Embed Size (px)
Citation preview

ARTICLE
Evidence that the C-Terminal Domain (CtD) AutoinhibitsNeural Repression by Drosophila E(spl)M8Bhaskar Kahali, Jee-Eun Kim, Umesh Karandikar, Clifton P. Bishop, and Ashok P. Bidwai*Department of Biology, West Virginia University, Morgantown, West Virginia
Received 10 August 2009; Accepted 5 October 2009
Summary: Analysis of the retinal defects of a CK2 phos-phomimetic variant of E(spl)M8 (M8S159D) and the trun-cated protein M8* encoded by the E(spl)D allele, suggestthat the nonphosphorylated CtD ‘‘autoinhibits’’ repres-sion. We have investigated this model by testing for inhi-bition (in ‘‘trans’’) by the CtD fragment in its nonphos-phorylated (M8-CtD) and phosphomimetic (M8SD-CtD)states. In N1 flies, ectopic M8-CtD compromises lateralinhibition, i.e., elicits supernumerary bristles as withloss of N signaling. This antimorphic activity of M8-CtDstrongly rescues the reduced eye and/or bristle lossphenotypes that are elicited by ectopic M8SD or wildtype M8. Additionally, the severely reduced eye of Nspl/Y;E(spl)D/1 flies is also rescued by M8-CtD. Rescue isspecific to the time and place, the morphogenetic fur-row, where ‘‘founding’’ R8 photoreceptors are specified.In contrast, the phosphomimetic M8SD-CtD that is pre-dicted to be deficient for autoinhibition, exhibits signifi-cantly attenuated or negligible activity. These studiesprovide evidence that autoinhibition by the CtD regu-lates M8 activity in a phosphorylation-dependent man-ner. genesis 48:44–55, 2010. VVC 2009 Wiley-Liss, Inc.
Key words: Notch; lateral inhibition; E(spl)D; Nspl;neurogenesis
INTRODUCTION
N signaling has been intensively studied during Drosoph-ila neurogenesis, a process leading to stereotyped pat-terning of the compound eye and bristles (Baonza andFreeman, 2001; Bray, 2006; Campos-Ortega, 1997). Neu-ral development initiates with the expression of atonal(ato) or the achaete-scute complex (ASC), whichencode basic-helix-loop-helix (bHLH) transcription fac-tors, the proneural activators (Cubas et al., 1991; Heit-zler et al., 1996; Jarman et al., 1994, 1995; Modolell andCampuzano, 1998; Skeath and Carroll, 1991). ato isrequired for eye development and ASC for the bristle.During early neurogenesis, these activators areexpressed in groups of cells called the proneural clusters[PNC’s (Calleja et al., 2002; Dambly-Chaudiere and Ver-voort, 1998; Frankfort and Mardon, 2002; Gibert andSimpson, 2003; Hsiung and Moses, 2002)]. However,
only a fixed number of cells from each PNC are selectedto adopt the neural fate, while the others are redirectedto an alternative fate. This selection begins when onecell of a PNC gains an advantage by expressing the high-est level of Ato/ASC. This cell is destined to form the‘‘founding’’ R8 photoreceptor or the bristle sensoryorgan precursor (SOP), and inhibits other PNC cellsfrom adopting the (default) R8/SOP fate. This N-depend-ent process is termed lateral inhibition (Lehmann et al.,1983; Simpson, 1990). The future R8/SOP expressesDelta (Dl) at a higher level, and activates N in adjoiningcells of the PNC. As a result, N is cleaved and its intracel-lular domain (NICD) then elicits transcription of theEnhancer of Split Complex [E(spl)C, (Bailey and Posak-ony, 1995; Lecourtois and Schweisguth, 1995; Schronset al., 1992)]. In cells receiving this inhibitory N signal,E(spl) repressors complex with the corepressor Grou-cho (Gro) and antagonize Ato/ASC, thereby preventingthem from adopting the R8/SOP fate.
Given the importance of E(spl) for lateral inhibition,numerous studies have sought to define their modes ofaction. These studies have turned out to be complicated,in part, because the E(spl)C encodes seven bHLH pro-teins with similar functional domains, and mutationsaffecting each transcription unit have been unavailable.Earlier studies that overexpression of E(spl) proteinselicits generalized bristle loss (Giebel and Campos-Ortega, 1997; Nakao and Campos-Ortega, 1996) raisedthe possibility of functional redundancy (Cooper et al.,2000). A more recent model, termed ‘‘the proteintether’’ (Giagtzoglou et al., 2003), proposes that, in addi-tion to DNA-binding, E(spl) proteins mediate repressionby directly interacting with enhancer-bound Ato/ASC.One prediction of this model is that repression shouldreflect E(spl) dosage. Studies in the eye have produced
* Correspondence to: Ashok P. Bidwai, Department of Biology, West
Virginia University, Morgantown, WV, USA. E-mail: [email protected]
Contract grant sponsor: National Institutes of Health, Contract grant
number: EY015718Published online 15 December 2009 in
Wiley InterScience (www.interscience.wiley.com).
DOI: 10.1002/dvg.20581
' 2009 Wiley-Liss, Inc. genesis 48:44–55 (2010)

mixed results. Three E(spl) members (M8, Mg, and Md)are expressed in the morphogenetic furrow (MF), andmediate R8 selection (Ligoxygakis et al., 1999). How-ever, overexpression of only Md antagonizes Ato andelicits loss of the R8’s, no such effects were seen withM8 or Mg (Ligoxygakis et al., 1998, 1999; Nagel et al.,1999). This does not reflect sub-threshold expressionlevels, because ectopic M8 elicits potent bristle loss(Giebel and Campos-Ortega, 1997). A predominant rolefor M8 in R8 selection and eye development is high-lighted by E(spl)D, a unique dominant allele of m8 thatabrogates eye development in the presence of N
spl. Itwas, in fact, this genetic interaction that led to the identi-fication of the E(spl)C (Welshons, 1956).
The E(spl)D allele harbors two lesions; one stabilizesthe m8 transcript, and the second results in a truncatedprotein called M8* (Nagel et al., 1999; Tietze et al.,1992). M8* retains the basic, HLH and Orange (also anHLH) domains (see Fig. 1), but lacks 56 C-terminal resi-dues (CtD) including the penultimate WRPW tetra-pep-tide, which abolishes Gro-binding (Paroush et al., 1994).Despite this critical defect, E(spl)D dominantly elicitsloss of R8 cells by exacerbated interaction with, and an-tagonism of, Ato (Nagel et al., 1999). Importantly, theR8/eye defects of E(spl)D are mimicked by expressionof a UAS-m8* construct, but not by full length M8,strongly suggesting that the truncation triggers dominantbehavior. However, removal of just the WRPW motifdoes not elicit dominant activity on otherwise full lengthM8 (Giebel and Campos-Ortega, 1997; Nagel et al.,1999). These paradoxical findings, led Giebel and Cam-pos-Ortega (1997) to presciently suggest that ‘‘the regionbetween Orange and WRPW may have regulatory influ-ence on repressor activity.’’ Our previous studies indicatethat this regulatory influence involves phosphorylationof M8.
We had uncovered that protein kinase CK2 phospho-rylates M8 at Ser159 (Trott et al., 2001) in its CtD, aregion missing in M8*. Expression of a CK2 phosphomi-metic variant, M8S159D (M8SD), led to a severelyreduced eye due to exacerbated antagonism of Ato(Karandikar et al., 2004). These effects closely mimickedthe eye defects of M8* previously described by others(Nagel et al., 1999). In addition, the strength of bindingof M8SD with Ato was virtually identical to that of M8*and Ato. This result suggested that the CtD in its phos-phorylated state (M8SD) does not contribute to Ato-bind-ing, consistent with the role of Orange in Ato-binding(Nagel et al., 1999). In contrast, wild type M8 did notbind Ato. These findings and the suggestion of Giebeland Campos-Ortega (see above), led us to propose thatnonphosphorylated M8 was ‘‘autoinhibited’’ by its CtD.In this case, an intra-molecular (‘‘cis’’) interaction of thenonphosphorylated CtD with HLH/Orange preventedbinding of wild type M8 to Ato (see Fig. 1). Phosphoryla-tion by CK2 would displace the CtD to expose Orange,and permit binding to Ato, an interaction that is requiredfor repression of the R8 fate. The absence of the CtD inM8* would bypass autoinhibition and trigger dominant
activity against Ato and the R8 fate. However, direct evi-dence for autoinhibition by the nonphosphorylated CtDwas lacking.
We describe here studies employing Gal4-UAS drivenoverexpression of the CtD peptides, and their effects onneural patterning. Since phosphorylation of M8 isrequired for R8 and SOP selection (Bose et al., 2006),we hypothesized that ectopic CtD might bind (in‘‘trans’’) to the noninhibited state of M8 and impairrepression. To distinguish the effects of phosphoryla-tion, we used two CtD-variants; M8-CtD retains the phos-phorylation site and should possess autoinhibitory activ-ity, whereas M8SD-CtD should lack such activity byvirtue of the phosphomimetic Asp at the CK2 site. Ourstudies on rescue of the eye and bristle defects of ec-topic M8 and M8SD, and the reduced eye of N
spl/Y;
E(spl)D/1 are consistent with autoinhibition. The impli-cations of these findings are discussed.
RESULTS
The CtD Fragment is Phosphorylated by CK2 andBinds Gro In Vitro
As stated above (see Introduction), studies on M8SDand M8* suggested that phosphorylation of the CtDwould overcome its ability to ‘‘autoinhibit,’’ perhaps, bypreventing interaction with the HLH or Orange domains(see Fig. 1a). If so, over-expressed CtD might bind tophosphorylated (noninhibited) M8 and impair repres-sion, even though such a ‘‘trans’’ interaction is expectedto display first-order, rather than zero-order kinetics (aswith full-length M8). We generated two variants of theCtD (Fig. 1b). M8-CtD is phosphorylatable and shouldretain autoinhibitory activity, whereas the phosphomi-metic variant M8SD-CtD should exhibit lower potencyor lack such activity.
We first characterized the in vitro interactions of these56 residue peptides with CK2 and Gro. Phosphorylationof CtD-peptides (as GST-fusions) was tested using CK2(the a2b2 holoenzyme) purified from Drosophilaembryos (Kahali et al., 2008). In assays employing limit-ing amounts of CK2, we find that M8-CtD is readily phos-phorylated, whereas M8SD-CtD or GST-alone is not (Fig.1c, and data not shown). Phosphorylation was notobserved in (mock) reactions lacking the enzyme (datanot shown), and is specific because this purified enzymeis potently inhibited by heparin, a CK2-specific inhibitor(Kahali et al., 2008). It appears that the specificity ofCK2 for Ser159 is not altered in the CtD peptides. Wealso assessed and found that both CtD’s interact equiva-lently with Gro in yeast (Fig. 1d), consistent with therole of WRPW in mediating this interaction (Fisher et al.,1996). Therefore, the two motifs resident in the CtD,CK2-phosphorylation and Gro-binding (Fig. 1b), appearto retain function in the absence of the basic, HLH andOrange domains.
45CTD AUTOINHIBITS NEURAL REPRESSION

M8-CtD Elicits Stronger Ectopic Bristle DefectsThan Does M8SD-CtD
We next assessed if over-expression of M8-CtD, butnot M8SD-CtD, elicits ectopic macrochaetes (MC’s), aphenotype characteristic of impaired lateral inhibition(Brennan et al., 1997), and one seen with antimorphicor dominant-negative (DN) variants of M8 (Giebel andCampos-Ortega, 1997). For in vivo analysis, the UAS-m8-CtD and UAS-m8SD-CtD constructs were expressedusing the Gal4-UAS system (Brand and Perrimon, 1993).To eliminate position effects, multiple independent lines
harboring UAS-constructs were generated (Fig. 2a), andexpression was driven with the enhancer trap G455.2
that elicits Gal4 expression in the PNC’s that give rise tothe four scutellar MC’s (Giebel and Campos-Ortega,1997; Hinz et al., 1994).
In the case of G455.2/1 flies, by themselves, �11%display ectopic scutellar MC’s; this number was used asthe baseline for comparing the effects of ectopic M8-CtDor M8SD-CtD. We find that expression of M8-CtD elicitedectopic MC’s on the scutellum of 62–78% of the flies,with an average penetrance of �70% (Fig. 2b). Expres-
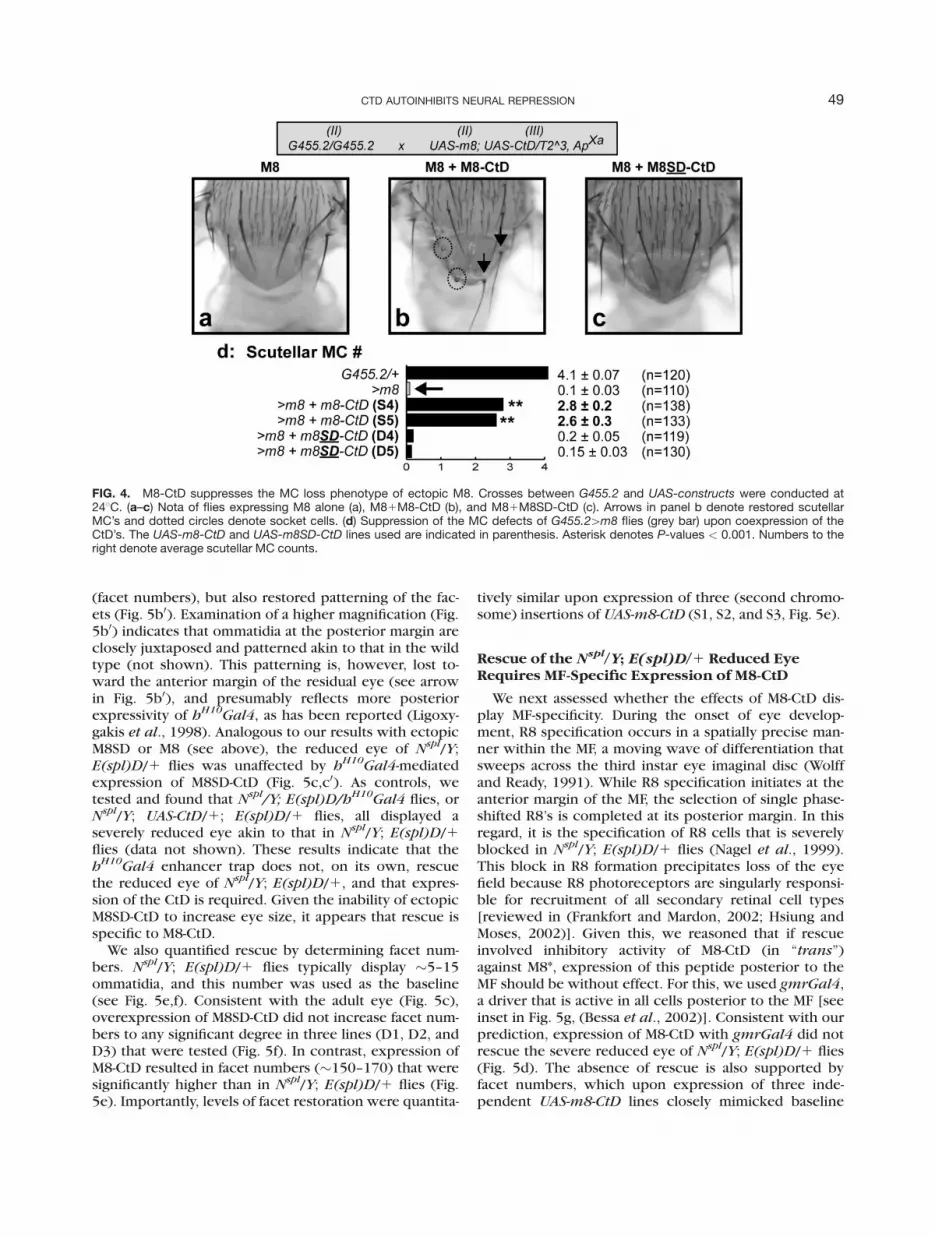
FIG. 2. M8-CtD elicits stronger ectopic bristle defects than does M8SD-CtD. Crosses between G455.2 and UAS-constructs were con-ducted at 248C. (a) Five independent insertions of UAS-m8-CtD (S1-S5) or UAS-m8SD-CtD (D1-D5) were analyzed. (b) The penetrance ofectopic scutellar macrochaetes (MC’s) in the absence of expression (solid bars) or upon expression with G455.2 (grey bars). The dashedline denotes the baseline MC defects of G455.2/1 flies, and the solid line denotes the average MC defect penetrance upon expression ofM8-CtD or M8SD-CtD.
FIG. 1. Role of phosphorylation of M8 and characterization of the CtD. (a) The CtD autoinhibits M8, and phosphorylation favors a confor-mational state permissive for antagonism of ASC/Ato (color of boxes corresponds to those in panel b). (b) Functional domains in M8 andM8*. The C-terminal domain (CtD) peptide encompasses residues 123–179 of full-length M8, and harbors the CK2 phosphorylation andGro-binding sites. (c). In vitro phosphorylation of GST-CtD fusions by Drosophila CK2 (dCK2). (d) Two hybrid interaction with Gro.
46 KAHALI ET AL.

sion of M8SD-CtD also led to ectopic scutellar MC’s,but with a generally lower penetrance; it ranged from31 to 54%, with an average of �41% (Fig. 2b). Ontheir own, the intrinsic MC defects in UAS-m8-CtD/1or UAS-m8SD-CtD/1 lines were �5% (Fig. 2b), indicat-ing that expression was required. Analyses of five inde-pendent lines revealed that the penetrance of the ec-topic MC phenotype of M8-CtD (Lines S1–S5) was gen-erally higher than those of M8SD-CtD (Lines D1–D5,Fig. 2b). These studies raised the possibility thatM8SD-CtD is less efficient at autoinhibition, perhapsreflecting kinetic effects. A direct assessment ofexpression levels of the CtD peptides was precludedby the absence of suitable antibodies. We thereforeused additional assays to compare the activities of theCtD peptides in vivo (see below).
M8-CtD More Strongly Suppresses the Eye andBristle Defects of Ectopic M8SD
We have previously reported that expression of M8SDwith 109-68Gal4 elicits a reduced eye and loss of theinterommatidial bristles [IOB’s, (Karandikar et al.,2004)]. This Gal4 driver elicits expression within the MF,where endogenous E(spl) mediates selection of R8 cells.The reduced eye of M8SD also displays defects in omma-tidial (facet) size and patterning (Fig. 3a,a0). Thesedefects are strongly rescued by coexpression of UAS-ato,an in vivo target of M8SD (Kahali et al., 2009). Impor-tantly, rescue does not involve competition betweentwo UAS-constructs for a limiting amount of Gal4,because coexpression of UAS-LacZ does not modulatethe eye defects of 109-68Gal4[UAS-m8SD flies. Wetherefore tested for rescue of the M8SD reduced eye by
FIG. 3. M8-CtD suppresses the eye and bristle defects of ectopic M8SD. Crosses between 109-68Gal4 and UAS-constructs were con-ducted at 248C. (a–c) Adult eye phenotypes. The proteins expressed are indicated, anterior is to the right, and magnification is 2003. (a0–c0)magnifications of regions of the eye in panels a–c; arrows denote fused ommatidia. (d) Rescue of the reduced eye of 109-68Gal4[m8SDflies (grey bar) upon coexpression of the CtD’s. The UAS-m8-CtD and UAS-m8SD-CtD lines used are indicated in parenthesis. Asteriskdenotes P-values < 0.001. (e) Suppression of the MC defects of 109-68[m8SD flies (grey bar) upon coexpression of the CtD’s. Numbers tothe right denote average scutellar MC counts.
47CTD AUTOINHIBITS NEURAL REPRESSION

coexpressing the CtD peptides. Balanced stocks of UAS-m8SD1UAS-CtD were crossed to 109-68Gal4 flies (seeinset in Fig. 3). We find that coexpression of M8-CtDstrongly rescued the reduced eye of 109-68Gal4[UAS-m8SD flies (Fig. 3b), and significantly restored the hex-agonal facet phasing and the positioning of the IOB’s(Fig. 3b0). In addition, M8-CtD largely restored the aber-rant ommatidial size that was seen throughout the eyesof 109-68Gal4[UAS-m8SD flies (compare Fig. 3a0,b0),although a few areas of perturbation remained (dottedcircle in Fig. 3b0). In contrast, coexpression of M8SD-CtD did not rescue the reduced and rough eye of 109-68Gal4[UAS-m8SD flies (Fig. 3c), and neither did it res-cue the aberrant ommatidial size, IOB patterning loss,and occasional ‘‘fused’’ ommatidia that are seen in M8SDeyes (Fig. 3a0,c0).
To quantitatively compare the effects of M8-CtD andM8SD-CtD on the reduced eye, we determined facetnumbers in �15 flies of the relevant genotypes, anapproach previously used by others and us (Kahali et al.,2009; Kunttas-Tatli et al., 2009; Shepard et al., 1989).Akin to wild type flies (not shown), 109-68Gal4/1 fliesdisplay �779 6 20 facets, whereas 109-68Gal4/1 UAS-m8SD/1 flies display �411 6 26 facets (Fig. 3d); the lat-ter number served as the baseline. We find that coex-pression of UAS-m8-CtD elicited facet numbers (�650–670) that were significantly higher than the baselinewhen tested with two independent insertions (Lines S4and S5, Fig. 3d). In contrast, coexpression of UAS-m8SD-CtD elicited facet numbers (�430) that were indistin-guishable from the baseline in the two independentinsertions that were examined (Lines D4 and D5, Fig.3d), suggesting that the phosphomimetic CtD wasimpaired for inhibitory activity in ‘‘trans.’’
In addition to the reduced eye, 109-68Gal4[UAS-m8SD flies display a strong loss of the four scutellarMC’s (Fig. 3e), because this enhancer trap is active inthe bristle PNC’s (Kahali et al., 2009; Powell et al.,2004). We, therefore, assessed whether this neuraldefect is modulated by the CtD-peptides. Indeed, coex-pression of M8-CtD restored �2 MC’s/scutellum,whereas the effects of M8SD-CtD appeared markedlyweaker (Fig. 3e). These effects were not line-specific; asthey were recapitulated with two independent inser-tions of UAS-m8-CtD or UAS-m8SD-CtD (see Fig. 3e). Ittherefore appears that the eye and bristle loss of ectopicM8SD are more potently suppressed by M8-CtD, as com-pared to M8SD-CtD. Since the reduced eye of M8SD isinsensitive to CK2- or E(spl)-dosage (Kahali and Bidwai,unpublished), the possibility arises that suppression byM8-CtD involves DN-effects mediated by its interactionwith ectopically expressed M8SD.
M8-CtD More Strongly Suppresses the BristleDefects of Ectopic M8
We sought to further assess the differences in activityof M8-CtD versus M8SD-CtD. We have recently foundthat loss of the MC’s elicited by ectopically expressed
wild type M8 is strongly rescued by coexpression of aUAS-CK2a-RNAi construct or UAS-Tik, a CK2-DN (Boseet al., 2006; Kahali et al., 2009). These results suggestedthat phosphorylation also regulates antagonism of ASCby M8 during SOP selection (Fig. 1a). Crosses were con-ducted as before (inset in Fig. 4) to test whether the CtDhas similar effects on the MC loss of ectopic M8, and if adifference between the two phospho-forms is evi-denced. This appears to be the case. In contrast to thealmost complete loss of the four scutellar MC’s inG455.2[UAS-m8 flies (Fig. 4a,d), coexpression of M8-CtD restored �2–3 MC’s/scutellum (Fig. 4b,d). Often,socket cells and misshapen MC’s were observed (seedotted circles in Fig. 4b), as have previously beenreported upon G455.2 driven expression of an antimor-phic variant of M8 (Giebel and Campos-Ortega, 1997).Importantly, and in contrast, coexpression of M8SD-CtDdid not restore scutellar MC’s of G455.2[UAS-m8 flies(Fig. 4c,d). This difference in activity of M8-CtD versusM8SD-CtD was observed with multiple independentinsertions (Fig. 4d).
It therefore appears that the phosphorylation status ofSer159 does influence the DN-activity of the CtD duringeye and MC development, i.e., M8-CtD suppresses theneural defects of ectopic M8SD or M8 more stronglythan does M8SD-CtD. The stronger rescue by M8-CtD isunlikely to involve sequestration of Gro per se, given itsequivalent interactions with both CtD-variants (Fig. 1d).Moreover, these effects are unlikely to involve competi-tion between two UAS-constructs for (rate) limitingamounts of Gal4 produced either by the G455.2 or 109-68Gal4 enhancer traps, because no modulation of theneural defects of ectopic M8SD or M8 is seen upon coex-pression of a UAS-LacZ construct [data not shown;(Kahali et al., 2009)].
M8-CtD, But Not M8SD-CtD, Rescues the SeverelyReduced Eye of Nspl/Y; E(spl)D/1 Flies
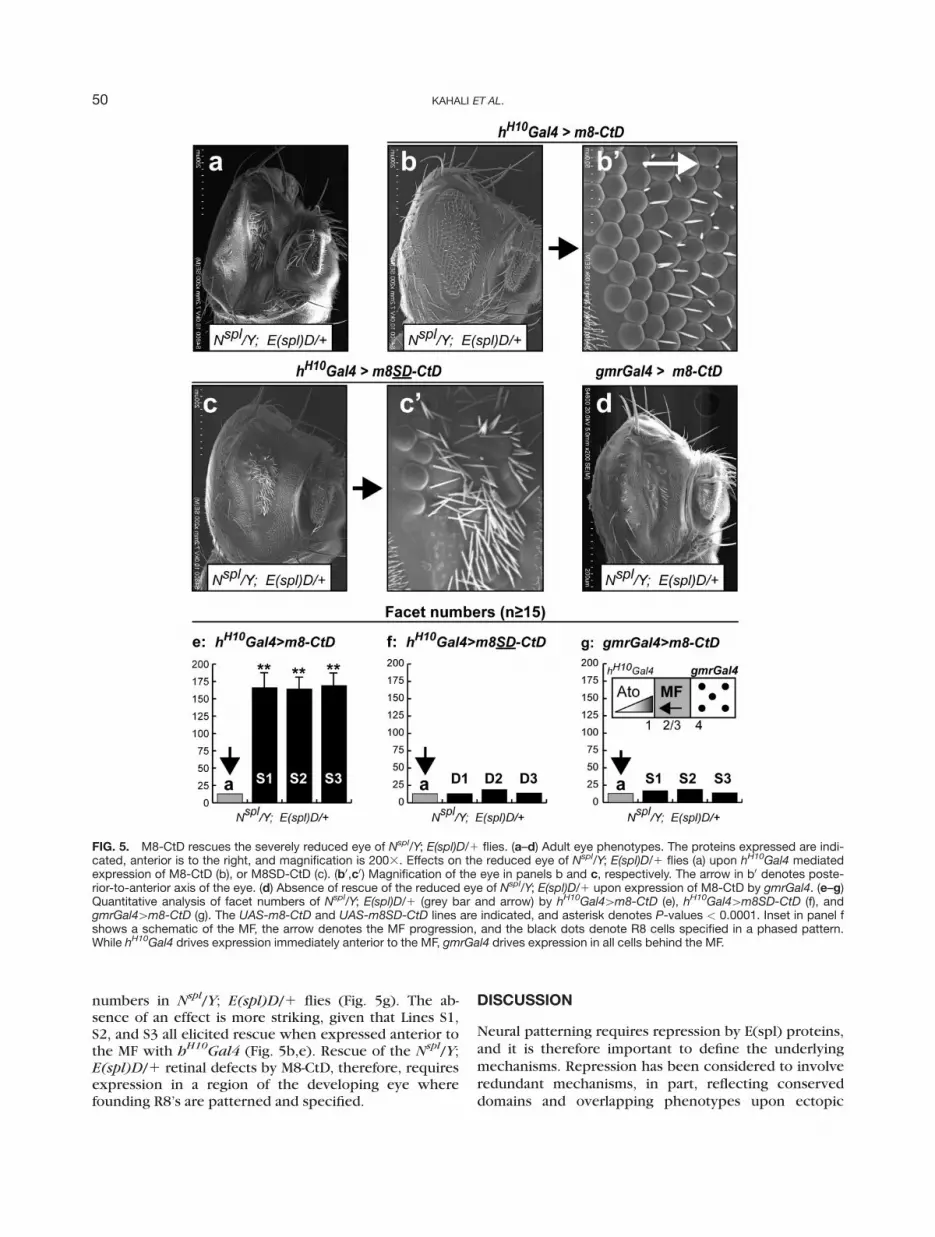
The severely reduced eye of Nspl/Y; E(spl)D/1 (Fig.
5a) has provided important mechanistic insights intorepression by M8 (Giebel and Campos-Ortega, 1997;Kahali et al., 2009; Karandikar et al., 2004; Nagel et al.,1999; Tietze et al., 1992). Given the ability of M8-CtD tonegate repression by ectopic M8SD or M8, we nexttested whether this peptide would display similar activ-ity in an E(spl)D mutant background.
Expression was driven immediately anterior to the MFwith h
H10Gal4, as has previously been used for analysis
of the activity of E(spl)-variants during eye development(Kahali et al., 2009; Ligoxygakis et al., 1998; Nagel et al.,1999). Indeed, the severe reduced eye of N
spl/Y;E(spl)D/1 flies was partially rescued upon overexpres-sion of M8-CtD (Fig. 5b). As previously reported (Karan-dikar et al., 2004; Nagel et al., 1999), the residual eyefield of N
spl/Y; E(spl)D/1 flies (Fig. 5a) is highly disor-ganized, and the few remaining facets are not juxta-posed; the clustering of the IOB’s might well reflect facetloss. Expression of M8-CtD not only increased eye size
48 KAHALI ET AL.
(facet numbers), but also restored patterning of the fac-ets (Fig. 5b0). Examination of a higher magnification (Fig.5b0) indicates that ommatidia at the posterior margin areclosely juxtaposed and patterned akin to that in the wildtype (not shown). This patterning is, however, lost to-ward the anterior margin of the residual eye (see arrowin Fig. 5b0), and presumably reflects more posteriorexpressivity of h
H10Gal4, as has been reported (Ligoxy-
gakis et al., 1998). Analogous to our results with ectopicM8SD or M8 (see above), the reduced eye of N
spl/Y;E(spl)D/1 flies was unaffected by h
H10Gal4-mediated
expression of M8SD-CtD (Fig. 5c,c0). As controls, wetested and found that N
spl/Y; E(spl)D/h
H10Gal4 flies, or
Nspl/Y; UAS-CtD/1; E(spl)D/1 flies, all displayed a
severely reduced eye akin to that in Nspl/Y; E(spl)D/1
flies (data not shown). These results indicate that theh
H10Gal4 enhancer trap does not, on its own, rescue
the reduced eye of Nspl/Y; E(spl)D/1, and that expres-
sion of the CtD is required. Given the inability of ectopicM8SD-CtD to increase eye size, it appears that rescue isspecific to M8-CtD.
We also quantified rescue by determining facet num-bers. N
spl/Y; E(spl)D/1 flies typically display �5–15ommatidia, and this number was used as the baseline(see Fig. 5e,f). Consistent with the adult eye (Fig. 5c),overexpression of M8SD-CtD did not increase facet num-bers to any significant degree in three lines (D1, D2, andD3) that were tested (Fig. 5f). In contrast, expression ofM8-CtD resulted in facet numbers (�150–170) that weresignificantly higher than in N
spl/Y; E(spl)D/1 flies (Fig.5e). Importantly, levels of facet restoration were quantita-
tively similar upon expression of three (second chromo-some) insertions of UAS-m8-CtD (S1, S2, and S3, Fig. 5e).
Rescue of the Nspl/Y; E(spl)D/1 Reduced EyeRequires MF-Specific Expression of M8-CtD
We next assessed whether the effects of M8-CtD dis-play MF-specificity. During the onset of eye develop-ment, R8 specification occurs in a spatially precise man-ner within the MF, a moving wave of differentiation thatsweeps across the third instar eye imaginal disc (Wolffand Ready, 1991). While R8 specification initiates at theanterior margin of the MF, the selection of single phase-shifted R8’s is completed at its posterior margin. In thisregard, it is the specification of R8 cells that is severelyblocked in N
spl/Y; E(spl)D/1 flies (Nagel et al., 1999).This block in R8 formation precipitates loss of the eyefield because R8 photoreceptors are singularly responsi-ble for recruitment of all secondary retinal cell types[reviewed in (Frankfort and Mardon, 2002; Hsiung andMoses, 2002)]. Given this, we reasoned that if rescueinvolved inhibitory activity of M8-CtD (in ‘‘trans’’)against M8*, expression of this peptide posterior to theMF should be without effect. For this, we used gmrGal4,a driver that is active in all cells posterior to the MF [seeinset in Fig. 5g, (Bessa et al., 2002)]. Consistent with ourprediction, expression of M8-CtD with gmrGal4 did notrescue the severe reduced eye of N
spl/Y; E(spl)D/1 flies(Fig. 5d). The absence of rescue is also supported byfacet numbers, which upon expression of three inde-pendent UAS-m8-CtD lines closely mimicked baseline
FIG. 4. M8-CtD suppresses the MC loss phenotype of ectopic M8. Crosses between G455.2 and UAS-constructs were conducted at248C. (a–c) Nota of flies expressing M8 alone (a), M81M8-CtD (b), and M81M8SD-CtD (c). Arrows in panel b denote restored scutellarMC’s and dotted circles denote socket cells. (d) Suppression of the MC defects of G455.2[m8 flies (grey bar) upon coexpression of theCtD’s. The UAS-m8-CtD and UAS-m8SD-CtD lines used are indicated in parenthesis. Asterisk denotes P-values < 0.001. Numbers to theright denote average scutellar MC counts.
49CTD AUTOINHIBITS NEURAL REPRESSION
numbers in Nspl/Y; E(spl)D/1 flies (Fig. 5g). The ab-
sence of an effect is more striking, given that Lines S1,S2, and S3 all elicited rescue when expressed anterior tothe MF with h
H10Gal4 (Fig. 5b,e). Rescue of the N
spl/Y;E(spl)D/1 retinal defects by M8-CtD, therefore, requiresexpression in a region of the developing eye wherefounding R8’s are patterned and specified.
DISCUSSION
Neural patterning requires repression by E(spl) proteins,and it is therefore important to define the underlyingmechanisms. Repression has been considered to involveredundant mechanisms, in part, reflecting conserveddomains and overlapping phenotypes upon ectopic
FIG. 5. M8-CtD rescues the severely reduced eye of Nspl/Y; E(spl)D/1 flies. (a–d) Adult eye phenotypes. The proteins expressed are indi-cated, anterior is to the right, and magnification is 2003. Effects on the reduced eye of Nspl/Y; E(spl)D/1 flies (a) upon hH10Gal4 mediatedexpression of M8-CtD (b), or M8SD-CtD (c). (b0,c0) Magnification of the eye in panels b and c, respectively. The arrow in b0 denotes poste-rior-to-anterior axis of the eye. (d) Absence of rescue of the reduced eye of Nspl/Y; E(spl)D/1 upon expression of M8-CtD by gmrGal4. (e–g)Quantitative analysis of facet numbers of Nspl/Y; E(spl)D/1 (grey bar and arrow) by hH10Gal4[m8-CtD (e), hH10Gal4[m8SD-CtD (f), andgmrGal4[m8-CtD (g). The UAS-m8-CtD and UAS-m8SD-CtD lines are indicated, and asterisk denotes P-values < 0.0001. Inset in panel fshows a schematic of the MF, the arrow denotes the MF progression, and the black dots denote R8 cells specified in a phased pattern.While hH10Gal4 drives expression immediately anterior to the MF, gmrGal4 drives expression in all cells behind the MF.
50 KAHALI ET AL.

expression of individual E(spl) members. As a result,repression has been thought to reflect their expressionand ensuing accumulation in the non-SOP’s/R8 cells inresponse to inhibitory N signaling. Accumulating evi-dence suggests that this model might obscure mechanis-tic diversity of these proteins. This is best exemplifiedduring R8 selection, where ectopic expression of wildtype Md (but not M8 or Mg) affects eye development, afinding interpreted to reflect ‘‘qualitative’’ differencesbetween these members (Ligoxygakis et al., 1998). A de-finitive role for M8 in R8 patterning is, however, illus-trated by the dominant effects of the E(spl)D mutation,and the finding that a similar outcome is elicited byM8SD, a phosphomimetic variant. These studies raisethe possibility that functional diversity involves differen-tial phosphorylation. We have focused on the M8 pro-tein because the E(spl)D mutation provides an excellentframework for structure/function studies to more clearlydefine the mechanisms by which this protein mediatesneural repression. As part of this effort, we have investi-gated the role of phosphorylation of the CtD in autoinhi-bition. The studies we present here provide more directevidence for this layer of regulation and, in addition, sug-gest that diversity of the CtD’s might underlie the ‘‘quali-tative’’ differences ascribed to by Ligoxygakis et al.,(1998).
Our studies more directly test the autoinhibitionmodel (see Fig. 1) in two relevant developmental con-texts, the eye and the bristle, where endogenous E(spl)mediates R8/SOP selection. In support of this model, weprovide multiple lines of evidence that M8-CtD displaysstronger (inhibitory) activity than does the phosphomi-metic variant M8SD-CtD. This difference in the activityof the CtD’s is recapitulated with multiple independentinsertions, and is best seen with their ability to rescuethe neural defects associated with ectopic expression ofM8SD or M8 (Figs. 3 and 4). Although expression levelsof the CtD peptides have not been determined, our find-ings collectively suggest that the differences we observelikely reflect inhibitory activity engendered by interac-tion of M8-CtD with M8SD or with M8 (in its phospho-rylated state) in vivo.
Rescue by M8-CtD is unlikely to reflect sequestrationof Gro, which is not rate limiting for N signaling (Nageland Preiss, 1999). Moreover, suppression by M8-CtD isunlikely to involve competition for, or sequestration of,endogenous CK2. The interaction of CK2 with M8 orM8-CtD is disrupted upon phosphorylation (Kahali andBidwai, unpublished; Karandikar et al., 2004), and whilehalved CK2 dosage is sufficient for proper N signaling,its further knockdown leads to supernumerary(‘‘twinned’’) SOP’s/R8’s, and to rough eyes due toimpaired lateral inhibition (Bose et al., 2006). Consistentwith the possibility that (endogenous) CK2 levels arenot rate limiting for lateral inhibition, the reduced eye ofM8SD is not suppressed by decreased CK2 levels orE(spl)-dosage (Kahali, unpublished). In accordance withthe autoinhibition model, the DN-activity of the M8-CtDpeptide likely reflects its ability to interact with and neu-
tralize repression by endogenous or ectopic E(spl). An al-ternative possibility is that the DN-effects of the CtD,instead, reflect its interaction with Ato/ASC. Since inter-actions between E(spl) and Ato/ASC underlie lateral inhi-bition, binding of CtD to Ato/ASC could also prevent M8or M8SD from mediating neural repression. A number oflines of evidence argue against this alternative possibil-ity. First, M8SD and M8* bind Ato with near identicalstrength, suggesting that the CtD does not contribute tothe Ato interaction. Second, the ability of M8* to interactwith Ato is, in fact, abolished by removal of the Orangedomain. This is best exemplified by E(spl)
BE25, a rever-tant allele of E(spl)D that lacks Orange and does notelicit a reduced eye in the presence of N
spl (Nagel et al.,1999). Third, if the M8SD-Ato interaction were toinvolve the CtD, the DN-effects would have been seenwith M8SD-CtD, rather than M8-CtD. Our findings thatonly M8-CtD displays potent DN-effects do not supportthis alternative possibility. The most parsimonious inter-pretation is that binding of M8-CtD to ectopic M8SD, orto the phosphorylated state of ectopic M8 (M8PO4, thenoninhibited state), attenuates repression.
The rescue of the severely reduced eye of Nspl/Y;
E(spl)D/1 by M8-CtD is the first such example of modu-lation by an ectopically expressed E(spl) sub-domain. Inthis case, M8-CtD rescues the reduced eye, albeit par-tially and significantly restores ommatidial numbers andpatterning, as well as the specification of the IOB’s atalternating positions of the ommatidial lattice. Impor-tantly, no such effects are seen when M8-CtD isexpressed posterior to the MF (with gmrGal4), and arenot observed with the phosphomimetic M8SD-CtD pep-tide that is predicted to bind with reduced strength (seeFig. 5).
How might one interpret the rescue of Nspl/Y;
E(spl)D/1? During development of the founding R8photoreceptors, N signaling occurs in a biphasic mannerin the MF of the developing third instar eye disc[reviewed in (Baonza and Freeman, 2001)]. At the ante-rior margin of the MF, this mediates ato expression,which is subject to a positive feedback loop (Sun et al.,1998), whereas in the MF it drives expression of E(spl)enabling refinement of single R8 cells from the PNC’s.N
spl renders R8 precursors hypersensitive to inhibitoryN signaling, and thus impairs R8 specification (Li et al.,2003). The loss of differentiated R8’s, in turn, impairsexpression of Hedgehog and Decapentaplegic, whoseactivities are necessary for ato expression (Li et al.,2003). In this sensitized background, M8* (E(spl)D) fur-ther decreases ato expression, by impairing the positivefeedback loop (Kahali et al., 2009). As a result, Ato levelsdrop below a threshold necessary for conferring the R8fate, and results in a loss of the eye field. Given its effectson Ato, the retinal defects of E(spl)D are enhanced bymutations in ato or its heteromeric partner, daughterless(da), whereas they are suppressed by mutations in Delta(Dl), which would reduce inhibitory N signaling in R8precursors (Nagel and Preiss, 1999; Shepard et al.,1989).
51CTD AUTOINHIBITS NEURAL REPRESSION

It would, therefore, seem to be the case that M8-CtDbinds to non-autoinhibited M8* (see Fig. 5), whichwould attenuate its interactions with Ato. This wouldattenuate that ability of M8* to impair the ato feedbackloop, and consequently Ato would rise to a level suffi-cient to confer the R8 fate, which would increase eyesize (ommatidial numbers). Our finding that the antimor-phic activity of M8-CtD is manifest only when expressedanterior to the MF, but not posterior to it (see Fig. 5), isconsistent with the findings that E(spl)D effects are MF-and R8-specific [discussed in (Kahali et al., 2009)].Therefore, the possibility arises that the inability ofM8SD-CtD to rescue the eye defects of N
spl/Y; E(spl)D/1 reflects a reduced capacity, or perhaps an inability, tobind M8*. Under these conditions, M8* retains its abilityto bind to Ato and block eye development. Collectively,the effects we see (Figs. 2–5) are consistent withautoinhibition.
The domains/motifs that are highly conserved in allE(spl) proteins are the b/HLH and Orange and WRPW.While the HLH domain appears to mediate homo/het-ero-dimerization, Orange is thought to be the specificitydeterminant (Dawson et al., 1995). In this regard, auto-inhibition by the CtD could involve interactions with ei-ther domain (Fig. 1a). If this involved the HLH domain,the autoinhibited state of M8 is predicted to be a mono-mer, a conformation that is non-permissive for repres-sion [discussed in (Giebel and Campos-Ortega, 1997)].In the yeast two-hybrid assay, dimerization of M8 doesappears weaker than that of other E(spl) proteins (Ali-fragis et al., 1997; Nagel et al., 1999), but it is not knownif this weak interaction reflects the native state or is aconsequence of misfolding. Alternatively, the CtD inter-acts with Orange. In this case, the CtD, in a phosphoryla-tion dependent manner, would regulate antagonism ofASC/Ato (Fig. 1a). The suppression of neural defects ofectopic M8 or M8SD, and the rescue of the reduced eyeof N
spl/Y; E(spl)D/1 (by ectopic M8-CtD) would be con-sistent with its interactions with either HLH or Orange.We attempted, but were unable to detect interactionsbetween M8-CtD and either M8* or M8SD (data notshown), even though these proteins are expressed inyeast (Fig. 1d, and data not shown). Given the small sizeof the CtD peptides, the possibility remains that expres-sion as a (two hybrid) fusion impedes protein-interac-tions. Alternative approaches on native proteins and iso-lated CtD peptides, such as chemical cross-linking andpeptide mapping, or surface plasmon resonance, will beneeded to identify the interaction site/interface.
Autoinhibition by the CtD, which represents a novelproposition in the regulation of repression by E(spl)-M8,is an established mechanism regulating other proteins.Well described in protein kinases, e.g., the MAPK’s andcyclin-dependent protein kinases (CDK’s), autoinhibi-tion and phosphorylation are increasingly found to regu-late transcription factors [reviewed in (Gardner andMontminy, 2005; Schlessinger, 2003; Smock and Gier-asch, 2009; Tokuriki and Tawfik, 2009)]. An excellentexample is IRF-3 (Interferon Regulatory Factor-3). Crys-
tal structure of IRF-3 shows that a highly disorderedregion, the autoinhibitory peptide, binds to a two-helixbundle preventing dimerization (Qin et al., 2003). Phos-phorylation of this peptide elicits displacement, whichthen permits dimerization and transcriptional activities.It has, therefore, been proposed that autoinhibition via a‘‘reversible intra-molecular latch’’ is dynamically coupledto signaling (Smock and Gierasch, 2009). Such regula-tion influences the activities of diverse transcription fac-tors such as Vav, Ets, Fos, Myc, and p53 [reviewed in(Garza et al., 2009)]. More recently, it has been sug-gested that such regulatory regions, often the sites forphosphorylation, are ‘‘intrinsically disordered’’ (ID), andthat the co-localization of these features underlies func-tional diversity (Smock and Gierasch, 2009; Tokuriki andTawfik, 2009). Consistent with this, these regions of IDhave evolved rapidly, are enriched in Ser residues, andoften contain motifs for phosphorylation as well as pro-tein interactions (Collins, 2009).
Structure and sequence analyses indicate that such IDregions exhibit a preference for certain amino acids[reviewed in (Dunker et al., 2008; Tompa, 2002)]. Basedon these data, computational approaches have beendeveloped to predict their presence [reviewed in (Leeet al., 2007)]. We reasoned that in the case of the E(spl)proteins, if regions of sequence divergence, phosphoryl-ation, and ID-propensity co-localize to the CtD, it wouldfurther implicate this sub-domain in the regulation ofrepression and in nonredundancy. Aside from small dif-ferences at the N-terminus, the CtD significantly contrib-utes to the length/sequence heterogeneity of E(spl)members (Fig. 6a). This is best illustrated by the conser-vation of the SPxS----SDxE/D motif. The former repre-sents a putative MAPK site, while the latter is the knownCK2 recognition/phosphorylation site in M8/5/7 (Fig.6a). A closer examination reveals the presence of theSPxS motif in Mg that is separated from a CK2 phospho-rylation site S195EDE (Fig. 6a,b). The latter site, whichfully conforms to the consensus for CK2, i.e., S/T-D/E-x-D/E (Kuenzel and Krebs, 1985), has remained invariantin Mg homologues through Drosophila evolution (seealignment in Fig. 6b). Moreover, the replacement and/orinsertions upstream of Ser195 in some Mg members donot affect the consensus requirements for CK2. Evolu-tionary principles would suggest that this conservationis, therefore, of consequence. It will be of interest todetermine if Mg is modified at Ser195 by CK2, andwhether its replacement with Asp generates an activerepressor that elicits a reduced eye, analogous to M8SD.
Given that M8, Mg, and Md are the only membersexpressed in the MF, we assessed whether the CK2 sitelocalizes to an ID-region. This appears to also be thecase for M8 and Mg (Fig. 6b). In contrast, Md, the onlymember that blocks eye development when overex-pressed (Ligoxygakis et al., 1998), is predicted to belargely globular and lacks an ID-region or CK2 phospho-rylation site. As shown in Figure 6b, the other ID-regionslocalize to the sequence between HLH and Orange (inMg and Md), or the loop connecting the two helices of
52 KAHALI ET AL.

Orange (in M8). These loops might, however, be struc-turally constrained by their positioning between two hel-ices. In the case of Hes6, the mammalian homolog ofDrosophila E(spl), an ID region and CK2 site also coloc-alize (not shown), suggesting that the stimulatory effectsof phosphorylation on Hes6 activity (Gratton et al.,2003) may reflect a similar regulation.
The possibility thus arises that, as with M8, repressionby Mg might also be regulated by phosphorylation,whereas Md is independent. If so, the ‘‘qualitative’’ differ-ences between these three E(spl) members, proposedby Ligoxygakis et al. (1998), may reflect diversity of theCtD’s, and their differential phosphorylation in an iso-form-dependent manner. Future studies on the regioncontacted by the nonphosphorylated CtD of M8, therelevance of this mechanisms to other E(spl) members,and analysis of chimeric proteins will be required tomore fully define the diverse mechanisms underlyingrepression by this group of bHLH proteins.
METHODS
Construction of CtD Variants
The region of the cDNA’s encoding residues 123–179of M8 and M8SD were amplified by PCR using customprimers, and contain BamH1 and Xho1 sites 50 and 30 to
the open reading frame, respectively. The PCR productswere verified by sequencing.
Phosphorylation and Protein Interactions
Phosphorylations were conducted using CK2 holo-enzyme purified to homogeneity from Drosophilaembryos (Karandikar et al., 2005). Bacterially expressedCtD peptides were purified as fusions with glutathione-S-transferase (GST). Following purification, the GST-fusion proteins were exchanged into storage buffer (50mM Tris, pH 8.0, 0.5 mM EDTA, 10% glycerol, 200 mMNaCl, 1 mM PMSF) using a Biomax-10K centrifugal filterdevice (Millipore). Phosphorylations were carried out in50 mM Tris, pH 8.5, 100 mM NaCl, 10 mM MgCl2, 10 lMATP, 5 lCi [g-32P]-ATP in a total volume of 40 ll, at 258Cfor 10 min. GST-fusions (2 lg each) were phosphoryl-ated with 40 ng of the CK2-holoenzyme for 15 min at258C, conditions that ensure an assessment of the initialrates (Bidwai et al., 1993). The reactions were termi-nated with 10 ll of 53 sample buffer, boiled for 5 min,and separated on 12% acrylamide gels containing sodiumdodecylsulfate. Gels were stained with Coomassie Blue,and exposed to Kodak XAR-5 film.
Interactions with Gro were assessed using a LexA ver-sion of the yeast two-hybrid system (Gyuris et al., 1993).LexA-CtD constructs were tested against a fusion of Growith the activation-domain of protein B42. Interactions
FIG. 6. Structure of the CtD. (a) Alignment of the CtD sequences of E(spl) members. Proteins phosphorylated by CK2 are highlighted inred. Mg is predicted to contain a CK2 consensus site (arrow). (b) GlobPlot-2 (http://globplot.embl.de/) based prediction of globular and IDregions in E(spl) members expressed in the eye. Grey inset shows effects of ectopic expression on eye development. Alignment of Mghomologs. Abbreviations are; mel, melanogaster; ere, erecta; sec, sechellia; sim, simulans; yak, yakuba; ana, ananassae; wil, willistoni, moj,mojavensis; hyd, hydei; vir, virilis; per, persimilis; pse, pseudoobscura; gri, grimshawi. The Ser of the predicted CK2 site is shown as a star;1 denotes conserved substitution; 2 denotes not conserved/insertion; black dot denotes invariant.
53CTD AUTOINHIBITS NEURAL REPRESSION

were assessed in yeast strain EGY048. LacZ reporter ac-tivity was determined for at least three independenttransformants, each in triplicate, as described. LacZ ac-tivity was determined using the formula 1,000 3 OD420/(T 3 V 3 OD600), where T is minutes and V is the con-centration factor of the assay.
Fly Stocks, Crosses, and Phenotypes
For in vivo expression, constructs encoding M8-CtDand M8SD-CtD were cloned into the plasmid pUAST(Brand and Perrimon, 1993). Both constructs are identi-cal in length and 50 and 30 ends, aside from the missensemutation at Ser159. Germline transformants were gener-ated using a commercial embryo injection facility (Best-Gene). w
1 progeny were identified and the location ofinsertions was determined via crosses to lines harboringchromosomes carrying dominant visible markers. Multi-ple independent insertions of each construct were usedin these studies.
Flies were raised at 248C on standard Yeast-Glucosemedium. The Gal4 drivers used in these studies areG455.2, 109-68Gal4, h
H10Gal4, and gmrGal4. Fly heads
were dehydrated by sequential passes through a gradedalcohol series (25–50–75–99%) and finally through Hex-amethyldisalizane. Heads were mounted on EM stubs,dried for 24 h, sputter coated with gold, and examinedwith a JEOL-6400 scanning electron microscope at anaccelerating voltage of 10–20 kV. For bristle phenotypes,newly eclosed adults were photographed. For quantita-tive analysis of the bristle phenotypes, multiple crosseswere established (�triplicates), and adults were scoredfor bristle artifacts. For quantitative analysis of the eyefield, �15 adults were photographed, and ommatidialnumbers were determined manually. In every case multi-ple independent insertions of UAS-constructs wereused.
ACKNOWLEDGMENTS
The authors thank Philip Keeting for comments, SophiaZhang for technical assistance, and Diane Schwegler-Berry (NIOSH, CDC) for assistance with electronmicroscopy.
LITERATURE CITED
Alifragis P, Poortinga G, Parkhurst SM, Delidakis C. 1997. A network ofinteracting transcriptional regulators involved in Drosophila neu-ral fate specification revealed by the yeast two-hybrid system. ProcNatl Acad Sci USA 94:13099–13104.
Bailey AM, Posakony JW. 1995. Suppressor of hairless directly activatestranscription of enhancer of split complex genes in response toNotch receptor activity. Genes Dev 9:2609–2622.
Baonza A, Freeman M. 2001. Notch signalling and the initiation of neu-ral development in the Drosophila eye. Development 128:3889–3898.
Bessa J, Gebelein B, Pichaud F, Casares F, Mann RS. 2002. Combinatorialcontrol of Drosophila eye development by eyeless, homothorax,and teashirt. Genes Dev 16:2415–2427.
Bidwai AP, Reed JC, Glover CVC. 1993. The phosphorylation of Cal-modulin by the catalytic subunit of casein kinase II is inhibited bythe regulatory subunit. Arch Biochem Biophys 300:265–270.
Bose A, Kahali B, Zhang S, Lin J-M, Allada R, Karandikar U, Bidwai A.2006. Drosophila CK2 regulates lateral-inhibition during eye andbristle development. Mech Dev 123:649–664.
Brand AH, Perrimon N. 1993. Targeted gene expression as a means ofaltering cell fates and generating dominant phenotypes. Develop-ment 118:401–415.
Bray SJ. 2006. Notch signalling: A simple pathway becomes complex.Nat Rev Mol Cell Biol 7:678–689.
Brennan K, Tateson R, Lewis K, Arias AM. 1997. A functional analysisof notch mutations in Drosophila. Genetics 147:177–188.
Calleja M, Renaud O, Usui K, Pistillo D, Morata G, Simpson P. 2002.How to pattern an epithelium: Lessons from achaete-scute regula-tion on the notum of Drosophila. Gene 292:1–12.
Campos-Ortega JA. 1997. Neurogenesis in Drosophila: A historical per-spective and some prospects. Perspect Dev Neurobiol 4:267–271.
Collins MO. 2009. Cell biology. Evolving cell signals. Science 325:1635–1636.
Cooper MTD, Tyler DM, Furriols M, Chalkiadaki A, Delidakis C, Bray SJ.2000. Spatially restricted factors cooperate with notch in the regu-lation of enhancer of split genes. Dev Biol 221:390–403.
Cubas P, de Celis JF, Campuzano S, Modolell J. 1991. Proneural clustersof achaete-scute expression and the generation of sensory organsin the Drosophila imaginal wing disc. Genes Dev 5:996–1008.
Dambly-Chaudiere C, Vervoort M. 1998. The bHLH genes in neural de-velopment. Int J Dev Biol 42:269–273.
Dawson SR, Turner DL, Weintraub H, Parkhurst SM. 1995. Specificityfor the hairy/enhancer of split basic helix-loop-helix (bHLH) pro-teins maps outside the bHLH domain and suggests two separablemodes of transcriptional repression. Mol Cell Biol 15:6923–6931.
Dunker AK, Silman I, Uversky VN, Sussman JL. 2008. Function andstructure of inherently disordered proteins. Curr Opin Struct Biol18:756–764.
Fisher AL, Ohsako S, Caudy M. 1996. The WRPW motif of the hairy-related bHLH repressor proteins acts as a 4-amino acid transcrip-tional repression and protein–protein interaction domain. MolCell Biol 16:2670–2677.
Frankfort BJ, Mardon G. 2002. R8 development in the Drosophila eye:A paradigm for neural selection and differentiation. Development129:1295–1306.
Gardner KH, Montminy M. 2005. Can you hear me now? Regulatingtranscriptional activators by phosphorylation. Sci STKE2005:pe44.
Garza AS, Ahmad N, Kumar R. 2009. Role of intrinsically disorderedprotein regions/domains in transcriptional regulation. Life Sci84:189–193.
Giagtzoglou N, Alifragis P, Koumbanakis KA, Delidakis C. 2003. Twomodes of recruitment of E(spl) repressors onto target genes. De-velopment 130:259–270.
Gibert JM, Simpson P. 2003. Evolution of cis-regulation of the proneuralgenes. Int J Dev Biol 47:643–651.
Giebel B, Campos-Ortega JA. 1997. Functional dissection of the Dro-sophila enhancer of split protein, a suppressor of neurogenesis.Proc Natl Acad Sci USA 94:6250–6254.
Gratton M-O, Torban E, Jasmin SB, Theriault FM, German MS, Stifani S.2003. Hes6 promotes cortical neurogenesis and inhibits Hes1 tran-scription repression activity by multiple mechanisms. Mol CellBiol 23:6922–6935.
Gyuris J, Golemis E, Chertkov H, Brent R. 1993. Cdi1, a human G1 andS phase protein phosphatase that associates with cdk2. Cell75:791–803.
Heitzler P, Bourouis M, Ruel L, Carteret C, Simpson P. 1996. Genes ofthe enhancer of split and achaete-scute complexes are requiredfor a regulatory loop between notch and delta during lateral signal-ling in Drosophila. Develop 122:161–171.
Hinz U, Giebel B, Campos-ortega JA. 1994. The basic-helix-loop-helixdomain of Drosophila lethal of scute protein is sufficient for pro-neural function and activates neurogenic genes. Cell 76:77–87.
Hsiung F, Moses K. 2002. Retinal development in Drosophila: Specify-ing the first neuron. Hum Mol Genet 11:1207–1214.
Jarman AP, Grell EH, Ackerman L, Jan LY, Jan YN. 1994. Atonal is theproneural gene for Drosophila photoreceptors. Nature 369:398–400.
54 KAHALI ET AL.

Jarman A, Sun Y, Jan L, Jan Y. 1995. Role of the proneural gene, atonal,in formation of Drosophila chordotonal organs and photorecep-tors. Development 121:2019–2030.
Kahali B, Trott R, Paroush Z, Allada R, Bishop CP, Bidwai AP. 2008. Dro-sophila CK2 phosphorylates Hairy and regulates its activity invivo. Biochem Biophys Res Commun 373:637–642.
Kahali B, Bose A, Karandikar U, Bishop CP, Bidwai A. 2009. On themechanism underlying the divergent retinal and bristle defects ofM8* (E(spl)D) in Drosophila. Genesis 47:456–468.
Karandikar U, Trott RL, Yin J, Bishop CP, Bidwai AP. 2004. DrosophilaCK2 regulates eye morphogenesis via phosphorylation ofE(spl)M8. Mech Dev 121:273–286.
Karandikar U, Shaffer J, Bishop CP, Bidwai AP. 2005. Drosophila CK2phosphorylates Deadpan, a member of the HES family of basic-he-lix-loop-helix (bHLH) repressors. Mol Cell Biochem 274:133–139.
Kuenzel EA, Krebs EG. 1985. A synthetic substrate specific for caseinkinase II. Proc Natl Acad Sci USA 82:737–741.
Kunttas-Tatli E, Bose A, Kahali B, Bishop CP, Bidwai AP. 2009. Functionaldissection of Timekeeper (Tik) implicates opposite roles for CK2and PP2A during Drosophila neurogenesis. Genesis 47:647–658.
Lecourtois M, Schweisguth F. 1995. The neurogenic suppressor of hair-less DNA-binding protein mediates the transcriptional activationof the enhancer of split complex genes triggered by Notch signal-ing. Genes Dev 9:2598–2608.
Lee D, Redfern O, Orengo C. 2007. Predicting protein function fromsequence and structure. Nat Rev Mol Cell Biol 8:995–1005.
Lehmann R, Jimenez F, Dietrich U, Campos-Ortega JA. 1983. On thephenotype and development of mutants of early development inDrosophila melanogaster. Roux’s Arch Dev Biol 192:62–74.
Li Y, Lei L, Irvine KD, Baker NE. 2003. Notch activity in neural cells trig-gered by a mutant allele with altered glycosylation. Develop130:2829–2840.
Ligoxygakis P, Yu SY, Delidakis C, Baker NE. 1998. A subset of Notchfunctions during Drosophila eye development require Su(H) andE(spl) gene complex. Development 125:2893–2900.
Ligoxygakis P, Bray SJ, Apidianakis Y, Delidakis C. 1999. Ectopic expres-sion of individual E(spl) genes has differential effects on differentcell fate decisions and underscores the biphasic requirement fornotch activity in wing margin establishment in Drosophila. Devel-opment 126:2205–2214.
Modolell J, Campuzano S. 1998. The achaete-scute complex as an inte-grating device. Int J Dev Biol 42:275–282.
Nagel AC, Preiss A. 1999. Notch spl is deficient for inductive processesin the eye, and E(spl)D enhances split by interfering with proneu-ral activity. Dev Biol 208:406–415.
Nagel A, Yu Y, Preiss A. 1999. Enhancer of Split [E(spl)D] is a Gro-inde-pendent, hypermorphic mutation in Drosophila. Dev Genet25:168–179.
Nakao K, Campos-Ortega JA. 1996. Persistent expression of genes ofthe enhancer of split complex suppress neural development inDrosophila. Neuron 16:275–286.
Paroush Z, Finley RL, Kidd T, Wainwright SM, Ingham PW, Brent R, Ish-Horowcz D. 1994. Groucho is required for Drosophila neurogene-sis, segmentation, and sex determination and interacts directlywith hairy related bHLH proteins. Cell 79:805–815.
Powell LM, Zur Lage PI, Prentice DR, Senthinathan B, Jarman AP. 2004.The proneural proteins Atonal and Scute regulate neural targetgenes through different E-box binding sites. Mol Cell Biol24:9517–9526.
Qin BY, Liu C, Lam SS, Srinath H, Delston R, Correia JJ, Derynck R, LinK. 2003. Crystal structure of IRF-3 reveals mechanism of autoinhi-bition and virus-induced phosphoactivation. Nat Struct Biol10:913–921.
Schlessinger J. 2003. Autoinhibition control. Science 300:750–752.
Schrons H, Knust E, Campos-Ortega JA. 1992. The enhancer of splitcomplex and adjacent genes in the 96F region of Drosophila mel-
anogaster are required for segregation of neural and epidermalcells. Genetics 132:481–503.
Shepard SB, Broverman SA, Muskavitch MA. 1989. A tripartite interac-tion among alleles of notch, delta, and enhancer of split duringimaginal development of Drosophila melanogaster. Genetics122:429–438.
Simpson P. 1990. Lateral inhibition and the development of the sensorybristles of the adult peripheral nervous system of Drosophila. De-velopment 109:509–519.
Skeath JB, Carroll SB. 1991. Regulation of achaete-scute gene expres-sion and sensory organ pattern formation in the Drosophila wing.Genes Dev 5:984–995.
Smock RG, Gierasch LM. 2009. Sending signals dynamically. Science324:198–203.
Sun Y, Jan L, Jan Y. 1998. Transcriptional regulation of atonal during de-velopment of the Drosophila peripheral nervous system. Develop125:3731–3740.
Tietze K, Oellers N, Knust E. 1992. Enhancer of SplitD, a dominantmutation of Drosophila, and its use in the study of functionaldomains of a helix-loop-helix protein. Proc Natl Acad Sci USA89:6152–6156.
Tokuriki N, Tawfik DS. 2009. Protein dynamism and evolvability. Sci-ence 324:203–207.
Tompa P. 2002. Intrinsically unstructured proteins. Trends Biochem Sci27:527–533.
Trott RL, Kalive M, Paroush Z, Bidwai AP. 2001. Drosophila mela-
nogaster casein kinase II interacts with and phosphorylates thebasic-helix-loop-helix (bHLH) proteins m5, m7, and m8 derivedfrom the enhancer of split complex. J Biol Chem 276:2159–2167.
Welshons WJ. 1956. Analysis of a gene in Drosophila. Science150:1122–1129.
Wolff T, Ready D. 1991. The beginning of pattern formation in the Dro-sophila compound eye: The morphogenetic furrow and the sec-ond mitotic wave. Development 113:841–850.
55CTD AUTOINHIBITS NEURAL REPRESSION





























