Embed Size (px)
Citation preview

RESEARCH ARTICLE
Zebrafish models of skeletal dysplasia induced by cholesterolbiosynthesis deficiencyRebecca A. Anderson1, Kevin T. Schwalbach2, Stephanie R. Mui2, Elizabeth E. LeClair4,Jolanta M. Topczewska2 and Jacek Topczewski1,2,3,*
ABSTRACTHuman disorders of the post-squalene cholesterol biosynthesispathway frequently result in skeletal abnormalities, yet ourunderstanding of the mechanisms involved is limited. In a forward-genetic approach, we have found that a late-onset skeletal mutant,named kolibernu7, is the result of a cis-acting regulatorymutation leadingto loss of methylsterol monooxygenase 1 (msmo1) expression withinpre-hypertrophic chondrocytes. Generated msmo1nu81 knockdownmutation resulted in lethality at larval stage. We demonstrated thatthis is a result of both cholesterol deprivation and sterol intermediateaccumulation by creating a mutation eliminating activity of Lanosterolsynthase (Lss). Our results indicate that double lssnu60;msmo1nu81 andsingle lssnu60 mutants survive significantly longer than msmo1nu81
homozygotes. Liver-specific restoration of either Msmo1 or Lss incorresponding mutant backgrounds suppresses larval lethality.Rescued mutants develop dramatic skeletal abnormalities, with a lossof Msmo1 activity resulting in amore-severe patterning defect of a near-complete loss of hypertrophic chondrocytes marked by col10a1aexpression. Our analysis suggests that hypertrophic chondrocytesdepend on endogenous cholesterol synthesis, and blocking C4demethylation exacerbates the cholesterol deficiency phenotype. Ourfindings offer new insight into the genetic control of bone developmentand provide new zebrafish models for human disorders of thecholesterol biosynthesis pathway.
KEYWORDS: Cholesterol, Chondrodysplasia punctata, Lss,Msmo1,Skeletal dysplasia, Zebrafish
INTRODUCTIONThe cholesterol biosynthesis pathway is one of the most complexbiochemical pathways and consists of over 30 enzymatic steps(A�cimovi�c et al., 2016; Sharpe and Brown, 2013). It can be dividedinto two major sections: the pre-squalene pathway, which isinvolved in isoprenoid synthesis and contains the rate-limitingenzyme of cholesterol biosynthesis, HMG-CoA reductase, and the
post-squalene pathway, which is devoted to sterol synthesis(A�cimovi�c et al., 2016; Sharpe and Brown, 2013) (Fig. S1). Atleast ten human disorders result from mutations in post-squalenepathway genes (Herman and Kratz, 2012; Porter and Herman, 2011;Rossi et al., 2015). These disorders are characterized by intellectualdisabilities, behavioral problems, heart and genital malformations,eye defects, skin conditions and skeletal deformities (Jira, 2013;Porter and Herman, 2011). Skeletogenesis defects vary frommild tosevere, and are seen in endochondral bones, which develop throughmineralization of cartilage, and intramembranous bones, whichdevelop directly from condensed mesenchymal cells (Kronenberg,2003; Ornitz and Marie, 2002).
In a subset of the post-squalene pathway disorders,chondrodysplasia punctata (CDP) is observed (Jurkiewicz et al.,2013). This rare skeletal phenotype is characterized by the observationof dot-like calcium deposits, or punctate, within cartilage onradiographs (Irving et al., 2008; Jurkiewicz et al., 2013; Lykissas,2013). A result of ectopic calcification, the punctate is most oftenobserved at the end of long bones and within cartilage around jointsand the vertebral column (Jurkiewicz et al., 2013). Our understandingof how mutations in the post-squalene cholesterol biosynthesispathway lead to abnormal skeletogenesis and CDP is limited.
Here, we show that the loss of methylsterol monooxygenase 1(msmo1) expression within pre-hypertrophic chondrocytes, owingto a cis-acting kolibernu7 (kolnu7) regulatory mutation, results indefective chondrocyte differentiation, irregular bone formation andectopic ossification within growth plates. We show that loss ofMsmo1, and that of Lanosterol synthase (Lss), is lethal in zebrafishlarvae. Restoration of hepatic Msmo1 or Lss activity is sufficient forpost-larval survival of corresponding mutants. Transgenicallyrescued mutants develop strong skeletal defects similar to thoseseen in kolnu7. We show that Msmo1 and Lss activity is needed forproper chondrocyte differentiation, especially in the formation ofhypertrophic chondrocytes. Our results suggest that the observedphenotypes are not a result of loss of Indian hedgehog (Ihh)signaling activity within growth plates. The msmo1nu81 mutantphenotype is likely to be the combined result of cholesteroldepletion and toxic intermediate accumulation as the lssnu60mutant,with blocked sterol synthesis, is epistatic to msmo1 and has a less-severe phenotype.
RESULTSThe kolnu7mutation results in late-onset skeletal deformitiesWe identified a novel mutation, referred to as kolnu7, based on thereduced body length and small head size of adult homozygote fish(Fig. 1A). The kolnu7 phenotype is first detectable by grossmorphological examination at ∼6 weeks of development and themutation is fully recessive. The kolnu7 homozygote mutants areviable and fertile. Bone and cartilage staining of adult kolnu7 usingAlizarin Red and Alcian Blue, respectively (Fig. 1B), reveals
Handling Editor: E. Elizabeth PattonReceived 25 September 2019; Accepted 27 April 2020
1Department of Pediatrics, Northwestern University Feinberg School of Medicine,Chicago, IL 60611, USA. 2Developmental Biology Program, Stanley ManneChildren’s Research Institute, Ann and Robert H. Lurie Children’s Hospital ofChicago, Chicago, IL 60611, USA. 3Department of Biochemistry and MolecularBiology, Medical University of Lublin, Lublin 20-093, Poland. 4Department ofBiological Sciences, DePaul University, Chicago, IL 60614, USA.
*Author for correspondence ( [email protected])
R.A.A., 0000-0001-9065-235X; K.T.S., 0000-0001-9136-0586; E.E.L., 0000-0002-4935-4577; J.M.T., 0000-0003-1876-5039; J.T., 0000-0001-6023-9556
This is an Open Access article distributed under the terms of the Creative Commons AttributionLicense (https://creativecommons.org/licenses/by/4.0), which permits unrestricted use,distribution and reproduction in any medium provided that the original work is properly attributed.
1
© 2020. Published by The Company of Biologists Ltd | Disease Models & Mechanisms (2020) 13, dmm042549. doi:10.1242/dmm.042549
Disea
seModels&Mechan
isms

defects in both endochondral and intramembranous bones (Bird andMabee, 2003; Cubbage and Mabee, 1996; Parichy et al., 2009).Specifically, the endochondral bones of adult kolnu7 mutants havedramatically reduced or missing growth plates. Interestingly, onlythe intramembranous bones located next to cartilaginous elementsappear affected. For example, the dentary bone, which developsaround Meckel’s cartilage (Cubbage and Mabee, 1996), issignificantly shortened in mutants relative to wild-type siblings(Fig. S2B,D) and the cartilage itself contains ectopic ossifications(Fig. S2D,D′). Similarly, the observable compressed bodyphenotype of kolnu7 (Fig. 1A,B) is a result of partial or completevertebral fusions (Fig. 1B; Fig. S3D,D′). In zebrafish, the vertebraedevelop through direct mineralization around a relatively largecartilaginous notochord (Pogoda et al., 2018). In contrast, isolatedintramembranous bones, such as the fin rays and operculum, appearrelatively unaffected in kolnu7 (Fig. 1B; Fig. S2C). Consistent withthe late-onset phenotype of kolnu7, initial cartilage formation andpatterning is normal (Fig. S4B), ossification is not prematurelyinitiated (Fig. 1C-F) and the initial patterning of vertebra centraossification is normal (Fig. S3A,B).Using positional cloning, we mapped the kolnu7 mutation to a
critical region located on chromosome 1 flanked by markerssegregating in either one or two out of 1844 meioses (Fig. 1G). Thecritical region physical distance is ∼457 kb, while the genetic
distance corresponds to 0.16 cM. As the average physical distancecorresponds to ∼650 Mb per 1 cM (Talbot and Schier, 1999), thisresult indicates a greater than four times reduction in recombinationfrequency in this region. The critical region overlaps with threeknown genes (Fig. 1G). Coding sequence analysis of genes withinthe critical region, as well as those genes neighboring the criticalregion, did not reveal any changes in protein-coding sequencebetween wild-type and kolnu7 siblings (data not shown). UsingCRISPR/Cas9 genome editing, we mutagenized all three knowngenes within the critical region and found that each fullycomplemented the kolnu7 mutation (Table 1; Fig. S5). Theseresults suggested that the kolnu7 mutation disrupts a regulatorysequence located within the critical region.
The kolnu7 mutation negatively regulates expression ofmsmo1, a gene involved in cholesterol biosynthesisTo test the prediction that the kolnu7 mutation disrupts a cis-actingregulatory element important for bone development, we assessedexpression of the genes located in a ∼1.2Mb region containing thekolnu7 locus (Fig. 1G,H). For quantitative real-time PCR (RT-PCR)analysis, we extracted total RNA from the hypural complexes ofwild-type and kolnu7 mutant fish. The hypural complex, composedof endochondral bones, is severely affected in kolnu7 mutants. Forour analysis, we selected fish that were ∼3 months old,corresponding to a standard length (SL) of ∼18 mm in wild-typesiblings. Of the 11 genes analyzed, expression of methylsterolmonooxygenase 1 (msmo1) was more than 10-fold downregulated(Fig. 1H). Similarly, using RNA isolated from the cranial vault,
Fig. 1. Late-onset skeletal defects observed in the kolibernu7 (kolnu7)mutant are the result of downregulation of msmo1 expression.(A) Compared to wild-type (wt) siblings (top), adult kolnu7 mutants display areduced body length and small head size. wt n=300, kolnu7 n=300. (B) Whole-mount skeletal preparations reveal gross malformations and hyperossificationthroughout the adult kolnu7 craniofacial and axial skeleton after Alcian Blue(cartilage) and Alizarin Red (ossified bone andmineralized tissues) staining. wtn=100, kolnu7 n=100. (C-F) Early larval mutants do not display patterningdefects or premature ossification. Whole-mount Alizarin Red staining of 4.7mm (∼8 dpf) wt (C,E) and kolnu7. (D,F). Ventral view (C,D) and lateral view(E,F). wt n=3, kolnu7 n=4. (G) Positional cloning reveals that the kolnu7 locus islocated to the ∼457 kb critical region flanked by polymorphic markers withone or two recombinants out of 1844 meioses, corresponding to a geneticdistance of 0.16 cM. (H) Screen of gene expression using quantitative RT-PCRfromRNA extracted from hypural complex of∼18mmSL kolnu7 and wt siblings.wt n=2, kolnu7 =2. Onlymsmo1 level was significantly different out of 11 testedgenes, located in the ∼1.2 Mb region encompassing the kolnu7 locus. Initialscreen results: grhprb not detected (ND); uba6mean difference 0.64, s.d. 0.36;abpp2 −1.62, s.d. 1.00; mettl14 1.16, s.d. 0.68; prss12 1.20, s.d. 0.72; ndst31.51, s.d. 1.50; ugt8 −1.03, s.d. 0.07; spock3 ND; tll1 0.67, s.d. 0.27; cpe 0.90,s.d. 0.42; msmo1 −10.07, s.d. 4.32. Confirmation test of msmo1 expression(msmo1 −11.47, s.d. 8.49; P=0.0012), wt n=3, kolnu7 =5. Three technicalreplicates were included for all assays. Gene expression was normalized to thereference gene eefla1. Fold change was calculated using Livak method (Livakand Schmittgen, 2001). P-value calculated using unpaired Student’s t-test ondCt values. (I) The msmo1nu81 mutant allele is not able to complement thekolnu7 mutation. Adult kolnu7/+:msmolnu81/+ transheterozygotes phenocopy thekolnu7mutant. wt n=100, kolnu7/+:msmolnu81/+ n=100. (J) Whole-mount skeletalpreparations reveal gross malformations throughout the kolnu7/+:msmolnu81/+
craniofacial and axial skeleton, similar to those observed in kolnu7. wt n=10,kolnu7/+:msmo1nu81/+ n=10. (K) The msmo1nu81 allele is the result of a 37 bpinsertion, allowing for allele-specific expression analysis between msmo1nu81
and kolnu7. PAM, protospacer adjacent motif (underlined in red). (L) Strongdownregulation of the kolnu7-linked allele (asterisks) in kolnu7/+:msmo1nu81/+
compared to the wt allele in msmo1nu81/+ suggests that the kolnu7 mutation iscis-acting. kolnu7/+:msmo1nu81/+ n=3, msmo1nu81/+ n=3. The top band is aheterodimer of wt and mutant strands.
2
RESEARCH ARTICLE Disease Models & Mechanisms (2020) 13, dmm042549. doi:10.1242/dmm.042549
Disea
seModels&Mechan
isms

which consists of both intramembranous and endochondral bones(Topczewska et al., 2016), we found a 3.8-fold downregulation ofmsmo1 in kolnu7. Finally, a 2-fold downregulation of msmo1expression was detected in total RNA isolated from evisceratedtrunks of kolnu7 mutants. These results indicate that the kolnu7
mutation induces a downregulation of msmo1 expression,particularly in bone-enriched tissues.The Msmo1 enzyme catalyzes the removal of a methyl group
from C4-methlysterols during the post-squalene cholesterolbiosynthesis pathway (Sharpe and Brown, 2013) (Fig. S1). Tocharacterize msmo1 expression in early zebrafish development, weused whole-mount in situ hybridization. Expression was firstdetected during early stages of somitogenesis in the yolk syncytiallayer (YSL) (Fig. 2D), an extraembryonic cell that expresses severalmarkers of the primitive liver (Li et al., 2007; Mudumana et al.,2004). Expression continued in the YSL at 3 days post-fertilization(dpf) (Fig. 2E), with a new domain appearing in the newly formedliver at 4 dpf and 5 dpf (Fig. 2F,F′). Consistent with the late onset ofthe kolnu7 early phenotype, msmo1 expression was not observed inskeletal elements during the first 5 days of development (Fig. 2E,F).Furthermore, therewas no difference in the in situ signals formsmo1expression between wild-type and kolnu7 siblings during the first 5days of development (data not shown).
We next characterized msmo1 expression during juveniledevelopment by RNAscope in situ hybridization. In ∼2-month-old fish, SL ∼15 mm, msmo1 expression was found withincartilaginous elements such as Meckel’s cartilage, but not withinintramembranous skeletal elements such as the dentary bone(Fig. S6H) (Bird and Mabee, 2003; Cubbage and Mabee, 1996).Importantly, expression of msmo1 was seen within endochondralgrowth plates, specifically in the region corresponding to pre-hypertrophic chondrocytes (Kronenberg, 2003; LeClair et al., 2009)(see Fig. 5C,C′). In addition, we observed msmo1 expression in theliver, kidney, intestine, brain, retina, spinal cord and skin (Fig. S6A-G). Comparison of msmo1 expression in juvenile wild-type andkolnu7 siblings revealed an undetectable level of msmo1 expressionwithin endochondral growth plates of kolnu7 mutants (Fig. 5F,F′).Our findings support the notion that a deficit in msmo1 expression,
Table 1. CRISPR/Cas9 mutations
Allele CRISPR target sequence (5′-3′) RE site lost Mutation
msmo1nu81 GGCTGTGCCGTTCACCTCCA HphI V3AfsX44lssnu60 GGACAGACCGCAGAGCATGC SphI D43IfsX36ndst3nu20 GGGGCAGCCCAGATCATCCC MboI I247AfsX28ugt8nu82 GGTTTCGTGGTGGTCTCATT BsaI S293F_F294insGFVspock3nu83 GAACCAGGAACATGGAGCCC BsaJI S45RfsX30
RE, restriction enzyme.
Fig. 2. The suppression of early lethality of the msmo1nu81 mutationyields adult fish with kolnu7-like phenotype. (A) Survival of larvae frommsmo1nu81/+ in-crosses, from 7 dpf to 70 dpf. Mostmsmo1nu81 mutants die by9 dpf. [7 dpf wild type (wt; black) n=19, heterozygotes (het; pale gray) n=49,knockout mutants (KO; dark gray) n=16, P=0.02794; 8 dpf wt n=21, het n=39,KO n=17, P=0.8065; 9 dpf wt n=12, het n=40, KO n=3, ***P=0.0008; 10 dpf wtn=37, het n=44, KO n=3, ****P<0.0001; 15 dpf wt n=26, het n=51, KO n=3,****P<0.0001; 70 dpf wt n=18, het n=35, KO n=0, ***P=0.001.] (B)Overexpression of msmo1 driven by daily heat shock of the transgenic lineTg(hsp70l:msmo1:IRESnlsGFP)nu99 rescued the lethality of msmo1nu8
mutants. Transgenic screening based on cardiac GFP. (Control non-transgenic siblings 14 dpf wt n=14, het n=32, KO n=2, ***P=0.0035; transgenicsiblings 14 dpf wt n=18, het n=44, KO n=28, P=0.3214.) cont, control; hs, heatshock. (C) Dietary cholesterol supplementation does not improve thesurvivability of msmo1nu81 mutants. Clutches from msmo1nu81/+ in-crosseswere fed either a high-cholesterol diet (hcd) or a control standardized diet(cont) beginning at 5 dpf until collection at 10 dpf. (HCD wt n=26, het n=50, KOn=9, P=0.0089; control diet wt n=22, het n=66, KO n=5, P<0.0001.) All two-tailed P-values were calculated using chi-squared test. (D-F′) Whole-mountin situ hybridization during the first 5 days of development shows msmo1expression predominately in the yolk syncytial layer (YSL) and liver.Expression is first detected during early somitogenesis in the YSL (D) andcontinues there at 3 dpf (E). At 4 dpf, strong expression is observed in thedifferentiated liver (F,F′). (G-I) Generation of msmo1nu81/wild-type chimerasusing endoderm replacement rescues early lethality of msmo1nu81 mutantsand reveals a strong kolnu7-like phenotype. (G) Schematic of the procedure.Wild-type Tg(ubi:Zebrabow-M)a131 donor embryos were injected with sox32RNA to force an endodermal fate. At high stage (∼3 hpf), cells weretransplanted from donor to host embryos collected from msmo1nu81/+ in-crosses. (H) Surviving msmo1nu81 mutants display a strong kolnu7-likephenotype. msmo1nu81 n=4. (I) The majority of organs of endodermal origindisplayed a high enrichment in transplanted Tg(ubi:Zebrabow-M)a131 cells(red). msmo1nu81 n=4. (J-L) Liver-specific msmo1 expression in msmo1nu81
mutants rescues early lethality and produces juvenilemsmo1nu81mutants withstrong kolnu7-like phenotype. (J) Liver-specific regulatory element fabp10awasused to drivemsmo1 expression inmsmo1nu81mutants. (K) Adult Tg(fabp10a:msmo1:pA)nu100;msmo1nu81 mutants phenocopy kolnu7 mutant. Tg(fabp10:msmo1:pA)nu100 n=50, Tg(fabp10a:msmo1:pA)nu100;msmo1nu81 n=50. (L)Whole-mount skeletal preparations reveal gross malformations throughoutTg(fabp10a:msmo1:pA)nu100;msmo1nu81 craniofacial and axial skeleton,similar to those observed in kolnu7. Tg(fabp10:msmo1:pA)nu100 n=3,Tg(fabp10a:msmo1:pA)nu100;msmo1nu81 n=7.
3
RESEARCH ARTICLE Disease Models & Mechanisms (2020) 13, dmm042549. doi:10.1242/dmm.042549
Disea
seModels&Mechan
isms

particularly in differentiating chondrocytes, underlies the kolnu7
phenotype.
The kolnu7 mutation perturbs a cis-acting msmo1 regulatoryelementTo confirm that loss of Msmo1 is solely responsible for thedevelopment of the kolnu7 phenotype, we mutagenized the msmo1gene using CRISPR/Cas9 genome editing. We isolated an allele,msmo1nu81, which results in a 37 bp insertion (Fig. 1K) in the firstcoding exon, resulting in a frameshift and premature proteintruncation. The msmo1nu81 heterozygotes, similar to kolnu7/+, arephenotypically normal. Next, to test genetic complementation, wecrossed msmo1nu81 and kolnu7 heterozygotes. Transheterozygotekolnu7/+:msmo1nu81/+ appeared phenotypically normal until∼5 weeks post-fertilization, after which they began to take on akolnu7-like appearance and became morphologically indistinguishablefrom kolnu7 mutants (Fig. 1I,J).Our results suggest that the kolnu7 mutation disrupts a regulatory
element driving msmo1 expression. We directly examined thispossibility by taking advantage of a relatively large insertion inmsmo1nu81 (Fig. 1K) that allows us to compare allele-specificexpression. Once again, we used total RNA isolated from the hypuralcomplexes for semi-quantitative RT-PCR analysis (Fig. 1L). Incontrast to msmo1nu81/+, in which both wild-type and mutantproducts can easily be identified after RT-PCR, the wild-typemsmo1 allele linked to the kolnu7 locus is under-represented, stronglysupporting the notion that the kolnu7 mutation disrupts a cis-actingregulatory element. In summary, based on the results of our positionalcloning, in situ hybridization and complementation testing, weconcluded that the kolnu7 mutant phenotype is a result of stronglyreduced msmo1 expression after loss of a positively acting regulatoryelement.
Loss of Msmo1 function is lethal in zebrafish larvaeWhile kolnu7 homozygote fish survive until adulthood, msmo1nu81
homozygote mutants show a decrease in growth at 6 dpf (data notshown) and die by 9 dpf (Fig. 2A). Overexpression of wild-typemsmo1 driven by the Tg(hsp70l:msmo1:IRESnlsEGFP)nu99
transgene was able to suppress the early lethality of msmo1nu81
homozygotes (Fig. 2B), indicating that loss of Msmo1 is responsiblefor the death of the msmo1nu81 mutants. Because Msmo1 activityplays a crucial role in cholesterol biosynthesis (Sharpe and Brown,2013) (Fig. S1), we tested whether a cholesterol-enriched diet couldextend the lifespan of msmo1nu81 mutants. However, even a 4%cholesterol-enriched diet, shown to induce hypercholesterolemia inzebrafish (Stoletov et al., 2009), did not significantly improvemutants’ survival (Fig. 2C).
Expression of msmo1 in liver rescues msmo1nu81 mutantsand produces kolnu7 phenotypeTo study the role of Msmo1 in bone formation, we needed tosuppress the early lethality of msmo1nu81 mutants. We observedstrongmsmo1 expression in the larval and juvenile liver (Fig. 2D-F′;Fig. S6A), an organ responsible for producing endogenouscholesterol (Turley, 2004). We therefore predicted that hepaticrestoration of Msmo1 activity might rescue the msmo1nu81 mutants,allowing us to study juvenile bone development in this geneticbackground. To test this prediction, we used two approaches. First,with the help of partial endoderm replacement, we createdmsmo1nu81 mutants with chimeric endodermal organs. We pushedcells of the Tg(ubi:Zebrabow-M)a131 (Pan et al., 2013) donorembryos to an endodermal fate by overexpressing sox32 at the one-
cell stage (Stafford et al., 2006). The red fluorescing donor cellswere transplanted from high-stage donor embryos into shield-stagehost embryos obtained from msmo1nu81 heterozygote in-crosses(Fig. 2G). We analyzed the host fish at ∼5 weeks of development.Four of 15 transplanted fish, with significant contribution of donorcells, appeared to have a kolnu7-like appearance (Fig. 2H,I).Genotyping revealed that all of the kolnu7-like fish were chimeraswith msmo1nu81 mutants. In a parallel effort, we employed atransgenic approach to drive hepatic msmo1 expression using theliver-specific regulatory element of the fatty acid binding protein,liver basic ( fabp10a) gene (Her et al., 2003; Kwan et al., 2007)(Fig. 2J). In contrast to the transplantation experiments, thetransgenic expression of msmo1 was much more efficient insuppression of lethality, and close to Mendelian ratios ofmsmo1nu81
mutants were found at 5 weeks in ∼700 fish studied from 13separate crosses. Rescued mutants grew to adulthood and displayeda severe kolnu7-like phenotype (Fig. 2K,L). These results indicatethat restoration of hepatic Msmo1 activity is sufficient for survivalof msmo1nu81 mutants beyond early larval stages. Interestingly,liver-derived cholesterol synthesis is unable to rescue the skeletalphenotype of msmo1nu81 mutants, indicating a requirement forchondrocyte-specific sterol production.
Loss of sterol synthesis partially suppresses msmo1nu81
mutant phenotypeIn most human syndromes resulting from mutations in genesinvolved in the post-squalene cholesterol biosynthesis pathway, asimultaneous decrease in cholesterol levels and an accumulation ofsterol intermediates is observed (Porter and Herman, 2011). Toelucidate the effects of cholesterol deprivation from sterolintermediates’ accumulation, we eliminated the activity of Lss.This enzyme catalyzes the cyclization of the first sterol in thecholesterol biosynthesis pathway (Huff and Telford, 2005)(Fig. S1). Mutation of the lss gene is predicted to result in adeficit of cholesterol biosynthesis, but not an accumulation of sterolintermediates (Sharpe and Brown, 2013). Using CRISPR/Cas9genome editing, we generated the lssnu60 allele, resulting in a 23 bpdeletion (Fig. 3A), causing a frameshift and premature proteintruncation. Heterozygote lssnu60 fish are phenotypically normal andfertile. In contrast to kolnu7/+:msmo1nu81/+ transheterozygotes, thestudy of over 180 fish from two separate crosses did not identify anysigns of skeletal abnormalities in adult kolnu7/+;lssnu60/+ animals(data not shown). This observation further supports the notion that areduced level of Msmo1 activity, and not a general deficit in thecholesterol biosynthesis pathway, is responsible for the kolnu7
phenotype.The growth of msmo1nu81 and lssnu60 homozygote mutants lags
behind that of wild-type siblings beginning at 6 dpf (data not shown).Unlike msmo1nu81 mutants, lssnu60 homozygotes live until 11 dpf(Fig. 3B). Ameasurement of total cholesterol levels at 8 dpf indicateda strong reduction in both mutants. Compared to wild-type siblings,total cholesterol levels of lssnu60 homozygousmutants were 43% (s.d.9.14, P=0.0011) of wild-type levels, whereas those of msmo1nu81
homozygous mutants were 22% (s.d. 10.59, P=0.0006) of wild-typelevels. To determinewhether better survival is a result of a less-severedefect in cholesterol biosynthesis or an accumulation of sterolintermediates, we conducted an epistatic analysis. We found thatdouble lssnu60;msmo1nu81 mutants survived to 10 dpf in close toexpected numbers, whereas mostmsmo1nu81 single mutants were lost(Fig. 3C). Our results strongly suggest that an accumulation of sterolintermediates is responsible for the more-severe msmo1nu81
phenotype, compared to that of lssnu60 mutants.
4
RESEARCH ARTICLE Disease Models & Mechanisms (2020) 13, dmm042549. doi:10.1242/dmm.042549
Disea
seModels&Mechan
isms

Loss of Msmo1 and Lss activity is associated with skeletaldefectsTo study juvenile bone development in the lssnu60 knockoutmutants, we again employed a transgenic approach to restore hepaticlss expression (Her et al., 2003; Kwan et al., 2007). Similar to theresults described for the rescued msmo1nu81 mutants, close toMendelian ratios of rescued lssnu60 were found at 5 weeks in ∼425fish studied from nine separate crosses. Rescued mutants grew toadulthood and displayed a severe kolnu7-like phenotype (Fig. 3D).Skeletal staining revealed compressions of the axial skeleton due tovertebral column fusions and deformations in the craniofacialskeleton (Fig. 3E).To better understand the skeletal phenotype associated with
loss of Msmo1 and Lss activity, we analyzed the skeletons of kolnu7,kolnu7/+:msmo1nu81/+, Tg(fabp10a:msmo1:pA)nu100;msmo1nu81 andTg(fabp10:lss:pA)nu101;lssnu60 mutants. We collected mutant andwild-type siblings at 4 months and compared bone formation,focusing on selected bones formed by cartilage replacementmechanisms: the hyomandibular, ceratohyal and hypural complex(Bird and Mabee, 2003; Cubbage and Mabee, 1996) (Fig. 4). Thebones of mutants and transheterozygotes were irregularly shaped(Fig. 4A,E,I,M,Q) with abnormal stratification of the growth plates(Fig. 4D,H,L,P,T). The persistent cartilage bands separating
individual bones (Fig. 4B), which normally never fuse in zebrafish(LeClair et al., 2009), were often closed as a result of ectopicossification (Fig. 4F,J,N,R). The endochondral bones of the kolnu7/+:msmo1nu81/+ (Fig. 4I-L), Tg(fabp10a:msmo1:pA)nu100;msmo1nu81
(Fig. 4M-P) and Tg(fabp10:lss:pA)nu101;lssnu60 mutants (Fig. 4Q-T)phenocopied those of the kolnu7 mutant (Fig. 4E-H), indicating thatthe loss of Msmo1 and Lss activity within cartilage-derived elementsproduces severely deformed, compressed endochondral bones withfrequent ectopic ossification of the growth plate.
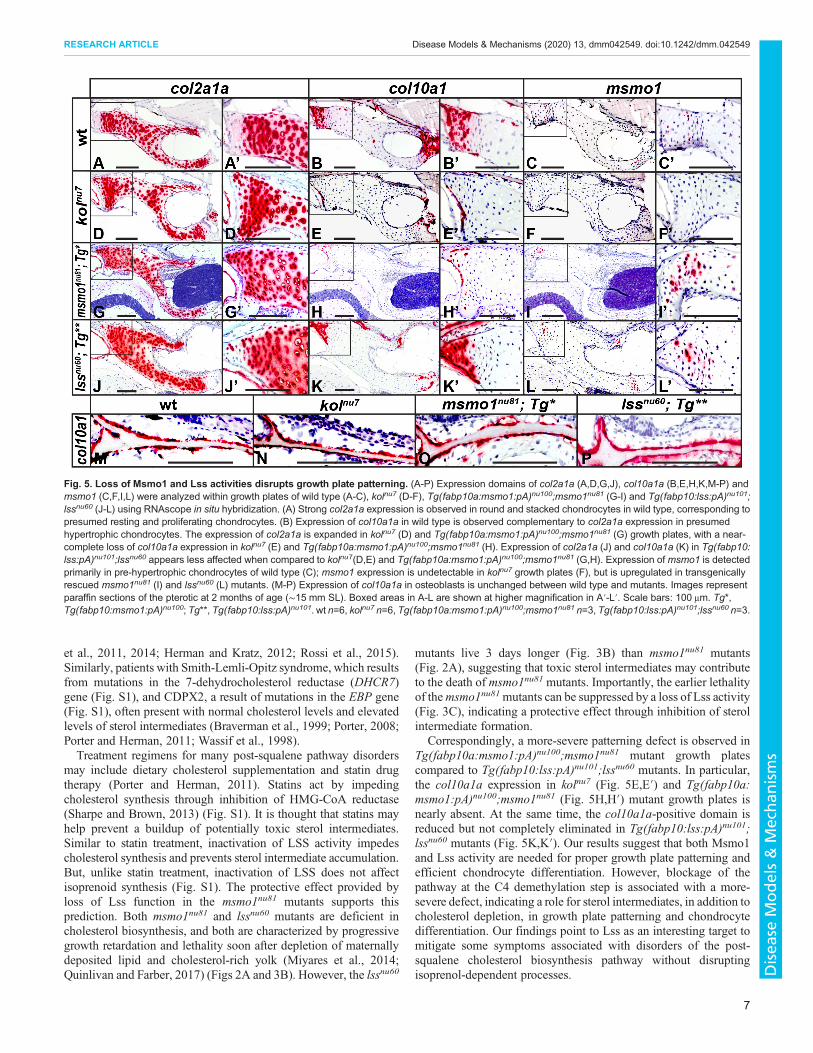
Msmo1 and Lss activity in the growth plate is needed fornormal chondrocyte differentiationTo examine changes in growth plate patterning after loss of Msmo1and Lss activity, we used RNAscope in situ hybridization to analyzethe gene expression of key markers in the growth plate of the pteroticbone, an element of the chondocranium (Cubbage and Mabee, 1996).We focused on collagen type II, alpha 1 (col2a1a), a marker of restingand proliferating chondrocytes (Dale and Topczewski, 2011; Zhaoet al., 1997), and collagen type 10, alpha 1a (col10a1a), a marker ofhypertrophic chondrocytes (Dale and Topczewski, 2011; Eameset al., 2012; Topczewska et al., 2016) and osteoblasts (Avaron et al.,2006; Eames et al., 2012; Kim et al., 2013). We analyzed wild-type,kolnu7, Tg(fabp10a:msmo1:pA)nu100;msmo1nu81 and Tg(fabp10:lss:pA)nu101;lssnu60 fish at ∼2 months of age, when wild-type fishhad an SL of ∼15 mm. In wild-type animals, strong col2a1aexpression was observed in round and stacked chondrocytes(Fig. 5A,A′), corresponding to presumed resting and proliferativezones of the growth plate. In a complementary pattern, col10a1awas detected in the presumed hypertrophic chondrocytes(Fig. 5B,B′). In the kolnu7 and Tg(fabp10a:msmo1:pA)nu100;msmo1nu81 mutants, col2a1a expression is expanded (Fig. 5D,D′,G,G′) and accompanied by near complete loss of col10a1aexpression (Fig. 5E,E′,H,H′). Although a similar expansion of theof col2a1a domain was observed in Tg(fabp10:lss:pA)nu101;lssnu60
mutants (Fig. 5J,J′), col10a1a-positive chondrocytes were presentin all growth plates studied (Fig. 5K,K′). These results indicate thatalthough Lss activity is needed for proper growth plate development,loss of Msmo1 activity has a more-severe effect on chondrocytedifferentiation. Importantly, the osteoblast expression of col10a1ain all mutants was similar to that in wild type (Fig. 5M-P), indicatinga defect specific to chondrocyte differentiation. Interestingly, thedomain ofmsmo1 expression in wild-type growth plates is localizedto the transition zone from the columnar to hypertrophicchondrocytes (Fig. 5C,C′). This domain is absent in kolnu7
mutants (Fig. 5F,F′), consistent with the proposed regulatorynature of the mutation. In contrast, the msmo1 expression domain isexpanded in the Tg(fabp10a:msmo1:pA)nu100;msmo1nu81 andTg(fabp10:lss:pA)nu101;lssnu60 mutants (Fig. 5I,I′,L,L′),suggesting de-repression of the pathway. Based on these results,we conclude that, during normal development,msmo1 is transientlyupregulated in pre-hypertrophic chondrocytes, and loss of theability to synthesize cholesterol impedes chondrocyte hypertrophicdifferentiation.
LossofMsmo1andLss activitywithin growthplates doesnotresult in loss of Ihh signalingIhh is a key ligand in cartilage and bone development. Ihh issynthesized by pre-hypertrophic chondrocytes and stimulateschondrocyte proliferation and hypertrophy, as well as osteoblastdifferentiation (Hammond andSchulte-Merker, 2009; St-Jacques et al.,1999; Vortkamp et al., 1996). Given the critical role of cholesterol inthe post-translational Hedgehog (Hh) modification (Chen et al., 2011;
Fig. 3. Loss of Lss activity phenotype is epistatic tomsmo1nu81mutation.(A) The lssnu60 allele. PAM, protospacer adjacent motif (underlined in red).(B) Survival analysis of clutches of lssnu60/+ in-crosses, from 9 dpf to 23 dpf,reveals that most lssnu60 mutants die by 12 dpf. [9 dpf wild type (wt; black)n=21, heterozygotes (het; pale gray) n=30, knockout mutants (KO; dark gray)n=15, P=0.4412; 10 dpf wt n=15, het n=27, KO n=16, P=0.8563; 11 dpf wtn=20, het n=44, KO n=14, P=0.3320; 12 dpf wt n=24, het n=48, KO n=10,*P=0.0277; 13 dpf wt n=33, het n=51, KO n=10, **P=0.0026; 23 dpf wt n=8,het n=27, KO n=0, ***P=0.0009.] (C) Survival analysis of clutches ofmsmo1nu81/+;lssnu60/+ in-crosses reveals that loss of Lss activity increasessurvivability ofmsmo1nu81mutants at 10 dpf. [wt observed (O) n=11, expected(E) n=11; msmo1nu81 O n=4, E n=11; msmo1nu81;lssnu60/+ O n=9, E n=22;msmo1nu81;lssnu60 O n=10, E n=11; lssnu60 O n=12, E n=11; msmo1nu81/+;lssnu60 O n=30, E n=22; msmo1nu81/+;lssnu60/+ O n=40, E n=44; msmo1nu81/+
O n=18, E n=22; lssnu60/+ O n=18, E n=22; *P=0.0431.] Two-tailed P-valueswere calculated using chi-squared test. (D,E) Suppression of early lethality oflssnu60 mutants by liver-specific expression of lss. (D) Adult Tg(fabp10:lss:pA)nu101;lssnu60 mutants phenocopy kolnu7. Tg(fabp10:lss:pA)nu101 n=12,Tg(fabp10:lss:pA)nu101;lssnu60 n=12. (E) Whole-mount skeletal preparationsreveal gross malformations throughout Tg(fabp10:lss:pA)nu101;lssnu60
craniofacial and axial skeleton, similar to those observed in kolnu7.Tg(fabp10:lss:pA)nu101 n=3, Tg(fabp10:lss:pA)nu101;lssnu60 n=5.
5
RESEARCH ARTICLE Disease Models & Mechanisms (2020) 13, dmm042549. doi:10.1242/dmm.042549
Disea
seModels&Mechan
isms

Porter et al., 1995),we investigatedwhat potential effect loss ofMsmo1and Lss activity may have on Ihh signaling within growth plates.Zebrafish have two Ihh paralogs, ihha and ihhb, with partiallyoverlapping expression domains (Avaron et al., 2006). The receptorpatched 1 (ptch1), a transcriptional target of Hh signaling, is expressedin chondrocytes and the perichondrium of endochondral bone (Felberet al., 2011; Hammond and Schulte-Merker, 2009; St-Jacques et al.,1999; Vortkamp et al., 1996). We analyzed expression of ihha, ihhband ptch1 by RNAscope in situ hybridization using the same stagesand anatomical location as described above. In wild-type fish, abroader ihha domain of expression in the hypertrophic chondrocyteszone and amore restricted ihhb domain of expression inmostly the pre-hypertrophic chondrocytes zone (Fig. 6A,B) was observed. At thesame time, strong ptch1 expression was observed in the adjacentcolumnar zone and in individual hypertrophic chondrocytes (Fig. 6C).Expression of ihha and ihhb was still observed in growth platesof kolnu7, Tg(fabp10a:msmo1:pA)nu100;msmo1nu81 and Tg(fabp10:lss:pA)nu101;lssnu60 mutants (Fig. 6D,E,G,H,J,K). Importantly, ptch1expression was also detected in all mutant growth plates (Fig. 6F,I,L),indicating that loss of cholesterol biosynthesis in the mutants doesnot abolish Ihh signaling activity.
DISCUSSIONZebrafish post-squalene mutants show phenotypicsimilarities to human CDPMany post-squalene pathway disorders with documented CDPresult from partial loss-of-function mutations, such as mutations in
the NAD(P)-dependent steroid dehydrogenase-like (NSDHL) gene(Fig. S1), which result in congenital hemidysplasia withichthyosiform erythroderma and limb defects (CHILD) syndrome(Bornholdt et al., 2005; Mi et al., 2015; Herman and Kratz, 2012),and mutations in the emopamil binding protein (EBP) gene(Fig. S1), which result in X-linked dominant chondrodysplasiapunctata (CDPX2), or Conradi-Hunermann-Happle syndrome(Cañueto et al., 2014; Herman and Kratz, 2012). Similarly, thekolnu7, kolnu7/+:msmo1nu81/+ and Tg(fabp10a:msmo1:pA)nu100;msmo1nu81 mutants result from reduction and/or absence ofMsmo1 activity within skeletal elements and display ectopicossification within growth plates similar to that seen in CDP(Figs 4 and 7) (Cañueto et al., 2014; Herman and Kratz, 2012; Rossiet al., 2015). As such, these mutants could prove helpful as animalmodels to help understand the mechanisms behind the development,and potential treatment, of CDP.
Sterol intermediates may play a role in msmo1nu81 mutantphenotypeA lack of cholesterol alone does not explain all of the defectsobserved in post-squalene cholesterol biosynthesis disorders, asmany patients present with normal cholesterol levels (Porter, 2008;Porter and Herman, 2011). Hypomorphic mutations in MSMO1result in the rare autosomal recessive disorder C4 methyloxidase-like deficiency (He et al., 2011). Since patients may not present withdecreased cholesterol levels, the definitive test for this disorder is anaccumulation of C4 sterol intermediates (Frisso et al., 2017; He
Fig. 4. Loss of Msmo1 and Lss activity isassociated with abnormal endochondralbone formation. (A-T) The hyomandibular (A,E,I,M,Q), ceratohyal (B,F,J,N,R) and hypuralcomplex (C,D,G,H,K,L,O,P,S,T) werecompared between wild type (A-D), kolnu7
(E-H), kolnu7/+:msmolnu81/+ (I-L), Tg(fabp10a:msmo1:pA)nu100;msmo1nu81 (M-P) andTg(fabp10:lss:pA)nu101;lssnu60 (Q-T). (E-T)The endochondral bones of the mutants arecompressed and irregularly shaped withectopic ossification and fusions throughout thegrowth plates, marked with arrows. Imagesrepresent disarticulated whole-mount adultskeletal preparations at ∼4 months of age.Scale bars: 100μm. Tg*, Tg(fabp10:msmo1:pA)nu100; Tg**, Tg(fabp10:lss:pA)nu101. wtn=4, kolnu7 n=4, kolnu7/+:msmolnu81/+ n=4,Tg(fabp10a:msmo1:pA)nu100;msmo1nu81 n=4,Tg(fabp10:lss:pA)nu101;lssnu60 n=4.
6
RESEARCH ARTICLE Disease Models & Mechanisms (2020) 13, dmm042549. doi:10.1242/dmm.042549
Disea
seModels&Mechan
isms
et al., 2011, 2014; Herman and Kratz, 2012; Rossi et al., 2015).Similarly, patients with Smith-Lemli-Opitz syndrome, which resultsfrom mutations in the 7-dehydrocholesterol reductase (DHCR7)gene (Fig. S1), and CDPX2, a result of mutations in the EBP gene(Fig. S1), often present with normal cholesterol levels and elevatedlevels of sterol intermediates (Braverman et al., 1999; Porter, 2008;Porter and Herman, 2011; Wassif et al., 1998).Treatment regimens for many post-squalene pathway disorders
may include dietary cholesterol supplementation and statin drugtherapy (Porter and Herman, 2011). Statins act by impedingcholesterol synthesis through inhibition of HMG-CoA reductase(Sharpe and Brown, 2013) (Fig. S1). It is thought that statins mayhelp prevent a buildup of potentially toxic sterol intermediates.Similar to statin treatment, inactivation of LSS activity impedescholesterol synthesis and prevents sterol intermediate accumulation.But, unlike statin treatment, inactivation of LSS does not affectisoprenoid synthesis (Fig. S1). The protective effect provided byloss of Lss function in the msmo1nu81 mutants supports thisprediction. Both msmo1nu81 and lssnu60 mutants are deficient incholesterol biosynthesis, and both are characterized by progressivegrowth retardation and lethality soon after depletion of maternallydeposited lipid and cholesterol-rich yolk (Miyares et al., 2014;Quinlivan and Farber, 2017) (Figs 2A and 3B). However, the lssnu60
mutants live 3 days longer (Fig. 3B) than msmo1nu81 mutants(Fig. 2A), suggesting that toxic sterol intermediates may contributeto the death of msmo1nu81 mutants. Importantly, the earlier lethalityof themsmo1nu81mutants can be suppressed by a loss of Lss activity(Fig. 3C), indicating a protective effect through inhibition of sterolintermediate formation.
Correspondingly, a more-severe patterning defect is observed inTg(fabp10a:msmo1:pA)nu100;msmo1nu81 mutant growth platescompared to Tg(fabp10:lss:pA)nu101;lssnu60 mutants. In particular,the col10a1a expression in kolnu7 (Fig. 5E,E′) and Tg(fabp10a:msmo1:pA)nu100;msmo1nu81 (Fig. 5H,H′) mutant growth plates isnearly absent. At the same time, the col10a1a-positive domain isreduced but not completely eliminated in Tg(fabp10:lss:pA)nu101;lssnu60 mutants (Fig. 5K,K′). Our results suggest that both Msmo1and Lss activity are needed for proper growth plate patterning andefficient chondrocyte differentiation. However, blockage of thepathway at the C4 demethylation step is associated with a more-severe defect, indicating a role for sterol intermediates, in addition tocholesterol depletion, in growth plate patterning and chondrocytedifferentiation. Our findings point to Lss as an interesting target tomitigate some symptoms associated with disorders of the post-squalene cholesterol biosynthesis pathway without disruptingisoprenol-dependent processes.
Fig. 5. Loss of Msmo1 and Lss activities disrupts growth plate patterning. (A-P) Expression domains of col2a1a (A,D,G,J), col10a1a (B,E,H,K,M-P) andmsmo1 (C,F,I,L) were analyzed within growth plates of wild type (A-C), kolnu7 (D-F), Tg(fabp10a:msmo1:pA)nu100;msmo1nu81 (G-I) and Tg(fabp10:lss:pA)nu101;lssnu60 (J-L) using RNAscope in situ hybridization. (A) Strong col2a1a expression is observed in round and stacked chondrocytes in wild type, corresponding topresumed resting and proliferating chondrocytes. (B) Expression of col10a1a in wild type is observed complementary to col2a1a expression in presumedhypertrophic chondrocytes. The expression of col2a1a is expanded in kolnu7 (D) and Tg(fabp10a:msmo1:pA)nu100;msmo1nu81 (G) growth plates, with a near-complete loss of col10a1a expression in kolnu7 (E) and Tg(fabp10a:msmo1:pA)nu100;msmo1nu81 (H). Expression of col2a1a (J) and col10a1a (K) in Tg(fabp10:lss:pA)nu101;lssnu60 appears less affected when compared to kolnu7(D,E) and Tg(fabp10a:msmo1:pA)nu100;msmo1nu81 (G,H). Expression of msmo1 is detectedprimarily in pre-hypertrophic chondrocytes of wild type (C); msmo1 expression is undetectable in kolnu7 growth plates (F), but is upregulated in transgenicallyrescued msmo1nu81 (I) and lssnu60 (L) mutants. (M-P) Expression of col10a1a in osteoblasts is unchanged between wild type and mutants. Images representparaffin sections of the pterotic at 2 months of age (∼15 mm SL). Boxed areas in A-L are shown at higher magnification in A′-L′. Scale bars: 100 μm. Tg*,Tg(fabp10:msmo1:pA)nu100; Tg**, Tg(fabp10:lss:pA)nu101. wt n=6, kolnu7 n=6, Tg(fabp10a:msmo1:pA)nu100;msmo1nu81 n=3, Tg(fabp10:lss:pA)nu101;lssnu60 n=3.
7
RESEARCH ARTICLE Disease Models & Mechanisms (2020) 13, dmm042549. doi:10.1242/dmm.042549
Disea
seModels&Mechan
isms

Growth plates of kolnu7 and Tg(fabp10a:msmo1:pA)nu100;msmo1nu81 mutants show distinct phenotypeThe growth plates of kolnu7 and Tg(fabp10a:msmo1:pA)nu100;msmo1nu81 are unique in that they display an enhanced and ectopicossification phenotype (Fig. 4E-H,M-P), yet patterning of thegrowth plates revealed loss of col10a1a expression and absence ofthe presumptive hypertrophic chondrocyte zone (Fig. 5E,E′,H,H′).Interestingly, Ihh genes are still expressed in the mutants,suggesting that the transition from proliferating to pre-hypertrophic zone is not inhibited. The enhanced ossificationphenotype seen in kolnu7 and Tg(fabp10a:msmo1:pA)nu100;msmo1nu81 mutants appears similar to that in zebrafish cyp26b1mutants, but these mutants do not show a loss of col10a1aexpression in endochondral bones (Laue et al., 2008; Spoorendonket al., 2008). The unique phenotype of kolnu7 and Tg(fabp10a:msmo1:pA)nu100;msmo1nu81mutants most closely resembles Sox9inactivation in mouse growth plates (Dy et al., 2012; Ikegami et al.,2011). Sox9 is a master regulator of chondrocyte differentiationand survival (Lefebvre et al., 2019), indicating that Msmo1activity within pre-hypertrophic chondrocytes might play a role incontrolling cell hypertrophy and chondrocyte differentiationwithin the growth plate.These results indicate that cholesterol depletion, and possibly
accumulation of sterol intermediates, within mutant chondrocytesexiting from the proliferation zone may prevent normaldifferentiation and proper growth plate patterning, leading to anaccelerated ossification. Intriguingly, CDP is observed in patients
with a deficit in the initial steps of the post-squalene cholesterolbiosynthesis pathway (Rossi et al., 2015), pointing to a potentialactive role of sterol intermediates in the first steps of biosynthesis inbone pathology. Recently, sterol intermediates were implicated ininteractions with numerous signaling pathways by acting as ligandsfor LXRs (also known as NR1Hs), RORγ (also known as RORC)and EGFR (Gabitova et al., 2015; Hu et al., 2015; Santori et al.,2015; Sukhanova et al., 2013; Urlep et al., 2017; Yang et al., 2006).These data highlight the importance of sterol intermediates in thecholesterol biosynthesis pathway and the role these intermediatesmay play in skeletal dysplasia.
Fig. 6. Ihh signaling within endochondral growth plates is present despiteloss of Msmo1 and Lss activity. (A-L) Expression domains of ihha (A,D,G,J),ihhb (B,E,H,K) and ptch1 (C,F,I,L) were analyzed within growth plates of wildtype (A-C), kolnu7 (D-F), Tg(fabp10a:msmo1:pA)nu100;msmo1nu81 (G-I) andTg(fabp10:lss:pA)nu101;lssnu60 (J-L) using RNAscope in situ hybridization.(A-C) In wild type, expression of ihha and ihhb is observed in the pre-hypertrophic chondrocyte zone, with ptch1 expression seen to either side ofthese domains. (D-L) Expression of ihha, ihhb and ptch1 is present withinmutant growth plates and similar to domains observed in wild type. Imagesrepresent paraffin sections of the pterotic at 2 months of age (∼15 mm SL).Scale bars: 100 μm. Tg*, Tg(fabp10:msmo1:pA)nu100; Tg**, Tg(fabp10:lss:pA)nu101. wt n=3, kolnu7 n=3, kolnu7/+:msmolnu81/+ n=3, Tg(fabp10a:msmo1:pA)nu100;msmo1nu81 n=3, Tg(fabp10:lss:pA)nu101;lssnu60 n=3.
Fig. 7. New zebrafish models for CDP. (A-D) Abnormal ossification, markedwith arrows, within endochondral growth plates of kolnu7 (B), kolnu7/+:msmolnu81/+ (C) and Tg(fabp10a:msmo1:pA)nu100;msmo1nu81 (D) mutantphenotypes resembles defects observed in patients with chondrodysplasiapunctata. Images represent lateral views of palatoquadrate growth plates ofcraniofacial bones from disarticulated whole-mount adult skeletal preparationsat ∼4 months of age. Scale bars: 100 μm. Tg*, Tg(fabp10:msmo1:pA)nu100. wtn=4, kolnu7 n=6, kolnu7/+:msmolnu81/+ n=4, Tg(fabp10a:msmo1:pA)nu100;msmo1nu81 n=4.
8
RESEARCH ARTICLE Disease Models & Mechanisms (2020) 13, dmm042549. doi:10.1242/dmm.042549
Disea
seModels&Mechan
isms

Msmo1 and Lss activity in growth plates is not essential forIhh signalingIt is currently proposed that defects observed in disorders of thepost-squalene pathway are due to two main mechanisms: abnormalHh signaling and accumulation of sterol intermediates (Porter andHerman, 2011). Cholesterol modification of the Hh protein isnecessary for proper Hh signaling and influences the spread of themorphogen through tissue (Briscoe and Thérond, 2013; Zeng,2001; Zeng et al., 2001). Recently, it has been shown thatcholesterol interacts with Smoothened (SMO) and the inability ofSMO to bind cholesterol negatively affects Hh signaling (Byrneet al., 2016).The role of Hh signaling in skeletogenesis is well known, with
Shh playing a pivotal role in early patterning and Ihh acting laterduring bone formation (Yang et al., 2015). Thus, defects resulting inimpaired cholesterol biosynthesis can affect Hh signaling, andmanybone defects seen in cholesterol biosynthesis disorders, such aspolydactyly, syndactyly and cleft palate, are hallmarks of abnormalShh signaling (Briscoe and Thérond, 2013; Cooper et al., 2003; Gaoet al., 2009; Hui, 1993; Hui and Joyner, 1993). It is interesting tonote that although the kolnu7, msmo1nu81 and lssnu60 mutationsnegatively affect cholesterol synthesis, these mutants do not displaythe classical patterning defects associated with defective Shhsignaling (Eberhart et al., 2006; Schwend and Ahlgren, 2009; Wadaet al., 2005) (Fig. S4). This is likely to be caused by maternallydeposited cholesterol within the yolk of the zebrafish that allows fornormal embryonic and early larval development (Miyares et al.,2014; Quinlivan and Farber, 2017). Zebrafish have a large yolk thatis slowly absorbed over the first 5 days of development (Miyareset al., 2014; Quinlivan and Farber, 2017). Therefore, zebrafishlarvae do not require an endogenous biosynthesis of cholesteroluntil ∼6 dpf.Although juvenile kolnu7 mutants display an absence of msmo1
expression in growth plates (Fig. 5F,F′), Ihh signaling within kolnu7growth plates is not lost (Fig. 6D-F). Similarly, juvenileTg(fabp10a:msmo1:pA)nu100;msmo1nu81 and Tg(fabp10:lss:pA)nu101;lssnu60 mutants exhibit loss of Msmo1 and Lss activity,respectively, yet expression of Ihh ligands and their transcriptionaltarget ptch1 can be detected (Fig. 6G-L). These results suggest thatthe level of cholesterol in chondrocytes depleted of Msmo1 and Lssactivity is sufficient to maintain Hh signaling.
The kolnu7 mutant highlights the significance of cis-actingregulatory mutations in diseaseThe importance of regulatory elements in human health anddisease has been well recognized (Venter et al., 2001; Wray,2007). With protein-coding exons making up less than 2% of thehuman genome (Venter et al., 2001), cis-regulatory elements arenow believed to be more important than gene-encoding sequencesin determining human traits and susceptibility to disease (Niuet al., 2018). In fact, many skeletal malformations are the result ofmutations in regulatory elements that control expression of genessuch as bone morphogenetic protein 2 (BMP2) (Dathe et al., 2009),SOX9 (Kurth et al., 2009), distal-less homeobox 5/6 (DLX5/6)(Kouwenhoven et al., 2010) and SHH (Albuisson et al., 2011;Wieczorek et al., 2010). Given the importance of regulatoryelements in disease, the identification of new cis-regulatorymutations and the genes they regulate is crucial. The regionharboring the kolnu7 locus and msmo1 gene is highly syntenic witha corresponding region of human chromosome 4 (data not shown),raising the possibility that similar long-distance regulationmechanisms operate in higher vertebrates.
Mutations in msmo1 and lss reveal an important role ofcholesterol biosynthesis genes in bone developmentPreviously, very little was known of the role MSMO1 and LSSplayed in bone development. Of the five reported cases of C4methyloxidase-like deficiency, symptoms reported includecataracts, intellectual delays, skin abnormalities, immunedysfunction, shortened stature and microcephaly (Frisso et al.,2017; He et al., 2011, 2014; Herman and Kratz, 2012; Rossi et al.,2015). Of the seven published articles on MSMO1 gene function,none show its role in bone development. Similarly, very fewpublished articles show a link between LSS gene function and bonedevelopment. In humans, hypomorphic mutations in LSS result incongenital cataracts (Chen and Liu, 2017; Zhao et al., 2015),hypotrichosis simplex (Romano et al., 2018) and alopecia withmental retardation syndrome (Besnard et al., 2019).
Here, for the first time, we show that msmo1 and lss are neededfor proper chondrocyte differentiation, growth plate formation andendochondral ossification. To our knowledge, the msmo1nu81 andlssnu60mutants reported here are the first-described loss-of-functionfish mutants in Msmo1 and Lss, respectively. The kolnu7 andmsmo1nu81 mutants are the first-reported animal models for thehuman disorder C4 methyloxidase-like deficiency. These mutants,along with the stable transgenic mutant lines Tg(fabp10a:msmo1:pA)nu100;msmo1nu81 and Tg(fabp10:lss:pA)nu101;lssnu60 describedabove, provide essential tools to further elucidate the roles ofmsmo1and lss in skeletal dysplasia. Zebrafish are an excellent tool for pre-clinical drug testing (MacRae and Peterson, 2015; Tan and Zon,2011); thus, the lines presented here may provide invaluable insightinto treatment options for those suffering from C4 methyloxidase-like deficiency, as well as other disorders of the post-squalenecholesterol biosynthesis pathway.
MATERIALS AND METHODSPositional cloningThe kolnu7 mutation was localized to zebrafish chromosome 1 using agenetic linkage map of sequence-length polymorphism markers (Knapiket al., 1998). Additional closely linked markers, based on either restrictionenzyme polymorphism or sequencing for single-nucleotide polymorphismwere used to narrow down the kolnu7 location to a ∼457 kb critical region(Fig. 1C).
MultAlin analysisAll protein sequences were obtained from UniProtKB (UniProt, 2019):UGT8 Homo sapiens (Entry ID: Q16880), UGT8 Gallus gallus (Entry ID:F1NH08) and Ugt8 Danio rerio (Entry ID: Q1ECX6). These sequenceswere then aligned using MultAlin (Corpet, 1988), using an absolute scoringmethod and Blosum62 symbol comparison table with a gap opening defaultvalue of 12 and gap extension default value of 2.
RNA extractionTo extract bone-enriched total RNA, the hypural complexes from kolnu7 andwild-type siblings were dissected after the SL for each animal was recorded.Each hypural complex was placed in a 1.5-ml RINONavy Bead tube (RINOBulletBlender Navy Bead Lysis kit; Next Advance, Troy, NY, USA) with600 μl TRIzol (Invitrogen). Tissue was homogenized using the BeadBugMicrotube Homogenizer (Benchmark Scientific, Sayreville, NJ, USA).Samples were extracted using five cycles of 1 min homogenization followedby 1 min cooling in a wet ice bath. RNA was purified from the TRIzolsolution using Directzol RNA Minipreps (Zymo Research, Irvine, CA,USA).
Quantitative RT-PCRAll assays were performed using an EXPRESS One-Step Superscript qRT-PCR Kit, with premixed ROX (#11791200; Thermo Fisher Scientific,
9
RESEARCH ARTICLE Disease Models & Mechanisms (2020) 13, dmm042549. doi:10.1242/dmm.042549
Disea
seModels&Mechan
isms

Waltham, MA, USA), Applied Biosystems MicroAmp Fast Optical reactionplates and 8-cap strips (Thermo Fisher Scientific) on an Applied BiosystemsStepOnePlus Real-Time PCR System. For each reaction, 8 ng total RNAwas used. All assays consisted of three technical replicates and a minimumof two biological replicates. The following Made to Order Taqman probes(Thermo Fisher Scientific) were used:mettl14 (Assay ID: Dr03438758_g1),tll1 (Assay ID: Dr03138096_m1), cpe (Assay ID: Dr03188311_s1), grhprb(Assay ID: Dr03088390_g1), ugt8 (Assay ID: Dr03427025) msmo1 (AssayID: Dr03133463_m1) and eef1a1l1 (Assay ID: Dr03432748_m1). Inaddition, the following Custom Plus Taqman probes (Thermo FisherScientific) were designed and used: ndst3 (Assay ID: AJY9Y10), spock3(Assay ID: AJ20TKF), prss12 (Assay ID: AJ5IPWS), apbb2 (Assay ID:AJT96CS) and uba6 (Assay ID: AJPADNW). The efficiency of each probewas tested prior to experimentation using the following equation: PCRefficiency (%)=(10(–1/S) – 1)×100, where S=slope of the standardized curve.
Semi-quantitative RT-PCRAll assays were performed using a Superscript III One-Step RT-PCRSystem with Platinum Taq DNA Polymerase. For each reaction, 30 ng RNAwas used. The wild-type, kolnu7 andmsmo1nu81 alleles were amplified usingthe primers 5′-TGGTCTTATGTGAACTTTCTTTACA-3′ and 5′-GATAAATCCAGGCAGGCAGA-3′. The PCR products were separatedusing a 3.5% Metaphor agarose (Lonza) gel. All assays consisted of threebiological replicates.
CRISPR/Cas9 genome editingGenome editing was performed as previously described (Hwang et al.,2013; Jao et al., 2013). We used linearized plasmid pCS2-nCas9n (Addgeneplasmid #47929) (Jao et al., 2013) as a template for Cas9 mRNA synthesis(mMessage mMachine; Thermo Fisher Scientific). The guide RNA (gRNA)template was made by PCR and RNA was synthesized using aMEGAshortscript T7 kit (Thermo Fisher Scientific). In all cases,30-40 pg gRNA was co-injected with 150 pg Cas9 mRNA per embryo atone-cell stage. Indels were identified by loss of restriction enzyme sites. TheG0 founders positive for mutation were outcrossed with wild-type fish toproduce F1 heterozygotes, which were then analyzed using sequencing toidentify the mutations. Individuals carrying desirable, identified mutationswere outcrossed with wild type to generate an F2 heterozygous family.
High-cholesterol diet (HCD)Beginning at 5 dpf, larvae were fed a HCD similar to that previouslydescribed (Stoletov et al., 2009). Fish were raised in 450 ml system water,6 parts per thousand (ppt) salinity, in 2.8-l tanks, and fed 15 mg of either anormal diet or HCD twice daily. Water was changed daily. Normal (0.12%cholesterol) and HCD (4% cholesterol) was provided by the Department ofBiology, University of Alabama at Birmingham, Birmingham, AL, USA(Watts et al., 2012).
Total cholesterol measurementMutant clutches were collected in Petri dishes filled with egg water at 0 dpf.Embryos were placed in a 28°C incubator and fasted until collection at 8 dpfwhen they were snap frozen. An eyeball of each larva was removed forgenotyping purposes. Once genotyping results were confirmed, individuallarvae were homogenized in 200 µl extraction buffer chloroform:isopropanol:IGEPAL CA-630 (7:11:0.1) using a micro-tube homogenizer.The extraction procedure and total cholesterol measurements were carriedout using a BioVision Total Cholesterol and Cholesteryl Ester Colorimetric/Fluorometric Assay Kit (#K603-100), as per the manufacturer’srecommendations, with 25 µl extracted larva sample used per assay, and25 µl Reaction Mix or 25 µl Background Control Mix used per well. Allassays consisted of three technical sample replicates, three technicalbackground control replicates and three biological replicates. Fluorescencewas detected using a CLARIOstar Microplate Reader (BMG Labtech,Ortenberg, Germany). Mean relative fluorescence units (RFUs) for eachsample were calculated by subtracting the mean background control valuefrom the mean sample value. From these data, the mean RFU for each groupwas calculated. Wild-type mean RFU=206081, s.d. 8150, s.e.m. 4705, n=3;
msmo1nu81 mean RFU=45401, s.d. 26629, s.e.m. 15374, n=3 (P=0.0006);lssnu60 mean RFU=89447, s.d. 22756, s.e.m. 13138, n=3 (P=0.0011).Mutant RFU levels were compared towild-type RFU levels to determine thepercentage of total cholesterol levels. P-values were calculated usingunpaired Student’s t-test.
Embryonic cell transplantationTransplantation was performed as previously described (Ho and Kane,1990; Stafford et al., 2006). Wild-type donor embryos were co-injected atone- to two-cell stage with 20 ng/μl sox32 mRNA and 40 kD fluoresceindextran to allow for visualization of donor cells. Approximately 20-30 cellsfrom high-stage donors [∼3 h post-fertilization (hpf)] were distributed alongthe blastoderm margin of shield-stage (∼6 hpf) hosts generated form amsmo1nu81 heterozygote in-cross. Then, 3 dpf embryos with a significantnumber of transplanted cells located in the intestines were selected forgrowth. At ∼6 weeks of age, fish were photographed using a Zeiss V-8stereomicroscope equipped with a Zeiss Axiocam camera. To detect Tg(ubi:Zebrabow-M)a131 donor cells, a Texas Red filter was used. Host genotypewas determined based on DNA extracted from a fin clip devoid of red,transplanted cells.
Creation of transgenic linesThe genomic and coding sequences of msmo1 and lss were obtained fromWellcome Trust Sanger Ensembl database,Danio rerio (GRCz11). The open-reading frame (ORF) ofmsmo1was amplified fromwild-type RNA; the ORFof lsswas amplified from cDNA clone (NM_001083567.1) and cloned usingGateway BP reaction on the pDONOR221 plasmid (Invitrogen). The fabp10promoter was amplified from pHD157 (Addgene plasmid #84033) (Ni et al.,2012) using primers 5′-CTGAGCATCAGAATGGGGAAG-3′ and 5′-GCTCAACACAAAGTGAAGGTC-3′, and re-amplified with primers 5′-TGATATCGAATTCCTGCAGCCTGAGCATCAGAATGGGG-3′ and 5′-CGCTCTAGAACTAGTGGATCGCTCAACACAAAGTGAAG-3′ to directproper orientation of the insert. Using NEBuilder HiFi DNA AssemblyMaster Mix, this product was then subcloned into the p5E-MCS (Addgeneplasmid #26029) (Quillien et al., 2017) linearized with SmaI. Finally,plasmids were assembled using Gateway LR reaction and clones available inthe Tol2kit (Kwan et al., 2007). To create the hsp70l:msmo1:IRESnlsEGFPplasmid, the ORF ofmsmo1was cloned between the hsp70l promoter and theIRESnlsEGFP sequence in the pDestTol2CG2 vector. To create the fabp10:msmo1:pA and fabp10:lss:pA lines, the ORF of msmo1 or lss was clonedbetween the fabp10 promoter and a polyA sequence in the pDestTol2CG2vector. All transgenic lines were generated with the help of the Tol2transposase method of transgenesis (Kawakami and Shima, 1999; Kwanet al., 2007). As the transgene backbone contains a cmlc2:EGFP element, alltransgene-positive fish were identified at 2 dpf by EGFP-positive hearts.
Heat-shock protocolTo induce transgene expression in Tg(hsp70l:msmo1:IRESnlsEGFP)nu99
progeny of the identified msmo1nu81 heterozygote crosses, fish were heatshocked daily at 42°C for 15 min, beginning at 24 hpf until collection andgenotyping at 14 dpf.
Histological sectionsTwo-month old fish were fixed overnight in phosphate-buffered 10%formalin and treated for 8 h before processing with 0.5 M EDTA, pH8. Samples were processed using an STP 120 Spin Tissue Processor (ThermoFisher Scientific). Dehydration steps were conducted gradually with 10%,30%, 50%, and 70% ethanol, each for 1 h. Final ethanol treatments of 95%and 100%were each performed twice, both for 1 h each. Xylenewas appliedtwice, 45 min each time, and paraffin (Histoplast Paraffin LP 8332; ThermoFisher Scientific) applied three times for 60 min each time. Specimens wereembedded using a Microm EC350-2 tissue-embedding center. All tissuesamples were sectioned at 5 μm using a rotary microtome.
In situ hybridizationThe 918 bp template for the msmo1 anti-sense RNA probe was amplifiedfrom wild-type RNA using a primer tailed with the T7 RNA polymerase
10
RESEARCH ARTICLE Disease Models & Mechanisms (2020) 13, dmm042549. doi:10.1242/dmm.042549
Disea
seModels&Mechan
isms

promoter (5′-TGGTCTTATGTGAACTTTCTTTACA-3′ and 5′-TAATAC-GACTCACTATAGGGGTCTGTCCCACCAGGTGAAT-3′). Whole-mountin situ hybridization was performed as previously described (Thisse andThisse, 2008), using low-stringency conditions. Hybridization mix wassupplemented with 5% dextran sulfate to increase signal intensity (Lauteret al., 2011; Thisse and Thisse, 2014). RNAscope in situ hybridization wascarried out on formalin-fixed, paraffin-embedded (FFPE) 5-μm sections. AllRNA probes, hybridization kits and hybridization equipment, including ACDHybEZ Hybridization oven, were obtained from Advanced Cell Diagnostics(Newark, CA, USA). All FFPE sections were treated with an RNAscope 2.5Universal Pretreatment Reagents kit and RNAscope 2.5 High Definition(HD)-Red Assay kit, as per the manufacturer’s recommendations, with thefollowing adjustments to the tissue pre-treatment protocol. Slides were heatedto 98-102°C in 1× Target Retrieval Reagents for 8 min and incubated at 40°Cin Protease Plus for 15 min. Staining intensity for each probe was adjusted byvarying the incubation time of Hybridization AMP 5. Signal detection timefor each probe was as follows: 5 min for col2a1a (#409471) and col10a1a(#409481) or 30 min for msmo1 (#438981), ihha (#490681), ihhb (#490691)and ptch1 (#490701). The probe dapB (#310043) was included as a negativecontrol in each assay, and incubated for either 5 min or 30 min depending onthe assay. Each probe was tested in a minimum of three independentexperiments on multiple sections. Slides were counterstained with 50%Hematoxylin and 0.02% (w/v) ammonia water, as per the manufacturer’ssuggestions. Photographs were taken using a Zeiss Axioplan 2stereomicroscope equipped with a Zeiss Axiocam camera.
Bone and cartilage stainingLarvae and adult fish were euthanized by submersion in ice-cold systemwater and fixed in phosphate-buffered 10% formalin for 2 h (larvae) or 1-2 days (adults). After rinsing in PBS, specimens were dehydrated graduallyto 70% ethanol for long-term storage at 4°C. Whole-mount skeletal stainsfor adult bone and cartilage were performed as previously described (Taylorand Vandyke, 1985). Bone-only or cartilage-only protocols were performedfor larval fish as previously described (Detrich et al., 2004), with thefollowing modification: larvae were stained in a 0.005% solution of AlizarinRed dissolved in 1% KOH overnight. When not in use, specimens were keptin the dark to avoid fading. Photographs were taken using Zeiss Stemi 2000-CS and Zeiss Axioplan 2 stereomicroscopes, both equipped with a ZeissAxiocam camera.
GenotypingFor all genotyping protocols, small DNA fragments were amplified using theprimers listed in Table S1. The kolnu7 genotyping protocol was developedbased on the polymorphism in linked exon 5 of ndst3 that eliminates a SacIrestriction enzyme site. The genotyping protocols for ndst3nu20, ugt8nu82 andspock3nu83were based on the elimination of restriction enzyme sites (Table 1).The genotyping of msmo1nu81 and lssnu60 alleles takes advantage of a 37 bpinsertion and a 23 bp deletion, respectively. The PCR products were separatedusing either a 3.5% Metaphor agarose (Lonza) gel (kolnu7, spock3nu83,msmo1nu81, lssnu60) or 2.0% agarose gel (ndst3nu20 and ugt8nu82).
Statistical analysisAll statistical calculations were carried out with GraphPad Prism using theChi-square or t-test calculator as noted.
Zebrafish lines and maintenanceAll fish used in this study were raised and cared for in accordance with anapproved protocol by Ann and Robert H. Lurie Children’s Hospital ofChicago Institutional Animal Care and Use Committee (IACUC; #13-036),and complied with National Institutes of Health standards provided in the‘Guide for the Care and Use of Laboratory Animals’. The following wild-type, mutant and transgenic fish lines were used: AB (ZFIN ID: ZDB-GENO-960809-7), TU (ZFIN ID: ZDB-GENO-990623-3), Tg(ubi:Zebrabow-M)a131 (ZFIN ID: ZDB-ALT-130816-2) (Pan et al., 2013),kolnu7, msmo1nu81, lssnu60, ndst3nu20, ugt8nu82, spock3nu83, Tg(hsp70l:msmo1:IRESnlsEGFP)nu99, Tg(fabp10:msmo1:pA)nu100 and Tg(fabp10:lss:pA)nu101. All mutant fish described in the experiments are the results of
crosses between heterozygous parents. The kolnu7mutant line is the result ofan uncharacterized, naturally occurring mutation.
Fish harvestingFish were collected at various developmental stages and SL was measuredfrom snout to the end of the hypuralia, with any post-fixation adjustmentsapplied to SL as necessary (Parichy et al., 2009).
AcknowledgementsWe acknowledge the Stanley Manne Children’s Research Institute Microscopy andHistology Group.
Competing interestsThe authors declare no competing or financial interests.
Author contributionsConceptualization: R.A.A., J.T.; Methodology: R.A.A.; Validation: R.A.A., J.T.;Formal analysis: R.A.A.; Investigation: R.A.A., K.T.S., S.R.M.; Resources: R.A.A.,E.E.L., J.M.T., J.T.; Data curation: R.A.A.; Writing - original draft: R.A.A.; Writing -review & editing: R.A.A., E.E.L., J.T.; Visualization: R.A.A.; Supervision: E.E.L.,J.M.T., J.T.; Project administration: J.T.; Funding acquisition: R.A.A., J.T.
FundingThis work was supported by the National Institute of Dental and CraniofacialResearch (F31DE023481-03 to R.A.A.), Children’s Research Fund (OGS Award toR.A.A.), Lurie Children’s Hospital (to J.T.) and the Walter S. and Lucienne DriskillFoundation (to J.T.).
Supplementary informationSupplementary information available online athttp://dmm.biologists.org/lookup/doi/10.1242/dmm.042549.supplemental
ReferencesA�cimovi�c, J., Goyal, S., Kosir, R., Goli�cnik, M., Perse, M., Beli�c, A., Urlep, Z.,
Guengerich, F. P. and Rozman, D. (2016). Cytochrome P450 metabolism of thepost-lanosterol intermediates explains enigmas of cholesterol synthesis.Sci. Rep.6, 28462. doi:10.1038/srep28462
Albuisson, J., Isidor, B., Giraud, M., Pichon, O., Marsaud, T., David, A., LeCaignec, C. and Bezieau, S. (2011). Identification of two novel mutations in Shhlong-range regulator associated with familial pre-axial polydactyly. Clin. Genet.79, 371-377. doi:10.1111/j.1399-0004.2010.01465.x
Avaron, F., Hoffman, L., Guay, D. andAkimenko,M. A. (2006). Characterization oftwo new zebrafish members of the hedgehog family: atypical expression of azebrafish indian hedgehog gene in skeletal elements of both endochondral anddermal origins. Dev. Dyn. 235, 478-489. doi:10.1002/dvdy.20619
Besnard, T., Sloboda, N., Goldenberg, A., Kury, S., Cogne, B., Breheret, F.,Trochu, E., Conrad, S., Vincent, M., Deb, W. et al. (2019). Biallelic pathogenicvariants in the lanosterol synthase gene LSS involved in the cholesterolbiosynthesis cause alopecia with intellectual disability, a rare recessiveneuroectodermal syndrome. Genet. Med. 21, 2025. doi:10.1038/s41436-019-0445-x
Bird, N. C. and Mabee, P. M. (2003). Developmental morphology of the axialskeleton of the zebrafish, Danio rerio (Ostariophysi: Cyprinidae). Dev. Dyn. 228,337-357. doi:10.1002/dvdy.10387
Bornholdt, D., Konig, A., Happle, R., Leveleki, L., Bittar, M., Danarti, R.,Vahlquist, A., Tilgen, W., Reinhold, U., Poiares Baptista, A. et al. (2005).Mutational spectrum of NSDHL in CHILD syndrome. J. Med. Genet. 42, e17.doi:10.1136/jmg.2004.024448
Braverman, N., Lin, P., Moebius, F. F., Obie, C., Moser, A., Glossmann, H.,Wilcox, W. R., Rimoin, D. L., Smith, M., Kratz, L. et al. (1999). Mutations in thegene encoding 3 beta-hydroxysteroid-delta 8, delta 7-isomerase cause X-linkeddominant Conradi-Hunermann syndrome. Nat. Genet. 22, 291-294. doi:10.1038/10357
Briscoe, J. and Therond, P. P. (2013). The mechanisms of Hedgehog signallingand its roles in development and disease. Nat. Rev. Mol. Cell Biol. 14, 416-429.doi:10.1038/nrm3598
Byrne, E. F., Sircar, R., Miller, P. S., Hedger, G., Luchetti, G., Nachtergaele, S.,Tully, M. D., Mydock-McGrane, L., Covey, D. F., Rambo, R. P. et al. (2016).Structural basis of Smoothened regulation by its extracellular domains. Nature535, 517-522. doi:10.1038/nature18934
Canueto, J., Giros, M. and Gonzalez-Sarmiento, R. (2014). The role of theabnormalities in the distal pathway of cholesterol biosynthesis in the Conradi-Hunermann-Happle syndrome. Biochim. Biophys. Acta 1841, 336-344. doi:10.1016/j.bbalip.2013.09.002
11
RESEARCH ARTICLE Disease Models & Mechanisms (2020) 13, dmm042549. doi:10.1242/dmm.042549
Disea
seModels&Mechan
isms

Chen, X. and Liu, L. (2017). Congenital cataract with LSS gene mutations: a newcase report. J. Pediatr. Endocrinol. Metab. 30, 1231-1235. doi:10.1515/jpem-2017-0101
Chen, X., Tukachinsky, H., Huang, C.-H., Jao, C., Chu, Y.-R., Tang, H.-Y.,Mueller, B., Schulman, S., Rapoport, T. A. andSalic, A. (2011). Processing andturnover of the Hedgehog protein in the endoplasmic reticulum. J. Cell Biol. 192,825-838. doi:10.1083/jcb.201008090
Cooper, M. K., Wassif, C. A., Krakowiak, P. A., Taipale, J., Gong, R., Kelley, R. I.,Porter, F. D. and Beachy, P. A. (2003). A defective response to Hedgehogsignaling in disorders of cholesterol biosynthesis.Nat. Genet. 33, 508-513. doi:10.1038/ng1134
Corpet, F. (1988). Multiple sequence alignment with hierarchical clustering. NucleicAcids Res. 16, 10881–10890. doi:10.1093/nar/16.22.10881
Cubbage, C. C. and Mabee, P. M. (1996). Development of the cranium and pairedfins in the zebrafish danio rerio (Ostariophysi, Cyprinidae). J. Morphol. 229,121-160. doi:10.1002/(SICI)1097-4687(199608)229:2<121::AID-JMOR1>3.0.CO;2-4
Dale, R. M. and Topczewski, J. (2011). Identification of an evolutionarily conservedregulatory element of the zebrafish col2a1a gene.Dev. Biol. 357, 518-531. doi:10.1016/j.ydbio.2011.06.020
Dathe, K., Kjaer, K. W., Brehm, A., Meinecke, P., Nurnberg, P., Neto, J. C.,Brunoni, D., Tommerup, N., Ott, C. E., Klopocki, E. et al. (2009). Duplicationsinvolving a conserved regulatory element downstream of BMP2 are associatedwith brachydactyly type A2. Am. J. Hum. Genet. 84, 483-492. doi:10.1016/j.ajhg.2009.03.001
Detrich, H. W., Westerfield, M. and Zon, L. I. (2004). The Zebrafish: 2nd EditionCellular and Developmental Biology. 2 edn: Academic Press.
Dy, P.,Wang,W., Bhattaram, P.,Wang, Q., Wang, L., Ballock, R. T. and Lefebvre,V. (2012). Sox9 directs hypertrophic maturation and blocks osteoblastdifferentiation of growth plate chondrocytes. Dev. Cell 22, 597-609. doi:10.1016/j.devcel.2011.12.024
Eames, B. F., Amores, A., Yan, Y.-L. and Postlethwait, J. H. (2012). Evolution ofthe osteoblast: skeletogenesis in gar and zebrafish. BMC Evol. Biol. 12, 27.doi:10.1186/1471-2148-12-27
Eberhart, J. K., Swartz, M. E., Crump, J. G. and Kimmel, C. B. (2006). EarlyHedgehog signaling from neural to oral epithelium organizes anterior craniofacialdevelopment. Development 133, 1069-1077. doi:10.1242/dev.02281
Felber, K., Croucher, P. and Roehl, H. H. (2011). Hedgehog signalling is requiredfor perichondral osteoblast differentiation in zebrafish. Mech. Dev. 128, 141-152.doi:10.1016/j.mod.2010.11.006
Frisso, G., Gelzo, M., Procopio, E., Sica, C., Lenza, M. P., Dello Russo, A.,Donati, M. A., Salvatore, F. and Corso, G. (2017). A rare case of sterol-C4-methyl oxidase deficiency in a young Italian male: Biochemical and molecularcharacterization. Mol. Genet. Metab. 121, 329-335. doi:10.1016/j.ymgme.2017.06.013
Gabitova, L., Restifo, D., Gorin, A., Manocha, K., Handorf, E., Yang, D.-H., Cai,K. Q., Klein-Szanto, A. J., Cunningham, D., Kratz, L. E. et al. (2015).Endogenous sterol metabolites regulate growth of EGFR/KRAS-dependenttumors via LXR. Cell Rep 12, 1927-1938. doi:10.1016/j.celrep.2015.08.023
Gao, B., Hu, J., Stricker, S., Cheung, M., Ma, G., Law, K. F., Witte, F., Briscoe, J.,Mundlos, S., He, L. et al. (2009). A mutation in Ihh that causes digit abnormalitiesalters its signalling capacity and range. Nature 458, 1196-1200. doi:10.1038/nature07862
Hammond, C. L. and Schulte-Merker, S. (2009). Two populations of endochondralosteoblasts with differential sensitivity to Hedgehog signalling. Development 136,3991-4000. doi:10.1242/dev.042150
He, M., Kratz, L. E., Michel, J. J., Vallejo, A. N., Ferris, L., Kelley, R. I., Hoover,J. J., Jukic, D., Gibson, K. M., Wolfe, L. A. et al. (2011). Mutations in the humanSC4MOL gene encoding a methyl sterol oxidase cause psoriasiform dermatitis,microcephaly, and developmental delay. J. Clin. Invest. 121, 976-984. doi:10.1172/JCI42650
He, M., Smith, L. D., Chang, R., Li, X. and Vockley, J. (2014). The role of sterol-C4-methyl oxidase in epidermal biology. Biochim. Biophys. Acta 1841, 331-335.doi:10.1016/j.bbalip.2013.10.009
Her, G. M., Yeh, Y.-H. andWu, J.-L. (2003). 435-bp liver regulatory sequence in theliver fatty acid binding protein (L-FABP) gene is sufficient tomodulate liver regionalexpression in transgenic zebrafish. Dev. Dyn. 227, 347-356. doi:10.1002/dvdy.10324
Herman, G. E. and Kratz, L. (2012). Disorders of sterol synthesis: beyond Smith-Lemli-Opitz syndrome. Am. J. Med. Genet. C Semin. Med. Genet. 160C, 301-321.doi:10.1002/ajmg.c.31340
Ho, R. K. and Kane, D. A. (1990). Cell-autonomous action of zebrafish spt-1mutation in specific mesodermal precursors. Nature 348, 728-730. doi:10.1038/348728a0
Hu, X., Wang, Y., Hao, L.-Y., Liu, X., Lesch, C. A., Sanchez, B. M., Wendling,J. M., Morgan, R. W., Aicher, T. D., Carter, L. L. et al. (2015). Sterol metabolismcontrols T(H)17 differentiation by generating endogenous RORgamma agonists.Nat. Chem. Biol. 11, 141-147. doi:10.1038/nchembio.1714
Huff, M. W. and Telford, D. E. (2005). Lord of the rings–the mechanism foroxidosqualene:lanosterol cyclase becomes crystal clear. Trends Pharmacol. Sci.26, 335-340. doi:10.1016/j.tips.2005.05.004
Hui, C. (1993). A mouse model of Greig cephalopolysyndactyly syndrome: the extratoesJmutation contains an intragenic deletion of theGli3 gene.Nature 3, 241-246.doi:10.1038/ng0393-241
Hui, C.-C. and Joyner, A. L. (1993). Amousemodel of greig cephalopolysyndactylysyndrome: the extra-toesJ mutation contains an intragenic deletion of the Gli3gene. Nat. Genet. 3, 241-246. doi:10.1038/ng0393-241
Hwang, W. Y., Fu, Y., Reyon, D., Maeder, M. L., Tsai, S. Q., Sander, J. D.,Peterson, R. T., Yeh, J.-R. J. and Joung, J. K. (2013). Efficient genome editing inzebrafish using a CRISPR-Cas system. Nat. Biotechnol. 31, 227-229. doi:10.1038/nbt.2501
Ikegami, D., Akiyama, H., Suzuki, A., Nakamura, T., Nakano, T., Yoshikawa, H.and Tsumaki, N. (2011). Sox9 sustains chondrocyte survival and hypertrophy inpart through Pik3ca-Akt pathways. Development 138, 1507-1519. doi:10.1242/dev.057802
Irving, M. D., Chitty, L. S., Mansour, S. and Hall, C. M. (2008). Chondrodysplasiapunctata: a clinical diagnostic and radiological review. Clin. Dysmorphol. 17,229-241. doi:10.1097/MCD.0b013e3282fdcc70
Jao, L.-E., Wente, S. R. and Chen, W. (2013). Efficient multiplex biallelic zebrafishgenome editing using a CRISPR nuclease system. Proc. Natl. Acad. Sci. USA110, 13904-13909. doi:10.1073/pnas.1308335110
Jira, P. (2013). Cholesterol metabolism deficiency. Handb. Clin. Neurol. 113,1845-1850. doi:10.1016/B978-0-444-59565-2.00054-X
Jurkiewicz, E., Marcinska, B., Bothur-Nowacka, J. and Dobrzanska, A. (2013).Clinical and radiological pictures of two newborn babies with manifestations ofchondrodysplasia punctata and review of available literature. Pol. J. Radiol. 78,57-64. doi:10.12659/PJR.883947
Kawakami, K. and Shima, A. (1999). Identification of the Tol2 transposase of themedaka fish Oryzias latipes that catalyzes excision of a nonautonomous Tol2element in zebrafish Danio rerio. Gene 240, 239-244. doi:10.1016/S0378-1119(99)00444-8
Kim, Y.-I., Lee, S., Jung, S.-H., Kim, H.-T., Choi, J. H., Lee, M.-S., You, K.-H., Yeo,S.-Y., Yoo, K.-W., Kwak, S. A. et al. (2013). Establishment of a bone-specificcol10a1:GFP transgenic zebrafish. Mol. Cells 36, 145-150. doi:10.1007/s10059-013-0117-7
Knapik, E. W., Goodman, A., Ekker, M., Chevrette, M., Delgado, J., Neuhauss,S., Shimoda, N., Driever, W., Fishman, M. C. and Jacob, H. J. (1998). Amicrosatellite genetic linkage map for zebrafish (Danio rerio). Nat. Genet. 18,338-343. doi:10.1038/ng0498-338
Kouwenhoven, E. N., van Heeringen, S. J., Tena, J. J., Oti, M., Dutilh, B. E.,Alonso, M. E., de la Calle-Mustienes, E., Smeenk, L., Rinne, T., Parsaulian, L.et al. (2010). Genome-wide profiling of p63 DNA-binding sites identifies anelement that regulates gene expression during limb development in the 7q21SHFM1 locus. PLoS Genet. 6, e1001065. doi:10.1371/journal.pgen.1001065
Kronenberg, H. M. (2003). Developmental regulation of the growth plate. Nature423, 332-336. doi:10.1038/nature01657
Kurth, I., Klopocki, E., Stricker, S., van Oosterwijk, J., Vanek, S., Altmann, J.,Santos, H. G., van Harssel, J. J., de Ravel, T., Wilkie, A. O. et al. (2009).Duplications of noncoding elements 5′ of SOX9 are associated withbrachydactyly-anonychia. Nat. Genet. 41, 862-863. doi:10.1038/ng0809-862
Kwan, K. M., Fujimoto, E., Grabher, C., Mangum, B. D., Hardy, M. E., Campbell,D. S., Parant, J. M., Yost, H. J., Kanki, J. P. and Chien, C. B. (2007). The Tol2kit:a multisite gateway-based construction kit for Tol2 transposon transgenesisconstructs. Dev. Dyn. 236, 3088-3099. doi:10.1002/dvdy.21343
Laue, K., Janicke, M., Plaster, N., Sonntag, C. and Hammerschmidt, M. (2008).Restriction of retinoic acid activity by Cyp26b1 is required for proper timing andpatterning of osteogenesis during zebrafish development. Development 135,3775-3787. doi:10.1242/dev.021238
Lauter, G., Soll, I. and Hauptmann, G. (2011). Two-color fluorescent in situhybridization in the embryonic zebrafish brain using differential detection systems.BMC Dev. Biol. 11, 43. doi:10.1186/1471-213X-11-43
LeClair, E. E., Mui, S. R., Huang, A., Topczewska, J. M. and Topczewski, J.(2009). Craniofacial skeletal defects of adult zebrafish Glypican 4 (knypek)mutants. Dev. Dyn. 238, 2550-2563. doi:10.1002/dvdy.22086
Lefebvre, V., Angelozzi, M. and Haseeb, A. (2019). SOX9 in cartilagedevelopment and disease. Curr. Opin. Cell Biol. 61, 39-47. doi:10.1016/j.ceb.2019.07.008
Li, Z., Korzh, V. and Gong, Z. (2007). Localized rbp4 expression in the yolksyncytial layer plays a role in yolk cell extension and early liver development. BMCDev. Biol. 7, 117. doi:10.1186/1471-213X-7-117
Livak, K. J. and Schmittgen, T. D. (2001). Analysis of relative gene expression datausing real-time quantitative PCR and the 2(-delta delta C(T)) method.Methods 25,402–408. doi:10.1006/meth.2001.1262
Lykissas, M. (2013). Challenges of spine surgery in patients with chondrodysplasiapunctata. J. Pediatr. Orthop. 33, 685-693. doi:10.1097/BPO.0b013e31829e86a9
MacRae, C. A. and Peterson, R. T. (2015). Zebrafish as tools for drug discovery.Nat. Rev. Drug Discov. 14, 721-731. doi:10.1038/nrd4627
12
RESEARCH ARTICLE Disease Models & Mechanisms (2020) 13, dmm042549. doi:10.1242/dmm.042549
Disea
seModels&Mechan
isms

Mi, X. B., Luo, M. X., Guo, L. L., Zhang, T. D. and Qiu, X. W. (2015). CHILDsyndrome: case report of a Chinese patient and literature review of the NAD[P]Hsteroid dehydrogenase-like protein gene mutation. Pediatr. Dermatol. 32,e277–e282. doi:10.1111/pde.12701
Miyares, R. L., de Rezende, V. B. and Farber, S. A. (2014). Zebrafish yolk lipidprocessing: a tractable tool for the study of vertebrate lipid transport andmetabolism. Dis. Model. Mech. 7, 915-927. doi:10.1242/dmm.015800
Mudumana, S. P., Wan, H., Singh, M., Korzh, V. and Gong, Z. (2004). Expressionanalyses of zebrafish transferrin, ifabp, and elastaseB mRNAs as differentiationmarkers for the three major endodermal organs: liver, intestine, and exocrinepancreas. Dev. Dyn. 230, 165-173. doi:10.1002/dvdy.20032
Ni, T. T., Lu, J., Zhu, M., Maddison, L. A., Boyd, K. L., Huskey, L., Ju, B.,Hesselson, D., Zhong, T. P., Page-McCaw, P. S. et al. (2012). Conditionalcontrol of gene function by an invertible gene trap in zebrafish. Proc. Natl. Acad.Sci. USA 109, 15389-15394. doi:10.1073/pnas.1206131109
Niu, M., Tabari, E., Ni, P. and Su, Z. (2018). Towards a map of cis-regulatorysequences in the human genome. Nucleic Acids Res. 46, 5395-5409. doi:10.1093/nar/gky338
Ornitz, D. M. and Marie, P. J. (2002). FGF signaling pathways in endochondral andintramembranous bone development and human genetic disease. Genes Dev.16, 1446-1465. doi:10.1101/gad.990702
Pan, Y. A., Freundlich, T., Weissman, T. A., Schoppik, D., Wang, X. C.,Zimmerman, S., Ciruna, B., Sanes, J. R., Lichtman, J. W. and Schier, A. F.(2013). Zebrabow:multispectral cell labeling for cell tracing and lineage analysis inzebrafish. Development 140, 2835-2846. doi:10.1242/dev.094631
Parichy, D. M., Elizondo, M. R., Mills, M. G., Gordon, T. N. and Engeszer, R. E.(2009). Normal table of postembryonic zebrafish development: staging byexternally visible anatomy of the living fish. Dev. Dyn. 238, 2975-3015. doi:10.1002/dvdy.22113
Pogoda, H. M., Riedl-Quinkertz, I., Lohr, H., Waxman, J. S., Dale, R. M.,Topczewski, J., Schulte-Merker, S. and Hammerschmidt, M. (2018). Directactivation of chordoblasts by retinoic acid is required for segmented centramineralization during zebrafish spine development. Development 145,dev159418. doi:10.1242/dev.159418
Porter, F. D. (2008). Smith-Lemli-Opitz syndrome: pathogenesis, diagnosis andmanagement. Eur. J. Hum. Genet. 16, 535–541. doi:10.1038/ejhg.2008.10
Porter, F. D. and Herman, G. E. (2011). Malformation syndromes caused bydisorders of cholesterol synthesis. J. Lipid Res. 52, 6-34. doi:10.1194/jlr.R009548
Porter, J. A., von Kessler, D. P., Ekker, S. C., Young, K. E., Lee, J. J., Moses, K.and Beachy, P. A. (1995). The product of hedgehog autoproteolytic cleavageactive in local and long-range signalling. Nature 374, 363-366. doi:10.1038/374363a0
Quillien, A., Abdalla, M., Yu, J., Ou, J., Zhu, L. J. and Lawson, N. D. (2017).Robust identification of developmentally active endothelial enhancers in zebrafishusing FANS-assisted ATAC-Seq. Cell Rep 20, 709-720. doi:10.1016/j.celrep.2017.06.070
Quinlivan, V. H. and Farber, S. A. (2017). Lipid uptake, metabolism, and transportin the larval zebrafish. Front. Endocrinol. (Lausanne) 8, 319. doi:10.3389/fendo.2017.00319
Romano, M. T., Tafazzoli, A., Mattern, M., Sivalingam, S., Wolf, S., Rupp, A.,Thiele, H., Altmuller, J., Nurnberg, P., Ellwanger, J. et al. (2018). Bi-allelicmutations in LSS, encoding lanosterol synthase, cause autosomal-recessivehypotrichosis simplex. Am. J. Hum. Genet. 103, 777-785. doi:10.1016/j.ajhg.2018.09.011
Rossi, M., Hall, C. M., Bouvier, R., Collardeau-Frachon, S., Le Breton, F.,Bucourt, M., Cordier, M. P., Vianey-Saban, C., Parenti, G., Andria, G. et al.(2015). Radiographic features of the skeleton in disorders of post-squalenecholesterol biosynthesis. Pediatr. Radiol. 45, 965-976. doi:10.1007/s00247-014-3257-9
Santori, F. R., Huang, P., van de Pavert, S. A., Douglass, E. F., Jr., Leaver, D. J.,Haubrich, B. A., Keber, R., Lorbek, G., Konijn, T., Rosales, B. N. et al. (2015).Identification of natural RORgamma ligands that regulate the development oflymphoid cells. Cell Metab. 21, 286-297. doi:10.1016/j.cmet.2015.01.004
Schwend, T. and Ahlgren, S. C. (2009). Zebrafish con/disp1 reveals multiplespatiotemporal requirements for Hedgehog-signaling in craniofacial development.BMC Dev. Biol. 9, 59. doi:10.1186/1471-213X-9-59
Sharpe, L. J. and Brown, A. J. (2013). Controlling cholesterol synthesis beyond 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR). J. Biol. Chem. 288,18707-18715. doi:10.1074/jbc.R113.479808
Spoorendonk, K. M., Peterson-Maduro, J., Renn, J., Trowe, T., Kranenbarg, S.,Winkler, C. and Schulte-Merker, S. (2008). Retinoic acid and Cyp26b1 arecritical regulators of osteogenesis in the axial skeleton. Development 135,3765-3774. doi:10.1242/dev.024034
St-Jacques, B., Hammerschmidt, M. and McMahon, A. P. (1999). Indianhedgehog signaling regulates proliferation and differentiation of chondrocytesand is essential for bone formation.Genes Dev. 13, 2072-2086. doi:10.1101/gad.13.16.2072
Stafford, D., White, R. J., Kinkel, M. D., Linville, A., Schilling, T. F. and Prince,V. E. (2006). Retinoids signal directly to zebrafish endoderm to specify insulin-expressing beta-cells. Development 133, 949-956. doi:10.1242/dev.02263
Stoletov, K., Fang, L., Choi, S. H., Hartvigsen, K., Hansen, L. F., Hall, C., Pattison,J., Juliano, J., Miller, E. R., Almazan, F. et al. (2009). Vascular lipid accumulation,lipoprotein oxidation, and macrophage lipid uptake in hypercholesterolemiczebrafish. Circ. Res. 104, 952-960. doi:10.1161/CIRCRESAHA.108.189803
Sukhanova, A., Gorin, A., Serebriiskii, I. G., Gabitova, L., Zheng, H., Restifo, D.,Egleston, B. L., Cunningham, D., Bagnyukova, T., Liu, H. et al. (2013).Targeting C4-demethylating genes in the cholesterol pathway sensitizes cancercells to EGF receptor inhibitors via increased EGF receptor degradation. CancerDiscov 3, 96-111. doi:10.1158/2159-8290.CD-12-0031
Talbot, W. S. and Schier, A. F. (1999). Positional cloning of mutated zebrafishgenes. Methods Cell Biol. 60, 259-286. doi:10.1016/S0091-679X(08)61905-6
Tan, J. L. and Zon, L. I. (2011). Chemical screening in zebrafish for novel biologicaland therapeutic discovery.Methods Cell Biol. 105, 493-516. doi:10.1016/B978-0-12-381320-6.00021-7
Taylor, W. R. and Vandyke, G. C. (1985). Revised procedures for staining andclearing small fishes and other vertebrates for bone and cartilage study.Cybium 9,107-120.
Thisse, B. and Thisse, C. (2014). In situ hybridization on whole-mount zebrafishembryos and young larvae. Methods Mol. Biol. 1211, 53-67. doi:10.1007/978-1-4939-1459-3_5
Thisse, C. and Thisse, B. (2008). High-resolution in situ hybridization to whole-mount zebrafish embryos. Nat. Protoc. 3, 59-69. doi:10.1038/nprot.2007.514
Topczewska, J. M., Shoela, R. A., Tomaszewski, J. P., Mirmira, R. B. andGosain, A. K. (2016). The morphogenesis of cranial sutures in zebrafish. PLoSONE 11, e0165775. doi:10.1371/journal.pone.0165775
Turley, S. D. (2004). Cholesterol metabolism and therapeutic targets: rationale fortargeting multiple metabolic pathways. Clin. Cardiol. 27, III16-III21. doi:10.1002/clc.4960271506
Urlep, Z., Lorbek, G., Perse, M., Jeruc, J., Juvan, P., Matz-Soja, M., Gebhardt,R., Bjorkhem, I., Hall, J. A., Bonneau, R. et al. (2017). Disrupting HepatocyteCyp51 from Cholesterol Synthesis Leads to Progressive Liver Injury in theDeveloping Mouse and Decreases RORC Signalling. Sci. Rep. 7, 40775. doi:10.1038/srep40775
Venter, J. C., Adams, M. D., Myers, E. W., Li, P. W., Mural, R. J., Sutton, G. G.,Smith, H. O., Yandell, M., Evans, C. A., Holt, R. A. et al. (2001). The sequence ofthe human genome. Science 291, 1304-1351. doi:10.1126/science.1058040
Vortkamp, A., Lee, K., Lanske, B., Segre, G. V., Kronenberg, H. M. and Tabin,C. J. (1996). Regulation of rate of cartilage differentiation by Indian hedgehog andPTH-related protein. Science 273, 613-622. doi:10.1126/science.273.5275.613
Wada, N., Javidan, Y., Nelson, S., Carney, T. J., Kelsh, R. N. and Schilling, T. F.(2005). Hedgehog signaling is required for cranial neural crest morphogenesisand chondrogenesis at the midline in the zebrafish skull. Development 132,3977-3988. doi:10.1242/dev.01943
Wassif, C. A., Maslen, C., Kachilele-Linjewile, S., Lin, D., Linck, L. M., Connor,W. E., Steiner, R. D. and Porter, F. D. (1998). Mutations in the human steroldelta7-reductase gene at 11q12-13 cause Smith-Lemli-Opitz syndrome. Am. J.Hum. Genet. 63, 55–62. doi:10.1086/301936
Watts, S. A., Powell, M. and D’Abramo, L. R. (2012). Fundamental approaches tothe study of zebrafish nutrition. ILAR J. 53, 144-160. doi:10.1093/ilar.53.2.144
Wieczorek, D., Pawlik, B., Li, Y., Akarsu, N. A., Caliebe, A., May, K. J.,Schweiger, B., Vargas, F. R., Balci, S., Gillessen-Kaesbach, G. et al. (2010). Aspecific mutation in the distant sonic hedgehog (SHH) cis-regulator (ZRS) causesWerner mesomelic syndrome (WMS) while complete ZRS duplications underlieHaas type polysyndactyly and preaxial polydactyly (PPD) with or withouttriphalangeal thumb. Hum. Mutat. 31, 81-89. doi:10.1002/humu.21142
Wray, G. A. (2007). The evolutionary significance of cis-regulatory mutations. Nat.Rev. Genet. 8, 206-216. doi:10.1038/nrg2063
Yang, C., McDonald, J. G., Patel, A., Zhang, Y., Umetani, M., Xu, F., Westover,E. J., Covey, D. F., Mangelsdorf, D. J., Cohen, J. C. et al. (2006). Sterolintermediates from cholesterol biosynthetic pathway as liver X receptor ligands.J. Biol. Chem. 281, 27816-27826. doi:10.1074/jbc.M603781200
Yang, J., Andre, P., Ye, L. and Yang, Y. Z. (2015). The Hedgehog signallingpathway in bone formation. Int. J. Oral. Sci. 7, 73-79. doi:10.1038/ijos.2015.14
Zeng, X. (2001). A freely diffusible form of Sonic hedgehog mediates long-rangesignalling. Nature 411, 716-720. doi:10.1038/35079648
Zeng, X., Goetz, J. A., Suber, L. M., Scott, W. J., Jr., Schreiner, C. M. andRobbins, D. J. (2001). A freely diffusible form of Sonic hedgehog mediates long-range signalling. Nature 411, 716-720. doi:10.1038/35079648
Zhao, Q., Eberspaecher, H., Lefebvre, V. and De Crombrugghe, B. (1997).Parallel expression of Sox9 and Col2a1 in cells undergoing chondrogenesis.Dev.Dyn. 209, 377-386. doi:10.1002/(SICI)1097-0177(199708)209:4<377::AID-AJA5>3.0.CO;2-F
Zhao, L., Chen, X.-J., Zhu, J., Xi, Y.-B., Yang, X., Hu, L.-D., Ouyang, H., Patel,S. H., Jin, X., Lin, D. et al. (2015). Lanosterol reverses protein aggregation incataracts. Nature 523, 607-611. doi:10.1038/nature14650
13
RESEARCH ARTICLE Disease Models & Mechanisms (2020) 13, dmm042549. doi:10.1242/dmm.042549
Disea
seModels&Mechan
isms





























