Embed Size (px)
Citation preview

American Journal of Medical Genetics 120A:157–168 (2003)
Somatic and Germline Mosaicism for a R248CMissense Mutation in FGFR3, Resulting in a SkeletalDysplasia Distinct From Thanatophoric Dysplasia
Valentine J. Hyland,1,2 Stephen P. Robertson,3 Simon Flanagan,1,4 Ravi Savarirayan,3,5
Tony Roscioli,4,6 John Masel,7 Mark Hayes,4 and Ian A. Glass4,6*1Department of Surgery, University of Queensland, Royal Brisbane Hospital, Herston Hospitals Campus,Brisbane, Australia2Molecular Genetics Laboratory, Queensland Health Pathology Service, Royal Brisbane Hospital, Brisbane, Australia3Victorian Clinical Genetics Services, Murdoch Children’s Research Institute, Royal Children’s Hospital,Parkville, Australia4Department of Paediatrics and Child Health, University of Queensland, Royal Children’s Hospital,Herston Hospitals Campus, Brisbane, Australia5Department of Paediatrics, University of Melbourne, Parkville, Victoria, Australia6Queensland Clinical Genetics Service, Herston Hospitals Campus, Brisbane, Australia7Department of Radiology, Royal Children’s Hospital, Brisbane, Australia
In this communication, we report the identi-fication of a mosaic R248C missense muta-tion in the IgII–III linker region of thegene encoding the fibroblast growth factorreceptor-3 (FGFR3), in an individual whomanifests a skeletal dysplasia and epider-mal hyperplasia. By means of DenaturingHigh Performance Liquid Chromatography(DHPLC), we determined that 25% of herlymphocytes are heterozygous for this par-ticular missense mutation in FGFR3, andthat 12.5% of her lymphocyte-derived geno-mic DNA encodes a cysteine residue at thisposition. The proposita has disproportion-ate short stature, radial head dislocation,coxa vara, and bowing of some of the longbones, associated with an S-shaped de-formity of the humerus, accompanied bywidespread acanthosis nigricans in the inte-gument. These features do not match any
previously described skeletal dysplasia.Further, the proposita’s only pregnancyended in the delivery of a fetus manifestinga lethal short-limbed dwarfism with pulmon-ary hypoplasia, strongly suggestive of an un-diagnosed thanatophoric dysplasia. Thesefindings confirm the proposita to be asomatic and germline mosaic for this partic-ular missense mutation in FGFR3. Thus far,all reported FGFR3 R248C mutations haveresulted in thanatophoric dysplasia type I(TDI). � 2003 Wiley-Liss, Inc.
KEY WORDS: fibroblast growth factor re-ceptor; mutation; skeletaldysplasia; acanthosis nigri-cans; epidermal hyperplasia;thanatophoric dysplasia;mosaicism
INTRODUCTION
The four known fibroblast growth factor receptor(FGFR) molecules comprise a family of closely relatedreceptors that share the common structural motifs ofthree extracellular immunoglobulin (Ig)-like bindingdomains, a single transmembrane domain (TMD), and asplit intra-cytoplasmic tyrosine kinase (TK) domain[Burke et al., 1998]. FGFRs appear to play a significantrole in limb and craniofacial development. In particular,tightly regulated FGFR3 activity appears to be integralto achieving normal skeletal growth [Naski and Ornitz,1998]. FGFR3 receptors are expressed in proliferat-ing chondrocytes [Colvin et al., 1996] and activated
Grant sponsor: Royal Children’s Hospital Foundation, Brisbane,Australia; Grant number: 724, 603; Grant sponsor: Institute forCraniofacial Studies, Adelaide, Australia: Collaborative Cranio-facial Research 2000.
*Correspondence to: Dr. Ian A. Glass, M.D., Division ofGenetics and Development, Department of Pediatrics andMedicine, University of Washington, CH-25, CHRMC, 4800 SandPoint Way NE, Seattle, WA 98112. E-mail: [email protected]
Received 5 April 2001; Accepted 10 December 2002
DOI 10.1002/ajmg.a.20012
� 2003 Wiley-Liss, Inc.

receptors appear to modulate bony developmentalpathways [Orr-Urtreger et al., 1993] by restraint ofchondrocyte proliferation and inhibition of endochon-dral bone growth [Deng et al., 1996; Naski et al., 1996;Segev et al., 2000].
Mutations in FGFR3 are responsible for the skeletaldysplasias, achondroplasia (ACH), hypochondroplasia(HCH), and thanatophoric dysplasia types I and II (TDIand TDII) [Rousseau et al., 1994; Bellus et al., 1995;Tavormina et al., 1995], and severe achondroplasiawithdevelopmental delay and acanthosis nigricans (SAD-DAN) [Tavormina et al., 1999], as well as the coronalcraniosynostosis syndrome [Moloney et al., 1997;Muenke et al., 1997], and Crouzon syndrome with acan-thosis nigricans [Meyers et al., 1995]. Causative muta-tions in FGFR3 skeletal dysplasias appear to lead toligand-independent FGFR3 activation [Webster andDonoghue, 1996; d’Avis et al., 1998] and in vitro at least,the severity of the mutant phenotype appears to cor-relate with the extent of autologous receptor activation[Naski et al., 1996].
In this communication,we report the characterizationat the clinical and molecular level of a patient manifest-ing an unusual skeletal dysplasia and epidermalhyperplasia, as the result of mosaicism for an R248Cmissense mutation in FGFR3.
MATERIALS AND METHODS
Molecular Analyses
Genomic DNA was isolated from peripheral lympho-cytes by standardmethods [Miller et al., 1988] andPCRswere performed in 50 ml volumes, containing 100 ng ofextracted genomic DNA, 0.4 mM of each primer, 20 mMTris/HCl pH 8.4, 50 mM KCl, 0.2 mM each dNTP,1.5 mM MgCl2, and 2.5 U of Platinum Taq polymerase(InVitrogen, Mount Waverley, Victoria, Australia). ThePCR products were purified using QIAquick PCR puri-fication kit (Qiagen, Clifton Hills, Victoria, Australia)and then bidirectionally sequenced, using the BigDyeterminator cycle sequencing ready reaction kit (AppliedBiosystems, Scoresby, Victoria, Australia). Restrictionanalysis was carried out using HhaI (New EnglandBiolabs, Arundel, Queensland, Australia).
FGFR2 Analyses
The FGFR2–TMD and adjacent FGFR2 IgIII–TMDregion were PCR amplified, using intronic primers andconditions as previously described [Przylepa et al.,1996]. The FGFR3–TMD, encoded by exon 10 wasPCR amplified, using the intronic primers: 50-GCCAGGCCT CAA CGC CCA TG-30 (forward) and 50-CGG GCAGGC AGC TCA GAA CC-30 (reverse) through 30 cycles,each consisting of denaturation at 948C for 30 sec,annealing at 608C for 30 sec, and extension for 728C for30 sec.
FGFR3 Analyses
The intra-cellular region of FGFR3 was amplified inthree overlapping PCR fragments. A 1991 bp fragmentencompassing most of the intracellular region (exons
12–17 inclusive) was generated, using previouslydescribed primers [Tavormina et al., 1995]. The cyclingparameters employed for this purpose were 30 cycles ofdenaturation at 948C for 30 sec, annealing at 648C for30 sec, andextensionat 728 for 90 sec.Exon11ofFGFR3,was PCR amplified in a 523 bp fragment using theprimers: 50-CAA CCT GCC CCT GCT GAC CC-30
(forward) and 50-AAC TCG CCC ACT CTC AGC CC-30
(reverse) in a two-step PCR protocol, consisting of de-naturation at 948C for 30 sec and annealing/extension at728C for 30 sec for 30 cycles. The 30 exons (17, 18) and thecoding region of exon 19 were amplified in a 1050 bpfragment using the primers: 50-GAA GTG GAT GGCGCC TGA GG-30 (forward) and 50-CAC CAG CAG CAGGGT GGG CTG CTA G-30 (reverse), employing cyclingparameters of denaturationat 948Cfor 30 sec, annealingat 678C for 30 sec, and extension at 728C for 30 sec for30 cycles. TheFGFR3 IgII–IgIII linker region, spanningexon 7 of FGFR3 was PCR amplified using primers andconditions, as previously described [Moloney et al.,1997].
Denaturing High Performance LiquidChromatography Analyses
Denaturing high performance liquid chromatography(DHPLC) was used to fractionate and determine theproportion of wild type and mutant FGFR3 alleles forthe proposita. This analysis was performed on a Trans-genomic WAVE DNA fragment analysis system with aDNASep column, in an oven at 65.98C. Prior to loading,the PCR products were denatured at 948C for 5min, andthen allowed to reanneal gradually over 30 min, from948C to 658C. The PCRproducts were then cooled to 48C,prior to their loading on the column. The PCR productswere eluted from the column using a linear gradientof acetonitrile in 0.1 M triethylamine acetate buffer(TEAA) pH 7, at a constant flow rate of 0.9 ml/min.Mixing of buffer A (0.1 M TEAA at pH 7) and buffer B(25% acetonitrile in 0.1 M TEAA, at pH 7) was under-taken to create the gradient. At 0 min, 8 ml of PCRproduct was loaded. The acetonitrile gradient at 0.5minwas 45% buffer A/55% buffer B, and at 5.0 min wascomposed of 36% buffer A/64% buffer B. At 5.0 min, afinalwashwith buffer Bwas performed. The elutedPCRproducts were then detected using a UV detector at260nm.TheheteroduplexDNAcontaining one strand ofDNA from the R248 allele hybridized to its complemen-tary strand of 248Cwere eluted first, followed by elutionof the homoduplex DNAs, R248 and 248C.
A plot of time (min) versus absorbance (mV) wasgenerated and plots derived from controls and theproposita were overlaid. The peaks were analyzed inorder to estimate the DNA either as amount in mg or as apercentage of the total. These values were determinedby the WAVE instrument and derived from theestimated proportion of the area under the peaksattributable to the heteroduplex and homoduplex con-tributions, respectively.Only thepercentageDNAin theheteroduplex peaks is presented. The percentage DNAin the heteroduplex peak is the area of the heteroduplexpeak, divided by the total area of the heteroduplex and
158 Hyland et al.

the homoduplex peak, expressed as a percentage value.Similarly, the percentage DNA in the homoduplex peakis the area of the homoduplex peak divided by the totalarea of the heteroduplex and the homoduplex peak,expressed as a percentage value.
The level of mosaicism was estimated by mixinggenomicDNA fromawild type control with TDI controls
previously identified to be heterozygous for the R248Cmutation in their lymphocyte extractedDNA.The ratiosof wild type tomutant DNAmixes were 1:1, 2:1, and 3:1.The DNA samples were mixed both before and after thePCR; these details are outlined in Table I. Prior toDHPLC analysis, the PCR reactions were denaturedand allowed to reanneal as described above to maximize
TABLE I. Percentage Heteroduplex Analysis Using DHPLC
Mix ratio, wild type:TD1 control
Typeof mix
%Mosaicism
Observed,mean�SD
Expectedvalue
1:1 PrePCR 50 43.82�0.77 37.51:1 PostPCR 50 39.98�1.52 37.52:1 PrePCR 33.33 32.73�3.13 27.772:1 PostPCR 33.33 29.15�0.16 27.773:1 PrePCR 25 28.23�4.81 21.873:1 PostPCR 25 26.83�3.13 21.87
Both pre- and post-PCRmixings of DNAwere used to ensure PCR products containing the R248 or 248C amplifiedat the same rate.
Fig. 1. A, B: A-P and lateral view of the proposita in hermid 20s (exact age not recorded), showing her disproportionate short stature and rhizomelic limbshortening, pelvic tilt, and asymmetric lower limbs.
A Mosaic R248C Missense Mutation in FGFR3 159

the amount of DNA in the heteroduplex. Both pre- andpost-PCR mixings of DNA were used to ensure PCRproducts containing the R248 or 248C amplified at thesame rate.
RESULTS
Clinical and Radiological Findings
The proposita is the first-born child of unrelatedwhiteAustralian parents, and has one normal male siblingand two normal half siblings. At the age of 3 months,detection of leg length asymmetry led to the discoveryof bilateral congenital hip dislocation which requirednon-operative reduction in the first 2-years of her life.Short-limbed disproportionate stature was suspectedfrom 6months of age and at this time, abnormal flexuralskin hyperpigmentation, overlying the neck region wasfirst noted. By the age of 12 months, skin hyperpigmen-tation and thereafter, skin redundancy in the form ofloose skin and folds extended to the flexor aspects of thelimbs. The proposita’s appearance in her mid 20s isdepicted in Figures 1–3. The abnormal skin extended inits coverage up until the age of 5 years but has remainedrelatively stable over the last 20 years. Laser debride-ment surgery for cosmetic relief has been undertakenperiodically. Delayed neurodevelopmental milestoneswere recorded in childhood, as well as a lesser degree ofeducational and career achievement as compared to hersibs but the proposita graduated from high school, readsand writes fluently and has been employed as a factoryworker.
On review at the age of 47 years, the propositamanifested disproportionate short stature, with herheight being 139.8 cm (�3.7 SDS) and her US/LS ratiobeing 1.3 (Normal female adult ratio �1.0), along withmoderate obesity. Maternal and paternal heights were157 cm (�0.87 SDS) and 165 cm (�1.61 SDS), respec-tively. She has (relative) macrocephaly (OFC¼ 58 cm,þ2 SD) mild frontal bossing and a depressed bridge ofnose,butotherwiseher facialmeasurementsarenormal.Shehas rhizomelic shortening of her limbs, small hands,and brachydactyly. Her middle finger lengths are 6/6.5 cm (<3rd centile), palm lengths are 8/8.5 cm (<3rdcentile), and she has radial deviation of the right 3rdfinger. Asymmetric bilateral fixed flexion deformities ofthe elbows (R¼45 degrees, L¼65 degrees) and bilateralradial headdislocationarepresent.Despite thepartiallydislocated right femoral head, a right upward pelvic tilt,due to gross lower limb asymmetry is present. The leftleg length is 6.5 cm less than the right leg and onlypartial compensation is achieved from a shoe-raiseorthotic. Bilateral genu valgum is present, more severeon the left, associated with a fixed left knee flexiondeformity. The spine is normal with a full range ofmovements. There is widespread hyperpigmentation,redundancy and roughened thickening of the integu-ment, which hangs in loose folds. The skin lesions arepresent on the anterior chest, breast folds, posteriortrunk, the flexor aspects of most of the large joints(notably the axillae and elbows), themalar facial region,posterior to the ears, the neck, the groin creases, and theintracrural region. The legs and feet are relatively
Fig. 2. A-P facial view of the proposita in her mid 20s, depicting herfacial features, including frontal bossing and depressed nasal bridge.Redundant, thickened, and pigmented skin is present in folds on the neck,and the upper thoracic region.
Fig. 3. A-P hand view of the proposita, in her mid 20s, showing herbrachydactyly, with lateral and medial curving of the 2nd and 3rd digits,respectively (most obvious in her right hand).
160 Hyland et al.
spared in comparison to the upper limbs, as are theextensor surfaces (including the dorsum of the handsand forearms), the buttocks, the scalp, and upper facialregions. The proposita wears bifocal lenses, has normalhearing, is currently premenopausal and is otherwisein good health. At the age of 41 years, a mildly elevatedinsulin level in the context of fasting normoglycemia(insulin¼13 IU/L: NR< 10 IU/L) indicated a borderlineinsulin resistance state to be present, but a morepronounced pattern of insulin resistance is now evident(insulin¼88 IU/L, NR< 10 IU/L). The proposita’sappearance at the age of 47 years is shown in Figure 4.
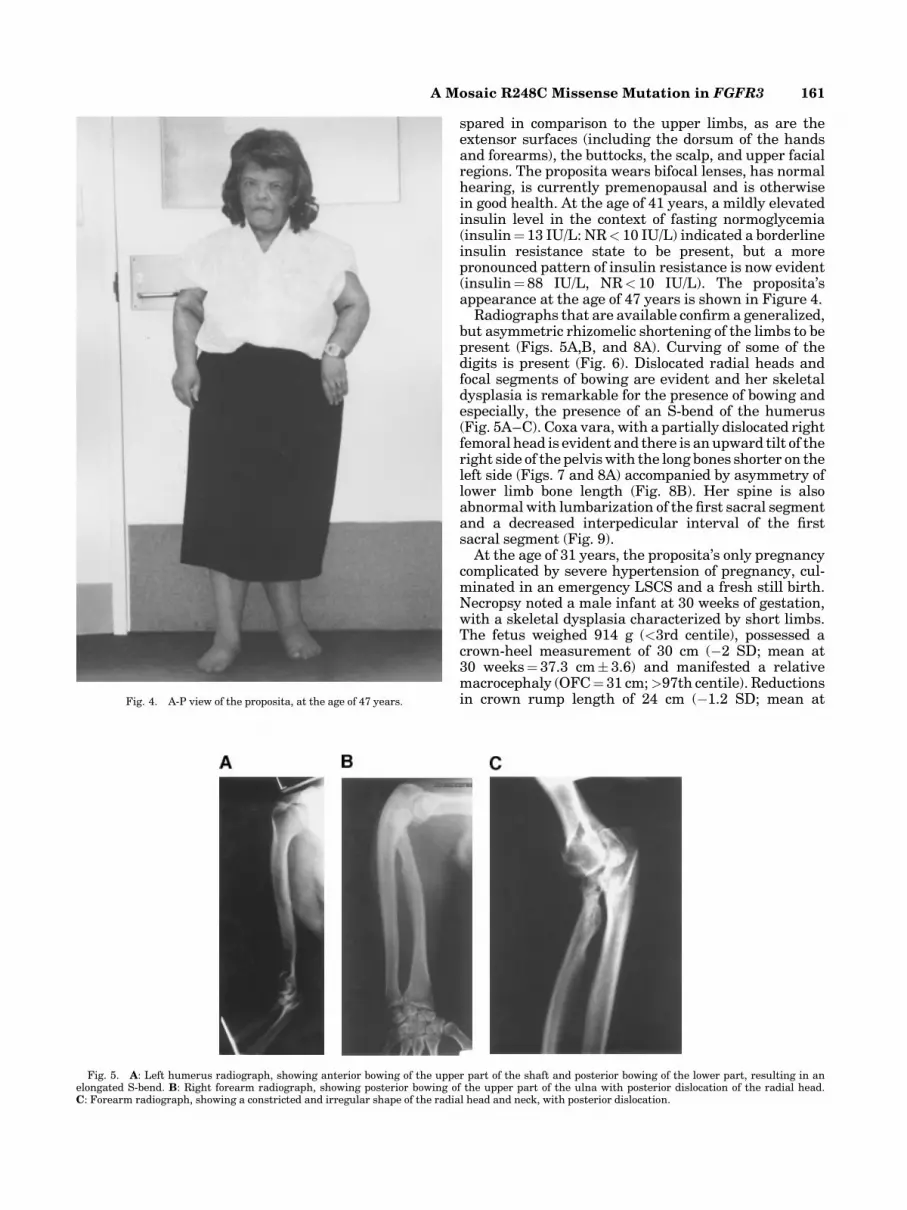
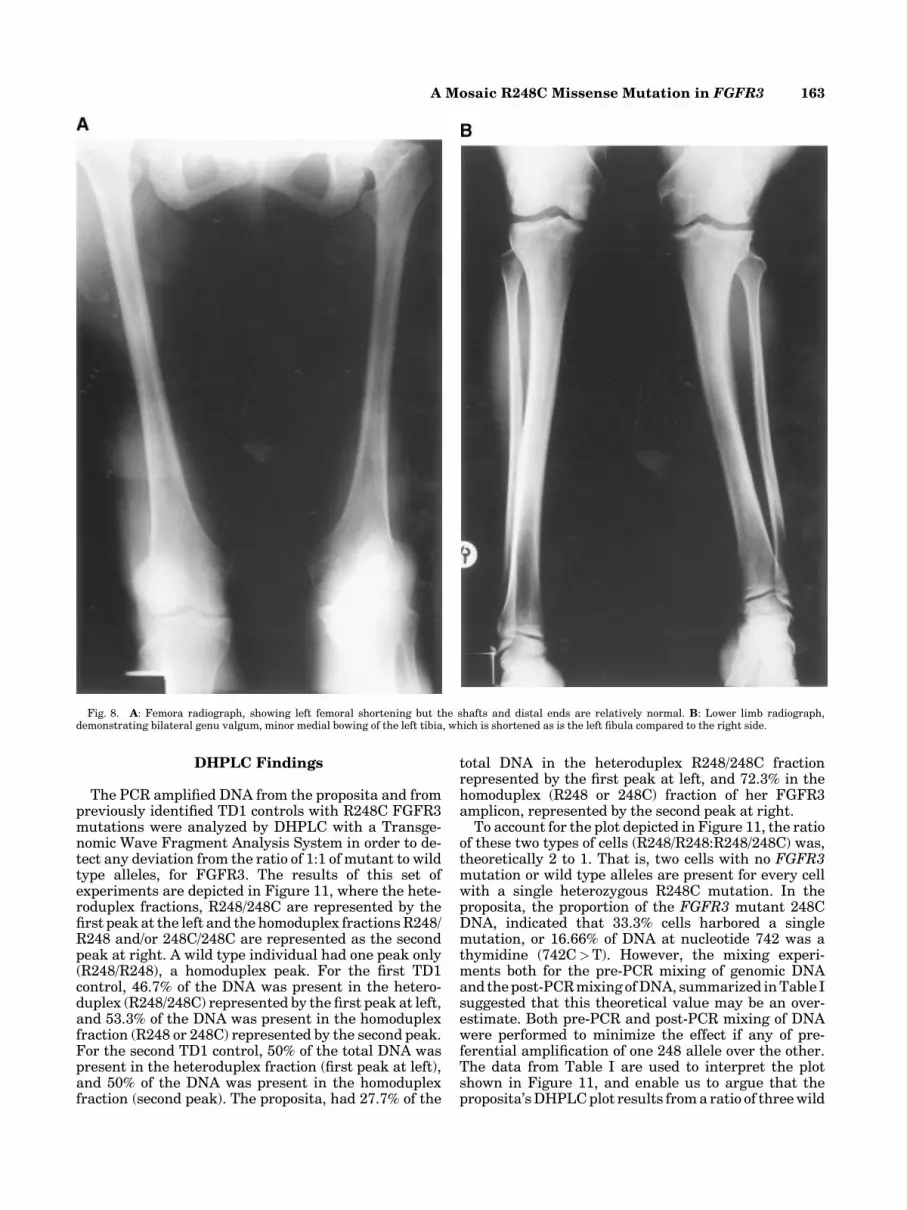
Radiographs that are available confirm a generalized,but asymmetric rhizomelic shortening of the limbs to bepresent (Figs. 5A,B, and 8A). Curving of some of thedigits is present (Fig. 6). Dislocated radial heads andfocal segments of bowing are evident and her skeletaldysplasia is remarkable for the presence of bowing andespecially, the presence of an S-bend of the humerus(Fig. 5A–C). Coxa vara, with a partially dislocated rightfemoral head is evident and there is an upward tilt of theright side of the pelviswith the long bones shorter on theleft side (Figs. 7 and 8A) accompanied by asymmetry oflower limb bone length (Fig. 8B). Her spine is alsoabnormal with lumbarization of the first sacral segmentand a decreased interpedicular interval of the firstsacral segment (Fig. 9).
At the age of 31 years, the proposita’s only pregnancycomplicated by severe hypertension of pregnancy, cul-minated in an emergency LSCS and a fresh still birth.Necropsy noted a male infant at 30 weeks of gestation,with a skeletal dysplasia characterized by short limbs.The fetus weighed 914 g (<3rd centile), possessed acrown-heel measurement of 30 cm (�2 SD; mean at30 weeks¼37.3 cm�3.6) and manifested a relativemacrocephaly (OFC¼31 cm;>97th centile). Reductionsin crown rump length of 24 cm (�1.2 SD; mean at
Fig. 5. A: Left humerus radiograph, showing anterior bowing of the upper part of the shaft and posterior bowing of the lower part, resulting in anelongated S-bend. B: Right forearm radiograph, showing posterior bowing of the upper part of the ulna with posterior dislocation of the radial head.C: Forearm radiograph, showing a constricted and irregular shape of the radial head and neck, with posterior dislocation.
Fig. 4. A-P view of the proposita, at the age of 47 years.
A Mosaic R248C Missense Mutation in FGFR3 161

30 weeks¼ 26.6 cm�2.4), upper limb length of 7 cm(�5 SD; mean at 30 weeks¼ 15.6 cm� 1.7), and lowerlimb length of 8 cm (�4.4 SD; mean at 30 weeks¼13.7 cm�1.3) were recorded. In addition to short limbedshort stature, pulmonary hypoplasia was discovered(combined weight of lungs¼10.5 g, �4 SD, mean at30 weeks¼32.7 g/�5.5). The skeletal films were re-ported to resemble ACH and the long bone histopathol-ogy that was performed was reputed to confirm thisdiagnosis (original reports not available).
Molecular Findings
Analysis of the bidirectional sequencing data forthe exons encoding the FGFR3 TMD, the intracellularportion of FGFR3, aswell as theDNAsequence obtainedfrom the FGFR2 TMD and adjacent FGFR2 IgIII–TMDlinker in the proposita demonstrated the presence ofwild type sequence only. However, analysis of the pro-posita’s FGFR3 exon 7, which encodes the IgII–IgIIIlinker domain of FGFR3, identified a heterozygous C toT changeatnucleotide 742 [Tavormina et al., 1995]. Thistransition results in the substitution of an arginine atresidue 248, by cysteine (Fig. 10A). Restriction analysisof the PCR product confirmed loss of the HhaI site,conferred by this particular point mutation (Fig. 10B).Thereafter,HhaI digestion of the PCR amplifiedDNA ofthe proposita and two other TD1 individuals, hetero-zygous for an R248C mutation was compared. Thisrevealed a diminished intensity/dosage of the mutantfragments between the proposita and the two TD1controls. However, no clear value could be assigned tothe amount of mutant versus normal fragment in theproposita (data not shown).
Fig. 7. Pelvic radiograph, illustrating tilting of the pelvis. The iliac wings are mildly squared in shape. There are bilateral foreshortened femoral necksand a flattened oval shape of the right femoral head, which is partially subluxed. There is localized osteoarthritis in the right upper acetabulum.
Fig. 6. Right hand radiograph demonstrating lateral and medialcurving of the 2nd and 3rd digits, respectively with mild shortening of the5th metacarpal.
162 Hyland et al.
DHPLC Findings
The PCR amplified DNA from the proposita and frompreviously identified TD1 controls with R248C FGFR3mutations were analyzed by DHPLC with a Transge-nomic Wave Fragment Analysis System in order to de-tect any deviation from the ratio of 1:1 of mutant to wildtype alleles, for FGFR3. The results of this set ofexperiments are depicted in Figure 11, where the hete-roduplex fractions, R248/248C are represented by thefirst peak at the left and the homoduplex fractionsR248/R248 and/or 248C/248C are represented as the secondpeak at right. A wild type individual had one peak only(R248/R248), a homoduplex peak. For the first TD1control, 46.7% of the DNA was present in the hetero-duplex (R248/248C) represented by the first peak at left,and 53.3% of the DNA was present in the homoduplexfraction (R248 or 248C) represented by the second peak.For the second TD1 control, 50% of the total DNA waspresent in the heteroduplex fraction (first peak at left),and 50% of the DNA was present in the homoduplexfraction (second peak). The proposita, had 27.7% of the
total DNA in the heteroduplex R248/248C fractionrepresented by the first peak at left, and 72.3% in thehomoduplex (R248 or 248C) fraction of her FGFR3amplicon, represented by the second peak at right.
To account for the plot depicted in Figure 11, the ratioof these two types of cells (R248/R248:R248/248C) was,theoretically 2 to 1. That is, two cells with no FGFR3mutation or wild type alleles are present for every cellwith a single heterozygous R248C mutation. In theproposita, the proportion of the FGFR3 mutant 248CDNA, indicated that 33.3% cells harbored a singlemutation, or 16.66% of DNA at nucleotide 742 was athymidine (742C>T). However, the mixing experi-ments both for the pre-PCR mixing of genomic DNAand thepost-PCRmixing ofDNA, summarized inTable Isuggested that this theoretical value may be an over-estimate. Both pre-PCR and post-PCR mixing of DNAwere performed to minimize the effect if any of pre-ferential amplification of one 248 allele over the other.The data from Table I are used to interpret the plotshown in Figure 11, and enable us to argue that theproposita’sDHPLCplot results froma ratio of threewild
Fig. 8. A: Femora radiograph, showing left femoral shortening but the shafts and distal ends are relatively normal. B: Lower limb radiograph,demonstrating bilateral genu valgum, minor medial bowing of the left tibia, which is shortened as is the left fibula compared to the right side.
A Mosaic R248C Missense Mutation in FGFR3 163

type cells to each cell with a single heterozygousmutantR248C, in FGFR3. This ratio indicates that 25% of lym-phocytes bear a single heterozygous R248C mutation,and that 12.5% of the DNA at nucleotide 742 isthymidine in this tissue.
DISCUSSION
Common features of FGFR3-skeletal dysplasias com-prise varying degrees of disproportionate rhizomelicshort stature, macrocephaly, brachydactyly, and facialanomalies, which our proposita also manifested. How-ever, the presence of additional distinctive skeletalfeatures, notably radial head dislocation and long bonebowing suggested a distinct bone dysplasia was present,
with no match to known skeletal dysplasias. We hy-pothesized that our proposita’s combination of anunusual skeletal dysplasia and acanthosis nigricanswould be due to amutation located in a region ofFGFR3,where mutations responsible for other skeletal dyspla-sias and/or epidermal hyperplasia had been previouslyidentified [Webster and Donoghue, 1997; Burke et al.,1998; Tavormina et al., 1999; Roscioli et al., 2001].
Following, molecular exclusion of either a causativeACH mutation or the Crouzon–acanthosis nigricansmutation from our findings of wild type FGFR3–TMDsequence [Rousseau et al., 1994; Meyers et al., 1995;Webster and Donoghue, 1996], we also determined thatonly wild type sequence for the intracellular portion ofthe FGFR3 molecule was present in the proposita. Thislatter finding excluded mutations of the common HCHcodon (residue 540) [Bellus et al., 1995] and at theSADDAN locus at residue 650 [Tavormina et al., 1999],as well as any other potential novel point mutationslocated in this region of FGFR3. Furthermore, DNAsequence obtained from the FGFR2 region known to beinvolved in a proportion of Beare–Stevenson syndromepatients [Przylepa et al., 1996; Krepelova, 1998], inwhich epidermal hyperplasia (acanthosis nigricans andcutis gyrata) is such a prominent feature, also provedunremarkable.
We then reasoned that the IgII–IgIII linker region ofFGFR3 could potentially harbor a causative mutationleading to epidermal hyperplasia [Roscioli et al., 2001].Thereafter, DNA sequencing identified a heterozygousC to T transition at nucleotide 742 in FGFR3. In thisparticular point mutation, the non-conservative sub-stitution of a charged polar amino acid (arginine) with aneutral polar amino acid (cysteine), promotes disulfidecross-linking resulting in ligand independent dimeriza-tion and constitutive activation of the receptor [Naskiet al., 1996; Burke et al., 1998]. The R248C mutation inFGFR3 accounts for about 50% of all TD cases and 60%of TDI cases [Wilcox et al., 1998]. Until very recently,documentation of phenotypes other than TDI as theresult of the R248C missense mutation in FGFR3 werelacking [Tavormina et al., 1995; Wilcox et al., 1998].However, two newly reported cases with ACH werefound to be due to R248C and N540K missensemutations in FGFR3 [Camera et al., 2001]. Although,evaluation of mosacism was incomplete for the TDImutation in that report, when taken with our casesuggests that this particular phenomenon may be morecommon than previously thought.
Acanthosis nigricans has also observed in the skin oflong-term survivors of TDI, due to the same FGFR3R248Cmutation [Baker et al., 1997]. This suggests thatepidermal hyperplasia may be an invariable outcome ofthis particular FGFR3 mutation, given sufficient long-evity. Although, we have also documented an age andobesity-related hyperinsulinism in our patient the
Fig. 9. A-P lumbar spine radiograph, which shows a localized scoliosiswith a left convexity in the lumbosacral region, associated with a leftdownward tilt of the pelvis. The interpedicular distance of S1 is reduced, andlumbarization of the 1st segment of the sacrum is evident.
Fig. 10. A: FGFR3 Ig II–IgIII linker (exon 7) sequencing results. The proposita’s forward and reverse sequence is depicted in the top and intermediatepanels, respectively. This demonstratesaheterozygousC toT transitionatnucleotide 742, indicated by the ‘‘N’’. This results in the substitution of anarginineat residue 248 with cysteine. ‘‘Wild type’’ reverse sequence from a normal control is depicted in the lower panel C. B: Restriction analysis of FGFR3.HhaIdigestion ofFGFR3 exon 7 amplicon. This demonstrates heterozygous abolition of this restriction site, conferred by the C to T transition at nucleotide 742 ofFGFR3 in the proposita. An uncutmutant band of 337 bp is seen in the proposita, which is not present in the DNA of the normal control. TheDNAmarker ispUC19 digested with HpaII. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
164 Hyland et al.

Fig. 10.
A Mosaic R248C Missense Mutation in FGFR3 165

etiology in other cases of FGFR-related acanthosisnigricans appears unrelated to hyperinsulinism. More-over and as is documented in our proposita, FGFR-related acanthosis nigricans appears to have an earlyage of onset [Beare et al., 1969; Rendon et al., 1989;Meyers et al., 1995; Bellus et al., 1999; Roscioli et al.,2001]. Thus, in our patient the hyperinsulinism iseither an independent adult onset event or possibly, atrue association of FGFR–acanthosis nigricans notpreviously observed due to the lack of long termfollow up.
It seems highly likely that the lethal skeletal dy-splasia and pulmonary hypoplasia documented in ourpatient’s 30-week fetus,wasdue to the inheritance of theR248Cmissense inFGFR3 resulting inTDI, rather thanthe assigned diagnosis of ACH. Unfortunately, we didnot have the opportunity to absolutely confirm this,
because of the unavailability of original radiographs,photographs, and tissues. The absence of the TDI phe-notype in the proposita raises questions as to the basis ofthe wide divergence in phenotype observed betweenthese two individuals. This could potentially be due todifferent levels ofmosaicism for an identicalmutation inFGFR3. Thus, reduced levels of expression from themutant allele in the proposita may account for herattenuated phenotype as the result of somatic mosai-cism for the R248C mutation. As the result of hergermline mosaicism, any offspring could thus be non-mosaic for the R248C mutation and consequently beseverely affected, manifesting as TD1. This parti-cular mechanism has been recently demonstrated tooccur in an unexpected sibling recurrence of ACH, bornto a clinically unaffected mother demonstrated tomanifest the causative mutation (G380R) in FGFR3, in
Fig. 11. DHPLC analysis of FGFR3, exon 7 PCR amplicon. Plots of time(min) versus absorbance at 260nm (mV) are depicted. Plot 1 (red tracing) is anormal wild type individual with a single peak of a homoduplex fraction ofDNA only (R248 sequence only). Plot 2 and 3 (green and blue tracings) arefrom the DNA of two unrelated individuals with TD1 due to a heterozygousR248C mutation in FGFR3. Both show two peaks, demonstrating theirapproximately equal fractions or comparable amounts of DNA, at close to50% for the R248/248C heteroduplex (the first peak at left), and R248 with
248C homoduplexes (the second peak at right). Plot 4 (black tracing) fromthe DNA of the proposita also manifests two peaks. The proposita’s peaksconfirm the deviation from the expected 1:1 ratio seen in the TD1 controls,with 27.8% of DNA appearing in the R248/248C heteroduplex fraction (thefirst peak at left) and 72.2% in the R248/R248 or 248C/248C homoduplexfraction (the second peak at right). [Color figure can be viewed in the onlineissue, which is available at www.interscience.wiley.com.]
166 Hyland et al.

approximately 30% of her peripheral lymphocytes[Henderson et al., 2000].
By means of DHPLC, the proportions of homoduplexand heteroduplex DNA were able to be estimated. Avalue of 50% of DNA concentration in the heteroduplexfraction was theoretically equivalent to zero level ofsomatic mosaicism, with every cell containing one copyof a wild type R248 allele and one copy of the mutant248C allele. A value of 37.5% of DNA concentration inthe heteroduplex fraction was theoretically equivalentto 50% somatic mosaicism, i.e., for every two cells, onecell bears no mutation and the other type of cell isheterozygous having one copy of the wild type R248allele and one copy of the mutant 248C allele. A value of27.8% of DNA concentration in the heteroduplex frac-tion was theoretically equivalent to 33.3% somaticmosaicism, i.e., for every three cells, two cells have nomutation and the third cell is heterozygous for the 248Callele. A value of 21.9% of DNA concentration in theheteroduplex fraction was theoretically equivalent to25% somatic mosaicism, i.e., for every four cells, threecells have no mutation and another cell is heterozygousfor the 248C allele. In practice, the theoretical valueswere less than those observed in themixing experimentsperformed.
Using this approach, we have confirmed that theproposita indeed had somatic mosaicism for the FGFR3R248C missense mutation in peripheral lymphocytes.The majority of cells from this tissue harbor normalDNA sequence at this particular codon in FGFR3, but aproportion of cells are heterozygotes, containing boththe R248 allele and the 248C alleles. To account for theplot seen in Figure 11, using data from Table I, the ratioof these two types of cells is proposed to be 3 to 1, and theoverall amount of the mutant 248C DNA to total DNAwas estimated to be 12.5%, in this tissue.
In conclusion, our proposita manifests an unusualskeletal dysplasia resulting from a FGFR3 R248Cmissense mutation thus far only reported in TDI,typically a lethal short limbed dwarfism. We concludethat the milder phenotype observed (notably in theskeleton) in our proposita that bears some resemblanceto ACH, is the result of somatic mosaicism, althoughdirect confirmation awaits analysis in that tissue.Furthermore, the retrospective diagnosis of TD in herfetus confirms that the proposita harbors an identicalmutation in her germline, thereby establishing her as asomatic and germline mosaic for the R248C missensemutation in FGFR3. Clearly, other individuals withdisproportionate short stature and/or a resemblance toACH, associatedwith coxa vara, radial head dislocation,and bowing or an S-like deformity of the long bones,also warrant exclusion of a (mosaic) FGFR3 mutation,especially if accompanied by signs of early onsetepidermal hyperplasia, a helpful indicator of the pre-sence of an FGFR mutation.
ACKNOWLEDGMENTS
The authors thank the willing participation andcooperation of the patient and her family and AngelaSharp and Nicola Marks for technical assistance with
the DHPLC, and in addition, Desiree du Sart, MurdochChildren’s Research Institute, and Andrew Wilkie,Oxford University for critical discussions.
REFERENCES
Baker K, Olson D, Harding C, Pauli R. 1997. Long-term survival in typicalthanatophoric dysplasia type 1. Am J Med Genet 70:427–436.
Beare J, Dodge J, Nevin N. 1969. Cutis gyratum, acanthosis nigricans andother congenital anomalies: A new syndrome. Br J Dermatol 81:241–247.
Bellus G, McIntosh I, Smith E, Aylsworth A, Kaitila I, Horton W,Greenhaw F, Hecht J, Francomano C. 1995. A recurrent mutation inthe tyrosine kinase domain of fibroblast growth factor receptor 3 causeshypochondroplasia. Nat Genet 10:357–359.
BellusG,BamshadM, PrzylepaK,Dorst J, LeeR,HurkoO, JabsE,CurryC,Wilcox W, Lachman R, Rimoin D, Francomano C. 1999. Severeachondroplasia with developmental delay and acanthosis nigricans(SADDAN): Phenotypic analysis of a new skeletal dysplasia caused by aLys650Met mutation in fibroblast growth factor receptor 3. Am J MedGenet 85:53–65.
Burke D, Wilkes D, Blundell T, Malcolm S. 1998. Fibroblast growth factorreceptors: Lessons from the genes. Trends Biol Sci 23:59–62.
Camera G, Baldi M, Strisciuglio G, Concolino D, Mastroiacovo P,Baffico M. 2001. Occurrence of thantophoric dysplasia type I (R248C)and hypochondroplasia (N540K) mutations in two patients withachondoplasia. Am J Med Genet 104:277–281.
Colvin J, Bohne B, Harding G, McEwen D, Ornitz D. 1996. Skeletalovergrowth and deafness in mice lacking fibroblast growth factorreceptor 3. Nat Genet 12:390–397.
d’Avis P, Robertson S,Meyer A, BardwellW,WebsterM,DonoghueD. 1998.Constitutive activation of fibroblast growth factor receptor 3 by mu-tations responsible for the lethal skeletal dysplasia thantophoricdysplasia type I. Cell Growth Differ 9:71–78.
Deng C, Wynshaw-Boris A, Zhou F, Kuo A, Leder P. 1996. Fibroblastgrowth factor receptor 3 is a negative regulator of bone growth. Cell84:911–921.
Henderson S, Sillence D, Loughlin J, Bennetts B, Sykes B. 2000.Germline and somatic mosacism in achondroplasia. J Med Genet 37:956–958.
Krepelova A. 1998. FGFR2 gene mutation (Tyr375Cys) in a new case ofBeare–Stevenson syndrome. Am J Med Genet 76:362–364.
Meyers GA, Orlow SJ, Munro IR, Przylepa KA, Jabs EW. 1995. Fibroblastgrowth factor receptor 3 (FGFR3) transmembrane mutation in Crouzonsyndrome with acanthosis nigricans. Nat Genet 11:462–464.
Miller SA, Dykes DD, Polesky HF. 1988. A simple salting out procedurefor extracting DNA from human nucleated cells. Nucleic Acids Res16:1215.
Moloney D, Wall S, Ashworth G, Oldridge M, Glass I, Francomano C,Muenke M, Wilkie A. 1997. Prevalence of Pro240Arg mutation of fi-broblast growth factor receptor 3 in coronal craniosynostosis. Lancet349:1059–1062.
Muenke M, Gripp K, McDonald-McGinn D, Gaudenz K, Whitaker L,Bartlett S, Markowitz R, Robin N, Nwokoro N, Mulvihill JJ, Losken H,Mulliken J,Guttmacher A,WilroyR, Clarke L,HollwayG,AdesL,HaanE, Mulley J, Cohen M, Bellus G, Francomano C, Moloney D, Wall S,Wilkie A, Zackai E. 1997. A unique point mutation in the fibroblastgrowth factor receptor 3 gene (FGFR3) defines a new craniosynostosissyndrome. Am J Hum Genet 60:555–564.
Naski M, Ornitz D. 1998. FGF signalling in skeletal development. FrontBiosci 3:781–794.
Naski M, Wang Q, Xu J, Ornitz D. 1996. Graded activation of fibroblastgrowth factor receptor 3 by mutations causing achondroplasia andthantophoric dysplasia. Nat Genet 13:233–237.
Orr-Urtreger A, Bedford M, Burakova T, Arman E, Zimmer Y, Yayon A,Givol D, Lonai P. 1993. Developmental localization of the splicingalternatives of fibroblast growth factor receptor-2 (FGFR2). Dev Biol158:475–486.
Przylepa K, PaznekasW, ZhangM, GolabiM, BiasW, BamshadM, Carey J,Hall B, Stevenson R, Orlow S, CohenM, Jabs E. 1996. Fibroblast growthfactor receptor 2 mutations in Beare–Stevenson cutis gyrata syndrome.Nat Genet 13:492–494.
A Mosaic R248C Missense Mutation in FGFR3 167

Rendon M, Cruz P, Sontheimer R, Bergstresser P. 1989. Acanthosisnigricans: A cutaneous marker of tissue resistance to insulin. J AmAcad Dermatol 31:461–469.
Roscioli T, Flanagan S, Mortimore R, Kumar P, Weedon D, Masel J,Lewandoski R, Hyland V, Glass I. 2001. Premature calvarial synostosisand epidermal hyperplasia (Beare Stevenson-like anomalies) resultingfrom a P250Rmissensemutation in the gene encoding fibroblast growthfactor receptor 3. Am J Med Genet 101:1–9.
Rousseau F,Bonaventure J, Legeai-Mallet L, Pelet A, Rozet J,MaroteauxP,Le Merrer M, Munnich A. 1994. Mutations in the gene encoding fibro-blast growth factor receptor-3 in achondroplasia. Nature 371:252–254.
Segev O, Chumakov I, Nevo Z, Givo D, Madar-Shapiro L, Sheinin Y,Weinreb M, Yayon A. 2000. Restrained chondrocyte proliferation andmaturationwith abnormal growth plate vascularisation and ossificationin human FGFR-3 G380R transgenic mice. Hum Mol Genet 9:249–258.
Tavormina PL, Shiang R, Thompson LM, Zhu Y, Wilkin DJ,Lachman RS, Wilcox WR, Rimoin DL, Cohn DH, Wasmuth JJ.
1995. Thanatophoric dysplasia (types I and II) caused by dis-tinct mutations in fibroblast growth factor receptor 3. Nat Genet 9:321–328.
Tavormina P, Bellus G, Webster M, Bamshad M, Fraley A, McIntosh I,Szabo J, Jiang W, Jabs E, Wilcox W, Wasmuth J, Donoghue D,Thompson L, Francomano C. 1999. A novel skeletal dysplasia withdevelopmental delay and acanthosis nigricans is caused by a Lys650Metmutation in the fibroblast growth factor 3 gene. Am J Hum Genet 64:722–731.
Webster M, Donoghue D. 1996. Constitutive activation of fibroblast growthfactor receptor 3 by the transmembrane domain point mutation foundin achondroplasia. EMBO J 15:520–527.
Webster MK, Donoghue DJ. 1997. FGFR activation in skeletal disorders;too much of a good thing. Trend Genet 13:178–182.
Wilcox W, Tavormina P, Krakow D, Kitoh H, Lachman R, Wasmuth J,Thompson L, Rimoin D. 1998. Molecular, radiological, and histo-pathological correlations in thantophoric dysplasia. Am J Med Genet78:274–281.
168 Hyland et al.





























