Embed Size (px)
Citation preview

Estuarine and Coastal Marine Science (1978) 6, 387-408
The Complexation of Metals with Humic Materials in Natural Waters
R. F. C. Mantoura,” Andrew Dickson and J. P. Riley Department of Oceanography, iJniversity of Liverpool, P.O. Box Iq7,
Liver@ol L6p 3BX, England Received 23 December 1976 and in revised form 20 April 1977
Keywords: humic substances; trace elements; ionic interactions; estuaries
Humic materials have been isolated from sea, river and lake waters by an adsorption technique, and the stability constants of their complexes with Ca, Mg, Mn, Co, Ni, Cu, Zn, Cd and Hg have been determined at pH 8.0 using a gel filtration chromatographic technique. In general, the order of increasing strength of binding of the metals followed the Irving-Williams series. The stability constant data have been used in conjunction with the HALTAFALL program of Ingri et aE. (1967) to compute speciation models for freshwaters and seawater which take into account metal-humic inter- actions. In freshwater more than 90% of the copper and mercury was found to be complexed by humic materials. However, < I I $& of the other metals was bound in this way. In seawater, >gg% of the humic material is complexed by calcium and magnesium because of their relatively high concentrations; metal chelation is only appreciable for copper (- 10%).
The pattern of trace element speciation with inorganic ligands in seawater differs somewhat from that computed by previous workers largely because of the use of more up-to-date stability constant data. Finally, an attempt has been made to compute the changes in metal speciation which would occur when river water becomes diluted with seawater as in an estuary.
Illtroductioll
The humic materials dissolved in natural waters have attracted increasing interest in recent years, not only because of their involvement in marine food chains and organic geochemical cycles (Nisaenbaum & Kaplan, 1972), but also because of their ability to form complexes with metals (Siegel, 1971; Stumm & Brauner, 1975). Although the purely inorganic species present in freshwater and particularly seawater have been extensively studied (see e.g. Whitfield, 1975a; Millero, 1974, 1975)~ the interactions of metals with the humic materials present in such waters have not been investigated. There are two principal reasons for this; first, the difficulty of isolating and characterizing the humic material, and second, the lack until recently, of techniques for the examination of the interactions between metals and polydentate ligands under conditions of pH and metal concentration approximating to those in natural waters. Because of this, some chemists interested in metal speciation in natural waters have disregarded the formation of organic complexes (see e.g. Zirino & Yamamoto,
‘Present address: Institute for Marine Environmental Research, Prospect Place, The Hoe, Plymouth, Devon, PLI 3DH, U.K.
387

388 R. F. C. Mantoura, A. Dickson &i .r. P. Riley
1972; Dyrssen & Wedborg, 1974). Others have based their models on synthetic complexing agents, such as EDTA (Duursma, 1970; Florence & Batley, 1976), salicylic acid (Morel & Morgan, 1972), nitrilotriacetic acid (Childs, 1971) and citric acid (Stumm & Brauner, 1975).
However, models such as these cannot realistically predict the extent of metal-organic interactions in natural waters, since the stability constants of the hypothetical complexes may differ considerably from those of the humic materials which are the principal metal binding agents in most waters (Schnitzer, 1971; Perdue et al., 1976).
The present study was undertaken with the intention of obtaining information about the extent to which metals are organically bound in natural waters. It had two principal aims. Firstly, to use a gel filtration technique (Mantoura & Riley, 19753) to determine the stability constants of the complexes formed between humic materials and metals under realistic conditions of pH and free metal ion concentration. Secondly, to use these constants in a multi-element multi-ligand interaction model to investigate the extent to which metals are organically bound in various types of aquatic environment.
Materials and methods
Freshwater humic compounds were isolated from the surface waters of the northern Welsh Lakes Celyn (samples CEL I (3/11/74), CEL 2 (12/12/p+) and CEL 3 (4/3/75)) and Bala (sample BAL (10/3/75)) and f rom the waters of the rivers Dee (DEE; 4/3/75) and Conway
(CON; 4/3/75). M arine humic compounds were isolated from the surface waters of the Irish Sea (IRI and IR2; 4/2/74) and the sea loch Loch Etive (ET1 and ETz). The com- pounds were recovered from 50-200 1 of the filtered (Whatman GF/F) samples by hydro- phobic adsorption at pH 2-2 onto Amberlite XAD-2 resin and eluted with a I : I v/v
mixture of methanol and 2 M-ammonia (Mantoura & Riley, 1975~).
The method of Schnitzer & Khan (1972) was used to extract humic compounds from samples of sediment from Loch Etive (ET3-ET6) and humic (HA) and fulvic (FA) acids from a sample of horticultural grade peat. A portion of the humic acid HA was subjected to fractional precipitation at 21 “C and pH 7.0 using ammonium sulphate (Theng et al., 1968). This yielded 14 fractions (FP I-FP 14) having narrow ranges of polydispersity, ranging in number average molecular weight (membrane osmometry) from 32 ooo for FP 2 to <4600 for FP 14.
After isolation, all the humic compounds were freed from trace metals by passage of a 0.04 ok solution of the ammonium salt through an 18 cm x 1.7 cm2 column of the ammonium form of the strongly acidic cation exchanger Amberlite IR-120. After washing the column with water, the combined percolate and washings from the column were evaporated in a rotary-film evaporator at a temperature not exceeding 40 “C. The residue, whichconsisted of the ammonium derivative of the humic material was dried overnight in vacua over phosphorus pentoxide. The stability constants of the complexes which it formed with metals were determined at 20 “C and pH 8 by means of the gel filtration complexometric technique developed by Mantoura & Riley (1975b). The concentrations of the various metals in the effluent from the gel filtration column were determined by atomic absorption spectro- photometry.
The compositional data required for the construction of the Lake Celyn water model were obtained as follows: sodium and potassium by flame spectrometry; calcium and magnesium directly by atomic absorption spectrometry (AAS); manganese, nickel, cobalt, zinc, copper and cadmium by preconcentration by chelating ion exchange followed by AAS (Riley & Taylor, 1968); iron calorimetrically with bathophenanthroline (Lewis & Goldberg, 1954);
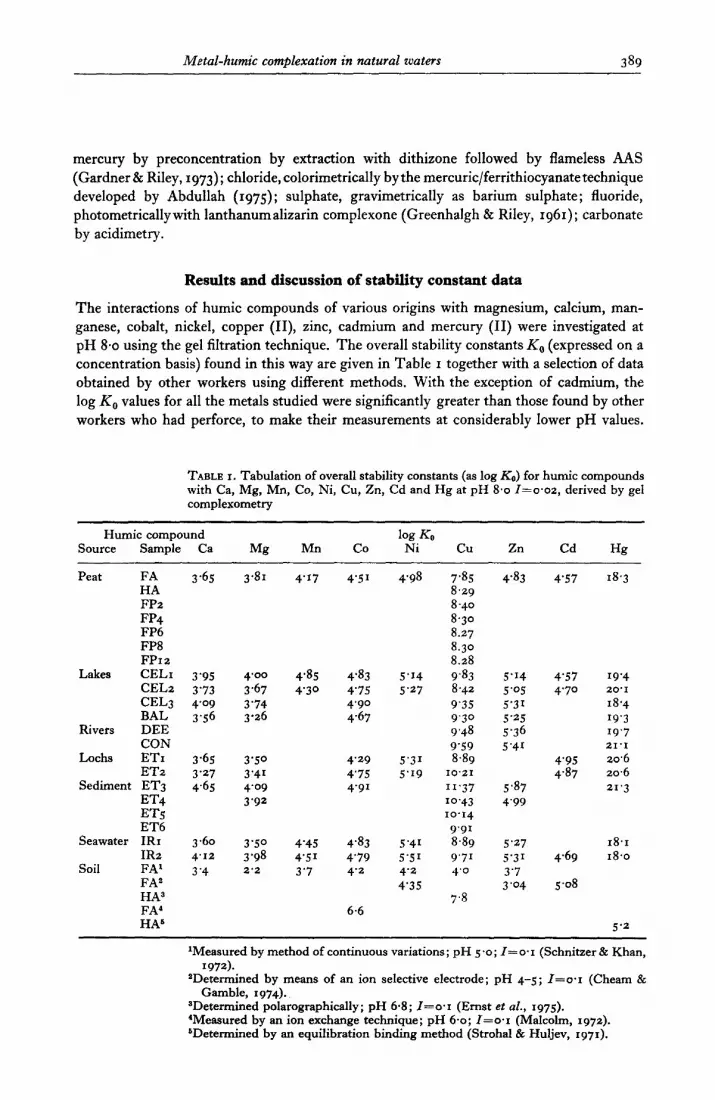
Metal-humic complexation in natural waters 389
mercury by preconcentration by extraction with dithizone followed by flameless AAS (Gardner & Riley, 1973); hl c ori d e, calorimetrically by the mercuric/ferrithiocyanatetechnique developed by Abdullah (1975); sulphate, gravimetrically as barium sulphate; fluoride, photometricallywith lanthanumalizarin complexone (Greenhalgh & Riley, 1961); carbonate by acidimetry.
Results and discussion of stability constant data
The interactions of humic compounds of various origins with magnesium, calcium, man- ganese, cobalt, nickel, copper (II), zinc, cadmium and mercury (II) were investigated at pH 8.0 using the gel filtration technique. The overall stability constants K,, (expressed on a concentration basis) found in this way are given in Table I together with a selection of data obtained by other workers using different methods. With the exception of cadmium, the log K, values for all the metals studied were significantly greater than those found by other workers who had perforce, to make their measurements at considerably lower pH values.
TABLE I. Tabulation of overall stability constants (as log &) for humic compounds with Ca, Mg, Mn, Co, Ni, Cu, Zn, Cd and Hg at pH 8.0 I=o.oz, derived by gel complexometry
Humic compound Source Sample Ca Mg Mn
Peat FA 3.65 3.81 4’17 HA FP2 FP4 FP6 FP8 FP12
Lakes CELI CEL2 CEL3 BAL
Rivers DEE CON
IX&S ETr ET2
Sediment ET3 ET4 ET<
3'95 4.00 4.85 3'73 3.67 4'30 4'09 3'74 3.56 3.26
3.65 3'50 3'27 3'41 4.65 4.09
3'92
ET6 Seawater IRr 3.60 3'50 4'45
IR2 4.12 3'98 4'51 Soil FA’ 3.4 2’2 3’7
FAa HA3 FA’ HA6
co log KS
Ni cu Zn Cd
4'51 4.98 7.85 4.83 4'57 8.29 8.40 8.30 8.27 8.30 8.28
4'83 5'14 9.83 5'14 4'57 4'75 5'27 8.42 5'05 4'70 4'90 9‘35 5'31 4.67 9'30 5’25
9.48 5.36 9'59 5'41
4'29 5'31 8.89 4'95 4'75 5'19 10'21 4.87 4'91 II.37 5.87
IO.43 4'99 10.14
9'91 4.83 5'41 8.89 5'27 4'79 5'51 9'71 5'31 4.69 4'2 4'2 4'0 3'7
4'3.5 3'04 5.08
7.8 6.6
Hg
18.3
19'4 20'1
18.4
19.3 19'7 21'1
20.6 20.6 21.3
18.1 18.0
5’2
‘Measured by method of continuous variations; pH 5.0; I=o.I (Schnitzer & Khan, 1972).
*Determined by means of an ion selective electrode; pH 4-5; I=o*I (Cheam & Gamble, I 974).
SDetermined polarographically; pH 6.8; I=o.I (Ernst et al., 1975). ‘Measured by an ion exchange technique; pH 6.0; I=o.I (Malcolm, 1972).
&Determined by an equilibration binding method (Strohal & Huljev, 1971).

390 R. F. C. Mantoura, A. Dickson WY. P. Riley
The log K, for the copper-peat fulvic acid complex was less than that of the corresponding peat humic acid complex. However, no consistent trends with molecular weight were observed for the log K,, values of the products obtained by fractional precipitation of humic acid. The stability constants of the complexes of copper with humic materials from different environments followed the sequence:
soil FA<soil HA<peat FA<peat HA<seawater HM4ake HMwriver HM <marine sedimentary FA<marine sedimentary HA.
In general, the stabilities of the humic complexes of the various metals follow the well- known Irving-Williams order of stabilities of chelates formed by metal ions with ligands
Mg<Ca<CdmMn<Co<Zn%Ni<Cu<Hg.
Attempts were made to use the gel filtration method to study the binding of iron (III) with the humic compounds. However, they were unsuccessful because of the formation of colloidal hydroxy species which resulted in the production of anomalous bimodal elution curves similar to those observed by Henry & Rogers (1968).
Modelling of natural waters
The stability constant data which were obtained have been used in speciation models to determine the humic-metal interactions in natural waters. In deriving the models it is necessary to take account of the complex network of interactions which results from the fact that any cation can interact with any ligand, yielding a complex. Thus, at equilibrium the jth complex formation reaction can be written as
Iz M %J 4 = K4mlJ &hJ. * * @Y)rnM,l (j=I, . . . N) I-l
where the Al terms are the M reactant species, and the mll terms are the stoichiometric coefficients of reactant i in complex j. The equilibrium concentration (cJ of this complex is given by
M
CJ = b, n [blm(J f=l
(j=r,...N)
where /3, is the conditional stability constant of complex j and [A,] is the concentration of the free (uncomplexed) reactant i.
A mass balance condition can be formulated for each of the various reactant species
ATOT, = [tzl] + $ m,JcJ (i=z, . . ..M) j=l
= fAil +,I, hJ /%fi, fAl1 mfJ*
The resultant set of non-linear equations can then be solved for [A,] provided that the following information is available.
(i) The total concentration of the various reactant species (ATOT,) (ii) The composition of each complex considered (i.e. the values of mLJ) and the con-
ditional stability constants of the complexes. Several general computer programs have been developed for the solution of this problem
(see for example Ingri et aL, 1967; Perrin & Sayce, 1967; Morel & Morgan, 1972). A

Metal-humic complexation in natural waters 391
FORTRAN version of the HALTAFALL program devised by Lars Gunnar SillCn’s group (Ingri et al., 1967) has been used throughout the present study. A detailed description of the application of this program to the study of chemical equilibria has been published in a monograph by Dyrssen et al. (1968).
Values of ATOT,
The data shown in Table 2, which are based on actual analyses, were used in developing a model for the speciation in Lake Celyn water. The transition from a typical river water to seawater (S=35*00%,) was also studied using the following model to determine the reactant concentrations corresponding to various mixing ratios (salinities).
(i) The major components (sodium, potassium, calcium, magnesium, chloride, sulphate, carbonate alkalinity(Ac) and total inorganic carbon (C,)were considered to mix conservatively between the composition of average river water (Livingstone 1963) and that of sea water (Wilson, 1975) as given in Table 2.
TABLE 2. Compositions of waters used in construction of models
Species Lake Celyn’
mol 1-l
[mol (kg solution) -I] Average
River water” Seawater’
Na+ K+ Mga + Ca’ + cl- so:- AC
CT
7’3 x 10-d
1’9 x 10-s
1'0 x 10-K
2.6 x 10-s
8.4 x 10-s
3’9 x 10-b
5.8 x 10-d -
2.83 x row4 5’9 x 10-e
x.69 x IO-~
3’74 x IO-’
2’20 x IO-’
I.17 x 10-a
9’54 x 10-d
%.02x 10-s
0.46847 0’01020
0.05307 0.01028
o.s459o 0.02823 0.0024 0’0022
cu I’OX 10-8
Zn I.8 x IO-’
Cd 3.6 x 10-O
Mn 3’5 x IO-’
Hg 3’5 x x0-10 Ni 2’OX 10-s
if&M
1.7x 10-n
1*2x 10-I
PH 6.2
‘Analytical Data. bLivingstone (1963). Wilson (1975). ‘Calculated from Ac and pH of 7.5.
10-a
10-e
10-e
10-8
10-O
IO-~
10-O -8
See &we I
(ii) The concentrations of the various trace elements were assumed to be constant over the whole range of salinities (Table 2). This assumption (based on lack of reliable analytical data) will have no appreciable effect on the distribution of a particular trace metal between its various species as the ligand concentrations in natural waters are in great excess over any expected total metal concentration.
(iii) The concentration of the ‘humic ligand’ (HUM) was considered to be constant at a value of IO --6 mol kg -1 (values of log K,, of metal-humic complexes were assumed to be constant over the restricted pH range considered; this was, in fact,

392 R. F. C. Mantoura, A. Dickson &31J. P. Riley
so for the mixing of Lake Celyn water with Irish Sea water. The possibility of fractionation of humic materials in the estuary has not been taken into account). The concentrations of HCO,- and COs2 -, and the activity of OH - (Figure I) were calculated using the expression given by Mook & Koene (1975). It would have been possible to take account of the relevant ionization reactions in the speciation model. However, to do this it would have been necessary to first calculate the hydrogen ion concentration as a function of A,, CT and the apparent first and second dissociation constants of carbonic acid before superimposing this ‘pH’ distribution on the model.
-4 - -4 - .A”/ ./-“/ oq;$ oq;~”
/ / -5- cd -5- cd
/ / f//+--~;~~ f//+--~;~~
I I I I I I I I I I I I 0 5 IO 15 20 25 30 35 0 5 IO 15 20 25 30 35
Salinity (‘/..I
Figure I. pH, [HCOII-IT, [COs+]r and (IOH profiles for an ‘estuary’, obtained by conservative mixing of World Average River Water (Living&one, 1963) and sea- water of salinity 3$‘&, (Wilson, 1975). Values of carbonic acid dissociation constants have been taken from Mook & Koene (1975).
Composition of complexes and choice of stability constants
The decision on which complexes should be considered in the interaction model is, perforce, a rather arbitrary one since the choice is limited to those species for which stability constants are available, rather than to those which are necessarily the most important. The difficulty of selecting probable species is exemplified by the observation by Bilinski et al. (1976) that the data from which bicarbonate complexes have been evaluated could be as easily explained in terms of carbonate complexes. The selection of values for the stability constants of the complexes is also somewhat arbitrary as the published data for a given complex often vary widely, and there is indeed no critical compilation of such constants. The values published by Truesdell & Jones (1974) [see Table 3(a)] were employed for the interactions of the major ions (Naf, K+, Caa f, MgB +, SO, 2 -, HCO, -, COs2-), even though some workers (e.g. Reardon, 1975) have expressed reservations about them. The stability constant data of Zirino&Yamamoto (1972) were mainly used for the interactions of copper, zinc and cadmium with inorganic ligands [Tables 3(b) and 3(c)]. The data for the remaining elements were

-- Metal-humic complexation in natural waters 393
chosen principally from the compilations by SillCn & Mar-tell (1964, 1971), mostly on the basis of selecting those which had already been corrected to zero ionic strength.
Estimation of activity corrections for the evaluation of the conditional stability constants
In any natural water appreciable interactions exist between the various charged species. For this reason, the stability constant for any particular interaction will depend on the ionic strength and composition of the solution.
For a general reaction involving the reacting species B for which the mass balance condition is
0= EB vB B,
the thermodynamic equilibrium constant at infinite dilution in pure water IP is given by:
Km= n B (aB) vB.
This can be split into two terms, one the concentration product, or conditional constant, and the other the activity coefficient product (which accounts for the various electrostatic interactions), i.e.
P= II B (cd “I3 n B (YB) vB
=B rIB (YB) vg*
Hence, if the activity coefficients can be evaluated, the conditional constant (i.e. that for the selected conditions) can be calculated.
For hydroxide complexes it was more convenient to use mixed stability constants defined by:
K’ = ~~~~~~~~~~~~~~~~ a VOOHH),
these can also be evaluated from the thermodynamic stability constant, using the relation- ship:
K’=Py 1 metal Ycomplex~
Evaluation of conventional free ion activity coeficients
The evaluation of the various free ion activity coefficients required to make the necessary activity corrections in the model is fraught with difficulties. Whitfield (1975a, b) has provided a detailed and cogent account of the problems involved, and to aid discussion it will be useful to summarize some of the major points below.
In the traditional approach used in marine chemistry (see e.g. Garrels & Thompson, 1962) free ion activities are evaluated by extending the MacInnes (1919) convention beyond the originally suggested limit of a molal ionic strength of O-I. This convention involves three assumptions.
(i) That there are some solutions in which no ion association occurs. (ii) That the free ion activity coefficients of K+ and Cl- are equal in solutions of
potassium chloride. (iii) That these free ion activity coefficients are a function of ionic strength only, and
hence are independent of the composition of the solution. The validity of these assumptions is questionable especially at higher ionic strenghts (see
Hogfeldt, 1975), and unfortunately the convention cannot be validly extended to those

394 R. F. C. Mantoura, A. Dickson &“J. P. Riley
components for which no unassociated single salt solution data are available. For the latter, it is necessary to adopt an alternative approach; the only one that has been employed, to date (by Zirino Sz Yamamoto, 1972), is the Davies (1962) equation, viz:
log Ylon = 0*51 22 (
A*- 0’3 I 1
.
This equation is based on the assumption that the charge (z) of the ion is the only factor which must be considered. Although this may well be true if the ionic strength (I) is low, the differing effects of the various ions on the water structure make it unlikely that such an electrostatic equation will be valid at higher concentrations.
5 IO 15 20 25 30 35 Sali nify P&J
Figure 2. The equilibrium speciation of magnesium as a function of salinity along a model estuary. - Activity coefficients calculated using the Davies equation; -----, activity coefficients for major ion pairs using expression for dipoles (log y= - BI). The difference between the two lines is probably a maximum as the value of y for a 2-2 ion pair used at 1~0.7 (S=357&,) is appreciably lower than that suggested for the ion pair MgSO,” by Kester & F’ytkowicz (1975).
Further difficulties are encountered when attempts are made to evaluate the conventional activity coefficients of the various complex species formed. In developing their model, Garrels & Thompson (1962) evaluated the activity coefficients of neutral complexes from a Setchenow-type relationship, log yIp=kI, in which the value of K was taken as that of carbonic acid in seawater. The activity coefficients of singly charged complex species were assumed to be the same as that of the bicarbonate ion under the same conditions. In contrast, Zirino & Yamamoto (1972) calculated the activity coefficients of complexes from the Davies equation (see above).

Metal-humic complexution in natural waters 395
In the present study the activity coefficients of all species have, for consistency, been calculated using the Davies equation. This approach is, however, not completely satisfactory in that the total activity coefficients which result from the use of such a model do not agree very well with the experimental data. Despite this, the use of y-values derived in this way does give a reasonable representation for the various chemical species because electrostatic effects predominate even up to the ionic strength of seawater. It is therefore suitable for a preliminary survey of speciation. However, ion-pair species (e.g. CaSO,O) cannot be validly treated in the same way as charged particles since they are, in fact, dipoles and will have considerably lower activity coefficients (yIP) in accordance with the semi-theoretical equation:
log YIP = - BI
in which B=o*I, 0.3 and 0.5 for I-I, 1-2 and 2-2 ion pairs respectively (Reardon & Langmuir, 1976). Throughout the various calculations the ratio of the activity coefficients yhumic/ Y metal-h,,mic has been assumed to be unity.
I o co Hum o-
-o- COCO$
O.lo I I -0’1 I I 5 IO 15 20 25 30 :
Salinity (%.I
Figure 3. The equilibrium speciation of calcium as a function of salinity along a model estuary.
The models
In the preparation of the models 83 species were considered (see Table 3). Because of the lack of the relevant stability constant data the possibility of the formation of mixed ligand complexes (e.g. Zn(OH)Cl)) and polynuclear complexes [e.g. Cu,(OH)a2+] has generally been discounted. In addition, interphase reactions such as precipitation, dissolution and sorption were assumed to be absent. Data for the various species were calculated to the

396 R. F. C. Mantoura, A. Dickson WJ. P. Riley
TABLE 3. Stability constants of the various species considered in the model (expressed as log& at infinite dilution)
(a) Major ions (from Truesdell & Jones, 1974, except as indicated)
Chloride Sulphate Bicarbonate Carbonate Hydroxide
Na+ - 0.82 -0.25 I '27 -0.7b K+ - 0.85 - - -
Mgs + - 2.24 0.93 3'40 2.60 Caa + - 2.30 1.26 3.20 I'40
‘This value from Sill&r & Martell (1964, rg7r) agrees better with recent work by Reardon (1975).
%ilkI & Martell (x964, 1971).
(b) Copper species (from ZirinO & YamamotO, 1972)
Ligand log 81 1w 88 1% 88 log 84
Cl- 0 -0.7 -2.2 -4.4 so:- 2'3 - - - HCO; 2.7 - - - co:- 6.77 10'00 - -
OH- 6.3 14'3 15 16
(c) Zinc and cadmium species (from Zirino & Yamamoto, 1972, except as indicated)
Ligand 1% 81 log BP loti! 88 log 84
Zinc Cl- so:- HCO; co:- OH-
Cadmium cl- so:- HCO; co;- OH-
0.40 0.61 0.53 0’20 2.3 - - - 1‘9' - - - 4.8* - - -
4'40 10*0= I4 15
2’0 2'7 2’1 -
2.3 3'5 - - I $8” - - - 4.4b - - - 5 10.6 IO 10
OEstimated from carbonate stability constant using log Bsrcos/log BhI nCoII= 2.5. “From Bilinski et al. (1976) corrected to zero ionic strength using the Davies
equation (see text). ‘From Bradford (1972).
(d) Manganese species (from Sill&-r & Martell, x964, 1971, except as indicated)
Ligand log 81
Cl- 0’1 so:- 2.26 HCOi 1.8 co:- 5.0* OH- 3’41’
‘Although Fontana & Brito (1968) have suggested that Mn(OH): exists, the evidence for it is unconvincing and Perrin (1962) has denied its importance.
bEstimated from a log /9 versus electronegativity plot.

Metal-humic comglexation in natural waters 397
(e) Mercury species (from Sill&r & Martell, 1964, 1971 except as indicated), but corrected from values in 0.5 M-sodium perchlorate
Ligand log 81 log 83 lot3 Bs loi3 86
cl- 7.28 14.01 14.98 15.76 SO:- 2.42 3’52 - - HCO, . (I - - - CO!- ;.:* - - - OH- 10.86 12.24 - -
Bromo and mixed halogen complexes
log B HgBr:
'18*14 HgClBr“ HgCIBrt- HgCltBr- HgCl%Br:-HgC1,Bra- 16.67 19.14 17’25 19.18 17.69
“Estimated from log &coa/log /IwHco~=~.~. bEstimated from a log /3 were electronegativity plot. “From Dryssen & Wedborg (1974), corrected to I=o using the Davies equation.
(f) Nickel species (from SillCn and Martell, 1964, 1971, except as indicated)
Ligand
Cl- SOi- HCO, co:-
OH-
log Bl log I% log Bs hi! B4
o.73a 0.72’ - -
2.32 - - - . b - - -
;.;= - - -
4.16 - - -
“Corrected to I=o from 0.69 M-HCIO~ using the Davies equation. “Estimated from log j?~~o~/log & ncoI = 2.5. ‘Estimated from log B versus electronegativity plot.
(g) Cobalt species (from Sillen & Martell, 1964, 1971, except as indicated)
Ligand
cl- soi- HCO, co;- OH-
log 81 loI3 Bs log Bs
-1.28 - -
2.36 - - . (I - -
Z.fb - -
4’15 -4.7 -
“Estimated from log /3Hcos/log /TM “co8 = 2.5.
*Estimated from log /I oersus electronegativity plot.
nearest 0.1% of the total metal, and for this reason species accounting for less than 0.1%
of the total metal were disregarded. It should be pointed out that, as has already been noted by Bernhard et al. (1975), specia-
tion models are very dependent upon the values which are chosen for the various stability constants. Two problems are involved in making this choice. First, and more importantly, the selection of reliable data from the often conflicting mass of figures available. Secondly, the choice of the activity function used to correct the values from those at infinite dilution to those applicable to the solution under consideration. The effect of the former factor is clearly shown by a comparison of the principal zinc species predicted in the present work for seawater having a salinity of 35x0 and a pH of 8.1 (47% Zn2 +, 20% ZnCl +, 10% ZnCl,O and 10% ZnSO,O) with those suggested by Zirino & Yamamoto (60% Zn(OH),O, 17% Zn*+,

398 R. F. C. Mantoura, A. Dickson N.7. P. Riley
Cu Hum
b-o CuHCQ;
-0-o cuso~ cuct l
I -P----o-o-8 cuccn&
5 IO 15 20 25 30 35 Salinity k.)
Figure 4. The equilibrium speciation of copper (II) as a function of salinity along a model estuary. -, Activity coefficients calculated using the Davies equation; - - - - -, activity coefficients for major ion pairs using expression for dipoles (log y= -BI). The difference between the two lines is probably a maximum as the value of y for a 2-2 ion pair used at 1~0.7 (S=35y&,) is appreciably lower than that sug- gested for the ion pair MgSO$ by Kester & Pytkowicz (1975).
7% ZnCl+, 5% ZnCOs”). These differences are obviously caused by the use in the model of more recent, lower, values for ~(Zn(OH)a”) and @nCO,O). In contrast to errors produced in this way, those introduced by the use of incorrect estimates of activity coefficients are comparatively minor. If, as seems theoretically desirable, ion-pairs formed between the major ions are considered to be dipoles (see above), their amounts will increase (see e.g.
TABLE 4. Speciation for Lake Celyn water (mole per cent) (using analytical data given in Table 2)
Component Free ion Chloro sulphato bicarbonate carbonato hydroxy Humic
Magnesium Calcium Ckwr Zinc Cadmium Manganese Mercury Nickel Cobalt
98.2 97’8 -
88.8 92.8 90.6
69.6 76.0
- 0.5 - 0.6 - -
- 0.5 0.7 o-5 - 0.5 - -
- 0.4 - 0.5
0.5 - 0.9 - - -
3’1 0.6
3’0 0’2 2’9 0.9 - -
11’1 11’2
9.6 9.8
cu Zn 0.8 1’0
- 0.8
- 0.7 - 99’97 - 7’0 0’1 2.7 - 5’1 - 100’0
- 7’7 - 4’0
free Mg Ca 75’2 6-4 15’0
Mn Ni I’S 0’1

Metal-humic complexation in natural waters 399
Figure 2). The concentrations of the unassociated ligands (SO,a - and COs2 -) will therefore drop and this will cause a decrease in the proportion of trace metals complexed with these ligands. The extent of this effect on copper speciation is indicated diagrammatically in Figure 4.
Freshwaters
A model was derived for an analysed sample of Lake Celyn water (Table 4). In this, although the concentrations of competing ligands are low, complexation with humic compounds is only of major importance (>gg*g%) f or copper and mercury. For the remaining trace metals such interactions only bind 8% or lessof the individual metals, and these metals occur dominantly (>70%) as free ions. In contrast to saline media (see below), the humic com- pounds occur principally in the unbound state. Of the complexes with inorganic ligands, only those of bicarbonate, and for cobalt and nickel, those of carbonate, play any appreciable role.
The results of the model of Lake Celyn water may be compared with those for one derived using World Average River Water data (Table 2) which were employed for the freshwater component of the estuarine mixing model together with stability constant data for CELI humic (see Figures 2-12). Although the concept of an ‘average’ river water is artificial and perhaps unrealistic, it is of interest to note the differences in speciation resulting from the different compositions of these two freshwaters. The major factor producing these differences is the change in composition reflected by pH. In Lake Celyn water the pH is lower, thus decreasing the total carbonate ion concentration, and therefore the relative importance of carbonato species. A secondary factor is the higher concentrations of magnesium and calcium in World Average River Water. This leads to a decrease in the proportion of free, and thus available, humic ligand. The consequence of both of these factors is that humic compounds play a greater role in speciation in Celyn water than they do in ‘river’ water.
Sea and estuurim waters
Speciation calculations were carried out for both an average seawater (having the composition shown in Table 2) and also, to simulate estuarine conditions, for mixtures of this with World Average River water.
(I) Organic speciation. With the exception of copper, and to a lesser extent, mercury, there is a rapid decrease in the proportion of trace metals bound with the humic components, as the salinity increases (see e.g. Stumm & Brauner, 1975, who used model organic ligands). Only copper exists to a significant extent (N 10%) as such a complex when a salinity of 35x,, is reached (Figure 4). This drop is principally the result of competition for the humic ligand from calcium and magnesium which, despite the relatively low stability constants of their complexes, are sufficiently abundant to utilize >gg% of the complexing capacity of the humic ligand (see Figure I I). An additional factor for copper is the increased importance of carbonato and hydroxy species due to the rapid pH increase associated with the initial mixing. With mercury (Figure 7), competition from chloride ion is the principal factor, since not only are the stability constants of its chlorocomplexes very high, but also because the concentration of the nth complex is dependent on the nth power of the chloride concen- tration. (2) Inorganic speciation (see Figures Z--IO). In saline waters, of the metals considered only the major components (Na, K, Mg and Ca), manganese and zinc exist predominantly (>46%)

400 R. F. C. Mantoura, A. Dickson &‘J. P. Riley
Salinity (%A
Figure 5. The equilibrium speciation of zinc (II) as a function of salinity along a model estuary.
Salinity I%.)
Figure 6. The equilibrium speciation of cadmium (II) as a function of salinity along a model estuary.

- Metal-humic complexation in natural waters 40’
as the free ion. The free ions are also important species for cobalt and nickel. In waters having salinities of > IO%, chloro species are significant (>5%) for all trace metals except copper and cobalt. They are particularly so for mercury and cadmium which are present in such complexes to an extent of N 84%. The proportion of metal bound as chloro complexes increases with salinity (chloride concentration), that present as higher complexes (e.g. HgC1,2-) becoming more important at high salinities. The latter effect is due to the depen- dence of the concentration of the nth complex on the nth power of the ligand concentration.
Sulphato complexes are only significant for magnesium, calcium, zinc, manganese and cobalt, and, in general, only vary by a few per cent over the salinity range ~-35%~.
Salinity (%A
Figure 7. The equilibrium speciation of manganese (II) as a function of salinity along a model estuary.
In considering interactions of metals with the various protolytic ligands it is necessary to take into account the pH of the water. As discussed by Mook & Koene (1975) the conserva- tive mixing of river water and sea water does not lead to a linear change in the pH of the water. The computed variations of pH and hydroxide activity, along with those of the total concen- trations of bicarbonate and carbonate ions for the model are shown in Figure I. The carbonato species are only really important for cobalt and for nickel at all salinities, but play a minor role (the importance of which increases with increasing salinity-i.e. pH) for copper, manganese, and the major components: Na, Mg and Ca. Hydroxy species [except for Cu(OH),q are of little consequence in the model over the range of pH and salinity under consideration. However, at higher pH values they would become more significant especially in freshwaters.
The estuarine model shows interesting minima for certain species (e.g. MgCO,O, CoCOa”, Zn(OH) +) at salinity values of <$&. These result from the sudden drop in the value of the

402 R. F. C. Mantoura, A. Dickson @‘J. P. Riley
activity coefficient of ions (especially doubly charged ones) due to the increase in ionic strength. They are only found for complexes with ligands whose concentrations do not rise rapidly enough to mask this drop in activity; they are thus not observed for sulphato and chloro species since the concentrations of these ligands change by several orders of magnitude between river water and $& salinity estuarine water.
Discussion
Humic acids exert a significant influence on trace metal speciation in freshwaters, especially at low pH values. This importance increases with the stability constant of the particular complex; the extent of metal humic interaction thus follows the Irving-Williams series.
5 IO I5 20 25 30
Solinily (%J
Figure 8. The equilibrium speciation of mercury (II) as a function of salinity along a model estuary, allowing for the existence of mixed halogen complexes. Assuming mixing between river water containing no bromide and seawater (S=35%) with a bromide concentration of 8.42 x IO-’ mol kg-l, and using the stability constant data for bromo species given in Table 3(e).
Calculations have shown that if the stability constants for the various humic compounds shown in Table I are used in the Lake Celyn model there is little significant change in the overall pattern of humic speciation.
The intense binding of Hg by humics in the model freshwater systems may be a causal factor in (a) the humic acid mediated reduction and evolution of elemental Hg recently observed by Alberts et al, (1974)~ and (b) the inhibitory effect of humic acid on the spon- taneous electrodeposition of Hg on silver gauze (Millward & Burton, 1975). The observation that Cu is nearly completely associated with hurnic ligands in the freshwater models is in

Metal-humic complexation in natural waters 403
-0-O v-.
\,/- -10
NiCOg
IO
f .g 5 E 6 z
I.0
I I v A-O-~~- 0c’- tzs-
0.1 I I I I I I 0 5 IO 15 20 25 30 :
Salinity %I
Figure 9. The equilibrium speciation of nickel (II) as a function of salinity along a model estuary.
k:~~~” I 0-O
/
-O-coso; I o-o-0-0 COHCO;
/Co Hum
15 20
Salinity W&J
Figure IO. The equilibrium speciation of cobalt (II) as a function of salinity along a model estuary.

404 R. F. C. Mantoura, A. Dickson USJ. P. Riley
agreement with the known electroanalytical chemistry of this element in the presence of humics (Chau & Lum-Shue-Chan, 1974; Ernst et al., 1975). It may also explain why a significant part of the dissolved Cu in natural waters is extractable into hexanol (Stiff, 1971)
or chloroform (Slowey et al., 1967).
In considering trace metal-humic interactions in saline waters it is important to realize that the extent of complexatidn will vary markedly according to the nature of the humic material. This effect is illustrated by Figure 12 which shows the variation at different salinities of the proportion of copper complexed as a function of the stability constant of the copper-humic complex. The values for the constants found in the present work (Table I) for humic compounds from a variety of environments are also marked on the graph. The variation in copper-humic complexation corresponding to changes in salinity can be readily determined by making vertical transects across the iso-halines. It is thus evident that, dependent on the humic ligand, up to 8074 of the copper in seawater may be complexed by humic compounds, and an even greater proportion would be complexed in the pore waters of sediments.
\ o-0- o-o-o-o-
Co Hum
1 I I I I I 5 IO 15 20 25 30
Salinity WA
5
Figure I I. The equilibrium speciation of humic compound as a function of salinity along a model estuary.
The discrepancies between the present model and the various published models for sea- water speciation are due predominantly to the use of different values for the stability con- stants (at infinite dilution). For the major elements (i.e. Mg and Ca) the speciation predicted for seawater (S=35%J agrees well with previously reported models (see Stumm & Brauner, 1975). Any discrepancy for these elements is usually the result of a different choice of activity function (see e.g. Figure 2). For the various trace elements considered a less con- sistent picture emerges. The use of more recent values for the stability constants of species

Metal-humic complexation in natural waters 40s
0.
0.01
Cu-Hum,
/
Aqueous humics
Lag KC. ICu Humics)
Figure 12. The extent of copper-humic interaction, as a function of the magnitude of the stability constant, at various salinities. The annotation on the horizontal axis portrays the range of stability constants found for various natural samples (see text for explanation of abbreviations).
such as Zn(OH) so, ZnCOsO and CuCOs” results in speciation patterns which are different from those described for these elements by Zirino & Yamamoto (1972); however, that for cadmium remains essentially the same. It has been recognized by Dyrssen and Wedborg (1974) that bromo and chloro/bromo complexes are probably important species for mercury (II) in seawater. Such complexes have therefore been incorporated in the model for the estuarine speciation of this element. The model at S=2& (I=o*s) thus bears a close resemblance to that proposed by the former authors.
Conclusion
Although, in a real system, kinetic factors will obviously exert some control on the metal speciation pattern, the proposed models do give an indication of the main factors that control the extent of metal-humic interactions at equilibrium in a variety of natural aquatic environments. These are:
(i) the value of K for the metal-humic interaction, (ii) the pH, which provides a measure of the competition from carbonato and hydroxy
ligands, and (iii) the major ion concentrations which control the competition for humic acid by
magnesium and calcium, and the competition for trace metals by chloro and sulphato ligands.

406 R. F. C. Mantoura, A. Dickson N’J. P. Riley
Humic substances from the various natural environments show a wide range of values of K, in their interaction with any particular metal. For exactness it is therefore important when modelling any particular water to use data for K, for humic components isolated from that water.
The patterns of inorganic speciation given by the models are, of course, a function of the assumptions made. The weakest link in the development of reliable models for trace metal speciation is, however, the values of the various stability constants used, and there is an urgent need for a critical reassessment of the relevant stability constants.
Acknowledgement
The authors wish to thank Dr M. Whitfield for helpful discussion.
References
Abdullah, M. I. 1975 Personal communication. Alberts, J. J., Schindler, J. E., Miller, R. W. & Nutter, D. E. 1974 Elemental mercury evolution
mediated by humic acid. Science 184, 895-896. Bernhard, M., Goldberg, E. D. & Piro, A. 1975 Zinc in seawater-an overview 1975. In The Nature of
Sea B’ater (Goldberg, E. D., ed.) Physical and Chemical Sciences Research Report, Vol. I. Dahlem Konferenzen, Berlin, pp. 43-68.
Bilinski, H., Huston, R., & Stumm, W. 1976 Determination of the stability constants of some hydroxo and carbonato complexes of Pb(II), Cu(II), Cd(II) and Zn(I1) in dilute solutions by anodic stripping voltammetry and differential pulse polarography. Analytica Chimica Acta 84, 157-164.
Bradford, W. R. 1972 The determination of a stability constant for the aqueous complex Zn(OH)$ using anodic stripping voltammetry. Limnology and Oceanography 18, 757762.
Chau, Y. K. & Lum-Shue-Chan, K. 1974 Determination of labile and strongly bound metals in lake water. Water Research 8, 383388.
Cheam, V. & Gamble, D. S. 1974 Metal-fulvic acid chelation equilibrium in aqueous NaNOs solution. Canadian Journal of Soil Science 54,413-417.
Childs, C. W. 1971 Chemical equilibrium models for lake water which contains nitrilotriacetic acid and for normal lake water. Proceedings rqth Conference on Great Lakes Research. pp. 198-210.
Davies, C. W. 1962 Ion Association, 190 pp. International Association for Great Lakes Research. Butterworths, London.
Dyrssen, D. & Wedborg, M. 1974 Equilibrium calculations of the speciation of elements in seawater. In The Sea, Vol. 5 Marine Chemistry (Goldberg, E. D., ed.), Wiley-Interscience, New York. pp. 181-195.
Dyrssen, D., Jagner, D. & Wengelin, F. 1968 Computer calculation of ionic equilibria and titration procedures. Almqvist & Wiksell, Stockholm.
Duursma, E. K. 1970 Organic chelation of s°Co and 66Zn by leucine in relation to sorption by sediments. In Symposium on Organic Matter in Natural Waters (Hood, D. W., ed.) University of Alaska. PP. 387-397.
Ernst, R., Allen, H. E. & Mancy, K. H. 1975 Characteristics of trace metal species and measurements of trace metal stability constants by electrochemical techniques. Water Research 9,969-979.
Florence, T. M. & Batley, G. E. 1976 Trace metal species in sea water I, removal of trace metals from sea water by a chelating resin. TuZunta 23, 179-186.
Fontana, S. & Brito, F. 1968 On the hydrolysis of manganese (II) in I M-(Nar, Mn)SOI ionic medium at 25°C. Inorganica Chimica Acta 2, 179-180.
Gardner, D. & Riley, J. P. 1973 Mercury in the Atlantic around Iceland. Journal du Conseil, Consed International pour I’Exploration de la Mer 35,202-204.
Garrels, R. M. & Thompson, M. E. 1962 A chemical model for sea water at 25 “C and one atmosphere pressure. American Journal of Science 260, 57-66.
Greenhalgh, R. & Riley, J. P. 1961 Determination of fluoride in natural water with particular reference to sea water. Analytica Chimica Acta 25, 179-186.
Henry, R. A. & Rogers, L. B. 1968 Gel chromatographic behaviour of aqueous ferric nitrate. Separation Science 3, I 1-25.
Hiigfeldt, E. 1975 On the construction of single-ion activity functions and some comments on the formulation of conventions to describe sea water. In The nature of sea water (Goldberg, E. D., ed.) Physical and Chemical Sciences Research Report, Vol. I. Dahlem Konferenzen, Berlin, pp. 281-312.

Metal-humic complexatian in natural waters 407
Ingri, N., Kakolowicz, W., SillCn, L. G. & Wamqvist, B. 1967 High speed computers as a supplement to graphical methods, V HALTAFALL, a general program for calculating the composition of eauilibrium mixtures. TuZantu, 12, 1261-1286; Errata ibid. (1968) 15, xi-xii.
Kester; D. R. & Pytkowicz, R. M. 1975 Theoretical model for the formation of ion-pairs in sea water. Marine Chemistry 3,365-374.
Lewis, G. J. & Goldberg, E. D. 1954 Iron in marine waters. ~ouwzul of Murine Research If 183-197. Livingstone, D. A. 1963 Chemical composition of rivers and lakes. U.S. Geol. Survey Professional Paper
440-G. MacInnes, D. A., 1919 The activities of the ions of strong electrolytes.%urnal of the American Chemical
Society 41, 1086-1092. Malcolm, E. L. 1972 Comparison of conditional stability constants of North Carolina humic and fulvic
acids with Co II and Fe III. In Environmental Framework of Coastal Plain Estuaries (Nelson, E. B., ed.) Geological Society of America. Washington.
Mantoura, R. F. C. & Riley, J. P. 1975~ The analytical concentration of humic substances from natural waters. Analytica Chimica Acta 76, 97-106.
Mantoura, R. F. C. & Riley, J. P. 1975b The use of gel filtration in the study of metal binding by humic acids and related compounds. Analytica Chimica Acia 78, 193-200.
Millero, F. J. 1974 Seawater as a multicomponent electrolyte solution. In The Sea, Vol. 5, Marine Chemistry (Goldberg, E. D., ed.) Wiley-Interscience, New York, pp. 3-81.
Millero, F. J. 1975 The physical chemistry of estuaries. In Marine Chemistry in the CoastaZ Environment. (Church, T. M. ed.) American Chemical Society Symposium Series No. 18, pp. 25-55.
Millward, G. E. & Burton, J. D. 1975 Association of mercuric ions and humic acid in sodium chloride solution. Marine Science Communications I, 15-26.
Mook, W. G. & Koene, B. K. S. 1975 Chemistry of dissolved inorganic carbon in estuarine and coastal brackish waters. Estuarine and Coastal Marine Science 3, 325-336.
Morel, F. & Morgan, J. 1972 A numerical method for computing equilibria in aqueous chemical systems. Environmental Science and Technology 6, 58-67.
Nissenbaum, A. & Kaplan, I. R. 1972 Chemical and isotopic evidence for the in situ origin of marine humic substance. Limnology and Oceanography 17, 570-582.
Perdue, E. M., Beck, K. C. & Reuter, J. H. 1976 Organic complexes of iron and aluminium in natural waters. Nature 260,418-420.
Perrin, D. D. 1962 The hydrolysis of manganese (II), ion. journal of the ChemicaI Society, 2197-2200. Perrin, D. D. & Sayce, I. G. 1967 Computer calculation of equilibrium concentration in mixtures of
metal ions and complexing species. Tdunta 14, 833-842. Reardon, E. J. 1975 Dissociation constants of some monovalent sulphate ion pairs at 25 “C from
stoichiometric activity coefficients. &u?nal of Physicul Chemistry 79, 422-425. Reardon, E. J. & Langmuir, D. 1976 Activity coefficients of MgCOaO and CaSOlo ion pairs as a function
of ionic strength Geochimica et Cosmochimica Acta 40, 549-554. Riley, J. P. & Taylor, D. 1968 Chelating resins for the concentration of trace elements from sea water
and their analytical use in conjunction with atomic absorption spectrophotometry. Analytica Chimica Acta 40,479-485.
Riley, J. P. 1975 Tables of physical and chemical constants relevant to marine chemistry. In Chemical Oceunogruphy (Riley, J. P. & Skirrow, G., eds), 2nd Ed. Vol. 2. Academic Press, London, pp. 599-631.
Schnitzer, M. 1971 Metal-organic matter interactions in soils and waters. In Organic Compounds in Aquutic Environments (Faust, S. D. & Hunter, J. V., eds.) Marcel Dekker, New York, pp. 297-313.
Schnitzer, M. & Khan, S. U. 1972 Humic Subsstances in the Environment. Marcel Dekker, New York. Siegel, A. 1971 Metal-organic interactions in the marine environment. In organic Compounds in Aquatic
Environments (Faust, S. D. & Hunter, J. V., eds.) Marcel Dekker, New York. pp. 265-295. Sill&n, L. G. & Martell, A. E. 1964 Stability constants of metal ion complexes. Chemical Society Special
Publication No. 17 London. Sillen, L. G. & Martell, A. E. 1971 Stability constants of metal ion complexes. Chemical Society Special
Publication No. 25 London. Slowey, J. F., Jeffrey, L. M. & Hood, D. W. 1967 Evidence for organic complexed copper in sea water.
Nature 214, 373-378. Stiff, M. J. 1971 The chemical states of copper in polluted freshwater and a scheme of analysis to
differentiate these. Water Research 5, 585-599. Strohal, P. & Huljev, D. 1971 Investigation of mercury pollutant reaction with humic acid by means of
radio tracers. In Nuclear Techniques in Environmental Pollution. Internation Atomic Energy Agency Vienna.
Stumm, W. & Brauner, D. A. 1975 Chemical speciation. In Chemical Oceanography (Riley, J. P. & Skirrow, G., eds.) 2nd Ed., Vol. I, Academic Press, London. pp. 173-234.
Truesdell, A. H. & Jones, B. F. 1974. WATEQ. a computer program for calculating chemical equilibria of natural waters. Journal of Research U.S. Geological Survey 2, 233-248.

408 R. F. C. Mantoura, A. Dickson &‘J. P. Riley
Theng, B. K. G., Wake, J. R. H. & Posner, A. M. 1968 The fractional precipitation of soil humic acid by ammonium sulphate. Plant and Soil 29, 305-316.
Wilson, T. R. S. 1975 Salinity and the major elements of sea water. In Chemical Oceanography, 2nd Ed. Vol. I (J. P. Riley & G. Skirrow, eds.) Academic Press, London. pp. 364-413.
Whitfield, M. q75a Sea water as an electrolyte solution. In Chemical Oceanography, 2nd Ed., Vol. I (Riley, J. P. & Skirrow, G., eds.) Academic Press, London. pp. 44-162.
Whitfield, M. I975b Extension of chemical models for sea water to include trace components at 25 “C and I atm. pressure. Geochimica et Cosmochimica Acta 39, 1545-1557.
Zirino, A. & Yamamoto, S. 1972 A pH-dependent model for the chemical speciation of copper, zinc, cadmium and lead in sea water. Limnology and Oceanography 17,661-67x.





























