Dr. Mario Enrique Vega Carbó.
Endocrinólogo.
2015
Síndrome de Kallman.
Universidad de Ciencias Médicas de la Habana
Facultad de Ciencias Médicas “Comandante Manuel Fajardo”
Instituto Nacional de Endocrinología

Paciente: RLB Edad :22años
MasculinoHC: 955422
Egresado de sala 4D/3: 9julio 2012.
MI: “Baja estatura y no desarrollo genital”.HEA: Paciente masculino de 22 años que ingresa para estudio de Hipogonadismo hipogonadotropo de inicio prepuberal, baja talla e hiposmia. sin cefalea, ni trastornos visuales.APP: Agenesia renal derecha.APF: Hermano Hipogonadismo tratado.

INTERROGATORIO Sistema urogenital Deseo sexual y relaciones sexuales a 18 años, con erección, sin eyaculación. Sistema endocrinodesarrollo genital pobre, baja tallaAumento de peso 15 lbs en últimos mesesSistema nerviosoNo percibe olores de comidas, perfumes, aromas
fuertes como café ,vainilla.Estatura Madre:160cmPadre:172cm

Examen físico (+):General Ausencia de vello facial y corporal. Adrenales: Vello axilar ausente. Vello pubiano Tanner II.Panículo adiposo: aumentado en cintura pélvica
Regionaltiroides: grado 0mamas: No ginecomastia, ni galactorrea

Genitales externo:Teste I: V 2ml, en escroto, blando, no sensible,
superficie lisa.Teste D: V 1ml en canal inguinal que desciende al
agacharse retráctil, blando, no sensible, liso.Escrotos: poco desarrollo, hipopigmentados, poco
rugosos.Pene: Tanner I Largo: 3.5cm Circunsferencia: 5cm Glande: sin desarrolloSoma: Hipoatrofia muscular generalizada a predominio de
cintura escapular y miembros superiores.
S Nervioso Central: Hiposmia bilateral al café, vainilla, canela.

MesuracionesPeso:46.5kgTalla:1.53cm IMC:19.86Kg/m2
Brazada 165cm >12cm que la tallaVP:72cmPP:81cm PP > 9cm que VPDBA:32cmDBT:31.5C cintura:73cmC Cadera:87cm ICC =0.8

HPSocialPrenatales, Perinatales, Posnatales y DSM
Etapa escolar: Rendimiento intelectual medio. Comenzó a notar desde 8 años diferencias en su estatura con respecto a compañeros de escuela.
Adolescencia: Deprivación afectiva, aporte nutricional inadecuado, conflictos familiares.
Madre fallecida, padre internacionalista.

Bioquímica general EN PARÁMETROS NORMALES
Hormonales basales FSH=0,25UI/LLH =0,21UI/LPRL =298mU/LCORTISOL =400nmol/L17 OH PROGESTERONA =0,87ng/ml
Genéticos CARIOTIPO = 46XY 11 METAFASES

Prueba dinámica Test estimulación HCG Testosterona nmol/l y E2
basal 1.9924h 1.8748h 1.0272h 1.42

IMAGENRx selectivo de silla turca: Aumento del diámetro AP de
la silla turca.
RMN de cráneo: ausencia de surcos y giros rectos compatible con agenesia olfatoria.
Rx de manos izquierda y rodilla: edad ósea compatible con 13 años. Edad cronológica de 22 años.
Ultrasonido testicularTI y TD: hipoplasia de ambos bien vascularizados
localizados en escroto y canal inguinal respectivamente.
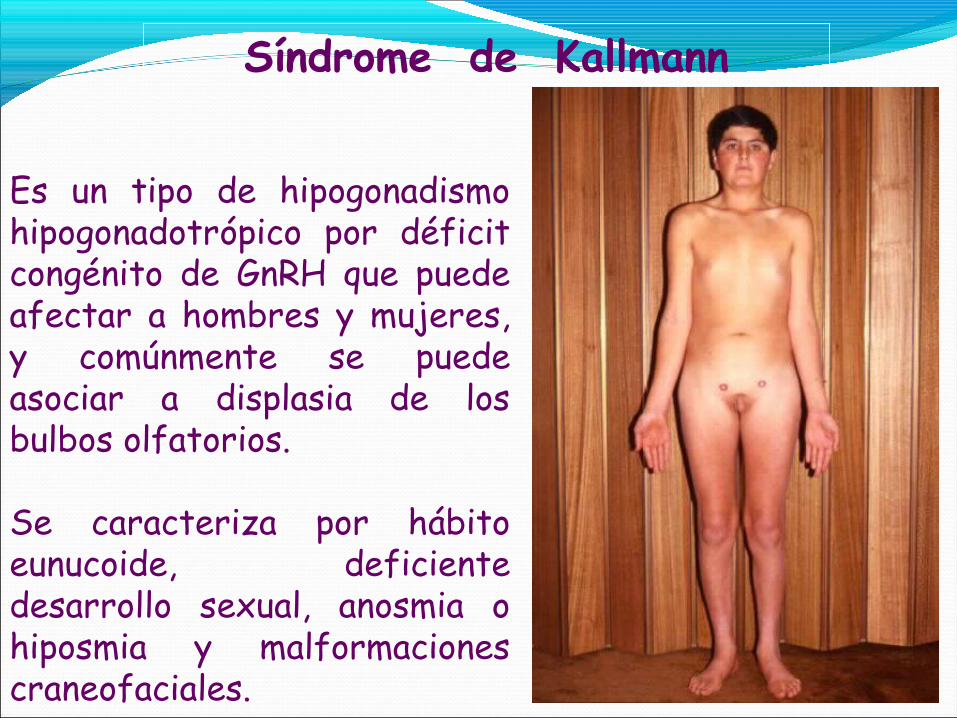
Síndrome de Kallmann
Es un tipo de hipogonadismo hipogonadotrópico por déficit congénito de GnRH que puede afectar a hombres y mujeres, y comúnmente se puede asociar a displasia de los bulbos olfatorios.
Se caracteriza por hábito eunucoide, deficiente desarrollo sexual, anosmia o hiposmia y malformaciones craneofaciales.

Síndrome de Kallmann
Formas clínicasEsporádica: 70%Familiar: 30%
Modelos familiares:Ligado al cromosoma X: 85%Autosómico dominante: 10%
Autosómico recesivo: 5%
Prevalencia1x10 mil en varones.
1x50 mil en hembras.

Patrón de herencia ligado al X
Gen KAL 1. Localizado en Xp 22.31
Codifica para la Anosmina-1.
Participa en la sinapsis bulbo-olfatoria.
Trasmiten mujeres portadoras a enfermos.
Síndrome de Kallmann

Patrón de herencia autosómico dominante
Gen KAL 2 FGFR1 8 p11.2-p12
Codifica para el receptor 1 del factor de crecimiento de fibroblastos, relacionados con la evaginación del bulbo olfatorio.
Síndrome de Kallmann

Patrón de herencia autosómico recesivo
Gen PROK 2 (KAL 3) Codifica para el receptor Procinecticina 2.
Gen PROK (KAL 4) Codifica para la Procinecticina 2.
Síndrome de Kallmann

Cuadro clínicoBebes: Micropene. Criptorquidea.Infancia: anosmia o hiposmia, no obligado
Adolescentes y adultos:Hipogonadismo. Desarrollo sexual incompleto.Volumen testicular: menor de 4 ml.Ausencia de caracteres sexuales secundarios.Aumento de masa muscular.Poca o ausencia de líbido.Disfunción eréctil.Infertilidad.Proporciones enucoides.

Otras manifestaciones clínicas:
Déficit olfatorio (anosmia o hiposmia).Agenesia renal unilateral (30%) es exclusivo del KAL 1.Perdida auditiva unilateral.Labio y/o paladar hendido.Agenesia de una o ambas piezas dentales.Sinequia de los dígitos.Braquidactilia o sindactiliaAgenesia del cuerpo calloso.Hipoplasia o aplasia del pabellón auricular.Pérdida del cartílago nasalSinquinesia bimanual o movimientos en espejo de miembros superiores.

Criterios diagnósticos del Síndrome de Kallmann
I. Clínicos:1.Hipogonadismo prepuberal2.Anosmia o hiposmia.3.Anomalías craneo-faciales.
II. Hormonales: Hipogonadismo hipogonadotrópico secundario.1.Testosterona total, FSH y LH baja.2.Se elevan FSH y LH con estimulación con GnRH.

III. Imagenológicos.1. Resonacia magnética de hipófisis con o sin gadolineo.Para descartar lesión hipotalamo-hipofisaria.Ausencia o hipoplasia del bulbo olfatorio.Ausencia del surco del giro recto.2. Ultrasonido de vías urinarias: Malformaciones.
IV. Examenes genéticos.1. Cariotipo normal.2. Prueba tipo FISH o por hibridación genómica comparativa. (Detecta delección de genes: KAL1-4)
Criterios diagnósticos del Síndrome de Kallmann

Tratamiento del Síndrome de Kallmann
Objetivos del tratamiento. Lograr desarrollo de caracteres sexuales secundarios y fertilidad.
Bases del tratamiento hormonal.1.Reemplazo hormonal con testosterona en prepuberes.2.Mejorar volumen testicular con HCG y HMG que pueda desarrollar espermatogénesis.3.Conseguir fertilidad con análogos de GnRH.
Psicoterapia de apoyo a pacientes y familiares.
Recommended