Embed Size (px)
Citation preview

Welcome to
The Otesaga Hotel and Resort
60 Lake St.
Cooperstown, NY
October 15-18, 2018
Supported by NIH/NIAID
Grant Funding
21st Annual Upstate New York
Immunology Conference
American Association of Immunologists
Young investigator Awards
ThermoFisher
Trainee Travel Awards
Major Corporate Sponsors
BD Biosciences
BioLegend, Inc.

2.

3.
Conference and Venue ............................................................................... 5
Schedule of Events .................................................................................... 6
Platinum Corporate Sponsors
BD Biosciences ..................................................................................... 14
BioLegend, Inc. .................................................................................... 16
Silver-Plus Corporate Sponsors
Krackeler Scientific .............................................................................. 18
ThermoFisher Scientific .............................................................................. 20
Silver Corporate Sponsors .......................................................................... 22
Agilent Technologies ............................................................................ 23
Leinco Technologies ............................................................................ 24
Lonza ................................................................................................. 25
MilliporeSigma ..................................................................................... 26
Shenandoah Biotechnology .................................................................. 27
StemCell Technologies ......................................................................... 28
Taconic Bioscience .............................................................................. 29
American Association of Immunologists ....................................................... 30
Institutional Financial Supporters ................................................................ 31
NYIC Scientific Advisory Board .................................................................... 32
Grant Support ........................................................................................... 34
Keynote Speaker—Sponsored by BD Biosciences
Thomas A Wynn, Ph.D. ......................................................................... 35
Symposium I: B-cells and Humoral Immunity ............................................... 36
Symposium II: Immunology at the Host-Pathogen Interface .......................... 40
Corporate Presentation: BioLegend, Inc. ...................................................... 44
Table of Contents

4.
Oral Poster Presentations
Session A: Lymphocyte Biology .............................................................. 45
Session B: Innate Immunity .................................................................. 51
Session C: Tumor Biology ...................................................................... 57
Session D: Infection and Vaccines .......................................................... 63
Workshop I - Dr. Jeremy Boss
“Getting and Negotiating an Academic Faculty Position” ..................... 69
Corporate Presentation: BD Biosciences ....................................................... 70
Symposium III: Impact of Microenvironment on the Immune Response ......... 71
Workshop II - Dr. Thomas Wynn
“The Merits (and Perils) of Transitioning from
an Academic to Industry Career” ......................................................................... 75
Poster Listing ............................................................................................ 76
Poster Abstracts ........................................................................................ 77
Symposium IV: Immunoregulation and Homeostasis .................................... 105
Keynote Speaker—Sponsored by BioLegend
Jeremy M. Boss, Ph.D. .......................................................................... 109
Author Index ............................................................................................. 110
Attendee Contact Information ..................................................................... 117

5.
UPSTATE NEW YORK IMM UNOLOGY CONFERENCE (NYIC)
We’ve come a long way from Garnet Hill! This meeting started in 1997 as a small retreat to facili-
tate interactions among young scientists, institutions, and renowned experts in the field of Immunology.
In just a few short years, the number of attendees grew and a larger venue was needed to meet the fu-
ture needs of the Conference.
We are happy to announce the American Association of Immunologists (AAI) is once
again providing ten (10) Young Investigator Awards. Ther-
moFisher Scientific is also proving ten (10) Trainee Travel
Awards. All award winners will give Oral Poster Presenta-
tions. There will also be two Workshops by Keynote speakers, Dr. Thomas A. Wynn
(Pfizer) and Dr. Jeremy M. Boss (Emory University).
We are also excited for the change of venue. We know there are many participants who drive a
long distance to meet with colleagues and friends when attending the NYIC meeting. In order to provide
a more central location for everyone, we have moved this year’s meeting to The Otesaga Resort Hotel
(see below) in Cooperstown, NY. There’s even a couple of new activities planned, or you can take the
planned time to explore Cooperstown on your own. It’s just a short stroll from the hotel. Weather per-
mitting, you can take advantage of the fire bar, and engage fellow researchers in informal discussions.
Trainees will also have an opportunity to win an iPad during one of two drawings. You must be present
at the drawing to win!
While all these elements lend to the atmosphere, one simple principle goal of this Conference re-
mains. To provide an opportunity for young and senior scientists to gather in a setting that is diverse
enough to meet the needs of all attendees while remaining small enough to allow for personal interac-
tions. While always challenging, it is the goal of the NYIC Scientific Advisory Board and the NYIC Confer-
ence Organizers to give graduate students and postdoctoral fellows the opportunity to present their re-
search and engage in conversations that will stimulate further discussions, collaborations, and interest in
pursuing a new or different way of looking at their research.
We hope you share our enthusiasm and enjoy your time with us!
THE OTESAGA
Since its opening in 1909, The Otesaga
Resort Hotel has been a destination people
couldn’t wait to see and experience. The Clark
family, who owns the hotel to this day, commis-
sioned architect Percy Griffin to design The
Otesaga. The 400 windows that wrap around
the Otesaga provide stunning views of Lake Otsego. An architectural feature common today that was
considered exciting when built is the width of the driveway. It was uncommon in those days for drive-
ways to be wide enough for two cars (or carriages) to easily pass one another. The hotel has continued
to maintain its original aura of charm and gracious hospitality, while also growing and changing with the
times. The Otesaga is now an AAA Four-Diamond hotel and belongs to the Historic Hotels of America.

6.
Upstate New York Immunology Conference
Schedule of Events
Monday, October 15th
3:00-5:00 p.m. Hotel Check-in (Main Hotel Lobby) (Main Level) and Conference Registration (Oak Room) 5:00-6:00 p.m. Welcome Reception (Lower Level) Abner Double Day/Fire Bar 6:15-7:45 p.m. Plated Dinner (Main Level) Glimmerglass
7:30 p.m. Welcome and Introductions
Keynote Presentation
Sponsored by BD Biosciences Introduction: Beth Wohlfert
Thomas A. Wynn, Ph.D. Vice President Discovery Inflammation & Immunology Pfizer
“The Role of Inflammation in Tissue Regeneration and Fibrosis” 8:30 p.m. Hawkeye Bar and Grille/Fire Bar (Lower Level)

7.
Tuesday, October 16th 7:00-8:15 a.m. Breakfast Buffet at Leisure (Glimmerglass)
8:25-8:30 a.m. Morning Announcements (Ballroom)
8:30-10:00 a.m. Symposium I: B-cells and Humoral Immunity (Ballroom) Chair: Dr. Jeremy Boss 8:30-9:00 Gary Winslow, Ph.D. (SUNY Upstate) Development, Differentiation, and Maintenance of T-bet+ IgM Memory C Cells” 9:00-9:30 Sean Diehl, Ph.D. (University of Vermont) “Human B Cell Responses to Dengue and Zika Viruses” 9:30-10:00 Adam Matson, M.D. (University of Connecticut) “Transplacental Allergic Sensitization and Immune Development” 10:00-10:15 a.m. Break (Iroquois Room) 10:15-11:45 p.m. Symposium II: Immunity at the Host-Pathogen Interface (Ballroom) Chair: Dr. Thomas Wynn 10:15-10:45 Brent Berwin, Ph.D. (Dartmouth College) “Host Interactions with Bacterial Pathogenesis: Heads I Win, Tails You Lose” 10:45-11:15 Bibhuti Mishra, Ph.D. (Albany Medical College)
“Inflammation in Tuberculosis: Good, Bad, and the Ugly”
11:15-11:45 Troy Sutton, Ph.D. (Penn State University) “Sequential Airborne Transmission of Pandemic, Seasonal, and Emerging Influenza Viruses—A Potential Strategy to Evaluate Vaccine Efficacy and Non-sterilizing Immunity”

8.
12:00-1:15 p.m. Lunch Buffet (Glimmerglass)
12:45-1:15 p.m. Platinum Corporate Sponsor—BioLegend, Inc. (Glimmerglass) Nathan Lucas 1:30-2:45 p.m. Oral Poster Presentations (Lower Level—Kingfisher) Chairs: Jorg Fritz and Qi Yang
Session A: Lymphocyte Biology
1:30-1:45 Kristel Yee Mon, B.S. (Cornell University)
“MicroRNA-29 Alters the CD8+ T cell Response to Infection In an Age Dependent Manner” (#2)
1:45-2:00 Barbara C. Mindt, M.S. (McGill University)
“Essential Role of c-Rel in IL-33-mediated Activation of Group 2 Innate Lymphoid Cells” (#41) 2:00-2:15 Shanti D’Souza, M.S. (Albany Medical College)
“Commensal Microbes Induce Serum IgA Responses That Protect Against Polymicrobial Sepsis” (#33) 2:15-2:30 Shivana M. Lightman, B.S. (Roswell Park)
“A New Role of Idoleamine 2,3-dioxygenase (IDO): Supporting the Survival of Bone Marrow Resident Long Lived Plasma Cells” (#35) 2:30-2:45 Catherine G. Burke, M.S. (University of Rochester)❖
“Developmental Activation of the Aryl Hydrocarbon Receptor Reduces CD4+ T cell Responses by Altering DNA Methylation Patterns” (#35)
1:30-2:45 p.m. Oral Poster Presentations (Lower Level—Council Rock) Chairs: Kate MacNamara and Timothy LaRocca
Session B: Inflammation & Innate Immunity 1:30-1:45 Allison N. Seyfried, B.S. (Albany Medical College)
“CCR5 Signaling Reduces Platelet-biased Hematopoietic Stem Cells and Drives Thrombocytopenia in a Model of Severe ”Aplastic Anemia” (#25)
1:45-2:00 Abhinit Nagar, M.S. (Albany Medical College)❖
“NLRP3 Inflammasome Activity and Speck Formation:

9.
Mutually Exclusive Outcomes of NLRP3 Activation” (#1)
2:00-2:15 Stacey Ceron, M.S. (Dartmouth College)❖
“The STING Agonist 5,6 Dimethylxanthenone-4-acetic acid (DMXAA) Stimulates an Antiviral State and Protects Mice Against Herpes Simplex Virus-(#30)
2:15-2:30 Janelle Veazey, M.S. (University of Rochester)❖
“Multifaceted Coordination of Airway Epithelial Barrier Integrity and Inflammation by Protein Kinase D” (#42) 2:30-2:45 Oyebola Oyesola, M.S. (Cornell University)
“The Prostaglandin D2 Receptor CRTH2 Suppresses Epithelial Cell Responses During Intestinal Helminth Infection” (#9)
2:45-3:00 p.m. Beverage Break (Kingfisher Foyer) 3:00-4:15 p.m. Oral Poster Presentations (Lower Level Kingfisher) Chairs: Scott Gerber & Yasmin Thanavala
Session C: Tumor Biology
3:00-3:15 Guanxi Qiao, M.S. (Roswell Park)
“Adrenergic Signaling Impairs Activation of CD*+ T-cells by Blocking Metabolic Reprogramming” (#26)
3:15-3:30 Bradley N. Mills, Ph.D. (University of Rochester)
“Stereotactic Body Radiation and Interleukin 12 Combination Therapy Eradicates Pancreatic Tumors by Composite Repolarization of the Tumor Microenvironment” (#33) 3:30-3:45 Riddhi Falk-Mahapatra, M.S. (Roswell Park)❖
“Treatment Induced PGE2 Plays an Unexpected Beneficial Role in the Generation of Anti-tumor Immunity“ (#21) 3:45-4:00 G. Aaron Holling, B.S. (Roswell Park)❖
“The CD28-Ars2 Axis Primes T-cells for Expansion” (#19) 4:00-4:15 Minhui Chen, M.S. (Roswell Park)❖
“Impact of Adrenergic Stress on the “Abscopal Effect” Following Radiation Therapy and on the Anti-immune Response” (#38)

10.
3:00-4:15 p.m. Oral Poster Presentations (Lower Level Kingfisher) Chairs: Brent Berwin & Michael Robek
Session D: Infection & Vaccines
3:00-3:15 Sally Demirdjian, M.S. (Dartmouth College)
“PIP3 Induces Phagocytosis of Non-motile Pseudomonas aeruginosa” (#10)
3:15-3:30 Marija Landekic, M.S. (McGill University)
“The Role of the CARD9/GM-CSF Axis in Immunity to Candida albicans” (#34) 3:30-3:45 Amit K. Singh, Ph.D. (Albany Medical College)❖
“Single Dose Oral Administration of Yersinia pseudotuberculosis vaccine Induces Specific Immunity Against Pneumonic Y. pestis Infection” (#30) 3:45-4:00 Anthony Marchese, B.S. (Albany Medical College)❖
“Understanding Innate Immune Responses to a Replicating Virus-based Vaccine Platform” (#18) 4:00-4:15 Americo Lopez-Yglesias, Ph.D. (University of Rochester)❖ “T-bet-dependent ILC1-derived IFN-γ is Required for Sustaining
Inflammatory DCs During T. gondii Infection” (#27)
AAI Young Investigator Award Winner
❖ThermoFisher Trainee Travel Award Winner
4:30 - 5:30 p.m. Workshop I - Dr. Jeremy Boss (Ballroom) “Getting and Negotiating an Academic Faculty Position” 5:45-6:30 p.m. SAB Meeting (Main Level—Lake Room) 6:30-7:30 p.m. Plated Dinner (Glimmerglass)
7:30-8:00 p.m. Platinum Corporate Sponsor-BD Biosciences (Glimmerglass) “TBD”

11.
8:00-8:30 p.m. American Association of Immunologists (Glimmerglass) Young Investigator Awards and ThermoFisher Travel Awards (Award Presentation & Photos) 8:30 p.m. Hawkeye Bar and Grille/Fire Bar (Lower Level) Wednesday, October 17th 7:00-8:15 a.m. Breakfast at Leisure (Glimmerglass)
8:25-8:30 a.m. Morning Announcements (Ballroom)
8:30-10:00 a.m. Symposium III: Impact of Microenvironment on the (Ballroom) Immune Response Chair: Eyal Amiel 8:30-9:00 David C. Linehan, M.D. (University of Rochester) “Targeting Immunosuppressive Myeloid Cells to Treat Pancreatic Cancer” 9:00-9:30 Yasmin Thanavala, Ph.D. (Roswell Park Cancer Institute)
“β-AR Signaling Suppresses Immunity to Infection and
Vaccination by Blocking Immunometabolism” 9:30-10:00 Elia Tait Wojno, Ph.D. (Cornell University) “Comparative Immunology to Dissect Innate Immune Responses During Type 2 Inflammation” 10:00-10:15 a.m. Beverage Break (Iroquois Room)
10:15-11:15 a.m. Workshop II - Dr. Thomas Wynn (Ballroom) “The Merits (and Perils) of Transitioning from an Academic to an Industry Career”

12.
11:30-12:30 p.m. Lunch Buffet (Glimmerglass) 1:00-4:00 p.m. Pre-arranged Activities or Free Time (Meet at 12:45 p.m. in front of Hotel Entrance for Trolley) 4:00-4:30 p.m. Display Posters (Ballroom and Iroquois) 4:30-6:00 p.m. Vendor/Poster Mixer (Passport Stamps) Poster Viewing and Questions (Odd Numbers) 6:00-7:30 p.m. Plated Dinner (Glimmerglass)
7:30-9:00 p.m. Vendor/Poster Mixer (Passport Stamps)
Poster Viewing and Questions (Even Numbers) iPad drawing during this event. Must be present to win. 9:00-9:30 p.m. Remove Posters (Posters left behind will be discarded) (Ballroom and Iroquois)
9:30 p.m. Hawkeye Bar and Grill/Fire Bar (Lower Level) Thursday, October 18th 7:00-8:15 a.m. Breakfast at Leisure (Glimmerglass)
8:25-8:30 a.m. Morning Announcements (Ballroom)
8:30-10:00 a.m. Symposium IV: Immunoregulation and Homeostasis (Nirvana) Chair: Steve Szczepanek 8:30-9:00 Timothy LaRocca, Ph.D. (Wadsworth/ACPHS) “Hyperglycemia Potentiates a Shift From Apoptosis to RIP1-dependent Necroptosis”

13.
9:00-9:30 Jorg Fritz, Ph.D. (McGill University) “Group 2 Innate Lymphoid Cells Regulate Pulmonary Immunity and Tissue Homeostasis” 9:30-10:00 Elsa Bou Ghanem, Ph.D. (University at Buffalo) “Extracellular Adenosine Shapes PMN Phenotype and Their Ability to Kill Streptococcus pneumoniae by Blunting IL-10 Production” 10:00-10:15 Beverage Break & Check-Out (Iroquois & Hotel Lobby)
10:15-11:15 p.m. Keynote Presentation – Sponsored by BioLegend (Ballroom) Introduction: Dr. Michael Robek Jeremy M. Boss, Ph.D. Emory Chair in Basic Sciences Research Professor and Chair, Dept. of Microbiology & Immunology Emory University “Epigenetic Regulation of B-Cell Differentiation” 11:15-11:30 a.m. Closing Remarks (Ballroom) iPad drawing during this event. Must be present to win. 11:45-12:45 p.m. Lunch Buffet (Glimmerglass)
Departure
Please join us next year for the
22nd Annual Upstate New York Immunology Conference
October 28-31, 2019
(Monday-Thursday)
The Otesaga Resort Hotel, Cooperstown, NY

14.
bdbiosciences.comBD Life Sciences, San Jose, CA, 95131, USA
23-20468-00Class 1 Laser Product.For Research Use Only. Not for use in diagnostic or therapeutic procedures.© 2018 BD. BD, the BD Logo and BD FACSMelody are trademarks of Becton, Dickinson and Company.
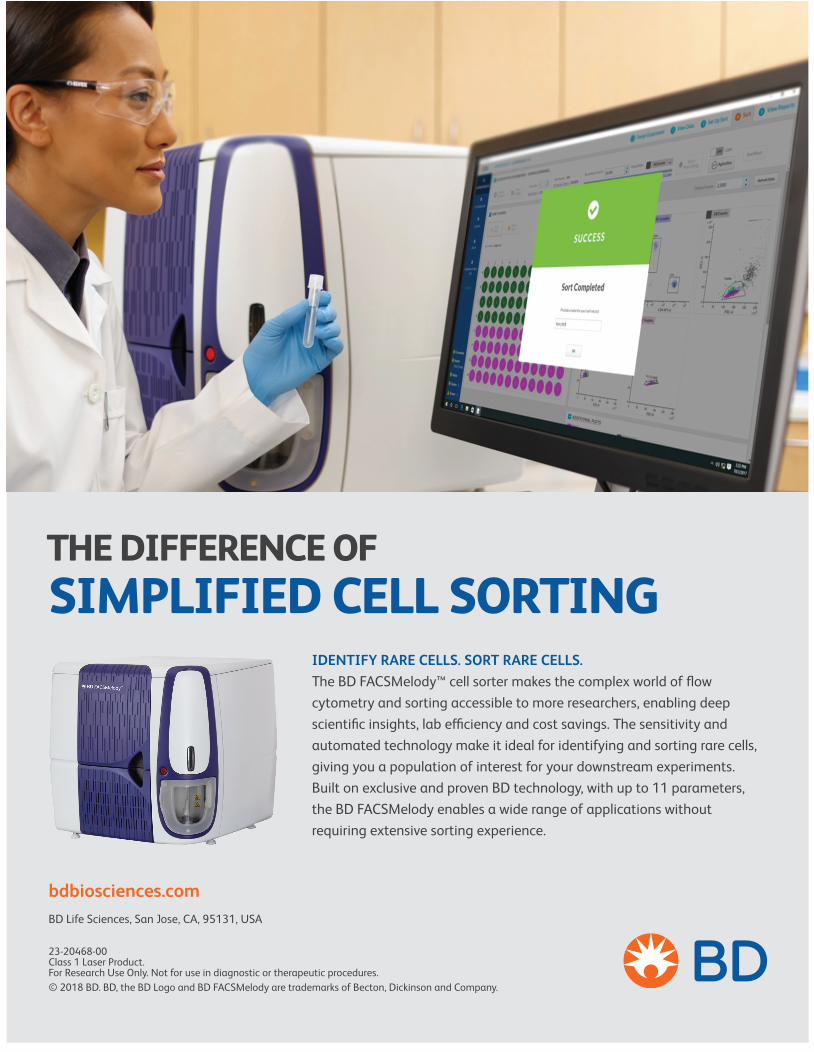
IDENTIFY RARE CELLS. SORT RARE CELLS. The BD FACSMelody™ cell sorter makes the complex world of flow cytometry and sorting accessible to more researchers, enabling deep scientific insights, lab efficiency and cost savings. The sensitivity and automated technology make it ideal for identifying and sorting rare cells, giving you a population of interest for your downstream experiments. Built on exclusive and proven BD technology, with up to 11 parameters, the BD FACSMelody enables a wide range of applications without requiring extensive sorting experience.
THE DIFFERENCE OFSIMPLIFIED CELL SORTING

16.
World-Class Quality | Superior Customer Support | Outstanding Value
Toll-Free Tel: (US & Canada): 1.877.BIOLEGEND (246.5343)Tel: 858.768.5800biolegend.com
08-0074-12
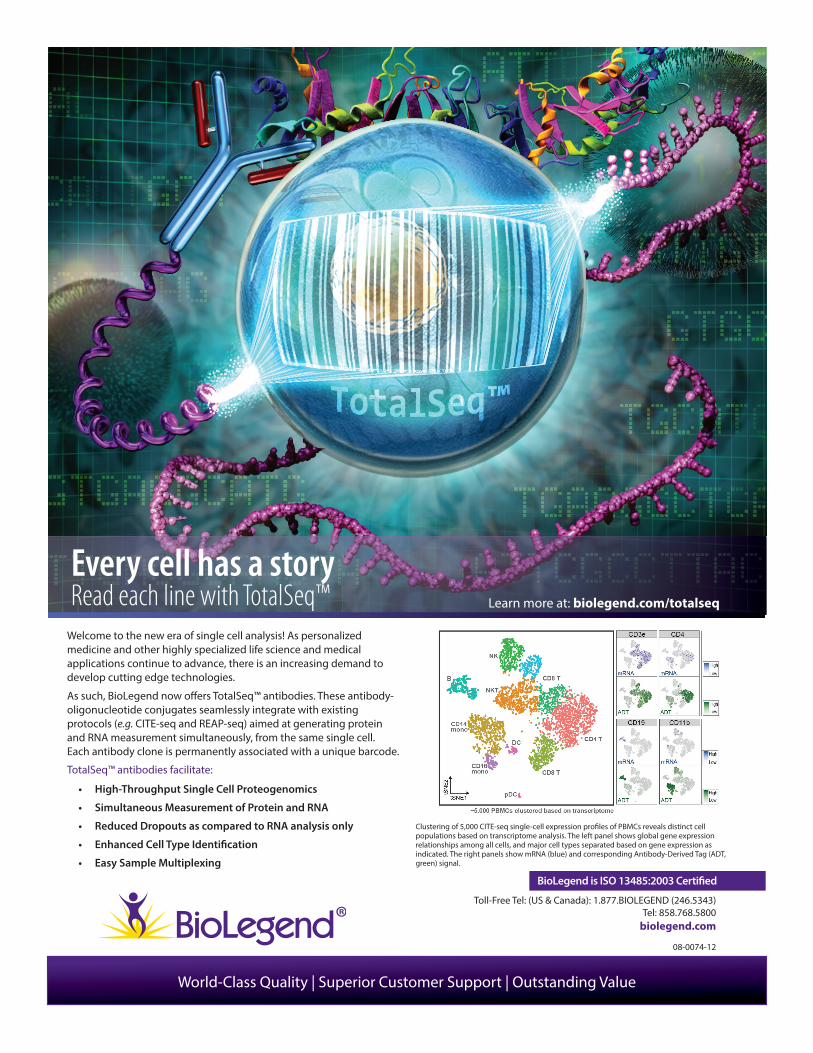
Welcome to the new era of single cell analysis! As personalized medicine and other highly specialized life science and medical applications continue to advance, there is an increasing demand to develop cutting edge technologies.
As such, BioLegend now o� ers TotalSeq™ antibodies. These antibody-oligonucleotide conjugates seamlessly integrate with existing protocols (e.g. CITE-seq and REAP-seq) aimed at generating protein and RNA measurement simultaneously, from the same single cell. Each antibody clone is permanently associated with a unique barcode.
TotalSeq™ antibodies facilitate:
• High-Throughput Single Cell Proteogenomics
• Simultaneous Measurement of Protein and RNA
• Reduced Dropouts as compared to RNA analysis only
• Enhanced Cell Type Identi� cation
• Easy Sample Multiplexing
BioLegend is ISO 13485:2003 Certi� ed
Clustering of 5,000 CITE-seq single-cell expression pro� les of PBMCs reveals distinct cell populations based on transcriptome analysis. The left panel shows global gene expression relationships among all cells, and major cell types separated based on gene expression as indicated. The right panels show mRNA (blue) and corresponding Antibody-Derived Tag (ADT, green) signal.
Every cell has a story Read each line with TotalSeq™ Learn more at: biolegend.com/totalseq

We are proud to sponsor
The 21st Annual Upstate New YorkImmunology Conference 2018
Krackeler Scientific is a leading distributor of quality scientific products. We serve customers nationwide in biotechnology, nanotechnology,
life sciences, pharmaceutical, biomedical, environmental, & industrial sectors.
Our comprehensive product line represents all of the leading manufacturers including Corning, Sigma, Agilent, Eppendorf & Celltreat. Our home is Albany, NY.
www.krackeler.com • [email protected] • 800-334-7725

Let’s get to the science
Decide for yourself at thermofisher.com/compareflow
For Research Use Only. Not for use in diagnostic procedures. © 2018 Thermo Fisher Scientific Inc. All rights reserved. All trademarks are the property of Thermo Fisher Scientific and its subsidiaries unless otherwise specified. COL06215 0318
Our flow cytometer evaluation guide will help you gain a solid understanding of how to objectively compare system components and capabilities from several manufacturers’ models.
This 40-page guide was developed with input from: • Flow cytometry core lab directors
• Researchers with informed perspectives on comparing instrument options
• Mechanical, systems, and software engineers
Instrument evaluation and comparison guide Researcher demand is driving the evolution of instrumentation, resulting in flow cytometers designed to fulfill the needs of different applications, sample states, cells of interest, and research goals.
Flow cytometry
Stimulation antibody
Conjugated antibodies
Cell health Flow RNA Buffers
COL06215-2018-BnW-Ad.indd 1 3/8/18 1:40 PM

22.
Silver Corporate Sponsor
Agilent Biosciences
Leinco Technologies
Lonza Biosciences
MilliporeSigma
Shenandoah Biotechnologies
StemCell Technologies
Taconic Biosciences

For Research Use Only. Not for use in diagnostic procedures.
© Agilent Technologies, Inc. 2018
Discover the Power of Real-Time Live Cell AnalysisAgilent Seahorse XF Analysis uses label-free technology to detect discrete changes in cell bioenergetics in real-time, providing a window into the critical functions driving cell signaling, proliferation, activation, toxicity and biosynthesis.
Seahorse instruments, sensor cartridges, assay kits and software combine seamlessly to provide powerful metabolic data from live cells in real-time for the most comprehensive assessment of cell metabolism.
Learn more about Agilent Seahorse XF products at: agilent.com/chem/discoverxf
Discover the Power of RTLC Analysis 8.5x11 nobleed.indd 1 8/21/18 10:59 AM


CytoSMART Ad – NYIC 2015
BioResearch
CytoSMART™ System Live Cell Imaging – The Smart Way
View your cell culture via cloud access on smart phone, tablet or computer.© 2015 Lonza Walkersville, Inc.
Small, Easy and AffordableSized and priced for virtually any lab and budget, the CytoSMART™ System has been developed for live cell imaging and monitoring. The system is set up within minutes. Via innovative cloud technology, your cell culture is just one click away – monitor your cells anytime, anywhere.
Watch CytoSMART™ live in action!www.lonza.com/cytosmart


FOR A TEST DRIVE
SHENANDOAH’S SAMPLE POLICY
TAKE SHENANDOAHCYTOKINES
Order one of our human, mouse or rat cytokines in a size that fits your needs. The Mini is just $60 for enough material to put through the paces and see for yourself why we are the Protein Pros!
Order a larger size of the same protein within 3 months and receive a full credit for the Test Drive Product. Simply ask for the Test Drive Credit
by phone,
email, or in the notes section of your online shopping cart. It’s that simple!
CONTACT US TODAY TO GET A TEST DRIVE QUOTE OR FOR MORE INFORMATION
Credit is not automatic. You must reference the Test Drive Credit at the time of your order.
Credit will equal the amount actually paid for the Test Drive product (not including shipping).
A larger size of the same product must be purchased within 3 months of the Test Drive purchase.
One trial credit is valid per product. Interested in trying other cytokines? Take them for a Test Drive too!
This policy is subject to be discontinued or changed at any time.
www.shenandoah-bt.com
21
•
•
•
•
•

C
M
Y
CM
MY
CY
CMY
K
AD349CS-NY Immunology Conference-Final.pdf 1 8/30/2018 1:36:03 PM

Get in touch for more information about our services.
US: 1-888-822-6642 | EU: +45 70 23 04 05 | [email protected] | TACONIC.COM
Leading the Industry in Animal Model Solutions
f Global footprint with the capability to ship worldwide
f Only globally harmonized health standard platform
f Superior selection and quality of traditional and spontaneous mutants
f Only comprehensive provider of microbiome product and service solutions
f Severely immunodeficient and humanized models
f Custom model design solution platform
f Comprehensive neuroscience portfolio
f Unique breeding pathway design
f Unparalleled scientific leadership
Models for
Life
© Taconic Biosciences, Inc. All rights reserved. Contents of this publication may
not be reproduced in any form without prior permission.
Superior Products
Expertise
Unique Platforms

30.
In recognition of the significance
of this meeting and work being done by
Graduate Students
and Postdoctoral Fellows,
the
American Association
Of Immunologists
has provided
Ten(10) Young Investigator Awards.
Each will receive a monetary award,
as well as the opportunity
to present their research both in poster format and brief talks.

31.
Institutional Financial Supporters
Albany Medical College
Alumni Association
Cornell University
Microbiology & Immunology
Dartmouth College
Department of Microbiology & Immunology
McGill University
Pennsylvania State University
Roswell Park Cancer Institute
Department of Immunology
SUNY Upstate Medical University
Microbiology & Immunology Program
University at Buffalo
Buffalo School of Medicine
Department of Microbiology & Immunology
University of Connecticut
Department of Molecular & Cellular Biology
Center of Excellence for Vaccine Research
University of Rochester Medical Center
Department of Microbiology & Immunology
University of Vermont
Vermont Center for Immunology & Infectious Diseases
Wadsworth Center

32.
NYIC Scientific Advisory Board Institutional Representatives
Albany Medical College
Jim Drake and Kate MacNamara
(NYIC Conference Organizers)
Cornell University
Margaret Bynoe
Dartmouth College
Brent Berwin
McGill University
Jorg Fritz
Penn State University
Girish Kirimanjeswara

33.
Roswell Park Cancer Institute
Yasmin Thanavala
SUNY Upstate Medical University
Gary Winslow
University of Connecticut
Steven Szczepanek
University of Rochester Medical Center
Scott Gerber
University of Vermont
Eyal Amiel
Wadsworth Center/ACPHS
Nicholas Mantis

34.
Grant support provided to
Graduate Students
and
Postdoctoral Fellows
by the
National Institutes of Health
National Institute of
Allergy and Infectious Diseases
R13AI051522
“Thank You”

35.
Keynote Speaker
Thomas A. Wynn, Ph.D. Vice President
Discovery Inflammation and Immunology
Pfizer
“The Role of Inflammation in Tissue Regeneration and Fibrosis”
Dr. Wynn is VP of Discovery in the Inflammation and Immunology Research Unit at
Pfizer, Cambridge, MA. Prior to joining Pfizer in 2017, he was a Senior Investigator
and Chief of the Immunopathogenesis Section of the Laboratory of Parasitic Dis-
ease, in the National Institute of Allergy and Infectious Diseases, NIH in Bethesda,
MD. He received his Ph.D. from the Department of Medical Microbiology and Im-
munology at the University of Wisconsin, in Madison, Wisconsin. He has published
over 300 scholarly research papers, reviews, and book chapters in many prestig-
ious journals including Nature, Science, and Nature Medicine and has contributed
substantially to our understanding of the role of cytokines in the progression and
resolution of chronic inflammation and fibrosis. He also investigates the role of
macrophages, fibroblasts and tissue progenitor cells in tissue regeneration. For the
past four years, Thomson Reuters/Clarivate Analytics included him among their list
of Highly Cited Researchers in the field of Immunology.
After 26 years as a senior investigator at the National Institutes of Health (NIH), he decided to take a posi-
tion to lead Pfizer’s inflammation and immunology unit. His major motivation for coming to Pfizer is to
have collaborations in drug development, which can happen at a much higher level because there are ex-
perts in all these areas at Pfizer. As a basic scientist, he has lots of great ideas. With the varied experts
here, he can have meetings and target the best for the project in mind.

36.
Symposium I
B-Cells and
Humoral Immunity
Chair : Dr. Jeremy Boss

37.
Development, Differentiation, and Maintenance of T-bet+ IgM Memory B cells
Gary Winslow* Russell Levack, Kevin Kenderes, Berenice Cabrera-Martinez,
Maria Popescu, and Rebecca Harris
Upstate Medical University, Syracuse, NY
Our work is focused on a population of CD11c+ B cells that we first described in 2008. This population
was later shown to be characterized by expression of the T helper cell lineage-specific transcription fac-
tor, T-bet. Although B cell-specific T-bet expression had been shown in the study that first characterized
the transcription factor, CD11c+ T-bet+ B cells had previously not been observed in vivo. T-bet+ B cells
have now also been identified in a range of immunological contexts, including acute viral and parasitic
infections, aging, and autoimmunity. The cells have been nominally described as ABCs (Age-related B
Cells) by other investigators. As part of our initial studies, we showed that CD11c+ (T-bet+) B cells were
generated early following infection with the tick-transmitted bacterium, Ehrlichia muris. The population
at that time following infection consists largely of IgM plasmablasts. Within 30 days T-bet+ B cells are
found as spleen IgM memory cells. These memory cells are multi-potent, as they can populate all effector
B cell lineages following challenge infection, and self-renew. We have proposed that T-bet+ B cells are
maintained as memory cells under inflammatory conditions associated with chronic infection, or with
autoimmunity. Our current work, which will be discussed, centers on 1) how T-bet+ IgM memory B cells
are generated in the absence of germinal centers, 2) how they differentiate following secondary chal-
lenge, 3) how they are maintained indefinitely, and on 4) the properties of the antibodies the cells pro-
duce. Targeting T-bet+ B cells for vaccinations, or for disease treatment, offers an attractive strategy for
the clinic.
This work was supported by U.S. Department of Health and Human Services grant R01AI064678.

38.
Human B Cell Responses to Dengue and Zika Viruses
Sean A. Diehl
Department of Microbiology and Molecular Genetics and Vaccine Testing Center,
Larner College of Medicine, University of Vermont, Burlington, VT, USA;
correspondence: [email protected]
Mosquito-borne members of the flavivirus family including dengue virus (DENV), zika virus
(ZIKV), and yellow fever virus (YFV) have widely plagued human health since the beginning of recorded
history. These related viruses often co-circulate and can elicit both cross-reactive and virus-specific humor-
al responses. Antibodies are a critical component in protection from flaviviruses, yet mechanisms of anti-
body development to flavivirus infection or vaccination are not completely understood. In my presentation
I will cover our approach to dissect the B cell response to DENV vaccination and controlled DENV infec-
tion of humans, highlighting our findings from early plasmablast responses and Ig diversification to devel-
opment of DENV-specific B cell frequencies, and profiling of serotype specificity. I will also describe our
findings on ZIKV-specific B cell and antibody responses to natural infection. These studies have yielded
novel ultra-potent ZIKV neutralizing antibodies that are highly represented in serum from ZIKV-immune
patients. In sum, our studies have yielded novel tools that can be used as standards for evaluating patient
immune responses and studying antibody:virus interactions and modes of virus blockade. In addition our
results quantitating and characterizing virus-specific memory B cells in humans offers another parameter
that could be used to evaluate vaccines.

39.
Transplacental Allergic Sensitization and Immune Development
Sara Paveglio1,2, Karim Rezaul1 and Adam Matson1,2. 1University of Connecticut Health, Farmington, CT; 2
Connecticut Children's Medical Center, Hartford, CT
The propensity to develop asthma and allergies, like many chronic diseases of childhood, may be influ-
enced by events that occur during the fetal and perinatal periods. Previous work from our laboratory pro-
vided compelling evidence that the placental transport of maternal IgE is mediated by IgG autoantibodies
directed against IgE. The autoantibody-IgE complexes bind to the FcRn receptor, which mediates their
transport across the placenta. Strikingly, essentially all the IgE in cord blood (CB) serum was found to be
complexed to IgG, suggesting that this is the predominant mechanism by which the fetus acquires IgE.
Furthermore, the transport and ensuing binding of IgE complexes to FcεRI-expressing fetal cells, such as
basophils, appeared dependent on the subclass and epitope specificity of the anti-IgE autoantibodies, which
in turn was influenced by maternal allergic status. We hypothesize that following entry into the fetal circu-
lation maternal IgE/IgG immune complexes (ICs) have the capacity to sensitize, and in some cases sponta-
neously activate, FcεRI-expressing cells such as basophils. Preliminary evidence to support such a process
was found by demonstrating upregulation of the basophil activation marker CD63 on CB basophils ob-
tained from infants of allergic vs. non-allergic mothers. Further investigation demonstrated that purified
maternal ICs added to culture-derived basophils could prime the cells for either anti-IgE- or anti-IgG-
induced activation. Additional studies using human FcɛRI-expressing RBL-SX38 cells demonstrated that
maternal ICs spontaneously induced β-hexosaminidase release, which was limited by the humanized anti-
IgE omalizumab. These data are significant as we previously showed that secreted basophil products, such
as histamine, impairs the ability of CB myeloid dendritic cells (mDCs) to respond optimally to the TLR4
ligand lipopolysaccharide. Ongoing studies are evaluating the impact of FcεRI activation and secreted ba-
sophil products on mDC development and innate-adaptive crosstalk. Specifically, we speculate that in
utero FcεRI activation alters mDC-driven T cell differentiation resulting in an increased propensity for de-
veloping Th2-type responses after birth.

40.
Symposium II
Immunology at the
Host-Pathogen Interface
Chair : Dr. Thomas Wynn

41.
Host Interactions with Bacterial Pathogens: Heads I Win, Tails You Lose
Sally Demirdjian1, Hector Sanchez1, Dan Hopkins1, Yash Patankar1,3, Rustin Lovewell1,3, Eyal Amiel1,2,
Brent Berwin1 1 Microbiology and Immunology Dept., Dartmouth College, Lebanon, NH 03756
2 Current address: Medical Laboratory and Radiation Sciences,
University of Vermont, Burlington, VT 05405, USA 3 Current address: Dept. of Microbiology, University of Massachusetts Medical School, Worcester, MA
01655
Phagocytosis of the bacterial pathogen Pseudomonas aeruginosa is the primary means by which the host
prevents and controls infections: compromise of macrophage or neutrophil numbers or function results in
extreme susceptibility to infection. We previously identified flagellar swimming motility as a key patho-
gen-associated molecular pattern (PAMP) recognized by phagocytes to initiate engulfment of several gen-
era of bacteria. Correspondingly, loss of flagellar motility is observed during chronic infections with P.
aeruginosa, and this likely reflects a selection for bacteria resistant to phagocytic clearance. However, the
mechanisms underlying the preferential phagocytic response to motile bacteria are poorly understood. Our
recent data support that the phagocytic susceptibility for swimming bacteria is proportional to flagellar ro-
tation since complementary genetically- and biochemically-modulated incremental decreases in flagellar
motility result in corresponding and proportional phagocytic evasion. Interrogation of the mechanisms re-
vealed that flagellar motility enhances both the association and the uptake of bacteria by phagocytes. Spe-
cifically, bacterial association with phagocytes is mediated by cell-surface polyanions, can be enhanced by
the integration of the anionic lipid PIP3 into the plasma membrane, and can be competed by soluble poly-
anions. Bacterial association with macrophage plasma membranes also facilitates inflammatory responses
and, accordingly, nonmotile P. aeruginosa evasion of productive cell-surface interactions with macrophag-
es results in reduced IL-1β host responses in vitro and in vivo in comparison to those elicited by wild-type
P. aeruginosa. Subsequent uptake is mediated through activation of the phagocyte PI3K/Akt pathway
which is specifically and proportionally activated in response to the bacterial flagellar motility. These find-
ings contribute to understanding the mechanism behind motility-dependent phagocytosis of extracellular
bacteria and support a model whereby phagocytic clearance exerts a selective pressure on P. aeruginosa
populations in vivo, which contributes to changes in pathogenesis during infections.

42.
Inflammation during TB: good, bad and the ugly
Bibhuti Mishra
Department of Immunology and Microbial Disease
Albany Medical College, Albany, NY
Mycobacterium tuberculosis (Mtb) infects nearly 10.8 million and kills 2 million people every year, mak-
ing Tuberculosis (TB) the single largest cause of mortality and morbidity worldwide. Tuberculosis (TB)
disease is often marked by high pathogen burden and inflammation in infected tissues. It is often believed
that the inability of the host to control bacterial replication, underlies susceptibility to TB. Nitric oxide syn-
thase 2 (Nos2) is critical for protecting against TB for its ability to generate nitric oxide (NO) derived radi-
cals that can directly kill Mtb. Mice lacking Nos2 succumb to TB disease within four weeks after infection,
with high bacterial burden and severe lung damage, both cardinal features of TB disease. We used Nos2
deficient mice as a model of active TB and found that instead of directly inhibiting Mtb growth, Nos2 pri-
marily protects mice by repressing an interleukin 1- and 12/15-lipoxygenase dependent neutrophil recruit-
ment cascade. Parallel clinical studies indicate that a similar inflammatory pathway promotes TB in hu-
mans. The human 12/15 lipoxygenase ortholog, ALOX12, is expressed in cavitary TB lesions, the abun-
dance of its products correlates with the number of airway neutrophils and bacterial burden. Finally, we
employed a saturated library of Mtb transposon mutants to demonstrate that the neutrophilic inflammation
provides a conducive environment for Mtb growth. Together, these data suggest that Mtb induces neutro-
philic inflammation to preferentially replicate at sites of tissue damage, and the ability to restrain this path-
ogenic host response is central to immune protection against TB. In sum, neutrophilic inflammation being
protective against a number of extracellular bacterial pathogens, plays a pathological role during TB.

43.
Sequential Airborne Transmission of Pandemic, Seasonal, and Emerging Influenza
Viruses – A Potential Strategy to Evaluate Vaccine Efficacy and Non-sterilizing Immunity
Troy C Sutton*, Elaine W Lamirande, Rita Czako, Renuka E Joseph, Kanta Subbarao**
Laboratory of Infectious Diseases, Division of Intramural Research, National Institute of Allergy and In-
fectious Diseases, USA
Current Affiliation: * Department of Veterinary Medicine, and the Huck Institutes of Life Sciences, The
Pennsylvania State University
** WHO Collaborating Centre for Reference and Research on Influenza, Australia
A hallmark of pandemic influenza viruses is their ability to spread from person-to-person by the
airborne route. The pandemic potential of emerging avian influenza viruses is assessed by evaluating air-
borne transmission between ferrets. Some avian influenza viruses that have crossed the species barrier
transmit in ferrets but have not spread among humans. In contrast, seasonal and pandemic influenza virus-
es transmit efficiently from experimentally infected ferrets to contacts via the airborne route. Therefore, we
evaluated whether onward transmission from ferrets infected by respiratory contact would be more in-
formative. We hypothesized pandemic and seasonal viruses would transmit over two sequential rounds of
respiratory transmission in ferrets, while emerging avian viruses would fail to transmit during a second
round of transmission.
Influenza A/California/07/2009 (H1N1pdm09), A/Texas/50/2012 (H3N2), A/Anhui/1/2013 (H7N9)
and A/seal/ New Hampshire/179629/2011 (H3N8) viruses were evaluated as representative pandemic, sea-
sonal, and emerging viruses, respectively. Donor ferrets (n=5-6) were inoculated and housed adjacent to a
respiratory contact ferret (RC-1). When the RC-1 ferret became infected, this animal was housed adjacent
to a second RC (RC-2) ferret, and nasal washes were collected every other day for viral titration.
All four viruses transmitted over two rounds of respiratory contact. For the H1N1pdm09 and H3N8
viruses, 5/6 RC-1 ferrets shed virus, followed by transmission to 5/5 RC-2 ferrets. For the H3N2, 4/5 RC-1
ferrets and 3/4 RC-2 ferrets shed virus, while for the H7N9 virus, 3/6 RC-1 and 2/3 RC-2 ferrets shed vi-
rus. We found that influenza viruses that transmit by the airborne route transmit onward to new respiratory
contact ferrets with similar efficiency.
Importantly, these are the first studies to reproduce chains of sequential airborne transmission in an
animal model. With the possible introduction of hemagglutinin stem and neuraminidase antibody inducing
vaccines that do not induce sterilizing immunity or completely block transmission, we propose to develop
a new correlate of protection by examining the ability of these vaccines to disrupt chains of transmission.
We anticipate that this will provide an additional measure of vaccine efficacy and may more closely repre-
sent the ability for these novel approaches to prevent transmission. This research was funded by the Intra-
mural Research Program of the NIH. Future studies will be performed at the Pennsylvania State Universi-
ty.

44.
Platinum Corporate Sponsor
TotalSeq™: Standardized Oligonucleotide Barcode Antibody Conjugates for
Multiplex Immunophenotyping
Nathan Lucas
BioLegend
The CITE-Seq (Cellular Indexing of Transcriptomes and Epitopes by Sequencing) platform is a recent ad-
vancement in single cell analysis, based on high-throughput single cell sequencing (scSeq). CITE-Seq pro-
vides simultaneous unbiased measurements of cellular surface proteins and transcriptomes. This platform
will potentially transform how complex cell populations (e.g., lineage differentiation or tumor infiltrating
lymphocytes) are studied. Current published data has indicated that scSeq analysis on cell surface protein
expression is comparable to multi-color flow cytometry, and in addition, enables superior multiplexing ca-
pabilities. Currently, individual investigators use their choice of oligo barcodes for different surface pro-
tein markers, and there is no standardized method of control available, making comparison of data from
independent studies difficult. The availability of standardized barcode-labeled antibodies enables reliable
comparisons of data across longitudinal and multi-site studies. After assigning a unique oligo barcode to
each of our monoclonal antibodies, we prepared directly conjugated reagents: TotalSeq™ products. Our
standardized barcoding system and ready-to-use oligo-antibody conjugates support scSeq based multiplex
immunophenotyping and demonstrate that these conjugates meet the rigorous manufacturing standards and
perform as expected to classical cytometry methodologies.

45.
Oral Poster Presentations 1:30-2:45 p.m.
Kingfisher
Category A
Lymphocyte Biology
Chairs: Jorg Fritz and Qi Yang

46.
Poster #2
MicroRNA-29 alters the CD8+ T cell Response to Infection in an Age Dependent Manner
Kristel Yee Mon1, Norah Smith1, Ravi Patel2, Andrew Grimson2, Brian Rudd1
Department of Microbiology & Immunology, College of Veterinary Medicine, Cornell University, Ithaca,
NY1, Department of Molecular Biology and Genetics, Cornell University, Ithaca, NY2
Unlike adults, neonates fail to generate complete immunity against viruses and bacteria. As CD8+
T cells play a critical role in protecting the host against these pathogens, it is important to understand how
CD8+ T cells from neonates and adults respond differently to infection. Published studies indicate that ne-
onatal CD8+ T cells fail to form memory cells because they are inherently more proliferative and rapidly
become terminally differentiated during infection, at the expense of forming memory. To understand the
basis of these age-related differences, we examined miRNA expression and found that one miRNA in par-
ticular (miR-29), was selectively upregulated in adult CD8+ T cells (mice and humans) and acting on its
target genes in a biologically-active manner. However, whether miR-29 alters the CD8+ T cell behavior
remains an open question. Our hypothesis is that upregulation of miR-29 in adult CD8+ T cells promotes
memory formation by targeting genes that are typically associated with effector cell differentiation. To test
this hypothesis, we first validated target gene activity in human adult CD8+ T cells via electroporation of
mir-29 mimics or incubation with mir-29 antagomirs and observed that mir-29 target expression (T-bet &
Eomes – T cell regulators) was downregulated and upregulated respectively.
To further test this hypothesis, we compared the CD8+ T cell response to Vaccinia virus in WT and
miR-29KO mice and observed fewer memory CD8+ T cells in miR-29KO mice, same phenotype observed
in neonates. We next performed adoptive transfer experiments and found that mir29KO donor CD8+ T
cells preferentially differentiate into short-lived effectors, suggesting miR-29 alters the response in a cell-
intrinsic manner. The donor mir-29KO CD8+ T cells also secreted more effector molecules (IFNg, gzmB)
and expressed higher amounts of effector associated transcription factors (T-bet, Eomes, blimp-1). To de-
termine if miR-29 alters the behavior of CD8+ T cells by altering cytokine thresholds, we used a dendritic
cell immunization approach, so T cells are stimulated with an equivalent amount of cognate peptide, while
increasing the dose of IL-12. The results indicated that WT CD8+ T cells require more IL-12 than miR-
29KO CD8+ T cells to undergo a similar amount of effector cell differentiation so absence of mir-29 low-
ers the cytokine induced activation threshold. Collectively, these results provide key insight into how to
improve T cell immunity in early life since a single genetic regulatory element can reverse the memory
phenotype of an adult CD8+ T cell into an effector neonate-like phenotype.

47.
Poster #41
Essential Role of c-Rel in IL-33-mediated activation of group 2 innate lymphoid cells
Barbara C. Mindt1,2,3, Claudia U. Duerr1,2,3,4, Mathieu Mancini1,5, Silvia Vidal1,5, Steve Gerondakis6,
Philippe Gros1,7, David Langlais1,5,8, Jörg H. Fritz1,2,3,9
1McGill University Research Centre on Complex Traits, Montréal, QC 2FOCiS Centre of Excellence in
Translational Immunology, Montréal, QC 3Department of Microbiology and Immunology, McGill Univer-
sity, Montréal, QC 4Institut für Mikrobiologie und Infektionsbiologie, Charité – Universitätsmedizin, Ber-
lin, Germany 5Department of Human Genetics, McGill University, Montréal, QC 6Biomedicine Discovery
Institute and Department of Biochemistry and Molecular Biology, Monash University, Clayton, Australia 7Department of Biochemistry, McGill University, Montréal, QC 8McGill University and Genome Quebec
Innovation Centre, Montréal, QC 9Department of Physiology, McGill University, Montréal, QC
Group 2 innate lymphoid cells (ILC2) are a recently described cell population that play a key role
in the initiation and orchestration of early type 2 immune responses. Upon tissue damage ILC2 are activat-
ed by the alarmins IL-25, IL-33 and/or TSLP and rapidly secrete large amounts of type 2 signature cyto-
kines such as IL-5 and IL-13. While it is known that activating IL-33 receptor signalling results in down-
stream NF-κ(kappa)B activation, the underlying molecular mechanisms remain elusive. The NF-κ(kappa)B
subunit c-Rel has been shown to promote airway hyperreactivity and allergic inflammation in a murine
asthma model. Since ILC2 are main drivers of asthma-mediated type 2 immune responses we hypothesized
that c-Rel positively regulates ILC2 activation and function.
We initially observed that after intranasal challenge with IL-33, wild-type mice mounted a substan-
tial type 2 immune response including increased lung ILC2 numbers and eosinophilia whereas c-Rel defi-
cient (Rel-/-) mice failed to respond to challenge. To further investigate the role of c-Rel in IL-33-mediated
activation and function of ILC2, bone marrow ILC2 of wild-type and Rel-/- mice were isolated, expanded
and stimulated with IL-33 ex vivo. We observed that after stimulation with IL-33, c-Rel mRNA and protein
levels were induced and that c-Rel translocates to the nucleus upon ILC2 activation. Importantly, ILC2 ef-
fector functions were impaired in Rel-/- ILC2. To further elucidate the underlying molecular mechanisms, c
-Rel chromatin interaction sites in ILC2 after IL-33-mediated activation were determined by ChIP-Seq and
compared to RNA-Seq data of activated wild-type and Rel-/- ILC2 in order to identify target genes that are
directly regulated by c-Rel and deregulated in Rel-/- ILC2 after activation. Selected target genes were vali-
dated by ChIP-qPCR and their potential role(s) will be further investigated ex vivo and in vivo.
Altogether, our data indicate that c-Rel positively regulates IL-33-mediated ILC2 activation and
function ex vivo as well as in vivo. Thus, we show for the first time that c-Rel plays an essential role in
ILC2 biology.

48.
Poster #15
The effect of aging on tissue-resident lymphocytes at homeostasis and during pulmonary infection
Shanti D’Souza and Qi Yang
Albany Medical College, Albany NY
Aging is paradoxically associated with increased inflammation and decreased immune cell func-
tion, leaving the elderly more susceptible to infection. More than any other age group, people aged 65
years or older have the highest susceptibility to influenza infection. We propose that this predisposition to
infection is compounded by a defect in a recently described immune cell population known as innate lym-
phoid cells (ILC). ILC are a long-lived tissue resident innate lymphocyte population that can be further dif-
ferentiated into ILC1, ILC2 and ILC3 displaying unique effector functions. These cells can be stimulated
in an antigen-independent manner, resulting in rapid proliferation and cytokine production, important for
mounting an early response to pathogen. ILC2 are enriched in lungs and play an important protective role
in homeostasis and pulmonary infection. We observed that the numbers of ILC2 were drastically reduced
in lungs of aged mice (>20 months) at homeostasis. This was compounded with decreased protein expres-
sion of GATA3, the key transcription factor required for ILC2 development and function. We also ob-
served that aged ILC2 were functionally compromised, producing less of a characteristic ILC2 cytokine, IL
-5. In addition, old ILC2 took up less fatty acid, suggesting a difference in their metabolic function. RNA
sequencing of ILC2 from young and old mice provided multiple signaling pathways that were compro-
mised with aging, including metabolic pathways. Reverse chimera studies indicated that the lung environ-
ment also affects ILC2. Young ILC2 introduced into old mice resulted in decreased frequency, GATA3
expression and fatty acid uptake compared to those cells introduced into young mice. We hypothesized that
this reduction in ILC2 number and function in old mice would increase their susceptibility to infection and
this was confirmed using a mouse model of pulmonary influenza infection. Old mice were susceptible to
low doses of Influenza A Virus compared to young mice. Pulmonary ILC2 were further reduced during
infection. To counteract this, ILC2 from young mice were adoptively transferred into old mice. Weight
loss and survival were improved in old mice that received young ILC2. However, this improvement was
not observed when old ILC2 were transferred into old mice, suggesting that defects in old ILC2 are medi-
ated through cell-intrinsic and cell-extrinsic mechanisms. Together, our studies revealed a novel effect of
aging on ILC that predispose the elderly to infection. Targeting these cells may provide new avenues to
improve prevention and treatment of infections and other airway diseases in the elderly.

49.
Poster #18
A new role for Indoleamine 2,3-dioxygenase (IDO): supporting the survival of bone marrow resident
long lived plasma cells (LLPC)
Shivana M. Lightman, Louise M. Carlson, and Kelvin P. Lee
Roswell Park Cancer Institute, Department of Immunology, Buffalo, NY
Long lived plasma cells (LLPC) are essential for sustained antibody responses and protective hu-
moral immunity. How these cells maintain longevity and a durable antibody response is largely dependent
on the complex nature of the bone marrow microenvironment in which these cells reside, and the pro sur-
vival factors produced in this niche. Work done by our lab has demonstrated that the CD28 receptor is es-
sential for survival of LLPC and the maintenance of durable titers. Additionally, our lab and others have
found that LLPC are in direct contact with dendritic cells (DC) in the BM, and that in vitro co-culture with
bone marrow derived DC promotes LLPC survival and immunoglobulin (Ig) production. Furthermore, we
and others have shown that when CD28 ligates with its ligands CD80/CD86 expressed on DC, it induces
upregulation of the enzyme Indoleamine 2,3-dioxygenase (IDO), a tryptophan catabolizing enzyme, in the
same fashion as T cells. IDO activity catabolizes tryptophan (Trp) into L-kynurenine (Kyn), which is a
well-known ligand of the aryl hydrocarbon receptor (AhR), a nuclear transcription factor that has been
demonstrated to modulate immune responses and is highly expressed in plasma cells. Upon investigation
of the role of IDO in plasma cell survival and function, our preliminary data reveals that IDO knock-out
(KO) mice have a decrease in bone marrow resident plasma cells in comparison to wild type (Wt) mice,
and a decrease in overall Ig levels. Additionally, depletion of DC, an IDO producing cell, significantly de-
creases antigen-specific antibody secreting cells (ASCs) post vaccination. Furthermore, upon treatment
with the metabolite Kyn, the well-known AhR gene CYP1A1, is upregulated in BM-derived LLPC. This
leads us to propose a model where CD28, through back signaling to CD80/86 induces IDO production in
DC, a mechanism to promote LLPC survival and sustained antibody production. These findings challenge
the paradigm of IDO as an inhibitory molecule and provide the rationale for investigating its undefined, but
essential role in maintaining LLPC survival and sustained antibody responses. Filling this gap in
knowledge will not only further our understanding of diseases that involve LLPC, but also has direct impli-
cations in future vaccine and therapeutic drug development.
Support: T32 CA085183, R01CA121044-08

50.
Poster #35
Developmental activation of the aryl hydrocarbon receptor reduces CD4+ T cell responses by
altering DNA methylation patterns
Catherine G. Burke1, Jason R. Myers2, and B. Paige Lawrence1,3
Department of 1Microbiology and Immunology, 2Genomics Research Center, and 3Department of Environ-
mental Medicine University of Rochester Medical Center, Rochester, NY
Early life environmental exposures can have lasting effects on the function of the immune system
and contribute to disease later in life. Yet, the mechanisms by which exposures at one point in time alter
immune cell responses at another point in time remain unclear. Studies in animal models and human popu-
lations suggest the aryl hydrocarbon receptor (AHR) provides a link between developmental exposure to
xenobiotics and dysregulated immune responses later in life. Despite this association, little is known about
the cell types and underlying mechanisms driving these durable changes. Recently, we showed that activa-
tion of the AHR during development, by maternal exposure to 2,3,7,8-tetrachloroibenzo-p-dioxin (TCDD,
the prototype AHR agonist), impairs CD4+ T cell responses to influenza A virus (IAV) infection in adult
offspring. In contrast, naïve animals do not have detectable changes in lymphoid organ cellularity or distri-
bution of T cell subpopulations. Moreover, the dampened CD4+ T cell response to IAV can be transferred
to mice that were not exposed during development. This suggests that these changes are laid down during
development, are long lasting, and implicates altered epigenetic regulation as a potential mechanism. Since
DNA methylation is an epigenetic system that influences CD4+ T cell proliferation and differentiation, we
hypothesize that developmental AHR activation alters immune responses via changes to DNA methylation.
We utilized whole genome bisulfite sequencing (WGBS) to map how developmental exposure impacts
DNA methylation in CD4+ T cells before and after infection. Developmental exposure results in differen-
tial methylation patterns across the entire CD4+ T cells genome. Differentially methylated regions (DMRs)
reflect a combination of hyper- and hypo-methylated regions, and span all genomic features; including pro-
moters, exons, introns, enhancers, and intergenic regions. To determine whether altered DNA methylation
is responsible for impaired CD4+ T cell responses, we treated developmentally exposed mice with S-
adenosylmethionine (SAM) or zebularine to enhance or decrease DNA methylation, respectively. Follow-
ing infection, SAM restored the expansion of CD4+ T cells in TCDD exposed offspring. It also reversed
the dampened Th1 cell response, but did not restore the reduced Tfh response. Zebularine rescued the di-
minished frequency of Th1 and Tfh, but it did not alleviate suppression of CD4+ T cell expansion. Taken
together, these results indicate that altered DNA methylation is a potential mechanism by which AHR acti-
vation during development causes durable changes in antiviral immunity, and that hyper- and hypo-
methylation may regulate distinct aspects of CD4+ T cell responses to infection.

51.
Oral Poster Presentations 1:30-2:45 p.m. Council Rock
Category B
Inflammation
& Innate Immunity
Chairs: Kate MacNamara & Tim LaRocca

52.
Poster #25
CCR5 signaling reduces platelet-biased hematopoietic stem cells and drives thrombocytopenia in a
model of severe aplastic anemia
Allison N. Seyfried, Jackson Maloney, HuiJin Jo, Katherine C. MacNamara, PhD
Albany Medical College, 47 New Scotland Avenue, Albany, NY 12208
Severe Aplastic Anemia (SAA) is a bone marrow failure (BMF) disease caused by the inability to main-
tain blood production due to loss of hematopoietic stem cells (HSCs). BMF diseases develop from either a
genetic predisposition, known as inherited BMF, or as a result of environmental insults such as radiation
or infection, known as acquired BMF. Acquired BMF is associated with inflammation and elevated levels
of interferon-gamma (IFN-γ). Treatment for BMF includes immunosuppressive therapy or HSC trans-
plant, though these therapies often fail or are not possible for all patients. To investigate mechanisms driv-
ing disease we utilized a murine model of SAA induced by splenocyte infusion 4 hours post sub-lethal
irradiation of C57BL/6 x BALB/c F1 hybrids. We demonstrated that the interferon-gamma (IFN-γ)-
dependent loss of HSCs required bone marrow macrophages. Ablation of IFN-γ (gamma) signaling in
macrophages or macrophage depletion with clodronate-loaded liposomes resulted in increased HSCs, pro-
tection against thrombocytopenia, and significantly improved mouse survival. Rescued HSCs were en-
riched for CD41 expression and displayed robust platelet production. During SAA, macrophages charac-
terized by low CD11b expression persisted and comprised an increasing percentage of bone marrow cells
through 15 days post splenocyte transfer (dpst), despite loss of HSCs. We noted a marked increase in
RANTES (Regulated on Activation Normal T-cell Expressed and Secreted), or CCL5, production during
SAA that required both IFN-γ (gamma) and macrophages, prompting our hypothesis that CCL5 was im-
portant for pathogenesis. T cells are a main producer CCL5 and express high Ccl5 in the bone marrow
during SAA. However, T cell transfer studies demonstrated that T cell-derived CCL5 was not necessary
for disease induction or progression. CD11blo/- macrophages also exhibited high Ccl5 levels during SAA
and CD11blo/- macrophages exhibited a striking increase in CCR5 expression during SAA. CCL5 signal-
ing through CCR5 can provide anti-apoptotic signals to adipose tissue macrophages and megakaryocytes
via the Akt/Erk pathways, suggesting CCR5 signaling may be necessary for maintaining CD11blo/- macro-
phages during SAA. To determine whether CCR5 signaling was critical for macrophage survival and per-
sistence, we treated mice with the CCR5 antagonist, Maraviroc. CCR5 antagonism lead to increased
apoptosis of macrophages, increased platelet output 12 dpst, and significantly increased CD41hi HSCs 8
dpst. Our data demonstrate that IFN-γ (gamma)-dependent CCL5 production contributes to SAA patholo-
gy via CCR5 signaling, potentially through its ability to maintain macrophages and drive CD41hi HSC
loss.

53.
Poster #1
NLRP3 inflammasome activity and speck formation: Mutually exclusive outcomes
of NLRP3 activation
Abhinit Nagar, Tabassum Rahman, Jonathan A. Harton
Department of Immunology and Microbial Disease, Albany Medical College
Albany, NY 12208, USA
Inflammasomes are multi-protein complexes that regulate the activation of caspase-1 leading to initiation
of inflammatory cascades and cell death. Inflammasome dysregulation is a hallmark of various inflamma-
tory diseases. The best-studied inflammasome, NLRP3, is activated by structurally divergent agonists of
microbial, environmental, and host origin indicating its highly significant clinical relevance. NLRP3 acti-
vation is characterized by formation of singular perinuclear speck, concomitant activation of caspase-1 and
release of IL-1β. The speck is often referred to as the ternary inflammasome structure, but no substantial
evidence exists to support this idea. Moreover, the stoichiometry and morphology of the speck and ternary
inflammasome complexes are distinct arguing whether these two structures represent the same event. We
used multiple NLRP3 activators and studied their ability to induce speck and activate caspase-1/IL-1β. In
the absence of NLRP3 F.novicida U112 induced ASC specks which did not process IL-1β. In the presence
of NLRP3, sterile agonists, H2O2 and MSU, did not induce specks but still resulted in IL-1β maturation.
Time-course analysis of speck-formation revealed that the failure to induce speck was not because of its
rapid degradation. In colchicine-treated cells, inflammasome activity, but not speck formation, was re-
lieved by increased doses of nigericin. Moreover, inflammasome activation was associated with decreased
speck size, which requires NLRP3 and active caspase-1. Finally, active caspase-1 does not colocalize with
speck. Active caspase-1 is primarily distributed throughout the cytoplasm suggesting that the inflam-
masome and speck might function as separate signaling platforms. These results demonstrate that the speck
is not necessary for inflammasome function and suggest that this large, highly-organized, structure may
serve a distinct function.

54.
Poster #39
The STING agonist 5,6 dimethylxanthenone-4-acetic acid (DMXAA) stimulates an antiviral state
and protects mice against herpes simplex virus-induced neurological disease
Stacey Ceron1,2, Brian J. North2, Sean A. Taylor2, David A. Leib2 1 Guarini School of Graduate and Advance Studies, 2 Geisel School of Medicine at
Dartmouth, Lebanon, New Hampshire
Found in over eighty animal species, members of the Herpesviridae are enveloped DNA viruses that
establish lytic and latent life cycles in their respective hosts. Herpes simplex virus (HSV)-1 is the causative
agent of cold sores, genital sores, corneal keratitis and herpes simplex encephalitis (HSE) in humans and
establishes latent infection in neurons. The innate immune response to HSV-1 is dependent, in part, upon
the ability of the host to generate an interferon response through the stimulator of interferon genes
(STING) pathway. The aim of these studies was to explore the usage of 5,6-dimethylxanthenone-4-acetic
acid (DMXAA), a STING agonist, as a potential therapeutic agent for HSE and other herpetic infections.
We hypothesized that DMXAA stimulates interferon production in a STING-dependent manner thereby
establishing an antiviral state. In-vitro experiments using C57BL/6J fibroblasts demonstrated that treatment
with DMXAA was able to reduce viral replication through increased production of type I interferon. Fur-
thermore, administration of DMXAA to HSV-1 infected mice resulted in a reduction of viral burden in the
peripheral and central nervous systems. This reduced viral burden also correlated with increased survival
of DMXAA-treated infected mice. These results therefore demonstrate the potential of STING agonists for
immunotherapy against HSV-1
.

55.
Poster #42
Multifaceted Coordination of Airway Epithelial Barrier Integrity and Inflammation by
Protein Kinase D
Janelle Veazey1, Timothy Chapman2, Timothy Smyth3, Sara Hillman2, Sophia Eliseeva2,
Steve Georas
Department of Microbiology and Immunology1, Department of Pulmonary and Critical Care
Medicine2, Department of Environmental Medicine3, University of Rochester, Rochester NY
The serine/threonine kinase protein kinase D (PKD) is expressed in most cell types. The three
isoforms (PKD1-3) have been implicated in a wide array of cellular functions, including golgi vesicle
formation, differentiation, proliferation, and cellular migration. We and others have published that PKD,
and PKD3 in particular, mediate barrier disruption in the human bronchial epithelial cell line 16HBE.
Here we used a small molecule PKD inhibitor (CRT0066101) and a classic measure of pulmo-
nary permeability (total protein leak into the airspace) to demonstrate PKD promotes barrier disruption
in vivo. We confirm this relationship using a novel method of ‘outside-in’ barrier integrity: loss of in-
haled FITC-dextran from the airspace into circulation. We further report that TLR3, but not MDA5, pro-
motes barrier disruption in this model. Furthermore, CRT- treated, but not PKD3-/- mice are protected
from barrier disruption, indicating that PKD1 or PKD2 is the isoform responsible for barrier regulation
in mice. Current work investigates the molecular mechanism by which TLR3 engagement activates PKD
to promote barrier disruption.
Our work further reveals that PKD, and PKD3 in particular, upregulates proinflammatory cyto-
kines via mRNA transcription both in human cell lines and in vivo. Furthermore, the effect
of PKD3 on proinflammatory cytokines is largely dependent on MDA5, and not TLR3. Current work is
aimed at elucidating the molecular pathway by which PKD3 activity promotes proinflammatory cytokine
transcription. Additionally, a bone marrow chimera of PKD3-/- and wild type mice revealed that PKD3
activity in both epithelial cells and leukocytes is critical for neutrophil infiltration following dsRNA
(polyI:C) stimulation. Current work aims to determine the molecular mechanism of PKD3 in epithelial
cytokine production and in neutrophil migration and extravasation.
We conclude that barrier integrity and inflammation are distinctly regulated in this
model. Furthermore, the segregation of PKD1/2 and PKD3 into these two aspects of pathogen response
allows the potential for development of highly specific therapeutic targets for a wide range of respirato-
ry pathologies. For instance, a PKD1/2 antagonist may ameliorate cytokine storm during severe influen-
za infection, while a PKD3 antagonist may lessen COPD or asthma.
Funding: The project described was supported by Award Number R01 HL12424 from
NIH/NHLBI, F31 HL14079501 from NIH/NHLBI, T32 HL066988 from NIH/NHLBI, and T32
ES007026 from NIH/NIEHS.

56.
Poster #9
The prostaglandin D2 receptor CRTH2 suppresses epithelial cell responses during
intestinal helminth infection
Oyebola Oyesola1, Lauren M. Webb1, Sabrina Solouki1, Duc Pham1, Pamela Campioli1, Seth A. Peng1,
Rebecca L. Cubitt1, and Elia D. Tait Wojno1 1Baker Institute for Animal Health and Department of Microbiology and Immunology, Cornell Uni-
versity College of Veterinary Medicine, Ithaca, NY, USA
Intestinal helminth infections induce Type 2 inflammatory responses, characterized by immune cell activa-
tion, Type 2 cytokine production and increased epithelial cell responses that drive helminth expulsion.
While previous studies show that cytokines play key roles in regulating Type 2 inflammation, the role of
non-protein biochemical species such as lipid mediators in regulating Type 2 inflammation is less clear.
Previous studies show that the bioactive lipid mediator prostaglandin D2 (PGD2) is increased during Type 2
inflammation. PGD2 binds its receptor, chemoattractant receptor-homologous molecule expressed on Th2
cells (CRTH2), to promote Type 2 inflammation in the allergic lung. However, how CRTH2 influences
helminth-induced Type 2 inflammation in the intestine was unclear. In this study, we show that Nip-
postrongylus brasiliensis infection leads to increased expression of PGD2 synthase, suggesting increased
levels of PGD2. Surprisingly, despite the known pro-inflammatory role of CRTH2 in the lung, following N.
brasiliensis infection, CRTH2-deficient mice had lower worm burdens and increased intestinal mucin re-
sponses compared to wild type mice. In addition, chimeric mice with CRTH2 deficiency isolated to the
non-hematopoietic system had increased worm expulsion and goblet cell responses compared to wild type
mice. Critically, small intestinal epithelial cells expressed the gene that encodes for CRTH2. In vitro, mu-
rine small intestinal organoid cultures had decreased expression of Type 2 cytokine-induced goblet cell
associated genes following PGD2 stimulation. Together, these data suggest that the PGD2-CRTH2 pathway
acts directly on small intestinal epithelial cells to suppress anti-helminth inflammatory responses during
Type 2 inflammation. This study has improved the understanding of the role of the PGD2-CRTH2 pathway
during helminth-induced Type 2 intestinal inflammation and may inform the development and use of thera-
pies for treatment of Type 2 intestinal inflammatory diseases.

57.
Oral Poster Presentations 3:00-4:15 p.m.
Kingfisher
Category C
Tumor Biology
Chairs: Scott Gerber & Yasmin Thanavala

58.
Poster #26
Adrenergic signaling impairs activation of CD8+ T-cells by blocking metabolic reprogramming
Guanxi Qiao, Minhui Chen, Mark J. Bucsek, Hemn Mohammadpour, Cameron R MacDonald
Bonnie L. Hylander, Elizabeth A. Repasky
Department of Immunology
Roswell Park Comprehensive Cancer Center; Buffalo, NY
Adrenergic stress promotes tumor progression by several mechanisms. We have previously pub-
lished that the anti-tumor immune response is significantly suppressed by adrenergic stress (Bucsek et al,
Can Res 2018). Treatment of tumor-bearing mice with a beta-adrenergic receptor (beta-AR) antagonist
(propranolol) reverses immunosuppression and slows tumor growth. Furthermore, tumor infiltrating CD8+
T-cells from 4T1 and B16-OVA tumors grown in propranolol treated mice have increased expression of
markers of activation (CD69) and effector function (IFN-gamma, Gramzyme B). Notably, we also find in-
creased cell surface expression of the glucose transporter (GLUT1) on tumor-infiltrating CD8+T cells in
propranolol treated mice. Our recent data demonstrate that, in vitro, adrenergic signaling during activation
(using the beta-AR agonist isoproterenol- ISO) inhibits CD8+ T-cell GLUT1 expression on cell surface.
Based on the fact that T-cell activation is associated with metabolic reprogramming, requiring increased
glucose uptake for upregulation of glycolysis, we hypothesize that adrenergic signaling impairs the an-
titumor efficacy of CD8+ T-cells by impairing metabolic reprogramming. Analysis of T-cell metabo-
lism by Seahorse using cells treated with ISO revealed a significant impairment in glycolysis when T-cells
are activated in the presence adrenergic signaling. Additionally, beta-AR signaling inhibits the increased
mitochondrial respiration and which also occurs during T-cell activation as indicated by the observations
that both mitochondrial membrane potential and mitochondrial mass are decreased in cells activated the
presence of ISO. We also found that metabolic fitness of T-cells decreased in the presence of beta-AR as
indicated by a decrease in spare respiratory capacity. Additional data indicates that the impairments seen in
vitro also occur in vivo. We find that the mitochondrial mass of CD8+ T-cells isolated from tumors is de-
creased compared to CD8+ T-cells from tumor draining lymph nodes, however, when tumor bearing mice
are treated with propranolol, tumor infiltrating CD8+T cells have higher mitochondrial mass than tumor
infiltrating CD8+T cells from non-treated mice (higher MFI by flow cytometry). Additionally, CD8+ T-
cells from the spleen of tumor bearing mice treated with propranolol have a trend towards increased gly-
colysis, which supports the idea that in vivo, adrenergic signaling inhibits the antitumor immune response
through inhibition of CD8+ T-cell metabolic reprogramming.
This work was supported by grants from the Breast Cancer Coalition of Rochester, The New York
State Department of Health Peter T. Rowley Breast Cancer Research Grant (C028252), National Institute
of Health Grant R01CA205246, The Roswell Park Alliance Foundation, and Roswell Park Comprehensive
Cancer Center and National Cancer Institute (NCI) grant P30CA016056.

59.
Poster #33
Stereotactic Body Radiation and Interleukin 12 Combination Therapy Eradicates Pancreatic Tumors by
Composite Repolarization of the Tumor Microenvironment
Bradley N. Mills1, Kelli A. Connolly1, Jian Ye1, Taylor P. Uccello1, Joseph Murphy1, Tony Zhao1,
Booyeon Han1, Nejat K. Egilmez2, David C. Linehan1 and Scott A. Gerber1
1University of Rochester, Rochester NY; 2University of Louisville, Louisville KY
The only curative treatment for pancreatic cancer (PC) is surgical resection, however, most patients
(>80%) are diagnosed with non-resectable late stage disease. While standard therapies including conven-
tional radiotherapy are rarely capable of downsizing locally advanced lesions to surgical candidacy, the
recent emergence of stereotactic body radiation therapy (SBRT) has shown promise. This targeted ap-
proach minimizes toxicity to surrounding tissues, thereby affording the delivery of higher dose fractions.
Given the fundamental role of immune effectors in tumoricidal radioresponses, we hypothesized that an
adjuvant immunotherapy would enhance the antitumor potency of SBRT in advanced pancreas tumors. To
test this, we developed a syngeneic mouse model of PC for treatment with clinically relevant SBRT and
immunotherapy regimens. Mouse pancreas tumor cells stably expressing luciferase were orthotopically
implanted six days prior to SBRT treatment (6 Gy x 4 days) on a Small Animal Radiation Research Plat-
form (SARRP). Twenty-four hours after the final radiation treatment, biodegradable polymer microspheres
(MS) containing the pleiotropic cytokine interleukin 12 (IL12) were injected intratumorally. Remarkably,
whereas SBRT and IL12 MS treatments alone caused moderate reductions in tumor burden, combined
treatments demonstrated tumor eradication and cures. To assess the dependence of SBRT/IL12 MS anti-
tumor activity on downstream Ifng function, we repeated orthotopic modeling in Ifng-/- mice and confirmed
nearly complete therapeutic dependence on the effector cytokine. Cytometric analysis revealed Ifng-
dependent increases of Cd8+ T cells following SBRT, however, concurrent infiltration of immunosuppres-
sive T regulatory and monocyte-derived lineages was also observed. Interestingly, RNA-seq analysis on
intratumoral lymphoid and myeloid cell types demonstrated that the addition of IL12 MS to SBRT resulted
in marked repolarization of both suppressor populations toward antitumor Th1 and M1 forms, respectively.
Antibody depletion studies identified Cd8+ T cells as the sole dependent of the SBRT/IL12 MS response,
and RNA-seq confirmed synergistic increases in Cd8+ T cell activation. In summary, our studies illustrate
a comprehensive repolarization of the immune microenvironment following SBRT and IL12 MS interven-
tion, and advocate this combination immunotherapy as a prospective treatment for locally advanced PC.

60.
Poster #21
Treatment induced PGE2 plays an unexpected beneficial role in the generation
of anti-tumor immunity
Riddhi Falk-Mahapatra1 and Sandra O. Gollnick1, 2
Departments of Immunology1 and Cell Stress Biology2
Roswell Park Cancer Institute; Buffalo, NY
Photodynamic Therapy (PDT) is the treatment of solid malignancies with visible light following
the systemic administration of a tumor-localizing photosensitizer. In addition to generating reactive oxygen
species (ROS) for tumoricidal effect, PDT enhances anti-tumor immunity. Seminal studies from our lab
have demonstrated that PDT-induced acute inflammation is critical for augmentation of anti-tumor immun-
ity. However, the mechanism(s) by which PDT-induced inflammation enhances anti-tumor immunity is
poorly understood. Here we report a beneficial regulatory role of Prostaglandin E2 (PGE2), an inflammato-
ry mediator, in the induction of anti-tumor immunity following PDT. Our studies using a colon carcinoma
model (CT26) and a HPV+ Head and Neck Squamous Cell Carcinoma model (HNSCC) model (MTERL)
demonstrates a rapid increase of PGE2 in in tumor draining lymph nodes (TDLNs) immediately after PDT;
enhanced PGE2 levels diminished to baseline within 24 hours. Administration of NS398, a small-molecule
inhibitor of COX2, to tumor-bearing mice immediately prior to PDT, eliminated the treatment-induced
PGE2 surge and resulted in significantly faster tumor regrowth to endpoint. Our studies using NS398
demonstrate that PGE2 surge following PDT regulates PDT-induced acute inflammation. Most important-
ly, we observe that PGE2 imparts a superior quality to TDLNs for activation of anti-tumor immune effec-
tors (T cells and NK cells) by facilitating accumulation of multiple subpopulations of activated dendritic
cells.
Defining a beneficial role of PDT-induced PGE2 in regulating PDT-enhanced anti-tumor immunity is
of crucial importance for clinical PDT. Patients treated with PDT are routinely administered nonsteroidal
anti-inflammatory drugs (NSAIDs) to reduce pain and swelling that occur both during and post treatment.
Thus, the anti-tumor immune activation function of PDT might be lost. Studies proposed here aim to intro-
duce the argument of shifting the timing of NSAID administration to 24 hours post treatment in order to
take advantage of the anti-tumor immune activating role of PDT.

61.
Poster #19
The CD28-Ars2 axis primes T-cells for expansion
G. Aaron Holling, Shawn M. Egan, Rachel Kandefer, Guanxi Qiao, Jianmin Wang, Kelvin P. Lee,
Elizabeth A. Repasky, and Scott H. Olejniczak
Roswell Park Comprehensive Cancer Institute, Buffalo, NY
It has become increasingly evident that CD28 does much more than amplify TCR signaling, and affects a
variety of intracellular processes that serve to modulate gene expression during priming of naive T-cells.
Despite early studies demonstrating that major effects of CD28 co-stimulation are mediated through RNA,
how regulation of RNA maturation processes contributes to CD28-driven T-cell priming has only recently
begun to be explored. The current study set out to address this significant gap in knowledge by examining
the influence of CD28 on the earliest regulatory events to occur during maturation of RNA polymerase II
transcripts, namely RNA capping and association of the cap-binding complex (CBC) with nascent tran-
scripts. We reasoned that since these early co-transcriptional events are essential to protect nascent tran-
scripts and to guide them to appropriate maturation pathways, their modulation by CD28 could have signif-
icant effects on gene expression in T-cells. We found that, among genes coding for proteins involved in
RNA capping and the CBC, only Srrt, the gene coding for Ars2, was dynamically upregulated in T-cells
exposed to in vitro and in vivo conditions where CD28 signaling is critical for T-cell responses. In vitro
stimulation of T-cells from CD28 knockout mice, or mice harboring knockin mutations of the CD28 cyto-
plasmic tail, revealed that the membrane distal domain responsible for recruitment of the adaptor proteins
Grb2 and Lck was of particular importance for Ars2 upregulation. Functionally, deletion of Ars2 from ma-
ture immune cells resulted in loss of CD8+ T-cell dependent protection from in vivo tumor challenge, likely
as a result of defects in activation-induced metabolic programming and proliferative expansion of T-cells
lacking Ars2. Mechanistically, data suggest a model in which Ars2 supports T-cell activation-induced met-
abolic reprogramming by regulating alternative splicing of mRNAs encoding key metabolic enzymes, such
as pyruvate kinase (PKM), and proliferation by enhancing replication-dependent histone (RDH) mRNA
biogenesis. Importantly, the distal CD28 signaling domain necessary for Ars2 induction also enhanced
RDH mRNA biogenesis and alternative splicing of PKM, suggesting Ars2 may be a critical downstream
mediator of CD28-driven co-transcriptional events that contribute to the well-known ability of CD28 to
stimulate CD8+ T-cell anti-tumor responses.

62.
Poster #38
Impact of adrenergic stress on the “Abscopal Effect” following radiation therapy and on
the anti-tumor response
Minhui Chen, Guanxi Qiao, Bonnie Hylander, Anurag Singh, and Elizabeth Repasky
Roswell Park Comprehensive Cancer Center, Buffalo, NY
Tumor-bearing mice experience a significant amount of adrenergic stress due to mandated mildly
cool housing temperatures, which results in increased norepinephrine production by nerves of the sympa-
thetic nervous system. Our recent publications show that blockade of adrenergic receptors (using beta-
blockers) or reduction of stress through housing at thermoneutral temperatures significantly slows tumor
growth and improves anti-tumor immunity. Thus, we tested here the hypothesis that combining beta-
blockers with radiation or housing mice at thermoneutral temperatures prior to, and subsequent to radia-
tion, would improve its efficacy, not only for tumors in the irradiated field, but also of distant, non-
irradiated tumors. In this study, we treated mice (housed at the standard temperature of ~22 °C) implanted
with colon carcinoma (CT26) cells with a combination of the pan beta-blocker, propranolol and radiation
(6Gy x 1 Day) and found that the combination significantly improved both the response of tumors in the
irradiated field as well as distant, non-irradiated tumors. These effects were lost in immunodeficient SCID
mice, and in mice in which depleted CD8+ T cells, supporting a role for the adaptive immune system. In
addition, when mice were housed at a thermoneutral temperature (32°C), the tumor growth of irradiated
and non-irradiated tumors is much slower than that of mice housed at standard cool temperature (22°C).
Additional experiments in beta2 adrenergic receptor knockout (KO) mice specifically identify a role for
the beta2 adrenergic receptor in mediating control of these effects. Furthermore, we found that intra-
tumoral effector T-cells, including IFN-gamma positive, GzmB positive and T-bet positive CD8+ T-cells,
as well as the ratio of CD8+ T-cells to Tregs, increased while numbers of MDSC and Tregs decreased, in
mice given beta-blockers with radiation. Finally, cured mice treated with propranolol and radiation reject-
ed local and distant rechallenge with tumor in a tumor-specific manner requiring CD8+ T cells. In conclu-
sion, these data suggest that adrenergic stress plays a major role in regulating the efficacy of radiation.
Blockade of beta2 adrenergic signaling could be a useful new strategy in the radiation oncology clinic.
Further, these data highlight the importance of environmental factors in assessment of anti-tumor immuni-
ty following radiation in mouse models. T
This research was supported by: NIH grants (R01 CA099326 and R01 CA205246); The Roswell
Park Alliance Foundation; The National Cancer Institute (NCI) grant P30CA016056 involving the use of
Roswell Park Comprehensive Cancer Center’s Flow and Image Cytometry and Immune Analysis Shared
Resources.

63.
Oral Poster Presentations 3:00-4:15 p.m. Council Rock
Category D
Infection & Vaccines
Chairs: Brent Berwin & Michael Robek

64.
Poster #10
PIP3 Induces Phagocytosis of Non-motile Pseudomonas aeruginosa
Sally Demirdjian, Daniel Hopkins, Hector Sanchez, and Brent Berwin
Geisel School of Medicine at Dartmouth, Lebanon, NH
Pathogenic bacteria that establish chronic infections in immunocompromised patients frequently
undergo adaptation or selection for traits that are advantageous for their growth and survival. Clinical
isolates of Pseudomonas aeruginosa, a Gram-negative, opportunistic bacterial pathogen, exhibit a tem-
poral transition from a motile to a non-motile phenotype through loss of flagellar motility during the
course of chronic infection. This progressive loss of motility is associated with increased resistance to
both antibiotic and immune clearance. We have previously shown that loss of bacterial motility enables
P. aeruginosa to evade phagocytic clearance both in vitro and in vivo and fails to activate the phosphati-
dylinositol 3-kinase
(PI3K)/Akt-dependent phagocytic pathway. Therefore, we tested the hypothesis that clearance of phago-
cytosis-resistant bacteria could be induced by exogenously pretreating innate immune cells with the Akt-
activating molecule phosphatidylinositol-(3,4,5)-trisphosphate (PIP3). Our data demonstrate that PIP3
induces the uptake of non-motile P. aeruginosa by primary human neutrophils >25-fold, and this effect
is phenocopied with the use of murine phagocytes.
However, surprisingly, mechanistic studies revealed that the induction of phagocytosis by PIP3
occurs because polyphosphoinositides promote bacterial binding by the phagocytes rather than bypass-
ing the requirement for PI3K. Moreover, this induction was selective since the uptake of other non-
motile Gram-negative, but not Gram-positive, bacteria can also be induced by PIP3. Since there is cur-
rently no treatment that effectively eradicates chronic P. aeruginosa infections, these findings provide
novel insights into a potential methodology by which to induce clearance of non-motile pathogenic bac-
teria and into the endogenous determinants of phagocytic recognition of P. aeruginosa.

65.
Poster #34
The Role of the CARD9/GM-CSF Axis in Immunity to Candida albicans
M. Landekic, M.Sc.1, I. Angers, M.Sc.3, S. Qureshi, M.D.2,3, M. Divangahi, PhD,
1,3, D. Vinh, M.D.
1Dept of Microbiology and Immunology, 2Dept of Medicine, McGill University; Meakins-Christie
Institue, Montreal, Canada;4Infectious Diseases in Global Health, RI-MUHC, Montreal, Canada
Introduction: Candida albicans is an opportunistic fungal pathogen with a propensity for systemic
disease as well as a high mortality rate. The recently identified p.Y91H mutation in the gene for Caspase
Recruitment Domain-containing protein 9 (CARD9) causes invasive candidiasis in the central nervous sys-
tem, termed “spontaneous CNS candidiasis” (sCNSc). How this mutation leads to invasive candidiasis is
poorly understood. CARD9 is selectively expressed by myeloid cells, especially macrophages and mono-
cytes. Monocyte-derived cells from p.Y91H patients showed impaired expression of the gene csf2, which
codes for granulocyte and macrophage colony stimulating factor (GM-CSF). Patients with sCNSc treated
with adjunt GM-CSF cleared the infection where anti-fungal therapy alone was insufficient. The mecha-
nism of CARD9-mediated GM-CSF protection during anti- fungal immunity remains enigmatic.
Methods: A knock-in mouse model of CARD9Y91H was generated using CRISPR/Cas9 gene edit-
ing. Disseminated candidiasis was modelled using I.V. inoculation with C. albicans SC5413. Mortality was
evaluated by Kaplan-Meier analysis. Histopathology of brain tissue was done to assess extent of fungal
growth and in vivo host response. Immunophenotyping of the myeloid compartment was done by multi-
parametric flow cytometry. Bone-marrow derived macrophages were challenged with C. albicans yeast,
and qPCR performed to assess csf2 expression levels.
Results: When compared to age/sex-matched infected CARD9-wild-type controls, mice homozy-
gous for the p.Y91H mutation demonstrate significantly earlier morbidity and mortality. The immune re-
sponse to infection is impaired in the brain, with a specific defect in the microglia compartment of
CARD9p.Y91H mice. BMDM derived from CARD9p.Y91H mice showed impaired csf2 expression upon
C. albicans challenge.
Conclusions: This model of CARD9 deficiency can now be used to assess the role of GM-CSF and
myeloid cells in the response to disseminated candidiasis, helping us to understand the mechanisms of
CARD9-mediated GM-CSF protection during anti-fungal immunity.

66.
Poster #30
Single dose oral administration of Yersinia pseudotuberculosis vaccine induces specific immunity
against pneumonic Y. pestis infection
Amit K Singh, Xiuran Wang, Wei Sun
Department of Immunology and Microbial Disease,
Albany Medical Center, Albany New York 12208
Email: [email protected], [email protected], [email protected]
In the present study, we assessed the immunogenicity and protective efficacy of a live attenuated Y.
pseudotuberculosis strain in Swiss Webster mouse model. For the construction of live attenuated strain
(χ10069), the pathogenic Y. pseudotuberculosis PB1+ was disrupted with yopJ and yopK virulence genes
as well a selective marker asd gene. The χ10069 mutant was further modified with the introduction of
additional Y. pestis caf1 operon (caf1R-caf1A-caf1M-caf1) and labelled as χ10070. The chimeric protein
YopENt138-LcrV contriving Y. pestis YopENt (1 to 138 amino acids) spliced with LcrV was inserted in
pYA3332 plasmid (pYA5199: yopENt138-lcrV). Constitutive production and T3SS (type 3 secretion system)
mediated delivery of recombinant protein (YopENt138-LcrV) were assessed using in-vitro (Western blot)
and in-vivo (Specific antibody titer and T cell responses) mouse models. Results obtained from the in-vivo
survival study demonstrated the substantial reduction (>100 folds) in virulence of mutant Y. pseudotuber-
culosis infections via oral route of administration. Single-dose oral immunization with 109 CFU of both
mutants χ10069 (pYA5199) and χ10070 (pYA5199) elicited robust LcrV specific antibody titers at 14th
day and stayed stable until 28th day of administration in mice. Antibody isotype profile of serum and
flowcytometric analysis of lung cells demonstrated concurrent induction of CD4+ and CD8+ T cell mediat-
ed Th1 and Th2 immune responses, even after 42nd day of immunization. The comprehensive protection
(>90% survival) achieved against lethal intranasal dose (1.28 × 103) of Y. pestis, with no conspicuous signs
of infection in lung, liver, spleen and Peyer’s patch of immunized mice showed prophylactic abilities of
these recombinant mutants against pneumonic plague. Findings in this study suggested that Y. pseudotu-
berculosis mutants χ10069 (pYA5199) and χ10070 (pYA5199) were effective vaccine, precursors for fur-
ther improvement.

67.
Poster #7
Understanding innate immune responses to a replicating virus-based vaccine platform
Anthony Marchese, Carolina Chiale, Safiehkhatoon Moshkani, and Michael D. Robek
Department of Immunology and Microbial Disease, Albany Medical College, Albany NY
Virus-like vesicles (VLV) are a novel RNA virus-based vaccine platform with the potential to be
used in vaccines for a wide range of human diseases. VLV are an infectious, self-propagating alphavirus-
vesiculovirus hybrid vector that can be engineered to express foreign antigens to elicit a protective immune
response. Unlike viruses and virus-like particles, VLV do not contain capsid structural proteins. Previous
work has shown VLV to be highly immunogenic and non-pathogenic in vivo. We hypothesize that the
unique replication and structural characteristics of VLV create an adjuvant-like effect by acting as potent
inducers of innate immunity, and that these vectors lack pathogenicity due to their sensitivity to the antivi-
ral cytokines they induce. We found that VLV replication is inhibited by interferon (IFN)-alpha, IFN-
gamma, and IFN-lambda, but not by TNF-alpha. In primary human cells and human cell lines, VLV infec-
tion induced the production of IFN-lambda and expression of interferon stimulated genes (ISGs) with anti-
viral activity such as MXA, ISG15, and IFI27. This response was dependent on VLV RNA replication, as
IFN-lambda and ISG expression were abrogated by UV-inactivation of the vesicles. Knockdown of the
pattern recognition receptors RIG-I and MDA5 or the intermediary signaling protein MAVS blocked IFN
production following VLV infection. ISG expression was dependent on IFN receptor signaling through the
tyrosine kinases Jak1 and Jak2, and phosphorylation of the STAT-1 protein. VLV have previously been
shown to be non-pathogenic in wild-type and IFN-alpha/beta receptor knockout mice. In STAT-1 deficient
mice that lack all IFN signaling, we found that intranasal VLV infection spreads to the brain and is lethal,
highlighting the importance of innate immune control of this vector. These results provide new insight into
the safety, immunogenicity, and intrinsic adjuvant-like effects of a novel virus-based vaccine platform
through a mechanism of innate immune activation mediated by a capsid-free, self-replicating RNA.

68.
Poster #27
T-bet-dependent ILC1-derived IFN-γ is required for sustaining inflammatory DCs during
T. gondii infection
Américo López-Yglesias, Ellie T. Camanzo, Elise Burger, Alessandra Araujo, Andrew T. Martin,
Felix Yarovinsky
Center for Vaccine Biology and Immunology, Department of Microbiology and Immunology,
University of Rochester Medical Center, Rochester, New York
Mounting a robust type I immune response is essential for resistance against a variety of intracellu-
lar pathogens, including Mycobacterium, Leishmania, Salmonella, and Toxoplasma gondii. Classically, the
type I immune response has been characterized by the transcription factor T-bet, encoded by Tbx21, which
is considered the master regulator for determining the lineage commitment of CD4+ TH1 cells and their
production of the effector molecule IFN-γ. However, recent investigations by our own group and others
have demonstrated that T-bet is dispensable for a T. gondii-mediated TH1-derived IFN-γ response; yet,
despite robust IFN-γ responses, Tbx21-/- mice rapidly succumbed to parasite infection. Moreover, T-bet-
deficient animals remained significantly more susceptible to T. gondii infection than mice lacking both T
and B cells (RAG2-/-), suggesting that T-bet expression in innate immune cells is paramount to host surviv-
al. It has been demonstrated that T. gondii-mediated recruitment of innate myeloid cells such as mono-
cytes, macrophages, and DCs is critical for host defense against the parasite. Our results demonstrated that
during infection in both Tbx21-/- and RAGγc-/- mice, local DC recruitment was significantly reduced. T-bet-
dependent group 1 innate lymphoid cells (ILC1s) are known to be a critical source of innate IFN-γ
production and pathogen restriction. Therefore, we hypothesized that the absence of ILC1-derived IFN-γ
would result in abrogated recruitment of inflammatory DCs during T. gondii infection. Using Tbx21-/- and
RAGγc-/- mice, our results revealed that ILC1-derived IFN-γ is required for the recruitment of inflammatory
DCs during parasite infection. These data demonstrate that T-bet-dependent ILC1-derived IFN-γ is a cru-
cial cytokine regulating inflammatory DCs during T. gondii infection.

69.
Workshop I
“Getting and Negotiating an Academic Faculty Position”
Dr. Jeremy Boss
NOTES:

70.
Platinum Corporate Sponsor
“TBD”
BD Biosciences
Unavailable at time of printing.

71.
Symposium III
Impact of Microenvironment on the
Immune Response
Chair : Dr. Eyal Amiel

72.
Targeting Immunosuppressive Myeloid Cells to Treat Pancreatic Cancer
David C. Linehan
Center for Tumor Immunology, University of Rochester Medical Center, Rochester, NY, USA
Department of Surgery, University of Rochester Medical Center, Rochester, NY, USA
Wilmot Cancer Institute, University of Rochester Medical Center, Rochester, NY, USA
The incidence of pancreatic ductal adenocarcinoma (PDAC) is rising yet patient survival rates following
conventional interventions remain abysmal, therefore more effective targeted therapies represent a significant un-
met medical need. A unique feature of PDAC tumors is the presence of a prominent mass of non-malignant cells
called the stroma. The PDAC tumor stroma consists of a dense network of fibrotic material including fibroblasts,
extracellular matrix, and soluble factors with an abundant inflammatory immune infiltrate. Bone marrow derived
myeloid cells of both monocytic and granulocytic origin, including immunosuppressive tumor associated macro-
phages (TAM) and neutrophils (TAN) respectively, are the most prevalent immune cells found within the tumor
microenvironment (TME) of PDAC. PDAC tumors co-opt chemokine signaling pathways to attract myeloid cells
from the bone marrow to the TME where they dampen anti-tumor immune responses, support tumor progression,
and confer resistance to therapy. In a previous clinical study, we demonstrated that targeted blockade of CCR2+
TAM with a small molecule inhibitor (PF-04136309) nearly doubled responses to the FOLFIRINOX chemotherapy
regimen in patients with locally advanced PDAC and was associated with augmented anti-tumor T cell responses.
Additionally, we demonstrated that tumor infiltrating CXCR2+ TAN compensate for the loss of TAM following
CCR2 blockade. Therefore, we hypothesized that dual targeting of both CCR2+ TAM and CXCR2+TAN would
further enhance the efficacy of chemotherapy for treating PDAC. In the murine model that recapitulated human
disease, dual targeting of both CCR2+ TAM and CXCR2+ TAN with FOLFIRINOX generated a more robust anti-
tumor response than vehicle, FOLFIRINOX alone, or individual inhibitors with FOLFIRINOX. These data high-
light that the CXC/CXCR2 chemokine pathway plays a key role in PDAC tumor progression, and dual targeting of
both CCR2+ TAM and CXCR2+ TAN represents a novel immunotherapeutic approach to enhance the efficacy of
chemotherapy responses in PDAC.

73.
Β-AR Signaling Suppresses Immunity to Infection And Vaccination By
Blocking Immunometabolism
Yasmin Thanavala
Department of Immunology, Roswell Park Comprehensive Cancer Institute, Buffalo, NY USA
Stress responses evolved as “fight or flight” reactions to improve survival under adverse conditions of
threat from predators, infections and injury. The neuroendocrine system responds to and translates stress
into either beneficial or harmful endpoint effects. For instance, acute stress is considered beneficial as
an immuno-enhancer; contrarily chronic stress is immunosuppressive. While stress impacts host immuni-
ty by neuroendocrine-immune system cross-talk via neurotransmitters, hormones or cytokines, its role in
immune-modulation and disease outcome, although recognized, has not been extensively explored.
While acute stress can enhance immunity, chronic stress is immunosuppressive. Mandated standard hous-
ing temperature of 20-26oC (ST) induces mild, chronic cold stress in laboratory mice stimulating sympa-
thetic response-induced immunosuppressive β-adrenergic pathway in an attempt to activate
thermogenesis to maintain normal body temperature at 37oC. Murine models are routinely used to inves-
tigate the pathophysiology of human disorders to gain critical insights or to evaluate the safety and effi-
cacy of therapeutic approaches intended for human application. Therefore, efforts are being made to bet-
ter understand murine models so that their usefulness as pre-clinical models of human diseases is im-
proved.
We exploited the physiologic model of mild, chronic cold stress to explore the role of chronic adrenergic
system as a physiological regulator of immunosuppression in a model of respiratory infection and
vaccination. Our novel finding establishes for the first time, that housing-stress-induced increased adren-
ergic signaling suppressed CD28-mediated plasma cell fitness/survival, and is likely the mechanism
through which sympathetic stress represses immunity to infection or vaccination. Furthermore, we find
that this immune suppression could be reversed by blocking adrenergic activation using pan-β-AR block-
er propranolol or housing mice at thermoneutral conditions (TT, 30°C) and the improved immunity cor-
related with augmented pulmonary inflammation and rapid bacterial clearance from the lungs of mice.
Importantly our results establish that adrenergic stress reduces the magnitude of inflammatory and im-
mune responses to infection and vaccination by blocking B and T cell metabolic fitness.

74.
Comparative Immunology to Dissect Innate Immune Responses During
Type 2 Inflammation
Elia D. Tait Wojno1 1Baker Institute for Animal Health and Department of Microbiology and Immunology, Cornell University
College of Veterinary Medicine, Ithaca, NY, USA
The Tait Wojno laboratory investigates the cellular and molecular pathways that control type 2 immune
responses during helminth infection and allergic disease. Helminth infection and allergy are associated
with a polarized type 2 inflammatory response. In helminth infection, this response is host-protective,
while during allergy, this response causes pathology and is a result of inappropriate activation against
harmless allergens. Type 2 inflammation is characterized by activation of innate and adaptive immune cells
such as group 2 innate lymphoid cells, basophils, and CD4+ T helper type 2 (Th2) cells; production of the
cytokines interleukin (IL)-4, IL-5, IL-9, and IL-13; and activation of epithelial cells that results in goblet
cell hyperplasia and enhanced mucus production. While previous work has highlighted many pathways
that regulate adaptive Th2 responses, our understanding of innate immune and epithelial cell responses
during type 2 inflammation remains incomplete. Studies in the Tait Wojno laboratory take a unique com-
parative immunology approach to address this gap in knowledge. Ongoing studies focus on unraveling how
prostaglandins and their receptors intersect with cytokine pathways to promote and resolve type 2 inflam-
mation. Additional work investigates the role of direct cell-cell interactions mediated through the Notch
signaling pathway in promoting host-protective innate immune responses during helminth infection. Em-
ploying murine models of parasite infection and allergy alongside analysis of human and canine patient
samples allows us to dissect how cytokines, lipids, and cell-cell interactions shape type 2 inflammatory re-
sponses at mucosal tissues during helminth infection and allergy.

75.
Workshop II
“The Merits (and Perils) of Transitioning from an
Academic to Industry Career”
Dr. Thomas Wynn
NOTES:
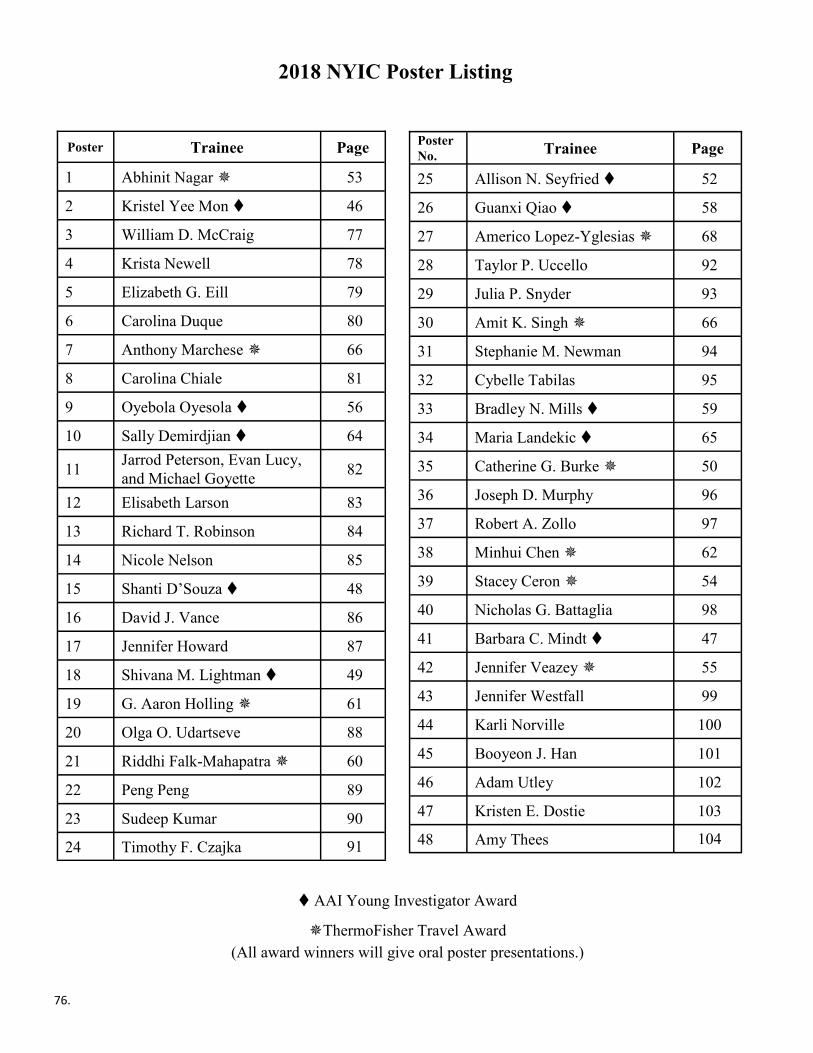
76.
2018 NYIC Poster Listing
⧫ AAI Young Investigator Award
ThermoFisher Travel Award
(All award winners will give oral poster presentations.)
Poster Trainee Page
1 Abhinit Nagar 53
2 Kristel Yee Mon ⧫ 46
3 William D. McCraig 77
4 Krista Newell 78
5 Elizabeth G. Eill 79
6 Carolina Duque 80
7 Anthony Marchese 66
8 Carolina Chiale 81
9 Oyebola Oyesola ⧫ 56
10 Sally Demirdjian ⧫ 64
11 Jarrod Peterson, Evan Lucy,
and Michael Goyette 82
12 Elisabeth Larson 83
13 Richard T. Robinson 84
14 Nicole Nelson 85
15 Shanti D’Souza ⧫ 48
16 David J. Vance 86
17 Jennifer Howard 87
18 Shivana M. Lightman ⧫ 49
19 G. Aaron Holling 61
20 Olga O. Udartseve 88
21 Riddhi Falk-Mahapatra 60
22 Peng Peng 89
23 Sudeep Kumar 90
24 Timothy F. Czajka 91
Poster
No. Trainee Page
25 Allison N. Seyfried ⧫ 52
26 Guanxi Qiao ⧫ 58
27 Americo Lopez-Yglesias 68
28 Taylor P. Uccello 92
29 Julia P. Snyder 93
30 Amit K. Singh 66
31 Stephanie M. Newman 94
32 Cybelle Tabilas 95
33 Bradley N. Mills ⧫ 59
34 Maria Landekic ⧫ 65
35 Catherine G. Burke 50
36 Joseph D. Murphy 96
37 Robert A. Zollo 97
38 Minhui Chen 62
39 Stacey Ceron 54
40 Nicholas G. Battaglia 98
41 Barbara C. Mindt ⧫ 47
42 Jennifer Veazey 55
43 Jennifer Westfall 99
44 Karli Norville 100
45 Booyeon J. Han 101
46 Adam Utley 102
47 Kristen E. Dostie 103
48 Amy Thees 104

77.
Poster #3
Mitochondrial ROS prime cells for the hyperglycemic shift to necroptosis
William D. McCaig, Matthew A. Deragon, Payal S. Patel, Nicole L. Shakerley, Tori A. Smiraglia, and
Timothy J. LaRocca
Albany College of Pharmacy and Health Sciences, Albany NY
Programmed cell death (PCD) is most commonly characterized by the non-inflammatory, caspase-
dependent apoptosis and the inflammatory, caspase-independent necroptosis. Necroptosis is driven by a
complex known as the necrosome, which is comprised of the kinases RIP1, RIP3, and MLKL. Previously,
we observed a novel shift from apoptosis to necroptosis in a hyperglycemic environment. We have shown
that this PCD shift is relevant to neonatal hypoxia-ischemia (HI) brain injury as it was exacerbated by hy-
perglycemia in a RIP1-dependent manner. Moreover, during this injury we observed decreases in down-
stream effectors and components of apoptosis and increases in RIP1 and MLKL. Here we highlight an es-
sential role for reactive oxygen species (ROS) and mitochondrial factors in the hyperglycemic shift of
apoptosis to necroptosis. We show that glycolysis and ROS are critical for the hyperglycemic shift to
necroptosis and that ROS alone are sufficient to produce the cell death shift. ROS were shown to enhance
the oligomerization and activation of RIP1 while they antagonized caspases. Translocation of RIP1,
MLKL, Drp1, and Bax to the mitochondria was enhanced in hyperglycemic conditions. Moreover, inhibi-
tion of these factors prevents the hyperglycemic shift to necroptosis to varying degrees.

78.
Poster #4
Separation of GVHD from GVL Effects by Modulation of TCR Signaling
Krista Newell1, Weishan Huang2, Christian Pacheco1, Avery August2 and Mobin Karimi 1 1Department of Microbiology and Immunology, SUNY Upstate Medical University, Syracuse, NY;
2Department of Microbiology and Immunology, Cornell University, Ithaca, NY
Allogeneic hematopoietic stem cell transplantation (allo-HSCT) for the treatment of hematological
malignancy is burdened by the deleterious effects of T cell-mediated graft-versus-host disease (GVHD).
The key to the future of allo-HSCT as cancer immunotherapy lies in the ability to enhance the beneficial
graft-versus-tumor (GVT) effects of the donor T cells while minimizing their capacity to cause GVHD.
Despite recent progress in developing therapeutic interventions, GVHD remains a significant cause of mor-
bidity and mortality worldwide (20-80%).
Activation and expansion of T cells in response to alloantigens is central to GVHD pathophysi-
ology. Donor T cell receptor interaction with host antigen presenting cells (APC) activates Lck, lead-
ing to ITAM phosphorylation, Zap-70 activation, and subsequent phosphorylation of the adaptors LAT
and SLP-76. IL-2-inducible T cell kinase (ITK) is required for subsequent phosphorylation of phos-
pholipase C–gamma 1 (PLC-g1), a key regulator of calcium mobilization and extracellular signal-
regulated kinase (ERK) activation, as well as its downstream effects. ITK not only influences cytokine
-producing T-cell populations in the periphery, but also regulates differentiation and the development
of distinct innate-type cytokine-producing T-cell populations in the thymus.
To separate GVT from GVHD, our lab has been investigating the impact of manipulating TCR sig-
nal transduction pathways in allo-HSCT settings. We demonstrated that T cells from ITK KO mice do not
induce GVHD but maintain beneficial GVT effects in allo-HSCT recipient mice (B6àBALB/c) challenged
with tumor cells. The attenuated GVHD correlates with reduced proinflammatory cytokine production, de-
fective chemokine receptor upregulation, defects in donor T cell migration to GVHD target organs, and
higher expression of levels of the transcription factor Eomes by ITK KO T cells compared to wildtype
cells. Our data indicate that this defective migration into GVHD target organs contributed to the separation
of GVHD and GVT effects, as ITK KO CD8+ T cells cleared intravenously injected, but not subcutaneous-
ly injected tumor cells in allo-HSCT mice.
Moreover, pharmacological ITK inhibition with 10n attenuated GVHD and preserved GVT func-
tion in wildtype CD8+ T cells. Lastly, ITK inhibition of TCR-activated human T cells also led to reduced
cytokine production and chemokine receptor expression. Together, our data suggest that ITK inhibition
could be used as therapy after allo-HSCT to reduce GVHD while preserving beneficial donor T cell GVT
effects. Future studies aim to elucidate the downstream mechanisms involved in GVT maintenance, migra-
tory defects, and transcriptional regulation of effector cell fate.

79.
Poster #5
M1- and M2-like polarization in macrophages leads to differential expression of Cx43
and gap junction formation
Elizabeth G Eill and Steven M Taffet
SUNY Upstate Medical University, Syracuse, NY
Gap junctions are six-membered protein aggregates that can form a channel, facilitating the intercellular
exchange of small materials between adjacent cells. These junctions act as cytoplasmic bridges and are in-
volved in numerous types of communication. Connexin43 (Cx43), a gap junction protein, is ubiquitously
expressed by many cells of the immune system and has been implicated in various functions. In antigen-
presenting cells, Cx43 is thought to be involved in antigen transfer and cross-presentation. In mononuclear
phagocytes such as macrophages, the function of gap junctions is well characterized but the requirements
for Cx43 expression and gap junction formation are not well understood. Here we demonstrate how M1-
and M2-like polarization stimuli differentially influence Cx43 expression and gap junction plaque for-
mation in bone marrow-derived macrophages (BMDMs) and RAW264.7 macrophage-like cells. In
BMDMs, LPS and IFN-g (gamma), M1-like mediators, are potent inducers of Cx43 expression but demon-
strate little Cx43 trafficking to the membrane. Uniquely, the addition of IL-10, an M2-polarization cyto-
kine, encourages Cx43 trafficking to the membrane. RAW264 macrophage-like cells were transfected with
Cx43 tagged with GFP (green fluorescent protein) expressed under the CMV promoter. We have deter-
mined that expression from that promoter is induced by LPS. LPS stimulation of transfected RAW264
cells leads to significant increase in GFP signal with a significant amount of gap junction plaque formation.
The addition of IL-10 does not increase the expression from the CMV promoter but does increase the
amount of Cx43-GFP protein while significantly increasing gap junction plaques. The increase in Cx43-
GFP is presumably through stabilization of the protein plaque. In contrast to IL-10, the addition of a sec-
ond M2 polarizing cytokine, TGF-b (beta), does not alter the level of Cx43 protein and reduces the pres-
ence of Cx43 plaques. These results suggest a complex regulation of Cx43 gap junction formation in M2
macrophage populations.

80.
Poster #6
IL-33 and the prostaglandin D2 receptor CRTH2 regulate ILC2 accumulation in tissues during
type 2 inflammation
Oyebola O. Oyesola, Carolina Duque, and Elia D. Tait Wojno
Cornell University, Ithaca NY
ILC2s are rare innate immune cells that produce large amounts of type 2 cytokines and play an im-
portant role in allergic inflammation and helminth expulsion. These cells accumulate at sites of type 2 in-
flammation, but the mechanisms that regulate ILC2 population expansion during inflammation are not ful-
ly understood. Previous studies show that a variety of biochemical mediators may contribute to the regula-
tion of ILC2 population size in tissues. Current models suggest that during type 2 inflammation, interleu-
kin (IL)-33 mediates ILC2 proliferation of tissue-resident ILC2s, whereas prostaglandin D2 (PGD2) medi-
ates migration of new ILC2s into the tissue from the periphery or other sites via its receptor chemoattract-
ant receptor-homologous molecule expressed on TH2 cells (CRTH2). However, how these two pathways
coordinate to regulate ILC2 population size in the tissue in vivo remains undefined. In this study, we show
that exogenous IL-33 treatment increases PGD2 synthase mRNA levels, suggesting that IL-33 elicits an
increase in PGD2 production. Following exogenous treatment with IL-33, we observed a partial ablation in
IL-33-elicited ILC2 accumulation in the CRTH2-deficient mice compared to wild type (WT) mice, indicat-
ing that CRTH2-dependent ILC2 accumulation in tissues occurs downstream of IL-33. The decreased
ILC2 accumulation in CRTH2-deficient mice in response to IL-33 was not a result of reduced proliferation
or increased apoptosis or cell death. Rather, we observed that ILC2s in CRTH2-deficient mice treated with
exogenous IL-33 had decreased expression of chemokine receptors, including CCR4, which is expressed
on human and murine ILC2s. Together, these data suggest a model in which ILC2 population size in tis-
sues during type 2 inflammation is coordinated by the IL-33 and PGD2-CRTH2 pathways, with IL-33 pro-
moting ILC2 proliferation and the production of PGD2 and PGD2-CRTH2 interactions regulating ILC2
migration and chemokine receptor expression. An increased understanding of the networks of biochemical
mediators that regulate, ILC2 accumulation will inform the use and development of drugs that target these
mediators during type 2 inflammatory conditions such as allergic disease and helminth infection.

81.
Poster #8
Differential infection of dendritic cell subsets by virus-based vaccine platforms
Carolina Chiale and Michael D. Robek
Department of Immunology and Microbial Disease, Albany Medical College, Albany NY
The use of virus-based vectors as vaccine platforms represents an attractive alternative to conven-
tional immunization with non-replicating antigens. Virus infection of cells leads to the production of path-
ogen- and damage-associated molecular patterns, which promote inflammation, cytokine production, and
the potent induction of immune responses. However, the precise mechanism by which many virus-based
vaccine vectors prime the immune response remains unknown. Dendritic cells (DCs) are powerful antigen
presenting cells, and different DC subtypes are characterized by their specialized functions. Conventional
dendritic cells (cDCs) efficiently present antigens to T cells via direct and/or cross-presentation, plasmacy-
toid dendritic cells (pDCs) produce significant amounts of antiviral type I IFN, and monocyte-derived den-
dritic cells (moDCs) are induced under inflammatory conditions and promote initial T cell activation in
draining lymph nodes. These DC subtypes play key roles in priming immune responses and controlling
virus replication, but their functions in generating protective immunity following vaccination with viral
vectors are not well understood. We hypothesize that highly immunogenic viral vectors infect dendritic
cells and promote direct antigen presentation that leads to the efficient induction of antigen-specific T
cells. We found that distinct DC subtypes are differentially infected with specific virus-based vaccine vec-
tors, and display differences in activation following infection. Bone marrow derived DCs that are produced
by GM-CSF stimulation and resemble moDCs rapidly succumb to infection. In contrast, bone marrow de-
rived DCs that are produced by Flt3L stimulation and resemble cDCs and pDCs are efficiently infected but
survive. DC activation following virus infection as measured by expression of the cell surface markers
CD80, CD86, and MHC class II was more pronounced in the Flt3L-derived DCs, and showed distinctive
requirements for further virus replication. Although Flt3L-derived DCs produced significant amounts of
type I IFN when infected with virus-based vaccine platforms, the mechanism of DC infection and activa-
tion was independent of the ability of the cells to signal through the type I IFN receptor. These results
highlight the differential survival and activation of specific DC subsets following infection with viral vac-
cine vectors, and indicate potentially unique roles of DC subtypes in activating the immune response fol-
lowing immunization.

82.
Poster #11
Characterization of macrophage growth, maturation and polarization: best practices
for the Thp-1 cell line
Jarrod Peterson, Evan Lucey, Michael Goyette, and Kristen Porter, Ph.D.
Westfield State University, Westfield Ma
Macrophages are a vital piece of the innate immune system, which can be divided into different
subtypes with a broad spectrum of functions. M1 (classically activated) macrophages are involved in clear-
ance of infection, while M2 (alternatively macrophages) and M2 subtypes (alternatively activated macro-
phages) are linked to Th-2 like responses, chronic inflammation, and wound healing. While there are copi-
ous information on phenotypes and effector functions of these macrophage subtypes, the data is conflicting
at best, in both cell lines and in vivo systems. We posit that different methods for growth conditions, mono-
cyte maturation, and differentiation between laboratories are responsible for the conflicting literature.
Thus, using the laboratory standard Thp-1 monocytic cell line, common culturing and maturation condi-
tions were used and subsequent macrophage phenotypes were compared to each condition and primary
macrophage data. Differences in morphology, cytokine and protein expression, HIV susceptibilityi and
wound healing capabilities clearly demonstrate the dramatic difference culturing and treatment conditions
make toward macrophage polarization. These results may provide standardization of protocols for macro-
phage plasticity studies, in order to reduce conflicts in the literature and provide an in vitro model that re-
flects what have been observed among in vivo systems.

83.
Poster #12
Phenotyping IgE-binding monocytes in seasonal allergic disease of Icelandic Horses
Elisabeth Larson, Susanna Babasyan, Fahad Raza, Christiane Schnabel, Bettina Wagner
Cornell University, College of Veterinary Medicine, Ithaca NY
During an allergic reaction, Fc epsilon RI (FcεRI) bound IgE is responsible for the initial signaling
in response to allergen. In most species, FcεRI is predominantly found on mast cells, basophils and eosino-
phils. However, in humans and horses (but not rodents) this receptor is also present on a subpopulation of
monocytes (IgE+ monocytes). Human studies report that IgE+ monocytes produce IL-10, but the down-
stream effect on allergic response has yet to be described. Human and equine basophils express a three
subunit IgE receptor (αβɣ2 – alpha/beta/gamma) while human IgE+ monocytes only express an αɣ2 (alpha/
gamma) variant. Previously, the IgE+ receptor on equine IgE+ monocytes had not been characterized.
In this study, our Icelandic horse allergy model is a model for human IgE-mediated seasonal allergy
because they 1) experience naturally occurring seasonal IgE-mediated allergic disease called seasonal Culi-
coides hypersensitivity, and 2) have a population of IgE+ monocytes. Icelandic horses are also a model for
equine IgE-mediated seasonal allergy because Culicoides hypersensitivity occurs more readily in Icelandic
horses imported from Iceland to the United States compared to other breeds.
We hypothesize that these IgE+ monocytes promote the development of allergen tolerance. Here
we show the novel findings that, as in humans, IgE+ monocytes in horses express the αɣ2 variant of FcεRI
and produce interleukin-10 (IL-10) upon receptor cross-linking. In addition, we have characterized the sur-
face expression of IgE+ monocytes in horses: MHCII+ CD14lo CD16- CD163-. These results support a reg-
ulatory role for IgE+ monocytes and are consistent with IgE+ monocytes in humans. Further study of the
downstream effects of equine IgE+ monocyte stimulation will have a translational benefit for both human
and equine allergy medicine. Specifically, we hope to use IgE+ monocytes and their IgE-mediated secreted
factors as biomarkers for immunotherapy treatment effectiveness and success.

84.
Poster #13
Attenuated TH1 and TH17 differentiation underlies susceptibility to nontuberculous mycobacteria
infection in otherwise healthy toddlers
Richard T. Robinson
The Ohio State University, Columbus OH
Nontuberculous mycobacteria (NTM) cause disease with increasing frequency in the non-AIDS popula-
tion, including otherwise healthy toddlers. We took advantage of our being in a region with higher than
expected prevalence of NTM infection (NTMI), and have identified geographical and immunological vari-
ables that associate with NTMI. Geographical data from a first-of-its-kind database created by the Wiscon-
sin Mycobacteriology Laboratory Network (WMLN) demonstrate that NTMI prevalence is geographically
biased, as new cases occur primarily within “hot spot” clusters of urban and rural counties. The etiological
agents of NTMI in hot spot clusters were more diverse than those which caused NTMI in “cold spot” clus-
ters. NTM-infected toddlers living in these clusters—who were otherwise healthy prior to infection and
undergoing surgical treatment for NTM lymphadenitis—donated blood and lymph node (LN) tissue that
we assayed for defects in innate recognition and TH cell differentiation. Although innate recognition of my-
cobacterial products was intact, naïve TH cells from the blood and LN of NTMI children had an attenuated
capacity for TH1 and TH17 differentiation. Non-naïve TH cells likewise expressed less IFNgamma and IL17
following activation, and the levels of IFNgamma and IL17 in the NTMI LNs were unchanged compared
to uninfected control LNs. Attenuated TH1/TH17 differentiation was not observed in controls (i.e. children
undergoing tonsillectomy for obstructed sleep apnea), nor children with lymphadenitis due to an unrelated
bacterial species (Staphylococcus aureus). Consistent with attenuated TH17 responsiveness, neutrophil in-
flux into the site of infection was undetectable in NTMI patients. Collectively, our data demonstrate that
NTMI susceptibility is associated with both geographical and immunological factors, the latter being an
attenuated capacity for TH1/TH17 differentiation.

85.
Poster #14
Optimization of a fusion protein-based vaccine strategy via analysis of Fc receptor-induced
dendritic cell maturation
Nicole Nelson, Sudeep Kumar, Ph.D., Victoria Cahill, and Edmund Gosselin, Ph.D.
Albany Medical College, Albany, NY
We previously showed, using immune complexes (mAb-iFt) and a novel fusion protein (FP), that
targeting immunogens to Fc receptors intranasally generates protection against mucosal pathogens. How-
ever, multiple boosts of the FP are required for optimal protection. We therefore sought to improve the FP
design to reduce the number of doses required. While stimulation of bone marrow-derived dendritic cells
(BMDCs) with LPS or mAb-iFt increased maturation marker expression, administration of FP resulted in
no change. We hypothesized that designing the FP to generate BMDC maturation equivalent to that of
mAb-iFt-stimulation would lead to a more effective vaccine. To test this, we focused on three major differ-
ences between the mAb-iFt and FP design: toll-like receptor (TLR) engagement, Fc gamma receptor
(FcγR) crosslinking, and the particulate nature of the mAb-iFt.
Studies with TLR4-/- BMDCs, TLR2-/- BMDCs, and a TLR2/4 inhibitor demonstrated that neither
TLR is required for the maturation of mAb-iFt-stimulated cells. In addition, BMDCs stimulated with both
iFt and FP did not exhibit an increase in maturation markers comparable to mAb-iFt-stimulated BMDC,
further implying a requirement for something other than TLR engagement. We next examined FcγR cross-
linking by adding another human FcγRI binding region to the original FP (tFP). However, stimulation of
BMDCs with the tFP also failed to induce maturation. To further increase the FP’s FcγR crosslinking po-
tential, and mimic the particulate nature of mAb-iFt, magnetic beads were coated with FP. These FP-beads
did not induce maturation either. We therefore hypothesized that TLR ligation in combination with FcγR
crosslinking would induce DC maturation. Hence, we engineered a particle that contains both the FP and a
synthetic TLR2 ligand. Indeed, this particle design does result in increased DC maturation marker expres-
sion. Future studies will test the efficacy of this particle in vivo, as well as examine cytokine secretion from
BMDCs stimulated with the soluble FP or particulate forms of the FP with and without TLR ligand.

86.
Poster #16
Contribution of An Unusual CDR2 Element of a Single Domain Antibody in Ricin Toxin Binding
Affinity and Neutralizing Activity
Michael J. Rudolph1,*, David J. Vance2,*, Simon Kelow3,4, Siva Krishna Angalakurthi5, Sophie Nguyen1,
Simon A. Davis1, Yinghui Rong2, C. Russell Middaugh5, David D. Weis6, Roland Dunbrack, Jr.4,
John Karanicolas4, and Nicholas J. Mantis2 *, These authors contributed equally to this study.
1New York Structural Biology Center, New York, New York 10027; 2Division of Infectious Diseases,
Wadsworth Center, New York State Department of Health, Albany, NY 12208; 3Dept. of Biochemistry
and Molecular Biophysics, University of Pennsylvania, Philadelphia, PA 19104; 4Institute for Cancer Re-
search, Fox Chase Cancer Center, Philadelphia, PA 19111; 5Department of Pharmaceutical Chemistry and
Macromolecule and Vaccine Stabilization Center, University of Kansas, Lawrence, KS 66045; 6Department of Chemistry and Ralph Adams Institute for Bioanalytical Chemistry, University of Kansas,
Lawrence, KS 66045
Ricin toxin’s enzymatic subunit (RTA) has been subjected to intensive B cell epitope mapping
studies using a combination of competition ELISAs, hydrogen exchange-mass spectrometry, and X-ray
crystallography. Those studies identified four spatially distinct clusters (I-IV) of toxin-neutralizing
epitopes on the surface of RTA. Here we describe A9, a new single domain camelid antibody (VHH) that
was proposed to recognize a novel epitope on RTA that straddles clusters I and III. The X-ray crystal struc-
ture of A9 bound to RTA (2.6 A resolution) revealed extensive antibody contact with RTA’s beta-strand h
(732 A2 buried surface area; BSA), along with limited engagement with alpha-helix D (90 A2) and alpha-
helix C (138 A2). Collectively, these contacts explain the overlap between epitope clusters I and III, as
identified by competition ELISA. However, considerable binding affinity, and, consequently, toxin-
neutralizing activity of A9 is mediated by an unusual CDR2 containing five consecutive Gly residues that
interact with alpha-helix B (82 A2), a known neutralizing hotspot on RTA. Removal of a single Gly residue
from the penta-glycine stretch in CDR2 reduced A9’s binding affinity by 10-fold and eliminated toxin-
neutralizing activity. Computational modeling indicates that removal of a Gly from CDR2 does not perturb
contact with RTA per se, but results in the loss of an intramolecular hydrogen bond network involved in
stabilizing CDR2 in the unbound state. These results reveal a novel configuration of a CDR2 element in-
volved in neutralizing ricin toxin.

87.
#17
Type I Interferon-dependent IL-18 Suppresses Hematopoiesis In Severe Shock-Like
Ehrlichial Infection
Jennifer Howard, Hui Jin Jo, Julianne Smith, and Katherine MacNamara
Department of Immunology & Microbial Disease, Albany Medical College, Albany, NY
Ehrlichiosis is an emerging tick-borne disease that can range from mild to severe and lethal. In a
model of severe ehrlichial infection caused by Ixodes ovatus ehrlichia (IOE), type I interferons and IL-18
induce severe pathology. Here we show that IL-18 production is significantly reduced in IFNαR-/-
(interferon alpha receptor knock-out) mice, thus suggesting IFNα/β (interferon alpha/beta) causes patholo-
gy, in part, via induction of IL-18. Based on our previous observation that type I IFNs drive a loss of hema-
topoietic stem cell and progenitor cell populations (HSC/HSPC) via both direct and indirect mechanisms,
we hypothesized that type I IFN-dependent IL-18 reduced HSC/HSPCs during IOE infection, thus impair-
ing emergency hematopoiesis. Emergency hematopoiesis allows for an enhanced immune cell production
rate to respond to infection; thus, IL-18-/- mice are expected to respond better to IOE infection. In response
to IOE infection we observed an increase in bone marrow cellularity in IL-18-/- mice, relative to WT mice.
IL-18-/- mice experienced a decrease in levels of quiescent (G0) ST (short-term) HSCs when infected with
IOE, while WT mice saw a slight increase in G0 ST HSCs following IOE infection at both 6 and 7dpi, sug-
gesting IL-18 suppresses proliferation of ST-HSCs. The change in bone marrow cellularity may have also
been influenced by cell death, as IOE infection increased necrotic cell death in WT mice, relative to infect-
ed IL-18-/- mice. Therefore, in the absence of IL-18, ST-HSCs are more active and protected from cell
death, potentially contributing to the overall increase in BM cellularity in IL-18-/- mice. Our data support a
model wherein type I interferons drive hematopoietic suppression via direct signaling in progenitors and
through the induction of IL-18.

88.
Poster #20
Interleukin-1: hidden in the prostate cancer biology
Olga O. Udartseva1 and Sandra O. Gollnick1,2
Departments of Cell Stress Biology1 and Immunology2, Roswell Park Comprehensive Cancer Center,
Buffalo NY
The inflammatory microenvironment has been shown to play an important role in cancer progres-
sion and metastasis. Interleukin-1 (IL-1) is a potent pro-inflammatory cytokine essential for host defense
against pathogens. The contribution of IL-1 in cancer progression remains controversial and understudied.
In particular, it has been demonstrated that IL-1β levels are elevated in tumors and serum of prostate can-
cer patients with advanced disease. However, the cellular and molecular basis underlying IL-1-mediated
tumorigenesis is unclear. Recently our lab identified the IL-1α pathway as a molecular mechanism respon-
sible for myeloid-driven prostate cancer development. Here, we report that IL-1 signaling in tumor cells
promotes prostate cancer progression.
this questionTo identify the contribution of IL-1 signaling in prostate cancer progression, we gener-
ated androgen-sensitive shIL1R1-LNCaP and androgen-independent shIL1R1-PC-3M prostate tumor cell
lines lacking IL1R1 – the functional receptor for IL-1α and IL-1β. As expected, cells knockdown for
IL1R1 demonstrated impaired responsiveness to IL-1. IL-1α stimulation decreased expression of androgen
receptor in prostate cancer cells (LNCaP) while downregulation of IL-1 signaling abrogated this effect. At
the same time knocking down of IL1R1 significantly inhibited expression of neuron-specific enolase
(marker of neuroendocrine differentiation ) in androgen-insensitive PC-3M cells while neuroendocrine dif-
ferentiation was shown to be associated with poor survival outcome. More importantly, downregulation of
IL-1 signaling in cancer cells resulted in significant inhibition of prostate tumor growth. We established
this phenomenon and found several possible mechanisms underlying it. No difference was found either in
tumor cell proliferation or in myeloid cell infiltration between shIL1R1-PC-3M and control tumors. How-
ever, tumors lacking IL1R1 demonstrated significant reduction of cyclooxygenase-2 (COX2) expression;
COX2 immunosuppressive activity has been implicated in the development and progression of prostate
cancer. Furthermore, we found that downregulation of IL-1 signaling inhibited tumor angiogenesis. ItIm-
paired vascularization of tumors lacking IL1R1 was accompanied by increased expression of angiopoietin-
2 while VEGF levels remained unchanged. TUNEL staining revealed increased number of apoptotic cells
in shIL1R1-PC-3M tumors indicating that knocking down of IL1R1 made tumor cells more susceptible to
cell death in vivo.
Taken together, our findings indicate that downregulation of IL-1 signaling modifies prostate can-
cer cell phenotype and suppresses tumor growth. Further studies will identify if combination of IL-1 target-
ed and anti-androgen therapies could be beneficial for prostate cancer treatment and improve advanced
prostate cancer outcome.

89.
Poster #22
CD28 Mediates Pro-Survival Response Through Autophagy In Multiple Myeloma
Peng Peng, Adam Utley, Louise Carlson, Colin Chavel and Kelvin Lee
Immunology Department, Roswell Park Comprehensive Cancer Center, Buffalo, NY 14203
Multiple myeloma (MM) is a proliferative plasma cell malignancy in the bone marrow. Despite ad-
vances in MM treatment, developed resistance to chemotherapy and subsequent relapse are current thera-
peutic challenges that demonstrate the urgent need for novel therapeutic targets. Advanced knowledge of
the pro-survival response in MM is critical to develop new therapeutic approaches. We have previously
demonstrated that MM and its non-malignant counterpart, bone marrow resident plasma cells (BMPC), ex-
press the T cell co-stimulation receptor CD28, and that CD28 signaling plays a central role in support of
stress adaptation and survival of both MM and BMPC. However, how CD28 mediates a pro-survival re-
sponse remains largely undetermined. Recent studies have shown that autophagy is crucial for BMPC sur-
vival and long-lived humoral immunity, suggesting its intrinsic role in pro-survival response. Autophagy is
a conserved self-digestive recycling process in response to multiple stresses. It has also been demonstrated
that autophagy mediates resistance in MM and inhibition of pro-survival autophagy enhances MM death.
However, little is known in MM as to which pro-survival signals regulate autophagy and how they may do
so. In this study, we found CD28 mediates a pro-survival response through autophagy in MM. Crosslink-
ing CD28 by activating antibody or myeloid dendritic cells (DC), which express the CD28 ligands CD80
and CD86, prevent chemotherapy or serum deprivation induced death in MM. Blocking of autophagy by
chemical inhibitors or genetic manipulation abrogates the pro-survival effects of CD28. We also found in-
creased autophagy upon CD28 crosslinking by antibody or binding to its ligands on DC, elucidated by in-
creased LC3 lipidation or elevated autophagosome number per cell. By using a dual fluorescence reporters
system, we have shown that CD28 ligation in MM induces autophagic cargo degradation. Given that CD28
signaling has been shown to increase cellular metabolic fitness, we found inhibition of autophagy minimiz-
es mitochondrial respiratory capacity. This might indicate that CD28-mediated autophagy maintains meta-
bolic homeostasis to improve MM viability and drug resistance. Taken together, our findings suggest au-
tophagy is important in CD28-mediated pro-survival response and can be targeted in combination of with
CD28 treatment for MM patients.

90.
Poster #23
Optimization of an Antigen Presenting Cell Targeting Approach
for Improved Mucosal Vaccine Delivery
Sudeep Kumar, Raju Sunagar, Edmund J Gosselin
Albany Medical College, Albany NY, 12208, USA
Subunit vaccines are preferred over whole-cell-killed or live-attenuated vaccines due to their enhanced
safety profile. However, purified microbial products used as vaccine antigens exhibit poor immunogenicity
and require adjuvants to elicit adequate immune protection. Thus far there is no adjuvant licensed for mu-
cosal immunization. Our lab has previously demonstrated that targeting the Streptococcus pneumonia (Sp),
protective antigen PspA, to antigen presenting cells (APCs) generate antibody-mediated protection against
Sp. This protection, however, requires a prime and two booster immunizations. In this investigation, we
have modified the APC-targeting component to enhance the efficacy of our mucosal vaccine platform. The
modified vaccine construct trivalent-anti-h-FcgRI-PspA (Trivalent-FP) has three human-FcgRI binding
antibody moieties, compared to the previously tested bivalent-anti-h-FcgRI-PspA (Bivalent-FP), which has
only two human- FcgRI binding antibody moieties. When groups of human FcgRI expressing mice were
immunized with both the vaccine constructs, the Trivalent-FP group induced significantly higher levels of
Sp-specific IgG and protection against a lethal Sp challenge compared to the Bivalent-FP group. The triva-
lent-FP also induced higher levels of mucosal IgA and better protected against Sp infection compared to
the Bivalent-FP. In addition, only one booster immunization was required to generate more than 90% pro-
tection. We are further characterizing the immune mechanism of this enhanced protection.

91.
Poster #24
Single Domain Antibodies that Target Ribosome Binding Surfaces on Ricin's A Chain
Neutralize Enzymatic Activity
Timothy F. Czajka1,2, Michael J. Rudolph3, David J. Vance1, and Nicholas J. Mantis1,2
1Division of Infectious Diseases, New York State Department of Health, Albany NY 12208; 2Department
of Biomedical Sciences, University at Albany, Albany NY 12201; 3New York Structural Biology Center,
New York, NY 10027
Ricin toxin, derived from the ubiquitous castor plant, is a highly lethal potential future biothreat
agent with no available cure. It is an AB ribosome inactivating protein (RIP), which enters mammalian
cells via endocytosis and retrograde transport to the endoplasmic reticulum as a holotoxin, after which the
two subunits dissociate and the enzymatic A subunit (RTA) is secreted into the cytosol. In the cytosol, the
acidic P-stalk proteins of the 60S ribosome bind RTA and guide it to the sarcin-ricin loop (SRL), where-
upon it cleaves a critical adenine residue, thus halting protein synthesis. We have isolated a library of al-
paca-derived, heavy chain-only antibody VH domains (VHHs) directed against RTA. Using a combination
of X-ray crystallography and hydrogen-deuterium exchange mass spectrometry (HX-MS) analyses, we
have identified VHHs that specifically bind both of RTA’s ribosome-interacting surfaces. In this study, we
demonstrate using a cell-free translation assay broad range of neutralizing activity for this subset of VHHs
against RTA. The ability to neutralize RTA’s enzymatic activity is strongly correlated with binding affini-
ty, as measured by surface plasmon resonance, with affinities ranging from 0.227 to 17.8 nM. We rea-
soned that highest affinity VHHs might have potential to neutralize RTA intracellularly by blocking RTA-
ribosome interactions. To test this, we generated mammalian expression vectors encoding V9E1 and
V2A11, which target the P-stalk binding site and the active site of RTA, respectively, and transfected them
into Vero cells prior to ricin challenge. While these studies are ongoing, preliminary results suggest ex-
pression of either VHH increases cell viability after ricin treatment, as compared to control cells transfected
with a botulinum neurotoxin-specific VHH, ciA-H7. Our results are consistent with V9E1 exerting its neu-
tralizing effects in the cytosol, considering that V9E1’s epitope is only accessible on RTA, not ricin holo-
toxin. We postulate that the use of high affinity intracellular VHHs targeting the ribosome interfaces on
RTA may not only provide a powerful therapeutic option for ricin intoxication, but also an invaluable tool
for better understanding the elusive mechanisms of RTA after it has exited the ER.

92.
Poster #28
Cross-Talk between the Immune and Nervous Systems: Effect of the Beta-Blocker,
Propranolol, on the Immune Response Generated after Stereotactic Body Radiation
Therapy (SBRT) in an Orthotopic Pancreatic Cancer Model
Taylor P. Uccello1, Bradley N. Mills1, Elizabeth A. Repasky2, David C. Linehan1,
Scott A.Gerber 1University of Rochester School of Medicine and Dentistry, Rochester NY
2University of Buffalo Roswell Park Comprehensive Cancer Center, Buffalo NY
Pancreatic cancer (PC) is a leading cause of cancer related deaths worldwide with a dismal 5- year
survival rate of only 8%. Unfortunately, the incidence of PC is predicted to increase exponential-
ly partially due to lack of effective therapies. In light of remarkable advances in treating other malignan-
cies, therapies for PC have yielded unpromising clinical results. Therefore, a considerable amount of re-
search is needed to understand more about this disease and develop innovative treatment options.
To address this, we have focused on the immune and sympathetic nervous systems; two
systems commonly thought to have divergent functions, but now understood to be intertwined. The nerv-
ous system is important in regulating many processes such as the well documented “fight or flight re-
sponse”, and recently a novel role in maintaining immune homeostasis has been recognized. For exam-
ple, interactions between the nervous and immune systems mediated by beta- adrenergic receptor (BAR)
signaling have been shown to dampen immune cell activity and augment tumor cell proliferation.
Therefore, we hypothesized that altering the communication between these two systems by suppress-
ing BAR signaling would enhance the anti-tumor immune response and inhibit tumor growth and metas-
tasis. To test this, we developed a preclinical PC model where KCKO pancreatic tumor cells, derived from
KC mice (p48cre/LSL-KRASG121D) that develop spontaneous PC, are injected orthotopically in the
pancreas and monitored noninvasively using the IVIS Spectrum in vivo Imaging System (IVIS). To
model standard-of-care therapy commonly used clinically, established pancreatic tumors were treated
with stereotactic body radiation therapy (SBRT) with or without a beta-blocker that inhibits BAR signal-
ing, propranolol. Interestingly, administration of propranolol coupled with SBRT resulted in a potent re-
duction of tumor burden and increased long term survival, with 67% of mice cured. To explore mechanism
of action, in vitro experiments using various murine PC cell lines were carried out. These experiments
suggest the response observed in vivo is not a direct result of the drug acting as a radiosensitizer or in an
exclusively toxic manner.
With these preliminary studies, we have begun to understand more about the complex interac-
tion between the nervous and immune systems. By modifying the cross-talk between nerves and immune
cells, we hope to repurpose the nervous system to mediate a more robust immune response in the tumor
microenvironment.

93.
Poster #29
Syk-dependent Glycolytic Reprogramming in Dendritic Cells Regulates IL-1b (beta)
production to Fungal-associated Ligands in a TLR-independent Manner
Julia P. Snyder1, Phyu M. Thwe1, Daniel Fritz2, Alexandra Ojemann3, Nicholas A. Galasso3, Leslie Sepa-
niac1, Benjamin J. Adamik3, Laura E. Hoyt2,3, Matthew E. Poynter4, and Eyal Amiel5
1Predoctoral student or graduate of the Cellular, Molecular, and Biomedical (CMB) Sciences Graduate
Program at the University of Vermont
2Research Technician at the University of Vermont 3Undergraduate student at the University of Vermont
4 Vermont Lung Center, University of Vermont, Burlington, VT 05405, USA; Division of Pulmonary Dis-
ease and Critical Care, Department of Medicine, University of Vermont, Burlington, VT 05405, USA;
Cellular, Molecular, and Biomedical Sciences Graduate Program, University of Vermont, Burlington,
VT 05405, USA 5 Department of Biomedical and Health Sciences, University of Vermont, Burlington, VT 05405, USA;
Cellular, Molecular, and Biomedical Sciences Graduate Program, University of Vermont, Burlington,
VT 05405, USA
Dendritic cells (DCs) activated via TLR ligation experience metabolic reprogramming, in which the
cells are heavily dependent on glucose and glycolysis for the synthesis of molecular building blocks essen-
tial for maturation, cytokine production, and the ability to stimulate T cells. Although the TLR-driven met-
abolic reprogramming events are well documented, fungal-mediated metabolic regulation via C-type Lec-
tin Receptors such as Dectin-1 is not clearly understood. Here, we show that activation of DCs with fungal-
associated ligands induces acute glycolytic reprogramming that supports the production of IL-1b (beta) via
inflammasome formation. This acute glycolytic induction to fungal-associated ligands is depending on Syk
signaling in a TLR-independent manner, suggesting now that different classes of innate immune receptors
functionally induce conserved metabolic responses to support immune cell activation. These studies pro-
vide new insight into the complexities of metabolic regulation of DCs immune effector function regarding
cellular activation associated with protection against fungal microbes.

94.
Poster #31
Exploring the Role of Neuritin:
A Potential Marker of Activation in Regulatory T cells
Stephanie M. Newman, Stephanie N. Sass, Baiju Sharda, Robert A. Zollo, Joseph Barbi
Roswell Park Comprehensive Cancer Center, Buffalo NY
The immune system is comprised of diverse cells whose effector functions need to be tightly regu-
lated in order to avoid the aberrant targeting of self (i.e. autoimmunity), or collateral damage resulting from
excessive immune activation in response to infectious threats or innocuous commensal microbes. One ma-
jor mechanism to restrain immune function and prevent these pathologies are the Foxp3 expressing CD4+
Regulatory T Cells (Tregs). These cells are known to exhibit multiple characteristic suppressive mecha-
nisms such as anti-inflammatory cytokines and modulation of antigen presenting cells in order to promote
tolerance. In normal physiology, Treg mediated immune suppression enforces immune homeostasis. How-
ever, immunosuppression, by means of Tregs, can negatively impact the desirable tumor immune response
and result in poor patient prognosis and ineffective response to immunotherapies or tumor vaccines. Thus,
understanding of the mechanisms that control the activation and stabilization of Tregs is crucial for poten-
tially improving immunotherapeutic interventions for a broad range of diseases. Our work has demonstrat-
ed that neuritin, a cell surface protein previously shown to modulate nerve growth, is uniquely expressed
by Tregs in the immune system, and its expression is upregulated following activation. Using both the in-
trinsic spectrum of neuritin expression and genetic ablation of neuritin models, our goal was to elucidate
the properties of Tregs expressing distinct levels of neuritin. Multi-color flow cytometric analysis of acti-
vation markers and molecules involved in Treg function implicated a role of neuritin as a marker for deter-
mining the activation status of Tregs and their potential suppressive ability. Furthermore, our findings sug-
gest a role of neuritin on the phenotype of peripheral tissue resident Tregs, such as those infiltrating the
bone marrow and tumor, which are known to be sites of immune privilege and enhanced immune toler-
ance. This apparent link between expression of neuritin and an activated or effector cell-like phenotype in
Tregs suggests that this neurotrophin may be relevant to the detrimental restraint of the antitumor response
and the necessary protection of the hematopoietic niche site. Thus, neuritin may constitute a novel, poten-
tially targetable, marker of tumor-immunity obstructing Tregs or a factor that, if supplemented, may have
therapeutic potential for specific autoimmune diseases. Altogether, our findings may impact the future de-
velopment of Treg modulating therapies.

95.
Poster #32
Microbial exposure in early life alters the CD8+ T cell response to infection in adulthood
Cybelle Tabilas1, Norah L. Smith1, Brian D. Rudd1
Department of Microbiology and Immunology1
During the past decade, there has been increased interest in understanding how microbial exposure
can impact our immune system. Multiple lines of evidence have shown that the degree of microbial expo-
sure early in life has long-lasting effects on the health status of an individual. However, the underlying ba-
sis of how environmental microbes shape the immune compartment is not well understood. With the recent
development and popularization of the pet-shop “dirty” mouse model, researchers have started to appreci-
ate the interplay between environmental microbiota and host immunity. However, it remains unclear how
timing of microbial exposure impacts the immune compartment. In this study, we examined how high mi-
crobial exposure early in development impacts the CD8+ T cell compartment. To do this, we developed an
approach to make laboratory mice dirty for the entirety of an animal’s early development. We then com-
pared the behavior of CD8+ T cells from mice developed in the dirty environment to CD8+ T cells derived
from mice raised in specific pathogen free conditions. Consistent with previously reported data, we found
CD8+ T cells in the steady state appear to adopt a more reactive effector phenotype and accumulate in
higher numbers. We next asked if this phenotype would translate to better host protection against intracel-
lular infection. Surprisingly, unlike adult-exposed mice, which showed improved immune control, mice
exposed to a dirty environment in utero and early stages of development had an impaired ability to clear
listeria infection. Collectively, these data suggest the timing of microbial exposure is critical in shaping the
development and behavior of the CD8+ T cell compartment both in the steady state and during infection.
In the future, we plan to use a fate-mapping approach to understand how microbial exposure alters the on-
togeny of the CD8+ T cell compartment at the population and individual cell level.

96.
Poster #36
CXCR1/2 inhibition with checkpoint blockade enhances chemotherapy responses in
pancreatic ductal adenocarcinoma
Joseph D Murphy2,3, Booyeon J Han1,2,3, Brian A Belt2,3, Jian Ye2,3, Scott A Gerber1,2,3, and David C Line-
han2,3,4 1Department of Microbiology and Immunology, University of Rochester, Rochester, NY, USA. 2Center for
Tumor Immunology, University of Rochester Medical Center, Rochester, NY, USA. 3Department of Sur-
gery, University of Rochester Medical Center, Rochester, NY, USA. 4Wilmot Cancer Institute, University
of Rochester Medical Center, Rochester, NY, USA.
Pancreatic ductal adenocarcinoma (PDAC) is a highly recalcitrant and lethal disease that remains
the fourth leading cause of cancer deaths. With increasing incidence and abysmal patient survival rates fol-
lowing conventional treatment, PDAC is a compelling candidate for different treatment modalities. The
PDAC tumor microenvironment (TME) has a characteristic dense stroma and abundant inflammatory im-
mune infiltrate. Most notably, bone marrow derived myeloid cells are prevalent immune cells in the TME
of PDAC. PDAC tumors co-opt chemokine signaling pathways to attract myeloid cells from the bone mar-
row to the TME where they suppress anti-tumor immune responses, support tumor progression, and confer
resistance to therapy. Previous data show that immune suppressive myeloid cells of granulocytic origin, or
tumor associated neutrophils (TAN), are elevated after chemotherapy and thus remain targets for immuno-
therapy. Here, we show tumor infiltrating CXCR2+ TAN depletion by CXCR1/2 blockade and T cell influx
by áPD-1/áCTLA-4, so we hypothesized that the combination therapy of both CXCR1/2 and checkpoint
inhibition with conventional chemotherapy would further enhance PDAC tumor regression and survival.
Murine PDAC cell lines derived from spontaneous tumors were orthotopically implanted into
C57BL/6 mice. Tumor bearing mice were randomized and treated with vehicle, FOLFIRINOX, CXCR1/2i
(SX-682), CXCR1/2i + FOLFIRINOX, á(alpha)PD-1/á(alpha)CTLA-4 + FOLFIRINOX, or CXCR1/2i + á
(alpha)PD-1/á(alpha)CTLA-4 + FOLFIRINOX. Mice were followed for survival and tumor growth by bio-
luminescence. Resected tissues from mice sacrificed at a fixed time point were fixed in formalin for histo-
logical staining and immunohistochemistry, or digested into single cell suspensions for flow cytometry
analysis.
Orthotopically implanted PDAC tumors in mice upregulated CXCR2+ TAN in the peripheral blood
and TME compared to normal pancreas controls. The TAN levels increased with FOLFIRINOX treatment,
as expected. Immune modulation with either CXCR1/2i or áPD-1/áCTLA-4 in conjunction with FOLFIRI-
NOX resulted in reduced overall tumor burden. CXCR1/2i significantly decreased infiltrating CXCR2+
TAN and checkpoint blockade enhanced cytotoxic T cell influx and activation. However, dual targeting of
both CXCR2+ TAN and anti-tumor cytotoxic CD8+ T cells reprogrammed the tumor immune microenvi-
ronment by blocking immune suppressive myeloid cells and thus sustaining a robust anti-tumor T cell re-
sponse for improved tumor regression and survival.
In conclusion, combination therapy with CXCR1/2 inhibition and checkpoint blockade further en-
hances the efficacy of chemotherapy responses in PDAC. Thus, further studies exploring complementary

97.
Poster #37
The Impact of Neuritin on the Tumor-Leukocyte Interface
Robert A. Zollo, Stephanie M. Newman, Stephanie N. Sass, Baiju Sharda, Joseph Barbi
Roswell Park Comprehensive Cancer Center
The important role played by T regulatory cells (Tregs) in maintaining immune homeostasis has
been extensively studied and repeatedly demonstrated. Furthermore, the presence or absence of these cells
as well as their fitness and characteristic function can be crucial in determining patient outcomes in a num-
ber of diseases. Examples of these include the disparate settings of immune dysregulation observed in au-
toimmunity and cancer. Treg-mediated immune suppression is achieved through and array of secreted anti
-inflammatory cytokines and membrane bound molecules that have immunosuppressive properties. De-
spite the undeniable importance of Treg function across a range of diseases, the mechanisms responsible
for their ability to maintain tolerance and shape the immunological character/composition of diverse mi-
croenvironments are not well understood. Neuritin is a novel immunomodulatory neurotropin originally
discovered and almost exclusively studied in the nervous system. It was also recently shown to be selec-
tively expressed by Tregs. Prior studies revealed that neuritin expression by Tregs is important for their
suppressive function in mouse models of cancer and autoimmune disease. Notably, in the tumor microen-
vironment (TME), Tregs dramatically upregulate neuritin expression, and production of neuritin by tumor
cells themselves has been reported as well. These observations suggest that this factor contributes to tumor
progression and a reduced anti-tumor immune response within the TME. Therefore, we set out to deter-
mine the impact of neuritin expression and exposure on the properties and behavior of Tregs and other
cells at the interface of the tumor and the immune system. Our preliminary flow cytometric analysis and in
vitro cellular assays, suggest that neuritin may foster immune suppression by altering the proliferative
phenotypes of T cell subsets including Tregs, as well as their metabolic preferences. These data point to
potential roles of neuritin in the restructuring of immune responses in favor of tolerance in the cancer set-
ting. They also suggest that augmenting neuritin-mediated functions may be therapeutic in the setting of
autoimmune disease or transplant biology.

98.
Poster #40
Response to fractionated and ablative radiotherapy does not depend on RIPK1
mediated necroptosis
Nicholas G. Battaglia, Scott A. Gerber, and Edith M. Lord
University of Rochester Medical Center, Rochester NY
Hypofractionated radiotherapy (RT) is often used clinically to treat solid tumor malignancies. Alt-
hough RT was previously thought to work solely by inducing cell death through excessive DNA damage,
recent work by our lab and others has demonstrated the importance of the anti-tumor immune response
elicited by RT, generating interest in combining RT with immunotherapy. Our previous work has exam-
ined the immune response following high dose, ablative RT and found that CD8 T cells producing IFN-γ
(gamma) were essential for reduction in tumor volume. Ablative doses are rarely used clinically as they
result in excessive damage to healthy tissue surrounding the tumor, thus we sought to compare the efficacy
of our ablative schedule to a hypofractionated schedule of an equivalent biologically effective dose.
Through comparing these different RT schedules, we have observed that ablative RT results in more dura-
ble local tumor control whereas tumor regression following hypofractionated RT is transient. Interestingly,
both schedules result in similar levels of tumor-infiltrating CD8 T cells that are necessary for response to
RT, suggesting either inadequate activation and/or suppression of the anti-tumor immune response follow-
ing hypofractionated compared to ablative RT. While both RT schedules result in similar levels of direct
tumor cell killing, different RT doses can result in different mechanisms of cell death. In vitro, immuno-
logically quiescent apoptosis has been shown to be the predominant cell death mechanism following low
dose RT, whereas higher doses of radiation can induce more immunostimulatory necroptosis. Thus, we
hypothesized that ablative RT induced more necroptotic tumor cell death, creating a more inflammatory
environment that resulted in better efficacy of therapy. Using the necroptosis inhibitor Nec-1S, we have
observed that inhibition of necroptosis has no effect on response to either fractionated or ablative RT, sug-
gesting that RIPK1 mediated necroptosis does not dictate response to either schedule. By further analyzing
other forms of necroptosis and other mechanisms of cell death in these schedules, we hope to elucidate
mechanisms by which we can improve the immune response to hypofractionated RT, resulting in more
efficacious treatment clinically.

99.
Poster #43
TRAIL (CD253) Sensitizes Human Airway Epithelial Cells to Ricin-Induced Cell Death
Yinghui Rong1, Jennifer Westfall1, Dylan Ehrbar1, Timothy LaRocca2,
and Nicholas J. Mantis1
1Division of Infectious Diseases, New York State Department of Health, Albany NY 12208; and 2Albany College of Pharmacy and Health Sciences, Albany NY 12208
Inhalation of ricin toxin is associated with the onset of acute respiratory distress syndrome (ARDS),
characterized by hemorrhage, inflammatory exudates, and tissue edema, as well as the near complete de-
struction of the lung epithelium. Here we report that the Calu-3 human airway epithelial cell line is rela-
tively impervious to the effects of ricin, with little evidence of cell death even upon exposure to microgram
amounts of toxin. However, the addition of exogenous soluble TNF-Related Apoptosis Inducing Ligand
(TRAIL; CD253) dramatically sensitized Calu-3 cells to ricin-induced apoptosis. Calu-3 cell killing in re-
sponse to ricin and TRAIL was partially inhibited by caspase-8 and caspase-3/7 inhibitors, consistent with
involvement of extrinsic apoptotic pathways in cell death. We employed nCounter Technology to define
the transcriptional response of Calu-3 cells to ricin, TRAIL, and the combination of ricin plus TRAIL. An
array of genes associated with inflammation- and cell death were significantly up regulated upon treatment
with ricin toxin, and further amplified upon addition of TRAIL. Of particular note was IL-6, whose expres-
sion in Calu-3 cells increased 300-fold upon ricin treatment and more than 750-fold upon ricin and TRAIL
treatment. IL-6 secretion by Calu-3 cells was confirmed by cytometric bead array. Based on these finding,
we speculate that the severe airway epithelial cell damage observed in animal models following ricin expo-
sure is a result of a positive feedback loop driven by pro-inflammatory cytokines like TRAIL and IL-6.

100.
Poster #44
Synergistic effects of cytokine combinations (IL-2, IL-12 and IL-18) and their potential for
tumor immunotherapy
Karli Norville, Denise Skrombolas, and John G. Frelinger
Department of Microbiology and Immunology, University of Rochester School of
Medicine and Dentistry, Rochester, NY
Cytokines are potent immune mediators that often act in concert between immune cells that are in
close proximity. They are involved in many immune processes, but their role in T cell proliferation and
differentiation during the initiation of an immune response has been a major research focus. However, a
number of studies have shown that cytokines can also act at later stages of an immune response, includ-
ing the effector stage. Intriguingly, some cytokines, including IL-2, appear to activate viral antigen spe-
cific T cells, inducing IFN-γ (gamma) production, in a T-cell receptor independent fashion. In the current
work, we used a Luminex assay to examine the ability of IL-2, IL-12, and IL-18 alone or in pairwise
combinations, to stimulate the expression of a panel of key cytokines and chemokines (IFN-γ (gamma),
IL-4, IL-5, IL-10, IP-10, MIG, TNF- (alpha) and IL-13) from a mixed population of spleen cells. We sur-
veyed multiple time-points for a kinetics analysis of early vs late expression of this panel of analytes in
order to get a better understanding of how IL-2, IL-12, and IL-18 work in concert to activate immune
cells. All three cytokines when administered individually showed a robust activation of IFN-γ (gamma)
and the downstream chemokines MIG and IP-10, indicating a strong type 1 response. Moreover, pairwise
combinations of the three cytokines revealed a striking synergy in the ability of spleen cells to produce
these same cytokines and chemokines. Intriguingly, only the combination of IL-2 and IL-18 induced ex-
pression of IL-5 and IL-13, type 2 cytokines, indicating that each combination may differentially polarize
the responses. Further, our initial flow data showed that spleen cultures treated with cytokine combina-
tions (IL-2 and IL-12) resulted in a higher percentage of activated effector CD8 and NK cells and a re-
duced percentage of Tregs in these cultures. Importantly, the combination of IL-2 and IL-12 shifted the
balance in favor of effector CD8 T cells over Tregs. These data suggest that cytokine combinations could
have synergistic effects in enhancing immune effectors at the sites of tumors as well as promoting an IFN
-γ (gamma) dominant anti-tumor response. While, systemic delivery of cytokines has shown promise,
severe toxicity has limited their use. To overcome this limitation, we will present a novel strategy to sys-
temically deliver combinations of inactive cytokine fusion proteins, while targeting their biological activ-
ity and synergistic effects at the sites of tumors.

101.
Poster #45
Targeting the Tumor Draining Lymph Node to Enhance Anti-tumor Responses in
Pancreatic Ductal Adenocarcinoma
Booyeon J. Han, Bradley N. Mills, Brian A. Belt, David C. Linehan, Scott A. Gerber
University of Rochester Medical Center, Rochester, NY
Pancreatic ductal adenocarcinoma (PDAC) is a solid malignancy with abysmal patient surviv-
al rates for which little is known about the role of the tumor draining lymph node (tdLN)
in tumor immunity. The tdLN is essential in establishing immune surveillance and tolerance, and
thus is a possible mechanism of resistance for immune-based therapies. Previous studies in several
cancer models have shown tdLNs to initially mount an anti-tumor response but eventually establish
systemic tolerance resulting in tumor progression. This transition may arise from inadequate antigen
presentation by dendritic cells to stimulate anti-tumor effector T cells within the tdLN. In a mouse
model of PDAC, we observe that the pancreatic tdLN loses its potential to reduce tumor burden and
increase host survival after prolonged exposure to tumor factors. We therefore hypothesize that the
immune microenvironment in the tdLN is adaptable with both anti-tumor and pro-tumor potential that
can be modulated by tdLN-targeted immunotherapy. Recent work has demonstrated that the PDAC
tdLN generates a tumor-promoting immune response that suppresses anti-tumor immune cells, includ-
ing Type 1 T helper cells (TH1 cells) and cytotoxic T lymphocytes (CTLs). To activate and mobilize
this anti-tumor potential, we aim to reprogram the immunosuppressive tdLN and tumor by targeting
the tdLN with an immune-stimulating strategy. Preliminary data demonstrate that administration of the
immune stimulatory cytokine interleukin 12 (IL-12) via microspheres (IL-12MS) decreased pancreatic
tumor burden and increased survival, especially in combination with standard stereotactic body radia-
tion therapy. In addition, with fluorescence microscopy, fluorescently- labeled MS can be visualized in
the tdLN as early as 30 minutes after intratumoral injection. Therefore, we hypothesize that the IL-
12MS traffic to the PDAC tdLN and increase intranodal IL-12 levels. Abundant intranodal IL-12 can
enhance tumor-associated antigen presentation and induce substantial TH1 differentiation and activa-
tion. The combination of both events results in more impressive stimulation of CD8+ CTLs and subse-
quently increases cytotoxic activity in the TME for tumor eradication. The most recent data confirm
that CD8+ T-cell activation is specific to the tdLN, whereas activation of CD4+ T cells is more sys-
temic as evidenced by presence in the non-tdLN as well. A thorough investigation of the tdLN in the
context of PDAC will further inform the therapeutic value of targeting the pancreatic lymph node to
mobilize anti-tumor potential and reverse immunosuppression in the TME for dramatic tumor regres-
sion and improved host survival.

102.
Poster #46
CD28 Induces Mitochondrial Respiration Dependent Reactive Oxygen Species (ROS) Signaling for
Metabolic Fitness and Survival in Long-Lived Plasma Cells
Adam Utley1, James Cooper1, Peng Peng1, Wensheng Liu1, Shivana Lightman1, Colin Chavel1,
Louise Carlson1, Kelvin Lee1
1Roswell Park Comprehensive Cancer Center, Immunology Department, Buffalo, NY
Durable humoral immunity is dependent upon antigen-specific antibody production by bone mar-
row resident long-lived plasma cells (LLPCs). We have published that CD28, the canonical T cell costimu-
latory molecule, is required for LLPC survival both in vitro and in vivo; however, the mechanistic basis for
this is unclear.
Upon T cell activation, CD28 induces glycolysis at the expense of mitochondrial respiration. CD28
activation in LLPCs induces PI3k-dependent expression of the glucose transporter glut1 as well as in-
creased glucose uptake. Interestingly, CD28 does not increase the extracellular acidification rate (ECAR),
a direct readout of glycolysis. After the first steps of glycolysis, 6-carbon glucose molecules are processed
into 3-carbon pyruvate molecules that can be used in mitochondrial respiration. CD28 activation in LLPCs
increases the basal oxygen consumption rate (OCAR), a direct readout of mitochondrial respiration, as well
as the maximal respiratory capacity. CD28 also increases mitochondrial mass in LLPCs and in human plas-
ma cell lines. This leads to a model wherein CD28-mediated glucose uptake is required for pyruvate de-
pendent mitochondrial respiration and enhanced metabolic fitness in LLPCs.
A major byproduct of mitochondrial respiration is the production of reactive oxygen species (ROS).
Interestingly, CD28-mediated survival is dependent upon mitochondrial respiration-derived ROS. How
ROS are able to provide a pro-survival signal to LLPCs, however, is unknown. Although well character-
ized as cell damaging agents, ROS are also known to induce both PI3k signaling via inhibition of PTEN as
well as NFkB activation. ROS inhibition leads to decreased Akt phosphorylation, suggesting that respira-
tion-derived ROS are augmenting PI3k-Akt signaling for enhanced glucose uptake and metabolic fitness.
Indeed, inhibition of PI3k prevents CD28-mediated LLPC survival.
Recently published data demonstrate an absolute requirement for the Irf4 transcription factor in
plasma cell survival. CD28 activation induces Irf4 expression in LLPCs in an NFkB dependent manner.
Interestingly, inhibition of ROS leads to decreased NFkB activity, similar to its effect on PI3k signaling.
When we inhibited NFkB, it diminished CD28-induced metabolic fitness in mitochondrial mass, basal
OCR and the maximal respiratory capacity. Furthermore, in Irf4 heterozygous mice, bone marrow resident
LLPCs with high mitochondrial mass are absent. LLPCs in CD28 knock-out mice have a similar mitochon-
drial defect. This leads to a global model wherein CD28 activation induces glucose uptake in LLPCs for
mitochondrial respiration-dependent production of ROS. ROS then reinforce survival signaling through
augmentation of both PI3k and NFkB signaling to increase Irf4 expression and metabolic fitness for LLPC
survival.

103.
Poster #47
Metallothionein, a small stress response protein, acts as a potential co-stimulatory molecule in
mitogen-induced lymphoproliferation
Kristen E. Dostie, Michael A. Lynes.
University of Connecticut, Storrs CT
Metallothioneins (MTs) are small metal-binding proteins that are upregulated in response to various
stressors such as heavy metal exposure, reactive oxygen and reactive nitrogen species, acute proinflamma-
tory signals, and glucocorticoids. MTs are biochemically unique proteins due to their unusually high cyste-
ine content, which accounts for ~33 mol% of the total amino acid composition. These cysteines provide
sulfhydryl groups which bind essential metal cations such as zinc and copper and can also serve to regulate
the local redox environment. Although MT has conventionally been considered an intracellular protein, it
has been found in extracellular compartments where it can act as a potential “danger signal” that can medi-
ate both innate and adaptive immune responses. There is evidence that the various roles of MT in the im-
mune response may be implicated in the pathogenesis of various autoimmune diseases.
Previous work in our lab has shown that exogenous metallothionein has a mild proliferative effect
on some lymphocyte populations and has a synergistic effect on the proliferative response when co-
administered with B or T cell mitogen. This suggests MT may act as a costimulatory signal that enhances
the vigor of the response to proliferative stimuli in the context of cellular stressors.
To characterize the role MT-mediated enhancement of mitogen-induced proliferation, two possible
mechanisms were addressed. First, the antioxidant activity conferred by the abundant free thiols in the MT
structure may provide a redox remodeling system to promote proliferative signaling by redox-sensitive sur-
face molecules. Alternatively, MT may provide a costimulatory signal for proliferation by interacting with
a surface receptor, as it has been shown that MT binds to the surface of murine lymphocytes. To determine
the effect of MT’s antioxidant capacity on mitogen-induced proliferation, mitogen-treated lymphocytes
were administered with various thiol-containing molecules including 2-mercaptoethanol, glutathione, and
cysteine-containing MT peptides at concentrations that provide equal reactive thiols. These thiol-
containing molecules yielded inconsistent effects on mitogen-induced proliferation. These results suggest
that MT-mediated enhancement of mitogenic proliferation may only partly be due to thiol-mediated redox
remodeling and may also involve a receptor-mediated signaling component. Future experiments will ad-
dress the potential role of MT interaction with signaling receptors on the lymphocyte surface to initiate or
propagate proliferative signaling events. Determining MT’s mechanism of action may elucidate the role of
endogenous MT in the context of the chronic inflammatory stress associated with autoimmune disease and
thus lead to novel therapeutic opportunities.

104.
Poster #48
The role of PmtA in Pseudomonas aeruginosa infections
Amy V. Thees, Michael Lynes
University of Connecticut, Storrs, CT
Metallothioneins are highly conserved proteins and well-studied in mammals as inflammatory mediators.
Bacterial metallothioneins share sequence homology and heavy metal binding role with their eukaryotic
counterparts, but their potential role in host-microbe interactions is not well-defined. Pseudomonas aeru-
ginosa PAO1, a chronic cystic fibrosis pathogen, produces a bacterial metallothionein, known as PmtA.
Here we show that PmtA modulates the production of redox-active phenazine compounds. Compared to
wild-type, expression of phzH, phenazine-1-carboxamide enzyme, and phzM, pyocyanin precursor en-
zyme, were decreased in PmtA mutant. Deletion of PmtA also results in decreased biofilm production
compared to wild-type. We also demonstrate PmtA’s ability to influence immune cell proliferation and
block SDF-1α-mediated chemotaxis in Jurkat T cells and primary splenocytes. In collaboration with
StressMarq Inc., we developed an antibody to target PmtA. Targeting PmtA may present a new type of
therapeutic strategy in the treatment of antibiotic-resistant P. aeruginosa infections.

105.
Symposium IV
Immunoregulation and
Homeostasis
Chair : Dr. Steve Szczepanek

106.
Hyperglycemia potentiates a shift from apoptosis to RIP1-dependent necroptosis
William D. McCaig1*, Payal S. Patel1*, Sergey A. Sosunov2, Nicole L. Shakerley1, Tori A. Smiraglia1,
Miranda M. Craft1, Katharine M. Walker1, Matthew A. Deragon1, Vadim S. Ten2,
and Timothy J. LaRocca1,3
1Department of Basic and Clinical Sciences, Albany College of Pharmacy and Health Sciences, Albany,
NY, 12208; 2Department of Pediatrics, Columbia University, New York, NY, 10032; *These authors contributed equally to this work
Apoptosis and necroptosis are the primary modes of eukaryotic cell death, with apoptosis being non-
inflammatory while necroptosis is highly inflammatory. We previously demonstrated that, once activated,
necroptosis is enhanced by hyperglycemia in several cell types. Here, we determine if hyperglycemia af-
fects apoptosis similarly. We show that hyperglycemia does not enhance extrinsic apoptosis but potentiates
a shift to RIP1-dependent necroptosis. This is due to increased levels and activity of RIP1, RIP3, and
MLKL, as well as decreased levels and activity of executioner caspases under hyperglycemic conditions
following stimulation of apoptosis. Cell death under hyperglycemic conditions was classified as necropto-
sis via measurement of markers and involvement of RIP1, RIP3, and MLKL. The shift to necroptosis was
driven by RIP1, as mutation of this gene using CRISPR-Cas9 caused cell death to revert to apoptosis under
hyperglycemic conditions. The shift of apoptosis to necroptosis depended on glycolysis and production of
mitochondrial ROS. Importantly, the shift in PCD was observed in primary human T-cells. Levels of RIP1
and MLKL increased, while executioner caspases and PARP1 cleavage decreased, in cerebral tissue from
hyperglycemic neonatal mice that underwent hypoxia-ischemia (HI) brain injury, suggesting that this cell
death shift occurs in vivo. This is significant as it demonstrates a shift from non-inflammatory to inflamma-
tory cell death which may explain the exacerbation of neonatal HI-brain injury during hyperglycemia.
These results are distinct from our previous findings that hyperglycemia enhanced necroptosis under condi-
tions where apoptosis was artificially inhibited. Here we demonstrate a shift from apoptosis to necroptosis
under hyperglycemic conditions while both pathways are fully active. Therefore, while our previous work
documented that intensity of necroptosis is responsive to glucose, this work sheds light on the molecular
balance between apoptosis and necroptosis and identifies hyperglycemia as a condition that pushes cells to
undergo necroptosis despite the initial activation of apoptosis.

107.
Group 2 Innate Lymphoid Cells Regulate Pulmonary Immunity and Tissue Homeostasis
Jörg H. Fritz
McGill University
Montreal, Canada
Group 2 innate lymphoid cells (ILC2) represent an evolutionary old member of the family of ILCs that
orchestrate innate as well as adaptive immune responses as they interact with and influence several im-
mune and non-immune cell populations at mucosal surfaces. ILC2 rapidly secrete large amounts of type 2
cytokines upon activation that contribute to protective as well as detrimental immune responses depend-
ing on kinetic, location, and physiological context. Despite their scarcity ILC2 are the dominant ILC pop-
ulation in the lung, highlighting a key role as first responders and amplifiers upon immune challenge. In
my presentation I will provide an overview of known and potential ILC2 interactions within the unique
pulmonary environment and will further outline our recent observations regarding pulmonary ILC2 dur-
ing immune challenge including respiratory infections. In addition, I will discuss our recent findings stud-
ying the molecular requirements of IL-33-mediated ILC2 activation and its effects on pulmonary inflam-
mation.

108.
Extracellular Adenosine Shapes PMN Phenotype and Their Ability to Kill Streptococcus
Pneumoniae by Blunting IL-10 Production
Nalat Siwapornchai*, James N. Lee*, Essi Tchalla†, Sara E. Roggensack*, John M. Leong*
and Elsa N. Bou Ghanem†
*Department of Molecular Biology and Microbiology at Tufts University School of Medicine,
Boston, MA 02111 USA †Department of Microbiology and Immunology, University at Buffalo School of Medicine,
Buffalo, NY 14203, USA
PMNs are crucial for initial control of Streptococcus pneumoniae (pneumococcus) early on during
lung infection, however, as the infection progresses their persistence in the lungs is detrimental to the host.
CD73 is surface enzyme that is required for production of extracellular adenosine (EAD), which we previ-
ously showed is required for host resistance against pneumococcal pneumonia in mice. It is now appreciat-
ed that PMNs in tissues can be heterogeneous, so we wanted to explore if the inability of PMNs to control
pneumococci later in infection correlated with phenotypic differences, particularly in CD73 expression,
over time. We found that 18 hours after intratracheal inoculation of C57BL/6 mice with S. pneumoniae, a
time point at which PMN presence in the lungs no longer correlated with control of bacterial numbers, both
the percentage of pulmonary PMNs expressing CD73 and the amount of CD73 expressed on PMNs de-
creased dramatically. PMNs from CD73-/- mice failed to kill pneumococci ex vivo and supplementation
with exogenous EAD was sufficient to reverse this defect, indicating that EAD production was crucial for
PMN anti-microbial activity. Adoptive transfer of PMNs from wild type mice prior to lung challenge was
sufficient to boost the resistance of CD73-/- mice to infection. Further, EAD-mediated resistance could be
largely attributed to its effects on IL-10 secretion. PMNs from CD73-/-, but not wild type mice, up-
regulated IL-10 production upon pneumococcal infection ex vivo and during lung challenge, responses that
were blunted by exogenous EAD. Addition of recombinant IL-10 impaired the ability of PMNs from wild
type mice to kill pneumococci ex vivo, and treatment with anti-IL-10 boosted the anti-bacterial activity of
CD73-/- PMNs. Importantly, administering anti-IL-10 also significantly boosted the resistance of CD73-/-
mice to pneumococcal lung infection. This study demonstrates that EAD, produced by CD73, enhances
PMN anti-microbial function by blunting IL-10 responses. These findings further suggest that there are
changes in PMNs in the lungs over the course of bacterial pneumonia and that CD73 expression on PMNs
may be indicative of a PMN phenotype that is associated with lower anti-microbial activity.

109.
Keynote Speaker
Jeremy M. Boss, Ph.D. Emory Chair in Basic Sciences Research
Professor and Chair
Department of Microbiology & Immunology
Emory University School of Medicine
“Epigenetic Regulation of B Cell Differentiation”
Abstract not provided.
Memory T and B cells constitute our primary system of defense against reoccurring infectious disease and
the ability to form these cells is the ultimate goal of vaccination. Dr. Kaech’s laboratory aims to under-
stand how memory T cells are generated during infection and vaccination, and why, in some circumstanc-
es, an immunization fails to induce long-term T cell immunity. We are also learning how T cells are regu-
lated in tumor microenvironments to better understand how their functions become suppressed as they
infiltrate tumors in order to develop new methods of immunotherapy that enhance anti-tumor responses.

110.
Authors Index
CS – Corporate Speaker P# - Poster Number
KS - Keynote Speaker S – Speaker
O - Oral Poster Presentation
Page (s)
*************************************************************************************
Adamik, Benjamin J. ........................................................................................................... 93
Amiel, Eyal .......................................................................................................................... 41, 93
Angalakurthi, Siva Krishna ................................................................................................. 86
Angers, I. ............................................................................................................................. 65
Araujo, Alessandra .............................................................................................................. 68
August, Avery ...................................................................................................................... 78
Babasyan, Susanna .............................................................................................................. 83
Barbi, Joseph ....................................................................................................................... 94, 97
Battaglia, Nicholas G. (P40) ................................................................................................ 98
Belt, Brian A. ....................................................................................................................... 96, 101
Berwin, Brent (S) ................................................................................................................. 41, 64
Bucsek, Mark J. ................................................................................................................... 58
Burger, Elise ........................................................................................................................ 68
Burke, Catherine G. (P35, O) .............................................................................................. 50
Cabrera-Martinez, Berenice ................................................................................................. 37
Cahill, Victoria .................................................................................................................... 85
Camanzo, Ellie T. ................................................................................................................ 68
Campioli, Pamela ................................................................................................................. 56
Carlson, Louise M. .............................................................................................................. 49, 89, 102
Ceron, Stacey (P39, O) ........................................................................................................ 54
Chapman, Timothy .............................................................................................................. 55
Chavel, Colin ....................................................................................................................... 89, 102
Chen, Minhui (P38, O) ........................................................................................................ 58, 62
Chiale, Carolina (P8) ........................................................................................................... 67, 81

111.
Connolly, Kelli A. ............................................................................................................... 59
Cooper, James ...................................................................................................................... 102
Craft, Miranda M. ................................................................................................................ 106
Cubitt, Rebecca L. ............................................................................................................... 56
Czajka, Timothy F. (P24) .................................................................................................... 91
Czako, Rita .......................................................................................................................... 43, 91
Davis, Simon A. ................................................................................................................... 86
Demirdjian, Sally (P10, O) .................................................................................................. 41, 64
Deragon, Matthew A. .......................................................................................................... 77, 106
Diehl, Sean A. (S) ................................................................................................................ 38
Divangahi, M. ...................................................................................................................... 65
Dostie, Kristen E. (P47) ....................................................................................................... 103
D’Souza, Shanti (P15, O) .................................................................................................... 48
Duerr, Claudia U. ................................................................................................................. 47
Dunbrack, Jr., Roland .......................................................................................................... 86
Duque, Carolina (P6) ........................................................................................................... 80
Egan, Shawn M. ................................................................................................................... 61
Egilmez, Nejat K. ................................................................................................................ 59
Ehrbar, Dylan ....................................................................................................................... 99
Eill, Elizabeth G. (P5) ........................................................................................................... 79
Eliseeva, Sophia ................................................................................................................... 55
Falk-Mahapatra, Riddhi (P21, O) ........................................................................................ 60
Frelinger, John G. ................................................................................................................ 100
Fritz, David .......................................................................................................................... 93
Fritz, Jörg H. (S) .................................................................................................................. 47, 107
Galasso, Nicholas A. ............................................................................................................ 93
Georas, Steve ....................................................................................................................... 55
Gerber, Scott A. ................................................................................................................... 59, 92, 96, 98, 101
Gerondakis, Steve ................................................................................................................ 47
Ghanem, Elsa N. Bou (S) .................................................................................................... 108

112.
Gollnick, Sandra O. ............................................................................................................. 60, 88
Gosselin, Edmund J. ............................................................................................................ 85, 90
Goyette, Michael (P11) ....................................................................................................... 82
Grimson, Andrew ................................................................................................................ 46
Gros, Philippe ...................................................................................................................... 47
Han, Booyeon J. (P45) ........................................................................................................ 59, 96, 101
Harris, Rebecca ................................................................................................................... 37
Harton, Jonathan A. ............................................................................................................. 53
Hillman, Sara ...................................................................................................................... 55
Holling, G. Aaron (P19, O) ................................................................................................. 61
Hopkins, Daniel ................................................................................................................... 41, 64
Howard, Jennifer (P17) ....................................................................................................... 87
Hoyt, Laura E. ..................................................................................................................... 93
Huang, Weishan .................................................................................................................. 78
Hylander, Bonnie L. ............................................................................................................ 58, 62
Jo, Hui Jin ........................................................................................................................... 52, 87
Joseph, Renuka E. ............................................................................................................... 43
Kandefer, Rachel ................................................................................................................. 61
Karanicolas, John ................................................................................................................ 86
Karimi, Mobin ..................................................................................................................... 78
Kelow, Simon ...................................................................................................................... 86
Kenderes, Kevin .................................................................................................................. 37
Kumar, Sudeep (P23) .......................................................................................................... 85, 90
Lamirande, Elaine W. ......................................................................................................... 43
Landekic, Marija (P34, O) ................................................................................................... 65
Langlais, David ................................................................................................................... 47
LaRocca, Timothy J. (S) ..................................................................................................... 77, 99, 106
Larson, Elisabeth (P12) ....................................................................................................... 83
Lawrence, B. Paige ............................................................................................................. 50
Lee, James N. ...................................................................................................................... 108

113.
Lee, Kelvin P. ...................................................................................................................... 49, 61, 89, 102
Leib, David A. ..................................................................................................................... 54
Leong, John M. .................................................................................................................... 108
Levack, Russell .................................................................................................................... 37
Lightman, Shivana (P18, O) ................................................................................................ 49, 102
Linehan, David C. (S) .......................................................................................................... 59, 72, 92, 96, 101
Liu, Wensheng ..................................................................................................................... 102
López-Yglesias, Américo (P27, O) ...................................................................................... 68
Lord, Edith M. ..................................................................................................................... 98
Lovewell, Rustin .................................................................................................................. 41
Lucas, Nathan (CS) .............................................................................................................. 44
Lucy, Evan (P11) ................................................................................................................. 82
Lynes, Michael A. ................................................................................................................ 103, 104
MacDonald, Cameron R. ..................................................................................................... 58
MacNamara, Katherine C. .................................................................................................... 52, 87
Maloney, Jackson ................................................................................................................ 52
Mancini, Mathieu ................................................................................................................. 47
Mantis, Nicholas J. ............................................................................................................... 86, 91, 99
Marchese, Anthony (P7, O) ................................................................................................. 67
Martin, Andrew T. ............................................................................................................... 68
Matson, Adam (S) ................................................................................................................ 39
McCaig, William D. (P3) ...................................................................................................... 77, 106
Middaugh, C. Russell ........................................................................................................... 86
Mills, Bradley N. (P33, O) ................................................................................................... 59, 92, 101
Mindt, Barbara C. (P41, O) .................................................................................................. 47
Mishra, Bibhuti (S) .............................................................................................................. 42
Mohammadpour, Hemm ...................................................................................................... 58
Mon, Kristel Yee (P2, O) ..................................................................................................... 46
Moshkani, Safiehkhatoon .................................................................................................... 67
Murphy, Joseph D. (P36) ..................................................................................................... 59, 96

114.
Myers, Jason R. ................................................................................................................... 50
Nagar, Abhinit (P1, O) ........................................................................................................ 53
Nelson, Nicole (P14) ........................................................................................................... 85
Newell, Krista (P4) .............................................................................................................. 78
Newman, Stephanie M. (P31) ............................................................................................... 94, 97
Nguyen, Sophie ................................................................................................................... 86
North, Brian J. ..................................................................................................................... 54
Norville, Karli (P44) ............................................................................................................ 100
Ojemann, Alexandra ............................................................................................................ 93
Olejniczak, Scott H. ............................................................................................................. 61
Oyesola, Oyebola O. (P9, O) ............................................................................................... 56, 80
Pacheco, Christian ............................................................................................................... 78
Patankar, Yash ..................................................................................................................... 41
Patel, Payal S. ...................................................................................................................... 77, 106
Patel, Ravi ........................................................................................................................... 46
Paveglio, Sara ...................................................................................................................... 39
Peng, Peng (P22) ................................................................................................................. 89
Peng, Seth A. ....................................................................................................................... 56
Petersen, Jarrod (P11) .......................................................................................................... 82
Pham, Duc ........................................................................................................................... 56
Popescu, Maria .................................................................................................................... 37
Porter, Kristen ...................................................................................................................... 82
Poynter, Mattew E. .............................................................................................................. 93
Qiao, Guanxi (P26, O) ......................................................................................................... 58, 61, 62
Qureshi, S. ........................................................................................................................... 65
Rahman, Tabassum .............................................................................................................. 53
Raza, Fahad ......................................................................................................................... 83
Repasky, Elizabeth A. .......................................................................................................... 58, 61, 62, 92
Rezaul, Karim ...................................................................................................................... 39
Robek, Michael D. ............................................................................................................... 67, 81

115.
Robinson, Richard T. (P13) ................................................................................................. 84
Roggensack, Sara E. ............................................................................................................ 108
Rong, Yinghui ..................................................................................................................... 86, 99
Rudd, Brian D. .................................................................................................................... 46, 95
Rudolph, Michael J. ............................................................................................................ 86, 91
Sanchez, Hector .................................................................................................................. 41, 64
Sass, Stephanie N. ............................................................................................................... 94, 97
Schnabel, Chrisiane ............................................................................................................. 83
Sepaniac, Leslie .................................................................................................................. 93
Seyfried, Allison N. (P25, O) .............................................................................................. 52
Shakerly, Nicole L. ............................................................................................................. 77, 106
Sharda, Baiju ....................................................................................................................... 94, 97
Singh, Amit K. (P30, O) ...................................................................................................... 66
Singh, Anurag ..................................................................................................................... 62
Siwapornchai, Nalat ............................................................................................................ 108
Skrombolas, Denise ............................................................................................................. 100
Smiraglia, Tori A. ............................................................................................................... 77, 106
Smith, Julianne .................................................................................................................... 87
Smith, Norah L. ................................................................................................................... 46, 95
Smyth, Timothy .................................................................................................................. 55
Snyder, Julia P. (P29) .......................................................................................................... 93
Solouki, Sabrina .................................................................................................................. 56
Sosunov, Sergey A. ............................................................................................................. 106
Subbarao, Kanta .................................................................................................................. 43
Sun, Wei .............................................................................................................................. 66
Sunagar, Raju ...................................................................................................................... 90
Sutton, Troy C. (S) .............................................................................................................. 43
Tabilas, Cybelle (P32) ......................................................................................................... 95
Taffet, Steven M. ................................................................................................................ 79
Taylor, Sean A. ................................................................................................................... 54

116.
Tchalla, Essi ........................................................................................................................ 108
Ten, Vadim S. ...................................................................................................................... 106
Thanavala, Yasmin (S) ........................................................................................................ 73
Thees, Amy V. (P48) ........................................................................................................... 104
Thwe, Phyu M. .................................................................................................................... 93
Uccello, Taylor P. (P28) ...................................................................................................... 59, 92
Udartseva, Olga O. (P20) .................................................................................................... 88
Utley, Adam (P46) .............................................................................................................. 89, 102
Vance, David J. (P16) .......................................................................................................... 86, 91
Veazey, Janelle (P42, O) ..................................................................................................... 55
Vidal, Silvia ......................................................................................................................... 47
Vinh, D. ............................................................................................................................... 65
Wagner, Bettina ................................................................................................................... 83
Walker, Katharine M. .......................................................................................................... 106
Wang, Jianmin ..................................................................................................................... 61
Wang, Xiuran ...................................................................................................................... 66
Webb, Lauren M. ................................................................................................................ 56
Weis, David D. .................................................................................................................... 86
Westfall, Jennifer (P43) ....................................................................................................... 99
Winslow, Gary (S) ............................................................................................................... 37
Wojno, Elia D. Tait (S) ....................................................................................................... 56, 74, 80
Wynn, Thomas A. (KS) ....................................................................................................... 35
Yang, Qi .............................................................................................................................. 48
Yarovinsky, Felix ................................................................................................................ 68
Ye, Jian ................................................................................................................................ 59, 96
Zhao, Tony .......................................................................................................................... 59
Zollo, Robert A. (P37) ......................................................................................................... 94, 97

117.
Amiel, Eyal
University of Vermont
802-656-0522
Barman, Tarani Kanta
Albany Medical College
518-262-6226
Battaglia, Nicholas
University of Rochester
716-471-6114
Bellville, Dawn
Albany Medical College
518-262-5365
Berwin, Brent
Dartmouth College
603-208-7446
Jesse Bonin
Albany Medical College
518-262-6226
Busby, William
Lonza BioSciences
203-615-4168
Burke, Catherine
University of Rochester
716-949-2802
Campbell, Heather
Roswell Park Cancer Institute
651-208-3127
Ceron, Stacey
Dartmouth College
718-704-4351
Chen, Minchu
Roswell Park Cancer Institute
412-961-4869
Chiale, Carolina
Albany Medical College
412-218-4411
Cordero, Doris
BD Biosciences
917-743-7118
Czajka, Timothy
Wadsworth Center
518-766-7339
Demirdjian, Sally
Dartmouth College
603-650-6899
Diehl, Sean
University of Vermont
860-486-3648
Attendee Contact Information

118.
Dostie, Kristen
University of Connecticut
860-486-3648
Drake, James (Jim)
Albany Medical College
518-262-9337
D’Souza, Shanti
Albany Medical College
518-935-7368
Duque, Carolina
Cornell University
905-745-8780
Echandy, Victor Hugo
Krackeler Scientific
518-423-7009
Eill, Elizabeth
SUNY Upstate
585-771-7259
Falk-Mahapatra, Riddhi
Roswell Park Cancer Institute
716-704-2326
Fritz, Jörg
McGill University
514-398-1707
Gerber, Scott
University of Rochester
585-275-7380
Ghanem, Elsa Bou
University at Buffalo
716-829-2422
Goyette, Michael
Westfield State University
413-575-8194
Green, Kathy
Dartmouth College
603-650-5056
Green, William
Dartmouth College
603-650-8607
Grieco, Anthony
Taconic Biosciences
518-567-3767
Han, Booyeon
University of Rochester
480-570-1796
Holling, George
Roswell Park Cancer Institute
716-845-5774

119.
Houser, Cassandra
University of Rochester
717-875-0663
Howard, Jennifer
Albany Medical College
551-427-8857
Karimi, Mobin
SUNY Upstate
315-464-2344
Kumar, Sudeep
Albany Medical College
518-262-6972
Landekic, Marija
McGill University
514-835-0550
LaRocca, Timothy
ACPHS/Wadsworth
631-879-7904
Larson, Elisabeth
Cornell University
360-739-6314
Lightman, Shivana
Roswell Park Cancer Institute
716-380-0223
Linehan, David
University of Rochester
585-275-2725
Lopez-Yglesias, Americo
University of Rochester
206-852-2707
Lotze, Michael
University of Pittsburgh
412-478-3316
Lucas, Nathan
BioLegend
309-370-7468
Lucey, Evan
Westfield State University
978-337-3244
Machikas, Alexa
StemCell Technologies
203-725-4719
MacNamara, Kate
Albany Medical College
518-262-0921
Marchese, Anthony
Albany Medical College
518-209-7420

120.
Matson, Adam
University of Connecticut
860-679-3096
McCaig, William
Wadsworth/ACPHS
917-548-3047
McCarthy, Amy
MilliporeSigma
978-715-1380
McLaughlin, Fran
ThermoFisher Scientific
860-334-3081
Metzger, Dennis
Albany Medical College
518-262-6750
Mills, Bradley
University of Rochester
585-813-3445
Mindt, Barbara
McGill University
541-398-6417
Mishra, Bibhuti
Albany Medical College
518-264-3816
Mon, Kristel Yee
Cornell University
347-462-7190
Murphy, Joseph
University of Rochester
585-503-0406
Nagar, Abhinit
Albany Medical College
518-262-4447
Napoli, Lauren
Krackeler Scientific
800-334-7725
Nelson, Nicole
Albany Medical College
518-262-1650
Newman, Stephanie
Roswell Park Cancer Institute
716-544-5379
Newell, Krista
SUNY Upstate
315-464-7652
Norville, Karli
University of Rochester
661-902-3681

121.
Oyesola, Oyebola
Cornell University
607-379-7745
Peng, Peng
Roswell Park Cancer Institute
716-845-4348
Petersen, Jarrod
Westfield State University
508-942-2748
Porter, Kristen
Westfield State University
603-233-0304
Qiao, Guanxi
Roswell Park Cancer Institute
716-364-8252
Qin, Shuyang
University of Rochester
201-680-9788
Riley, Kevin
Taconic Biosciences
518-257-0494
Robek, Michael
Albany Medical Center
518-264-2580
Robinson, Richard
Ohio State University
614-293-4164
Savage, Michael
Agilent
646-634-9903
Serafino, Lauren
Leinco Technologies
440-429-5749
Sethuraman, Jyothi
Northeastern University
617-373-8993
Seyfried, Allison
Albany Medical Collge
518-262-0922
Singer, Cassandra
MilliporeSigma
724-799-9726
Singh, Amit
Albany Medical College
518-258-8222
Snyder, Julia
University of Vermont
802-309-3739

122.
Schneider, Olivia
Shenandoah Biotechnology
513-659-1309
Sutton, Troy
Penn State University
814-863-0693
Szczepanek, Steven
University of Connecticut
860-486-8101
Tabilas, Cybelle
Cornell University
530-867-2880
Taffet, Steven
SUNY Upstate Medical University
315-464-5419
Thacker, Robert
MilliporeSigma
513-284-6606
Thanavala, Yasmin
Roswell Park Cancer Institute
716-845-8536
Thees, Amy
University of Connecticut
732-232-4739
Uccello, Taylor
University of Rochester
978-621-5409
Udartseva, Olga
Roswell Park Cancer Institute
716-544-1784
Utley, Adam
Roswell Park Cancer Institute
336-847-4725
Vance, David
Wadsworth Center
518-486-4394
Veazey, Janelle
University of Rochester
585-519-8472
Warren, Rachel
University of Rochester
260-705-5646
Westfall, Jennifer
Wadsworth Center
518-755-1886
Winslow, Gary
SUNY Upstate Medical University
315-464-7658

123.
Wohlfert, Beth
University at Buffalo
716-829-3969
Wojno, Elia Tait
Cornell University
607-256-5635
Yang, Qi
Albany Medical College
518-264-2582
Yates, Jennifer
Wadsworth Center
518-339-2556
Zollo, Robert
Roswell Park Cancer Institute
603-630-3064



















![[Visiting immunology]](https://img.dokumen.tips/doc/110x75/63453fbb38eecfb33a06782b/visiting-immunology.jpg)











