Embed Size (px)
Citation preview

NLRP7 mutations in women with diploidandrogenetic and triploid moles: a proposedmechanism for mole formation
Catherine Deveault1,2,{, Jian Hua Qian1,2,4,{, Wafaa Chebaro1,2, Asangla Ao1,2, Lucy Gilbert2,
Amira Mehio3, Rabia Khan1, Seang Lin Tan2, Anita Wischmeijer5, Philippe Coullin6, Xing Xie4
and Rima Slim1,2,�
1Department of Human Genetics, 2Department of Obstetrics and Gynecology, 3Department Pathology, McGill
University Health Center, Montreal H3G 1A4, Canada, 4Women’s Reproductive Health Laboratory, Women’s Hospital,
Zhejiang University School of Medicine, Hangzhou 310006, Peoples’ Republic of China, 5Department of Medical
Genetics, Policlinico Sant’Orsola-Malpighi-University of Bologna, Bologna, Italy and 6INSERM U 782, Endocrinologie
et Genetique de la Reproduction et du Developpement, 32 rue des carnets, F 92140 Clamart, France
Received August 4, 2008; Revised November 5, 2008; Accepted December 5, 2008
Hydatidiform mole is an aberrant pregnancy with abnormal embryonic development and hydropic placentalvilli. Common moles are sporadic, not recurrent and affect one in every 1500 pregnancies in Westernsocieties. Approximately, half of common moles are complete and mostly diploid androgenetic, whereasthe remaining are partial and mostly triploid diandric. NLRP7 has been found to be responsible for a recurrentform of molar pregnancies. Recently, we showed that patients with NLRP7 mutations have an impairedinflammatory response to various stimuli. To date, molar tissues analyzed from patients with NLRP7mutations have been found to be diploid and biparental. In this study, we report 10 new non-synonymousvariants and one stop codon found in patients and not in controls. We demonstrate the presence of differenttypes of moles, diploid biparental, diploid androgenetic, triploid and tetraploid conceptions, in patients withNLRP7 variants. We document in vitro and in vivo early embryo cleavage abnormalities in three patients. Wepropose a two-hit mechanism at the origin of androgenetic moles. This mechanism consists of variabledegrees of early embryo cleavage abnormalities leading to chaotic mosaic aneuploidies, with haploid,diploid, triploid and tetraploid blastomeres. Surviving embryonic cells that reach implantation are then sub-ject to the maternal immune response. Because of the patients’ impaired inflammatory response, androge-netic cells, which are complete allograft, are able to grow and proliferate. In women with normal immunesystem, chaotic mosaic aneuploidies may also occur during early cleavage, however, androgenetic cellswould die after implantation or stay undetected, confined to a small portion of the placenta.
INTRODUCTION
Hydatidiform mole (HM) is an abnormal human pregnancycharacterized by absence of, or abnormal embryonic developmentand hydropic degeneration of chorionic villi. The common formof this condition is sporadic, not recurrent, and occurs once inevery 1000–1500 pregnancies in western countries, but at twoto 10 times higher frequencies in underdeveloped and developing
countries (1). Among women with sporadic moles, 1–6% willhave a second mole (2–7) and about 10–20% will have asecond non-molar reproductive wastage, mostly as spontaneousabortion (5,7–9). The frequency of familial recurrent HMs(RHMs) is not known, but such cases are believed to be veryrare. So far, approximately 20 familial cases have been reportedin the recent English literature in PubMed.
†The authors wish it to be known that, in their opinion, the first two authors should be regarded as joint First Authors.
�To whom correspondence should be addressed at: Montreal General Hospital Research Institute, L3-121, 1650 Cedar Avenue, Montreal P.Q. H3G1A4, Canada. Tel: þ1 5149341934 (ext. 44550); Fax: þ1 5149348261; Email: [email protected]
# The Author 2008. Published by Oxford University Press. All rights reserved.For Permissions, please email: [email protected]
Human Molecular Genetics, 2009, Vol. 18, No. 5 888–897doi:10.1093/hmg/ddn418Advance Access published on December 9, 2008

At the histopathological level, HMs are divided into twotypes, complete (CHM) and partial (PHM) based on the extentof trophoblast proliferation and the absence or presence ofembryonic tissues other than the chorionic villi. At the karyotypeand genotype levels, most sporadic CHMs are diploid androge-netic, but may also have any of the following genotypes, diploidbiparental, tetraploid androgenetic or biparental, aneuploid (non-triploid/tetraploid aneuploid), triploid diandric or dygenic, ormosaic with two cellular populations (10–15). Among androge-netic moles, the majority is monospermic and 5–20% are disper-mic. PHMs are mostly triploid diandric, but may also be diploidbiparental, triploid digynic or aneuploid (11,13,15). To date, allcharacterized recurrent HMs (RHMs) from patients with nofamily history of moles have been found to be diploid and bipar-ental with the exception of two cases where one mole in a patientand three in another were shown to be diploid androgenetic (16).Also, all analyzed molar tissues from familial cases have beenfound diploid biparental (17–23).
By studying rare families of RHMs, a defective gene, NLRP7,responsible for this condition has been identified (24) andmutations in this gene have been found in women from severalethnic groups. Eleven NLRP7 variants found only in patientsare now listed in INFEVERS (http://fmf.igh.cnrs.fr/ISSAID/inf-evers/) and several others are in the process of being reported byvarious groups. NLRP7 is a cytoplasmic protein whose tran-scripts were found in a wide range of normal human tissuesincluding oocytes at the germinal vesicle and metaphase IIstages, Fallopian tubes (unpublished data) and endometrium(24,25). A recent study has shown that NLRP7 transcriptiondeclines during development from oocytes to day 3 but increasesagain by day 5 (25). To date, it is not clear how NLRP7 defectslead to RHMs and associated reproductive wastage. As amaternal effect gene, NLRP7 could cause recurrent fetal lossby leading to defective oocytes or/and by creating a hostilematernal environment for embryonic development in the Fallo-pian tubes and the uterine cavity. We recently demonstrated thatpatients with NLRP7 mutations have impaired inflammatoryresponse against various stimuli, such as lipopolysaccharides,various microbial products and synthetic compounds, whichinduce strong inflammatory response in normal individuals(Deveault, manuscript in preparation).
Here we report 11 new NLRP7 variants found only in patientswith recurrent reproductive wastage and the characterization ofsome of their products of conception. We show that NLRP7 var-iants are present, not only in patients with diploid biparentalmoles as previously shown, but also in patients with diploidandrogenetic moles, triploid moles and tetraploid spontaneousabortions. Furthermore, we document in vivo and in vitro earlyembryo cleavage abnormalities in three patients with NLRP7 var-iants. Our findings shed new lights on the mechanism of mole for-mation and expand the involvement of NLRP7 to a wider range ofrecurrent and non-recurrent reproductive wastage.
RESULTS
New NLRP7 variants in patients with recurrentreproductive wastage
The search for mutations revealed two previously reportedmutations, a missense, c.2248C.G, p.Leu750Val (26), in a
homozygous state and a splice mutation, c.2471þ1G.A,p.L825X (24), in a heterozygous state in sisters from familiesMoUs99 and MoCh77, respectively (Table 1). Eleven other var-iants, 10 leading to non-synonymous amino acid changes andone leading to a stop codon were also found. These variantswere not found in 50–289 control subjects from variousethnic groups indicating their association with the disease phe-notype (Supplementary Material). Patient 428 had also a rarevariant, c.1460G.A, p.Gly487Glu, in a heterozygous statethat was found in trans with p.Cys399Tyr. Variantp.Gly487Glu is reported in public databases and we found itin heterozygous state in 4.6% of control women (with five tonine children) of European descent and at lower frequencies insubjects from other ethnic groups (Supplementary Material).All the other variants that we found are known frequent poly-morphisms in the general population. In five patients, 492,501, 647, 565 and 636, only one variant which is not presentin controls was found. Available parents were tested for theiroffspring variants. This analysis revealed the inheritance ofp.Cys84Tyr to patient 492 and of p.Ala719Val to patient 636from their fathers. Variants p.Arg156Gln and p.Lys511Argwere inherited to patients 647 and 565, respectively, fromtheir mothers. For patient 501, only the mother was availablefor genetic testing and does not carry p.Lys379Asn (Fig. 1).Haplotype establishment for coding and intronic DNA variantsfound in NLRP7 demonstrated the absence of a common haplo-type in patients, 492, 501 and 636 (Supplementary Material),which is not in favor of a common rearrangement in the threepatients. In patients 492 and 428, long range polymerase chainreaction (PCR) was also used to amplify the entire genomicregion from the start to the stop codons, in five fragments, anddid not reveal any rearrangement. In patient 492, the five ampli-fied fragments contained heterozygous single nucleotide poly-morphisms (SNPs) demonstrating the amplification of the twoparental alleles. cDNA sequencing from patient 428, fromwhich an Epstein-Barr virus (EBV)-transformed cell line isavailable, revealed normal splicing of all exons. The variantfound in patient 662, p.Leu964Pro, is the first in exon 10,present only in splice isoform v2 (27), and highlights the import-ance of screening the 11 NLRP7 exons (NM_001127255.1) inthe search for mutations.
NLRP7 variants are responsible for severaltypes of hydatidiform moles
To better understand the mechanism(s) responsible for molarpregnancies caused by NRLP7 mutations/variants, we usedseveral methods to characterize available products of conception(POC) from six patients (Fig. 1). Ploidy analysis was performedeither by karyotyping or by flow cytometry and demonstratedthat nine moles were diploid, one had a triploid peak, one spon-taneous abortion had a tetraploid peak and one mole was incon-clusive because of strong fixation of the tissues (Fig. 1 andSupplementary Material). We note that flow cytometry analysison embedded tissues does not allow distinguishing whether thePOC are only triploid or tetraploid or whether they are mosaicand contain other diploid cells because of the presence of endo-metrial diploid maternal tissues. Immunohistochemistry withp57KIP2 antibody demonstrated the absence of its expression infour diploid moles, two CHMs (Fig. 2) and two PHMs. DNA
Human Molecular Genetics, 2009, Vol. 18, No. 5 889
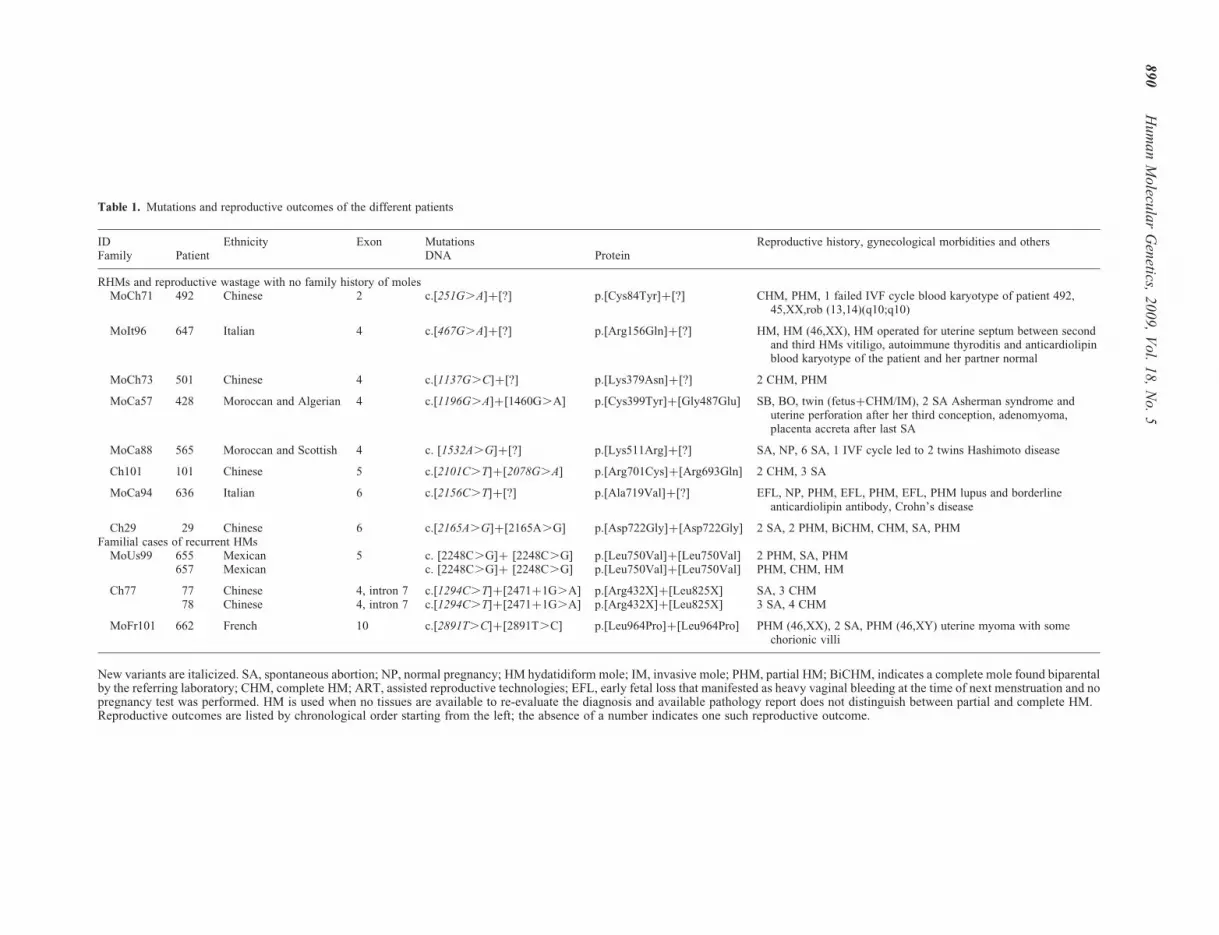
Table 1. Mutations and reproductive outcomes of the different patients
ID Ethnicity Exon Mutations Reproductive history, gynecological morbidities and othersFamily Patient DNA Protein
RHMs and reproductive wastage with no family history of molesMoCh71 492 Chinese 2 c.[251G.A]þ[?] p.[Cys84Tyr]þ[?] CHM, PHM, 1 failed IVF cycle blood karyotype of patient 492,
45,XX,rob (13,14)(q10;q10)
MoIt96 647 Italian 4 c.[467G.A]þ[?] p.[Arg156Gln]þ[?] HM, HM (46,XX), HM operated for uterine septum between secondand third HMs vitiligo, autoimmune thyroditis and anticardiolipinblood karyotype of the patient and her partner normal
MoCh73 501 Chinese 4 c.[1137G.C]þ[?] p.[Lys379Asn]þ[?] 2 CHM, PHM
MoCa57 428 Moroccan and Algerian 4 c.[1196G.A]þ[1460G.A] p.[Cys399Tyr]þ[Gly487Glu] SB, BO, twin (fetusþCHM/IM), 2 SA Asherman syndrome anduterine perforation after her third conception, adenomyoma,placenta accreta after last SA
MoCa88 565 Moroccan and Scottish 4 c. [1532A.G]þ[?] p.[Lys511Arg]þ[?] SA, NP, 6 SA, 1 IVF cycle led to 2 twins Hashimoto disease
Ch101 101 Chinese 5 c.[2101C.T]þ[2078G.A] p.[Arg701Cys]þ[Arg693Gln] 2 CHM, 3 SA
MoCa94 636 Italian 6 c.[2156C.T]þ[?] p.[Ala719Val]þ[?] EFL, NP, PHM, EFL, PHM, EFL, PHM lupus and borderlineanticardiolipin antibody, Crohn’s disease
Ch29 29 Chinese 6 c.[2165A.G]þ[2165A.G] p.[Asp722Gly]þ[Asp722Gly] 2 SA, 2 PHM, BiCHM, CHM, SA, PHMFamilial cases of recurrent HMs
MoUs99 655 Mexican 5 c. [2248C.G]þ [2248C.G] p.[Leu750Val]þ[Leu750Val] 2 PHM, SA, PHM657 Mexican c. [2248C.G]þ [2248C.G] p.[Leu750Val]þ[Leu750Val] PHM, CHM, HM
Ch77 77 Chinese 4, intron 7 c.[1294C.T]þ[2471þ1G.A] p.[Arg432X]þ[Leu825X] SA, 3 CHM78 Chinese 4, intron 7 c.[1294C.T]þ[2471þ1G.A] p.[Arg432X]þ[Leu825X] 3 SA, 4 CHM
MoFr101 662 French 10 c.[2891T.C]þ[2891T.C] p.[Leu964Pro]þ[Leu964Pro] PHM (46,XX), 2 SA, PHM (46,XY) uterine myoma with somechorionic villi
New variants are italicized. SA, spontaneous abortion; NP, normal pregnancy; HM hydatidiform mole; IM, invasive mole; PHM, partial HM; BiCHM, indicates a complete mole found biparentalby the referring laboratory; CHM, complete HM; ART, assisted reproductive technologies; EFL, early fetal loss that manifested as heavy vaginal bleeding at the time of next menstruation and nopregnancy test was performed. HM is used when no tissues are available to re-evaluate the diagnosis and available pathology report does not distinguish between partial and complete HM.Reproductive outcomes are listed by chronological order starting from the left; the absence of a number indicates one such reproductive outcome.
89
0H
um
an
Mo
lecula
rG
enetics,
20
09
,V
ol.
18
,N
o.
5

genotyping of the two CHMs, from patients 428 and 492, usingvariable number of tandem repeats (VNTRs) and microsatellitemarkers from several chromosomes demonstrated their androge-netic monospermic origin (Tables 2 and 3). In patient 428, the
molar and normal placentae had identical paternal alleles at 17informative markers from 10 different chromosomes demon-strating their common origin from a single sperm and zygote(Table 2). Available parents, siblings, offsprings and materials
Figure 1. Characterization of molar tissues and conceptions from six patients with NLRP7 variants. The reproductive outcomes of the patients are listed bychronological order from left to right. Gestational age is from the last menstrual period. CHM, complete HM; PHM, partial HM; HM was used when the path-ology report does not specify the type of moles and the available material is not sufficient to establish the diagnosis; BO, blighted ovum; SB, stillbirth; SA,spontaneous abortion; n, ploidy; hap, indicates biparental contribution based on haplotype analysis of several SNPs in NLRP7; þ and 2 indicate, respectively,presence and absence of p57KIP2 expression in most of the villi; gray circles indicate women with unknown reproductive outcomes; asterisk (�) indicates that theproducts of conception with triploid and tetraploid peaks may also be mosaic with other diploid cells.
Human Molecular Genetics, 2009, Vol. 18, No. 5 891

from POC were genotyped at several SNPs in NLRP7. Haplotypeanalysis of the last mole of patient 501 is in favor of her biparen-tal origin. In the two moles from patient 636, haplotype data andmicrosatellite genotyping at three to four markers indicate thepresence of the maternal genome in both moles (SupplementaryMaterial). Microsatellite data combined with the flow cytometry
Figure 2. P57KIP2 immunohistochemistry of two products of conception from patients 428 (A) and 492 (B). (A) Microphotograph of P57KIP2 stain on the twinpregnancy consisting of a mole and a normal placenta attached to a fetus. The large molar chorionic villous from the hydatidiform mole (HM), on the top-right,does not express p57KIP2 in the cytotrophoblast (CT, arrows) while the small chorionic villi from the normal placenta display strong p57KIP2 nuclear stain. B.Microphotograph showing the absence of p57KIP2 stain in the computed tomography of a chorionic villous from the complete HM of patient 492. Extravilloustrophoblast (ET), which are positive in all types of moles serve as an internal positive control (arrowheads).
Table 3. Genotype data of molar placenta from patient 492
Marker Chromosome Patient 492 First CHM Partner
D1S235 1 187/187 185/185 181/185D1S199 1 100/108 100/100 100/100D1S419 1 172/172 172/172 172/188D1S490 1 203/205 203/203 199/203D1S213 1 114/118 106/106 106/110IL-1ra 2 1/1 1/1 1/1ApoB 2 1/2 2/2 n.a.D2S149 2 218/228 210/210 210/224D2S113 2 208/226 212/212 206/212D2S151 2 225/227 223/223 223/225D3S1265 3 226/234 226/226 226/236D3S1307 3 1/2 3/3 3/4D3S1292 3 148/156 150/150 150/156D5S426 5 193/203 201/201 201/203D5S407 5 113/127 123/123 123/123D5S393 5 164/178 172/172 172/176D5S400 5 210/228 228/228 224/228D6S89 6 2/3 1/1 1/2D6S282 6 112/124 112/112 112/112D6S265 6 126/132 128/128 128/132D7S2250 7 143/149 149/149 145/149D8S1745 8 131/133 131/131 131/133D8S1819 8 215/221 215/215 215/221D9S169 9 237/245 247/247 245/247D9S153 9 145/153 153/153 151/153D11S1888 11 213/213 211/211 211/213D11S902 11 153/157 157/157 155/157D11S4175 11 175/193 151/151 151/181D11S1890 11 1/1 2/2 2/3D13S328 13 275/279 281/281 281/283D14S61 14 215/215 215/215 197/215D15S974 15 124/129 127/127 127/136D15S1005 15 114/120 126/126 124/126TP53 17 1/1 2/2 n.a.NFATC 18 1/1 1/1 1/2D18S1153 18 205/219 205/205 205/219KIR2DS4 19 1/1 1/1 1/1AMEL X and Y XX XX XY
Italicized genotypes demonstrate the androgenetic origin of the mole. n.a.indicates not available.
Table 2. Genotype data of molar and normal placentae from patient 428
Marker Chromosome Patient428
Normal placentaof twins
Mole Partner 635
D1S199 1 108/110 96/110 96/96 96/100D1S213 1 104/108 104/104 104/104 104/108D1S419 1 178/192 180/192 180/180 180/184D1S255 1 80/80 80/74 74/74 74/84D1S207 1 148/148 148/158 158/158 158/158D2S125 2 88/94 94/92 92/92 92/100D2S143 2 124/132 n.a. 114/114 114/130D3S1292 3 148/158 148/166 166/166 164/166D3S1262 3 118/126 124/126 124/124 124/116D4S1426 4 183/183 183/185 185/185 181/185D5S407 5 110/110 110/110 110/110 n.a.D5S392 5 97/111 101/111 101/101 99/101D5S393 5 168/172 162/168 162/162 162/164D5S424 5 118/124 118/130 130/130 130/134D6S1717 6 124/124 112/124 n.a. 112/114D6S282 6 108/108 108/112 112/112 110/112D7S2250 7 149/157 159/149 159/159 159/161D8S1810 8 201/201 185/201 185/185 185/185D8S1745 8 125/127 131/131 131/131 127/131D8S261 8 136/136 136/140 140/140 140/140D9S162 9 176/188 176/184 184/184 184/184D12S329 12 155/161 159/161 159/159 159/159D13S1246 13 201/205 201/203 203/203 203/207D15S1033 15 158/166 164/166 164/164 164/168D15S974 15 117/138 117/136 136/136 136/136D15S1005 15 116/118 116/116 116/116 106/116AMEL X & Y XX XX XX XY
Italicized genotypes indicate those demonstrating the androgenetic originof the mole. Note the presence of the same allele in the normal placentaattached to the fetus. n.a., stands for not available.
892 Human Molecular Genetics, 2009, Vol. 18, No. 5

result indicate that the second PHM from patient 636 is diploidbiparental but do not allow clarifying the origin of the triploidy(diandric or digynic) of her first mole. A recapitulation of all dataon the different moles is summarized along with the pedigreestructures in Figure 1. Altogether our data demonstrate thatNLRP7 mutations/variants are responsible not only for diploidbiparental moles as previously demonstrated in several familialcases (18–23, 26), but also for diploid androgenetic and triploidmoles, and for spontaneous abortions with tetraploid genome.
Segregation of NLRP7 mutations in the haploid oocytesdoes not lead to hydatidiform moles
Mutation analysis of biparental POC from patients 428 and636 demonstrated that in patient 428, the placenta of the still-born baby and the zygote that led to a mole, both inheritedfrom their mother the rare variant, p.Gly487Glu, found in upto 4.6% of the general population and not variant,p.Cys399Tyr that was not found in controls. Also, in patient636, a healthy 6-year-old boy inherited his mother’s mutation,p.Ala719Val (Fig. 1). These findings are in agreement with ourprevious report, in which we documented a healthy daughterheterozygous for her mother’s mutation (p.Gly118fs) (24),and confirm further that the development of moles is notassociated with the segregation of a mutated NLRP7 allelein the haploid oocyte.
In vitro development of embryos from patientswith NLRP7 mutations
Three patients described in this study have tried various typesof assisted reproductive technologies (ART), patients 492 and565 with their own oocytes and patient 517 with donated
oocytes. In patient 492, seven oocytes were retrieved afterovulation induction; six were in metaphase II and were ferti-lized by sperm microinjection. Normal fertilization was docu-mented in three, based on the presence of two pronuclei (PN)and two polar bodies. The three zygotes cleaved to the six-cell,but the embryos were all of poor quality (grade IV), fragmen-ted and none was suitable for transfer (Table 4). In patient 565,38 oocytes were collected after ovulation induction, 33 weremetaphase II and were inseminated. Evidence of fertilizationwas found in 23, one embryo had one PN, one had three PNand 21 had two PN. Pre-implantation genetic diagnosis(PGD) was performed on three fresh embryos using fluor-escent in situ hybridization (FISH) with probes specific forchromosomes 13, 15, 16, 18, 21, 22, X and Y chromosomes.One embryo was diploid but could not be transferredbecause of the patient’s hyperstimulation. All the embryoswere frozen, thawed 3 months later, and 16 embryos werebiopsied, on day 3. Three embryos displayed a single signalfor more than three chromosomes and two signals for theremaining chromosomes and were therefore haploid–diploid.One embryo displayed a single signal for more than threechromosomes, but was chaotic because different blastomeresdisplayed different chromosome complements and was there-fore classified as haploid–aneuploid. The presence of threeembryos with haploid blastomeres for at least three chromo-somes (12.9%) (Table 4) is higher than what is usuallyobserved in in vitro fertilization (IVF) patients (3%) (28).Three embryos with diploid blastomeres were transferred,two implanted, and the patient delivered at term two dichorio-nic diamniotic twins in good health. In patient 517, whose sisterhad diploid biparental moles (19), seven donated oocyteswere fertilized, two embryos were of good quality and weretransferred 48 h post-insemination (PI). Subsequent failure
Table 4. In vitro fertilization data from patients and review of previous cases
Edwards et al. (40) Pal et al. (39) Reubinoff et al. (38) Current studyCycle 1 Cycle 2 492 565
Reproductive history 5 HM NP, 2 HM 2 GTD 2 HM 1 NP, 7 SAOocytes (own) 14 16 17 11 7 38Metaphase II 6 33Fertilized 13 14 17 9ICSI 6ICSI 2313–21 h PI
2 PN 7 (54%) 6 3 (6c) 211 PN 4 (31%) 13 PN 3 (23%) 3 (21%) 8 (47%) 14 PN 1
Frozen embryo 3 no no no no 18PGD-FISH no no no X, Y, 18 no X, Y, 13, 15, 16, 18, 21, 22Mosaic
1n-aneuploid 1 (4.3%)2n-haploid 2 (8.6%)2n-triploid 12n-aneuploid/chaotic� 4 (19%)2n 5 (55%) 9 (35%)
Embryo transfer no 4 5 2 no 3b-hCG –Pregnancy Failed Failed Failed Healthy twins
HM, indicates hydatidiform mole; NP, normal pregnancy; SA, spontaneous abortion; GTD, gestational trophoblastic disease; ICSI, intracytoplasmicsperm injection; PN, pronucleus; n, ploidy; haploid, indicates embryos with a single signal for at least three of the eight tested chromosomes; aneuploid,indicates monosomy or trisomy for one to two chromosomes; PI, post-insemination; hCG, human chorionic gonadotrophin.�Chaotic indicates random loss or gain of some chromosomes in different blastomeres.
Human Molecular Genetics, 2009, Vol. 18, No. 5 893

was documented by negative b-human chorionic gonadotrophin14 days PI. Because of the small number of transferredembryos in patient 517, this case does not allow to concludewhether ovum donation fails in patients with NLRP7mutations.
DISCUSSION
Mutations in NLRP7 have been shown to be responsible forrecurrent RHMs associated with other forms of reproductivewastage. Here we report 11 new variants in NLRP7 inwomen with RHMs and various forms of reproductivewastage. Our data expand further the spectrum of reproductivewastage associated with NLRP7 mutations/variants to patientswith very early fetal loss, 4 weeks from last menstrual period,and to patients with recurrent spontaneous abortions and nomolar pregnancies.
Four autoimmune conditions – Hashimoto disease, lupusantibody, vitiligo and Crohn’s disease, were found in some ofour patients. Two of these features, vitiligo and Crohn’sdisease are associated with variants in other genes from theCATERPILLER family, NLRP1 and NOD2, respectively.With the exception of an old report of ulcerative colitis, vitiligo,and HM (29), no previous associations between molar pregnan-cies and autoimmune conditions have so far been reported.However, ulcerative colitis, coeliac and Crohn’s diseases areknown risk factors for recurrent spontaneous abortions (30–32). Our data indicate that RHMs, believed so far to be an auto-inflammatory condition, has also an autoimmune componentand some patients have abnormal autoantibodies and overlap-ping clinical features with other autoimmune diseases. Our find-ings indicate that patients with RHMs should be screened foradrenal, thyroid and ovarian autoantibodies and in the presenceof such autoantibodies, the patients should be closelyfollowed-up to prevent adrenal, thyroid and ovarian failure. Inaddition, we note the occurrence of uterine problems in threeof the reported patients, which have also been previouslyobserved in patients with HMs (16,33).
In five of the patients included in this study, 647, 636, 501,565 and 492, we found only a single variant that was not foundin controls, by genomic DNA sequencing of the 11 NLRP7exons in the two directions. The presence of a single NLRP7variant associated with the disease phenotype in five patientsindicates: (i) the presence of undetected mutations, deletions,rearrangements in the exons, introns, or promoter and regulat-ory regions; (ii) the contribution of one or several other genesas well as environmental factors to the disease phenotype; and(iii) the possibility that a single variant may be associated withthe disease phenotype. Furthermore, the presence of a non-synonymous rare variant, p.Gly487Glu, in one patient, thatis present in up to 4.6% of women from the general populationindicates that this variant could confer susceptibility for spora-dic moles and reproductive wastage.
In the current study, we demonstrate that NLRP7 mutationsor variants are responsible not only for diploid biparentalmoles, but also for diploid androgenetic moles, triploidmoles and for tetraploid spontaneous abortions. After the dem-onstration by Kajii and Ohama in 1977 that most sporadicCHMs are diploid and androgenetic, a hypothetical mechan-ism was postulated to explain their formation (34). This
mechanism consisted of the fertilization of an oocytewithout nucleus or with an inactivated nucleus followed bythe duplication of the paternal chromosomes without cytokin-esis (34). Later studies have endorsed this postulate (35) andsome investigators raised other possibilities involving theexclusion or the elimination of the egg nucleus before fertili-zation (36). To date, we do not have any experimental evidenceof neither of the above proposed mechanisms and we still donot know how androgenesis occurs and when the maternalnucleus or genome is eliminated. Furthermore, in more than30 years of assisted reproductive technologies, during whichtime oocytes from women with various medical conditionshave been cultured in vitro and examined, nobody has reportedempty oocytes without nucleus that fertilize, cleave andimplant. Consequently, scientists have started questioning theconcept of the ‘empty oocyte’ and its existence at the originof androgenetic moles in twin conceptions (37). These datahave led Golubovsky to propose an interesting hypothesissuggesting that post-zygotic diploidization of triploid zygotesis at the origin of androgenetic moles, which is definitelymore convincing than the empty oocyte scenario (37).
In vitro development of embryos from patient 565 showed ahigher rate of mosaic, haploid–diploid and haploid–aneuploidembryos when compared with patients undergoing IVF forvarious medical reasons (28). In patient 492, all the embryoswere degenerated by the 6-cell stage. Our data on these twopatients demonstrate in vitro early cleavage abnormalities inpatients with NLRP7 variants. To date, three other cases ofIVF in women with RHMs have been documented and nopregnancies were achieved in any of them (Table 4) (38–40). In the first case, higher rates of triploid and haploidembryos were observed based on the number of pronuclei13–21 h PI (40); in the second, higher rates of triploidembryos were noted and one embryo was tetraploid, againbased on the number of pronuclei several hours PI (39);while in the third, no aneuploidies were documented basedon PGD and FISH with probes that detect three chromosomes(38). Unfortunately, these three cases were studied before thecurrent knowledge about moles and consequently the parentalcontribution to the different moles and the presence of NLRP7mutations in the patients were not investigated. Our currentstudy confirms previous observations but in two patientswith NLRP7 variants and documented parental contributionto their moles from natural conceptions. Interestingly, embryo-nic arrest during early cleavage has also been reported in amouse knockout for Nlrp5, where embryos from nullfemales start to degenerate from the 2-cell stage and nonereaches the blastocyst stage (41).
Data on patient 428 demonstrate, in a natural conception,the occurrence of an androgenetic mole and a fetus derivingfrom the same zygote. This demonstrates in vivo post-zygoticabnormalities and their causal role in the diploid androgeneticmole in this patient. Five similar cases of androgenetic molesand fetuses deriving from the same zygotes have so far beenreported (42–44), but our finding is the first in a patientwith NLRP7 variants. Our data on POC from five patientswith different NLRP7 variants (Fig. 1) demonstrate the alter-nation, in the same patient, of several types of moles/con-ceptions, diploid biparental, triploid (or 3n þ 2n) andtetraploid (or 4n þ 2n), which somehow mimic the spectrum
894 Human Molecular Genetics, 2009, Vol. 18, No. 5

of in vitro abnormalities observed in patient 565 and inpreviously reported women with recurrent moles undergoingART services (38–40). Analyses of large cohorts of normallyin vitro-fertilized embryos with two pronuclei have demon-strated that post-zygotic errors leading to mosaic embryos isthe major abnormality observed during in vitro cleavage.These studies have shown that the rate of mosaic embryosincreases significantly at sequential stages of pre-implantationdevelopment and reaches 90.9% at the blastocyst stage (28).Among these embryos, those with haploid blastomeres arethe least frequent (0.5%), and when present, they are in themosaic state with other diploid blastomeres (28). Altogether,our data lead us to propose a two-hit mechanism that mayexplain androgenetic mole manifestation. NLRP7 mutationslead, directly or indirectly, to an increased rate of stochasticand mosaic aneuploidies during early embryo cleavage withall possible combinations of haploid, diploid, triploid and tet-raploid blastomeres. The resulting embryos may, or may not,survive until implantation depending on the severity of theaneuploidies for embryonic development and on the percen-tage of diploid cells in the chimeric embryos. After implan-tation, surviving embryonic cells are subject to an additionaltype of selection that depends on the maternal immunesystem and its tolerance to the abnormal conception. Recently,we found that patients with NLRP7 mutations, including 428and 636, are not able to mount an appropriate inflammatoryresponse against various antigens (Deveault et al., manuscriptin preparation). Consequently, in such patients, androgeneticblastomeres, which are complete allograft, may thrive, growand proliferate without being rejected by the patients. It isalso possible that androgenetic blastomeres occur, de novo,in an important number of women in the general population.However, in women with active immune system, androgeneticcells most likely die or stay confined, and undetected, to asmall portion of the placenta. A similar mechanism mayunderlie triploid diandric moles, which have a higher antigeni-city than diploid biparental embryos; such cells may onlysurvive in women with some forms of immune deficiencies.Our suggestion is corroborated by the high degree of mosaicaneuploidies and chromosomal abnormalities, 50–100%,observed in pre-implantation embryos (45,46).
The rate of embryo cleavage abnormalities in patients withNLRP7 mutations seems to be mutation-dependent with somealleles being more severe than others. For instance, patient492, who had an androgenetic mole from a natural conception,all her in vitro-fertilized embryos fragmented before themorula stage while in patient 565, who had had a normal preg-nancy and several spontaneous abortions from natural con-ceptions, 35% of her in vitro-fertilized embryos were ofgood quality, diploid and the patient achieved a secondnormal pregnancy. Among described patients with NLRP7mutations, 492 and 428, are the first with reported androge-netic moles and the first with cysteine-to-tyrosine changes.Mutations affecting cysteine residues have been reported inanother inflammatory disease, the TNF receptor-associatedperiodic syndrome (TRAPS), to be more penetrant than non-cysteine mutations and are associated with different clinicalfeatures (47). Analyzing more molar tissues from patientswith different NLRP7 mutations may or may not allow validat-ing our observation.
In the case of patients with NLRP7 mutations, the use ofPGD does not cure their genetic defect, but it helps selectingdiploid embryos, if any, for transfer and therefore increasingthe likelihood of the patients to have normal pregnancies.Taking into consideration our PGD data and those of othergroups (38–40), we believe that the selection of diploidembryos by PGD is currently the best strategy for patientswith NLRP7 mutations who wish to conceive.
MATERIALS AND METHODS
Patients, histopathology analysis and mutation analysis
The reproductive outcomes of 13 new patients are summarizedin Table 1 and Figure 1. Five patients are from familial casesand eight have no family history of moles. Each patient hashad either RHMs or recurrent reproductive wastage includingat least one mole with the exception of patient 565, who didnot have any molar pregnancy. Patient 565 has had twonormal pregnancies, and seven spontaneous abortions; onlyone of them required dilatation, suction and curettage.Patient 636 has had three very early fetal losses (EFL) thatmanifested as heavy vaginal bleeding at the time of next men-struation. Patient 428 had a molar pregnancy that was part of atwin pregnancy originating from a natural conception. At 8weeks, a normal fetus with heart activity and measurementswas detected and a small region of 8 mm of placental detach-ment was noted. Echogenic structures in the placenta started tomanifest on ultrasound around week 10, but the fetus contin-ued to have normal growth parameters. The death of thefetus was noted at 15 weeks. Dilatation and suction revealeda female fetus with a supra-umbilical defect attached to anormal placenta and a molar placenta. The fetal foot-lengthwas normal for gestational age. The normal placenta was kar-yotyped and had normal female karyotype. Histopathologyrevealed a CHM with abundant fibrin and focal necrosis.An admixture of non-hydatidiform villous structures withdysmature features was noted in some regions. Bowel tissueswere identified among the normal placenta. The histopathol-ogy diagnosis was established according to Szhulman’scriteria (48) (Supplementary Material). Three patients hadother immunological manifestations, Hashimoto disease,autoimmune thyroditis, lupus antibodies, Crohn’s diseaseand vitiligo (Table 1), two patients had uterine myomas, 428and 662, and one, 647, was operated for a uterine septum.Patient 492 has an abnormal blood karyotype, 45,XX,rob(13,14)(q10;q10), carrying one of the most frequent Robertso-nian translocations.
Patient 517 is from a previously reported family, MoCh76,and is compound heterozygote for p.Glu99X and p.Asp657Val(19). Mutation analysis in the new patients was performed aspreviously described by genomic DNA sequencing of the 11exons of NLRP7 in the two directions (19,24). Otherapproaches were used in patients where a single mutationwas found and appropriate material was available. Theseapproaches were: (i) long range PCR amplification andsequencing of the entire genomic region from the start to thestop codons (17.9 kb) in five overlapping fragments usingthe TAKARA kit (Fisher Scientific); (ii) haplotype establish-ment at NLRP7 variants on available parents, siblings and
Human Molecular Genetics, 2009, Vol. 18, No. 5 895

products of conception; and (iii) cDNA sequencing, which wasperformed only on one patient, 428, from which an EBV-lymphoblastoid cell line was available.
Parental contribution to the moles
Ploidy was determined either by karyotype analysis on freshchorionic villi or by flow cytometry on one paraffin-embeddedtissue containing several chorionic villi from each availablePOC. P57KIP2 immunohistochemistry was performed on oneparaffin-embedded tissue from each POC and was used to dis-tinguish between CHM, PHM and hydropic abortions. P57KIP2
is an imprinted, maternally expressed gene that is used as acomplementary diagnostic marker; its expression in the cyto-tropblast and the villous mesenchyme indicates the presenceof the maternal genome (49) and consequently the non-androgenetic nature of the POC, which could be in this case,triploid, tetraploid, or diploid biparental (50). However, theabsence of p57KIP2 expression does not allow concludingthat the maternal genome is absent since several diploid bipar-ental moles do not express p57KIP2. P57KIP2 immunohisto-chemistry slides were screened independently by twoobservers and the conclusion was based on the majority ofchorionic villi. DNA genotyping was performed on DNAextracted from freshly dissected or paraffin-embedded chorio-nic villi along with parental blood DNA using microsatellites,VNTRs and NLRP7 SNPs.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG online.
ACKNOWLEDGEMENT
We thank the patients, their families and control subjects fortheir cooperation. We also thank Drs Kevin Watters, Alexan-dra Duncan, Serge Allard, and the Department of Pathology atthe St Alexius and Sherman Hospitals for providing biologicalmaterials. We also thank Kisha Johnson for her excellent ded-ication to patients and Abbas Kerim-Dikeni for his assistanceand cooperation.
Conflict of Interest statement. The authors declare that theyhave no conflict of interest.
FUNDING
This study was supported by the Canadian Institute of HealthResearch (grant number MOP 86546); the ‘Fonds de laRecherche en Sante du Quebec’ (grant number 5539) andthe National Natural Science Foundation of China (grantnumber 30511120178). R.S. is supported by a Fraser, Monatand McPherson Scholarship Salary Award.
REFERENCES
1. Grimes, D.A. (1984) Epidemiology of gestational trophoblastic disease.Am. J. Obstet. Gynecol., 150, 309–318.
2. Sebire, N.J., Fisher, R.A., Foskett, M., Rees, H., Seckl, M.J. andNewlands, E.S. (2003) Risk of recurrent hydatidiform mole and
subsequent pregnancy outcome following complete or partialhydatidiform molar pregnancy. BJOG, 110, 22–26.
3. Sand, P.K., Lurain, J.R. and Brewer, J.I. (1984) Repeat gestationaltrophoblastic disease. Obstet. Gynecol., 63, 140–144.
4. Kronfol, N.M., Iliya, F.A. and Hajj, S.N. (1969) Recurrent hydatidiformmole: a report of five cases with review of the literature. J. Med. Liban.,22, 507–520.
5. Kim, J.H., Park, D.C., Bae, S.N., Namkoong, S.E. and Kim, S.J. (1998)Subsequent reproductive experience after treatment for gestationaltrophoblastic disease. Gynecol. Oncol., 71, 108–112.
6. Horn, L.C., Kowalzik, J., Bilek, K., Richter, C.E. and Einenkel, J. (2006)Clinicopathologic characteristics and subsequent pregnancy outcome in139 complete hydatidiform moles. Eur. J. Obstet. Gynecol. Reprod. Biol.,128, 10–14.
7. Berkowitz, R.S., Im, S.S., Bernstein, M.R. and Goldstein, D.P. (1998)Gestational trophoblastic disease. Subsequent pregnancy outcome,including repeat molar pregnancy. J. Reprod. Med., 43, 81–86.
8. Van Thiel, D.H., Ross, G.T. and Lipsett, M.B. (1970) Pregnancies afterchemotherapy of trophoblastic neoplasms. Science, 169, 1326–1327.
9. Lan, Z., Hongzhao, S., Xiuyu, Y. and Yang, X. (2001) Pregnancyoutcomes of patients who conceived within 1 year after chemotherapy forgestational trophoblastic tumor: a clinical report of 22 patients. Gynecol.
Oncol., 83, 146–148.10. Vejerslev, L.O., Dissing, J., Hansen, H.E. and Poulsen, H. (1987)
Hydatidiform mole: genetic markers in diploid abortuses withmacroscopic villous enlargement. Cancer Genet. Cytogenet., 26,143–155.
11. Paradinas, F.J., Browne, P., Fisher, R.A., Foskett, M., Bagshawe, K.D.and Newlands, E. (1996) A clinical, histopathological and flow cytometricstudy of 149 complete moles, 146 partial moles and 107 non-molarhydropic abortions. Histopathology, 28, 101–110.
12. Lawler, S.D., Fisher, R.A. and Dent, J. (1991) A prospective genetic studyof complete and partial hydatidiform moles. Am. J. Obstet. Gynecol., 164,1270–1277.
13. Jacobs, P.A., Szulman, A.E., Funkhouser, J., Matsuura, J.S. and Wilson,C.C. (1982) Human triploidy: relationship between parental origin of theadditional haploid complement and development of partial hydatidiformmole. Ann. Hum. Genet., 46, 223–231.
14. Fukunaga, M., Endo, Y. and Ushigome, S. (1995) Flow cytometric andclinicopathologic study of 197 hydatidiform moles with special referenceto the significance of cytometric aneuploidy and literature review.Cytometry, 22, 135–138.
15. Fukunaga, M. (2001) Flow cytometric and clinicopathologic study ofcomplete hydatidiform moles with special reference to the significance ofcytometric aneuploidy. Gynecol. Oncol., 81, 67–70.
16. van der Smagt, J.J., Scheenjes, E., Kremer, J.A., Hennekam, F.A. andFisher, R.A. (2006) Heterogeneity in the origin of recurrent completehydatidiform moles: not all women with multiple molar pregnancies havebiparental moles. BJOG, 113, 725–728.
17. Zhao, J., Moss, J., Sebire, N.J., Cui, Q.C., Seckl, M.J., Xiang, Y. andFisher, R.A. (2006) Analysis of the chromosomal region 19q13.4 in twoChinese families with recurrent hydatidiform mole. Hum. Reprod., 21,536–541.
18. Sensi, A., Gualandi, F., Pittalis, M.C., Calabrese, O., Falciano, F., Maestri,I., Bovicelli, L. and Calzolari, E. (2000) Mole maker phenotype: possiblenarrowing of the candidate region. Eur. J. Hum. Genet., 8, 641–644.
19. Qian, J., Deveault, C., Bagga, R., Xie, X. and Slim, R. (2007) Womenheterozygous for NALP7/NLRP7 mutations are at risk for reproductivewastage: report of two novel mutations. Hum. Mutat., 28, 741.
20. Panichkul, P.C., Al-Hussaini, T.K., Sierra, R., Kashork, C.D., Popek, E.J.,Stockton, D.W. and Van den Veyver, I.B. (2005) Recurrent biparentalhydatidiform mole: additional evidence for a 1.1-Mb locus in 19q13.4 andcandidate gene analysis. J. Soc. Gynecol. Investig., 12, 376–383.
21. Judson, H., Hayward, B.E., Sheridan, E. and Bonthron, D.T. (2002)A global disorder of imprinting in the human female germ line. Nature,416, 539–542.
22. Helwani, M.N., Seoud, M., Zahed, L., Zaatari, G., Khalil, A. and Slim, R.(1999) A familial case of recurrent hydatidiform molar pregnancies withbiparental genomic contribution. Hum. Genet., 105, 112–115.
23. Fisher, R.A., Hodges, M.D., Rees, H.C., Sebire, N.J., Seckl, M.J.,Newlands, E.S., Genest, D.R. and Castrillon, D.H. (2002) The maternallytranscribed gene p57(KIP2) (CDNK1C) is abnormally expressed in both
896 Human Molecular Genetics, 2009, Vol. 18, No. 5

androgenetic and biparental complete hydatidiform moles. Hum. Mol.Genet., 11, 3267–3272.
24. Murdoch, S., Djuric, U., Mazhar, B., Seoud, M., Khan, R., Kuick, R.,Bagga, R., Kircheisen, R., Ao, A., Ratti, B. et al. (2006) Mutations inNALP7 cause recurrent hydatidiform moles and reproductive wastage inhumans. Nat. Genet., 38, 300–302.
25. Zhang, P., Dixon, M., Zucchelli, M., Hambiliki, F., Levkov, L.,Hovatta, O. and Kere, J. (2008) Expression analysis of the NLRP genefamily suggests a role in human preimplantation development. PLoSONE, 3, e2755.
26. Kou, Y.C., Shao, L., Peng, H.H., Rosetta, R., del Gaudio, D.,Wagner, A.F., Al-Hussaini, T.K. and Van den Veyver, I.B. (2008)A recurrent intragenic genomic duplication, other novel mutations inNLRP7 and imprinting defects in recurrent biparental hydatidiform moles.Mol. Hum. Reprod., 14, 33–40.
27. Okada, K., Hirota, E., Mizutani, Y., Fujioka, T., Shuin, T., Miki, T.,Nakamura, Y. and Katagiri, T. (2004) Oncogenic role of NALP7 intesticular seminomas. Cancer Sci., 95, 949–954.
28. Bielanska, M., Tan, S.L. and Ao, A. (2002) Chromosomal mosaicismthroughout human preimplantation development in vitro: incidence, type,and relevance to embryo outcome. Hum. Reprod., 17, 413–419.
29. Hovdenak, N. (1979) [Ulcerative colitis, vitiligo and hydatidiform mole].Tidsskr Nor Laegeforen, 99, 1089–1090.
30. Lejeune, V. (2006) [Early recurrent spontaneous abortion: How to takecare in 2006?]. Gynecol. Obstet. Fertil., 34, 927–937.
31. Hudson, M., Flett, G., Sinclair, T.S., Brunt, P.W., Templeton, A. andMowat, N.A.G. (1997) Fertility and pregnancy in inflammatory boweldisease. Int. J. Gynecol. Obstet., 58, 229.
32. Gasbarrini, A., Torre, E.S., Trivellini, C., De Carolis, S., Caruso, A. andGasbarrini, G. (2000) Recurrent spontaneous abortion and intrauterinefetal growth retardation as symptoms of coeliac disease. Lancet, 356,399–400.
33. Novachkov, L. and Koleva, Z. (1984) [Combined hydatidiform mole anduterine myoma in the climacteric]. Akush Ginekol (Sofiia), 23, 187–189.
34. Kajii, T. and Ohama, K. (1977) Androgenetic origin of hydatidiformmole. Nature, 268, 633–634.
35. Jacobs, P.A., Wilson, C.M., Sprenkle, J.A., Rosenshein, N.B. and Migeon,B.R. (1980) Mechanism of origin of complete hydatidiform moles.Nature, 286, 714–716.
36. Wake, N., Takagi, N. and Sasaki, M. (1978) Androgenesis as a cause ofhydatidiform mole. J. Natl Cancer Inst., 60, 51–57.
37. Golubovsky, M.D. (2003) Postzygotic diploidization of triploids as asource of unusual cases of mosaicism, chimerism and twinning. Hum.Reprod., 18, 236–242.
38. Reubinoff, B.E., Lewin, A., Verner, M., Safran, A., Schenker, J.G. andAbeliovich, D. (1997) Intracytoplasmic sperm injection combined withpreimplantation genetic diagnosis for the prevention of recurrentgestational trophoblastic disease. Hum. Reprod., 12, 805–808.
39. Pal, L., Toth, T.L., Leykin, L. and Isaacson, K.B. (1996) High incidenceof triploidy in in vitro fertilized oocytes from a patient with a previoushistory of recurrent gestational trophoblastic disease. Hum. Reprod., 11,1529–1532.
40. Edwards, R.G., Crow, J., Dale, S., Macnamee, M.C., Hartshorne, G.M.and Brinsden, P. (1992) Pronuclear, cleavage and blastocyst histories inthe attempted preimplantation diagnosis of the human hydatidiform mole.Hum. Reprod., 7, 994–998.
41. Tong, Z.B., Gold, L., Pfeifer, K.E., Dorward, H., Lee, E., Bondy, C.A.,Dean, J. and Nelson, L.M. (2000) Mater, a maternal effect gene requiredfor early embryonic development in mice. Nat. Genet., 26, 267–268.
42. Niemann, I., Bolund, L. and Sunde, L. (2008) Twin pregnancies withdiploid hydatidiform mole and co-existing normal fetus may originatefrom one oocyte. Hum. Reprod., 23, 2031–2035.
43. Makrydimas, G., Sebire, N.J., Thornton, S.E., Zagorianakou, N., Lolis, D.and Fisher, R.A. (2002) Complete hydatidiform mole and normal livebirth: a novel case of confined placental mosaicism: case report. Hum.
Reprod., 17, 2459–2463.
44. Kaiser-Rogers, K.A., McFadden, D.E., Livasy, C.A., Dansereau, J., Jiang,R., Knops, J.F., Lefebvre, L., Rao, K.W. and Robinson, W.P. (2006)Androgenetic/biparental mosaicism causes placental mesenchymaldysplasia. J. Med. Genet., 43, 187–192.
45. Wells, D. and Delhanty, J.D. (2000) Comprehensive chromosomalanalysis of human preimplantation embryos using whole genomeamplification and single cell comparative genomic hybridization. Mol.
Hum. Reprod., 6, 1055–1062.
46. Malmgren, H., Sahlen, S., Inzunza, J., Aho, M., Rosenlund, B., Fridstrom,M., Hovatta, O., Ahrlund-Richter, L., Nordenskjold, M. and Blennow, E.(2002) Single cell CGH analysis reveals a high degree of mosaicism inhuman embryos from patients with balanced structural chromosomeaberrations. Mol. Hum. Reprod., 8, 502–510.
47. Aksentijevich, I., Galon, J., Soares, M., Mansfield, E., Hull, K., Oh, H.H.,Goldbach-Mansky, R., Dean, J., Athreya, B., Reginato, A.J. et al. (2001)The tumor-necrosis-factor receptor-associated periodic syndrome: newmutations in TNFRSF1A, ancestral origins, genotype-phenotype studies,and evidence for further genetic heterogeneity of periodic fevers.Am. J. Hum. Genet., 69, 301–314.
48. Szulman, A.E. and Surti, U. (1978) The syndromes of hydatidiform mole.II. Morphologic evolution of the complete and partial mole. Am. J. Obstet.
Gynecol., 132, 20–27.
49. Castrillon, D.H., Sun, D., Weremowicz, S., Fisher, R.A., Crum, C.P. andGenest, D.R. (2001) Discrimination of complete hydatidiform mole fromits mimics by immunohistochemistry of the paternally imprinted geneproduct p57KIP2. Am. J. Surg. Pathol., 25, 1225–1230.
50. Crisp, H., Burton, J.L., Stewart, R. and Wells, M. (2003) Refining thediagnosis of hydatidiform mole: image ploidy analysis and p57KIP2immunohistochemistry. Histopathology, 43, 363–373.
Human Molecular Genetics, 2009, Vol. 18, No. 5 897





























