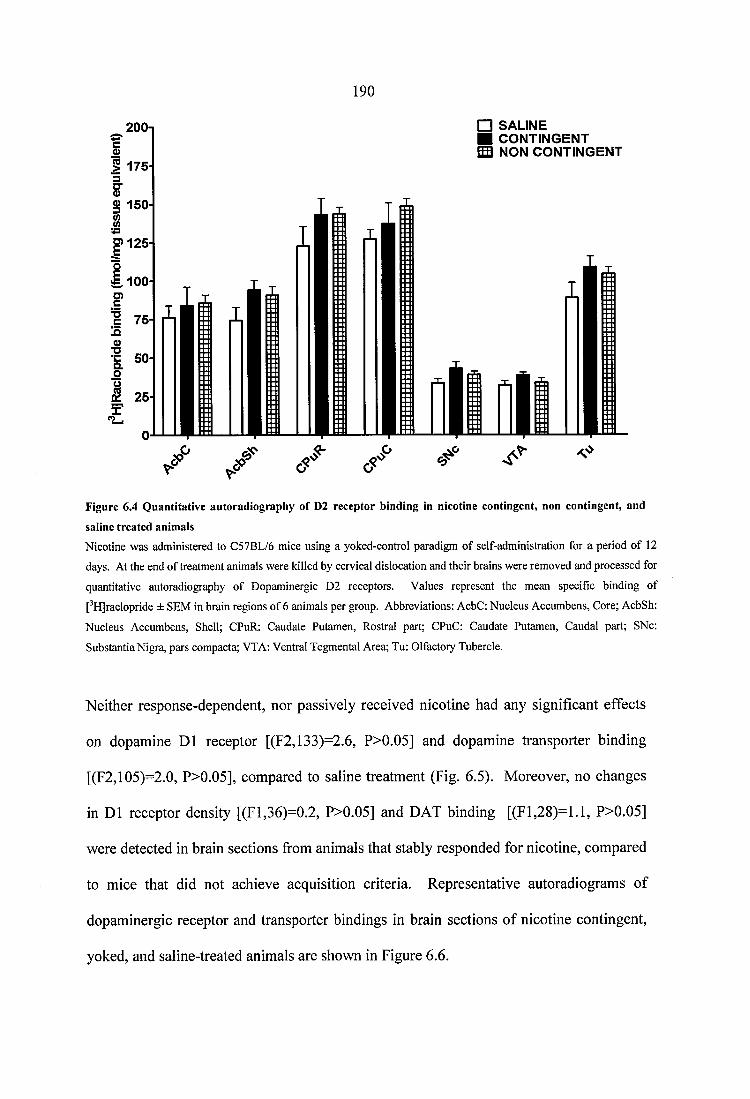
190
200 i D SALINE ■ CONTINGENT H NON CONTINGENT
3: 175
o 1 so
i t 100
Figure 6.4 Q uantitative autoradiography o f D2 receptor binding in nicotine contingent, non contingent, and
saline treated animals
Nicotine was administered to C57BL/6 mice using a yoked-control paradigm of self-administration for a period of 12
days. At the end of treatment animals were killed by cervical dislocation and their brains were removed and processed for
quantitative autoradiography of Dopaminergic D2 receptors. Values represent the mean specific binding of
[^Hjraclopride ± SEM in brain regions of 6 animals per group. Abbreviations: AcbC: Nucleus Accumbens, Core; AcbSh:
Nucleus Accumbens, Shell; CPuR: Caudate Putamen, Rostral part; CPuC: Caudate Putamen, Caudal part; SNc:
Substantia Nigra, pars compacta; VTA: Ventral Tegmental Area; Tu: Olfactory Tubercle.
Neither response-dependent, nor passively received nicotine had any significant effects
on dopamine D1 receptor [(F2,133)=2.6, P>0.05] and dopamine transporter binding
[(F2,105)=2.0, P>0.05], compared to saline treatment (Fig. 6.5). Moreover, no changes
in D1 receptor density [(Fl,36)=0.2, P>0.05] and DAT binding [(FI,28)= 1.1, P>0.05]
were detected in brain sections from animals that stably responded for nicotine, compared
to mice that did not achieve acquisition criteria. Representative autoradiograms of
dopaminergic receptor and transporter bindings in brain sections of nicotine contingent,
yoked, and saline-treated animals are shown in Figure 6.6.
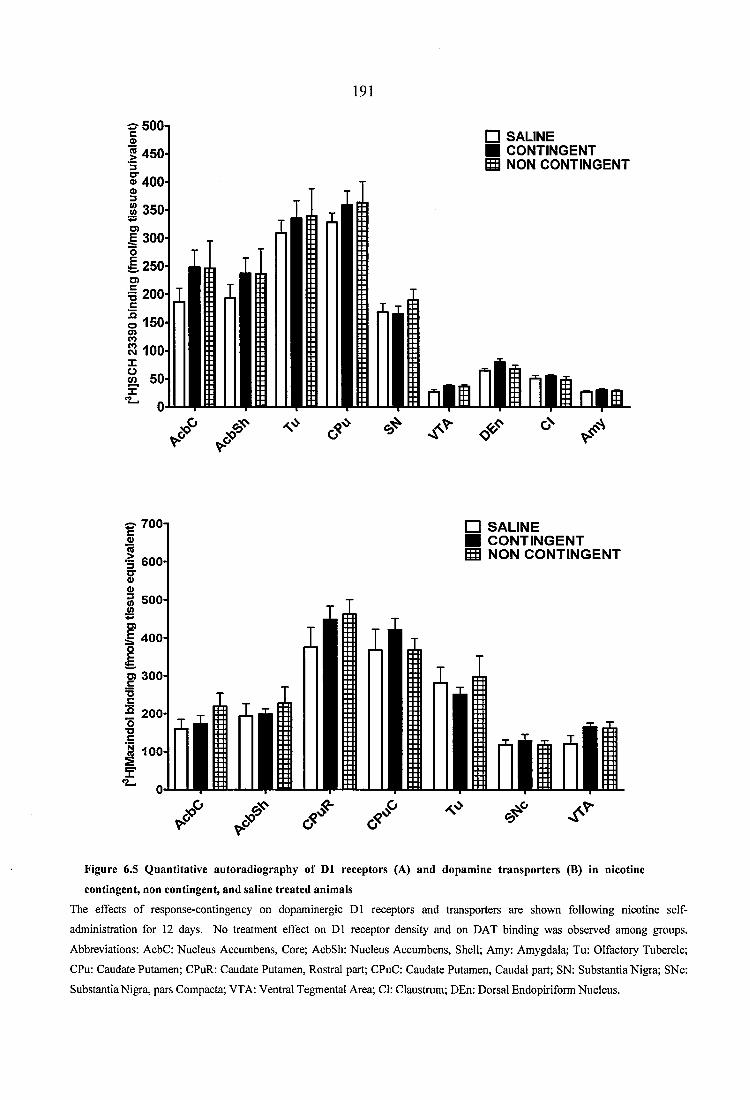
191
□ SALINE ■ CONTINGENT M NON CONTINGENT
3='500
5 450-
o 400-
O)E 300-
I 200-
R 100 linilni□ SA LIN E ■ C O N T IN G E N T H N O N C O N T IN G E N T
23* 700-
5 600-
o) 300-
Figure 6.5 Q uantitative autoradiography o f D1 reeeptors (A) and dopam ine transporters (B) in nicotine
contingent, non contingent, and saline treated animals
The effects of response-contingency on dopaminergic D1 receptors and transporters are shown following nicotine self
administration for 12 days. No treatment effect on D1 receptor density and on DAT binding was observed among groups.
Abbreviations: AcbC: Nucleus Accumbens, Core; AcbSh: Nucleus Accumbens, Shell; Amy: Amygdala; Tu: Olfactory Tubercle;
CPu: Caudate Putamen; CPuR: Caudate Putamen, Rostral part; CPuC: Caudate Putamen, Caudal part; SN: Substantia Nigra; SNc:
Substantia Nigra, pars Compacta; VTA: Ventral Tegmental Area; Cl: Claustrum; DEn: Dorsal Endopiriform Nucleus.
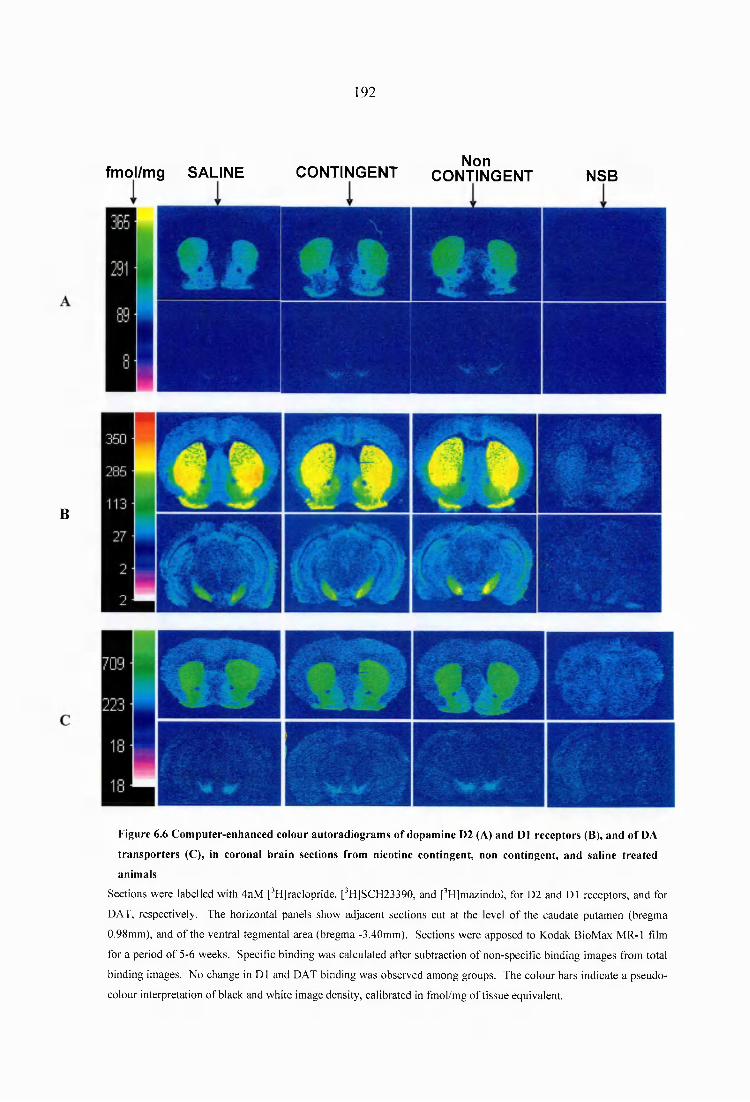
192
fmol/mg SALINE CONTINGENTNon
CONTINGENT NSB
B
Figure 6.6 Com puter-enhanced colour autoradiogram s o f dopam ine D2 (A) and D1 receptors (B), and o f DA
transporters (C), in coronal brain sections from nicotine contingent, non contingent, and saline treated
animals
Sections were labelled with 4nM [^HJraclopride, [^H]SCH23390, and [^H]mazindol, for D2 and DI receptors, and for
DAT, respectively. The horizontal panels show adjacent sections cut at the level of the caudate putamen (bregma
0.98mm), and of the ventral tegmental area (bregma -3.40mm). Sections were apposed to Kodak BioMax MR-1 film
for a period of 5-6 weeks. Specific binding was calculated after subtraction of non-specific binding images from total
binding images. No change in D 1 and DAT binding was observed among groups. The colour bars indicate a pseudo
colour interpretation of black and white image density, calibrated in fmol/mg of tissue equivalent.

193
6.4 Discussion
Previous studies have shown that nicotine, at a concentration range between 0.01-0.05
mg/kg/infusion, supports self-administration in diverse species, including humans
(Henningfield et al., 1983), non-human primates (Goldberg et al., 1981) and rodents
(Martellotta et al., 1995; Donny et al., 1998). In agreement with this literature, it is
shown here that freely moving C57BL/6 mice will chronically self-administer an optimal
dose of response-dependent nicotine, using nose-poking as the operating procedure. The
yoked-control model of self-administration allows discriminating the neurobiological
response that occurs as a result o f the direct pharmacological effects of a drug, from
neuroplasticity that is due to the contingency between a behavioural reaction and drug
delivery. By applying this operant paradigm in mice, it is revealed that differential
neuroadaptation takes place in the nicotinic cholinergic system, depending on whether
nicotine is administered actively or passively. Indeed, cytisine-sensitive [^^^I]epibatidine
binding differed between self-administering and nicotine-yoked animals in the ventral
tegmental area (VTA) and the dorsal lateral geniculate nucleus (DLG), suggesting that
these brain regions are critically involved in the processes relating to nicotine self
administration.
The increased rate of self-administration observed on days 1 and 2 may be partly
attributed to the novelty of the experimental procedure. Similar patterns of increased
intake during the initial phase of drug exposure have also been observed elsewhere
(Mendizabal et al., 2006). As early as on day 1, however, contingent mice were
discriminating between the active and inactive holes, suggesting that the enhanced initial
consumption may partly constitute a specific, nicotine-maintained effect on behaviour.
In line with this, a pattern of escalated drug intake that levels off in subsequent sessions

194
has also been observed in rats that chronically self-administer nicotine (O'Dell et al.,
2007). Out of a wide range of doses, acute self-administration of nicotine in mice occurs
around a narrow window of approximately 0.03 mg/kg/infusion, indicating that rodents
will tightly regulate their amount of nicotine intake (Paterson et al., 2003; Pons et al.,
2008). Indeed, titration seems to be an important feature of nicotine self-administration
across species, as both adult (Benowitz and Jacob, 1985) and adolescent (Kassel et al.,
2007) smokers carefiilly control their nicotine levels in order to experience the
reinforcing effects of the drug. The observed enhanced initial rate of nicotine
consumption may thus reflect a learning response at a given level of titration, which
rapidly occurred within the first two experimental sessions.
Following day 7, reliable drug-taking behaviour was observed in animals receiving
response-dependent nicotine, which was maintained for the duration of the experiment.
Response-contingent animals adjusted their behaviour to obtain a mean drug intake of
0.25 mg/kg/session, which approximates the range of nicotine chronically self
administered by rats (Donny et al., 1995; Kenny and Markou, 2006). As demonstrated by
cytisine-sensitive [^^^Ijepibatidine autoradiography, exposure to this dose of nicotine lead
to region-specific increases of a4p2* nAChR density in the brains of self-administering
animals. The upregulation displayed marked regional variability, with several areas of
the brain participating and showing different sensitivity to upregulation. In this respect,
these findings are consistent with previous observations that experimenter-administered
nicotine (Marks et al., 1983; Schwartz and Kellar, 1983; Nguyen et al., 2003), as well as
nicotine self-administration by rats (Parker et al., 2004), produces regional-specific
increases in the density of nicotinic receptors. A plethora of experimental work has
established nAChR upregulation as the hallmark of chronic nicotine treatment. Using

195
intravenous nicotine infusion, it has been consistently demonstrated that nicotine-induced
receptor upregulation is a dose and time dependent phenomenon, with the half maximal
dose required to induce 50% of region-specific nAChR upregulation in mice being
approximately 0.5 mg/kg/hr (Marks et al., 1991; Pauly et al., 1991; Marks et al., 2004).
In the present study, the mean daily amount of se lf administered nicotine was at 0.25
mg/kg/hr. Therefore, it is not surprising that this dose failed to produce pronounced
nAChR upregulation in animals passively receiving nicotine. Equal doses of self
administered nicotine, however, produced the typical pattern of dose-dependent, region-
specific nAChR upregulation across several brain regions. Therefore, this study
underlies the significance of contingency in nicotine-induced neuroadaptation, since
a4p2* nAChR upregulation was observed with smaller, albeit behaviourally relevant
doses of nicotine. The finding that cytisine-sensitive [^^^I]epibatidine binding sites were
increased in response-dependent mice that were stably se lf administering nicotine, as
well as in animals that did not achieve acquisition criteria, further supports that it is the
contingent relationship between a behavioural response and drug delivery that crucially
mediates nicotine-induced neuroplasticity.
Among the regions that responded differently to active and passive nicotine
administration, the VTA is the biological substrate most likely to be involved in the
initiation and maintenance of drug-taking behaviour. The VTA is source of the
dopaminergic projection to the nucleus accumbens (NAc), and it has been implicated in
mediating the reinforcing properties of many drugs of abuse, including nicotine (Di
Chiara, 2000). Rats will self-administer nicotine directly into the VTA through
intracranial injections, a behaviour that is attenuated by co-infusion of the p2-subunit
antagonist DhOE, indicating that nicotine’s rewarding effects are exerted through

196
nAChRs in this brain region (Corrigall et al., 1994). Moreover, evidence from studies on
gain-of-function a4 nAChR subunit knock-in animals (Tapper et al., 2004), as well as
data from p2 subunit knock-out mice (Picciotto et al., 1998), further reveals the critical
role of VTA a4p2* receptors in mediating nicotine reinforcement. Therefore, the present
data are not only in keeping with several other lines of evidence, but further suggest that
a4p2* nAChRs in the VTA are particularly sensitive to small doses of nicotine, when the
drug is self-administered rather than passively received.
A large body of literature suggests that nicotine reinforcement is not only the result of the
drug’s primary rewarding effects, but also stems from nicotine’s ability to establish
concurrent stimuli as conditioned reinforcers (reviewed in Chaudhri et al., 2006). In rat
studies, environmental stimuli promote the rapid acquisition of drug-seeking behaviour
(Caggiula et al., 2002), and the maintenance of nicotine self-administration during saline
substitution (Donny et al., 1999). Although the dissociation of nicotine’s primary
reinforcing, from its reinforcement-enhancing effects was not an aim of this study, the
autoradiography data are suggestive of a role for the visually paired stimulus in the
acquisition of nicotine self-administration. Increased cytisine-sensitive [^^^Ijepibatidine
binding was observed in self-administering mice compared to nicotine yoked animals in
the DLG, a brain region that is dedicated to the processing of visual information. In cells
of the DLG, P2 subunits have been associated with fast latencies of visual responding,
and high visually evoked firing rates (Grubb and Thompson, 2004). Moreover, mouse
DLG neurons are capable of switching from tonic to burst firing, in order to facilitate
signal detection (Grubb and Thompson, 2005). The enhanced density of a4p2* nAChRs
in these brain areas may therefore underlie intense visual information processing, which
might contribute to the acquisition of self-administration.

197
Neuronal nAChRs constitute a heterogeneous family of receptors, which, depending on
their subunit composition, exhibit distinct physiological and pharmacological properties
(reviewed in Dani and Bertrand, 2007). From the present results, the possibility that
contingent nicotine preferentially increases cytisine-sensitive [^^^IJepibatidine binding,
while its passive administration regulates other nicotinic subtypes in a similar manner
cannot be excluded. Cytisine-resistant nAChRs correspond to structurally diverse
receptor subtypes, which require expression of a3, a4, a6, a2, p2, and p4 subunits (Gotti
et al., 2005; Marks et al., 2006). Although cytisine-resistant nAChR density did not
differ between animals receiving nicotine in a response-dependent or passive manner,
subtle alterations in subunit-specific responses to contingent or passive nicotine remain to
be examined. a6 subunits, for instance, have been shown to be crucially involved in
acute nicotine self-administration (Pons et al., 2008). However, autoradiographic studies
on the effects of chronic nicotine administration on a6* nAChRs have not yet produced
conclusive results (Nguyen et al., 2003; Parker et al., 2004; Mugnaini et al., 2006). In
addition, a3p4 nAChRs are relatively resistant to up-regulation, both in vivo and in vitro
(Wang et al., 1998; Davila-Garcia et al., 2003; Nguyen et al., 2003). Moreover, evidence
argues against the role of a7 nAChRs in nicotine reinforcement (Walters et al., 2006;
Pons et al., 2008). Therefore, it seems probable that the effects of operant contingency
are primarily depicted as changes in cytisine-sensitive nAChR density in self-
administering mice.
Whatever produces neurobiological differences between response-dependent and
passively received nicotine, it does not seem to involve dopaminergic DI and D2
receptors or the dopamine transporters. In the current study, drug administration did not

198
alter DI receptor or DAT density, and it equally upregulated D2 receptors in contingent
and non-contingent animals. As shown by in vivo microdialysis, extracellular dopamine
levels do not differ between rats self-administering nicotine and nicotine-yoked animals,
further suggesting that dopaminergic responses to chronic nicotine administration are
independent of response contingency (Rahman et al., 2004). D2 receptors have been
implicated in nicotine dependence in both human (McEvoy et al., 1995) and animal
experiments (Ikemoto et al., 2006). Radioligand studies, however, have produced
conflicting results as to the effect of chronic nicotine on D2 receptor density. When
delivered through osmotic minipumps, nicotine has been shown to decrease (Janson et al.,
1992) or produce no differences (Kirch et al., 1992) in D2 receptor density. Therefore,
D2 receptor regulation seems to depend on the paradigm of nicotine administration
employed, as well as on the drug dose and the duration of treatment.
In conclusion, these results demonstrate the influence of response-dependency on the
neurobiological output of nicotine. The acquisition of self-administration specifically
involved a4p2* nAChRs of the visual and reward circuitry, whereas the administration of
equal doses of nicotine in a passive manner produced little change in the density of
cytisine-sensitive receptors. These data add to the increasing evidence that experimenter
and self-administered drugs of abuse produce different effects (Hemby et al., 1997;
Kuzmin and Johansson, 1999; Donny et al., 2000; Lecca et al., 2007), and suggest that
nicotine-induced neuroadaptation crucially depends on the contingent relationship
between a subject’s response and the delivery of the reinforcer.

199
CHAPTER
SEVEN

2 0 0
7 CONCLUSIONS AND FUTURE WORK
In the present thesis, in vivo and in vitro experiments have been carried out to further
characterize the mechanism of nicotine’s abuse related potential, and the interactions that
take place between nicotinic cholinergic with dopaminergic, opioidergic, and
adenosinergic systems. A large body of published research indicates that nicotine is a
weak primary reinforcer, and that the predominance of nicotine addiction stems from the
drug’s reward conveying properties upon nicotine-associated stimuli (reviewed in
Caggiula et al., 2009). As nicotine exerts its effects through neuronal nAChRs, it was
hypothesized that changes in the number of heteromeric and/or homomeric binding sites
occur following the administration of drugs of abuse, which could be indicative of the
interactions that render nicotine so widely abused.
7.1 Neuronal nicotinic receptor subtypes differentially influence cocaine-induced
behaviour, and are differentially regulated by chronic cocaine treatment and
withdrawal
By employing a ‘binge’ cocaine administration protocol, it is shown here for the first
time that the two most abundantly expressed nAChR subtypes, namely a4p2* and a7,
mediate distinct behavioural effects of chronic cocaine administration. a4p2* nAChRs
enhance the stimulating properties of the drug, whereas a7 nAChRs protect against the
incidence of an intense cocaine-induced grooming phenotype, which is reminiscent of
animal models of stress-induced or compulsive disorders. Moreover, cytisine-sensitive
[^^^I]epibatidine binding is region-specifically increased in the VTA and SNc of cocaine-
treated animals, whereas withdrawal from chronic cocaine results in a decrease of a7
nAChR density in the VTA of abstinent mice, compared to controls. As there is currently
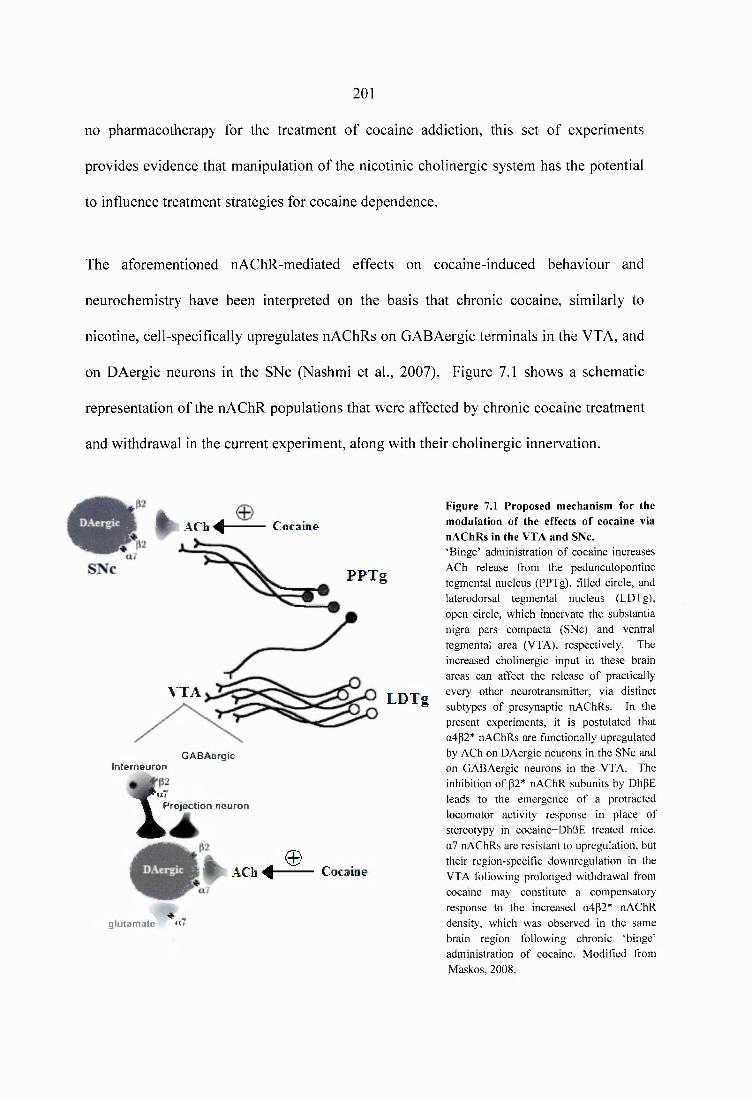
2 0 1
no pharmacotherapy for the treatment of cocaine addiction, this set of experiments
provides evidence that manipulation of the nicotinic cholinergic system has the potential
to influence treatment strategies for cocaine dependence.
The aforementioned nAChR-mediated effects on cocaine-induced behaviour and
neurochemistry have been interpreted on the basis that chronic cocaine, similarly to
nicotine, cell-specifically upregulates nAChRs on GABAergic terminals in the VTA, and
on DAergic neurons in the SNc (Nashmi et al., 2007). Figure 7.1 shows a schematic
representation of the nAChR populations that were affected by chronic cocaine treatment
and withdrawal in the current experiment, along with their cholinergic innervation.
Cocaine
PPTg
# A C h 4
VTA LDTg
Interneuroit
1
GABAergic
Projection neuron
# ACb 4 -©
Cocaine
glutamate * " 7
Figure 7.1 Proposed mechanism for the modulation of the effects of cocaine via nAChRs in the VTA and SNc.‘Binge’ administration of cocaine increases ACh release from the pedunculopontine tegmental nucleus (PPTg), filled circle, and laterodorsal tegmental nucleus (LDTg), open circle, which innervate the substantia nigra pars compacta (SNc) and ventral tegmental area (VTA), respectively. The increased cholinergic input in these brain areas can affect the release of practically every other neurotransmitter, via distinct subtypes of presynaptic nAChRs. In the present experiments, it is postulated that a4p2* nAChRs are functionally upregulated by ACh on DAergic neurons in the SNc and on GABAergic neurons in the VTA. The inhibition of p2* nAChR subunits by DhpE leads to the emergence of a protracted locomotor activity response in place of stereotypy in cocaine+DhBE treated mice. a7 nAChRs are resistant to upregulation, but their region-specific downregulation in the VTA following prolonged withdrawal from cocaine may constitute a compensatory response to the increased a4p2* nAChR density, which was observed in the same brain region following chronic ‘binge’ administration of cocaine. Modified from Maskos, 2008.

202
Altough the current data strongly imply that the upregulation of a4p2* nAChRs in the
SNc and the VTA is a neurobiological commonality that is shared by chronic nicotine
and cocaine, the exact subunit composition of the nAChR subtypes that mediate the
prolonged cocaine+DhBE induced locomotor activity, or the intense grooming phenotype
following cocaine and MLA administration, cannot be determined from the present study.
A confounding factor in the analyses of nAChR involvement in the behavioural effects of
cocaine is the lack of pharmacological tools that selectively block individual nAChR
subtypes. MLA, for instance, which was used to block a7 receptors, has been shown to
have an affinity for nAChRs containing the a4/p2 (Buisson et al., 1996), a3/a6 (Mogg et
al., 2002), and a3/p4 (Free et al., 2003) subunits. Therefore, the wide variety of nicotinic
subtypes expressed in the CNS, coupled with the lack of subtype-selective ligands,
renders the attribution of specific cocaine-induced behavioural phenotypes to specific
nAChRs on individual neuronal populations extremely optimistic. To more accurately
investigate the exact location and contribution of nAChRs to cocaine-induced behaviour,
nAChR antagonists can be administered to the postulated brain regions of cocaine-treated
mice intracerebrally, rather than systemically. This would allow a variety of selective
nAChR antagonists to be tested for their effects on ‘binge’ cocaine-induced behaviour,
without the confounding factors of blood brain barrier penetration or peripheral toxicity.
In addition, several nAChR subunit gene knockout mice are currently available (see
Section 1.1.4.2), which can be used to reproduce the phenotypes that were observed in
the present study. The application of transgenic animal models would confirm that lower
a7 nAChR availability predisposes to cocaine-induced behavioural abnormalities,
whereas the availability of P2* subunits determines cocaine-induced stimulation.

203
7.2 The density of distinct neuronal nicotinic receptor subtypes is not altered by
escalating doses of morphine, and following the acute and long-term withdrawal
from ‘intermittent’ morphine administration
The results from these studies reveal that morphine, contrary to cocaine, does not directly
interact with nAChRs at the receptor density level. Several explanations have been
offered for the lack of morphine-induced changes in nAChR binding sites, including the
fact that there are multiple levels of synergy between the effects of opiates and nicotine,
which range from the behavioural to the molecular levels (see Section 4.4). It should be
noted that escalating doses of morphine were used in this study, compared to the steady
dosing protocol that was employed in the cocaine experiments. As dosing regimens have
been shown to influence the effects of cocaine and morphine (Schlussman et al., 2005;
Dighe et al., 2009), it could be that changes in nAChR density are predominantly seen
under treatment with steady dose protocols. While this remains to be investigated, the
present results suggest that cocaine-induced effects on the regulation of nAChR binding
sites do not generalize to opiates. Future experiments using pshychostimulants and
opiates other than cocaine and morphine (amphetamines, heroin etc), will confirm that
the two classes of drugs exert different effects on nAChR density, following their
repeated administration.
7.3 The effects of adenosine A2A receptors on nicotine-induced neuroadaptation are
exerted via interactions with the a7 nAChR
The concurrent consumption of nicotine with caffeine, a non-selective adenosine Ai and
A2A receptor antagonist, prompted an investigation into the mechanism of interactions
between nicotine and A2A receptors. This was performed using naïve and nicotine-treated

204
adenosine A2A receptor knockout mice, as highly selective tools for the analysis of the
contribution of individual receptors to physiological function. The results of the present
study show that in the absence of adenosine A2A receptors nicotine fails to upregulate a7
nAChR density, suggesting that the effects of the neuromodulator on nicotine-induced
neuroadaptation are exerted via this nicotinic receptor subtype.
As there is a general consensus that the effects of nicotine on dopaminergic
neurotransmission are partly exterted via a7 nAChRs located on glutamatergic nerve
terminals (reviewed in Wonnacott, 1997), the current data suggest that nicotine-adenosine
A2A receptor interactions primarily involve glutamatergic, rather than dopaminergic
mechanisms. Indeed, no differences in DI and D2 dopaminergic receptor and transporter
binding were observed between nicotine-treated wild type and mutant mice. However,
this finding needs to be interpreted with caution. For instance, the small upregulation in
the constitutive expression of D2 receptors that was detected in naïve A2A knockout mice
(Figure 5.6) was not reproduced in saline-treated animals (Figure 5.13). Power analysis,
performed after the completion of these experiments, showed that the study might indeed
have missed a small effect of adenosine A2A receptor gene deletion on D2 receptors.
Calculations based on the mean difference of D2 receptor density between the naïve WT
and KO groups revealed that the nicotine treatment experiment had a power o f 60% to
detect a statistically significant difference of 15.0 fmol/mg tissue equivalent between D2
receptors in saline-treated WT and adenosine A2A receptor KO mice. Therefore, the
possiblility of a Type II error in this study cannot be excluded, and increased n numbers
are required to unambiguously investigate the regulation of D2 receptors after deletion of
the adenosine A2A receptor gene. Alternatively, the discrepancy between the D2 receptor
binding results from the naïve and nicotine-treatment studies may reflect increased inter-

205
individual variability in the regulation of D2 receptors in mice of the outbred CDl strain,
or could even be the result of surgical anaesthesia on the DAergic system.
7.4 The acquisition of nicotine self-administration induces region specific increases
in the density of a4p2* nAChRs, which are not observed following passive
administration of nicotine
Neuronal nAChR upregulation is the hallmark of chronic nicotine exposure.
Neuroplasticity to abused drugs, however, depends on whether their administration is
forced by the experimenter or is under the control of the experimental animal (reviewed
in Jacobs et al., 2003). In this set of experiments, nicotine self-administration specifically
upregulated a4(32* nAChRs of the visual and reward circuitry, whereas the
administration of equal doses of the drug in a passive manner produced little change in
the density of cytisine-sensitive [^^^I]epibatidine binding sites. Hence, these results add
to a large body of literature, documenting that contingent and non-contingent drug
administration produces different neurochemical effects, and extends the published
research on this topic to include nicotine.
Nevertheless, it is difficult to draw any further conclusions from the data, as a number of
fundamental questions arise from this study. First, is there a difference in the sensitivity
(i.e. function) of the upregulated receptors between the contingent and non-contingent
conditions? Second, would passively delivered nicotine also upregulate nAChRs
following longer drug exposure? If so, what is the interpretation of the results in relation
to the establishment and maintenance of nicotine self-administration? Whatever the case
may be, the current results suggest that nicotine-induced neuroadaptation crucially

206
depends on the contingent relationship between a subject’s response and the delivery of
the reinforcer, rather than on the amount of nicotine intake.
7.5 Quantitative autoradiography measurements
Quantitative autoradiography of nAChRs was conducted across all chapters of the present
thesis. For heteromeric nAChRs, the quantification of total, cytisine-resistant, and
cytisine-sensitive [^^^Ijepibatidine binding in brain sections from naïve or saline-treated
controls was reproducible across chapters. [^^^Ijepibatidine binding showed extremely
high selectivity for nAChRs; non-specific binding in the presence of excess nicotine as a
‘cold’ displacement ligand was literally indistinguishable from autoradiographic film
background. Extremelly high levels of total [^^^Ijepibatidine binding were observed in
the medial habenula, fasciculus retroflexus, and the interpeduncular nucleus, followed by
high to moderate levels of binding in the superficial gray layer of the superior colliculus,
certain thalamic nuclei, the ventral tegmental area, and the substantia nigra. Low levels
of binding were detected in the striatum, and in areas of the cortex. High levels of
cytisine-resistant [^^^Ijepibatidine binding were observed in the medial habenula,
fasciculus retroflexus, and interpeduncular nucleus, indicating that the nAChRs expressed
in these brain regions are not of the cytisine-sensitive, a4|32* nAChR subtype. In general,
these results are in good agreement with several rodent studies, which have used
comparable amounts of [^^^Ijepibatidine for the visualisation of nAChRs (Nguyen et ah,
2003; Whiteaker et al., 2002).
For the a? nAChR, [^^^I]a-bungarotoxin binding was higher in the superficial gray layer
of the superior colliculus and the zona incerta, followed by lower levels of binding in
nuclei of the amygdale, hypothalamus, hippocampus, and certain cortical brain regions.

207
This pattern of distribution is consistent with the literature (Rasmussen and Perry, 2006;
Sparks and Pauly, 1999). It should be noted that variation in the amount of specific a7
nAChR binding was noted between chapter 3 and chapters thereafter, which the author
attributes to the use of radioligands from different suppliers (GE Healthcare vs. Perkin
Elmer), producing [^^^I]a-bungarotoxin at different specific activities (GE Healthcare,
specific activity 250 Ci/mmol; PerkinElmer, specific activity 159 Ci/mmol), using
different means of production (mono- vs. di- iodinated products). This change was
necessary, however, as GE Healthcare announced the closure of its radiochemical
product line in December 2008. Although the change of supplier does not affect the
distribution pattern of a7 nAChRs, it underlines the importance of conducting
quantitative autoradiography using paired binding protocols, in which all sections are
processed together in an identical manner, in a carefully controlled environment.
7.6 Concluding remarks
The present work has generated further evidence for the presence of interactions that
might underlie why nicotine is one of the most widely abused substances in the world.
With the exception of morphine, all other types of interactions that were examined could
be depicted at the nicotinic receptor level, as changes in the density of distinct nAChR
subtypes. Importantly, this set of studies has prompted new questions concerning the
nature and mechanism of interactions between the nicotinic cholinergic with
dopaminergic, opioidergic, and adenosinergic systems, which remain to be answered.

208
REFERENCES

209
Abreu-Villaca Y, Seidler FJ, Slotkin TA (2003) Impact of adolescent nicotine exposure on adenylyl cyclase-mediated cell signaling: enzyme induction, neurotransmitter- specific effects, regional selectivities, and the role of withdrawal. Brain Res 988:164-172.
Acquas E, Tanda G, Di Chiara G (2002) Differential effects of caffeine on dopamine and acetylcholine transmission in brain areas of drug-naive and caffeine-pretreated rats. Neuropsychopharmacology 27:182-193.
Albuquerque EX, Pereira EF, Alkondon M, Rogers SW (2009) Mammalian nicotinic acetylcholine receptors: from structure to function. Physiol Rev 89:73-120.
Aldridge JW, Berridge KG (1998) Coding of serial order by neostriatal neurons: a "natural action" approach to movement sequence. J Neurosci 18:2777-2787.
Aliane V, Pérez S, Nieoullon A, Deniau JM, Kemel ML (2009) Cocaine-induced stereotypy is linked to an imbalance between the medial prefrontal and sensorimotor circuits of the basal ganglia. European Journal of Neuroscience 30:1269-1279.
Alkondon M, Albuquerque EX (1993) Diversity of nicotinic acetylcholine receptors in rat hippocampal neurons. I. Pharmacological and functional evidence for distinct structural subtypes. J Pharmacol Exp Ther 265:1455-1473.
Alkondon M, Pereira EF, Albuquerque EX (1998) alpha-bungarotoxin- and methyllycaconitine-sensitive nicotinic receptors mediate fast synaptic transmission in intemeurons of rat hippocampal slices. Brain Res 810:257-263.
Alkondon M, Rocha ES, Maelicke A, Albuquerque EX (1996) Diversity of nicotinic acetylcholine receptors in rat brain. V. alpha-Bungarotoxin-sensitive nicotinic receptors in olfactory bulb neurons and presynaptic modulation of glutamate release. J Pharmacol Exp Ther 278:1460-1471.
Alkondon M, Pereira EF, Barbosa CT, Albuquerque EX (1997) Neuronal nicotinic acetylcholine receptor activation modulates gamma-aminobutyric acid release from CAl neurons of rat hippocampal slices. J Pharmacol Exp Ther 283:1396- 1411.
Amalric M, Koob GF (1993) Functionally selective neurochemical afférents and efferents of the mesocorticolimbic and nigrostriatal dopamine system. Prog Brain Res 99:209-226.
Anand R, Nelson ME, Gerzanich V, Wells GB, Lindstrom J (1998) Determinants of channel gating located in the N-terminal extracellular domain of nicotinic alpha? receptor. J Pharmacol Exp Ther 287:469-479.

2 1 0
Anderson DJ, Americ SP (1994) Nicotinic receptor binding of [3H]cytisine, [3H]nicotine and [3H]methylcarbamylcholine in rat brain. Eur J Pharmacol 253:261-267.
Anderson SM, Pierce RC (2005) Cocaine-induced alterations in dopamine receptor signaling: implications for reinforcement and reinstatement. Pharmacol Ther 106:389-403.
Antonelli T, Beani L, Bianchi C, Rando S, Simonato M, Tanganelli S (1986) Cortical acetylcholine release is increased and gamma-aminobutyric acid outflow is reduced during morphine withdrawal. Br J Pharmacol 89:853-860.
Aosaki T, Tsubokawa H, Ishida A, Watanabe K, Graybiel AM, Kimura M (1994) Responses of tonically active neurons in the primate's striatum undergo systematic changes during behavioral sensorimotor conditioning. J Neurosci 14:3969-3984.
Araki H, Kawakami KY, Jin C, Suemaru K, Kitamura Y, Nagata M, Futagami K, Shibata K, Kawasaki H, Gomita Y (2004) Nicotine attenuates place aversion induced by naloxone in single-dose, morphine-treated rats. Psychopharmacology (Berl) 171:398-404.
Arenas E, Alberch J, Sanchez Arroyos R, Marsal J (1990) Effect of opioids on acetylcholine release evoked by K+ or glutamic acid from rat neostriatal slices. Brain Res 523:51-56.
Azam L, Winzer-Serhan UH, Chen Y, Leslie FM (2002) Expression of neuronal nicotinic acetylcholine receptor subunit mRNAs within midbrain dopamine neurons. J Comp Neurol 444:260-274.
Azar MR, Acar N, Erwin VG, Barbato GF, Morse AC, Heist CL, Jones BC (1998) Distribution and clearance of cocaine in brain is influenced by genetics. Pharmacol Biochem Behav 59:637-640.
Badio B, Daly JW (1994) Epibatidine, a potent analgetic and nicotinic agonist. Mol Pharmacol 45:563-569.
Bahk JY, Li S, Park MS, Kim MO (2002) Dopamine DI and D2 receptor mRNA up- regulation in the caudate-putamen and nucleus accumbens of rat brains by smoking. Prog Neuropsychopharmacol Biol Psychiatry 26:1095-1104.
Bailey A, Ledent C, Kelly M, Hourani SM, Kitchen I (2002) Changes in spinal delta and kappa opioid systems in mice deficient in the A2A receptor gene. J Neurosci 22:9210-9220.
Bailey A, Metaxas A, Yoo JH, McGee T, Kitchen I (2008) Decrease of D2 receptor binding but increase in D2-stimulated G-protein activation, dopamine transporter binding and behavioural sensitization in brains of mice treated with a chronic

2 1 1
escalating dose ’binge' cocaine administration paradigm. Eur J Neurosci 28:759- 770.
Bailey A, Weber D, Zimmer A, Zimmer AM, Hourani SM, Kitchen 1 (2004a) Quantitative autoradiography of adenosine receptors and NBTl-sensitive adenosine transporters in the brains of mice deficient in the preproenkephalin gene. Brain Res 1025:1-9.
Bailey A, Davis L, Lesscher HM, Kelly MD, Ledent C, Hourani SM, Kitchen 1 (2004b) Enhanced morphine withdrawal and micro -opioid receptor G-protein coupling in A2A adenosine receptor knockout mice. JNeurochem 88:827-834.
Baker A, Lee NK, Claire M, Lewin TJ, Grant T, Pohlman S, Saunders JB, Kay-Lambkin F, Constable P, Jenner L, Carr VJ (2004) Drug use patterns and mental health of regular amphetamine users during a reported 'heroin drought'. Addiction 99:875- 884.
Barik J, Wonnacott S (2006) Indirect modulation by alpha7 nicotinic acetylcholine receptors of noradrenaline release in rat hippocampal slices: interaction with glutamate and GABA systems and effect of nicotine withdrawal. Mol Pharmacol 69:618-628.
Baskin DG, Wimpy TH (1989) Calibration of [14C]plastic standards for quantitative autoradiography of [125]labeled ligands with Amersham Hyperfilm beta max. Neurosci Lett 104:171-177.
Bayer HM, Glimcher PW (2005) Midbrain dopamine neurons encode a quantitative reward prediction error signal. Neuron 47:129-141.
Beani L, Bianchi C, Siniscalchi A (1982) The effect of naloxone on opioid-induced inhibition and facilitation of acetylcholine release in brain slices. Br J Pharmacol 76:393-401.
Bechtholt AJ, Mark GP (2002) Enhancement of cocaine-seeking behavior by repeated nicotine exposure in rats. Psychopharmacology (Berl) 162:178-185.
Benowitz NL, Jacob P, 3rd (1985) Nicotine renal excretion rate influences nicotine intake during cigarette smoking. J Pharmacol Exp Ther 234:153-155.
Benwell ME, Balfour DJ, Anderson JM (1988) Evidence that tobacco smoking increases the density of (-)-[3H]nicotine binding sites in human brain. J Neurochem 50:1243-1247.
Berke JD, Hyman SE (2000) Addiction, dopamine, and the molecular mechanisms of memory. Neuron 25:515-532.

2 1 2
Berrendero F, Kieffer BL, Maldonado R (2002) Attenuation of nicotine-induced antinociception, rewarding effects, and dependence in mu-opioid receptor knockout mice. J Neurosci 22:10935-10940.
Berrendero F, Mendizabal V, Robledo P, Galeote L, Bilkei-Gorzo A, Zimmer A, Maldonado R (2005) Nicotine-induced antinociception, rewarding effects, and physical dependence are decreased in mice lacking the preproenkephalin gene. J Neurosci 25:1103-1112.
Berridge KG, Whishaw IQ (1992) Cortex, striatum and cerebellum: control of serial order in a grooming sequence. Exp Brain Res 90:275-290.
Berridge KG, Aldridge JW (2000) Super-stereotypy I: enhancement of a complex movement sequence by systemic dopamine DI agonists. Synapse 37:194-204.
Berridge KG, Fentress JG, Parr H (1987) Natural syntax rules control action sequence of rats. Behav Brain Res 23:59-68.
Berridge KG, Aldridge JW, Houchard KR, Zhuang X (2005) Sequential super-stereotypy of an instinctive fixed action pattern in hyper-dopaminergic mutant mice: a model of obsessive compulsive disorder and Tourette’s. BMG Biol 3:4.
Besson M, Granon S, Mameli-Engvall M, Gloez-Tayarani I, Maubourguet N, Cormier A, Gazala P, David V, Ghangeux JP, Faure P (2007) Long-term effects of chronic nicotine exposure on brain nicotinic receptors. Proc Natl Acad Sci U S A 104:8155-8160.
Bhargava HN, Way EL (1975) Brain acetylcholine and choline following acute and chronic morphine treatment and during withdrawal. J Pharmacol Exp Ther 194:65-73.
Biala G, Weglinska B (2004) Calcium channel antagonists attenuate cross-sensitization to the rewarding and/or locomotor effects of nicotine, morphine and MK-801. J Pharm Pharmacol 56:1021-1028.
Biala G, Budzynska B, Kruk M (2005) Naloxone precipitates nicotine abstinence syndrome and attenuates nicotine-induced antinociception in mice. Pharmacol Rep 57:755-760.
Birrell GE, Balfour DJ (1998) The influence of nicotine pretreatment on mesoaccumbens dopamine overflow and locomotor responses to D-amphetamine. Psychopharmacology (Berl) 140:142-149.
Bjorklund A, Dunnett SB (2007) Dopamine neuron systems in the brain: an update. Trends Neurosci 30:194-202.

213
Blokhina EA, Kashkin VA, Zvartau EE, Danysz W, Bespalov AY (2005) Effects of nicotinic and NMDA receptor channel blockers on intravenous cocaine and nicotine self-administration in mice. Eur Neuropsychopharmacol 15:219-225.
Borycz J, Pereira MF, Melani A, Rodrigues RJ, Kofalvi A, Panlilio L, Pedata F, Goldberg SR, Cunha RA, Ferre S (2007) Differential glutamate-dependent and glutamate- independent adenosine A1 receptor-mediated modulation of dopamine release in different striatal compartments. J Neurochem 101:355-363.
Bouthenet ML, Souil E, Martres MP, Sokoloff P, Giros B, Schwartz JC (1991) Localization of dopamine D3 receptor mRNA in the rat brain using in situ hybridization histochemistry: comparison with dopamine D2 receptor mRNA. Brain Res 564:203-219.
Brecht ML, Greenwell L, Anglin MD (2007) Substance use pathways to methamphetamine use among treated users. Addict Behav 32:24-38.
Breese CR, Marks MJ, Logel J, Adams CE, Sullivan B, Collins AC, Leonard S (1997) Effect of smoking history on [3H]nicotine binding in human postmortem brain. J Pharmacol Exp Ther 282:7-13.
Brejc K, van Dijk WJ, Klaassen RV, Schuurmans M, van Der Cost J, Smit AB, Sixma TK (2001) Crystal structure of an ACh-binding protein reveals the ligand-binding domain of nicotinic receptors. Nature 411:269-276.
Breslau N (1995) Psychiatric comorbidity of smoking and nicotine dependence. Behav Genet 25:95-101.
Britt JP, McGehee DS (2008) Presynaptic opioid and nicotinic receptor modulation of dopamine overflow in the nucleus accumbens. J Neurosci 28:1672-1681.
Brockwell NT, Beninger RJ (1996) The differential role of A1 and A2 adenosine receptor subtypes in locomotor activity and place conditioning in rats. Behav Pharmacol 7:373-383.
Brockwell NT, Eikelboom R, Beninger RJ (1991) Caffeine-induced place and taste conditioning: production of dose-dependent preference and aversion. Pharmacol Biochem Behav 38:513-517.
Brown CR, Benowitz NL (1989) Caffeine and cigarette smoking: behavioral, cardiovascular, and metabolic interactions. Pharmacol Biochem Behav 34:565- 570.
Brown SJ, James S, Reddington M, Richardson PJ (1990) Both A1 and A2a purine receptors regulate striatal acetylcholine release. J Neurochem 55:31-38.

214
Brunzell DH, Russell DS, Picciotto MR (2003) In vivo nicotine treatment regulates mesocorticolimbic CREB and ERK signaling in C57B1/6J mice. J Neurochem 84:1431-1441.
Brunzell DH, Mineur YS, Neve RE, Picciotto MR (2009) Nucleus accumbens CREB activity is necessary for nicotine conditioned place preference. Neuropsychopharmacology 34:1993-2001.
Budney AJ, Higgins ST, Hughes JR, Bickel WK (1993) Nicotine and caffeine use in cocaine-dependent individuals. J Subst Abuse 5:117-130.
Buisson B, Bertrand D (2001) Chronic exposure to nicotine upregulates the human (alpha)4((beta)2 nicotinic acetylcholine receptor function. J Neurosci 21:1819- 1829.
Buisson B, Gopalakrishnan M, Americ SP, Sullivan JP, Bertrand D (1996) Human alpha4beta2 neuronal nicotinic acetylcholine receptor in HEK 293 cells: A patch- clamp study. J Neurosci 16:7880-7891.
Burghaus L, Schütz U, Krempel U, Lindstrom J, Schroder H (2003) Loss of nicotinic acetylcholine receptor subunits alpha4 and alpha? in the cerebral cortex of Parkinson patients. Parkinsonism Relat Disord 9:243-246.
Cadet JL, Bolla KI (1996) Chronic cocaine use as a neuropsychiatrie syndrome: a model for debate. Synapse 22:28-34.
Caggiula AR, Donny EC, Chaudhri N, Perkins KA, Evans-Martin FF, Sved AF (2002) Importance of nonpharmacological factors in nicotine self-administration. Physiol Behav 77:683-687.
Caggiula AR, Donny EC, Palmatier MI, Liu X, Chaudhri N, Sved AF (2009) The role of nicotine in smoking: a dual-reinforcement model. Nebr Symp Motiv 55:91-109.
Caine SB, Koob GF (1993) Modulation of cocaine self-administration in the rat through D-3 dopamine receptors. Science 260:1814-1816.
Campbell KM, McGrath MJ, Burton FH (1999) Behavioral effects of cocaine on a transgenic mouse model of cortical-limbic compulsion. Brain Res 833:216-224.
Canals M, Marcellino D, Fanelli F, Ciruela F, de Benedetti P, Goldberg SR, Neve K, Fuxe K, Agnati LF, Woods AS, Ferre S, Lluis C, Bouvier M, Franco R (2003) Adenosine A2A-dopamine D2 receptor-receptor heteromerization: qualitative and quantitative assessment by fluorescence and bioluminescence energy transfer. J Biol Chem 278:46741-46749.

215
Carbon! E, Bortone L, Giua C, Di Chiara G (2000) Dissociation of physical abstinence signs from changes in extracellular dopamine in the nucleus accumbens and in the prefrontal cortex of nicotine dependent rats. Drug Alcohol Depend 58:93-102.
Carrera MR, Meijler MM, Janda KD (2004) Cocaine pharmacology and current pharmacotherapies for its abuse. Bioorg Med Chem 12:5019-5030.
Castane A, Soria G, Ledent C, Maldonado R, Valverde O (2006) Attenuation of nicotine- induced rewarding effects in A2A knockout mice. Neuropharmacology 51:631- 640.
Castro NG, Albuquerque EX (1993) Brief-lifetime, fast-inactivating ion channels account for the alpha-bungarotoxin-sensitive nicotinic response in hippocampal neurons. Neurosci Lett 164:137-140.
Celik E, Uzbay IT, Karakas S (2006) Caffeine and amphetamine produce crosssensitization to nicotine-induced locomotor activity in mice. Prog Neuropsychopharmacol Biol Psychiatry 30:50-55.
Champtiaux N, Kalivas PW, Bardo MT (2006) Contribution of dihydro-beta-erythroidine sensitive nicotinic acetylcholine receptors in the ventral tegmental area to cocaine-induced behavioral sensitization in rats. Behavioural Brain Research 168:120-126.
Champtiaux N, Han ZY, Bessis A, Rossi FM, Zoli M, Marubio L, McIntosh JM, Changeux JP (2002) Distribution and pharmacology of alpha 6-containing nicotinic acetylcholine receptors analyzed with mutant mice. J Neurosci 22:1208- 1217.
Champtiaux N, Gotti C, Cordero-Erausquin M, David DJ, Przybylski C, Lena C, Clement! F, Moretti M, Rossi FM, Le Novere N, McIntosh JM, Gardier AM, Changeux JP (2003) Subunit composition of functional nicotinic receptors in dopaminergic neurons investigated with knock-out mice. J Neurosci 23:7820- 7829.
Changeux J-P, Edelstein SJ (1998) Allosteric Receptors after 30 Years. Neuron 21:959- 980.
Chaudhri N, Caggiula AR, Donny EC, Palmatier MI, Liu X, Sved AF (2006) Complex interactions between nicotine and nonpharmacological stimuli reveal multiple roles for nicotine in reinforcement. Psychopharmacology (Berl) 184:353-366.
Chefer SI, Horti AG, Lee KS, Koren AO, Jones DW, Gorey JG, Links JM, Mukhin AG, Weinberger DR, London ED (1998) In vivo imaging of brain nicotinic acetylcholine receptors with 5-[123I]iodo-A-85380 using single photon emission computed tomography. Life Sci 63:PL355-360.

216
Chen D, Patrick JW (1997) The alpha-bungarotoxin-binding nicotinic acetylcholine receptor from rat brain contains only the alpha? subunit. J Biol Chem 272:24024- 24029.
Chen JF, Huang Z, Ma J, Zhu J, Moratalla R, Standaert D, Moskowitz MA, Fink JS, Schwarzschild MA (1999) A(2A) adenosine receptor deficiency attenuates brain injury induced by transient focal ischemia in mice. J Neurosci 19:9192-9200.
Chen JF, Beilstein M, Xu YH, Turner TJ, Moratalla R, Standaert DG, Aloyo VJ, Fink JS, Schwarzschild MA (2000) Selective attenuation of psychostimulant-induced behavioral responses in mice lacking A(2A) adenosine receptors. Neuroscience 97:195-204.
Chen JF, Xu K, Petzer JP, Staal R, Xu YH, Beilstein M, Sonsalla PK, Castagnoli K, Castagnoli N, Jr., Schwarzschild MA (2001a) Neuroprotection by caffeine and A(2A) adenosine receptor inactivation in a model of Parkinson's disease. J Neurosci 21:RC143.
Chen JF, Moratalla R, Impagnatiello F, Grandy DK, Cuellar B, Rubinstein M, Beilstein MA, Hackett E, Fink JS, Low MJ, Ongini E, Schwarzschild MA (2001b) The role of the D(2) dopamine receptor (D(2)R) in A(2A) adenosine receptor (A(2A)R)- mediated behavioral and cellular responses as revealed by A(2A) and D(2) receptor knockout mice. Proc Natl Acad Sci U S A 98:1970-1975.
Chen R, Tilley MR, Wei H, Zhou F, Zhou FM, Ching S, Quan N, Stephens RL, Hill ER, Nottoli T, Han DD, Gu HH (2006) Abolished cocaine reward in mice with a cocaine-insensitive dopamine transporter. Proc Natl Acad Sci U S A 103:9333- 9338.
Chergui K, Suaud-Chagny MF, Gonon F (1994) Nonlinear relationship between impulse flow, dopamine release and dopamine elimination in the rat brain in vivo. Neuroscience 62:641-645.
Ciliax BJ, Heilman C, Demchyshyn LL, Pristupa ZB, Ince E, Hersch SM, Niznik HB, Levey AT (1995) The dopamine transporter: immunochemical characterization and localization in brain. J Neurosci 15:1714-1723.
Ciruela F, Casado V, Rodrigues RJ, Lujan R, Burgueno J, Canals M, Borycz J, Rebola N, Goldberg SR, Mallol J, Cortes A, Canela El, Lopez-Gimenez JF, Milligan G, Lluis C, Cunha RA, Ferre S, Franco R (2006) Presynaptic control of striatal glutamatergic neurotransmission by adenosine A1-A2A receptor heteromers. J Neurosci 26:2080-2087.
Clarke PB, Hommer DW, Pert A, Skirboll LR (1985a) Electrophysiological actions of nicotine on substantia nigra single units. Br J Pharmacol 85:827-835.

217
Clarke PB, Schwartz RD, Paul SM, Pert CB, Pert A (1985b) Nicotinic binding in ratbrain: autoradiographic comparison of [3H]acetylcholine, [3H]mcotme, and [125I]-alpha-bungarotoxin. J Neurosci 5:1307-1315.
Clarke S, Chen Z, Hsu MS, Pintar J, Hill R, Kitchen I (2001) Quantitative autoradiographic mapping of the ORLl, mu-, delta- and kappa-rec^ors m the brains of knockout mice lacking the ORLl receptor gene. Brain Res 906:13-24.
Coe JW Brooks PR, Vetelino MG, Wirtz MC, Arnold EP, Huang J, Sands SB, Davis Tl, Lebel LA, Fox CB, Shrikhande A, Heym JH, Schaeffer E, Rollema H, Lu Y Mansbach RS, Chambers LK, Rovetti CC, Schulz DW, Tingley FD, 3rd, O'Neill BT (2005) Varenicline: an alpha4beta2 nicotinic receptor partial agonist tor smoking cessation. J Med Chem 48:3474-3477.
Cohen C, Welzl H, Battig K (1991) Effects of nicotine, caffeine, and their combination on locomotor activity in rats. Pharmacol Biochem Behav 40:121-123.
Collins AC, Bhat RV, Pauly JR, Marks MJ (1990) Modulation of nicotine receptors by chronic exposure to nicotinic agonists and antagonists. Ciba Found Symp 152:68- 82; discussion 82-66.
Collins SL, Izenwasser S (2004) Chronic nicotine differentially alters cocaine-induced locomotor activity in adolescent vs. adult male and female rats. Neuropharmacology 46:349-362.
Connolly JG, Gibb AJ, Colquhoun D (1995) Heterogeneity o f neuronal niœtinic acetylcholine receptors in thin slices of rat medial habenula. J Physiol 484 ( Pt 1);87-105.
Conroy WG, Berg DK (1995) Neurons can maintain multiple classes of nicotinic acetylcholine receptors distinguished by different subunit compositions. J Bio Chem 270:4424-4431.
Cordero-Erausquin M, Marubio LM, Klink R, Changeux J-P (2000) Nicotinic receptor function: new perspectives from knockout mice. Trends in Pharmacological Sciences 21:211-217.
Corrigall WA, Coen KM (1991) Selective dopamine antagonists reduce nicotine selfadministration. Psychopharmacology (Berl) 104:171-176.
Corrigall WA, Herling S, Coen KM (1988) Evidence for opioid mechanisms in the behavioral effects of nicotine. Psychopharmacology (Berl) 96:29-35.
Corrigall WA, Coen KM, Adamson KL (1994) Self-administered nicotine activates the mesolimbic dopamine system through the ventral tegmental area. Brain Res 653:278-284.

218
Corringer P-J, Sallette J, Changeux J-P (2006) Nicotine enhances intracellular nicotinic receptor maturation: A novel mechanism of neural plasticity? Journal of Physiology-Paris 99:162-171.
Corringer PJ, Galzi JL, Eisele JL, Bertrand S, Changeux JP, Bertrand D (1995) Identification of a new component of the agonist binding site of the nicotinic alpha 7 homooligomeric receptor. J Biol Chem 270:11749-11752.
Corringer PJ, Bertrand S, Bohler S, Edelstein SJ, Changeux JP, Bertrand D (1998) Critical elements determining diversity in agonist binding and desensitization of neuronal nicotinic acetylcholine receptors. J Neurosci 18:648-657.
Court JA, Lloyd S, Thomas N, Piggott MA, Marshall EF, Morris CM, Lamb H, Perry RH, Johnson M, Perry EK (1998) Dopamine and nicotinic receptor binding and the levels of dopamine and homovanillic acid in human brain related to tobacco use. Neuroscience 87:63-78.
Couturier S, Bertrand D, Matter JM, Hernandez MC, Bertrand S, Millar N, Valera S, Barkas T, Ballivet M (1990) A neuronal nicotinic acetylcholine receptor subunit (alpha 7) is developmentally regulated and forms a homo-oligomeric channel blocked by alpha-BTX. Neuron 5:847-856.
Covey LS, Glassman AH, Stetner F (1999) Naltrexone effects on short-term and longterm smoking cessation. J Addict Dis 18:31-40.
Cragg SJ (2003) Variable dopamine release probability and short-term plasticity between functional domains of the primate striatum. J Neurosci 23:4378-4385.
Crawley JN, Belknap JK, Collins A, Crabbe JC, Frankel W, Henderson N, Hitzemann RJ, Maxson SC, Miner LL, Silva AJ, Wehner JM, Wynshaw-Boris A, Paylor R (1997) Behavioral phenotypes of inbred mouse strains: implications and recommendations for molecular studies. Psychopharmacology (Berl) 132:107-124.
Creese 1, Sibley DR (1981) Receptor adaptations to centrally acting drugs. Annu Rev Pharmacol Toxicol 21:357-391.
Crespo JA, Sturm K, Saria A, Zemig G (2006) Activation of muscarinic and nicotinic acetylcholine receptors in the nucleus accumbens core is necessary for the acquisition of drug reinforcement. J Neurosci 26:6004-6010.
Crum RM, Anthony JC (1993) Cocaine use and other suspected risk factors for obsessive-compulsive disorder: a prospective study with data from the Epidemiologic Catchment Area surveys. Drug Alcohol Depend 31:281-295.
Cui C, Booker TK, Allen RS, Grady SR, Whiteaker P, Marks MJ, Salminen O, Tritto T, Butt CM, Allen WR, Stitzel JA, McIntosh JM, Boulter J, Collins AC, Heinemann

219
SF (2003) The beta3 nicotinic receptor subunit: a component of alpha-conotoxin Mil-binding nicotinic acetylcholine receptors that modulate dopamine release and related behaviors. J Neurosci 23:11045-11053.
Curtis L, Buisson B, Bertrand S, Bertrand D (2002) Potentiation of human alpha4beta2 neuronal nicotinic acetylcholine receptor by estradiol. Mol Pharmacol 61:127-135.
Daly JW, Fredholm BB (1998) Caffeine-an atypical drug of dependence. Drug Alcohol Depend 51:199-206.
Damaj MI, Kao W, Martin BR (2003) Characterization of Spontaneous and Precipitated Nicotine Withdrawal in the Mouse. J Pharmacol Exp Ther 307:526-534.
Dani JA (2001) Overview of nicotinic receptors and their roles in the central nervous system. Biological Psychiatry 49:166-174.
Dani JA, Bertrand D (2007) Nicotinic Acetylcholine Receptors and Nicotinic Cholinergic Mechanisms of the Central Nervous System. Annual Review of Pharmacology and Toxicology 47.
Dani JA, Radcliffe KA, Pidoplichko VI (2000) Variations in desensitization of nicotinic acetylcholine receptors from hippocampus and midbrain dopamine areas. Eur J Pharmacol 393:31-38.
Dasgupta S, Ferre S, Kull B, Hedlund PB, Finnman UB, Ahlberg S, Arenas E, Fredholm BB, Fuxe K (1996) Adenosine A2A receptors modulate the binding characteristics of dopamine D2 receptors in stably cotransfected fibroblast cells. Eur J Pharmacol 316:325-331.
Dassesse D, Massie A, Ferrari R, Ledent C, Parmentier M, Arckens L, Zoli M, Schiffmann SN (2001) Functional striatal hypodopaminergic activity in mice lacking adenosine A(2A) receptors. J Neurochem 78:183-198.
Davies AR, Hardick DJ, Blagbrough IS, Potter BV, Wolstenholme AJ, Wonnacott S (1999) Characterisation of the binding of [3H]methyllycaconitine: a new radioligand for labelling alpha 7-type neuronal nicotinic acetylcholine receptors. Neuropharmacology 38:679-690.
Davila-Garcia MI, Musachio JL, Kellar KJ (2003) Chronic nicotine administration does not increase nicotinic receptors labeled by [I25I]epibatidine in adrenal gland, superior cervical ganglia, pineal or retina. Journal of Neurochem 85:1237-1246.
Davila-Garcia MI, Musachio JL, Perry DC, Xiao Y, Horti A, London ED, Dannals RF, Kellar KJ (1997) [125I]IPH, an epibatidine analog, binds with high affinity to neuronal nicotinic cholinergic receptors. J Pharmacol Exp Ther 282:445-451.

2 2 0
Davis JA, Gould TJ (2006) The effects of DHBE and MLA on nicotine-inducedenhancement of contextual fear conditioning in C57BL/6 mice.Psychopharmacology (Berl) 184:345-352.
Davis JA, James JR, Siegel SJ, Gould TJ (2005) Withdrawal from chronic nicotineadministration impairs contextual fear conditioning in C57BL/6 mice. J Neurosci25:8708-8713.
Day JC, Piazza PV, Le Moal M, Maccari S (1997) Cocaine-induced increase in cortical acetylcholine release: interaction with the hypothalamo-pituitary-adrenal axis. Eur J Neurosci 9:1130-1136.
de Rover M, Lodder JC, Kits KS, Schoffelmeer AN, Brussaard AB (2002) Cholinergic modulation of nucleus accumbens medium spiny neurons. Eur J Neurosci 16:2279-2290.
De Wit H, Wise RA (1977) Blockade of cocaine reinforcement in rats with the dopamine receptor blocker pimozide, but not with the noradrenergic blockers phentolamine or phenoxybenzamine. Can J Psychol 31:195-203.
Desai RI, Terry P (2003) Evidence of cross-tolerance between behavioural effects of nicotine and cocaine in mice. Psychopharmacology (Berl) 166:111-119.
Desai RI, Barber DJ, Terry P (1999) Asymmetric generalization between the discriminative stimulus effects of nicotine and cocaine. Behav Pharmacol 10:647- 656.
Descarries L (1998) The hypothesis of an ambient level of acetylcholine in the central nervous system. J Physiol Paris 92:215-220.
Devine DP, Leone P, Pocock D, Wise RA (1993) Differential involvement of ventral tegmental mu, delta and kappa opioid receptors in modulation of basal mesolimbic dopamine release: in vivo microdialysis studies. J Pharmacol Exp Ther 266:1236-1246.
Dhatt RK, Gudehithlu KP, Wemlinger TA, Tejwani GA, Neff NH, Hadjiconstantinou M (1995) Preproenkephalin mRNA and methionine-enkephalin content are increased in mouse striatum after treatment with nicotine. J Neurochem 64:1878-1883.
Di Chiara G (2000) Role of dopamine in the behavioural actions of nicotine related to addiction. European Journal of Pharmacology 393:295-314.
Di Chiara G, Imperato A (1988) Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. Proc Natl Acad Sci U S A 85:5274-5278.

2 2 1
DiFranza JR, Guerrera MP (1990) Alcoholism and smoking. J Stud Alcohol 51:130-135.
Dighe SV, Madia PA, Sirohi S, Yobum BC (2009) Continuous morphine produces more tolerance than intermittent or acute treatment. Pharmacol Biochem Behav 92:537- 542.
Dixon AK, Gubitz AK, Sirinathsinghji DJ, Richardson PJ, Freeman TC (1996) Tissue distribution of adenosine receptor mRNAs in the rat. Br J Pharmacol 118:1461- 1468.
Donny EC, Caggiula AR, Knopf S, Brown C (1995) Nicotine self-administration in rats. Psychopharmacology (Berl) 122:390-394.
Donny EC, Caggiula AR, Mielke MM, Jacobs KS, Rose C, Sved AF (1998) Acquisition of nicotine self-administration in rats: the effects of dose, feeding schedule, and drug contingency. Psychopharmacology (Berl) 136:83-90.
Donny EC, Caggiula AR, Rose C, Jacobs KS, Mielke MM, Sved AF (2000) Differential effects of response-contingent and response-independent nicotine in rats. Eur J Pharmacol 402:231-240.
Donny EC, Caggiula AR, Mielke MM, Booth S, Gharib MA, Hoffman A, Maldovan V, Shupenko C, McCallum SE (1999) Nicotine self-administration in rats on a progressive ratio schedule of reinforcement. Psychopharmacology (Berl) 147:135- 142.
Dourmap N, Clero E, Costentin J (1997) Involvement of cholinergic neurons in the release of dopamine elicited by stimulation of mu-opioid receptors in striatum. Brain Res 749:295-300.
Dunwiddie TV, Masino SA (2001) The role and regulation of adenosine in the central nervous system. Annu Rev Neurosci 24:31-55.
Emurian HH, Nellis MJ, Brady JV, Ray RL (1982) Event time-series relationship between cigarette smoking and coffee drinking. Addict Behav 7:441-444.
Endo T, Yanagawa Y, Obata K, Isa T (2005) Nicotinic acetylcholine receptor subtypes involved in facilitation of GABAergic inhibition in mouse superficial superior colliculus. J Neurophysiol 94:3893-3902.
Fallon JH, Moore RY (1978) Catecholamine innervation of the basal forebrain. IV. Topography of the dopamine projection to the basal forebrain and neostriatum. J Comp Neurol 180:545-580.

222
Fehr C, Yakushev I, Hohmann N, Buchholz HG, Landvogt C, Deckers H, Eberhardt A, Klager M, Smolka MN, Scheurich A, Dielentheis T, Schmidt LG, Rosch F, Bartenstein P, Grander G, Schreckenberger M (2008) Association of low striatal dopamine d2 receptor availability with nicotine dependence similar to that seen with other drugs of abuse. Am J Psychiatry 165:507-514.
Fenster CP, Rains MF, Noerager B, Quick MW, Lester RAJ (1997) Influence of Subunit Composition on Desensitization of Neuronal Acetylcholine Receptors at Low Concentrations of Nicotine. J Neurosci 17:5747-5759.
Fenster CP, Whitworth TL, Sheffield EB, Quick MW, Lester RA (1999) Upregulation of surface alpha4beta2 nicotinic receptors is initiated by receptor desensitization after chronic exposure to nicotine. J Neurosci 19:4804-4814.
Ferrari R, Le Novere N, Picciotto MR, Changeux JP, Zoli M (2002) Acute and long-term changes in the mesolimbic dopamine pathway after systemic or local single nicotine injections. Eur J Neurosci 15:1810-1818.
Ferre S (2008) An update on the mechanisms of the psychostimulant effects of caffeine. J Neurochem 105:1067-1079.
Ferre S, von Euler G, Johansson B, Fredholm BB, Fuxe K (1991) Stimulation of high- affmity adenosine A2 receptors decreases the affinity of dopamine D2 receptors in rat striatal membranes. Proc Natl Acad Sci U S A 88:7238-7241.
Ferre S, Fredholm BB, Morelli M, Popoli P, Fuxe K (1997) Adenosine-dopamine receptor-receptor interactions as an integrative mechanism in the basal ganglia. Trends Neurosci 20:482-487.
Ferre S, Diamond I, Goldberg SR, Yao L, Hourani SM, Huang ZL, Urade Y, Kitchen I (2007) Adenosine A2A receptors in ventral striatum, hypothalamus and nociceptive circuitry implications for drug addiction, sleep and pain. Prog Neurobiol 83:332-347.
Ferre S, Karcz-Kubicha M, Hope BT, Popoli P, Burgueno J, Gutierrez MA, Casado V, Fuxe K, Goldberg SR, Lluis C, Franco R, Ciruela F (2002) Synergistic interaction between adenosine A2A and glutamate mGlu5 receptors: implications for striatal neuronal function. Proc Natl Acad Sci U S A 99:11940-11945.
Feyerabend C, Russell MA (1990) A rapid gas-liquid chromatographic method for the determination of cotinine and nicotine in biological fluids. J Pharm Pharmacol 42:450-452.
Fischer H, Orr-Urtreger A, Role LW, Huck S (2005) Selective deletion of the alpha5 subunit differentially affects somatic-dendritic versus axonally targeted nicotinic ACh receptors in mouse. J Physiol 563:119-137.

223
Fiserova M, Consolo S, Krsiak M (1999) Chronic morphine induces long-lasting changes in acetylcholine release in rat nucleus accumbens core and shell: an in vivo microdialysis study. Psychopharmacology (Berl) 142:85-94.
Flores CM, DeCamp RM, Kilo S, Rogers SW, Hargreaves KM (1996) Neuronal nicotinic receptor expression in sensory neurons of the rat trigeminal ganglion: demonstration of alpha3beta4, a novel subtype in the mammalian nervous system.J Neurosci 16:7892-7901.
Foltin RW, Fischman MW (1991) Smoked and intravenous cocaine in humans: acute tolerance, cardiovascular and subjective effects. J Pharmacol Exp Ther 257:247- 261.
Franco R, Ferre S, Agnati L, Torvinen M, Gines S, Hillion J, Casado V, Lledo P, Zoli M, Lluis C, Fuxe K (2000) Evidence for adenosine/dopamine receptor interactions: indications for heteromerization. Neuropsychopharmacology 23:S50-59.
Franklin KBJ, Paxinos G (2001) The mouse brain in stereotaxic coordinates, 2"" Edition. San Diego: Academic Press.
Frazier CJ, Buhler AV, Weiner JL, Dunwiddie TV (1998) Synaptic potentials mediated via alpha-bungarotoxin-sensitive nicotinic acetylcholine receptors in rat hippocampal intemeurons. J Neurosci 18:8228-8235.
Fredholm BB, Battig K, Holmen J, Nehlig A, Zvartau EE (1999) Actions of caffeine in the brain with special reference to factors that contribute to its widespread use. Pharmacol Rev 51:83-133.
Free RB, von Fischer ND, Boyd RT, McKay DB (2003) Pharmacological characterization of recombinant bovine alpha3beta4 neuronal nicotinic receptors stably expressed in HEK 293 cells. Neurosci Lett 343:180-184.
Frosch DL, Shoptaw S, Nahom D, Jarvik ME (2000) Associations between tobacco smoking and illicit drug use among methadone-maintained opiate-dependent individuals. Exp Clin Psychopharmacol 8:97-103.
Funk D, Marinelli PW, Le AD (2006) Biological processes underlying co-use of alcohol and nicotine: neuronal mechanisms, cross-tolerance, and genetic factors. Alcohol Res Health 29:186-192.
Fuxe K, Ferre S, Genedani S, Franco R, Agnati LF (2007) Adenosine receptor-dopamine receptor interactions in the basal ganglia and their relevance for brain function. Physiol Behav 92:210-217.
Galeote L, Kieffer BL, Maldonado R, Berrendero F (2006) Mu-opioid receptors are involved in the tolerance to nicotine antinociception. J Neurochem 97:416-423.

224
Galzi JL, Changeux JP (1995) Neuronal nicotinic receptors: molecular organization and regulations. Neuropharmacology 34:563-582.
Garris PA, Ciolkowski EL, Pastore P, Wightman RM (1994) Efflux of dopamine from the synaptic cleft in the nucleus accumbens of the rat brain. J Neurosci 14:6084- 6093.
Gasior M, Jaszyna M, Peters J, Goldberg SR (2000) Changes in the ambulatory activity and discriminative stimulus effects of psychostimulant drugs in rats chronically exposed to caffeine: effect of caffeine dose. J Pharmacol Exp Ther 295:1101-1 111.
Gasior M, Jaszyna M, Munzar P, Witkin JM, Goldberg SR (2002) Caffeine potentiates the discriminative-stimulus effects of nicotine in rats. Psychopharmacology (Berl) 162:385-395.
Gerasimov MR, Franceschi M, Volkow ND, Rice O, Schiffer WK, Dewey SL (2000) Synergistic interactions between nicotine and cocaine or methylphenidate depend on the dose of dopamine transporter inhibitor. Synapse 38:432-437.
Gerfen CR (1992) The neostriatal mosaic: multiple levels of compartmental organization. Trends Neurosci 15:133-139.
Gerzanich V, Peng X, Wang F, Wells G, Anand R, Fletcher S, Lindstrom J (1995) Comparative pharmacology of epibatidine: a potent agonist for neuronal nicotinic acetylcholine receptors. Mol Pharmacol 48:774-782.
Girod R, Barazangi N, McGehee D, Role LW (2000) Facilitation of glutamatergic neurotransmission by presynaptic nicotinic acetylcholine receptors. Neuropharmacology 39:2715-2725.
Click SD, Maisonneuve IM, Kitchen BA, Fleck MW (2002) Antagonism of alpha 3 beta 4 nicotinic receptors as a strategy to reduce opioid and stimulant selfadministration. Eur J Pharmacol 438:99-105.
Click SD, Ramirez RL, Livi JM, Maisonneuve IM (2006) 18-Methoxycoronaridine acts in the medial habenula and/or interpeduncular nucleus to decrease morphine selfadministration in rats. Eur J Pharmacol 537:94-98.
Click SD, Kuehne ME, Maisonneuve IM, Bandarage UK, Molinari HH (1996) 18- Methoxycoronaridine, a non-toxic iboga alkaloid congener: effects on morphine and cocaine self-administration and on mesolimbic dopamine release in rats. Brain Res 719:29-35.
Coldberg SR, Spealman RD, Goldberg DM (1981) Persistent behavior at high rates maintained by intravenous self-administration of nicotine. Science 214:573-575.

225
Goldstein A, Fischli W, Lowney LI, Hunkapiller M, Hood L (1981) Porcine pituitary dynorphin: complete amino acid sequence of the biologically activeheptadecapeptide. Proc Natl Acad Sci U S A 78:7219-7223.
Gotti C, Clementi F (2004) Neuronal nicotinic receptors: from structure to pathology. Progress in Neurobiology 74:363.
Gotti C, Zoli M, Clementi F (2006) Brain nicotinic acetylcholine receptors: native subtypes and their relevance. Trends in Pharmacological Sciences 27:482-491.
Gotti C, Moretti M, Zanardi A, Gaimarri A, Champtiaux N, Changeux JP, Whiteaker P, Marks MJ, Clementi F, Zoli M (2005) Heterogeneity and selective targeting of neuronal nicotinic acetylcholine receptor (nAChR) subtypes expressed on retinal afférents of the superior colliculus and lateral geniculate nucleus: identification of a new native nAChR subtype alpha3beta2(alpha5 or beta3) enriched in retinocollicular afférents. Mol Pharmacol 68:1162-1171.
Gray R, Rajan AS, Radcliffe KA, Yakehiro M, Dani JA (1996) Hippocampal synaptic transmission enhanced by low concentrations of nicotine. Nature 383:713-716.
Greer JM, Capecchi MR (2002) Hoxb8 is required for normal grooming behavior in mice. Neuron 33:23-34.
Grubb MS, Thompson ID (2004) Visual response properties in the dorsal lateral geniculate nucleus of mice lacking the beta2 subunit of the nicotinic acetylcholine receptor. J Neurosci 24:8459-8469.
Grubb MS, Thompson ID (2005) Visual response properties of burst and tonic firing in the mouse dorsal lateral geniculate nucleus. J Neurophysiol 93:3224-3247.
Gruber SA, Silveri MM, Yurgelun-Todd DA (2007) Neuropsychological consequences of opiate use. Neuropsychol Rev 17:299-315.
Grucza RA, Wang JC, Stitzel JA, Hinrichs AL, Saccone SF, Saccone NL, Bucholz KK, Cloninger CR, Neuman RJ, Budde JP, Fox L, Bertelsen S, Kramer J, Hesselbrock V, Tischfield J, Numberger JI, Jr., Almasy L, Porjesz B, Kuperman S, Schuckit MA, Edenberg HJ, Rice JP, Goate AM, Bierut LJ (2008) A Risk Allele for Nicotine Dependence in CHRNA5 Is a Protective Allele for Cocaine Dependence. Biol Psychiatry.
Gubitz AK, Widdowson L, Kurokawa M, Kirkpatrick KA, Richardson PJ (1996) Dual signalling by the adenosine A2a receptor involves activation of both N- and P- type calcium channels by different G proteins and protein kinases in the same striatal nerve terminals. J Neurochem 67:374-381.

226
Guo JZ, Tredway TL, Chiappinelli VA (1998) Glutamate and GABA release are enhanced by different subtypes of presynaptic nicotinic receptors in the lateral geniculate nucleus. J Neurosci 18:1963-1969.
Haberstock-Debic H, Wein M, Barrot M, Colago EE, Rahman Z, Neve RL, Pickel VM, Nestler EJ, von Zastrow M, Svingos AL (2003) Morphine acutely regulates opioid receptor trafficking selectively in dendrites of nucleus accumbens neurons. J Neurosci 23:4324-4332.
Haghighi AP, Cooper E (2000) A molecular link between inward rectification and calcium permeability of neuronal nicotinic acetylcholine alpha3beta4 and alpha4beta2 receptors. J Neurosci 20:529-541.
Haghparast A, Khani A, Naderi N, Alizadeh AM, Motamedi F (2008) Repeated administration of nicotine attenuates the development of morphine tolerance and dependence in mice. Pharmacol Biochem Behav 88:385-392.
Hansen SB, Taylor P (2007) Galanthamine and non-competitive inhibitor binding to ACh-binding protein: evidence for a binding site on non-alpha-subunit interfaces of heteromeric neuronal nicotinic receptors. J Mol Biol 369:895-901.
Hansen ST, Mark GP (2007) The nicotinic acetylcholine receptor antagonist mecamylamine prevents escalation of cocaine self-administration in rats with extended daily access. Psychopharmacology (Berl) 194:53-61.
Harkness PC, Millar NS (2002) Changes in Conformation and Subcellular Distribution of alpha 4beta 2 Nicotinic Acetylcholine Receptors Revealed by Chronic Nicotine Treatment and Expression of Subunit Chimeras. J Neurosci 22:10172-10181.
Harvey SC, Maddox FN, Luetje CW (1996) Multiple determinants of dihydro-beta- erythroidine sensitivity on rat neuronal nicotinic receptor alpha subunits. J Neurochem 67:1953-1959.
Hatsukami DK, Stead LF, Gupta PC (2008) Tobacco addiction. Lancet 371:2027-2038.
Hatton GI, Yang QZ (2002) Synaptic potentials mediated by alpha 7 nicotinic acetylcholine receptors in supraoptic nucleus. J Neurosci 22:29-37.
Heidmann T, Changeux JP (1980) Interaction of a fluorescent agonist with the membrane-bound acetylcholine receptor from Torpedo marmorata in the millisecond time range: resolution of an "intermediate" conformational transition and evidence for positive cooperative effects. Biochem Biophys Res Commun 97:889-896.
Hemby SE, Martin TJ, Co C, Dworkin SI, Smith JE (1995) The effects of intravenous heroin administration on extracellular nucleus accumbens dopamine

227
concentrations as determined by in vivo microdialysis. J Pharmacol Exp Ther 273:591-598.
Hemby SE, Co C, Koves TR, Smith JE, Dworkin SI (1997) Differences in extracellular dopamine concentrations in the nucleus accumbens during response-dependent and response-independent cocaine administration in the rat. Psychopharmacology (Berl) 133:7-16.
Henningfield JE, Miyasato K, Jasinski DR (1983) Cigarette smokers self-administer intravenous nicotine. Pharmacol Biochem Behav 19:887-890.
Henningfield JE, Clayton R, Pollin W (1990) Involvement of tobacco in alcoholism and illicit drug use. Br J Addict 85:279-291.
Henningfield JE, Stapleton JM, Benowitz NL, Grayson RF, London ED (1993) Higher levels of nicotine in arterial than in venous blood after cigarette smoking. Drug Alcohol Depend 33:23-29.
Hernandez SC, Vicini S, Xiao Y, Davila-Garcia MI, Yasuda RP, Wolfe BB, Kellar KJ(2004) The nicotinic receptor in the rat pineal gland is an alpha3beta4 subtype. Mol Pharmacol 66:978-987.
Hettinger BD, Lee A, Linden J, Rosin DL (2001) Ultrastructural localization of adenosine A2A receptors suggests multiple cellular sites for modulation of GABAergic neurons in rat striatum. J Comp Neurol 431:331-346.
Higgins ST, Budney AJ, Hughes JR, Bickel WK, Lynn M, Mortensen A (1994) Influence of cocaine use on cigarette smoking. Jama 272:1724.
Hikida T, Kitabatake Y, Pastan I, Nakanishi S (2003) Acetylcholine enhancement in the nucleus accumbens prevents addictive behaviors of cocaine and morphine. Proc Natl Acad Sci U S A 100:6169-6173.
Hikida T, Kaneko S, Isobe T, Kitabatake Y, Watanabe D, Pastan I, Nakanishi S (2001) Increased sensitivity to cocaine by cholinergic cell ablation in nucleus accumbens. Proc Natl Acad Sci U S A 98:13351-13354.
Hildebrand BE, Nomikos GG, Bondjers C, Nisell M, Svensson TH (1997) Behavioral manifestations of the nicotine abstinence syndrome in the rat: peripheral versus central mechanisms. Psychopharmacology (Berl) 129:348-356.
Hillion J, Canals M, Torvinen M, Casado V, Scott R, Terasmaa A, Hansson A, Watson S, Olah ME, Mallol J, Canela El, Zoli M, Agnati LF, Ibanez CF, Lluis C, Franco R, Ferre S, Fuxe K (2002) Coaggregation, cointemalization, and codesensitization of adenosine A2A receptors and dopamine D2 receptors. J Biol Chem 277:18091- 18097.

228
Hirose N, Murakawa K, Takada K, Oi Y, Suzuki T, Nagase H, Cools AR, Koshikawa N(2005) Interactions among mu- and delta-opioid receptors, especially putative deltal- and delta2-opioid receptors, promote dopamine release in the nucleus accumbens. Neuroscience 135:213-225.
Horger BA, Giles MK, Schenk S (1992) Preexposure to amphetamine and nicotine predisposes rats to self-administer a low dose of cocaine. Psychopharmacology (Berl) 107:271-276.
Houdi AA, Dasgupta R, Kindy MS (1998) Effect of nicotine use and withdrawal on brain preproenkephalin A mRNA. Brain Res 799:257-263.
Houghtling RA, Davila-Garcia MI, Kellar KJ (1995) Characterization of (+/-)(- )[3H]epibatidine binding to nicotinic cholinergic receptors in rat and human brain. Mol Pharmacol 48:280-287.
Howell LL, Byrd LD (1995) Serotonergic modulation of the behavioral effects of cocaine in the squirrel monkey. J Pharmacol Exp Ther 275:1551-1559.
Hughes J, Smith TW, Kosterlitz HW, Fothergill LA, Morgan BA, Morris HR (1975) Identification of two related pentapeptides from the brain with potent opiate agonist activity. Nature 258:577-580.
Hukkanen J, Jacob P, 3rd, Benowitz NL (2005) Metabolism and disposition kinetics of nicotine. Pharmacol Rev 57:79-115.
Hussain RJ, Taraschenko OD, Glick SD (2008) Effects of nicotine, methamphetamine and cocaine on extracellular levels of acetylcholine in the interpeduncular nucleus of rats. Neurosci Lett.
Hyland BI, Reynolds JN, Hay J, Perk CG, Miller R (2002) Firing modes of midbrain dopamine cells in the freely moving rat. Neuroscience 114:475-492.
Hyman SE, Malenka RC, Nestler EJ (2006) Neural mechanisms of addiction: the role of reward-related learning and memory. Annu Rev Neurosci 29:565-598.
Ikemoto S, Qin M, Liu ZH (2006) Primary reinforcing effects of nicotine are triggered from multiple regions both inside and outside the ventral tegmental area. J Neurosci 26:723-730.
Imperato A, Mulas A, Di Chiara G (1986) Nicotine preferentially stimulates dopamine release in the limbic system of freely moving rats. Eur J Pharmacol 132:337-338.

229
Imperato A, Obinu MC, Demontis MV, Cessa GL (1992) Cocaine releases limbic acetylcholine through endogenous dopamine action on DI receptors. Eur J Pharmacol 229:265-267.
Imperato A, Obinu MC, Casu MA, Mascia MS, Carta G, Gessa GL (1996) Chronic morphine increases hippocampal acetylcholine release: possible relevance in drug dependence. Eur J Pharmacol 302:21-26.
Ise Y, Narita M, Nagase H, Suzuki T (2000) Modulation of opioidergic system on mecamylamine-precipitated nicotine-withdrawal aversion in rats. Psychopharmacology (Berl) 151:49-54.
Isola R, Zhang H, Duchemin AM, Tejwani GA, Neff NH, Hadjiconstantinou M (2002) Met-enkephalin and preproenkephalin mRNA changes in the striatum of the nicotine abstinence mouse. Neurosci Lett 325:67-71.
Istvan J, Matarazzo JD (1984) Tobacco, alcohol, and caffeine use: a review of their interrelationships. Psychol Bull 95:301-326.
Itzhak Y, Martin JL (1999) Effects of cocaine, nicotine, dizocipline and alcohol on mice locomotor activity: cocaine-alcohol cross-sensitization involves upregulation of striatal dopamine transporter binding sites. Brain Res 818:204-211.
Jackisch R, Geppert M, Brenner AS, Illes P (1986) Presynaptic opioid receptors modulating acetylcholine release in the hippocampus of the rabbit. Naunyn Schmiedebergs Arch Pharmacol 332:156-162.
Jacobs EH, Smit AB, de Vries TJ, Schoffelmeer AN (2003) Neuroadaptive effects of active versus passive drug administration in addiction research. Trends Pharmacol Sci 24:566-573.
Jacobs I, Anderson DJ, Surowy CS, Puttfarcken PS (2002) Differenflal regulaflon of nicotinic receptor-mediated neurotransmitter release following chronic (-)- nicotine administration. Neuropharmacology 43:847-856.
Jage J (2005) Opioid tolerance and dependence - do they matter? Eur J Pain 9:157-162.
Janson AM, Hedlund PB, Hillefors M, von Euler G (1992) Chronic nicotine treatment decreases dopamine D2 agonist binding in the rat basal ganglia. Neuroreport 3:1117-1120.
Javitch JA, Blaustein RO, Snyder SH (1984) [3H]mazindol binding associated with neuronal dopamine and norepinephrine uptake sites. Mol Pharmacol 26:35-44.

230
Johnson SW, North RA (1992) Opioids excite dopamine neurons by hyperpolarization of local intemeurons. J Neurosci 12:483-488.
Jones HE, Garrett BE, Griffiths RR (1999) Subjective and physiological effects of intravenous nicotine and cocaine in cigarette smoking cocaine abusers. J Pharmacol Exp Ther 288:188-197.
Jones IW, Wonnacott S (2004) Precise localization of alpha7 nicotinic acetylcholine receptors on glutamatergic axon terminals in the rat ventral tegmental area. J Neurosci 24:11244-11252.
Jones IW, Bolam JP, Wonnacott S (2001) Presynaptic localisation of the nicotinic acetylcholine receptor beta2 subunit immunoreactivity in rat nigrostriatal dopaminergic neurones. J Comp Neurol 439:235-247.
Jozwiak K, Moaddel R, Yamaguchi R, Maciuk A, Wainer IW (2006) Non-competitive inhibitory activities of morphinan and morphine derivatives at the alpha 3 beta 4 Neuronal nicotinic acetylcholine receptor determined using nonlinear chromatography and chemometric techniques. Pharm Res 23:2175-2182.
Justinova Z, Ferre S, Barnes C, Wertheim CE, Pappas LA, Goldberg SR, Le Foil B (2009) Effects of chronic caffeine exposure on adenosinergic modulation of the discriminative-stimulus effects of nicotine, methamphetamine, and cocaine in rats. Psychopharmacology (Berl) 203:355-367.
Kaiser S, Wonnacott S (2000) alpha-bungarotoxin-sensitive nicotinic receptors indirectly modulate [(3)H]dopamine release in rat striatal slices via glutamate release. Mol Pharmacol 58:312-318.
Kalueff AV, Tuohimaa P (2005) Mouse grooming microstructure is a reliable anxiety marker bidirectionally sensitive to GABAergic drugs. Eur J Pharmacol 508:147- 153.
Kane JK, Hwang Y, Konu O, Loughlin SE, Leslie FM, Li MD (2005) Regulation of Homer and group 1 metabotropic glutamate receptors by nicotine. Eur J Neurosci 21:1145-1154.
Karlin A (2002) EMERGING STRUCTURE OF THE NICOTINIC ACETYLCHOLINE RECEPTORS. Nature Reviews Neuroscience 3:102-114.
Karlin A, Akabas MH (1995) Toward a structural basis for the function of nicotinic acetylcholine receptors and their cousins. Neuron 15:1231-1244.
Karras A, Kane JM (1980) Naloxone reduces cigarette smoking. Life Sci 27:1541-1545.

231
Kassel JD, Greenstein JE, Evatt DP, Wardle MC, Yates MC, Veilleux JC, Eissenberg T (2007) Smoking topography in response to denicotinized and high-yield nicotine cigarettes in adolescent smokers. J Adolesc Health 40:54-60.
Ke L, Eisenhour CM, Bencherif M, Lukas RJ (1998) Effects of Chronic Nicotine Treatment on Expression of Diverse Nicotinic Acetylcholine Receptor Subtypes.I. Dose- and Time-Dependent Effects of Nicotine Treatment. J Pharmacol Exp Ther 286:825-840.
Kedmi M, Beaudet AL, Orr-Urtreger A (2004) Mice lacking neuronal nicotinic acetylcholine receptor beta4-subunit and mice lacking both alpha5- and beta4- subunits are highly resistant to nicotine-induced seizures. Physiol Genomics 17:221-229.
Kelley BM, Rowan JD (2004) Long-term, low-level adolescent nicotine exposure produces dose-dependent changes in cocaine sensitivity and reward in adult mice. Int J Dev Neurosci 22:339-348.
Kelly PH, Iversen SD (1976) Selective 60HDA-induced destruction of mesolimbic dopamine neurons: abolition of psychostimulant-induced locomotor activity in rats. Eur J Pharmacol 40:45-56.
Kenny PJ, Markou A (2006) Nicotine self-administration acutely activates brain reward systems and induces a long-lasting increase in reward sensitivity. Neuropsychopharmacology 31:1203-1211.
Khan ZU, Gutierrez A, Martin R, Penafiel A, Rivera A, de la Calle A (2000) Dopamine D5 receptors of rat and human brain. Neuroscience 100:689-699.
Kieffer BL (1999) Opioids: first lessons from knockout mice. Trends Pharmacol Sci 20:19-26.
King AC, Meyer PJ (2000) Naltrexone alteration of acute smoking response in nicotine- dependent subjects. Pharmacol Biochem Behav 66:563-572.
King GR, Ellinwood EH, Jr., Silvia C, Joyner CM, Xue Z, Caron MG, Lee TH (1994) Withdrawal from continuous or intermittent cocaine administration: changes in D2 receptor function. J Pharmacol Exp Ther 269:743-749.
Kirch DG, Taylor TR, Creese I, Xu SX, Wyatt RJ (1992) Effect of chronic nicotine treatment and withdrawal on rat striatal D1 and D2 dopamine receptors. J Pharm Pharmacol 44:89-92.
Kitabatake Y, Hikida T, Watanabe D, Pastan I, Nakanishi S (2003) Impairment of reward-related learning by cholinergic cell ablation in the striatum. Proc Natl Acad Sci U S A 100:7965-7970.

232
Kitchen I, Slowe SJ, Matthes HW, Kieffer B (1997) Quantitative autoradiographic mapping of mu-, delta- and kappa-opioid receptors in knockout mice lacking the mu-opioid receptor gene. Brain Res 778:73-88.
Klink R, de Kerchove d'Exaerde A, Zoli M, Changeux JP (2001) Molecular and physiological diversity of nicotinic acetylcholine receptors in the midbrain dopaminergic nuclei. J Neurosci 21:1452-1463.
Konings E, Dubois-Arber F, Narring F, Michaud PA (1995) Identifying adolescent drug users: results of a national survey on adolescent health in Switzerland. J Adolesc Health 16:240-247.
Koob G, Kreek MJ (2007) Stress, dysregulation of drug reward pathways, and the transition to drug dependence. Am J Psychiatry 164:1149-1159.
Koob GF (2000) Neurobiology of addiction. Toward the development of new therapies. Ann N Y Acad Sci 909:170-185.
Koopmans JR, van Doomen LJ, Boomsma DI (1997) Association between alcohol use and smoking in adolescent and young adult twins: a bivariate genetic analysis. Alcohol Clin Exp Res 21:537-546.
Kozlowski LT, Henningfield JE, Keenan RM, Lei H, Leigh G, Jelinek LG, Pope MA, Haertzen CA (1993) Patterns of alcohol, cigarette, and caffeine and other drug use in two drug abusing populations. J Subst Abuse Treat 10:171-179.
Kranzler HR, Satel S, Apter A (1994) Personality disorders and associated features in cocaine-dependent inpatients. Compr Psychiatry 35:335-340.
Kreek MJ, LaForge KS, Butelman E (2002) Pharmacotherapy of addictions. Nat Rev Drug Discov 1:710-726.
Krishnan-Sarin S, Rosen MI, O'Malley SS (1999) Naloxone challenge in smokers. Preliminary evidence of an opioid component in nicotine dependence. Arch Gen Psychiatry 56:663-668.
Kulak JM, Nguyen TA, Clivera BM, McIntosh JM (1997) Alpha-conotoxin Mil blocks nicotine-stimulated dopamine release in rat striatal synaptosomes. J Neurosci 17:5263-5270.
Kurokawa M, Kirk IP, Kirkpatrick KA, Kase H, Richardson PJ (1994) Inhibition by KF17837 of adenosine A2A receptor-mediated modulation of striatal GAB A and ACh release. Br J Pharmacol 113:43-48.

233
Kurokawa M, Koga K, Kase H, Nakamura J, Kuwana Y (1996) Adenosine A2a receptor- mediated modulation of striatal acetylcholine release in vivo. J Neurochem 66:1882-1888.
Kuryatov A, Luo J, Cooper J, Lindstrom J (2005) Nicotine acts as a pharmacological chaperone to up-regulate human alpha4beta2 acetylcholine receptors. Mol Pharmacol 68:1839-1851.
Kuzmin A, Johansson B (1999) Expression of c-fos, NGFI-A and secretogranin II mRNA in brain regions during initiation of cocaine self-administration in mice. Eur J Neurosci 11:3694-3700.
Lapchak PA, Araujo DM, Collier B (1989) Regulation of endogenous acetylcholine release from mammalian brain slices by opiate receptors: hippocampus, striatum and cerebral cortex of guinea-pig and rat. Neuroscience 31:313-325.
Le Novere N, Zoli M, Changeux JP (1996) Neuronal nicotinic receptor alpha 6 subunit mRNA is selectively concentrated in catecholaminergic nuclei of the rat brain. Eur J Neurosci 8:2428-2439.
Lecca D, Cacciapaglia F, Valentini V, Acquas E, Di Chiara G (2007) Differential neurochemical and behavioral adaptation to cocaine after response contingent and noncontingent exposure in the rat. Psychopharmacology (Berl) 191:653-667.
Ledent C, Vaugeois JM, Schiffmann SN, Pedrazzini T, El Yacoubi M, Vanderhaeghen JJ, Costentin J, Heath JK, Vassart G, Parmentier M (1997) Aggressiveness, hypoalgesia and high blood pressure in mice lacking the adenosine A2a receptor. Nature 388:674-678.
Lee CY (1972) Chemistry and pharmacology of polypeptide toxins in snake venoms. Annu Rev Pharmacol 12:265-286.
Lejoyeux M, Mourad I, Ades J (2000) [Psychiatric disorders induced by drug dependence other than alcohol]. Encephale 26:21-27.
Lena C, Changeux JP, Mulle C (1993) Evidence for "preterminal" nicotinic receptors on GABAergic axons in the rat interpeduncular nucleus. J Neurosci 13:2680-2688.
Lena I, Matthes H, Kieffer B, Kitchen I (2004) Quantitative autoradiography of dopamine receptors in the brains of micro-opioid receptor knockout mice. Neurosci Lett 356:220-224.
Lester RA, Dani JA (1995) Acetylcholine receptor desensitization induced by nicotine in rat medial habenula neurons. J Neurophysiol 74:195-206.

234
Levin ED, Mead T, Rezvani AH, Rose JE, Gallivan C, Gross R (2000) The nicotinic antagonist mecamylamine preferentially inhibits cocaine vs. food selfadministration in rats. Physiol Behav 71:565-570.
Levin ED, Tizabi Y, Rezvani AH, Caldwell DP, Petro A, Getachew B (2005) Chronicnicotine and dizocilpine effects on regionally specific nicotinic and NMDAglutamate receptor binding. Brain Res 1041:132-142.
Levin ED, Petro A, Rezvani AH, Pollard N, Christopher NC, Strauss M, Avery J,Nicholson J, Rose JE (2009) Nicotinic alpha7- or beta2-containing receptorknockout: effects on radial-arm maze learning and long-term nicotine consumption in mice. Behav Brain Res 196:207-213.
Li CH, Chung D (1976) Isolation and structure of an untriakontapeptide with opiate activity from camel pituitary glands. Proc Natl Acad Sci U S A 73:1145-1148.
Lichtensteiger W, Hefti F, Felix D, Huwyler T, Melamed E, Schlumpf M (1982) Stimulation of nigrostriatal dopamine neurones by nicotine. Neuropharmacology 21:963-968.
Lindsay GB, Rainey J (1997) Psychosocial and pharmacologic explanations of nicotine's ''gateway drug'' function. J Sch Health 67:123-126.
Liu YF, Civelli O, Zhou QY, Albert PR (1992) Cholera toxin-sensitive 3',5'-cyclic adenosine monophosphate and calcium signals of the human dopamine-D 1 receptor: selective potentiation by protein kinase A. Mol Endocrinol 6:1815-1824.
Ljungberg T, Ungerstedt U (1985) A rapid and simple behavioural screening method for simultaneous assessment of limbic and striatal blocking effects of neuroleptic drugs. Pharmacol Biochem Behav 23:479-485.
Lloyd GK, Williams M (2000) Neuronal nicotinic acetylcholine receptors as novel drug targets. J Pharmacol Exp Ther 292:461-467.
Lowenstein PR, Castro MG (2001) Genetic engineering within the adult brain: implications for molecular approaches to behavioral neuroscience. Physiol Behav 73:833-839.
Luetje CW, Patrick J (1991) Both alpha- and beta-subunits contribute to the agonist sensitivity of neuronal nicotinic acetylcholine receptors. J Neurosci 11:837-845.
MacDonald RL, Skerritt JH, Werz MA (1986) Adenosine agonists reduce voltage- dependent calcium conductance of mouse sensory neurones in cell culture. J Physiol 370:75-90.

235
Maggos CE, Tsukada H, Kakiuchi T, Nishiyama S, Myers JE, Kreuter J, Schlussman SD, Unterwald EM, Ho A, Kreek MJ (1998) Sustained withdrawal allows normalization of in vivo [llC]N-methylspiperone dopamine D2 receptor binding after chronic binge cocaine: a positron emission tomography study in rats. Neuropsychopharmacology 19:146-153.
Maisonneuve IM, Click SD (1999) Attenuation of the reinforcing efficacy of morphine by 18-methoxycoronaridine. Eur J Pharmacol 383:15-21.
Maldonado R, Blendy JA, Tzavara E, Gass P, Roques BP, Hanoune J, Schütz G (1996) Reduction of morphine abstinence in mice with a mutation in the gene encoding CREB. Science 273:657-659.
Malin DH, Lake JR, Carter VA, Cunningham JS, Wilson OB (1993) Naloxone precipitates nicotine abstinence syndrome in the rat. Psychopharmacology (Berl) 112:339-342.
Mansour A, Fox CA, Akil H, Watson SJ (1995) Opioid-receptor mRNA expression in the rat CNS: anatomical and functional implications. Trends Neurosci 18:22-29.
Mansvelder HD, McGehee DS (2000) Long-term potentiation of excitatory inputs to brain reward areas by nicotine. Neuron 27:349-357.
Mansvelder HD, Keath JR, McGehee DS (2002) Synaptic Mechanisms Underlie Nicotine-Induced Excitability of Brain Reward Areas. Neuron 33:905-919.
Marco EM, Granstrem O, Moreno E, Llorente R, Adriani W, Laviola G, Viveros MP (2007) Subchronic nicotine exposure in adolescence induces long-term effects on hippocampal and striatal cannabinoid-CB 1 and mu-opioid receptors in rats. Eur J Pharmacol 557:37-43.
Marinelli M, Piazza PV (2002) Interaction between glucocorticoid hormones, stress and psychostimulant drugs. Eur J Neurosci 16:387-394.
Mark GP, Hajnal A, Kinney AE, Keys AS (1999) Self-administration of cocaine increases the release of acetylcholine to a greater extent than response- independent cocaine in the nucleus accumbens of rats. Psychopharmacology (Berl) 143:47-53.
Markou A, Paterson NE (2001) The nicotinic antagonist methyllycaconitine has differential effects on nicotine self-administration and nicotine withdrawal in the rat. Nicotine Tob Res 3:361-373.
Marks MJ, Burch JB, Collins AC (1983) Effects of chronic nicotine infusion on tolerance development and nicotinic receptors. J Pharmacol Exp Ther 226:817-825.

236
Marks MJ, Stitzel JA, Collins AC (1985) Time course study of the effects of chronic nicotine infusion on drug response and brain receptors. J Pharmacol Exp Ther 235:619-628.
Marks MJ, Smith KW, Collins AC (1998) Differential agonist inhibition identifies multiple epibatidine binding sites in mouse brain. J Pharmacol Exp Ther 285:377- 386.
Marks MJ, Whiteaker P, Collins AC (2006) Deletion of the alpha7, beta2, or beta4 nicotinic receptor subunit genes identifies highly expressed subtypes with relatively low affinity for [3H]epibatidine. Mol Pharmacol 70:947-959.
Marks MJ, Campbell SM, Romm E, Collins AC (1991) Genotype influences the development of tolerance to nicotine in the mouse. J Pharmacol Exp Ther 259:392-402.
Marks MJ, Stitzel JA, Romm E, Wehner JM, Collins AC (1986) Nicotinic binding sites in rat and mouse brain: comparison of acetylcholine, nicotine, and alpha- bungarotoxin. Mol Pharmacol 30:427-436.
Marks MJ, Whiteaker P, Grady SR, Picciotto MR, McIntosh JM, Collins AC (2002) Characterization of [(125) I]epibatidine binding and nicotinic agonist-mediated (86) Rb(+) efflux in interpeduncular nucleus and inferior colliculus of beta2 null mutant mice. J Neurochem 81:1102-1115.
Marks MJ, Rowell PP, Cao JZ, Grady SR, McCallum SE, Collins AC (2004) Subsets of acetylcholine-stimulated 86Rb+ efflux and [125I]-epibatidine binding sites in C57BL/6 mouse brain are differentially affected by chronic nicotine treatment. Neuropharmacology 46:1141-1157.
Marks MJ, Pauly JR, Gross SD, Deneris ES, Hermans-Borgmeyer I, Heinemann SF, Collins AC (1992) Nicotine binding and nicotinic receptor subunit RNA after chronic nicotine treatment. J Neurosci 12:2765-2784.
Marshall WR, Epstein LH, Green SB (1980) Coffee drinking and cigarette smoking: I. Coffee, caffeine and cigarette smoking behavior. Addict Behav 5:389-394.
Martellotta MC, Kuzmin A, Zvartau E, Cossu G, Gessa GL, Fratta W (1995) Isradipine inhibits nicotine intravenous self-administration in drug-naive mice. Pharmacology Biochemistry and Behavior 52:271-274.
Martinez-Ortega JM, Jurado D, Martinez-Gonzalez MA, Gurpegui M (2006) Nicotine dependence, use of illegal drugs and psychiatric morbidity. Addict Behav 31:1722-1729.

237
Marubio LM, Changeux J (2000) Nicotinic acetylcholine receptor knockout mice as animal models for studying receptor function. Eur J Pharmacol 393:113-121.
Marubio LM, del Mar Arroyo-Jimenez M, Cordero-Erausquin M, Lena C, Le Novere N, de Kerchove d’Exaerde A, Huchet M, Damaj Ml, Changeux JP (1999) Reduced antinociception in mice lacking neuronal nicotinic receptor subunits. Nature 398:805-810.
Maskos U (2008) The cholinergic mesopontine tegmentum is a relatively neglected nicotinic master modulator of the dopaminergic system: relevance to drugs of abuse and pathology. Br J Pharmacol 153 Suppl 1 :S438-445.
Maskos U, Molles BE, Pons S, Besson M, Guiard BP, Guilloux JP, Evrard A, Cazala P, Cormier A, Mameli-Engvall M, Dufour N, Cloez-Tayarani I, Bemelmans AP, Mallet J, Gardier AM, David V, Faure P, Granon S, Changeux JP (2005) Nicotine reinforcement and cognition restored by targeted expression of nicotinic receptors. Nature 436:103-107.
Mathew SV, Law AJ, Lipska BK, Davila-Garcia MI, Zamora ED, Mitkus SN, Vakkalanka R, Straub RE, Weinberger DR, Kleinman JE, Hyde TM (2007) Alpha7 nicotinic acetylcholine receptor mRNA expression and binding in postmortem human brain are associated with genetic variation in neuregulin 1. Hum Mol Genet 16:2921-2932.
Mathie A, Colquhoun D, Cull-Candy SG (1990) Rectification of currents activated by nicotinic acetylcholine receptors in rat sympathetic ganglion neurones. J Physiol 427:625-655.
McEvoy JP, Freudenreich O, Levin ED, Rose JE (1995) Haloperidol increases smoking in patients with schizophrenia. Psychopharmacology (Berl) 119:124-126.
McGehee DS, Heath MJ, Gelber S, Devay P, Role LW (1995) Nicotine enhancement of fast excitatory synaptic transmission in CNS by presynaptic receptors. Science 269:1692-1696.
McKay BE, Placzek AN, Dani JA (2007) Regulation of synaptic transmission and plasticity by neuronal nicotinic acetylcholine receptors. Biochem Pharmacol 74:1120-1133.
Meador-Woodruff JH, Mansour A, Grandy DK, Damask SP, Civelli O, Watson SJ, Jr. (1992) Distribution of D5 dopamine receptor mRNA in rat brain. Neurosci Lett 145:209-212.
Mendizabal V, Zimmer A, Maldonado R (2006) Involvement of kappa/dynorphin system in WIN 55,212-2 self-administration in mice. Neuropsychopharmacology 31:1957-1966.

238
Meunier JC, Mollereau C, Toll L, Suaudeau C, Moisand C, Alvinerie P, Butour JL, Guillemot JC, Ferrara P, Monsarrat B, et al. (1995) Isolation and structure of the endogenous agonist of opioid receptor-like ORLl receptor. Nature 377:532-535.
Miao H, Liu C, Bishop K, Gong ZH, Nordberg A, Zhang X (1998) Nicotine exposure during a critical period of development leads to persistent changes in nicotinic acetylcholine receptors of adult rat brain. J Neurochem 70:752-762.
Miller JA, Zahniser NR (1987) The use of 14C-labeled tissue paste standards for the calibration of 1251-labeled ligands in quantitative autoradiography. Neurosci Lett 81:345-350.
Miner LL, Collins AC (1989) Strain comparison of nicotine-induced seizure sensitivity and nicotinic receptors. Pharmacol Biochem Behav 33:469-475.
Missale C, Nash SR, Robinson SW, Jaber M, Caron MG (1998) Dopamine receptors: from structure to function. Physiol Rev 78:189-225.
Modrow HE, Holloway FA, Carney JM (1981) Caffeine discrimination in the rat. Pharmacol Biochem Behav 14:683-688.
Mogenson GJ, Jones DL, Yim CY (1980) From motivation to action: functional interface between the limbic system and the motor system. Prog Neurobiol 14:69-97.
Mogg AJ, Whiteaker P, McIntosh JM, Marks M, Collins AC, Wonnacott S (2002) Methyllycaconitine is a potent antagonist of alpha-conotoxin-MII-sensitive presynaptic nicotinic acetylcholine receptors in rat striatum. J Pharmacol Exp Ther 302:197-204.
Montague PR, McClure SM, Baldwin PR, Phillips PE, Budygin EA, Stuber GD, Kilpatrick MR, Wightman RM (2004) Dynamic gain control of dopamine delivery in freely moving animals. J Neurosci 24:1754-1759.
Morris G, Arkadir D, Nevet A, Vaadia E, Bergman H (2004) Coincident but distinct messages of midbrain dopamine and striatal tonically active neurons. Neuron 43:133-143.
Mugnaini M, Garzotti M, Sartori 1, Pilla M, Repeto P, Heidbreder CA, Tessari M (2006) Selective down-regulation of [( 125)1]Y0-alpha-conotoxin Mil binding in rat mesostriatal dopamine pathway following continuous infusion of nicotine. Neuroscience 137:565-572.
Mukhin AG, Gundisch D, Horti AG, Koren AO, Tamagnan G, Kimes AS, Chambers J, Vaupel DB, King SL, Picciotto MR, Innis RB, London ED (2000) 5-Iodo-A- 85380, an alpha4beta2 subtype-selective ligand for nicotinic acetylcholine receptors. Mol Pharmacol 57:642-649.

239
Muller DL, Unterwald EM (2004) In vivo regulation of extracellular signal-regulated protein kinase (ERK) and protein kinase B (Akt) phosphorylation by acute and chronic morphine. J Pharmacol Exp Ther 310:774-782.
Nashmi R, Lester HA (2006) CNS localization of neuronal nicotinic receptors. J Mol Neurosci 30:181-184.
Nashmi R, Xiao C, Deshpande P, McKinney S, Grady SR, Whiteaker P, Huang Q, McClure-Begley T, Lindstrom JM, Labarca C, Collins AC, Marks MJ, Lester HA (2007) Chronic nicotine cell specifically upregulates hmctional alpha4* nicotinic receptors: basis for both tolerance in midbrain and enhanced long-term potentiation in perforant path. J Neurosci 27:8202-8218.
Nellis MJ, Emurian HH, Brady JV, Ray RL (1982) Behavior analysis of cigarette smoking. Pavlov J Biol Sci 17:140-149.
Nemeth-Coslett R, Griffiths RR (1986) Naloxone does not affect cigarette smoking. Psychopharmacology (Berl) 89:261-264.
Nestler EJ, Aghajanian GK (1997) Molecular and cellular basis of addiction. Science 278:58-63.
Nguyen HN, Rasmussen BA, Perry DC (2003) Subtype-selective up-regulation by chronic nicotine of high-affmity nicotinic receptors in rat brain demonstrated by receptor autoradiography. J Pharmacol Exp Ther 307:1090-1097.
Nguyen HN, Rasmussen BA, Perry DC (2004) Binding and functional activity of nicotinic cholinergic receptors in selected rat brain regions are increased following long-term but not short-term nicotine treatment. J Neurochem 90:40-49.
Nikodijevic O, Jacobson KA, Daly JW (1993) Locomotor activity in mice during chronic treatment with caffeine and withdrawal. Pharmacol Biochem Behav 44:199-216.
Nisell M, Nomikos GG, Svensson TH (1994) Infusion of nicotine in the ventral tegmental area or the nucleus accumbens of the rat differentially affects accumbal dopamine release. Pharmacol Toxicol 75:348-352.
O'Brien CP (2005) Benzodiazepine use, abuse, and dependence. J Clin Psychiatry 66 Suppl 2:28-33.
O'Brien CP, Childress AR, Ehrman R, Robbins SJ (1998) Conditioning factors in drug abuse: can they explain compulsion? J Psychopharmacol 12:15-22.
O'Dell LE, Chen SA, Smith RT, Specio SE, Balster RL, Paterson NE, Markou A, Zorrilla EP, Koob GF (2007) Extended access to nicotine self-administration leads to

240
dependence; Circadian measures, withdrawal measures, and extinction behavior in rats. J Pharmacol Exp Ther 320:180-193.
Orejarena MJ, Berrendero F, Maldonado R, Robledo P (2009) Differential changes m mesolimbic dopamine following contingent and non-contingent MDMA selfadministration in mice. Psychopharmacology (Berl) 205:457-466.
Orr-Urtreger A, Goldner FM, Saeki M, Lorenzo I, Goldberg L, De Biasi M, Dani JA, Patrick JW, Beaudet AL (1997) Mice deficient in the alpha7 neuronal nicotinic acetylcholine receptor lack alpha-bungarotoxin binding sites and hippocampal fast nicotinic currents. J Neurosci 17:9165-9171.
Pabreza LA, Dhawan S, Kellar KJ (1991) [3H]cytisine binding to nicotinic cholinergic receptors in brain. Mol Pharmacol 39:9-12.
Panchal V, Taraschenko OD, Maisonneuve IM, Glick SD (2005) Attenuation of morphine withdrawal signs by intracerebral administration of 18- methoxycoronaridine. Eur J Pharmacol 525:98-104.
Parikh VS, Apparsundaram S, Kozak R, Richards JB, Sarter M (2006) Reduced expression and capacity of the striatal high-afflnity choline transporter in hyperdopaminergic mice. Neuroscience 141:379-389.
Parker SL, Fu Y, McAllen K, Luo J, McIntosh JM, Lindstrom JM, Sharp BM (2004) Up- regulation of brain nicotinic acetylcholine receptors in the rat during long-term self-administration of nicotine: disproportionate increase of the alpha6 subunit. Mol Pharmacol 65:611-622.
Parsons WD, Neims AH (1978) Effect of smoking on caffeine clearance. Clin Pharmacol Ther 24:40-45.
Paterson NE, Semenova S, Gasparini F, Markou A (2003) The m01uR5 antagonist MPEP decreased nicotine self-administration in rats and mice. Psychopharmacology (Berl) 167:257-264.
Patkar AA, Lundy A, Leone FT, Weinstein SP, Gottheil E, Steinberg M (2002) Tobacco and alcohol use and medical symptoms among cocaine dependent patients. Subst Abus 23:105-114.
Pauly JR, Stitzel JA, Marks MJ, Collins AC (1989) An autoradiographic analysis of cholinergic receptors in mouse brain. Brain Res Bull 22:453-459.
Pauly JR, Marks MJ, Gross SD, Collins AC (1991) An autoradiographic analysis of cholinergic receptors in mouse brain after chronic nicotine treatment. J Pharmacol Exp Ther 258:1127-1136.

241
Pauly JR, Marks MJ, Robinson SF, van de Kamp JL, Collins AC (1996) Chronic nicotine and mecamylamine treatment increase brain nicotinic receptor binding without changing alpha 4 or beta 2 mRNA levels. J Pharmacol Exp Ther 278:361-369.
Paylor R, Nguyen M, Crawley JN, Patrick J, Beaudet A, Orr-Urtreger A (1998) Alpha7 nicotinic receptor subunits are not necessary for hippocampal-dependent learning or sensorimotor gating: a behavioral characterization of Acra7-deficient mice. Leam Mem 5:302-316.
Peng X, Gerzanich V, Anand R, Whiting PJ, Lindstrom J (1994) Nicotine-induced increase in neuronal nicotinic receptors results from a decrease in the rate of receptor turnover. Mol Pharmacol 46:523-530.
Perry DC, Davila-Garcia MI, Stockmeier CA, Kellar KJ (1999) Increased nicotinic receptors in brains from smokers: membrane binding and autoradiography studies.J Pharmacol Exp Ther 289:1545-1552.
Picciotto MR (1998) Common aspects of the action of nicotine and other drugs of abuse. Drug Alcohol Depend 51:165-172.
Picciotto MR, Caldarone BJ, King SL, Zachariou V (2000) Nicotinic receptors in the brain. Links between molecular biology and behavior. Neuropsychopharmacology 22:451-465.
Picciotto MR, Zoli M, Rimondini R, Lena C, Marubio LM, Pich EM, Fuxe K, Changeux JP (1998) Acetylcholine receptors containing the beta2 subunit are involved in the reinforcing properties of nicotine. Nature 391:173-177.
Picciotto MR, Zoli M, Lena C, Bessis A, Lallemand Y, Le Novere N, Vincent P, Pich EM, Brulet P, Changeux JP (1995) Abnormal avoidance learning in mice lacking functional high-affmity nicotine receptor in the brain. Nature 374:65-67.
Pidoplichko VI, DeBiasi M, Williams JT, Dani JA (1997) Nicotine activates and desensitizes midbrain dopamine neurons. Nature 390:401-404.
Pierce RC, Kumaresan V (2006) The mesolimbic dopamine system: The final common pathway for the reinforcing effect of drugs of abuse? Neuroscience & Biobehavioral Reviews 30:215-238.
Pietila K, Salminen O, Leikola-Pelho T, Ahtee L (1996) Tolerance to nicotine's effects on striatal dopamine metabolism in nicotine-withdrawn mice. Eur J Pharmacol 318:17-22.
Pomerleau OF (1998) Endogenous opioids and smoking: a review of progress and problems. Psychoneuroendocrinology 23:115-130.

242
Pons S, Fattore L, Cossu G, Tolu S, Porcu E, McIntosh JM, Changeux JP, Maskos U, Fratta W (2008) Crucial role of alpha4 and alphaô nicotinic acetylcholine receptor subunits from ventral tegmental area in systemic nicotine self-administration. J Neurosci 28:12318-12327.
Pontieri FE, Tanda G, Orzi F, Di Chiara G (1996) Effects of nicotine on the nucleus accumbens and similarity to those of addictive drugs. Nature 382:255-257.
Prentice DJ, Kelly MD, Ledent C, Hourani SM (2002) Relaxation of the mouse isolated aorta and carotid artery in response to adenosine analogues in genetically- modified mice lacking the adenosine A(2A) receptor. Naunyn Schmiedebergs Arch Pharmacol 366:127-133.
Preston Z, Lee K, Widdowson L, Freeman TC, Dixon AK, Richardson PJ (2000) Adenosine receptor expression and function in rat striatal cholinergic intemeurons. B rJ Pharmacol 130:886-890.
Prince RJ, Sine SM (1996) Molecular dissection of subunit interfaces in the acetylcholine receptor. Identification of residues that determine agonist selectivity. J Biol Chem 271:25770-25777.
Quarta D, Ferre S, Solinas M, You ZB, Hockemeyer J, Popoli P, Goldberg SR (2004a) Opposite modulatory roles for adenosine A1 and A2A receptors on glutamate and dopamine release in the shell of the nucleus accumbens. Effects of chronic caffeine exposure. J Neurochem 88:1151-1158.
Quarta D, Borycz J, Solinas M, Patkar K, Hockemeyer J, Ciruela F, Lluis C, Franco R, Woods AS, Goldberg SR, Ferre S (2004b) Adenosine receptor-mediated modulation of dopamine release in the nucleus accumbens depends on glutamate neurotransmission and N-methyl-D-aspartate receptor stimulation. J Neurochem 91:873-880.
Quick MW, Lester RA (2002) Desensitization of neuronal nicotinic receptors. J Neurobiol 53:457-478.
Quik M, Vailati S, Bordia T, Kulak JM, Fan H, McIntosh JM, Clementi F, Gotti C (2005) Subunit composition of nicotinic receptors in monkey striatum: effect of treatments with 1 -methyl-4-phenyl-1,2,3,6-tetrahydropyridine or L-DOPA. Mol Pharmacol 67:32-41.
Rada P, Mark GP, Pothos E, Hoebel BG (1991) Systemic morphine simultaneously decreases extracellular acetylcholine and increases dopamine in the nucleus accumbens of freely moving rats. Neuropharmacology 30:1133-1136.

243
Rada PV, Mark GP, Taylor KM, Hoebel BG (1996) Morphine and naloxone, i.p. or locally, affect extracellular acetylcholine in the accumbens and prefrontal cortex. Pharmacol Biochem Behav 53:809-816.
Raehal KM, Bohn LM (2005) Mu opioid receptor regulation and opiate responsiveness. Aaps J 7:E587-591.
Rahman S, Zhang J, Engleman EA, Corrigall WA (2004) Neuroadaptive changes in the mesoaccumbens dopamine system after chronic nicotine self-administration: a microdialysis study. Neuroscience 129:415-424.
Ramirez-Latorre J, Yu OR, Qu X, Perin F, Karlin A, Role L (1996) Functional contributions of alpha5 subunit to neuronal acetylcholine receptor channels. Nature 380:347-351.
Rasmussen BA, Perry DC (2006) An autoradiographic analysis of [ 1251] [alpha]- bungarotoxin binding in rat brain after chronic nicotine exposure. Neuroscience Letters 404:9-14.
Reid MS, Mickalian JD, Delucchi KL, Berger SP (1999) A nicotine antagonist, mecamylamine, reduces cue-induced cocaine craving in cocaine-dependent subjects. Neuropsychopharmacology 20:297-307.
Reid MS, Mickalian JD, Delucchi KL, Hall SM, Berger SP (1998) An acute dose of nicotine enhances cue-induced cocaine craving. Drug Alcohol Depend 49:95-104.
Reinscheid RK, Nothacker HP, Bourson A, Ardati A, Henningsen RA, Bunzow JR, Grandy DK, Langen H, Monsma FJ, Jr., Civelli O (1995) Orphanin FQ: a neuropeptide that activates an opioidlike G protein-coupled receptor. Science 270:792-794.
Reuben M, Clarke PB (2000) Nicotine-evoked [3H]5-hydroxytryptamine release from rat striatal synaptosomes. Neuropharmacology 39:290-299.
Ribeiro FM, Black SA, Prado VF, Rylett RJ, Ferguson SS, Prado MA (2006) The "ins" and "outs" of the high-affinity choline transporter CHTl. J Neurochem 97:1-12.
Rice ME, Cragg SJ (2004) Nicotine amplifies reward-related dopamine signals in striatum. Nat Neurosci 7:583-584.
Ritz MC, Lamb RJ, Goldberg SR, Kuhar MJ (1987) Cocaine receptors on dopamine transporters are related to self-administration of cocaine. Science 237:1219-1223.

244
Roberts DC, Koob GF (1982) Disruption of cocaine self-administration following 6- hydroxydopamine lesions of the ventral tegmental area in rats. Pharmacol Biochem Behav 17:901-904.
Roberts DC, Corcoran ME, Fibiger HC (1977) On the role of ascending catecholaminergic systems in intravenous self-administration of cocaine. Pharmacol Biochem Behav 6:615-620.
Roberts DC, Koob GF, Klonoff P, Fibiger HC (1980) Extinction and recovery of cocaine self-administration following 6-hydroxydopamine lesions of the nucleus accumbens. Pharmacol Biochem Behav 12:781-787.
Rodrigues RJ, Alfaro TM, Rebola N, Oliveira CR, Cunha RA (2005) Co-localization and functional interaction between adenosine A(2A) and metabotropic group 5 receptors in glutamatergic nerve terminals of the rat striatum. J Neurochem 92:433-441.
Roerig B, Nelson DA, Katz LC (1997) Fast synaptic signaling by nicotinic acetylcholine and serotonin 5-HT3 receptors in developing visual cortex. J Neurosci 17.8353- 8362.
Role LW, Berg DK (1996) Nicotinic receptors in the development and modulation of CNS synapses. Neuron 16:1077-1085.
Roll JM, Higgins ST, Budney AJ, Bickel WK, Badger GJ (1996) A comparison of cocaine-dependent cigarette smokers and non-smokers on demographic, drug use and other characteristics. Drug Alcohol Depend 40:195-201.
Romanelli MN, Gratteri P, Guandalini L, Martini E, Bonaccini C, Gualtieri F (2007) Central nicotinic receptors: structure, function, ligands, and therapeutic potential. ChemMedChem 2:746-767.
Rose JE, Behm FM, Levin ED (1993) Role of nicotine dose and sensory cues in the regulation of smoke intake. Pharmacol Biochem Behav 44:891-900.
Rosecrans JA (1989) Nicotine as a discriminative stimulus: a neurobiobehavioral approach to studying central cholinergic mechanisms. J Subst Abuse 1:287-300.
Rosin DL, Hettinger BD, Lee A, Linden J (2003) Anatomy of adenosine A2A receptors in brain: morphological substrates for integration of striatal function. Neurology 6LS12-18.
Ross SA, Wong JY, Clifford JJ, Kinsella A, Massalas JS, Horne MK, Scheffer IE, Kola I, Waddington JL, Berkovic SF, Drago J (2000) Phenotypic characterization of an alpha 4 neuronal nicotinic acetylcholine receptor subunit knock-out mouse. J Neurosci 20:6431-6441.

245
Rowell PP, Li M (1997) Dose-response relationship for nicotine-induced up-regulation of rat brain nicotinic receptors. J Neurochem 68:1982-1989.
Rupniak NM, Patel S, Marwood R, Webb J, Traynor JR, Elliott J, Freedman SB, Fletcher SR, Hill RG (1994) Antinociceptive and toxic effects of (+)-epibatidine oxalate attributable to nicotinic agonist activity. Br J Pharmacol 113:1487-1493.
Russell MA, Jarvis M, Iyer R, Feyerabend C (1980) Relation of nicotine yield of cigarettes to blood nicotine concentrations in smokers. Br Med J 280:972-976.
Sakmann B, Patlak J, Neher E (1980) Single acetylcholine-activated channels show burst- kinetics in presence of desensitizing concentrations of agonist. Nature 286:71-73.
Salas R, Fieri F, De Biasi M (2004) Decreased signs of nicotine withdrawal in mice null for the beta4 nicotinic acetylcholine receptor subunit. J Neurosci 24:10035-10039.
Salas R, Main A, Gangitano D, De Biasi M (2007) Decreased withdrawal symptoms but normal tolerance to nicotine in mice null for the alpha7 nicotinic acetylcholine receptor subunit. Neuropharmacology.
Salas R, Fieri F, Fung B, Dani JA, De Biasi M (2003a) Altered anxiety-related responses in mutant mice lacking the beta4 subunit of the nicotinic receptor. J Neurosci 23:6255-6263.
Salas R, Orr-Urtreger A, Broide RS, Beaudet A, Paylor R, De Biasi M (2003b) The nicotinic acetylcholine receptor subunit alpha 5 mediates short-term effects of nicotine in vivo. Mol Pharmacol 63:1059-1066.
Salim H, Ferre S, Dalai A, Peterfreund RA, Fuxe K, Vincent JD, Lledo PM (2000) Activation of adenosine A1 and A2A receptors modulates dopamine D2 receptor- induced responses in stably transfected human neuroblastoma cells. J Neurochem 74:432-439.
Salminen O, Whiteaker P, Grady SR, Collins AC, McIntosh JM, Marks MJ (2005) The subunit composition and pharmacology of alpha-Conotoxin Mll-binding nicotinic acetylcholine receptors studied by a novel membrane-binding assay. Neuropharmacology 48:696-705.
Salminen O, Murphy KL, McIntosh JM, Drago J, Marks MJ, Collins AC, Grady SR (2004) Subunit composition and pharmacology of two classes of striatal presynaptic nicotinic acetylcholine receptors mediating dopamine release in mice. Mol Pharmacol 65:1526-1535.
Schenk S, Snow S, Horger BA (1991) Preexposure to amphetamine but not nicotine sensitizes rats to the motor activating effect of cocaine. Psychopharmacology 103:62-66.

246
Schlussman SD, Zhou Y, Bailey A, Ho A, Kreek MJ (2005) Steady-dose and escalating- dose "binge" administration of cocaine alter expression of behavioral stereotypy and striatal preprodynorphin mRNA levels in rats. Brain Res Bull 67:169-175.
Schlussman SD, Zhang Y, Kane S, Stewart CL, Ho A, Kreek MJ (2003) Locomotion, stereotypy, and dopamine DI receptors after chronic 'binge' cocaine in C57BL/6J and 129/J mice. Pharmacol Biochem Behav 75:123-131.
Schoepfer R, Conroy WG, Whiting P, Gore M, Lindstrom J (1990) Brain alpha- bungarotoxin binding protein cDNAs and MAbs reveal subtypes of this branch of the ligand-gated ion channel gene superfamily. Neuron 5:35-48.
Schoffelmeer AN, De Vries TJ, Wardeh G, van de Ven HW, Vanderschuren LJ (2002) Psychostimulant-induced behavioral sensitization depends on nicotinic receptor activation. J Neurosci 22:3269-3276.
Schorling JB, Gutgesell M, Klas P, Smith D, Keller A (1994) Tobacco, alcohol and other drug use among college students. J Subst Abuse 6:105-115.
Schultz W (2007) Multiple dopamine functions at different time courses. Annu Rev Neurosci 30:259-288.
Schultz W, Dayan P, Montague PR (1997) A neural substrate of prediction and reward. Science 275:1593-1599.
Schwartz RD, Kellar KJ (1983) Nicotinic cholinergic receptor binding sites in the brain: regulation in vivo. Science 220:214-216.
Semenova S, Markou A (2003) Cocaine-seeking behavior after extended cocaine-free periods in rats: role of conditioned stimuli. Psychopharmacology (Berl) 168:192- 200.
Sharma G, Vijayaraghavan S (2003) Modulation of presynaptic store calcium induces release of glutamate and postsynaptic firing. Neuron 38:929-939.
Shimo Y, Hikosaka O (2001) Role of tonically active neurons in primate caudate in reward-oriented saccadic eye movement. J Neurosci 21:7804-7814.
Shippenberg TS, Heidbreder C, Lefevour A (1996) Sensitization to the conditioned rewarding effects of morphine: pharmacology and temporal characteristics. Eur J Pharmacol 299:33-39.
Shoaib M, Swanner LS, Yasar S, Goldberg SR (1999) Chronic caffeine exposure potentiates nicotine self-administration in rats. Psychopharmacology (Berl) 142:327-333.

247
Short JL, Ledent C, Drago J, Lawrence AJ (2006a) Receptor crosstalk: characterization of mice deficient in dopamine DI and adenosine A2A receptors. Neuropsychopharmacology 31:525-534.
Short JL, Ledent C, Borrelli E, Drago J, Lawrence AJ (2006b) Genetic interdependence of adenosine and dopamine receptors: evidence from receptor knockout mice. Neuroscience 139:661-670.
Sinha R, Garcia M, Paliwal P, Kreek MJ, Rounsaville BJ (2006) Stress-induced cocaine craving and hypothalamic-pituitary-adrenal responses are predictive of cocaine relapse outcomes. Arch Gen Psychiatry 63:324-331.
Sloan JW, Martin WR, Bostwick M, Hook R, Wala E (1988) The comparative binding characteristics of nicotinic ligands and their pharmacology. Pharmacol Biochem Behav 30:255-267.
Slotkin TA (2002) Nicotine and the adolescent brain: insights from an animal model. Neurotoxicol Teratol 24:369-384.
Slowe SJ, Simonin F, Kieffer B, Kitchen I (1999) Quantitative autoradiography of mu- ,delta- and kappal opioid receptors in kappa-opioid receptor knockout mice. Brain Res 818:335-345.
Smith JE, Vaughn TC, Co C (2004) Acetylcholine turnover rates in rat brain regions during cocaine self-administration. J Neurochem 88:502-512.
Snell BJ, Short JL, Drago J, Ledent C, Lawrence AJ (2000) Visualisation of AMPA binding sites in the brain of mice lacking the adenosine A(2a) receptor. Neurosci Lett 291:97-100.
Sokoloff P, Giros B, Martres MP, Bouthenet ML, Schwartz JC (1990) Molecular cloning and characterization of a novel dopamine receptor (D3) as a target for neuroleptics. Nature 347:146-151.
Solinas M, Ferre S, You ZB, Karcz-Kubicha M, Popoli P, Goldberg SR (2002) Caffeine induces dopamine and glutamate release in the shell of the nucleus accumbens. J Neurosci 22:6321-6324.
Sorenson EM, Shiroyama T, Kitai ST (1998) Postsynaptic nicotinic receptors on dopaminergic neurons in the substantia nigra pars compacta of the rat. Neuroscience 87:659-673.
Soria G, Castane A, Ledent C, Parmentier M, Maldonado R, Valverde O (2006) The lack of A2A adenosine receptors diminishes the reinforcing efficacy of cocaine. Neuropsychopharmacology 31:978-987.

248
Soria G, Castane A, Berrendero F, Ledent C, Parmentier M, Maldonado R, Valverde O (2004) Adenosine A2A receptors are involved in physical dependence and place conditioning induced by THC. Eur J Neurosci 20:2203-2213.
Soria G, Mendizabal V, Tourino C, Robledo P, Ledent C, Parmentier M, Maldonado R, Valverde O (2005) Lack of CBl cannabinoid receptor impairs cocaine selfadministration. Neuropsychopharmacology 30:1670-1680.
Spanagel R (1995) Modulation of drug-induced sensitization processes by endogenous opioid systems. Behav Brain Res 70:37-49.
Sparks JA, Pauly JR (1999) Effects of continuous oral nicotine administration on brain nicotinic receptors and responsiveness to nicotine in C57B1/6 mice Psychopharmacology (Berl) 141:145-153.
Stewart J, Wise RA (1992) Reinstatement of heroin self-administration habits: morphine prompts and naltrexone discourages renewed responding after extinction. Psychopharmacology (Berl) 108:79-84.
Sutherland G, Stapleton JA, Russell MA, Feyerabend C (1995) Naltrexone, smoking behaviour and cigarette withdrawal. Psychopharmacology (Berl) 120:418-425.
Svenningsson P, Lindskog M, Ledent C, Parmentier M, Greengard P, Fredholm BB, Fisone G (2000) Regulation of the phosphorylation of the dopamine- and cAMP- regulated phosphoprotein of 32 kDa in vivo by dopamine D I, dopamine D2, and adenosine A2A receptors. Proc Natl Acad Sci U S A 97:1856-1860.
Svingos AL, Periasamy S, Pickel VM (2000) Presynaptic dopamine D(4) receptor localization in the rat nucleus accumbens shell. Synapse 36:222-232.
Svingos AL, Colago EE, Pickel VM (2001) Vesicular acetylcholine transporter in the rat nucleus accumbens shell: subcellular distribution and association with mu-opioid receptors. Synapse 40:184-192.
Swanson JA, Lee JW, Hopp JW (1994) Caffeine and nicotine: a review of their joint use and possible interactive effects in tobacco withdrawal. Addict Behav 19:229-256.
Szumlinski KK, Maisonneuve IM, Glick SD (2000) The potential anti-addictive agent, 18-methoxycoronaridine, blocks the sensitized locomotor and dopamine responses produced by repeated morphine treatment. Brain Res 864:13-23.
Taly A, Corringer PJ, Guedin D, Lestage P, Changeux JP (2009) Nicotinic receptors: allosteric transitions and therapeutic targets in the nervous system. Nat Rev Drug Discov 8:733-750.

249
Tanda G, Goldberg SR (2000) Alteration of the behavioral effects of nicotine by chronic caffeine exposure. Pharmacol Biochem Behav 66:47-64.
Tang YL, Kranzler HR, Gelemter J, Farrer LA, Cubells JF (2007) Comorbid psychiatric diagnoses and their association with cocaine-induced psychosis in cocaine- dependent subjects. Am J Addict 16:343-351.
Tapper AR, McKinney SL, Nashmi R, Schwarz J, Deshpande P, Labarca C, Whiteaker P, Marks MJ, Collins AC, Lester HA (2004) Nicotine activation of alpha4* receptors: sufficient for reward, tolerance, and sensitization. Science 306:1029- 1032.
Taraschenko OD, Shulan JM, Maisonneuve IM, Glick SD (2007a) 18-MC acts in the medial habenula and interpeduncular nucleus to attenuate dopamine sensitization to morphine in the nucleus accumbens. Synapse 61:547-560.
Taraschenko OD, Rubbinaccio HY, Shulan JM, Glick SD, Maisonneuve IM (2007b) Morphine-induced changes in acetylcholine release in the interpeduncular nucleus and relationship to changes in motor behavior in rats. Neuropharmacology 53:18- 26.
Tepper JM, Sawyer SF, Groves PM (1987) Electrophysiologically identified nigral dopaminergic neurons intracellularly labeled with HRP: light-microscopic analysis. J Neurosci 7:2794-2806.
Tepper JM, Sun BC, Martin LP, Creese I (1997) Functional roles of dopamine D2 and D3 autoreceptors on nigrostriatal neurons analyzed by antisense knockdown in vivo. J Neurosci 17:2519-2530.
Thomsen M, Hall FS, Uhl GR, Caine SB (2009) Dramatically decreased cocaine selfadministration in dopamine but not serotonin transporter knock-out mice. J Neurosci 29:1087-1092.
Tobler PN, Fiorillo CD, Schultz W (2005) Adaptive coding of reward value by dopamine neurons. Science 307:1642-1645.
Tredway TL, Guo JZ, Chiappinelli VA (1999) N-type voltage-dependent calcium channels mediate the nicotinic enhancement of GABA release in chick brain. J Neurophysiol 81:447-454.
Trigo JM, Zimmer A, Maldonado R (2009) Nicotine anxiogenic and rewarding effects are decreased in mice lacking beta-endorphin. Neuropharmacology 56.1147-1153.
Trussell LO, Jackson MB (1985) Adenosine-activated potassium conductance in cultured striatal neurons. Proc Natl Acad Sci U S A 82:4857-4861.

250
Turek JW, Kang CH, Campbell JE, Americ SP, Sullivan JP (1995) A sensitive technique for the detection of the alpha 7 neuronal nicotinic acetylcholine receptor antagonist, methyllycaconitine, in rat plasma and brain. J Neurosci Methods 61:113-118.
Tzavara ET, Monory K, Hanoune J, Nomikos GG (2002) Nicotine withdrawal syndrome: behavioural distress and selective up-regulation of the cyclic AMP pathway in the amygdala. Eur J Neurosci 16:149-153.
Undie AS, Weinstock J, Sarau HM, Friedman E (1994) Evidence for a distinct Dl-like dopamine receptor that couples to activation of phosphoinositide metabolism in brain. J Neurochem 62:2045-2048.
Unterwald EM, Ho A, Rubenfeld JM, Kreek MJ (1994) Time course of the development of behavioral sensitization and dopamine receptor up-regulation during binge cocaine administration. J Pharmacol Exp Ther 270:1387-1396.
Unwin N (2005) Refined Structure of the Nicotinic Acetylcholine Receptor at 4 A Resolution. Journal of Molecular Biology 346:967.
Unwin N, Toyoshima C, Kubalek E (1988) Arrangement of the acetylcholine receptor subunits in the resting and desensitized states, determined by cryoelectron microscopy of crystallized Torpedo postsynaptic membranes. J Cell Biol 107:1123-1138.
Vaccarino AL, Kastin AJ (2001) Endogenous opiates: 2000. Peptides 22.2257-2328.
Valera S, Ballivet M, Bertrand D (1992) Progesterone modulates a neuronal nicotinic acetylcholine receptor. Proc Natl Acad Sci U S A 89:9949-9953.
Vallejo YF, Buisson B, Bertrand D, Green WN (2005) Chronic nicotine exposure upregulates nicotinic receptors by a novel mechanism. J Neurosci 25:5563-5572.
Vaupel DB, Mukhin AG, Kimes AS, Horti AG, Koren AO, London ED (1998) In vivo studies with [125I]5-I-A-85380, a nicotinic acetylcholine receptor radioligand. Neuroreport 9:2311-2317.
Vemallis AB, Conroy WG, Berg DK (1993) Neurons assemble acetylcholine receptors with as many as three kinds of subunits while maintaining subunit segregation among receptor subtypes. Neuron 10:451-464.
Vetter DE, Liberman MC, Mann J, Barhanin J, Boulter J, Brown MC, Saffiote-Kolman J, Heinemann SF, Elgoyhen AB (1999) Role of alpha9 nicotinic ACh receptor subunits in the development and function of cochlear efferent innervation. Neuron 23:93-103.

251
Vetter DE, Katz E, Maison SF, Taranda J, Turcan S, Ballestero J, Liberman MC, Elgoyhen AB, Boulter J (2007) The alphalO nicotinic acetylcholine receptor subunit is required for normal synaptic function and integrity of the olivocochlear system. Proc Natl Acad Sci U S A 104:20594-20599.
Vihavainen T, Mijatovic J, Piepponen TP, Tuominen RK, Ahtee L (2006) Effect of morphine on locomotor activity and striatal monoamine metabolism in nicotine- withdrawn mice. Behav Brain Res 173:85-93.
Vihavainen T, Piltonen M, Tuominen RK, Korpi ER, Ahtee L (2008a) Morphine-nicotine interaction in conditioned place preference in mice after chronic nicotine exposure. Eur J Pharmacol 587:169-174.
Vihavainen T, Relander TR, Leiviska R, Airavaara M, Tuominen RK, Ahtee L, Piepponen TP (2008b) Chronic nicotine modifies the effects of morphine on extracellular striatal dopamine and ventral tegmental GABA. J Neurochem 107:844-854.
Volz TJ, Schenk JO (2005) A comprehensive atlas of the topography of functional groups of the dopamine transporter. Synapse 58:72-94.
Wada E, Wada K, Boulter J, Deneris E, Heinemann S, Patrick J, Swanson LW (1989) Distribution of alpha 2, alpha 3, alpha 4, and beta 2 neuronal nicotinic receptor subunit mRNAs in the central nervous system: a hybridization histochemical study in the rat. J Comp Neurol 284:314-335.
Waldhoer M, Bartlett SE, Whistler JL (2004) Opioid receptors. Annu Rev Biochem 73:953-990.
Walters CL, Cleck JN, Kuo YC, Blendy JA (2005) Mu-opioid receptor and CREB activation are required for nicotine reward. Neuron 46:933-943.
Walters CL, Brown S, Changeux JP, Martin B, Damaj MI (2006) The beta2 but not alpha7 subunit of the nicotinic acetylcholine receptor is required for nicotine- conditioned place preference in mice. Psychopharmacology (Berl) 184:339-344.
Wang F, Chen H, Steketee JD, Sharp BM (2007) Upregulation of ionotropic glutamate receptor subunits within specific mesocorticolimbic regions during chronic nicotine self-administration. Neuropsychopharmacology 32:103-109.
Wang F, Gerzanich V, Wells GB, Anand R, Peng X, Keyser K, Lindstrom J (1996) Assembly of human neuronal nicotinic receptor alpha5 subunits with alpha3, beta2, and beta4 subunits. J Biol Chem 271:17656-17665.
Wang F, Nelson ME, Kuryatov A, Olale F, Cooper J, Keyser K, Lindstrom J (1998) Chronic nicotine treatment up-regulates human alpha3 beta2 but not alpha3 beta4

252
acetylcholine receptors stably transfected in human embryonic kidney cells. J Biol Chem 273:28721-28732.
Ward JM, Cockcroft VB, Lunt GG, Smillie FS, Wonnacott S (1990) Methyllycaconitine: a selective probe for neuronal alpha-bungarotoxin binding sites. FEBS Lett 270:45-48.
Warpman U, Friberg L, Gillespie A, Hellstrom-Lindahl E, Zhang X, Nordberg A (1998) Regulation of nicotinic receptor subtypes following chronic nicotinic agonist exposure in MIO and SH-SY5Y neuroblastoma cells. J Neurochem 70:2028-2037.
Watkins SS, Stinus L, Koob GF, Markou A (2000) Reward and somatic changes during precipitated nicotine withdrawal in rats: centrally and peripherally mediated effects. J Pharmacol Exp Ther 292:1053-1064.
Wedzony K, Chocyk A, Mackowiak M, Fijal K, Czyrak A (2000) Cortical localization of dopamine D4 receptors in the rat brain-immunocytochemical study. J Physiol Pharmacol 51:205-221.
Weinberger AH, Sofuoglu M (2009) The impact of cigarette smoking on stimulant addiction. Am J Drug Alcohol Abuse 35:12-17.
Weiner DM, Levey AI, Sunahara RK, Niznik HB, ODowd BF, Seeman P, Brann MR (1991) DI and D2 dopamine receptor mRNA in rat brain. Proc Natl Acad Sci U S A 88:1859-1863.
Werner P, Hussy N, Buell G, Jones KA, North RA (1996) D2, D3, and D4 dopamine receptors couple to G protein-regulated potassium channels in Xenopus oocytes. Mol Pharmacol 49:656-661.
Wevers A, Monteggia L, Nowacki S, Bloch W, Schütz U, Lindstrom J, Pereira EF, Eisenberg H, Giacobini E, de Vos RA, Steur EN, Maelicke A, Albuquerque EX, Schroder H (1999) Expression of nicotinic acetylcholine receptor subunits in the cerebral cortex in Alzheimer's disease: histotopographical correlation with amyloid plaques and hyperphosphorylated-tau protein. Eur J Neurosci 11:2551- 2565.
Wewers ME, Dhatt RK, Snively TA, Tejwani GA (1999) The effect of chronic administration of nicotine on antinociception, opioid receptor binding and met- enkelphalin levels in rats. Brain Res 822:107-113.
Whiteaker P, Sharpies CG, Wonnacott S (1998) Agonist-induced up-regulation of alpha4beta2 nicotinic acetylcholine receptors in MIO cells: pharmacological and spatial definition. Mol Pharmacol 53:950-962.

253
Whiteaker P, Garcha HS, Wonnacott S, Stolerman IP (1995) Locomotor activation and dopamine release produced by nicotine and isoarecolone in rats. Br J Pharmacol 116:2097-2105.
Whiteaker P, McIntosh JM, Luo S, Collins AC, Marks MJ (2000a) 1251-alpha-conotoxin Mil identifies a novel nicotinic acetylcholine receptor population in mouse brain. Mol Pharmacol 57:913-925.
Whiteaker P, Jimenez M, McIntosh JM, Collins AC, Marks MJ (2000b) Identification of a novel nicotinic binding site in mouse brain using [(125)I]-epibatidine. Br J Pharmacol 131:729-739.
Whiteaker P, Peterson CG, Xu W, McIntosh JM, Paylor R, Beaudet AL, Collins AC, Marks MJ (2002) Involvement of the alpha3 subunit in central nicotinic binding populations. J Neurosci 22:2522-2529.
Widnell KL, Russell DS, Nestler EJ (1994) Regulation of expression of cAMP response element-binding protein in the locus coeruleus in vivo and in a locus coeruleus- like cell line in vitro. Proc Natl Acad Sci U S A 91:10947-10951.
Wilkie GI, Hutson P, Sullivan JP, Wonnacott S (1996) Pharmacological characterization of a nicotinic autoreceptor in rat hippocampal synaptosomes. Neurochem Res 21:1141-1148.
Williams M, Robinson JL (1984) Binding of the nicotinic cholinergic antagonist, dihydro-beta-erythroidine, to rat brain tissue. J Neurosci 4:2906-2911.
Wilson GG, Karlin A (2001) Inaugural Article: Acetylcholine receptor channel structure in the resting, open, and desensitized states probed with the substituted-cysteine- accessibility method. PNAS 98:1241-1248.
Wise RA, Murray A, Bozarth MA (1990) Bromocriptine self-administration and bromocriptine-reinstatement of cocaine-trained and heroin-trained lever pressing in rats. Psychopharmacology (Berl) 100:355-360.
Wiseman EJ, McMillan DE (1998) Rationale for cigarette smoking and for mentholation preference in cocaine- and nicotine-dependent outpatients. Compr Psychiatry 39:358-363.
Wong GY, Wolter TD, Croghan GA, Croghan IT, Offord KP, Hurt RD (1999) A randomized trial of naltrexone for smoking cessation. Addiction 94:1227-1237.
Wonnacott S (1997) Presynaptic nicotinic ACh receptors. Trends Neurosci 20:92-98.

254
"“'"ZEISjST "" «-«I p»s
~ a S S H S = = - = :
■■■figpSSHSiSS!'■ s a a s s s s s“ ssH S S " S “ -
" - = e f = = : B a = : = =Psychopharmacology (Berl) 190:189-199. mice.

255
Zhou FM, Liang Y, Dani JA (2001) Endogenous nicotinic cholinergic activity regulates dopamine release in the striatum. Nat Neurosci 4:1224-1229.
Zocchi A, Pert A (1994) Alterations in striatal acetylcholine overflow by cocaine, morphine, and MK-801: relationship to locomotor output. Psychopharmacology (Berl) 115:297-304.
Zoli M, Lena C, Picciotto MR, Changeux JP (1998) Identification of four classes of brain nicotinic receptors using beta2 mutant mice. J Neurosci 18:4461-4472.
Zoli M, Picciotto MR, Ferrari R, Cocchi D, Changeux JP (1999) Increased neurodegeneration during ageing in mice lacking high-affmity nicotine receptors. Em boJ 18:1235-1244.
Zoli M, Moretti M, Zanardi A, McIntosh JM, Clementi F, Gotti C (2002) Identification of the nicotinic receptor subtypes expressed on dopaminergic terminals in the rat striatum. J Neurosci 22:8785-8789.
JITY OF SUKSEY iffiRâSY





























