Embed Size (px)
Citation preview

HUMAN MUTATION 28(3), 273^283,2007
RESEARCH ARTICLE
Schimke Immunoosseous Dysplasia: Suggestionsof Genetic Diversity
J. Marietta Clewing,1 Helen Fryssira,2 David Goodman,3 Sarah F. Smithson,4 Emily A. Sloan,1 Shu Lou,1
Yan Huang,1 Kunho Choi,1 Thomas Lucke,5 Harika Alpay,6 Jean-Luc Andre,7 Yumi Asakura,8 NathalieBiebuyck-Gouge,9 Radovan Bogdanovic,10 Dominique Bonneau,11 Caterina Cancrini,12 Pierre Cochat,13
Sandra Cockfield,14 Laure Collard,15 Isabel Cordeiro,16 Valerie Cormier-Daire,17 Karlien Cransberg,18
Karel Cutka,19 Georges Deschenes,20 Jochen H.H. Ehrich,5 Stefan Frund,21 Helen Georgaki,22 EncarnaGuillen-Navarro,23 Barbara Hinkelmann,24 Maria Kanariou,25 Belde Kasap,26 Sara Sebnem Kilic,27
Guiliana Lama,28 Petra Lamfers,29 Chantal Loirat,30 Silvia Majore,31 David Milford,32 Denis Morin,33
Nihal Ozdemir,6 Bertram F. Pontz,34 Willem Proesmans,35 Stavroula Psoni,2 Herbert Reichenbach,36 SilkeReif,37 Cristina Rusu,38 Jorge M. Saraiva,39 Onur Sakallioglu,40 Beate Schmidt,41 Lawrence Shoemaker,42
Sabine Sigaudy,43 Graham Smith,44 Flora Sotsiou,45 Natasa Stajic,9 Anja Stein,46 AsbjørgStray-Pedersen,47 Doris Taha,48 Sophie Taque,49 Jane Tizard,50 Michel Tsimaratos,51 Newton A.C.S.Wong,52 and Cornelius F. Boerkoel1�
1Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, Texas; 2Department of Medical Genetics, AthensUniversity Medical School, ‘‘Aghia Sophia’’ Children’s Hospital, Athens, Greece; 3Department of Pathology, University of North Carolina School ofMedicine, Chapel Hill, North Carolina; 4Department of Clinical Genetics, St Michael’s Hospital, Bristol, United Kingdom; 5Department ofPediatrics, Hannover Medical School, Hannover, Germany; 6Pediatric Nephrology, Marmara University, Istanbul, Turkey; 7NephrologiePediatrique, Hopital d’Enfants, Centre Hospitalier Universitaire de Nancy, Vandoeuvre les Nancy Cedex, France; 8Department of Endocrinologyand Metabolism, Kanagawa Children’s Medical Center, Yokohama, Japan; 9Nephrologie Pediatrique, Necker-Enfant Malades, Paris, France;10Institute of Mother and Child Health Care of Serbia, Belgrade, Serbia; 11Service de Genetique Medicale, Centre Hospitalier Universitaired’Angers, Angers, France; 12Children’s Hospital - Bambino Gesu, Unita Operativa di Immunoinfettivologia, Rome, Italy; 13Departement dePediatrie, Hopital Edouard Herriot, Lyon, France; 14University of Alberta, Edmonton, Alberta, Canada; 15Paediatric Nephrology, CentreHospitalier Regional-Centre Hospitalier (CHU-CHR-CHC) Liege, Belgium; 16Unidade de Genetica, Hospital de Santa Maria, Servico dePediatria, Lisbon, Portugal; 17Group Hospitalier, Necker-Enfants Malades, Paris, France; 18Department of Pediatric Nephrology, Erasmus MedischCentrum (MC) Sophia, Rotterdam, The Netherlands; 19Faculty Hospital of Prague-Charles University, Ceske Budejovice, Czech Republic;20Department de Pediatre, Hopital Robert Debre, Paris, France; 21Kuratorium fur Hemidialyse (KfH) Kinderdialyse, Munster, Germany;22Department of Nephrology, Athens University Medical School, ‘‘Aghia Sophia’’ Children’s Hospital, Athens, Greece; 23Unidad de GeneticaMedica, Servicio de Pediatrıa, Hospital Universitario Virgen de La Arrixaca, Murcia, Spain; 24Karolinska University Hospital, Stockholm, Sweden;25Department of Immunology, Athens University Medical School, ‘‘Aghia Sophia’’ Children’s Hospital, Athens, Greece; 26Department of Pediatrics,Division of Nephrology, Dokuz Eylul University, Izmir, Turkey; 27Department of Pediatrics, Division of Immunology, Uludag University MedicalFaculty, Gorukle-Bursa, Turkey; 28Department of Pediatrics, University of Naples, Naples, Italy; 29Mercy Pediatrics and Adolescent Clinic, ClearLake, Iowa; 30Assistance Publique-Hopitaux de Paris, Hopital Robert Debre, Paris, France; 31Medical Genetics, Department of ExperimentalMedicine and Pathology, University La Sapienza, S. Camillo-Forlanini Hospital, Rome, Italy; 32Department of Nephrology, Birmingham Children’sHospital, Birmingham, United Kingdom; 33Service de Pediatrie, Centre Hospitalier Universitaire de Montpellier, Montpellier, France; 34Children’sHospital, Technical University, Munich, Germany; 35Renal Unit, Department of Pediatrics, University of Leuven, Leuven, Belgium; 36Facharztfuer Humangenetik, Halle, Germany; 37Institut fur Humangenetik und Medizinische Biologie, Halle/Saale, Germany; 38Medical Genetics Centre,St. Mary Hospital of Iasi, Iasi, Romania; 39Hospital Pediatrico de Coimbra, Consulta de Genetica, Coimbra, Portugal; 40GATA PediatricNephrology Unite, Ankara, Turkey; 41Children’s Hospital, University of Cologne, Cologne, Germany; 42Department of Pediatrics, Division ofNephrology, University of Louisville School of Medicine and Kosair Children’s Hospital, Louisville, Kentucky; 43Departement de GenetiqueMedicale, Hopital Timone Enfant, Marseille, France; 44Kidney Research Unit for Wales Foundation (KRUF) Children’s Kidney Centre,University Hospital of Wales, Heath Park, Cardiff, United Kingdom; 45Renal Pathology Department, ‘‘Evangelismos’’ Hospital, Athens, Greece;
Published online 6 November 2006 in Wiley InterScience (www.interscience.wiley.com).yThis article is a US Government work and, as such, is in the public
domain in the United States of America.
DOI10.1002/humu.20432
The Supplementary Material referred to in this article can be ac-cessed at http://www.interscience.wiley.com/jpages/1059-7794/suppmat.Received 30 June 2006; accepted revised manuscript 5 September
2006.
Grant sponsor: NIH National Institute of Diabetes, Digestive, andKidney Diseases; Grant sponsor: Gillson Longenbaugh Foundation;Grant sponsor: Dana Foundation.
J. Marietta Clewing and Helen Fryssira contributed equally to thiswork.
�Correspondence to:Cornelius F. Boerkoel, M.D., Ph.D., ProvincialMedical Genetics Program, Department of Medical Genetics, Chil-dren’s and Women’s Health Centre of BC, 4500 Oak St., Rm. C234,Vancouver, B.C. V6H 3N1Canada.E-mail: [email protected]
PUBLISHED 2006 WILEY-LISS, INC.

46Universitatsklinikum Essen, Kinderklinik, Essen, Germany; 47Department of Medical Genetics, Rikshospitalet, Oslo, Norway; 48Department ofPediatrics, King Faisal Specialist Hospital and Research Centre-Jeddah, Jeddah, Saudi Arabia; 49Centre Hospitalier Universitaire (CHU)Rennes, Pediatrie Hopital Sud, Rennes, France; 50Department of Paediatric Nephrology, Bristol Childrens Hospital, Bristol, United Kingdom;51Pediatric Dialysis and Nephrology, Hopital Timone Enfants, Marseille, France; 52Department of Histopathology, Bristol Royal Infirmary, Bristol,United Kingdom
Communicated by Iain McIntosh
Schimke immunoosseous dysplasia (SIOD), which is characterized by prominent spondyloepiphyseal dysplasia,T-cell deficiency, and focal segmental glomerulosclerosis, is a panethnic autosomal recessive multisystemdisorder with variable expressivity. Biallelic mutations in switch/sucrose nonfermenting (swi/snf) related,matrix-associated, actin-dependent regulator of chromatin, subfamily a-like 1 (SMARCAL1) are the onlyidentified cause of SIOD. However, among 72 patients from different families, we identified only 38 patientswith biallelic mutations in the coding exons and splice junctions of the SMARCAL1 gene. This observation, thevariable expressivity, and poor genotype–phenotype correlation led us to test several hypotheses includingmodifying haplotypes, oligogenic inheritance, or locus heterogeneity in SIOD. Haplotypes associated with thetwo more common mutations, R820H and E848X, did not correlate with phenotype. Also, contrary tomonoallelic SMARCAL1 coding mutations indicating oligogenic inheritance, we found that all these patientsdid not express RNA and/or protein from the other allele and thus have biallelic SMARCAL1 mutations. Wehypothesize therefore that the variable expressivity among patients with biallelic SMARCAL1 mutations arisesfrom environmental, genetic, or epigenetic modifiers. Among patients without detectable SMARCAL1 codingmutations, our analyses of cell lines from four of these patients showed that they expressed normal levels ofSMARCAL1 mRNA and protein. This is the first evidence for nonallelic heterogeneity in SIOD. From analysisof the postmortem histopathology from two patients and the clinical data from most patients, we propose theexistence of endophenotypes of SIOD. Hum Mutat 28(3), 273–283, 2007. Published 2006 Wiley-Liss, Inc.
y
KEY WORDS: genocopy; immunodeficiency; proteinuria; skeletal dysplasia; locus heterogeneity; SMARCAL1
INTRODUCTION
The classical model for describing inheritance of single-genedisorders assumes that the inheritance of a trait in familiesis synonymous with the transmission of a single molecular defect.However, progressively fewer diseases can be explained bymutations at a single locus [Badano and Katsanis, 2002; Dippleand McCabe, 2000; Scriver and Waters, 1999]. Increasinglydisorders previously attributed to a defect in a single gene arecaused by a combination of mutant alleles at more than one locus.This suggests that perhaps many other genetic disorderstraditionally considered to be single-gene Mendelian traits,particularly those displaying multiple and varying phenotypicfeatures, may be better understood with an oligogenic model. Forexample, digenic triallelic inheritance has been described forBardet-Biedl syndrome [Katsanis et al., 2001], familial hyperch-olanemia [Carlton et al., 2003], cortisone reductase deficiency[Draper et al., 2003], and hemochromatosis [Merryweather-Clarkeet al., 2003]. In addition, digenic biallelic inheritance has beendescribed for retinitis pigmentosa [Kajiwara et al., 1994].
Schimke immunoosseous dysplasia (SIOD; MIM] 242900)displays multiple phenotypic features that differ in penetranceand expressivity within and among families [Boerkoel et al., 2000;Lucke et al., 2005]. SIOD is characterized by renal disease,spondyloepiphyseal dysplasia, T-cell deficiency, and characteristicdysmorphic features [Boerkoel et al., 2000; Ehrich et al., 1988;Lucke et al., 2006; Spranger et al., 1991]. Other features observedin some but not all patients include atherosclerosis with cerebralischemia, migraine-like headaches, deficiency of other blood celllineages, corneal opacities, dental anomalies, hyperpigmentedmacules, opportunistic infections, autoimmune enteropathy,pulmonary hypertension, and hypothyroidism [Boerkoel et al.,2000; Kilic et al., 2005; Lucke et al., 2004].
SIOD has been shown to be caused by biallelic putative loss-
of-function mutations in swi/snf–related, matrix-associated,
actin-dependent regulator of chromatin, subfamily a-like 1
(SMARCAL1) (MIM] 606622) [Boerkoel et al., 2002].
SMARCAL1 encodes a protein homologous to the sucrosenonfermenting type 2 (SNF2) family of chromatin remodelingproteins [Coleman et al., 2000; Muthuswami et al., 2000]. SNF2-related proteins restructure DNA-histone interactions and thusmediate the chromatin remodeling necessary for DNA transcrip-tion, replication, repair, recombination, and covalent modification[Havas et al., 2001; Pazin and Kadonaga, 1997]. However, SIODpatients do not exhibit hypersensitivity to ultraviolet radiation,genomic instability, increased cancer incidence, or defective DNArepair following exposure to gamma radiation [Boerkoel et al.,2000]. This suggests that SMARCAL1 is not a regulator of DNAreplication, repair, or recombination.
Although SIOD segregates as a classic recessive trait, itspleiotropism has been particularly intriguing and challengingto reconcile within a Mendelian paradigm of a single-genedefect. Identifying genotype–phenotype correlations has beenparticularly difficult. Suggestive of locus heterogeneity, nearlyhalf of SIOD patients do not have identifiable mutations in thecoding exons of SMARCAL1. Also, suggestive of oligogenicinheritance, a few patients have only detectable monoallelicSMARCAL1 coding mutations. To investigate these conceptsfurther, we examined the cDNA sequence and SMARCAL1mRNA and protein expression in a subset of patients meeting thediagnostic criteria for SIOD.
MATERIALSANDMETHODSHuman Subjects
Patients referred to this study gave informed consent approvedby the Institutional Review Board of Baylor College of Medicine(H-9669, Houston, TX). The clinical data for patients wereobtained from questionnaires completed by the referring physicianas well as from medical records and summaries provided by thatphysician.
274 HUMAN MUTATION 28(3), 273^283,2007
Human Mutation DOI 10.1002/humu

Selection of Patients for Mutation Screening
The criterion used for selection and analysis of patient samplesis shown in Figure 1. We reviewed clinical data for similarity tothe signs and symptoms previously described for SIOD [Boerkoelet al., 2000] and sequenced the coding exons of SMARCAL1 fromsuch patients. From the patients in whom we did not identifySMARCAL1 coding mutations, we selected a subset for furtheranalysis based on a point system reflecting the severity ofsymptoms associated with SIOD. Linear growth failure before 10years of age received one point; renal failure received one point;lymphopenia received one point; recurrent infections received one
point; cerebral ischemia received one point; death before 10 yearsof age received two points; and death after 10 years of age receivedone point. Patients with four or more points and their affectedsiblings were selected for further analysis.
Cell Culture
Fibroblasts were isolated and cultured from the patient’s skin.They were maintained in Dulbecco’s modified Eagle’s medium(DMEM) cell culture media with 20% fetal bovine serum at 371C,5% CO2. Lymphoblastoid cell lines were obtained throughisolation of mononuclear cells from patients. The cells were
Phenotypic similarity to SIOD
Yes No No further investigation
Sequencing of coding exons
Biallelic mutation
Segregation of mutation
Monoallelic mutationUndetectable mutation
Severity score < 4
Severity score > 4
No further investigation Detectable mutation
Segregation of mutationBiallelic expression
Yes No
Monoallelic expression
mRNA expression
Protein expression
NormalReduced Absent
Normal Reduced Absent
Noncoding mutation or mRNA decay
Reduced protein stability or impaired translation
Undetectable or no alteration
Possible genocopy or phenocopy
Cell line
No Yes
Sequencing of cDNAReexamine diagnosis
FIGURE 1. Algorithm for SMARCAL1mutation detection andmolecular analysis in SIODpatients.
HUMANMUTATION 28(3), 273^283,2007 275
Human Mutation DOI 10.1002/humu

Epstein-Barr virus (EBV)-transformed and cultured and grown inRoswell Park Memorial Institute (RPMI) cell culture media with10% fetal bovine serum at 371C, 5% CO2.
DNA Sequencing
We identified mutations in the SMARCAL1 gene as previouslydescribed [Boerkoel et al., 2002]. Briefly, we amplified the cDNAor coding exons and intronic splice junctions from genomic DNAby PCR amplification, purified the PCR products using the QiagenPCR purification kit (Qiagen, Valencia, CA; www1.qiagen.com),and sequenced them with dye-terminator chemistry (AppliedBiosystems, Foster City, CA; www.appliedbiosystems.com). Theprimer pairs used for amplifying and sequencing the SMARCAL1cDNA were CCTTGCCTCTTACAGAGGAGCAGAGGAAAAAand GGCTTATGAGTTGGGTTAGCAAAGGGTGTG, CCCAAGAGACACCAGCTCAT and GCTCCACTTCCTGGTCTTGA,CAGGACCTTATTGCGCTTTT and CGGGCAGTCCTACTGTTTTT, ACAGCTGGCCTGATCAACAT and TCCACTTTCCAGTAGGTCCAA, and AGTTCCAACTGTCGGAGAGGCATGCTGTG and CTGTGGACTGGCTGTCAATGAGCTTTCATC.Using sequence AF432223.1 as a reference, we numbered theSMARLCAL1 cDNA sequence such that the adenine of the ATGtranslation initiation codon is 11. Those mutations that arenumbered based on the cDNA sequence are preceded by ‘‘c.’’ Theprotein reference sequence was derived from the open readingframe in AF432223.1. We numbered the SMARLCAL1 proteinsequence such that the methionine initiating translation is 11,and those mutations that are numbered based on the amino-acidsequence are preceded by ‘‘p.’’ Those mutations that are numberedbased on the genomic sequence are preceded by a ‘‘g.’’ Themutations are described according to standard nomenclature [denDunnen and Antonarakis, 2000].
RT-PCR
Using RNA extracted from patient-derived lymphoblastoid cellsor fibroblasts, we measured the relative amount of SMARCAL1mRNA by RT-PCR. We harvested the RNA with TRIzol(Invitrogen, Carlsbad, CA; www.invitrogen.com) according tothe manufacturer’s protocol. We used random hexamer primers(Invitrogen) and Super Script III (Invitrogen) for cDNAproduction and Platinum Taq DNA Polymerase (Invitrogen) forPCR amplification. For each reverse-transcriptase reaction, weused 1,000 ng of total RNA. Following PCR amplification using600 ng of the cDNA per reaction, we quantified the amount ofPCR product by fractionation on a 1% agarose gel andmeasurement of ethidium bromide fluorescence. We quantifiedthe fluorescence using a Kodak Gel Logic 200 Imaging System andthe Kodak 1D Analysis Software version 3.6 (Kodak, Rochester,NY; www.kodak.com). We made all measures relative to amplifi-cation of glyceraldehyde-3-phosphate dehydrogenase (GAPDH).The primers pairs used for SMARCAL1 PCR were CAGGACCTTATTGCGCTTTT and CGGGCAGTCCTACTGTTTTT and forGAPDH were TTAGCACCCCTGGCCAAG and CTTACTCCTTGGAGGCCATG.
Western Blotting
We measured SMARCAL1 protein in patient-derived lympho-blastoid cells or fibroblasts by Western blot analysis as previouslydescribed [Elizondo et al., 2006]. Following chemiluminescentdevelopment of the Western blot, we quantified the amount ofprotein by measuring band intensity on the film using the Kodak1D Analysis Software version 3.6.
SNPAnalysis
To determine segregation of SMARCAL1 alleles with SIOD, wefollowed segregation of the SNPs rs6711457, rs10498053,rs6744504, and rs1110998. To detect these polymorphisms, theMicroarray Core Facility at Baylor College of Medicine hybridizedgenomic DNA to the Affymetrix GeneChip Mapping 100K Arrayset in accordance with the standard protocol (see the GeneChipMapping 100K Assay Manual for full protocol, Affymetrix, SantaClara, CA; www.affymetrix.com). The GeneChips were washedusing a Fluidics Station 450 (Affymetrix). Each microarray wasscanned using the GeneChip scanner 3000 and GeneChipOperating software (GCOS) v1.1.1 with patch 5. Cell intensity(.cel) files were generated and subsequently saved and transportedas Cabinet (.CAB) files (using Data Transfer Tool v1.1) to aworkstation that contained GCOS software v1.2. Using GCOSv1.2, new .cel files were generated and analyzed using GeneChipDNA Analysis Software (GDAS) v3.0.
For characterization of the SMARCAL1 haplotype on which thec.2459A4G (R820H) and c.2542G4T (p.E848X) mutationsarose, we followed segregation of the following SNPs in eachfamily: 1-rs3770491, 2-rs2449774, 3-rs2449773, 4-rs6708185,5-rs2666290, 7-rs3770487, 8-rs2738281, 9-rs2738282, 10-rs284559,11-rs2271334, 12-rs2066521, 13-rs484375, 14-rs10498053,15-rs284544, 16-rs184028, 17-rs284547, 18-rs284548, 19-rs284551,20-rs2738286, 21-rs6744504, and 22-rs755625. These SNPs spanthe SMARCAL1 promoter and 50 half of the gene. We detectedthese polymorphisms by PCR amplification of genomic DNA anddye terminator sequencing of the PCR products.
CLINICAL REPORT
Patient SD74 was a 3-year-old boy with a younger healthysibling. He was the eldest child of healthy nonconsanguineousGreek parents. His prenatal history was remarkable for intrauterinegrowth retardation and oligohydramnios. He was deliveredby Cesarean section at 36 weeks of gestation. He grew poorlypostnatally, and at 3 years of age he was referred to the GeneticsService for evaluation of dysmorphic features, poor growth,microscopic hematuria, proteinuria, recurrent otitis media, andupper respiratory infections. He had normal motor, social,language, and cognitive development. His dysmorphic featuresincluded a triangular face, deep set eyes, broad nasal bridge andbulbous nasal tip, high arched palate, long thin upper lip, shortneck, barrel chest, lumbar lordosis, protruding abdomen,disproportion of trunk and limbs, and hyperpigmented macules.He also had hypermetropia and astigmatism and a high-pitchedvoice. His presentation met the diagnostic criteria for SIOD. ByDNA sequencing of the coding exons of SMARCAL1, he wasfound to be heterozygous for c.1736C4T. This paternally-derivedallele encodes the p.S579L missense mutation; we did not identifya maternally-derived mutation.
RESULTSMutations in the SMARCAL1Gene
Since the c.1736C4T mutation was previously detected in anunrelated SIOD patient from Spain [Boerkoel et al., 2002], therecurrence in another coastal Mediterranean population raised thequestion of whether recurring mutations in SMARCAL1 arisede novo or are attributable to founder mutations. A total of 43different mutations in the SMARCAL1 gene have been identifiedand include splice-site mutations, nonsense mutations, missensemutations, deletions, and insertions (Table 1). The mutations are
276 HUMANMUTATION 28(3), 273^283,2007
Human Mutation DOI 10.1002/humu
distributed along the whole gene with the exception of exon 18,the last exon. Many mutations are private, but like c.1736C4T,several mutations have been identified in more than one family.Interestingly, the expressivity of SIOD varies both among andwithin classes of mutations (Table 1 and Fig. 2). Compared topatients with biallelic nonsense, frameshift, or splicing mutations,SIOD patients with a missense mutation often have milder diseaseregardless of whether the second allele encodes a nonsense ora missense mutation (Fig. 2). Patients with biallelic missensemutations clustered into milder and more severely affected groups;
however, since patients for each recurrent mutation could befound in both groups, this clustering did not reflect a genotype-phenotype correlation.
Recurrent Mutations and Phenotypic Comparisons
One possible cause for such variable expressivity is therecurrence of mutations in the context of different cis promoterand chromatin regulatory elements. To test this hypothesis, weanalyzed the origins of three recurrent mutations. Mutations
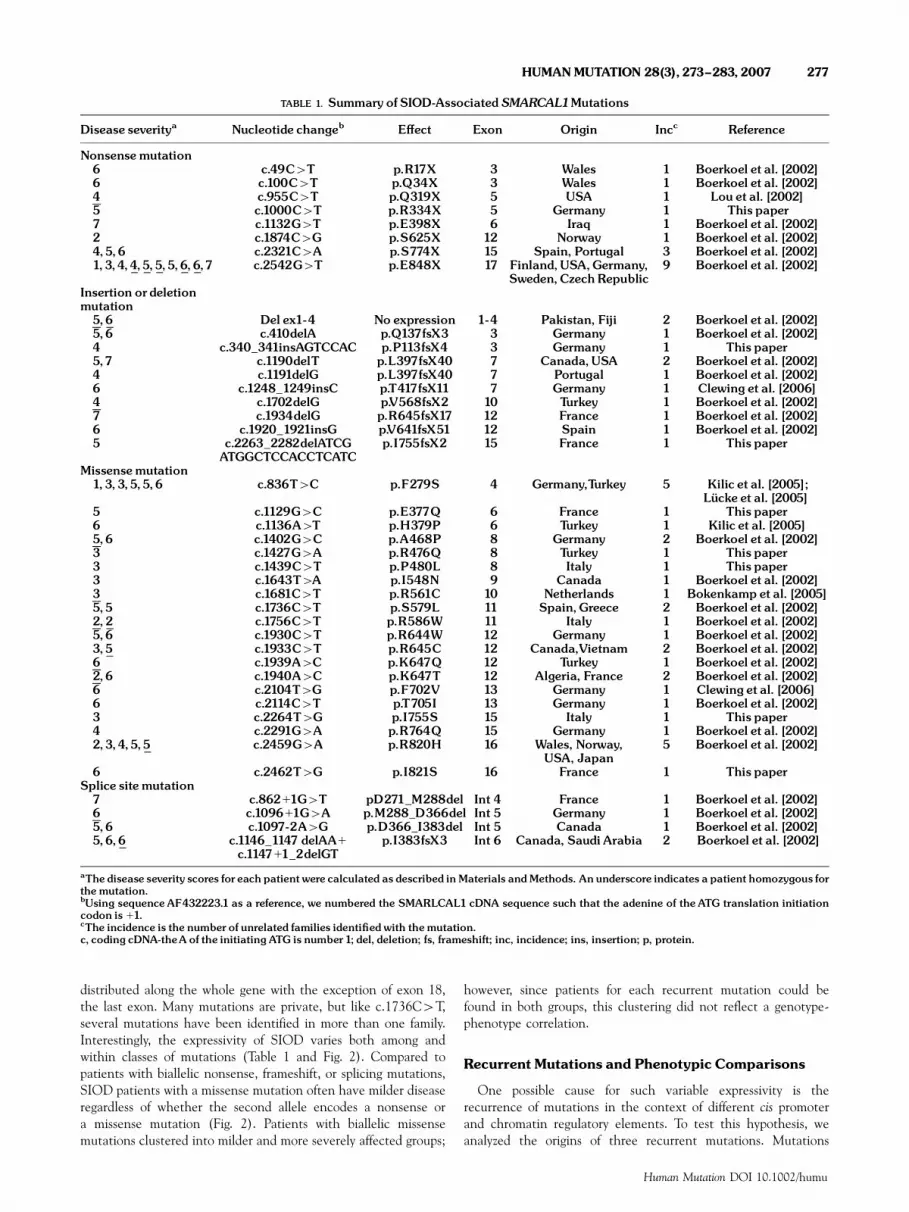
TABLE 1. Summary of SIOD-AssociatedSMARCAL1Mutations
Disease severitya Nucleotide changeb E¡ect Exon Origin Incc Reference
Nonsensemutation6 c.49C4T p.R17X 3 Wales 1 Boerkoel et al. [2002]6 c.100C4T p.Q34X 3 Wales 1 Boerkoel et al. [2002]4 c.955C4T p.Q319X 5 USA 1 Lou et al. [2002]5 c.1000C4T p.R334X 5 Germany 1 This paper7 c.1132G4T p.E398X 6 Iraq 1 Boerkoel et al. [2002]2 c.1874C4G p.S625X 12 Norway 1 Boerkoel et al. [2002]4,5,6 c.2321C4A p.S774X 15 Spain, Portugal 3 Boerkoel et al. [2002]1,3,4,4,5,5,5,6,6,7 c.2542G4T p.E848X 17 Finland,USA,Germany,
Sweden,CzechRepublic9 Boerkoel et al. [2002]
Insertion or deletionmutation5,6 Del ex1-4 No expression 1-4 Pakistan, Fiji 2 Boerkoel et al. [2002]5,6 c.410delA p.Q137fsX3 3 Germany 1 Boerkoel et al. [2002]4 c.340_341insAGTCCAC p.P113fsX4 3 Germany 1 This paper5,7 c.1190delT p.L397fsX40 7 Canada,USA 2 Boerkoel et al. [2002]4 c.1191delG p.L397fsX40 7 Portugal 1 Boerkoel et al. [2002]6 c.1248_1249insC p.T417fsX11 7 Germany 1 Clewing et al. [2006]4 c.1702delG p.V568fsX2 10 Turkey 1 Boerkoel et al. [2002]7 c.1934delG p.R645fsX17 12 France 1 Boerkoel et al. [2002]6 c.1920_1921insG p.V641fsX51 12 Spain 1 Boerkoel et al. [2002]5 c.2263_2282delATCG
ATGGCTCCACCTCATCp.I755fsX2 15 France 1 This paper
Missensemutation1,3,3,5,5,6 c.836T4C p.F279S 4 Germany,Turkey 5 Kilic et al. [2005];
Luº cke et al. [2005]5 c.1129G4C p.E377Q 6 France 1 This paper6 c.1136A4T p.H379P 6 Turkey 1 Kilic et al. [2005]5,6 c.1402G4C p.A468P 8 Germany 2 Boerkoel et al. [2002]3 c.1427G4A p.R476Q 8 Turkey 1 This paper3 c.1439C4T p.P480L 8 Italy 1 This paper3 c.1643T4A p.I548N 9 Canada 1 Boerkoel et al. [2002]3 c.1681C4T p.R561C 10 Netherlands 1 Bokenkamp et al. [2005]5,5 c.1736C4T p.S579L 11 Spain,Greece 2 Boerkoel et al. [2002]2,2 c.1756C4T p.R586W 11 Italy 1 Boerkoel et al. [2002]5,6 c.1930C4T p.R644W 12 Germany 1 Boerkoel et al. [2002]3,5 c.1933C4T p.R645C 12 Canada,Vietnam 2 Boerkoel et al. [2002]6 c.1939A4C p.K647Q 12 Turkey 1 Boerkoel et al. [2002]2,6 c.1940A4C p.K647T 12 Algeria, France 2 Boerkoel et al. [2002]6 c.2104T4G p.F702V 13 Germany 1 Clewing et al. [2006]6 c.2114C4T p.T705I 13 Germany 1 Boerkoel et al. [2002]3 c.2264T4G p.I755S 15 Italy 1 This paper4 c.2291G4A p.R764Q 15 Germany 1 Boerkoel et al. [2002]2,3,4,5,5 c.2459G4A p.R820H 16 Wales, Norway,
USA, Japan5 Boerkoel et al. [2002]
6 c.2462T4G p.I821S 16 France 1 This paperSplice sitemutation7 c.86211G4T pD271_M288del Int 4 France 1 Boerkoel et al. [2002]6 c.109611G4A p.M288_D366del Int 5 Germany 1 Boerkoel et al. [2002]5,6 c.1097-2A4G p.D366_I383del Int 5 Canada 1 Boerkoel et al. [2002]5,6,6 c.1146_1147 delAA1
c.114711_2delGTp.I383fsX3 Int 6 Canada, Saudi Arabia 2 Boerkoel et al. [2002]
aThe disease severity scores for each patient were calculated as described inMaterials andMethods. Anunderscore indicates a patient homozygous forthemutation.bUsing sequenceAF432223.1 as a reference, we numbered the SMARLCAL1 cDNA sequence such that the adenine of the ATG translation initiationcodon is 11.cThe incidence is the number of unrelated families identi¢edwith themutation.c, coding cDNA-theA of the initiating ATG is number1; del, deletion; fs, frameshift; inc, incidence; ins, insertion; p, protein.
HUMANMUTATION 28(3), 273^283,2007 277
Human Mutation DOI 10.1002/humu

c.836T4C (p.F279S), c.2459G4A (p.R820H), and c.2542G4T(p.E848X) occurred in five or more families (Table 1). Weobserved the c.2459G4A mutation on alleles of Welsh, German,Norwegian, Irish, and Japanese origin (Table 1 and SupplementaryTable S1 [Supplementary material available online at http://www.interscience.wiley.com/jpages/1059-7794/suppmat]), andusing four families demonstrated that this mutation occurred onat least three different haplotypes (Supplementary Table S1).Similarly, although the geographical proximity of patients with thec.2542G4T mutation might suggest a common founder, thismutation also occurred on multiple haplotypes in the six testedfamilies (Supplementary Table S2). Thus we conclude that thesetwo mutations have arisen independently in different populationsand that c.2459G and c.2542G are hotspots for SIOD-associatedSMARCAL1 mutations.
To determine whether SMARCAL1 cis regulatory elementsunique to each haplotype had a major influence on phenotype, wecompared the phenotypes of the patients. We did not observephenotypic differences attributable to the haplotype (Supplemen-tary Tables S1 and S2). Further supporting this, the expressivity inbrothers with identical SMARCAL1 alleles was strikingly different(Patients 65a and 65b in Supplementary Table S2 [Lucke et al.,2005]). Based on these observations, we hypothesized that othermodifiers such as oligogenic inheritance would be majordeterminants of SIOD expressivity.
Identi¢cation of Noncoding SMARCAL1Mutations
Identification of SIOD-associated monoallelic SMARCAL1mutations would support the hypothesis that SIOD is anoligogenic disease. A review of data from DNA sequencing ofthe coding exons of SMARCAL1 identified four patients withmonoallelic mutations: Patients SD8, SD47, SD74, and SD79(Table 2). To exclude noncoding mutations, we sequenced thecDNA and analyzed SMARCAL1 mRNA and protein expressionusing skin fibroblast or lymphoblastoid cell lines from each patient.
For Patients SD47, SD74, and SD79, sequencing of the PCRamplification products derived from the SMARCAL1 cDNAdetected the mutant allele only (Fig. 3a). The absence of
heterozygosity in the cDNA suggested either that the other allelewas deleted or not transcribed, or that the RNA from that allelewas rapidly degraded. Quantitative RT-PCR (qRT-PCR) showedexpression of SMARCAL1 mRNA approximately half thatobserved in control cells (Fig. 3b). For Patients SD47 and SD74,Western blot analysis also showed SMARCAL1 protein expressedat approximately half that observed in control cells(Fig. 3c). In contrast, lymphoblastoid cells from Patient SD79expressed the SMARCAL1 protein at levels comparable to thatobserved in control cells (Fig. 3c); this could arise fromstabilization of protein expressed from the mutant allele.
For Patient SD8, sequencing of the PCR amplification productsderived from the SMARCAL1 cDNA detected the nonmutantallele only (Fig. 3a). Because the identified mutation (c.1190DT,p.L397fsX40) predicts a frameshift and a premature terminationcodon, we hypothesize that the mRNA from this allele is rapidlydegraded by nonsense-mediated RNA decay. Further sequencingof the expressed cDNA did not identify a mutation in the codingsequence or the 58 base pairs of the 50 untranslated region (UTR)preceding the coding sequence of the second allele. However, wedid not detect expression of the SMARCAL1 protein by Westernblot analysis, which suggests that this allele has a mutationimpairing translation or protein stability (Fig. 3c).
From these results, we conclude that patients with an identifiedmonoallelic SMARCAL1 coding mutation also have a mutationaffecting the expression of the other SMARCAL1 allele. Thus itappears less likely that the variable expressivity of SIOD arisesfrom inheritance patterns in which only one allele of SMARCAL1is mutated.
Genetic and Phenotypic Evidencefor Locus Heterogeneity of SIOD
Identification of other genetic causes for SIOD could furtherour understanding of the pathophysiology of SIOD as well asprovide insight into the variability of the SIOD phenotype. Of 72patients referred for molecular testing, 30 did not haveSMARCAL1 coding mutations in either allele. These 30 patientswere phenotypically very similar to SIOD patients with SMAR-CAL1 mutations, but as a group, they had a few notabledifferences (Table 3). The incidences of hyperpigmented macules,lymphopenia, focal segmental glomerulosclerosis, and cerebralischemic symptoms were nearly half that observed among patientswith SMARCAL1 mutations. The rate of developmental andschooling delay was nearly triple that observed among patientswith SMARCAL1 mutations. In addition, patients with SMAR-CAL1 mutations died on average a decade earlier. However,although these differences distinguish the groups, the variableexpressivity in both groups precludes the use of these features todistinguish individual patients with or without SMARCAL1mutations.
To analyze SMARCAL1 further among these patients, weselected representative patients, Patients SD52a, SD52b, SD63,and SD91, for further analyses according to the algorithm shownin Figure 1. Using mRNA and protein from skin fibroblast orlymphoblastoid cell lines, we detected no SMARCAL1 mutationsin the cDNA, and qRT-PCR and Western blot analysis,respectively, showed expression of SMARCAL1 mRNA andprotein at levels similar to that observed in control cells (Fig. 3dand e). Since these patients had many features of SIOD butundetectable SMARCAL1 mutations, we hypothesized that theymanifest locus heterogeneity for SIOD.
Sev
erity
sco
re
1
2
3
4
5
6
7
Fram
eshi
ft+
Non
sens
e
Spl
ice
+ N
onse
nse
Mis
sens
e+
Non
sens
e
Mis
sens
e+
Fra
mes
hift
Spl
ice
+ F
ram
eshi
ft
Spl
ice
+ S
plic
e
Non
sens
e+
Non
sens
e
Fram
eshi
ft+
Fra
mes
hift
Mis
sens
e+
Mis
sens
e
FIGURE 2. Plot of disease severity against mutation type. Eachdot indicates a patient. The bar indicates the average severityscore.The disease severity scores were calculated as described inMaterials andMethods.
278 HUMANMUTATION 28(3), 273^283,2007
Human Mutation DOI 10.1002/humu

Histopathology in SIOD PatientsWith UndetectableSMARCAL1Mutations
Locus heterogeneity in the context of profound clinical andhistological similarity implies a model of unitary physiologicderangement, whereas histological discordance implies either amodel of endophenotypes or of distinct physiologic perturbationsconverging on a common phenotype. To determine the histo-pathologic similarity of these two groups of patients, we comparedthe postmortem findings for Patients SD52a and SD91 to patientswith biallelic SMARCAL1 mutations (Supplementary Table S3)[Clewing et al., 2006]. These two patients had similar immuno-logic and renal pathology to that described for patients withbiallelic SMARCAL1 mutations. However, they did not have signsof premature atherosclerosis, brain ischemia, or pulmonaryhypertension. Prominent atherosclerosis has been observed in all
three of the published autopsies of SIOD patients [Clewing et al.,2006; Spranger et al., 1991]. Thus, although the small number ofpatients and the variable expressivity of the phenotype associatedwith SMARCAL1 mutations confound interpretation, we proposethat Patients SD52a and SD91 have an endophenotype of SIOD.
DISCUSSIONSMARCAL1Allelic Heterogeneity and SIODPhenotypic Heterogeneity
Factors underlying the variable expressivity of Mendeliandiseases include allelic heterogeneity, nonallelic or locusheterogeneity, exogenous or environmental factors, and stochasticfactors. In recessive diseases with allelic heterogeneity suchas SIOD, a classical phenotype frequently occurs when no
TABLE 2. Comparison of Patients withMonoallelic SMARCAL1CodingMutations
Feature
Patients
SD8 SD47 SD74 SD79
SMARCAL1mutationsa
Paternal allele c.1190DTp.L397fsX40 NomRNA c.1736 C4Tp.S579L c.2459 G4Ap.R820HMaternal allele No protein c.2459 G4A p.R820H NomRNA NomRNA
EthnicityPaternal UK French-German Greek JapaneseMaternal UK Irish Greek Japanese
Gender F M M FDysmorphismBroad low nasal bridge Yes Yes Yes NoBulbous nasal tip Yes Yes Yes YesMicrodontia Yes Yes Yes YesHyperpigmentedmacules Yes Yes Yes YesLumbar lordosis Yes Yes Yes YesProtuberant abdomen Yes No Yes Yes
Skeleton and growthOnset of kgrowth (age in years) 0 0 1 0Disproportionate short stature Yes Yes Yes YesSED NR Yes Yes Yes
DevelopmentDelayed development No No No NoSchooling delay NA No NA No
EndocrineSerologic hypothyroidism NR No No No
HematologyLymphopenia Yes Yes Yes NoNeutropenia NR Yes No NoAnemia Yes No No NoThrombocytopenia Yes No No No
ImmunologyRecurrent infections Yes No Yes NoCirculating T-cell de¢ciency Yes Yes Yes Yes
NephrologyRenal pathology FSGS FSGS FSGS FSGSRenal dysfunction Yes Yes Yes YesDialysis or graft No Yes No No
NeurologyTIAs No Yes No NoStrokes No Yes No NoMigraine-like headaches NR Yes Yes No
GeneticsNormal karyotype Yes Yes Yes NR
OutcomeCurrent age (years) 15 5.9 7.4Age of death (years) 5.7Cause of death Pn
aUsing sequenceAF432223.1 as a reference, we numbered the SMARLCAL1 cDNA sequence such that the adenine of the ATG translation initiationcodon is 11.c, coding cDNA-theA of the initiating ATG is number 1; F, female; FSGS, focal segmental glomerulosclerosis; M, male; NA, not applicable; NR, notreported; p, protein; Pn, bacterial pneumonia; SED, spondyloepiphyseal dysplasia;TIA, transient ischemic attack; UK,United Kingdom.
HUMANMUTATION 28(3), 273^283,2007 279
Human Mutation DOI 10.1002/humu

functional gene product is produced. In addition, as observed formany other inherited disorders, we show that SIOD patients withbiallelic mutations in SMARCAL1 also exhibit a continuumfrom mild to severe disease (Fig. 2). SIOD patients withSMARCAL1 biallelic missense mutations or a missense mutationand a nonsense mutation have milder disease as a group;however, this genotype–phenotype correlation does not extendto individual patients and therefore cannot be used as a prognosticindicator.
The large number of private mutations and the variableexpressivity associated with a genotype also complicate prognosticdeductions from the genotype. These two observations and the
geographic dispersal of recurrent mutations suggest that mostSMARCAL1 mutations arise independently and not from afounder effect. The multiple haplotypes that we observed withthe recurrent c.2459G4A and c.2542G4T mutations supportsthis proposal and could also suggest that variable expressivityamong patients with the same SMARCAL1 mutations might arisefrom effects within the SMARCAL1 locus. However, we foundthat the SMARCAL1 locus haplotype generally does not correlatewith phenotype. Thus, in contrast to the classic examples ofhereditary elliptocytosis, cystic fibrosis, and sickle cell disease[Scriver et al., 2001], we conclude that given the caveats of thesmall number of patients and compound heterozygosity, the
TABLE 3. Comparison of PatientsWithoutSMARCAL1Mutations toThoseWith Biallelic Mutations
Clinical characteristic
Patient subset without biallelicSMARCAL1mutations
Frequency amongpatients withoutSMARCAL1mutations (%)
Frequency amongpatients with
biallelic SMARCAL1mutations (%)SD52a SD52b SD63 SD91
EthnicityPaternal Eng Eng Afghan Lv-GerMaternal Eng Eng Afghan Ir-Eng
Gender M F M F 14F/15M 17F/25MDysmorphismBroad low nasal bridge Yes Yes Yes No 18/23 (78) 27/40 (68)Bulbous nasal tip Yes Yes Yes Yes 20/23 (87) 30/36 (83)Hyperpigmentedmacules
No No Yes No 11/24 (46) 34/40 (85)
Lumbar lordosis Yes Yes Yes No 18/25 (72) 32/38 (84)Protuberant abdomen Yes Yes Yes Yes 23/26 (88) 34/39 (87)
Skeleton and growthOnset kgrowth (years) 9 7 5 1.5 Range 50^11;
mean 53.6Range 50^13; mean 52
Disproportionateshort stature
Yes Yes Yes Yes 25/26 (96) 39/40 (98)
SED Yes Yes Yes No 16/25 (64) 32/37 (86)DevelopmentDelayed development Yes Yes No Yes 12/25 (48) 7/39 (18)Schooling delay Yes Yes No Yes 8/15 (53) 4/24 (17)
EndocrineSerologic hypothyroidism No No Yes Yes 11/25 (44) 14/33 (42)
HematologyLymphopenia Yes Yes Yes Yes 11/20 (55) 35/39 (90)Neutropenia Yes Yes Yes Yes 9/24 (38) 19/33 (58)Anemia Yes No Yes Yes 16/25 (64) 24/35 (69)Thrombocytopenia Yes No Yes Yes 7/25 (28) 13/37 (35)
ImmunologyRecurrent infections Yes No Yes Yes 19/27 (70) 19/38 (50)CirculatingT-cellde¢ciency
Yes Yes Yes No 12/12 (100) 32/33 (97)
NephrologyRenal pathology NR MPGN MCGN FSGS FSGS 59/13 (69) FSGS 524/26 (92)Renal dysfunction Yes Yes Yes Yes 23/24 (96) 43/43 (100)Dialysis or graft Yes Yes No Yes 11/24 (46) 27/42 (64)
NeurologyTIAs No No Yes No 5/23 (22) 20/43 (47)Strokes No No Yes No 6/21 (29) 18/41 (44)Migraine-likeheadaches
No No Yes No 3/14 (21) 21/35 (60)
GeneticsNormal karyotype Yes Yes Yes Yes 15/15 (100) 24/25 (96)
OutcomeCurrent age (years) 16.7 Range 51^32 Range 56^41Age of death (years) 18 12.5 33.9 Range 510^33;
mean 519.2Range 53^15;mean 59.2
Cause of death mes in ards pn
Afghan, Afghani; ards, acute respiratory distress syndrome; Eng, English; F, female; FSGS, focal segmental glomerulosclerosis; Ir, Irish; Lav, Latvian;M, male; mes in, mesenteric infarction; MCGN, minimal changeglomerulonephritis; MPGN, mesangioproliferative glomerulonephritis; NA, not applic-able; NR, not reported; p, protein; pn, bacterial pneumonia; SED, spondyloepiphyseal dysplasia;TIA, transient ischemic attack.
280 HUMAN MUTATION 28(3), 273^283,2007
Human Mutation DOI 10.1002/humu

Genomic cDNA
b
a
c
d
e
Cnt
SD
52a
SD
52b
SD
63
SD
91
Cnt
SMARCAL1
GAPDH
SMARCAL1
GAPDH
Cnt
SD
8
SD
47
SD
74
SD
79
Cnt
10
30
50
70
SM
AR
CA
L1 m
RN
A e
xpre
ssio
n (%
)
SD8SD47
SD74SD79
20406080
100120
SD52a
SD52b
SD63SD91
SM
AR
CA
L1 m
RN
A e
xpre
ssio
n (%
)
20406080
100120
SM
AR
CA
L1 p
rote
in e
xpre
ssio
n (%
)
SD52a
SD52b
SD63SD91
SD8
SD47SD74
SD79
20406080
100120
SM
AR
CA
L1 p
rote
in e
xpre
ssio
n (%
)
SD8
c.1190delT
SD47
c.2459G>A
SD74
c.1736C>T
SD79
c.2459G>A
FIGURE 3. Analyses to identify mutations not detected by DNA sequencing of SMARCAL1 coding exons. a:Comparison of genomicand cDNA sequence showing expression of a single allele for SD8, SD47, SD74, and SD79. Note that SD8 only expresses the allelewithout an identi¢ed codingmutation and SD47, SD74, andSD79 predominantly express the alleleswith an identi¢ed codingmuta-tion. Using sequenceAF432223.1as a reference, themutations are numbered such that the adenine of theATG translation initiationcodon is 11. b:Graphical presentation of qRT-PCR analysis showing SMARCAL1mRNA expression in SIOD patients with monoal-lelic SMARCAL1 codingmutations. SMARCAL1mRNA expressionwas normalized toGAPDHmRNA expression andwas expressedas the percent of that observed in a cell line from an una¡ected individual. c:Western blot analysis showing SMARCAL1protein ex-pression in SIODpatients withmonoallelic SMARCAL1codingmutations.The graph shows SMARCAL1protein expression normal-ized toGAPDHmRNAexpression and relative to that observed in a cell line fromanuna¡ected individual. d:Graphical presentationof qRT-PCR analysis showing SMARCAL1 mRNA expression in SIOD patients without detectable SMARCAL1 mutations. SMAR-CAL1mRNAexpressionwas normalized toGAPDHmRNAexpression andwasexpressed as the percent of that observed in a cell linefrom an una¡ected individual. e:Western blot analysis showing SMARCAL1protein expression in SIODpatients without detectableSMARCAL1mutations.The graph shows SMARCAL1 protein expression normalized to GAPDH mRNA expression and relative tothat observed in a cell line from an una¡ected individual. [Color ¢gure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
HUMANMUTATION 28(3), 273^283,2007 281
Human Mutation DOI 10.1002/humu

SMARCAL1 cis elements have a minor modifying effect on theSIOD phenotype. These conclusions are consistent with theintrafamilial variation in SIOD expressivity and indicate a greaterrole for locus-independent modifying factors [Lucke et al., 2005].
As a putative ATP-dependent chromatin remodeling enzyme[Coleman et al., 2000; Muthuswami et al., 2000], the SMAR-CAL1 enzyme could regulate biological processes sensitive toquantitative modulation. We hypothesize that SMARCAL1regulates gene expression, a quantitative trait, via whichepigenetic, environmental, stochastic, or other genetic factorscould contribute to the crossing of a transcriptional threshold togive rise to a phenotype or disease susceptibility in a cell or tissue.Thus we propose a multifactorial threshold model for thecausation of SIOD by SMARCAL1 mutations.
One manner in which other genetic factors could modify theSIOD phenotype would be through complex oligogenic inheri-tance. As observed for some patients with Bardet-Biedl syndrome[Katsanis et al., 2001], we hypothesized that some SIOD patientswould have only monoallelic SMARCAL1 mutations as well asmutations in an unidentified locus. However, we were unable toidentify any SIOD patients with only a monoallelic SMARCAL1mutation. Although these results do not exclude complexoligogenic inheritance, they do make it less likely that monoallelicSMARCAL1 mutations associate with biallelic mutations of othergenes to cause SIOD.
Locus Heterogeneity and SIOD
We have found that approximately half of the SIOD patientsreferred for molecular analysis have undetectable SMARCAL1coding mutations despite having the primary and often secondaryfeatures of SIOD. Our analysis of a subset of these patientsconfirmed the absence of detectable SMARCAL1 mutations.Although this SIOD-like disease might arise as a phenocopy, weproposed existence of nonallelic heterogeneity based on therecurrence of disease in siblings, the absence of disease in parents,and the geographic dispersal of patients.
Many monogenic disorders like SIOD exhibit nonallelicheterogeneity. Classical examples of this include phenylketonuria,hypercholesterolemia, hyperammonemia, and maturity-onsetdiabetes of youth [Scriver et al., 2001]. Subgroups of thesediseases not only exhibit profound clinical similarity but alsohistological similarity because mutations at the various loci cause acommon physiologic perturbation. However, other defects withinthese pathways result in endophenotypes. Often the defect at arate-limiting step or bottleneck produces the full phenotypewhereas other defects in the pathway produce the endophenotype.According to this model, we hypothesize that patients withSMARCAL1 mutations express the full phenotype becauseSMARCAL1 functions at the hierarchically highest or rate-limiting step, whereas patients without SMARCAL1 mutationsexpress an endophenotype because their defect is hierarchicallylower or at a non-rate-limiting step. Integrated with themultifactorial threshold hypothesis for the causation of SIODby SMARCAL1 mutations, this model explains the gradation ofpathology observed between patients with and without SMAR-CAL1 mutations.
As expected for such a model of disease and as exemplified byPatient SD63 (Table 3), some patients without detectableSMARCAL1 mutations were clinically indistinguishable frompatients with SMARCAL1 mutations. This suggests that theymight have disease secondary to defects at nearly the same
hierarchical level as SMARCAL1 and supports our hypothesis ofunitary physiologic disturbance.
Diagnostic Relevance
With this report, we affirm that identification of SMARCAL1mutations confirms the diagnosis of SIOD and that missensemutations are more frequently associated with less severe disease.Our study also shows for the first time that SIOD exhibits locusheterogeneity; however, inclusion of hyperpigmented macules,normal development, and migraine-like headaches into ourscoring system should improve identification of patients likely tohave SMARCAL1 mutations. Of particular relevance for physi-cians and families making treatment decisions, this study alsoemphasizes the influence of modifiers other than the SMARCAL1genotype on SIOD expressivity (Fig. 2). Contrary to our previousconclusion, this additional data shows that not all patients with anonsense mutation have a severe disease and poorer prognosiscompared to those with two missense mutations.
In summary, we have shown that at least 10% of SIOD-associated SMARCAL1 mutations cannot be detected by sequen-cing of the coding exons and that the variable expressivity amongSIOD patients with biallelic SMARCAL1 mutations cannot bereadily explained by allelic heterogeneity. Also, our results providethe first evidence for nonallelic heterogeneity in SIOD. Ultimatelyelucidation of the function of the SMARCAL1 enzyme will benecessary to understand the precise molecular mechanismswhereby loss of its function leads to SIOD and how the variableexpressivity of SIOD arises. Identification of modifiers and othergenes associated with SIOD will provide important clues for thefunction of SMARCAL1 and the pathomechanism underlyingSIOD.
ACKNOWLEDGMENTS
We thank V. Reid Sutton, Jennifer Northrop, and Millan Patelfor critical review of this manuscript. This work was supportedin part by grants from the NIH National Institute of Diabetes,Digestive, and Kidney Diseases, the Gillson LongenbaughFoundation, and the Dana Foundation (all to C.F.B).
REFERENCES
Badano JL, Katsanis N. 2002. Beyond Mendel: an evolving view of human
genetic disease transmission. Nat Rev Genet 3:779–789.
Boerkoel CF, O’Neill S, Andre JL, Benke PJ, Bogdanovic R, Bulla M,
Burguet A, Cockfield S, Cordeiro I, Ehrich JH, Frund S, Geary DF,
Ieshima A, Illies F, Joseph MW, Kaitila I, Lama G, Leheup B, Ludman
MD, McLeod DR, Medeira A, Milford DV, Ormala T, Rener-Primec Z,
Santava A, Santos HG, Schmidt B, Smith GC, Spranger J, Zupancic N,
Weksberg R. 2000. Manifestations and treatment of Schimke immuno-
osseous dysplasia: 14 new cases and a review of the literature. Eur J
Pediatr 159:1–7.
Boerkoel CF, Takashima H, John J, Yan J, Stankiewicz P, Rosenbarker L,
Andre JL, Bogdanovic R, Burguet A, Cockfield S, Cordeiro I, Frund S,
Illies F, Joseph M, Kaitila I, Lama G, Loirat C, McLeod DR, Milford DV,
Petty EM, Rodrigo F, Saraiva JM, Schmidt B, Smith GC, Spranger J,
Stein A, Thiele H, Tizard J, Weksberg R, Lupski JR, Stockton DW. 2002.
Mutant chromatin remodeling protein SMARCAL1 causes Schimke
immuno-osseous dysplasia. Nat Genet 30:215–220.
Bokenkamp A, deJong M, van Wijk JA, Block D, van Hagen JM, Ludwig
M. 2005. R561C missense mutation in the SMARCAL1 gene associated
with mild Schimke immuno-osseous dysplasia. Pediatr Nephrol 20:
1724–1728.
Carlton VE, Harris BZ, Puffenberger EG, Batta AK, Knisely AS, Robinson
DL, Strauss KA, Shneider BL, Lim WA, Salen G, Morton DH, Bull LN.
282 HUMANMUTATION 28(3), 273^283,2007
Human Mutation DOI 10.1002/humu

2003. Complex inheritance of familial hypercholanemia with associated
mutations in TJP2 and BAAT. Nat Genet 34:91–96.
Clewing JM, Antalfy BC, Lucke T, Najafian B, Marwedel KM, Hori A,
Powel RM Jr, Do AF, Najera L, Santacruz K, Hicks MJ, Armstrong DL,
Boerkoel CF. 2006. Schimke immuno-osseous dysplasia: a clinicopatho-
logical correlation. J Med Genet eprint July 13.
Coleman MA, Eisen JA, Mohrenweiser HW. 2000. Cloning
and characterization of HARP/SMARCAL1: a prokaryotic HepA-
related SNF2 helicase protein from human and mouse. Genomics
65:274–282.
den Dunnen JT, Antonarakis SE. 2000. Mutation nomenclature extensions
and suggestions to describe complex mutations: a discussion. Hum
Mutat 15:7–12.
Dipple KM, McCabe ER. 2000. Phenotypes of patients with ‘‘simple’’
Mendelian disorders are complex traits: thresholds, modifiers, and
systems dynamics. Am J Hum Genet 66:1729–1735.
Draper N, Walker EA, Bujalska IJ, Tomlinson JW, Chalder SM,
Arlt W, Lavery GG, Bedendo O, Ray DW, Laing I, Malunowicz E,
White PC, Hewison M, Mason PJ, Connell JM, Shackleton CH,
Stewart PM. 2003. Mutations in the genes encoding
11beta-hydroxysteroid dehydrogenase type 1 and hexose-6-phosphate
dehydrogenase interact to cause cortisone reductase deficiency. Nat
Genet 34:434–439.
Ehrich JHH, Offner G, Schirg E, Helmchen U, Brodehl J. 1988.
Minderwuchssyndrom mit Skelettdyplasien und focal segmental skler-
osierender Glomerulonephritis. Monatsschr Kinderheilkd 136:6.
[German]
Elizondo LI, Huang C, Northrop JL, Deguchi K, Clewing JM, Armstrong
DL, Boerkoel CF. 2006. Schimke immuno-osseous dysplasia: a cell
autonomous disorder? Am J Med Genet A 140:340–348.
Havas K, Whitehouse I, Owen-Hughes T. 2001. ATP-dependent
chromatin remodeling activities. Cell Mol Life Sci 58:673–682.
Kajiwara K, Berson EL, Dryja TP. 1994. Digenic retinitis pigmentosa due to
mutations at the unlinked peripherin/RDS and ROM1 loci. Science
264:1604–1608.
Katsanis N, Ansley SJ, Badano JL, Eichers ER, Lewis RA, Hoskins BE,
Scambler PJ, Davidson WS, Beales PL, Lupski JR. 2001. Triallelic
inheritance in Bardet-Biedl syndrome, a Mendelian recessive disorder.
Science 293:2256–2259.
Kilic SS, Donmez O, Sloan EA, Elizondo LI, Huang C, Andre JL,
Bogdanovic R, Cockfield S, Cordeiro I, Deschenes G, Frund S, Kaitila I,
Lama G, Lamfers P, Lucke T, Milford DV, Najera L, Rodrigo F, Saraiva
JM, Schmidt B, Smith GC, Stajic N, Stein A, Taha D, Wand D,
Armstrong D, Boerkoel CF. 2005. Association of migraine-like head-
aches with Schimke immuno-osseous dysplasia. Am J Med Genet A
135:206–210.
Lou S, Lamfers P, McGuire N, Boerkoel CF. 2002. Longevity in Schimke
immuno-osseous dysplasia. J Med Genet 39:922–925.
Lucke T, Marwedel KM, Kanzelmeyer NK, Hori A, Offner G, Kreipe HH,
Ehrich JH, Das AM. 2004. Generalized atherosclerosis sparing the
transplanted kidney in Schimke disease. Pediatr Nephrol 19:672–675.
Lucke T, Billing H, Sloan EA, Boerkoel CF, Franke D, Zimmering M,
Ehrich JH, Das AM. 2005. Schimke-immuno-osseous dysplasia: new
mutation with weak genotype-phenotype correlation in siblings. Am J
Med Genet A 135:202–205.
Lucke T, Franke D, Clewing JM, Boerkoel CF, Ehrich JHH, Das AM,
Zivicnjak M. 2006. Schimke versus non-Schimke chronic kidney disease:
an anthropometric approach. Pediatrics 118:e400–e407.
Merryweather-Clarke AT, Cadet E, Bomford A, Capron D, Viprakasit V,
Miller A, McHugh PJ, Chapman RW, Pointon JJ, Wimhurst VL, Livesey
KJ, Tanphaichitr V, Rochette J, Robson KJ. 2003. Digenic inheritance of
mutations in HAMP and HFE results in different types of haemochro-
matosis. Hum Mol Genet 12:2241–2247.
Muthuswami R, Truman PA, Mesner LD, Hockensmith JW. 2000. A
eukaryotic SWI2/SNF2 domain, an exquisite detector of double-
stranded to single-stranded DNA transition elements. J Biol Chem
275:7648–7655.
Pazin MJ, Kadonaga JT. 1997. SWI2/SNF2 and related proteins: ATP-
driven motors that disrupt protein-DNA interactions? Cell 88:737–740.
Scriver CR, Waters PJ. 1999. Monogenic traits are not simple: lessons from
phenylketonuria. Trends Genet 15:267–272.
Scriver CR, Sly WS, Childs B, Beaudet AL, Valle D, Kinzler KW,
Vogelstein B, editors. 2001. The metabolic and molecular bases of
inherited disease, 8th edition. New York: McGraw-Hill. p 4571–4636,
4665–4730, 5121–5188.
Spranger J, Hinkel GK, Stoss H, Thoenes W, Wargowski D, Zepp F. 1991.
Schimke immuno-osseous dysplasia: a newly recognized multisystem
disease. J Pediatr 119:64–72.
HUMANMUTATION 28(3), 273^283,2007 283
Human Mutation DOI 10.1002/humu





























