Embed Size (px)
Citation preview

Estrogen Receptor b Ligands: RecentAdvances and Biomedical Applications
Filippo Minutolo,1 Marco Macchia,1 Benita S. Katzenellenbogen,2
and John A. Katzenellenbogen3
1Dipartimento di Scienze Farmaceutiche, Universita di Pisa, Via Bonanno 6, 56126 Pisa, Italy2Department of Molecular and Integrative Physiology, University of Illinois, 407 S. Goodwin Avenue, Urbana,
Illinois 618013Department of Chemistry, University of Illinois, Urbana, Illinois 61801
Published online 4 December 2009 in Wiley Online Library (wileyonlinelibrary.com).
DOI 10.1002/med.20186
.
Abstract: Recent work elucidating the role that the estrogen receptor b (ERb), a member of the nuclear
receptor superfamily, plays in regulating various physiological functions has highlighted the potential of this
receptor subtype as a therapeutic target for several pathologies. In fact, molecules that are able to selectively
activate ERb hold promise for the treatment of certain cancers, as well as endometriosis, inflammatory
diseases including rheumatoid arthritis, and cardiovascular and CNS conditions. Nevertheless, ERbremains a challenging target because its ligand-binding cavity is very similar to that present in ERa, and this
makes it difficult to develop ligands having sufficient levels of ERb selectivity for therapeutic use. Never-
theless, considerable advances have recently been made in developing both nonsteroidal and steroidal ERb-selective agonists. These molecules constitute not only important tools to probe the biological effects of the
selective stimulation of ERb, but some of them appear to be agents with considerable therapeutic potential.
This study provides a detailed review of selective ERb ligands that have been developed recently. After a
brief introduction to the structure and nature of the two ERs and the biology of ERb and its isoforms, the
ligands are classified on the basis of their structures and activities. Common pharmacophore elements are
highlighted throughout the description of the various chemical classes analyzed, and these elements are
presented in a concluding summary overview along with a discussion of potential therapeutic applications
of these agents in biomedicine. & 2009 Wiley Periodicals, Inc. Med Res Rev, 31, No. 3, 364–442, 2011
Key words: estrogens; estrogen receptor b; ligand; binding; pharmacophore; agonist; antagonist
1. INTRODUCTION
A. Estrogen Receptor b (ERb)
1. Estrogens act through two receptors, ERa and ERbWhile estrogens are classically considered female reproductive hormones because theyplay key roles in many aspects of female reproductive development and physiology, it is
Correspondence to:Filippo Minutolo, Dipartimento di Scienze Farmaceutiche,UniversitaØ di Pisa,Via Bonanno 6, 56126 Pisa, Italy,
E-mail: [email protected]
Medicinal Research Reviews, 31,No. 3, 364--442, 2011
& 2009 Wiley Periodicals, Inc.

increasingly appreciated that estrogens also regulate reproductive processes in males, as wellas influencing many nonreproductive physiological and metabolic processes in both sexes.There are notable effects on bone, brain, the cardiovascular system, liver, lung, colon, skin,adipose tissue, and, most likely, other organs and tissues. They are also involved in manydisease states, including hormone-regulated cancers, such as breast and prostate cancers.
The effects of estrogens are mediated through two receptors, estrogen receptor a (ERa)and b (ERb), which are encoded by genes on different chromosomes and that function asligand-modulated transcription factors, up- and downregulating gene expression in targettissues. While the focus of this review is on the medicinal chemistry of ER ligands that showselectivity for one receptor subtype, ERb, and on associated structure–activity relationships,it is worthwhile to provide a brief overview of the distinct biological effects that are mediatedby ERa and ERb to understand the potential medical applications there might be for ERb-selective ligands and why there is great interest in ERb as a therapeutic target. These topicshave been the subject of a number of review articles,1–5 many of which are cited belowtogether with primary references.
2. Differences in the binding cavities of ERa and ERbThe ligand-binding domains (LBDs) of ERa and ERb are only 59% amino acid sequenceidentical, yet the ligand-binding pockets of the two subtypes have only minor differences instructure and composition.6 Thus, an overall structural analysis of the two receptor com-plexes with estradiol (E2) shows that both of them have a conserved high energy attractiveinteraction involving the phenolic OH of E2 in a hydrogen bond network that includes abound water molecule and two amino acid residues of the ER LBD (Glu353 and Arg394 inERa, Glu305 and Arg346 in ERb, Fig. 1). Furthermore, in both receptors, the 17b-hydroxygroup of E2 participates in an additional hydrogen bond donation to His524 (ERa) or His475(ERb). A more indepth look at the ERa/ERb-binding cavities shows that each is composedof 23 residues that are within 4 A of the ligand; of these 23, only at two positions are theamino acids different, and these differences represent conservative replacements: Leu384 andMet421 of ERa are, respectively, replaced by Met336 and Ile373 in ERb.7 The Leu384/Met336 residues are positioned above the B- and C-rings, near position 8b of E2, and theMet421/Ile373 residues are positioned below the D-ring, near position 16a and 17a, of thesame ligand (Fig. 1).
OH
HO
Me
H
HH
ER
Glu353
Arg394
His524
Leu384
Me S
Met421
ER
Glu305
Arg346
SMet336
Ile373
Me
OH
HO
Me
H
HH
His475
Figure 1. Principal interactions of estradiol with ERa and ERb conserved and nonconserved residues.
ESTROGENRECEPTOR bLIGANDS K 365
Medicinal Research Reviews DOI 10.1002/med

Finally, the ERb-binding pocket has a smaller volume than that of ERa, and there arealso slight differences in the shape of these cavities because of differences in the amino acidresidues lining the cavity borders. The high similarity between the ERa and ERb-bindingcavities in terms of sequence, size, and shape has made the production of highly ERb-selective ligands particularly challenging.
3. Tissue distribution and phenotypes of ER knockout animalsThe tissue distributions of ERa and ERb are very different, with high ERa levels being foundin the uterus, mammary gland, ovarian theca cells, and lower levels in bone, vascularendothelium, liver, prostate, pituitary gland, and regions of the brain. ERb is also found,together with ERa, in many of these tissues, but generally at lower levels. In addition, ERb isthe predominant and sometimes the exclusive subtype in certain other regions of the brain,and in lung, colon, and ovarian granulosa cells. These differences in the tissue distribution ofthe two ER subtypes suggest organ systems in which ERb-selective ligands might haveselective biological effects.8
Much of what is known about the distinctive biology of ERb versus ERa comes fromstudies in ERa, ERb, and ERa/b knockout mice.1,3,4 The phenotype of the ERa knockoutmouse is severe and what one might expect from abrogation of a major regulator of thereproductive system. In females, the uterus is atrophic and nonresponsive to estrogen, andthe mammary gland shows only prepubertal development. Both females and males areinfertile, with males having abnormal sperm and fluid accumulation in the testes. ERaknockout mice also become obese with age and exhibit metabolic changes.3,4
ERb knockout mice generated in different laboratories have exhibited different pheno-types.2 Although the basis for this is not fully clear, some may originate in different tech-nologies and gene deletion constructs that result in differences in alternative splice transcriptsproduced. Initial reports of ERb knockout mice showed a more limited phenotype; femaleshad ovarian pathologies with inefficient folliculogenesis and low fertility, while malesappeared to be largely normal.2 However, ERb-knockout mice, generated more recentlyusing Cre/LoxP technology, showed both male and female sterility, with little or no obviouseffects of ERb deletion on nonreproductive organs.9
There also appear to be behavioral changes in ER knockout mice, with ERa knockoutmales showing reduced aggressive behavior and ERb knockout males exhibiting exaggeratedaggression.3,4 Intriguingly, in the absence of ERb, some estrogen-responsive tissues (uterusand prostate) appeared in some studies to be more responsive to estrogens than in wild-typeanimals and to show evidence of hyperproliferative activity,10 although there is no universalagreement on this point.9
4. Contrasting activities of ERa and ERb in breast, breast cancer, and other targetsThe level of the two estrogen receptors in both normal tissues and cancers can vary, withinteresting effects on cell regulation and proliferative character. For example, while ERbpredominates in normal breast, ERa is the major subtype in most breast cancers, with ERblevels declining as breast cancer develops, dwindling to lower levels with increasing malig-nancy of the disease.11 These observations in the breast and in breast cancer, as well as theevidence for the hyperproliferative state of the prostate in ERb knockout mice,10 have led tothe articulation of a paradigm in which ERa is considered the driver of estrogen-mediatedcellular proliferation, with ERb being the partner that provides a restraint or brake on ERaaction. The relationship of these activities to the interdependent yet opposing forces of ‘‘yin’’and ‘‘yang’’ of Chinese philosophy has been noted, with ERa being the yin and ERb theyang.12 While useful as a generalization, this philosophical metaphor fails to capture all theactivities of both ER subtypes.
366 K MINUTOLOETAL.
Medicinal Research Reviews DOI 10.1002/med

The contrasting nature of ERa and ERb has also been evaluated in many cell culturesystems. In reporter gene assays in cells treated with saturating concentrations of E2, ERbshows a lower maximal activity, often being only ca. 15–20% as active as ERa.13 Conse-quently, ERb moderates the magnitude of gene expression regulation by ERa.14–17 Also,when ERb is added to ERa-containing cells, it moderates the proliferative stimulation ofestrogens.14–17 Thus, because it can reduce ERa activity, ERb can be considered an en-dogenous partial dominant negative receptor,18,19 again a useful generalization but not onethat is universally true.
Nevertheless, one might expect that ligands with preferential affinity or efficacy for ERbmight moderate the proliferative drive that estrogens have through ERa in various targettissues, at least in those tissues (or stages of hormone-regulated cancers) in which both ERaand ERb levels are significant. This thinking underlies many of the investigations of thetherapeutic utility of ERb-selective ligands.1,2,20
It has been reported that some synthetic estrogens can elicit some actions through amembrane-associated G-protein, GPR30. Because the ligand-binding selectivity of GPR30 isvery different from that of either ERa or ERb, and its role in many tissues is as yet unclear,we do not discuss it further but refer readers to recent review articles.21–25
B. Challenges and Opportunities with ERb as a Target for Pharmaceuticals:Effects in Normal versus Disease States, Receptor Isoforms, and Compound-Specific Activity
An intriguing ‘‘challenge hypothesis’’ has been advanced regarding the activities of ERb-selective ligands: Because they typically have relatively minor effects in normal, healthyanimals, yet are effective in a number of disease models, activity of ERb-selective ligands mayonly become apparent in cells that are ‘‘yinjured/stressed/or otherwise compromisedy’’1,2
While lack of an effect in normal tissues is a benefit for therapeutic agents developed fortreatment of disease states (see Section 5), it can, however, present a challenge to in-vestigations of the biology of ERb.1,2
An aspect that has added to the challenge of understanding the biology of ERb is theexistence of different isoforms of this protein. These result either from alternative start sitesin transcription of the ERb gene or in translation of the ERb mRNA, which gives variationsin the length of the N-terminal domain, as well as splice variants that have inserts in thesequence coding for the LBD or splice variants that are truncated in the C-terminus.26–28
While the longest form, ERb1, appears to be the one with the greatest activity, some of thevariant forms, even ones incapable of ligand binding, may interact with ERb1 and with ERaand, thereby, modulate their activity.26 Consequently, the biological result of the binding ofan ERb-selective ligand will depend, to some extent, on the relative levels of these differentERb isoforms in different cells and tissues. Generally in pharmacology, the existence of moretargets means more opportunities for selectivity, but, as a corollary, it requires broaderconsideration of where candidate pharmaceuticals are able to interact and the effects of theseinteractions.
One final intriguing aspect of estrogen receptor pharmacology is target-tissue selectivity,the fact that the same compound can act as an agonist in one target tissue and an antagonistin another. This target-tissue selectivity or target tissue-dependent intrinsic efficacy fornonsteroidal estrogens was initially considered a mechanistic curiosity;29 however, our ex-panded understanding that the ERs exert their gene-regulatory activities at many sites indifferent chromatin contexts and operate through a multitude of coregulatory partnersprovides many opportunities for target cell-specific effects based on differences in targetgene activities.29,30 Compounds having such tissue-dependent mixed agonist/antagonist
ESTROGENRECEPTOR bLIGANDS K 367
Medicinal Research Reviews DOI 10.1002/med

biocharacter are termed selective estrogen receptor modulators (SERMs), and the develop-ment of novel SERMs that have the best balance of agonist and antagonist activitiesin different estrogen target tissues has become a major activity in the pharmaceuticalindustry,31–33 and in research laboratories in academia as well, and is likely to herald a newage of better, more selective estrogen pharmaceuticals.
While the existence of two ER subtypes, ERa and ERb, provides a very crucialmechanism for generating tissue-selective activity, it appears that even among ERb-selectiveligands there is a spectrum of activity, so that not all ERb ligands are biologically the same,and both ERa and ERb-selective ligands can also have SERM-like cell and tissue selecti-vities. A combination of ER subtype selectivity with SERM-like behavior should lead toestrogens with greater pharmaceutical selectivity and greater therapeutic utility.
2. BACKGROUND
Estrogen receptor ligands have been extensively reviewed in the past, and some excellentreviews covering up to year 2005 may be found.32–39 More recent reviews cover only specificsubjects, such as an excellent one about SERMs developed at Merck,40 and another onediscussing some of the molecular requirements for ER subtype selectivity in a restrictednumber of ER ligands.41 To the best of our knowledge, no review has yet been completelydedicated to ERb-selective ligands. Therefore, our intent is to cover the literature on thissubject, with particular focus on the 2005–2008 period.
A. Relative-Binding Affinity
In the literature, two different systems are used to specify the affinity of a ligand for ERa orERb. Academic laboratories typically express ligand affinity relative to that of E2, as arelative-binding affinity (RBA) value. These RBA values come directly and conveniently fromIC50 values determined in competitive radiometric or fluorometric ligand-binding assays
RBAð%Þ ¼ ðICestradiol50 =ICligand
50 Þ � 100 ð1Þ
Thus, an RBA of 100% represents an affinity equivalent to that of E2 on either ERa or ERb.Industrial laboratories typically express ligand affinity as a Ki value, which also can be
calculated from the IC50 values obtained from competitive ligand-binding assays, using theCheng–Prusoff equation
Ki ¼ IC50=ð1þ ½tracer�=K tracerd Þ ð2Þ
In competitive radiometric ligand-binding assays, the tracer is generally [3H] E2, and goodconsensus Kd values for E2 are 0.2 nM for ERa and 0.5 nM for ERb. These E2 Kd values canbe used to estimate ligand Kd values from the RBA values
Kligandd ¼ Kestradiol
d =ðRBA=100Þ ð3Þ
If fluorometric assays are used, the concentration of the tracer and its Kd value need to besubstituted to obtain Ki values from IC50 values.
Rather than attempting to convert ligand-binding affinity values from one system to theother, we have chosen in this review to report the affinity values using the system that wasused in each publication. We did this is to avoid introducing errors, and also to make it easierto refer to the original data in the publications themselves. The reader should be aware ofsome consequences of using these two systems to express binding affinities: ERb-bindingselectivities, calculated as ERb-binding affinity/ERa-binding affinity ratios or simply b/avalues, are somewhat different using the two systems. Because the affinity of E2 for ERa is
368 K MINUTOLOETAL.
Medicinal Research Reviews DOI 10.1002/med

about 2-fold greater than for ERb (see E2 Kd values noted above), the b/a ratios calculatedfrom RBA values (i.e. RBAERb/RBAERa) will, systematically, be about 2-fold larger than b/avalues calculated from the ratio of Ki or IC50 values (i.e. KERa
i =KERbi ).
As a cautionary note, we acknowledge that the lack of standard deviations of the bindingdata makes it difficult to determine whether there actually is a statistically significant dif-ference between similar values.
B. Reference ERb-Selective Agonists
In spite of the fact that several estrogens have high ERa selectivity, only a limited number ofcompounds showing good levels of selectivity for ERb have, thus far, been reported. Amongnatural products, there are a few examples of ERb-selective agonists, such as coumestrol(1, Fig. 2) and genistein (2). Coumestrol 1, a benzofurano-condensed chromenone derivativefound in many plants and foods (soybeans), has a 7-fold ERb selectivity, with an ERb-RBAvalue of 140%. Genistein 2, another phytoestrogen, the isoflavone found in soybeans, ismuch more selective for the b subtype (b/a5 20).42
Among the synthetic derivatives so far developed, diarylpropionitrile (DPN, 3), dis-covered some time ago by Katzenellenbogen and co-workers at the University of Illinois,emerged as one of the most potent and selective ERb agonists, with a 70-fold selectivity inbinding assays (RBA of 0.25% for ERa and 18% for ERb), and a 78-fold selectivity intranscriptional assays (EC50 of 66 nM for ERa and 0.85 nM for ERb).43 In these studies case,despite the presence of a chiral center, only the racemic mixture was used.
A recent article has shown that the enantiomer assigned the S configuration is the mostactive on ERb.44 This is consistent with what was initially predicted by molecular modelinganalysis, which showed that the stereochemistry of S-DPN allows the CN group to interactpositively with Met336 of ERb. By contrast, the nitrile group in the R-enantiomer pointsaway from this residue and, consequently, does not form as productive interactions.45 Thetwo enantiomers were separated by chiral HPLC and separately submitted to binding assays.The enantiomer assigned as S-DPN displayed a greater affinity for ERb (Ki 5 0.27 nM) thandid that assigned as R-DPN (Ki 5 1.82 nM), although their selectivity levels were comparable(b/a�80 for both enantiomers). Furthermore, cell-based functional assays revealed that theS-enantiomer is a potent activator of transcription, whereas the R-enantiomer is not.
Some of the most effective ERb-selective agonists were found at Wyeth within the classof benzoxazoles. Among these derivatives, ERB-041 (4, Fig. 3) showed a 200-fold selectivityfor ERb with binding affinities corresponding to IC50 values of 1200 nM on ERa and 5.4 nMon ERb.46 The interactions of 4 within the binding pocket of ERa and ERb were studied bycombining X-ray crystallographic analysis with computational docking studies (Fig. 3).47 Inboth receptor subtypes, it turned out that the 3-fluoro-4-hydroxyphenyl substituent is in closeproximity to the glutamate/arginine amino acid couple (the portion hosting the A-ring of E2),so that the phenol OH group participates in this important hydrogen bonding network,
O
O
O
OH
HO
coumestrol(1)
O OH
OHOHO
genistein(2)
HOCN
OH
DPN(3)
Figure 2. Examples of natural (1and 2) and synthetic (3) ERb-selective agonists.
ESTROGENRECEPTOR bLIGANDS K 369
Medicinal Research Reviews DOI 10.1002/med

whereas the other hydroxy group, present in the benzoxazole scaffold, forms an H-bond withthe histidine residue at the opposite end of the binding pocket. The differential binding affinityfor each subtype may be explained by the interaction that the vinyl substituent has with thenonconserved residue Met421(ERa)/Ile 373(ERb). In fact, it was shown that the vinyl sub-stituent is close to the ‘‘long’’ side chain of Met421 in ERa, where a steric clash occurs. Thefluoro substituent makes a specific contribution to this effect. In fact, the close proximity of theF-atom with the backbone carbonyl group of Leu339, and the consequent repulsion occurringbetween them, seems to shift the ligand so that the steric clash between the vinyl substituentand Met421 is exacerbated. Therefore, 4 displays a poor binding affinity for this receptorsubtype. In contrast, the b subtype has a shorter residue in that position, which is Ile373; thisleaves enough space to host the vinyl substituent or even to form an additional attractivehydrophobic interaction; thus, leading to the high affinity levels found for 4 with ERb.
An extensive structure–activity relationship study was carried out with a large number ofanalogues of ERB-041, and some of them (5–7, Fig. 4) displayed appreciable levels of bselectivity.48
Benzoxazole 5 lacks the meta-fluoro atom on the 2-aryl group of 4, and this modificationcauses a 3-fold increase in its affinity for the a subtype, thus lowering ERb selectivity.Moreover, the removal of the vinyl group, as in 6, causes a significant decrease in bindingaffinity for both receptor subtypes. A certain recovery, in terms of ERb selectivity, resultedfrom replacing the vinyl group with a bromo substituent. This compound (7) showed thehighest ERb-binding affinity of this series (IC50 5 2 nM), and a good level of b selectivity(b/a5 67). All of these studies confirm that the simultaneous presence of the fluoro and vinylsubstituents, like in ERB-041 (4), sustain the maximum level of ERb selectivity within thisclass of compounds.
C. Reference ERb-Selective Antagonists
Very few examples of ERb antagonists have been reported in the past, and some of themost representative are shown in Figure 5. Among the triazine class discovered at
N
OHO
OH
F
stericclash
Glu353
Arg394
SMet421 Ile373Me
ER
His524
N
OHO
OH
F
Glu305
Arg346
ER
His475
44
Figure 3. Azole derivative ERB-041 (4) and its differential binding interactions with the two receptor subtypes.
WAY-659(5)
N
OHO
OH
WAY-818(6)
N
OHO
OH
WAY-200070(7)
N
OHO
OH
Br
Figure 4. Other highly b-selective benzoxazole derivatives (5--7).
370 K MINUTOLOETAL.
Medicinal Research Reviews DOI 10.1002/med

GlaxoSmithKline, compound 8 displayed b-selective binding properties, with Ki values of25 nM for ERb and 650 nM for ERa, together with a 30-fold selective ERb-antagonistactivity (IC50 5 5 nM) over the a subtype.49 Compound 9, belonging to the 10-aryl-sub-stituted benzo[b]fluorenes described by researchers at Organon, was reported to possessERb-selective antagonist properties (430-fold over ERa) by up to 100% bioactivity, withpotencies exceeding the antagonism observed for the nonselective steroidal full antagonistICI 164384.50 Finally, optically pure (R,R)-cis-tetrahydrochrysene 10, discovered at theUniversity of Illinois,51 showed an interesting dual profile, by behaving as a full agonist onERa and as full antagonist on ERb.13
This last compound (10) has proved to be a very valuable tool to obtain crystal structuresof its complexes with ERa- and ERb-LBDs.52
3. STRUCTURAL CLASSES OF ERb-SELECTIVE LIGANDS
A. Steroidal Derivatives
1. Steroid hormone metabolitesSome of the first steroidal b-selective ligands, belonging to the class of steroid hormonemetabolites, were reported as early as 1998 at the Karolinska Institute. Among severalcompounds assayed, 5-androstenediol (11), 3b-androstanediol (3b-Adiol, 12), and 3a-an-drostanediol (13), showed a certain preference for the b subtype (Fig. 6). The highest RBAfor ERb was found with compound 11 (ERb-RBA5 17%; ERa-RBA5 6%), whereascompound 13 showed the highest selectivity, although with rather poor binding affinity forERb (ERb-RBA5 0.3%; ERa-RBA5 0.07%). An intermediate profile was associated with3b-Adiol 12 (ERb-RBA5 7%; ERa-RBA5 3%).53
3b-Adiol 12 was later found to effectively stimulate ERb in functional assays, with aEC50 of 23 nM, comparable to that of an E2 metabolite, estriol (14), which showed an EC50
of 17 nM, although 14 has a very low ERb selectivity.54 A higher ERb-binding selectivity wasassociated with 17-epiestriol (15), which showed an RBA of 80% on ERb and 29% onERa.42 The main metabolite of dehydroepiandroserone (DHEA) in human prostate, namely,its CYP7B1-catalyzed hydroxylation product 7a-hydroxy-DHEA (7-HD, 16), proved toeffectively stimulate ERb, with a maximum activation at 20 mM of 60–70% with respect
N
N
N
N
CH3
N
N
N
S
NH3C
HO
ClH
8HO
OHEt
Et
10
OH
HO O N
9
Figure 5. Examples of ERb-antagonists (8--10).
ESTROGENRECEPTOR bLIGANDS K 371
Medicinal Research Reviews DOI 10.1002/med

to E2; it did not show any activation of either ERa or the androgen receptor (AR) at the sameconcentration. Nevertheless, in this study, 3b-Adiol (12) was still the most efficient ERbactivator among all of the DHEA metabolites.55 It is important to consider whether the invivo use of the androstene and androstane derivatives would likely be complicated by3-hydroxysteroid dehydrogenases that are able to oxidize the 3-OH (whether a or b) to the3-keto form; thus, eliminating binding to either ERa or ERb and greatly increasing ARbinding.56
2. Synthetic androstene derivativesExtensive studies have been carried out at Merck on androstene derivatives,40 starting fromthe observation that 5-androstenediol bearing a vinyl substituent in the 10b-position (17,Table I), a known AR activator, possessed a remarkable ERb-selective affinity. Conse-quently, many other 10b-substituted androstenediol derivatives were prepared and studied.57
Compound 17, in fact, showed an excellent level of ERb selectivity in binding affinity assays(with an IC50 of 11 nM on ERb and a b/a selectivity ratio of 212), together with an efficientand selective activation of the b subtype (EC50 5 4 nM, b/a selectivity ratio of 246). Un-fortunately, this derivative binds well to the AR, with an IC50 of 33 nM. On the basis of theseresults, the 10b vinyl group in 17 was replaced by various alkyl, alkenyl, and alkynyl group;the best ERb-selective derivatives (18–23) are reported in Table I.
Parent compound, androstenediol (18), bearing a methyl group in the 10b-position,showed a good binding affinity for ERb (IC50 of 10 nM), but its selectivity over the a subtypewas reduced (b/a selectivity ratio of 21). Interestingly, androstenediol showed a much weakerbinding affinity for the AR (IC50 5 212 nM) than for ERb, thus showing an ERb/ARselectivity ratio of 21. Homologation of the Me- to an Et-group, as in compound 19,decreased ERa-binding affinity, which increased ERb selectivity, although no functionalassay data were reported for this substance. The insertion of the (Z)-fluorovinyl group, as in20, caused a shift in the binding preference toward the AR (b/AR5 0.4), despite the fact thatits selectivity between the ER subtypes was remarkable, both in binding affinity (b/a5 171)and transcriptional potency (b/a5 164). The change of the double bond configuration, as inthe (E)-fluorovinyl-substituted compound 21, was beneficial in terms of selectivity over the
OHMe
HO
Me H
H H
OHMe
HO
Me H
H H
H
OHMe
HO
Me H
H H
H
5-androstenediol(11)
3β-androstanediol(3β-Adiol, 12)
3α-androstanediol(13)
estriol(14)
OHMe
HO
H
H H
OH
7α-hydroxy-DHEA(7-HD, 16)
OMe
HO
Me H
H H
OH
17-epiestriol(15)
OHMe
HO
H
H H
OH
Figure 6. Steroid hormonemetabolites showing ERb-preference.
372 K MINUTOLOETAL.
Medicinal Research Reviews DOI 10.1002/med
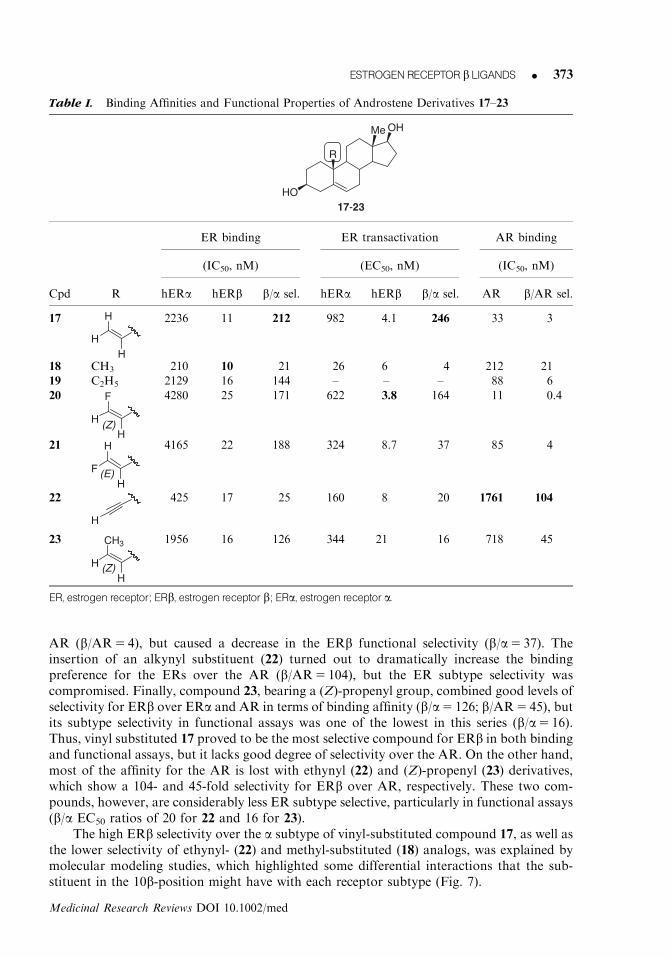
AR (b/AR5 4), but caused a decrease in the ERb functional selectivity (b/a5 37). Theinsertion of an alkynyl substituent (22) turned out to dramatically increase the bindingpreference for the ERs over the AR (b/AR5 104), but the ER subtype selectivity wascompromised. Finally, compound 23, bearing a (Z)-propenyl group, combined good levels ofselectivity for ERb over ERa and AR in terms of binding affinity (b/a5 126; b/AR5 45), butits subtype selectivity in functional assays was one of the lowest in this series (b/a5 16).Thus, vinyl substituted 17 proved to be the most selective compound for ERb in both bindingand functional assays, but it lacks good degree of selectivity over the AR. On the other hand,most of the affinity for the AR is lost with ethynyl (22) and (Z)-propenyl (23) derivatives,which show a 104- and 45-fold selectivity for ERb over AR, respectively. These two com-pounds, however, are considerably less ER subtype selective, particularly in functional assays(b/a EC50 ratios of 20 for 22 and 16 for 23).
The high ERb selectivity over the a subtype of vinyl-substituted compound 17, as well asthe lower selectivity of ethynyl- (22) and methyl-substituted (18) analogs, was explained bymolecular modeling studies, which highlighted some differential interactions that the sub-stituent in the 10b-position might have with each receptor subtype (Fig. 7).
Table I. Binding Affinities and Functional Properties of Androstene Derivatives 17–23
OHMe
HO17-23
R
ER binding ER transactivation AR binding
(IC50, nM) (EC50, nM) (IC50, nM)
Cpd R hERa hERb b/a sel. hERa hERb b/a sel. AR b/AR sel.
17
H
H
H
2236 11 212 982 4.1 246 33 3
18 CH3 210 10 21 26 6 4 212 21
19 C2H5 2129 16 144 – – – 88 6
20
H
F
H(Z)
4280 25 171 622 3.8 164 11 0.4
21
F
H
H(E)
4165 22 188 324 8.7 37 85 4
22
H
425 17 25 160 8 20 1761 104
23
H
CH3
H(Z)
1956 16 126 344 21 16 718 45
ER, estrogen receptor; ERb, estrogen receptor b; ERa, estrogen receptor a.
ESTROGENRECEPTOR bLIGANDS K 373
Medicinal Research Reviews DOI 10.1002/med

In fact, a docking analysis of 17 into ERa has shown that the terminal carbon of thevinyl substituent is too close to the Leu384 residue, whereas in ERb this residue is replaced byMet336, which accepts the vinyl group. In fact, leucine has a bulky and relatively rigid sidechain because of the preference of alkyl chains for extended conformations, whereas thesulfur atom in the methionine residue makes the side chain, though longer, much moreflexible.58 This explains the high ERb selectivity of 17. On the other hand, when the vinylsubstituent is replaced by a methyl (18) or an ethynyl group (22), these substituents fit muchmore comfortably in ERa, because they are further from the bulky Leu384 and, therefore,they are much less ERb selective than 17.
Further studies at Merck led to the development of bridged analogs of 17 in which the10b substituent is blocked by an additional covalent bond with C-4 (24–26, Fig. 8).59 In thiscase, the bridged androstenediols generally proved to be weaker ligands for ERb than werethe open-chain analogs. This was explained by a docking analysis, which shows that thebridging atoms establish a repulsive interaction with the Leu339 residue of ERb. This stericclash occurs to a greater extent with propylidene derivatives 24 and 25, whereas it appears tobe less severe with ethylidene derivative 26, which, in fact, has the highest ERb affinity of thisseries (IC50 value of 50 nM on ERb), although its binding selectivity for ERb over ERa wasonly 6-fold.
As noted before, the androstenediol derivatives 17–26 might undergo metabolic oxida-tion of the 3-OH group in vivo, catalyzed by 3-hydroxysteroid dehydrogenases. The resulting3-keto analogs are expected to be much weaker ligands for both ER subtypes and, at thesame, to stimulate AR.56 This research line was completed with the development of an-drostene-3,5-diene derivatives where the hydroxyl group in 3-position of androstenediolwas replaced by alkyl or aryl groups, and an additional double bond was inserted at the3,4-position. The members of this series showing the highest ERb-affinity values are the10b-methyl- (27) and 10b-vinyl-substituted (28) derivatives (Fig. 9).60
Compound 27 showed very good binding to ERb (IC50 5 9.4 nM), but its selectivity wasnot excellent (b/a5 12). As observed previously in the androstenediol series, replacement ofthe methyl with a vinyl group in the 10b-position (28) caused a dramatic reduction inERa-binding affinity (IC50 5 1440 nM), while the binding affinity for the b subtype wassubstantially maintained (IC50 5 9.4 nM), thus, increasing the ERb selectivity to a 160-fold
OHMe
HO
24
OHMe
HO
25
OHMe
HO
26
Leu339
H
Leu339 Leu339
Figure 8. Bridged analogs ofandrostenediol (24--26) and their steric clashwith Leu339 of ERb.
OHMe
HO
17
Glu353
Arg394
His524Leu384 SMet336 Me
ER ER
OHMe
HO
17
Glu305
Arg346
His475
repulsion
Me
18
Leu384
ER 22
Leu384
Figure 7. Interactions of androstenediol (18) and10b-substituted analogs 17 and 22 with ERa and ERb.
374 K MINUTOLOETAL.
Medicinal Research Reviews DOI 10.1002/med
value. A curious aspect of these compounds is the lack of the 3-OH group, which typicallyparticipates in the important interaction with the glutamate/arginine H-bond network inboth ER subtypes. The maintenance of high affinity when a polar group is replaced by anonpolar one, in a binding site in this case, is reminiscent of work by Bartlett on phos-phonamide inhibitors of proteases, where an amide N-H involved in a hydrogen bond couldbe replaced by a CH2 group had only a moderate effect on binding. The explanation offeredwas that the reduced energy of interaction between the inhibitor and the protein when CH2
was replaced by NH was balanced by the reduced solvation energy of the unbound CH2
inhibitor in water,61 although others have questioned this interpretation.62
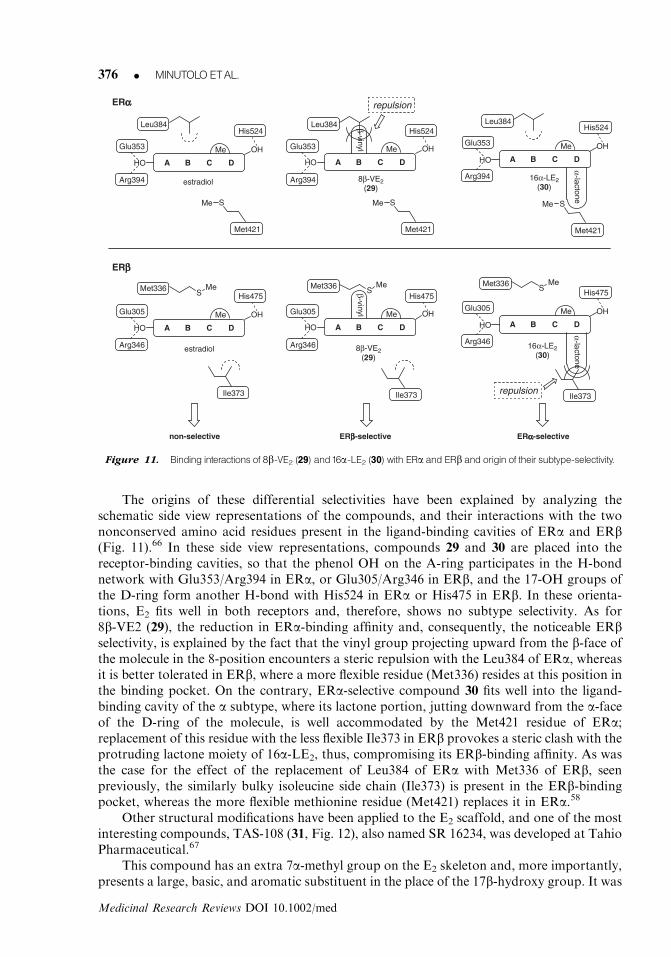
3. Synthetic estradiol analogsExtensive studies on synthetic steroidal structures have afforded several subtype selectiveligands. Some simple modifications to the E2 backbone at Schering AG have led to thedevelopment of a ERb subtype selective agonists, 8b-VE2 (29),63 as well as of another E2
analogue, 16a-LE2 (30),64 which is highly selective for ERa (Fig. 10). The insertion of a vinyl
substituent in the 8b-position of E2 caused a dramatic decrease of the transcriptional activityof the resulting compound (8b-VE2, 29) on ERa (EC50 5 2.3 vs. 0.0061 nM of E2), whereas itsagonist potency on ERb was substantially maintained (EC50 5 0.050 vs. 0.023 nM of E2),with an ERb/a functional selectivity ratio of 46. On the contrary, the insertion of a lactonering bridging the 16a- and 17a-positions, as in compound 16a-LE2 (30), caused the oppositeshift on the subtype selectivity, giving rise to a marked ERa selectivity (EC50 5 0.014 nM onERa and 23 nM on ERb).65
OHMe
Me
27
Me
OHMe
Me
28
Figure 9. Androstene-3,5-diene derivatives (27 and 28).
OHMe
HO
estradiol
A B
C D
A B C D
Me A B C D
Me
α-l a ct one
A B C D
Me
β-v iny l
OHMe
HO8β-VE2
(29)
A B
C D
Me
HO16α-LE2
(30)
A B
C D O
OH O
HO
OH HO
OH
HO
OH
Figure 10. Synthetic subtype-specific analogues of estradiol (29 and 30) and their side-view schematic representations.
ESTROGENRECEPTOR bLIGANDS K 375
Medicinal Research Reviews DOI 10.1002/med
The origins of these differential selectivities have been explained by analyzing theschematic side view representations of the compounds, and their interactions with the twononconserved amino acid residues present in the ligand-binding cavities of ERa and ERb(Fig. 11).66 In these side view representations, compounds 29 and 30 are placed into thereceptor-binding cavities, so that the phenol OH on the A-ring participates in the H-bondnetwork with Glu353/Arg394 in ERa, or Glu305/Arg346 in ERb, and the 17-OH groups ofthe D-ring form another H-bond with His524 in ERa or His475 in ERb. In these orienta-tions, E2 fits well in both receptors and, therefore, shows no subtype selectivity. As for8b-VE2 (29), the reduction in ERa-binding affinity and, consequently, the noticeable ERbselectivity, is explained by the fact that the vinyl group projecting upward from the b-face ofthe molecule in the 8-position encounters a steric repulsion with the Leu384 of ERa, whereasit is better tolerated in ERb, where a more flexible residue (Met336) resides at this position inthe binding pocket. On the contrary, ERa-selective compound 30 fits well into the ligand-binding cavity of the a subtype, where its lactone portion, jutting downward from the a-faceof the D-ring of the molecule, is well accommodated by the Met421 residue of ERa;replacement of this residue with the less flexible Ile373 in ERb provokes a steric clash with theprotruding lactone moiety of 16a-LE2, thus, compromising its ERb-binding affinity. As wasthe case for the effect of the replacement of Leu384 of ERa with Met336 of ERb, seenpreviously, the similarly bulky isoleucine side chain (Ile373) is present in the ERb-bindingpocket, whereas the more flexible methionine residue (Met421) replaces it in ERa.58
Other structural modifications have been applied to the E2 scaffold, and one of the mostinteresting compounds, TAS-108 (31, Fig. 12), also named SR 16234, was developed at TahioPharmaceutical.67
This compound has an extra 7a-methyl group on the E2 skeleton and, more importantly,presents a large, basic, and aromatic substituent in the place of the 17b-hydroxy group. It was
estradiol
A B C D
MeA B C D
Me
-lacto ne
A B C D
Me
-vinyl
8 -VE2(29)
16 -LE2(30)
HO
OHHO
OH
HO
OH
ER
Glu353
Arg394
His524
Glu353
Arg394
His524
Glu353
Arg394
His524Leu384 Leu384 Leu384
repulsion
Me S
Met421
Me S
Met421
Me S
Met421
estradiol
A B C D
MeA B C D
Me
-lacto ne
A B C D
Me-vinyl
8 -VE2(29)
16 -LE2(30)
HO
OHHO
OH
HO
OH
ER
Glu305
Arg346
His475
Glu305
Arg346
His475
Glu305
Arg346
His475SMet336
Ile373Ile373Ile373
Me Met336 SMe SMet336 Me
REevitceles-non REevitceles- -selective
repulsion
Figure 11. Binding interactions of 8b-VE2 (29) and16a-LE2 (30) with ERa and ERb and origin of their subtype-selectivity.
376 K MINUTOLOETAL.
Medicinal Research Reviews DOI 10.1002/med

not characterized by an ERb-selective binding affinity (IC50 5 11 nM on ERa and 5.6 nM onERb), but its selective ERb stimulation was due to its behavior as a pure ERa antagonist andERb partial agonist.
4. B-ring-modified estradiol analogsA study of conjugated equine estrogens revealed the structure of two ERb-selective estrogenderivatives, in which the B-ring is unsaturated, thus forming—together with the A-ring—anaphthalene system. The C-17 epimers, 17a- (17a-Eqn, 32) and 17b-dihydroequilenin (17b-Eqn, 33), are shown in Figure 13.68
The 17b-derivative (33) showed a higher degree of ERb selectivity than its a epimer 32, inboth binding affinity and transcriptional potency. In binding assays, 33 had an IC50 of 19 nMon ERb and 70 nM on ERa, with a 3.7-fold selectivity, and in transcriptional assays it hadEC50 values of 0.38 nM on ERb and 4.7 nM on ERa, corresponding to a 6.9 b/a-selectivityratio. The 17a epimer 32, despite having binding similar to the 17b epimer 33, was less potentin transcriptional assays (EC50 of 2.9 nM on ERb, with a 6.2 b/a-selectivity ratio).
Other studies, devoted to deconstructing E2, showed that the opening of the B-ringgenerated ACD pseudosteroids having interesting subtype-selective properties. Compound34, in particular, and its methyl-substituted analog 35 (Fig. 14), proved to be potent ERb-selective agonists.69 In fact, binding assays revealed very good affinity and selectivity for 34(RBA5 21.5% on ERb, b/a ratio5 14) and 35 (RBA5 33.6% on ERb, b/a ratio5 12), andfunctional assays confirmed their ERb-selective agonist activity, evaluated as transcriptionactivities relative to E2 at a single dose (RTAZ150 for both compounds with ERb, whereasno significant stimulation of ERa was observed).
A computational docking analysis revealed that the b selectivity, associated with thesecompounds, might be attributed to a closer contact that the ethylene portion of the D-ringhas with Met421 in ERa, when compared with E2, because of a different torsional anglebetween ring A and the two fused C and D rings. This would cause a greater repulsion in theERa-binding pocket, whereas in ERb, where this methionine residue is replaced by thesmaller Ile373, this repulsive interaction does not happen.
OHMe
HO
17α-Eqn(32)
OHMe
HO
BB
α β
17β-Eqn(33)
Figure 13. B-ringunsaturated estradiol analogs: 17a-Eqn (32) and17b-Eqn (33).
Me
HO Me
O
MeO
N Et
Et
TAS-108 (31)
Figure 12. Synthetic analog of estradiol (TAS-108) possessing ERa-antagonist and ERb-agonist properties.
ESTROGENRECEPTOR bLIGANDS K 377
Medicinal Research Reviews DOI 10.1002/med
5. Other natural steroidal derivativesA glycoconjugated steroid, ginsenoside Rb1 (36, Fig. 15), isolated from the root of ginseng(Panas ginseng), was reported to activate ERb and, consequently, to inhibit matrix-drivencapillary morphogenesis, thus exerting an antiangiogenic effect in vitro which may beexploited in antitumor therapy.70 Previous studies had, however, reported that 36 is a non-selective ERa/ERb agonist and that its ERs activation is independent of ligand binding.71
The latter fact is comforting, considering how divergent the structure of 36 is from that ofany other known ER ligand.
A derivative of cholesterol, namely, 24-methylenecholesterol (37, Fig. 15), a componentof royal jelly produced by honeybees (Apis mellifera), was reported to bind ERb, althoughwith a modest affinity (IC50 5 6.0 mM). Its binding affinity for ERa was too low to bedetected, which conferred to 37 a good level of selectivity for ERb.72
B. Nonsteroidal Derivatives
1. Natural compoundsIn addition to coumestrol (1) and genistein (2), mentioned above in Section 2, other phyto-chemicals of the flavone or isoflavone family have interesting ERb selectivity. Among fla-vones derivatives (Fig. 16), liquiritigenin (38), isolated from the root of Glycyrrhizae
OHMe
HO34
A
C D
HH
OHMe
HO35
A
C D
HH
Me
HOOH
AC
estradiol
OHC
B DHO
AD
ACD-pseudosteroids
B-ringremoval
Figure 14. B-ring deprived ACD-pseudosteroids 34 and 35.
H
O
36
Me
HMeMe
H Me
Me
OHH
OMe
OOH
OH
OH
O
O
HO
HO
HOOH
O
HO
HO
O
OH
OH
HO
OHHO
OH
HO
Me
Me
H H
H
37
Figure 15. Natural steroidal ERb-activators from ginseng (36) and royal jelly (37).
378 K MINUTOLOETAL.
Medicinal Research Reviews DOI 10.1002/med
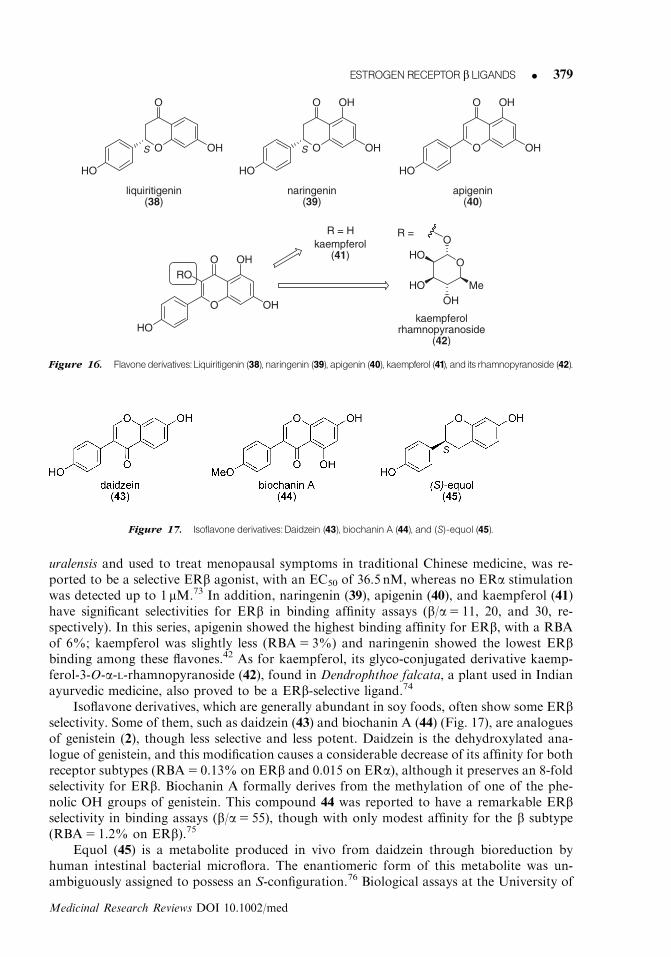
uralensis and used to treat menopausal symptoms in traditional Chinese medicine, was re-ported to be a selective ERb agonist, with an EC50 of 36.5 nM, whereas no ERa stimulationwas detected up to 1 mM.73 In addition, naringenin (39), apigenin (40), and kaempferol (41)have significant selectivities for ERb in binding affinity assays (b/a5 11, 20, and 30, re-spectively). In this series, apigenin showed the highest binding affinity for ERb, with a RBAof 6%; kaempferol was slightly less (RBA5 3%) and naringenin showed the lowest ERbbinding among these flavones.42 As for kaempferol, its glyco-conjugated derivative kaemp-ferol-3-O-a-L-rhamnopyranoside (42), found in Dendrophthoe falcata, a plant used in Indianayurvedic medicine, also proved to be a ERb-selective ligand.74
Isoflavone derivatives, which are generally abundant in soy foods, often show some ERbselectivity. Some of them, such as daidzein (43) and biochanin A (44) (Fig. 17), are analoguesof genistein (2), though less selective and less potent. Daidzein is the dehydroxylated ana-logue of genistein, and this modification causes a considerable decrease of its affinity for bothreceptor subtypes (RBA5 0.13% on ERb and 0.015 on ERa), although it preserves an 8-foldselectivity for ERb. Biochanin A formally derives from the methylation of one of the phe-nolic OH groups of genistein. This compound 44 was reported to have a remarkable ERbselectivity in binding assays (b/a5 55), though with only modest affinity for the b subtype(RBA5 1.2% on ERb).75
Equol (45) is a metabolite produced in vivo from daidzein through bioreduction byhuman intestinal bacterial microflora. The enantiomeric form of this metabolite was un-ambiguously assigned to possess an S-configuration.76 Biological assays at the University of
O
O
OH
OH
HO
S
naringenin(39)
O
O
OH
OH
HO
apigenin(40)
O
O
OH
HO
S
liquiritigenin(38)
O
O
OH
OH
HO
kaempferol(41)
ROO
O
OHMeHO
HO
R = H R =
kaempferolrhamnopyranoside
(42)
Figure 16. Flavonederivatives:Liquiritigenin (38), naringenin (39), apigenin (40), kaempferol (41), andits rhamnopyranoside (42).
Figure 17. Isoflavone derivatives:Daidzein (43), biochanin A (44), and (S)-equol (45).
ESTROGENRECEPTOR bLIGANDS K 379
Medicinal Research Reviews DOI 10.1002/med

Illinois of independently synthesized (R)- and (S)-enantiomers of equol, separated throughsemi-preparative HPLC using a b-cyclodextrin-based solid phase, confirmed that the ERb-selective form is (S)-equol, which has an RBA of 3.20% on ERb with a b/a selectivity ratio of32. By contrast, the (R)-enantiomer was completely nonselective and had poor bindingproperties for both receptor subtypes. However, transcriptional assays revealed that thefunctional selectivity of (S)-equol as a receptor agonist was not high, giving very similar EC50
values for ERa (85 nM) and ERb (65 nM).77 An enantioselective synthesis of (S)-equol hasbeen described.78
Modest levels of ERb-binding affinity were associated with three aliphatic acid deriva-tives, 10-hydroxy-trans-2-decenoic acid (10H2DA), 10-hydroxydecanoic acid (10HDA), andtrans-2-decenoic acid (2DEA). These compounds were isolated together with 24-methylene-cholesterol (37, Fig. 15) in royal jelly produced by honeybees (A. mellifera). Not surprisingly,considering their structure, their IC50 values for ERb were very poor (10H2DA: 90 mM,10HDA: 140 mM, 2DEA: 17 mM), but because they did not show any binding for the othersubtype, they were quite selective; they were agonists in transcription assays.72
More recently, a diaryl heptanoid, 1-(4-hydroxyphenyl)-7-phenyl-(6E)-6-hepten-3-one,extracted in Thailand from the rhizomes of Curcuma comosa Roxb, proved to be an efficientactivator of ERb in transcriptional assays, although neither its binding affinity nor itsfunctional subtype-selectivity values were reported.79
2. IndazolesOne of the authors, (J. A. K.), at the University of Illinois, recently described a pharma-cophore model for ERb-selective ligands,80 derived by modification of a previously proposedmodel for nonselective ER ligands (Fig. 18).81
The nonselective ER pharmacophore model81 contains two or three aryl substituents, ofwhich one or two are p-hydroxyphenyl rings attached to a central core, which may bearanother nonaromatic substituent. Starting from this model, a more ‘‘specialized’’ pharma-cophore model for ERb-selective ligands was proposed,80 taking into consideration thestructures of many nonsteroidal molecules showing selectivity for the b subtype, as well as thefact that ERb has a smaller ligand-binding pocket and contains two amino acid residuesdiffering from the a subtype. The derived model differs from the previous one because it lacksthe third aromatic substituent, and one of the two phenol rings is fused with the centralscaffold. This model was used as a guide to develop several ERb-selective agonists based onthe indazole scaffold (46–52, Table II). The simplest members of this series, possessing noadditional substituent in the 3-position of the indazole core (46 and 47), already showed somegood binding affinity levels for the b subtype, which was more pronounced in the derivativebearing the hydroxy group in the 5-position (46), rather than in the 6-OH-substituted analog(47). This trend was also confirmed in the 3-chloro-substituted analogs 48 and 49. In par-ticular, 48 showed the highest b selectivity in binding assays (higher than 100-fold), with a
OH
HO
centralcoreA
ERβ-selectivepharmacophoric model
substituent
NN OHHO
substituent
indazole-based ERβ ligands
OH
HO
centralcore
A
non-selectiveER pharmacophoric model
substituent
substituent(aromatic)
Figure 18. Development of a pharmacophoric model for ERb-selective ligands and its application to the indazole system
(Table II).
380 K MINUTOLOETAL.
Medicinal Research Reviews DOI 10.1002/med
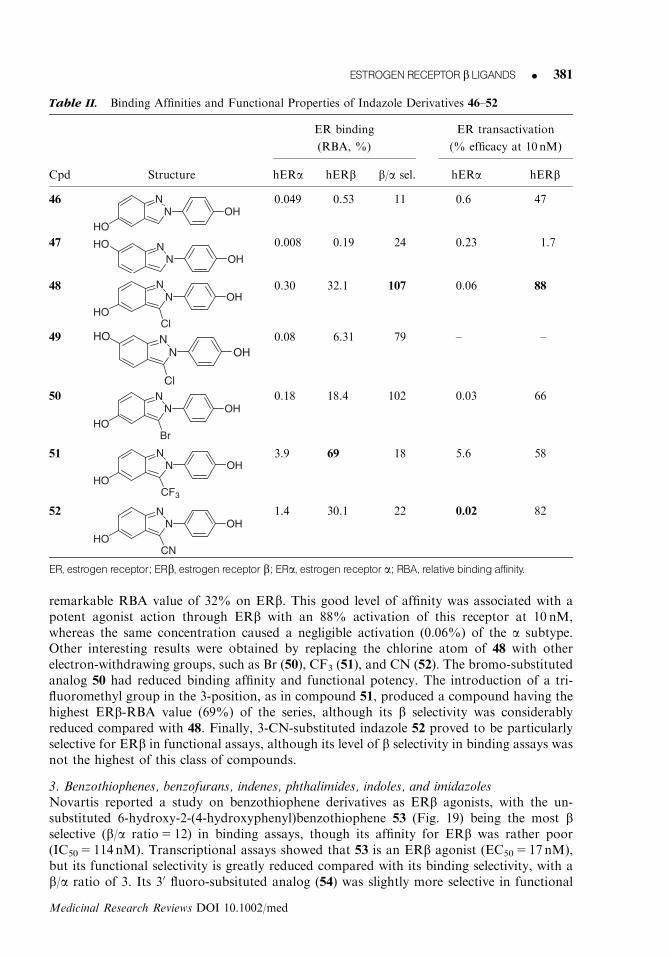
remarkable RBA value of 32% on ERb. This good level of affinity was associated with apotent agonist action through ERb with an 88% activation of this receptor at 10 nM,whereas the same concentration caused a negligible activation (0.06%) of the a subtype.Other interesting results were obtained by replacing the chlorine atom of 48 with otherelectron-withdrawing groups, such as Br (50), CF3 (51), and CN (52). The bromo-substitutedanalog 50 had reduced binding affinity and functional potency. The introduction of a tri-fluoromethyl group in the 3-position, as in compound 51, produced a compound having thehighest ERb-RBA value (69%) of the series, although its b selectivity was considerablyreduced compared with 48. Finally, 3-CN-substituted indazole 52 proved to be particularlyselective for ERb in functional assays, although its level of b selectivity in binding assays wasnot the highest of this class of compounds.
3. Benzothiophenes, benzofurans, indenes, phthalimides, indoles, and imidazolesNovartis reported a study on benzothiophene derivatives as ERb agonists, with the un-substituted 6-hydroxy-2-(4-hydroxyphenyl)benzothiophene 53 (Fig. 19) being the most bselective (b/a ratio5 12) in binding assays, though its affinity for ERb was rather poor(IC50 5 114 nM). Transcriptional assays showed that 53 is an ERb agonist (EC50 5 17 nM),but its functional selectivity is greatly reduced compared with its binding selectivity, with ab/a ratio of 3. Its 30 fluoro-subsituted analog (54) was slightly more selective in functional
Table II. Binding Affinities and Functional Properties of Indazole Derivatives 46–52
ER binding ER transactivation
(RBA, %) (% efficacy at 10 nM)
Cpd Structure hERa hERb b/a sel. hERa hERb
46 NN OH
HO
0.049 0.53 11 0.6 47
47 NN
HOOH
0.008 0.19 24 0.23 1.7
48 NN OH
HOCl
0.30 32.1 107 0.06 88
49 NN
HOOH
Cl
0.08 6.31 79 – –
50 NN OH
HOBr
0.18 18.4 102 0.03 66
51 NN OH
HOCF3
3.9 69 18 5.6 58
52 NN OH
HOCN
1.4 30.1 22 0.02 82
ER, estrogen receptor; ERb, estrogen receptor b; ERa, estrogen receptor a; RBA, relative bindingaffinity.
ESTROGENRECEPTOR bLIGANDS K 381
Medicinal Research Reviews DOI 10.1002/med

assays, with an EC50 value of 32 nM and a 4-fold selectivity for ERb, and its bindingproperties were basically the same as those of 53, with a slight decrease in b-binding se-lectivity (b/a5 9).82
Researchers at Wyeth have studied the ERb selectivity of a series of 5-hydroxy-2-(4-hy-droxyphenyl)benzofurans (55–61, Table III).83 The simplest member of this series (55) alreadyshowed a 30-fold selectivity for the b subtype in binding assays, with a IC50 value of 6 nM forERb. The introduction of a methoxy group at the 7-position of the benzofuran scaffold pro-duced three compounds (56–58) with enhanced b selectivities. In particular, compound 57,bearing an additional 4-bromo-substituent, displayed the highest ERb affinity (IC505 0.5 nM,b/a5 50); the other two methoxy-substituted derivatives (56 and 58) showed an almost 100-foldselectivity for the b receptor. Three other derivatives, bearing a 7-cyanomethyl substituent(59–61), also proved to be very selective for ERb. Within this group, the highest b selectivity wasreached by the 30 fluoro- (60) and 4 bromo-substituted (61) compounds, with b/a-selectivityratios of 108 and 104, respectively. All these compounds proved to be full agonists for ERb witha 100% activation (compared with 10nM E2) at a concentration of 1mM.
A docking analysis of these compounds in ERb revealed that the 40-OH phenol groupparticipates in the high energy H-bond network with Glu305 and Arg346, and the other OHgroup in the 5-position forms an H-bond with His475. Much of the selectivity appears to bedue to the substituent (OCH3 or CH2CN) at the 7-position which, in analogy to the vinylgroup of ERB-041 (4, see Fig. 3), is well tolerated by the binding cavity of the b subtype,where the Ile373 residue is the one close to this group. In ERa, this residue is replaced bya longer Met421, which comes too close to the 7-substituent, especially when this is a
Table III. Binding Affinities of Benzofuran Derivatives 55–61
OHO
HO
R3
R2
R1
23'
4
7
4'5
55-61
ER binding (IC50, nM)
Cpd R1 R2 R3 hERa hERb b/a sel.
55 H H H 176 6 30
56 OCH3 H F 990 10 99
57 OCH3 Br H 21 0.5 50
58 OCH3 Br F 335 3.3 99
59 CH2CN H H 1152 14 80
60 CH2CN H F 1056 10 108
61 CH2CN Br H 209 2.0 104
ER, estrogen receptor; ERb, estrogen receptor b; ERa, estrogen receptor a.
OHS
53
HO
OHS
F
54
HO
Figure 19. Benzothiophene-based ERb-agonists 53 and 54.
382 K MINUTOLOETAL.
Medicinal Research Reviews DOI 10.1002/med
cyanomethyl group as in 59–61, thus causing a considerable decrease in their binding affi-nities for the a subtype.
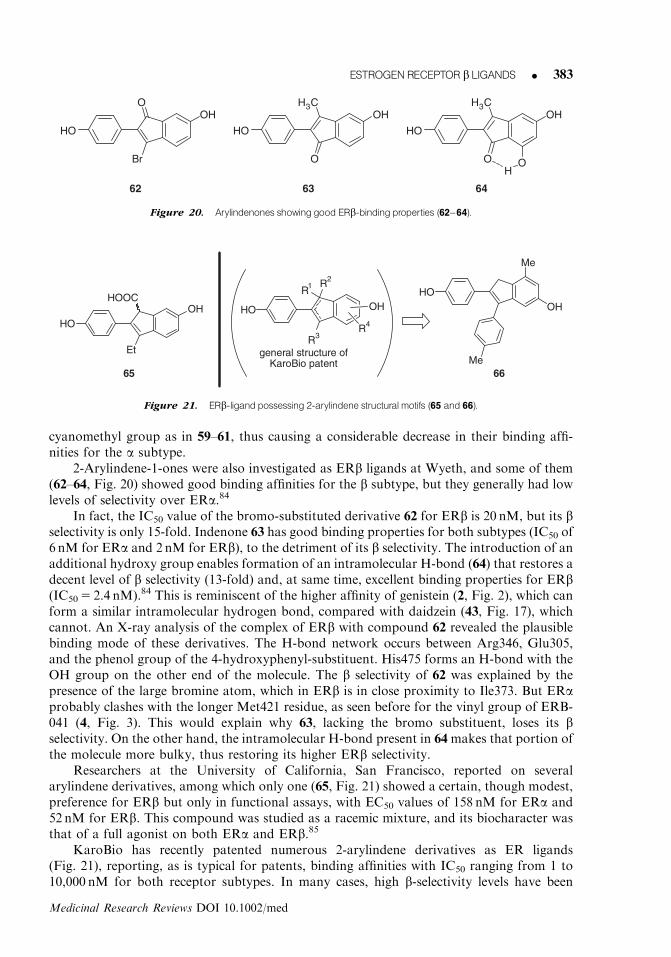
2-Arylindene-1-ones were also investigated as ERb ligands at Wyeth, and some of them(62–64, Fig. 20) showed good binding affinities for the b subtype, but they generally had lowlevels of selectivity over ERa.84
In fact, the IC50 value of the bromo-substituted derivative 62 for ERb is 20 nM, but its bselectivity is only 15-fold. Indenone 63 has good binding properties for both subtypes (IC50 of6 nM for ERa and 2 nM for ERb), to the detriment of its b selectivity. The introduction of anadditional hydroxy group enables formation of an intramolecular H-bond (64) that restores adecent level of b selectivity (13-fold) and, at same time, excellent binding properties for ERb(IC50 5 2.4 nM).84 This is reminiscent of the higher affinity of genistein (2, Fig. 2), which canform a similar intramolecular hydrogen bond, compared with daidzein (43, Fig. 17), whichcannot. An X-ray analysis of the complex of ERb with compound 62 revealed the plausiblebinding mode of these derivatives. The H-bond network occurs between Arg346, Glu305,and the phenol group of the 4-hydroxyphenyl-substituent. His475 forms an H-bond with theOH group on the other end of the molecule. The b selectivity of 62 was explained by thepresence of the large bromine atom, which in ERb is in close proximity to Ile373. But ERaprobably clashes with the longer Met421 residue, as seen before for the vinyl group of ERB-041 (4, Fig. 3). This would explain why 63, lacking the bromo substituent, loses its bselectivity. On the other hand, the intramolecular H-bond present in 64makes that portion ofthe molecule more bulky, thus restoring its higher ERb selectivity.
Researchers at the University of California, San Francisco, reported on severalarylindene derivatives, among which only one (65, Fig. 21) showed a certain, though modest,preference for ERb but only in functional assays, with EC50 values of 158 nM for ERa and52 nM for ERb. This compound was studied as a racemic mixture, and its biocharacter wasthat of a full agonist on both ERa and ERb.85
KaroBio has recently patented numerous 2-arylindene derivatives as ER ligands(Fig. 21), reporting, as is typical for patents, binding affinities with IC50 ranging from 1 to10,000 nM for both receptor subtypes. In many cases, high b-selectivity levels have been
HO
65
OHHOOC
Et
HO
66Me
OH
Me
HO
R3
R2
R1
R4
OH
general structure ofKaroBio patent
Figure 21. ERb-ligandpossessing 2-arylindene structural motifs (65 and 66).
HO
62
OHO
Br
HO
63
OHH3C
O
HO
64
OHH3C
O OH
Figure 20. Arylindenones showing good ERb-binding properties (62--64).
ESTROGENRECEPTOR bLIGANDS K 383
Medicinal Research Reviews DOI 10.1002/med

observed, but more precise specifications of molecular structure of the most b-selectiveligands are not disclosed in these patents. One of the examples contained in the patent isshown in Figure 21 (66).86
Some spiroindenes developed at Merck (67–69, Fig. 22) also showed a slight preferencefor ERb, but their selectivity levels are very low. The most ERb-selective compound is 67,which has a 3-fold preference for the b subtype in binding assays (IC50 5 5.9 nM). Theremoval of the methyl substituent of 67, leading to 68, causes an increase in the bindingaffinity for both receptors (IC50 5 4.3 on ERa and 2.1 on ERb), but this negatively affects bselectivity. The shift of the position of the two OH groups, as in 69, produced a compoundwith slightly weaker binding properties on both subtypes, and no gain of selectivity.87
More interesting results were obtained at Wyeth with the development of phthalimidoderivatives, some of which (70 and 71, Fig. 23) showed significant levels of b selectivity.Binding affinity values of the simplest analog 70 (IC50 5 1980 nM on ERa and 84 nM onERb) were considerably improved through the introduction of a bromo substituent into thephthalimido scaffold. In fact, the resulting analog 71 is twice as selective as 70, with a 45-foldpreference for the b subtype and an IC50 value of 36 nM. A computational modeling studyconfirmed that the large bromine atom in 71 has the same discriminating effect exerted bythe vinyl group of ERB-041 (4, Fig. 3), because it has a steric clash with Met421 in ERa,whereas it is well tolerated by the shorter Ile373 residue in ERb. This would explain why thebromo-substituted derivative (71) shows a higher ERb selectivity than its unsubstitutedcounterpart (70).88
Investigators at Pfizer described a series of imidazoles, benzimidazoles, and indolespossessing good ERb-binding properties.89 The benzimidazole series afforded four ERb-selective compounds (72–75, Table IV). Modest binding properties were found with the2-phenyl-substituted derivative 72, whereas ERb-binding affinity was considerably increasedwhen the phenyl group was replaced by 5-member ring heterocycles. In fact, the 2-thienylanalog 73 showed the highest b selectivity of this series (b/a4126), with a fairly good affinityfor ERb (IC50 5 49.7 nM). N-methylpyrrole-derivative 74 had a higher ERb-binding affinity(IC50 5 28.1 nM), while still preserving a 100-fold selectivity for the b subtype. A slight
HO N
70
OH
O
O
HO N
71
OH
O
O
Br
Figure 23. ERb-selective phthalimido-ligands (70 and 71).
HO
67
MeOH
HO
68
OH
69
HOOH
Figure 22. Spiroindene derivatives (67--69).
384 K MINUTOLOETAL.
Medicinal Research Reviews DOI 10.1002/med
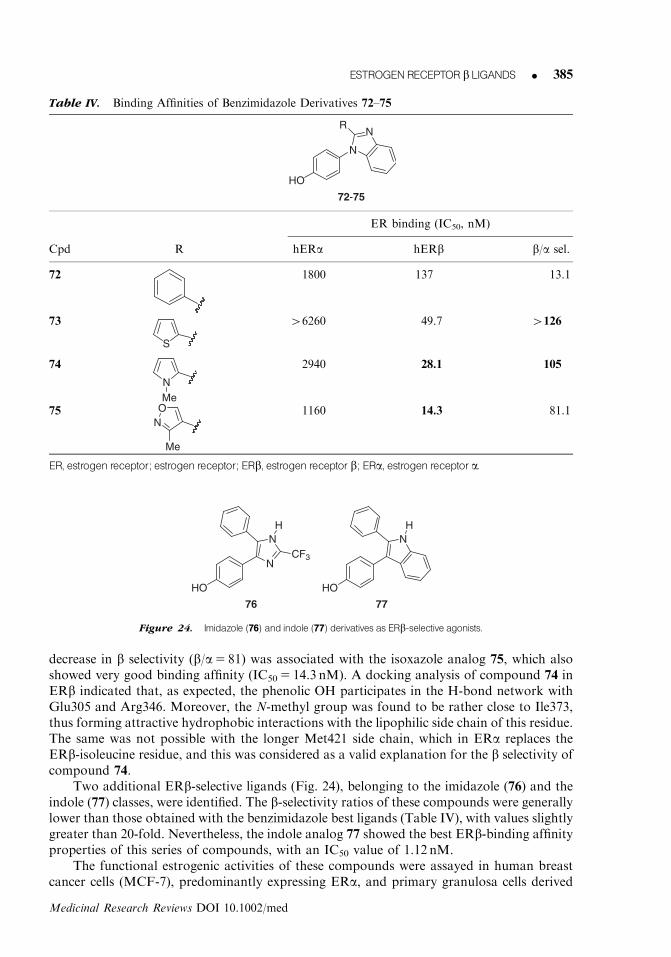
decrease in b selectivity (b/a5 81) was associated with the isoxazole analog 75, which alsoshowed very good binding affinity (IC50 5 14.3 nM). A docking analysis of compound 74 inERb indicated that, as expected, the phenolic OH participates in the H-bond network withGlu305 and Arg346. Moreover, the N-methyl group was found to be rather close to Ile373,thus forming attractive hydrophobic interactions with the lipophilic side chain of this residue.The same was not possible with the longer Met421 side chain, which in ERa replaces theERb-isoleucine residue, and this was considered as a valid explanation for the b selectivity ofcompound 74.
Two additional ERb-selective ligands (Fig. 24), belonging to the imidazole (76) and theindole (77) classes, were identified. The b-selectivity ratios of these compounds were generallylower than those obtained with the benzimidazole best ligands (Table IV), with values slightlygreater than 20-fold. Nevertheless, the indole analog 77 showed the best ERb-binding affinityproperties of this series of compounds, with an IC50 value of 1.12 nM.
The functional estrogenic activities of these compounds were assayed in human breastcancer cells (MCF-7), predominantly expressing ERa, and primary granulosa cells derived
76
N
NH
CF3
HO
77
NH
HO
Figure 24. Imidazole (76) and indole (77) derivatives as ERb-selective agonists.
Table IV. Binding Affinities of Benzimidazole Derivatives 72–75
72-75
N
NR
HO
ER binding (IC50, nM)
Cpd R hERa hERb b/a sel.
72 1800 137 13.1
73
S
46260 49.7 4126
74
NMe
2940 28.1 105
75 ON
Me
1160 14.3 81.1
ER, estrogen receptor; estrogen receptor; ERb, estrogen receptor b; ERa, estrogen receptor a.
ESTROGENRECEPTOR bLIGANDS K 385
Medicinal Research Reviews DOI 10.1002/med

from rat ovaries, which contain mostly endogenous ERb. In these assays, compounds 74, 76,and 77 were found to be full agonists on ERb, with EC50’s in the medium-to-low nanomolarrange and noticeable levels of b selectivity (25- to 35-fold), even though it should be notedthat the ERb receptor involved in these functional assays is not human, but rat. Althoughthese receptors are highly homologous, the selectivity ratios in this case might be affected byspecies selectivity rather than receptor selectivity.
4. Naphthalenes, quinolines, tetrahydroquinolines, tetrahydroisoquinolinesA series of 6-(4-hydroxyphenyl)-b-naphthols was investigated at Wyeth and five derivativeswere found to possess good ERb-binding selectivities (78–82, Fig. 25).90 Binding affinityassays indicate that the first example of this series, compound 78, has a 13-fold selectivity forERb with an IC50 value of 16.3 nM. The introduction of a chlorine atom into the 1-positionof the naphthalene nucleus, as in 79 (WAY-169122), causes an increase in b selectivity(b/a5 36), mostly by increasing the binding affinity for ERb (IC50 5 2.52 nM). A furtherimprovement was obtained by the addition of the 30-fluoro-substituent, which producedcompound 80, endowed with better binding affinity (IC50 5 1.2 nM) and selectivity (b/a5 48)for ERb. Compounds possessing substituents in the 8-position, such as 8-cyano-derivative 81(WAY-202196) or its 8-ethyl analog 82, showed even better b selectivities (b/a5 78 for 81
and 98 for 82). The key for their selectivity lies in their limited affinity for ERa(IC50’s4200 nM), accompanied by a good affinity for ERb (IC50 around 2.5 nM for bothcompounds). X-ray structural studies indicated that the 8-cyano group of 81 is, probably,mainly responsible for the high b selectivity of this compound, because it experiences a stericrepulsion with Met421 in ERa, whereas it fits well in ERb where Ile373 is present at this site.The same effect is likely exerted by the 8-ethyl substituent in 82.
Cell-based transcriptional assays revealed that compounds 79 and 81 are full agonists onERb, with 100% activation (reference: 10 nM E2) at concentrations of 1 mM. Furthermore, invivo assays, 81 was active in two animal models of inflammation (inflammatory bowel diseaseand rheumathoid arthritis).
In the naphthalene class, it is worth mentioning a compound discovered at Glaxo-SmithKline (83, Fig. 25), which contains only one phenol OH group and a peculiar acrylicacid portion.91 This functional group is also present in an analog of tamoxifen, also preparedby GlaxoSmithKline (GW5638),92 which has been the subject of considerable functional andpreclinical investigation.93,94 Compound 83 binds well to ERb (Ki 5 16 nM), but its selectivityis limited to about 4-fold over the a subtype. The authors noticed that when the cyclopropyl
HOOH
78
HOOH
79(WAY-169122)
HOOH
80
Cl Cl
F
HOOH
81(WAY-202196)
HOOH
82
F
CN Et
F
11
3'
88
'3'3 O
HOOC
HO
83
Figure 25. Naphthalene-based ERb-selective ligands (78--83).
386 K MINUTOLOETAL.
Medicinal Research Reviews DOI 10.1002/med

substituent of 83 was replaced by linear alkyl groups, such as n-butyl or n-pentyl, the affi-nities of the resulting compounds for ERb increase, but their b selectivities are reducedbecause of a greater increase in their ERa affinities.
The 2-arylquinolines, which are considered more synthetically accessible than theirnaphthalene analogs, were studied at Wyeth.95 Some of these quinoline-based ligands (84–88,Fig. 26) showed appreciable levels of b selectivity. Starting from unsubstituted derivative,which turned out to be the weakest ERb ligand in this series (IC50 5 171 nM, b/a5 10),insertion of a chlorine atom into the 4 position (84) caused a remarkable increase in b affinity(IC50 5 4.6 nM) and selectivity (b/a5 46). This trend was confirmed with the 4-bromo-sub-stituted analog 85 (IC50 5 4.3 nM, b/a5 50). As seen before, the addition of a fluorine atomin the 30-position, in this series, generated a further increase in b-selective binding, as shownby the IC50 value (3.4 nM) and b/a selectivity ratio (83) of compound 86, which was the mostpotent and most selective ERb ligand of this series. The 4-cyano-substituted derivatives 87and 88 showed lower binding affinity and selectivity than those of their bromo-substitutedanalogs (85 and 86, respectively). A computational docking study pointed out that theincrease in b selectivity associated with the insertion of the 4-bromo-substituent was due, asbefore, to the preferential accommodation that the bromine atom finds in ERb, where it isplaced close to the Ile373 residue, compared with the steric repulsions it suffers with thecorresponding Met421 in ERa. The most interesting compound in this series (86) was sub-mitted to cell-based transcriptional assays, where it proved to be a partial agonist forERb, with a 60% maximum activation at a concentration of 1 mM. It did not show anyERb-antagonist properties or any detectable ERa stimulation.
When the quinoline ring of 84 is partially reduced to a 1,2,3,4-tetrahydroquinoline core,the resulting compound (89) loses most of its b selectivity. This was demonstrated byresearchers at Eli Lilly, who found that 89 binds to ERb with a Ki of 65 nM, which is weakerthan that for 84 (Ki 5 35 nM). Consequently, the selectivity ratio was also reduced, droppingto 2.6 for 89, while 84 showed an almost 10-fold selectivity for ERb over ERa.96
Pfizer has described the preparation of several 1,2,3,4-tetrahydroisoquinolines, amongwhich some ERb-selective ligands were found.97 This is the case for compounds 90 and 91
(Fig. 27), both possessing two phenol moieties and a trifluoroacetyl group on the nitrogen atom.The two enantiomers of 90 were separated and independently assayed: one, tentatively
assigned by the authors as the R-form, showed an IC50 value of 8.9 nM on ERb and 53.6 onERa, thus being 6-fold b selective. The other enantiomer did not bind to either of thesubtypes (IC50 43,200 nM, in both cases). Compound 91 was instead assayed as a racemicmixture. Nevertheless, this compound showed a more pronounced preference for the bsubtype (b/a ratio5 10.8) and a fairly good ERb-binding affinity (IC50 5 11.4 nM).
HON OH
84
HON OH
85
HON OH
86
F
HON OH
87
HON OH
88
F
CN CN
3'
44
3'
89
Br Br44
HON OH
H
Cl4
Figure 26. ERb-ligands possessing quinoline (84--88) or tetrahydroquinoline (89) nuclei.
ESTROGENRECEPTOR bLIGANDS K 387
Medicinal Research Reviews DOI 10.1002/med

5. Aromatic aldoximesAldoximes are generally considered to be chemically unstable, and to undergo hydrolysisreadily, especially under acidic conditions, or to be rapidly metabolized in biological systems.However, this is not true for aromatic aldoximes, which typically undergo an extremely slowaqueous hydrolysis in buffer solutions, even at markedly acidic pH. In fact, their mainmetabolic transformation occurs in liver microsomes and involves a slow oxidation of theoxime moiety, which produces NO.98
A large number of compounds belonging to this category have been recently studied asER ligands. Minutolo and co-workers at the University of Pisa first introduced an oximegroup into the structure of ER ligands based on the diaryl-substituted salicylaldoximescaffold, as shown by compound 92 (Fig. 28).99
In this case, the pseudocycle (A0) formed by intramolecular H-bond was intended tobioisosterically replace the pharmacophoric phenol group (A-ring of E2). In fact, this ring(A’) presents several traits in common with the phenolic A-ring of estrogens: (1) the two ringshave roughly the same size and hexagonal geometry; (2) a similar planar p-conjugationcovers the atoms comprised in both systems (at least partially, in the case of the salicyl-aldoximes); (3) the oxime hydroxy group of this system has a pKa value of about 10, which isvery similar to typical pKa values of phenolic hydroxy groups; (4) the position of this OHgroup corresponds to the position 3 of the phenolic A-ring, commonly occupied by anhydroxyl in classical ER ligands, which participates in the strong H-bond network with theresidues arginine and glutamate in both subtypes.
This bioisosteric hypothesis was confirmed by the good binding affinity shown by 92 forboth ERs (RBA5 1.13% for ERa and 1.71% for ERb), although b selectivity was notobserved with this compound. The introduction of an additional OH group in either one ofthe two phenyl substituents caused a shift in the subtype selectivity of these molecules. Infact, compound 93, bearing a para-OH substituent in the ‘‘distal’’ phenyl group, displayed abinding preference for the a subtype (RBA5 2.59% for ERa and 1.44% for ERb), whereas94, where the p-OH is placed in the ‘‘proximal’’ ring, had the opposite subtype preference,
O
NHO
HA'
92
O
NHO
HA'
93
OH
O
NHO
HA'
94
OH
Figure 28. Diaryl-substituted salicylaldoximes 92--94.
N
O
CF3
HO
OH
90
NHO
OH
91O
CF3
Figure 27. N-Trifluoroacetyl-tetrahydroisoquinolines 90 and 91.
388 K MINUTOLOETAL.
Medicinal Research Reviews DOI 10.1002/med

showing a better affinity for ERb (RBA5 2.21%) than for ERa (RBA5 0.97%). The si-multaneous presence of two hydroxyls in both rings decreased binding affinity for both ERsubtypes, compared with 92.100
The chemical class of anthranylaldoximes was then explored, by replacing the endocyclicoxygen atom with an NH, NMe, or NEt moiety (95–97, Fig. 29), to see whether ER-bindingaffinities could be improved by modifying the stereoelectronic nature of the pseudocyclering.101
Within this series, the N-unsubstituted compound 95 proved to possess better bindingproperties than its salicylic analog 92, showing a 2-fold improvement for both receptorsubtypes (RBA5 2.2% for ERa and 2.8% for ERb). A possible explanation is that thepseudocycle of this anthranylic derivative has greater aromatic character, which makes it abetter mimic of the phenolic A-ring of E2 and other classical estrogen ligands. In fact, thenitrogen atom of 95 is of the aniline type and, therefore, its hybridization state has a greatersp2 character than its oxygen counterpart in 92. This result in a greater delocalization of thenitrogen lone pair through the aromatic central core to the oxime C-N double bond, givingthe pseudo cycle a greater degree of p-conjugation. Moreover, increased affinities were ob-served with the N-methylated derivative 96, which showed RBA values of 3.7 with ERa and5.2 with ERb, 3-fold higher affinity than 92 for both receptor subtypes. On the contrary, theN-ethylated analog 97 showed a significant drop in binding compared with its N-methylatedanalog 96, with almost a 10-fold reduction on ERa and over 100-fold reduction on ERb.Docking studies on both receptor subtypes revealed that the N-alkyl group of 96 and 97
occupies a small hydrophobic pocket whose boundaries are formed by three residues,Leu349, Ala350, and the peptide carbonyl portion of Leu346. This pocket has limited di-mensions; so, it is able to comfortably host only the N-Me group of 96 (Fig. 29), whereas itneeds to be deformed to a certain degree to accommodate even the slightly bulkier ethylmoiety in 97. Therefore, the low affinity values found with 97 are probably due to stericrepulsions from interactions within this small pocket. Nevertheless, despite the good bindingaccompanied by some ERb preference of 96, transcriptional assays revealed that this com-pound is only a partial antagonist for ERb, while it was able to stimulate ERa fully.
Selective insertion of an additional OH group was also effected in the N-methyl an-thranylaldoxime scaffold (98 and 99, Fig. 29).102 Compound 98, containing a para-hydroxy
N
NHO
H
95
N
NHO
H
96
N
NHO
H
97
H Me Et
N
NHO
H
98
N
NHO
H
99
Me Me
OH
OH
smallhydrophobic
pocketsteric clash
Figure 29. Diaryl-substituted anthranylaldoximes 95--99.
ESTROGENRECEPTOR bLIGANDS K 389
Medicinal Research Reviews DOI 10.1002/med

group on the ‘‘distal’’ phenyl group, displayed a binding preference for the a subtype(RBA5 5.38% for ERa and 1.11% for ERb), just like its salicylaldoxime analog 93 (Fig. 28)but with enhanced binding properties. The same parallelism with the salicylaldoxime serieswas not found with 99, however, where the OH group was present in the ‘‘proximal’’ phenylring. In fact, this compound showed a decrease in binding affinity with both receptor sub-types. Therefore, compound 96 remained the best ERb ligand of the anthranylaldoximecategory.
Subsequently, the molecular design of this series of aldoxime derivatives benefitedfrom the development of the previously mentioned ERb-selective pharmacophore model(Fig. 18).80 In fact, this model inspired the removal of one of the two aryl substituents fromthe salicylaldoxime scaffold, leading to the production of a series of monoaryl-substitutedsalicylaldoximes. The simplest member of this series, 100 (Fig. 30), showed a remarkable79-fold selectivity for the b subtype, although its binding affinity for ERb was rather low(RBA5 0.553%).103 A dramatic increase in ERb-binding affinity was achieved by introdu-cing a methyl group (101) or a chlorine atom (102) at the 3 position of the central core. Infact, methyl-substituted analog 101 showed an RBA of 0.249% on ERa and 4.10 % on ERb.A better selectivity was obtained with the chloro-substituted derivative 102, which gave a65-fold selectivity for ERb (RBA5 4.21%) over ERa (RBA5 0.065%). The best compoundof this series (102) also proved to be an efficient partial agonist for ERb, with a maximumstimulation of 60% and an EC50 value of 11 nM (roughly corresponding to its Ki value of12 nM derived from binding assays). However, much of the ERb-binding selectivity of 102was lost in these functional assays, in which it also showed efficient ERa stimulation(EC50 5 26 nM).
Further structural modification of these molecules involved an exchange of the relativepositions of the phenol OH and oxime groups of 100, leading to 103, and the introduction ofa meta-fluorine atom into the peripheral 4-aryl substituent of 102, thus producing compound104 (Fig. 30).104 Compound 103 showed a 5-fold increase in ERb-binding affinity (RBA5
2.64%) compared with 100, together with the maintenance of a notable level of b selectivity(b/a5 41). Compound 104, formally obtained by the simultaneous introduction of a3-chloro-substituent and a meta-fluorine atom into the 4-hydroxyphenyl ring of compound100, resulted in an increase in ERb-binding affinity. In fact, 104 reached the best value in this
O
NHO
H
OH
100
O
NHO
H
OH
101
O
NHO
H
OH
102
CH3 Cl
O
NHO
H
OH
104
Cl
F
OH
N
OH
HO
103
Figure 30. Monoaryl-substituted salicylaldoximes 100--104.
390 K MINUTOLOETAL.
Medicinal Research Reviews DOI 10.1002/med

series of compounds, a RBA value of 7.01% for ERb; in this case, a high level of subtypeselectivity was also preserved (ERb/a5 62).
Some explanations for the b selectivity of this class of monoaryl-substituted oximes werefound by docking studies of their molecular structures within the binding cavities of ERa andERb (Fig. 31).
As for compound 103, docking analysis in ERb showed that the pseudocycle/oximesystem is able to interact with the Arg346-Glu305 H-bond network, whereas the OH group ofthe p-phenol ring forms an H-bond with Thr299. This last interaction is only possible in ERb,where the aromatic ring of the phenol substituent nicely fits into a pocket surrounded byMet336; the same is not possible in ERa, where Met336 is replaced by a bulkier or less flexibleLeu384, which prevents formation of the H-bond with the threonine residue.58 A differentorientation was found for oximes 102 and 104 in the ERb-binding pocket. Both compoundsshow very similar binding modes, with the pseudocycle/oxime systems forming H-bonds withHis475, and the phenolic OH participating in an H-bond network that includes Glu305 andArg346. In these complexes with ERb, the chlorine atom is inserted into a small lipophilicpocket, containing Met336. The replacement of this methionine residue with Leu384 in ERacauses a steric repulsion between this residue and the chlorine atom of 102 and 104, thus,increasing their selectivity for the b-subtype. The higher affinity associated with the fluoro-substituted derivative 104, compared with 102, could be explained by considering the repul-sion between the fluorine atom of 104 and the carbonyl oxygen of Leu339, which results in ashift of the molecule by about 0.7 A along the long axis of the molecule. This allows a strongerinteraction of the pseudocycle/oxime system with His475, while the greater distance betweenthe phenol OH group and the Glu305-Arg346 H-bond network is balanced by the higherpolarization of the phenol hydroxy group due to the ortho-F substituent.
Functional assays of oxime 103 revealed that it is a potent full agonist for ERb, withan EC50 value of 10 nM; however, it also proved to efficiently activate transcriptionthrough ERa (EC50 5 17 nM), thus being a nonselective agonist, in spite of its ERb-bindingselectivity. Better results were obtained with chloro-fluoro-substituted oxime 104, whichshowed pronounced agonist properties for ERb, with an EC50 of 4.8 nM, a value close toits absolute affinity Ki (7.1 nM) found in the binding assay. This compound also had thebest b-selective profile within this series, because its activation of ERa has an EC50 valueof 19 nM.
OH
N
OH
OH
Arg346 Glu305
Thr299
ER : Met336
ER : Leu384
nice fit
steric clash
Arg346 Glu305
ER : Met336
ER : Leu384
nice fit
steric clash
His475
O
NOH
H
OH
Cl
R
201301 (R = H)104 (R = F)
Leu339
shift
Figure 31. Principal interactions ofoximes 102--104 with ERb and origins of their selectivity.
ESTROGENRECEPTOR bLIGANDS K 391
Medicinal Research Reviews DOI 10.1002/med

Work on aldoximes at Wyeth started from the observation that some 4-hydroxy-biphenyls, especially aldehyde 105 (Fig. 32), exhibited a good b selectivity in binding assays,although their ERb-binding affinities were generally not excellent.105 Biphenylic aldehyde105 showed an IC50 value of 69 nM on ERb, with a 72-fold selectivity over ERa. Dockingstudies revealed that the phenolic OH group interacts with the glutamate/arginine H-bondsystem, and the molecular portions responsible for the b selectivity seems to be the chlorosubstituents, which were found to be at a comfortable distance from Ile373 in ERb (4.1 A),although they suffer from steric repulsion in ERa, where they are too close (2.6 A) to thelonger Met421 residue. However, the authors noticed that the aldehyde group was too shortto interact with His475. Therefore, they decided to lengthen this molecular portion by in-serting an oxime group, so that it could interact with the histidine residue. This led to thedevelopment of a series of biphenylic aldoxime derivatives (106–108, Fig. 32).106
Oxime 106 (R5Cl, R05H) showed a 8-fold increase of ERb-binding affinity (IC50 5
8 nM) when compared with aldehyde 105, thus demonstrating the importance of the addi-tional attractive interaction of the oxime portion with His475. Unfortunately, though, itsselectivity for the b subtype was decreased (b/a5 8). As seen for other series of compounds,introduction of a 30 fluoro substituent in the phenol ring led to the production of compound107 (R5Cl, R05F) possessing a higher b selecivity (b/a5 31) than that of 106 and, at thesame time, a very good ERb-binding affinity (IC50 5 9 nM). Removal of one of the twochlorine atom surprisingly generated the most ERb-selective aldoxime of this series, 108(R5H, R05F), which showed a 49-fold selectivity over the a subtype, even though itsbinding affinity diminished (IC50 5 54 nM). Cell-based transcriptional assays conducted oncompound 107, revealed that this compound is a full agonist on ERb, with a 100% stimu-lation at the 1 mM concentration, whereas it is a partial agonist on ERa, with a 21% acti-vation at the same concentration. No antagonist activities were detected on either ER.
Further development of these derivatives consisted of replacing the phenyl ring bearingthe oxime functionality with a bicyclic aromatic system, such as a naphthalene or an indolemoiety (109–111, Fig. 33).107 The additional aromatic ring in these systems was intended toexert the same subtype discrimination seen before by the chloro substituents of oximes106–108, with a preferential accommodation by the Ile373 residue of ERb, and a coordinate
CHO
OH
Arg346 Glu305
aldehyde105
Cl Cl
ER β: Ile373
ER α: Met421
nice fit
steric clash
His475
no interaction
OH
Arg346 Glu305
oximes106 -108
Cl R
His475
N
OH
additionalattractive
interaction
R'
ER β: Ile373
ER α: Met421
nice fit
steric clash
Figure 32. Development of biphenylic oximes 106--108 from aldehyde 105.
392 K MINUTOLOETAL.
Medicinal Research Reviews DOI 10.1002/med
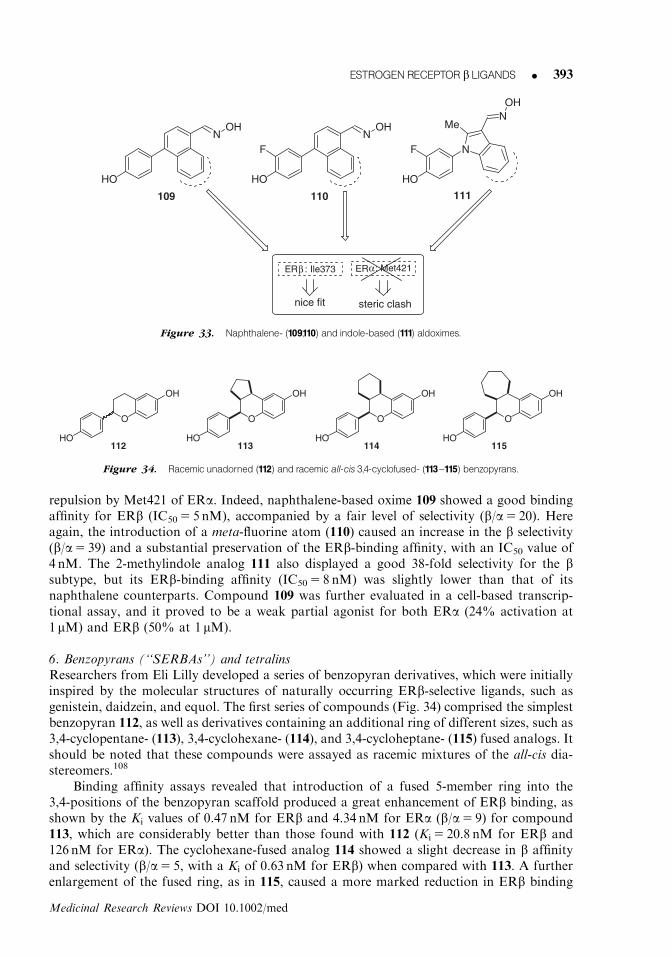
repulsion by Met421 of ERa. Indeed, naphthalene-based oxime 109 showed a good bindingaffinity for ERb (IC50 5 5 nM), accompanied by a fair level of selectivity (b/a5 20). Hereagain, the introduction of a meta-fluorine atom (110) caused an increase in the b selectivity(b/a5 39) and a substantial preservation of the ERb-binding affinity, with an IC50 value of4 nM. The 2-methylindole analog 111 also displayed a good 38-fold selectivity for the bsubtype, but its ERb-binding affinity (IC50 5 8 nM) was slightly lower than that of itsnaphthalene counterparts. Compound 109 was further evaluated in a cell-based transcrip-tional assay, and it proved to be a weak partial agonist for both ERa (24% activation at1 mM) and ERb (50% at 1 mM).
6. Benzopyrans (‘‘SERBAs’’) and tetralinsResearchers from Eli Lilly developed a series of benzopyran derivatives, which were initiallyinspired by the molecular structures of naturally occurring ERb-selective ligands, such asgenistein, daidzein, and equol. The first series of compounds (Fig. 34) comprised the simplestbenzopyran 112, as well as derivatives containing an additional ring of different sizes, such as3,4-cyclopentane- (113), 3,4-cyclohexane- (114), and 3,4-cycloheptane- (115) fused analogs. Itshould be noted that these compounds were assayed as racemic mixtures of the all-cis dia-stereomers.108
Binding affinity assays revealed that introduction of a fused 5-member ring into the3,4-positions of the benzopyran scaffold produced a great enhancement of ERb binding, asshown by the Ki values of 0.47 nM for ERb and 4.34 nM for ERa (b/a5 9) for compound113, which are considerably better than those found with 112 (Ki 5 20.8 nM for ERb and126 nM for ERa). The cyclohexane-fused analog 114 showed a slight decrease in b affinityand selectivity (b/a5 5, with a Ki of 0.63 nM for ERb) when compared with 113. A furtherenlargement of the fused ring, as in 115, caused a more marked reduction in ERb binding
HO
NOH
109
HO
NOH
110
F N
HO
111
F
NOH
Me
ERβ : Ile373 ERα: Met421
nice fit steric clash
Figure 33. Naphthalene- (109,110) and indole-based (111) aldoximes.
O
OH
HO112
O
OH
HO113
O
OH
HO114
O
OH
HO115
Figure 34. Racemic unadorned (112) and racemic all-cis 3,4-cyclofused- (113--115) benzopyrans.
ESTROGENRECEPTOR bLIGANDS K 393
Medicinal Research Reviews DOI 10.1002/med

(Ki 5 0.88 nM for ERb and 6.11 nM for ERa). So, the single enantiomers of the best com-pound of this series (113) were obtained in optically pure forms by chiral chromatographyand were separately submitted to biological assays. The (2R,3S,4R)-113 was named SERBA-1,whereas the other enantiomer (2S,3R,4S)-113 was referred to as SERBA-2.109 EnantiomerSERBA-1 displayed the highest levels of ERb affinity and selectivity (Ki 5 0.19 nM for ERband 2.68 nM for ERa, b/a5 14), whereas the other enantiomer, SERBA-2, showed a 8-foldlower binding affinity, together with a considerable loss of selectivity. Transcriptional assaysshowed that SERBA-1 is a full ERb agonist (EC50 5 0.66 nM), with a 30-fold selectivity overthe a subtype. Here again, SERBA-2 proved to be a less potent (EC50 5 3.61 nM) and lessselective (b/a5 10) ERb-agonist than SERBA-1. An X-ray structural analysis of SERBA-1,complexed to both ERa and ERb, revealed that this compound assumes two orientations,depending on the receptor subtype, differing by a rotation of about 1801 on its long axis(Fig. 35). The most efficient interactions of SERBA-1 occur with ERb, where the p-hydroxy-phenyl group participates in the Arg346/Glu305 H-bond network, the other phenol hydroxylforms an H-bond with His475, and the fused cyclopentane ring occupies a small hydrophobicpocket, close to the ERb-exclusive Ile373 residue. In ERa, the replacement of Ile373 withMet421 makes this pocket too small to host the cyclopentane portion. Hence, the moleculefinds its best accommodation in ERa by rotating along its bisphenol axis. As a consequence,the highest energy interaction with the Arg/Glu network is preserved, but the interaction withHis524 is likely weakened because of the longer distance between the OH group and thisresidue, when compared with its position in ERb. This would explain the b-selective prop-erties of SERBA-1. Furthermore, this molecule proved to be efficient in reducing prostateweight in mouse models of benign prostate hyperplasia, with no evidence of residual ERastimulation (e.g. no weight reduction of testes and seminal vesicles).
Subsequent series of benzopyrans, inspired by these encouraging results obtained withSERBA-1, have been produced by slight modifications of the cyclopentane ring. The mostinteresting compounds, derived from the introduction of the following groups (Fig. 36): acarbonyl (116), a gem-difluoro portion (117), a trifluoromethyl unit (118 and 119), and amethyl group (120).110 All of these compounds were obtained and tested as single en-antiomers and they all showed good levels of b selectivity, although none of them had ahigher binding affinity than unsubstituted 3,4-cyclopentane-fused derivative SERBA-1. Thehighest subtype-binding selectivity (b/a5 101) was achieved by carbonyl derivative 116, al-though this compound had diminished ERb-binding affinity, showing more than a 30-foldreduction (Ki 5 6.92 nM) with respect to SERBA-1. A better result was obtained by in-troducing two geminal fluorine atoms, giving a compound (117) with very high b-bindingaffinity (Ki 5 0.44 nM) and an improved selectivity level (b/a5 19) compared with SERBA-1.Similar results were obtained with the trifluoromethyl-substituted analogs 118 and 119,which both showed the same level of b selectivity (b/a5 17–18) and similar Ki values for ERb
O OH
HOO OH
HO
ER
His524
Met421
Arg394
Glu353
longerdistance
ER
His475
Ile373
Arg346
Glu305
Figure 35. Themost active enantiomerof 113 (SERBA-1) and its X-ray derived binding interactions.
394 K MINUTOLOETAL.
Medicinal Research Reviews DOI 10.1002/med
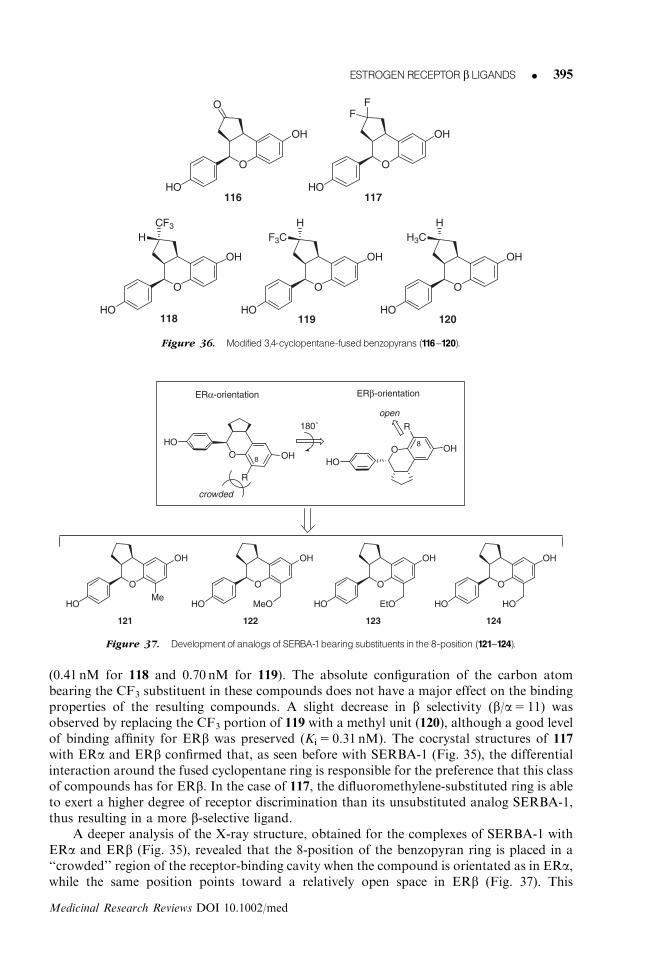
(0.41 nM for 118 and 0.70 nM for 119). The absolute configuration of the carbon atombearing the CF3 substituent in these compounds does not have a major effect on the bindingproperties of the resulting compounds. A slight decrease in b selectivity (b/a5 11) wasobserved by replacing the CF3 portion of 119 with a methyl unit (120), although a good levelof binding affinity for ERb was preserved (Ki 5 0.31 nM). The cocrystal structures of 117with ERa and ERb confirmed that, as seen before with SERBA-1 (Fig. 35), the differentialinteraction around the fused cyclopentane ring is responsible for the preference that this classof compounds has for ERb. In the case of 117, the difluoromethylene-substituted ring is ableto exert a higher degree of receptor discrimination than its unsubstituted analog SERBA-1,thus resulting in a more b-selective ligand.
A deeper analysis of the X-ray structure, obtained for the complexes of SERBA-1 withERa and ERb (Fig. 35), revealed that the 8-position of the benzopyran ring is placed in a‘‘crowded’’ region of the receptor-binding cavity when the compound is orientated as in ERa,while the same position points toward a relatively open space in ERb (Fig. 37). This
O
OH
HO116
O
O
OH
HO117
FF
O
OH
HO118
CF3
H
O
OH
HO119
HF3C
O
OH
HO120
HH3C
Figure 36. Modified 3,4-cyclopentane-fused benzopyrans (116--120).
O OH
HO
ERα-orientation
R
8
crowded
open
ERβ-orientation
O
OH
HO
121
8
MeO
OH
HO
122
MeO
O
OH
HO
123
EtO
O
OH
HO
124
HO
180˚
O OH
HO
R
Figure 37. Development of analogs of SERBA-1bearing substituents in the 8-position (121--124).
ESTROGENRECEPTOR bLIGANDS K 395
Medicinal Research Reviews DOI 10.1002/med
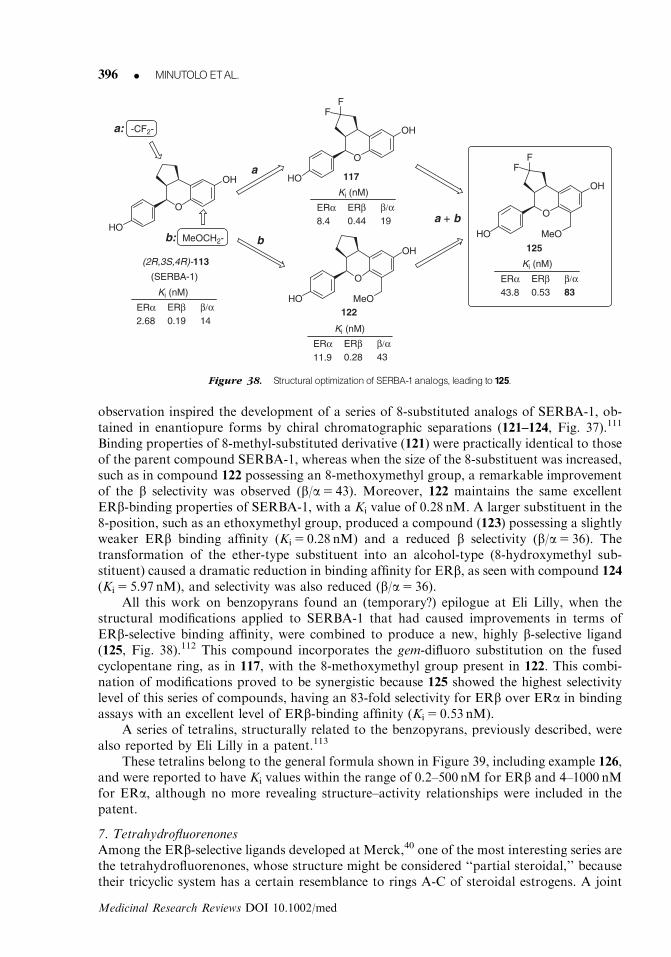
observation inspired the development of a series of 8-substituted analogs of SERBA-1, ob-tained in enantiopure forms by chiral chromatographic separations (121–124, Fig. 37).111
Binding properties of 8-methyl-substituted derivative (121) were practically identical to thoseof the parent compound SERBA-1, whereas when the size of the 8-substituent was increased,such as in compound 122 possessing an 8-methoxymethyl group, a remarkable improvementof the b selectivity was observed (b/a5 43). Moreover, 122 maintains the same excellentERb-binding properties of SERBA-1, with a Ki value of 0.28 nM. A larger substituent in the8-position, such as an ethoxymethyl group, produced a compound (123) possessing a slightlyweaker ERb binding affinity (Ki 5 0.28 nM) and a reduced b selectivity (b/a5 36). Thetransformation of the ether-type substituent into an alcohol-type (8-hydroxymethyl sub-stituent) caused a dramatic reduction in binding affinity for ERb, as seen with compound 124
(Ki 5 5.97 nM), and selectivity was also reduced (b/a5 36).All this work on benzopyrans found an (temporary?) epilogue at Eli Lilly, when the
structural modifications applied to SERBA-1 that had caused improvements in terms ofERb-selective binding affinity, were combined to produce a new, highly b-selective ligand(125, Fig. 38).112 This compound incorporates the gem-difluoro substitution on the fusedcyclopentane ring, as in 117, with the 8-methoxymethyl group present in 122. This combi-nation of modifications proved to be synergistic because 125 showed the highest selectivitylevel of this series of compounds, having an 83-fold selectivity for ERb over ERa in bindingassays with an excellent level of ERb-binding affinity (Ki 5 0.53 nM).
A series of tetralins, structurally related to the benzopyrans, previously described, werealso reported by Eli Lilly in a patent.113
These tetralins belong to the general formula shown in Figure 39, including example 126,and were reported to have Ki values within the range of 0.2–500 nM for ERb and 4–1000 nMfor ERa, although no more revealing structure–activity relationships were included in thepatent.
7. TetrahydrofluorenonesAmong the ERb-selective ligands developed at Merck,40 one of the most interesting series arethe tetrahydrofluorenones, whose structure might be considered ‘‘partial steroidal,’’ becausetheir tricyclic system has a certain resemblance to rings A-C of steroidal estrogens. A joint
O
OH
HO
(SERBA-1) O
OH
HO122
MeO
O
OH
HO 117
FF
-CF2-
MeOCH2-
O
OH
HO
125
FF
Ki (nM)
ERα ERβ β/α2.68 0.19 14
Ki (nM)
ERα ERβ β/α11.9 0.28 43
Ki (nM)
ERα ERβ β/α8.4 0.44 19
Ki (nM)
ERα ERβ β/α43.8 0.53 83
MeO
a:
b:
a
b
a + b
(2R,3S,4R)-113
Figure 38. Structural optimization of SERBA-1analogs, leading to 125.
396 K MINUTOLOETAL.
Medicinal Research Reviews DOI 10.1002/med

venture between Merck and Karo Bio produced a series of tetrahydrofluorenones that re-vealed the importance of the n-butyl substituent in the 9a-position (127–130, Table V); other9a-substituents (Me, Et, n-Pr, i-Pr, i-Bu, Ph, Bn, pentyl, and 2-hydroxyethyl) gave poorerligands.114 In this series, the effect of the substituent at the 4-position (R, Table V) wasanalyzed in racemic compounds. Selected molecules showing good levels of b selectivity inbinding assays were identified and their transcriptional activity through ERb was described,although comparable data on ERa were not available.
Bromo-substituted derivative 127 showed a remarkable 76-fold selectivity over ERa(IC50 5 1.8 nM on ERb) and a potent stimulation of ERb (EC50 5 4 nM, 81% of stimula-tion). A higher degree of b selectivity (b/a5 85) was obtained with the trifluoromethyl-substituted analog 128, which, however, was ten times less potent (EC50 5 40 nM) than 127 intranscriptional assays. Replacement of bromine with an iodine atom, leading to 129, caused afurther increase in the b/a selectivity ratio, reaching almost 100, despite a slight decrease inthe activation efficacy of ERb (70% activation). Finally, methyl-substituted compound 130
showed reduced ERb-binding affinity (IC50 5 16 nM) and selectivity (b/a5 39). This com-pound stimulated ERb very efficiently, with an activation level of 112% and an EC50 of20 nM. Resolution of racemic 127 and 130 by chiral HPLC provided enantiopure forms. Inboth cases, enantiomers of (S)-configuration showed ERb-binding affinities 100-fold higherthan those of (R)-configuration, with the bromo derivative (S)-127 showing the highestaffinity (IC50 5 1.5 nM) and b selectivity (b/a5 89); selectivities were lower with (S)-130(ERb-IC50 5 1.5 nM, b/a5 30). X-ray analysis of the cocrystallized complex of (S)-127 with
OH
HO
R2
HO
R1
R3
R4R5
R6
G
126
Figure 39. General formula of tetralin-based ERb-ligands represented by lead compound 126.
Table V. Binding Affinities and Functional Properties of Racemic Tetrahydrofluorenone Derivatives
127–130
HO
R O
Bun
127-130
ER binding (IC50, nM) ER transactivation (hERb)
Cpd R hERa hERb b/a sel. EC50 (nM) % activation
127 Br 141 1.8 76 4 81
128 CF3 128 1.5 85 40 81
129 I 124 1.3 95 2 70
130 CH3 630 16 39 20 112
ER, estrogen receptor; ERb, estrogen receptor b; ERa, estrogen receptor a.
ESTROGENRECEPTOR bLIGANDS K 397
Medicinal Research Reviews DOI 10.1002/med
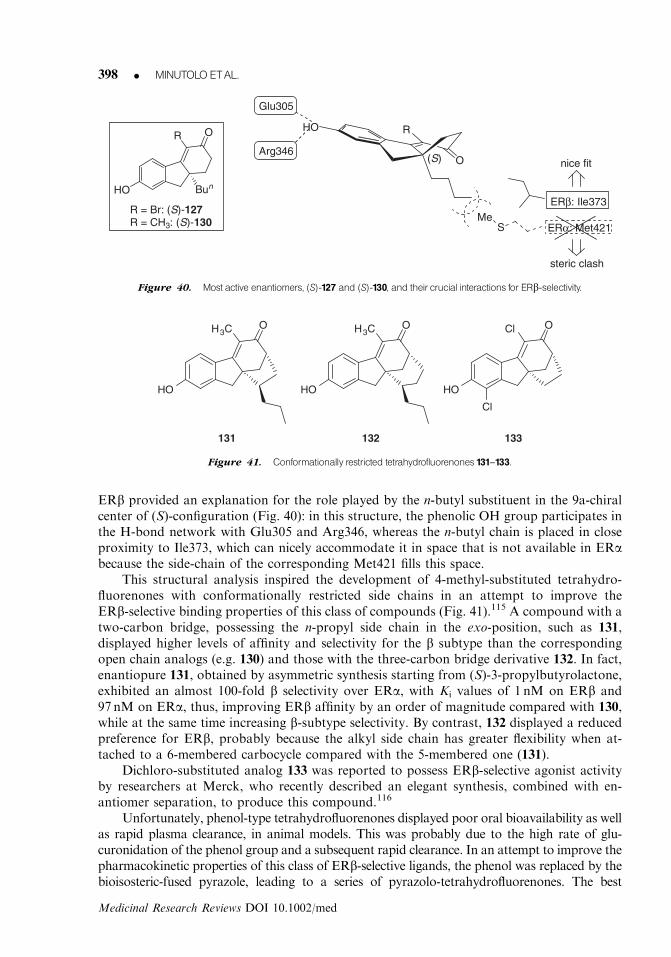
ERb provided an explanation for the role played by the n-butyl substituent in the 9a-chiralcenter of (S)-configuration (Fig. 40): in this structure, the phenolic OH group participates inthe H-bond network with Glu305 and Arg346, whereas the n-butyl chain is placed in closeproximity to Ile373, which can nicely accommodate it in space that is not available in ERabecause the side-chain of the corresponding Met421 fills this space.
This structural analysis inspired the development of 4-methyl-substituted tetrahydro-fluorenones with conformationally restricted side chains in an attempt to improve theERb-selective binding properties of this class of compounds (Fig. 41).115 A compound with atwo-carbon bridge, possessing the n-propyl side chain in the exo-position, such as 131,displayed higher levels of affinity and selectivity for the b subtype than the correspondingopen chain analogs (e.g. 130) and those with the three-carbon bridge derivative 132. In fact,enantiopure 131, obtained by asymmetric synthesis starting from (S)-3-propylbutyrolactone,exhibited an almost 100-fold b selectivity over ERa, with Ki values of 1 nM on ERb and97 nM on ERa, thus, improving ERb affinity by an order of magnitude compared with 130,while at the same time increasing b-subtype selectivity. By contrast, 132 displayed a reducedpreference for ERb, probably because the alkyl side chain has greater flexibility when at-tached to a 6-membered carbocycle compared with the 5-membered one (131).
Dichloro-substituted analog 133 was reported to possess ERb-selective agonist activityby researchers at Merck, who recently described an elegant synthesis, combined with en-antiomer separation, to produce this compound.116
Unfortunately, phenol-type tetrahydrofluorenones displayed poor oral bioavailability as wellas rapid plasma clearance, in animal models. This was probably due to the high rate of glu-curonidation of the phenol group and a subsequent rapid clearance. In an attempt to improve thepharmacokinetic properties of this class of ERb-selective ligands, the phenol was replaced by thebioisosteric-fused pyrazole, leading to a series of pyrazolo-tetrahydrofluorenones. The best
HO
R O
Bun
R = Br: (S)-127R = CH3: (S)-130
HO R
O
Glu305
Arg346
ERβ: Ile373
nice fit
steric clash
SMe
ERα: Met421
(S)
Figure 40. Most active enantiomers, (S)-127 and (S)-130, and their crucial interactions for ERb-selectivity.
HO
H3C O
HO
H3C O
132131
HO
Cl O
133
Cl
Figure 41. Conformationally restricted tetrahydrofluorenones 131--133.
398 K MINUTOLOETAL.
Medicinal Research Reviews DOI 10.1002/med
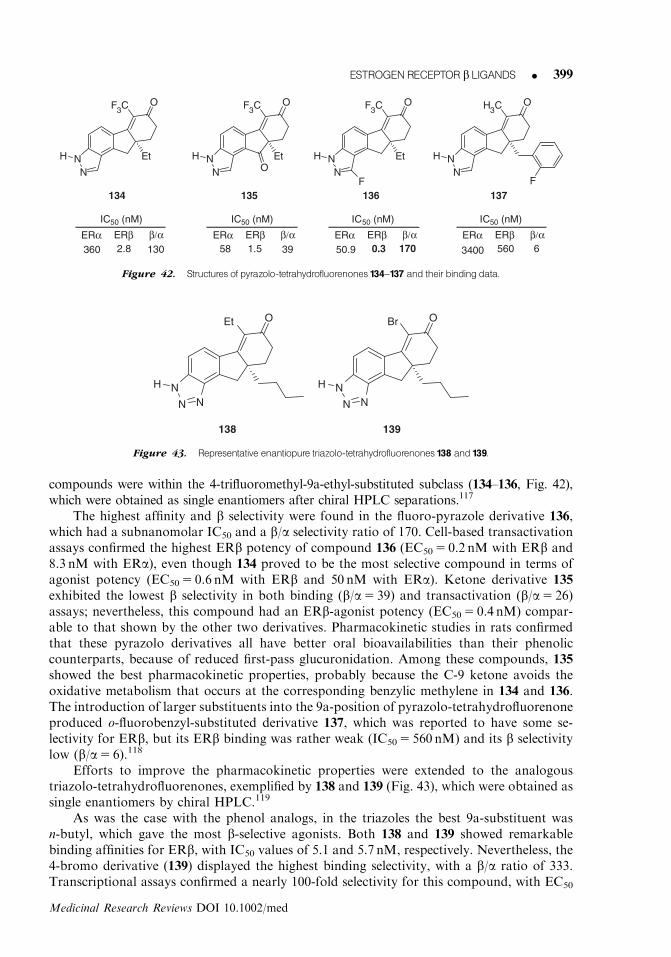
compounds were within the 4-trifluoromethyl-9a-ethyl-substituted subclass (134–136, Fig. 42),which were obtained as single enantiomers after chiral HPLC separations.117
The highest affinity and b selectivity were found in the fluoro-pyrazole derivative 136,which had a subnanomolar IC50 and a b/a selectivity ratio of 170. Cell-based transactivationassays confirmed the highest ERb potency of compound 136 (EC50 5 0.2 nM with ERb and8.3 nM with ERa), even though 134 proved to be the most selective compound in terms ofagonist potency (EC50 5 0.6 nM with ERb and 50 nM with ERa). Ketone derivative 135
exhibited the lowest b selectivity in both binding (b/a5 39) and transactivation (b/a5 26)assays; nevertheless, this compound had an ERb-agonist potency (EC50 5 0.4 nM) compar-able to that shown by the other two derivatives. Pharmacokinetic studies in rats confirmedthat these pyrazolo derivatives all have better oral bioavailabilities than their phenoliccounterparts, because of reduced first-pass glucuronidation. Among these compounds, 135showed the best pharmacokinetic properties, probably because the C-9 ketone avoids theoxidative metabolism that occurs at the corresponding benzylic methylene in 134 and 136.The introduction of larger substituents into the 9a-position of pyrazolo-tetrahydrofluorenoneproduced o-fluorobenzyl-substituted derivative 137, which was reported to have some se-lectivity for ERb, but its ERb binding was rather weak (IC50 5 560 nM) and its b selectivitylow (b/a5 6).118
Efforts to improve the pharmacokinetic properties were extended to the analogoustriazolo-tetrahydrofluorenones, exemplified by 138 and 139 (Fig. 43), which were obtained assingle enantiomers by chiral HPLC.119
As was the case with the phenol analogs, in the triazoles the best 9a-substituent wasn-butyl, which gave the most b-selective agonists. Both 138 and 139 showed remarkablebinding affinities for ERb, with IC50 values of 5.1 and 5.7 nM, respectively. Nevertheless, the4-bromo derivative (139) displayed the highest binding selectivity, with a b/a ratio of 333.Transcriptional assays confirmed a nearly 100-fold selectivity for this compound, with EC50
F3C O
EtNN
H
134
F3C O
EtNN
H
135
O
F3C O
EtNN
H
136F
IC50 (nM)
ERα ERβ β/α360 2.8 130
ERα ERβ β/α58 1.5 39
ERα ERβ β/α50.9 0.3 170
IC50 (nM) IC50 (nM)
H3C O
NN
H
137
ERα ERβ β/α3400 560 6
IC50 (nM)
F
Figure 42. Structures of pyrazolo-tetrahydrofluorenones 134--137 and their binding data.
Et O
N
N N
H
138
Br O
N
N N
H
139
Figure 43. Representative enantiopure triazolo-tetrahydrofluorenones 138 and 139.
ESTROGENRECEPTOR bLIGANDS K 399
Medicinal Research Reviews DOI 10.1002/med
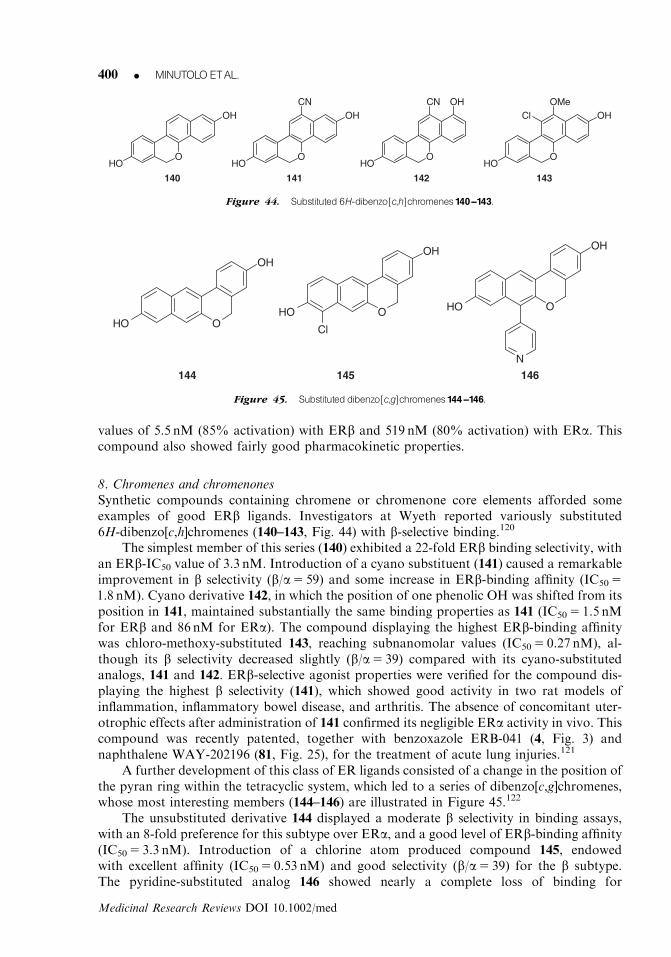
values of 5.5 nM (85% activation) with ERb and 519 nM (80% activation) with ERa. Thiscompound also showed fairly good pharmacokinetic properties.
8. Chromenes and chromenonesSynthetic compounds containing chromene or chromenone core elements afforded someexamples of good ERb ligands. Investigators at Wyeth reported variously substituted6H-dibenzo[c,h]chromenes (140–143, Fig. 44) with b-selective binding.120
The simplest member of this series (140) exhibited a 22-fold ERb binding selectivity, withan ERb-IC50 value of 3.3 nM. Introduction of a cyano substituent (141) caused a remarkableimprovement in b selectivity (b/a5 59) and some increase in ERb-binding affinity (IC50 5
1.8 nM). Cyano derivative 142, in which the position of one phenolic OH was shifted from itsposition in 141, maintained substantially the same binding properties as 141 (IC50 5 1.5 nMfor ERb and 86 nM for ERa). The compound displaying the highest ERb-binding affinitywas chloro-methoxy-substituted 143, reaching subnanomolar values (IC50 5 0.27 nM), al-though its b selectivity decreased slightly (b/a5 39) compared with its cyano-substitutedanalogs, 141 and 142. ERb-selective agonist properties were verified for the compound dis-playing the highest b selectivity (141), which showed good activity in two rat models ofinflammation, inflammatory bowel disease, and arthritis. The absence of concomitant uter-otrophic effects after administration of 141 confirmed its negligible ERa activity in vivo. Thiscompound was recently patented, together with benzoxazole ERB-041 (4, Fig. 3) andnaphthalene WAY-202196 (81, Fig. 25), for the treatment of acute lung injuries.121
A further development of this class of ER ligands consisted of a change in the position ofthe pyran ring within the tetracyclic system, which led to a series of dibenzo[c,g]chromenes,whose most interesting members (144–146) are illustrated in Figure 45.122
The unsubstituted derivative 144 displayed a moderate b selectivity in binding assays,with an 8-fold preference for this subtype over ERa, and a good level of ERb-binding affinity(IC50 5 3.3 nM). Introduction of a chlorine atom produced compound 145, endowedwith excellent affinity (IC50 5 0.53 nM) and good selectivity (b/a5 39) for the b subtype.The pyridine-substituted analog 146 showed nearly a complete loss of binding for
OH
HO
144
O
OH
HO
145
O
OH
HO
146
O
Cl
N
Figure 45. Substituted dibenzo[c,g]chromenes 144--146.
OHO
OH
140
OHO
OH
141
CN
142
OHO
143
CN OH
OHO
OHOMe
Cl
Figure 44. Substituted 6H-dibenzo[c,h]chromenes 140--143.
400 K MINUTOLOETAL.
Medicinal Research Reviews DOI 10.1002/med
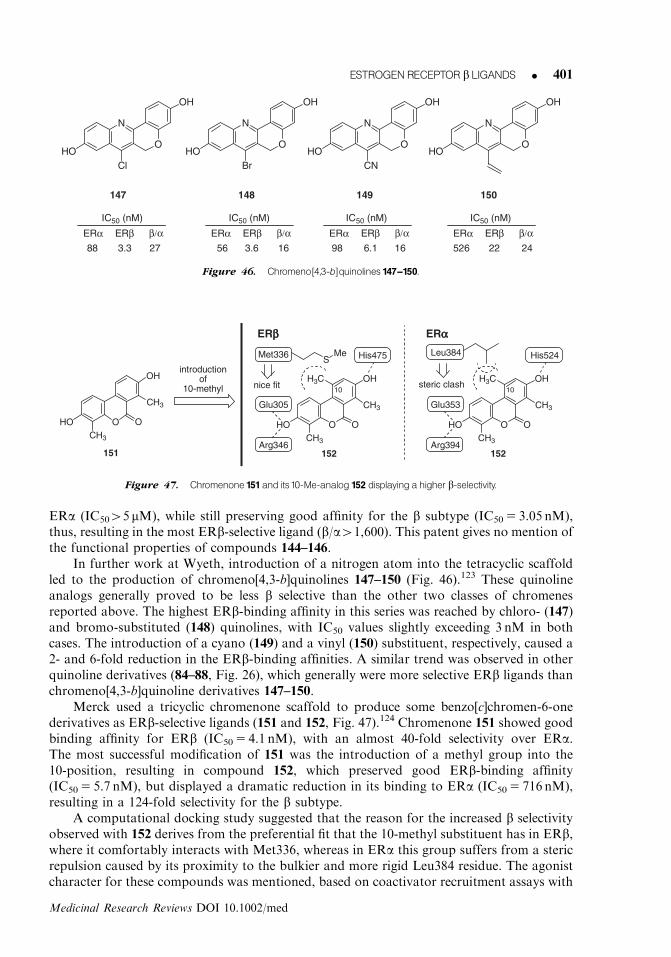
ERa (IC5045 mM), while still preserving good affinity for the b subtype (IC50 5 3.05 nM),thus, resulting in the most ERb-selective ligand (b/a41,600). This patent gives no mention ofthe functional properties of compounds 144–146.
In further work at Wyeth, introduction of a nitrogen atom into the tetracyclic scaffoldled to the production of chromeno[4,3-b]quinolines 147–150 (Fig. 46).123 These quinolineanalogs generally proved to be less b selective than the other two classes of chromenesreported above. The highest ERb-binding affinity in this series was reached by chloro- (147)and bromo-substituted (148) quinolines, with IC50 values slightly exceeding 3 nM in bothcases. The introduction of a cyano (149) and a vinyl (150) substituent, respectively, caused a2- and 6-fold reduction in the ERb-binding affinities. A similar trend was observed in otherquinoline derivatives (84–88, Fig. 26), which generally were more selective ERb ligands thanchromeno[4,3-b]quinoline derivatives 147–150.
Merck used a tricyclic chromenone scaffold to produce some benzo[c]chromen-6-onederivatives as ERb-selective ligands (151 and 152, Fig. 47).124 Chromenone 151 showed goodbinding affinity for ERb (IC50 5 4.1 nM), with an almost 40-fold selectivity over ERa.The most successful modification of 151 was the introduction of a methyl group into the10-position, resulting in compound 152, which preserved good ERb-binding affinity(IC50 5 5.7 nM), but displayed a dramatic reduction in its binding to ERa (IC50 5 716 nM),resulting in a 124-fold selectivity for the b subtype.
A computational docking study suggested that the reason for the increased b selectivityobserved with 152 derives from the preferential fit that the 10-methyl substituent has in ERb,where it comfortably interacts with Met336, whereas in ERa this group suffers from a stericrepulsion caused by its proximity to the bulkier and more rigid Leu384 residue. The agonistcharacter for these compounds was mentioned, based on coactivator recruitment assays with
N
OH
HOO
Cl
941841741
IC50 (nM)
ER ER
88 3.3 27
ER ER
56 3.6 16
ER ER
98 6.1 16
IC50 (nM) IC50 (nM)
150
ER ER
526 22 24
IC50 (nM)
N
OH
HOO
Br
N
OH
HOO
CN
N
OH
HOO
Figure 46. Chromeno[4,3-b]quinolines 147--150.
OHO O
151
OH
CH3
OHO O
152
OH
CH3
H3C
CH3CH3
Leu384SMet336 Me
Glu305
Arg346
His475
nice fit steric clash
OHO O
OH
CH3
H3C
CH3Glu353
Arg394
His524
ERER
152
0101
introductionof
10-methyl
Figure 47. Chromenone 151and its10-Me-analog 152 displayingahigher b-selectivity.
ESTROGENRECEPTOR bLIGANDS K 401
Medicinal Research Reviews DOI 10.1002/med

ERb, but no information on the other receptor subtype was mentioned, so the functionalselectivity cannot be estimated.
9. BenzoxepinsSeven-membered rings were introduced in the central scaffold of ERb ligands, such as thosebelonging to the benzoxepin class (153–155, Fig. 48), recently reported by researchers at theTrinity College of Dublin.125
These compounds were designed as selective modulators of the ERs. Compound 153
showed good binding affinity for ERb (IC50 5 2.9 nM), but its selectivity was not very high(b/a5 3). Replacement of one phenolic OH group with a fluorine atom led to compound 154,which had the highest affinity for ERb (IC50 5 0.70 nM) and the best b/a selectivity ratio (15-fold). A shift of the position of the remaining phenolic group of 154 from the para to the metaposition, afforded a compound (155) that lost some of its ERb-binding properties (IC50 5
6.0 nM) and was also less selective (b/a5 2.3). A computational docking analysis of thesecompounds revealed that their b selectivity may be attributed to a differential interaction thatthe ethylene portion of the oxazepine seven-member ring has with the two receptor subtypes,the steric interaction with the residue Met421 of ERa being more pronounced than with thecorresponding residue (Ile373) in ERb. An interesting observation was the interaction of thearyl-fluoride moiety of 154 with the two residues forming the H-bond network (Arg436 andGlu305). Remarkably, the fluorine atom proved to be not only tolerated, but even verybeneficial in terms of b selectivity. Attention should be given to this important findingbecause this replacement might also be important for metabolic stability, because an aro-matic F-atom generally retards metabolic oxidation, extending plasma half-life.
Cell-based proliferation assays were run on MCF-7 breast cancer cells, expressing mostlyERa, and Ishikawa cells, which are human endometrial adenocarcinoma cells having bothERa and ERb. The most interesting compound, 154, showed an antiproliferative effect onMCF-7 cells, with an IC50 value of 21.4 nM, most likely due to antagonism through ERa.The same compound antagonized E2 stimulation of Ishikawa cells with an IC50 of 194 nM,and had a residual stimulation of less than 10% at 1 mM. These data alone are not sufficientto establish the exact pharmacological character of 154, but selective ERb agonism may notbe excluded a priori.
10. BenzoxazinsInvestigators at Bristol-Myers Squibb reported some racemic 3-arylbenzoxazines (156–158,Table VI) possessing ERb-agonist properties.126
These compounds were not extremely strong binders for ERb (IC50490 nM), but theywere quite potent in transcriptional assays. In particular, the 2-ethyl-5-methyl-substitutedderivative 158 displayed an EC50 of 8 nM for ERb with a b/a selectivity ratio of 21. The effectof chirality on C2 was only investigated on compound 159, whose enantiomers were
OHO
OH
ON
OF
OH
ON
OF
ON
551451351
OH
Figure 48. ERb-ligands based ona 7-memberoxazepin ring 153--155.
402 K MINUTOLOETAL.
Medicinal Research Reviews DOI 10.1002/med

separated and independently assayed, although their absolute configurations were notidentified. Binding affinity depended on the absolute configuration of the 2-position to somedegree, as shown by compounds (R�)- and (S�)-159, but potency in transcriptional activationwas hardly affected.
A computational docking analysis of the most interesting compound (158) indicated thatthe OH group of the benzoxazine scaffold interacts with His524(a)/475(b), and the otherhydroxyl participates in the H-bond network with Glu353(a)/305(b) and Arg394(a)/346(b).The b selectivity appears to depend on the substituent in the 5-position and when R1 5Me,the 5-methyl group of 158 is too close to the Met421 residue of ERa, whereas it fits nicely inproximity of the Ile373 residue of ERb.
11. Quinazolines and quinazolinethionesA series of 3-aryl-quinazolinones and quinazolinethiones was developed at Bristol-Myers Squibb,including a pair of derivatives showing some preference for ERb (160 and 161, Fig. 49).127
Quinazolinone 160 showed moderate ERb-binding affinity (IC50 5 179 nM) with a good62-fold selectivity over the a subtype. Replacement of the oxygen with a sulfur atom, leadingto quinazolinethione 161, produced what the authors describe as the ‘‘thio effect,’’ resultingin an improvement in ERb binding (IC50 5 47 nM) and a substantial preservation of the bselectivity (b/a5 56). The ‘‘thio’’-derivative 161 also proved to be a selective b activator intransactivation assays, with an EC50 of 13 nM for ERb and 2793 nM for ERa; however, the
Table VI. Binding Affinities and Functional Properties of Benzoxazine Derivatives 156–159
O
NR1
OHR2
HO
156-159
ER binding (IC50, nM) ER transactivation (EC50, nM)
Cpd R1 R2 hERa hERb b/a sel. hERa hERb b/a sel.
156 OH Et 1200 120 10 170 26 6.5
157 Me Me 320 90 3.6 46 16 2.9
158 Me Et 180 90 2.0 170 8 21
159 OH Me 2900 310 9.4 150 47 3.2
(R�)-159 OH Me 2800 240 12 140 50 2.8
(S�)-159 OH Me 6300 1100 5.7 290 74 3.9
ER, estrogen receptor; ERb, estrogen receptor b; ERa, estrogen receptor a.
N
N
OH
OH
OHO
160
N
N
OH
OH
SHO
161
Figure 49. Quinazolinone 160 and quinazolinethione 161exhibiting ERb-binding preference.
ESTROGENRECEPTOR bLIGANDS K 403
Medicinal Research Reviews DOI 10.1002/med

EC50 value in ERb transactivation represents partial agonism, because this compound didnot reach 100% activity even at high concentrations (up to 10 mM).
The probable binding mode of these compounds with the ERs was deduced bymolecular modeling studies that revealed a mode similar to that found for benzoxazines(see Section B.10).126
12. IsocoumarinsA series of isocoumarins, inspired by the ERb-selective isoflavone phytoestrogens, was re-ported by Katzenellenbogen and co-workers, and some of them (162–165, Table VII) showednoticeable ERb selectivities in both binding affinity and transcriptional potency.128
These isocoumarins differ by the substituent in the 5-position. The 5-bromo-derivative(162) was the one showing the highest ERb-binding affinity (RBA5 129%), and its chloro-analogue 163 was only a slightly poorer b ligand. Introduction of a 5-methyl afforded themost b-selective ligand (164) in terms of binding affinity (ERb-RBA5 24%, b/a5 40). Theethyl-analogue 165 had lower ERb-binding affinity and selectivity. These compoundsbehaved as partial agonists for ERb with activation levels at 1 mM ranging from 70 to 90%when compared with 10 nM E2. Their functional selectivities were comparable to theirbinding selectivities.
Table VII. Relative Binding Affinities of Isocoumarins 162–165
ER binding (RBA, %)
Cpd hERa hERb b/a sel.
OOH
O
BrHO
162
16.2 129 7.9
OOH
O
ClHO
163
9.8 63 6.4
OOH
O
MeHO
164
0.59 23.9 40
OOH
O
EtHO
165
5.0 16.0 3.0
ER, estrogen receptor; ERb, estrogen receptor b; ERa, estrogen receptor a; RBA, relative bindingaffinity.
404 K MINUTOLOETAL.
Medicinal Research Reviews DOI 10.1002/med
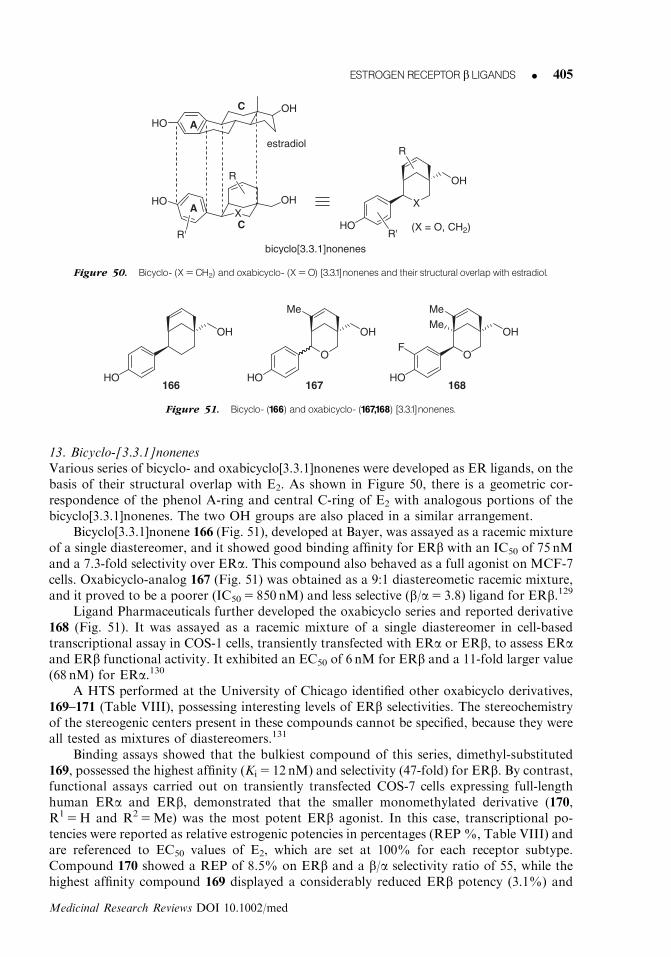
13. Bicyclo-[3.3.1]nonenesVarious series of bicyclo- and oxabicyclo[3.3.1]nonenes were developed as ER ligands, on thebasis of their structural overlap with E2. As shown in Figure 50, there is a geometric cor-respondence of the phenol A-ring and central C-ring of E2 with analogous portions of thebicyclo[3.3.1]nonenes. The two OH groups are also placed in a similar arrangement.
Bicyclo[3.3.1]nonene 166 (Fig. 51), developed at Bayer, was assayed as a racemic mixtureof a single diastereomer, and it showed good binding affinity for ERb with an IC50 of 75 nMand a 7.3-fold selectivity over ERa. This compound also behaved as a full agonist on MCF-7cells. Oxabicyclo-analog 167 (Fig. 51) was obtained as a 9:1 diastereometic racemic mixture,and it proved to be a poorer (IC50 5 850 nM) and less selective (b/a5 3.8) ligand for ERb.129
Ligand Pharmaceuticals further developed the oxabicyclo series and reported derivative168 (Fig. 51). It was assayed as a racemic mixture of a single diastereomer in cell-basedtranscriptional assay in COS-1 cells, transiently transfected with ERa or ERb, to assess ERaand ERb functional activity. It exhibited an EC50 of 6 nM for ERb and a 11-fold larger value(68 nM) for ERa.130
A HTS performed at the University of Chicago identified other oxabicyclo derivatives,169–171 (Table VIII), possessing interesting levels of ERb selectivities. The stereochemistryof the stereogenic centers present in these compounds cannot be specified, because they wereall tested as mixtures of diastereomers.131
Binding assays showed that the bulkiest compound of this series, dimethyl-substituted169, possessed the highest affinity (Ki 5 12 nM) and selectivity (47-fold) for ERb. By contrast,functional assays carried out on transiently transfected COS-7 cells expressing full-lengthhuman ERa and ERb, demonstrated that the smaller monomethylated derivative (170,R1 5H and R2 5Me) was the most potent ERb agonist. In this case, transcriptional po-tencies were reported as relative estrogenic potencies in percentages (REP %, Table VIII) andare referenced to EC50 values of E2, which are set at 100% for each receptor subtype.Compound 170 showed a REP of 8.5% on ERb and a b/a selectivity ratio of 55, while thehighest affinity compound 169 displayed a considerably reduced ERb potency (3.1%) and
HOOH
XOHHO
R
A
A
C
CR'
X
OH
HO
R
R'(X = O, CH2)
estradiol
bicyclo[3.3.1]nonenes
Figure 50. Bicyclo- (X5CH2) and oxabicyclo- (X5O) [3.3.1]nonenes and their structural overlap with estradiol.
OH
HO166
O
OH
HO167
Me
O
OH
HO168
MeMe
F
Figure 51. Bicyclo- (166) and oxabicyclo- (167,168) [3.3.1]nonenes.
ESTROGENRECEPTOR bLIGANDS K 405
Medicinal Research Reviews DOI 10.1002/med
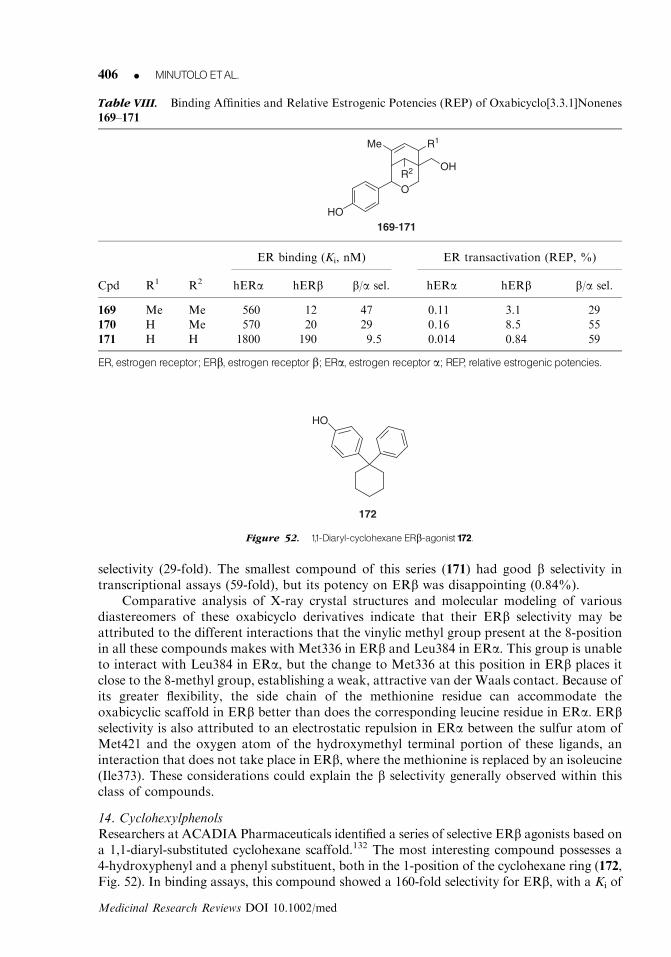
selectivity (29-fold). The smallest compound of this series (171) had good b selectivity intranscriptional assays (59-fold), but its potency on ERb was disappointing (0.84%).
Comparative analysis of X-ray crystal structures and molecular modeling of variousdiastereomers of these oxabicyclo derivatives indicate that their ERb selectivity may beattributed to the different interactions that the vinylic methyl group present at the 8-positionin all these compounds makes with Met336 in ERb and Leu384 in ERa. This group is unableto interact with Leu384 in ERa, but the change to Met336 at this position in ERb places itclose to the 8-methyl group, establishing a weak, attractive van der Waals contact. Because ofits greater flexibility, the side chain of the methionine residue can accommodate theoxabicyclic scaffold in ERb better than does the corresponding leucine residue in ERa. ERbselectivity is also attributed to an electrostatic repulsion in ERa between the sulfur atom ofMet421 and the oxygen atom of the hydroxymethyl terminal portion of these ligands, aninteraction that does not take place in ERb, where the methionine is replaced by an isoleucine(Ile373). These considerations could explain the b selectivity generally observed within thisclass of compounds.
14. CyclohexylphenolsResearchers at ACADIA Pharmaceuticals identified a series of selective ERb agonists based ona 1,1-diaryl-substituted cyclohexane scaffold.132 The most interesting compound possesses a4-hydroxyphenyl and a phenyl substituent, both in the 1-position of the cyclohexane ring (172,Fig. 52). In binding assays, this compound showed a 160-fold selectivity for ERb, with a Ki of
Table VIII. Binding Affinities and Relative Estrogenic Potencies (REP) of Oxabicyclo[3.3.1]Nonenes
169–171
O
OH
HO169-171
Me
R2
R1
ER binding (Ki, nM) ER transactivation (REP, %)
Cpd R1 R2 hERa hERb b/a sel. hERa hERb b/a sel.
169 Me Me 560 12 47 0.11 3.1 29
170 H Me 570 20 29 0.16 8.5 55
171 H H 1800 190 9.5 0.014 0.84 59
ER, estrogen receptor; ERb, estrogen receptor b; ERa, estrogen receptor a; REP, relative estrogenic potencies.
HO
172
Figure 52. 1,1-Diaryl-cyclohexane ERb-agonist 172.
406 K MINUTOLOETAL.
Medicinal Research Reviews DOI 10.1002/med

50nM. This b selectivity was even more pronounced (800-fold) in functional luciferase reportergene assays, where it showed an EC50 value for ERb of 20nM and a maximum stimulation of80%; for ERa, the EC50 was found to be around 16mM, with an efficacy of 58%.133
Compound 172 proved to be active in vivo as a modulator of neuropathic pain133 as wellas of inflammatory pain, presumably by stimulating ERb.134
15. FuransZimmermann and von Angerer at the University of Regensburg demonstrated that theycould transform an ERa-selective ligand, such as 1,5-diarylsubstituted furan 173 (Fig. 53),into b-selective ligands, simply by shifting the 5-aryl group to the 4-position.135 In thismanner, they were able to obtain two 1,4-diarylsubstituted furans (174 and 175, Fig. 53)exhibiting a preference for ERb. In particular, furan 174 showed the highest b selectivity inbinding assays (b/a5 21), although its ERb-binding affinity was only moderate (RBA5
1.5%). Introduction of two methyl groups at C3 and C5 of the furan ring produced acompound (175) having considerably increased ERb-binding affinity (RBA5 76%),although with very modest ER subtype selectivity (b/a5 3.6).
Overall, these diaryl-substituted furans did not reach the same level of ERb-selective bindingaffinity displayed by their benzo-condensed analogues (see benzofurans 55–61, Table III).83
16. PyrimidinesKatzenellenbogen and co-workers at the University of Illinois studied various series ofdiazenes and found some ERb-selective ligands within the 2,5-diaryl-substituted pyrimidineclass (176–178, Fig. 54).136
571471371
RBA (%)
ERα ERβ β/α23 7.1 0.31
ERα ERβ β/α0.07 1.5 21
ERα ERβ β/α21 76 3.6
RBA (%) RBA (%)
O
OH
HOO
OH
HO
Me
MeO
Et
HO
Et
OH
Figure 53. Furan-based ER-ligands: Switch of subtype-selectivity from1,5- (173) to1,4-diarylsubstituted (174,175) furans.
871771671
RBA (%)
ERα ERβ β/α0.04 0.30 7.5
ERα ERβ β/α0.93 2.5 2.7
ERα ERβ β/α5.1 11 2.1
)%(ABR)%(ABR
N
N
Me
Me
OH
HO
N
N
Prn
Me
OH
HO
N
N
Et
Et
OH
HO
Figure 54. 2,5-Diarylpyrimidines (176--178) endowed of affinity for ERb.
ESTROGENRECEPTOR bLIGANDS K 407
Medicinal Research Reviews DOI 10.1002/med

4,6-Dimethyl-substituted pyrimidine 176 possesses the highest degree of b selectivity (b/aratio5 7.5), but its affinity for ERb was only modest (RBA5 0.30%). Introduction of morelipophilic substituents, such as an n-propyl (177) or two ethyl groups (178), generally in-creased the binding affinity for both receptor subtypes, but reduced their subtype selectivities.In fact, compound 178, which had the highest ERb-binding affinity (RBA5 11%), showedonly a 2-fold preference for this receptor over ERa. These pyrimidines proved to be partialagonists for ERb, with 40-50% activation at 1 mM.
17. TriazolesA series of 1,4-diaryl-substituted 1,2,3-triazoles was recently synthesized by Tron and co-workers using an application of the Sharpless modification of the Huisgen reaction, whichinvolves a facile [312] cycloaddition between an azide and an alkyne in the presence of acopper(I)-based catalyst (Fig. 55).137 This method is also known as ‘‘click chemistry,’’ be-cause of its simplicity and efficiency. The triazoles produced were submitted to cell-basedassays to define their possible estrogenic character. The most interesting triazole possessestwo meta-OH groups in the aryl substituents (179, Fig. 55); a concentration as low as 100 pMof 179 was reported to promote transcriptional activation in HeLa cells, mostly expressingERb, at the same level as that caused by 100 nM E2. The same compound, however, alsostimulated proliferation of MCF-7 cells (mostly expressing ERa), although at higher con-centrations. These data show that 179 may be an ERb agonist, but its subtype selectivityneeds to be more clearly established. Moreover, receptor-binding assays have not been re-ported yet for these types of compounds, but the significant dipole moment of the triazolecore suggests that binding affinities might not be very high.138,139
18. DihydroisoxazolesA research group at the University of Kuopio (Finland) has developed a series of4,5-dihydroisoxazoles having some interesting estrogenic activity.140 The racemic form ofcompound 180 and the two enantiomers of compound 181 (Fig. 56), whose absolute con-figurations were not established, were assayed for ERa and ERb activation.
The activity of 10 mM racemic 180, relative to 10 nM E2, was 19.6% on ERa and 34.4%on ERb. A similar 2-fold activity preference for ERb was found with 10 mM of one of the twoenantiomers of 181, which showed an activation profile (24.3% on ERa and 41.5% on ERb)
NHO
NNOH
N
NNHO
OHN3
HO OH
Cu(I)
179
Figure 55. ‘‘Click chemistry’’ leading to1,4-diaryl-1,2,3-triazoles, including possible ERb-agonist 179.
ON
*
180
ON
181 (ent-a, ent-b)
HO
ON
HO
Figure 56. Dihydroisoxazoles 180 and 181 (separate enantiomers of unknown configuration).
408 K MINUTOLOETAL.
Medicinal Research Reviews DOI 10.1002/med

similar to that of racemic 180. The other enantiomer of 181 proved to be a more potentactivator for both subtypes (171 and 143% on ERa and ERb, respectively), but it lost activitypreference for ERb.
19. DiphenylaminesInvestigators at Tohoku Pharmaceutical University studied ER ligands based on a diphe-nylamine skeleton. The structural motif that guaranteed good binding affinities for the ERsincluded the presence of para-hydroxyls in both the aryl substituents (182–184, Fig. 57).141
N-benzyl-substituted derivative 182 showed the highest and most selective binding affinityfor ERb (RBA5 18%, b/a5 28). The replacement of the N-benzyl with an isopentyl sub-stituent (183) reduced ERb-binding affinity considerably, although reasonable ERb selectivitywas retained. ERb-binding affinity and selectivity were further reduced by transformation ofthe central nitrogen atom from an amino- into an amido-type, as in N-benzoyl derivative 184.
The functional estrogenic property of these compounds was measured in cell prolifera-tion assays using human breast cancer MCF-7 cells. Compounds having three aryl groupswere generally agonists (182, EC50 5 67 nM, EMAX 5 85% and 184, EC50 5 14 nM, EMAX 5 114%), whereas compound 183, having only two aryl substituents, had considerably reducedestrogenic activity (EC50 5 570 nM, EMAX 5 31 %). Because MCF-7 cells have only ERa, thisassay is not suitable for assessing ER subtype-selective activity.
20. SulfonamidesDiarylsubstituted benzenesulfonamides showing interesting estrogenic profiles were recentlyincluded in a patent by Katzenellenbogen and co-workers. Derivatives bearing two para-OHgroups in both aryl substituents displayed the highest levels of ER-binding affinities(185–188, Fig. 58).142
481381281
RBA (%)
ERα ERβ β/α
0.66 18.1 28
ERα ERβ β/α
0.13 2.34 18
ERα ERβ β/α
0.19 1.26 6.5
)%(ABR)%(ABR
N
OHHO
N
OHHO
N
OHHO
O
Figure 57. Diphenylamines bearing various N-substituents (182--184).
185
RBA (%)
ERα ERβ β/α0.009 0.058 6.4
ERα ERβ β/α0.026 2.56 98
ERα ERβ β/α0.006 0.97 162
RBA (%) RBA (%)
NS
CH3
HO
OH
O O
186
NS
HO
OH
O O
187
NS
HO
OH
O O
ERα ERβ β/α0.006 1.27 212
RBA (%)
188
NS
HO
OH
O O
*F3C
Figure 58. Benzenesulfonamides bearing various N-substituents (185--188).
ESTROGENRECEPTOR bLIGANDS K 409
Medicinal Research Reviews DOI 10.1002/med

Substantial differences were found in the receptor-binding affinities of members of thisclass of compounds, depending on the substituent on the nitrogen atom. The smallestmember of the series, N-methyl-substituted sulfonamide 185, was a weak ligand for bothERs, whereas elongation of the N-substituent to an n-propyl chain afforded a good ERbligand (186), having an RBA of 2.56% and a 100-fold selectivity over ERa. The introductionof a sec-butyl substituent gave compound 187, which was assayed as a racemic mixture andshowed greater b selectivity (b/a5 162), although its ERb-binding affinity was quite low(RBA5 0.97%). The highest ERb selectivity was found with the fluoroalkylated derivative188, which has a b/a selectivity ratio of 212 and a decent ERb-RBA value of 1.27%. This lastcompound also proved to be a selective agonist for ERb in functional assays: a 1.0 mMsolution of this compound activated ERb to a level 130% that of E2, with an approximateEC50 value in the low tens of nanomolar. Its functional ERb/ERa subtype potency selectivityis around 10-fold.
21. Bibenzyls, stilbenes, and phenethyl pyridinesDe Angelis and Katzenellenbogen found that deoxyhexestrol (189, Fig. 59) had a goodaffinity for ERb (RBA5 73%), being more ERb preferential (4-fold over ERa) than itsstructurally related nonsteroidal estrogen congeners hexestrol and diethylstilbestrol. Thisfinding inspired the development of nitrogen-containing analogs, derived by replacement ofthe phenol ring of 189 with pyridine and pyrimidine portions.143 The pyridine series containssome derivatives showing significant binding affinity preferences for the b subtype, such as190 and 191 (Fig. 59).
Pyridine derivative 190 was evaluated as a racemic mixture of the erythro-diastereomer,thus, closely resembling the relative configurations of deoxyhexestrol. However, it showed adramatic reduction in ERb-binding affinity. By contrast, its threo-analog 191 showed someaffinity for ERb (RBA5 0.684%) and, notably, its b/a selectivity ratio of 8.4 is considerablyhigher than that of 189.
The same research group also developed bibenzyl- and stilbene-based derivatives,structurally similar to hexestrol (192, Fig. 60). Monoethyl-substituted stilbene analog 193
proved to be the best and most selective ERb ligand, with RBA values of 76% for theb subtype and 4.09% for ERa (b/a5 19). Curiously, functional assays revealed an oppositesubtype potency selectivity for this compound (ERa: EC50 5 0.52 nM; ERb: EC50 5
2.21 nM). Racemic monopropyl-substituted bibenzyl derivative 194 had a very high affinityfor ERb (RBA5 68%, b/a ratio5 21). Nevertheless, it also was nonselective in functionalassays, having EC50 values of about 2 nM for both receptor subtypes. The most selectivebiphenyl analog was compound 195, possessing a gem-dimethyl unit. Its ERb-bindingselectivity was remarkably high (b/a5 125) and its affinity for the same subtype was quite
189
RBA (%)
ERα ERβ β/α1873 4.1
ERα ERβ β/α0.010 0.023 2.3
ERα ERβ β/α
0.081 0.684 8.4189
)%(ABR)%(ABR
HO
191091
H
HN
HO
H
HN
HO
H
H
deoxyhexestrol rac -erythro rac -threo
Figure 59. Pyridine analogs (190,191) of deoxyhexestrol (189).
410 K MINUTOLOETAL.
Medicinal Research Reviews DOI 10.1002/med
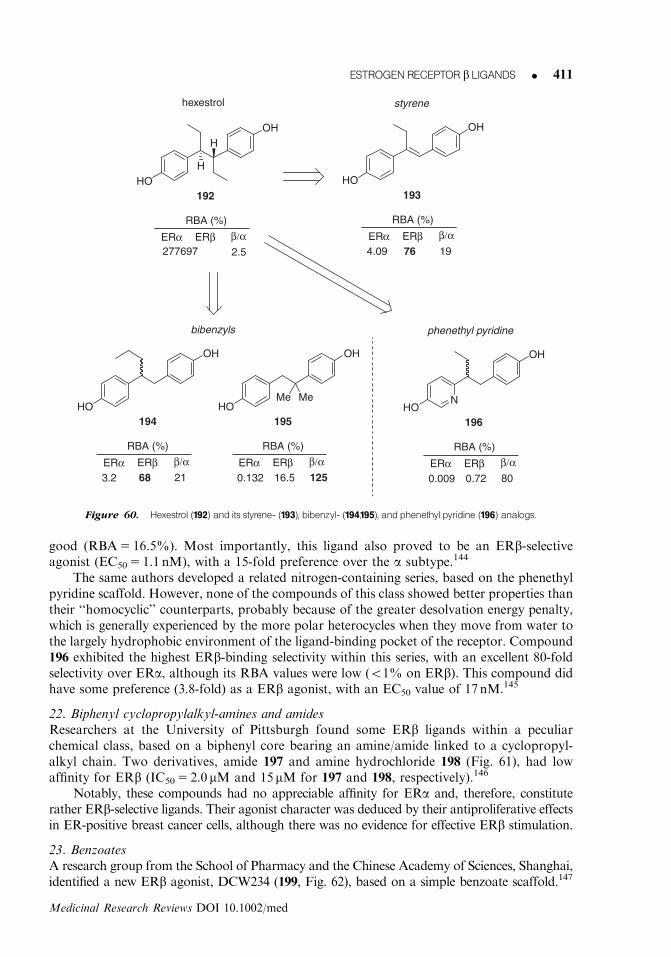
good (RBA5 16.5%). Most importantly, this ligand also proved to be an ERb-selectiveagonist (EC50 5 1.1 nM), with a 15-fold preference over the a subtype.144
The same authors developed a related nitrogen-containing series, based on the phenethylpyridine scaffold. However, none of the compounds of this class showed better properties thantheir ‘‘homocyclic’’ counterparts, probably because of the greater desolvation energy penalty,which is generally experienced by the more polar heterocycles when they move from water tothe largely hydrophobic environment of the ligand-binding pocket of the receptor. Compound196 exhibited the highest ERb-binding selectivity within this series, with an excellent 80-foldselectivity over ERa, although its RBA values were low (o1% on ERb). This compound didhave some preference (3.8-fold) as a ERb agonist, with an EC50 value of 17nM.145
22. Biphenyl cyclopropylalkyl-amines and amidesResearchers at the University of Pittsburgh found some ERb ligands within a peculiarchemical class, based on a biphenyl core bearing an amine/amide linked to a cyclopropyl-alkyl chain. Two derivatives, amide 197 and amine hydrochloride 198 (Fig. 61), had lowaffinity for ERb (IC50 5 2.0 mM and 15 mM for 197 and 198, respectively).146
Notably, these compounds had no appreciable affinity for ERa and, therefore, constituterather ERb-selective ligands. Their agonist character was deduced by their antiproliferative effectsin ER-positive breast cancer cells, although there was no evidence for effective ERb stimulation.
23. BenzoatesA research group from the School of Pharmacy and the Chinese Academy of Sciences, Shanghai,identified a new ERb agonist, DCW234 (199, Fig. 62), based on a simple benzoate scaffold.147
192
RBA (%)
ERα ERβ β/α277697 2.5
ERα ERβ β/α4.09 76 19
RBA (%)
HO193
H
HHO
hexestrol styrene
OH
ERα ERβ β/α3.2 68 21
RBA (%)
194HO
bibenzyls
ERα ERβ β/α0.132 16.5 125
RBA (%)
195HO
OH
OH OH
Me Me
ERα ERβ β/α0.009 0.72 80
RBA (%)
196
NHO
phenethyl pyridine
OH
Figure 60. Hexestrol (192) and its styrene- (193), bibenzyl- (194,195), and phenethyl pyridine (196) analogs.
ESTROGENRECEPTOR bLIGANDS K 411
Medicinal Research Reviews DOI 10.1002/med

The receptor-binding affinities of this compound were determined by surface plasmonresonance (SPR) technology, which showed some preference for ERb (KD 5 3.43 mM) overERa (KD 5 22.5 mM), although affinity levels were poor. Cell-based transcriptional assaysshowed that 199 was an agonist with good b selectivity, although potencies were weak, beingonly in the micromolar range (EC50 5 2.5 mM for ERb and 32.7 mM for ERa).
24. Compounds derived by direct modifications of natural productsA series of tetrahydroquinolines, structurally derived from equol (see also 45, Fig. 17), werereported to possess interesting ERb-binding properties.148 These compounds were formallyobtained by replacing the oxygen atom of the pyran ring of equol with a nitrogen atom(Fig. 63). These aza analogs of equol preserved the di-hydroxyphenyl pattern of the originalmolecule, but could be further functionalized on the newly inserted nitrogen atom, thus,producing a series of variously N-substituted derivatives, among which some interestingb-selective ligands, tested as racemic mixtures, emerged (200–203, Fig. 63). The N-ethyl-substituted derivative 200 showed the highest binding affinity for ERb (IC50 5 0.083 nM) anda good selectivity ratio (b/a5 17), whereas the most b-selective ligand was the N-mesylatedanalog 201, which displayed a 62-fold selectivity over ERa and a noticeable affinity for ERb(IC50 5 0.52 nM). Variations of the N-alkyl chain such as in 202 (n-propyl) and 203 (cyclo-propylmethyl) did not significantly improve their ERb-binding properties, compared withtheir N-ethylated counterpart 200. Functional assays of 200 revealed that this compound is aselective ERb agonist, with EC50 values of 0.58 mM for ERb and 24 mM for ERa, corre-sponding to a selectivity ratio of 41. Curiously, the other two N-alkylated derivatives 202 and203 showed agonist properties on ERb, with EC50 values in the low micromolar range,whereas they behave as antagonists on ERa (IC50 of 2.7 mM for 202 and 5.6 mM for 203).
A 7-hydroxycoumarin (204, Fig. 64), formally constituting an ‘‘open’’ version of cou-mestrol (see also 1, Fig. 2), had modest selectivity for ERb in binding assays (RBA5 0.1%for ERb and 0.02% for ERa) and considerably lower selectivity than that of coumestrolitself.149
Naringenin (39, Fig. 16) was also a starting point for structural modifications designed toimprove its ERb-binding properties. Some members of a series of variously 8-substitutedracemic analogs of naringenin reached good levels of ERb-binding affinity and selectivity
HO
F
NH
O
Ph
197HO
F
NH2·HCl
198
Figure 61. Biphenyl cyclopropylbutyl amide 197 andamine hydrochloride 198.
O
OO
199(DCW234)
Figure 62. Unusual structural motif for ERb-ligands: a benzoate (199).
412 K MINUTOLOETAL.
Medicinal Research Reviews DOI 10.1002/med
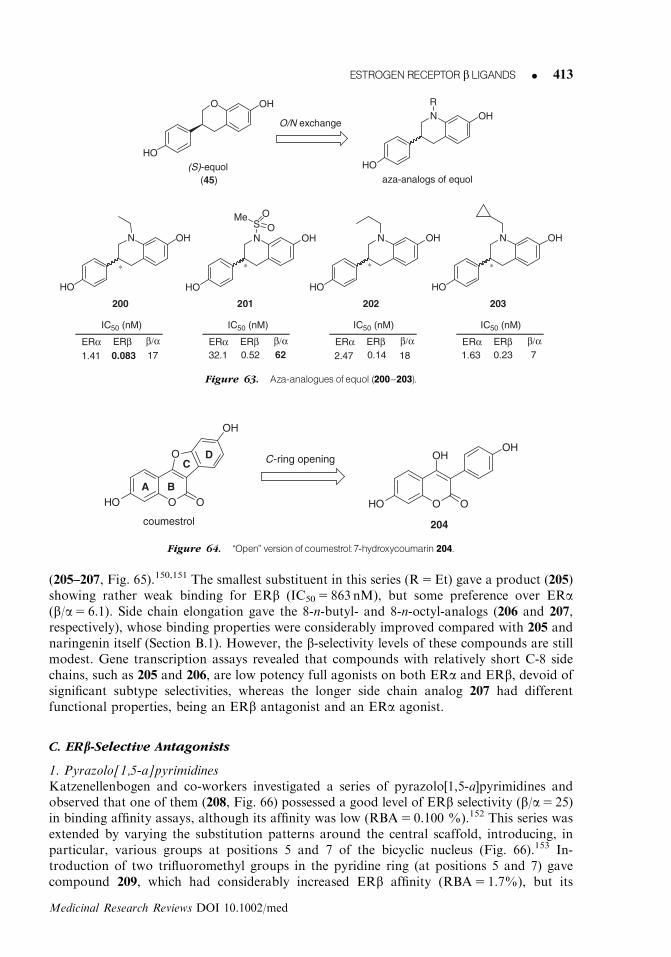
(205–207, Fig. 65).150,151 The smallest substituent in this series (R5Et) gave a product (205)showing rather weak binding for ERb (IC50 5 863 nM), but some preference over ERa(b/a5 6.1). Side chain elongation gave the 8-n-butyl- and 8-n-octyl-analogs (206 and 207,respectively), whose binding properties were considerably improved compared with 205 andnaringenin itself (Section B.1). However, the b-selectivity levels of these compounds are stillmodest. Gene transcription assays revealed that compounds with relatively short C-8 sidechains, such as 205 and 206, are low potency full agonists on both ERa and ERb, devoid ofsignificant subtype selectivities, whereas the longer side chain analog 207 had differentfunctional properties, being an ERb antagonist and an ERa agonist.
C. ERb-Selective Antagonists
1. Pyrazolo[1,5-a]pyrimidinesKatzenellenbogen and co-workers investigated a series of pyrazolo[1,5-a]pyrimidines andobserved that one of them (208, Fig. 66) possessed a good level of ERb selectivity (b/a5 25)in binding affinity assays, although its affinity was low (RBA5 0.100 %).152 This series wasextended by varying the substitution patterns around the central scaffold, introducing, inparticular, various groups at positions 5 and 7 of the bicyclic nucleus (Fig. 66).153 In-troduction of two trifluoromethyl groups in the pyridine ring (at positions 5 and 7) gavecompound 209, which had considerably increased ERb affinity (RBA5 1.7%), but its
200
IC50 (nM)
ERα ERβ β/α1.41 0.083 17
ERα ERβ β/α32.1 0.52 62
ERα ERβ β/α2.47 0.14 18
OH
HO
202102
ERα ERβ β/α1.63 0.23 7
203
N OH
HO
NS
MeO
O
OH
HO
N OH
HO
N
IC50 (nM) IC50 (nM) IC50 (nM)
OH
HO
OOH
HO
NR
(S)-equolaza-analogs of equol
O/N exchange
(45)
Figure 63. Aza-analogues of equol (200--203).
204
C-ring opening
O
O
O
OH
HO
coumestrol
A B
CD
O OHO
OHOH
Figure 64. ‘‘Open’’ version of coumestrol: 7-hydroxycoumarin 204.
ESTROGENRECEPTOR bLIGANDS K 413
Medicinal Research Reviews DOI 10.1002/med
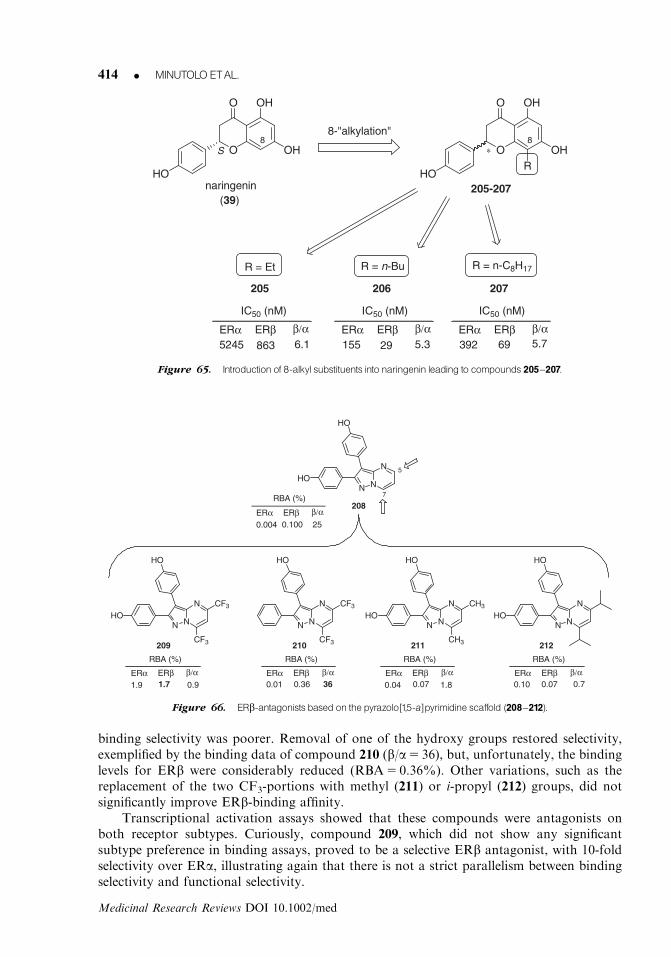
binding selectivity was poorer. Removal of one of the hydroxy groups restored selectivity,exemplified by the binding data of compound 210 (b/a5 36), but, unfortunately, the bindinglevels for ERb were considerably reduced (RBA5 0.36%). Other variations, such as thereplacement of the two CF3-portions with methyl (211) or i-propyl (212) groups, did notsignificantly improve ERb-binding affinity.
Transcriptional activation assays showed that these compounds were antagonists onboth receptor subtypes. Curiously, compound 209, which did not show any significantsubtype preference in binding assays, proved to be a selective ERb antagonist, with 10-foldselectivity over ERa, illustrating again that there is not a strict parallelism between bindingselectivity and functional selectivity.
205-207
8-"alkylation"
O
O
OH
OH
HO
S
naringenin
8O
O
OH
OH
HO
8
R
ERα ERβ β/α5245 863 6.1
ERα ERβ β/α155 29 5.3
205 206
ERα ERβ β/α392 69 5.7
207
IC50 (nM) IC50 (nM) IC50 (nM)
R = Et R = n-Bu R = n-C8H17
(39)
Figure 65. Introduction of 8-alkyl substituents into naringenin leading to compounds 205--207.
209
ERα ERβ β/α1.9 1.7 0.9
ERα ERβ β/α0.01 0.36 36
ERα ERβ β/α0.04 0.07 1.8
ERα ERβ β/α0.10 0.07 0.7
N
N
N
HO
HO
208RBA (%)
ERα ERβ β/α0.004 0.100 25
5
7
N
N
N
HO
HOCF3
CF3
)%(ABR)%(ABR)%(ABR)%(ABR
N
N
N
HO
CF3
CF3 112012
N
N
N
HO
HOCH3
CH3 212
N
N
N
HO
HO
Figure 66. ERb-antagonists based on the pyrazolo[1,5-a]pyrimidine scaffold (208--212).
414 K MINUTOLOETAL.
Medicinal Research Reviews DOI 10.1002/med

Computational docking analysis of 209 into the ERb ligand-binding pocket revealed thatthe phenol group in the 2-position (a to the pyrazole nitrogen atom) could participate in thestrong H-bond network with Glu305 and Arg346. The other phenol group could then bepositioned to interact with Thr299, in an area near the Asp303 residue that forms a saltbridge with the basic side chain of estrogen antagonists, such as raloxifene and tamoxifen, akey interaction that displaces helix-12 and prevents coactivator recruitment. This provides anexplanation for the antagonist character of these types of compounds, and, on this basis,further attempts to generate more potent and selective antagonists led to the introduction ofbasic side chains on one or the other of the two OH groups of 209.154 Although this approachdid not improve the ERb-selective antagonist properties of these molecules, it did affordcompounds that selectively antagonized the a subtype.
2. Bridged bicyclic derivativesThe Katzenellenbogen group developed several ERb antagonists containing cycloalkylene orbridged bicyclic scaffolds (Figs. 67 and 68). The first antagonists were obtained with com-pounds in which the 1,1-diarylethylene portion was either included in a cyclofenil scaffold(213, Fig. 67) or attached to a bicyclo[2.2.1]heptane unit (214).155 Cyclofenil derivative 213
exhibited a 4.9-fold preference for ERb over ERa, with high binding affinity for theb-subtype (RBA5 334%). The racemic bicyclo[2.2.1]heptane derivative 214 gave slightlylower values (RBA5 211% on ERb and 47% on ERa). Functional assays revealed that bothcompounds had ERa-partial antagonist and ERb-full antagonist character, and a compu-tational docking study suggested that the two OH groups of 213 establish the same polarinteractions reported above for pyrazolo[1,5-a]pyrimidine 209 (Glu305/Arg346 and Thr299).
The size of the central aliphatic ring was later modified to explore the effect of ring sizeon subtype selectivity.156 Cyclobutylene derivative 215 had the lowest binding affinity forERb (RBA 20.5%), and cyclopentylene analog 216 exhibited the highest b selectivity of thisseries (b/a5 7.7), together with a very good ERb affinity (RBA5 137%). Further increase inthe size of the central ring to a cycloheptane (217) improved ERb binding (RBA5 354%),but reduced b selectivity (b/a5 3.2).
In a study of estrogen–dendrimer conjugates, cyclofenil analog 213 was used to preparepoly(amido)amine (PAMAM) dendrimer conjugates.157 Binding affinities of both the
OH
HO213
OH
HO214
(racemic)
O
HO
218
HN
O
NH
~20
(NH2)n-20O
HO
HN
O
NH
NHAc
219
OH
HO215
OH
HO216
OH
HO217
Figure 67. Cycloalkylene and bridged bicyclic ERb-antagonists (213--219).
ESTROGENRECEPTOR bLIGANDS K 415
Medicinal Research Reviews DOI 10.1002/med

dendrimer conjugate (218) and its monomeric analog (219) were measured. The monomerhad a 3.6-fold b selectivity and a significant ERb-binding affinity (RBA5 10.2%); the cor-responding dendrimer conjugate had lower binding affinity for ERb (RBA5 5.8%) and areduced b selectivity (b/a5 2.0). Nevertheless, these studies hold important promise forfuture applications of polymer-bound estrogens.
Other modifications of the central ring of cyclofenil-type ER ligands produced bicy-clo[3.3.1]nonanes 220–222 (Fig. 68).158 Curiously, in this series, the bis-(para-hydroxyphenyl)-substituted derivative 220 did not show appreciable affinity for either of the ER subtypes,whereas removal of one phenol ring, as in compound 221, gave a ligand with better ERb-binding affinity (RBA5 1.33%) and a 10-fold selectivity over ERa. Further improvement bythe addition of a methyl group gave compound 222, which exhibited a b/a selectivity ratio of28 and an ERb-RBA value of 8.90%. All these compounds exhibited antagonist properties onboth ERs in cell-based transcriptional assays, but, unfortunately, no ERb-potency selectivitywas found in the functional assays for any of these bicyclo[3.3.1]nonane derivatives.
Further studies led to bridged oxabicyclic derivatives, as in compounds 223 and 224,which exhibited ERb-selective antagonist properties, despite having ERa-selective bindingaffinities. In fact, sulfonate 223 has RBA values of 9.3% on ERa and 1.7 % on ERb, yet itproved to be a rather b-selective (�10-fold) antagonist, with an IC50 in the submicromolarrange. Sulfone analog 224 had lower binding affinities and antagonist potencies, but itsoverall biological behavior was very similar to that of 223.159
3. Boron-nitrogen isosters of tri- and tetra-arylethylenesA very interesting approach to increase the chemical diversity of estrogen receptor mod-ulators in a novel way involves the elemental isosteric replacement of the central alkeneportion of typical diaryl- and triaryl-ethylene estrogenic agents (e.g. tamoxifen), with aboron-nitrogen unit (Fig. 69).160 The B–N bond is considered to be an isoelectronic surrogateof the CQC bond because boron, carbon, and nitrogen atoms have similar sizes, and theB–N bond has a strong double bond component that results from efficient overlap ofthe empty boron p-orbital, with the nitrogen p-orbital containing a lone electron pair.
OH
HO
220
HO
221
H
HO
222
CH3
O
HO
HO
S O
OO
223
O
HO
HO
S
OO
224
Figure 68. Bridged ERb-antagonists: bicyclo[3.3.1]nonanes (220--222) and oxabicyclic derivatives (223 and 224).
416 K MINUTOLOETAL.
Medicinal Research Reviews DOI 10.1002/med
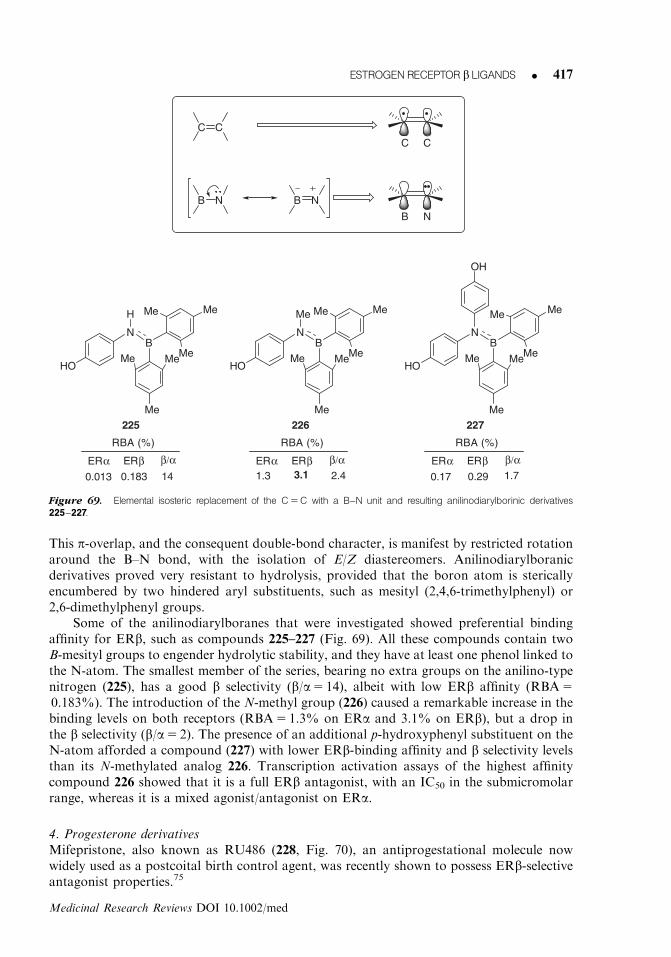
This p-overlap, and the consequent double-bond character, is manifest by restricted rotationaround the B–N bond, with the isolation of E/Z diastereomers. Anilinodiarylboranicderivatives proved very resistant to hydrolysis, provided that the boron atom is stericallyencumbered by two hindered aryl substituents, such as mesityl (2,4,6-trimethylphenyl) or2,6-dimethylphenyl groups.
Some of the anilinodiarylboranes that were investigated showed preferential bindingaffinity for ERb, such as compounds 225–227 (Fig. 69). All these compounds contain twoB-mesityl groups to engender hydrolytic stability, and they have at least one phenol linked tothe N-atom. The smallest member of the series, bearing no extra groups on the anilino-typenitrogen (225), has a good b selectivity (b/a5 14), albeit with low ERb affinity (RBA5
0.183%). The introduction of the N-methyl group (226) caused a remarkable increase in thebinding levels on both receptors (RBA5 1.3% on ERa and 3.1% on ERb), but a drop inthe b selectivity (b/a5 2). The presence of an additional p-hydroxyphenyl substituent on theN-atom afforded a compound (227) with lower ERb-binding affinity and b selectivity levelsthan its N-methylated analog 226. Transcription activation assays of the highest affinitycompound 226 showed that it is a full ERb antagonist, with an IC50 in the submicromolarrange, whereas it is a mixed agonist/antagonist on ERa.
4. Progesterone derivativesMifepristone, also known as RU486 (228, Fig. 70), an antiprogestational molecule nowwidely used as a postcoital birth control agent, was recently shown to possess ERb-selectiveantagonist properties.75
C C
B N B N
C C
B N
NB
HO
H Me
MeMe Me
Me
Me
225
NB
HO
Me Me
MeMe Me
Me
Me
226
NB
HO
Me
MeMe Me
Me
Me
227
OH
ERα ERβ β/α0.013 0.183 14
ERα ERβ β/α1.3 3.1 2.4
ERα ERβ β/α0.17 0.29 1.7
)%(ABR)%(ABR)%(ABR
Figure 69. Elemental isosteric replacement of the C5C with a B--N unit and resulting anilinodiarylborinic derivatives
225--227.
ESTROGENRECEPTOR bLIGANDS K 417
Medicinal Research Reviews DOI 10.1002/med
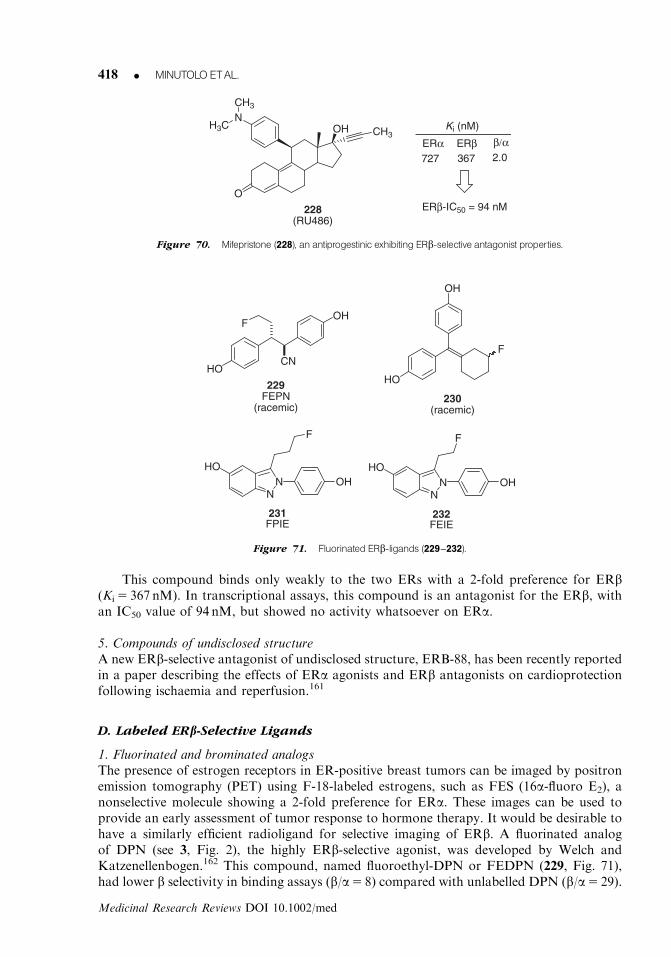
This compound binds only weakly to the two ERs with a 2-fold preference for ERb(Ki 5 367 nM). In transcriptional assays, this compound is an antagonist for the ERb, withan IC50 value of 94 nM, but showed no activity whatsoever on ERa.
5. Compounds of undisclosed structureA new ERb-selective antagonist of undisclosed structure, ERB-88, has been recently reportedin a paper describing the effects of ERa agonists and ERb antagonists on cardioprotectionfollowing ischaemia and reperfusion.161
D. Labeled ERb-Selective Ligands
1. Fluorinated and brominated analogsThe presence of estrogen receptors in ER-positive breast tumors can be imaged by positronemission tomography (PET) using F-18-labeled estrogens, such as FES (16a-fluoro E2), anonselective molecule showing a 2-fold preference for ERa. These images can be used toprovide an early assessment of tumor response to hormone therapy. It would be desirable tohave a similarly efficient radioligand for selective imaging of ERb. A fluorinated analogof DPN (see 3, Fig. 2), the highly ERb-selective agonist, was developed by Welch andKatzenellenbogen.162 This compound, named fluoroethyl-DPN or FEDPN (229, Fig. 71),had lower b selectivity in binding assays (b/a5 8) compared with unlabelled DPN (b/a5 29).
228(RU486)
ERα ERβ β/α727 367 2.0
Ki (nM)
O
OH CH3
NCH3
H3C
ERβ-IC50 = 94 nM
Figure 70. Mifepristone (228), an antiprogestinic exhibiting ERb-selective antagonist properties.
CN
OH
HO
F
229FEPN
(racemic)
NN
HOOH
231FPIE
NN
HO
F
OH
232FEIE
F
230(racemic)
OH
HO
F
Figure 71. Fluorinated ERb-ligands (229--232).
418 K MINUTOLOETAL.
Medicinal Research Reviews DOI 10.1002/med

Also, its affinity for ERb (RBA5 8.74%) is considered as too low for effective in vivo PET ofERb in breast tumors.
A racemic fluorinated cyclofenil derivative (230, Fig. 71) proved to be a good ligand forERb (RBA5 62%), but its ERb-binding selectivity was only about 2-fold.156
More recently, fluorinated indazoles were prepared and some of them, such as fluor-opropyl- 231 (FPIE) and fluoroethyl-derivatives 232 (FEIE), have several fold greater RBAvalues for ERb than for ERa.163 In fact, b/a selectivity ratios are 17.4 for 231 and 41.3 for232; nevertheless, their binding affinities for ERb are rather poor, with RBA values of0.087% (231) and 0.124% (232), and clearly not sufficient for in vivo imaging.
More fluorinated analogs of DPN were recently reported (233–235, Fig. 72).164
All these DPN derivatives are characterized by the presence of a fluorinated substituentin the 30-position, which proved to be well tolerated by ERb. None of the members of thisseries showed significant binding to ERa (RBA�0.02%), whereas the length of the fluori-nated group had a remarkable effect on ERb affinity, the longest group (3-fluoropropyl)being the poorest, with an ERb-RBA of 0.896% (235). Shortening the substituent to a2-fluoroethyl group provided some increase in binding affinity, with compound 234 havingan RBA of 1.58% for ERb. However, the most striking results were obtained with 233, wherethe fluorine atom is directly bound to the aromatic ring. This compound had high bindingaffinity for ERb (RBA5 6.25%) and an outstanding 272-fold selectivity over ERa. Never-theless, this binding affinity is still probably insufficient for in vivo imaging by PET tech-nology.
A different halogen radionuclide, 76Br, was used to label the corresponding ‘‘cold’’brominated azole derivative 7 (WAY-200070, Fig. 4), which had previously shown very highand very selective ERb-binding affinity (IC50 5 2 nM, b/a5 67).48 The radionuclide 76Br issuitable for PET imaging of slower physiologic processes, since its half-life (t1/2 5 16.2 h) ismuch longer than that of 18F (t1/2 5 110min). Therefore, an exploratory study on the syn-thetic accessibility at the tracer scale of 76Br-radiolabeled diacetate of 7 (236, Fig. 73) wasrecently reported.165
This radioligand 236 is a precursor of the real ERb-selective radiotracer [76Br]-7 in whichthe two acetate portions have been removed by hydrolysis.
CN
OH
HO
233
F
CN
OH
HO
234
CN
OH
HO
235
FF
'3'3'3
Figure 72. DPNanalogs bearing fluorinated substituents in the 30 -position (233--235).
[76Br]-7
N
OHO
OH
76Br
236
N
OAcO
OAc
76Br
Figure 73. Radiobrominated azole 236 and its foreseen conversion to ERb-selective radioligand [76Br]-7.
ESTROGENRECEPTOR bLIGANDS K 419
Medicinal Research Reviews DOI 10.1002/med

2. Metal complexesE2 was conjugated to tridentate pyridin-2-yl hydrazine Re/99mTc chelates, to generate a seriesof radiolabeled ER ligands.166 This chelate system was highly stable in aqueous solutions,with no evidence of decomplexation of the metal, and these compounds were also shown tobe cell-permeable and suitable for bioassays. The nature of the unit linking the steroid to themetal complex affected subtype-selective binding. Re-complex 237 (Fig. 74), possessing a(Z)-ethene linker, was the only conjugate showing any significant (2.4-fold) preference forERb, with a RBA of 24%. This complex has also proved a good ligand (RBA5 64%) forGPR30, a 7-transmembrane G-protein-coupled receptor located in the endoplasmicreticulum that binds estrogen with high affinity (Kd 5 6 nM) and mediates rapid cellularresponses, such as calcium mobilization and phosphatidylinositol 3,4,5-trisphosphate pro-duction in the nucleus. The role of this nonnuclear estrogen receptor in the progression ofestrogen-responsive tumors is currently under investigation, because it might represent asignificant target in ERa/b-negative breast cancers.167
Carborane-based Tc/Re organometallic complexes were developed at McMaster Uni-versity (Hamilton, Canada). Carboranes are carbon-containing polyhedral boron clusers,which were found to efficiently coordinate Tc/Re(I) tricarbonyl units. These complexes arereported to be stable and compatible with biological systems. Estrogen receptor-bindingassays revealed that complex 238 (Fig. 74), bearing a para-hydroxyphenyl substituent on oneof the two carbon atoms of the cluster, binds to ERb, although with a modest RBA of 0.55%and a b selectivity of 3.4-fold over ERa.168
Ferrocenyl estrogen receptor ligands were not designed for imaging purposes; rather,they were intended to add cytotoxic antiproliferative effect to ER-binding properties.Nevertheless, they are included in this section as metal complexes able to bind ERb. Re-searchers at the Ecole Nationale Superieure de Chimie (Paris) developed an extensive set offerrocenyl derivatives having good binding affinities for ERb (239–245, Fig. 75). Amongbis(para-hydroxyphenyl)butene-based complexes (239 and 240), compound 240, bearing aphenol substituent on two different vinyl carbon atoms, showed the highest ERb-bindingaffinity (RBA5 31%) and b selectivity (b/a5 3.6). Its close analog 239, where the twophenols are attached to the same vinylic carbon atom had a nearly 2-fold reduction in bothbinding affinity (RBA5 16.3%) and selectivity for ERb (b/a5 1.9).169 The other subclass,comprising mono- and di-arylmethylferrocenyls 241–245, had at least one para-hydroxy-phenyl substituent.170 Monoaryl-derivative 241 had a 3.2-fold selectivity for ERb, though itsbinding affinity was among the lowest of this class (RBA5 3.5%). The insertion ofan additional para-hydroxyphenyl group (244) improved binding (RBA5 28% on ERb),but practically destroyed subtype selectivity (b/a5 1.5). Regioisomers with an ortho- or a
OH
HO N
NN
(Z)
O
H Re
O
H
CO
CO
CO
237
C
C
Re
= BH
H
HO
COOC
OC
238
Figure 74. Rhenium complexes showing ERb-binding preference (237 and 238).
420 K MINUTOLOETAL.
Medicinal Research Reviews DOI 10.1002/med
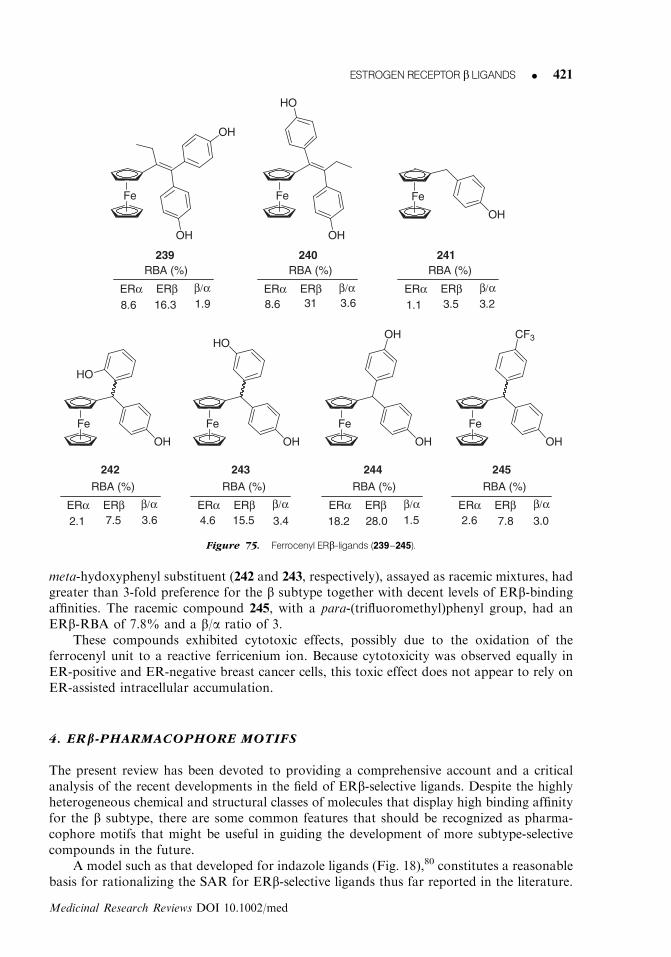
meta-hydoxyphenyl substituent (242 and 243, respectively), assayed as racemic mixtures, hadgreater than 3-fold preference for the b subtype together with decent levels of ERb-bindingaffinities. The racemic compound 245, with a para-(trifluoromethyl)phenyl group, had anERb-RBA of 7.8% and a b/a ratio of 3.
These compounds exhibited cytotoxic effects, possibly due to the oxidation of theferrocenyl unit to a reactive ferricenium ion. Because cytotoxicity was observed equally inER-positive and ER-negative breast cancer cells, this toxic effect does not appear to rely onER-assisted intracellular accumulation.
4. ERb -PHARMACOPHORE MOTIFS
The present review has been devoted to providing a comprehensive account and a criticalanalysis of the recent developments in the field of ERb-selective ligands. Despite the highlyheterogeneous chemical and structural classes of molecules that display high binding affinityfor the b subtype, there are some common features that should be recognized as pharma-cophore motifs that might be useful in guiding the development of more subtype-selectivecompounds in the future.
A model such as that developed for indazole ligands (Fig. 18),80 constitutes a reasonablebasis for rationalizing the SAR for ERb-selective ligands thus far reported in the literature.
Fe
OH
OH
239
Fe
OH
240
HO
Fe
OH
241
Fe
OH
242
HO
Fe
OH
243
HO
Fe
OH
244
Fe
OH
245
OH CF3
ERα ERβ β/α8.6 16.3 1.9
ERα ERβ β/α8.6 31 3.6
ERα ERβ β/α1.1 3.5 3.2
RBA (%) RBA (%) RBA (%)
ERα ERβ β/α2.1 7.5 3.6
ERα ERβ β/α4.6 15.5 3.4
ERα ERβ β/α18.2 28.0 1.5
RBA (%) RBA (%) RBA (%)
ERα ERβ β/α2.6 7.8 3.0
RBA (%)
Figure 75. Ferrocenyl ERb-ligands (239--245).
ESTROGENRECEPTOR bLIGANDS K 421
Medicinal Research Reviews DOI 10.1002/med

We elaborated the following structural features that emerge as fundamental for ERb-affinityand selectivity (Fig. 76):
Phenol-type hydroxyl. A phenolic OH is absolutely necessary to establish what is con-sidered to be the highest energy attractive interaction between the ligand and ER-bindingcavity, namely, the hydrogen bond network involving Arg346 and Glu305. In a few cases,this group may also be a ‘‘pseudo-phenol,’’ such as that present in some salicylaldoximederivatives. This interaction generally assures an efficient binding but does not guarantee anysubtype selectivity, because it is present in both ERa and ERb.
A second, antipodal additional hydroxyl. A second phenol/pseudophenol (oxime) oralcohol OH group, placed at a distance of about 12 A from the previously discussed phenolichydroxyl, is generally beneficial for receptor affinity because it may give rise to an additionalH-bond, similar to 17b-OH of E2. In most cases this interaction involves His475 in ERb or itscounterpart in ERa, His524, and, therefore, does not necessarily improve b selectivity.Nevertheless, if this second OH group is closer to the previous one (10–11 A or less) and it isappropriately oriented to establish an H-bond with Thr299 in ERb, this different interactionmay contribute to subtype discrimination, because the approach of a H-bond donor to the
OH
HO
OH
phenoltype
alcohol
or
phenol
mutuallyexclusive
?Met336
bulge-A
bulge-B
Ile373
inlet
Thr299
His475
Arg346
Glu305
ERβ
SMe
OH
HO
phenoltype
alcohol
or
phenol
Leu384
bulge-A
bulge-B
Arg394
Glu353
Met421
Me S
repulsion
repulsionERα
His524
Figure 76. Generic structural features and principles of selectivity for ERb-ligands.
422 K MINUTOLOETAL.
Medicinal Research Reviews DOI 10.1002/med

corresponding threonine residue in ERa (Thr347) is partially thwarted by Leu384, more sothan by Met336, the corresponding residue present in ERb. In fact, although the volume ofthe sulfur-containing methionine side chain (�86 A3) is slightly larger than that of thebranched amino acid side chain of leucine (�83 A3), the increased flexibility of the linearmethionine side chain is predicted to allow larger substituents to be accommodated. Aninteraction with ERb Thr299 was reasonably hypothesized to occur with ERb-agonist 103and ERb-antagonists 209, 213–217. At present, it is not clear whether these two interac-tions—His475 and Thr299—are mutually exclusive, because the presence of two antipodalOH groups able to bind simultaneously to both of these residues has not been reported in anyof the ER ligands known so far. Thus, this possibility might inspire the design of new ERb-selective ligands in the future. However, it should be noted that there are several exampleswhere this additional OH group is not present, and the ligands are still good ER-bindersshowing high selectivity for ERb. There are examples where this additional OH is replaced bya carbonyl group (estrone itself, aldehyde 105, tetrahydrofluorenones 127–130), by a fluorineatom (benzoxepins 154 and 155), or it is just absent (benzimidazoles 72–75, imidazole 76,indole 77, naphthalene 83, cyclohexylphenol 172, deoxyhexestrol 189). This suggests that thisinteraction may also be omitted and that it is not as energetically fundamental as the previousone, provided additional positive interactions (mostly lipophilic ones) take place.
Bulge A. The presence of a structural protrusion toward residue Met336 (ERb), indicatedhere as bulge A, seems to be one of the most significant selectivity-driving features found inmany ERb-selective ligands. In fact, the more flexible linear side chain of Met336 is able toaccommodate this molecular bulge in ERb, whereas Leu384, the bulkier and less con-formationally compliant corresponding residue in ERa, would not do the same. This mo-lecular protrusion is basically localized in the 8b-position of E2; in fact, 8b-vinylestradiol 29 isa very b-selective agonist, as are the structurally related steroids 10b-substituted androste-nediols 17–23. A reference nonsteroidal ERb-selective agonist, DPN (3), has a cyano-groupplaced in the area of bulge A, and many other nonsteroidal b-selective ligands possess smallgroups in this position. Some relevant examples are 3-substituted indazoles 48–52 (Cl, Br,CF3, CN, etc.), 3-chloro-substituted salicylaldoximes 102 and 104, 10-methyl-substitutedchromenone 152, and 8-methyl oxabicyclo[3.3.1]nonanes 169–171.
Bulge B. Another structural protrusion that generally increases the b selectivity hugs aside of Ile373 in ERb, represented here by bulge B. At this site, the length of the amino acidside chain determines the size of a substituent that can be accommodated: the shorter sidechain of Ile373 in ERb enables relatively small (and possibly hydrophobic) groups to behosted at this site, whereas the longer group of the ERa residue Met421 often clashes withsubstituents placed at this position in ligands. One of the most potent and selective agonistsfor ERb, benzoxazole ERB-041 (4), places its 7-vinyl substituent in this region, as doesWAY-200070 (7), a close analog with a 7-bromine group. There are many other examples ofERb-selective ligands that present substituents that occupy bulge B, mostly in the non-steroidal classes. Exceptions are the B-seco or pseudo-steroids lacking a B-ring, 34 and 35,whose particular torsional disposition of their D-ring places it in the bulge B portion of thecavity. Significant examples of nonsteroidal ligands containing molecular portions, usuallyrelatively small and linear, occupying bulge B are benzofurans bearing 7-OCH3 or 7-CH2CNsubstituents (56–61), 7-bromoindenone 62, bromo-substituted phthalimide 71, benzimida-zoles containing N-methyl-heterocycles (74 and 75), naphthalenes possessing 8-cyano (81) or8-ethyl (82) substituents, 4-substituted (Cl, Br, or CN) quinolines 84–88, benzaldehyde 105
and benzaldoximes 106–108 bearing ortho-chloro substituents, naphthalene- (109,110) andindole-based (111) aldoximes exposing a side of their bicyclic aromatic scaffold, SERBAscontaining a cyclopentane portion (113, 116–125), tetrahydro-fluorenones protrudingtheir 9a-n-butyl side chains (127–130), benzoxepines exposing the ethylene portion of their
ESTROGENRECEPTOR bLIGANDS K 423
Medicinal Research Reviews DOI 10.1002/med

7-member rings (153–155), and benzoxazines containing 5-methyl groups (157,158). Finally,it is worth mentioning again the oxabicyclo[3.3.1]nonenes 169–171, whose hydroxymethylsubstituent suffers from electrostatic repulsion with the sulfur atom of Met421 in ERa, butcan be more comfortably placed in the ERb cavity. These last compounds are the only ERligands that, up to now, exploit the simultaneous occupancy of bulge A (with 8-methylsubstituent) and bulge B (with the hydroxymethyl portion), although they do not appear totake advantage from the additional H-bond with the His475/Thr299 residues.
Inlet. In a position very close to bulge B, which corresponds roughly to the E2 16a-position (below the D ring), there appears to be the need for a molecular inlet that candevelop around the branched side chain of ERb Ile373. This feature is substantially respectedby all the ERb ligands reported so far. The importance of this structural feature for efficientbinding to ERb is indirectly confirmed by the dramatic loss of affinity for ERb experiencedby steroidal derivative 30, where a lactone ring bridging the 16a- and 17a-positions wasintroduced right below the D ring.
All of these pharmacophore considerations appear generally valid for ERb agonists. Atthis point, however, formulating a similar pharmacophore for ERb-selective antagonists ismore difficult, because, thus far, the relatively limited number of examples does not allow oneto make reliable structural generalizations.
It is of historical interest that many of the features noted in Figure 76, the hydrogenbonds, bulges and cavities, had actually been predicted in a review article by one of us in1997,171 even before crystal structures for ERa and ERb were available. The model at thattime was based on a careful, quantitative analysis of the effect that substituents on steroidalestrogens had on binding affinity to the only estrogen receptor known at that time, ERa. Ofcourse, the atomistic details of receptor structures and the comparisons that can now bemade between ERa and ERb, together with the prodigious array of structurally diverseligands, enable one to formulate much more refined pharmacophore models for the selectiveinteraction of ligands with the ERb subtype.
5. POTENTIAL CLINICAL USES OF ERb -SELECTIVE ESTROGENS
A. Breast Cancer
There has been extensive work on measuring the levels of ERb in normal breast cells and inbreast cancer, and in assessing the utility of ERb as a prognostic marker and a therapeutictarget. Although initially there was some discordance on technical points, perhaps becausethe antibodies used for assaying ERb protein levels were not well behaved, a general con-sensus has emerged more recently. This work has been covered in a number of recent,detailed reviews and articles to which the reader is referred.11,172–179
In the normal breast, ERb is found in both epithelial and stromal cells, whereas ERa islocalized to the former; in rodent mammary gland, it appears to be ERb that is associatedwith proliferating cells.180 The situation is very different in breast cancer, where ERa levelsincrease and ERb levels decline relative to normal breast, and continued reduction in ERblevels occurs as breast cancer progresses from localized to more invasive disease. Thesechanges in the relative levels of ERa and ERb in breast cancer fit the general paradigm ofestrogen action through ERb moderating the proliferative drive of estrogen through ERa. Itis also consistent with experiments in which the introduction of ERb into ERa-positive breastcancer cell lines reduces proliferation14 and retards the growth of tumor xenografts,15 andfrom results in cells containing both ERa and ERb, in which the regulation of proliferativemarkers and proliferation itself is increased with the ERa-selective ligand PPT (propyl
424 K MINUTOLOETAL.
Medicinal Research Reviews DOI 10.1002/med

pyrazole triol: 4,40,400-(4-propyl-[1H]-pyrazole-1,3,5-triyl)trisphenol),181 but reduced with theERb-selective ligand DPN (3, Fig. 2).182,183 The role of estrogen action through ERa andERb in breast cancer also involves effects on neoangiogenesis and the response of the im-mune system, where, again, ERb appears to be the mediator of beneficial effects.
Making accurate measurements of the protein levels of the various isoforms of ERb hasproved to be challenging184 but is important, because the isoforms have very different ac-tivities (see Section 1B). Nevertheless, ERb appears to be of value as a prognostic marker inbreast cancer, and tumors that contain both ERb and ERa are more likely to be responsive toendocrine therapies.172 On the contrary, however, ERb is found in some ERa-negative can-cers, where, in the absence of ERa, its level correlates with indices of proliferation.172,185,186
It is now well appreciated that cellular adaptations can profoundly alter the pathways bywhich breast cancer proliferation and tumor phenotype are regulated by hormones andgrowth factors, an area that is under intensive study. In terms of the utility of ERb as a targetfor endocrine therapies in breast cancer, one could imagine that a highly selective ERbagonist might be beneficial for disease in which both ERa and ERb are present, but that anER antagonist (perhaps not needing to be selective) might be more helpful when only ERb ispresent.186 Combinations of ER subtype-selective ligands with inhibitors of other signalingpathways in breast cancer might prove even more effective.
B. Prostate, Colon, and Lung Cancers
ERb appears to play a role in other cancers, although its prognostic value and potential as atherapeutic target are generally less well-defined. ERb is present in normal prostate, in benignprostatic hypertrophy,187 and in many prostate cancers;188 however, its regulatory effect onthe cancer appears to depend on tumor stage. In tumors confined to the prostate, ERbappears to be prodifferentiative and to moderate proliferation, and its levels decline withdisease progression.189,190 In cell transfection studies, introduction of ERb reduced growthand invasiveness.191 By contrast, ERb is high in prostate cancer metastases where it appearsto be the mediator of the malignant phenotype.188 The use of antiestrogens in prostate cancerhas been explored, but not the use of ER subtype-selective ligands.188,192,193
ERb is the predominant ER subtype in colon; it is found in many colon cancers and coloncancer cell lines, and ERb levels decrease with progression of colon cancer.194,195 ERb alsoappears to be the predominant ER in lung, and in lung cancers and lung cancer celllines;196,197 ERb levels in lung cancer are related to tumor histological grade,198 and they haveprognostic value,199 but the ERb-selective agonist DPN (3, Fig. 2) showed stimulatory effectsin various lung cancer cell models.200 The activity of an ERb antagonist was not suited.
C. Fertility
The predominance of ERb in ovarian granulosa cells suggests that ERb-selective estrogensmight be useful in treating infertility. Notably, the Schering steroidal ERb-selective ligand,8b-VE2 (29, Fig. 10) stimulated folliculogenesis in a hypophysectomised rat model.201 Effectsof ERb-selective ligands on male reproduction might also be expected, and the di-hydro-testosterone (DHT) metabolite, 5a-androstane-3a,17b-diol (13, Fig. 6), which has someaffinity for ERb, was found to increase spermatogonial DNA synthesis in rats.202
D. Reversing Menopausal Changes: Bone, Hot Flush, and Cardiovascular Effects
It was originally hoped that ERb might be an effective target for reversing the physiologicalchanges that occur in the menopause, without accruing the risk factors of estrogens that acteither through ERa or through both ERs.2 This has proved to be true only in part, however.
ESTROGENRECEPTOR bLIGANDS K 425
Medicinal Research Reviews DOI 10.1002/med

ERb-selective ligands do not seem to be active in models of prevention or reversal ofmenopausal bone loss.46,65 Although a desirable activity of estrogens used by menopausalwomen is suppression of hot flush, in animal models, some ERb-selective ligands were noteffective.46 ERa-selective ligands were active,203 although there is a report that E2 works inERa-knockout mice, suggesting a possible role for ERb in this response, at least when ERa iscompletely knocked out.204
Women derive cardiovascular benefit from premenopausal exposure to estrogens and fromhormone replacement therapy when initiated soon after the start of menopause.205 Therespective role of ERa and ERb in mediating vascular health is not clear,2 and some of thebenefits probably result from estrogen effects on liver, improving lipid profiles. Although studiesin knockout mice indicate that estrogen action through ERa appears responsible for blockingvascular smooth muscle overgrowth after injury and mediates vascular relaxation,2 ERbappears responsible for the important vascular reendothelialization response206 and for pro-tecting cardiomyocytes from ischemic injury.207 Other observations suggest that ERb-selectiveligands might provide cardiovascular benefits: ERb-knockout mice develop hypertension andhave abnormal vascular function,208 and ERb also appears to prevent chronic heart fail-ure.209,210 Thus, ERb-selective ligands could provide many aspects of cardiovascular benefit.
E. Diverse Antiinflammatory Activities
Estrogens are known to have antiinflammatory activity, presumably through suppression ofNF-kB signaling. Intriguingly, a number of ERb-selective estrogens repress the transcriptionof proinflammatory genes in cells after exposure to TNF-a,211 and they show promisingactivity in a number of animal models of inflammation. ERB-041 (4, Fig. 3) and WAY-202196 (81, Fig. 25), both from Wyeth, are effective in two animal models of inflammatorybowel disease and rheumatoid arthritis,46,90 and they also regularize changes in liver, lymphnodes, and spleen secondary to the disease. They also reduced lesion size in an animal modelof endometriosis, which is considered an inflammatory disease; the absence of ERb in theendometrial tissue, however, suggests that this is likely a host-mediated effect.212
An unexpected activity of the ERb-selective ligand WAY-202196 (81, Fig. 25) wasprotection against death in two experimental models of septic shock,213 and the ERb-selective ligand DPN (3, Fig. 2) was also effective in a model of inflammation following lunginjury.214 Curiously, despite the good activity of ERb-selective ligands in these models ofinflammation, compounds of this class have failed in a number of other models of in-flammation. Thus, the activity of these ERb ligands is rather compound and model specific.Moreover, antiinflammatory activity can also be mediated by estrogens acting through ERa.2
F. Neuroprotection and Behavioral Effects
Estrogens have neuroprotective activity in animal models of ischemic stroke, and in somemodels, ERb ligands appear to be protective,2,215,216 while studies in knockout animalsindicate a predominant role for ERa.217,218 It has been proposed that the neuroprotectiveeffect of ERb might be mediated through its action in mitochondria.219 ERB-041 (4, Fig. 3)was also effective in reducing inflammatory pain,220 and another ERb-selective ligand ofundisclosed structure (ERb-131) was reported to be active in reducing neuropathic pain inseveral animal models.133
Some unusual behaviors exhibited by ERb knockout mice have suggested that ERb-selective estrogens might have potential in the treatment of anxiety or depression,221–223 andin some animal models of these disorders, the ERb-selective ligand DPN (3, Fig. 2), but notthe ERa-selective ligand PPT, was effective.224–226 ERb also appears to play a role in synaptic
426 K MINUTOLOETAL.
Medicinal Research Reviews DOI 10.1002/med

plasticity, learning, and memory,222,227 and distinct effects on brain neurotransmitterreceptors have been observed with the ER subtype-selective ligands noted above.228
G. Other Activities in Metabolic Regulation
Studies in ER knockout mice have highlighted that estrogens play important roles in glucoseutilization, fat metabolism, and obesity.229–232 In certain behavioral assays, running wheelactivity in ovariectomized female rats, the Schering ERb-selective ligand 8b-VE2 (29, Fig. 10)actually reduced the stimulatory effect of the ERa-selective ligand.233
6. CONCLUSIONS
Since the discovery of ERb in 1996,6 this receptor has been the focus of the efforts of manyresearchers. Although much has been learned, there continues to be an evolving roster ofverified and potential therapeutic applications for ERb-selective ligands. The distinct target-tissue distribution and functional characteristics of this ER subtype—its activity alone and itsability to modulate the effect of estrogens through ERa—make ERb a promising yet tan-talizing target. Evolving phenotypic characterization of ER-subtype knockout animals andthe results of physiological studies in experimental animals and early stage results fromhuman clinical trials with subtype-selective ligands should provide additional insights. Thisprogress will be aided by a better understanding of what molecular characteristics underliethe ERb selectivity of certain ligands and the development of a panel of potent and highlyselective ligands, both as molecular probes to study the biology of ERb as well as potentialleads and clinical candidates for further studies. Therefore, future studies on ERb and ERb-selective ligands should continue to bear fruit in potential biomedical applications.
Finally, we wish to note that—as of yet—all the structural features we have indicated inFigure 76 for the selective interaction of ligands with ERb have not been exploited. Thus, wehope that the observations we have made and the generalizations we have gleaned throughour extensive review of this topic, which we have organized and presented in Section 4, will behelpful to researchers involved in this very exciting field of Medicinal Chemistry. We willbe pleased if these insights will help guide the development of ERb-selective ligands that willbe yet more potent and more selective, and, thus, will prove to be effective in addressingcurrently unmet health needs and clinical applications in the future through the selectiveregulation of the activity of the two estrogen receptors, ERa and ERb.
ACKNOWLEDGMENTS
J. A. K. and B. S. K. are grateful for the support of this research through grants from theNational Institutes of Health (PHS R37DK011556 and P01AG024387 [to J. A. K.] andR01CA018119 and P01AG024387 [to B. S. K.]); F. M. andM.M. thank the University of Pisa.
REFERENCES
1. Harris HA. The unexpected science of estrogen receptor-beta selective agonists: A new class of
anti-inflammatory agents? Nucl Recept Signal 2006;4:e012.
2. Harris HA. Estrogen receptor-beta: Recent lessons from in vivo studies. Mol Endocrinol
2007;21:1–13.
ESTROGENRECEPTOR bLIGANDS K 427
Medicinal Research Reviews DOI 10.1002/med

3. Hewitt SC, Harrell JC, Korach KS. Lessons in estrogen biology from knockout and transgenic
animals. Annu Rev Physiol 2005;67:285–308.
4. Pettersson K, Gustafsson J-A. Role of estrogen receptor beta in estrogen action. Annu Rev
Physiol 2001;63:165–192.
5. Zhao C, Dahlman-Wright K, Gustafsson J-A. Estrogen receptor beta: An overview and update.
Nucl Recept Signal 2008;6:e003.
6. Kuiper GGJM, Enmark E, Pelto-Huikko M, Nilsson S, Gustafsson J-A. Cloning of a novel
receptor expressed in rat prostate and ovary. Proc Natl Acad Sci USA 1996;93:5925–5930.
7. Brzozowski AM, Pike ACW, Dauter Z, Hubbard RE, Bonn T, Engstrom O, Ohman L,
Greene GL, Gustafsson J-A, Carlquist M. Molecular basis of agonism and antagonism in the
oestrogen receptor. Nature 1997;389:753–758.
8. Taylor AH, Al-Azzawi F. Immunolocalisation of oestrogen receptor beta in human tissues. J Mol
Endocrinol 2000;24:145–155.
9. Antal MC, Krust A, Chambon P, Mark, M. Sterility and absence of histopathological defects in
nonreproductive organs of a mouse ERbeta-null mutant. Proc Natl Acad Sci USA 2008;105:
2433–2438.
10. Imamov O, Morani A, Shim GJ, Omoto Y, Thulin-Andersson C, Warner M, Gustafsson J-A.
Estrogen receptor beta regulates epithelial cellular differentiation in the mouse ventral prostate.
Proc Natl Acad Sci USA 2004;101:9375–9380.
11. Fox EM, Davis RJ, Shupnik MA. ERbeta in breast cancer—onlooker, passive player, or active
protector? Steroids 2008;73:1039–1351.
12. Lindberg MK, Moverare S, Skrtic S, Gao H, Dahlman-Wright K, Gustafsson J-A, Ohlsson C.
Estrogen receptor (ER)-beta reduces ERalpha-regulated gene transcription, supporting a
‘‘ying yang’’ relationship between ERalpha and ERbeta in mice. Mol Endocrinol 2003;17:
203–208.
13. Sun J, Meyers MJ, Fink BE, Rajendran R, Katzenellenbogen JA, Katzenellenbogen BS. Novel
ligands that function as selective estrogens or antiestrogens for estrogen receptor-a or estrogen
receptor-b. Endocrinology 1999;140:800–804.
14. Chang EC, Frasor J, Komm B, Katzenellenbogen BS. Impact of estrogen receptor beta on gene
networks regulated by estrogen receptor alpha in breast cancer cells. Endocrinology 2006;147:
4831–4842.
15. Paruthiyil S, Parmar H, Kerekatte V, Cunha GR, Firestone GL, Leitman DC. Estrogen receptor
beta inhibits human breast cancer cell proliferation and tumor formation by causing a G2 cell
cycle arrest. Cancer Res 2004;64:423–428.
16. Strom A, Hartman J, Foster JS, Kietz S, Wimalasena J, Gustafsson J-A. Estrogen receptor beta
inhibits 17 beta-estradiol-stimulated proliferation of the breast cancer cell line T47D. Proc Natl
Acad Sci USA 2004;101:1566–1571.
17. Williams C, Edvardsson K, Lewandowski SA, Strom A, Gustafsson J-A. A genome-wide study of
the repressive effects of estrogen receptor beta on estrogen receptor alpha signaling in breast
cancer cells. Oncogene 2008;27:1019–1032.
18. Frasor J, Chang EC, Komm B, Lin CY, Vega VB, Liu ET, Miller LD, Smeds J, Bergh J,
Katzenellenbogen BS. Gene expression preferentially regulated by tamoxifen in breast cancer cells
and correlations with clinical outcome. Cancer Res 2006;66:7334–7340.
19. Chang EC, Charn, TH, Park SH, Helferich WG, Komm B, Katzenellenbogen JA,
Katzenellenbogen BS. Estrogen receptors alpha and beta as determinants of gene expression:
Influence of ligand, dose, and chromatin binding. Mol Endocrinol 2008;22:1032–1043.
20. Harris HA. Preclinical characterization of selective estrogen receptor beta agonists: New insights
into their therapeutic potential. Ernst Schering Found Symp Proc 2006;149–161.
21. Lin BC, Suzawa M, Blind RD, Tobias SC, Bulun SE, Scanlan TS, Ingraham HA. Stimulating the
GPR30 estrogen receptor with a novel tamoxifen analogue activates SF-1 and promotes
endometrial cell proliferation. Cancer Res 2009;69:5415–5423.
428 K MINUTOLOETAL.
Medicinal Research Reviews DOI 10.1002/med

22. Filardo EJ, Quinn JA, Sabo E. Association of the membrane estrogen receptor, GPR30, with
breast tumor metastasis and transactivation of the epidermal growth factor receptor. Steroids
2008;73:870–873.
23. Prossnitz ER, Arterburn JB, Smith HO, Oprea TI, Sklar LA, Hathaway HJ. Estrogen signaling
through the transmembrane G protein-coupled receptor GPR30. Annu Rev Physiol 2008;70:
165–190.
24. Prossnitz ER, Arterburn JB, Sklar LA. GPR30: A G protein-coupled receptor for estrogen. Mol
Cell Endocrinol 2007;265–266:138–142.
25. Filardo EJ, Thomas P. GPR30: A seven-transmembrane-spanning estrogen receptor that triggers
EGF release. Trends Endocrinol Metab 2005;16:362–367.
26. Leung YK, Mak P, Hassan S, Ho SM. Estrogen receptor (ER)-beta isoforms: A key to
understanding ER-beta signaling. Proc Natl Acad Sci USA 2006;103:13162–13167.
27. Treeck O, Juhasz-Boess I, Lattrich C, Horn F, Goerse R, Ortmann O. Effects of exon-deleted
estrogen receptor beta transcript variants on growth, apoptosis and gene expression of human
breast cancer cell lines. Breast Cancer Res Treat 2008;110:507–520.
28. Deroo BJ, Korach KS. Estrogen receptors and human disease. J Clin Invest 2006;116:561–570.
29. Katzenellenbogen JA, O’Malley BW, Katzenellenbogen BS. Tripartite steroid hormone receptor
pharmacology: Interaction with multiple effector sites as a basis for the cell- and promoter-specific
action of these hormones. Mol Endocrinol 1996;10:119–131.
30. McDonnell DP, Norris JD. Connections and regulation of the human estrogen receptor. Science
2002;296:1642–1644.
31. Bryant HU. Selective estrogen receptor modulators. Rev Endocr Metab Disord 2002;3:231–241.
32. Jordan VC. Antiestrogens and selective estrogen receptor modulators as multifunctional
medicines. 2. Clinical considerations and new agents. J Med Chem 2003;46:1081–1111.
33. Jordan VC. Antiestrogens and selective estrogen receptor modulators as multifunctional
medicines. 1. Receptor interactions. J Med Chem 2003;46:883–908.
34. Veeneman GH. Non-steroidal subtype selective estrogens. Curr Med Chem 2005;12:1077–1136.
35. Meegan MJ, Lloyd DG. Advances in the science of estrogen receptor modulation. Curr Med
Chem 2003;10:181–210.
36. Henke BR, Heyer D. Recent advances in estrogen receptor modulators. Curr Opin Drug Disc Dev
2005;8:437–448.
37. Hermkens PHH, Kamp S, Lusher S, Veeneman G. Non-steroidal steroid receptor modulators.
IDrugs 2006;9:488–494.
38. Redden PR. Selective oestrogen receptor modulators, pure antiestrogens and related oestrogen
receptor ligands. Expert Opin Ther Patents 2004;14:337–353.
39. Ullrich JW, Miller CP. Estrogen receptor modulator review. Expert Opin Ther Patents 2006;16:
559–572.
40. Blizzard TA. Selective estrogen receptor modulator medicinal chemistry at Merck. A review. Curr
Top Med Chem 2008;8:792–812.
41. Agatonovic-Kustrin S, Turner JV. Molecular structural characteristics of estrogen receptor
modulators as determinants of estrogen receptor selectivity. Mini-Rev Med Chem 2008;8:943–951.
42. Kuiper GGJM, Lemmen JG, Carlsson B, Corton JC, Safe SH, van der Saag PT, van der Burg B,
Gustafsson J-A. Interaction of estrogenic chemicals and phytoestrogens with estrogen receptor b.Endocrinology 1998;139:4252–4263.
43. Meyers MJ, Sun J, Carlson KE, Marriner GA, Katzenellenbogen BS, Katzenellenbogen JA.
Estrogen receptor b potency-selective ligands: Structure-activity relationship studies of
diarylpropionitriles and their acetylene and polar analogues. J Med Chem 2001;44:4230–4251.
44. Weiser MJ, Wu TJ, Handa RJ. Estrogen receptor-b agonist diarylpropionitrile: Biological
activities of R- and S-enantiomers on behavior and hormonal response to stress. Endocrinology
2009;150:1817–1825.
ESTROGENRECEPTOR bLIGANDS K 429
Medicinal Research Reviews DOI 10.1002/med

45. Sun J, Baudry J, Katzenellenbogen JA, Katzenellenbogen BS. Molecular basis for the subtype
discrimination of the estrogen receptor-b-selective ligand, diarylpropionitrile. Mol Endocrinol
2003;17:247–258.
46. Harris HA, Albert LM, Leathurby YL, Malamas MS, Mewshaw RE, Miller CP, Kharode YP,
Marzolf J, Komm BS, Winneker RC, Frail DE, Henderson RA, Zhu Y, Keith JC, Jr. Evaluation
of an estrogen receptor-b agonist in animal models of human disease. Endocrinology 2003;144:
4241–4249.
47. Manas ES, Unwalla RJ, Xu ZB, Malamas MS, Miller CP, Harris HA, Hsiao C, Akopian T,
Hum W-T, Malakian K, Wolfrom S, Bapat A, Bhat RA, Stahl ML, Somers WS, Alvarez JC.
Structure-based design of estrogen receptor-b selective ligands. J Am Chem Soc 2004;126:
15106–15119.
48. Malamas MS, Manas ES, McDevitt RE, Gunawan I, Xu ZB, Collini MD, Miller CP, Dinh T,
Henderson RA, Keith JC, Jr, Harris HA. Design and synthesis of aryl diphenolic azoles as potent
and selective estrogen receptor-b ligands. J Med Chem 2004;47:5021–5040.
49. Henke BR, Consler TG, Go N, Hale RL, Hohman DR, Jones SA, Lu AT, Moore LB, Moore JT,
Orband-Miller LA, Robinett RG, Shearin J, Spearing PK, Stewart EL, Turnbull PS, Weaver SL,
Williams SP, Wisely GB, Lambert MH. A new series of estrogen receptor modulators that display
selectivity for estrogen receptor b. J Med Chem 2002;45:5492–5505.
50. Veeneman GH, Loozen HJ, Mestres J, De Zwart EW. (Akzo Nobel NV) 10-Aryl-11-Hbenzo
[b]fluorene derivatives and analogs for medicinal use. WO0216316 [Chem Abstr 2002;136:
216545].
51. Meyers MJ, Sun J, Carlson KE, Katzenellenbogen BS, Katzenellenbogen JA. Estrogen receptor
subtype-selective ligands: Asymmetric synthesis and biological evaluation of cis- and trans-5,11-
Dialkyl-5,6,11,12-tetrahydrochrysenes. J Med Chem 1999;42:2456–2468.
52. Shiau AK, Barstad D, Radek J, Meyers MJ, Nettles KW, Katzenellenbogen BS,
Katzenellenbogen JA, Agard DA, Greene GL. Structural characterization of subtype-selective
ligand reveals a novel mode of estrogen receptor antagonism. Nat Struct Biol 2002;9:359–364.
53. Kuiper GGJM, Shughrue PJ, Merchenthaler I, Gustafsson J-A. The estrogen receptor b subtype:
A novel mediator of estrogen action in neuroendocrine systems. Front Neuroendocrin 1998;19:
253–286.
54. Mishra RG, Stanczyk FZ, Burry KA, Oparil S, Katzenellenbogen BS, Nealen ML,
Katzenellenbogen JA, Hermsmeyer RK. Metabolite ligands of estrogen receptor-beta reduce
primate coronary hyperreactivity. Am J Physiol Heart Circ Physiol 2006;290:H295–H303.
55. Petterson H, Holmberg L, Axelson M, Norlin M. CYP7B1-mediated metabolism of
dehydroepiandrosterone and 5a-androstane-3b,17b-diol—potential role(s) for estrogen signaling.
FEBS Journal 2008;275:1778–1789.
56. Gibb W, Hagerman DD. The specificity of the 3b-hydroxysteroid dehydrogenase activity of
bovine ovaries toward dehydroepiandrosterone and pregnenolone: Evidence for multiple enzymes.
Steroids 1976;28:31–41.
57. Blizzard TA, Gude C, Morgan II JD, Chan W, Birzin ET, Mojena M, Tudela C, Chen F,
Knecht K, Su Q, Kraker B, Mosley RT, Holmes MA, Sharma N, Fitzgerald PMD, Rohrer SP,
Hammond ML. Androstenediol analogs as ER-b-selective SERMs. Bioorg Med Chem Lett
2006;16:834–838.
58. Gellman SH. On the role of methionine residues in the sequence-independent recognition of
nonpolar protein surfaces. Biochemistry 1991;30:6633–6636.
59. Blizzard TA, Gude C, Chan W, Birzin ET, Mojena M, Tudela C, Chen F, Knecht K, Su Q,
Kraker B, Holmes MA, Rohrer SP, Hammond ML. Bridged androstenediol analogs as ER-
b-selective SERMs. Bioorg Med Chem Lett 2007;17:2944–2948.
60. Blizzard TA, Gude C, Morgan II JD, Chan W, Birzin ET, Mojena M, Tudela C, Chen F,
Knecht K, Su Q, Kraker B, Mosley RT, Holmes MA, Rohrer SP, Hammond ML. Androstene-
3,5-dienes as ER-b-selective SERMs. Bioorg Med Chem Lett 2007;17:6295–6298.
430 K MINUTOLOETAL.
Medicinal Research Reviews DOI 10.1002/med

61. Bartlett PA, Marlowe CK. Evaluation of intrinsic binding energy from a hydrogen bonding group
in an enzyme inhibitor. Science 1987;235:569–571.
62. Grobelny D, Goli UB, Galardy RE. Binding energetics of phosphorus-containing inhibitors of
thermolysin. Biochemistry 1989;28:4948–4951.
63. Peters O, Hillisch A, Thieme I, Elger W, Hegele-Hartung C, Kollenkirchen U, Fritzemeier K-H,
Patchev V. (Schering AG) Preparation of 8b-hydrocarbyl-substituted estratrienes for use as
selective estrogens. WO 2001077139 [Chem Abstr 2001;135:213936].
64. Mueller G, Kollenkirchen U, Kosemund D, Fritzemeier K-H. (Jenapharm G.m.b.H. & Co.)
Preparation of 19-nor-17a-pregna-1,3,5(10)-trien-17b-ols containing a 21,16a-lactone ring with a
preference for the estrogen alpha receptor. WO 2002026763 [Chem Abstr 2002;136:279608].
65. Hillisch A, Peters O, Kosemund D, Muller G, Walter A, Schneider B, Reddersen G, Elger W,
Fritzemeier KH. Dissecting physiological roles of estrogen receptor a and b with potent selective
ligands from structure-based design. Mol Endocrinol 2004;18:1599–1609.
66. Toschi L, Hilbig J, Wintermantel T, Engelhaupt A, Walter A, Fritzemeier KH, Hillisch A. Protein
structure-based prediction of animal model suitability for pharmacodynamic studies. Chem Med
Chem 2006;1:1237–1248.
67. Yamamoto Y, Shibata J, Yonekura K, Sato K, Hashimoto A, Aoyagi Y, Wierzba K, Yano S,
Asao T, Buzdar AU, Terada T. TAS-108, a novel oral steroidal antiestrogenic agent, is a pure
antagonist on estrogen receptor a and a partial agonist on estrogen receptor b with low
uterotrophic effect. Clin Cancer Res 2005;11:315–322.
68. Hsieh RW, Rajan SS, Sharma SK, Greene GL. Molecular characterization of a B-ring
unsaturated estrogen: Implications for conjugated equine estrogen components of Premarin.
Steroids 2008;73:59–68.
69. Asim M, El-Salfiti M, Qian Y, Choueiri C, Salari S, Cheng J, Shadnia H, Bal M, Pratt MAC,
Carlson KE, Katzenellenbogen JA, Wright JS, Durst T. Deconstructing estradiol: Removal of
B-ring generates compounds which are potent and subtype-selective estrogen receptor agonists.
Bioorg Med Chem Lett 2009;19:1250–1253.
70. Papapetropoulos A. A ginseng-derived oestrogen receptor b (ERb) agonist, Rb1 ginsenoside,
attenuates capillary morphogenesis. Br J Pharmacol 2007;152:172–174.
71. Cho J, Park W, Lee S, Ahn W, Lee Y. Ginsenoside-Rb1 from Panax ginseng C.A. Meyer activates
estrogen receptor-a and -b, independent of ligand binding. J Clin Endocrinol Metab 2004;89:
3510–3515.
72. Suzuki KM, Isohama Y, Maruyama H, Yamada Y, Narita Y, Ohta S, Araki Y, Miyata T,
Mishima S. Estrogenic activities of fatty acids and a sterol isolated from royal jelly. Evid-based
Complement Altern Med 2008;5:295–302.
73. Mersereau JE, Levy N, Staub RE, Baggett S, Zogovic T, Chow S, Ricke WA, Tagliaferri M,
Cohen I, Bjeldanes LF, Leitman DC. Liquiritigenin is a plant-derived highly selective estrogen
receptor b agonist. Mol Cell Endocrinol 2008;283:49–57. See also, erratum in Mol Cell Endocrinol
2008;295:121.
74. Mallavadhani UV, Narasimhan K, Sudhakar AVS, Mahapatra A, Li W, van Breemen RB. Three new
pentacyclic triterpenes and some flavonoids from the fruits of an Indian ayurvedic plantDendrophthoe
falcata and their estrogen receptor binding activity. Chem Pharm Bull 2006;54:740–744.
75. Escande A, Pillon A, Servant N, Cravedi JP, Larrea F, Muhn P, Nicolas JC, Cavailles V,
Balaguer P. Evaluation of ligand selectivity using reporter cell lines stably expressing estrogen
receptor alpha or beta. Biochem Pharmacol 2006;71:1459–1469.
76. Setchell KDR, Clerici C, Lephart ED, Cole SJ, Heenan C, Castellani D, Wolfe BE, Nechemias-
Zimmer L, Brown NM, Lund TD, Handa RJ, Heubi JE. S-Equol, a potent ligand for estrogen
receptor b, is the exclusive enantiomeric form of the soy isoflavone metabolite produced by human
intestinal bacterial flora. Am J Clin Nutr 2005;81:1072–1079.
77. Muthyala RS, Ju YH, Sheng S, Williams LD, Doerge DR, Katzenellenbogen BS, Helferich WG,
Katzenellenbogen JA. Equol, a natural estrogenic metabolite from soy isoflavones: Convenient
ESTROGENRECEPTOR bLIGANDS K 431
Medicinal Research Reviews DOI 10.1002/med

preparation and resolution of R- and S-equols and their differing binding and biological activity
through estrogen receptors alpha and beta. Bioorg Med Chem 2004;12:1559–1567.
78. Heemstra JM, Kerrigan SA, Doerge DR, Helferich WG, Boulanger WA. Total synthesis of
(S)-equol. Org Lett 2006;8:5441–5443. Erratum in: Org Lett 2007;9:379.
79. Suksamrarn A, Ponglikitmongkol M, Wongkrajang K, Chindaduang A, Kittidanairak S,
Jankam A, Yingyongnarongkul B, Kittipanumat N, Chokchaisiri R, Khetkam P, Piyachaturawat P.
Diarylheptanoids, new phytoestrogens from the rhizomes of Curcuma comosa: Isolation, chemical
modification and estrogenic activity evaluation. Bioorg Med Chem 2008;16:6891–6902.
80. De Angelis M, Stossi F, Carlson KE, Katzenellenbogen BS, Katzenellenbogen JA. Indazole
estrogens: Highly selective ligands for the estrogen receptor b. J Med Chem 2005;48:1132–1144.
81. Fink BE, Mortensen DS, Stauffer SR, Aron ZD, Katzenellenbogen JA. Novel structural templates
for estrogen-receptor ligands and prospects for combinatorial synthesis of estrogens. Chem Biol
1999;6:205–219.
82. Schopfer U, Schoeffter P, Bischoff SF, Nozulak J, Feuerbach D, Floersheim P. Toward selective
ERb agonists for central nervous system disorders: Synthesis and characterization of aryl
benzthiophenes. J Med Chem 2002;45:1399–1401.
83. Collini MD, Kaufman DH, Manas ES, Harris HA, Henderson RA, Xu ZB, Unwalla RJ,
Miller CP. 7-substituted 2-phenyl-benzofurans as ERb selective ligands. Bioorg Med Chem Lett
2004;14:4925–4929.
84. McDevitt RE, Malamas MS, Manas ES, Unwalla RJ, Xu ZB, Millere CP, Harris HA. Estrogen
receptor ligands: design and synthesis of new 2-arylindene-1-ones. Bioorg Med Chem Lett
2005;15:3137–3142.
85. Clegg NJ, Paruthiyil S, Leitman DC, Scanlan TS. Differential response of estrogen receptor
subtypes to 1,3-diarylindene and 2,3-diarylindene ligands. J Med Chem 2005;48:5989–6003.
86. Jernstedt H, Kruger L, Rhonnstad P, Noteberg D. Preparation of 2-phenyl indene derivatives as
estrogen receptor ligands. (KaroBio AB) WO 2008043567 [Chem Abstr 2008;148:471704].
87. Blizzard TA, Morgan II JD, Mosley RT, Birzin ET, Frisch K, Rohrer SP, Hammond ML.
2-phenylspiroindenes: A novel class of selective estrogen receptor modulators (SERMs). Bioorg
Med Chem Lett 2003;13:479–483.
88. Ullrich JW, Unwalla RJ, Singhaus RR, Jr, Harris HA, Mewshaw RE. Estrogen receptor b ligands:
Design and synthesis of new 2-phenyl-isoindole-1,3-diones. Bioorg Med Chem Lett
2007;17:118–122.
89. Chesworth R, Wessel MD, Heyden L, Mangano FM, Zawistoski MP, Gegnas L, Galluzzo D,
Lefker B, Cameron KO, Tickner J, Lu B, Castleberry TA, Petersen DN, Brault A, Perry P, Ng O,
Owen TA, Pan L, Ke H-Z, Brown TA, Thompson DD, DaSilva-Jardine P. Estrogen receptor bselective ligands: Discovery and SAR of novel heterocyclic ligands. Bioorg Med Chem Lett
2005;15:5562–5566.
90. Mewshaw RE, Edsall RJ, Jr, Yang C, Manas ES, Xu ZB, Henderson RA, Keith JC, Jr,
Harris HA. ERb ligands 3 exploiting two binding orientations of the 2-phenylnaphthalene scaffold
to achieve ERb selectivity. J Med Chem 2005;48:3953–3979.
91. Fang J, Akwabi-Ameyaw A, Britton JE, Katamreddy SR, Navas III F, Miller AB, Williams SP,
Gray DW, Orband-Miller LA, Shearin J, Heyer D. Synthesis of 3-alkyl naphthalenes as novel
estrogen receptor ligands. Bioorg Med Chem Lett 2008;18:5075–5077.
92. Willson TM, Henke BR, Momtahen TM, Charifson PS, Batchelor KW, Lubahn DB, Moore LB,
Oliver BB, Sauls HR, Triantafillou JA, Wolfe SG,Baer PG. 3-[4-(1,2-Diphenylbut-1-enyl)pheny-
l]acrylic acid: A non-steroidal estrogen with functional selectivity for bone over uterus in rats.
J Med Chem 1994;37:1550–1552.
93. Wittmann BM, Sherk A, McDonnell DP. Definition of functionally important mechanistic
differences among selective estrogen receptor down-regulators. Cancer Res 2007;67:9549–9560.
94. Wu YL, Yang X, Ren Z, McDonnell DP, Norris JD, Willson TM, Greene GL. Structural basis for
an unexpected mode of SERM-mediated ER antagonism. Mol Cell 2005;18:413–424.
432 K MINUTOLOETAL.
Medicinal Research Reviews DOI 10.1002/med

95. Vu AT, Cohn ST, Manas ES, Harris HA, Mewshaw RE. ERb ligands. Part 4: Synthesis and
structure–activity relationships of a series of 2-phenylquinoline derivatives. Bioorg Med Chem
Lett 2005;15:4520–4525.
96. Wallace OB, Lauwers KS, Jones SA, Dodge JA. Tetrahydroquinoline-based selective estrogen
receptor modulators (SERMs). Bioorg Med Chem Lett 2003;13:1907–1910.
97. Chesworth R, Zawistoski MP, Lefker BA, Cameron KO, Day RF, Mangano FM, Rosati RL,
Colella S, Petersen DN, Brault A, Lu B, Pan LC, Perry P, Ng O, Castleberry TA, Owen TA,
Brown TA, Thompson DD, DaSilva-Jardine P. Tetrahydroisoquinolines as subtype selective
estrogen agonists/antagonists. Bioorg Med Chem Lett 2004;14:2729–2733.
98. Kumpulainen H, Mahonen N, Laitinen M-L, Jaurakkajarvi M, Raunio H, Juvonen RO,
Vepsalainen J, Jarvinen T, Rautio J. Evaluation of hydroxyimine as cytochrome P450-selective
prodrug structure. J Med Chem 2006;49:1207–1211.
99. Minutolo F, Bertini S, Papi C, Carlson KE, Katzenellenbogen JA, Macchia M. Salicylaldoxime
moiety as a phenolic ‘‘A-ring’’ substitute in estrogen receptor ligands. J Med Chem 2001;44:
4288–4291.
100. Minutolo F, Antonello M, Bertini S, Rapposelli S, Rossello A, Sheng S, Carlson KE,
Katzenellenbogen JA, Macchia M. Synthesis, binding affinity, and transcriptional activity of
hydroxy- and methoxy-substituted 3,4-diarylsalicylaldoximes on estrogen receptors alpha and
beta. Bioorg Med Chem 2003;11:1247–1257.
101. Minutolo F, Antonello M, Bertini S, Ortore G, Placanica G, Rapposelli S, Sheng S, Carlson KE,
Katzenellenbogen BS, Katzenellenbogen JA, Macchia M. Novel estrogen receptor ligands based
on an anthranylaldoxime structure: Role of the phenol-type pseudocycle in the binding process.
J Med Chem 2003;46:4032–4042.
102. Tuccinardi T, Bertini S, Martinelli A, Minutolo F, Ortore G, Placanica G, Prota G, Rapposelli S,
Carlson KE, Katzenellenbogen JA, Macchia M. Synthesis of anthranylaldoxime derivatives as
estrogen receptor ligands and computational prediction of binding modes. J Med Chem 2006;49:
5001–5012.
103. Minutolo F, Bellini R, Bertini S, Carboni I, Lapucci A, Pistolesi L, Prota G, Rapposelli S, Solati F,
Tuccinardi T, Martinelli A, Stossi F, Carlson KE, Katzenellenbogen BS, Katzenellenbogen JA,
Macchia M. Monoaryl-substituted salicylaldoximes as ligands for estrogen receptor b. J Med Chem
2008;51:1344–1351.
104. Minutolo F, Bertini S, Granchi C, Marchitiello T, Prota G, Rapposelli S, Tuccinardi T, Martinelli A,
Gunther JR, Carlson KE, Katzenellenbogen JA, Macchia M. Structural evolutions of salicylald-
oximes as selective agonists for estrogen receptor b. J Med Chem 2009;52:858–867.
105. Edsall RJ, Jr, Harris HA, Manas ES, Mewshaw RE. ERb ligands. Part 1: The discovery of ERbselective ligands which embrace the 4-hydroxy-biphenyl template. BioorgMed Chem 2003;11:3457–3474.
106. Yang C, Edsall R, Jr, Harris HA, Zhang X, Manas ES, Mewshaw RE. ERb ligands. Part 2:
Synthesis and structure–activity relationships of a series of 4-hydroxy-biphenyl-carbaldehyde
oxime derivatives. Bioorg Med Chem 2004;12:2553–2570.
107. Mewshaw RE, Bowen SM, Harris HA, Xu ZB, Manas ES, Cohn ST. ERb ligands. Part 2:
Synthesis and structure–activity relationships of a series of 40-hydroxyphenyl-aryl-carbaldehyde
oxime derivatives. Bioorg Med Chem Lett 2007;17:902–906.
108. Richardson TI, Norman BH, Lugar CW, Jones SA, Wang Y, Durbin JD, Krishnan V, Dodge JA.
Benzopyrans as selective estrogen receptor b agonists (SERBAs). Part 2: Structure–activity
relationship studies on the benzopyran scaffold. Bioorg Med Chem Lett 2007;17:3570–3574.
109. Norman BH, Dodge JA, Richardson TI, Borromeo PS, Lugar CW, Jones SA, Chen K, Wang Y,
Durst GL, Barr RJ, Montrose-Rafizadeh C, Osborne HE, Amos RM, Guo S, Boodhoo A,
Krishnan V. Benzopyrans are selective estrogen receptor b agonists with novel activity in models
of benign prostatic hyperplasia. J Med Chem 2006;49:6155–6157.
110. Richardson TI, Dodge JA, Durst GL, Pfeifer LA, Shah J, Wang Y, Durbin JD, Krishnan V,
Norman BH. Benzopyrans as selective estrogen receptor b agonists (SERBAs). Part 3: Synthesis of
ESTROGENRECEPTOR bLIGANDS K 433
Medicinal Research Reviews DOI 10.1002/med

cyclopentanone and cyclohexanone intermediates for C-ring modification. Bioorg Med Chem Lett
2007;17:4824–4828.
111. Norman BH, Richardson TI, Dodge JA, Pfeifer LA, Durst GL, Wang Y, Durbin JD, Krishnan V,
Dinn SR, Liu S, Reilly JE, Ryter KT. Benzopyrans as selective estrogen receptor b agonists
(SERBAs). Part 4: Functionalization of the benzopyran A-ring. Bioorg Med Chem Lett
2007;17:5082–5085.
112. Richardson TI, Dodge JA, Wang Y, Durbin JD, Krishnan V, Norman BH. Benzopyrans as
selective estrogen receptor b agonists (SERBAs). Part 5: Combined A- and C-ring structure–
activity relationship studies. Bioorg Med Chem Lett 2007;17:5563–5566.
113. Krishnan V, Kroin JS, Norman BH, Thomas EM. Preparation of substituted tetralins as
selective estrogen receptor-b agonists. (Eli Lilly & Co.) WO 2006088716 [Chem Abstr 2006;145:
271511].
114. Wilkening RR, Ratcliffe RW, Tynebor EC, Wildonger KJ, Fried AK, Hammond ML,
Mosley RT, Fitzgerald PMD, Sharma N, McKeever BM, Nilsson S, Carlquist M, Thorsell A,
Locco L, Katz R, Frisch K, Birzin ET, Wilkinson HA, Mitra S, Cai S, Hayes EC, Schaeffer JM,
Rohrer SP. The discovery of tetrahydrofluorenones as a new class of estrogen receptor b-subtypeselective ligands. Bioorg Med Chem Lett 2006;16:3489–3494.
115. Wildonger KJ, Ratcliffe RW, Mosley RT, Hammond ML, Birzin ET, Rohrer SP. Tetrahydro-
fluorenones with conformationally restricted side chains as selective estrogen receptor beta
ligands. Bioorg Med Chem Lett 2006;16:4462–4466.
116. Scott JP, Ashwood MS, Brands KMJ, Brewer SE, Cowden CJ, Dolling UH, Emerson KM,
Gibb AD, Goodyear A, Oliver SF, Stewart GW, Wallace DJ. Development of a phase transfer
catalyzed asymmetric synthesis for an estrogen receptor beta selective agonist. Org Process Res
Dev 2008;12:723–730.
117. Wilkening RR, Ratcliffe RW, Fried AK, Meng D, Sun W, Colwell L, Lambert S, Greenlee M,
Nilsson S, Thorsell A, Mojena M, Tudela C, Frisch K, Chan W, Birzin ET, Rohrer SP,
Hammond ML. Estrogen receptor b-subtype selective tetrahydrofluorenones: Use of a fused
pyrazole as a phenol bioisostere. Bioorg Med Chem Lett 2006;16:3896–3901.
118. Kinzel O, Fattori D, Muraglia E, Gallinari P, Nardi MC, Paolini C, Roscilli G, Toniatti C,
Gonzalez Paz O, Laufer R, Lahm A, Tramontano A, Cortese R, De Francesco R, Ciliberto G,
Koch U. A structure-guided approach to an orthogonal estrogen-receptor-based gene switch
activated by ligands suitable for in vivo studies. J Med Chem 2006;49:5404–5407.
119. Parker DL, Jr, Meng D, Ratcliffe RW, Wilkening RR, Sperbeck DM, Greenlee ML, Colwell LF,
Lambert S, Birzin ET, Frisch K, Rohrer SP, Nilsson S, Thorsell AG, Hammond ML. Triazolo-
tetrahydrofluorenones as selective estrogen receptor beta agonists. Bioorg Med Chem Lett
2006;16:4652–4656.
120. Mewshaw RE, Edsall RJ, Cohn ST, Harris HA, Keith JC, Jr, Albert LM. Substituted 6H-
dibenzo[c,h]chromenes active as estrogenic agents, and their preparation, pharmaceutical
compositions, and use. (Wyeth Ltd) WO 2003051863 [Chem Abstr 2003;139:69149].
121. Keith JC, Vlasuk GP. Use of ER-beta selective ligands for treating acute lung injuries. (Wyeth
Ltd) US 2008182872 [Chem Abstr 2008;149:239347].
122. Mewshaw RE, Edsall RJ, Jr. Preparation of dibenzo[c,g]chromene derivatives as ERb selective
ligands. (Wyeth Ltd) WO 2005082880 [Chem Abstr 2005;143:286286].
123. Vu AT, Campbell AN, Harris HA, Unwalla RJ, Manas ES, Mewshaw RE. ERb ligands Part 6:
6H-Chromeno[4,3-b]quinolines as a new series of estrogen receptor b-selective ligands. Bioorg
Med Chem Lett 2007;17:4053–4056.
124. Sun W, Cama LD, Birzin ET, Warrier S, Locco L, Mosley R, Hammond ML, Rohrer SP. 6H-
Benzo[c]chromen-6-one derivatives as selective ERb agonists. Bioorg Med Chem Lett 2006;16:
1468–1472.
125. Barrett I, Meegan MJ, Hughes RB, Carr M, Knox AJS, Artemenko N, Golfis G, Zisterer DM,
Lloyd DG. Synthesis, biological evaluation, structural–activity relationship, and docking study for
434 K MINUTOLOETAL.
Medicinal Research Reviews DOI 10.1002/med

a series of benzoxepin-derived estrogen receptor modulators. Bioorg Med Chem 2008;16:
9554–9573.
126. Yang W, Wang Y, Ma Z, Golla R,Stouch T, Seethala R, Johnson S, Zhou R, Gungor T,
Feyen JHM, Dickson JK, Jr. Synthesis and structure–activity relationship of 3-arylbenzoxazines
as selective estrogen receptor b agonists. Bioorg Med Chem Lett 2004;14:2327–2330.
127. Gungor T, Chen Y, Golla R, Ma Z, Corte JR, Northrop JP, Bin B, Dickson JK, Stouch T,
Zhou R, Johnson SE, Seethala R, Feyen JHM. Synthesis and characterization of
3-arylquinazolinone and 3-arylquinazolinethione derivatives as selective estrogen receptor beta
modulators. J Med Chem 2006;49:2440–2455.
128. De Angelis M, Stossi F, Waibel M, Katzenellenbogen BS, Katzenellenbogen JA. Isocoumarins as
estrogen receptor beta selective ligands: Isomers of isoflavone phytoestrogens and their
metabolites. Bioorg Med Chem 2005;13:6529–6542.
129. Sibley R, Hatoum-Mokdad H, Schoenleber R, Musza L, Stirtan W, Marrero D, Carley W,
Xiao H, Dumas J. A novel estrogen receptor ligand template. Bioorg Med Chem Lett
2003;13:1919–1922.
130. Hamann LG, Meyer JH, Ruppar DA, Marschke KB, Lopez FJ, Allegretto EA, Karanewskya DS.
Structure–activity relationships and sub-type selectivity in an oxabicyclic estrogen receptor a/bagonist scaffold. Bioorg Med Chem Lett 2005;15:1463–1466.
131. Hsieh RW, Rajan SS, Sharma SK, Guo Y, DeSombre ER, Mrksich M, Greene GL. Identification
of ligands with bicyclic scaffolds provides insights into mechanisms of estrogen receptor subtype
selectivity. J Biol Chem 2006;281:17909–17919.
132. Olsson R, Hyldtoft L, Piu F, Gustafsson M. (ACADIA Pharmaceuticals) Compounds with
activity at estrogen receptor. WO2005108337 [Chem Abstr 2005;143:459876].
133. Piu F, Cheevers C, Hyldtoft L, Gardell LR, Del Tredici AL, Andersen CB, Fairbairn LC,
Lund BW, Gustafsson M, Schiffer HH, Donello JE, Olsson R, Gil DW, Brann MR. Broad
modulation of neuropathic pain states by a selective estrogen receptor beta agonist. Eur J
Pharmacol 2008;590:423–429.
134. Gardell LR, Hyldtoft L, Del Tredici AL, Andersen CB, Fairbairn LC, Lund BW, Gustafsson M,
Brann MR, Olsson R, Piu F. Differential modulation of inflammatory pain by a selective estrogen
receptor beta agonist. Eur J Pharmacol 2008;592:158–159.
135. Zimmermann J, von Angerer E. Estrogenic and antiestrogenic activities of 2,4-diphenylfuran-
based ligands of estrogen receptors a and b. J Steroid Biochem Mol Biol 2007;104:259–268.
136. Ghosh U, Ganessunker D, Sattigeri VJ, Carlson KE, Mortensen DJ, Katzenellenbogen BS,
Katzenellenbogen JA. Estrogenic diazenes: Heterocyclic non-steroidal estrogens of unusual
structure with selectivity for estrogen receptor subtypes. Bioorg Med Chem 2003;11:629–657.
137. Pirali T, Gatti S, Di Brisco R, Tacchi S, Zaninetti R, Brunelli E, Massarotti A, Sorba G,
Canonico PL, Moro L, Genazzani AA, Tron GC, Billington RA. Estrogenic analogues
synthesized by click chemistry. Chem Med Chem 2007;2:437–440.
138. Kolb HC, Sharpless KB. The growing impact of click chemistry on drug discovery. Drug Disc
Today 2003;8:1128–1137.
139. Begtrup M, Nielsen CJ, Nygaard L, Samdal S, Sjøgren CE, Sørensen GO. The molecular structure
and tautomer equilibrium of gaseous 1,2,3-triazole studied by microwave spectroscopy, electron
diffraction and Ab initio calculations. Acta Chem Scand 1988;A42:500–514.
140. Pulkkinen JT, Honkakoski P, Perakyla M, Berczi I, Laatikainen R. Synthesis and evaluation of
estrogen agonism of diaryl 4,5-dihydroisoxazoles, 3-hydroxyketones, 3-methoxyketones, and 1,3-
diketones: A compound set forming a 4D molecular library. J Med Chem 2008;51:3562–3571.
141. Ohta K, Chiba Y, Ogawa T, Endo Y. Promising core structure for nuclear receptor ligands:
Design and synthesis of novel estrogen receptor ligands based on diphenylamine skeleton. Bioorg
Med Chem Lett 2008;16:5050–5053.
142. Katzenellenbogen JA, Katzenellenbogen BS, Compton DR. Benzenesulfonamides as selective
estrogen receptor ligands. US2007021495 [Chem Abstr 2007;146:156287].
ESTROGENRECEPTOR bLIGANDS K 435
Medicinal Research Reviews DOI 10.1002/med

143. De Angelis M, Katzenellenbogen JA. Ring nitrogen-substituted non-steroidal estrogens: Pyridine
and pyrimidine analogs of the phenol in deoxyhexestrol experience resonance constraints on
preferred ligand conformation. Bioorg Med Chem Lett 2004;14:5835–5839.
144. Waibel M, De Angelis M, Stossi F, Kieser KJ, Carlson KE, Katzenellenbogen BS,
Katzenellenbogen JA. Bibenzyl- and stilbene-core compounds with non-polar linker atom
substituents as selective ligands for estrogen receptor beta. Eur J Med Chem 2009;44:3412–3424.
145. Waibel M, Kieser KJ, Carlson KE, Stossi F, Katzenellenbogen BS, Katzenellenbogen JA.
Phenethyl pyridines with non-polar internal substitutents as selective ligands for estrogen receptor
beta. Eur J Med Chem 2009;44:3560–3570.
146. Sarachine M, Janjic JM, Wipf P, Day BW. Biphenyl C-cyclopropylalkylamides: New scaffolds for
targeting estrogen receptor b. Bioorg Med Chem Lett 2009;19:2404–2408.
147. Lin Z, Shen H, Huang J, Chen S, Chen L, Chen J, Liu G, Jiang H, Shen X. Butyl
4-(butyryloxy)benzoate functions as a new selective estrogen receptor b agonist and induces
GLUT4 expression in CHO-K1 cells. J Steroid Biochem 2008;110:150–156.
148. Chen W, Lin Z, Ning M, Yang C, Yan X, Xie Y, Shen X, Wang M-W. Aza analogues of equol:
Novel ligands for estrogen receptor b. Bioorg Med Chem 2007;15:5828–5836.
149. Kirkiacharian S, Lormier AT, Chidiack H, Bouchoux F, Evelyne Cerede E. Synthesis and binding
affinity to human a and b estrogen receptors of various 7-hydroxycoumarins substituted at 4- and
3,4- positions. Farmaco 2004;59:981–986.
150. Roelens F, Heldring N, Dhooge W, Bengtsson M, Comhaire F, Gustafsson JA, Treuter E, De
Keukeleire D. Subtle side-chain modifications of the hop phytoestrogen 8-prenylnaringenin result
in distinct agonist/antagonist activity profiles for estrogen receptors a and b. J Med Chem
2006;49:7357–7365.
151. De Keukeleire D, Roelens F, Dhooge W. (Universiteit Gent) Preparation of naringenin
derivatives with selectivity on estrogen receptors. WO2007053915 [Chem Abstr 2007;146:521587].
152. Compton DR, Carlson KE, Katzenellenbogen JA. Pyrazolo[1,5-a]pyrimidines as estrogen
receptor ligands: Defining the orientation of a novel heterocyclic core. Bioorg Med Chem Lett
2004;14:5681–5684.
153. Compton DR, Sheng S, Carlson KE, Rebacz NA, Lee IY, Katzenellenbogen BS, Katzenellenbo-
gen JA. Pyrazolo[1,5-a]pyrimidines: Estrogen receptor ligands possessing estrogen receptor bantagonist activity. J Med Chem 2004;47:5872–5893.
154. Zhou HB, Sheng S, Compton DR, Kim Y, Joachimiak A, Sharma S, Carlson KE,
Katzenellenbogen BS, Nettles KW, Greene GL, Katzenellenbogen JA. Structure-guided
optimization of estrogen receptor binding affinity and antagonist potency of pyrazolopyrimidines
with basic side chains. J Med Chem 2007;50:399–403.
155. Muthyala RS, Sheng S, Carlson KE, Katzenellenbogen BS, Katzenellenbogen JA. Bridged
bicyclic cores containing a 1,1-diarylethylene motif are high-affinity subtype-selective ligands for
the estrogen receptor. J Med Chem 2003;46:1589–1602.
156. Seo JW, Comninos JS, Chi DY, Kim DW, Carlson KE, Katzenellenbogen JA. Fluorine-
substituted cyclofenil derivatives as estrogen receptor ligands: Synthesis and structure-affinity
relationship study of potential positron emission tomography agents for imaging estrogen
receptors in breast cancer. J Med Chem 2006;49:2496–2511.
157. Kim SH, Katzenellenbogen JA. Hormone–PAMAM dendrimer conjugates: Polymer
dynamics and tether structure affect ligand access to receptors. Angew Chem Int Ed 2006;45:
7243–7248.
158. Muthyala RS, Carlson KE, Katzenellenbogen JA. Exploration of the bicyclo[3.3.1]nonane system
as a template for the development of new ligands for the estrogen receptor. Bioorg Med Chem
2003;13:4485–4488.
159. Zhou H-B, Comninos JS, Stossi F, Katzenellenbogen BS, Katzenellenbogen JA. Synthesis and
evaluation of estrogen receptor ligands with bridged oxabicyclic cores containing a diarylethylene
motif: Estrogen antagonists of unusual structure. J Med Chem 2005;48:7261–7274.
436 K MINUTOLOETAL.
Medicinal Research Reviews DOI 10.1002/med

160. Zhou HB, Nettles KW, Bruning JB, Kim Y, Joachimiak A, Sharma S, Carlson KE, Stossi F,
Katzenellenbogen BS, Greene GL, Katzenellenbogen JA. Elemental isomerism: A boron-nitrogen
surrogate for a carbon-carbon double bond increases the chemical diversity of estrogen receptor
ligands. Chem Biol 2007;14:659–669.
161. Jeanes HL, Tabor C, Black D, Ederveen A, Gray GA. Oestrogen-mediated cardioprotection
following ischaemia and reperfusion is mimicked by an oestrogen receptor (ER) a agonist and
unaffected by an ERb antagonist. J Endocrinol 2008;197:493–501.
162. Yoo J, Dence CS, Sharp TL, Katzenellenbogen JA, Welch MJ. Synthesis of an estrogen receptor
b-selective radioligand: 5-[18F]fluoro-(2R�,3S�)-2,3-bis(4-hydroxyphenyl) pentanenitrile and
comparison of in vivo distribution with 16a-[18F]fluoro-17b-estradiol. J Med Chem 2005;48:
6366–6378.
163. Moon BS, Katzenellenbogen JA, Cheon GJ, Chi DY, Lee KC, An GI. Synthesis and evaluation of
estrogen receptor b-selective ligands: Fluoroalkylated indazole estrogens. Bull Korean Chem Soc
2008;29:1107–1114.
164. Moon BS, Carlson KE, Katzenellenbogen JA, Choi TH, Chi DY, Kim JY, Cheon GJ, Koh KY,
Lee KC, An G. Synthesis and evaluation of aryl-substituted diarylproprionitriles, selective ligands
for estrogen receptor b, as positron-emission tomographic imaging agents. Bioorg Med Chem
2009;17:3479–3488.
165. Zhou D, Zhou H, Jenks CC, Lewis JS, Katzenellenbogen JA, Welch MJ. Bromination from the
macroscopic level to the tracer radiochemical level: 76Br radiolabeling of aromatic compounds via
electrophilic substitution. Bioconj Chem 2009;20:808–816.
166. Ramesh C, Bryant B, Nayak T, Revankar CM, Anderson T, Carlson KE, Katzenellenbogen JA,
Sklar LA, Norenberg JP, Prossnitz ER, Arterburn JB. Linkage effects on binding affinity and
activation of GPR30 and estrogen receptors ERa/b with tridentate pyridin-2-yl hydrazine
tricarbonylRe/99mTc(I) chelates. J Am Chem Soc 2006;128:14476–14477.
167. Nayak TK, Hathaway HJ, Ramesh C, Arterburn JB, Dai D, Sklar LA, Norenberg JP,
Prossnitz ER. Preclinical development of a neutral, estrogen receptor-targeted, tridentate
99mTc(I)-estradiol-pyridin-2-yl hydrazine derivative for imaging of breast and endometrial
cancers. J Nucl Med 2008;49:978–986.
168. Causey PW, Besanger TR, Valliant JF. Synthesis and screening of mono- and di-aryl technetium
and rhenium metallocarboranes. A new class of probes for the estrogen receptor. J Med Chem
2008;51:2833–2844.
169. Vessieres A, Top S, Pigeon P, Hillard E, Boubeker L, Spera D, Jaouen G. Modification of the
estrogenic properties of diphenols by the incorporation of ferrocene. Generation of antiproli-
ferative effects in vitro. J Med Chem 2005;48:3937–3940.
170. Hillard E, Vessieres A, Le Bideau F, Plazuk D, Spera D, Huche M, Jaouen G. A series of
unconjugated ferrocenyl phenols: Prospects as anticancer agents. ChemMedChem 2006;1:551–559.
171. Anstead GM, Carlson KE, Katzenellenbogen JA. The estradiol pharmacophore: Ligand
structure–estrogen receptor binding affinity relationships and a model for the receptor binding
site. Steroids 1997;62:268–303.
172. Hartman J, Strom A, Gustafsson J-A. Estrogen receptor beta in breast cancer—diagnostic and
therapeutic implications. Steroids 2009;74:635–641.
173. Palmieri C, Cheng GJ, Saji S, Zelada-Hedman M, Warri A, Weihua Z, Van Noorden S,
Wahlstrom T, Coombes RC, Warner M, Gustafsson J-A. Estrogen receptor beta in breast cancer.
Endocr Relat Cancer 2002;9:1–13.
174. Roger P, Esslimani-Sahla M, Delfour C, Lazennec G, Rochefort H, Maudelonde T. Expression of
estrogen receptors alpha and beta in early steps of human breast carcinogenesis. Adv Exp Med
Biol 2008;617:139–148.
175. Roger P, Sahla ME, Makela S, Gustafsson J-A, Baldet P, Rochefort H. Decreased expression of
estrogen receptor beta protein in proliferative preinvasive mammary tumors. Cancer Res
2001;61:2537–2541.
ESTROGENRECEPTOR bLIGANDS K 437
Medicinal Research Reviews DOI 10.1002/med

176. Speirs V. Oestrogen receptor beta in breast cancer: Good, bad or still too early to tell? J Pathol
2002;197:143–147.
177. Speirs V, Carder PJ, Lane S, Dodwell D, Lansdown MR, Hanby AM. Oestrogen receptor beta:
What it means for patients with breast cancer. Lancet Oncol 2004;5:174–181.
178. Shaaban AM, O’Neill PA, Davies MP, Sibson R, West CR, Smith PH, Foster CS. Declining
estrogen receptor-beta expression defines malignant progression of human breast neoplasia. Am J
Surg Pathol 2003;27:1502–1512.
179. Ariazi EA, Ariazi JL, Cordera F, Jordan VC. Estrogen receptors as therapeutic targets in breast
cancer. Curr Top Med Chem 2006;6:181–202.
180. Cheng G, Weihua Z, Warner M, Gustafsson J-A. Estrogen receptors ER alpha and ER beta in
proliferation in the rodent mammary gland. Proc Natl Acad Sci USA 2004;101:3739–3746.
181. Stauffer SR, Coletta CJ, Tedesco R, Nishiguchi G, Carlson K, Sun J, Katzenellenbogen BS,
Katzenellenbogen JA. Pyrazole ligands: Structure-affinity/activity relationships and estrogen
receptor-a selective agonists. J Med Chem 2000;43:4934–4947.
182. Sotoca AM, van den Berg H, Vervoort J, van der Saag P, Strom A, Gustafsson J-A, Rietjens I,
Murk AJ. Influence of cellular ERalpha/ERbeta ratio on the ERalpha-agonist induced proli-
feration of human T47D breast cancer cells. Toxicol Sci 2008;105:303–311.
183. Helguero LA, Faulds MH, Gustafsson J-A, Haldosen LA. Estrogen receptors alfa (ERalpha) and
beta (ERbeta) differentially regulate proliferation and apoptosis of the normal murine mammary
epithelial cell line HC11. Oncogene 2005;24:6605–6616.
184. Green CA, Peter MB, Speirs V, Shaaban AM. The potential role of ER beta isoforms in the
clinical management of breast cancer. Histopathology 2008;53:374–380.
185. Skliris GP, Leygue E, Curtis-Snell L, Watson PH, Murphy LC. Expression of oestrogen receptor-
beta in oestrogen receptor-alpha negative human breast tumours. Br J Cancer 2006;95:616–626.
186. Skliris GP, Leygue E, Watson PH, Murphy LC. Estrogen receptor alpha negative breast cancer
patients: Estrogen receptor beta as a therapeutic target. J Steroid Biochem Mol Biol 2008;109:1–10.
187. Royuela M, de Miguel MP, Bethencourt FR, Sanchez-Chapado M, Fraile B, Arenas MI,
Paniagua R. Estrogen receptors alpha and beta in the normal, hyperplastic and carcinomatous
human prostate. J Endocrinol 2001;168:447–454.
188. Prins GS, Korach KS. The role of estrogens and estrogen receptors in normal prostate growth and
disease. Steroids 2008;73:233–244.
189. Carruba G. Estrogen and prostate cancer: An eclipsed truth in an androgen-dominated scenario.
J Cell Biochem 2007;102:899–911.
190. Fixemer T, Remberger K, Bonkhoff H. Differential expression of the estrogen receptor beta
(ERbeta) in human prostate tissue, premalignant changes, and in primary, metastatic, and
recurrent prostatic adenocarcinoma. Prostate 2003;54:79–87.
191. Cheng J, Lee EJ, Madison LD, Lazennec G. Expression of estrogen receptor beta in prostate
carcinoma cells inhibits invasion and proliferation and triggers apoptosis. FEBS Lett 2004;566:169–172.
192. Lai JS, Brown LG, True LD, Hawley SJ, Etzioni RB, Higano CS, Ho SM, Vessella RL, Corey E.
Metastases of prostate cancer express estrogen receptor-beta. Urology 2004;64:814–820.
193. Bonkhoff H, Motherby H, Fixemer T. New insights into the role of estogens and their receptors in
prostate cancer. Urologe A 2003;42:1594–1601.
194. Wong NA, Malcomson RD, Jodrell DI, Groome NP, Harrison DJ, Saunders PT. ERbeta isoform
expression in colorectal carcinoma: An in vivo and in vitro study of clinicopathological and
molecular correlates. J Pathol 2005;207:53–60.
195. Campbell-Thompson M, Lynch IJ, Bhardwaj B. Expression of estrogen receptor (ER) subtypes
and ERbeta isoforms in colon cancer. Cancer Res 2001;61:632–640.
196. Ivanova MM, Mazhawidza W, Dougherty SM, Minna JD, Klinge CM. Activity and intracellular
location of estrogen receptors alpha and beta in human bronchial epithelial cells. Mol Cell
Endocrinol 2009;305:12–21.
438 K MINUTOLOETAL.
Medicinal Research Reviews DOI 10.1002/med

197. Stabile LP, Siegfried JM. Estrogen receptor pathways in lung cancer. Curr Oncol Rep 2004;6:
259–267.
198. Ali G, Donati V, Loggini B, Servadio A, Dell’Omodarme M, Prati MC, Camacci T, Lucchi M,
Melfi F, Mussi A, Fontanini G. Different estrogen receptor beta expression in distinct histologic
subtypes of lung adenocarcinoma. Hum Pathol 2008;39:1465–1473.
199. Wu CT, Chang YL, Shih JY, Lee YC. The significance of estrogen receptor beta in 301 surgically
treated non-small cell lung cancers. J Thorac Cardiovasc Surg 2005;130:979–986.
200. Hershberger PA, Stabile LP, Kanterewicz B, Rothstein ME, Gubish CT, Land S, Shuai Y,
Siegfried JM, Nichols M. Estrogen receptor beta (ERbeta) subtype-specific ligands increase
transcription, p44/p42 mitogen activated protein kinase (MAPK) activation and growth in human
non-small cell lung cancer cells. J Steroid Biochem Mol Biol 2009;116:102–109.
201. Hegele-Hartung C, Siebel P, Peters O, Kosemund D, Muller G, Hillisch A, Walter A,
Kraetzschmar J, Fritzemeier KH. Impact of isotype-selective estrogen receptor agonists on
ovarian function. Proc Natl Acad Sci USA 2004;101:5129–5134.
202. Wahlgren A, Svechnikov K, Strand ML, Jahnukainen K, Parvinen M, Gustafsson J-A, Soder O.
Estrogen receptor beta selective ligand 5alpha-Androstane-3beta, 17beta-diol stimulates sperma-
togonial deoxyribonucleic acid synthesis in rat seminiferous epithelium in vitro. Endocrinology
2008;149:2917–2922.
203. Harris HA, Katzenellenbogen JA, Katzenellenbogen BS. Characterization of the biological roles
of the estrogen receptors, ERalpha and ERbeta, in estrogen target tissues in vivo through the use
of an ERalpha-selective ligand. Endocrinology 2002;143:4172–4177.
204. Opas EE, Gentile MA, Kimmel DB, Rodan GA, Schmidt A. Estrogenic control of
thermoregulation in ERalphaKO and ERbetaKO mice. Maturitas 2006;53:210–216.
205. Grodstein F, Manson JE, Stampfer MJ. Hormone therapy and coronary heart disease: The role of
time since menopause and age at hormone initiation. J Womens Health (Larchmt) 2006;15:35–44.
206. Brouchet L, Krust A, Dupont S, Chambon P, Bayard F, Arnal JF. Estradiol accelerates
reendothelialization in mouse carotid artery through estrogen receptor-alpha but not estrogen
receptor-beta. Circulation 2001;103:423–428.
207. Zhai P, Eurell TE, Cooke PS, Lubahn DB, Gross DR. Myocardial ischemia-reperfusion injury in
estrogen receptor-alpha knockout and wild-type mice. Am J Physiol Heart Circ Physiol
2000;278:H1640–H1647.
208. Zhu Y, Bian Z, Lu P, Karas RH, Bao L, Cox D, Hodgin J, Shaul PW, Thoren P, Smithies O,
Gustafsson J-A, Mendelsohn ME. Abnormal vascular function and hypertension in mice deficient
in estrogen receptor beta. Science 2002;295:505–508.
209. Pelzer T, Loza PA, Hu K, Bayer B, Dienesch C, Calvillo L, Couse JF, Korach KS, Neyses L,
Ertl G. Increased mortality and aggravation of heart failure in estrogen receptor-beta knockout
mice after myocardial infarction. Circulation 2005;111:1492–1498.
210. Skavdahl M, Steenbergen C, Clark J, Myers P, Demianenko T, Mao L, Rockman HA,
Korach KS, Murphy E. Estrogen receptor-beta mediates male-female differences in the development
of pressure overload hypertrophy. Am J Physiol Heart Circ Physiol 2005;288:H469–H476.
211. Cvoro A, Tatomer D, Tee MK, Zogovic T, Harris HA, Leitman DC. Selective estrogen receptor-
beta agonists repress transcription of proinflammatory genes. J Immunol 2008;180:630–636.
212. Harris HA, Bruner-Tran KL, Zhang X, Osteen KG, Lyttle CR. A selective estrogen receptor-beta
agonist causes lesion regression in an experimentally induced model of endometriosis. Hum
Reprod 2005;20:936–941.
213. Cristofaro PA, Opal SM, Palardy JE, Parejo NA, Jhung J, Keith JC, Jr, Harris HA. WAY-
202196, a selective estrogen receptor-beta agonist, protects against death in experimental septic
shock. Crit Care Med 2006;34:2188–2193.
214. Yu HP, Hsieh YC, Suzuki T, Shimizu T, Choudhry MA, Schwacha MG, Chaudry IH. Salutary
effects of estrogen receptor-beta agonist on lung injury after trauma-hemorrhage. Am J Physiol
Lung Cell Mol Physiol 2006;290:L1004–L1009.
ESTROGENRECEPTOR bLIGANDS K 439
Medicinal Research Reviews DOI 10.1002/med

215. Miller NR, Jover T, Cohen HW, Zukin RS, Etgen AM. Estrogen can act via estrogen receptor
alpha and beta to protect hippocampal neurons against global ischemia-induced cell death.
Endocrinology 2005;146:3070–3079.
216. Carswell HV, Macrae IM, Gallagher L, Harrop E, Horsburgh KJ. Neuroprotection by a selective
estrogen receptor beta agonist in a mouse model of global ischemia. Am J Physiol Heart Circ
Physiol 2004;287:H1501–H1504.
217. Dubal DB, Rau SW, Shughrue PJ, Zhu H, Yu J, Cashion AB, Suzuki S, Gerhold LM,
Bottner MB, Dubal SB, Merchanthaler I, Kindy MS, Wise PM. Differential modulation of
estrogen receptors (ERs) in ischemic brain injury: a role for ERalpha in estradiol-mediated
protection against delayed cell death. Endocrinology 2006;147:3076–3084.
218. Dubal DB, Zhu H, Yu J, Rau SW, Shughrue PJ, Merchenthaler I, Kindy MS, Wise PM. Estrogen
receptor alpha, not beta, is a critical link in estradiol-mediated protection against brain injury.
Proc Natl Acad Sci USA 2001;98:1952–1957.
219. Simpkins JW, Yang SH, Sarkar SN, Pearce V. Estrogen actions on mitochondria—physiological
and pathological implications. Mol Cell Endocrinol 2008;290:51–59.
220. Leventhal L, Brandt MR, Cummons TA, Piesla MJ, Rogers KE, Harris HA. An estrogen
receptor-beta agonist is active in models of inflammatory and chemical-induced pain. Eur J
Pharmacol 2006;553:146–148.
221. Rocha BA, Fleischer R, Schaeffer JM, Rohrer SP, Hickey GJ. 17 Beta-estradiol-induced
antidepressant-like effect in the forced swim test is absent in estrogen receptor-beta knockout
(BERKO) mice. Psychopharmacology (Berl) 2005;179:637–643.
222. Krezel W, Dupont S, Krust A, Chambon P, Chapman PF. Increased anxiety and synaptic
plasticity in estrogen receptor beta-deficient mice. Proc Natl Acad Sci USA 2001;98:12278–12282.
223. Choleris E, Clipperton AE, Phan A, Kavaliers M. Estrogen receptor beta agonists in
neurobehavioral investigations. Curr Opin Investig Drugs 2008;9:760–773.
224. Walf AA, Rhodes ME, Frye CA. Antidepressant effects of ERbeta-selective estrogen receptor
modulators in the forced swim test. Pharmacol Biochem Behav 2004;78:523–529.
225. Lund TD, Rovis T, Chung WC, Handa RJ. Novel actions of estrogen receptor-beta on anxiety-
related behaviors. Endocrinology 2005;146:797–807.
226. Weiser MJ, Foradori CD, Handa RJ. Estrogen receptor beta in the brain: From form to function.
Brain Res Rev 2008;57:309–320.
227. Rissman EF. Roles of oestrogen receptors alpha and beta in behavioural neuroendocrinology:
Beyond Yin/Yang. J Neuroendocrinol 2008;20:873–879.
228. Morissette M, Le Saux M, D’Astous M, Jourdain S, Al Sweidi S, Morin N, Estrada-Camarena E,
Mendez P, Garcia-Segura LM, Di Paolo T. Contribution of estrogen receptors alpha and beta to
the effects of estradiol in the brain. J Steroid Biochem Mol Biol 2008;108:327–338.
229. Ropero AB, Alonso-Magdalena P, Quesada I, Nadal A. The role of estrogen receptors in the
control of energy and glucose homeostasis. Steroids 2008;73:874–879.
230. Cooke PS, Heine PA, Taylor JA, Lubahn DB. The role of estrogen and estrogen receptor-alpha in
male adipose tissue. Mol Cell Endocrinol 2001;178:147–154.
231. Heine PA, Taylor JA, Iwamoto GA, Lubahn DB, Cooke PS. Increased adipose tissue in male and
female estrogen receptor-alpha knockout mice. Proc Natl Acad Sci USA 2000;97:12729–12734.
232. Naaz A, Zakroczymski M, Heine P, Taylor J, Saunders P, Lubahn D, Cooke PS. Effect of
ovariectomy on adipose tissue of mice in the absence of estrogen receptor alpha (ERalpha):
A potential role for estrogen receptor beta (ERbeta). Horm Metab Res 2002;34:758–763.
233. Hertrampf T, Seibel J, Laudenbach U, Fritzemeier KH, Diel P. Analysis of the effects of oestrogen
receptor alpha (ERalpha)- and ERbeta-selective ligands given in combination to ovariectomized
rats. Br J Pharmacol 2008;153:1432–1437.
440 K MINUTOLOETAL.
Medicinal Research Reviews DOI 10.1002/med

Filippo Minutolo was born in Lanciano (Abruzzo), Italy, on October 1, 1967. He received hisB.S./M.S. in Chemistry and Pharmaceutical Technology from the University of Pisa, Italy, in1992, under the supervision of Prof. Paolo Crotti. After the degree, he obtained a grant for agraduate research period working on the synthesis of bioactive carbohydrates in the group ofProf. Giancarlo Berti and Prof. Giorgio Catelani. Among his honors are two ‘‘Dow’’ prizes(1991 and 1992), a ‘‘Bracco’’ honor mention (1994), and the National ‘‘Federchimica’’ (1994)and ‘‘Lepetit Group’’ (1994) awards. In 1993, he was the recipient of an ENI fellowship toattend a triennial graduate school at the Scuola Normale Superiore in Pisa, working in thefield of enantioselective organic synthesis under the supervision of Prof. Piero Salvadori.During his PhD, he spent a research period (1994– 1995) in the group of Prof. Ben L. Feringaat the University of Groningen (The Netherlands). In 1996, he obtained his PhD from theScuola Normale Superiore, Pisa. He then got a two-year postdoctoral appointment(1997– 1999) at the University of Illinois at Urbana-Champaign (USA), working on thesynthesis of ligands and radio-ligands for the estrogen receptors in the group of Prof. John A.Katzenellenbogen. In 2000, he joined the Department of Pharmaceutical Sciences—Faculty ofPharmacy of the University of Pisa as assistant professor. In 2006, he was appointed associateprofessor of Medicinal Chemistry by the University of Pisa. Over the past ten years, he has beenteaching courses of Medicinal Chemistry to undergraduate and graduate students. Since 2007,he is a member of the Steering Committee of the Graduate School in ‘‘Science of Drugs andBioactive Substances’’ at the University of Pisa. He is also the chairman of the Didactics/Training Committee of the same Graduate School. Since 2009, he is a member of the EuropeanOrganization for Research and Treatment of Cancer (EORTC, Brussels, Belgium). Hisresearch interests include the design and synthesis of biologically active small organic moleculeswith particular regard to hormone receptor ligands and antitumor agents.
Marco Macchia received B.S./M.S. in Chemistry and Pharmaceutical Technology from theUniversity of Pisa, Italy, in 1991, after a research working experience at Smithkline BeechamPharmaceutical in Great Burgh, Epsom, Surrey, United Kingdom, where he also took hismedicinal chemistry thesis in the field of antiviral drugs. In 1994, he obtained the PhD degree inMedicinal Chemistry at the Department of Pharmaceutical Sciences of the University of Pisa,where he was then appointed as assistant professor in 1994, as associate professor in 1998, andas full professor of Medicinal Chemistry in 2001. Over the past 15 years, he has been teachingcourses of Medicinal Chemistry to undergraduate and graduate students. From 2002 to 2008, hewas a Member of the Scientific Commitee of the ‘‘European School of Medicinal Chemistry(ESMEC),’’ an European Federation for Medicinal Chemistry (EFMC) Accredited Schoolorganized from the Division of Medicinal Chemistry of the Italian Chemical Society in Urbino(Italy). From 2007, he is the vice-director of the Department of Pharmaceutical Sciences of theUniversity of Pisa. Since 2002, he is the president of the degree course in ‘‘Analytical andEnvironmental Toxicology’’ of the Faculty of Pharmacy of the University of Pisa. He is theauthor of 100 publications in prestigious international journals and his research work has dealtmainly with antitumor agents active on signal transduction of cell proliferation and molecularmechanisms of apoptosis induction, selective estrogen receptors ligands, compounds with a highaffinity and selectivity for D4 dopaminergic receptors, a- and b-adrenergic receptors, andcannabinoid receptors.
Benita S. Katzenellenbogen received a B.S. degree in biology from the City University ofNew York, and M.A. and PhD degree in biology (1970) from Harvard University. Hergraduate studies, done with Professor Fotis Kafatos, involved a study of hormone-regulated
ESTROGENRECEPTOR bLIGANDS K 441
Medicinal Research Reviews DOI 10.1002/med

processes in insect development. She then worked as an NIH Postdoctoral Fellow with ProfessorJack Gorski, at the University of Illinois (1970– 1971), on early responses to estrogens in targettissues. In 1971, she was appointed as an assistant professor of Physiology in the College ofMedicine at the University of Illinois at Urbana-Champaign. She was promoted to associateprofessor in 1976 and full professor in 1982. She also holds a faculty appointment in theDepartment of Cell and Developmental Biology, and she currently holds the Swanlund Chair ofMolecular and Integrative Physiology and is a Member of the Center for Advanced Studies. Herresearch covers investigations of the cellular and molecular functions of estrogens and otherhormones and growth factors in reproduction and breast cancer. She has extensivecollaborations, and has trained more than 70 graduate students and postdoctoral fellows. Shehas served on many editorial and grant review boards, award panels, and conference programcommittees, and she has been active in and served The Endocrine Society in many capacities,most notably as president in 2000– 2001. Among her numerous awards and honors are The ErnstOppenheimer Award (The Endocrine Society, 1984), MERIT Award (NIH, 1991– 1999),Fellow, American Academy of Arts and Sciences (elected 1993), Faculty Member of the YearAward (University of Illinois College of Medicine, 1994), Scientific Distinction Award (SusanG. Komen Breast Cancer Foundation, 1996), Jill Rose Award for Breast Cancer Research (TheBreast Cancer Research Foundation, 1998), City University of New York Distinguished AlumniAward (2002), the Roy O. Greep Lecture Award (jointly with John Katzenellenbogen, TheEndocrine Society, 2006), and Laurea ad Honorem (Honorary Degree, University of Milan,Italy, 2007).
John A. Katzenellenbogen received his A.B. degree in 1966, a M.A. degree in 1967, and a PhDdegree in chemistry, in 1969, from Harvard University. His graduate studies, done with ProfessorE. J. Corey, involved the development of stereospecific methods for the synthesis of isoprenoidalkenes and bioactive natural product syntheses. He began his academic career as an assistantprofessor at the University of Illinois at Urbana-Champaign in 1969, where he was promoted toassociate professor in 1975 and full professor in 1979. He was appointed as the Roger AdamsProfessor of Chemistry (1992– 1996), and he now holds the Swanlund Chaired Professor ofChemistry (since 1996). He also holds appointments in Bioengineering and in the BeckmanInstitute for Advanced Studies. His research spans chemistry, biology, and medicine, and involvesanalysis of steroid receptor structure and function, and the use of receptors and their ligands invarious biochemical, biological, bioanalytical, and biomedical applications. He collaboratesextensively with other researchers, both in the United States and internationally, and he hastrained more than 100 doctoral students and postdoctoral fellows, and has published more than450 articles. He is a member of a number of Editorial Boards and is currently an associate editorof Steroids and the Journal of Nuclear Medicine. He has chaired and served on many grantreview panels for federal agencies. His honors include Fellow of the American Association for theAdvancement of Science (elected 1989), Fellow of the American Academy of Arts and Sciences(elected 1992; Chair of Midwest Committee, since 2007; Vice-Chair of the National Council,since 2005), four NIH MERIT awards, the Paul C. Aebersold Award (The Society of NuclearMedicine, 1995), Cope Scholar Award (American Chemical Society, 1999), the Roy O. GreepLectureship Award (jointly with Benita Katzenellenbogen, The Endocrine Society, 2006), the E.B. Hershberg Award for Important Discoveries in Medicinal Chemistry (American ChemicalSociety, 2007), the Esselen Award for Chemistry in the Public Service (Northeastern Section ofthe American Chemical Society, 2008), The Royal Society of Chemistry Centenary Award(2009), and the Leading Edge Award in Basic Science (Society of Toxicology, 2009).
442 K MINUTOLOETAL.
Medicinal Research Reviews DOI 10.1002/med





























