Embed Size (px)
Citation preview

Urinary Kallikrein Activity and Renal VascularResistance in the Antihypertensive Response to
Thiazide Diuretics
DANIEL T. O'CONNOR, M.D., RICHARD A. PRESTON, M.D., JOHN A. MITAS, II, M.D.,
RONALD P. FRIGON, PH.D. , AND RICHARD A. STONE, M.D.
SUMMARY To evaluate the mechanism of chronic thiazide diuretic action in hypertension, we treated 19essential hypertensive white men for 1-month periods on placebo alone and hydrochlorothiazlde alone. Duringtherapy, mean arterial pressure (MAP) fell, but radioisotopically determined intrarascular volume remainedunchanged, suggesting other mechanisms of thiazide action upon blood pressure. In the renal circulation,thiazides did not change renal plasma flow or glomerular filtration rate, but renovascuiar resistance wasdiminished, probably at the afferent arteriole. Concomitant with the decline in blood pressure and renovascuiarresistance, urinary kallikrein excretion increased, from subnormal (hypertensive) levels back into the normalrange. The kallikrein increase did not correlate with changes in plasma aldosterone. In addition, patients withblood pressure responses (reduction £ 10%) to thiazides (n = 12) had greater increases in kallikrein excretionthan those without such a blood pressure decrement (n = 7), suggesting a role for renal kallikrein in thehypotensive response to thiazide diuretics. (Hypertension 3: 139-147, 1981)
KEY WORDS • kallikrein • thiazide diuretics • renal blood flowrenal vascular resistance • hypertension • plasma volume
BENZOTHIADIAZINE diuretics have longbeen regarded as effective "first line" anti-hypertensive drugs, but their mode of action
in lowering blood pressure remains in doubt, es-pecially during chronic antihypertensive therapy.1
Proposed mechanisms for thiazide action in hyper-tension have included: 1) intravascular volume deple-tion secondary to its natriuretic action1"4 and 2) reduc-tion in systemic vascular resistance by mechanisms asyet unspecified, with or without concomitant volumedepletion, especially during chronic administration.5"*
With these ideas in mind, we decided to re-examinethe mechanism of hydrochlorothiazide antihyper-tensive action in essential hypertensive humans. Ourinvestigation focused on intravascular volume, the
From the Departments of Medicine, Division of Nephrology,Veterans Administration Medical Center, and University ofCalifornia, San Diego, California.
Supported by the Veterans Administration and by Grant HL-18095 from the National Institutes of Health.
Presented, in part, at the annual national meetings of theAmerican Society of Nephrology, Washington, D.C. (November,1977) and the American Federation for Clinical Research, SanFrancisco, California (May, 1978).
Address for reprints: Daniel T. O'Connor, M.D., NephrologyIl l -H, Veterans Administration Medical Center, 3350 La JollaVillage Drive, San Diego, California 92161.
Received April 17, 1979; revision accepted March 28, 1980.
renal vascular bed (as a likely determinant of thiazidehypotensive effect), and the kallikrein-kinin system asa possible mediator of changes in renal and systemicvascular resistance. One proposed index of the renalkallikrein-kinin system is the 24-hour urinary excre-tion of the enzyme kallikrein (kininogenin; E.C.3.4.21.8), a tissue kallikrein derived from kidney, hav-ing the ability to generate vasoactive peptides calledkinins. This enzyme system may be important inblood pressure regulation*"11 and the determination ofrenal blood flow," and some evidence suggests it mayinfluence systemic kinin production,1814 although thisremains controversial.18"1"
Materials and Methods
Patients
139
We studied 19 white men with essential hyper-tension. They were patients who had exhibited supinemean arterial pressures (MAP; diastolic pressure plus1/3 pulse pressure) greater than 105 mm Hg in theoutpatient clinic. The mean age was 50.1 ± 2.6 years.Hypertensives were screened for known secondarycauses of hypertension with determination of theblood urea nitrogen concentration (BUN) and serumconcentrations of sodium, potassium, chloride, andbicarbonate; a hemogram, electrocardiogram, and in-
by guest on May 24, 2018
http://hyper.ahajournals.org/D
ownloaded from

140 HYPERTENSION VOL 3, No 1, JANUARY-FEBRUARY 1981
travenous urogram and roentgenogram of the chestand a 24-hour urinary collection for excretion ofcatecholamines, metanephrines, vanillylmandelicacid, and 17 hydroxycorticosteroids were also ob-tained. All subjects were found to be essentialhypertensives. Hypertensive patients' medicationswere discontinued for at least 1 month prior to study.In addition, all patients with evidence of end organdamage from hypertension (e.g., congestive heartfailure, myocardial infarction, nephrosclerosis,stroke) were excluded.
To establish a normal range for urinary kallikreinexcretion, 13 healthy white male normotensives werealso studied. They were paid volunteers on no medica-tion, and matched to the hypertensives for sex (male),race (white), and dietary sodium intake (unrestricted;149 ± 11 mEq/day for the normotensives versus 162± 20 mEq/day for hypertensives on placebo,/? > 0.1,ns). The mean age of the normotensives was 36.9 ±3.2 years, lower than that of the hypertensives (50.1 ±2.6 years, p < 0.01). However, we have shownpreviously that the depression of urinary kallikrein ex-cretion seen in essential hypertension is independent ofage.11
Every subject gave informed written consent, andthe Committee on Human Experimentation of theUniversity of California, San Diego, approved theprotocol.
Procedures
In randomized order, the patients were placedeither on placebo for 1 month or hydrochlorothiazideorally for 1 month, at a dose individually titrated untilblood pressure normalized or a maximum dose of 100mg/day was reached. The median dose was 50 mg/day with a mean ± SEM of 56.6 ± 4.6 mg/day and arange of 25-100 mg/day. No other medications wereutilized, and the diet was unrestricted in fluid and salt.The study was performed in crossover fashion; hence,all patients received both placebo and hydrochloro-thiazide, and acted as their own controls. Because ofthe lengthy placebo period (1 month), no "washout"period was deemed necessary between phases of thestudy.
At the end of each 1-month study period (bothplacebo and drug), patients were admitted to theDiagnostic and Treatment Unit of the San DiegoVeterans Administration Medical Center for a 2-dayprotocol. During the admission, each patient was on adiet unrestricted in sodium intake ( > 100 mEq/24hrs), which contained approximately 80 mEq ofpotassium. Multiple measurements of blood pressurewere done in the supine position upon admission, dur-ing both placebo and hydrochlorothiazide treatmentphases, with a manual sphygmomanometer. Thesevalues were averaged and the mean arterial pressure(MAP) was calculated. This MAP value as well asboth systolic and diastolic blood pressure values wereused for further statistical analysis. The MAP wasused as a criterion for degree of response to therapy
(see below). Diastolic blood pressure was taken asphase 5 Korotkoff sound (disappearance).
On Day 1 of each admission, a 24-hour urine collec-tion was begun for the measurement of volume,sodium, potassium, chloride, and kallikrein activity.During collection, urine was kept refrigerated at 4° C,and at the end of 24 hours, the volume was measuredand an aliquot frozen at -30° for later analysis.
In the morning of Day 2, blood was obtained fordetermination of BUN, serum creatinine, serum elec-trolytes, hemogram, supine and upright plasma reninactivity (SPRA, UPRA), and plasma aldosteroneconcentration. Renal plasma flow was measured usingthe method of constant infusion of para-amino-hippurate (PAH) (13 patients) without urine collec-tion.17 Whole blood volume was then measured in thesupine position.
The 13 normotensive white males underwenthospitalization and various measurements exactly asdescribed above, except that they were studied onlyonce and received neither placebo nor hydrochloro-thiazide. We report here only these patients' urinarykallikrein activities for purposes of establishing acomparative norm for urinary kallikrein excretion inwhite normotensive males on unrestricted sodium diet.The other parameters of these 13 patients have beenreported by us previously.13
Chemical Assays
Routine chemistries were performed in the clinicallaboratory of the San Diego Veterans AdministrationHospital. Blood for renin and aldosterone determina-tion was drawn into chilled EDTA tubes which werekept on ice until the samples could be centrifuged andthe plasma frozen at -30°C. The PRA was deter-mined by the radioimmunoassay method of Haber etal.18 with reproducibility in our laboratory as pre-viously described.12 Results are expressed as nano-grams of angiotensin I generated per milliliter ofplasma per hour (ng A^ml/hr). Plasma aldosteroneconcentration was determined by radioimmunoassayutilizing sH-aldosterone as described by Sampson etal.,19 utilizing reagents purchased from DiagnosticProducts Corporation, Los Angeles, California. Re-sults are expressed as pg/ml.
Urinary kallikrein activity was measured for p-tosylarginine methyl ester hydrolysis using the radio-chemical method of Beaven et al.20 and Margolius etal.21 under our conditions for the assay, pH 8.5 and37°C11> M with a purified13 human internal kallikreinstandard for recovery. Results are expressed as es-terase units (EU) per 24 hours. In separate ex-periments, hydrochlorothiazide at concentrations upto 100 Mg/ml urine did not affect our assay.
Whole blood volume was determined using bothMCr tagged autologous erythrocytes for red cell mass,and I12* labelled albumin for plasma volume. Theisotopes were injected intravenously, and blood wascollected at 10 and 20 minutes for counting of wholeblood and plasma samples in the Volmetron ap-paratus.
by guest on May 24, 2018
http://hyper.ahajournals.org/D
ownloaded from

URINARY KALLIKREIN ACTIVITY /O'Connor et al. 141
Statistics
All results are expressed as the mean value ± thestandard error of the mean (± SEM). Paired and(where appropriate) unpaired two-tailed Student's /tests and linear least squares correlation coefficientswere performed with standard techniques." The nullhypothesis was rejected for p values of 0.05 or less.
Results
Systemic and Renal Hemodynamic Parameters
Changes in these parameters during hydrochloro-thiazide therapy are shown in table 1. The MAP fellsignificantly (p > 0.01) in the total treatment group vsthe placebo group, from 106.7 ± 2.7 mm Hg down to92.4 ± 2.0 mm Hg. The blood pressure decline wasalso significant for both systolic pressure and diastolicpressure (from 140.0 ± 3.4/90.0 ± 2.6 down to 120.7±3.0/78.1 ± 1.9mmHg, both wi thp< 0.01). Also ofnote was a significant decline in systolic (from 167.5 ±4.3 down to 140.0 ± 3.4 mm Hg, p < 0.01), diastolic(from 108.4 ± 2.6 down to 90.0 ± 2.6 mm Hg, p <0.01), and mean (from 128.1 ± 2.8 down to 106.7 ±2.7 mm Hg, p < 0.01) arterial pressures, going fromoutpatient clinic values before treatment to placebophase, underscoring the necessity for a placebo periodas an appropriate control.
There were, however, no associated changes inwhole blood volume or plasma volume (table 1). Bodyweight declined slightly from 87.2 ± 3.0 kg down to86.4 ± 3.3 kg (p = 0.05).
Evaluation of renal hemodynamic parametersrevealed no significant changes in creatinine clearance,PAH clearance, renal blood flow, or renal filtrationfraction (table 1). Renal blood flow was calculatedfrom PAH clearance and hematocrit; renal filtrationfraction was calculated from the quotient of creatinineclearance/PAH clearance.
Renal vascular resistance (RVR), calculated fromsimultaneous repeated measurements of MAP andrenal blood flow," declined significantly (p = 0.03) onthiazide therapy, from 7815 ± 728 to 6827 ± 582dyne'sec'cnr5.
Biochemical Parameters
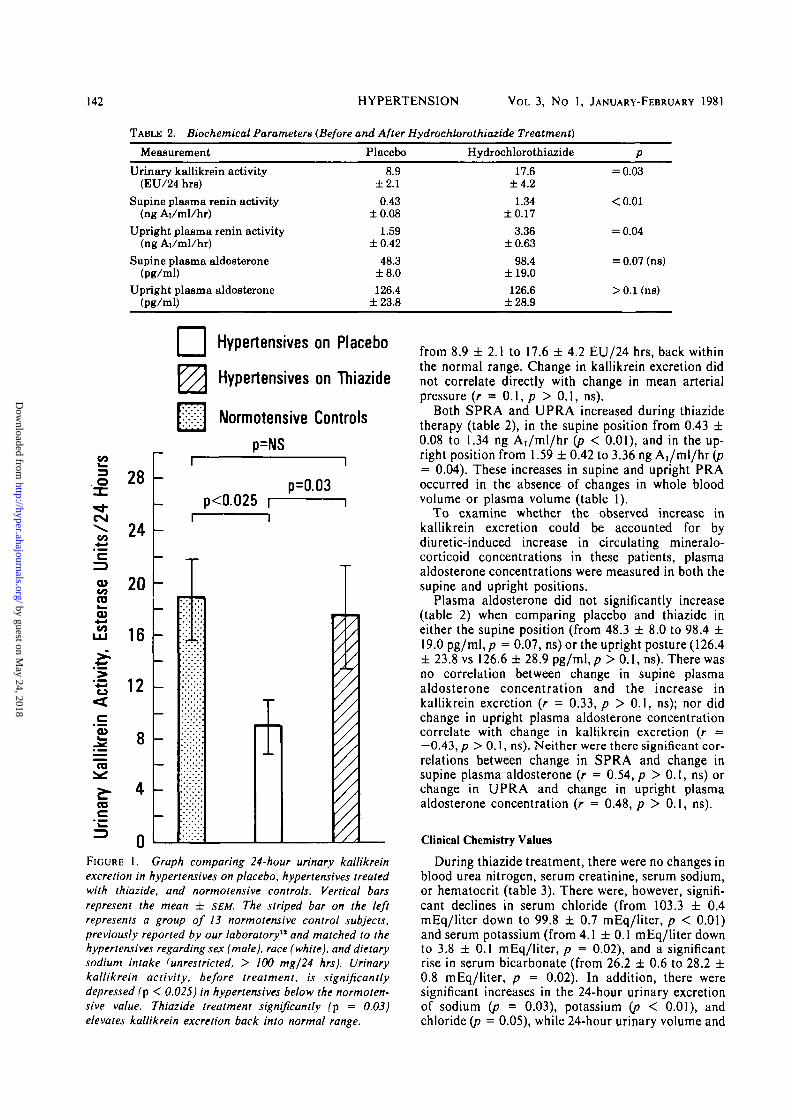
Hydrochlorothiazide effects upon the biochemicalparameters studied are shown in table 2. The 24-hoururinary kallikrein excretion (table 2, fig. 1) wassignificantly (p < 0.025) depressed in the placebo-treated essential hypertensives, when compared to agroup of 13 normotensive controls previously reportedfrom our laboratory12 and matched for sex (male),race (white), and dietary sodium intake (unrestricted,> 100 mEq/24 hrs). Thiazide treatment, however,significantly (p = 0.03) elevated kallikrein excretion
TABLE 1. Systemic and RenalTreatment)
Hemodynamic Parameters (Before and After Hydrochlorothiazide
Measurement Placebo Hydrochlorothiazide
Blood pressure (mm Hg)Systolic
Diastolic
Mean
Blood volume (ml)
Blood volume bodysurface area (ml/m2)
Plasma volume (ml)
Plasma volume bodysurface area (ml/m2)
Weight (kg)
Creatinine clearance (ml/min)
PAH clearance (ml/min)
Filtration fraction
Renal blood flow (ml/min)
Renal vascular resistance(dvne • sec • cm"6)
140.0±3.490.0
±2.6
106.7±2.75318
±310
2564±138
2812±162
1350±72
87.2±3.0
105.3±8.4
597±43
0.20±0.02
1093±90
7815±728
120.7±3.078.1
±1.9
92.4±2.0
5578±352
2792±1743013
±212
1403±83
86.4±3.3
107.7±8.7
621±56
0.19±0.02
1119±104
6827±582
<0.01
<0.01
<0.01
>0.1 (ns)
>0.1(ns)
= 0.08 (ns)
= 0.08 (ns)
= 0.05 (ns)
>0.1(ns)
>0.1(ns)
>0.1 (ns)
>0.1(ns)
= 0.03
by guest on May 24, 2018
http://hyper.ahajournals.org/D
ownloaded from
142 HYPERTENSION VOL 3, No 1, JANUARY-FEBRUARY 1981
TABLE 2. Biochemical Parameters (Before and After Hydrochlorothiazide Treatment)
Measurement Placebo Hydrochlorothiazide
Urinary kallikrein activity(EU/24 hrs)
Supine plasma renin activity(ng Ai/ml/hr)
Upright plasma renin activity(ng Ai/ml/hr)
Supine plasma aldosterone(pg/ml)
Upright plasma aldosterone(pg/ml)
8.9±2.10.43
±0.081.59
±0.4248.3
±8.0126.4
±23.8
17.6±4.21.34
±0.173.36
±0.6398.4
±19.0126.6
±28.9
= 0.03
<0.01
= 0.04
= 0.07 (ns)
>0.1(ns)
O J
28
24
ase
03
COUJ
i Act
ivity
'53_>£
ary
Kail
16
12
8
4
0
Hypertensives on Placebo
Hypertensives on Thiazide
Normotensive Controls
p=NS
p<0.025p=0.03
7/
FIGURE 1. Graph comparing 24-hour urinary kallikreinexcretion in hypertensives on placebo, hypertensives treatedwith thiazide, and normotensive controls. Vertical barsrepresent the mean ± SEM. The striped bar on the leftrepresents a group of 13 normotensive control subjects,previously reported by our laboratory" and matched to thehypertensives regarding sex (male), race (white), and dietarysodium intake (unrestricted, > 100 mg/24 hrs). Urinarykallikrein activity, before treatment, is significantlydepressed (p < 0.025) in hypertensives below the normoten-sive value. Thiazide treatment significantly (p = 0.03)elevates kallikrein excretion back into normal range.
from 8.9 ± 2.1 to 17.6 ± 4.2 EU/24 hrs, back withinthe normal range. Change in kallikrein excretion didnot correlate directly with change in mean arterialpressure (r = 0.1, p > 0.1, ns).
Both SPRA and UPRA increased during thiazidetherapy (table 2), in the supine position from 0.43 ±0.08 to 1.34 ng Aj/ml/hr (p < 0.01), and in the up-right position from 1.59 ± 0.42 to 3.36 ng A^ml/hr (p= 0.04). These increases in supine and upright PRAoccurred in the absence of changes in whole bloodvolume or plasma volume (table 1).
To examine whether the observed increase inkallikrein excretion could be accounted for bydiuretic-induced increase in circulating mineralo-corticoid concentrations in these patients, plasmaaldosterone concentrations were measured in both thesupine and upright positions.
Plasma aldosterone did not significantly increase(table 2) when comparing placebo and thiazide ineither the supine position (from 48.3 ± 8.0 to 98.4 ±19.0 pg/ml, p = 0.07, ns) or the upright posture (126.4± 23.8 vs 126.6 ± 28.9 pg/ml, p > 0.1, ns). There wasno correlation between change in supine plasmaaldosterone concentration and the increase inkallikrein excretion (r = 0.33, p > 0.1, ns); nor didchange in upright plasma aldosterone concentrationcorrelate with change in kallikrein excretion (r =—0.43, p > 0.1, ns). Neither were there significant cor-relations between change in SPRA and change insupine plasma aldosterone (r = 0.54, p > 0.1, ns) orchange in UPRA and change in upright plasmaaldosterone concentration (r = 0.48, p > 0.1, ns).
Clinical Chemistry Values
During thiazide treatment, there were no changes inblood urea nitrogen, serum creatinine, serum sodium,or hematocrit (table 3). There were, however, signifi-cant declines in serum chloride (from 103.3 ± 0.4mEq/liter down to 99.8 ± 0.7 mEq/liter, p < 0.01)and serum potassium (from 4.1 ±0 .1 mEq/liter downto 3.8 ± 0.1 mEq/liter, p = 0.02), and a significantrise in serum bicarbonate (from 26.2 ± 0.6 to 28.2 ±0.8 mEq/liter, p = 0.02). In addition, there weresignificant increases in the 24-hour urinary excretionof sodium {p = 0.03), potassium {p < 0.01), andchloride (p = 0.05), while 24-hour urinary volume and
by guest on May 24, 2018
http://hyper.ahajournals.org/D
ownloaded from

URINARY KALLIKREIN ACTIVITY /O'Connor et al. 143
24-hour urinary sodium/potassium ratio were un-changed (p > 0.1, ns). Change in kallikrein excretiondid not correlate with changes in either sodium excre-tion (r = —0.24,/? > 0.1, ns), potassium excretion (r =0.11, p > 0.1, ns), or chloride excretion (r = -0.24, p> 0.1, ns).
Degree of Response to Hydrochlorothiazide:Involvement of Kallikrein
Although the blood pressure decrement did notcorrelate directly with the increase in kallikrein excre-tion on thiazides (r = 0.1, p > 0.1, ns), we soughtfurther evidence of kallikrein involvement by dividingthe subjects into those with a MAP fall on therapy ofi> 10% vs those who did not experience such a fall;2* 12patients had a MAP fall of ;> 10%, while seven didnot. The 12 with a greater MAP fall had asignificantly greater increase in kallikrein excretionthan the seven others (12.9 ± 4.5 vs 1.4 ± 6.3 EU/24hrs, p = 0.05; fig. 2). During placebo phase, the 12with a greater MAP response could not be dis-tinguished from the seven others with respect to age,weight, body surface area, blood volume, plasmavolume, serum creatinine, creatinine clearance, 24-hour urinary sodium excretion, 24-hour urinarykallikrein excretion, supine or upright PRA, or supineor upright plasma aldosterone concentration (all p >0.1, ns).
While the 12 with a MAP response ^ 10% had out-patient clinic blood pressures (systolic/dia-stolic/mean pressures of 166.7 ± 6.7/112.2 ±2.5/129.8 ± 3.7 mm Hg) prior to entry into the study
that were similar to those of the other seven subjects(166.6 ± 6.0/105.1 ± 2.3/125.6 ± 3.2 mm Hg) priorto entry, there were differences between respondersand nonresponders in placebo phase blood pressures.
The 12 responders had systolic/diastolic/meanblood pressures on placebo of 143.9 ± 4.5/94.3 ±2.6/110.8 ± 3.1 mm Hg vs values for the seven non-responders of 133.1 ± 4.0/82.7 ± 4.4/99.6 mm Hg onplacebo (p < 0.05 for diastolic and mean arterialpressures;/? > 0.1, ns, for systolic arterial pressure).That is, many subjects had a substantial fall in bloodpressure even on transition from outpatient clinic (onno treatment) to placebo phase. A relative lack ofresponse to thiazides occurred in subjects with lowerdiastolic and mean blood pressures on placebo alone.
Discussion
Our patient group as a whole experienced a signifi-cant decline in MAP during thiazide therapy, whichwas unassociated with any decline in intravascularvolume or plasma volume. The modest decline inweight (87.2 ± 3.0 kg down to 86.4 ± 3.3 kg, p =0.005) with treatment, in the face of unchanged bloodvolume, has been observed previously." It might beargued that significant weight loss (0.8 kg), coupledwith an elevation in plasma renin activity (table 2) andan increase in urinary sodium excretion (table 3),suggests intravascular volume contraction onthiazides, of a magnitude insufficient to be detected byour volume techniques. While this possibility must beadmitted, other interpretations of the increase in PRA
TABLE 3. Clinical Chemistry Values (Before and After Hydrochlorothiazide Treatment)
Measurement Placebo Hydrochlorothiazide
Blood urea nitrogen (mg/dl)
Serum creatinine (mg/dl)
Serum sodium (mEq/liter)
Serum potassium (mEq/liter)
Serum chloride (mEq/liter)
Serum bicarbonate (mEq/liter)
Hematocrit (volume %)
Urinary sodium excretion(mEq/24 hre)
Urinary potassium excTetion(mEq/24 hrs)
Urinary chloride excretion(mEq/24 hrs)
Urinary sodium/potassium ratio
Urinary volume (ml/24 hrs)
15.9±0.9
1.10±.04
140.6±0.6
4.1±0.1
103.3±0.4
26.2±0.6
44.2±0.8
162.4±19.7
69.1±4.6
126.6±23.5
2.34±0.28
1636±180
15.8±1.11.14
±.03
140.9±0.5
3.8±0.1
99.8±0.7
28.2±0.8
44.9±0.7
225.4±22.9
94.8±6.3
216.9±30.5
2.43±0.26
1993±202
>0.1(ns)
> 0.1 (ns)
> .1 (ns)
= 0.02
<0.01
= 0.02
>0.1(ns)
= 0.03
<0.01
= 0.05
>0.1(ns)
>0.1(ns)
by guest on May 24, 2018
http://hyper.ahajournals.org/D
ownloaded from

144 HYPERTENSION VOL 3, No 1, JANUARY-FEBRUARY 1981
and urinary sodium excretion exist (discussed below)and an alternative explanation for the weight loss isthe diuretic-induced fluid loss from the extravascularextracellular fluid compartments, as has. beensuggested previously."
Although thiazide effects upon blood pressure havebeen much studied, these effects remain incompletelyunderstood. After acute administration, it seems clearthat the hypotensive action of these agents isassociated with and may be explained by diminutionin extracellular fluid volume, plasma volume, and ex-changeable sodium.*'' During chronic antihyper-tensive therapy, however, many studies show agradual return of extracellular fluid volume andplasma volume toward (though not quite equalling)pretreatment values.4- *• M Some studies report com-plete normalization of plasma volume' and extra-
p=0.05
CD
OX
—CO
CO
cz
c
ease
i
CNI
iits
/
CD
CO
CD
COLJJ
18
16
14
12
10
8
6
4
2
0
-2
- 4
MAP Response(n=12)
MAP Response(n=7)
FIGURE 2. Graph showing the increase in urinarykallikrein excretion during thiazide treatment of essentialhypertension: comparison of subjects with a blood pressureresponse ofZ. 10% (n = 12) vs those without such a response(n = 7). Those with a fall in MAP on therapy of~2. 10% dis-played a significantly (p = 0.05) greater increment inkallikrein excretion than did those without such an MAPreduction.
cellular fluid volume and total body sodium19 duringsuccessful antihypertensive therapy with thiazides,although more recent studies4- • have demonstratedpersistent slight volume contraction. In our subjects,plasma volume and blood volume did not decline onthiazide vs placebo, regardless of whether placebo orthiazide was given first in the randomized study order.This suggests that there was no bias introduced by an"order effect."
Although persistent volume reduction cannot beneglected as a thiazide antihypertensive mechanism,5
several lines of evidence suggest that othermechanisms besides intravascular volume depletionmay play a role in chronic thiazide effects upon bloodpressure, namely, diminished systemic vascularresistance after chronic thiazide treatment;8"' ordiminished vascular response to infused pressoragents;81"" or antihypertensive superiority of hydro-chlorothiazide to furosemide, a more potentnatriuretic agent;8* or the potent antihypertensiveeffect of the benzothiadiazine derivative diazoxide,40
even in the face of its characteristic effects of renalsodium retention;41'42 or the early finding that thehypotensive effect of intravenous chlorothiazideprecedes any natriuresis."
Our approach to thiazide effects on vascularresistance focused upon the renal circulation and onthe renal kallikrein-kinin system. We chose the renalcirculation because: 1) renal vascular resistance(RVR) is elevated in established essential hyperten-sion, with some evidence indicating the site of in-creased resistance to be the afferent arteriole;41 and 2)since the renal circulation represents 25% of total car-diac output,44 alterations in renal vascular resistancealone may modulate systemic arterial pressure.45"47
Our study showed a significant (p = 0.03) decline inRVR on thiazide therapy, suggesting that the an-tihypertensive effect of the drug may be mediated, atleast in part, by diminution in RVR. In addition, con-stancy of renal plasma flow, glomerular filtration rate,and filtration fraction during treatment point to theafferent arteriole as the site of diminished resistance,43
suggesting a thiazide-mediated correction of a fun-damental renal hemodynamic defect in established es-sential hypertension.
It is of note that the decline in RVR occurred inspite of significant elevations in both supine (p < 0.01)and upright (p = 0.04) PRA.
What mediated the effects of thiazide on the renal(and presumably systemic) circulation? To answer thisquestion, we examined the urinary excretion of the en-zyme kallikrein. This enzyme is derived from the dis-tal renal tubule48'4* and can act on a substrate,kininogen,50'51 to yield peptide products called kinins.The principal kinin produced by renal kallikrein islysyl-bradykinin, or kallidin. These kinins, in the localrenal circulation, have vasodilatory and natriureticproperties.52"" In man, some evidence suggests thatthe renal kallikrein-kinin system, as reflected by the24-hour urinary excretion of the enzyme kallikrein,correlates with renal blood flow and may modulaterenal vascular resistance.13 The reported decrease in
by guest on May 24, 2018
http://hyper.ahajournals.org/D
ownloaded from

URINARY KALLIKREIN ACTIVITY /O'Connor et al. 145
urinary kallikrein excretion in essential hyperten-sion912 may be of etiologic significance in the disease.
In our hypertensive patients on placebo, kallikreinexcretion was significantly (p < 0.025) diminished ascompared to a control group matched for race, sex,and dietary sodium intake.12 Following hydrochloro-thiazide, however, urinary kallikrein excretion in-creased (8.9 ± 2.1 to 17.6 ± 4.2 EU/24 hrs) back intothe normal range, in association with diminution ofrenovascular resistance and normalization of systemicarterial pressure.
We have not established a cause and effect relationbetween the increase in kallikrein and the decline inboth RVR and the systemic arterial pressure.However, it is of note that, while the decrement inMAP on thiazides did not correlate directly with theincrement in kallikrein excretion (r = 0.10, p > 0.1,ns), the 12 subjects with a larger (> 10%) MAP decre-ment on thiazides had a significantly greater incre-ment in kallikrein excretion than the seven otherswithout such a MAP fall (fig. 2), suggesting a role forrenal kallikrein in the blood pressure response tothiazides. The lack of a direct correlation betweenblood pressure decrement and kallikrein increment in-dicates that, if kallikrein is involved, it is not the onlyfactor governing MAP response to thiazides.
Localization of the RVR decrement to the afferentarteriole also suggests involvement of kallikrein, sincein animal studies infused kinins diminish RVR princi-pally at the afferent arteriole.56 Urinary kallikrein ex-cretion does correlate with renal blood flow in man,12
and local changes in RVR, induced in experimentalanimals, may be sufficient to alter systemic arterialpressure, although other humoral factors may be in-volved.4647
The additional possibility that changes in renal kal-likrein may contribute to systemic kinin genera-tion,13- u and thus alter resistance in other vascularbeds, further supports a role for changes in kallikreinexcretion in the normalization of our patients' bloodpressure, although this remains speculative16'16 sincewe have not documented a relationship between renalkallikrein and either plasma kinins or systemicvascular resistance.
We were unable to account for the kallikrein in-crease by changes in circulating mineralocorticoidlevels (table 2). Lack of change in plasma aldosteroneconcentration57'5S or urinary aldosterone excretion27
during thiazide treatment has been noted by other in-vestigators, especially in subjects on unlimited salt in-take,27' " as were our subjects. Lack of aldosteronerise may reflect unchanged adrenal aldosterone secre-tion in the face of the opposing stimuli of increasedPRA (tending to increase plasma aldosterone) anddiminished serum potassium (tending to decreaseplasma aldosterone), since recent evidence69 indicatesthe importance of both the renin-angiotensin systemand potassium ion as modulators of aldosterone secre-tion. However, we did not measure aldosteronesecretory rates, and we cannot exclude the possibilitythat endogenous mineralocorticoid influenced ourkallikrein results.60
Our results pertain to patients treated with thiazidediuretics who are on unlimited dietary salt and waterintake. It is conceivable that other antihypertensivemechanisms may come into play in thiazide-treatedpatients also subjected to dietary sodium restriction,but our data do not bear directly on such patients. Forexample, Leth4 demonstrated mild persistent volumecontraction during thiazide antihypertensive therapyin patients on dietary salt restriction of 6 g salt (ap-proximately 100 mEq sodium) per day, while otherstudies6'28 in patients with unrestricted dietary sodiumfound normalization of plasma volume after long-term thiazides.
Our subjects on unrestricted salt intake excreted162 ± 20 mEq sodium/day on placebo, suggesting adaily intake of approximately 4 g sodium or 10 g salt/day. This increased on therapy to 225 ± 23 mEqsodium/day (table 3), suggesting a daily intake of ap-proximately 5 g sodium or 13 g salt/day. Thisphenomenon has been observed previously in thiazide-treated hypertensive animals61 and man27 even in theabsence of measureable depletion of total bodysodium assessed by exchangeable radiosodium.27 Theunrestricted salt intake in our patients with enhancedsalt ingestion following thiazides may, in part, explainthe lack of plasma volume diminution in our study. Inthis regard, it should be restated that our study wasnot a balance study and that dietary sodium was un-restricted although the patients did not report anyconscious increase in sodium appetite or intake. It isalso conceivable that the increase in kallikrein, with itsnatriuretic capacity, may have played a role in the in-creased sodium excretion,62 although this relationshipremains speculative. One must also consider thepossibility that the observed kallikrein excretionchanges are purely secondary to changes in systemicor renal hemodynamics, since a cause and effectrelationship between augmentation of kallikrein ex-cretion and the blood pressure decrement cannot beestablished on the basis of our data. Likewise, changesin electrolyte excretion may be at least partially con-sequent on systemic and renal hemodynamics, as wellas direct tubular actions of thiazides.
We are unable to offer an immediate explanationfor the observed increases in SPRA and UPRA (table2) in our thiazide-treated patients, in the absence ofdiminution in intravascular volume. Other factorsbesides volume, however, modulate renin release.Possible explanations for the renin increases thus in-clude: the fall in blood pressure itself, activating renalor systemic baroreceptors resulting in renin release;63
changes in sympathetic nervous function, independentof volume, during thiazide therapy;64 and perhaps thePRA increase may have been related to the fall inserum chloride during thiazide treatment (table 3),since some recent evidence66 indicates that chlorideitself, as well as sodium, may be a modulator of reninrelease. An elevation of PRA during thiazide treat-ment, despite normalization of plasma volume, hasalso been reported by other investigators.66
In summary, we have treated chronically a group ofessential hypertensive men with hydrochlorothiazide
by guest on May 24, 2018
http://hyper.ahajournals.org/D
ownloaded from

146 HYPERTENSION VOL 3, No 1, JANUARY-FEBRUARY 1981
and observed the expected fall in blood pressure. In-travascular volume was unchanged on thiazides,suggesting other mechanisms of thiazide action. Con-comitant with the fall in blood pressure, we observed adecrease in RVR with an associated increase inurinary excretion of kallikrein, a substance withknown vasodilatory and hemodynamic effects. Thus,the kallikrein-kinin system may be involved in theantihypertensive response to thiazide diuretics.
Acknowledgments
The authors wish to thank Justine Cervenka for technicalassistance and Marta Zekan for secretarial support.
References
1. Tobian L: Why do thiazide diuretics lower blood pressure in es-sential hypertension? Ann Rev Pharm 7: 399, 1967
2. Wilson IM, Freis ED: Relationship between plasma and ecfvolume depletion and the antihypertensive effect ofchlorothiazide. Circulation 20: 1028, 1959
3. Dustan HP, Cumming GR, Corcoran AC, Page 1H: Amechanism of chlorothiazide enhanced effectiveness of an-tihypertensive ganglioplegic drugs. Circulation 19:360, 1959
4. Leth A: Changes in plasma and ecf volumes on patients with es-sential hypertension during long term treatment with hydro-chlorothiazide. Circulation 42: 479, 1970
5. Tarazi RC, Dustin HP, Frohlich ED: Long term thiazidetherapy in essential hypertension. Circulation 41: 709, 1970
6. Conway J, Lauwers P: Hemodynamic and hypotenisve effectsof long term therapy with chlorothiazide. Circulation 21: 21,1960
7. Lund-Johansen P: Hemodynamic changes in long term diuretictherapy of essential hypertension. Acta Med Scand 187: 509,1970
8. deCarvalho JGR, Dunn FG, Lohmaller G, Frohlich ED:Hemodynamic correlates of prolonged thiazide treatment: com-parison of responders and non-rcsponders. Clin Pharm Therap22: 875, 1977
9. Elliot AH, Nazum FR: The urinary excretion of a depressorsubstance (kallikrein of Frey and Kraut) in arterial hyperten-sion. Endocrinology 18: 462, 1934
10. Margolius HS, Geller R, Pisano JJ, Sjoerdsma A: Alteredurinary kallikrein excretion in human hypertension. Lancet 2:1063, 1065, 1971
11. Margolius HS, Horwitz D, Pisano JJ, Keiser HR: Urinarykallikrein excretion in hypertension man: relationship tosodium intake and sodium retaining steroids. Circ Res 35: 820,1974
12. Levy SB, Lilley JJ, Frigon RP, Stone RA: Urinary kallikreinand plasma renin activity as determinants of renal blood flow:the influence of race and dietary sodium intake. J Clin Invest60: 129, 1977
13. Wong PY, Talamo RC, Williams GH, Colman RW: Responseof the kallikrein-kinin and renin angiotensin systems to salineinfusion and upright posture. J Clin Invest 55: 691, 1975
14. Vinci JM, Zusman R, Bowden R, et al: The kallikrein-kininsystem in Banter's syndrome and its response to prostaglandinsynthetic inhibition. J Clin Invest 61: 1671, 1978
15. Mersey JH, Williams GH, Dluhy RG, Wong PY, Moore TJ:Plasma bradykinin levels and urinary kallikrein excretion innormal renin essential hypertension. J Clin Endo Metab 48:642, 1979
16. Vinci JM, Zusman RM, Izzo JL, Bowden RE, Horwitz D,Pisano JJ, Keiser HR: Human urinary and plasma kinins:relationship to sodium-retaining steroids and plasma renin ac-tivity. Circ Res 44: 228, 1979
17. Cole BR, Giangiacomo J, Ingelfinger JR, Robson AM:Measurement of renal function without urine collection. Acritical evaluation of the constant infusion technique for deter-mination of inulin and para-amino-hippurate. N Engl J Med287: 1109, 1972
18. Haber E, Koerner T, Page LB, Kliman B: Application of aradioimmunoassay for angiotensin I to the physiologicmeasurements of plasma renin activity in normal human sub-jects. J Clin Endocrinol Metab 29: 1349, 1969
19. Sampson EJ, Derek DD, Demers LM: Comparison of tworadioimmunoassay kits for aldosteronc determination. ClinBiochem 9: 216, 1976
20. Beaven VH, Pierce JV, Pisano JJ: A sensitive isotopicprocedure for the assay of esterase activity: measurement ofhuman urinary kallikrein. Clin Chem Acta 32: 67, 1971
21. Margolius HS, Horwitz D, Geller RG, Alexander RW, Gill JR,Pisano JJ, Keiser HR: Urinary kallikrein excretion in normalman: relationship to sodium intake and sodium retainingsteroids. Circ Res 35: 812, 1974
22. Levy SB, Frigon RP, Stone RA: The relationship of urinarykallikrein activity to renal salt and water excretion. Clin SciMol Med 54: 39, 1978
23. Pierce JV: Purification of mammalian kallikreins, kininogens,and kinins. In Handbook of Experimental Pharmacology:Bradykinin, Kallidin and Kallikrein, edited by Erdos EG.Berlin, Springer-Verlag, 1970, pp 21-51
24. Bliss CI: Statistics in Biology. New York, McGraw Hill, 196725. Schalekamp MADH, Schalekamp-Kuyken MPA, Birkenhager
WH: Abnormal renal hemodynamics and renin suppression inhypertensive patients. Clin Sci 38: 101, 1970
26. Weber MA, Drayer JIM, Rev A, Laragh JH: Disparatepatterns of aldosterone response during diuretic treatment ofhypertension. Ann Int Med 87: 558, 1977
27. Gifford RW, Mattox VR, Orvis AL, Stones DA, Rosevear JW:Effect of thiazide diuretics on plasma volume, body electrolytes,and excretion of aldosterone in hypertension. Circulation 24:1197, 1961
28. Hansen J: Hydrochlorothiazide in the treatment of hyperten-sion: the effects on blood volume, exchangeable sodium andblood pressure. Acta Med Scand 183: 317, 1968
29. Wilkins RM, Hollander W, Chobanian AV: Chlorothiazide inhypertension: studies on its mode of action. Ann NY Acad Sci71:465, 1958
30. Freis ED, Wanko AM, Schnaper HW, Frohlich ED:Mechanism of altered blood pressure responsiveness producedby chlorothiazide. J Clin Invest 39: 1277, 1960
31. Weinberger MH: Effect of chlorothiazide and sodium onvascular responsiveness to angiotensin II. Am J Physiol 223:1049, 1972
32. Aleksandrow D, Wysznacka W, Gajewski J: Studies on themechanisms of hypotensive action of hydrochlorothiazide. NEngl J Med 260: 51, 1959
33. Feisal KA, Eckstein JW, Horsley AW, Keasling HH: Effects ofchlorothiazide on forearm vascular response to norepinephrine.J Appl Physiol 16: 549, 1961
34. Mcndelowitz M, Naftchi N, Gitlow SE, Weinreb HL, WolfRL: The effect of chlorothiazide and its congeners on the digitalcirculation in normotensive subjects and in patients with essen-tial hypertension. Ann NY Acad Sci 88: 964, 1960
35. Ogilvie RI, Schlieper E: The effect of hydrochlorothiazide onvenous reactivity in hypertensive man. Clin Pharm Therap 11:589, 1970
36. Eckstein JW, Wendling H, Abboud FM: Circulatory responsesto norepinephrine after prolonged treatment withchlorothiazide. Circ Res 18 (suppl I): 48, 1966
37. GillenwaterJY, Scott JB, Frohlich ED: Effect of chlorothiazideupon the response of the renal bed to vasoactive substances.Circ Res 11: 283, 1962
38. Frohlich ED, Thurman WE, Pfeffer MA, Brobmann GF,Jacobson ED: Altered vascular responsiveness: initial hypoten-sive mechanism of thiazide diuretics. Proc Soc Exp Biol Med140: 1190, 1972
39. Anderson J, Godfrey BE, Hill DM, Munro-Faure AD, SheldonJ: A comparison of the effects of hydrochlorothiazide and offurosemide in the treatment of hypertensive patients. Quart JMed 40: 541, 1971
by guest on May 24, 2018
http://hyper.ahajournals.org/D
ownloaded from

URINARY KALLIKREIN ACTIVITY /O'Connor et al. 147
40. Rubin AA, Zitowitz L, Hausler L: Acute circulatory effects ofdiazoxide and sodium nitrite. J Pharmacol Exp Therap 140:46,1963
41. Thomson AE, Nickerson M, Gaskell P. GrahameGR: Clinicalobservations on an antihypertensive chlorothiazide analoguedevoid of diuretic activity. Can Med Assn J 78: 1306, 1962
42. Bartorelli C, Gargano N, Leonetti G, Zanchetti A: Hypotensiveand renal effects of diazoxide, a sodium retaining ben-zothiadiazine compound. Circulation 27: 895, 1963
43. Gomez DM: Evaluation of renal resistances, with specialreference to changes in essential hypertension. J Clin Invest 30:1143, 1951
44. Pitts RF: Physiology of the Kidney and Body Fluids, 2nd ed.Chicago, Yearbook Medical Publications, 1971, p 144
45. Kottke FJ, Kubicek WG, Visscher MB: The production ofarterial hypertension by chronic renal artery-nerve stimulation.Am J Physiol 145: 38, 1945
46. Katholi RE, Carey RW, Ayers CR, Vaughan ED, Yancey MR,Morton CL: Production of sustained hypertension by chronicintrarenal norepinephrine infusions in conscious dogs. Circ Res40 (suppl 1): 1-118, 1977
47. Fink GD, Brody MJ: Neurogenic control of the renal circula-tion in hypertension. Fed Proc 37: 1202, 1978
48. Orstavik TB, Nustad K, Brandtzalg P, Pierce JV: Cellularorigin of urinary kallikreins. J Histochem Cytochem 24: 1037,1976
49. Scili AG, Carretero OA, Hampton A, Cortes P: Site ofkininogenase secretion in the dog nephron. Am J Physiol 230:533, 1976
50. Webster WE, Pierce JV: The nature of kallidins released fromhuman plasma by kallikreins and other enzymes. Ann NYAcad Sci 104: 91, 1963
51. Kial V, Keiser HR, Pisano JJ: Origin and content of methionyl-lysyl-bradykinin, lysyl-bradykinin, and bradykinin in humanurine. Biochem Pharmacol 25: 2499, 1976
52. Webster ME, Gilmore JP: Influence of kallidin-10 on renalfunction. Am J Physiol 206: 714, 1964
53. Barraclogh MA, Hills IH: Effect of bradykinin on renal func-
tion. Clin Sci 28: 69, 196554. Stein JH, Congbalay RC, Karsh DL, Osgood RW: Effect of
bradykinin on proximal tubular sodium reabsorption in thedog. Evidence for functional nephron heterogeneity. J ClinInvest 51: 1709, 1972
55. Willis LR, Ludens JH, Hook JB, Williamson HE: Mechanismsof natriuretic action of bradykinin. Am J Physiol 217: 1, 1969
56. Baylis C, Deen WM, Myers BD, Brenner BM: Effects of somevasodilator drugs on transcapillary fluid exchange in renal cor-tex. Am J Physiol 230: 1148, 1976
57. van Brummelen P, Schalekamp M, de Graeff J: Influence ofsodium intake on hydrochlorothiazide-induced changes inblood pressure, serum electrolytes, rcnin and aldosterone in es-sential hypertension. Acta Med Scand 204: 151, 1978
58. Bourgoignie JJ, Catanzaro FJ, Perry HM: Renin-angiotensin-aldosterone system during chronic thiazide therapy of benignhypertension. Circulation 37: 27, 1968
59. Dluhy RG: Effects of simultaneous potassium and salineloading on plasma aldosterone levels. J Clin Endo Metab 45:141, 1977
60. Horton R: Laboratory determinations of aldosterone and theirclinical significance. Am J Clin Pharm 54: 297, 1970
61. Iwai J, Ohanian EV, Dahl LK: Influence of thiazide on salthypertension. Circ Res 40 (suppl I): 1-131, 1977
62. Carretero OA, Scicli AG: Renal kallikrein: its localization andpossible role in renal function. Fed Proc 35: 194, 1976
63. Davis JO, Freeman RH: Mechanisms regulating renin release.Physiol Rev 56: 1, 1976
64. Aoki VS, Brody MJ: The effect of thiazide on the sympatheticnervous system of hypertensive rats. Arch Int Pharmacodyn177: 423, 1969
65. Kotchcn TA, Galla JH, Luke RG: Contribution of chloride tothe inhibition of plasma renin by sodium chloride in the rat.Kidney Int 13: 201, 1978
66. van Brummelen P, Woerlcf M, Schalekamp MADH: Long-term versus short-term effects of hydrochlorothiazide on renalhemodynamics in essential hypertension. Clin Sci Mol Med 56:463, 1979
by guest on May 24, 2018
http://hyper.ahajournals.org/D
ownloaded from

D T O'Connor, R A Preston, J A Mitas, 2nd, R P Frigon and R A Stonethiazide diuretics.
Urinary kallikrein activity and renal vascular resistance in the antihypertensive response to
Print ISSN: 0194-911X. Online ISSN: 1524-4563 Copyright © 1981 American Heart Association, Inc. All rights reserved.
is published by the American Heart Association, 7272 Greenville Avenue, Dallas, TX 75231Hypertension doi: 10.1161/01.HYP.3.1.139
1981;3:139-147Hypertension.
http://hyper.ahajournals.org/content/3/1/139World Wide Web at:
The online version of this article, along with updated information and services, is located on the
http://hyper.ahajournals.org//subscriptions/
is online at: Hypertension Information about subscribing to Subscriptions:
http://www.lww.com/reprints Information about reprints can be found online at: Reprints:
document. Permissions and Rights Question and Answer process is available in the
Request Permissions in the middle column of the Web page under Services. Further information about thisOffice. Once the online version of the published article for which permission is being requested is located, click
can be obtained via RightsLink, a service of the Copyright Clearance Center, not the EditorialHypertension Requests for permissions to reproduce figures, tables, or portions of articles originally published inPermissions:
by guest on May 24, 2018
http://hyper.ahajournals.org/D
ownloaded from





























