Embed Size (px)
Citation preview

Boom in the Development ofNon-Peptidic b-Secretase (BACE1)Inhibitors for theTreatment of
Alzheimer’s Disease
Romano Silvestri
Istituto Pasteur, Fondazione Cenci Bolognetti, Dipartimento di Chimica e Tecnologie del Farmaco,
Sapienza Universita di Roma, Piazzale Aldo Moro 5, I-00185 Roma, Italy
Published online 23 July 2008 in Wiley InterScience (www.interscience.wiley.com).
DOI 10.1002/med.20132
!
Abstract: b-Amyloid cleaving enzyme-1 (BACE1) has become a significant target for the therapy
of Alzheimer’s disease. After the discovery of the first non-peptidomimetic b-secretase inhibitorsby Takeda Chemicals in 2001, several research teams focused on SAR development of these
agents. The non-peptidic BACE1 inhibitors may potentially overcome the classical problems
associated with the peptide structure of first generation, such as blood–brain barrier crossing,
poor oral bioavailability and susceptibility to P-glycoprotein transport. In the past 6 years a
boom in research of non-peptidic BACE1 inhibitors has disseminated findings over hundreds
of publications and patents. The rapidly growing literature has been reviewed with particular
emphasis on literature of pharmaceutical companies. � 2008 Wiley Periodicals, Inc. Med Res Rev, 29,
No. 2, 295–338, 2009
Key words: Alzheimer’s disease; b-secretase; BACE1; non-peptidic inhibitors
1 . I N T R O D U C T I O N
Alzheimer’s disease (AD) is a degenerative brain syndrome first described by Alois Alzheimer in
1906, that affectsmore than 37million peopleworldwide.1 Social cognition is an early impairment of
AD. Dementia of the Alzheimer type leads to a progressive decline in memory with concomitant
abatement of thinking, comprehension, and learning capabilities.2 AD accounts for most cases of
dementia that are diagnosed after the age of 60.3,4
Two main theories are invoked for AD treatment. The ‘‘cholinergic hypothesis’’ relates the
neurodegeneration with the loss of cholinergic neurotransmission; the ‘‘amyloid hypothesis’’
Correspondence to:RomanoSilvestri,DipartimentodiChimicaeTecnologiedel Farmaco,SapienzaUniversita' di Roma,Piazzale
AldoMoro 5, I-00185 Roma, Italy.E-mail: [email protected]
Medicinal Research Reviews, Vol. 29, No. 2, 295^338, 2009
� 2008 Wiley Periodicals, Inc.

correlates the formation of amyloid b (Ab) peptides with the progression of the disease. According tothe ‘‘cholinergic hypothesis,’’ the deterioration of cholinergic neurons in the basal forebrain and the
associated loss of cholinergic neurotransmission in the cerebral cortex and other areas, significantly
contribute to the neurodegeneration in AD.5 On the other hand, the involvement of the amyloid
cascade in the progression of AD is supported by many evidences. The b-amyloid precursor protein
(APP) is a multifunctional protein associated with the viability, growth and functions of the central
neuronal cells. According to the ‘‘amyloid hypothesis,’’ disruption of APP would be correlated to
the emergence of AD.6,7 a-Secretase enzyme usually cleaves APP to form sAPPa and CTF83
membrane-bound fragment; then g-secretase enzyme cleaves CTF83 into peptide p3 and CTFg. ThesAPPa fragment activates NF-kB which is correlated to a neuroprotective effect (Fig. 1). In AD
pathological condition, APP is first cleaved by b-secretase to form sAPPb and membrane-bound
Figure 1. The cholinergichypothesis andamyloidhypothesis in AD.
296 * SILVESTRI
Medicinal Research Reviews DOI 10.1002/med

CTF99 fragments. The CTF99 fragment is then cleaved by g-secretase to form soluble Ab peptides.
In hydrophilic medium the hydrophobic Ab peptides spontaneously polymerize into neurotoxic
oligomers.8 Ab40 is the major secreted fragment, but the longer Ab42 aggregates more readily to
toxic oligomers and is increased by Familial ADmutations suggesting that it is the pathological form;
some oligomeric strands are involved in the hyperphosphorylation of tau protein.9,10 The effect of
AChon tau phosphorylation at specific siteswas recognized.11Mutant types at positions 31–23 ofAbfragments are associated with the higher incidence in developingAD.12 Interference of the oligomers
with neurotransmission at the synapses results in cell death and memory loss.13–17
Aggregation of Ab peptides leads to neuritic plaques made of a central oligomeric core and
surrounded by dystrophic neurities, activated microglia and reactive astrocytes.18 The poorly soluble
plaques would trigger the neurodegenerative cascade responsible of AD.5,19
A. AD Therapies
In the past few years many research laboratories evaluated potential drugs for AD. However,
currently effective drugs for the treatment of AD are not available.2 Historically, acetylcholinesterase
inhibitors (AChEIs) were the first group of drugs marketed for AD treatment. AChEIs increase both
concentration and duration of action of acetylcholine (ACh) in the synaptic cleft. Consequently, the
cholinergic receptors decreased inAD are stimulated, and the growing ofAb aggregates is reduced.20
The AChEIs tacrine (First Horizon Pharmaceuticals*), donepezil (Eisai/Pfizer), rivastigmine
(Novartis), and galantamine (Johnson& Johnson) are on themarket for the symptomatic treatment of
AD,21 and their patent expiration is upcoming.1 New generation (also dual binding site) AChEIs
(phenserine, cymserine, and also AP2238 and tolserine, and the APP metabolism modulators
TV3326 and its enantiomer TV3279) and ACh release enhancers (coluracetam) are currently under
investigation.6,22–25 There is a correlation between ‘‘cholinergic’’ and ‘‘amyloid hypothesis.’’ Rapid
Ab deposition and consequent development of plaques occur in the presence of AChE (AChE acts as
mediator of plaque formation via peripheral anionic side (PAS)/Ab interaction).26,27 An Ab AChE-
cycle would favor Ab deposition in AD through expression of AChE.28 Accordingly, AchE and Abwould be co-localized in neuritic plaques where they boost transformation of Ab into fibrils. Both
cholinergic agonists and AChEIs can stimulate the non-amyloidogenic pathway and reduce the
biosynthesis of amyloidogenic compounds in the brain.21,29,30
2 . A D I N T E R V E N T I O N S T R A T E G I E S
Ab peptides and senile plaques derived from oligomers, as well as neurofibrillary tangles composed
of tau protein,31 are involved in AD pathogenesis.32 The b-amyloid production/deposit may be
potentially lowered using copper-zinc chelating agents (clioquinol),33 Ab-aggregation blockers,
disassembling amyloid fibrils agents (melatonin, AZD-103)34,35 and monoclonal antibody favor the
peripheral clearance of Ab from the brain.36 Tramiprosate is glycosaminoglycan mimetic that binds
to Ab peptides, thereby stopping the amyloid plaque growth.37
The characteristic hallmarks of AD include neuritic plaques and neurofibrillary tangles (NFTs).
NFTs are composed of tau protein in an abnormally hyperphosphorylated form. The pathological
hyperphosphorylation of tau protein is an early event in AD-related neurofibrillary changes.38 The
inhibition of tau-related neurofibrillary degeneration is a novel promising approach in AD therapy.
The tau protein inhibition may be achieved by (i) targeting one or more tau kinase(s) (GSK3-b,CDK5, ERK2, MARK, PKA, SAPK, calmodulin, casein kinase inhibitors), (ii) increasing the
activity of protein phosphatase 2A (PP2A), (iii) inhibiting the presumed toxic properties of
pathological tau proteins (huntingtin).33
*Fordetailed informationoncompaniesmentioned in this article, see the Appendix.
DEVELOPMENT OF NON-PEPTIDIC b-Secretase * 297
Medicinal Research Reviews DOI 10.1002/med

Protein kinase C may induce a-secretase activation in the non-amyloidogenic pathway.39
MuscarinicM1-agonists (talsaclidine, NGX267) andM3-antagonists can activate protein kinaseC.40
Interestingly, M1-agonists can enhance the ACh concentration at synaptic level, thereby reinforcing
the cholinergic system. Ab plaques were reduced using the enzyme adamalysin-10 which stimulates
the expression of a-secretase.41 3-Hydroxyl-3-methylglutaryl coenzyme A (HMG CoA) inhibitors
(lovastatine, simvastatine) proved to activate adamalysin-10.42–44
The g-secretase enzyme is a transmembrane complex made mainly by presenilin-1.45
g-Secretase inhibitors are generally hydrophobic non-transition-state analogues. Inhibitors of
g-secretase should have little effect on notch activity.46,47 Bristol-Myers Squibb Co., Merck and
Schering applied for sulfone and sulfone-related g-secretase inhibitors, Eli Lilly, DuPont and Merck
for benzodiazepines or benzocaprolactams.44,48–50 Currently three g-secretase inhibitors are
ongoing Phase II (LY450139, Eli Lilly; MK 0752, Merck) or Phase I (E2012, Eisai) clinical trials.1
The development of the amyloid vaccine AN-1792 (Elan/Wyeth) was discontinued. A newer
vaccine (ACC-001, Elan/Wyeth) with an improved safety profile is now in a Phase I trial. Three anti-
Ab monoclonal antibodies (mAbs) against various domains of Ab are currently ongoing Phase II
(bapineuzumab as symptom-management drug, Elan/Wyeth; LY2062430, Eli Lilly) or Phase I
(RN1219, Pfizer) clinical trials. Neuronal nicotinic ACh receptor agonists in Phase II trials are
AZD3480 (AstraZeneca/Targacept), MEM 3454 (Roche/Memory Pharmaceuticals), and GTS-21
(CoMentis) symptom-management drugs.1
A. bb-Secretase Inhibitors
b-Amyloid cleaving enzyme-1 (BACE1, Asp2 or memapsin) and BACE2 (Asp1) are the two
isoforms of the amyloid b-secretase identified so far. BACE1 belongs to the aspartyl protease family
and is the major isoform present in the brain; BACE2 plays a marginal role in AD.51 The early
discovery of BACE1 inhibitors of the Swedish mutant APP which significantly reduced Ab40 and
Ab42 levels, triggered the search for new BACE1 inhibitors for AD treatment.52 Several academic
(i.e., Ghosh and Tang, Shuto, Kimura, Fujii, Hu, and Hanessian) and industrial (i.e., Elan, Pfizer, Eli
Lilly, Merck, and Bachem) teams synthesized new peptidomimetic b-secretase inhibitors. This
strategy was based on the design of truncated polypetides bearing non-cleavable transition state
mimicking groups. Elan Pharmaceuticals discovered one of the early potent BACE1 inhibitors by
replacing the P1 leucine with a statin moiety.53 Ghosh and Tang used X-ray crystallographic data of
OM99-2 in complex with recombinant enzyme54 to develop a peptidomimetic b-secretase inhibitorbinding pocket that served as basis to design more potent BACE1 inhibitors (OM00-3, GT-1017).55
To overcome the problem of BACE1/BACE2 selectivity56 this team designed potent cyclic
derivatives based on the crystallographic data of OM99-2.57 Shuto and co-workers synthesized
hydroxymethylcarbonyl and phenylnorstatin transition state analogues and Manzenrieder et al.
incorporated a phosphino dipeptide isostere, all strategies leading to remarkable BACE1 inhibitory
activity.58–60 The not-isomerizable tetrazolyl derivative KMI-429 showed improved stability and
potent enzyme inhibitory activity.61 KMI-429 reduced the level of soluble and insoluble Ab40, andsoluble and insoluble Ab42, in both APP transgenic and wild-type (WT) mice (2 mg injected into thehippocampus of a WT mouse led to 30–40% reduction of Ab levels).62 Kimura and co-workers63
obtained potent BACE1 inhibitors (i.e., KMI-570 and KMI-684) by bioisosteric replacement of the
carboxylic group. Following the statin approach, Pfizer,64 Eli Lilly,65 and Elan Pharmaceuticals66
discovered highly potent BACE1 inhibitors. Merck and Bachem improved BACE1 selectivity by
designing a derivative capable to interact with the Arg235 residue.67 The development of BACE1
inhibitors was summarized by recent excellent reviews.68–72
Inhibitors based on the peptidomimetic strategy suffer from well-known difficulties
associated with polypeptides, such as blood–brain barrier crossing, poor oral bioavailability and
susceptibility to P-glycoprotein transport.73 Efforts to overcome these problems led to design new
298 * SILVESTRI
Medicinal Research Reviews DOI 10.1002/med

non-peptidomimetic b-secretase inhibitors. The first non-peptidomimetic inhibitors were discovered
by Takeda Chemicals in 2001.74,75 In the subsequent 5 years, a boom in research work on non-
peptidomimetic BACE1 inhibitors has disseminated findings over hundreds of publications and
patents. We here attempted to review this rapidly growing literature.
3 . D E V E L O P M E N T O F N O N - P E P T I D I C bb- S E C R E T A S E I N H I B I T O R S
In 2001, Takeda Chem. Ind. reported the first non-peptidomimetic BACE1 inhibitors.74,76 Derivative
5was found themost potent inhibitor among a series of tetralines 1–9, and showed IC50 ¼ 0.349 mMas determined by a fluorescence assay. Compounds 3, 6, 7, and 9 also showed IC50 values in the
submicromolar range of concentration (Table I). Afterwards, Takeda have discontinued publishing
non-peptidomimetic inhibitors and applied for peptidic BACE inhibitors.77,78
Neurologic, Inc. described non-peptidomimetic phosphinylmethyl and phosphorylmethyl
succinic and glutaric acid analogues 10–12 along with some phosphorous-containing tetrapep-
tides.79,80 Noteworthy, the b-secretase inhibitory activity of 10–12was indirectly associated throughmeasurements of the compounds ability to increase sAPPa (it was expected to increase the pool
of non-amyloidogenic derivatives). The activity of 12 (LQ b-3) on sAPPa secretion was dose-
proportional at � 1 nM concentration (Chart 1).
Table I. BACE1 Inhibitory Activity of Derivatives 1–9 (Takeda)
Chart 1. Structure ofcompounds10–12 (Neurologic).
DEVELOPMENT OF NON-PEPTIDIC b-Secretase * 299
Medicinal Research Reviews DOI 10.1002/med
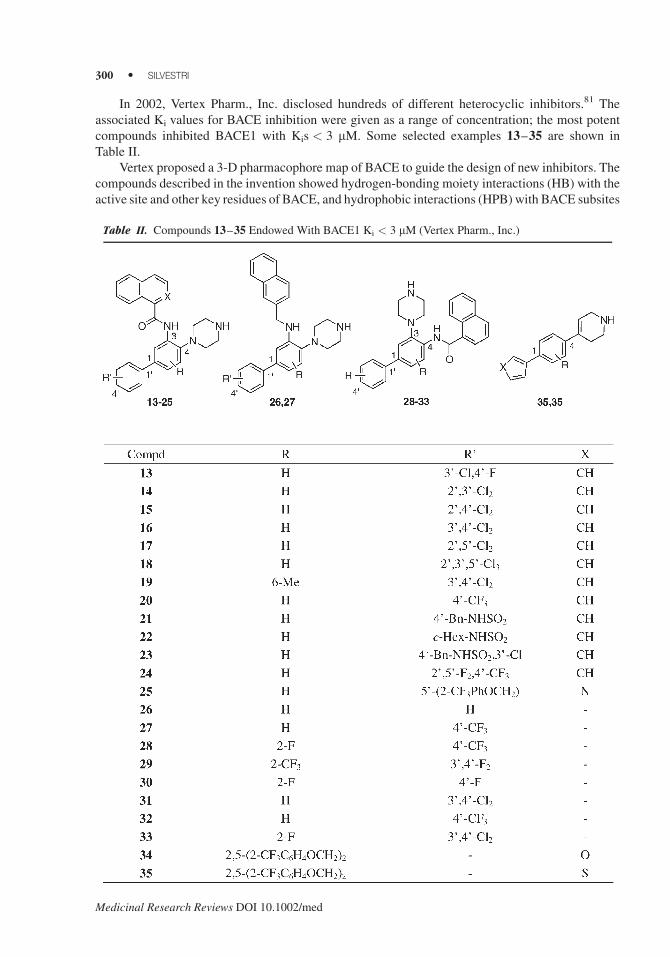
In 2002, Vertex Pharm., Inc. disclosed hundreds of different heterocyclic inhibitors.81 The
associated Ki values for BACE inhibition were given as a range of concentration; the most potent
compounds inhibited BACE1 with Kis < 3 mM. Some selected examples 13–35 are shown in
Table II.
Vertex proposed a 3-D pharmacophore map of BACE to guide the design of new inhibitors. The
compounds described in the invention showed hydrogen-bonding moiety interactions (HB) with the
active site and other key residues of BACE, and hydrophobic interactions (HPB) with BACE subsites
Table II. Compounds 13–35 Endowed With BACE1 Ki < 3 mM (Vertex Pharm., Inc.)
300 * SILVESTRI
Medicinal Research Reviews DOI 10.1002/med

(no crystallographics is provided). Five different BACE/inhibitor binding modes were hypothesized
involving four, five, six, six and an additional H-bond, or seven features of the inhibitor, respectively.
As an example of these binding modes, the seven features binding mode with BACE is shown in
Figure 2.
A. Isophthalamide-Based Inhibitors
1. Optimization of N-Terminus
Elan Pharm., Inc. found that the peptidic nature of potent and selective BACE inhibitors limited their
utility for drug development.82–84 In an effort to overcome this problem, Elan moved away from the
statin-like transition mimic, and selected the hydroxyethylene (HE) isostere because of fewer
peptidic bonds (Chart 2).85
Replacing the statin moiety with the HE core in a series of heptapeptide analogues resulted in a
10-fold increase of BACE inhibitory activity. Based on these findings, Elan synthesized a variety of
HEs bearing the optimal isophthalamideN-terminus of the statin series and different C termini at the
P1 0 substituent. Molecular modeling studies revealed that HE- and statin-based peptidic inhibitors
showed nearly identical binding modes for the N-terminal residues. All the peptidic amide groups of
the HE dipeptide isosteres formed H-bonds to the enzyme. However, the shorter dipeptide backbone
led to divergent conformations between the two series at the C terminus and showed the inability of
the statins to fill the S1 0 subsite. Structure andBACE1 inhibitory activity of selected examples 36–41
are shown in Table III.
Figure 2. Seven featuresbindingmodewith BACE.
Chart 2. The statinandhydroxyethylene cores (Elan).
DEVELOPMENT OF NON-PEPTIDIC b-Secretase * 301
Medicinal Research Reviews DOI 10.1002/med
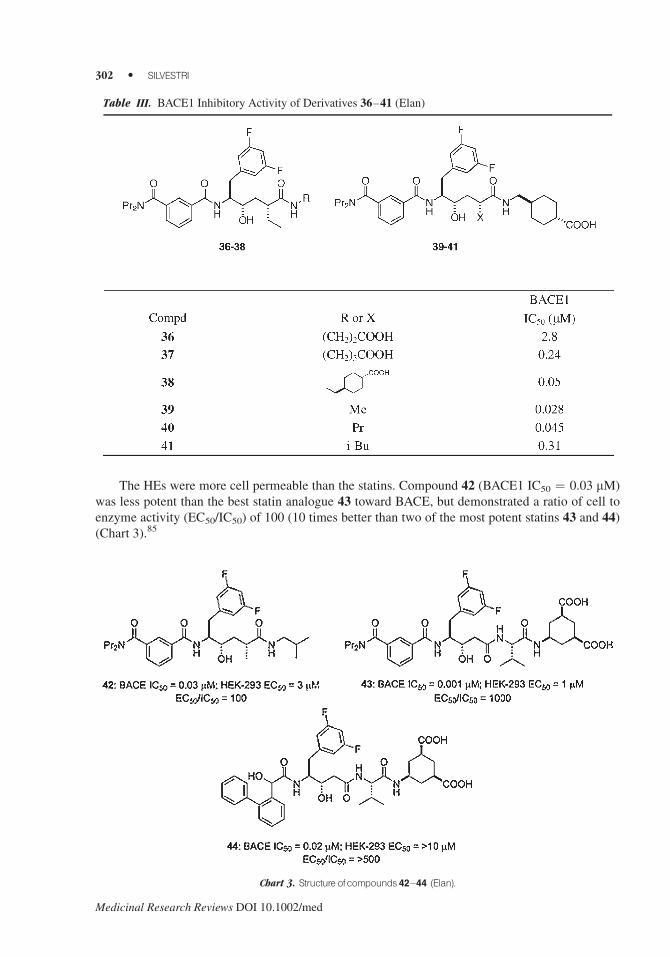
The HEs were more cell permeable than the statins. Compound 42 (BACE1 IC50 ¼ 0.03 mM)
was less potent than the best statin analogue 43 toward BACE, but demonstrated a ratio of cell to
enzyme activity (EC50/IC50) of 100 (10 times better than two of the most potent statins 43 and 44)
(Chart 3).85
Table III. BACE1 Inhibitory Activity of Derivatives 36–41 (Elan)
Chart 3. Structure ofcompounds 42–44 (Elan).
302 * SILVESTRI
Medicinal Research Reviews DOI 10.1002/med

Elan improved the unfavorable cell-to-enzyme potency ratios (C/E� 100) of statins and
HE transition state isostere (TSI)-based inhibitors.86 The hydroxyethyl secondary amine (HEA)
isostere was an alternative of HE because of the strict preference of BACE for acyclic residues
at the S1 0 subsite, and considering that the majority of CNS drugs contain a basic amine.87 HE
inhibitor 45 was converted into the HEA inhibitors 46 and 47 by insertion of a secondary
nitrogen between the methylene and the P1 0 substituent. The preferred configuration for the CH–OHfunctionalitywas opposite to the stereochemistry of otherBACETSIs previously described85 (the (R)
isomer was 1,000-fold more active than (S) isomer) (Chart 4, the differences in the structures
are highlighted in bold). Compound 47 was a potent cellular inhibitor of Ab production
(more potent than the statin and the HE BACE inhibitors with comparable enzyme potency).
Compound 51 bearing a C-terminal benzyl amine exhibited submicromolar BACE enzyme activity
and cell-to-enzyme ratio (C/E) of 0.3. The meta-iodo analog 52 was 25-fold more potent than the
parent analog 51. Compounds 52 and 53were enzymatically more active than 45; their improved cell
potency was correlated to the reduced number of amide bonds and introduction of a basic amine
(Table IV).87
Chart 4. Comparisonof HEandHEAcores (Elan).
DEVELOPMENT OF NON-PEPTIDIC b-Secretase * 303
Medicinal Research Reviews DOI 10.1002/med
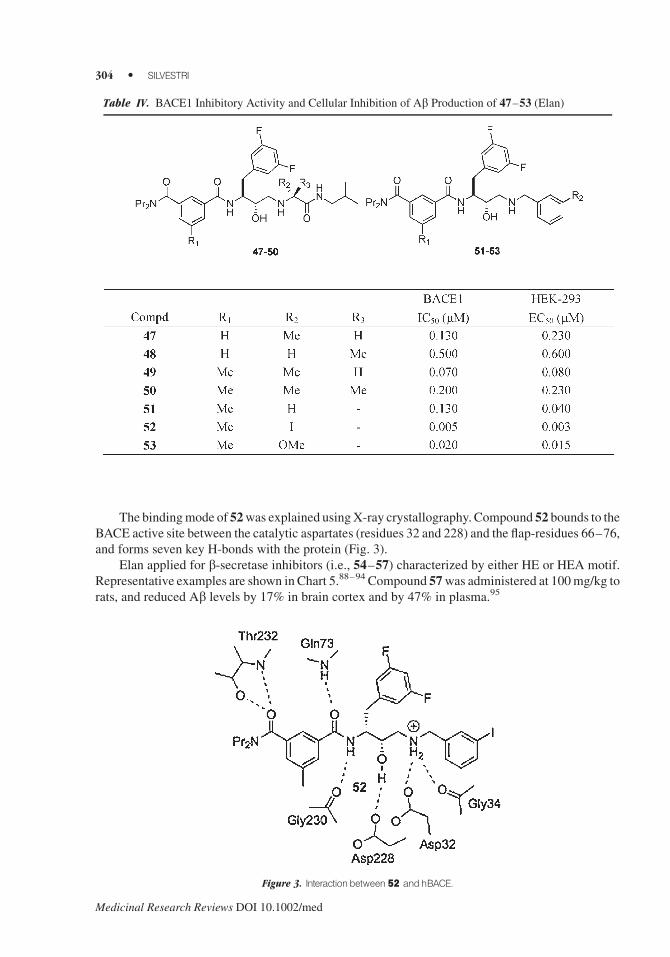
The bindingmode of 52was explained usingX-ray crystallography. Compound 52 bounds to the
BACE active site between the catalytic aspartates (residues 32 and 228) and the flap-residues 66–76,
and forms seven key H-bonds with the protein (Fig. 3).
Elan applied for b-secretase inhibitors (i.e., 54–57) characterized by either HE or HEA motif.
Representative examples are shown in Chart 5.88–94 Compound 57was administered at 100mg/kg to
rats, and reduced Ab levels by 17% in brain cortex and by 47% in plasma.95
Table IV. BACE1 Inhibitory Activity and Cellular Inhibition of Ab Production of 47–53 (Elan)
Figure 3. Interactionbetween52 andhBACE.
304 * SILVESTRI
Medicinal Research Reviews DOI 10.1002/med

Elan and also Elan in collaboration with Pharmacia & Upjohn applied for b-secretase inhibitorshaving different structures (some representative examples are shown in Chart 6): 3,4-disubstituted
piperidines,96 azahydroxylated ethylamines.97 macrocycles (58),98,99 aminodiols100–103 (59),
bicyclo compounds,104 diaminodiols105 (60), N-(3-amino-2-hydroxy-propyl) substituted alkyla-
mides (61),106 benzamide 2-hydroxy-3-diaminoalkanes,107 substituted amines,108 hydroxypropyl-
amines,109 allylamides,110 hydroxy substituted indanylamides (62),111 1,3-diamino-2-
hydroxypropanes,112,113 substituted hydroxyethylamines114 (63), acetyl 2-hydroxy-1,3-diaminoal-
kanes (64),115 phenacyl 2-hydroxy-3-diaminoalkanes (65),116 piperidines and piperazines,117
hydroxyethylamines (66),118 aminocarboxamides,119 amino-alkanoic acid amides and alkanoic acid
diamides,120 aryl alkanoic acid amides,121 hydroxyaminopropyl amides,122,123 ureas (67) and
carbamates,124,125 acetyl 1,3-diamino-2-hydroxyspirocyclohexanes,126 bicyclic compounds,127
2-amino- and 2-thio-substituted 1,3-diaminopropanes (68),128 oximes,129,130 ethanolcyclic-
amines,131 and cyclopropyl derivatives (69).132
2. Introduction of Sulfonates at P2 Position
Merck Research Laboratories set up a research project to identify non-peptide inhibitors of
b-secretase which might overcome the historical problems associated with peptide-like structures.
High throughput screening (HTS) of a multimillion compound library led to identify the N-(5-
aminopentyl)oxyacetamide (70). Compound 70 proved to be a reversible and selective BACE1
inhibitor, and reproducibly inhibited b-secretase in five different enzymatic assays at IC50 ¼25 mM.133,134 Replacing the tertiary amide group with either 2-cyanophenyl group (71) or the
sulfonate ester (72) resulted in improvement of inhibitory activity. The (R)-stereochemistry at the
chiral methyl group was essential for good enzymatic activity. In an electrochemiluminescence
(ECL) enzyme inhibition assay compound 74 displayed IC50 ¼ 1.4 mM (Table V).
Crystallographic examination of the BACE1/inhibitor complex revealed that 74 occupied
the S4–S1 subsites of the enzyme without any direct contact with the catalytic aspartic acids.
Compound 74 formed a H-bond between the oxyacetamide NH and a water molecule situated
between the aspartyl dyad. The authors observed that this water-mediated binding mode is almost
unique among aspartyl protease inhibitors. The P3 R-methyl group packs firmly against Ile110,
while unexpectedly orienting the para-fluorophenyl ring toward S3 creating a novel S3 subpocket
(S3 sp) (Fig. 4).
Chart 5. Structure ofcompounds 54–57 (Elan, and Elanand Pharmacia &Upjohn).
DEVELOPMENT OF NON-PEPTIDIC b-Secretase * 305
Medicinal Research Reviews DOI 10.1002/med
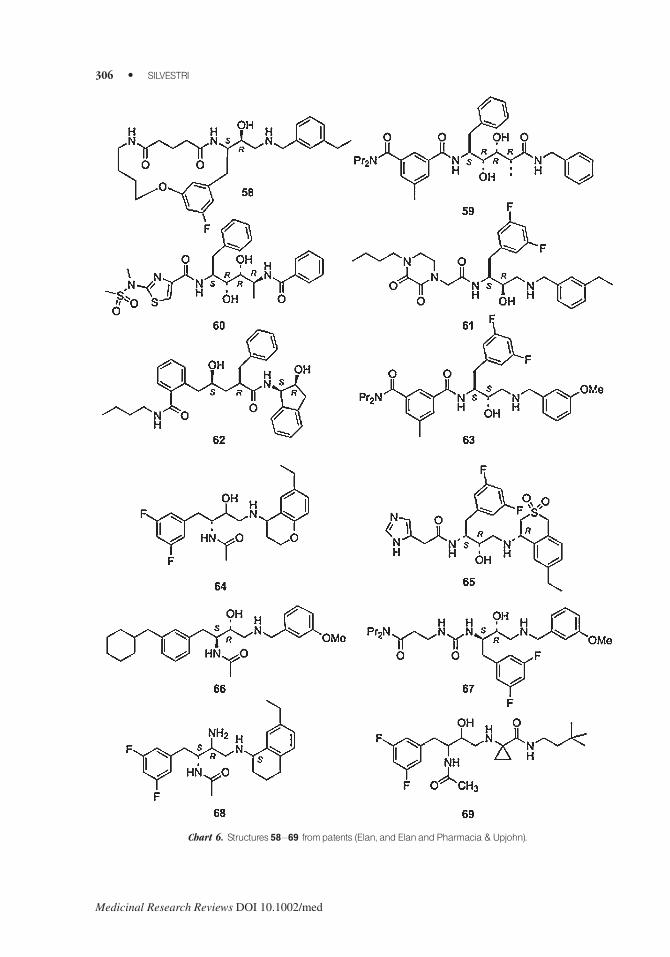
Chart 6. Structures 58–69 frompatents (Elan, and ElanandPharmacia &Upjohn).
306 * SILVESTRI
Medicinal Research Reviews DOI 10.1002/med
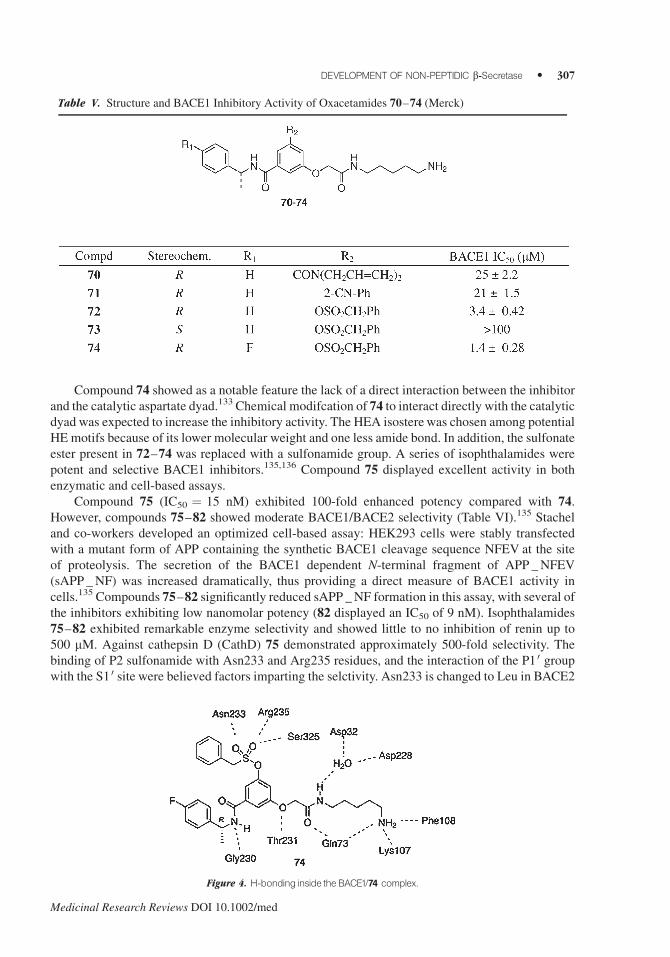
Compound 74 showed as a notable feature the lack of a direct interaction between the inhibitor
and the catalytic aspartate dyad.133 Chemical modifcation of 74 to interact directly with the catalyticdyad was expected to increase the inhibitory activity. The HEA isostere was chosen among potential
HE motifs because of its lower molecular weight and one less amide bond. In addition, the sulfonate
ester present in 72–74 was replaced with a sulfonamide group. A series of isophthalamides were
potent and selective BACE1 inhibitors.135,136 Compound 75 displayed excellent activity in both
enzymatic and cell-based assays.
Compound 75 (IC50 ¼ 15 nM) exhibited 100-fold enhanced potency compared with 74.
However, compounds 75–82 showed moderate BACE1/BACE2 selectivity (Table VI).135 Stachel
and co-workers developed an optimized cell-based assay: HEK293 cells were stably transfected
with a mutant form of APP containing the synthetic BACE1 cleavage sequence NFEV at the site
of proteolysis. The secretion of the BACE1 dependent N-terminal fragment of APP _NFEV
(sAPP _NF) was increased dramatically, thus providing a direct measure of BACE1 activity in
cells.135 Compounds 75–82 significantly reduced sAPP _NF formation in this assay, with several of
the inhibitors exhibiting low nanomolar potency (82 displayed an IC50 of 9 nM). Isophthalamides
75–82 exhibited remarkable enzyme selectivity and showed little to no inhibition of renin up to
500 mM. Against cathepsin D (CathD) 75 demonstrated approximately 500-fold selectivity. The
binding of P2 sulfonamide with Asn233 and Arg235 residues, and the interaction of the P1 0 groupwith the S1 0 site were believed factors imparting the selctivity. Asn233 is changed to Leu in BACE2
Table V. Structure and BACE1 Inhibitory Activity of Oxacetamides 70–74 (Merck)
Figure 4. H-bonding inside the BACE1/74 complex.
DEVELOPMENT OF NON-PEPTIDIC b-Secretase * 307
Medicinal Research Reviews DOI 10.1002/med

and cathD, and to Tyr in renin; Arg235 is changed to Val in cathD and Ser in renin. The more
hydrophilic S1 0 site of BACE1 better accommodates a small hydrophobic group, whereas cathD and
renin have longer, primarily hydrophobic S1 0 regions. The difference between BACE1 and BACE2consists of only two amino acid differences (Gln326 to Pro and Thr329 toAsn). Thus, this regionmay
primarily affect selectivity over cathD and renin rather than differentiate between BACE1 and
BACE2 activity.135
An overlay of 74with 75 showed that they occupy the samegeneral space in the active site: (i) the
(R)-a-methylbenzamide occupies the S3 subpocket andR-stereochemistry at themethyl is preferred;
(ii) the sulfonamide of 75 occupies the S2 site as the sulfonate ester of 74, and in both series each
sulfone oxygenmakes important H-bonding interactions with the enzyme; (iii) the alkyl group on the
nitrogen of the sulfonamide serves mainly to orient the binding conformation; (iv) in 75 the
phenylalanyl group occupies the S1 pocket in amanner similar to that of known peptide inhibitors (the
hydroxylamine motif is involved in direct interactions with the catalytic dyad); (v) the cyclopropyl
group of 75 is oriented toward the S 01 site of the enzyme.
The observation that the P2/P3 amide group of 75 serves mainly to orient the peptide backbone,
suggested the synthesis of the S3-truncated analogues.137 The feasibility of removing the
S3 amide bond was investigated by synthesis of the truncated analogues 83 and 84. The only
twofold loss of activity of 84 versus 83 (Table VII) demonstrated that amide removal was tolerated,
without significant loss of activity. Modeling studies showed that the Z-alkenyl compound
95would orient itself in the S3 pocket, thusmimicking the s-strand amide bond geometry. 95 showed
excellent binding activity. The apparent permeability (Papp) associated with 75 was low
(0.6� 10�6 cm/sec) and P-glycoprotein (P-gp) efflux values were indeterminable. In contrast, while
the olefine congeners (95) were still found to be moderate substrates for P-gp efflux, the associated
Papps (14� 10�6 cm/sec) were significantly increased as to permit measurement. The hydroxyethyl
amine moiety, amide bond, and sulfonamide of 95were thought to be all contributors to P-gp efflux.
This study demonstrated that analogues of 75 with both lower polar surface area and less H-bond
donors and acceptors possess better pharmacodynamic properties.
Table VI. Structure and BACE1 Inhibitory Activity of Isophthalamide 75–82 (Merck)
308 * SILVESTRI
Medicinal Research Reviews DOI 10.1002/med
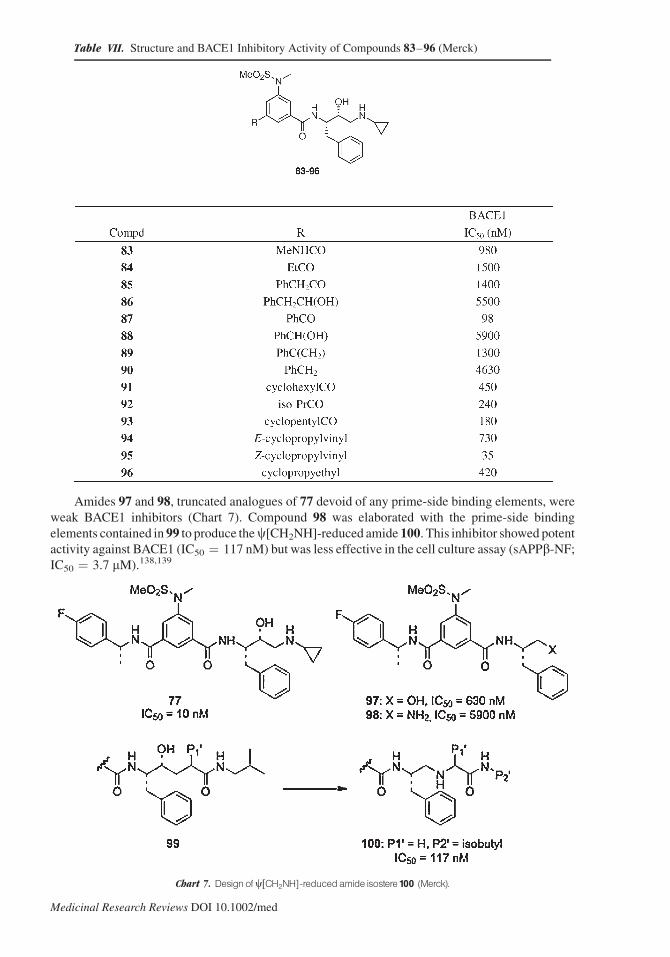
Amides 97 and 98, truncated analogues of 77 devoid of any prime-side binding elements, were
weak BACE1 inhibitors (Chart 7). Compound 98 was elaborated with the prime-side binding
elements contained in 99 to produce thec[CH2NH]-reduced amide 100. This inhibitor showed potentactivity against BACE1 (IC50 ¼ 117 nM) but was less effective in the cell culture assay (sAPPb-NF;IC50 ¼ 3.7 mM).138,139
Table VII. Structure and BACE1 Inhibitory Activity of Compounds 83–96 (Merck)
Chart 7. Designofc[CH2NH]-reducedamide isostere100 (Merck).
Medicinal Research Reviews DOI 10.1002/med

Either small alkyl groups (101–103) or longer hydrophilic substituents at the P1 0
region produced compounds with enhanced inhibitory activities (Table VIII). Compounds
101–103 also showed a dramatic increase in cellular potency and exhibited IC50 values in
the 17–22 nM range of concentration. Inhibitor 104 bearing a polar substituent showed
diminished cell-based activity as a result of the lower cell permeability. Derivatives 100–
104 showed some selectivity over BACE2 (5- to 10-fold) and the IC50 values versus human
renin were greater than 20 mM. Compound 105 was potent inhibitor in both the enzyme and
cell-based assays (BACE1; IC50 ¼ 20 nM; sAPPb _ NF; IC50 ¼ 23 nM). This modification
also improved the cell permeability of the series (Papp ¼ 24� 10�6 cm/sec) when compared to
either HEA inhibitor 77 (Papp ¼ 0.6� 10�6 cm/sec) or reduced amide 101 (Papp ¼ 10�10�6 cm/sec). However, both compounds were susceptible to P-gp transport. SAR at the P2 0
region showed that small alkyl, cycloalkyl and benzyl groups were tolerated but only at the
expense of both cellular and enzymatic potency (106–109). The presence of a secondary NH
group was also needed to align the H-bonding network between the enzyme and the inhibitor
(Table IX).
X-ray structure of 101 at 1.8 A resolution showed that when compared to compound 77,
both inhibitors occupied the same general space in the active site with a few key differences
(Fig. 5). The binding mode of 101 was consistent with the known binding properties of
c[CH2NH]-pseudopeptides to Asp proteases. The nitrogen atom at the scissile bond of
reduced amide 101 shared an H-bond with only half of the dyad, specifically Asp228 (typically,
the hydroxyl and/or amino group of transition state analogues is in close contact with both
catalytic Asps).
3. Macrocyclization
X-ray crystallographic data of the 75/BACE1 complex revealed the presence of space in proximity of
the P1 and P3 groups. This observation prompted the design of macrocycles that would link these
Table VIII. Structure and BACE1 Inhibitory Activity for P1 0 Derivatives 100–105 (Merck)
310 * SILVESTRI
Medicinal Research Reviews DOI 10.1002/med
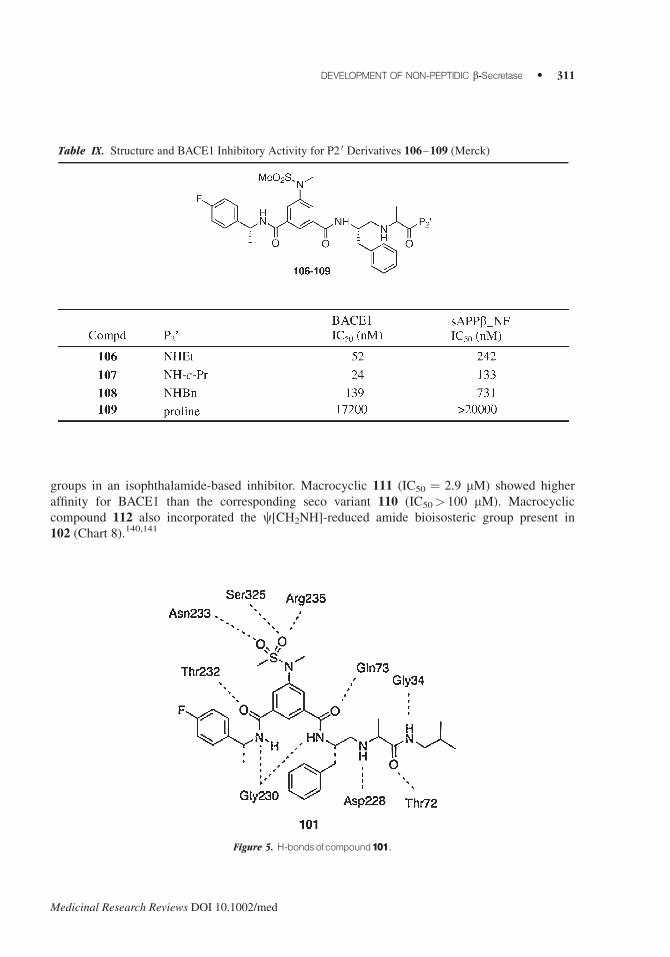
groups in an isophthalamide-based inhibitor. Macrocyclic 111 (IC50 ¼ 2.9 mM) showed higher
affinity for BACE1 than the corresponding seco variant 110 (IC50> 100 mM). Macrocyclic
compound 112 also incorporated the c[CH2NH]-reduced amide bioisosteric group present in
102 (Chart 8).140,141
Table IX. Structure and BACE1 Inhibitory Activity for P2 0 Derivatives 106–109 (Merck)
Figure 5. H-bondsofcompound101.
DEVELOPMENT OF NON-PEPTIDIC b-Secretase * 311
Medicinal Research Reviews DOI 10.1002/med
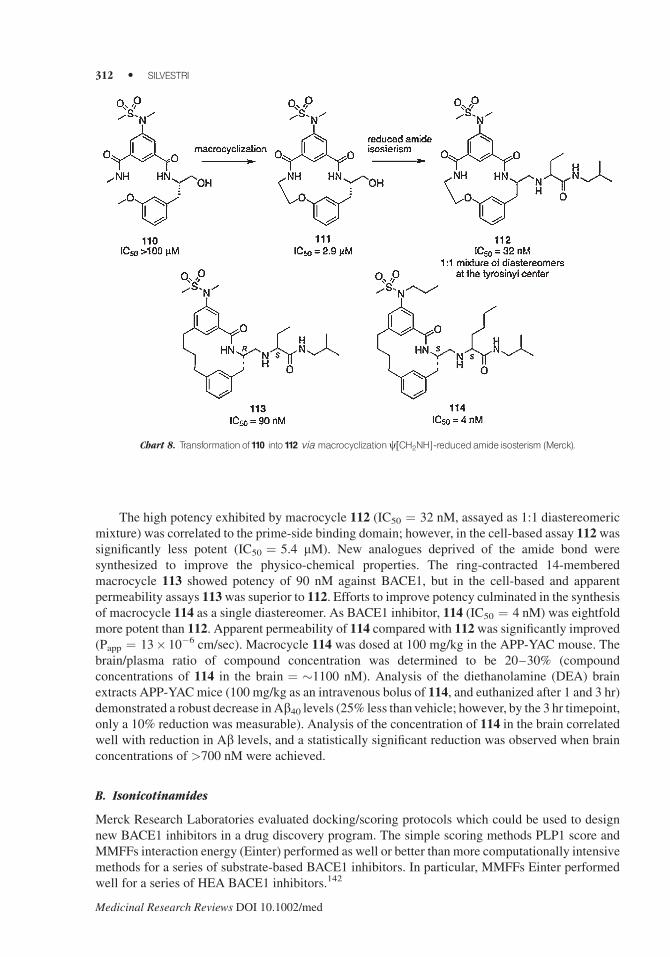
The high potency exhibited by macrocycle 112 (IC50 ¼ 32 nM, assayed as 1:1 diastereomeric
mixture) was correlated to the prime-side binding domain; however, in the cell-based assay 112wassignificantly less potent (IC50 ¼ 5.4 mM). New analogues deprived of the amide bond were
synthesized to improve the physico-chemical properties. The ring-contracted 14-membered
macrocycle 113 showed potency of 90 nM against BACE1, but in the cell-based and apparent
permeability assays 113was superior to 112. Efforts to improve potency culminated in the synthesis
of macrocycle 114 as a single diastereomer. As BACE1 inhibitor, 114 (IC50 ¼ 4 nM) was eightfold
more potent than 112. Apparent permeability of 114 compared with 112was significantly improved
(Papp ¼ 13� 10�6 cm/sec). Macrocycle 114 was dosed at 100 mg/kg in the APP-YAC mouse. The
brain/plasma ratio of compound concentration was determined to be 20–30% (compound
concentrations of 114 in the brain ¼ �1100 nM). Analysis of the diethanolamine (DEA) brain
extracts APP-YACmice (100 mg/kg as an intravenous bolus of 114, and euthanized after 1 and 3 hr)
demonstrated a robust decrease in Ab40 levels (25% less thanvehicle; however, by the 3 hr timepoint,
only a 10% reduction was measurable). Analysis of the concentration of 114 in the brain correlated
well with reduction in Ab levels, and a statistically significant reduction was observed when brain
concentrations of >700 nM were achieved.
B. Isonicotinamides
Merck Research Laboratories evaluated docking/scoring protocols which could be used to design
new BACE1 inhibitors in a drug discovery program. The simple scoring methods PLP1 score and
MMFFs interaction energy (Einter) performed as well or better than more computationally intensive
methods for a series of substrate-based BACE1 inhibitors. In particular, MMFFs Einter performed
well for a series of HEA BACE1 inhibitors.142
Chart 8. Transformationof110 into112 via macrocyclizationc[CH2NH]-reducedamide isosterism (Merck).
312 * SILVESTRI
Medicinal Research Reviews DOI 10.1002/med
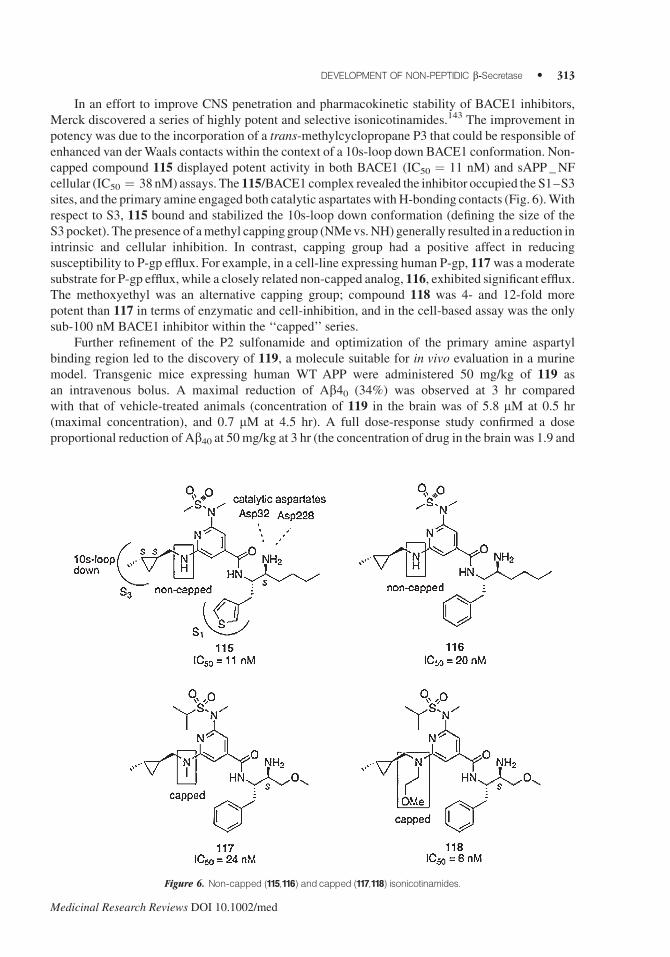
In an effort to improve CNS penetration and pharmacokinetic stability of BACE1 inhibitors,
Merck discovered a series of highly potent and selective isonicotinamides.143 The improvement in
potency was due to the incorporation of a trans-methylcyclopropane P3 that could be responsible of
enhanced van derWaals contacts within the context of a 10s-loop down BACE1 conformation. Non-
capped compound 115 displayed potent activity in both BACE1 (IC50 ¼ 11 nM) and sAPP _NF
cellular (IC50 ¼ 38 nM) assays. The 115/BACE1 complex revealed the inhibitor occupied the S1–S3
sites, and the primary amine engaged both catalytic aspartateswith H-bonding contacts (Fig. 6).With
respect to S3, 115 bound and stabilized the 10s-loop down conformation (defining the size of the
S3pocket). The presence of amethyl capping group (NMevs.NH) generally resulted in a reduction in
intrinsic and cellular inhibition. In contrast, capping group had a positive affect in reducing
susceptibility to P-gp efflux. For example, in a cell-line expressing human P-gp, 117was a moderate
substrate for P-gp efflux, while a closely related non-capped analog, 116, exhibited significant efflux.The methoxyethyl was an alternative capping group; compound 118 was 4- and 12-fold more
potent than 117 in terms of enzymatic and cell-inhibition, and in the cell-based assay was the only
sub-100 nM BACE1 inhibitor within the ‘‘capped’’ series.
Further refinement of the P2 sulfonamide and optimization of the primary amine aspartyl
binding region led to the discovery of 119, a molecule suitable for in vivo evaluation in a murine
model. Transgenic mice expressing human WT APP were administered 50 mg/kg of 119 as
an intravenous bolus. A maximal reduction of Ab40 (34%) was observed at 3 hr compared
with that of vehicle-treated animals (concentration of 119 in the brain was of 5.8 mM at 0.5 hr
(maximal concentration), and 0.7 mM at 4.5 hr). A full dose-response study confirmed a dose
proportional reduction of Ab40 at 50 mg/kg at 3 hr (the concentration of drug in the brain was 1.9 and
Figure 6. Non-capped (115,116) andcapped (117,118) isonicotinamides.
DEVELOPMENT OF NON-PEPTIDIC b-Secretase * 313
Medicinal Research Reviews DOI 10.1002/med
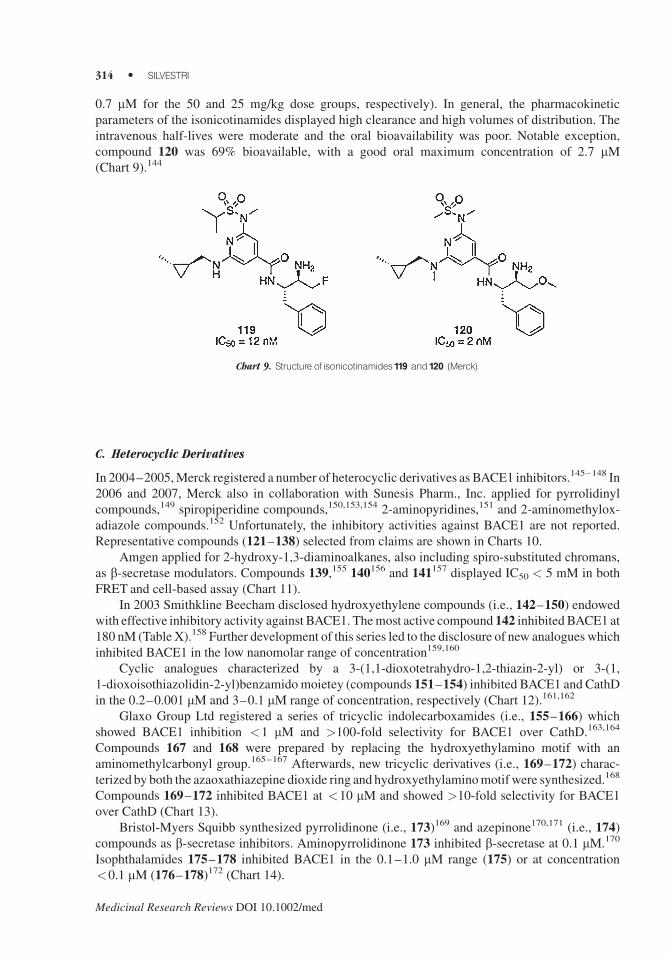
0.7 mM for the 50 and 25 mg/kg dose groups, respectively). In general, the pharmacokinetic
parameters of the isonicotinamides displayed high clearance and high volumes of distribution. The
intravenous half-lives were moderate and the oral bioavailability was poor. Notable exception,
compound 120 was 69% bioavailable, with a good oral maximum concentration of 2.7 mM(Chart 9).144
C. Heterocyclic Derivatives
In 2004–2005,Merck registered a number of heterocyclic derivatives as BACE1 inhibitors.145–148 In
2006 and 2007, Merck also in collaboration with Sunesis Pharm., Inc. applied for pyrrolidinyl
compounds,149 spiropiperidine compounds,150,153,154 2-aminopyridines,151 and 2-aminomethylox-
adiazole compounds.152 Unfortunately, the inhibitory activities against BACE1 are not reported.
Representative compounds (121–138) selected from claims are shown in Charts 10.
Amgen applied for 2-hydroxy-1,3-diaminoalkanes, also including spiro-substituted chromans,
as b-secretase modulators. Compounds 139,155 140156 and 141157 displayed IC50 < 5 mM in both
FRET and cell-based assay (Chart 11).
In 2003 Smithkline Beecham disclosed hydroxyethylene compounds (i.e., 142–150) endowedwith effective inhibitory activity against BACE1. Themost active compound 142 inhibited BACE1 at
180 nM (Table X).158 Further development of this series led to the disclosure of new analogues which
inhibited BACE1 in the low nanomolar range of concentration159,160
Cyclic analogues characterized by a 3-(1,1-dioxotetrahydro-1,2-thiazin-2-yl) or 3-(1,
1-dioxoisothiazolidin-2-yl)benzamido moietey (compounds 151–154) inhibited BACE1 and CathD
in the 0.2–0.001 mM and 3–0.1 mM range of concentration, respectively (Chart 12).161,162
Glaxo Group Ltd registered a series of tricyclic indolecarboxamides (i.e., 155–166) which
showed BACE1 inhibition <1 mM and >100-fold selectivity for BACE1 over CathD.163,164
Compounds 167 and 168 were prepared by replacing the hydroxyethylamino motif with an
aminomethylcarbonyl group.165–167 Afterwards, new tricyclic derivatives (i.e., 169–172) charac-
terized by both the azaoxathiazepine dioxide ring and hydroxyethylaminomotif were synthesized.168
Compounds 169–172 inhibited BACE1 at <10 mM and showed >10-fold selectivity for BACE1
over CathD (Chart 13).
Bristol-Myers Squibb synthesized pyrrolidinone (i.e., 173)169 and azepinone170,171 (i.e., 174)
compounds as b-secretase inhibitors. Aminopyrrolidinone 173 inhibited b-secretase at 0.1 mM.170
Isophthalamides 175–178 inhibited BACE1 in the 0.1–1.0 mM range (175) or at concentration<0.1 mM (176–178)172 (Chart 14).
Chart 9. Structure of isonicotinamides119 and120 (Merck).
314 * SILVESTRI
Medicinal Research Reviews DOI 10.1002/med
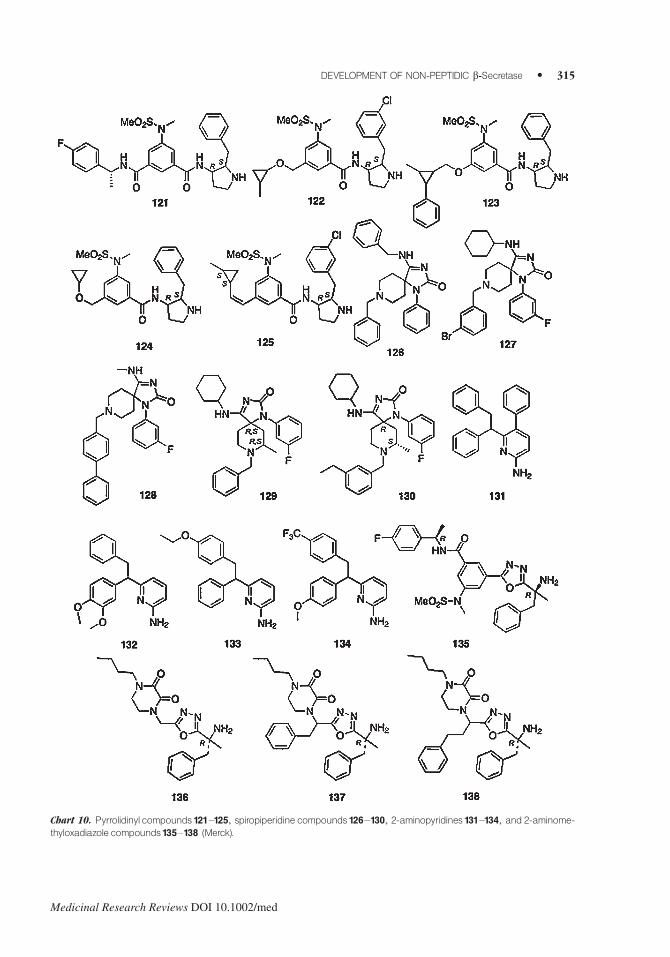
Chart 10. Pyrrolidinyl compounds121–125, spiropiperidine compounds126–130, 2-aminopyridines 131–134, and 2-aminome-thyloxadiazole compounds135–138 (Merck).
DEVELOPMENT OF NON-PEPTIDIC b-Secretase * 315
Medicinal Research Reviews DOI 10.1002/med
D. Guanidines
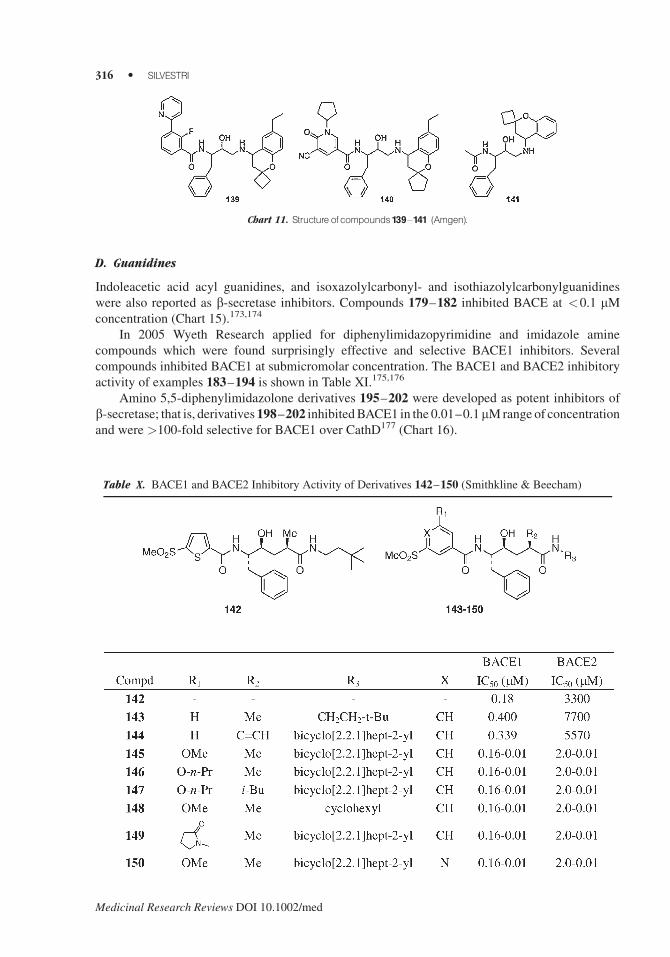
Indoleacetic acid acyl guanidines, and isoxazolylcarbonyl- and isothiazolylcarbonylguanidines
were also reported as b-secretase inhibitors. Compounds 179–182 inhibited BACE at <0.1 mMconcentration (Chart 15).173,174
In 2005 Wyeth Research applied for diphenylimidazopyrimidine and imidazole amine
compounds which were found surprisingly effective and selective BACE1 inhibitors. Several
compounds inhibited BACE1 at submicromolar concentration. The BACE1 and BACE2 inhibitory
activity of examples 183–194 is shown in Table XI.175,176
Amino 5,5-diphenylimidazolone derivatives 195–202 were developed as potent inhibitors of
b-secretase; that is, derivatives198–202 inhibitedBACE1 in the 0.01–0.1mMrange of concentration
and were >100-fold selective for BACE1 over CathD177 (Chart 16).
Chart 11. Structure ofcompounds139–141 (Amgen).
Table X. BACE1 and BACE2 Inhibitory Activity of Derivatives 142–150 (Smithkline & Beecham)
316 * SILVESTRI
Medicinal Research Reviews DOI 10.1002/med

2-Aminopyridine derivatives (i.e., 203–205) inhibited BACE1 in the 0.01–0.1 mM range of
concentration178 (Chart 17).
High-throughput screening (HTS) of Wyeth’s compound library by means of a FRET assay
identified the acylguanidine 206 (BACE1 IC50 ¼ 3.7 mM) as a prototype of a new class of BACE1
inhibitors.179 X-ray crystal structure of the BACE1/206 complex revealed that two nitrogen groups
on the acylguanidine moiety formed H-bonds with the catalytic aspartic acids Asp32 and Asp228.
The third nitrogen faced away from the catalytic residues toward the S1 0 pocket. In contrast
to complexes with peptidomimetic inhibitors, the flap region adopted an ‘‘open conformation’’ that
hosted the diarylpyrrole portion of the inhibitor (such a conformation would be stabilized by
p-stacking interactions between the pyrrole and the phenyl ring of Tyr71). The two aryl groups of theBACE1/206 complex extended into the S1 (large and spherical) and S2 0 pockets. The P2 0-phenyl wasinvolved in p-stacking interaction with Trp76 (Fig. 7)
Wyeth synthesized analogues of 206.179 Compound 207 (IC50 ¼ 0.6 mM) bearing the more
spherical adamantyl group was sixfold more potent than 206 (Table XII). BACE1/207 cocrystal
structure confirmed that the adamantyl group occupied the S1 pocket. The cocrystal structure of
208 (IC50 ¼ 160 nM) showed that the 4-(4-acetoxyphenoxy)phenyl group extended through the S1pocket into the S3 pocket as predicted by the models. Substitution of the guanidine nitrogen with a
3-propanol group gave 209 (IC50 ¼ 240 nM), which was approximately 2.5-fold more potent
than the corresponding unsubstituted acylguanidine 207. Compound 210 bearing the para-
propyloxyphenyl group extending from S1-S3 also showed potent BACE1 inhibitory activity
(IC50 ¼ 110 nM), and demonstrated that a large group was not required for an optimal S3 pocket
binding. These compoundswere generally highly selective for BACE1 overCathD (>50-fold, except
206which is 16-fold) and pepsin (IC50> 50 mM). Compounds with diaryl substituents on the pyrrole
generally had lower BACE1/BACE2 selectivity, while compounds with the adamantyl substituent
show greater BACE1 over BACE2 selectivity. Substitution on the guanidine nitrogen led to lower
BACE1/BACE2 selectivity. Several compounds had low micromolar activity in the cellular Abtotallowering ELISA assay.
Wyeth applied for thienyl (211–214) and terphenyl acylguanidines (215–218) structurally
correlated with compounds 206–210, which inhibited the BACE1 in the submicromolar range of
concentration (Chart 18).180–182
AstraZeneca andAstex applied for 2-aminopyrimidin-4-ones (219),183–185 2-aminopyrimidines
(220),186 2-aminoimidazol-4-ones (221a),187,188 and dihydroisoquinolin-1-amines (221b)189 for the
treatment of Alzheimer’s disease and related pathologies. As a common structural feature, the
heterocyclic moieties of 219–221a show a guanidino motif (Chart 19).
Janssen Pharm. applied for 2-aminoquinazoline derivatives which inhibited b-secretase in threedifferent assays (i.e., compounds 222–225, Table XIII.190–192
Chart 12. Dioxothiazobenzamides151–154 (GlaxoGroup Ltd).
DEVELOPMENT OF NON-PEPTIDIC b-Secretase * 317
Medicinal Research Reviews DOI 10.1002/med
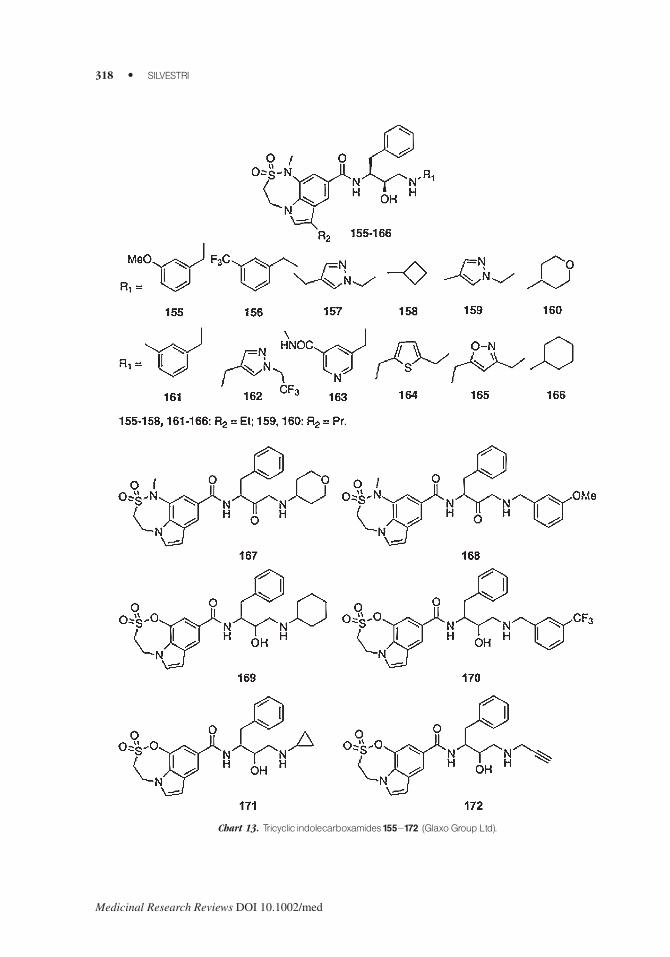
Chart 13. Tricyclic indolecarboxamides155–172 (GlaxoGroup Ltd).
318 * SILVESTRI
Medicinal Research Reviews DOI 10.1002/med
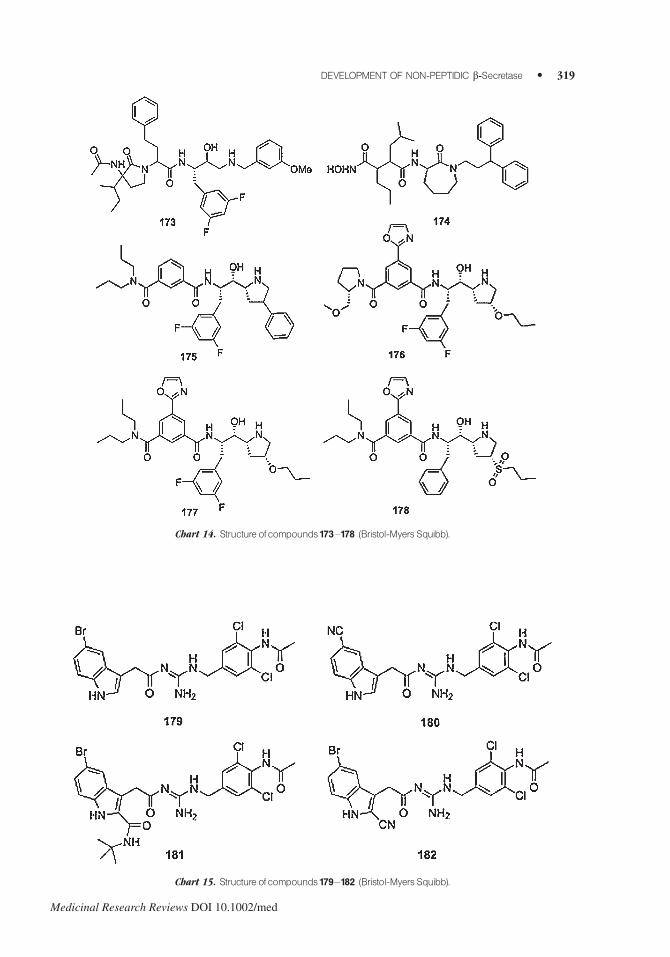
Chart 14. Structure ofcompounds173–178 (Bristol-Myers Squibb).
Chart 15. Structure ofcompounds179–182 (Bristol-Myers Squibb).
DEVELOPMENT OF NON-PEPTIDIC b-Secretase * 319
Medicinal Research Reviews DOI 10.1002/med
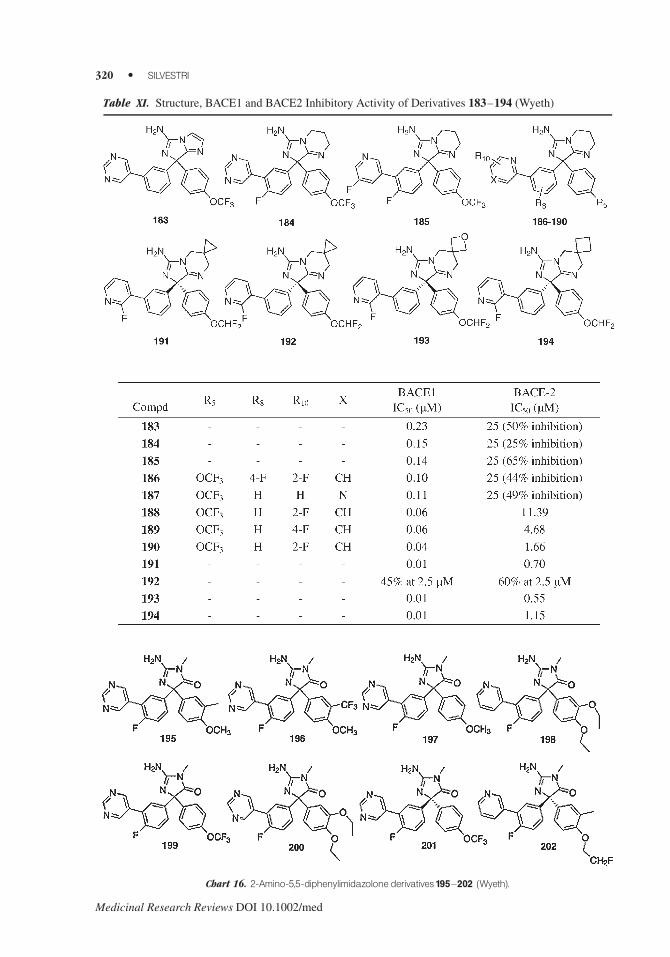
Table XI. Structure, BACE1 and BACE2 Inhibitory Activity of Derivatives 183–194 (Wyeth)
Chart 16. 2-Amino-5,5-diphenylimidazolone derivatives195–202 (Wyeth).
320 * SILVESTRI
Medicinal Research Reviews DOI 10.1002/med
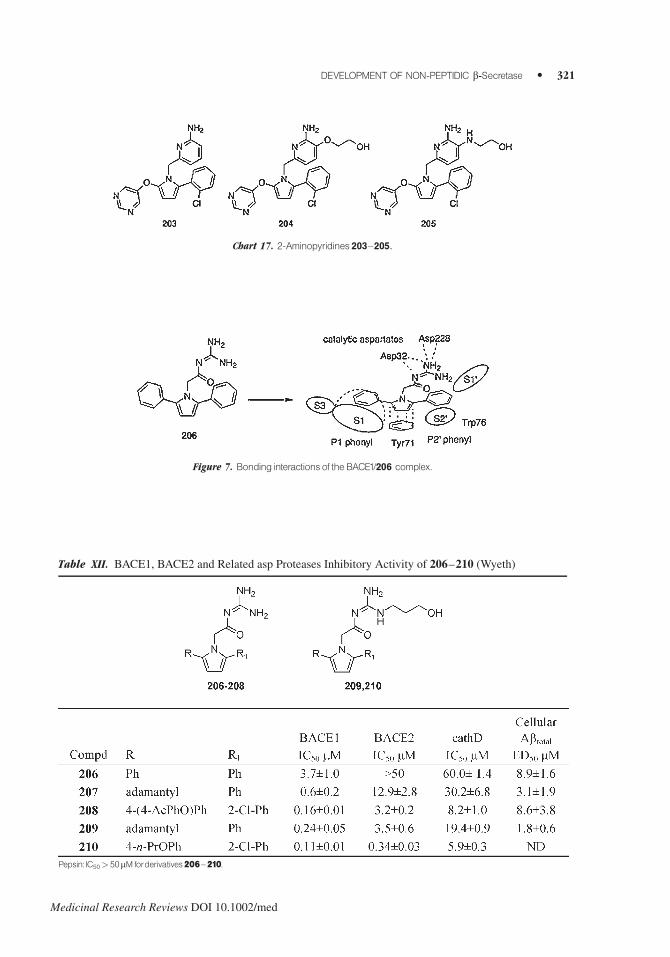
Chart 17. 2-Aminopyridines 203–205.
Figure 7. Bonding interactions of the BACE1/206 complex.
Table XII. BACE1, BACE2 and Related asp Proteases Inhibitory Activity of 206–210 (Wyeth)
Pepsin: IC50> 50mMforderivatives206^210.
DEVELOPMENT OF NON-PEPTIDIC b-Secretase * 321
Medicinal Research Reviews DOI 10.1002/med
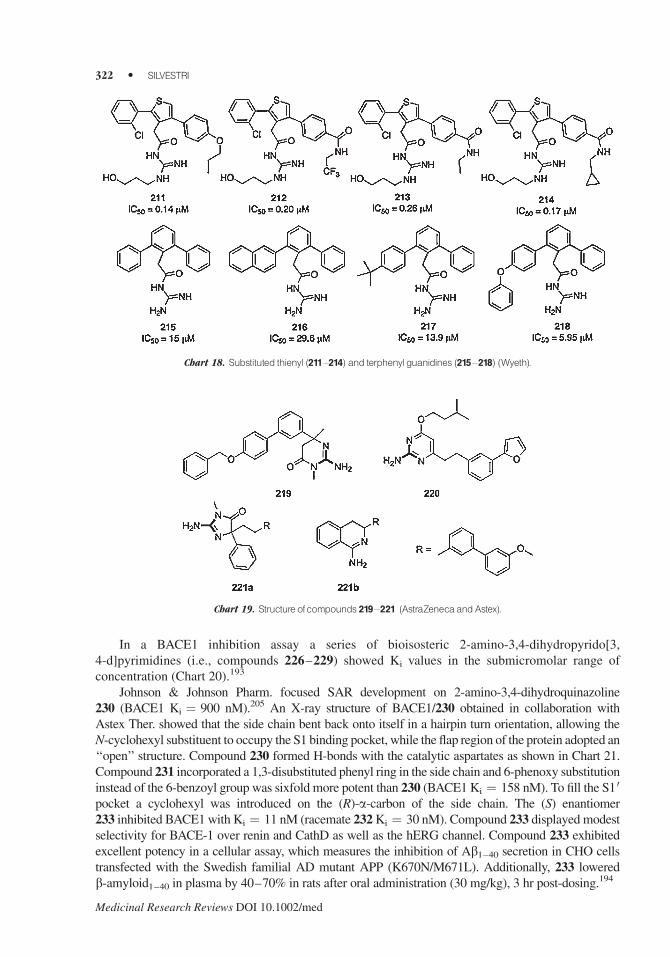
In a BACE1 inhibition assay a series of bioisosteric 2-amino-3,4-dihydropyrido[3,
4-d]pyrimidines (i.e., compounds 226–229) showed Ki values in the submicromolar range of
concentration (Chart 20).193
Johnson & Johnson Pharm. focused SAR development on 2-amino-3,4-dihydroquinazoline
230 (BACE1 Ki ¼ 900 nM).205 An X-ray structure of BACE1/230 obtained in collaboration with
Astex Ther. showed that the side chain bent back onto itself in a hairpin turn orientation, allowing the
N-cyclohexyl substituent to occupy the S1 binding pocket, while the flap region of the protein adopted an
‘‘open’’ structure. Compound 230 formed H-bonds with the catalytic aspartates as shown in Chart 21.
Compound 231 incorporated a 1,3-disubstituted phenyl ring in the side chain and 6-phenoxy substitution
instead of the 6-benzoyl group was sixfold more potent than 230 (BACE1 Ki ¼ 158 nM). To fill the S1 0
pocket a cyclohexyl was introduced on the (R)-a-carbon of the side chain. The (S) enantiomer
233 inhibited BACE1 with Ki ¼ 11 nM (racemate 232Ki ¼ 30 nM). Compound 233 displayed modest
selectivity for BACE-1 over renin and CathD as well as the hERG channel. Compound 233 exhibited
excellent potency in a cellular assay, which measures the inhibition of Ab1–40 secretion in CHO cells
transfected with the Swedish familial AD mutant APP (K670N/M671L). Additionally, 233 lowered
b-amyloid1–40 in plasma by 40–70% in rats after oral administration (30 mg/kg), 3 hr post-dosing.194
Chart 18. Substituted thienyl (211–214) and terphenyl guanidines (215–218) (Wyeth).
Chart 19. Structure ofcompounds 219–221 (AstraZenecaandAstex).
322 * SILVESTRI
Medicinal Research Reviews DOI 10.1002/med
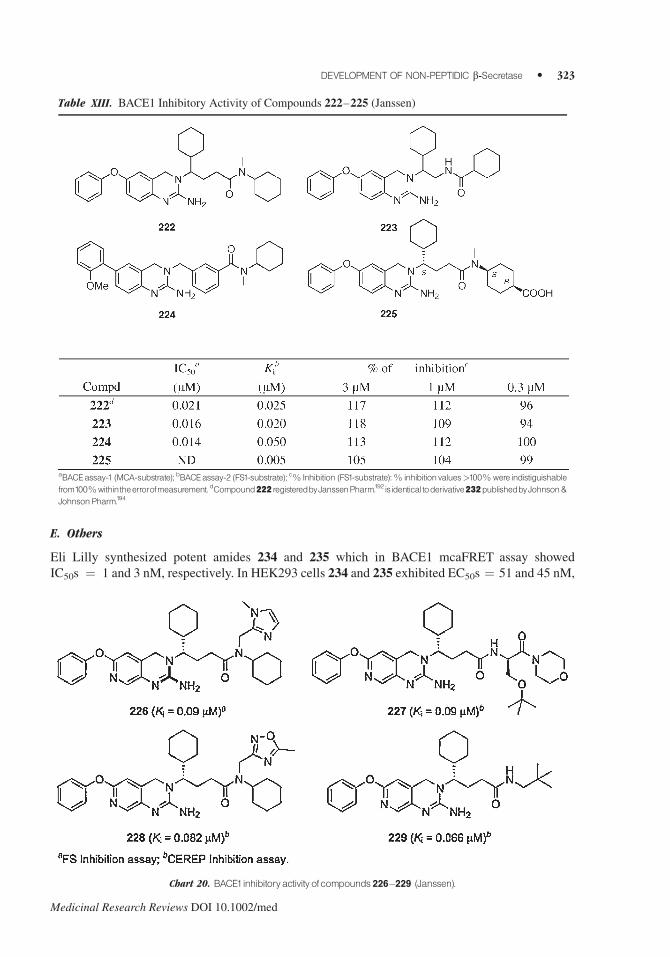
E. Others
Eli Lilly synthesized potent amides 234 and 235 which in BACE1 mcaFRET assay showed
IC50s ¼ 1 and 3 nM, respectively. In HEK293 cells 234 and 235 exhibited EC50s ¼ 51 and 45 nM,
Table XIII. BACE1 Inhibitory Activity of Compounds 222–225 (Janssen)
aBACEassay-1 (MCA-substrate);
bBACEassay-2 (FS1-substrate);
c% Inhibition (FS1-substrate):% inhibitionvalues>100%were indistiguishable
from100%withintheerrorofmeasurement.dCompound222 registeredbyJanssenPharm.192 isidenticaltoderivative232publishedbyJohnson&
JohnsonPharm.194
Chart 20. BACE1inhibitoryactivityofcompounds 226–229 (Janssen).
DEVELOPMENT OF NON-PEPTIDIC b-Secretase * 323
Medicinal Research Reviews DOI 10.1002/med
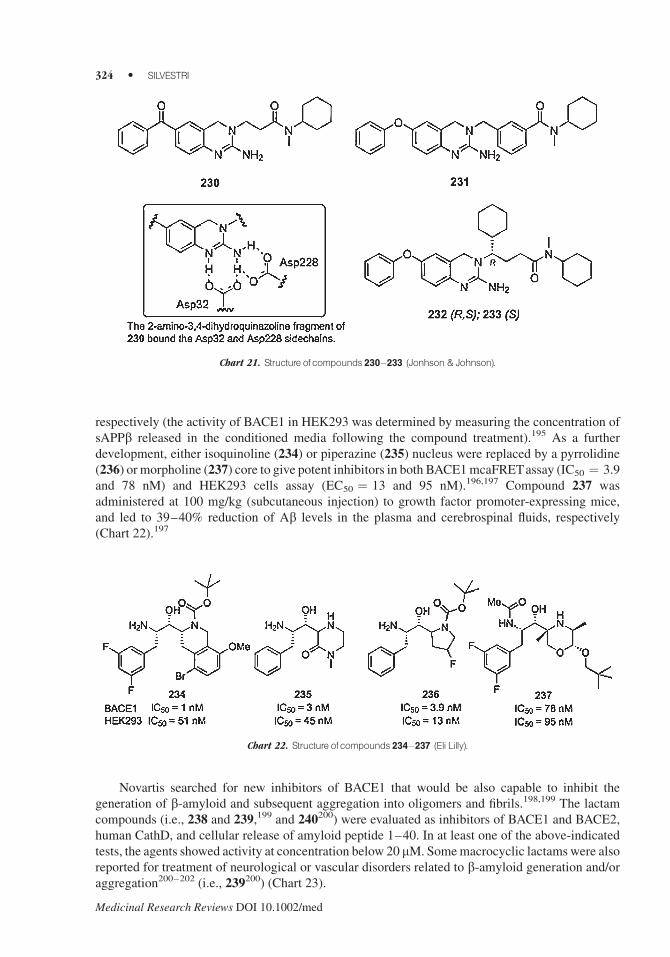
respectively (the activity of BACE1 in HEK293 was determined by measuring the concentration of
sAPPb released in the conditioned media following the compound treatment).195 As a further
development, either isoquinoline (234) or piperazine (235) nucleus were replaced by a pyrrolidine
(236) or morpholine (237) core to give potent inhibitors in both BACE1mcaFRETassay (IC50 ¼ 3.9
and 78 nM) and HEK293 cells assay (EC50 ¼ 13 and 95 nM).196,197 Compound 237 was
administered at 100 mg/kg (subcutaneous injection) to growth factor promoter-expressing mice,
and led to 39–40% reduction of Ab levels in the plasma and cerebrospinal fluids, respectively
(Chart 22).197
Novartis searched for new inhibitors of BACE1 that would be also capable to inhibit the
generation of b-amyloid and subsequent aggregation into oligomers and fibrils.198,199 The lactam
compounds (i.e., 238 and 239,199 and 240200) were evaluated as inhibitors of BACE1 and BACE2,
human CathD, and cellular release of amyloid peptide 1–40. In at least one of the above-indicated
tests, the agents showed activity at concentration below 20 mM. Somemacrocyclic lactams were also
reported for treatment of neurological or vascular disorders related to b-amyloid generation and/or
aggregation200–202 (i.e., 239200) (Chart 23).
Chart 21. Structure ofcompounds 230–233 (Jonhson& Johnson).
Chart 22. Structure ofcompounds 234–237 (Eli Lilly).
324 * SILVESTRI
Medicinal Research Reviews DOI 10.1002/med
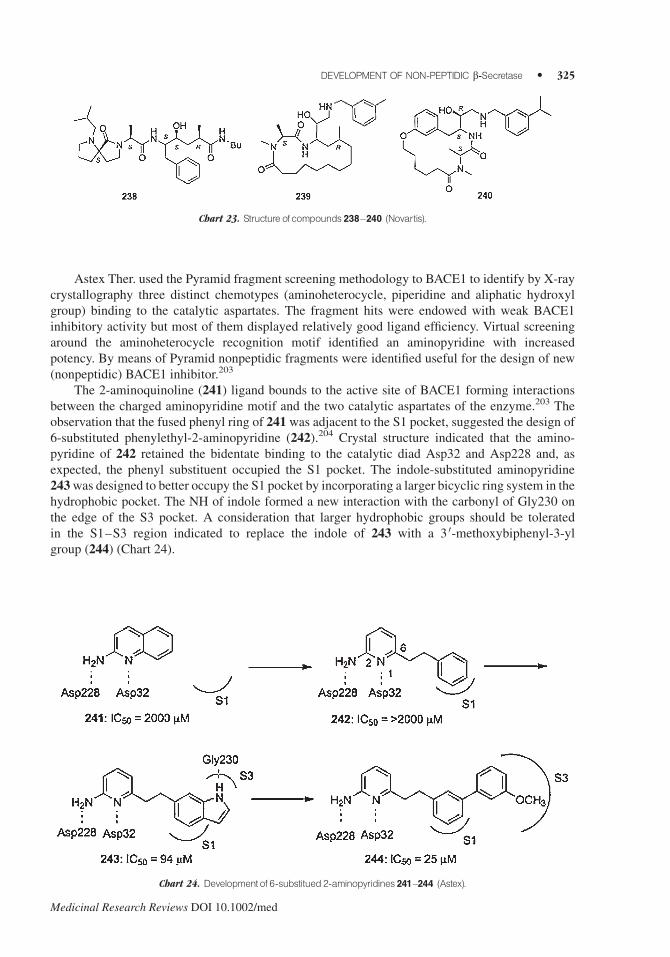
Astex Ther. used the Pyramid fragment screening methodology to BACE1 to identify by X-ray
crystallography three distinct chemotypes (aminoheterocycle, piperidine and aliphatic hydroxyl
group) binding to the catalytic aspartates. The fragment hits were endowed with weak BACE1
inhibitory activity but most of them displayed relatively good ligand efficiency. Virtual screening
around the aminoheterocycle recognition motif identified an aminopyridine with increased
potency. By means of Pyramid nonpeptidic fragments were identified useful for the design of new
(nonpeptidic) BACE1 inhibitor.203
The 2-aminoquinoline (241) ligand bounds to the active site of BACE1 forming interactions
between the charged aminopyridine motif and the two catalytic aspartates of the enzyme.203 The
observation that the fused phenyl ring of 241 was adjacent to the S1 pocket, suggested the design of
6-substituted phenylethyl-2-aminopyridine (242).204 Crystal structure indicated that the amino-
pyridine of 242 retained the bidentate binding to the catalytic diad Asp32 and Asp228 and, as
expected, the phenyl substituent occupied the S1 pocket. The indole-substituted aminopyridine
243was designed to better occupy the S1 pocket by incorporating a larger bicyclic ring system in the
hydrophobic pocket. The NH of indole formed a new interaction with the carbonyl of Gly230 on
the edge of the S3 pocket. A consideration that larger hydrophobic groups should be tolerated
in the S1–S3 region indicated to replace the indole of 243 with a 3 0-methoxybiphenyl-3-yl
group (244) (Chart 24).
Chart 23. Structure ofcompounds 238–240 (Novartis).
Chart 24. Developmentof 6-substitued 2-aminopyridines 241–244 (Astex).
DEVELOPMENT OF NON-PEPTIDIC b-Secretase * 325
Medicinal Research Reviews DOI 10.1002/med

The 2,3-diaminopyridine 245 was identified as a hit in silico by virtual screening of
aminopyridines.204 Compound 245 formed the charged bidentate interaction with the catalytic diad;
however, it changed its orientation relative to the fragment 241, and formed an additional H-bond
between the 3-amino group and Asp32. The switch in orientation of the aminopyridine ring allowed
the N-benzyl group to occupy the S1 pocket. Introduction of a pyridin-3-yl ring at position 3 of the
benzyl group provided ligands 246–248 which formed favorable contacts with the binding pocket
and showed improved aqueous solubility. Interestingly, the pyridyl nitrogen of 246 formed anH-bond
with a water molecule at the bottom of the S3 pocket. The oxygen of the methoxy group of
247 displaced this water molecule, and the alkyl group of 248 was positioned within the narrow
opening at the bottom of the S3 pocket. The phenyl ring of 249 bounds the S1 pocket with
concomitant small rotation of the 2,3-diaminopyridine moiety without loss of interaction with the
catalytic diad. The indolyl group occupied S3 pocket as expected, and formed a new interaction
between the NH of the heterocycle and the backbone carbonyl of Gly230. Compounds 250 and
251 were prepared to explore under ‘‘flap’’ toward the S2 0 region. Compound 251 occupied the S2 0
pocket of the enzyme, making hydrophobic contacts and a H-bond between the side chain NH of
Trp76 and the pyridyl nitrogen atom (Chart 25).
Sunesis Pharm., Inc. considered the primary amine group as a possible bioisostere of the
hydroxyl group present in HE-containing BACE1 inhibitors.205 Replacement of the HE motif of
252with the aminoethylene (AE) bioisostere afforded compound 253 endowedwith slightly reduced
enzymatic inhibitory activity but distinctly improved cell potency; the (S) isomer was preferred for
activity (compare 253 with 254) (Table XIV). Replacement of the benzylvaline amide group with a
4-fluoroaniline (compounds 255 and 256) caused loss of activity. Incorporation of a benzyl R1
substituent (257 and 258) resulted in a neat increase in inhibitory potency relative to the isobutyl
analogue. The three-dimensional cocrystal structure of 258 in complex with BACE1 showed that the
AE isostere had similar binding affinity for the catalytic site as the HE counterpart. The cell-based
activity of AE inhibitors wasmarkedly better than the HE-containing compounds. This result may be
due to themore hydrophilic character of theAE inhibitors at neutral pH and to themore acidic cellular
compartments in which BACE1 is localized.
Chart 25. Developmentof 3-substitued 2-aminopyridines 245–251 (Astex).
326 * SILVESTRI
Medicinal Research Reviews DOI 10.1002/med

4 . C O N C L U S I O N S
The aspartyl protease BACE1 has become a very attractive approach to develop new effective agents
for the treatment of Alzheimer’s disease. The research towards the discovery of non-peptidic BACE1
inhibitors has received a great impulse in the past 6 years. A wide number of structurally different
classes of compounds were found to be potent BACE1 inhibitors; however, some patents did not
disclose the BACE1 inhibitory concentrations. Isophthalamide-based compounds displayed
remarkable BACE1 inhibitory activity; chemical modification of this scaffold by introduction of a
sulfonate, macrocycle, isonicotinamide, guanidine and heterocycle (indole, imidazole, piperidine,
morpholine, isoquinoline) moiety provided BACE1 inhibitors which showed IC50s < 10 nM
concentration. The non-peptidic inhibitors showed high selectivity over BACE2 (BACE1/BACE2
selectivity>100) and other human proteases (cathD, pepsin, and renin). Their weak or non-peptidic
character favors CNS penetration and oral bioavailability. The non-peptidic structures showed potent
activity in both an in vitro assay and in a cellular assay (HEK2983 cells). In experiments in animal
models, the non-peptidic inhibitors (i.e., 57, 114, 225, and 258) led to signficant reductions of Ablevels in animal models. These inhibitors are valuable drug-candidates and might become first-line
drugs for the treatment of Alzheimer’s disease in the near future.
R E F E R E N C E S
1. Melnikova I. Therapies for Alzheimer’s disease. Nat Rev Drug Disc 2007;6:341–342.2. Citron M. Strategies for disease modification in Alzheimer’s disease. Nat Rev Neur 2004;5:677–685.
Table XIV. BACE1 Inhibitory Activity and Cellular Inhibition of Ab Production of 252–258
(Sunesis)
aCell-basedassay for thereleaseofthesolubleN-terminal fragmentofanAPP variant fromculturedHEK293Tcells.
bConcentrationat which � 20%cytotoxicitywasobservedviaTrypanBlueassay.
DEVELOPMENT OF NON-PEPTIDIC b-Secretase * 327
Medicinal Research Reviews DOI 10.1002/med

3. WHO: Alzheimer’s disease. http://www.who.int/en/.4. VerdonC-M, Fossati P,VernyM,DieudonneB,Teillet L,Nadel J. Social cognition:An early impairment in
dementia of the Alzheimer type. Alzh Dis Ass Dis 2007;21:25–30.5. Bartus RT, Dean RL III, Beer B, Lippa AS. The cholinergic hypothesis of geriatric memory dysfunction.
Science 1982;217:408–417.6. Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to
therapeutics. Science 2002;297:353–357.7. Nguyen J-T, Yamani A, Kiso Y. Views on amyloid hypothesis and secretase inhibitors for treating
Alzheimer’s disease: Progress and problems. Curr Pharm Des 2006;12:4295–4312.8. Stine WB Jr, Dahlgren KN, Krafft GA, LaDuMJ. In vitro characterization of conditions for amyloid-beta
peptide oligomerization and fibrillogenesis. J Biol Chem 2003;278:11612–11622.9. Espeseth AS, XuM,HuangQ, Coburn CA, Jones KLG, FerrerM, Zuck PD, Strulovici B, Price EA,WuG,
Wolfe AL, Lineberger JE, Sardana M, Tugusheva K, Pietrak BL, Crouthamel M-C, Lai M-T, Dodson EC,Bazzo R, Shi X-P, Simon AJ, Li Y, Hazuda DJ. Compounds that bind APP and inhibit a processing in vitrosuggest a novel approach to Alzheimer’s disease therapeutics. J Biol Chem 2005;280:17792–17797.
10. Jarrett JT, Berger EP, Lansbury PT Jr. The carboxy terminus of the b-amyloid protein is critical for theseeding of amyloid formation: Implications for the pathogenesis of Alzheimer’s disease. Biochemistry1993;32:4693–4697.
11. Rubio A, Avila J, Perez M. Effect of acetylcholine on tau phosphorylation in human neuroblastoma cells.J Mol Neurosci 2006;30:185–188.
12. Hardy J. The Alzheimer family of diseases: Many etiologies, one pathogenesis? Proc Natl Acad Sci USA1997;94:2095–2097.
13. Lacor PN, Buniel MC, Chang L, Fernandez SJ, Gong Y, Viola KL, Lambert MP, Velasco PT, Bigio EB,Finch CE, Krafft GA, Klein WL. Synaptic targeting by Alzheimer’s-related amyloid b oligomers.J Neurosci 2004;24:10191–10200.
14. Lacor PN, Buniel MC, Furlow PW, Clemente AS, Velasco PT, Wood M, Viola KL, Klein WL. Aboligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis forloss of connectivity in Alzheimer’s disease. J Neurosci 2007;27:796–807.
15. BashaMR,WeiW, Bakheet SA, Benitez N, Siddiqi HK, Ge Y-W, Lahiri DK, Zawia NH. The fetal basis ofamyloidogenesis: Exposure to lead and latent overexpression of amyloid precursor protein and amyloid inthe aging brain. J Neurosci 2005;25:823–829.
16. Buckner RL, Snyder AZ, Shannon BJ, LaRossa G, Sachs R, Fotenos AF, Sheline YI, Klunk WE, MathisCA,Morris JC,MintunMA.Molecular, structural, and functional characterization ofAlzheimer’s disease:Evidence for a relationship between default activity, amyloid, and memory. J Neurosci 2005;25:7709–7717.
17. Shankar GM, Bloodgood BL, TownsendM,Walsh DM, Selkoe DJ, Sabatini BL. Natural oligomers of theAlzheimer amyloid-b protein induce reversible synapse loss by modulating an NMDA-type glutamatereceptor-dependent signaling pathway. J Neurosci 2007;27:2866–2875.
18. Selkoe DJ. The molecular pathology of Alzheimer’s disease. Neuron 1991;6:487–498.19. Montalto MC, Farrar G, Hehir CT. Fibrillar and oligomeric b-amyloid as distinct local biomarkers for
Alzheimer’s disease. Ann New York Acad Sci 2007;1097:239–258.20. Alvarez A, Opazo C, Alarcon R, Garrido J, Inestrosa NC. Acetylcholinesterase promotes the aggregation
of amyloid-beta-peptide fragments by forming a complex with the growing fibrils. J Mol Biol 1997;272:348–361.
21. Castro A, Martinez A. Targeting beta-amyloid pathogenesis through acetylcholinesterase inhibitors. CurrPharm Des 2006;12:4377–4387.
22. Du D-M, Carlier PR. Development of bivalent acetylcholinesterase inhibitors as potential therapeuticdrugs for Alzheimer’s disease. Curr Pharm Design 2004;10:3141–3156.
23. Tezer N. Ab initio molecular structure study of alkyl substitute analogues of Alzheimer drug phenserine:Structure-activity relationships for acetyl- and butyrylcholinesterase inhibitory action. Theochem 2005;714:133–136.
24. Del Monte-Millan M, Gracıa-Palomero E, Valenzuela R, Usan P, de Austria C, Munoz-Rulz P, Rubio L,Dorronsoro I, Martınez A, Medina M. Dual binding site acetylcholinesterase inhibitors: Potential newdisease-modifying agents for AD. J Mol Neurosci 2006;30:85–88.
25. Bai D. Development of huperzine A and B for treatment of Alzheimer’s disease. Pure Appl Chem2007;79:469–479.
26. Inestrosa NC, Alvarez A, Perez CA, Moreno RD, Vicente M, Linker C, Casanueva OI, Soto C, Garrido J.Acetylcholinesterase accelerates assembly of amyloid-beta-peptides intoAlzheimer’s fibrils: Possible roleof the peripheral site of the enzyme. Neuron 1996;16:881–891.
328 * SILVESTRI
Medicinal Research Reviews DOI 10.1002/med

27. De Ferrari GV, Canales MA, Shin I, Weiner LM, Silman I, Inestrosa NC. A structural motif ofacetylcholinesterase that promotes amyloid-peptide fibril formation. Biochemistry 2001;40:10447–10457.
28. Giacobini E. Cholinesterase inhibitors stabilize Alzheimer’s disease. Neurochem Res 2000;25:1185–1190.
29. LahiriDK, Lewis S, FarlowMR.Tacrine alters the secretion of beta-amyloid precursor protein in cell lines.J Neurosci Res 1994;37:777–787.
30. Rees TM, Brimijoin S. The role of acetylcholinesterase in the pathogenesis of Alzheimer’s disease. DrugsToday 2003;39:75–83.
31. Gozes I. Tau as a drug target in Alzheimer’s disease. J Mol Neurosci 2002;19:337–338.32. BushAI.Metal complexing agents as therapies for Alzheimer’s disease. Neur Aging 2002;23:1031–1038.33. Klafki H-W, Staufenbiel M, Kornhuber J, Wiltfang J. Therapeutic approaches to Alzheimer’s disease.
Brain 2006;129:2840–2855.34. Permanne BM, Saborio GP, Soto C. In vivo disrupting of amyloid deposition after injection of b-sheet
breaker peptide into cerebral ventricle. Neur Aging 2000;21(S1):S87.35. McLaurin J, Kierstead ME, Brown ME, Hawkes CA, Lambermon MHL, Phinney AL, Darabie AA,
Cousins JE, French JE, Lan MF, Chen F, Wong SSN, Mount HTJ, Fraser PE, Westaway D, StGeorge-Hyslop P. Cyclohexanehexol inhibitors of A aggregation prevent and reverse Alzheimer phenotype in amouse model. Nature Med 2006;12:801–808.
36. de Mattos RB, Bales KR, Cummins DJ, Dodart JC, Paul SM, Holtzman DM. Peripheral anti-Ab antibodyalters CNS and plasma Ab clearance and decreases brain Ab burden in a mouse model of Alzheimer’sdisease. Proc Natl Acad Sci USA 2001;98:8850–8855.
37. (a)Revill P, Serradell N, Bolos J. Tramiprosate: Antiamyloidogenic agent for treatment of Alzheimer’sdisease and hemorrhagic stroke. Drugs Fut 2006;31:498–501;37.(b)Wright TM. Tramiprosate. Drugs Today 2006;42:291–298.
38. Braak H, Braak E. Staging of Alzheimer’s disease-related neurofibrillary changes. Neur Aging 1995;16:271–278.
39. Hung AY, Haass C, Nitsch RM, Qiu WQ, Citron M, Wurtman PJ, Growdon JH, Selkoe DJ. Activation ofprotein kinase C inhibits cellular production of the amyloid beta-protein. J Biol Chem 1993;268:22959–22962.
40. Nitsch RM, Slack BE, Wurtman RJ, Growdon JH. Release of Alzheimer amyloid precursor derivativesstimulated by activation of muscarinic acetylcholine receptors. Science 1992;258:304–307.
41. Postina R, Schroeder A, Dewachter I, Bohl J, Schmitt U, Kojiro E. A disintegrin-metalloproteinaseprevents amyloid plaque formation and hippocampal defects in an Alzheimer’s disease mouse model.J Clin Invest 2004;113:1456–1464.
42. Fassbender K, SimonsM, Bergmann C, StroickM, Lutjohann D, Keller P, Runz H, Kuhl S, Bertsch T, VonBergmannK,HennericiM,BeyreutherK,HartmannT. Simvastatin strongly reduces levels ofAlzheimer’sdisease b-amyloid peptides Ab42 and Ab40 in vitro and in vivo. Proc Natl Acad Sci USA 2001;98:5856–5861.
43. Kojiro E, Gimpl G, Lammich S,MarzW, Fahrenholz F. Low cholesterol stimulates the nonamyloidogenicpathway by its effect on the a-secretase ADAM10. Proc Natl Acad Sci USA 2001;98:5815–5820.
44. Larner AJ. Secretases as therapeutic targets in Alzheimer’s disease: Patents 2000–2004. Expert Opin TherPat 2004;14:1403–1420.
45. De Strooper B, Saftig P, Craessaerts K, Vanderstichele H, GuhdeG,AnnaertW,Von FiguraK, Van LeuvenF. Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature 1998;391:387–390.
46. Petit A, Bihel F, Alves da Costa C, Pourquie O, Checler F, Kraus JL. New protease inhibitors preventgamma-secretase-mediated production of Ab40/42 without affecting Notch cleavage. Nat Cell Biol2001;3:507–511.
47. CampbellWA, ReedML, Strahle J,WolfeMS, XiaW. Presenilin endoproteolysis mediated by an aspartylprotease activity pharmacologically distinct from gamma-secretase. J Neurochem 2003;85:1563–1574.
48. Wolfe MS. Therapeutic strategies for Alzheimer’s disease. Nat Rev Drug Disc 2002;1:859–865.49. TeallM,Oakley P,Harrison T, ShawD,KayE, Elliott J, GerhardU, Castro JL, ShearmanM,Ball RG,Tsou
NN. Aryl sulfones: A new class of g-secretase inhibitors. Bioorg Med Chem Lett 2005;15:2685–2688.50. Churcher I, Beher D. g-Secretase as a therapeutic target for the treatment of Alzheimer’s disease. Curr
Pharm Des 2005;11:3363–3382.51. Cai H, Wang Y, McCarthy D, Wen H, Borchelt DR, Price DL, Donald L, Wong PC. BACE1 is the major
b-secretase for generation of Ab peptides by neurons. Nature Neurosci 2001;4:233–234.
DEVELOPMENT OF NON-PEPTIDIC b-Secretase * 329
Medicinal Research Reviews DOI 10.1002/med

52. Nilsberth C,Westlind-DanielssonA, EckmanCB,CondronMM,AxelmanK, Forsell C, StenhC,LuthmanJ, Teplow DB, Younkin SG, Naeslund J, Lannfelt L. The ‘Arctic’ APP mutation (E693G) causesAlzheimer’s disease by enhanced Ab protofibril formation. Nature Neurosci 2001;4:887–893.
53. Sinha S, Anderson JP, Barbour R, Basi GS, Caccavello R, Davis D, DoanM, DoveyHF, Frigon N, Hong J,Jacobson-Croak K, Jewett N, Keim P, Knops J, Lieberburg I, Power M, Tan H, Tatsuno G, Tung J,Schenk D, Seubert P, Suomensaari SM, Wang S, Walker D, Zhao J, McConlogue L, John V. Purificationand cloning of amyloid precursor protein beta-secretase from human brain. Nature 1999;402:537–540.
54. Hong L, Koelsch G, Lin X, Wu S, Terzyan S, Ghosh AK, Zhang XC, Tang J. Structure of the proteasedomain of memapsin 2 (b-secretase) complexed with inhibitor. Science 2000;290:150–153.
55. Ghosh AK, Bilcer G, Harwood C, Kawahama R, Shin D, Hussain KA, Hong L, Loy JA, Nguyen C, KoelshG, Ermolieff J, Tang J. Structure-based design: Potent inhibitors of human brainmemapsin 2 (b-secretase).J Med Chem 2001;44:2865–2868.
56. GhoshAK,Tang J, BilcerG,ChangW,HongL,KoelschG,Loy J, TurnerRT III.b-Secretase inhibitors andmethods of use. PCT Int Appl 2003; WO03039454.
57. Ghosh AK, Devasamudram T, Hong L, Dezuter C, Xu X, Weerasena V, Koelsch G, Bilcer G, Tang J.Structure-based design of cycloamide-urethane-derived novel inhibitors of human brain memapsin2 b-secretase. Bioorg Med Chem Lett 2005;15:15–20.
58. Shuto D, Kasai S, Kimura T, Liu P, Hidaka K, Hamada T, Shibakawa S, Hayashi Y, Hattori C, Szabo B,Ishiura S, Kiso Y. KMI-008, a novel beta-secretase inhibitor containing a hydroxymethylcarbonyl isostereas a transition-state mimic: Design and synthesis of substrate-based octapeptides. BioorgMed Chem Lett2003;13:4273–4276.
59. Kimura T, ShutoD, Kasai S, Liu P, HidakaK, Hamada T, Hayashi Y, Hattori C, AsaiM, Kitazume S, SaidoTC, Ishiura S, KisoY.KMI-358 andKMI-370, highly potent and small-sizedBACE1 inhibitors containingphenylnorstatine. Bioorg Med Chem Lett 2004;14:1527–1531.
60. Manzenrieder F, Frank AO, Huber T, Dorner-Ciossek C, Kessler H. Synthesis and biological evaluation ofphosphino dipeptide isostere inhibitor of human b-secretase (BACE1). BioorgMedChem 2007;15:4136–4143.
61. Kimura T, Shuto D, Hamada Y, Igawa N, Kasai S, Liu P, Hidaka K, Hamada T, Hayashi Y, Kiso Y. Designand synthesis of highly active Alzheimer’s beta-secretase (BACE1) inhibitors, KMI-420 and KMI-429,with enhanced chemical stability. Bioorg Med Chem Lett 2005;15:211–215.
62. AsaiM, Hattori C, Iwata N, Saido TC, SasagawaN, Szabo B, Hashimoto Y,MaruyamaK, Tanuma S, KisoY, Ishiura S. The novel b-secretase inhibitor KMI-429 reduces amyloid b peptide production in amyloidprecursor protein transgenic and wild-type mice. J Neurochem 2006;96:533–540.
63. Kimura T, Hamada Y, Stochaj M, Ikari H, Nagamine A, Abdel-Rahman H, Igawa N, Hidaka K, NguyenJ-T, Saito K, Hayashi Y, Kiso Y. Design and synthesis of b-secretase (BACE1) inhibitors with P1 0carboxylic acid bioisosteres. Bioorg Med Chem Lett 2006;16:2380–2386.
64. Boyd JG, Singleton DH. Novel inhibitor of b-amyloid cleavage enzyme. US Pat Appl 2002;US2002115616.
65. Chen S-H, Lamar J, Guo D, Kohn T, Yang H-C, McGee J, TimmD, Erickson J, Yip Y,May P, McCarthy J.P3 cap modified Phe*-Ala series BACE inhibitors. Bioorg Med Chem Lett 2004;14:245–250.
66. Tung JS, Davis DL, Anderson JP, Walker DE, Mamo S, Jewett N, Hom RK, Sinha S, Thorsett ED, John V.Design of substrate-based inhibitors of human b-secretase. J Med Chem 2002;45:259–262.
67. Bradly F, Singh S, Crouthamel MC, Holloway MK, Coburn CA, Grasky VM, Bogusky M, PenningtonMW, Vacca JP, Hazuda D, Lai M-T. Rational design and synthesis of selective BACE1 inhibitors. BioorgMed Chem Lett 2004;14:601–614.
68. Hills DH,Vacca JP. Progress toward a practical BACE1 inhibitor. Curr OpinDrugDisc Dev 2007;10:383–391.
69. Guo T, Hobbs DW. Development of BACE1 inhibitors for Alzheimer’s disease. Curr Med Chem 2006;13:1811–1829.
70. Ziora Z, Kimura T, Kiso Y. Small-sized BACE1 inhibitors. Drug Fut 2006;31:53–63.71. Limongelli V, Marinelli L, Cosconati S, Braun HA, Schmidt B, Novellino E. Ensemble-docking approach
on BACE1: Pharmacophore perception and guidelines for drug design. Chem Med Chem 2007;2:667–678.
72. Eder J, Hommel U, Cumin F, Martoglio B, Gerhartz B. Aspartic proteases in drug discovery. Curr PharmDes 2007;11:271–285.
73. Gao J, Winslow SL, VanderVelde D, Aube J, Borchardt RT. Transport characteristics of peptides andpeptidomimetics: II. Hydroxyethylamine bioisostere-containing peptidomimetics as substrates for theoligopeptide transporter and P-glycoprotein in the intestinal mucosa. J Pept Res 2001;57:361–371.
330 * SILVESTRI
Medicinal Research Reviews DOI 10.1002/med

74. Miyamoto M, Matsui J, Fukumoto H, Tarui N. (Takeda Chem Ind) Preparation of 2-[2-amino- or2-(N-heterocyclyl)ethyl]-6-(4-biphenylylmethoxy)tetralin derivatives as b-secretase inhibitors. PCT IntAppl 2001; WO2001087293.
75. MiyamotoM,Matsui J, Fukumoto H, Tarui N. (Takeda Chem Ind) Beta-secretase inhibitors. Jpn Pat Appl2002; JP2002037731.
76. MiyamotoM,Matsui J, FukumotoH,TaruiN. (TakedaChem Ind)-Secretase inhibitors. Eur PatAppl 2003;EP1283039.
77. Uchikawa O, Aso K, Koike T, Tarui N, Hirai K. (Takeda Chem Ind) Preparation of benzamide derivativesas b-secretase inhibitors. PCT Int Appl 2004; WO 2004014843.
78. Watanabe H, Kurasawa O, Tarui N, Yorimoto T, Hirai K. (Takeda Chem Ind) Preparation of indoles asinhibitors against aspartate protease, b-secretase, and amyloid b protein for treatment of nerve disordersand myopathy. Jpn Pat Appl 2004; JP2004149429.
79. Qiao L, Etchenberrigaray R. (Neurologic Inc.) Preparation of phosphinylmethyl and phosphorylmethylsuccinic and glutaric acid analogs as b-secretase inhibitors useful in the treatment of Alzheimer’s disease.PCT Int Appl 2002; WO2002096897.
80. Etchenberrigaray R, Qiao L. (Neurologic Inc.) Phosphinylmethyl and phosphorylmethyl succinic andglutaric acid analogs as b-secretase inhibitors. PCT Int Appl 2003; US2003078240.
81. Bhisetti GR, Saunders JO, Murcko MA, Lepre CA, Britt SD, Come JH, Deninger DD, Wang T. (VertexPharm. Inc.) Preparation of b-carbolines and other inhibitors of BACE1 aspartic proteinase useful againstAlzheimer’s and other BACE-mediated diseases. PCT Int Appl 2002; WO2002088101.
82. John V, Beck JP, Bienkowski MJ, Sinha S, Heinrikson RL. Human b-secretase (BACE) and BACEinhibitors. J Med Chem 2003;46:4625–4630.
83. Elan Pharm, Inc. Statine-derived tetrapeptide inhibitors of b-secretase. ExpOpin Ther Pat 2001;11:1047–1050.
84. John V, Tung J, Fang L, Mamo SS. (Elan Pharm. Inc.) Preparation of statine-derived tetrapeptides asinhibitors of b-secretase. PCT Int Appl 2000; WO2000077030.
85. Hom RK, Gailunas AF, Mamo S, Fang LY, Tung JS, Walker DE, Davis D, Thorsett ED, Jewett NE, MoonJB, John V. Design and synthesis of hydroxyethylene-based peptidomimetic inhibitors of human b-secretase. J Med Chem 2004;47:158–164.
86. Tung JS, Davis DL, Anderson JP,Walker DE,Mamo S, Jewett N, HomRK, Sinha S, Thorsett ED, John V.Design of substrate-based inhibitors of human b-secretase. J Med Chem 2002;45:259–262.
87. Maillard MC, Hom RK, Benson TE, Moon JB, Mamo S, Bienkowski M, Tomasselli AG, Woods DD,Prince DB, Paddock DJ, Emmons TL, Tucker JA, DappenMS, Brogley L, Thorsett ED, Jewett N, Sinha S,John V. Design, synthesis, and crystal structure of hydroxyethyl secondary amine-based peptidomimeticinhibitors of human b-secretase. J Med Chem 2007;50:776–781.
88. Hom R, Mamo S, Tung J, Gailunas A, John V, Fang LY. (Elan Pharm. Inc.) Preparation ofhydroxyethylenes with peptide subunits for pharmaceutical use in the treatment of Alzheimer’s disease.PCT Int Appl 2001; WO2001070672.
89. Fang LY, Hom R, John V, Maillaird M. (Elan Pharm. Inc.) Preparation of substituted amines for thetreatment of Alzheimer’s disease. PCT Int Appl 2002; WO2002002505.
90. Maillard M, Hom C, Gailunas A, Jagodzinska B, Fang LY, John V, Freskos JN, Pulley SR, Beck JP,Tenbrink RE. (Elan Pharm. Inc.) Preparation of substituted amines to treat Alzheimer’s disease. PCT IntAppl 2002; WO2002002512.
91. Beck JP,Gailunas A,HomR, JagodzinskaB, JohnV,MaillairdM. (Elan Pharm. Inc., Pharmacia&UpjohnComp.) Preparation of disubstituted amines for treating Alzheimer’s disease. Eur Pat Appl 2001;EP1586556. PCT Int Appl 2002; WO2002002518.
92. JohnV, HomR, Tucker J. (Elan Pharm. Inc., Pharmacia&UpjohnComp.) Preparation of b-hydroxyaminederivatives for the treatment of Alzheimer’s disease. PCT Int Appl 2003; WO2003002122.
93. JohnV. (Elan Pharm. Inc.) Preparation of peptide isosteres containing a heterocycle useful in the treatmentof Alzheimer’s disease. PCT Int Appl 2003; WO2003047576.
94. JohnV,MaillardM, Tucker J, Aquino J, JagodzinskaB, BrogleyL, Tung J, Bowers S, DressenD, Probst G,ShahN. (Elan Pharm. Inc.) Preparation of hydroxyethylamines as aspartyl protease inhibitors for treatmentof amyloidosis. PCT Int Appl 2005; WO2005087751. and 2005; WO2005087752.
95. John V, Hom R, Tucker J. Methods of treatment of amyloidosis using bi-aryl aspartyl protease inhibitors.US Pat Appl 2006; US20060014737.
96. Nieman JA, FangL, JagodzinskaB. (Elan Pharm. Inc., Pharmacia&UpjohnComp.)Methods of treating orpreventing Alzheimer’s disease using 4-aryl-3-aralkoxypipiperidines and azabicyclooctanes. PCT IntAppl 2002; WO2002076440.
DEVELOPMENT OF NON-PEPTIDIC b-Secretase * 331
Medicinal Research Reviews DOI 10.1002/med

97. Schostarez H, Chrusciel RA, Cenko RS. (Elan Pharm. Inc., Pharmacia & Upjohn Comp.)Acylaminopropylhydrazines as b-secretase inhibitors. PCT Int Appl 2002; WO2002094768.
98. PulleySR,Beck JP, TenbrinkRE, Jacobs JS. (Elan Pharm. Inc. Pharmacia&UpjohnComp.) Preparation ofmacrocycles useful in the treatment of Alzheimer’s disease. PCT Int Appl 2002; WO2002100399.
99. Pulley SR, Beck JP, Tenbrink RE. (Elan Pharm. Inc., Pharmacia & Upjohn Comp.) Macrocycles useful inthe treatment of Alzheimer’s disease. PCT Int Appl 2002; WO2002100856.
100. Schostarez HJ, Chrusciel RA. (Elan Pharm. Inc., Pharmacia & Upjohn Comp.) Preparation of aminediolsas b-secretase inhibitors for the treatment ofAlzheimer’s and other diseases characterized by deposition ofAb peptide. PCT Int Appl 2002; WO2002100818.
101. Schostarez HJ, Hanson GJ. (Elan Pharm. Inc., Pharmacia & Upjohn Comp.) Aminodiols useful in thetreatment of Alzheimer’s disease and similar diseases. PCT Int Appl 2003; WO2003043618.
102. Romero AG, Schostarez H, Roels CM. (Elan Pharm. Inc., Pharmacia & Upjohn Comp.) Preparation ofamine 1,2- and 1,3-diol alditols and their use for treatment of Alzheimer’s disease. PCT Int Appl 2003;WO2003043975.
103. Beck JP. (Elan Pharm. Inc., Pharmacia & Upjohn Comp.) Preparation and use of aza-bicyclononanes forthe treatment of Alzheimer’s disease. PCT Int Appl 2003; WO2003000261.
104. Schostarez HJ, Chrusciel RA. (Elan Pharm. Inc., Pharmacia & Upjohn Comp.) Preparation of diaminediols as b-secretase inhibitors for the treatment of Alzheimer’s disease. PCT Int Appl 2003;WO2003006013.
105. Schostarez HJ, Chrusciel RA. (Elan Pharm. Inc., Pharmacia & Upjohn Comp.) Preparation of diaminediols as b-secretase inhibitors for the treatment of Alzheimer’s disease. PCT Int Appl 2003;WO2003006453.
106. Gailunas A, Hom R, John V, Mailard M, Chrusciel RA, Fisher J, Jacobs J, Freskos JN, Brown DL, FobianYM. (Elan Pharm. Inc., Pharmacia & Upjohn Comp.) Preparation of N-(3-amino-2-hydroxypropyl)substituted alkanamides as inhibitors of the beta secretase enzyme for treating Alzheimer’s disease. PCTInt Appl 2003; WO2003006423.
107. Aquino J, John V, Tucker JA, Hom R, Pulley S, Tenbrink R. (Elan Pharm. Inc., Pharmacia & UpjohnComp.) Preparation of 2-hydroxy-3-aminoalkylbenzamides as b-secretase inhibitors for the treatment ofAlzheimer’s disease. PCT Int Appl 2004; WO2004094384.
108. Gailunas AT, Tucker JA, JohnV. Preparation of phenethylamines for the treatment of Alzheimer’s disease.(Elan Pharm. Inc.). PCT Int Appl 2003; WO2003027068.
109. Fisher JF, Jacobs JS, Scherer BA. (Elan Pharm. Inc., Pharmacia & Upjohn Comp.) Preparation ofarylhydroxypropylamines used for treatment of Alzheimer’s disease and to reduce amyloid beta peptideformation. PCT Int Appl 2003; WO2003029169.
110. Roy H. (Elan Pharm. Inc.) Preparation of allylamides useful in the treatment of Alzheimer’s disease. PCTInt Appl 2003; WO2003030886.
111. Beck JP. (Elan Pharm. Inc., Pharmacia & Upjohn Comp.) Preparation of hydroxy substitutedindanylamides for the treatment of Alzheimer’s disease. PCT Int Appl 2003; WO2003037325.
112. John V, Maillard M, Jagodzinska B, Beck JP, Gailunas A, Fang L, Sealy J, Tenbrink R, Freskos J,Mickelson J, Samala L, Hom R. (Elan Pharm. Inc., Pharmacia & Upjohn Comp.) Preparation of N 0,N 0-susbstituted-1,3-diamino-2-hydroxypropanes for treating Alzheimer’s disease. PCT Int Appl 2003;WO2003040096.
113. Jagodzinska B, Maillard M, Beck JP, Tenbrink R, Getman D. (Elan Pharm. Inc., Pharmacia & UpjohnComp.) Preparation of substituted amine as prodrugs useful in treating Alzheimer’s disease. PCT Int Appl2003; WO2003072535.
114. Fobian YM, Freskos JN, Jagodzinska B. (Elan Pharm. Inc., Pharmacia &Upjohn Comp.) A preparation of1,3-diamino-2-hydroxypropane derivatives as beta-secretase enzyme inhibitors. PCT Int Appl 2004;WO2004022523.
115. Maillard M, Baldwin ET, Beck JT, Hughesm R, John V, Pulley SR, Tenbrink R. (Elan Pharm. Inc., Pfizer,Inc., Pharmacia & Upjohn Comp.) Preparation of ring-containing N-acetyl 2-hydroxy-1,3-diamonoal-kanes as b-secretase inhibitors for treating Alzheimer’s disease and other diseases characterized bydeposition of Ab-peptide. PCT Int Appl 2004; WO2004024081.
116. Aquino J, John V, Tucker JA, Hom R, Pulley S, Tenbrink R. (Elan Pharm. Inc., Pharmacia & UpjohnComp.) Preparation of phenacyl-substituted 2-hydroxy-3-diaminoalkanes as inhibitors of b-secretase.PCT Int Appl 2004; WO2004094413.
117. John V, Moon JB, Pulley SR, Rich DH, Brown D. (Elan Pharm. Inc., Pharmacia & Upjohn Comp.)Preparation of substituted piperidines and piperazines useful as b-secretase inhibitors against Alzheimer’sdisease. PCT Int Appl 2003; WO2003043987.
332 * SILVESTRI
Medicinal Research Reviews DOI 10.1002/med

118. Tenbrink R, Maillard M, Warpehoski M. (Elan Pharm. Inc., Pharmacia & Upjohn Comp.) Preparation ofsubstituted hydroxyethylamines as b-secretase inhibitors. PCT Int Appl 2003; WO2003050073.
119. Jagodzinska B, Warpehoski MA. (Elan Pharm. Inc., Pharmacia & Upjohn Comp.) Preparation ofsubstituted amino carboxamides for the treatment of Alzheimer’s disease. PCT Int Appl 2003;WO2003057721.
120. Maillard M, Varghese J. (Elan Pharm. Inc) Methods of treating Alzheimer’s disease using aromaticallysubstituted o-aminoalkanoic acid amides and alkanoic acid diamides. PCT Int Appl 2003;WO2003103652.
121. John V, Maillard M. (Elan Pharm. Inc) Methods of treating Alzheimer’s disease and methods of preparingd-amino-g-hydroxy-o-arylalkanoic acid amides. PCT Int Appl 2003; WO2003103653.
122. Hom R, Varghese J. (Elan Pharm. Inc.) Preparation of hydroxyaminopropyl benzamides for the treatmentof Alzheimer’s disease. PCT Int Appl 2004; WO2004029019.
123. Tucker JA. (Elan Pharm. Inc., Pharmacia & Upjohn Comp.) Preparation of hydroxypropyl amide peptideanalogs for the treatment of Alzheimer’s disease. PCT Int Appl 2005; WO2005042472.
124. Pulley SR, Tucker JA. (Elan Pharm. Inc., Pharmacia & Upjohn Comp.) Preparation of peptide-relatedsubstituted ureas and carbamates for the treatment of Alzheimer’s disease. PCT Int Appl 2004;WO2004050609.
125. John V, Maillard M, Tucker J, Aquino J, Hom R, Tung J, Dressen D, Shah N, Neitz RJ. (Elan Pharm. Inc.)Preparation of substituted ureas and carbamates, phenacyl-2-hydroxy-3-diaminoalkane and benzamide-2-hydroxy-3-diaminoalkane aspartyl protease and b-secretase inhibitors for treating conditions associatedwith amyloidosis such Alzheimer’s disease. PCT Int Appl 2005; WO2005087215.
126. JohnV,HomR, Saely J, Aquino J, ProbstG, Tung J, FangL. (Elan Pharm. Inc.) Preparation ofN-(3-amino-2-hydroxypropyl)acetamides as aspartyl protease and beta-secretase inhibitors for treating conditionsassociated with amyloidosis such Alzheimer’s disease. PCT Int Appl 2005; WO2005070407.
127. JohnV,MaillardM, FangL, Tucker J, Brogley L,Aquino J, Bowers S, Probst G, Tung J. (Elan Pharm. Inc.)Preparation of bicyclic compounds as aspartyl protease and beta-secretase inhibitors for treatingconditions associated with amyloidosis such Alzheimer’s disease. PCT Int Appl 2005; WO2005087714.
128. HomR,Tucker J, JohnV, ShahN. (Elan Pharm. Inc., Pharmacia&UpjohnComp.) Preparation of 2-amino-and 2-thio-substituted 1,3-diaminopropanes as b-secretase inhibitors for treating Alzheimer’s disease andother diseases characterized by deposition of Ab-peptide. PCT Int Appl 2005; WO2005095326.
129. JohnV,MaillardM, Jagodzinska B, Aquino J, Probst G, Tung JS. (Elan Pharm. Inc.) Preparation of oxime,hydrazone and other derivative substituted hydroxyethylamine selective b-secretase inhibitors for treatingamyloidosis. PCT Int Appl 2006; WO2006010095.
130. Sealy J, Hom R, John V, Probst G, Tung JS. (Elan Pharm. Inc.) Preparation of oxime-containingN-(3-amino-1-arylmethyl-2-hydroxypropyl) carboxamides and related selectiveb-secretase inhibitors fortreating amyloidosis. PCT Int Appl 2006; WO2006010094.
131. Hom R, Fang L, John V. (Elan Pharm. Inc.) Preparation of ethanolcyclicamines selective b-secretaseinhibitors for treatment of amyloidosis. PCT Int Appl 2006;WO2006026533 and 2006;WO2006026532.
132. Hom R, Toth G, Probst G, Bowers S, Truong A, Tung JS. (Elan Pharm. Inc.) Preparation of arylcyclopropyl(heterocyclyl)aminohydroxybutylamides as aspartyl protease inhibitors for treatment ofamyloidosis. PCT Int Appl 2007; WO2007047306 and 2007; WO2007047305.
133. Coburn CA, Stachel SJ, Li Y-M, Rush DM, Steele TG, Chen-Dodson E, Holloway MK, Xu M, Huang Q,LaiM-T, DiMuzio J, CrouthamelM-C, Shi X-P, SardanaV, Chen Z,Munshi S, KuoL,Makara GM,AnnisDA, Tadikinda PK, Nash HM, Vacca JP, Wang T. Identification of a small molecule nonpeptide active siteb-secretase inhibitor that displays a nontraditional binding mode for aspartyl proteases. J Med Chem2004;47:6117–6119.
134. CoburnCA, Steele TG,Vacca JP,AnnisDA Jr,MakaraGM,NashHM,Tadikonda PK, PraveenK,WangT.(Merck & Co.) Preparation of aminopently-aminooxoethylphenylcarboxamides as b-secretase inhibitorsfor treatment of Alzheimer’s disease. PCT Int Appl 2005; WO2005113484.
135. Stachel SJ, Coburn CA, Steele TG, Jones KG, Loutzenhiser EF, Gregro AR, Rajapakse HA, Lai M-T,CrouthamelM-C, XuM, TugushevaK, Lineberger JE, Pietrak BL, Espeseth AS, Shi X-P, Chen-Dodson E,Holloway M-K, Munshi S, Simon AJ, Kuo L, Vacca JP. Structure-based design of potent and selectivecell-permeable inhibitors of human b-secretase (BACE1). J Med Chem 2004;47:6447–6450.
136. Coburn CA, Stachel SJ, Vacca JP. Preparation of phenylcarboxamide derivatives as b-secretase inhibitorsfor treatment of Alzheimer’s disease. PCT Int Appl 2004; WO2004043916.
137. Stachel SJ, Coburn CA, Steele TG, Crouthamel M-C, Pietrak BL, Lai M-T, Holloway MK, Munshi SK,Graham SL, Vacca JP. Conformationally biased P3 amide replacements of b-secretase inhibitors. BioorgMed Chem Lett 2006;16:641–644.
DEVELOPMENT OF NON-PEPTIDIC b-Secretase * 333
Medicinal Research Reviews DOI 10.1002/med

138. Coburn CA, Stachel SJ, Jones KJ, Steele TJ, Rush DM, Di Muzio J, Pietrak BL, Lai M-T, Huang Q,Lineberger J, Jin L, Munshi S, Holloway MK, Espeseth A, Simon A, Hazuda D, Graham SL, Vacca JP.BACE-1 inhibition by a series of c[CH2NH] reduced amide isosteres. Bioorg Med Chem Lett 2006;16:3635–3638.
139. Coburn CA, Stachel SJ, Vacca JP. (Merck & Co.) N-alkyl phenylacetamides beta-secretase inhibitors fortreatment of Alzheimer’s disease. PCT Int Appl 2005; WO2005004802.
140. Stachel SJ, Coburn CA, Sankaranarayanan S, Price EA, Pietrak BL, Huang Q, Lineberger J, Espeseth AS,Jin L, Ellis J, Holloway MK, Munshi S, Allison T, Hazuda D, Simon AJ, Graham SL, Vacca JP.Macrocyclic inhibitors of b-secretase: Functional activity in an animal model. J Med Chem 2006;49:6147–6150.
141. Coburn CA, Stachel SJ, Vacca JP. (Merck&Co.) Preparation of macrocyclic b-secretase inhibitors for thetreatment of Alzheimer’s disease. PCT Int Appl 2005; WO2005018545.
142. Holloway MK, McGaughey GB, Coburn CA, Stachel SJ, Jones KG, Stanton EL, Gregro AR, Lai M-T,Crouthamel M-C, Pietrak BL, Munshi SK. Evaluating scoring functions for docking and designingb-secretase inhibitors. Bioorg Med Chem Lett 2007;17:823–827.
143. Stauffer SR, Stanton MG, Gregro AG, Steinbeiser MA, Shaffer JR, Nantermet PG, Barrow JC, Rittle KE,Collusi D, Espeseth AS, LaiM-T, Pietrak BL, HollowayMK,McGaughey GB,Munshi SK, Hochman JH,Simon AJ, Selnick HG, Graham SL, Vacca JP. Discovery and SAR of isonicotinamide BACE-1 inhibitorsthat bindb-secretase in aN-terminal 10s-loop down conformation. BioorgMedChemLett 2007;17:1788–1792.
144. StantonMG, Stauffer SR, Gregro AR, Steinbeiser M, Nantermet P, Sankaranarayanan S, Price EA,WuG,Crouthamel M-C, Ellis J, Lai M-T, Espeseth AS, Shi X-P, Jin L, Colussi D, Pietrak B, Huang Q, Xu M,Simon AJ, Graham SL, Vacca JP, Selnick H. Discovery of isonicotinamide derived b-secretase inhibitors:in vivo reduction of b-amyloid. J Med Chem 2007;50:3431–3433.
145. Barrow JC,McGaugheyGB,Nantermet PG, RajapakseHA, SelnickHG, Stauffer SR, CoburnCA. (Merck& Co.) Preparation of 1,3,5-substituted phenyl derivatives containing five-membered heterocycles usefulas b-secretase inhibitors for treatment of Alzheimer’s disease. PCT Int Appl 2005; WO2005103020.
146. Barrow JC, McGaughey GB, Nantermet PG, Rajapakse HA, Selnick HG, Stauffer SR, Shaun R, Vacca JP,Stachel SJ, Coburn CA, Stanton MG. (Merck & Co.) Preparation of amino oxadiazolyl pyridinilsulfonamides as b-secretase inhibitors for treatment of Alzheimer’s disease. PCT Int Appl 2005;WO2005103043.
147. Coburn CA, Espeseth AS, Stachel SJ, Olsen DB, Hazuda D, HollowayMK. (Merck &Co.) Preparation of2-aminothiazole compounds as aspartyl protease inhibitors. PCT Int Appl 2005; WO200597767.
148. Barrow JC, Coburns CA, Nantermet PG, Selnick HG, Stachel SJ, Shawn J, Stanton MG, Stauffer SR,Zhuang L, Davis JR. (Merck & Co.) Preparation of phenylamides and pyridylamides as b-secretaseinhibitors. PCT Int Appl 2005; WO2005065195.
149. Coburn CA. (Merck&Co.) Pyrrolidin-3-yl compounds, synthesis, pharmaceutical compositions and theiruse as b-secretase inhibitors for treatment of Alzheimer’s disease. PCT Int Appl 2006; WO2006002004.
150. Barrow JC, Coburn CA, Egbertson MS, McGaughey GB, McWheter MA, Neilson LA, Selnick HG,Stauffer SR, Yang Z-Q, YangW, LuW, Fahr B, Rittle KE. (Merck&Co., and Sunesis Pharm.) Preparationof spiropiperidine compounds as b-secretase inhibitors for treatment ofAlzheimer’s disease. PCT IntAppl2006; WO2006044497.
151. Coburn CA, Holloway MK, Stachel SJ. (Merck & Co.) Preparation of aminopyridines as b-secretaseinhibitors for treatment of Alzheimer’s disease. PCT Int Appl 2006; WO2006060109.
152. Coburn CA, Nantermet PG, Rajapakse HA, Selniuck HG, Stauffer SR. (Merck & Co.) Aminomethylb-secretase inhibitors for treatment of Alzheimer’s disease. PCT Int Appl 2006; WO2006078576.
153. Coburn CA, Egberston MS, Graham SL, Mcgaughey GB, Stauffer SR, Yang W, LuW, Fahr B. (Merck &Co., and Sunesis Pharm.) Spiropiperidine as b-secretase inhibitors for treatment of Alzheimer’s disease.PCT Int Appl 2007; WO2007011810.
154. Coburn CA, Egberston MS, Graham SL, Mcgaughey GB, Stauffer SR, Rajapakse HA, Nantermet PG,Stachel SR, Yang W, Lu W, Fahr B. (Merck & Co. and Sunesis Pharm.) Spiropiperidine as b-secretaseinhibitors for treatment ofAlzheimer’s disease b-secretase inhibitors for treatment of Alzheimer’s disease.PCT Int Appl 2007; WO2007011833.
155. XueQ,Albrecht BK,AndersenDL,BartbergerM,Brown J, BrownR,Chaffee SC,ChengY,CroghanmM,Graceffa R, Harried S, Hitchcock S, Hungate R, Judd T, Kaller M, Kreiman C, La D, Lopez P, Masse CE,Monenschein H, Nguyen T, Nixwy T, Patel VF, Pennington L, Weiss M, Yang B, ZhongW. (Amgen Inc.)Preparation of 2-hydroxy-1,3-diaminoalkanes including spiro substituted chroman derivatives asb-secretase modulators and their use for treatment Alzheimer’s disease and related condition. PCT IntAppl 2007; WO2007061930.
334 * SILVESTRI
Medicinal Research Reviews DOI 10.1002/med

156. Albrecht BK,AndersenDL,BartbergerM,Brown J, BrownR,Chaffee SC,ChengY,CroghanM,GraceffaR, Harried S, Hitchcock S, Hungate R, Judd T, Kaller M, Kreiman C, La D, Lopez P, Masse CE,MonenscheinH, Nguyen T, Nixey T, Patel VF, Pennington L,WeissM,XueQ, Yang B, ZhongW. (AmgenInc.) Preparation of 2-hydroxy-1,3-diaminoalkanes including spiro substituted chroman derivatives asb-secretase modulators and their use for treatment Alzheimer’s disease and related condition. PCT IntAppl 2007; WO2007062007.
157. ZhongW,Hitchcock S, Albrecht BK, BartbergerM,Brown J, BrownR, Chaffee SC, ChengY, CroghanM,Graceffa R, Harried S, Hickman D, Horne D, Hungate R, Judd T, Kaller M, Kreiman C, La D, Lopez P,MasseCE,MonenscheinH,NguyenT,NixeyT, PatelVF, PenningtonL,WeissM,XueQ,YangB. (AmgenInc.) Preparation of 2-hydroxy-1,3-diaminoalkanes including spiro substituted chroman derivatives asb-secretase modulators and their use for treatment Alzheimer’s disease and related condition. PCT IntAppl 2007; WO2007061670.
158. Faller A, Milner PH, Waard JG. (Smithkline Beecham P.L.C.) Hydroxyethylene compounds with Asp-2inhibitory activity. PCT Int Appl 2003; WO2003045903.
159. Faller A, MacPherson DT,Milner PH, Stanway SJ, Trow LS. (Smithkline Beecham P.L.C.) Preparation ofbenzamide derivatives as inhibitors of Asp-2. PCT Int Appl 2003; WO2003045913.
160. Demont EH, Faller A,MacPhersonDT,Milner PH,Naylor AR,RedshawS, Stanway SJ,VeseyDR,WalterDR. (Glaxo Group Ltd) Preparation of hydroxyethylamine derivatives for the treatment of Alzheimer’sdisease. PCT Int Appl 2004; WO2004050619.
161. RedshawS,Demont EH,Walter DS. (GlaxoGroupLtd) Preparation of tricyclic indole hydroxyethylaminederivatives and their use in the treatment of Alzheimer’s disease. PCT Int Appl 2005; WO2005058615.
162. Demont EH, Redshaw S, Walter DS. (Glaxo Group Ltd) Preparation of 3-(1,1-dioxotetrahydro-1,2-thiazin-2-yl) or 3-(1,1-dioxoisothiazolidin-2-yl) benzamido compounds for treatment of Alzheimer’sdisease. PCT Int Appl 2004; WO2004111022.
163. Demont EH, Redshaw S,Walter DS. (Glaxo Group Ltd) Preparation of hydroxyethylamine derivatives forthe treatment of Alzheimer’s disease. PCT Int Appl 2004; WO2004080376.
164. Demont EH, Redshaw S, Walter DS. (Glaxo Group Ltd) Preparation of hydroxydiaminopropyl tricyclicindolecarboxamides for treatment of b-amiloid related disease. PCT Int Appl 2004; WO2004094430.
165. Demont EH, Redshaw S, Walter DS. (Glaxo Group Ltd) Preparation of N,N 0-substituted-1,3-diamino-2-oxopropane derivatives as Asp2 inhibitors for use against diseases characterized by elevated b-amyloiddeposits, particularly Alzheimer’s disease. PCT Int Appl 2005; WO2005113525.
166. Demont EH, Redshaw S, Walter DS. (Glaxo Group Ltd) Tricyclic indole derivatives for use in thetreatment of Alzheimer’s disease. PCT Int Appl 2006; WO2006040148.
167. Demont EH, Redshaw S, Walter DS. (Glaxo Group Ltd) Heterocyclic ketone compounds for treatingAlzheimer’s disease. PCT Int Appl 2006; WO2006040149.
168. Demont EH, RedshawS,Walter DS. (GlaxoGroup Ltd) Novel hydroxyethylamine and ketone compoundshaving Asp2 inhibitory activity. PCT Int Appl 2006; WO2006103088.
169. Decicco CP, Tebben AJ, Thompson LA, Combs AP. (Bristol-Myers Squibb Co.) Preparation of novela-amino-g-lactams as b-secretase inhibitors. PCT Int Appl 2004; WO2004013098.
170. Olson RE, Maduskuie TP, Thompson LA. (Bristol-Myers Squibb Co.) Preparation of (succinylamino)a-zepinones as inhibitors of Ab protein. US Pat Appl 2004; US6794381.
171. Olson RE. (Bristol-Myers Squibb Co.) Preparation of succinoylaminobenzodiazepines as inhibitors of Abprotein production for treating neurological disorders. US Pat Appl 2006; US7053084.
172. Thompson ILA,BoyKM,Shi JM,Macor JE. (Bristol-Myers SquibbCo.) Preparation of isophthalates asb-secretase inhibitors . PCT Int Appl 2006; WO2006099352.
173. Thompson LA, Shi J, Zusi FC, DeeMF,Macor JE. (Bristol-Myers Squibb Co.) Preparation of indoleaceticacid acyl guanidines as b-secretase (BACE) inhibitors. US Pat Appl 2007; US2007049589.
174. Gerritz S, Zhai W, Shi S, Zhu S, Good AC, Thompson LA III. (Bristol-Myers Squibb Co.) Preparation ofisoxazolylcarbonyl- and isothiazolyl-carbonylguanidines as b-secretase (BACE). PCT Int Appl 2007;WO2007002214.
175. Malamas MS, Erdei JJ, Gunawan IS, Barnes KD, Johnson MR, Hui Y. (Wyeth, John & Br.)Diphenylimidazopyrimidine and imidazole amine as inhibitors of b-secretase. US Pat Appl 2005;US20050282826.
176. Malamas MS, Erdei JJ, Nawan IS, Npwak P, Harrison BL. (Wyeth, John & Br.) Amino-imidazolones forthe inhibition of b-secretase. PCT Int Appl 2006; WO2006076284.
177. Malamas MS, Erdei JJ, Gunawan IS, Zhou P, Yan Y, Quagliato DA. (Wyeth, John & Br.) Amino5,5-diphenylimidazolone derivatives for the inhibition of b-secretase. PCT Int Appl 2006;WO2006009653.
DEVELOPMENT OF NON-PEPTIDIC b-Secretase * 335
Medicinal Research Reviews DOI 10.1002/med

178. Malamas MS, Fobare WF, Solvibile WR, Lovering FE, Condon JS, Robichaud AJ. (Wyeth, John & Br.)Amino-pyridines as inhibitors of b-secretase. PCT Int Appl 2006; WO2006083760.
179. Cole DC, Manas ES, Stock JR, Condon JS, Jennings LD, Aulabaugh A, Chopra R, Cowling R, EllingboeJW, Fan KY, Harrison BL, HuY, Jacobsen S, Jin G, Lin L, Lovering FE,MalamasMS, StahlML, Strand J,SukhdeoMN, Svenson K, Turner MJ,Wagner E,Wu J, Zhou P, Bard J. Acylguanidines as small-moleculeb-Secretase inhibitors. J Med Chem 2006;49:6158–6161.
180. Cole DC, Manas ES, Jennigs LD, Lovering FE, Stock JR, MooreWJ, Ellingboe JW, Condon JS, SukhdeoMN, Zhou P, Wu J, Morris KM. (Wyeth, John & Br.) Preparation of azolylacylguanidines as b-secretaseinhibitors. PCT Int Appl 2006; WO2006088711.
181. FobareWF, SolvibileWR. (Wyeth, John&Br.) Substituted thienyl and furyl acylguanidines as b-secretasemodulators. PCT Int Appl 2006; WO2006088694.
182. BaihuaHU. (Wyeth, John&Br.) Terphenyl acylguanidines as b-secretasemodulators. PCT Int Appl 2006;WO2006088705.
183. Albert JS, Andrisk D, Arnold J, BrownD, Callaghan O, Campbell J, Carr RAE, Chessari G, CongreveMS,Edwards P, Empfield JR, Frederickson M, Koether GM, Krumrine J, Mauger R, Murray CW, Patel S,Sylvester M, Throner S. (AstraZeneca AB and Astex Ther.) Preparation of substituted 2-aminopyrimidin-4-ones for treating or preventing Ab-related pathologies. PCT Int Appl 2006; WO2006041404 and 2006;WO2006041405.
184. Albert J, Edwards P, Empfield J. (AstraZeneca AB and Astex Ther.) Preparation of substituted2-aminopyrimidin-4-ones for treating or preventing Ab-related pathologies. PCT Int Appl 2007;WO2007058582.
185. Albert J, AndisikD, Edwards P, SylvesterM. (AstraZenecaAB andAstexTher.) Preparation of substituted2-aminopyrimidin-4-ones for treating or preventing Ab-related pathologies. PCT Int Appl 2007;WO2007058580.
186. Albert J, Chessari G, Edwards P. (AstraZeneca AB and Astex Ther.) Preparation of substituted 2-aminopyrimidines for treating or preventingAb-related pathologies. PCT IntAppl 2007;WO2007058581.
187. Albert J, Arnold J, Chessari G, Congreve MS, Edwards P, Murrai C, Patel S. (AstraZeneca AB and AstexTher.) Preparation of 2-aminoimidazole-4-ones for treatment of cognitive impairment, Alzheimer’sdisease, neurodegeneration and dementia. PCT Int Appl 2007; WO2007058601.
188. Berg S, Burrows J, Chessari G, Congreve MS, Hedstroem J, Hellberg S, Hoegdin K, Kihlstroem J,Kolmodin K, Lindstroem J, Murray C, Patel S. (AstraZeneca AB) New 2-amino-3,5-dihydro-4H-imidazol-4-one derivatives and their use as BACE inhibitors for treatment cognitive impairment,Alzheimer’s disease, neurodegeneration and dementia. PCT Int Appl 2007; WO2007058602.
189. Albert J, Chessari G, Congreve MS, Edwards P, Murray C, Patel S, Sylvester M. (AstraZeneca AB andAstex Ther.) Preparation of dihydroisoquinolin-1-amines for treating or preventing Ab-relatedpathologies, such as cognitive impairment, Alzheimer’s disease, neurodegeneration and dementia. PCTInt Appl 2007; WO2007058583.
190. Baxter E, Boyd R, Coats S, Jordan A, Reitz A, Reynolds CH, Scott M, SchulzM, DeWinter HLJ. (JanssenPharm.) Novel 2-aminoquinazoline derivatives, their preparation and use as inhibitors of b-secretase fortreating Alzheimer’s disease and related disorders. PCT Int Appl 2006; WO2006017844.
191. Baxter E, Bischoff FP, BraekenM, Coats S, Huang Y, Jordan A, Luo C,MerckenMH, Reynolds CH, RossTM, Tounge BA, Schulz M, De Winte HLJ, Pieters SMA, Reitz AB. (Janssen Pharm.) Novel 2 -aminoquinazoline derivatives, their preparation and use as inhibitors of b-secretase for treatingAlzheimer’s disease and related disorders. PCT Int Appl 2006; WO2006017836.
192. Bishoff FP, Bracken M, Pieters SMA, Mercken MH, De Winter HLJ, Berthelot DJ-C. (Janssen Pharm.)Novel 2 -aminoquinazoline derivatives, their preparation and use as inhibitors of b-secretase for treatingAlzheimer’s disease and related disorders. PCT Int Appl 2006; WO2006024932.
193. Reitz AB, Luo C, Huang Y, Ross TM, Baxter EE, Tounge BA, Parker MH, Strobel ED, Reynolds CH.(Janssen Pharm.) Preparation of 2-amino-3,4-dihydropyrido[3,4-d]pyrimidines as inhibitors ofb-secretase (BACE). PCT Int Appl 2007; WO2007050612.
194. Baxter EW, Conway KA, Kennis L, Bischoff F, Mercken MH, DeWinter HL, Reynolds CH, Tounge BA,Luo C, Scott MK, Huang Y, Braeken M, Pieters SMA, Berthelot DJC, Masure S, Bruinzeel WD,Jordan AD, Parker MH, Boyd RE, Qu J, Alexander RS, Brenneman DE, Reitz B. (Johnson & JohnsonPharm.) 2-Amino-3,4-dihydroquinazolines as inhibitors of BACE-1 (b-Site APP cleaving enzyme): use ofstructure based design to convert a micromolar hit into a nanomolar lead. J Med Chem 2007;50:4261–4264.
195. BuenoMelendoAB,Chen S-H, Erickson JA,Gonzalez-GarciaMR,GuoD,Marcos Llorente A,McCarthyJR, Shepherd TA, Sheehan SM, Yip YYM. (Eli Lilly & Co.) Preparation of amides as BACE inhibitors fortreating Alzheimer’s. PCT Int Appl 2005; WO2005108391.
336 * SILVESTRI
Medicinal Research Reviews DOI 10.1002/med

196. Dally RD, Shepherd TAB, DavidM, Rojo GarciaMI. (Eli Lilly &Co.) Preparation of acylated 2-amino-1-(pyrrolidin-2-yl)ethanols and derivatives as BACE inhibitors for treatingAlzheimer’s. PCT IntAppl 2005;WO2005108358.
197. DurhamTB, Hahn PJ, Kohn TJ,McCarthy JR, Broughton HB, Dally RD, Gonzalez-GarciaMR, Henry KJJr, Shepherd TA, Erickson JA, Ana B. (Eli Lilly & Co.) Preparation of acylated 2-amino-1-(morpholin-3-yl)ethanols and derivatives as BACE inhibitors for treating Alzheimer’s. PCT Int Appl 2006;WO2006034093.
198. Auberson Y, Betschart C, Flohr S, Glatthar R, Simic O, Tintelnot-BlomleyM, Troxler TJ, VangrevelingheE, Veenstra SJ. (Novartis AG and Novartis Pharma) A preparation of dibenz[b,f]oxepincarboxamidederivatives, useful for the treatment of neurological and vascular disorders related to b-amyloid generationand aggregation. PCT Int Appl 2005; WO2005014517.
199. Auberson Y, Glatthar R, Salter R, Simic O, Tintelnot-Blomley M. (Novartis AG and Norvatis Pharma)Preparation of substituted spirocyclic lactams as inhibitors of proteinase BACE1. PCT Int Appl 2005;WO2005035535.
200. Auberson Y, Betschart C, Glatthar R, LaumenK,Machauer R, Tintelnot-BlomleyM, Troxler TJ, VeenstraSJ. Novartis AG and Novartis Pharma) Preparation of macrocyclic lactams for treatment of neurologicalor vascular disorders related to b-amyloid generation and/or aggregation. PCT Int Appl 2005;WO2005049585.
201. Lerchner A,Machauer R, Simic O, Tintelnot-BlomleyM. (Novartis AG andNovartis Pharma) Preparationof macrocyclic compounds as BACE inhibitors for treating vascular and neurological disorders related tob-amyloid generation. PCT Int Appl 2006; WO2006074940.
202. Betschart C, KollerM, LaumenK, Lerchner A,Machauer R,McCarthy C, Tintelnot-BlomleyM, VeenstraSJ. (Novartis AG andNovartis Pharma)Macrocyclic compounds useful as BACE inhibitors. PCT Int Appl2007; WO2007077004.
203. Murray CW, Callaghan O, Chessari C, Cleasby A, Congreve M, Frederickson M, Hartshorn MJ,McMenamin R, Patel S, Wallis N. Application of fragment screening by X-ray crystallography to b-secretase. J Med Chem 2007;50:1116–1123.
204. Congreve M, Aharony D, Albert A, Callaghan O, Campbell J, Carr RAE, Chessari G, Cowan S, EdwardsPD, FredericksonM,McMenaminR,Murray CW, Patel S,Wallis N. Application of fragment screening byX-ray crystallography to the discovery of aminopyridines as inhibitors of b-secretase. J Med Chem2007;50:1124–1132.
205. YangW, LuW, LuY, ZhongM, Sun J, Thomas AE,Wilkinson JM, Fucini RV, LamM, RandalM, Shi X-P,Jacobs JW, McDowell RS, Gordon EM, Ballinger MD. (Sunesis Pharm. Inc.) Aminoethylenes: Atetrahedral intermediate isostere yielding potent inhibitors of the aspartyl protease BACE-1. J Med Chem2006;49:839–842.
A P P E N D I X
Bristol-Myers Squibb, PO Box 4000, Route 206 and Provinceline road, Princeton, NJ 08453-4000
(US)
CoMentis, Inc., 280 Utah Avenue, Suite 275, South San Francisco, CA 94080 (US)
Eisai R&D Management Co., Ltd, 6-10, Koishikawa 4-chome, Bunkyo-ku, Tokyo, 1128088 (JP)
Elan Pharmaceuticals, Inc., 800 Gateway Boulevard, So. San Francisco, CA 94080 (US)
Eli Lilly and Company, Lilly Corporate Center, Indianapolis, Indiana 46285 (US)
First Horizon Pharmaceutical Corporation, 6195 Shiloh Road, Alpharetta, Georgia 30005 (US)
Glaxo Group Limited, Glaxo Wellcome House, Berkeley Avenue, Greenford, Middlesex UB6 0NN
(UK)
Johnson & Johnson Pharmaceutical Research and DeVelopment, LLC, Spring House, Pennsylvania
19477 (US), and Turnhoutseweg 30, 2340 Beerse, Belgium
Memory Pharmaceuticals Corporation, 100 Philips Parkway, Montvale, New Jersey 07645 (US)
Merck & Co., Inc., 126 East Lincoln Avenue, Rahway, NJ 07065-0907 (US)
Neurologic, Inc., 15010 Broschart Road, Suite 200, Rockville, MD 20850 (US)
Roche Laboratories, 340 Kingsland Street, Nutley, New Jersey, 07110 (US)
Sunesis Pharmaceuticals, Incorporated, 341Oyster Point Boulevard, South San Francisco, California
94080 (US)
Vertex Pharmaceuticals Incorporated, 130 Waverly Street, Cambridge, MA 02139-4242 (US)
DEVELOPMENT OF NON-PEPTIDIC b-Secretase * 337
Medicinal Research Reviews DOI 10.1002/med

Romano Silvestri is an Associate Professor in Medicinal Chemistry at the Dipartimento di Chimica e
Tecnologie del Farmaco (Department of Medicinal Chemistry and Technologies) of the Faculty of Pharmacy at
the Sapienza University of Rome (Italy). He was born in 1959 in Revo (Trento, Italy). He studied medicinal
chemistry (1983, Degree in Pharmacy) at the Faculty of Pharmacy in Rome. From 1986 to 1989 he worked on
his PhD inMedicinal Chemistry. From 1994 to1997 he was an Assistant Professor. He is the author of more than
130 publications and 6 patents in the field of antiviral agents (discovery of novel classes of HIV NNRTI classes:
IAS, PAS, PBTD, DAMNI), antifungal agents (analogues of bifonazole (Ph.D. thesis), ketoconazole and
miconazole), antitumor agents (antimitotic agents, ellipticine andCC-1065 analogues), antiinflammatory drugs
(analogues of lonazolac, tolmetin, lefetamine, and sulindac), CNS agents (antidepressant drugs: mianserine
analogues and MAO inhibitors), and heterocyclic chemistry (pyrrole, indole, tri- and tetracyclic systems).
338 * SILVESTRI
Medicinal Research Reviews DOI 10.1002/med





























