Embed Size (px)
Citation preview

www.elsevier.com/locate/braindev
Brain & Development 30 (2008) 53–58
Original article
Focal cortical dysplasia type 1b as a causeof severe epilepsy with multiple independent spike foci
Sabrina Buoni a, Raffaella Zannolli a,*, Clelia Miracco b, Francesca Macucci a,Joseph Hayek c, Luca Burroni d, Giovanni di Pietro e, Luigi Sardo e,
Federico Mussa e, Flavio Giordano e, Lorenzo Genitori e
a Department of Pediatrics, Section of Pediatric Neurology, Policlinico Le Scotte, University of Siena, Siena, Italyb Department of Human Pathology and Oncology, Section of Pathological Anatomy and Istology, Policlinico Le Scotte, University of Siena, Siena, Italy
c Pediatrics Neuropsychiatric Unit, Azienda Ospedaliera Universitaria Senese, Policlinico Le Scotte, Siena, Italyd UOC Nuclear Medicine, Azienda Ospedaliera Universitaria Senese, Policlinico Le Scotte, Siena, Italy
e Department of Pediatrics, Section of Pediatric Neurosurgery, Policlinico Le Scotte, University of Siena, Siena, Italy
Received 11 December 2006; received in revised form 23 April 2007; accepted 7 May 2007
Abstract
To investigate the clinical picture, the neurophysiological pattern, and neuropathological features of a young woman with severedrug-resistant epilepsy of unknown cause. We used the patient’s clinical records from the age of 2 to 20 years including neurophysi-ological patterns recorded via both scalp and cortex electrodes and results of studies conducted on the brain neuropathological spec-imen. The patient, with severe mental/psychomotor retardation, suffered from severe epilepsy from an early age, characterized bydaily seizures of multiple types (atypical absences, tonic, and complex partial seizures), high frequency, and intractability. Theneurophysiological pattern indicated multiple independent spike foci (SE-MISF). When she was 16, a vagal nerve stimulator wasimplanted without success. Neither neuroimaging (brain MRI and ictal SPECT) nor surface EEGs identified unique loci of seizureonset, establishing her as a candidate for a complete callosotomy. When the patient was 19, before the callosotomy, invasive EEG(i.e., electrocorticography) using just a few electrodes in different lobes showed the presence of a distinctive pattern. The surgicalspecimen, taken very close to one of the activity sites, showed architectural abnormalities and neurons that were giant or immaturebut not dysmorphic, indicative of focal cortical dysplasia (FCD) type 1b. Twelve months after the callosotomy, according to theEngel score, the patient exhibited a large improvement in quality of life, without permanent complications from the interhemisphericdisconnection. (1) Hidden FCD type 1b could represent a missing diagnosis in patients with SE-MISF in the absence of other causesfor their seizures. (2) Complete callosotomy can be efficacious in patients with SE-MISF with hidden FCD type 1b.� 2007 Elsevier B.V. All rights reserved.
Keywords: Electrocorticography; Focal cortical dysplasia type 1b; Multifocal refractory epilepsy; Mental/psychomotor retardation
1. Introduction
Focal cortical dysplasias (FCDs) are increasinglydiagnosed as a cause of symptomatic focal epilepsy inpaediatric and adult patients, but little is known about
0387-7604/$ - see front matter � 2007 Elsevier B.V. All rights reserved.
doi:10.1016/j.braindev.2007.05.010
* Corresponding author. Tel.: +39 (0)577 586514; fax: +39 (0)577586143.
E-mail address: [email protected] (R. Zannolli).
the clinical characteristics of epilepsy in these patients[1]. To elucidate these characteristics, we report aninvestigation of the clinical picture, neurophysiologicalpattern, and neuropathological features in a youngpatient we followed from the age of 2 to 20 years, withsevere epilepsy with multiple independent spike foci(SE-MISF) [2], severe mental/psychomotor retardation,and no identifiable cause for her seizures. Her final diag-nosis was hidden FCD type 1b.

54 S. Buoni et al. / Brain & Development 30 (2008) 53–58
2. Methods and results
2.1. Patient history
The patient was a 19-year-old girl whom we hadobserved since she was 2-years-old for delayed psycho-motor development and severe epilepsy, characterizedby daily seizures of multiple types (atypical absences,tonic, and complex partial seizures), high frequency,and intractability. There was no relevant family history,and the gestational period was uneventful. The child wasborn at 38 weeks of gestational age by caesarean deliv-ery because of foetal hypoxia. Her birth weight was2200 g; length and head circumference were notrecorded. The immediate neonatal period was character-ized by apnoeic spells, cyanosis, and seizures. Prolongedjaundice with a bilirubin level of 15 mg/dl was noted upto the end of the first week of life. She presented withhypotonia and weak sucking reflex. The patient was inan intensive-care unit for 45 days, then discharged.Delayed psychomotor development was apparent at anearly stage. At 3 months of age, however, she had a nor-mal EEG. At 1 year of age, she presented with a pro-longed, generalized tonic–clonic seizure. At the age of18 months, neuromotor development was severelyimpaired. A second prolonged generalized convulsiveepisode developed. At the age of 2 years, she presentedfor our observation.
At that time, head circumference was 48 cm (>2 SD),suggestive of macrocephaly. Weight (12.8 kg) and height(88 cm) were appropriate for age. Language was poor,as was neuromotor performance. She presented severaldysmorphisms (reduced bi-temporal diameter, ptosis,low-set ears and hairline, ‘fish’-like rima oris, smallhands and feet, generalized hypotonia, and ataxic gait).The EEG showed a mild diffuse paroxysmal activity.Electrocardiography, cardiac ultrasound, genitourinarysystem, and abdominal examinations were normal. AnX-ray survey of the skeleton was normal, and thepatient’s karyotype was 46, XX. Ammonia, lactate andpyruvate, biotinidase, amino acids, acylcarnitine profile,and sialotransferrin pattern in the plasma were normal.Amino acids, organic acids, oligosaccharides, muco-polysaccharides, and purine and pyrimidine analysis inthe urine were also normal. A biochemical examinationof the cerebrospinal fluid was not informative. Cortisollevels in serum and lysosomal enzymes in leukocytes,including cerebroside-beta-galactosidase, were also nor-mal. Nerve conduction velocity and electromyographywere unremarkable.
At the age of 3 years, the patient exhibited an EEG thathad worsened and was seriously impaired because of thepresence of a multifocal paroxysmal activity. Seizures(atypical absences, tonic, and complex partial seizures)became very frequent (three to five episodes daily) and dif-ficult to control. Since that time, no drug treatment (val-
proate, clobazam, phenobarbital, carbamazepine,primidone, phenytoin and ethosuximide, vigabatrin, lam-otrigine, gabapentin, topiramate, felbamate, and tiaga-bine) has been effective for the patient, except for atransient response to ACTH followed by felbamate, asadd-on therapies and raised to the maximum tolerateddoses. When the patient was 15-years-old, brain com-puted tomography (CT) provided evidence of a globalvolumetric reduction of both hemispheres with anincrease in the size of the lateral ventricles. A brain1.5 T MRI confirmed the brain CT findings and showedalso a thinning of the corpus callosum. When she was16-years-old, vagus nerve stimulation (VNS) wasattempted, using a Neurocyberonics pacemaker (Model100, Cyberonics Inc., Webster, TX, USA) implanted ina standard VNS procedure. There was no seizureimprovement, and the device was removed after 2 years.When the patient was 19-years-old, we planned a surgicaltreatment of her refractory epilepsy.
2.1.1. Preoperative evaluation
In the preoperative evaluation, a new brain MRI,performed using a high-field-strength magnet (1.5 T)with high-resolution thin slices, showed no substantialchanges and no apparent areas of cortical dysplasia orperiventricular heterotopia. The patient epileptogenicdata (video EEG monitoring) (Fig. 1A and B) showedan interictal multifocal bioelectrical activity (Fig. 1A)and an ictal activity starting both from the bifrontaland from the right frontal lobes (Fig. 1B). SPECTshowed significantly increased perfusion in the lowerregion of the right superior temporal gyrus (Fig. 1C)and spreading in the medial and lower regions of theright occipital lobe. A normal perfusion was revealedin both frontal lobes.
2.2. The first surgery
In a last effort to use a lobectomy instead of callosot-omy, electrocorticography (EcoG) was used to directlytest the epileptogenic activity of the right frontal lobe.The EcoG recording was performed with two frontal1- · -8 subdural strips. Moreover, a standard recordingwith 10–20 scalp electrodes was simultaneously made.Surgery was done with the patient in a supine decubitusposition and the head fixed in a neutral position. An ita-lic ‘‘S’’ shaped skin incision was made above the coronalsuture, and a bifrontal 4- · -4 craniotomy was per-formed. After the dura were opened, two 1- · -8 sub-dural strips were placed over the frontal regions,between the prefrontal cortex (Brodmann’s area 9) andthe premotor cortex (Brodmann’s area 6), so that con-tacts 1–2 were sampling the prefrontal cortex, and con-tacts 7–8 were sampling the premotor cortex. Aftersubcutaneous tunnelization and fixation of the elec-trodes, the dura was closed as usual.

Fig. 1. Patient epileptogenic data before performing electrocorticography (EcoG). The overall data clearly indicate a multifocal epilepsy. (A and B)Video-EEG monitoring. (C) Coronal slices of ECD-Tc99m ictal brain SPECT. (A) Interictal EEG. Multifocal sharp waves and spikes more evidentin the bifrontal regions. (B) Ictal EEG. Initial bioelectrical correlate of a seizure consisting of discharge of diffuse polyspikes followed (asterisks) byright frontal theta activity. (C) Significant increased perfusion in the right superior temporal gyrus (arrows).
S. Buoni et al. / Brain & Development 30 (2008) 53–58 55
The correct and final position of the strips wasassessed by lateral and antero-posterior skull radio-graphs (Fig. 2A). After the end of the surgical interven-tion, video EEG recording was started and performedfor 3 days, with the patient in a supine position or sit-ting, while awake and asleep, in her recovery room.
At the end of the 3 days of recordings, one of us(S.B.) analyzed the data, using a split-screen video, thusallowing optimal visualization of the patient and theEEG tracing simultaneously. The interictal patternshowed the presence of a multifocal, repetitive �2 Hzspike (±waves) independently in the right and left hemi-spheres and the presence of sporadic spreads of spikeand wave activity to the cortical surface (arrows) withamplitude much larger at the frontopolar (Fp1 andFp2) electrodes (Fig. 2B). The ictal data showed a dis-tinctive pattern starting only from two electrodes inthe left frontal lobe (L3 and L5) and in the first rightfrontal lobe electrode (R1) (Fig. 2, C–E). These findingsindicated the likely presence of multiple, independentmicrodysgenetic loci widespread within the brain cortex,and left no doubt about the widespread origin of the epi-leptogenicity of the cortex and the appropriateness ofcallosotomy as therapy for this patient with a multifocalepilepsy with no apparent cause. To clarify the aetiology
of this patient’s SE-MISF, a brain cortex biopsy wasscheduled to be performed during the second surgery.The parents gave informed consent.
2.3. The second surgery
After 2 weeks, the second surgery was completedthrough the previous reopened craniotomy, with the fol-lowing aims, listed in the sequence in which they werescheduled in pre-surgery planning: (1) to perform a cor-tex biopsy; (2) to remove the cortical electrodes; and (3)to perform callosotomy, preserving the two pericallosalarteries.
Prior to the removal of the two frontal 1- · -8 sub-dural strips, a brain cortex biopsy was performed bythe excision of a 2- · -2 cm surgical specimen of appar-ently normal brain parenchyma. The excision of the sur-gical specimen was made centred at the exact point ofthe third left frontal lobe subdural electrode (L3). Therationale for performing the cortex biopsy in this loca-tion was suggested by the presence of a distinctive pat-tern (fast activity at 10–12 Hz) starting from only twoelectrodes (L3 and L5) in the left frontal lobe and grad-ually decreasing in frequency and increasing in ampli-tude, and propagating over the subdural contiguous

Fig. 2. (A) Skull radiogram showing the position of the subdural electrodes: R1–8, right frontal lobe; L1–8, left frontal lobe. The market white bullet,at L3, indicates the exact point where the surgical specimen was taken to perform the cortex biopsy after the end of the electrocorticography (EcoG).(B–E) Patient’s epileptogenic data when performing EcoG. Recording was done via subdural strip electrodes (R1–5, L1–6) and simultaneously viastandard 10–20 scalp electrodes (Fp1, Fp2, F7, F8, T3, T4, C3, C4). Referential recording was done with reference electrodes on both ears. (B)Interictal EcoG recordings: presence of repetitive �2 Hz multifocal spike discharges (±waves) independently in the right (R1, R2, R3, R5) and left(L1, L2, L3, L5, L6) electrodes (arrows). Note the presence of sporadic, independent spreads of spike and wave activity to the cortical surface(asterisks) with an amplitude much larger at the frontopolar (Fp1 and Fp2) electrodes. (C–E) Ictal EcoG recordings: bioelectrical correlate of seizuresconsisting of fast activity at 10–12 Hz involving non-contiguous regions (i.e., respectively, arrows at subdural electrodes L3 (C), L5 (D), and R1 (E)).Note that this clear subdural ictal activity was poorly detected only by frontopolar (Fp1 and Fp2) cortical surface electrodes, showing only thepresence of irregular delta sharp waves, with a lack of epileptogenic significance.
56 S. Buoni et al. / Brain & Development 30 (2008) 53–58
electrode or over the correspondent contralateral sub-dural electrode (Fig. 2B–E). For ethical reasons, exci-sion of a further surgical specimen at the exact pointof the first right frontal lobe subdural electrode (R1),which also was epileptogenic, was not performed. Afterfixation for 12–24 h in 10% buffered formalin, tissuefragments were embedded in paraffin. Deparaffinizedsections were stained with haematoxylin and eosin(HE), thionin, Luxol fast blue, and Bielschowsky.Immunohistochemistry was also performed using anti-glial fibrillary acid protein (GFAP; Boehringer Mann-heim, Germany) and anti-neurofilament antibodies(SMI 311, Sternberger Monoclonal; 2F11 Monoclonal,DAKO, Glostrup, Denmark).
Following callosotomy and during the post-surgicaloutcome, the patient showed an incomplete left hemipa-resis that partially recovered after 6 months of follow-up, and completely at 12 months of follow-up. At thesame time, as regards epilepsy, the outcome accordingto the Engel score (1 = seizure free and 4 = no improve-
ment) was 2 [3], with a great improvement of quality oflife. Although the surface EEG was substantiallyunchanged, we concluded that our patient experiencedan improved quality of life, with a notable reductionin her seizure frequency and without permanent compli-cations from the interhemispheric separation.
2.4. Neuropathological findings
At the cut surface of the surgical specimen, a fewareas of the brain cortex showed a poor demarcationfrom the underlying white matter. At histological exam-ination, in a few areas of different non-consecutive sec-tions, the brain cortex showed disorganization of thenormal layering (Fig. 3A), and a poorly demarcatedboundary between gray and white matter. In all the cor-tical layers, except for layer V, we observed isolated,abnormally large, although morphologically normal,neurons (Fig. 3C). These neurons exhibited a greaterthan normal neurofilament content, as evidenced with
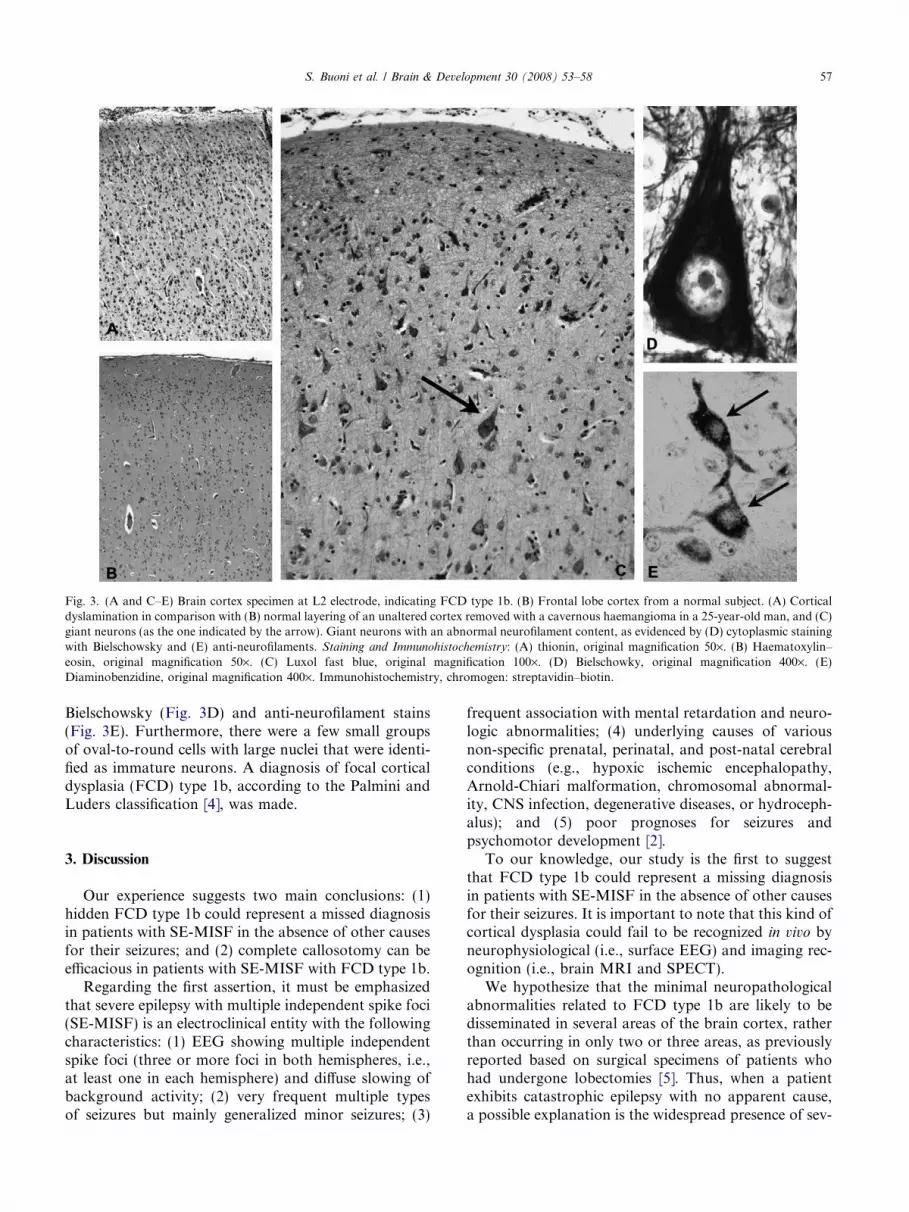
Fig. 3. (A and C–E) Brain cortex specimen at L2 electrode, indicating FCD type 1b. (B) Frontal lobe cortex from a normal subject. (A) Corticaldyslamination in comparison with (B) normal layering of an unaltered cortex removed with a cavernous haemangioma in a 25-year-old man, and (C)giant neurons (as the one indicated by the arrow). Giant neurons with an abnormal neurofilament content, as evidenced by (D) cytoplasmic stainingwith Bielschowsky and (E) anti-neurofilaments. Staining and Immunohistochemistry: (A) thionin, original magnification 50·. (B) Haematoxylin–eosin, original magnification 50·. (C) Luxol fast blue, original magnification 100·. (D) Bielschowky, original magnification 400·. (E)Diaminobenzidine, original magnification 400·. Immunohistochemistry, chromogen: streptavidin–biotin.
S. Buoni et al. / Brain & Development 30 (2008) 53–58 57
Bielschowsky (Fig. 3D) and anti-neurofilament stains(Fig. 3E). Furthermore, there were a few small groupsof oval-to-round cells with large nuclei that were identi-fied as immature neurons. A diagnosis of focal corticaldysplasia (FCD) type 1b, according to the Palmini andLuders classification [4], was made.
3. Discussion
Our experience suggests two main conclusions: (1)hidden FCD type 1b could represent a missed diagnosisin patients with SE-MISF in the absence of other causesfor their seizures; and (2) complete callosotomy can beefficacious in patients with SE-MISF with FCD type 1b.
Regarding the first assertion, it must be emphasizedthat severe epilepsy with multiple independent spike foci(SE-MISF) is an electroclinical entity with the followingcharacteristics: (1) EEG showing multiple independentspike foci (three or more foci in both hemispheres, i.e.,at least one in each hemisphere) and diffuse slowing ofbackground activity; (2) very frequent multiple typesof seizures but mainly generalized minor seizures; (3)
frequent association with mental retardation and neuro-logic abnormalities; (4) underlying causes of variousnon-specific prenatal, perinatal, and post-natal cerebralconditions (e.g., hypoxic ischemic encephalopathy,Arnold-Chiari malformation, chromosomal abnormal-ity, CNS infection, degenerative diseases, or hydroceph-alus); and (5) poor prognoses for seizures andpsychomotor development [2].
To our knowledge, our study is the first to suggestthat FCD type 1b could represent a missing diagnosisin patients with SE-MISF in the absence of other causesfor their seizures. It is important to note that this kind ofcortical dysplasia could fail to be recognized in vivo byneurophysiological (i.e., surface EEG) and imaging rec-ognition (i.e., brain MRI and SPECT).
We hypothesize that the minimal neuropathologicalabnormalities related to FCD type 1b are likely to bedisseminated in several areas of the brain cortex, ratherthan occurring in only two or three areas, as previouslyreported based on surgical specimens of patients whohad undergone lobectomies [5]. Thus, when a patientexhibits catastrophic epilepsy with no apparent cause,a possible explanation is the widespread presence of sev-

58 S. Buoni et al. / Brain & Development 30 (2008) 53–58
eral foci containing microareas of scattering and abnor-mal morphology intermingled within the normal braincortex.
Of course, the validity of our conclusion in the cur-rent case could be limited by the lack of multiple cortexbiopsies demonstrating widespread occurrence of ourneuropathological findings and corresponding to severalloci of independent, multifocal EEG abnormalities.Obviously, for ethical reasons, we could not performmore than one biopsy; however, the limits of resultsfrom the single biopsy specimen could be expanded bythe fact that a similar EcoG abnormal pattern was iden-tified both on the biopsy side and on the contralateralside.
In asserting the efficacy of complete callosotomy inpatients with SE-MISF with hidden FCD type 1b, wemust note that classically, the main indication for callos-otomy remains the occurrence of generalized seizureswith falling (drop attacks). Most recently, corpus callos-otomy has been indicated in patients with refractory idi-opathic generalized epilepsy [6], particularly when theEEG shows intense secondary bilateral synchrony [7].Moreover, some reports have found that epilepticpatients with severe drug-resistant epilepsy due tobihemispheric cortical dysplasias could be suitable can-didates for callosotomy [8]; however, severe mentalretardation may be a contraindication for callosotomy,associated with an incompletely satisfactory outcomefor seizures and the onset of impaired visuo-motorresponses in the affected patient. Based on our experi-
ence, severe mental retardation was not a contraindica-tion for callosotomy: in fact, we found that our patient’squality of life improved, with a strong reduction in thefrequency of her seizures and no permanent complica-tions from the interhemispheric disconnection.
References
[1] Fauser S, Huppertz HJ, Bast T, et al. Clinical characteristics infocal cortical dysplasia: a retrospective evaluation in a series of 120patients. Brain 2006;129:1907–16.
[2] Yamatogi Y, Ohtahara S. Severe epilepsy with multiple indepen-dent spike foci. J Clin Neurophysiol 2003;20:442–8.
[3] Engel J, Rasmussen TB. Outcome with respect to seizures. In:Engel J, editor. Surgical treatment of the epilepsies. NewYork: Raven Press; 1993. p. 609–21.
[4] Palmini A, Luders HO. Classification issues in malformationscaused by abnormalities of cortical development. Neurosurg ClinN Am 2002;13:1–16.
[5] Widdess-Walsh P, Kellinghaus C, Jeha L, Kotagal P, Prayson R,Bingaman W, et al. Electro-clinical and imaging characteristics offocal cortical dysplasia: correlation with pathological subtypes.Epilepsy Res 2005;67:25–33.
[6] Jenssen S, Sperling MR, Tracy JI, Nei M, Joyce L, David G, et al.Corpus callosotomy in refractory idiopathic generalized epilepsy.Seizure 2006;15:621–9.
[7] Cukiert A, Burattini JA, Mariani PP, et al. Extended, one stagecallosal section for treatment of refractory secondarily generalizedepilepsy in patient with Lennox-Gastaut and Lennox-like syn-dromes. Epilepsia 2006;47:371–4.
[8] Pallini R, Aglioti S, Tassinari G, Berlucchi G, Colosimo C, RossiGF. Callosotomy for intractable epilepsy from bihemisphericcortical dysplasias. Acta Neurochir 1995;132:79–86.





























