Embed Size (px)
Citation preview

Subscriber access provided by ALBANY STATE UNIV
Molecular Pharmaceutics is published by the American Chemical Society. 1155Sixteenth Street N.W., Washington, DC 20036Published by American Chemical Society. Copyright © American Chemical Society.However, no copyright claim is made to original U.S. Government works, or worksproduced by employees of any Commonwealth realm Crown government in the courseof their duties.
Article
Cathepsin B Degradable Star Shaped PeptidicMacromolecules for Delivery of 2-methoxyestradiol
Ravi Shankar, Abhilash Samykutty, Corinne Riggin, Sneha Kannan, Ursula Wenzel, and Rohit KolhatkarMol. Pharmaceutics, Just Accepted Manuscript • DOI: 10.1021/mp400261h • Publication Date (Web): 23 Aug 2013
Downloaded from http://pubs.acs.org on August 27, 2013
Just Accepted
“Just Accepted” manuscripts have been peer-reviewed and accepted for publication. They are postedonline prior to technical editing, formatting for publication and author proofing. The American ChemicalSociety provides “Just Accepted” as a free service to the research community to expedite thedissemination of scientific material as soon as possible after acceptance. “Just Accepted” manuscriptsappear in full in PDF format accompanied by an HTML abstract. “Just Accepted” manuscripts have beenfully peer reviewed, but should not be considered the official version of record. They are accessible to allreaders and citable by the Digital Object Identifier (DOI®). “Just Accepted” is an optional service offeredto authors. Therefore, the “Just Accepted” Web site may not include all articles that will be publishedin the journal. After a manuscript is technically edited and formatted, it will be removed from the “JustAccepted” Web site and published as an ASAP article. Note that technical editing may introduce minorchanges to the manuscript text and/or graphics which could affect content, and all legal disclaimersand ethical guidelines that apply to the journal pertain. ACS cannot be held responsible for errorsor consequences arising from the use of information contained in these “Just Accepted” manuscripts.

1
Cathepsin B Degradable Star Shaped Peptidic Macromolecules for Delivery of 2-
methoxyestradiol
Ravi Shankar1, 2*, Abhilash Samykutty1*, Corinne Riggin2, Sneha Kannan2, Ursula Wenzel1 and Rohit Kolhatkar1,2†
1 Department of Biopharmaceutical Sciences, University of Illinois Chicago, Rockford, IL 61107, USA
2 Department of Pharmaceutical Sciences, University of Maryland, Baltimore, Baltimore, MD, 21201, USA
* Both authors contributed equally.
† Corresponding author:
Rohit Kolhatkar
Department of Biopharmaceutical Sciences
University of Illinois Chicago
1601 Parkview Ave, Rm N302
Rockford, IL 61107
Tel: (815) 395 5922
Email: [email protected]
Page 1 of 53
ACS Paragon Plus Environment
Molecular Pharmaceutics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

2
Abstract
2-methoxyestradiol (2ME), a natural metabolite of estradiol has antiproliferative and
antiangiogenic activity. However, its clinical success is limited due to poor water solubility and
poor pharmacokinetic parameters suggesting the need for a delivery vehicle. In this study we
evaluated cathepsin B degradable star shaped peptidic macromolecules (SPMs) that can
potentially be used to create higher generation and high molecular weight peptidic polymer as
delivery vehicle of 2ME. Two peptidic macromolecules having positively charged amine
(ASPM) or negatively charged carboxyl surface groups (CSPM) were synthesized and evaluated
for their degradation in the presence of cathepsin B and stability in the presence of neutral or
acidic buffer and serum. Both ASPM and CSPM degraded rapidly in the presence of cathepsin
B. Both were stable in neutral and acidic buffer whereas only CSPM exhibited substantial
stability in the presence of serum. Both macromolecules were nontoxic towards breast cancer
cells whereas 2ME-containing macromolecules exhibited antiproliferative activity in the
micromolar range. Overall, results from current study indicate that tetrapeptide GFLG can be
used to create star-shaped macromolecules that are degraded in the presence of cathepsin B and
have the potential to be developed as delivery vehicle of 2ME.
KEY WORDS: 2-Methoxyestradiol, Cathepsin B, Peptidic dendrimer, Degradable polymer.
Page 2 of 53
ACS Paragon Plus Environment
Molecular Pharmaceutics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

3
INTRODUCTION
2-methoxyestradiol (2ME, Figure 1, I), has been identified as a potential antitumor agent
due to its ability to inhibit angiogenesis and proliferation of the cancer cells in various human
cell lines. This includes cell lines that are resistant to chemotherapeutic agents [1-4]. It inhibits
the activation of HIF-1α [5], which plays a key role in developing resistance of tumor cells to
chemotherapeutic agents. Several in-vivo studies demonstrate the effectiveness of 2ME in
inhibiting tumor growth [1, 3-5]. The results from the Phase I clinical trials demonstrate the
safety of 2ME [6, 7]. However, low plasma levels detected, despite higher drug administration, is
limiting its clinical success. Lower solubility and extensive metabolism are responsible for its
lower plasma levels [1, 6]. The therapeutic benefit of 2ME can be improved by using a delivery
vehicle that can increase its solubility, decrease metabolism and release the drug at the site of
action.
Delivery vehicles in the nanometer size range such as polymers [8-10], liposomes [11,
12], micelles, and inorganic nanomaterials [13] can overcome several limitations associated with
small molecular weight anticancer therapeutics like 2ME. One of the prerequisites for the
effectiveness of such delivery vehicle is to release the free drug at tumor sites. The site specific
release of the free drug can be achieved by using stimuli-sensitive linker that will respond to the
differences between, normal and tumor tissue, or intracellular and extracellular environment
[14]. Two extensively studied stimuli-sensitive linkers are hydrolysable [15] or reducible linkers
[16]. However, these approaches provide limited control over the site-specific degradation after
in-vivo administration. In contrast, the release of the drug triggered by the presence of specific
enzyme entails higher chemical stability and concurrent specificity associated with the enzyme
Page 3 of 53
ACS Paragon Plus Environment
Molecular Pharmaceutics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

4
selected [17]. Therefore, we designed peptidic macromolecules that can be completely degraded
in the presence of an enzyme cathepsin B.
Cathepsin B is one of the most prominent proteases present in normal cells and tissues.
The expression of cathepsin B is substantially increased in human tumors such as prostate,
breast, colon, esophageal, gastric, lung, ovarian, and thyroid carcinomas as well as in gliomas
and melanomas [18-20]. This has been observed at mRNA and protein level. The higher
expression level can be the result of oncogene contribution, gene amplification, alternative
splicing, or post-translational modification [20, 21]. The differences in cathepsin B expression
levels in normal and cancerous tissues can be exploited to achieve higher drug release at tumor
sites using two types of approaches. An extensively studied approach is the use of cathepsin B
sensitive linker for drug release. This has been reported to reduce toxicities associated with small
molecular weight therapeutic molecules like doxorubicin [10, 22-24]. One disadvantage for the
linker approach is the high reliance on kidneys for the clearance of non-degradable polymeric
backbone. Increasing evidence from preclinical studies suggest the induction of intracellular
vacuolation in animal models raising the concerns about biosafety of nondegradable polymers
after chronic administration at high doses [25, 26]. Completely degradable polymers can be
cleared easily and can potentially release their entire therapeutic payload at tumor sites compared
to lower drug release associated with linker strategy. It has been reported that less than 5% of the
injected polymeric conjugate reaches tumor [27]. Furthermore, most polymeric conjugates have
only 10% wt/wt of the therapeutic component. Therefore, it is important for the small
percentage of the drug-containing polymeric conjugates to release its entire drug load after
reaching its destination. We envision a completely degradable delivery system can be developed
to achieve higher drug release at tumor sites. In this manuscript we report the first generation of a
Page 4 of 53
ACS Paragon Plus Environment
Molecular Pharmaceutics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

5
completely degradable system. We report the synthesis of star shaped peptidic macromolecules
(SPMs) that can be completely degraded within a short time period in the presence of cathepsin
B. Using 2ME we then demonstrate that 2ME-conjugated star shaped macromolecule (MESPM)
is as effective as a 2ME-containing peptidic monomer in inhibiting the growth of three triple
negative breast cancer cell lines.
Page 5 of 53
ACS Paragon Plus Environment
Molecular Pharmaceutics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

6
MATERIALS AND METHODS:
Reagents and chemicals:
Estradiol was purchased from Ochem Inc. (Des Plaines, IL), Amino acids were purchased from
Novabiochem (Darmstadt, Germany). Coupling Reagents dicyclohexylcarbodiimide (DCC), 1-
hydroxybenzotriazole (HOBT), and O-(Benzotriazol-1-yl)-N,N,N’,N’-tetramethyluronium
hexafluorophosphate (HBTU) were obtained from Oakwood Chemicals (West Columbia, SC).
PAMAM dendrimer (Generation 0), N,N-Diisopropylethylamine (DIPEA), trifluoroacetic acid
(TFA), di-tert-butyl dicarbonate ((Boc)2O), dichloromethane (DCM), dimethylsulfoxide
(DMSO), deutrited DMSO-d6, chloroform (CDCl3), D2O and other chemicals were ACS grade
and purchased from Sigma Chemical Co. (St. Louis, MO). Cathepsin B (CPB) and model
substrate N-benzoyl-Phe-Val-Arg-p-nitroanilide hydrochloride were also purchased from Sigma
Chemical Co. (St. Louis, MO). 2ME [28], Gly-2ME [29], and BocGFLGOMe [30] were
synthesized as described before. All commercial reagents and anhydrous solvents were used
without further purification or distillation unless otherwise stated. High-performance liquid
chromatography (HPLC) grade solvents were purchased from Fisher Scientific (Pittsburgh, PA).
All small molecules were analyzed using TLC, NMR, MS and HPLC for the purity. Polymers
were purified using HPLC before analysis. Compounds showing >95 % purity form HPLC
analysis were used for biological characterization. Analytical thin layer chromatography was
performed on Whatman silica gel 60 Å with fluorescent indicator (Partisil K6F). Compounds
were visualized by UV light and/or stained with ninhydrin solution followed by heating. Flash
column chromatography was performed on Whatman silica gel 60 Å (230-400 mesh). NMR (1H,
13C) spectra were recorded on a Varian 300/400 MHz or a Bruker 400 MHz spectrometer and
calibrated using an internal reference. ESI mode mass spectra were recorded on a Shimatzu
Page 6 of 53
ACS Paragon Plus Environment
Molecular Pharmaceutics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

7
LCMS 2020. MALDI analysis were performed using Voyager-DE PRO mass spectrometer
(Applied Biosystems, Foster City, CA, USA) equipped with a 337 nm pulsed nitrogen laser.
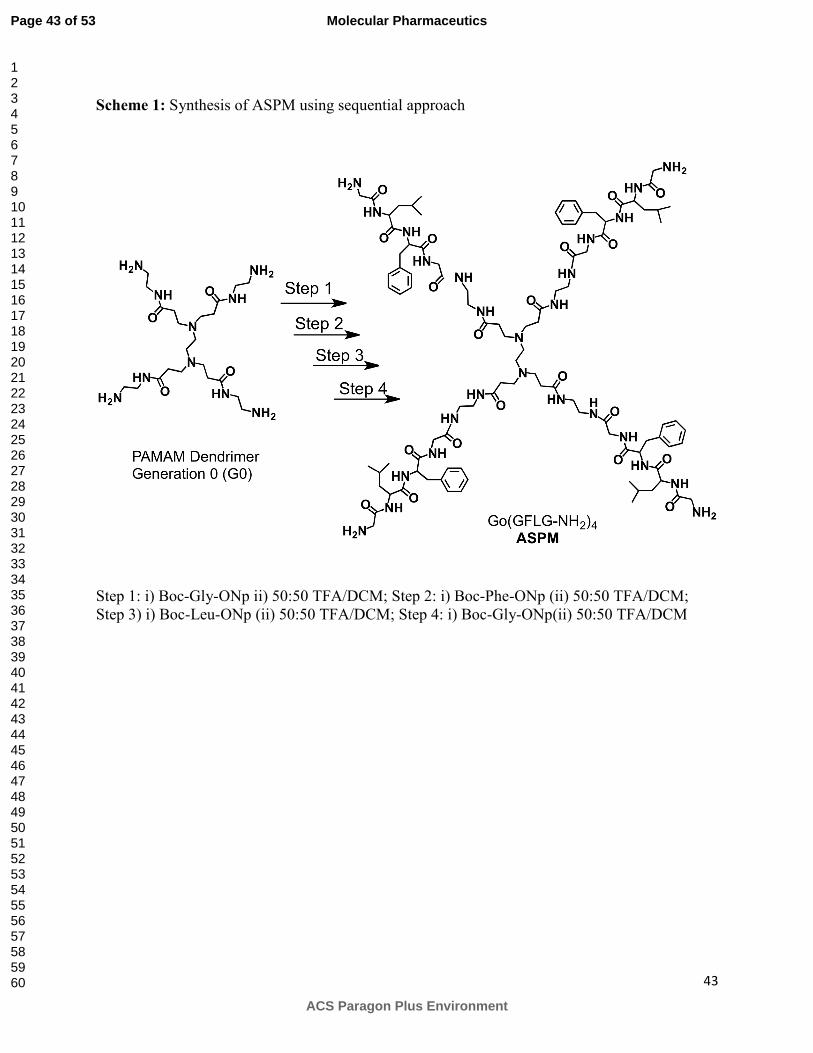
Synthesis of positively charged amine-terminated star-shaped peptidic macromolecule
(ASPM):
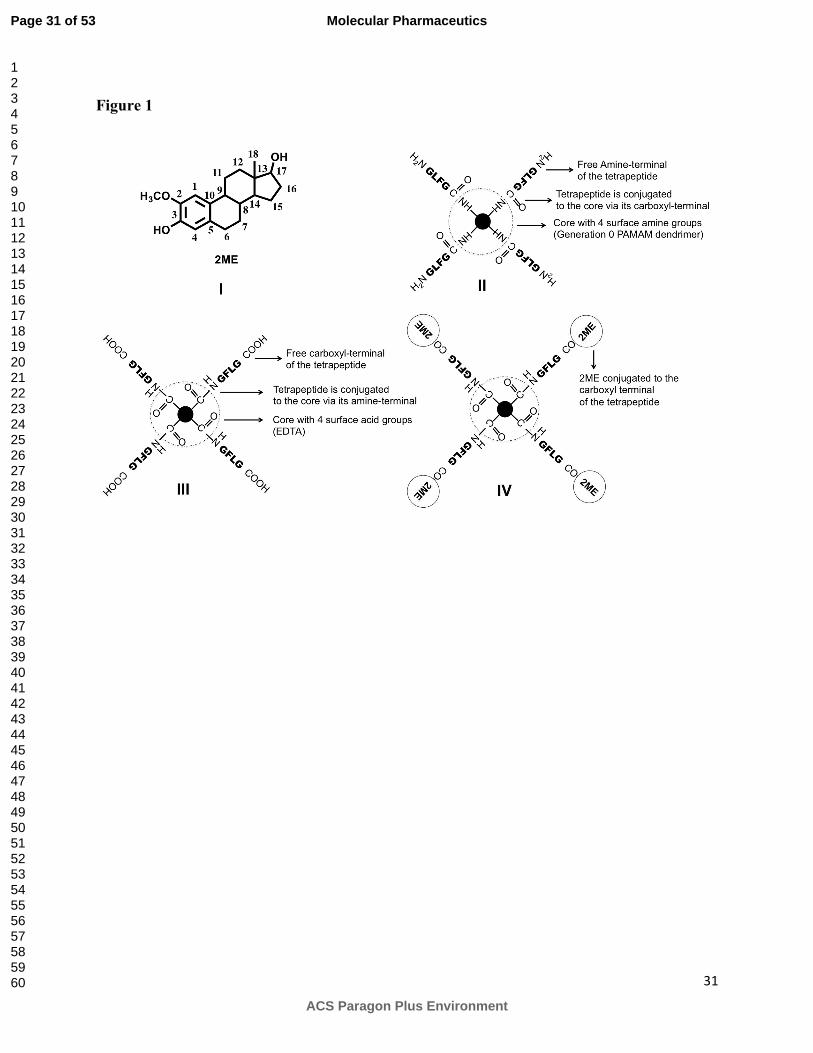
Positively charged amine-terminated star-shaped peptidic macromolecule (ASPM, II, Figure 1)
was synthesized using a sequential approach where a reactive ester of each amino acid was
added to the PAMAM dendrimer (Generation 0, G0) in a stepwise fashion (Scheme 1). In each
step, one amino acid was conjugated to the four surface amine groups through two reactions. In
the first reaction, p-nitrophenylester (ONp) of boc-protected amino acid was conjugated to the
free terminal amines present in PAMAM dendrimer. In the second step, boc groups on amino
acids were deprotected under acidic conditions to get positively charged amine-terminated star-
shaped macromolecule. Typical procedure for the conjugation of amino acid to four arms is
described below using glycine as an example. Briefly, into the solution of PAMAM dendrimer
(G0) (0.1 g, 0.19 mmol) in DMSO was added Boc-Gly-ONp (0.34 g, 1.1 mmol) and the reaction
mixture was stirred overnight. Solvent was evaporated in vacuo and the crude mixture was
dissolved in acetone and precipitated in cold ether to obtain gummy compound that was
dissolved in the mixture of dichloromethane and TFA (50:50, 6 mL). The solution was stirred
for 6 hours. Solvent was evaporated and the crude mixture was dissolved in methanol and
precipitated in cold ether to obtain G0(Gly)4 (0.23 mg, yield 78%). Repetitions of conjugation
and deprotection reactions using p-nitrophenyl esters of phenylalanine (Step 2, Scheme 1),
leucine (Step 3, Scheme 1) and glycine (Step 4, Scheme 1) yielded ASPM with four positively
charged surface amine groups. The crude ASPM was purified using preparative HPLC using
Waters HPLC system equipped with 600E multisolvent delivery system, 717 plus autosampler
Page 7 of 53
ACS Paragon Plus Environment
Molecular Pharmaceutics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

8
and 996 PDA detector. Varian Dynamex column (250×10 mm) was used for preparative
purification using water (0.1 % TFA) as mobile phase A and mixture of 80 % acetonitrile (0.1 %
TFA) and 20 % water (0.1 % TFA) as mobile phase B at the flow rate of 5 ml/min with gradient
of 0 to 60 % B over the period of 60 min. Fractions containing product were pooled together,
solvent was evaporated under vacuo followed by lyophilization to obtain pure ASPM as white
solid. The final product obtained was > 97 % pure as analyzed by HPLC. Dynamex column (250
x 4.6 mm) was used for analytical HPLC run using water (0.1 % TFA) as mobile phase A and
mixture of 80 % acetonitrile (0.1 % TFA) and 20 % water (0.1 % TFA) as mobile phase B at the
flow rate of 1.5 ml/min with gradient of 0 to 100 % B over the period of 45 min followed by a
100% B for 10 min. The same instrument method was used for analysis of all the polymers using
HPLC and retention time reported in Table 1. MS Anal. MALDI Mol wt. Calc 2014.42;
observed 2038 (Figure S1A) 1H NMR (500MHz, D2O): 0.81-0.87 (m, 24H, 8XCH3, -Leu), 1.43-
1.6 (m, 12H, β-Leu, γ-Leu), 2.40-2.53 (m, 12H, -NCH2CH2N-, CH2CO), 2.84-3.10 (m, 16H, β-
Phe, CH2N(CH2)2), 3.13-3.35 (m, 16H, CH2NHCO), 3.61-3.83 (m, 16H, α-Gly), 4.12-4.32 (m,
4H, α-Leu), 4.58-4.66 (m, 4H, α-Phe ), 7.11-7.36 (m, 20H, ArH).
Synthesis of negatively charged carboxyl-terminated star-shaped peptidic macromolecule
(CSPM) and 2ME containing star-shaped peptidic macromolecule (MESPM):
Carboxyl-terminated star-shaped peptidic macromolecule (CSPM, III, Figure 1) and 2ME
containing star-shaped peptidic macromolecule (MESPM, IV, Figure 1) were synthesized using
convergent approach where a tetrapeptide GFLG or GFLGG2ME was concurrently conjugated
to the all four arms of the central core. The synthesis involved three steps i) synthesis of core ii)
synthesis of monomers and iii) conjugation of monomers to core.
Page 8 of 53
ACS Paragon Plus Environment
Molecular Pharmaceutics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

9
Synthesis of core:
Tetrakis-(p-nitrophenyl ester) (EDTA(ONp)4) was used as central core for CSPM and MESPM.
EDTA(ONp)4 was synthesized from ethylene diamine tetraacetic acid and p-nitrophenol using
DCC as a coupling agent (Scheme 2). Briefly, EDTA (1 g, 3.34 mmol) was dissolved in
anhydrous DMF and the solution was cooled down to -10 °C. Into the solution was then added
solution of p-nitrophenol (2.08 g, 14.9 mmol) and solution of DCC (3.08 g, 14.9 mmol) and the
reaction mixture was stirred for 3 h at -10°C. The reaction mixture was then stirred overnight at
4°C followed by stirring for 12 h at room temp. Diclyclohexylurea (DCU) was precipitated,
solvent evaporated and the product was crystalized using ethanol. After two crystallizations pale
yellow color product was obtained (1.7 g, yield 64 %) which was found to be pure according to
TLC. 1H NMR (500MHz, CDCl3): δ 3.1 (s, 4H, N-CH2CH2-N), 4.9 (s, 8H, COCH2N), 7.4 (d,
8H, ArH), 8.5 (d, 8H, ArH); MS Anal.; Mol wt calculated 776.62; observed 777.62 (M+1).
Synthesis of monomers:
Synthesis of Boc-GFLGOH (3): BocGFLGOMe was synthesized according to previously
reported method [30]. Deprotection of methyl ester under basic conditions yielded BocGFLGOH
(3, Scheme 3). Briefly, Boc-GFLGOMe (1 g, 1.9 mmol) was dissolved in THF (10 mL) and
mixed with a NaOH solution in water (0.15 g in 10 mL). After 1 h of stirring, the reaction
mixture was concentrated in vacuo. The crude product was dissolved in ethyl acetate (50 mL)
and the solution was acidified with dilute acetic acid (10%). The ethyl acetate layer was washed
with water (2 x 10 mL) and brine (10 mL), dried over Na2SO4 and concentrated in vacuo to
obtain solid compound that was crystallized from ethyl acetate / diethyl ether to give 0.65 g
(67% yield) as a white crystalline solid. 1H NMR (DMSO; d6): 0.83 (d, 3H, CH3-Leu), 0.88 (d,
Page 9 of 53
ACS Paragon Plus Environment
Molecular Pharmaceutics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

10
3H, CH3-Leu), 1.3 (s, 9H, Boc), 1.45-1.48 (m, 2H, β-Leu), 1.56-1.63 (m, 1H, γ-Leu), 2.78 (dd,
1H, β-Phe), 3.01(dd, 1H, β-Phe), 3.35-3.45 (m, 2H, α-Gly), 3.71(t, 2H, α-Gly), 4.32-4.34 (m,
1H, α-Leu), 4.53-4.54 (m, 1H, α-Phe), 6.92 (t, 1H, NH-Boc) 7.20-7.22 (m, 5H, ArH Phe), 7.83
(d, 1H, -CONH-), 8.04-8.07(t, 1H, -CONH-), 8.14(d, 1H, -CONH-), 12.5(s, 1H, COOH) ); MS
Anal.; Mol wt calculated 492.57; observed 493.26 (M+1).
Synthesis of GFLGOH (4): Deprotection of Boc group in BocGFLGOH (3) under acidic
conditions yielded GFLGOH (4, Scheme 3). Briefly, Boc-GFLGOH (0.1 g, 0.2 mmol) was
dissolved in 50:50 mixtures of TFA and DCM (1 mL). After 1h solvent was evaporated under
vacuum. The residue was dissolved in minimum volume of methanol and precipitated in diethyl
ether to obtain white solid. (TFA salt of GFLGOH; 0.1g, 98% yield). 1H NMR (500MHz,
DMSO-d6): δ 0.85 (d, 3H, CH3-Leu), 0.9 (d, 3H, CH3-Leu), 1.46-1.50 (m, 2H, β-Leu), 1.61-
1.63 (m, 1H, γ-Leu), 2.70-2.76 (m, 1H, β-Phe), 3.03-3.07 (dd, 1H, β-Phe), 3.48-3.54 (m, merged
with water impurities in DMSO, 2H, α-Gly), 3.76 (t, 2H, α-Gly ), 4.35 (dd, 1H, α-Leu), 4.64-
4.67 (m, 1H, α-Phe), 7.18-7.25 (m, 5H, ArH Phe), 7.95 (bs, 2H, NH2), 8.15 (t,1H, NH-), 8.33 (d,
1H, NH), 8.59 (d, 1H, NH) ); MS Anal.; Mol wt calculated 392.45; observed 393.20 (M+1).
Synthesis of GFLGG2ME (5): 2ME containing monomer was synthesized according to scheme
3. Briefly, Boc-GFLG-OH (3) (0.1 g, 0.20 mmol) and HBTU (0.11 g, 0.30 mmol) were
dissolved in 10 mL dry DMF. The solution was cooled to 0 0C and into it was added 2 mL DMF
containing Gly-2ME (72.96 mg, 0.20 mmol). The temperature was maintained at 0 0C for 30 min
after which reaction mixture was allowed to warm to room temperature followed by stirring for
overnight. The reaction mixture was then quenched by 30 mL distilled water, solid precipitated
was filtered and purified by flash column chromatography over silica gel using ethyl
Page 10 of 53
ACS Paragon Plus Environment
Molecular Pharmaceutics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

11
acetate/hexane (50:50) as eluent to obtain BocGFLGG2ME as a white solid (60% yield, 100
mg). The corresponding Boc-GFLGG2ME (100 mg, 0.12 mmol) was dissolved in 50:50
mixtures of TFA and DCM (1 mL). After 1h solvent was evaporated under vacuum. The residue
was dissolved in minimum volume of methanol and precipitated in diethyl ether to obtain white
solid which was purified by flash column chromatography using methanol/ethyl acetate (2:98) as
eluent to obtain GFLGG2ME (5) (TFA salt, 98% yield, 100 mg). 1H NMR (500MHz, DMSO-
d6): δ = 0.77 (s, 3H, 2ME H-18, CH3), 0.85 (d, 3H, CH3-Leu), 0.90 (d, 3H, CH3-Leu), 1.23-1.32
(m, 6H, 2ME H-14(1H), H-7(2H), H-15(2H), H-8(1H)), 1.47-1.52 (m, 3H, β-Leu, 2ME H-12
(1H), ) 1.57-1.65(m, 2H, γ-Leu(1H) & 2ME H-12(1H), 1.74-1.77(m, 2H, 2ME H-16), 2.05-2.11
(m, 2H, 2ME H-11), 2.21-2.24 (m, 1H, 2ME H-9), 2.62-2.66 (m, 1H, 2ME H-6), 2.73-2.75(m,
1H, β-Phe), 3.02 (dd, 1H, β-Phe), 3.37-3.43(m, 1H, 2ME H-17), 3.57-3.58(m, 1H, 2ME H-6),
3.74 (s, 3H, OCH3), 3.75 (d, 2H, α-Gly), 3.86 (d, 2H, α-Gly), 4.0 (br, 2H Gly), 4.30-4.35 (m,
1H, α-Leu), 4.59-4.67 (m, 1H, α-Phe), 6.44 (s, 1H, 2ME H-1), 6.7(s, 1H, 2ME H-4), 7.17-7.20
(m, 5H, ArH Phe), 8.07 (t, 1H, -CONH-), 8.24 (t, 1H, -CONH-), 8.37(d, 1H, CONH), 8.59(d,
1H, CONH) ); MS Anal.; Mol wt calculated 834.38; observed 835.40 (M+1).
Synthesis of SPMs:
Synthesis of CSPM (III): Conjugation of GFLGOH (4) with EDTA(ONp)4 (2) yielded CSPM
(III), EDTA(GFLGOH)4) (Scheme 3). Briefly, EDTA(ONp)4 (0.1 g, 0.12 mmol) was dissolved
in anhydrous DMSO (1mL). Into the solution was then added solution of GFLGOH (4) (0.4 g,
1.02 mmol) in DMSO (0.5 mL) and triethylamine (150 µL, 1.02 mmol). After 8h, reaction
mixture was concentrated in vacuo to obtain crude product which was purified by preparative
HPLC as described before for purification of ASPM. Fractions containing product were pooled
Page 11 of 53
ACS Paragon Plus Environment
Molecular Pharmaceutics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

12
together, solvent was evaporated under vacuo followed by lyophilization to obtain pure
EDTA(GFLGOH)4 (CSPM) as white solid. 1H NMR (500MHz, DMSO-d6): 0.81-0.87 (m, 24H,
CH3, Leu), 1.23-1.25 (m, 4H, γ-Leu), 1.46-1.47 (m, 8H, β-Leu), 1.57-1.60 (m, 4H, β-Leu), 2.73-
2.76 (m, 4H, -NCH2CH2N-), 3.00-3.07 (m, 8H, β-Phe), 3.67-3.72 (m, 12H, α-Gly & -
COCH2NCH2CH2), 3.74-3.80 (m, 12H, α-Gly & -COCH2NCH2CH2 ), 4.33-4.35 (m, 4H, α-Leu),
4.59-4.60 (m, 4H, α-Phe ), 7.15-7.20 (m, 20H, ArH), 8.11-8.19(m, 12H, CONH-), 8.32-8.34 (m,
4H, CONH). MS Anal. Mol wt 1787.9 [M-2], MALDI Mol wt Calc 1788.87 observed 1791.87
(Figure S1B).
Synthesis of MESPM (IV): MESPM (EDTA(GFLGG2ME)4) (IV) was synthesized using
GFLGG2ME (5) as a monomeric unit and EDTA(ONP)4 (2) as central core (Scheme 3). Briefly,
EDTA(ONp)4 (2) (0.1 g, 0.12 mmol) was dissolved in anhydrous DMSO (1 mL). Then solutions
of GFLGG2ME.TFA (5) (0.85 g, 1.02 mmol) in DMSO (0.5 mL) and triethyl amine (0.15 mL,
1.02 mmol) were added dropwise. The reaction was allowed to proceed for 8h at room
temperature. Then reaction mixture was concentrated by vacuum pump with 550C temperature to
obtain yellowish mixture which was washed with ether (2 X 5 mL) and purified by preparative
HPLC using the same column and conditions as described above for purification of ASPM to
obtain EDTA(GFLGG2ME)4 as a white solid (57% yield, 23 mg). 1H NMR (500MHz, DMSO-
d6): 0.76 (s, 12H, 2ME H-18), 0.81 (d, 12H, CH3-Leu), 0.87 (d, 12H, CH3-Leu), 1.28-1.32 (m,
24H, 2ME H-14(4H), H-7(8H), H-15(8H), H-8(4H)), 1.46-1.50 (m, 8H, β-Leu) 1.53-1.66 (m,
8H, 2ME H-12(4H), γ-Leu (4H)), 1.73-1.77 (m, 8H, 2ME H-16), 2.05-2.11 (m, 8H, 2ME H-11),
2.21-2.24 (m, 4H, 2ME H-9), 2.57-2.66 (m, 4H, 2ME H-6), 2.74-2.78 (m, 4H, β-Phe), 2.99-3.04
(m, 4H, β-Phe), 3.16-3.17 (m, 4H, -NCH2CH2N-), 3.70 (s, 12H, OCH3), 3.72-3.75 (m, 8H, α-
Gly), 3.62-3.84 (d, 8H, α-Gly), 4.21-4.29 (m, 8H, α-Leu (4H), -COCH2N (4H)), 4.50-4.56 (m,
Page 12 of 53
ACS Paragon Plus Environment
Molecular Pharmaceutics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

13
4H, α-Phe), 4.59-4.63 (t, 4H, COCH2N-), 6.4 (s, 4H, 2ME H-1), 6.7 (s, 4H, 2ME H-4), 7.14-7.24
(m, 20H, ArH), 8.16 - 8.21 (m, 20H, CONH), 8.5 (s, 4H, OH). MS Anal. MALDI Mw Calc
3155.67 Mw observed 3178.39 (M+23) (Figure 2).
Stability studies:
0.24 mg of the polymer was dissolved in 150 µL of the PBS or PBS (10 % FBS) or acetate buffer
and the solutions were incubated for the respective incubation period. At the end of the
incubation period solutions were placed in boiling water (10 min) followed by addition of cold
acetone to precipitate proteins. Supernatant was collected, dried, reconstituted in 100 µL of
mobile phase and centrifuged. 70 µL of the supernatant was store at -70 °C until further analysis.
At the time of analysis 10 µL of Gly-Phe (4 mg/mL) was added to the supernatant solution to
serve as internal standard and the sample was analyzed using reverse phase HPLC (Waters
Corporation, Milford, MA). Percentage of intact polymer remaining was calculated based on the
calibration curve generated for the intact polymer.
Cathepsin B degradation studies:
The degradation of peptidic dendrimer in the presence of lysosomal enzyme cathepsin B (CPB)
was evaluated according to previously described procedures [31-33] with minor modifications.
Enzyme incubation mixture consisted of 100 µL of CPB stock solution (0.98 mg/mL or 0.49
mg/mL) in 0.1 M ammonium acetate buffer pH=5, 1 mM EDTA) and 50 µL of reduced
glutathione solution (250 mM in acetate buffer pH=5, 1 mM EDTA). The mixture was incubated
at 37 °C for 5 min before addition of polymer solution. 0.24 mg of polymer to be evaluated was
dissolved in ammonium acetate buffer (50 µL) and was added to the preincubated mixture. After
the incubation period the mixture was placed in boiling water (10 min) followed by addition of
Page 13 of 53
ACS Paragon Plus Environment
Molecular Pharmaceutics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

14
cold acetone to precipitate proteins. Supernatant was collected, dried, reconstituted in 100 µL of
mobile phase and centrifuged. 70 µL of the supernatant was stored at -70 °C until further
analysis. At the time of analysis 10 µL of Gly-Phe solution (4 mg/mL) was added to the
supernatant solution to serve as internal standard and the sample was analyzed using reverse
phase HPLC (Waters Corporation, Milford, MA). Percentage of intact polymer remaining was
calculated based on the calibration curve generated for the intact polymer. The activity of
enzyme was determined spectrophotometrically using N-benzoyl-Phe-Val-Arg-p-nitroanilide
hydrochloride as a substrate. The recovery of the polymer from the experimental procedure was
determined by using acetate buffer without cathepsin B and was found to be more than 90 %.
Samples obtained from degradation studies of amine-terminated polymer were also analyzed by
MALDI to determine degradation profile of the polymer.
Cell lines
Breast cancer cell lines MDA-MB-231 human adenocarcinoma (ATCC, Manassas, VA) and BT-
549 (ATCC, Manassas, VA) were cultured in RPMI 1640 media (Invitrogen, Carlsbad, CA)
supplemented with 4 mM L-glutamine, 10% (v/v) heat-inactivated fetal bovine serum (FBS) and
1% 100x antibiotic-antimycotic (Invitrogen) at 37°C in a humidified atmosphere of 5% CO2
(v/v). Breast cancer cell line Hs 578T (ATCC, Manassas, VA) was cultured in Dulbecco’s
modified eagle medium (DMEM) supplemented with 10 % (v/v) non-heat-inactivated FBS and
0.01 mg/mL of bovine insulin. For all experimental procedures, sub-confluent cells were
harvested with 0.05% trypsin/0.02% EDTA in PBS.
Solubility studies:
Page 14 of 53
ACS Paragon Plus Environment
Molecular Pharmaceutics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

15
Stock solutions of 2ME and MESPM were prepared by dissolving 10 mg of drug in 1 mL
DMSO. Five working solutions (1,5, 10, 30, 50 ug/mL for 2ME and 1, 2, 3.1, 6.25, 12.5 ug/mL
for MESPM) were prepared by dilution in DMSO from the stock solution. Solubility was
determined using LCMS. Chromatography was performed on Shimatzu LCMS 2020 (Shimatzu
Scientific Instrument Inc., Addison, IL) which included a quaternary pump with an online
degasser and an autosampler (SIL-20A UFLC). Analysis was performed at ambient temperature
on Atlantis dC18 column (3 µm; 2.1 x 150 mm) using mobile phase composed of water (0.1 %
acetic acid) and acetonitrile (0.1 % acetic acid). The mobile phase was delivered as an isocratic
run (50 % water and 50 % acetonitrile) for 2ME and as a gradient run for MESPM
(Supplementary data Table T1) at a flow rate of 0.4 mL/min. A single quadrupole detector with
electrospray interface operated in negative mode (for 2ME) and positive ionization mode (for
MESPM) was used for ion detection. Data processing and analysis was performed using
LCMSsolution software. Retention time for 2ME and MESPM was found to be 2.88 and 7.98
respectively. A calibration curve was constructed by least-squares linear regression analysis of
peak area ratio of 2ME or MESPM versus the drug concentration (Supplementary data Figure
S4A and S4B). To determine solubility in water, stock solutions of 2ME or MESPM were
diluted with water, kept for 24h, centrifuged at 3000 rpm for 5 min and the supernatant was
filtered through syringe filter (0.45 µM, Whatman) before analysis using LCMS to obtain peak
area ratio that was used to determine the concentration of 2ME or MESPM in solution. Two
independent studies were performed starting from preparing stock solutions and running the
samples in triplicate.
In vitro growth inhibition assay:
Page 15 of 53
ACS Paragon Plus Environment
Molecular Pharmaceutics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

16
The WST (water soluble tetrazolium salt) assay was used to determine the antiproliferative effect
of 2ME, Gly2ME, GFLGG2ME, CSPM, ASPM and MESPM using three triple negative breast
cancer cell lines MDA-MB-231, BT-549 and Hs 578T. Cells were plated at predetermined
densities (2500 cells/well for all cell line) in 96-well plates and allowed to attach and grow for 24
h. Control and various treatments were added and the cells were incubated for another 48 h, 72 h,
4 days or 5 days (depending on the treatment). Medium was aspirated and cells were incubated
in the presence of WST-1 reagent for 4 h and absorbance was read at 440 nm. Cell growth
inhibition was determined by subtracting the cell viability at day 0 from the cell viability after
treatment and expressed as % cell viability compared to untreated cells. Experiments were
routinely conducted in the exponential growth phase. Seven different concentrations of each
compound were used for the experiment. GI50 values were determined by nonlinear regression
analysis using GraphPad Prism software. Mean GI50 values are reported for at least two
independent experiments with n=3 for each concentration.
RESULTS:
Synthesis and characterization of degradable star shaped peptidic macromolecules
The characteristics of SPMs used for biological studies are summarized in Table 1. A single
molecular peak was observed for all the macromolecules in MALDI (Figure 2 B, Figure S1),
indicating the presence of a single molecular entity as opposed to a polymer with polydispersity.
MALDI and NMR analysis for all three SPMs confirmed the conjugation of monomer to all four
branches. NMR spectra for MESPM is shown in Figure 2A. The characteristic peaks at δ 1.2-
1.3, δ 3.6-3.8, and δ 7.2-7.5 in the NMR spectra were used to confirm the number of leucines,
glycines, and phenylalanines, respectively, whereas peaks at δ 6.4 and δ 6.7 were used to
confirm the number of 2ME molecules present in a macromolecule. Based on NMR and MALDI
Page 16 of 53
ACS Paragon Plus Environment
Molecular Pharmaceutics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

17
analysis, 2ME content was calculated to be 38% wt/wt. The absence of the monomeric
tetrapeptide and the free 2ME in MESPM was confirmed by performing HPLC analysis.
Stability and in-vitro degradation of SPMs:
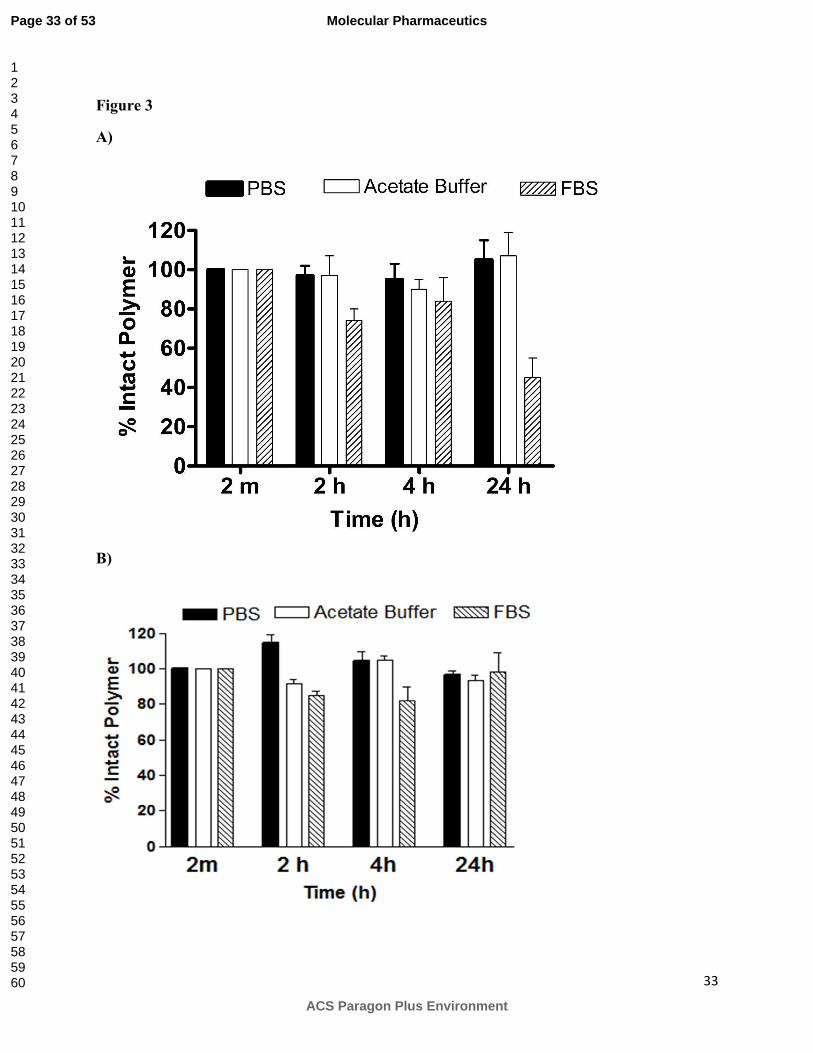
To evaluate stability, SPMs were incubated with PBS (pH =7.4), 10% FBS in PBS, and
acetate buffer (pH = 5) and percentage of intact SPMs remaining is shown in Figure 3A (ASPM)
and Figure 3B (CSPM). Both ASPM and CSPM were quite stable in PBS. No degradation was
observed for either of SPMs at earlier time points (2 and 4 h) and more than 95% of the intact
SPMs were detected even after longer incubation (24 h). Degradation profiles of SPMs were
different in the presence of serum. ASPM showed signs of degradation at earlier time points (74-
84% intact ASPM detected after 2 h and 4 h) with 50% intact macromolecule remaining after 24
h (Figure 3A). In contrast, CSPM was stable in 10% FBS over 24 h period (97% of intact CSPM
detected after 24 h incubation, Figure 3B). Cathepsin B has its maximum activity at pH 5.0.
Thus, degradation studies in the presence of cathepsin B were conducted at acidic pH. To
determine if the pH has any effect on degradation, SPMs were incubated in an acetate buffer
(without cathepsin B) and evaluated for stability. None of the polymers showed any degradation
in acidic pH (Figure 3A,B).
Degradation studies after incubation with cathepsin B revealed the presence of 10%, 1%
and 0 % intact ASPM after 2 h, 4 h, and 24 h respectively (Figure 4A). The lower concentration
of enzyme (0.12 units/µmol of peptide vs 0.25 units/µmol peptide used in the initial studies) used
in subsequent experiments decreased the rate of degradation as indicated by detection of higher
percentages of the intact ASPM (35% and 5% vs 10% and 0% at 2 and 4 h respectively) (Figure
4B). Figure 5 illustrates the percentage of intact CSPM after incubation with cathepsin B (0.25
Page 17 of 53
ACS Paragon Plus Environment
Molecular Pharmaceutics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

18
units/µmol peptide). None of the intact CSPM was detected after 2 h while 46%, 83%, 91%,
93%, and 94% degradation were seen after 4, 10,15, 30, and 60 min, respectively (Figure 5).
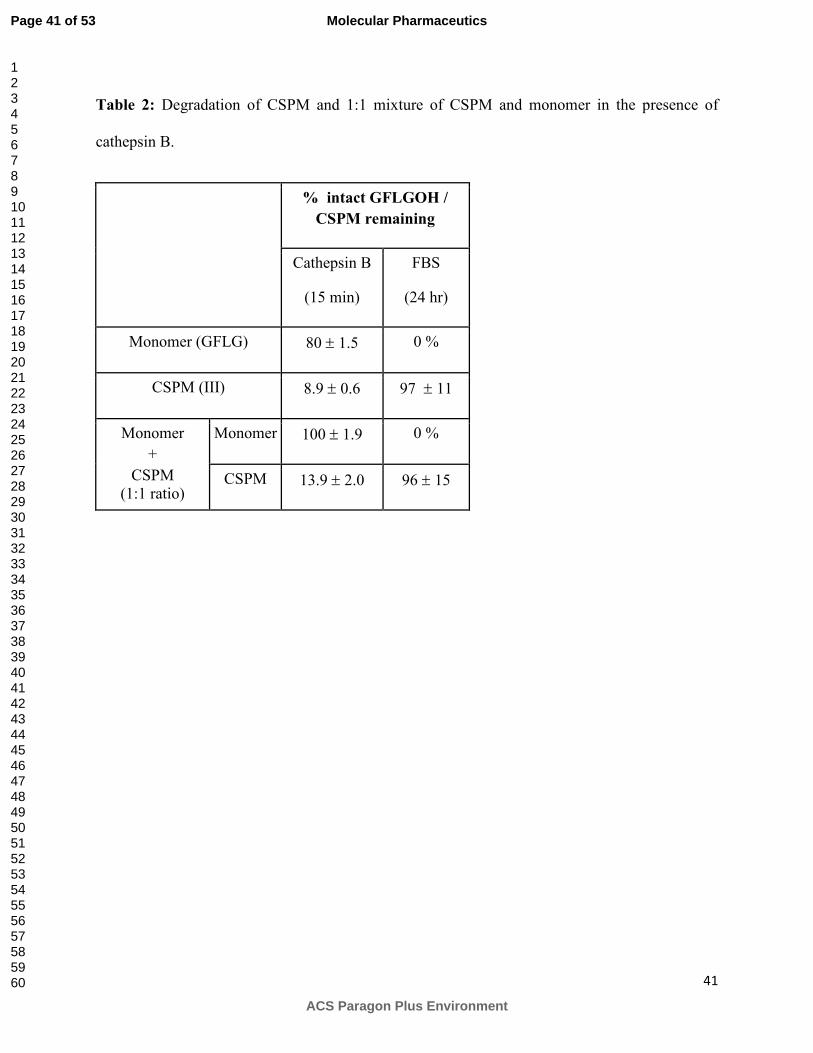
Table 2 illustrates the percentages of intact monomer GFLG after incubation with cathepsin B or
FBS. In contrast to macromolecules, 80% of the intact monomer was detected in the presence of
cathepsin B whereas it degraded completely in the presence of FBS. Degradation behavior of
single entities was retained within a mixture (1:1) of monomer and CSPM. Thus, incubation of a
mixture with cathepsin B degraded only CSPM whereas incubation of a mixture with FBS
degraded only monomer (Table 2).
Solubility and in-vitro cytotoxicity of SPMs and MESPM
Solubility of ASPM and CSPM was found to be more than 5mg/mL. Solubility of 2ME
and MESPM in water was determined using LCMS and was found to be 0.3 ug/mL and 8 ug/mL
respectively. Figure 7 shows % cell viability of MDA-MB-231 after treatment with SPMs for
various time periods. After 48 h exposure, cell viability was more than 90% for both SPMs at the
highest concentrations tested indicating their non-toxic behavior. Our previous reports and
literature suggest that surface charge and density affects cell viability [34-36]. Positively
charged amine groups exhibit higher toxicity compared to their negatively charged or neutral
counterparts. Thus, ASPM was incubated for longer periods (5 days) to evaluate cytotoxicity.
No toxicity was seen even after these longer incubation times. GI50 values could not be
calculated for any of the treatments since cell viability was always more than 50%.
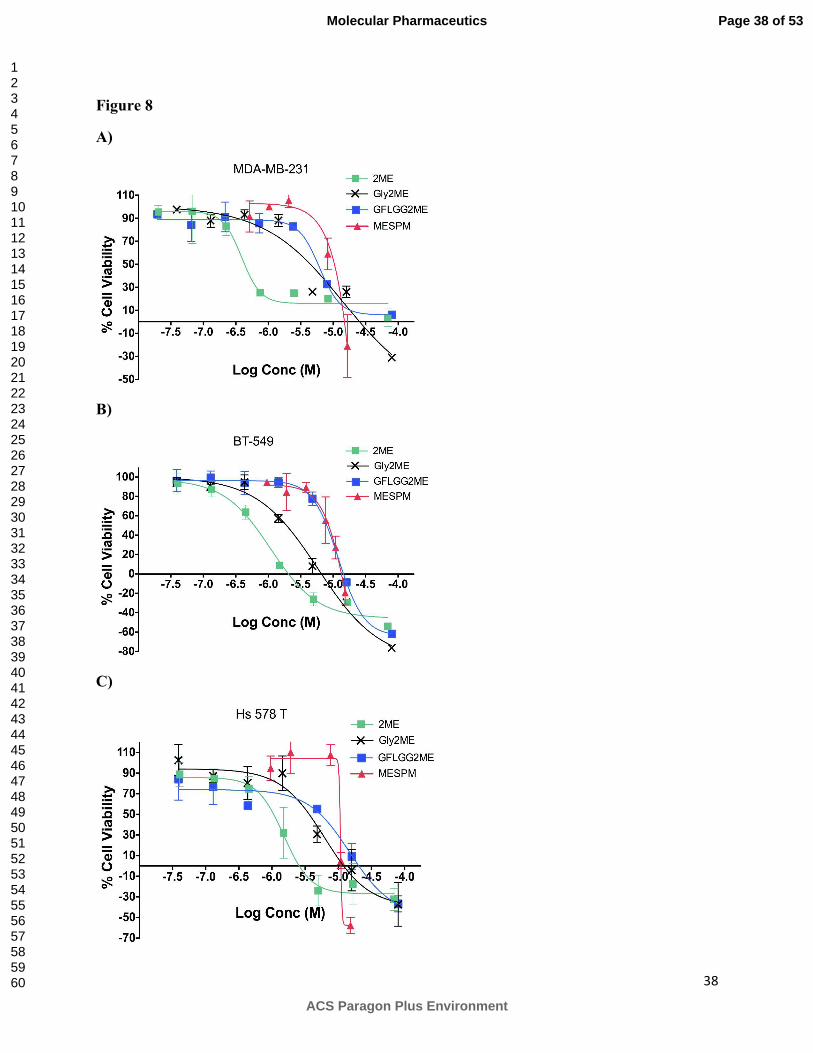
Antiproliferative activity of 2ME, Gly2ME, GFLGG2ME, CSPM and MESPM was further
evaluated in three triple negative breast cancer cell lines viz MDA-MB-231, BT-549 and Hs578
T after 48 and 72 h treatment. Representative cell viability graphs after 48 h treatment are shown
in Figure 8A for MDA-MB-231, Figure 8B for BT-549 and Figure 8C for Hs578 T cell line. GI50
Page 18 of 53
ACS Paragon Plus Environment
Molecular Pharmaceutics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

19
values for all drug-containing treatments are listed in Table 3. The rank order for the
antiproliferative effect of 2ME was found to be BT-549 > MDA-MB-231 > Hs 578T. BT-549
cell line was 1.6- and 2-fold more sensitive to 2ME than MBA-MB-231 and Hs 578T cell line
respectively. Irrespective of the treatment GI50 values remained mostly the same after 48 and 72
h. Cell viability for three cell lines, after incubation with MESPM for 48 h at the highest
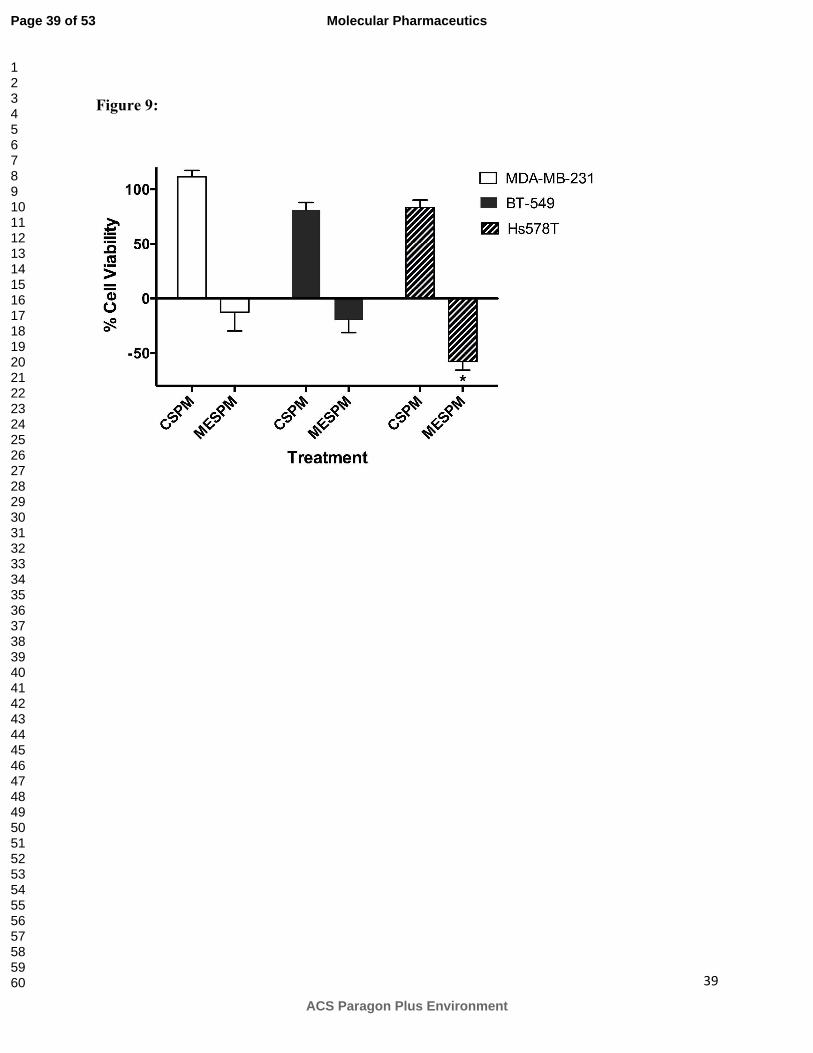
concentration tested, is reported in Figure 9. MESPM showed significantly higher toxicity in
Hs578T cell line compared to other two cell lines after 48h treatment. As indicated in Figure 9
no significant toxicity was detected for CSPM in all cell lines used in the study.
DISCUSSION
Our aim is to develop a degradable delivery system and trigger its degradation at tumor sites. We
decided to use enzymatic trigger due to the specificity that can be imparted by using enzyme
substrate as a building block. We chose cathepsin B as a trigger because of its higher expression
at tumor sites. The tetrapeptide GFLG was used as a building block because previously it has
been used to conjugate anticancer drugs to the polymeric backbone [8, 23, 37] and as a linker
between low molecular weight HPMA copolymers to create high molecular weight degradable
system [38-40]. The major reason for selection of this tetrapeptide was the stability it entails to
the polymeric conjugates in the presence of serum, whereas the degradability it imparts in the
presence of cathepsin B. Two star-shaped peptidic macromolecules (SPMs) were synthesized.
One in which tetrapeptide was conjugated to the core thorough its carboxyl-terminus leaving free
amines at the surface to yield amine-terminated star shaped peptidic macromolecule (ASPM,
Figure 1, II) and one in which tetrapeptide was conjugated to the core through its amine terminus
to yield carboxyl-terminated star-shaped peptidic macromolecule (CSPM, Figure 1, III). CSPM
was also used to conjugate model drug 2ME to yield 2ME containing star shaped peptidic
Page 19 of 53
ACS Paragon Plus Environment
Molecular Pharmaceutics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

20
macromolecule (MESPM, Figure 1, III). ASPM was synthesized by building the tetrapeptide
sequence on PAMAM dendrimer (G0, ethylenediamine core) by sequential addition of one
amino acid (G, F, L, G) at a time (Scheme 1). During the synthesis, we discovered that coupling
agents could be used instead of reactive ester to conjugate the first and second amino acid to the
central core. However, they failed to give quantitative yield during the conjugation of the third
and fourth amino acid. Sequential approach could not be used for CSPM and MESPM due to the
requirement of cumbersome synthesis of reactive esters at each step. Instead, a convergent
approach was used, where tetrapeptides (GFLG or GFLGG2ME) were conjugated to the central
core (EDTA) in one step. Our initial attempts to conjugate the tetrapeptides to the central EDTA
core using various coupling reagents yielded a mixture of compounds having one to four
branches of tetrapeptides attached to the central core. In contrast, the use of tetrakis-p-
nitrophenyl ester of EDTA was efficient with better yields and less side products. The use of the
convergent approach for the MESPM also precluded the possibility of the presence of free 2ME
in the conjugate.
Both SPMs showed substantial stability in the presence of serum at earlier time points.
CSPM was stable even after longer incubation period (24 h) but only 50 % of intact ASPM was
detected at that time point. The detection of lower amount of ASPM can in part be due to its
binding to plasma proteins or due to its degradation. The stability of GFLG-containing-HPMA
copolymers in the presence of serum has been reported previously and been attributed to
specificities of the enzymes present in serum, or the presence of the protease inhibitors, or
combination of both [41]. The degradation profile of SPMs in the presence of cathepsin B was
evaluated under in-vitro conditions, which mimic the conditions of lysosomal degradation as
described in previous reports [31-33]. Time-dependent degradation was observed for both ASPM
Page 20 of 53
ACS Paragon Plus Environment
Molecular Pharmaceutics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

21
and CSPM. Degradation was complete for CSPM within 2 h whereas 10% of ASPM was
detected at the same time point indicating differences in their rates of degradations that can be
attributed to their surface charge. Previously, HPMA copolymers having GFLG linker have been
shown to release the model-drug or drug (anthracyclin, campthothecin, and ansamycin analogs)
within few hours [31, 42-46]. Approximately, 60-70 % of doxorubicin release within 10 h time
period has been reported for doxorubicin containing HPMA copolymer [42]. In contrast, rapid
and complete degradation of CSPMs observed incurrent study is expected to facilitate the drug
release in an in-vivo setting after its encounter with cathepsin B.
Cathepsin B is a cysteine protease and can act as an endopeptidase [47, 48] or an
exopeptidase [49, 50] depending on the pH of the environment [47, 51, 52]. As an exopeptidase,
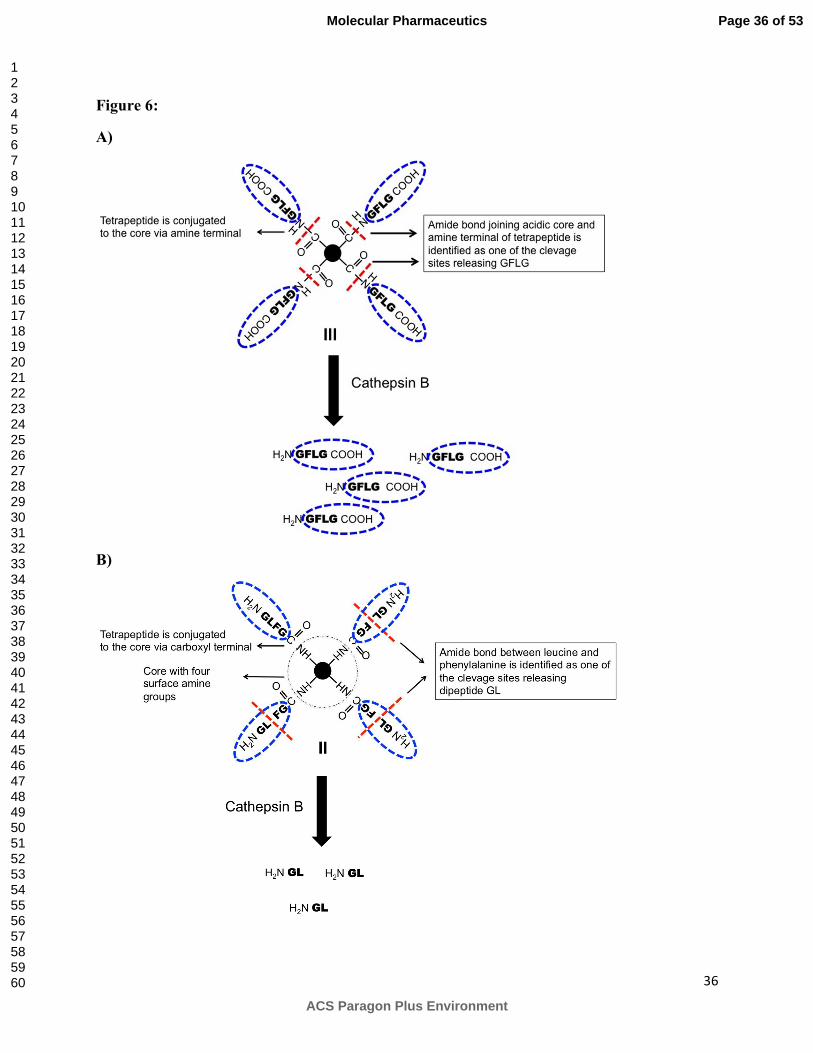
it has been reported to cleave a dipeptide bond at the carboxyl terminus. HPLC chromatogram of
incubation mixture of CSPM indicated the release of tetrapeptide GFLGOH (Fig S2) suggesting
that enzyme is acting as an endopeptidase cleaving the GFLG branch from the EDTA core. The
schematic representation of such type of cleavage and degradation is shown in figure 6A.
However, it is not the only pathway for degradation of CSPM since various peaks between 20-25
min of retention time suggests the presence of other fragments formed due to cleavage at other
sites. In contrast to CSPM, HPLC chromatogram of the incubation mixture of ASPM revealed
the absence of the peak corresponding to the monomeric tetrapeptide and the appearance of
various peaks at higher retention time (20-25 min) indicating the presence of other molecular
fragments. MALDI analysis of these samples indicated the presence of fragments corresponding
to the cleavage of dipeptide (GL) from one, two and three branches (Figure S3). No product
corresponding to the cleavage of all four dipeptides from the dendrimer was detected, even after
24 h of incubation. It is unclear at this point why one GFLG branch remains attached to the core.
Page 21 of 53
ACS Paragon Plus Environment
Molecular Pharmaceutics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

22
Perhaps higher incubation time is required to cleave the last branch from the core. Schematic
representation for the degradation of ASPM is shown in Figure 6B. The differences in the
degradation behavior of CSPM and ASPM can be attributed to the surface charge and the
difference in the sequence. CSPM has GFLG as core-to-surface sequence while ASPM has
GFLG as surface-to-core sequence. Very few studies have been conducted where large protein
substrates have been used to understand the mechanism for endopeptidase activity of cathepsin
B. The peptide segment (Ile105-Pro126) termed the occluding loop in the primed side of the active
site of cathepsin B is known to play a critical role in determining an endo- or exopeptidase
activity of the enzyme [48, 53, 54]. Disruption of two salt-bridge interactions between the loop
and the main body of the enzyme (Asp22-His110 and Arg116-Asp224) has been shown to result in
dramatic increase in the endopeptidase activity of the enzyme [48, 50, 54]. Surface acidic groups
present on CSPM might disrupt these salt-bridge interactions resulting in higher endopeptidase
activity. It is surprising, however, not uncommon to report that irrespective of surface positive
charges, cathepsin B starts cleaving terminal dipeptide GL from the first branch of ASPM
followed by the second and the third branch. Cathepsin B has been shown to cleave amide
terminated peptides [53]. To gain more knowledge about the degradative mechanism, we
conducted degradation studies using monomeric tetrapeptide GFLG. Surprisingly, no
degradation was seen for the monomeric tetrapeptide (Table 2), which suggests that the
tetrapeptide itself does not act as a substrate for cathepsin B supporting the specific sequence
requirements for small peptides that must be met in order to behave as cathepsin B substrates.
However, degradation of SPMs indicates the lack of stringent substrate requirements for
macromolecules. Next, we evaluated the degradation of a 1:1 mixture of monomeric tetrapeptide
and CSPM in the presence of cathepsin B and 10% FBS solutions. Table 2 illustrates the
Page 22 of 53
ACS Paragon Plus Environment
Molecular Pharmaceutics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

23
percentages of intact CSPM or tetrapeptide detected after incubation with cathepsin B or FBS.
As expected, within a mixture, CSPM was preferentially degraded in the presence of cathepsin B
while being stable in FBS. Conversely, monomeric tetrapeptide was completely stable in the
presence of cathepsin B and degraded completely in the presence of serum. These results
emphasize the importance of molecular weight and architecture in serum stability and enzymatic
degradation.
Next, we evaluated the potential of SPMs for delivery of 2ME. CSPM was selected due
to its serum stability and complete degradation in the presence of cathepsin B. A glycine
derivative of 2ME (Gly-2ME) was conjugated to the terminal acidic groups to get MESPM.
Solubility studies indicated modest improvement (26-fold) in the solubility of MESPM
compared to 2ME. Growth inhibition studies were performed using three triple negative breast
cancer cell lines viz MDA-MB-231, BT-549 and Hs 578T. Xing et al has reported higher
expression levels of cathepsin B in Hs 578T compared to MDA-MB-231 and BT-549 cell lines
[55]. We wanted to observe if these expression levels could be translated into differential toxicity
profile of MESPM. Although GI50 values for MESPM were quite similar in Hs 578T and BT-
549, MESPM showed significantly higher toxicity at the highest concentration studied in Hs
578T compared to BT-549 (Figure 9). This is in spite of 2-fold lower sensitivity of Hs 578T to
2ME. Comparison of Hs 578 T and MDA-MB-231 cell line proved difficult due to differential
activity of MESPM in MDA-MB-231 cell lines after 48 and 72 h treatment. Overall
antiproliferative activity of MESPM in all cell lines was almost similar to the monomer
GFLGG2ME indicating that GFLGG2ME is probably getting released. In-vitro release studies
can only confirm that hypothesis. Nevertheless, antiproliferative activity of MESPM indicates
that either 2ME or 2ME containing peptide is getting released from MESPM. Increasing the
Page 23 of 53
ACS Paragon Plus Environment
Molecular Pharmaceutics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

24
incubation time from 48 to 72 hours did not change GI50 values significantly for monomers or for
MESPM (except for MESPM in MDA-MB-231). Decrease in the antiproliferative activity of
polymeric conjugate compared to free drug is expected and well known. MESPM exhibited
higher GI50 compared to free drug. However, the loss of activity is significantly less compared
to the loss of activity observed after conjugation of drugs to the water-soluble polymers using
GFLG tetrapeptide as a linker. More importantly drug content (wt/wt) of the peptidic dendrimer
was much higher (38%) compared to other drug containing polymeric conjugates reported in the
literature.
Low solubility of 2ME along with its rapid metabolism is reported to limit its
bioavailability. In the current study solubility of 2ME in water was found to be 0.3 ug/mL,
which is in agreement with the reported value in the literature [56]. The molecular weight of
MESPM is low and will allow for rapid renal clearance. However, a modest increase in solubility
of MESPM (26-fold more than 2ME) along with the potential of lowering the metabolism (due
to the conjugation of the SPM through 17-OH group on the steroidal backbone of 2ME) is
expected to achieve higher plasma levels compared to 2ME alone. The primary intention of this
work was to evaluate if cathepsin B is able to recognize peptide GFLG in the star-like
architecture and if such a system would exhibit any antiproliferative property when conjugated to
the drug. Confirmatory results from current study set a stage for building a high molecular
weight dendritic systems using GFLG as a building block. Such a system that will respond to the
presence of cathepsin B can be benefitted by its higher molecular weight to achieve better in-
vivo profile such as longer circulation time and tumor accumulation. Structural optimization can
also be achieved at this stage to make the system highly water soluble while maintaining its
antiproliferative property.
Page 24 of 53
ACS Paragon Plus Environment
Molecular Pharmaceutics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

25
CONCLUSION
Here, we report the use of degradable SPMs as a potential platform for building delivery
vehicle for 2ME. SPMs were stable in the presence of serum but degraded completely within
short period of time in the presence of cathepsin B, which indicates their potential as stimuli-
sensitive drug carriers. Surface charge affected both serum stability as well as degradation
behavior of SPMs in the presence of cathepsin B. Higher drug loading (38% wt/wt) was achieved
with good antiproliferative activity in a panel of triple negative breast cancer cell lines.
Page 25 of 53
ACS Paragon Plus Environment
Molecular Pharmaceutics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

26
ABBREVIATIONS
ATCC American type culture collection
BOC tert-Butyloxycarbonyl
CCK cell counting kit
DCM dichloromethane
DCC dicyclohexylcarbodiimide
DCU dicyclohexylurea
DIPEA N,N-diisopropyl ethylamine
DMAP 4-dimethyl amino pyridine
DMSO dimethyl sulfoxide
EDTA Ethylenediaminetetraacetic acid
FBS fetal bovine serum
G0 Generation 0
GFLG Gly-Phe-Leu-Gly
HOBt Hydroxybenzotriazole
HBTU O-Benzotriazole-N,N,N’,N’-tetramethyl-uronium-hexafluoro-phosphate
HPLC high performance liquid chromatography
IC50 half maximal inhibitory concentration
MW molecular weight
ONp p-nitrophenol
PAMAM poly (amido amine)
Page 26 of 53
ACS Paragon Plus Environment
Molecular Pharmaceutics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

27
PBS phosphate buffered saline
TFA trifluoroacetic acid
THF tetrahydrofuran
TLC thin layer chromatography
UV ultraviolet
Wt weight
ACKNOWLEDGEMENTS:
Authors would like to thank Dr. Lee, Director of Protein Research lab, Research Resource
Center at UIC and Dr. Campbell from University of Maryland, Baltimore for their assistance
with MALDI analysis. This work was supported by Department of Defense Multidisciplinary
Postdoctoral fellowship W81XWH-06-1-0698 and Department of Defense Breast Cancer
Research Program Concept Award (W81XWH-09-1-0687) to RK. The work was also supported
by start-up funds to RK from Department of Biopharmaceutical Sciences, University of Illinois
Chicago.
Page 27 of 53
ACS Paragon Plus Environment
Molecular Pharmaceutics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

28
Legends to figures:
Figure 1:
Chemical structure of 2-methoxyestradiol (I), schematic representation of amine-terminated star-
shaped peptidic macromolecule (II, ASPM), carboxyl-terminated star-shaped peptidic
macromolecule (III, CSPM) and 2-methoxyestradiol containing peptidic macromolecule (IV,
MESPM).
Figure 2:
A) NMR spectra of MESPM (IV) showing peaks corresponding to all amino acids and 2ME B)
MALDI analysis of MESPM.
Figure 3:
Stability of A) ASPM and B) CSPM in the presence of PBS (pH = 7.4), acetate buffer (pH = 5.0)
and 10% FBS in PBS over the period of 24 hours. Percentage of the intact polymer remaining
after incubation in respective buffers is reported and was determined using HPLC. Area under
curve (AUC) for polymer was normalized with the AUC obtained for Gly-Phe that was used as
an internal standard. (n = 3, mean ± SD).
Figure 4:
Degradation profile of ASPM (II) in the presence of A) Cathepsin B (0.25 units/µmol of peptide)
and B) Cathepsin B (0.12 units/µmol of peptide). Percentage of the intact polymer remaining
after incubation with cathepsin B is reported and is determined using HPLC. Area under curve
Page 28 of 53
ACS Paragon Plus Environment
Molecular Pharmaceutics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

29
(AUC) for polymer was normalized with the AUC obtained for Gly-Phe that was used as an
internal standard. (n = 3, mean ± SD).
Figure 5:
Degradation profile of CSPM (III) in the presence of Cathepsin B (0.25 units/µmol of peptide).
Percentage of the intact polymer remaining after incubation with cathepsin B is reported and is
determined using HPLC. Area under curve (AUC) for polymer was normalized with the AUC
obtained for Gly-Phe that was used as an internal standard. (n = 3, mean ± SD).
Figure 6:
Schematic representation for the degradation of A) CSPM and B) ASPM in the presence of
cathepsin B. CSPM and ASPM degraded differently. HPLC analysis indicated that one of the
cleavage sites during degradation of CSPM is the amide bond between the acidic core and amine
terminal of tetrapeptide releasing GFLG. ASPM degraded by releasing terminal dipeptide GL. ‘-
------‘ indicates cleavage sites.
Figure 7:
Growth inhibition effect of A) CSPM and B) ASPM on breast cancer cell line (MDA-MB-231)
after incubation for 48, 72 h and 5 days. Cell viability was determined using WST-1. Results are
mean ± SD (n=3).
Figure 8:
Growth inhibition effect of 2ME, Gly2ME, GFLGG2ME and MESPM on breast cancer cell
lines. A) MDA-MB-231 B) BT-549 C) Hs 578T. Cell viability after 48 h treatment was
Page 29 of 53
ACS Paragon Plus Environment
Molecular Pharmaceutics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

30
determined using WST-1. Results are mean ± SD (n=6, two independent experiments in
triplicates). 2ME; Gly2ME; GFLGG2ME; MESPM
Figure 9:
Growth inhibition effect of CSPM and MESPM on MDA-MB-231, BT-549 and Hs 578T cells
after incubation for 48 at 15 µM 2ME equivalent concentration. Cell viability was determined
using WST-1. Results are mean ± SD (n=6, two independent experiments in triplicates).
*Statistical difference between cell viabilities of different cell lines was compared using student t
test where * denotes significant difference between cell viability of Hs578T and MDA-MB-231
(p < 0.05) and cell viability of Hs 578T and BT-549 (p < 0.05).
Page 30 of 53
ACS Paragon Plus Environment
Molecular Pharmaceutics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960
31
Figure 1
Page 31 of 53
ACS Paragon Plus Environment
Molecular Pharmaceutics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

32
Figure 2:
Page 32 of 53
ACS Paragon Plus Environment
Molecular Pharmaceutics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960
33
Figure 3
A)
B)
Page 33 of 53
ACS Paragon Plus Environment
Molecular Pharmaceutics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

34
Figure 4:
A)
B)
Page 34 of 53
ACS Paragon Plus Environment
Molecular Pharmaceutics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

35
Figure 5
Page 35 of 53
ACS Paragon Plus Environment
Molecular Pharmaceutics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960
36
Figure 6:
A)
B)
Page 36 of 53
ACS Paragon Plus Environment
Molecular Pharmaceutics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

37
Figure 7
A)
B)
Page 37 of 53
ACS Paragon Plus Environment
Molecular Pharmaceutics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960
38
Figure 8
A)
B)
C)
Page 38 of 53
ACS Paragon Plus Environment
Molecular Pharmaceutics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960
39
Figure 9:
Page 39 of 53
ACS Paragon Plus Environment
Molecular Pharmaceutics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

40
Table 1: Physicochemical characteristics of peptidic dendrimers
Polymer Mw
Calc.
Mw
Observed by
MALDI
Retention
Time
(HPLC)
(Min)
G0(GLFGNH2)4
(ASPM) 2014.42 2038
(M+Na) 21.77
EDTA(GFLGOH)4
(CSPM)
1788.87 1791.87 27.08
EDTA(GFLGG2ME)4
MESPM 3155.67 3178.39
(M+Na) 46.42
Page 40 of 53
ACS Paragon Plus Environment
Molecular Pharmaceutics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960
41
Table 2: Degradation of CSPM and 1:1 mixture of CSPM and monomer in the presence of
cathepsin B.
% intact GFLGOH /
CSPM remaining
Cathepsin B
(15 min)
FBS
(24 hr)
Monomer (GFLG) 80 ± 1.5 0 %
CSPM (III) 8.9 ± 0.6 97 ± 11
Monomer
+
CSPM (1:1 ratio)
Monomer 100 ± 1.9 0 %
CSPM 13.9 ± 2.0 96 ± 15
Page 41 of 53
ACS Paragon Plus Environment
Molecular Pharmaceutics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

42
Table 3: Growth inhibition (GI50) values of 2ME, Gly2ME, GFLGG2ME and MESPM in breast
cancer cell lines MDA-MB-231, BT-549 and Hs 578T
Sample GI50 (µM)
MDA-MB-231 BT-549 Hs578T
48 h 72 h 48 h 72 h 48 h 72 h
2ME 1.0 ± 0.2 1.1 ± 0.4 0.6 ± 0.08 0.66 ± 0.04 1.22 ± 0.50 1.47 ± 0.42
Gly-2ME 2.7 ± 0.8 - 1.9 ± 0.35 1.77 ± 0.11 3.7 ± 0.6 2.44 ± 0.70
GFLGG2ME 7.8 ± 0.3 7.6 ± 0.6 7.9 ± 0.5 7.70 ± 0.10 7.03 ± 1.12 8.22 ± 0.87
MESPM 8.5 ± 0.3 13.8 ± 4 10.3 ± 2.4 10.81 ± 0.62 10.2 ± 0.7 10.93 ± 1.27
GI50 values are calculated from dose response curves obtained by performing nonlinear
regression analysis using GraphPad Software. Values are calculated for at least two
independent experiments with n=3 for each concentration (mean ± SD).
Page 42 of 53
ACS Paragon Plus Environment
Molecular Pharmaceutics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960
43
Scheme 1: Synthesis of ASPM using sequential approach
Step 1: i) Boc-Gly-ONp ii) 50:50 TFA/DCM; Step 2: i) Boc-Phe-ONp (ii) 50:50 TFA/DCM; Step 3) i) Boc-Leu-ONp (ii) 50:50 TFA/DCM; Step 4: i) Boc-Gly-ONp(ii) 50:50 TFA/DCM
Page 43 of 53
ACS Paragon Plus Environment
Molecular Pharmaceutics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

44
Scheme 2: Synthesis of Tetrakis(p-nitrophenyl) ester of EDTA (central reactive core)
Page 44 of 53
ACS Paragon Plus Environment
Molecular Pharmaceutics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

45
Scheme 3: Synthesis of CSPM (III) and MESPM (IV) using convergent approach.
Page 45 of 53
ACS Paragon Plus Environment
Molecular Pharmaceutics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

46
TOC figure
Page 46 of 53
ACS Paragon Plus Environment
Molecular Pharmaceutics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

47
References
[1] Verenich S, Gerk PM. Therapeutic promises of 2-methoxyestradiol and its drug disposition
challenges. Mol Pharm. 2010;7:2030-9.
[2] Sutherland TE, Anderson RL, Hughes RA, Altmann E, Schuliga M, Ziogas J, et al. 2-
Methoxyestradiol--a unique blend of activities generating a new class of anti-tumour/anti-
inflammatory agents. Drug discovery today. 2007;12:577-84.
[3] Klauber N, Parangi S, Flynn E, Hamel E, D'Amato RJ. Inhibition of angiogenesis and breast
cancer in mice by the microtubule inhibitors 2-methoxyestradiol and taxol. Cancer research.
1997;57:81-6.
[4] Fotsis T, Zhang Y, Pepper MS, Adlercreutz H, Montesano R, Nawroth PP, et al. The
endogenous oestrogen metabolite 2-methoxyoestradiol inhibits angiogenesis and suppresses
tumour growth. Nature. 1994;368:237-9.
[5] Mueck AO, Seeger H. 2-Methoxyestradiol--biology and mechanism of action. Steroids.
2010;75:625-31.
[6] Dahut WL, Lakhani NJ, Gulley JL, Arlen PM, Kohn EC, Kotz H, et al. Phase I clinical trial
of oral 2-methoxyestradiol, an antiangiogenic and apoptotic agent, in patients with solid tumors.
Cancer biology & therapy. 2006;5:22-7.
[7] James J, Murry DJ, Treston AM, Storniolo AM, Sledge GW, Sidor C, et al. Phase I safety,
pharmacokinetic and pharmacodynamic studies of 2-methoxyestradiol alone or in combination
with docetaxel in patients with locally recurrent or metastatic breast cancer. Investigational new
drugs. 2007;25:41-8.
[8] Kopecek J. Polymer-drug conjugates: Origins, progress to date and future directions.
Advanced drug delivery reviews. 2013;65:49-59.
Page 47 of 53
ACS Paragon Plus Environment
Molecular Pharmaceutics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

48
[9] Zhou Y, Kopecek J. Biological rationale for the design of polymeric anti-cancer
nanomedicines. Journal of drug targeting. 2013;21:1-26.
[10] Duncan R, Vicent MJ. Polymer therapeutics-prospects for 21st century: The end of the
beginning. Advanced drug delivery reviews. 2013;65:60-70.
[11] Allen TM, Cullis PR. Liposomal drug delivery systems: From concept to clinical
applications. Advanced drug delivery reviews. 2013;65:36-48.
[12] Koshkaryev A, Sawant R, Deshpande M, Torchilin V. Immunoconjugates and long
circulating systems: Origins, current state of the art and future directions. Advanced drug
delivery reviews. 2013;65:24-35.
[13] Kim CS, Tonga GY, Solfiell D, Rotello VM. Inorganic nanosystems for therapeutic
delivery: Status and prospects. Advanced drug delivery reviews. 2013;65:93-9.
[14] Hoffman AS. Stimuli-responsive polymers: Biomedical applications and challenges for
clinical translation. Advanced drug delivery reviews. 2013;65:10-6.
[15] Felber AE, Dufresne MH, Leroux JC. pH-sensitive vesicles, polymeric micelles, and
nanospheres prepared with polycarboxylates. Advanced drug delivery reviews. 2012;64:979-92.
[16] Graf N, Lippard SJ. Redox activation of metal-based prodrugs as a strategy for drug
delivery. Advanced drug delivery reviews. 2012;64:993-1004.
[17] de la Rica R, Aili D, Stevens MM. Enzyme-responsive nanoparticles for drug release and
diagnostics. Advanced drug delivery reviews. 2012;64:967-78.
[18] Gondi CS, Rao JS. Cathepsin B as a cancer target. Expert opinion on therapeutic targets.
2013;17:281-91.
[19] Roshy S, Sloane BF, Moin K. Pericellular cathepsin B and malignant progression. Cancer
metastasis reviews. 2003;22:271-86.
Page 48 of 53
ACS Paragon Plus Environment
Molecular Pharmaceutics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

49
[20] Mohamed MM, Sloane BF. Cysteine cathepsins: multifunctional enzymes in cancer. Nature
reviews Cancer. 2006;6:764-75.
[21] Reiser J, Adair B, Reinheckel T. Specialized roles for cysteine cathepsins in health and
disease. The Journal of clinical investigation. 2010;120:3421-31.
[22] Duncan R, Vicent MJ. Do HPMA copolymer conjugates have a future as clinically useful
nanomedicines? A critical overview of current status and future opportunities. Advanced drug
delivery reviews. 2010;62:272-82.
[23] Vasey PA, Kaye SB, Morrison R, Twelves C, Wilson P, Duncan R, et al. Phase I clinical
and pharmacokinetic study of PK1 [N-(2-hydroxypropyl)methacrylamide copolymer
doxorubicin]: first member of a new class of chemotherapeutic agents-drug-polymer conjugates.
Cancer Research Campaign Phase I/II Committee. Clinical cancer research : an official journal
of the American Association for Cancer Research. 1999;5:83-94.
[24] Seymour LW, Ferry DR, Kerr DJ, Rea D, Whitlock M, Poyner R, et al. Phase II studies of
polymer-doxorubicin (PK1, FCE28068) in the treatment of breast, lung and colorectal cancer.
International journal of oncology. 2009;34:1629-36.
[25] Duncan R. Polymer therapeutics as nanomedicines: new perspectives. Current opinion in
biotechnology. 2011;22:492-501.
[26] Rob Webster VE, B. Kevin Park, Donald Walker, Mark Hankin, Philip Taupin. PEG and
PEG conjugates toxicity: towards an understanding of the toxicity of PEG and its relevance to
PEGylated biologicals. In: Veronese FM, editor. PEGylated Protein Drugs: Basic Science and
Clinical Applications: Birkhäuser Basel; 2009. p. 127-46.
[27] Park K. The optimal formulation variables for tumor targeting. Journal of controlled release
: official journal of the Controlled Release Society. 2012;157:315-6.
Page 49 of 53
ACS Paragon Plus Environment
Molecular Pharmaceutics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

50
[28] Rao PN, Cessac JW. A new, practical synthesis of 2-methoxyestradiols. Steroids.
2002;67:1065-70.
[29] Ho A, Kim YE, Lee H, Cyrus K, Baek SH, Kim KB. SAR studies of 2-methoxyestradiol
and development of its analogs as probes of anti-tumor mechanisms. Bioorganic & medicinal
chemistry letters. 2006;16:3383-7.
[30] Pechar M, Ulbrich K, Subr V, Seymour LW, Schacht EH. Poly(ethylene glycol) multiblock
copolymer as a carrier of anti-cancer drug doxorubicin. Bioconjugate chemistry. 2000;11:131-9.
[31] Borgman MP, Ray A, Kolhatkar RB, Sausville EA, Burger AM, Ghandehari H. Targetable
HPMA Copolymer-Aminohexylgeldanamycin Conjugates for Prostate Cancer Therapy. Pharm
Res-Dord. 2009;26:1407-18.
[32] Kasuya Y, Lu ZR, Kopeckova P, Tabibi SE, Kopecek J. Influence of the structure of drug
moieties on the in vitro efficacy of HPMA copolymer-geldanamycin derivative conjugates.
Pharm Res. 2002;19:115-23.
[33] Wang D, Kopeckova JP, Minko T, Nanayakkara V, Kopecek J. Synthesis of starlike N-(2-
hydroxypropyl)methacrylamide copolymers: potential drug carriers. Biomacromolecules.
2000;1:313-9.
[34] Kolhatkar RB, Kitchens KM, Swaan PW, Ghandehari H. Surface acetylation of
polyamidoamine (PAMAM) dendrimers decreases cytotoxicity while maintaining membrane
permeability. Bioconjugate chemistry. 2007;18:2054-60.
[35] Fischer D, Li Y, Ahlemeyer B, Krieglstein J, Kissel T. In vitro cytotoxicity testing of
polycations: influence of polymer structure on cell viability and hemolysis. Biomaterials.
2003;24:1121-31.
Page 50 of 53
ACS Paragon Plus Environment
Molecular Pharmaceutics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

51
[36] Ryser HJ. A membrane effect of basic polymers dependent on molecular size. Nature.
1967;215:934-6.
[37] Seymour LW, Ferry DR, Anderson D, Hesslewood S, Julyan PJ, Poyner R, et al. Hepatic
drug targeting: phase I evaluation of polymer-bound doxorubicin. Journal of clinical oncology :
official journal of the American Society of Clinical Oncology. 2002;20:1668-76.
[38] Pan H, Sima M, Yang J, Kopecek J. Synthesis of Long-Circulating, Backbone Degradable
HPMA Copolymer-Doxorubicin Conjugates and Evaluation of Molecular-Weight-Dependent
Antitumor Efficacy. Macromolecular bioscience. 2013;13:155-60.
[39] Shiah JG, Dvorak M, Kopeckova P, Sun Y, Peterson CM, Kopecek J. Biodistribution and
antitumour efficacy of long-circulating N-(2-hydroxypropyl)methacrylamide copolymer-
doxorubicin conjugates in nude mice. Eur J Cancer. 2001;37:131-9.
[40] Dvorak M, Kopeckova P, Kopecek J. High-molecular weight HPMA copolymer-adriamycin
conjugates. Journal of controlled release : official journal of the Controlled Release Society.
1999;60:321-32.
[41] Rejmanova P, Kopecek J, Duncan R, Lloyd JB. Stability in rat plasma and serum of
lysosomally degradable oligopeptide sequences in N-(2-hydroxypropyl) methacrylamide
copolymers. Biomaterials. 1985;6:45-8.
[42] Etrych T, Chytil P, Jelinkova M, Rihova B, Ulbrich K. Synthesis of HPMA copolymers
containing doxorubicin bound via a hydrazone linkage. Effect of spacer on drug release and in
vitro cytotoxicity. Macromolecular bioscience. 2002;2:43-52.
[43] Caiolfa VR, Zamai M, Fiorino A, Frigerio E, Pellizzoni C, d'Argy R, et al. Polymer-bound
camptothecin: initial biodistribution and antitumour activity studies. Journal of controlled release
: official journal of the Controlled Release Society. 2000;65:105-19.
Page 51 of 53
ACS Paragon Plus Environment
Molecular Pharmaceutics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

52
[44] Duncan R, Kopeckova-Rejmanova P, Strohalm J, Hume I, Cable HC, Pohl J, et al.
Anticancer agents coupled to N-(2-hydroxypropyl)methacrylamide copolymers. I. Evaluation of
daunomycin and puromycin conjugates in vitro. Br J Cancer. 1987;55:165-74.
[45] Kopecek J, Rejmanova P, Duncan R, Lloyd JB. Controlled release of drug model from N-
(2-hydroxypropyl)-methacrylamide copolymers. Annals of the New York Academy of Sciences.
1985;446:93-104.
[46] Rejmanova P, Kopecek J, Pohl J, Baudys M, Kostka V. Polymers Containing Enzymatically
Degradable Bonds .8. Degradation of Oligopeptide Sequences in N-(2-
Hydroxypropyl)Methacrylamide Co-Polymers by Bovine Spleen Cathepsin-B. Makromol Chem.
1983;184:2009-20.
[47] Khouri HE, Plouffe C, Hasnain S, Hirama T, Storer AC, Menard R. A model to explain the
pH-dependent specificity of cathepsin B-catalysed hydrolyses. The Biochemical journal.
1991;275 ( Pt 3):751-7.
[48] Nagler DK, Storer AC, Portaro FC, Carmona E, Juliano L, Menard R. Major increase in
endopeptidase activity of human cathepsin B upon removal of occluding loop contacts.
Biochemistry. 1997;36:12608-15.
[49] Musil D, Zucic D, Turk D, Engh RA, Mayr I, Huber R, et al. The refined 2.15 A X-ray
crystal structure of human liver cathepsin B: the structural basis for its specificity. The EMBO
journal. 1991;10:2321-30.
[50] Illy C, Quraishi O, Wang J, Purisima E, Vernet T, Mort JS. Role of the occluding loop in
cathepsin B activity. The Journal of biological chemistry. 1997;272:1197-202.
Page 52 of 53
ACS Paragon Plus Environment
Molecular Pharmaceutics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

53
[51] Renko M, Pozgan U, Majera D, Turk D. Stefin A displaces the occluding loop of cathepsin
B only by as much as required to bind to the active site cleft. The FEBS journal. 2010;277:4338-
45.
[52] Ruzza P, Quintieri L, Osler A, Calderan A, Biondi B, Floreani M, et al. Fluorescent,
internally quenched, peptides for exploring the pH-dependent substrate specificity of cathepsin
B. Journal of peptide science : an official publication of the European Peptide Society.
2006;12:455-61.
[53] Krupa JC, Hasnain S, Nagler DK, Menard R, Mort JS. S2' substrate specificity and the role
of His110 and His111 in the exopeptidase activity of human cathepsin B. The Biochemical
journal. 2002;361:613-9.
[54] Quraishi O, Nagler DK, Fox T, Sivaraman J, Cygler M, Mort JS, et al. The occluding loop
in cathepsin B defines the pH dependence of inhibition by its propeptide. Biochemistry.
1999;38:5017-23.
[55] Xing R, Addington AK, Mason RW. Quantification of cathepsins B and L in cells. The
Biochemical journal. 1998;332 ( Pt 2):499-505.
[56] Cho JK, Hong KY, Park JW, Yang HK, Song SC. Injectable delivery system of 2-
methoxyestradiol for breast cancer therapy using biodegradable thermosensitive
poly(organophosphazene) hydrogel. Journal of drug targeting. 2011;19:270-80.
Page 53 of 53
ACS Paragon Plus Environment
Molecular Pharmaceutics
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960





























