Embed Size (px)
Citation preview

Uniwersytet Warszawski
Wydział Biologii
Łukasz Michał Szafron Nr albumu: 195473
I. Dysrupcja genu mcmA Aspergillus nidulans
II. Analiza wpływu mutacji mcmAI70A
na regulację katabolizmu argininy u A. nidulans
Praca magisterska
na kierunku Biotechnologia
w zakresie Biologii Molekularnej
Praca wykonana pod kierunkiem
Dr Agnieszki Dzikowskiej
Wydział Biologii Uniwersytetu Warszawskiego
Warszawa, listopad 2005

Oświadczenie kierującego pracą Oświadczam, że niniejsza praca została przygotowana pod moim kierunkiem i stwierdzam, że spełnia ona warunki do przedstawienia jej w postępowaniu o nadanie tytułu zawodowego. Data Podpis kierującego pracą Oświadczenie autora pracy Świadom odpowiedzialności prawnej oświadczam, że niniejsza praca dyplomowa została napisana przez mnie samodzielnie i nie zawiera treści uzyskanych w sposób niezgodny z obowiązującymi przepisami. Oświadczam również, że przedstawiona praca nie była wcześniej przedmiotem procedur związanych z uzyskaniem tytułu zawodowego w wyższej uczelni. Oświadczam ponadto, że niniejsza wersja pracy jest identyczna z załączoną wersją elektroniczną. Data Podpis autora pracy

Szczególne, z serca płynące podziękowania składam moim
kochanym rodzicom, których miłość, wsparcie i nieustająca
wiara we mnie zawsze pomagały mi przezwyciężać
wszelkie trudności i dodawały sił, niezbędnych do
realizacji życiowych planów.
Dziękuję wszystkim moim Koleżankom i Kolegom z
Zakładu Genetyki UW za pomoc i duchowe wsparcie w
chwilach zwątpienia.
Dziękuję Pani Danucie Ślepowrońskiej za wyrozumiałość
i cierpliwość.
Dziękuję Pani Dr Agnieszce Dzikowskiej za opiekę
promotorską, cierpliwość, fachową pomoc i cenne rady.
Dziękuję Panu Prof. Piotrowi Węgleńskiemu za
powierzenie mi tematu niniejszej pracy.


SPIS TREŚCI STRESZCZENIE ....................................................................................................................... 7
WSTĘP....................................................................................................................................... 9
1. Czynniki transkrypcyjne z domeną MADS ......................................................................... 9 1.1. Budowa czynników transkrypcyjnych z rodziny MADS............................................ 9 1.2. Roślinne białka MADS stanowią dużą i bardzo różnorodną grupę czynników
transkrypcyjnych ....................................................................................................... 11 1.3. Białka z domeną MADS wiążą się do specyficznych sekwencji DNA .................... 12 1.4. Charakterystyczną cechą czynników transkrypcyjnych typu MADS jest zdolność
zaginania cząsteczki DNA ........................................................................................ 13 1.5. Czynniki transkrypcyjne z domeną MADS pełnią w komórkach wiele różnych
funkcji........................................................................................................................ 13 2. Czynnik Mcm1p – drożdżowe białko typu MADS............................................................ 15
2.1. Czynnik Mcm1p uczestniczy w kontroli cyklu komórkowego................................. 16 2.2. Białko Mcm1p odgrywa kluczową rolę w procesie zmiany typu płciowego u
drożdży ...................................................................................................................... 19 2.3. Czynnik Mcm1p w kompleksie z białkami z grupy ArgRp reguluje metabolizm
argininy...................................................................................................................... 23 3. Regulacja katabolizmu argininy u A. nidulans................................................................... 28 4. Wydajna, kilkuetapowa metoda dysrupcji genów u A. nidulans ....................................... 30 CEL PRACY............................................................................................................................ 33
MATERIAŁY .......................................................................................................................... 34
1. Szczepy............................................................................................................................... 34 1.1. Szczepy A. nidulans .................................................................................................. 34 1.2. Szczepy bakterii Escherichia coli ............................................................................. 34
2. Podłoża ............................................................................................................................... 34 2.1. Podłoża do hodowli A. nidulans................................................................................ 34 2.2. Podłoża do hodowli bakterii...................................................................................... 35 2.3. Antybiotyki i inne uzupełnienia ................................................................................ 35
3. Enzymy............................................................................................................................... 35 3.1. Enzymy restrykcyjne................................................................................................. 35 3.2. Inne enzymy .............................................................................................................. 36
4. Gotowe zestawy komercyjne ............................................................................................. 36 5. Syntetyczne oligonukleotydy i fragmenty cDNA .............................................................. 36
5.1. Startery do reakcji PCR, wykonanej w celu uzyskania kasety zeo/pyrG do dysrupcji genu mcmA na kosmidzie W02D09 .......................................................... 36
5.2. Startery do amplifikacji regionu, otaczającego miejsce dysrupcji genu mcmA na kosmidzie W02D09................................................................................................... 37
5.3. Fragmenty cDNA ...................................................................................................... 37 6. Odczynniki chemiczne ....................................................................................................... 37 7. Plazmidy i kosmid.............................................................................................................. 37 8. Standardy wielkości ........................................................................................................... 40 9. Inne materiały..................................................................................................................... 40

METODY................................................................................................................................. 41
1. Sprawdzanie fenotypu szczepów A. nidulans .................................................................... 41 2. Otrzymywanie diploidalnego szczepu A. nidulans ............................................................ 41
2.1. Otrzymywanie heterokarionu (metoda I) .................................................................. 41 2.2. Otrzymywanie heterokarionu (metoda II)................................................................. 42 2.3. Otrzymanie szczepu diploidalnego ........................................................................... 42
3. Izolacja DNA genomowego z A. nidulans ......................................................................... 43 4. Elektroforeza ...................................................................................................................... 44 5. Izolacja plazmidowego DNA z bakterii ............................................................................. 44 6. Precypitacja białek 2,5 M NH4Ac ...................................................................................... 44 7. Izolacja DNA z żelu agarozowego..................................................................................... 45 8. Hybrydyzacja typu Southern (wg Sambrook i wsp., 1989) ............................................... 45
8.1. Transfer DNA z żelu na filtr nylonowy .................................................................... 45 8.2. Prehybrydyzacja i hybrydyzacja ............................................................................... 45 8.3. Wyznakowanie sondy radioaktywnym izotopem fosforu 32P ................................... 46
9. Dysrupcja genu mcmA na kosmidzie W02D09.................................................................. 46 9.1. Transformacja bakterii kompetentnych KS272 (pKOBEG) kosmidem W02D09.... 46 9.2. Przygotowanie bakterii elektrokompetentnych KS272 (pKOBEG + W02D09) ...... 47 9.3. Przygotowanie kasety zeo/pyrG do dysrupcji genu mcmA z A. nidulans ................. 48 9.4. Oczyszczanie produktu PCR..................................................................................... 49 9.5. Transformacja komórek elektrokompetentnych KS272 (pKOBEG + W02D09)
kasetą dysrupcyjną zeo/pyrG .................................................................................... 49 10. Oznaczanie stężenia białek w ekstrakcie z grzybni A. nidulans ........................................ 49 11. Oznaczanie arginazy i OTA u A. nidulans ......................................................................... 50 12. Analiza komputerowa ........................................................................................................ 50 WYNIKI................................................................................................................................... 52
I. Dysrupcja genu mcmA A. nidulans .................................................................................... 52 1. Uzyskanie diploidalnego szczepu A. nidulans .......................................................... 52
1.1. Otrzymanie heterokarionu................................................................................... 52 1.2. Otrzymanie szczepu diploidalnego ..................................................................... 53 1.3. Kontrola hybrydyzacyjna uzyskanego szczepu diploidalnego A. nidulans. ....... 55
2. Analiza hybrydyzacyjna kosmidu W02D09, zawierającego gen mcmA................... 56 3. Dysrupcja genu mcmA na kosmidzie W02D09......................................................... 58
3.1. Transformacja szczepu KS272 (pKOBEG) kosmidem W02D09 ....................... 58 3.2. Przygotowanie kasety dysrupcyjnej zeo/pyrG .................................................... 60 3.3. Transformacja szczepu KS272 (pKOBEG + W02D09) kasetą dysrupcyjną zeo/pyrG ....................................................................................................................... 63 3.4. Kontrola hybrydyzacyjna kosmidu W02D09 z dysrupcją genu mcmA............... 65
II. Analiza wpływu mutacji mcmAI70A na regulację katabolizmu argininy u A. nidulans....... 68 1. Analiza wpływu mutacji mcmAI70A na indukcję arginazy i OTA przez argininę ...... 68 2. Analiza wpływu mutacji mcmAI70A na wykorzystanie różnych źródeł azotu przez
A. nidulans................................................................................................................. 70 DYSKUSJA ............................................................................................................................. 73
I. Dysrupcja genu mcmA A. nidulans .................................................................................... 73 II. Analiza wpływu mutacji mcmAI70A na regulację katabolizmu argininy u A. nidulans....... 78 LITERATURA......................................................................................................................... 84

STRESZCZENIE
STRESZCZENIE
Czynniki transkrypcyjne, posiadające domenę MADS, stanowią dużą rodzinę białek,
zaangażowanych w regulację wielu istotnych procesów w komórkach eukariotycznych.
Sekwencja aminokwasowa domeny MADS jest silnie konserwowana ewolucyjnie. Jedną z
najlepiej poznanych funkcji białek MADS jest ich udział w regulacji metabolizmu argininy u
drożdży (Saccharomyces cerevisiae). W regulacji tego procesu uczestniczy białkowy
kompleks Mcm1p-ArgRp. Dwa z czterech białek, budujących ten kompleks (Mcm1p i
ArgRIp) należą do rodziny MADS. Pozostałe dwa – to: ArgRIIp (białko z domeną
dwujądrowego palca cynkowego (Zn2Cys6)) oraz ArgRIIIp (spełniające rolę białka
opiekuńczego, a ponadto wykazujące aktywność kinazy). W regionach promotorowych
genów, związanych z metabolizmem argininy u drożdży, stwierdzono obecność swoistych
sekwencji, tzw. UASarg. Wiadomo, że do sekwencji UASarg wiążą się białka, tworzące
kompleks Mcm1p-ArgRp.
U kropidlaka (Aspergillus nidulans), który, tak jak drożdże, jest przedstawicielem
workowców (Ascomycetes), w promotorach genów katabolizmu argininy zauważono
obecność sekwencji podobnych do UASarg, które nazwano AnUASarg. Wiadomo również, że
aktywatorem ekspresji tych genów jest białko ARCA. Podobnie jak drożdżowe białko
ArgRIIp, posiada ono domenę dwujądrowego palca cynkowego (Zn2Cys6) i podobnie jak
ArgRIIp in vitro nie wiąże się samo do DNA. W genomie A. nidulans zidentyfikowano dwa
geny, kodujące białka z domeną MADS: mcmA oraz rlmA. Znana jest kompletna sekwencja
cDNA obu tych genów.
Głównym celem niniejszej pracy było określenie funkcji białka MCMA poprzez
dysrupcję genu mcmA w haploidalnym i diploidalnym szczepie A. nidulans, a następnie
określenie fenotypu szczepów z dysrupcją. Zastosowano dwuetapową metodę dysrupcji
genów A. nidulans, w której najpierw otrzymuje się dysrupcję badanego genu na kosmidzie, a
następnie kosmidem z dysrupcją transformuje się komórki A. nidulans. W niniejszej pracy
zidentyfikowano kosmid W02D09, zawierający gen mcmA oraz uzyskano dysrupcję genu
mcmA na tym kosmidzie.
Drugim celem tej pracy była analiza wpływu mutacji mcmAI70A na regulację
katabolizmu argininy u A. nidulans. Wiadomo, że analogiczna mutacja w drożdżowym genie
MCM1 wpływa negatywnie na funkcje aktywatorowe białka Mcm1p oraz na jego zdolność do
zaginania cząsteczki DNA. Wyniki uzyskane w niniejszej pracy wskazują, że mutacja
7

STRESZCZENIE
8
mcmAI70A obniża aktywność właściwą arginazy o ok. 40-50%, zarówno na poziomie
podstawowym, jak i po indukcji argininą. Aktywność właściwa transaminazy ornitynowej
(OTA) w zmutowanym szczepie również ulega obniżeniu, lecz różnice są mniejsze i nie
przekraczają 15%. Ponadto zauważono, że mutacja mcmAI70A wpływa negatywnie na tempo
wzrostu i konidiowanie A. nidulans. Na podstawie uzyskanych wyników można wnioskować,
że białko MCMA odgrywa istotną rolę w regulacji katabolizmu argininy u A. nidulans i że
jest też zaangażowane w kontrolę innych procesów fizjologicznych u tego organizmu.
Słowa kluczowe
Aspergillus nidulans; domena MADS; metabolizm argininy; dysrupcja; gen mcmA
Dziedzina pracy (kody wg programu Socrates-Erasmus)
13.4 – Biotechnologia

WSTĘP
WSTĘP
1. Czynniki transkrypcyjne z domeną MADS
Białka z domeną MADS (ang. MADS-box) – to liczna rodzina czynników
transkrypcyjnych, regulujących wiele ważnych procesów w komórkach eukariotycznych.
Białka te są szeroko rozpowszechnione w świecie ożywionym. Można je spotkać zarówno u
zwierząt, jak i u roślin i grzybów. Stwierdzono, że domena MADS, mająca kluczowe
znaczenie dla aktywności tych czynników transkrypcyjnych, wykazuje duże podobieństwo
sekwencji aminokwasowej u organizmów należących do wszystkich królestw
eukariotycznych (Alvarez-Buylla i wsp., 2000; Shore i Sharrocks, 1995). Zatem przetrwała w
postaci prawie niezmienionej ponad miliard lat ewolucji.
Czynniki transkrypcyjne z domeną MADS podzielono na dwie odrębne podrodziny.
Do pierwszej zalicza się m.in. ludzki czynnik odpowiedzi na surowicę krwi (SRF) i
drożdżowe czynniki transkrypcyjne: Mcm1p oraz ArgRIp (Arg80p). Do tej podrodziny
zaklasyfikowano również niektóre roślinne czynniki typu MADS, np. AGL34-like, AGL30 i
AGL33, pochodzące z rzodkiewnika (Arabidopsis thaliana) (Alvarez-Buylla i wsp., 2000).
Białka te nazwano czynnikami MADS typu I (SRF-like) (Shore i Sharrocks, 1995).
Do drugiej podrodziny należą liczne roślinne czynniki transkrypcyjne, a także niektóre
białka zwierzęce, np. MEF2A-D. W literaturze białka te określa się wspólną nazwą MADS II
(MEF2-like) (Shore i Sharrocks, 1995; Hasebe i Banks, 1997; Theissen i wsp., 1996).
Niektórych czynników transkrypcyjnych, zawierających domenę MADS, nie można
zaklasyfikować do żadnej z powyższych grup (Alvarez-Buylla i wsp., 2000; Johansen i wsp.,
2002).
1.1. Budowa czynników transkrypcyjnych z rodziny MADS
Nazwa domeny MADS pochodzi od nazw 4 najwcześniej poznanych białek,
posiadających tę domenę: drożdżowego Mcm1p, roślinnych AG i DEFA oraz znalezionego w
komórkach zwierzęcych SRF (Shore i Sharrocks, 1995). Domena ta jest zbudowana z ok. 80
aminokwasów. Szczególnie silnie konserwowanych jest pierwszych 56 reszt
aminokwasowych, licząc od końca N– tzw. rdzeń domeny MADS. Wykazano, że sam rdzeń
9

WSTĘP
domeny MADS ma cechy typowe dla
czynników transkrypcyjnych, tzn. jest
zdolny do oddziaływania z DNA,
dimeryzacji oraz wiązania innych
białek kompleksu inicjującego
transkrypcję (Norman i wsp., 1988;
Chai i Tarnawski, 2002). W obrębie
domeny MADS można wyróżnić 2
regiony o odmiennych funkcjach.
Region znajdujący się przy końcu N
ma budowę α-helikalną i odpowiada za
wiązanie się czynnika do cząsteczki
DNA, a region położony bliżej końca
C jest zbudowany z hydrofobowej β-
kartki i uczestniczy w dimeryzacji dwóch białek typu MADS. W miejscu, gdzie stykają się ze
sobą oba regiony, występuje silnie konserwowana reszta glicyny. Jej obecność stwierdzono
we wszystkich znanych czynnikach transkrypcyjnych typu MADS (Rys. 1) (Sharrocks i wsp.,
1993). Lokalizacja domeny MADS w obrębie białka jest zmienna, lecz najczęściej jest ona
położona przy końcu N łańcucha aminokwasowego (Norman i wsp., 1988; Shore i Sharrocks,
1995; Chai i Tarnawski, 2002).
MADS
Region hydrofobowy
helisa α
Gly
Region zmienny
N
C
Oddziaływania białko - białko
Wiązanie DNA
Dimeryzacja
Rys. 1. Schemat budowy domeny wiążącej DNA, charakterystycznej dla białek z rodziny MADS. W regionie zmiennym, położonym na końcu C, zależnie od typu białka, może występować domena SAM, MEF2 lub I (szczegóły w tekście). (Rys. wg Shore i Sharrocks, 1995).
Udowodniono, że różne białka typu MADS mogą tworzyć struktury homo- bądź
heterodimeryczne, np. ludzki SRF potrafi dimeryzować z drożdżowymi czynnikami ArgRIp i
Mcm1p, dając z tymi białkami trwałe kompleksy (Mueller i Nordheim, 1991). Z tego powodu
region zmienny, położony za domeną MADS, charakterystyczny dla trzech wymienionych
białek, nazywany jest czasami domeną SAM (Rys. 2) (Shore i Sharrocks, 1995).
Jak się wydaje, precyzyjna kontrola ekspresji wielu genów jest możliwa głównie
dzięki temu, że czynniki transkrypcyjne typu MADS nie oddziałują z DNA pojedynczo, lecz
tworzą złożone kompleksy z innymi białkami, zwłaszcza że sekwencje DNA rozpoznawane
przez różne czynniki MADS są do siebie bardzo podobne (Sprague, 1990; Dolan i Fields,
1991; Treisman i Ammerer, 1992; Mead i wsp., 2002; Dubois i Messenguy, 1991; Messenguy
i Dubois, 2000).
10

WSTĘP
11
1.2. Roślinne białka MADS stanowią dużą i bardzo różnorodną grupę
czynników transkrypcyjnych
1.2. Roślinne białka MADS stanowią dużą i bardzo różnorodną grupę
czynników transkrypcyjnych
Drożdże (Mcm1p, ArgRIp); Zwierzęta (SRF)
Rośliny (AGL30, AGL33, AGL39)
Drożdże (Rlm1p, Smp1p); Zwierzęta (MEF2)
Roślinne białka: (Agamous, Apetala 1, 3)
Rys. 2. Różnice w budowie białek z domeną MADS. (Rys. wg Alvarez-Buylla i wsp., 2000). „?” – budowa domeny znajdującej się na końcu C roślinnych białek MADS typu I jest słabo poznana.
Część roślinnych białek z domeną MADS zalicza się do podrodziny pierwszej
(MADS I), a inne zaklasyfikowano do podrodziny MADS II. Istnieje też cała grupa
czynników transkrypcyjnych, których nie sposób przypisać do żadnej z wymienionych
podrodzin (Johansen i wsp., 2002; Alvarez-Buylla i wsp., 2000). Sekwencjonowanie genomu
rzodkiewnika (A. thaliana) (Arabidopsis Genome Initiative, 2000), ukończone kilka lat temu,
umożliwiło zidentyfikowanie w genomie tego organizmu 107 genów kodujących białka typu
MADS. Funkcja 84% z nich nie jest znana (Parenicová i wsp., 2003).
Część roślinnych białek z domeną MADS zalicza się do podrodziny pierwszej
(MADS I), a inne zaklasyfikowano do podrodziny MADS II. Istnieje też cała grupa
czynników transkrypcyjnych, których nie sposób przypisać do żadnej z wymienionych
podrodzin (Johansen i wsp., 2002; Alvarez-Buylla i wsp., 2000). Sekwencjonowanie genomu
rzodkiewnika (A. thaliana) (Arabidopsis Genome Initiative, 2000), ukończone kilka lat temu,
umożliwiło zidentyfikowanie w genomie tego organizmu 107 genów kodujących białka typu
MADS. Funkcja 84% z nich nie jest znana (Parenicová i wsp., 2003).
Roślinne białka MADS typu II są zbudowane nieco inaczej, niż opisane powyżej
białka typu SRF. Przede wszystkim, oprócz domeny MADS, występuje tu często inny
konserwowany motyw sekwencji, nazywany domeną K (ang. K-box). Nazwa tej domeny
wynika z jej podobieństwa do domeny typu „coiled coil” ze zwierzęcej keratyny (Shore i
Sharrocks, 1995). Domena K jest zbudowana z około 80 reszt aminokwasowych i zapewne
uczestniczy w dimeryzacji roślinnych białek typu MADS (Davies i Schwarz-Sommer, 1994).
Domeny MADS i K są rozdzielone krótką, 30 aminokwasową sekwencją, którą nazwano
regionem I (ang. intervening region) (Rys. 2) (Alvarez-Buylla i wsp., 2000; Johansen i wsp.,
2002). Domena MADS jest zwykle położona przy końcu N łańcucha aminokwasowego,
domena K leży w środkowej jego części, a motyw, znajdujący się bliżej końca C łańcucha
Roślinne białka MADS typu II są zbudowane nieco inaczej, niż opisane powyżej
białka typu SRF. Przede wszystkim, oprócz domeny MADS, występuje tu często inny
konserwowany motyw sekwencji, nazywany domeną K (ang. K-box). Nazwa tej domeny
wynika z jej podobieństwa do domeny typu „coiled coil” ze zwierzęcej keratyny (Shore i
Sharrocks, 1995). Domena K jest zbudowana z około 80 reszt aminokwasowych i zapewne
uczestniczy w dimeryzacji roślinnych białek typu MADS (Davies i Schwarz-Sommer, 1994).
Domeny MADS i K są rozdzielone krótką, 30 aminokwasową sekwencją, którą nazwano
regionem I (ang. intervening region) (Rys. 2) (Alvarez-Buylla i wsp., 2000; Johansen i wsp.,
2002). Domena MADS jest zwykle położona przy końcu N łańcucha aminokwasowego,
domena K leży w środkowej jego części, a motyw, znajdujący się bliżej końca C łańcucha

WSTĘP
12
aminokwasowego, pełni być może rolę domeny transaktywacyjnej (Riechmann i Meyerowitz,
1997). Ze względu na budowę, roślinnym czynnikom typu MADS nadano wspólną nazwę
MIK lub MIKC (Johansen i wsp., 2002). Ostatnio wykazano, że dotychczasowa klasyfikacja
roślinnych białek z domeną MADS może być niewystarczająca, dlatego też niektórzy badacze
zaczęli je zaliczać do kilku różnych grup: MIKC, Mα, Mβ, Mγ i Mδ (Parenicová i wsp.,
2003).
1.3. Białka z domeną MADS wiążą się do specyficznych sekwencji DNA
Sekwencja DNA rozpoznawana przez białka z rodziny MADS ma długość ok. 10 par
zasad i znaleziono ją w regionach promotorowych i wzmacniaczach wielu różnych genów
(Sprague, 1990; Dolan i Fields, 1991; Treisman, 1990). W przypadku kilku czynników
transkrypcyjnych typu MADS udało się ustalić sekwencje najwyższej zgodności dla miejsc, w
których wiążą się one z DNA (Pollock i Treisman, 1990; Pollock i Treisman, 1991; Wynne i
Treisman, 1992). Na przykład dla SRF wspomniana sekwencja najwyższej zgodności
przedstawia się następująco:
Litery N w powyższych wzorach oznaczają, że w danej pozycji ze zbliżonym
prawdopodobieństwem może wystąpić każdy nukleotyd. Małe litery wskazują, iż
prawdopodobieństwo pojawienia się takiej zasady azotowej w opisanym położeniu wynosi
8-24%, czyli jest niższe niż prawdopodobieństwo losowe. Z kolei użycie wielkich liter
świadczy o tym, że przedstawiony nukleotyd znaleziono w ponad 30% naturalnych sekwencji
wiążących dane białko (Shore i Sharrocks, 1995).
Niektórym sekwencjom DNA, rozpoznawanym przez czynniki typu MADS, nadano
specyficzne dla nich nazwy, np. sekwencja, do której wiąże się SRF, bywa nazywana
sekwencją CArG (ang. CArG-box) lub SRE (ang. Serum Response Element), zaś region
wiążący Mcm1p nazwano sekwencją P (ang. P-box) (Shore i Sharrocks, 1995). Sekwencja
najwyższej zgodności dla czynników typu MEF2 wygląda nieco inaczej i można ją opisać
ogólnym wzorem: CTA(A/T)4TAG (Pollock i Treisman, 1991).

WSTĘP
13
1.4. Charakterystyczną cechą czynników transkrypcyjnych typu MADS jest
zdolność zaginania cząsteczki DNA
Zdolność zaginania cząsteczki kwasu nukleinowego zauważono dla różnych
czynników transkrypcyjnych (Brown, 2001; Kerppola i Curran, 1997), w tym także dla białek
typu MADS (West i wsp., 1997; West i Sharrocks, 1999). Dowiedziono, że zdolność
zaginania DNA ma kluczowe znaczenie dla ich funkcji. Stwierdzono też, iż poszczególne
białka MADS zaginają DNA w różnym stopniu, wykorzystując do tego celu odmienne
mechanizmy (Shore i Sharrocks, 1995; West i Sharrocks, 1999). Wiadomo, że jedne białka z
domeną MADS, jak SRF czy Mcm1p, zaginają DNA znacznie silniej niż inne, np. MEF2
(Pellegrini i wsp., 1995; Tan i wsp., 1988; West i wsp., 1997). Co ciekawe, o zdolności
zaginania cząsteczki DNA decydują pojedyncze reszty aminokwasowe, położone w obrębie
domeny MADS, np. lizyna 154 w SRF (K154) czy też reszta kwasu glutaminowego (E14) w
MEF2A (West i wsp., 1997). Na uwagę zasługuje również fakt, że niektóre czynniki typu
MADS, np. Mcm1p czy ArgRIp zaginają DNA w różnym stopniu, w zależności od sekwencji
nukleotydowej, podczas gdy inne, np. SRF, zaginają tak samo silnie każdą cząsteczkę kwasu
nukleinowego (West i Sharrocks, 1999). Jak się wydaje, ma to związek z różnym sposobem
oddziaływania tych białek z DNA. Udowodniono, że pewne reszty aminokwasowe silnie
konserwowane w SRF i mające kluczowe znaczenie dla jego funkcji (np. lizyna K154), w
przypadku białka Mcm1p nie odgrywają tak ważnej roli, choć również występują w
sekwencji domeny MADS (West i Sharrocks, 1999).
1.5. Czynniki transkrypcyjne z domeną MADS pełnią w komórkach wiele
różnych funkcji
Ustalono, że czynniki transkrypcyjne typu MADS kontrolują proces powstawania
kwiatów u roślin wyższych. W tym przypadku spełniają więc funkcję homeotyczną (Schwarz-
Sommer i wsp., 1992; Bowman i Meyerowitz, 1991; Bowman i wsp., 1991; Pelaz i wsp.,
2000). Inne białka tego typu, występujące u zwierząt i grzybów, są zaangażowane w kontrolę
różnych procesów, niejednokrotnie o kluczowym znaczeniu dla całego organizmu (Shore i
Sharrocks, 1995).
Jednym z najlepiej poznanych i najszerzej opisanych czynników typu MADS jest,
występujący w ludzkich komórkach, SRF (ang. Serum Response Factor), czyli czynnik

WSTĘP
14
odpowiedzi na surowicę krwi. Nazwa upamiętnia sposób, w jaki czynnik odkryto. Było to w
1984 roku (Greenberg i Ziff, 1984; Rollins i Stiles, 1989; Chai i Tarnawski, 2002). Do dziś
udało się poznać ok. 30 ssaczych genów, których ekspresja zależy od czynnika SRF. Należą
do nich m.in. protoonkogeny: c-fos, fosB, junB, egr1, egr2, geny neuronalne: nurr77 i nurr1,
geny kodujące: białko szoku cieplnego: HSP70, liczne białka mięśniowe, np. α-aktynę,
ciężkie i lekkie łańcuchy miozynowe, SM22α, telokinę, troponinę, tropomiozynę, kalponinę,
dystrofinę, hormon: natriuretyczny czynnik przedsionkowy (ANF) i wiele innych genów
(Chai i Tarnawski, 2002). Kolejnym, obecnym u człowieka, białkiem typu MADS jest MEF2
(ang. M
odpowiedzi na surowicę krwi. Nazwa upamiętnia sposób, w jaki czynnik odkryto. Było to w
1984 roku (Greenberg i Ziff, 1984; Rollins i Stiles, 1989; Chai i Tarnawski, 2002). Do dziś
udało się poznać ok. 30 ssaczych genów, których ekspresja zależy od czynnika SRF. Należą
do nich m.in. protoonkogeny: c-fos, fosB, junB, egr1, egr2, geny neuronalne: nurr77 i nurr1,
geny kodujące: białko szoku cieplnego: HSP70, liczne białka mięśniowe, np. α-aktynę,
ciężkie i lekkie łańcuchy miozynowe, SM22α, telokinę, troponinę, tropomiozynę, kalponinę,
dystrofinę, hormon: natriuretyczny czynnik przedsionkowy (ANF) i wiele innych genów
(Chai i Tarnawski, 2002). Kolejnym, obecnym u człowieka, białkiem typu MADS jest MEF2
(ang. Myocyte Enhancer Factor 2). W rzeczywistości jest to cała grupa białek (MEF2A-D),
należących do podrodziny MADS II. Kontrolują one rozwój mięśni u ssaków (McDermott i
wsp., 1993; Martin i wsp., 1994).
U drożdży (S. cerevisiae) opisano 4 białka należące do rodziny MADS: Mcm1p,
ArgRIp, Rlm1p oraz Smp1p. Białko Rlm1p jest czynnikiem transkrypcyjnym
zaangażowanym w kontrolę integralności ściany komórkowej drożdży (Garcia i wsp., 2004),
a Smp1p jest zapewne związane z regulacją odpowiedzi komórki na stres osmotyczny (de
Nadal i wsp., 2003). Czynniki transkrypcyjne Rlm1p oraz Smp1p zaliczono do podrodziny
Rys. 3. Procesy regulowane przez drożdżowe białka z domeną MADS (Mcm1p, ArgRIp, Rlm1p, Smp1p). (Rys. wg Messenguy i Dubois, 2003).

WSTĘP
15
MEF2-like (Alvarez-Buylla i wsp., 2000). Białka Mcm1p i ArgRIp należą do podrodziny
SRF-like i m.in. współuczestniczą w regulacji metabolizmu argininy (Rys. 3).
MEF2-like (Alvarez-Buylla i wsp., 2000). Białka Mcm1p i ArgRIp należą do podrodziny
SRF-like i m.in. współuczestniczą w regulacji metabolizmu argininy (Rys. 3).
2. Czynnik Mcm1p – drożdżowe białko typu MADS 2. Czynnik Mcm1p – drożdżowe białko typu MADS
Czynnik Mcm1p pełni w
komórkach drożdży wiele ważnych
funkcji. Najlepiej poznane to:
regulacja cyklu komórkowego
(Althoefer i wsp., 1995, McInerny i
wsp., 1997; Boros i wsp., 2003),
kontrola zmian typu płciowego (Shore
i Sharrocks, 1995; Herskowitz i wsp.,
1992), utrzymywanie w komórce
minichromosomów (Passmore i wsp.,
1988), udział w rekombinacji (Elble i
Tye, 1992), regulacja transkrypcji
retrotranspozonu Ty1 (Errede, 1993;
Yu i Fassler, 1993), udział w
procesach osmoregulacyjnych (Kuo i
wsp., 1997), a także regulacja
metabolizmu argininy (Messenguy i Dubois, 2000).
Czynnik Mcm1p pełni w
komórkach drożdży wiele ważnych
funkcji. Najlepiej poznane to:
regulacja cyklu komórkowego
(Althoefer i wsp., 1995, McInerny i
wsp., 1997; Boros i wsp., 2003),
kontrola zmian typu płciowego (Shore
i Sharrocks, 1995; Herskowitz i wsp.,
1992), utrzymywanie w komórce
minichromosomów (Passmore i wsp.,
1988), udział w rekombinacji (Elble i
Tye, 1992), regulacja transkrypcji
retrotranspozonu Ty1 (Errede, 1993;
Yu i Fassler, 1993), udział w
procesach osmoregulacyjnych (Kuo i
wsp., 1997), a także regulacja
metabolizmu argininy (Messenguy i Dubois, 2000).
Mcm1p składa się z 286 aminokwasów, z czego 80, położonych przy końcu N białka,
tworzy domenę MADS. Silnie konserwowany rdzeń domeny MADS jest zbudowany w
sposób charakterystyczny dla wszystkich czynników, zaliczanych do tej rodziny i zawiera 56
aminokwasów (schemat budowy przedstawiono na Rys. 1). Mcm1p ma na końcu N 17-
nukleotydową sekwencję, która poprzedza domenę MADS, zaś przy końcu C stwierdzono
obecność regionu bogatego w reszty glutaminy. Jak się wydaje oba regiony spełniają w białku
dość ważną rolę. N-końcowy fragment ulega w komórce fosforylacji, a jego mutacje lub
delecje prowadzą do spowolnienia wzrostu komórek drożdży na podłożach o wysokich
stężeniach soli (Kuo i wsp., 1997; Yu i Fassler, 1993). Leżący na końcu C region bogaty w
glutaminę, jest zapewne zaangażowany w proces zmiany typu płciowego (Primig i wsp.,
1991; Bruhn i wsp. 1992).
Mcm1p składa się z 286 aminokwasów, z czego 80, położonych przy końcu N białka,
tworzy domenę MADS. Silnie konserwowany rdzeń domeny MADS jest zbudowany w
sposób charakterystyczny dla wszystkich czynników, zaliczanych do tej rodziny i zawiera 56
aminokwasów (schemat budowy przedstawiono na Rys. 1). Mcm1p ma na końcu N 17-
nukleotydową sekwencję, która poprzedza domenę MADS, zaś przy końcu C stwierdzono
obecność regionu bogatego w reszty glutaminy. Jak się wydaje oba regiony spełniają w białku
dość ważną rolę. N-końcowy fragment ulega w komórce fosforylacji, a jego mutacje lub
delecje prowadzą do spowolnienia wzrostu komórek drożdży na podłożach o wysokich
stężeniach soli (Kuo i wsp., 1997; Yu i Fassler, 1993). Leżący na końcu C region bogaty w
glutaminę, jest zapewne zaangażowany w proces zmiany typu płciowego (Primig i wsp.,
1991; Bruhn i wsp. 1992).
Helisa α -
Kartka β -
DNA -
Rys. 4. Model struktury krystalicznej kompleksu α2-Mcm1p-DNA. Model wykonano w oparciu o dane z bazy PDB.

WSTĘP
2.1. Czynnik Mcm1p uczestniczy w kontroli cyklu komórkowego
Rola białka Mcm1p w regulacji cyklu komórkowego u drożdży jest niebagatelna,
ponieważ nadzoruje ono dwa ważne procesy: przejście z fazy M do G1 oraz z G2 do M
(Althoefer i wsp., 1995; Oehlen i wsp., 1996; McInerny i wsp., 1997).
Tab. 1. Funkcje czynnika Mcm1p i kofaktory, z którymi oddziałuje wg (Messenguy i Dubois, 2003).
Białka, z którymi oddziałuje Mcm1p
Regulowane geny
Funkcja kofaktora
Cykl komórkowy
Yox1, Yhp1 (oba białka posiadają homeodomenę)
Geny specyficzne dla przełomu faz M i G1:
CDC6, CDC47, SWI4, CLN3
represory (oba)
Fkh2p+Ndd1p
Geny specyficzne dla przełomu
faz G2 i M: 26 genów, tzw. klaster CLB2
aktywator
Różnicowanie typu płciowego α1
Geny specyficzne dla typu płciowego α:
MFα1, STE3, SAG1, MFα2
aktywator
Geny specyficzne dla
typu płciowego a: α2
(białko posiadające homeodomenę) MFA1, STE2, STE6, MFA2, BAR1,
AGA2, DPS1, ASG7, DPS2
represor
Ste12 Geny specyficzne dla obu typów płciowych (α i a)
aktywator
Metabolizm argininy ArgRIp (Arg80p) – białko typu MADS ArgRIIp (Arg81p) – białko posiadające domenę dwujądrowego palca cynkowego (Zn2Cys6) ArgRIIIp (Arg82p) – kinaza polifosforanów inozytolu
Geny anaboliczne:
aktywator lub represor1 ARG5,6, ARG8, ARG3, ARG1
Geny kataboliczne: CAR1, CAR2
1) Kompleks Mcm1p−ArgRIp−ArgRIIp hamuje biosyntezę i aktywuje katabolizm argininy. Białko ArgRIIIp stabilizuje powstały kompleks, pełni więc rolę białka opiekuńczego. (Rys. wg Messenguy i Dubois, 2003).
W komórkach drożdży opisano element promotorowy, nazwany ECB (ang. early cell-
cycle box), który jest obecny w promotorach 4 genów (CLN3, SWI4, CDC6 oraz CDC47).
Najwyższy poziom transkrypcji tych genów zaobserwowano na przełomie faz M i G1. W
regionie ECB znajduje się miejsce wiązania Mcm1p (sekwencja P) (McInerny i wsp., 1997).
Udowodniono, że czynnik Mcm1p jest stale związany do ECB (Mai i wsp., 2002), a mimo to
16

WSTĘP
17
ekspresja genów znajdujących się pod kontrolą tych promotorów nie jest konstytutywna. Od
niedawna wiadomo, że dzieje się tak za sprawą dwóch białek: Yox1 i Yhp1 (Pramila i wsp.,
2002). Oba posiadają homeodomenę, oba też wiążą się do DNA oraz oddziałują z Mcm1p,
pełniąc funkcję represorów (Tab. 1). Białka Yox1 i Yhp1 wiążą się do DNA w pobliżu
sekwencji P, rozpoznawanej przez Mcm1p. By nastąpiła represja, wystarczy przyłączenie
jednego z tych białek do Mcm1p. Zatem wydaje się, że role obu białek się pokrywają.
Czynnik Yox1 jest produkowany jedynie w pewnych fazach cyklu komórkowego.
Przypuszcza się, że w przypadku Yhp1 jest podobnie. Wyniki doświadczeń skłaniają do
postawienia tezy, że okresowa aktywność transkrypcyjna genów zależnych od ECB wynika
wyłącznie ze skoordynowanej ekspresji genów kodujących białka Yox1 i Yhp1 (Messenguy i
Dubois, 2003). Oba białka zachowują funkcję represorów nawet w przypadku usunięcia
rozpoznawanych przez nie sekwencji DNA.
Wykonanie szczegółowych badań dotyczących regulacji transkrypcji w genomie S.
cerevisiae umożliwiło znalezienie 28 genów, których ekspresja zależy od elementów ECB i
związanego z nimi czynnika Mcm1p (Pramila i wsp., 2002). Są to geny aktywne w późnej
mitozie, uczestniczące w formowaniu kompleksu pre-replikacyjnego (PRC) i rozpoczynające
fazę S. Ostatnio dowiedziono, że czynnik Mcm1p może również bezpośrednio regulować
replikację DNA, wiążąc się w kompleksach z innymi białkami do miejsc startu replikacji (ori)
(Chang i wsp., 2003). Zauważono, że w przypadku wielu ori inicjacja replikacji DNA
następuje po przyłączeniu czynnika Mcm1p. Postuluje się, iż wykorzystanie miejsc startu
replikacji w komórkach drożdży zależy od aktywności Mcm1p (Chang i wsp., 2004).
Mcm1p, oddziałując z promotorami genów, tworzących klaster CLB2, reguluje
również inny ważny etap cyklu komórkowego – przejście z fazy G2 do M (Tab. 1). Klaster
CLB2 to grupa 26 genów, których ekspresja jest uzależniona od obecności białka Mcm1p
(Zhu i wsp., 2000). Najwyższy poziom transkrypcji genów tworzących klaster obserwuje się
na przełomie faz G2 i M. Najlepiej poznanymi genami z tej grupy są CLB2 oraz SWI5. Gdy
na początku lat 90-tych badano strukturę klastera CLB2, stwierdzono, że do promotorów
tworzących go genów przyłącza się białko Mcm1p oraz nieznany czynnik, który nazwano
SFF (ang. SWI five-factor) (Lydall i wsp., 1991; Althoefer i wsp., 1995; Maher i wsp., 1995).
Dziś wiadomo, że na promotorach powstaje w rzeczywistości kompleks złożony z wielu
białek – kompleks SFF. Niedawno udało się zidentyfikować jeden z czynników, budujących
ten kompleks, który nazwano Fkh2p (Kumar i wsp., 2000; Pic i wsp., 2000, Zhu i wsp., 2000;
Boros i wsp., 2003). Dowiedziono, że białko Mcm1p w kompleksie z Fkh2p inicjuje
transkrypcję genów, wchodzących w skład klastera CLB2. Ustalono też, że konieczne jest

WSTĘP
18
wcześniejsze związanie się z kwasem nukleinowym czynnika Mcm1p, by do DNA mogło się
przyłączyć białko Fkh2p (Pic i wsp., 2000). Zjawisko to nazwano kooperatywnością
wiązania. Wydaje się, że do wystąpienia kooperatywności wiązania niezbędna jest sekwencja
aminokwasowa w białku Fkh2p, położona przed regionem oddziałującym z DNA (Boros i
wsp., 2003). Dla blisko spokrewnionego z Fkh2p czynnika Fkh1p, w którym brak
wspomnianej sekwencji, nie zaobserwowano kooperatywnego wiązania z DNA (Hollenhorst i
wsp., 2001). Do inicjacji transkrypcji genów w klasterze CLB2, oprócz kompleksu Fkh2p-
Mcm1p, wymagana jest obecność jeszcze jednego białka Ndd1p (Koranda i wsp., 2000).
Białko Fkh1p, przyłączając się do DNA, wywołuje efekt przeciwny – hamuje inicjację
transkrypcji genów klastera CLB2, choć udowodniono, że nie wiąże się ono z Mcm1p.
Przypuszczalnie jego rola polega na osłabianiu aktywności kompleksu Fkh2p-Mcm1p-DNA
na zasadzie rywalizacji z białkiem Fkh2p o miejsce wiązania w DNA (Hollenhorst i wsp.,
2000; Hollenhorst i wsp., 2001).
Przeprowadzone ostatnio badania, polegające na ukierunkowanej mutagenezie
różnych regionów białka Fkh2p, pokazują, że czynnik ten wprawdzie nie potrafi się sam
związać z cząsteczką kwasu nukleinowego, ale główną tego przyczyną nie jest brak białka
Mcm1p, lecz obecność specyficznej sekwencji inhibitorowej (między 254 a 292
aminokwasem). Jej usunięcie sprawia, że Fkh2p może się wiązać z DNA, nawet wtedy, gdy
brakuje czynnika Mcm1p (Boros i wsp., 2003). W toku dalszych analiz ustalono, że do
wystąpienia kooperatywności wiązania są niezbędne pewne reszty aminokwasowe zarówno w
jednym, jak i w drugim białku. W czynniku Mcm1p kluczową rolę odgrywają aminokwasy 1-
98 (domena MADS), a w szczególności walina 69 (V69). W Fkh2p najważniejsze są niektóre
aminokwasy aromatyczne z przedziału: 312-458, a zwłaszcza: tyrozyna 315 (Y315) (Shore i
Sharrocks, 1995; Boros i wsp., 2003).
Sekwencje DNA, wiążące Mcm1p i Fkh2p, są oddalone zaledwie o kilka
nukleotydów. W regionie łączącym te sekwencje szczególnie silnie konserwowane są 2
nukleotydy (AA) (Boros i wsp., 2003). Jak wspomniano powyżej, czynnik Fkh2p musi
bezpośrednio oddziaływać z Mcm1p, by mógł przyłączyć się do DNA (kooperatywność
wiązania), dlatego ważne jest, by oba białka po związaniu się z DNA znajdowały się
dostatecznie blisko siebie (Boros i wsp., 2003). Gdy do sekwencji łączącej motywy
rozpoznawane przez białka Mcm1p i Fkh2p wprowadzono insercję 10 nukleotydów (tyle
przypada na jeden skręt helisy DNA), zaobserwowano osłabienie wzajemnych interakcji obu
białek, lecz kompleks nie uległ rozpadowi. Taka trwałość kompleksu ma zapewne związek ze
zdolnością zaginania cząsteczki DNA przez Mcm1p (Boros i wsp., 2003). Warto zaznaczyć,

WSTĘP
19
że sekwencja DNA rozpoznawana przez białko Fkh2p jest niesymetryczna. Zatem właściwa
orientacja motywów, z którymi się wiążą białka Mcm1p oraz Fkh2p, ma duże znaczenie dla
ich wzajemnych oddziaływań (Boros i wsp., 2003).
Część badaczy uważa, że kompleks Fkh2p-Mcm1p-DNA spełnia rolę nieco odmienną
od wyżej opisanej. Twierdzą oni, że omawiany kompleks jest stale związany z promotorami
odpowiednich genów, a inicjacja transkrypcji odbywa się poprzez przyłączenie do kompleksu
białka Ndd1p (Koranda i wsp., 2000). Inni dowodzą, że Fkh2p ulega fosforylacji w sposób
zależny od fazy cyklu komórkowego, a znaczenie tej modyfikacji nie jest znane (Pic i wsp.,
2000). Fosforylacja Fkh2p może więc odpowiadać za regulację oddziaływań białka Ndd1p z
kompleksem Fkh2p-Mcm1p-DNA (Messenguy i Dubois, 2003).
2.2. Białko Mcm1p odgrywa kluczową rolę w procesie zmiany typu płciowego
u drożdży
Drożdże, choć zwykle rozmnażają się przez pączkowanie (czyli wegetatywnie),
zdolne są też do wytwarzania form generatywnych. Nie powstają jednak osobniki odmiennej
płci, lecz komórki należące do 2 różnych haploidalnych typów płciowych: α i a. Na
powierzchni obu typów komórek tworzą się swoiste receptory. Jednocześnie produkowane są
feromony, oddziałujące na komórki odmiennego typu. Dzięki temu, w komórkach
haploidalnych, wystawionych na działanie feromonów, następuje taka zmiana profilu
ekspresji genów, że możliwa się staje fuzja komórek o różnym typie płciowym i wytworzenie
komórek diploidalnych (α/a) (Mead i wsp., 2002).
Za wykształcenie jednego lub drugiego typu płciowego odpowiada gen MAT,
zlokalizowany na 3 chromosomie. Zależnie od typu płciowego komórki, w jej genomie
występuje jeden z dwóch alleli genu MAT (MATα lub MATa odpowiednio w komórkach α
lub a). Zmiana typu płciowego jest możliwa dlatego, że obok genu MAT w genomie S.
cerevisiae obecne są też trwale wyciszone allele tego genu: HMLα i HMLa. Z tego powodu
allele HMLα i HMLa nazywa się czasami milczącymi kasetami typu płciowego. Za sprawą,
obecnej w komórkach drożdży, endonukleazy HO dokonuje się proces konwersji genów. W
tej reakcji milczące kasety spełniają rolę matryc, na których tworzone są nowe cząsteczki
genu MAT. Zmiana typu płciowego następuje, gdy nowo powstałe cząsteczki genu MAT
różnią się od obecnych w genomie przed konwersją (Brown, 2001). Białko Mcm1p odgrywa

WSTĘP
w opisanym procesie kluczową rolę,
ponieważ odpowiada za kontrolę
ekspresji genów właściwych dla
każdego typu płciowego (Rys. 5).
Dotychczas znaleziono 9
genów specyficznych dla typu
płciowego a, które w regionach
promotorowych mają sekwencje
rozpoznawane przez czynnik Mcm1p
(sekwencje P) (Tab. 1) (Messenguy i
Dubois, 2003). Białko Mcm1p,
przyłączając się do tych sekwencji,
aktywuje transkrypcję odpowiednich
genów (Ammerer, 1990). U typu
płciowego α występują przynajmniej
4 geny, których ekspresja zależy od
białka Mcm1p (Tab. 1), lecz w tym
przypadku regulacja przebiega nieco
inaczej. By umożliwić inicjację
ekspresji genów α-specyficznych, do
czynnika Mcm1p musi się
przyłączyć białko α1. Mcm1p wiąże się wówczas do zdegenerowanej sekwencji P, tzw. P’, a
α1 do sąsiadującej z nią sekwencji Q. Dzięki temu ekspresja genów typu α staje się możliwa.
Ponadto w komórkach typu α oraz w diploidalnych komórkach α/a tworzy się kompleks α2-
Mcm1p-α2, który przyłącza się do promotorów genów specyficznych dla typu płciowego a.
Do powstałego kompleksu wiążą się następnie białka Ssn6-Tup1, co powoduje zahamowanie
ekspresji genów a-specyficznych (Rys. 5). Białka α1 i α2 nie występują w komórkach typu a.
U drożdży poddanych działaniu feromonów dochodzi do charakterystycznych zmian
morfologicznych. Zmiany te zapoczątkowuje pobudzenie receptorów powierzchniowych
STE2 i STE3 przez feromony. Potem następuje aktywacja szlaku przekazywania sygnału z
powierzchni komórki do jądra. Wywołana zostaje kaskada reakcji, która prowadzi do
powstania białka Ste12. W komórkach typu a białko Ste12 wiąże się do specyficznych
sekwencji DNA (PRE) i do Mcm1p, a w komórkach typu α oddziałuje z α1. W rezultacie
Rys. 5. Regulacja procesu zmiany typu płciowego u drożdży. Białko Mcm1p, oddziałując ze swoimi kofaktorami, reguluje proces zmiany typu płciowego u drożdży (szczegóły w tekście). W obecności feromonów, zarówno w komórkach α jak i a, do kompleksu inicjującego transkrypcję przyłącza się białko Ste12 (zaznaczone na rysynku linią przerywaną). Rys. wg (Messenguy i Dubois, 2003).
20

WSTĘP
21
następuje zatrzymanie podziałów komórkowych, fuzja komórek i wytworzenie form
diploidalnych α/a (Rys. 5) (Oehlen i wsp., 1996; Messenguy i Dubois, 2003).
następuje zatrzymanie podziałów komórkowych, fuzja komórek i wytworzenie form
diploidalnych α/a (Rys. 5) (Oehlen i wsp., 1996; Messenguy i Dubois, 2003).
Rys. 6. Rola aminokwasów, tworzących domenę MADS, w oddziaływaniu białka Mcm1p z DNA. Na rysunku zaznaczono położenie poszczególnych struktur drugorzędowych, a pod spodem zamieszczono odpowiadający im fragment sekwencji aminokwasowej. Różnymi figurami geometrycznymi oznaczono reszty aminokwasowe kontaktujące się z DNA, wiążące się do DNA i zaangażowane w proces zaginania cząsteczki kwasu nukleinowego (patrz: legenda). Kwadratami zaznaczono aminokwasy o kluczowym znaczeniu dla życia komórki. Letalny efekt niektórych substytucji może być zniesiony przez nadekspresję białka Mcm1p (jasne kwadraty). Warto zauważyć, że reszty aminokwasowe, kontaktujące się z cząsteczką kwasu nukleinowego i niezbędne do wytworzenia kompleksu Mcm1p-DNA, zlokalizowane są w obrębie N-końcowego regionu sekwencji oraz w helisie α I. Powstawanie zagięcia cząsteczki DNA również zależy głównie od reszt aminokwasowych budujących helisę α I. Dane pochodzą z prac: (Acton i wsp., 1997, 2000; Mead i wsp., 2002). (Rys. wg Messenguy i Dubois, 2003).
By w pełni zrozumieć, jak czynnik Mcm1p oddziałuje z DNA i z innymi białkami
(kofaktorami), wykonano szereg substytucji aminokwasów w sekwencji Mcm1p, a następnie
przebadano efekty fenotypowe in vitro oraz in vivo (Acton i wsp., 2000; Jamai i wsp., 2002;
Mead i wsp., 2002). Udowodniono, że Mcm1p (podobnie jak SRF), przyłączając się do
cząsteczki kwasu nukleinowego, wywołuje jej silne zagięcie (Acton i wsp., 1997; West i
wsp., 1997). Dowiedziono też, że stopień zagięcia zależy od sekwencji DNA (West i
Sharrocks, 1999). Wykonując liczne substytucje reszt aminokwasowych w białku Mcm1p,
udało się zidentyfikować 6 o krytycznym znaczeniu dla procesu zaginania DNA: K29, V34,
S37, K40, T66 oraz L68 (Rys. 6) (Acton i wsp., 2000; Lim i wsp., 2003). Zaburzenia tego
procesu skutkowały takimi efektami fenotypowymi, jak problemy z transkrypcją czy
spowolniony wzrost komórek. Wyniki te sugerują, że zagięcie DNA, wywołane
By w pełni zrozumieć, jak czynnik Mcm1p oddziałuje z DNA i z innymi białkami
(kofaktorami), wykonano szereg substytucji aminokwasów w sekwencji Mcm1p, a następnie
przebadano efekty fenotypowe in vitro oraz in vivo (Acton i wsp., 2000; Jamai i wsp., 2002;
Mead i wsp., 2002). Udowodniono, że Mcm1p (podobnie jak SRF), przyłączając się do
cząsteczki kwasu nukleinowego, wywołuje jej silne zagięcie (Acton i wsp., 1997; West i
wsp., 1997). Dowiedziono też, że stopień zagięcia zależy od sekwencji DNA (West i
Sharrocks, 1999). Wykonując liczne substytucje reszt aminokwasowych w białku Mcm1p,
udało się zidentyfikować 6 o krytycznym znaczeniu dla procesu zaginania DNA: K29, V34,
S37, K40, T66 oraz L68 (Rys. 6) (Acton i wsp., 2000; Lim i wsp., 2003). Zaburzenia tego
procesu skutkowały takimi efektami fenotypowymi, jak problemy z transkrypcją czy
spowolniony wzrost komórek. Wyniki te sugerują, że zagięcie DNA, wywołane

WSTĘP
22
przyłączeniem się do niego białka Mcm1p, ma duże znaczenie dla zachowania prawidłowej
funkcji Mcm1p w komórce.
przyłączeniem się do niego białka Mcm1p, ma duże znaczenie dla zachowania prawidłowej
funkcji Mcm1p w komórce.
Rys. 7. Rola aminokwasów, tworzących domenę MADS, w oddziaływaniu białka Mcm1p z kofaktorami. Figurami geometrycznymi zaznaczono reszty aminokwasowe, uczestniczące w oddziaływaniach z kofaktorami (patrz: legenda). Kwadratami oznaczono aminokwasy, oddziałujące z α2 w kompleksie α2-Mcm1p-DNA. Znana jest dokładna struktura krystaliczna tego kompleksu (Rys. 4) (Tan i Richmond, 1998). Ciemne gwiazdki wskazują reszty aminokwasowe, niezbędne białku Mcm1p do utworzenia aktywnego kompleksu z Ste12, a jasne gwiazdki – reszty, które po wprowadzeniu do SRF, pozwalają mu oddziaływać z Ste12 (Mueller i Nordheim, 1991). Do sporządzenia schematu wykorzystano dane z prac: (El Bakkoury i wsp., 2000; Mead i wsp., 2002). (Rys. wg Messenguy i Dubois, 2003).
Przyczyną opisanych zaburzeń może być brak możliwości utworzenia tzw. kompleksu
potrójnego (ang. ternary complex), w którym dwa białka wiążą się ze sobą i z DNA, np.
kompleks α1-Mcm1p-DNA. W tym przypadku rola Mcm1p polega zapewne na takim
odkształceniu łańcucha DNA, by umożliwić obu białkom oddziaływanie ze sobą (Lim i wsp.,
2003). Udowodniono, że tworzenie kompleksów Mcm1p z białkami α1 i Ste12 rzeczywiście
zależy od obecności zagięć w DNA i że pojedyncze mutacje punktowe, w regionach
odpowiedzialnych za powstawanie zagięć, wyraźnie wpływają na aktywność wspomnianych
kompleksów (Rys. 7). W przypadku kompleksu α2-Mcm1p-DNA nie stwierdzono, by
pojedyncze mutacje punktowe w opisywanych regionach miały nań jakikolwiek wpływ.
Dopiero zmutowanie dwóch kluczowych aminokwasów (R87F, S51F lub R87F, V69F)
Przyczyną opisanych zaburzeń może być brak możliwości utworzenia tzw. kompleksu
potrójnego (ang. ternary complex), w którym dwa białka wiążą się ze sobą i z DNA, np.
kompleks α1-Mcm1p-DNA. W tym przypadku rola Mcm1p polega zapewne na takim
odkształceniu łańcucha DNA, by umożliwić obu białkom oddziaływanie ze sobą (Lim i wsp.,
2003). Udowodniono, że tworzenie kompleksów Mcm1p z białkami α1 i Ste12 rzeczywiście
zależy od obecności zagięć w DNA i że pojedyncze mutacje punktowe, w regionach
odpowiedzialnych za powstawanie zagięć, wyraźnie wpływają na aktywność wspomnianych
kompleksów (Rys. 7). W przypadku kompleksu α2-Mcm1p-DNA nie stwierdzono, by
pojedyncze mutacje punktowe w opisywanych regionach miały nań jakikolwiek wpływ.
Dopiero zmutowanie dwóch kluczowych aminokwasów (R87F, S51F lub R87F, V69F)

WSTĘP
23
wywoływało oczekiwany efekt – osłabienie aktywności kompleksu α2-Mcm1p-DNA. Wyniki
te wyraźnie wskazują, że różne kofaktory odmienne oddziałują z białkiem Mcm1p, pomimo
iż przyłączają się do tej samej domeny MADS (Messenguy i Dubois, 2003).
wywoływało oczekiwany efekt – osłabienie aktywności kompleksu α2-Mcm1p-DNA. Wyniki
te wyraźnie wskazują, że różne kofaktory odmienne oddziałują z białkiem Mcm1p, pomimo
iż przyłączają się do tej samej domeny MADS (Messenguy i Dubois, 2003).
Ostatnio udało się ustalić funkcję, jaką spełnia białko α1 w kompleksie z Mcm1p.
Stwierdzono, że przy braku białka α1 czynnik Mcm1p zbyt słabo zagina cząsteczkę kwasu
nukleinowego, by było możliwe utworzenie aktywnego kompleksu inicjacji transkrypcji (Carr
i wsp., 2004). Jest to kolejny dowód na to, że zaginanie DNA ma kluczowe znaczenie dla
aktywności białek z domeną MADS.
Ostatnio udało się ustalić funkcję, jaką spełnia białko α1 w kompleksie z Mcm1p.
Stwierdzono, że przy braku białka α1 czynnik Mcm1p zbyt słabo zagina cząsteczkę kwasu
nukleinowego, by było możliwe utworzenie aktywnego kompleksu inicjacji transkrypcji (Carr
i wsp., 2004). Jest to kolejny dowód na to, że zaginanie DNA ma kluczowe znaczenie dla
aktywności białek z domeną MADS.
2.3. Czynnik Mcm1p w kompleksie z białkami z grupy ArgRp reguluje
metabolizm argininy
2.3. Czynnik Mcm1p w kompleksie z białkami z grupy ArgRp reguluje
metabolizm argininy
Kontrola metabolizmu argininy u drożdży jest procesem bardzo złożonym i
wielokierunkowym (Rys. 8). Ustalono, że zarówno anabolizm, jak i katabolizm argininy
podlegają specyficznej regulacji, zależnej od dostępności tego aminokwasu. Oba procesy są
również regulowane przez mechanizmy ogólne. Anabolizm argininy jest powiązany z
odpowiedzią komórki na głód aminokwasowy. Dowiedziono, że wysokie stężenie argininy w
komórce hamuje ekspresję genów, kodujących dwa pierwsze enzymy szlaku jej biosyntezy.
Kontrola metabolizmu argininy u drożdży jest procesem bardzo złożonym i
wielokierunkowym (Rys. 8). Ustalono, że zarówno anabolizm, jak i katabolizm argininy
podlegają specyficznej regulacji, zależnej od dostępności tego aminokwasu. Oba procesy są
również regulowane przez mechanizmy ogólne. Anabolizm argininy jest powiązany z
odpowiedzią komórki na głód aminokwasowy. Dowiedziono, że wysokie stężenie argininy w
komórce hamuje ekspresję genów, kodujących dwa pierwsze enzymy szlaku jej biosyntezy.
+ + −
Anabolizm Katabolizm Arginina
Arg81 Arg80 Mcm1
Arg82
Aktywacja Represja
ARG5,6 ARG3 ARG8 ARG1 ARG2 ARG7 ARG4 CPA1 CPA2
+ CAR2 CAR1
Nil1
Gln3
+ − Ure2
Ume6 Sin3 Rpd3
+ dostępność azotu
Dal81 + Dal82 +
sól kwasu allofanowego (ang. allophanate)
+
−
peptyd
liderowy +
arginina
oraz
Upf1 Upf2 Upf3
najlepsze źródło azotu −
+ Gcn4
dostępność aminokwasu
Rys. 8. Sieć zależności między różnymi czynnikami regulującymi metabolizm argininy w komórkach drożdży. (Rys. wg Messenguy i Dubois, 2000).

WSTĘP
24
Katabolizm argininy jest uzależniony od obecności argininy oraz braku w podłożu
łatwiej przyswajalnych źródeł azotu, takich jak: amoniak, asparagina czy glutamina. W
przypadku, gdy są dostępne łatwiej przyswajalne źródła azotu, hamowana jest ekspresja
genów CAR1 i CAR2, kodujących odpowiednio arginazę i transaminazę ornitynową (OTA).
Zjawisko to, nazywane represją kataboliczną azotową, jest zależne od czynników Gln3p,
Nil1p, Ure2p (Courchesne i Magasanik, 1988; Stanbrough i wsp., 1995; Coffman i wsp.,
1996; Smart i wsp., 1996; Dubois i Messenguy, 1997), od kompleksu białkowego
Ume6(CargRI)-Sin3(CargRII)-Rpd3(CargRIII) (Messenguy i wsp., 2000), a także od białek
Dal81p i Dal82p (Park i wsp., 1999) (Rys. 8).
Precyzyjną koordynację katabolizmu i anabolizmu argininy u drożdży zapewnia
kompleks czterech białek: Mcm1p-ArgRIp-ArgRIIp-ArgRIIIp. Czynniki ArgRIp i ArgRIIp są
specyficzne tylko dla tego procesu, podczas gdy ArgRIIIp i Mcm1p pełnią w komórce wiele
różnych funkcji. Dotychczas nie stwierdzono, by omawiane białka wzajemnie regulowały
swoją ekspresję (Bercy i wsp., 1987).
Czynnik ArgRIp (Arg80p) jest zbudowany ze 177 aminokwasów. Podobnie jak
Mcm1p należy do pierwszej podrodziny białek z domeną MADS (SRF-like), lecz w
przeciwieństwie do niego, wiąże się do DNA z bardzo niskim powinowactwem, przez co nie
jest w stanie sam aktywować transkrypcji genów (Qiu i wsp., 1990; Jamai i wsp., 2002). Poza
tym jest zaangażowany jedynie w regulację metabolizmu argininy i nie uczestniczy w
regulacji innych procesów, czym również różni się od Mcm1p. Także półokresy życia obu
białek bardzo się różnią i wynoszą odpowiednio: kilka minut dla ArgRIp i kilka godzin dla
Mcm1p (El Bakkoury i wsp., 2000). Podobieństwo domen MADS obu czynników jest
znaczne (56 identycznych aminokwasów z 81), mimo to każdy pełni w komórce odmienne
funkcje (Qiu i wsp., 1990; Bruhn i wsp., 1992).
W cząsteczce ArgRIp znaleziono kilka reszt aminokwasowych o kluczowym
znaczeniu dla oddziaływania czynnika z jego kofaktorami (Y87, Y110, A118, N119, T135).
Ich zastąpienie powoduje poważne zaburzenia w aktywności białka. Ponadto zidentyfikowano
dwa regiony, które mogą służyć czynnikowi ArgRIp do wiązania się z kofaktorami:
hydrofobową kieszeń na powierzchni białka oraz bruzdę, powstającą na styku dwóch
podjednostek, tworzących dimer z Mcm1p. Wydaje się, że niektóre z reszt aminokwasowych,
wymienionych powyżej, mogą być częścią jednej i drugiej struktury. Istnienie dwóch
regionów oddziaływania, pozwalałoby czynnikowi ArgRIp wiązać się jednocześnie z dwoma
kofaktorami, co ułatwiałoby tworzenie kompleksów złożonych z wielu białek (Jamai i wsp.,
2002).

WSTĘP
25
Białko ArgRIIp (Arg81p) jest czynnikiem transkrypcyjnym, posiadającym domenę
dwujądrowego palca cynkowego – Zn2Cys6, złożonym z 880 aminokwasów. ArgRIIp, w
przeciwieństwie do innych białek z tej rodziny, nie tworzy homodimerów, a do związania z
DNA wymaga obecności czynników Mcm1p i ArgRIp. W kompleksie Mcm1p-ArgRp spełnia
rolę czujnika argininy (Messenguy i Dubois, 2000).
Białko ArgRIIIp (Arg82p) (kinaza polifosforanów inozytolu) jest zbudowane z 355
aminokwasów. W komórkach drożdży uczestniczy w regulacji wielu procesów, takich jak:
zmiana typu płciowego, wzrost komórek i sporulacja (Dubois i wsp., 1987; Dubois i
Messenguy, 1994). W przeciwieństwie do trzech pozostałych białek, ArgRIIIp nie ma
zdolności wiązania się z DNA, a w procesie regulacji metabolizmu argininy pełni rolę białka
opiekuńczego, stabilizującego heterodimer dwóch białek MADS: ArgRIp-Mcm1p. Jego
aktywność kinazowa nie odgrywa w tym żadnej roli, jest jednak niezbędna do prawidłowego
przebiegu innych procesów zależnych od ArgRIIIp: transportu mRNA z jądra do cytoplazmy,
odporności na stres osmotyczny, zachowania ciągłości ściany komórkowej, formowania
wodniczek, remodelowania chromatyny, i kontroli procesów metabolicznych komórki, w
zależności od stężenia fosforanów i źródła azotu w podłożu (York i wsp., 1999; Dubois i
wsp., 2002; El Alami i wsp., 2003; Steger i wsp., 2003). Udowodniono, że po wywołaniu
nadekspresji białka Mcm1p lub ArgRIp obecność czynnika ArgRIIIp, podczas tworzenia
heterodimeru Mcm1p-ArgRIp, staje się zbędna, tak in vitro, jak in vivo (El Bakkoury i wsp.,
2000).
Przypuszcza się, że białka z grupy ArgRp i czynnik Mcm1p mogą ze sobą nawzajem
oddziaływać w jądrze komórkowym w sposób niezależny od obecności argininy. Sekwencję
zdarzeń, która prowadzi do powstania kompleksu Mcm1p-ArgRp, przedstawiono na Rys. 9.
Kompleks Mcm1p-ArgRp hamuje ekspresję genów związanych z biosyntezą argininy
(ARG1, ARG3, ARG5,6 oraz ARG8) oraz aktywuje ekspresję genów zaangażowanych w
katabolizm tego aminokwasu (CAR1 i CAR2) (Tab. 1) (Béchet i wsp., 1970; Messenguy i
Dubois, 1993; Messenguy i Dubois, 2000).
Dzięki zastosowaniu mutagenezy ukierunkowanej, w regionach promotorowych
każdego genu, regulowanego przez kompleks Mcm1p-ArgRp, udało się zlokalizować dwa
motywy sekwencji homologiczne do rozpoznawanej przez Mcm1p sekwencji P, które
nazwano „arginine box”. W genach katabolicznych: CAR1, CAR2, jak również w genie
anabolicznym ARG1, sekwencje „arginine box” są położone powyżej sekwencji TATA. Z
kolei w pozostałych genach anabolicznych: ARG5,6, ARG3, ARG8 znaleziono je poniżej
sekwencji TATA (Messenguy i wsp., 1991; DeRijcke i wsp., 1992; Crabeel i wsp., 1995;

WSTĘP
26
Dubois i Messenguy, 1997). Oznacza to, że położenie „arginine box” względem sekwencji
TATA nie decyduje o represji bądź indukcji genu. Pomiędzy dwiema sekwencjami typu
„arginine box” stwierdzono obecność regionu bogatego w pary G-C. Przypuszcza się, że z
tym regionem oddziałuje białko ArgRIIp i dlatego jest on niezbędny do właściwej regulacji
ekspresji genów (Dubois i Messenguy, 1997). Obszar złożony z dwóch sekwencji „arginine-
box” połączonych regionem bogatym w G-C nazwano UASarg (ang. U
Dubois i Messenguy, 1997). Oznacza to, że położenie „arginine box” względem sekwencji
TATA nie decyduje o represji bądź indukcji genu. Pomiędzy dwiema sekwencjami typu
„arginine box” stwierdzono obecność regionu bogatego w pary G-C. Przypuszcza się, że z
tym regionem oddziałuje białko ArgRIIp i dlatego jest on niezbędny do właściwej regulacji
ekspresji genów (Dubois i Messenguy, 1997). Obszar złożony z dwóch sekwencji „arginine-
box” połączonych regionem bogatym w G-C nazwano UAS (ang. arg Upstream Activation
Sequence) lub URS (ang. arg Upstream Repression Sequence), zależnie od spełnianej funkcji.
Co ciekawe, w promotorze genu ARG8 jest obecna tylko jedna sekwencja typu „arginine box”
(Messenguy i Dubois, 2000).
Kompleks białek ArgRp-Mcm1p powstaje w kilku etapach (Rys. 9):
Rys. 9. Kompleks Mcm1p-ArgRIp(Arg80)-ArgRIIp(Arg81)-DNA, regulujący metabolizm argininy u drożdży,powstaje w kilku etapach (szczegóły w tekście). (Rys. wg Messenguy i Dubois, 2000).
Tworzy się heterodimer dwóch białek z domeną MADS (ArgRIp-Mcm1p)
stabilizowany przez czynnik ArgRIIIp. Pewne badania wskazują, że heterodimer
może być cały czas związany z sekwencją URSarg genu ARG1 (Yoon i wsp., 2004).
Ustabilizowany heterodimer łączy się z ArgRIIp – czujnikiem argininy.
Białko ArgRIIIp odłącza się.

WSTĘP
27
Gdy stężenie argininy w komórce jest wysokie, powstały kompleks 3 białek wiąże
się z sekwencjami UASarg i URSarg odpowiednich genów, aktywując lub
reprymując ich ekspresję.
Rola białek typu MADS polega zapewne na takim zagięciu cząsteczki kwasu
nukleinowego, by w obecności argininy powstawał stabilny kompleks białka-DNA, w którym
czynnik ArgRIIp może się związać z regionem bogatym w pary G-C i pełnić funkcję czujnika
argininowego. Ponadto stwierdzono, że żadne z białek, budujących opisany kompleks, nie
potrafi się osobno związać do UASarg lub URSarg (Messenguy i Dubois, 2000).
Zastanawiający jest fakt, że kontrola metabolizmu argininy u drożdży jest jedynym
znanym procesem u tych organizmów, którego regulacja uzależniona jest od dwóch białek z
domeną MADS. Wydaje się, że może to mieć związek z możliwością tworzenia
heterodimerów Mcm1p-ArgRIp in vivo. Dzięki badaniom struktury krystalicznej
powstających kompleksów i wykorzystaniu metody dwuhybrydowej wiadomo, iż Mcm1p z
łatwością tworzy homo- i heterodimery z innymi białkami typu MADS (Tan i Richmond,
1998; El Bakkoury i wsp., 2000). Co prawda nie zaobserwowano, by czynnik ArgRIp tworzył
homodimery in vivo, lecz w pewnych warunkach udało się in vitro uzyskać heterodimery
ArgRIp z białkami SRF oraz Mcm1p (Mueller i Nordheim, 1991). Przypuszcza się, że
powstały dimer białek ArgRIp i Mcm1p może wydajniej i z większą specyficznością
oddziaływać z ArgRIIp i DNA niż pojedyncze białka. Niektóre badania in vitro zdają się
potwierdzać to założenie (Amar i wsp., 2000). Teoria o powstawaniu heterodimerów
wyjaśniałaby, dlaczego do regulacji metabolizmu argininy u drożdży są niezbędne aż dwa
czynniki typu MADS (Messenguy i Dubois, 2003).
Jednym z lepiej poznanych genów biosyntezy argininy jest ARG1. W przypadku
niedoboru argininy, transkrypcja tego genu jest aktywowana przez czynnik Gcn4p. Białko to
powstaje w warunkach głodu aminokwasowego (Hinnebusch, 1992). Regulacja aktywacji
transkrypcji genu ARG1 polega na tym, że czynnik Gcn4p przyłącza lub odłącza od jego
promotora kompleks Mcm1p-ArgRIp-ArgRIIp-ArgRIIIp. Najnowsze badania wykazały, że
heterodimer Mcm1p-ArgRIp jest stale związany z sekwencją promotora genu ARG1. W takiej
postaci jest jednak nieaktywny i dopiero dołączenie do niego białek ArgRIIp i ArgRIIIp
przywraca kompleksowi pełną funkcjonalność. Gdy stężenie argininy w komórce jest
wysokie, czynnik Gcn4p przyłącza białka ArgRIIp i ArgRIIIp do heterodimeru Mcm1p-
ArgRIp, hamując w ten sposób syntezę argininy (Yoon i wsp., 2004). Przedstawiony powyżej
mechanizm stanowi wyraźne potwierdzenie tezy, zakładającej możliwość powstawania
heterodimerów Mcm1p-ArgRIp w komórkach drożdży.

WSTĘP
3. Regulacja katabolizmu argininy u A. nidulans
Kropidlak (A. nidulans), tak jak drożdże, jest przedstawicielem workowców
(Ascomycetes). Wiele procesów metabolicznych, w tym biosynteza i katabolizm argininy,
przebiega u obu gatunków bardzo podobnie (Griffin, 1994; Cybis i wsp., 1972) (Rys. 10).
kwas Δ1pirolino-5-karboksylowy
GluORNITYNA
CYTRULINA
OTC CP GlnGlu
CPS-A
PROLINA
PROLINA
kwas Δ1pirolino-5-karboksylowy
γ-semialdehyd kwasu glutaminowego
ORNITYNA
CYTRULINA
ARGININA
mocznikarginaza
ORNITYNA
OTA
arginino-bursztynian
Glu
mitochondrium cytoplazma
Rys. 10. Szlaki przemian argininy u grzybów nitkowatych, np. A. nidulans. U drożdży (S. cerevisiae) zachodzą takie same przemiany metaboliczne, a różnica polega na cytoplazmatycznej lokalizacji enzymów OTC i CPS-A (Davis, 1986; Griffin, 1994). Użyte oznaczenia: OTA –transaminaza ornitynowa, Glu – glutaminian, Gln – glutamina, CP – karbamoilofosforan, CPS-A – syntetaza karbamoilofosforanu, OTC – karbamoilotransferaza ornitynowa.
28

WSTĘP
29
Podobnie jak u drożdży, katabolizm argininy u A. nidulans jest uzależniony od dwóch
białek enzymatycznych: arginazy i transaminazy ornitynowej (OTA), kodowanych
odpowiednio przez geny agaA i otaA. Transkrypcja obu genów jest indukowana przez
argininę (Borsuk i wsp., 1999; Dzikowska i wsp., 1999) oraz regulowana przez systemy
regulacji ogólnej: represję węglową i azotową (Bartnik i Węgleński, 1973; Dzikowska i wsp.,
1999, 2003). Indukcja transkrypcji genów agaA i otaA u A. nidulans zależy od aktywatora
ARCA, kodowanego przez gen arcA (Bartnik i Węgleński, 1974). Wykazano, że białko
ARCA posiada domenę dwujądrowego palca cynkowego (Zn2Cys6) (Empel i wsp., 2001),
podobnie jak drożdżowe białko ArgRIIp, które w kompleksie Mcm1p-ArgRIp-ArgRIIp
spełnia rolę czujnika argininowego (Messenguy i Dubois, 2000).
W białku ARCA, podobnie jak u innych przedstawicieli tej rodziny (Todd i
Andrianopoulos, 1997), motyw wiążący się z DNA położony jest na N-końcu, a domena
aktywatorowa, zbudowana z aminokwasów o charakterze kwasowym, znajduje się bliżej
końca C łańcucha aminokwasowego. W ARCA znaleziono też potencjalną sekwencję
sygnałową NLS, kierującą białko do jądra oraz potencjalne miejsce fosforylacji dla kinazy
ck2. Fosforylacja łańcucha aminokwasowego może zwiększać wydajność transportu białka
ARCA do jądra, a także wpływać na jego aktywność. Ustalono, że czynnik ARCA nie
reguluje ekspresji genu arcA i że ekspresja tego genu zachodzi na bardzo niskim poziomie,
niezależnie od obecności argininy w komórce (Empel i wsp., 2001).
Charakterystyczną cechą białek z domeną dwujądrowego palca cynkowego (Zn2Cys6)
jest wiązanie się do DNA w formie homodimerów. Region DNA rozpoznawany przez taki
dimer składa się z dwóch trójkowych sekwencji CGG, rozdzielonych kilkoma nukleotydami
(CGG(Nn)CGG lub CGG(Nn)CCG) (Schjerling i Holmberg, 1996; Todd i Andrianopoulos,
1997). W przypadku niektórych białek z domeną dwujądrowego palca cynkowego
zaobserwowano odstępstwa od tego schematu. Jak opisano wcześniej, drożdżowe białko
ArgRIIp nie tworzy homodimerów i wiąże się do DNA w obrębie sekwencji UASarg,
niezawierającej powtórzonej trójki nukleotydów CGG. Do związania z DNA wymaga jednak
obecności dwóch białek typu MADS (Mcm1p i ArgRIp), z którymi tworzy kompleks
(Messenguy i Dubois, 2000).
Ponieważ czynnik ARCA prawdopodobnie nie tworzy dimerów (nie posiada domeny
dimeryzacyjnej typu „coiled-coil”), przypuszcza się, że wiąże się on do DNA jako monomer,
tak jak aktywator ALCR z A. nidulans (Lenouvel i wsp., 1997) lub tworzy kompleksy z
innymi białkami (Empel i wsp., 2001). Ustalono, że in vitro białko ARCA nie potrafi się
samo związać do regionu promotorowego genów agaA i otaA (Dzikowska i Empel, dane

WSTĘP
30
niepublikowane), czym przypomina drożdżowy czynnik ArgRIIp, który jedynie w
kompleksie Mcm1p-ArgRp może się przyłączyć do UASarg (Messenguy i Dubois, 2000). O
możliwości formowania kompleksów ARCA z białkami typu MADS, świadczy fakt, że w
promotorze genu otaA u A. nidulans stwierdzono obecność sekwencji podobnej do
drożdżowej UASarg, którą nazwano AnUASarg. Delecje w obrębie tej sekwencji powodują, że
in vivo, w obecności argininy nie następuje aktywacja ekspresji genu otaA, nawet jeżeli w
komórce jest eksprymowane zmienione białko ARCA o właściwościach superaktywatora
(efekt mutacji arcAd47). Świadczy to o tym, że miejsca wiązania białka ARCA znajdują się w
obrębie sekwencji AnUASarg i, że sekwencja AnUASarg (tak jak UASarg u drożdży)
uczestniczy w zależnej od argininy regulacji ekspresji genu otaA (Dzikowska i wsp., 2003).
W promotorze genu agaA również zidentyfikowano sekwencję podobną do UASarg, którą
nazwano ABS (ang. ARCA binding site). Delecja sekwencji ABS powoduje brak indukcji
arginazy przez argininę (Grzelak, 2003).
Analiza sekwencji genomowej A. nidulans doprowadziła do zidentyfikowania dwóch
genów, kodujących białka z rodziny MADS: mcmA i rlmA. Określono kompletną sekwencję
cDNA genu mcmA (Walczuk, 2005; Dzikowska i wsp., 2005) i genu rlmA (Fujioka i wsp.,
2003). Gen mcmA koduje białko MCMA, które być może jest czynnikiem transkrypcyjnym z
domeną MADS typu I (SRF-like). Z kolei gen rlmA prawdopodobnie koduje białko RLMA –
potencjalny czynnik transkrypcyjny MADS typu II (MEF2-like). Wydaje się, że czynniki
MCMA i RLMA mogą być homologami drożdżowych białek Mcm1p i Rlm1p.
Obecność sekwencji AnUASarg i ABS w genach katabolizmu argininy, a także
występowanie białka MCMA, skłania do postawienia pytania, czy u A. nidulans działa
mechanizm regulacji metabolizmu argininy analogiczny do obserwowanego u drożdży.
4. Wydajna, kilkuetapowa metoda dysrupcji genów u A. nidulans
Jednym ze sposobów określenia funkcji genu jest jego delecja, a następnie analiza
efektów fenotypowych tej delecji. Częściej jednak, zamiast delecji, wykonuje się dysrupcję
genu, tzn. zastąpienie go genem markerowym, pozwalającym na późniejszą selekcję
komórek, w których zaszła dysrupcja.
W niniejszej pracy, w celu ustalenia funkcji genu mcmA, zastosowano nową metodę
dysrupcji genów u A. nidulans (Chaveroche i wsp., 2000). Jest to metoda dwuetapowa. W
pierwszym etapie wykonuje się dysrupcję badanego genu na kosmidzie, pochodzącym z

WSTĘP
31
banku genomowego A. nidulans, przy użyciu kasety dysrupcyjnej zeo/pyrG. Drugi etap
polega na transformacji szczepu A. nidulans o fenotypie pyr− kosmidem po dysrupcji. W
wyniku rekombinacji homologicznej pomiędzy kosmidem a chromosomem A. nidulans
dochodzi do dysrupcji badanego genu w komórkach grzyba (Rys. 11). Kaseta zeo/pyrG
umożliwia selekcję kosmidu z dysrupcją zarówno w bakteriach (gen Sh ble (zeo) warunkuje
oporność na zeocynę), jak i w grzybie (gen pyrG z Aspergillus fumigatus komplementuje
mutację w genie pyrG z A. nidulans).
W tradycyjnej metodzie dysrupcji genów u A. nidulans przeważnie używa się kasety
dysrupcyjnej skonstruowanej na plazmidzie, homologicznej do regionów otaczających
miejsce dysrupcji na obszarze 1–2 kb. Rekombinacja homologiczna zachodzi wówczas z
niską wydajnością (ok. 5–10%). Opisywana tu metoda dysrupcji jest znacznie wydajniejsza.
Wykorzystuje się w niej system rekombinacyjny faga λ (operon Redαβγ na plazmidzie
pKOBEG), pozwalający na dysrupcję badanego genu na kosmidzie, z wykorzystaniem dwóch
50-nukleotydowych regionów homologii pomiędzy kosmidem a kasetą zeo/pyrG.
Transformacja komórek A. nidulans kosmidem po dysrupcji pozwala na wydajną
rekombinację (do 60%), co wynika z obecności na kosmidzie dużego obszaru homologii do
genomu grzyba (do kilkudziesięciu tysięcy par zasad). Metoda ta może być także stosowana
w celu dysrupcji genów u innych grzybów nitkowatych, takich jak: Aspergillus niger, A.
fumigatus, Neurospora crassa.

WSTĘP
32
1
2
3
Rys. 11. Metoda dysrupcji genów wg Chaveroche i wsp. (2000). 1 – Kasetą dysrupcyjną zeo/pyrG transformuje się bakterie, które posiadają kosmid z genem ulegającym dysrupcji oraz plazmid pKOBEG z systemem rekombinacyjnym faga λ (operon Redαβγ). Po transformacji bakterie hoduje się w 30°C. W tych warunkach następuje rekombinacja homologiczna pomiędzy kasetą zeo/pyrG a kosmidem, prowadząca do dysrupcji genu na kosmidzie. 2 – Następnie bakterie hoduje się w 42°C, w celu zgubienia plazmidu pKOBEG (pKOBEG jest replikonem wrażliwym na temperaturę). 3 – Kosmidem po dysrupcji transformuje się komórki A. nidulans. W wyniku rekombinacji homologicznej pomiędzy kosmidem a chromosomem następuje dysrupcja badanego genu w grzybie.

CEL PRACY
CEL PRACY
1. Celem niniejszej pracy było ustalenie funkcji, jaką spełnia u A. nidulans czynnik MCMA
– potencjalne białko z rodziny MADS. Czynnik ten jest kodowany przez gen mcmA. By
osiągnąć zamierzony cel, podjęto próbę wykonania dysrupcji genu mcmA w haploidalnym
i diploidalnym szczepie A. nidulans.
2. Ponieważ wiadomo, że mutacja V34A w drożdżowym białku Mcm1p powoduje znaczny
spadek jego aktywności, polegający m.in. na zmniejszeniu zdolności do aktywacji
transkrypcji genów in vivo, kolejnym celem tej pracy było ustalenie, jaki wpływ na
katabolizm argininy u A. nidulans będzie miała analogiczna mutacja w białku MCMA.
33

MATERIAŁY
MATERIAŁY
1. Szczepy
1.1. Szczepy A. nidulans
1. proA1(6), pyrG89, adF9, phenA2, yA2
2. proA6, pabaA9, biA1
3. proA1(6, 7), uaZ11, prnA432, pyrG89, pabaA1, yA2
4. (D6B-11) biA1, pyrG89, wA3, argB2::eglAp-taaG2::argB+, pyroA4
5. (mcmAI70A) biA1, pyrG89, wA3, argB2::eglAp-taaG2::argB+, pyroA4, pyr4+,
mcmAI70A
Szczepy 4 i 5 otrzymano od Prof. Tetsuo Kobayashi z Uniwersytetu w Nagoya w Japonii.
1.2. Szczepy bakterii Escherichia coli
BW19610 [DE3(lac)X74 Δ uidA::pir-116 recA1 Δ phoA532 Δ (phnC?D-P)33-30]
(Metcalf i wsp., 1994)
KS272 (pKOBEG) [F–ΔlacX74 galE galK thi rpsL ΔphoA (PvuII)]. Szczep ten zawiera
plazmid pKOBEG (Chaveroche i wsp., 2000).
DH5α [supE44 ΔlacU169 (Φ80lacZ ΔM15) hsdR17 recA1 endA1 gyrA96 thi-1 relA1]
(Sambrook i wsp., 1989)
2. Podłoża
2.1. Podłoża do hodowli A. nidulans
CM – podłoże kompletne
MM – podłoże minimalne z 1% glukozą (źródło węgla) i 10 mM azotanem sodu
(źródło azotu)
34

MATERIAŁY
MM−N – podłoże minimalne bez źródła azotu (bez azotanu sodu)
(−N+Arg) − pożywka MM bez azotanów, w której jedynym źródłem azotu jest arginina
o stężeniu 10 mM
(MM+Arg) − pożywka MM, do której po 8 h hodowli grzybni A. nidulans dodano
argininy do ostatecznego stężenia 10 mM
Podłoże minimalne do krzyżówek z 2% glukozą i 12 mM azotanem sodu
2.2. Podłoża do hodowli bakterii
LB – (wg Sambrook i wsp., 1989)
LBLS – podłoże do selekcji z zeocyną (1% bakto-trypton, 0,5% ekstrakt drożdżowy, 0,5%
NaCl, pH 7.5)
SOB – ( 2.0% bakto-trypton, 0.5% ekstrakt drożdżowy, 10 mM NaCl, 2.5 mM KCl,
10 mM MgCl2, 10 mM MgSO4)
2.3. Antybiotyki i inne uzupełnienia
ampicylina (Amp) (ICN Biomedicals, Inc.) o stężeniu końcowym 50 μg/ml
kanamycyna (Km) (Sigma-Aldrich) o stężeniu końcowym 50 μg/ml
chloramfenikol (Cm) (Boehringer Mannheim GmbH) o stężeniu końcowym 25
μg/ml
zeocyna (Zeo) (CAYLA) o stężeniu końcowym 50 μg/ml
tetracyklina (Tc) o stężeniu końcowym 50 μg/ml
L (+) arabinoza (Sigma) o stężeniu końcowym 0,2%
3. Enzymy
3.1. Enzymy restrykcyjne
• EcoRI, HincII, BamHI (MBI Fermentas). Używano firmowych buforów.
Trawienia wykonywano zgodnie z zaleceniami producenta.
35

MATERIAŁY
3.2. Inne enzymy
• Agaraza (MBI Fermentas)
• Rnaza A z trzustki bydlęcej (Sigma)
• Proteinaza K
• Polimeraza exo- (MBI Fermentas)
• Taq polimeraza (MBI Fermentas)
• Mieszanina polimeraz DNA Taq i Tgo z zestawu Expand Long Template PCR
System (Roche)
4. Gotowe zestawy komercyjne
Plasmid Mini (A&A Biotechnology) – zestaw do izolacji plazmidów z komórek
bakteryjnych
Gel-Out (A&A Biotechnology) – zestaw do izolacji DNA z żelu agarozowego
Expand Long Template PCR System (Roche) – zestaw do amplifikacji dużych
genomowych fragmentów DNA
DecaLabel™ DNA Labeling Kit (MBI Fermentas) – zestaw do radioaktywnego
znakowania cząsteczek DNA metodą „random priming”
5. Syntetyczne oligonukleotydy i fragmenty cDNA
5.1. Startery do reakcji PCR, wykonanej w celu uzyskania kasety zeo/pyrG do
dysrupcji genu mcmA na kosmidzie W02D09
o starter: AN_st1b (72 bp) 5’CCAATCATACAGCTCGCCAAGAGCGCTGAATGGTCTGGACAAACTTAGTCCCGGAATT
CTCAGTCCTGCTCC3’
o starter: AN_st2 (70 bp) 5’GGTCAAGGTTGCTCAAGGCCTACATCAACCAACCACCAACCCGGACATCCGAATTCGC
CTCAAACAATGC3’
36

MATERIAŁY
37
Na czerwono – oznaczono regiony komplementarne do sekwencji otaczającej gen mcmA
w genomie A. nidulans (52 i 50 par zasad).
Na zielono – oznaczono regiony komplementarne do sekwencji kasety zeo/pyrG (20 par
zasad).
5.2. Startery do amplifikacji regionu, otaczającego miejsce dysrupcji genu
mcmA na kosmidzie W02D09
o zp_dysr1 (19 bp) 5' GGAGACAAGCATACCAACC 3'
o zp_dysr2 (19 bp) 5' GCTGATAGACAACTCTGCC 3'
5.3. Fragmenty cDNA
Fragment cDNA o długości 168 bp, uzyskany w wyniku reakcji RT-PCR na całkowitym
RNA A. nidulans (A. Dzikowska), którego sekwencja odpowiada fragmentowi mRNA
genu mcmA, kodującemu domenę typu MADS.
6. Odczynniki chemiczne
Do doświadczeń użyto odczynników najwyższej czystości różnych firm.
7. Plazmidy i kosmid
W niniejszej pracy wykorzystano plazmidy: pKOBEG (Rys. 12), pCDA21 (Rys. 13),
bAN649 (Rys. 14), pBR322 (Rys. 15).

MATERIAŁY
Rys. 12. Mapa restrykcyjna plazmidu pKOBEG. Plazmid pKOBEG wyizolowano ze szczepu KS272. Jest on pochodną wektora pSC101 i niesie oporność na chloramfenikol. Na plazmidzie jest wklonowany operon Redα(exo)βγ z faga λ, ułatwiający rekombinację homologiczną w komórkach bakteryjnych. Operon ten znajduje się pod kontrolą promotora pBAD. Ekspresja genów będących pod kontrolą tego promotora jest indukowana przez arabinozę. Plazmid pKOBEG jest replikonem wrażliwym na temperaturę (replikuje się w 30°C). (Chaveroche i wsp., 2000).
Rys. 13. Mapa restrykcyjna plazmidu pCDA21. Plazmid pCDA21 jest pochodną wektora pGP704Not. Wyizolowano go ze szczepu BW19610. Na plazmidzie wklonowanokonstrukt złożony z dwóch genów: Sh ble(zeo) ze Streptoalloteichus hindustanusoraz pyrG z A. fumigatus. Plazmid pCDA21 niesie oporność na ampicylinę (gen bla), zeocynę (gen Sh ble) i replikuje się jedynie w szczepie, z którego go wyizolowano (ma ori replikacji R6Kγ)(Chaveroche i wsp., 2000). Na czerwono wyróżniono miejsca cięcia dla enzymu EcoRI oraz podano wielkości fragmentów, powstających po cięciu tym enzymem.
1,9 kb 3,6 kb
0,5 kb
Rys. 14. Mapa restrykcyjna plazmidu bAN649. Plazmid bAN649 jest pochodną wektora pBluescript® II KS+ i zawiera fragment ClaI – XhoI genu uaZ z A. nidulans(Oestreicher i wsp., 1993a).
uaZ
38

MATERIAŁY
W niniejszej pracy wykorzystano kosmid W02D09 (ok. 48 kb), pochodzący z banku
genomowego A. nidulans, utworzonego na wektorze pWE15 (Brody i wsp., 1991; Prade i
wsp., 1997) (Rys. 16). Kosmid W02D09 zawiera fragment genomu A. nidulans o wielkości
ok. 39 kb z genem mcmA.
Rys. 15. Mapa restrykcyjna plazmidu pBR322. Plazmid pBR322 jest wektorem powszechnie używanym do klonowania w E. coli. Posiada miejsce startu replikacji z plazmidu pMB1. Warunkuje oporność na ampicylinę (gen bla) oraz tetracyklinę (gen tet) (Sutcliffe, 1979).
Rys. 16. Mapa restrykcyjna wektora kosmidowego pWE15. Wektor pWE15 ma wielkość 8,8 kb. Posiada pojedynczą sekwencję cos faga lambda, bakteryjne miejsce startu replikacji ColE1 oraz miejsce startu replikacji małpiego wirusa SV40 i warunkuje oporność na ampicylinę (gen bla) orazkanamycynę i neomycynę (gen kodujący fosfotransferazę II neomycyny (NPTII) pod kontrolą promotora SV40). Dzięki temu selekcja kosmidów, pochodzących od wektora pWE15, jest możliwa zarówno w komórkach prokariotycznych, jak i eukariotycznych. Na wektorze pWE15 znajdują się promotory fagów T3 i T7, otaczające pojedyncze miejsce klonowania BamHI (CS) (Wahl i wsp., 1987;Evans i Wahl, 1987).
39

MATERIAŁY
8. Standardy wielkości
GeneRuler™ DNA Ladder Mix (MBI Fermentas)
GeneRuler™ 1kb DNA Ladder (MBI Fermentas)
GeneRuler™ DNA Ladder Mix, #SM0331/2/3
GeneRuler™ 1kb DNA Ladder, #SM0311/2/3
9. Inne materiały
Agaroza (Sigma)
Agaroza o niskiej temperaturze krzepnięcia (Sea Plaque® FMC®)
Sephacryl S-200 HR (Amersham Pharmacia Biotech)
Filtry nylonowe Nytran N (Schleicher & Schuell Bioscience)
Klisze rentgenowskie Agfa Medical X-Ray Film Blue
Izotop [α – 32P] dATP, (3000 Ci/mmol, 10μCi/μl) (ICN Biomedicals, Inc.)
Ekrany wzmacniające Kodak BioMax MS Intensifying Screen
Kolumienki do oczyszczania DNA SPIN Module (Bio 101)
Bibuła Whatmann 3MM (Schleicher & Schuell Bioscience)
Bio-Rad Protein Assay (Bio-Rad Laboratories)
40

METODY
METODY
1. Sprawdzanie fenotypu szczepów A. nidulans
Do pożywek dodawano uzupełnień w następujących ilościach:
• Jedyne źródło azotu: 5 mM kwas moczowy; 5 mM lub 10 mM prolina; 10 mM arginina;
10 mM ornityna
• Uzupełnienia – 0,2 mM uracyl; 0,1 mM urydyna; 0,5 mM prolina;
adenina; fenyloalanina; PABA; biotyna; pirydoksyna
Roztwory wodne uracylu i urydyny sterylizowano przez filtrację, ponieważ w wyniku
autoklawowania związki te ulegają rozkładowi.
2. Otrzymywanie diploidalnego szczepu A. nidulans
Do otrzymania szczepu diploidalnego użyto 2 szczepów haploidalnych, różniących się
wymaganiami pokarmowymi oraz barwą konidiów.
2.1. Otrzymywanie heterokarionu (metoda I)
Do 20 ml probówki szklanej nalano ok. 10 ml płynnej pożywki kompletnej (CM) +
ewentualne uzupełnienia.
Do probówki przeniesiono przy pomocy sterylnej ezy równą ilość konidiów
każdego ze szczepów i naniesiono je na powierzchnię pożywki.
Probówkę ułożono ukośnie w cieplarce (37ºC) i inkubowano przez 24 h.
Biały kożuszek grzybni obu szczepów przeniesiono ezą do szklanej probówki ze
sterylną wodą miliRo i wstawiono do cieplarki na ok 2 h (37ºC).
Używając ezy, grzybnię wyjęto z wody i przeniesiono na wieczko sterylnej szalki
Petriego z bibułą.
Przy pomocy 2 sterylnych igieł grzybnię pofragmentowano na jak najmniejsze
41

METODY
kawałki.
Kawałki grzybni przeniesiono igłą na szalki ze stałą pożywką minimalną (MM) +
uzupełnienia wspólne dla obu szczepów.
Szalki wstawiono do cieplarki (37ºC).
Po 3 – 5 dniach inkubacji można zaobserwować charakterystyczny wzrost
heterokarionu w formie „drzewkowatej” grzybni.
By oczyścić heterokarion, igłą wycinano agar z fragmentami nie konidiującej
heterokariotycznej grzybni i przenoszono na pożywkę minimalną (MM) +
uzupełnienia wspólne dla obu szczepów, a następnie hodowano w cieplarce (37ºC)
przez 3 – 5 dni.
2.2. Otrzymywanie heterokarionu (metoda II)
Na tej samej szalce z podłożem kompletnym (CM) + ewentualne uzupełnienia
zaszczepiano punktowo igłą w 4 rogach szalki oba szczepy A. nidulans (po 2
nakłucia dla każdego ze szczepów).
Szalki inkubowano w 37ºC przez 3 – 5 dni, czekając, aż grzybnie obu szczepów
się zetkną.
Przy pomocy sterylnej igły wycinano z szalki agar z fragmentami nie konidiującej
grzybni w miejscach, gdzie grzybnie obu szczepów się zetknęły i przenoszono na
podłoże minimalne (MM) + uzupełnienia wspólne dla obu szczepów.
Po 3 – 5 dniach wyrastały grzybnie heterokariotyczne (wzrost w formie
„drzewkowatej” grzybni), które dalej oczyszczano w sposób opisany w metodzie I.
2.3. Otrzymanie szczepu diploidalnego
1. 5 dużych szalek z 5 – dniową grzybnią heterokariotyczną zalewano ok. 5 ml
Tweenu każdą.
2. Przy pomocy miękkiej ezy delikatnie zawieszano konidia w Tweenie.
3. Roztwór konidiów przesączano przez siatkę nylonową do sterylnej kolbki (50 ml).
4. Na 5 dużych szalek wylewano po 20 ml stałego podłoża selekcyjnego, o takim
składzie, by tylko szczep diploidalny był zdolny do wzrostu na nim (dolna
warstwa). Resztę podłoża (ok. 100 ml) wstawiono do 65ºC.
42

METODY
43
5. Roztwór konidiów przelewano z kolbki do pozostałych 100 ml ciepłego podłoża,
mieszano i wylewano w postaci górnej warstwy po 20 ml na 5 przygotowanych
wcześniej szalek.
6. Po zastygnięciu, szalki wstawiano do cieplarki ( 37ºC) i inkubowano przez 3 – 5
dni.
7. Prowadzono obserwacje pod binokularem i wybierano kolonie o zielonych
konidiach.
8. Wybrane kolonie wysiewano na pojedyncze konidia, a następnie testowano na
podłożach selekcyjnych.
3. Izolacja DNA genomowego z A. nidulans
1. Każdą grzybnię po dokładnym utarciu w moździeżu z ciekłym azotem, przenoszono do
2 probówek o pojemności 2 ml (ok. 1/3 probówki).
2. Dodano 1 ml buforu do ekstrakcji: (0,2 M Tris, pH 8,0, 1% SDS, 1 mM EDTA).
3. Inkubowano 5 min. w temp. pokojowej.
4. Inkubowano 20 minut w lodzie.
5. Dodano 100 μl 3 M NaAc, pH 5,2.
6. Dodano 500 μl mieszaniny fenol:chloroform:oktanol (25:24:1) – (2x).
7. Próbki dokladnie wymieszano przez worteksowanie i wstawiono na 5 min do lodu.
8. Zwirowano 5 min. w temp. pokojowej.
9. Górną, wodną warstwę podzielono na 2 części i przeniesiono do nowych probówek.
10. Dodano 2 obj. EtOH 96 %.
11. Inkubowano w lodzie przez 15 min.
12. Zwirowano 10 min., 15000 obr/min., 4°C.
13. Zlano supernatant, a osad przemyto 1ml 70% EtOH.
14. Zwirowano jak wyżej.
15. Osad DNA zawieszono w 20 μl TE, pH 8,0.
16. Dodano 1 μl Rnazy o stężeniu 10 μg/μl i inkubowano w temp. 37°C przez 30 min.
17. Oczyszczono na kolumnie S-200:
Na kolumnę SPIN Module (Bio 101) naniesiono 500 μl złoża Sephacryl.
Odwirowano przez 3 min., 3500 obr/min., 4°C.
Złoże przemyto 400 μl TE, pH 8,0.

METODY
Odwirowano przez 3 min., 3,500 obr/min., 4°C.
Na złoże naniesiono roztwór DNA.
Odwirowano przez 4 min., 4000 obr/min., 4°C.
Eluent (zawierający oczyszczone DNA genomowe) zebrano do nowej probówki.
4. Elektroforeza
Rozdział elektroforetyczny prowadzono w żelach agarozowych: 0,8 %, 1 % i 1,5 % i buforze
1 x TAE (0,04M TRIS-octan; 0,001M EDTA, pH 8,0) (wg Sambrook i wsp. 1989).
5. Izolacja plazmidowego DNA z bakterii
DNA plazmidowe izolowano metodą lizy alkalicznej (Sambrook i wsp., 1989). W celu
odbiałczenia preparatów stosowano mieszaninę: fenol (pH ok. 8):chloroform:oktanol w
proporcjach 25:24:1 lub precypitację białek 2,5 M NH4Ac.
6. Precypitacja białek 2,5 M NH4Ac
1. Do oczyszczanego roztworu DNA dodano 1/2 objętości 7,5 M NH4Ac.
2. Probówki wstawiono do lodu na 20 minut.
3. Zwirowano przez 15 min, 10000 obr./min., 4°C.
4. Zebrano supernatant i dodano do niego 2 – 3 obj. 96 % EtOH lub 0,6 – 1 obj.
izopropanolu.
5. Probówki odstawiono do lodu na 15 min. (jeśli użyto izopropanolu, probówki
pozostawiono w temp. pokojowej).
6. Zwirowano przez 15 min, 10000 obr./min., 4°C.
7. Osad DNA przepłukano 70 % EtOH.
8. Zwirowano przez 5 min, 10000 obr./min., 4°C.
9. Osad DNA zawieszono w 500 μl TE.
10. Dodano 5 μl Rnazy o stężeniu 1 μg/μl i inkubować przez 1h w 37°C.
11. Dodano 50 μl NH4Ac.
44

METODY
12. Wykonano fenolowanie.
13. Wytrącono DNA z roztworu, dodając etanolu lub izopropanolu.
7. Izolacja DNA z żelu agarozowego
W celu izolacji DNA z żelu agarozowego, wykonywano elektroforezę w żelu, używając
agarozy o niskiej temperaturze topnienia (ang. low melting point). Następnie pod
transiluminatorem wycinano właściwy prążek z żelu przy pomocy skalpela. Wycięty fragment
żelu przenoszono do zważonej wcześniej probówki Eppendorfa.
Izolację wykonywano dwoma sposobami:
Wykorzystując agarazę
Używając zestawu Gel-Out do izolacji DNA z żelu agarozowego
W obu przypadkach izolację przeprowadzono zgodnie z zaleceniami producentów.
8. Hybrydyzacja typu Southern (wg Sambrook i wsp., 1989)
8.1. Transfer DNA z żelu na filtr nylonowy
1. Po wykonaniu elektroforezy żel naświetlano na transiluminatorze przez 3 – 5 min., żeby
ułatwić transfer DNA na filtr nylonowy.
2. Żel denaturowano w roztworze: (0,5M NaOH, 1,5M NaCl) przez 20 min. (1x).
3. Żel neutralizowano w roztworze: (1M NH4Ac, 0,02M NaOH) przez 10 min (2x).
4. Transfer DNA z żelu na filtr nylonowy prowadzono przez noc w buforze: (1M NH4Ac,
0,02M NaOH).
5. Po zakończeniu transferu filtr naświetlano promieniami UV o energii 70000 µJ/cm2 w
urządzeniu Crosslinker Hoefer.
8.2. Prehybrydyzacja i hybrydyzacja
Prehybrydyzację prowadzono w 5 – 10 ml buforu hybrydyzacyjnego: (0,5 M NaPi; 1 mM
EDTA; 7 % SDS, 1 % BSA; pH 7,0), w piecu HYBAID w temp. 65°C przez 2 godziny.
45

METODY
46
Hybrydyzację z sondą prowadzono w 5 – 10 ml świeżego buforu hybrydyzacyjnego w
warunkach jak wyżej przez 12 – 24 godziny.
Ekspozycję filtru po hybrydyzacji prowadzono od 20 min. do 24 h w -70°C, z użyciem klisz
fotograficznych i ekranów wzmacniających. Czas ekspozycji i dobór klisz zależą od stopnia
radioaktywności filtru.
8.3. Wyznakowanie sondy radioaktywnym izotopem fosforu 32P
1. Sondę wyznakowano przy użyciu zestawu do znakowania DecaLabelTM DNA
Labeling Kit, zgodnie z zaleceniami producenta. Do znakowania używano
promieniotwórczego izotopu fosforu [α – 32P] dATP.
2. Wyznakowany fragment DNA oczyszczano z niewbudowanych nukleotydów,
nanosząc roztwór sondy na kolumnę ze złożem Sephacryl S-200, przygotowaną, jak w
punkcie 3 (podpunkt 17).
3. Sondę denaturowano chemicznie, dodając 1/10 obj. 3M NaOH, a następnie
inkubowano przez 5 min. w temp. pokojowej.
4. Roztwór zdenaturowanej sondy ostrożnie dodawano do buforu hybrydyzacyjnego,
uważając, by nie nanieść sondy bezpośrednio na filtr.
9. Dysrupcja genu mcmA na kosmidzie W02D09
9.1. Transformacja bakterii kompetentnych KS272 (pKOBEG) kosmidem
W02D09
Wykonano dializę DNA kosmidu W02D09 na krążku do dializy przez 30 min.
Kuwety do transformacji, DNA i bakterie kompetentne umieszczono w lodzie.
Do bakterii dodano ok. 200 ng kosmidu W02D09 po dializie, zamieszano i wstawiono
do lodu na 1 min.
Wykonano elektroporację w elektroporatorze Electroporator 2510 (Eppendorf) prądem
o napięciu 2500V.
Po elektroporacji do bakterii dodano 1 ml płynnej pożywki SOB i inkubowano w 30ºC

METODY
przez 1 h.
Bakterie wysiano na szalki z podłożem LB+Amp+Km+Cm i inkubowano w 30ºC
przez 24 h.
Szczep bakterii, uzyskany w wyniku transformacji szczepu KS272 (pKOBEG) kosmidem
W02D09, nazwano: KS272 (pKOBEG + W02D09).
9.2. Przygotowanie bakterii elektrokompetentnych KS272 (pKOBEG +
W02D09)
Materiałem z pojedynczej, jednodniowej kolonii zaszczepiono 5 ml pożywki płynnej:
(LB+Amp+Km+Cm+0,2% arabinoza).
Bakterie inkubowano przez 16 h z wytrząsaniem w 30ºC (przez noc).
Hodowlą nocną zaszczepiono 500 ml płynnej pożywki LB (wcześniej ogrzanej do
30ºC), z tymi samymi uzupełnieniami, a następnie inkubowano z wytrząsaniem w
30ºC przez ok. 4 – 5 h, mierząc OD600 co 20 min., aż do momentu, gdy OD600 = 0,35 –
0,4.
Hodowlę szybko schłodzono przenosząc kolbę do chłodni do naczynia z lodem (15
min.), po czym przelano ją do 2 schłodzonych probówek wirówkowych (250 ml).
Bakterie zwirowano w rotorze Beckman JA14, przy 3000 rpm przez 15 min, w 4°C.
Osad bakterii delikatnie zawieszono szklaną pipetą w 300 – 400 ml sterylnej,
schłodzonej do 4°C, wody miliQ, potem zwirowano (3500 rpm, 15 min.).
/ powtórzono 3x /
Osad zawieszono w 300 ml zimnego 10% glicerolu i zwirowano jak wyżej. / 1x /
Osad zawieszono w 100 ml zimnego 10% glicerolu i zwirowano jak wyżej. / 1x /
Supernatant dokładnie usunięto przy pomocy pipety, a osad bakteryjny zawieszono w
500µl 10% glicerolu, delikatnie obracając probówki (bez użycia pipety).
Absorbancja tak uzyskanej zawiesiny bakterii, rozcieńczonej 100x, powinna wynosić
ok. 1 przy długości fali λ = 600 nm (OD600 ≈ 1).
Następnie wykonywano próbną elektroporację, żeby sprawdzić czy zawiesina bakterii
jest dobrze odsolona. (W przypadku zbyt silnego zasolenia osad należy ponownie
przepłukać wodą miliQ, a potem glicerolem).
Zawiesinę bakterii rozporcjowywano po 40 µl do schłodzonych probówek typu
Eppendorf, schładzano w ciekłym azocie i przenoszono do -70ºC.
47

METODY
48
Porcję bakterii kompetentnych rozmrażano i wysiewano w całości na szalkę z zeocyną
lub tertacykliną, w celu wykluczenia ewentualnych zakażeń opornych na te
antybiotyki.
Porcję bakterii kompetentnych rozmrażano i wysiewano w całości na szalkę z zeocyną
lub tertacykliną, w celu wykluczenia ewentualnych zakażeń opornych na te
antybiotyki.
Po upewnieniu się, że uzyskane bakterie kompetentne są wrażliwe zarówno na
zeocynę, jak i tetracyklinę, sprawdzano wydajność transformacji plazmidem pBR322.
Transformanty selekcjonowano na podłożu z tetracykliną.
Po upewnieniu się, że uzyskane bakterie kompetentne są wrażliwe zarówno na
zeocynę, jak i tetracyklinę, sprawdzano wydajność transformacji plazmidem pBR322.
Transformanty selekcjonowano na podłożu z tetracykliną.
9.3. Przygotowanie kasety zeo/pyrG do dysrupcji genu mcmA z A. nidulans 9.3. Przygotowanie kasety zeo/pyrG do dysrupcji genu mcmA z A. nidulans
Wykonano reakcję PCR w termocyklerze GeneAmp PCR System 2400 (Applied
Biosystems), zgodnie z poniższą procedurą:
Wykonano reakcję PCR w termocyklerze GeneAmp PCR System 2400 (Applied
Biosystems), zgodnie z poniższą procedurą:
Do probówek do PCR dodano:Do probówek do PCR dodano:
• 1 µl plazmidu pCDA21 (200 ng)
• 2,5 µl dNTP (25 nmoli)
• 2 x 1 µl rozcieńczonych starterów: An_st1b oraz An_st2 (po 100 pmoli
każdego startera)
• 5 µl buforu nr 1
• 0,75 µl (3,75 U) polimeraz z zestawu Expand Long Template Enzyme Mix
• Dopełniono wodą miliQ do 50 µl (38,75 µl)
Warunki reakcji PCR:
• Denaturacja 93ºC 5 min.
• Denaturacja 93ºC 30 s.
• Przyłączanie starterów 58ºC 2 min.
• Elongacja 68ºC 3 min.
4 cykle
• Denaturacja 93ºC 30s.
• Przyłączanie starterów 60ºC 2 min.
• Elongacja 68ºC 3 min.
24 cykle
• Elongacja 68ºC 10 min.
• Przechowywanie 4ºC ∞

METODY
9.4. Oczyszczanie produktu PCR
Produkt reakcji PCR oczyszczono poprzez rozdział w 1% żelu agarozowym, a następnie
wycięcie z żelu „prążka” o wielkości odpowiadającej kasecie zeo/pyrG (ok. 2,5 kb). Do
izolacji DNA z żelu użyto zestawu Gel-Out.
9.5. Transformacja komórek elektrokompetentnych KS272 (pKOBEG +
W02D09) kasetą dysrupcyjną zeo/pyrG
Transformację bakterii wykonano metodą opisaną w punkcie 9.1.
• Do transformacji użyto ok. 200 – 300 ng DNA kasety zeo/pyrG, oczyszczonej
przez izolację z żelu agarozowego.
• Po transformacji bakterie hodowano w płynnej pożywce SOB w 30°C z
wytrząsaniem przez 2h.
• Następnie bakterie wysiano na szalkę z podłożem selekcyjnym:
LBLS+Amp+Km+Cm+Zeo i inkubowano przez 24 h., w 30°C.
• By bakterie utraciły plazmid pKOBEG, przesiano je na podłoże
LBLS+Amp+Km+Zeo i inkubowano w 42°C przez 24 h.
• Wyrosłe bakterie wysiano na pojedyncze kolonie, wykonując posiew redukcyjny
(Skład podłoża i warunki inkubacji jak wyżej).
• Materiałem z pojedynczych kolonii zaszczepiano 2 szalki, różniące się składem
podłoża. Bakterie wysiane na szalkę z podłożem: LBLS+Amp+Km+Zeo
inkubowano w temp. 42°C, a bakterie wysiane na szalkę z podłożem:
LBLS+Amp+Km+Cm+Zeo inkubowano w temp. 30°C.
• Po 24 h inkubacji poszukiwano bakterii, które utraciły oporność na chloramfenikol
(nie rosły na podłożu z chloramfenikolem (Cm), a na podłożu bez Cm rosły
normalnie).
10. Oznaczanie stężenia białek w ekstrakcie z grzybni A. nidulans
Stężenie białek w ekstrakcie z grzybni A. nidulans oznaczano metodą Bradforda (Bradford,
1976).
49

METODY
11. Oznaczanie arginazy i OTA u A. nidulans
Grzybnie A. nidulans do oznaczeń enzymatycznych hodowano przez 10,5 h w temp. 37°C z
wytrzasaniem. Po 8 godzinach od momentu założenia hodowli do pożywek, w których
badano indukcję genów katabolicznych przez argininę (MM+Arg), dodano argininy do
ostatecznego stężenia 10 mM. Następnie wszystkie hodowle prowadzono jeszcze przez 2,5 h.
Aktywność właściwą arginazy oznaczano wg Cybis i Węgleński (Cybis i Węgleński, 1972).
Aktywność właściwą OTA oznaczono wg Albrecht i Vogel (Albrecht i Vogel, 1964).
W niniejszej pracy aktywność właściwą arginazy i OTA wyrażono w jednostkach (u).
Za jednostkę aktywności właściwej arginazy (1 u) uznaje się taką ilość enzymu, która w
czasie 1 min. w reakcji hydrolizy argininy katalizuje wytworzenie 1 μmola mocznika w
przeliczeniu na 1 mg białka.
Za jednostkę aktywności właściwej transaminazy ornitynowej (OTA) (1 u) uznaje się taką
ilość enzymu, która w czasie 1 min. katalizuje wytworzenie z ornityny 1 μmola γ-
semialdehydu kwasu glutaminowego w przeliczeniu na 1 mg białka.
Wymiar jednostki (u) można opisać wzorem: [u] = [μmol×min-1×mg-1].
12. Analiza komputerowa
W niniejszej pracy wykorzystano dane z bazy genomowej A. nidulans BROAD (Genome
Sequencing and Analysis Program Broad Institute of MIT and Harvard)
(http://www.broad.mit.edu/).
Informacje o sekwencjach DNA zaczerpnięto z baz: EMBL-EBI: (http://srs.ebi.ac.uk/) oraz
NCBI: (http://www.ncbi.nlm.nih.gov/). Informacji o białkach poszukiwano w bazach:
ExPASy (http://www.expasy.ch/), CATH (http://cathwww.biochem.ucl.ac.uk/) oraz SCOP
(http://scop.mrc-lmb.cam.ac.uk/scop/). Dane o strukturze krystalicznej białek pochodzą z
bazy PDB (http://www.rcsb.org/pdb/). Do wykonania analiz bioinformatycznych posłużyły
programy z pakietu EMBOSS, np. Transeq (http://www.ebi.ac.uk/emboss/transeq/), Revseq
50

METODY
51
(http://humpback.bii.a-star.edu.sg/cgi-bin/emboss/emboss.pl?_action=input&_app=revseq), a
także serwisy bioinformatyczne dostępne „on-line”, np.:
Webcutter 2.0 (http://rna.lundberg.gu.se/cutter2/),
Nebcutter V.2.0 (http://tools.neb.com/NEBcutter2/),
BLAST (http://www.ncbi.nlm.nih.gov/BLAST/).
Do projektowania doświaczeń i obróbki uzyskanych wyników użyto programów DNATools
6.0, Swiss-PdbViewer 3.7, Clone Manager 4.0, ClustalX (1.83), GEL (program do kalibracji
żeli autorstwa P. Golika).
Do sporządzenia niniejszej pracy wykorzystano programy pakietu Microsoft Office firmy
Microsoft Corporation, pakietu OpenOffice.org firmy Sun Microsystems Inc., Adobe
Photoshop firmy Adobe, Corel Draw i Corel Photo-Paint firmy Corel Corporation.

WYNIKI
WYNIKI
I. Dysrupcja genu mcmA A. nidulans
1. Uzyskanie diploidalnego szczepu A. nidulans
Aby otrzymać diploidalny szczep A. nidulans do dysrupcji genu mcmA, należało
najpierw wyselekcjonować szczep z grzybnią heterokariotyczną (w takim szczepie strzępki
grzybni zawierają dwa genetycznie różne haploidalne jądra komórkowe). Charakterystyczną
cechą grzybni heterokariotycznej jest wytwarzanie dwóch rodzajów haploidalnych konidiów
o fenotypach specyficznych dla każdego z tworzących ją szczepów. Z kolei w szczepie
diploidalnym powstają konidia diploidalne o jednakowym fenotypie. Fakt ten można
wykorzystać do łatwiejszego odróżnienia diploida od heterokarionu, selekcjonując
heterokarion z dwóch szczepów haploidalnych, różniących się barwą konidów. W takim
przypadku grzybnia heterokariotyczna będzie wytwarzać różnobarwne konidia, a w szczepie
diploidalnym konidia będą jednobarwne.
Diploid do dysrupcji genu mcmA powinien mieć zmutowne oba allele genu proA i
genu pyrG. Mutacja w genie proA umożliwia wykorzystanie diploida do badania genów,
uczestniczących w regulacji metabolizmu argininy, które w formie zmutowanej mogą
wywoływać supresję mutacji prolinowej. Ponieważ w niniejszej pracy wykorzystano nową
metodę dysrupcji genów A. nidulans, wg (Chaveroche i wsp., 2000), mutacja w genie pyrG
była niezbędna, gdyż umożliwia ona selekcję szczepu A. nidulans, w którym nastąpiła
dysrupcja określonego genu.
1.1. Otrzymanie heterokarionu
Dysponowano szczepem:
proA1(6, 7), uaZ11, prnA432, pyrG89, pabaA1, yA2 (żółte konidia)
Należało otrzymać szczep z mutacjami w genach proA i pyrG, o zielonej barwie konidiów.
W tym celu wykonano krzyżówkę szczepu:
52

WYNIKI
proA1(6), pyrG89, adF9, phenA2, yA2 (żółte konidia)
ze szczepem:
proA6, pabaA9, biA1 (zielone konidia).
W wyniku tej krzyżówki otrzymano dwa szczepy o właściwych fenotypach:
proA1(6), pyrG89, adF9, biA1 (zielone konidia)
proA1(6), pyrG89, adF9, biA1, phenA2 (zielone konidia)
Heterokarion selekcjonowano dwiema metodami (patrz: Metody, punkty 2.1 i 2.2)
ze szczepów:
proA1(6), pyrG89, adF9, biA1 i proA1(6, 7), uaZ11, prnA432, pyrG89, pabaA1, yA2
oraz
proA1(6), pyrG89, adF9, biA1, phenA2 i proA1(6, 7), uaZ11, prnA432, pyrG89,
pabaA1, yA2
Udało się wyselekcjonowano tylko jeden rodzaj grzybni heterokariotycznej:
proA1(6), pyrG89, adF9, biA1 +
proA1(6, 7), uaZ11, prnA432, pyrG89, pabaA1, yA2
Heterokarion miał fenotyp: pro−, pyr− oraz dwa rodzaje konidiów: zielone i żółte (Rys. 17).
Drugiego heterokarionu nie udało się otrzymać, mimo wielu prób.
1.2. Otrzymanie szczepu diploidalnego
Z heterokarionu, opisanego w punkcie 1.2., zebrano konidia, które następnie wysiano na
podłoża selekcyjne, w celu znalezienia konidiów diploidalnych (Metody, punkt 2.3).
Uzyskano szczep diploidalny:
proA1(6), pyrG89, adF9, biA1
proA1(6, 7), pyrG89, uaZ11, prnA432, pabaA1, yA2
o fenotypie: pro−, pyr−, (zielone konidia), (Rys. 17).
53

WYNIKI
Szczep haploidalny: proA1(6, 7), pyrG89, uaZ11,
prnA432, pabaA1, yA2
Szczep haploidalny: proA1(6), pyrG89, adF9, biA1
Heterokarion + sektor diploidalny
Szczep diploidalny:
proA1(6), pyrG89, adF9, biA1
proA1(6, 7), pyrG89, uaZ11, prnA432, pabaA1, yA2
Heterokarion: proA1(6), pyrG89, adF9, biA1
+ proA1(6,7), pyrG89, uaZ11, prnA432, pabaA1, yA2
Rys. 17. Otrzymywanie diploida. Na czerwono zaznaczono sektor diploidalny.
54

WYNIKI
ndy.
1.3. Kontrola hybrydyzacyjna uzyskanego szczepu diploidalnego A. nidulans.
Rys. 18. Mapa restrykcyjna fragmentów chromosomów I i VIII A. nidulans. Na mapie przedstawiono zmiany w budowie chromosomów, wywołane translokacją uaZ11. Kolor biały –chromosom I, kolor czarny – chromosom VIII, kolor zielony – gen uaZ, kolor żółty – sonda użyta do hybrydyzacji (734 bp). Na mapie zaznaczono tylko dwa miejsca rozpoznawane przez enzym HincII, między którymi znajduje się punkt pęknięcia chromosomów w translokacji uaZ11. (Rys. wg Oestreicher i wsp., 1993b).
Jeden ze szczepów haploidalnych, użytych do otrzymania diploida
posiadał mutację uaZ11, uniemożliwiającą wzrost na podłożu, w którym
jedynym źródłem azotu jest kwas moczowy. Mutacja ta jest aberracją
chromosomalną, polegającą na wzajemnej translokacji fragmentów
chromosomów I i VIII (Rys. 18). Obecność mutacji uaZ11 pozwoliła na
udowodnienie diploidalności badanego szczepu metodami biologii
molekularnej.
1kb
W celu uzyskania sondy do hybrydyzacji Southern, wykonano
trawienie plazmidu bAN649 enzymem HincII, a następnie wyizolowano z
żelu agarozowego fragment DNA o wielkości 734 par zasad (Rys. 19),
który obejmował część genu uaZ11 z A. nidulans. Fragmentu tego użyto
jako so
Rys. 19. Żel z plazmidem bAN649 trawionym enzymem HincII. Elipsą zaznaczono fragment DNA o wielkości 734 bp, którego użyto jako sondy do hybrydyzacji. 1kb – standard wielkości 1kb DNA Ladder.
55
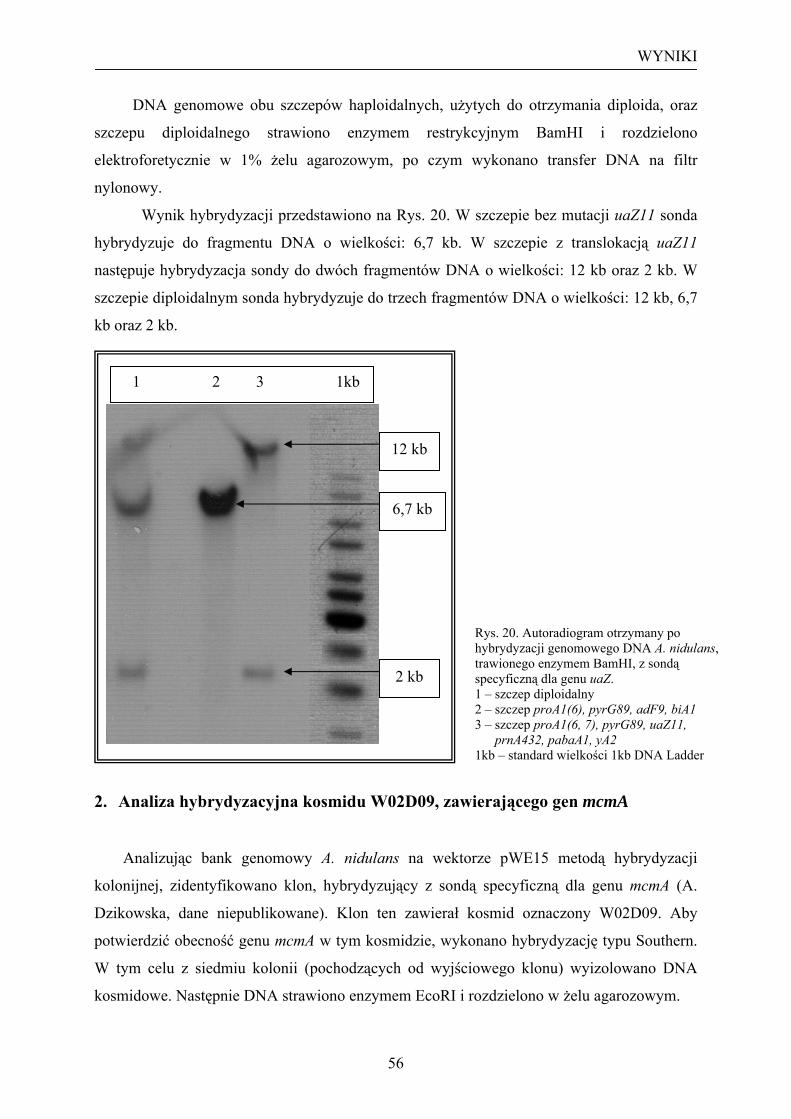
WYNIKI
56
DNA genomowe obu szczepów haploidalnych, użytych do otrzymania diploida, oraz
szczepu diploidalnego strawiono enzymem restrykcyjnym BamHI i rozdzielono
elektroforetycznie w 1% żelu agarozowym, po czym wykonano transfer DNA na filtr
nylonowy.
Wynik hybrydyzacji przedstawiono na Rys. 20. W szczepie bez mutacji uaZ11 sonda
hybrydyzuje do fragmentu DNA o wielkości: 6,7 kb. W szczepie z translokacją uaZ11
następuje hybrydyzacja sondy do dwóch fragmentów DNA o wielkości: 12 kb oraz 2 kb. W
szczepie diploidalnym sonda hybrydyzuje do trzech fragmentów DNA o wielkości: 12 kb, 6,7
kb oraz 2 kb.
1 2 3 1kb
12 kb
6,7 kb
Rys. 20. Autoradiogram otrzymany po hybrydyzacji genomowego DNA A. nidulans, trawionego enzymem BamHI, z sondą specyficzną dla genu uaZ. 2 kb 1 – szczep diploidalny 2 – szczep proA1(6), pyrG89, adF9, biA1 3 – szczep proA1(6, 7), pyrG89, uaZ11, prnA432, pabaA1, yA2 1kb – standard wielkości 1kb DNA Ladder
2. Analiza hybrydyzacyjna kosmidu W02D09, zawierającego gen mcmA
Analizując bank genomowy A. nidulans na wektorze pWE15 metodą hybrydyzacji
kolonijnej, zidentyfikowano klon, hybrydyzujący z sondą specyficzną dla genu mcmA (A.
Dzikowska, dane niepublikowane). Klon ten zawierał kosmid oznaczony W02D09. Aby
potwierdzić obecność genu mcmA w tym kosmidzie, wykonano hybrydyzację typu Southern.
W tym celu z siedmiu kolonii (pochodzących od wyjściowego klonu) wyizolowano DNA
kosmidowe. Następnie DNA strawiono enzymem EcoRI i rozdzielono w żelu agarozowym.

WYNIKI
Po przeniesieniu DNA na filtr nylonowy, wykonano hybrydyzację typu Southern. Jako sondy
użyto fragmentu cDNA (168 bp) genu mcmA, kodującego domenę typu MADS. Wynik
A
1 2 3 4 5 6 7 MIX kb
11,59,6 8,8
4,5
3
2,6
2,1
1,8
1,4
11,5 kb
wektor
1 2 3 4 5 6 7 MIX
B
11,5 kb
Rys. 21. Analiza hybrydyzacyjna kosmidów W02D09. A. Rozdział elektroforetyczny kosmidów W02D09, trawionych enzymem EcoRI. W wyniku trawienia powstają fragmenty DNA o wielkości: 11,5 kb; 9,6 kb; 8,8 kb; 4,5 kb; 3 kb; 2,6 kb (podwójny); 2,1 kb; 1,8 kb; 1,4 kb. B. Autoradiogram otrzymany po hybrydyzacji w/w kosmidów z fragmentem cDNA genu mcmA. MIX – standard wielkości DNA Ladder Mix. Czarna strzałka wskazuje fragment DNA (11,5 kb), do którego hybrydyzuje sonda specyficzna dla genu mcmA. Białą strzałką zaznaczono fragment (ok. 8,8 kb), odpowiadający wielkością wektorowi pWE15.
57

WYNIKI
58
hybrydyzacji zamieszczono na Rys. 21. Potwierdził on obecność genu mcmA we wszystkich
analizowanych klonach we fragmencie DNA o wielkości 11,5 kb. Na podstawie analizy
restrykcyjnej przy użyciu enzymów EcoRI oraz BamHI oszacowano wielkość kosmidu
W02D09 na ok. 48 kb (Rys. 21; Rys. 30; Rys. 32). Wektor pWE15 ma wielkość ok. 8,8 kb
(Rys. 16). Wynika z tego, że na kosmidzie W02D09 znajduje się fragment genomu A.
nidulans o wielkości ok. 39 kb, zawierający gen mcmA. Mapę restrykcyjną tego fragmentu
zamieszczono na Rys. 22.
hybrydyzacji zamieszczono na Rys. 21. Potwierdził on obecność genu mcmA we wszystkich
analizowanych klonach we fragmencie DNA o wielkości 11,5 kb. Na podstawie analizy
restrykcyjnej przy użyciu enzymów EcoRI oraz BamHI oszacowano wielkość kosmidu
W02D09 na ok. 48 kb (Rys. 21; Rys. 30; Rys. 32). Wektor pWE15 ma wielkość ok. 8,8 kb
(Rys. 16). Wynika z tego, że na kosmidzie W02D09 znajduje się fragment genomu A.
nidulans o wielkości ok. 39 kb, zawierający gen mcmA. Mapę restrykcyjną tego fragmentu
zamieszczono na Rys. 22.
(33815) (34722) TAA stop ATG start
Sekwencja kodująca (ORF)
genu mcmA
1 kb
Rys. 22. Mapa restrykcyjna fragmentu genomu A. nidulans, sklonowanego na kosmidzie W02D09. Sklonowany fragment genomu ma wielkość ok. 39 kb. Sekwencja DNA tego fragmentu jest dostępna on-line w bazie genomowej A. nidulans Broad Institute, jako część kontigu 1.159 (58286 bp). Liczby w nawiasach odpowiadają numeracji nukleotydów w tym kontigu. Do utworzenia mapy wykorzystano wyniki analizy restrykcyjnej kosmidu W02D09 in vitro oraz wyniki komputerowej analizy restrykcyjnej kontigu 1.159. Na czarno zaznaczono fragment genomu z sekwencją kodującą (ORF) genu mcmA.
3. Dysrupcja genu mcmA na kosmidzie W02D09 3. Dysrupcja genu mcmA na kosmidzie W02D09
3.1. Transformacja szczepu KS272 (pKOBEG) kosmidem W02D09 3.1. Transformacja szczepu KS272 (pKOBEG) kosmidem W02D09
Aby uzyskać dysrupcję genu mcmA na kosmidzie W02D09, bakterie ze szczepu E.
coli KS272 (pKOBEG) stransformowano kosmidem W02D09. Do transformacji użyto ok.
200 ng DNA kosmidu W02D09. Bakterie po transformacji selekcjonowano na podłożu
LB+Amp+Km+Cm w 30°C. Transformacja zaszła z niską wydajnością (ok. 2,5×102
transformantów / μg DNA kosmidu). Po 24 h na w/w podłożu wyrosło ok. 50 kolonii
Aby uzyskać dysrupcję genu mcmA na kosmidzie W02D09, bakterie ze szczepu E.
coli KS272 (pKOBEG) stransformowano kosmidem W02D09. Do transformacji użyto ok.
200 ng DNA kosmidu W02D09. Bakterie po transformacji selekcjonowano na podłożu
LB+Amp+Km+Cm w 30°C. Transformacja zaszła z niską wydajnością (ok. 2,5×102
transformantów / μg DNA kosmidu). Po 24 h na w/w podłożu wyrosło ok. 50 kolonii

WYNIKI
59
bakteryjnych. W celu potwierdzenia obecności kosmidu W02D09 w uzyskanych
transformantach, wyizolowano pozachromosomalne DNA z 17 kolonii, opornych na
ampicylinę (Amp), kanamycynę (Km) i chloramfenikol (Cm). Następnie otrzymane DNA
poddano analizie restrykcyjnej przy użyciu enzymu EcoRI. Oczekiwano, że obok fragmentów
charakterystycznych dla kosmidu W02D09, na żelu pojawią się też fragmenty świadczące o
obecności w komórkach plazmidu pKOBEG. Wynik omawianej analizy restrykcyjnej
zamieszczono na Rys. 23. W przypadku wszystkich 17 trawień stwierdzono obecność dwóch
fragmentów DNA, których wielkość odpowiadała trawionemu plazmidowi pKOBEG (3,5 kb
oraz 3 kb). Jedynie w dwóch próbkach wynik trawienia był zgodny z oczekiwaniami i
wskazywał na obecność kosmidu W02D09 (próbki: 14 i 16, Rys. 23). W pozostałych
próbkach stwierdzono obecność fragmentu DNA o wielkości ok. 5 – 6 kb, niewiadomego
pochodzenia. Fragment o wielkości 5 – 6 kb nie występował w próbkach 14 i 16. Szczep
bakterii, otrzymany w wyniku transformacji szczepu KS272 (pKOBEG) kosmidem W02D09,
nazwano: KS272 (pKOBEG + W02D09). Do dalszych doświadczeń użyto klonu nr 14.
bakteryjnych. W celu potwierdzenia obecności kosmidu W02D09 w uzyskanych
transformantach, wyizolowano pozachromosomalne DNA z 17 kolonii, opornych na
ampicylinę (Amp), kanamycynę (Km) i chloramfenikol (Cm). Następnie otrzymane DNA
poddano analizie restrykcyjnej przy użyciu enzymu EcoRI. Oczekiwano, że obok fragmentów
charakterystycznych dla kosmidu W02D09, na żelu pojawią się też fragmenty świadczące o
obecności w komórkach plazmidu pKOBEG. Wynik omawianej analizy restrykcyjnej
zamieszczono na Rys. 23. W przypadku wszystkich 17 trawień stwierdzono obecność dwóch
fragmentów DNA, których wielkość odpowiadała trawionemu plazmidowi pKOBEG (3,5 kb
oraz 3 kb). Jedynie w dwóch próbkach wynik trawienia był zgodny z oczekiwaniami i
wskazywał na obecność kosmidu W02D09 (próbki: 14 i 16, Rys. 23). W pozostałych
próbkach stwierdzono obecność fragmentu DNA o wielkości ok. 5 – 6 kb, niewiadomego
pochodzenia. Fragment o wielkości 5 – 6 kb nie występował w próbkach 14 i 16. Szczep
bakterii, otrzymany w wyniku transformacji szczepu KS272 (pKOBEG) kosmidem W02D09,
nazwano: KS272 (pKOBEG + W02D09). Do dalszych doświadczeń użyto klonu nr 14.
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 K M P
Rys. 23. Analiza restrykcyjna DNA niechromosomalnego, wyizolowanego z transformantów po transformacjiszczepu KS272 (pKOBEG) kosmidem W02D09. (1 – 17) – DNA wyizolowane z transformantów trawione EcoRI; K – kosmid W02D09 trawiony EcoRI; M –standard wielkości DNA Ladder Mix; P – plazmid pKOBEG. Podkreślono numery ścieżek (14, 16), w których wzór restrykcyjny jest zgodny z oczekiwanym i wskazuje na obecność zarówno plazmidu pKOBEG, jak i kosmidu W02D09. Białymi elipsami zaznaczono jeden z fragmentów DNA plazmidu pKOBEG, świadczący o obecności tego plazmidu w badanych klonach. Czarna strzałka wskazuje prążek o wielkości ok. 5 – 6 kb, niewiadomego pochodzenia (szczegóły w tekście).

WYNIKI
3.2. Przygotowanie kasety dysrupcyjnej zeo/pyrG
Kaseta zeo/pyrG do dysrupcji genu mcmA została tak zaprojektowana, by w wyniku
rekombinacji homologicznej z kosmidem W02D09, powodowała dysrupcję fragmentu
genomu A. nidulans o długości 1773 bp. Fragment ten obejmuje całą sekwencję genu mcmA
transkrybowaną na mRNA. Sekwencję kasety zeo/pyrG przedstawiono na Rys. 26, a fragment
sekwencji genomu A. nidulans z genem mcmA zamieszczono na Rys. 27.
Kasetę zeo/pyrG uzyskano w wyniku reakcji PCR na plazmidzie pCDA21, z
wykorzystaniem długich starterów: AN_st1b (72 bp) oraz AN_st2 (70 bp). Startery
zaprojektowano w taki sposób, by na końcach 5’ miały 50 lub 52 nukleotydy komplementarne
do sekwencji DNA regionu otaczającego gen mcmA w genomie A. nidulans, a 20-
nukleotydowe sekwencje, znajdujące się na końcach 3’, umożliwiały amplifikację kasety.
Aby zapewnić jak największą dokładność amplifikacji DNA, do reakcji PCR użyto zestawu
komercyjnego do powielania długich cząsteczek DNA Expand Long Template PCR System.
Rys. 25. Analiza restrykcyjna plazmidu pCDA21 (1) i kasety zeo/pyrG (2), trawionych enzymem EcoRI. MIX – standard wielkości DNA Ladder Mix.
Rys. 24. Rozdział elektroforetyczny w żelu agarozowym produktów reakcji PCR, wykonanej w celu otrzymania kasety zeo/pyrG. (1, 2, 3) – produkty reakcji PCR, uzyskane przy użyciu buforów: 1, 2, 3 (szczegóły w tekście). Czarną elipsą zaznaczono fragmenty DNA, odpowiadające kasecie zeo/pyrG (ok. 2,5 kb). 1kb – standard wielkości 1kb DNA Ladder.
1 2 1kb 1 2 3 MIX
3,6 kb
1,9 kb
0,5 kb
60

WYNIKI
sekwencja AN_st1b (72 bp)
CCAATCATACAGCTCGCCAAGAGCGCTGAATGGTCTGGACAAACTTAGTCCCGGAATTCT CAGTCCTGCTCCTCGGCCACGAAGTGCACGCAGTTGCCGGCCGGGTCGCGCAGGGCGAAC TCCCGCCCCCACGGCTGCTCGCCGATCTCGGTCATGGCCGGCCCGGAGGCGTCCCGGAAG TTCGTGGACACGACCTCCGACCACTCGGCGTACAGCTCGTCCAGGCCGCGCACCCACACC CAGGCCAGGGTGTTGTCCGGCACCACCTGGTCCTGGACCGCGCTGATGAACAGGGTCACG TCGTCCCGGACCACACCGGCGAAGTCGTCCTCCACGAAGTCCCGGGAGAACCCGAGCCGG TCGGTCCAGAACTCGACCGCTCCGGCGACGTCGCGCGCGGTGAGCACCGGAACGGCACTG GTCAACTTGGCCATGGTTTAGTTCCTCACCTTGTCGTATTATACTATGCCGATATACTAT GCCGATGATTAATTGTCAACACGTGCTCGAGCTAGCGATATCGCGGCCGCTCTAGAACTA GTGGATCCCCCGGGCTGCAGGAATTCTGTCTGAGAGGAGGCACTGATGCGTGATGCCAAG CTGTTATCTCTTCTGCACAGGTCGACTTGTCCCTGAGGGACCTTCCATATTAAGCTATCA GTCCACTTTGCTTATCGACTACATAGATAAAGCCTTGGCGTTTCTGGCAAAGCTTACCTC ACTTGCTAGATGACTGGTAGGAATCTAACAAAGGGAAAACCTTGACGATTGGGGGCGGAA AACATGGCATTACAGGGGCCAATACGGACGAGCTCATCGTTTAATGACAGGCACAATGTA TGCAGCCTTCCTCCACAACACTCGTACTGGCACTACACTTTTGCTCCAAGAGGAAATCAT GACTTGCCGCATACTCTGGCCATATAAGCTTCCCAGCCTTCTTTCTGGTACCGCTGTGCA GCTTCAACCGGGTCGGGAGCAGCGTAGATGCCTCGACCGGCGATGATAAAGTCGGCACCG CGTCCAATAGCCGATGCAGGAGTCTGGTATTGCTGTCCAAGCTTATCTCCTTTGGAAGAG AGGTTCACACCCGTCGTGAAGACCACGAAATCTTCATCCTCCGAGGCTGAAGACACATCC GACTGCACTTCCGTCAGGGCCCGCGTCGACACGAAACCCATAACGAAGTTCTTGTATTTG CGAGCGTAGTCAACCGATGCCTTGGTATACTCGCCCGTAGCCAGCGATCCTTTGGAGGTC ATCTCTGCCAGGACCAACAGTCCTCTCTCAGGACCATAGGGGAAGTCTTGCGCAGATGCG GTCTGGGCCAGAGCCTCGACGATGCCCTCGCCAGGGAGAACGCTGCAGTTGATAATGTGG GCCCATTCGGAGATCCTCAGAGCACCGCCGTGGTATTGCTTCTGGACGGTATTGCCGATG TCGATGAATTTGCGGTCCTCGAAGATCAAAAAGTTGTGCTTTTGAGCCAGCACATTCAGG CCATTGATAGTGTCGACGCTGAAATCGGTGAGGATGTCGATGTGTGTCTTGATGACGGCG ATGTAGGGACCGAGACCTGTATCATCAAAAGTTGATTAGTCCATTGCTAGACGGCATATG TATTGGATCCAACAGCTTCCGTACGGTCAGCGAGGTCCAGGAGTTCTCGGGTTGTCGTCA CATCAGCAGAGACGGTAACGTTTGTCTTCTTTGCTTCGGCAATCTCAAAAAGTCTCTTTG CCAGAGGATTGGGGTGCTTGCTGGCTCGAGCACCGTAAGTCAATTGCGACTTGGACGACA TCGTGGGAATGGAGGGTTATATGCGGGGTATGACGGTGATTGATGAGTTGAAGTAGCCGA GCAATGAGGTATATTATCCAATTGAGGGTGACCCAACCAAATCAATTGCTTGAGAATCAA ATCTCAAATATTCTAAAATAGAAGCCGCCGGGGACTAAATTTAAAGTCGTACTCTATCTC GATCTTTTCCGCGTGTAAAAATTGCGTGCCAATCTGGATGGAGACAGGCCACATCGGTGC TGTATTCCTCCGCCTCCGACTGTGGGGTCAACCACAATTAAACTATTCTGTAGTCAGGTA CAGCTAGAATGGGGTAGACAGGCAGAACTGTAGAAGATAAAACATTGGTCAATCACTGGT AACTCCACGGAACTTTTAACGTACATATATATATATACACAACATATTTCGTCAGACACA GAATAACTCTCAGTAAGAGATCATGTCTTTGGCAACACCAAACACACTCAGAGCCCACAG AGCGCCTTGAGAAAACCAGAAGAAACCTCCCAGCAATCTGCCCTTGCCTTGTGGCTTACC CGAGTACACATAGCCGTACAAGTAGAGAGATCGGAAAAACAGCCATGAAGCGCCAATTGC TGTTGCCAGGTGAGGGTATTTCAGACCCGCGAAGAGGGTGAAGAGCATTGTTTGAGGCGA
ATTCGGATGTCCGGGTTGGTGGTTGGTTGATGTAGGCCTTGAGCAACCTTGACC Użyte oznaczenia: kolor zielony – miejsce cięcia EcoRI
kolor różowy – miejsce cięcia BamHI
kolor żółty – miejsce cięcia NotI
kolor błękitny – gen pyrG z A. fumigatus (1,9 kb)
kolor szary – gen Sh ble (warunkujący oporność na zeocynę) ze Streptoalloteichus hindustanus (0,5 kb)
kolor czerwony – sekwencje komplementarne do genomu A. nidulans w regionie otaczającym gen mcmA,
dołączone w wyniku reakcji PCR (52 i 50 bp)
czcionka pogrubiona – fragment polilinkera z wektora pBluescript
61
sekwencja komplementarna do sekwencji AN_st2 (70 bp)
Rys. 26. Sekwencja kasety zeo/pyrG, uzyskanej w wyniku reakcji PCR na plazmidzie pCDA21. Do kasety są dołączone sekwencje komplementarne do genomu A. nidulans w regionie otaczającym gen mcmA(52 i 50 bp). Sekwencję kasety ustalono na podstawie informacji zawartych w pracach: (Chaveroche i wsp., 2000; Weidner i wsp., 1998). Sekwencję genu pyrG pobrano z bazy EMBL, a sekwencja genu Sh ble pochodzi z plazmidu pEM7/Zeo (Invitrogen). Sekwencję plazmidu pBluescript pobrano ze strony: www.stratagene.com.

WYNIKI
1 AACTGTGGGC GTGAATAATG GGGGTCTCGT GGGACGGAGT ACTACCGGTA ATATTACGTG 61 GTATTAGCGG GGAGTGACTT TCTAGAGTCG ACCAATCATA CAGCTCGCCA AGAGCGCTGA 121 ATGGTCTGGA CAAACTTAGT CCCGGCCGAT GCCAGTCGTC TATCTCGCTT TTGTCCTTGA 181 CAGCTTCAGG AAGAATGTGT AAATGCTCAT TAAGGCCGTA AAGGTGATAT AACATGATAA 241 TTTAAGCTTC TGGTCCTTGA GATGAAAGGT GAGTTCTAGA TGATTGTCTT CCAAGCGGAG 301 AGTATGAGGA GTTGACTGGA ACTGTGGGGC GTACCATACC GTAAGCGCAC GTGAGCCACC 361 GCCTTCCCAA CTGCACCGAC ACTTATCCTT TTCGAACTAC TTCCCAGCTC CTCCCCGTTT 421 TCCTCTTTCT CTCGTCCTGA ACCCAAACCC TGCGAAGCCC AAACGCGTTA CTCGATTTGG 481 TCATCTTCCC TTCCCCTCAG AAGCATCATT CCTCTTTGGG CATTCGTCCT CAAGTCCCCC 541 AGATCCTTCC CTTTACTGCT CATCCACTCA ATCGATACAA TTCTGCTCGT CGCCTCTTCG 601 TTTCTATTTC CATCATGGCC GACATCACCG CCGTTGGTGA GGAGAACCCT TCGCCCACCC 661 AGGACGACCT GCAGCAGGCC GCCGGTAACG GCGCTACGGA CAACCGCGGT ACTAAGCGCA 721 ACCGCATGAG CGCCGACGAC GACGATGACG ATGAGGACAA GACCGGTCGC GAGCGCAGGA 781 AGATTGAGAT CAAGTTCATC CAAGATAAGT CGCGTCGCCA CATTACCTTC TCCAAGCGGA 841 AGGCGGGTAT CATGAAGAAG GTGCGTACCT CCGCGTCAGA CTCAAGACGC CCTTTTCTCT 901 CTGTACCACG CGCACTGTGA TTTGCGAATC GTTGATTCCT TCTTCAATTA CCTTGTCGTG 961 TTATTTTCTC TTTTAGCAAA ATTGATCATG GATGGTCGTC GACTTACAAT TCAATGCGGT 1021 CGCGTAACAG GCATACGAGC TCTCCGTCCT CACAGGCACT CAAGTGCTCC TTCTAGTCGT 1081 GTCCGAGACT GGCCTTGTCT ACACCTTCAC AACCCCCAAG CTGCAGCCCT TGGTCACGAA 1141 GCCCGAAGGA AAGAACCTCA TCCAGGTATG TCTCGTCCCC CGAGCCGACT GATTTCGTAC 1201 GAGACTAACA AGGCCCCTGA ACCTATAGGC CTGCCTTAAT GCTCCTGATC CAGCTACCAA 1261 CGAGAATGGC GTCGACGCCC CCGAGGTTCC TGCCGAGACA CCGGAGGATG TGAACCACAG 1321 CAATGTTAGT GCCCAGCAGG CGAACATCCC TCGGCCTGGT GGCATGCACC CCGCCTACAT 1381 GACAAGCGAA CAACAACAGC AAATGGCCTA CTATCAGAAT CTTCAACAAC AGCAGCAGCA 1441 ACAGCAGCAG CAGGCTGGCG GACAGTACCC CGGAATGCCT GTAGGCAATC GGATGCCGAC 1501 CCAGCACCAA CCCACTGCTT AAATTCGCGC TTAAGCGCTT GCATAGTCGC CCAGAAGAAC 1561 AAAGGAGCTC AAGTTGTAGC TCCGCAAGCA ACTAGCTTCT TTTCGCGCAA CTGGGCATAC 1621 TCTCTCGGCC TTCTCTTGAT ACCCAGCGAA TCCGTTCCAA ACGATTGTTC CACCAAAATC 1681 CGGTCTTGAT TGTATACCAC TTTTCCGCGC AAGACAAGAA CCTTTCCCCA CAACCTGTGT 1741 ATATCTTGTG CCTTTTGGTT CTGAATGTTG CAGGACACCG GATACTTGAT ATCGATGTGG 1801 CAATATCTCG TTTACCCTGC TATCTATCTA TCCCCTTTAA TGACGTCGCT TTCCCTCTCT 1861 TTCCCCGAGA AAATGGCCAA AAAAAAATGC AGAAAAAGAA ACTAGAGAAA GGAACATGGA 1921 TGTCCGGGTT GGTGGTTGGT TGATGTAGGC CTTGAGCAAC CTTGACCTGG ATTTCGACAT 1981 CGTTGTTCGA TCATATCGAC GATCGCCTCA TAATCATTTC CTAGACGATG TGGGAAGATG Użyte oznaczenia:
kolor zielony – sekwencje kodujące 5’ i 3’ UTR
kolor żółty – sekwencja kodująca białko MCMA (ORF)
kolor czerwony – sekwencja kodująca domenę typu MADS
kolor szary – sekwencje intronów
kolor błękitny – sekwencje otaczające gen mcmA, komplementarne do sekwencji, dołączonych do kasety
zeo/pyrG w wyniku reakcji PCR (por. Rys. 26)
czcionka pogrubiona – określa miejsce startu transkrypcji oraz początek i koniec otwartej ramki odczytu
Rys. 27. Sekwencja fragmentu genomu A. nidulans z genem mcmA. Sekwencja pochodzi z bazy genomowej A. nidulans Broad Institute. Informacje o genie mcmA pochodzą z w/w bazy oraz z pracy magisterskiej B. Walczuka (Walczuk, 2005).
62

WYNIKI
63
Z zestawem dostarczane są trzy bufory dla polimeraz DNA, różniące się stężeniem jonów
Mg2+ i obecnością lub brakiem detergentów. Najwyższą wydajność amplifikacji DNA
uzyskano po zastosowaniu buforu nr 1 (tzn. przy stężeniu MgCl2 równym 1,75 mM),
mniejszą stosując bufor nr 2, a po zastosowaniu buforu nr 3 amplifikacja DNA kasety nie
nastąpiła (Rys. 24). W każdym przypadku do reakcji PCR użyto ok. 200 ng DNA plazmidu
pCDA21.
Aby mieć pewność, że transformanty, otrzymane po transformacji bakterii KS272
(pKOBEG + W02D09) kasetą zeo/pyrG, nabędą oporność na zeocynę na drodze rekombinacji
homologicznej między kasetą a kosmidem W02D09, a nie w wyniku transformacji
plazmidem pCDA21 (matrycą w reakcji PCR), produkt reakcji PCR oczyszczono poprzez
izolację z żelu agarozowego fragmentu DNA o wielkości odpowiadającej kasecie zeo/pyrG
(ok. 2,5 kb).
W celu potwierdzenia, że w reakcji PCR powstał produkt o właściwej sekwencji,
oczyszczoną kasetę zeo/pyrG strawiono enzymem EcoRI, a otrzymane wyniki porównano z
mapą restrykcyjną plazmidu pCDA21 (Rys. 25, Rys. 13). Enzym EcoRI trawi plazmid
pCDA21 na fragmenty o wielkości 3,6 kb, 1,9 kb oraz 0,5 kb. Jeśli tym samym enzymem
strawi się kasetę zeo/pyrG, powstają fragmenty DNA o wielkości 1,9 kb (pyrG) oraz 0,5 kb
(zeo), ale nie powstaje fragment 3,6 kb.
3.3. Transformacja szczepu KS272 (pKOBEG + W02D09) kasetą dysrupcyjną
zeo/pyrG
Ze szczepu KS272 (pKOBEG + W02D09), uzyskanego w punkcie 3.1, przygotowano
komórki elektrokompetentne, a następnie wykonano transformację kontrolną plazmidem
pBR322. Transformanty selekcjonowano na podłożu z tetracykliną. Wydajność transformacji
kontrolnej była niska i wynosiła ok. 103 – 104 stransformowanych komórek / μg DNA
plazmidu pBR322.
Szczep KS272 (pKOBEG + W02D09) stransformowano kasetą dysrupcyjną zeo/pyrG.
Do transformacji użyto ok. 200 ng DNA kasety. Po transformacji bakterie selekcjonowano na
podłożu LBLS+Amp+Km+Cm+Zeo w 30°C. Mimo niskiej wydajności transformacji
kontrolnej plazmidem pBR322, po transformacji kasetą otrzymano ok. 100 transformantów.
Dla 40 losowo wybranych transformantów przeprowadzono gubienie plazmidu pKOBEG,
warunkującego oporność na chloramfenikol (patrz: Metody, punkt 9.5). Wydajność gubienia

WYNIKI
64
plazmidu pKOBEG wyniosła 100% (wszystkie analizowane transformanty utraciły oporność
na chloramfenikol).
plazmidu pKOBEG wyniosła 100% (wszystkie analizowane transformanty utraciły oporność
na chloramfenikol).
Z 15 kolonii, które utraciły oporność na chloramfenikol, wyizolowano DNA
kosmidowe. Następnie wykonano analizę restrykcyjną przy użyciu enzymu EcoRI. Wyniki
analizy restrykcyjnej zamieszczono na Rys. 28. Spodziewano się, że w kosmidzie W02D09
po dysrupcji genu mcmA nie będzie fragmentu DNA o wielkości ok. 11,5 kb, (we fragmencie
tym stwierdzono wcześniej doświadczalnie obecność genu mcmA (Rys. 21)). Jednocześnie
oczekiwano, że w trawionym kosmidzie po dysrupcji pojawią się fragmenty DNA o wielkości
ok. 6,7 kb oraz ok. 3 kb, a także fragmenty kasety zeo/pyrG, odpowiadające genom: pyrG
(ok. 1,9 kb) i Sh ble (zeo) (ok. 0,5 kb). Nie wszystkie kosmidy dawały po strawieniu
oczekiwany wzór restrykcyjny, jednakże dla 10 kosmidów (oznaczonych numerami: 2, 3, 4,
5, 6, 7, 9, 10, 11, 13), otrzymano wzór restrykcyjny, który wskazywał na dysrupcję genu
mcmA. Kosmidy z dysrupcją genu mcmA nazwano: W02D09::zeo/pyrG, a szczep, który je
zawiera: KS272 (W02D09::zeo/pyrG).
Z 15 kolonii, które utraciły oporność na chloramfenikol, wyizolowano DNA
kosmidowe. Następnie wykonano analizę restrykcyjną przy użyciu enzymu EcoRI. Wyniki
analizy restrykcyjnej zamieszczono na Rys. 28. Spodziewano się, że w kosmidzie W02D09
po dysrupcji genu mcmA nie będzie fragmentu DNA o wielkości ok. 11,5 kb, (we fragmencie
tym stwierdzono wcześniej doświadczalnie obecność genu mcmA (Rys. 21)). Jednocześnie
oczekiwano, że w trawionym kosmidzie po dysrupcji pojawią się fragmenty DNA o wielkości
ok. 6,7 kb oraz ok. 3 kb, a także fragmenty kasety zeo/pyrG, odpowiadające genom: pyrG
(ok. 1,9 kb) i Sh ble (zeo) (ok. 0,5 kb). Nie wszystkie kosmidy dawały po strawieniu
oczekiwany wzór restrykcyjny, jednakże dla 10 kosmidów (oznaczonych numerami: 2, 3, 4,
5, 6, 7, 9, 10, 11, 13), otrzymano wzór restrykcyjny, który wskazywał na dysrupcję genu
mcmA. Kosmidy z dysrupcją genu mcmA nazwano: W02D09::zeo/pyrG, a szczep, który je
zawiera: KS272 (W02D09::zeo/pyrG).
Rys. 28. Analiza restrykcyjna DNA kosmidów, wyizolowanych z transformantów po transformacji szczepu KS272 (pKOBEG + W02D09) kasetą zeo/pyrG i zgubieniu plazmidu pKOBEG. (1 – 15) – kosmidy wyizolowane z transformantów, trawione enzymem EcoRI; K – kosmid W02D09 bez dysrupcji genu mcmA trawiony EcoRI; MIX – standard wielkości DNA Ladder Mix. Biała strzałka wskazuje fragment DNA o wielkości 11,5 kb, którego brak w kosmidach po dysrupcji. Elipsami zaznaczono fragmenty, pochodzące z kasety zeo/pyrG (gen pyrG (1,9 kb), gen Sh ble (zeo) (0,5 kb)). Podkreślono numery ścieżek, w których wzór restrykcyjny kosmidu jest wyraźny i zgodny z oczekiwanym.
MIX MIX 1 2 3 4 5 6 7 9 10 11 12 13 14 15 kb
11,59,6 8,8 6,7
4,5
3
2,6
2,1 1,9 1,8
1,4
K kb
10 8
6
5
4 3,5
3
2,5
2
1,5
1,2
1,031
0,9 0,8 0,7 0,6 0,5 0,5

WYNIKI
3.4. Kontrola hybrydyzacyjna kosmidu W02D09 z dysrupcją genu mcmA
Aby się upewnić, że otrzymano kosmid z dysrupcją właściwego regionu DNA,
zaprojektowano doświadczenie hybrydyzacyjne z regionem otaczającym miejsce dysrupcji
genu mcmA na kosmidzie W02D09.
A
In silico, używając różnych enzymów, wykonano analizę restrykcyjną kasety
zeo/pyrG (Rys. 26) oraz sekwencji kontigu 1.159 (58286 bp), pobranej z bazy genomowej A.
nidulans Broad Institute, obejmującej fragment genomu z genem mcmA. Do dalszych
doświadczeń wybrano enzym BamHI, który inaczej trawi kosmid W02D09 z dysrupcją genu
mcmA, a inaczej bez dysrupcji. Komputerowa analiza restrykcyjna pokazała, że po strawieniu
genomu A. nidulans enzymem BamHI gen mcmA znajduje się we fragmencie DNA o
wielkości 13,2 kb oraz, że sąsiednie fragmenty mają wielkość 230 bp i 306 bp (Rys. 29).
B
13,2 kb 306 bp 230 bp
Gen pyrG z A. fumigatus
230 bp 9,2 kb 1,1 kb 3,6 kb 306 bp
1 kb
Gen Sh ble (zeo) z S. hindustanus
Rys. 29. Mapa restrykcyjna fragmentu kosmidu W02D09, obejmująca region dysrupcji genu mcmA wraz z sekwencjami przylegającymi do tego regionu. A). Przed dysrupcją sonda wiąże się z fragmentem BamHI – BamHI o wielkości 13,2 kb, do którego przylegają fragmenty o wielkości 230 bp oraz 306 bp. B). Po dysrupcji sonda wiąże się z fragmentem BamHI – BamHI o wielkości 3,6 kb.
65
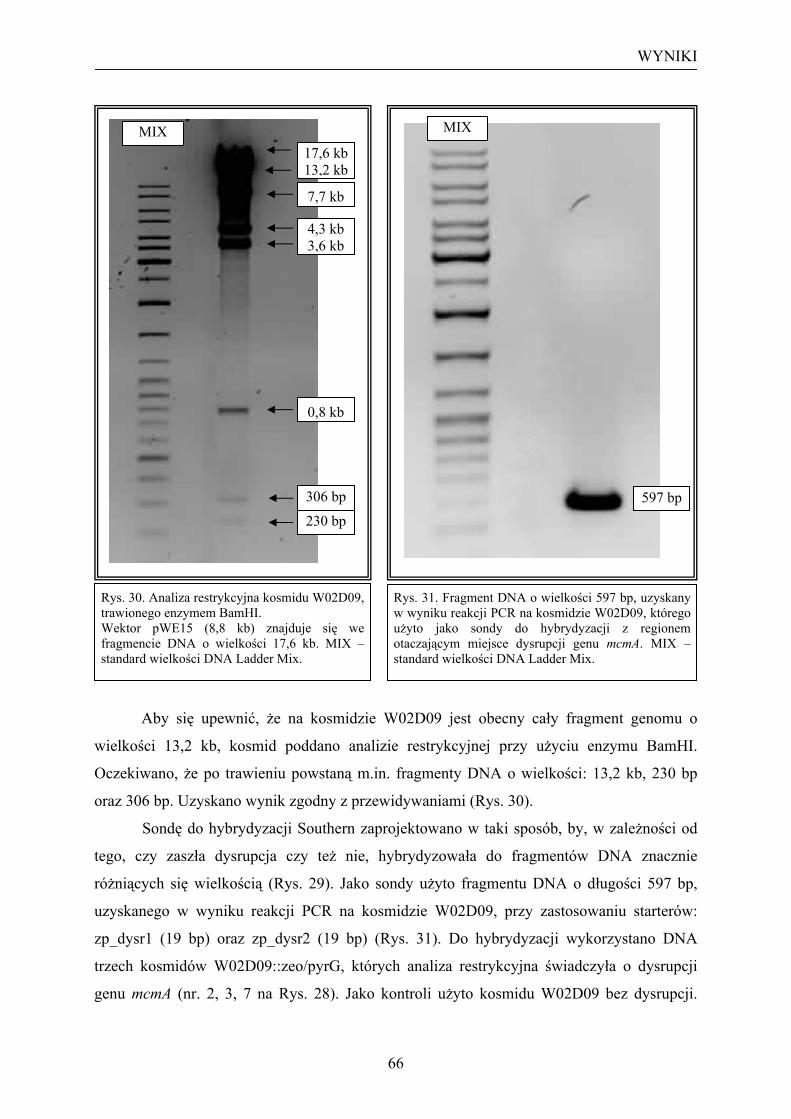
WYNIKI
Rys. 31. Fragment DNA o wielkości 597 bp, uzyskany w wyniku reakcji PCR na kosmidzie W02D09, którego użyto jako sondy do hybrydyzacji z regionem otaczającym miejsce dysrupcji genu mcmA. MIX –standard wielkości DNA Ladder Mix.
Rys. 30. Analiza restrykcyjna kosmidu W02D09, trawionego enzymem BamHI. Wektor pWE15 (8,8 kb) znajduje się we fragmencie DNA o wielkości 17,6 kb. MIX –standard wielkości DNA Ladder Mix.
Aby się upewnić, że na kosmidzie W02D09 jest obecny cały fragment genomu o
wielkości 13,2 kb, kosmid poddano analizie restrykcyjnej przy użyciu enzymu BamHI.
Oczekiwano, że po trawieniu powstaną m.in. fragmenty DNA o wielkości: 13,2 kb, 230 bp
oraz 306 bp. Uzyskano wynik zgodny z przewidywaniami (Rys. 30).
Sondę do hybrydyzacji Southern zaprojektowano w taki sposób, by, w zależności od
tego, czy zaszła dysrupcja czy też nie, hybrydyzowała do fragmentów DNA znacznie
różniących się wielkością (Rys. 29). Jako sondy użyto fragmentu DNA o długości 597 bp,
uzyskanego w wyniku reakcji PCR na kosmidzie W02D09, przy zastosowaniu starterów:
zp_dysr1 (19 bp) oraz zp_dysr2 (19 bp) (Rys. 31). Do hybrydyzacji wykorzystano DNA
trzech kosmidów W02D09::zeo/pyrG, których analiza restrykcyjna świadczyła o dysrupcji
genu mcmA (nr. 2, 3, 7 na Rys. 28). Jako kontroli użyto kosmidu W02D09 bez dysrupcji.
306 bp
230 bp
17,6 kb 13,2 kb
MIX
7,7 kb
4,3 kb 3,6 kb
0,8 kb
MIX
597 bp
66

WYNIKI
67
Wszystkie badane próbki strawiono enzymem BamHI, a następnie rozdzielono w żelu
agarozowym. W wyniku trawienia kosmidów po dysrupcji otrzymano fragmenty DNA o
następujących wielkościach: 17,6 kb; 9,2 kb; 7,7 kb; 4,3 kb; 3,6 kb (podwójny); 1,1 kb; 0,8
kb; 306 bp; 230 bp. DNA przeniesiono na filtr nylonowy i hybrydyzowano z sondą 597 bp.
Wynik hybrydyzacji zamieszczono na Rys. 32. W kosmidzie W02D09 bez dysrupcji sonda
hybrydyzuje do fragmentu DNA o wielkości ok. 13,2 kb, a w kosmidach, w których
oczekiwano dysrupcji genu mcmA, nastąpiła hybrydyzacja sondy do fragmentu o wielkości
ok. 3,6 kb. Uzyskany wynik jest zgodny z wcześniejszymi przewidywaniami.
Wszystkie badane próbki strawiono enzymem BamHI, a następnie rozdzielono w żelu
agarozowym. W wyniku trawienia kosmidów po dysrupcji otrzymano fragmenty DNA o
następujących wielkościach: 17,6 kb; 9,2 kb; 7,7 kb; 4,3 kb; 3,6 kb (podwójny); 1,1 kb; 0,8
kb; 306 bp; 230 bp. DNA przeniesiono na filtr nylonowy i hybrydyzowano z sondą 597 bp.
Wynik hybrydyzacji zamieszczono na Rys. 32. W kosmidzie W02D09 bez dysrupcji sonda
hybrydyzuje do fragmentu DNA o wielkości ok. 13,2 kb, a w kosmidach, w których
oczekiwano dysrupcji genu mcmA, nastąpiła hybrydyzacja sondy do fragmentu o wielkości
ok. 3,6 kb. Uzyskany wynik jest zgodny z wcześniejszymi przewidywaniami.
1 2 3 K MIX 1 2 3 K MIX
Rys. 32. Analiza hybrydyzacyjna kosmidów W02D09::zeo/pyrG. A. Rozdział elektroforetyczny kosmidów W02D09::zeo/pyrG (1-3) oraz kosmidu W02D09 bez dysrupcji (K), trawionych enzymem BamHI. B. Autoradiogram po hybrydyzacji sondy 597 bp z regionem przylegającym do miejsca dysrupcji genu mcmAna kosmidzie W02D09. (1-3) – kosmidy W02D09::zeo/pyrG z dysrupcją genu mcmA; K – kosmid W02D09 bez dysrupcji; MIX – standard wielkości DNA Ladder Mix. Na rysunku widoczne są fragmenty DNA większe niż 1,2 kb. Nie widać fragmentów o wielkości: 1,1 kb (obecny tylko w kosmidzie po dysrupcji); 0,8 kb; 306 bp; 230 bp (por. Rys. 30).
A B
13,2 kb
3,6 kb
17,6 kb 13,2 kb
9,2 kb 7,7 kb
4,3 kb
3,6 kb

WYNIKI
68
II. Analiza wpływu mutacji mcmAI70A na regulację katabolizmu argininy u A. nidulans
1. Analiza wpływu mutacji mcmAI70A na indukcję arginazy i OTA przez argininę
Aby stwierdzić, jaki wpływ na indukcję ekspresji genów agaA i otaA u A. nidulans
mają mutacje w białku MCMA – potencjalnym czynniku transkrypcyjnym typu MADS,
zbadano aktywność właściwą arginazy i OTA w szczepie z substytucją jednej reszty
aminokwasowej w obrębie domeny MADS białka MCMA. Do doświadczenia użyto dwóch
szczepów A. nidulans: szczepu kontrolnego D6B-11 oraz mutanta mcmAI70A. Szczep
rodzicielski (D6B-11) posiada dziki allel genu mcmA. Z kolei szczep potomny (mcmAI70A),
posiada gen mcmA z mutacją punktową, powodującą substytucję I70A w łańcuchu
aminokwasowym białka MCMA.
Hodowle grzybni A. nidulans do oznaczeń enzymatycznych prowadzono w trzech
różnych płynnych podłożach: minimalnym z azotanem jako jedynym źródłem azotu,
nieindukowanym argininą (MM) lub indukowanym argininą (MM+Arg) oraz na podłożu, w
którym jedynym źródłem azotu była arginina (−N+Arg). Sposób hodowli grzybni opisano w
rozdziale „Metody”, punkt 11.
W zebranej grzybni oznaczono aktywności arginazy i OTA. Doświadczenie
powtórzono 3 razy, za każdym razem używając do oznaczeń świeżo wyhodowanej grzybni.
Uśrednione wyniki trzech niezależnych oznaczeń zamieszczono na Rys. 33 i w Tab. 2.
Stwierdzono, że poziom podstawowy aktywności arginazy (MM) w szczepie mcmAI70A
wynosi 0,16 ± 0,02 jednostki (u) i jest o ok. 40 - 50% niższy niż w szczepie dzikim (D6B-11).
Aktywność właściwa arginazy w szczepie mcmAI70A po indukcji argininą (MM+Arg)
wynosiła ok. 1,10 ± 0,11 u, a po hodowli na podłożu z argininą jako jedynym źródłem azotu
(−N+Arg), wyniosła ok. 1,02 ± 0,13 u. W obu przypadkach uzyskano wartości o 40 - 50%
niższe niż w szczepie dzikim.
W przypadku aktywności właściwej OTA nie stwierdzono tak istotnych różnic
pomiędzy szczepami mcmAI70A i D6B-11. Aktywność właściwa OTA na poziomie
podstawowym (MM) w szczepie mcmAI70A wynosiła 6 ± 1 u, a w szczepie dzikim 3 ± 3 u.
Aktywność właściwa OTA w szczepach mcmAI70A i D6B-11 po indukcji argininą (MM+Arg)
również była podobna i wynosiła 113 ± 9 u dla szczepu mcmAI70A oraz 121 ± 1 u dla szczepu
dzikiego. Jedynie w przypadku, gdy szczepy mcmAI70A i D6B-11 hodowano na podłożu z
argininą jako jedynym źródłem azotu (−N+Arg), różnice w aktywności właściwej OTA były

WYNIKI
69
zauważalne i wynosiły ok. 15%. Aktywność OTA w szczepie mcmAI70A wynosiła 127 ± 9 u,
a w szczepie dzikim 150 ± 5 u.
zauważalne i wynosiły ok. 15%. Aktywność OTA w szczepie mcmA
I70A wynosiła 127 ± 9 u,
a w szczepie dzikim 150 ± 5 u.
Oznaczenie aktywności arginazy w szczepach D6B-11 i MCMI70A-4
0,0
0,5
1,0
1,5
2,0
2,5
D6B-11 (MM) D6B-11(MM+Arg)
D6B-11 (-N+Arg) MCMI (MM) MCMI (MM+Arg) MCMI (-N+Arg)
Nazwa próby
Akty
wność
właśc
iwa
argi
nazy
[u]
Aktywność właściwa arginazy w szczepach D6B-11 i mcmAI70A
D6B-11 (MM)
D6B-11 (MM+Arg)
D6B-11 (−N+Arg)
mcmAI70A (MM)
mcmAI70A (MM+Arg)
mcmAI70A (−N+Arg)
Oznaczenie aktywności OTA w szczepach D6B-11 i MCMI70A-4
0
20
40
60
80
100
120
140
160
180
D6B-11 (MM) D6B-11(MM+Arg)
D6B-11 (-N+Arg) MCMI (MM) MCMI (MM+Arg) MCMI (-N+Arg)
Nazwa próby
Akty
wność
właśc
iwa
OTA
[u]
Aktywność właściwa OTA w szczepach D6B-11 i mcmAI70A
D6B-11 (MM)
D6B-11 (MM+Arg)
D6B-11 (−N+Arg)
mcmAI70A (MM)
mcmAI70A (MM+Arg)
mcmAI70A (−N+Arg)
Rys. 33. Wpływ mutacji mcmAI70A na aktywność arginazy i OTA. Do doświadczenia użyto grzybni dwóch szczepów: D6B-11 oraz mcmAI70A, hodowanych na podłożu minimalnym, nieindukowanym argininą (MM); na podłożu minimalnym po indukcji argininą (MM+Arg) oraz na podłożu z argininą jako jedynym źródłem azotu (−N+Arg). Wartości aktywności właściwej arginazy i OTA pochodzą z uśrednienia wyników trzech odrębnych doświadczeń. Słupki błędów wyznaczają wartość jednego odchylenia standardowego.

WYNIKI
Aktywność właściwa arginazy w szczepach D6B-11 i mcmAI70A [u]
Nazwa próby 1 2 3 Średnia SD D6B-11 (MM) 0,25 0,28 0,31 0,28 0,03 D6B-11 (MM+Arg) 1,83 1,85 2,13 1,94 0,17 D6B-11 (−N+Arg) 1,83 1,78 2,04 1,88 0,14 mcmAI70A (MM) 0,16 0,14 0,18 0,16 0,02
mcmAI70A (MM+Arg) 1,07 1,01 1,22 1,10 0,11 mcmAI70A (−N+Arg) 0,96 0,92 1,17 1,02 0,13
2. Analiza wpływu mutacji mcmAI70A na wykorzystanie różnych źródeł azotu
przez A. nidulans
Aktywność właściwa OTA w szczepach D6B-11 i mcmAI70A [u]
Nazwa próby 1 2 3 Średnia SD D6B-11 (MM) 1 2 7 3 3 D6B-11 (MM+Arg) 121 121 120 121 1 D6B-11 (−N+Arg) 155 150 145 150 5 mcmAI70A (MM) 7 6 5 6 1 mcmAI70A (MM+Arg) 121 115 103 113 9 mcmAI70A (−N+Arg) 137 125 120 127 9
Tab. 2. Aktywność właściwa arginazy i OTA w szczepach: D6B-11 i mcmAI70A.
1, 2, 3 – kolejne oznaczenia aktywności właściwej arginazy i OTA SD – wartość odchylenia standardowego
Aby stwierdzić, czy mutacja mcmAI70A wpływa na zdolność wykorzystania różnych
źródeł azotu przez A. nidulans, zbadano wzrost szczepów D6B-11 i mcmAI70A na podłożach
stałych z różnymi źródłami azotu: 10 mM azotanem sodu (MM); 10 mM argininą (−N+Arg);
10 mM ornityną (−N+Orn); 10 mM proliną (−N+Pro). Podłoża na szalkach zaszczepiono
konidiami w tym samym wieku, a następnie inkubowano w 37°C przez 4 – 5 dni. Wyniki
przedstawiono na Rys. 34. i w Tab. 3.
Wzrost zmutowanego szczepu mcmAI70A jest wolniejszy na wszystkich testowanych
podłożach minimalnych. Jeżeli hodowlę prowadzi się na podłożu kompletnym (CM), tempo
wzrostu szczepu mcmAI70A jest podobne jak szczepu dzikiego. Zaobserwowano też, że szczep
mcmAI70A wyraźnie gorzej konidiuje. Upośledzenie konidiowania jest najbardziej widoczne,
gdy zmutowany szczep hoduje się na podłożach z azotanem, argininą i ornityną, jako
jedynymi źródłami azotu. W przypadku, gdy jedynym źródłem azotu jest prolina, mutacja
mcmAI70A w mniejszym stopniu wpływa negatywnie na konidiowanie.
70
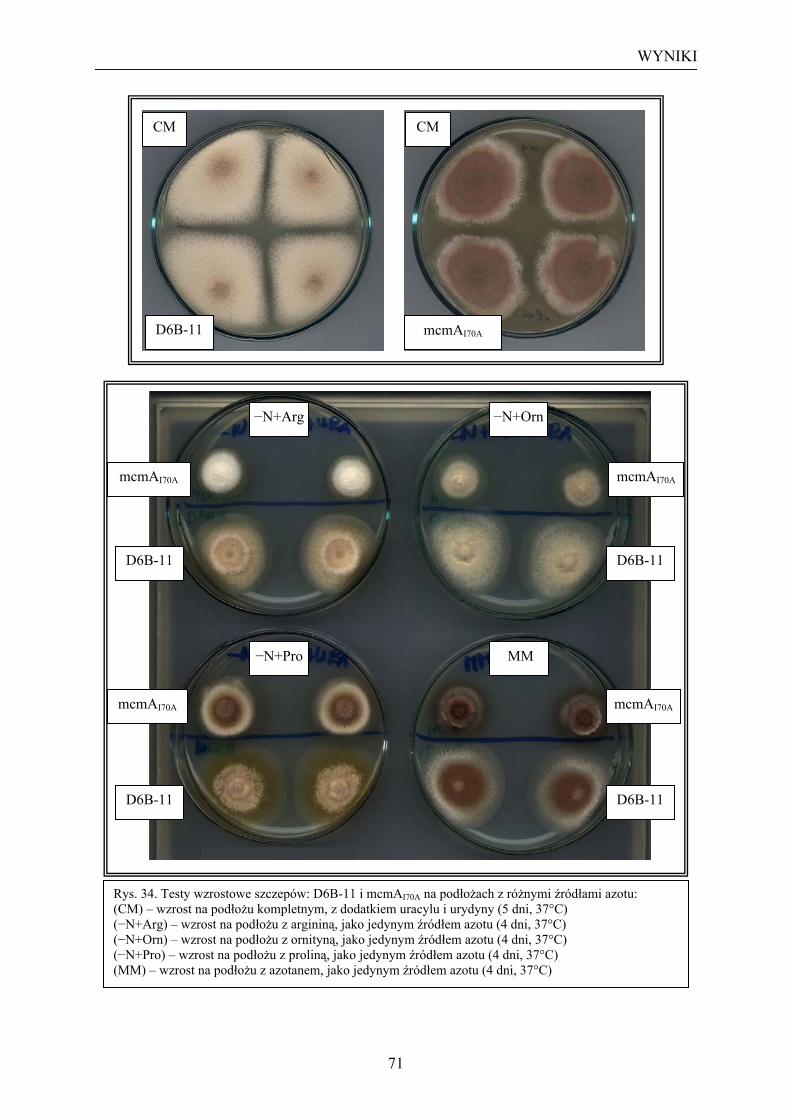
WYNIKI
CM CM
D6B-11 mcmAI70A
−N+Arg −N+Orn
mcmAI70A mcmAI70A
D6B-11 D6B-11
−N+Pro MM
mcmAI70A mcmAI70A
D6B-11 D6B-11
Rys. 34. Testy wzrostowe szczepów: D6B-11 i mcmAI70A na podłożach z różnymi źródłami azotu: (CM) – wzrost na podłożu kompletnym, z dodatkiem uracylu i urydyny (5 dni, 37°C) (−N+Arg) – wzrost na podłożu z argininą, jako jedynym źródłem azotu (4 dni, 37°C) (−N+Orn) – wzrost na podłożu z ornityną, jako jedynym źródłem azotu (4 dni, 37°C) (−N+Pro) – wzrost na podłożu z proliną, jako jedynym źródłem azotu (4 dni, 37°C) (MM) – wzrost na podłożu z azotanem, jako jedynym źródłem azotu (4 dni, 37°C)
71

WYNIKI
Źródło azotu:
azwa szczepu:
Podłoże kompletne
(CM)
10 mM azotan (MM)
10 mM arginina
(−N+Arg)
10 mM ornityna
(−N+Orn)
10 mM prolina
(−N+Pro)
Tab. 3. Wzrost szczepów D6B-11 oraz mcmAI70A na podłożach z różnymi źródłami azotu.
N
D6B-11
+++
+++ gorzej konidiuje
+++
gorzej konidiuje
+++
++
źle konidiuje
mcmAI70A+++
gorzej konidiuje +
źle konidiuje +
źle konidiuje +
źle konidiuje ++
gorzej konidiuje
W tabeli użyto następujących oznaczeń: (+++) – normalny wzrost, (++) – wzrost nieznacznie spowolniony, (+) – wzrost wyraźnie wolniejszy. Do wszystkich podłoży (również do CM) dodano uracylu w stężeniu 0,2 mM i urydyny w stężeniu 0,1 mM.
72

DYSKUSJA
DYSKUSJA
I. Dysrupcja genu mcmA A. nidulans
Głównym celem tej pracy było zbadanie funkcji genu mcmA z A. nidulans, w
szczególności zaś ustalenie, czy gen ten bierze udział w regulacji katabolizmu argininy,
podobnie jak jego drożdżowy odpowiednik – gen MCM1. By osiągnąć zamierzony cel,
postanowiono wykonać dysrupcję genu mcmA, wykorzystując nową metodę dysrupcji genów
u A. nidulans (Chaveroche i wsp., 2000). Kropidlak (A. nidulans), podobnie jak drożdże (S.
cerevisiae), należy do klasy workowców (Ascomycetes). Oba gatunki ze swej natury są
haploidalne, choć mogą również wytwarzać formy diploidalne. Wykonanie dysrupcji
jakiegokolwiek genu w haploidalnym szczepie A. nidulans niesie ze sobą pewne
niebezpieczeństwo. W przypadku, gdy analizowany gen jest niezbędny do życia, jego
dysrupcja może się okazać letalna, co w konsekwencji uniemożliwia selekcję szczepów z taką
mutacją i ich dalszą hodowlę.
Gen mcmA koduje białko MCMA – potencjalny czynnik transkrypcyjny blisko
spokrewniony z drożdżowym białkiem Mcm1p. Wykazano, że delecja białka Mcm1p jest
letalna (Giaever i wsp., 2002). Jeżeli czynnik MCMA spełnia u A. nidulans podobne funkcje,
jak Mcm1p u drożdży, jego dysrupcja również może powodować śmierć organizmu. By temu
zapobiec, postanowiono wykonać dysrupcję genu mcmA w dwóch szczepach A. nidulans:
haploidalnym i diploidalnym, oczekując, że u diploida dysrupcja może w mniejszym stopniu
upośledzić funkcje życiowe, a fenotyp szczepu z dysrupcją może dostarczyć informacji o roli
białka MCMA.
Diploid do dysrupcji genu mcmA powinien mieć zmutowne oba allele genów proA i
pyrG, ponieważ mutacja w genie proA pozwala na wykorzystanie diploida do badania genów,
uczestniczących w regulacji metabolizmu argininy, które w formie zmutowanej wywołują
supresję mutacji prolinowej, a mutacja w genie pyrG umożliwia selekcję szczepu A. nidulans
z dysrupcją określonego genu (Chaveroche i wsp., 2000). By było możliwe wyhodowanie
diploida o takim genotypie, każdy ze szczepów haploidalnych, użytych do jego uzyskania,
musi posiadać obie mutacje.
Wstępnym etapem selekcji szczepu diploidalnego u A. nidulans jest otrzymanie
heterokarionu. W niniejszej pracy otrzymano heterokarion z dwóch szczepów haploidalnych
73

DYSKUSJA
o różnej barwie konidiów:
proA1(6), pyrG89, adF9, biA1 (zielone konidia)
oraz
proA1(6, 7), uaZ11, prnA432, pyrG89, pabaA1, yA2 (żółte konidia)
Następnie wyselekcjonowano szczep diploidalny o genotypie:
proA1(6), pyrG89, adF9, biA1
proA1(6, 7), pyrG89, uaZ11, prnA432, pabaA1, yA2
i fenotypie: pro−, pyr−, (zielone konidia).
Diploidalność szczepu A. nidulans można potwierdzić, obserwując konidia w
mikroskopie świetlnym, ponieważ konidia diploidalne są wyraźnie większe od haploidalnych
(Hfagy i Roper, 1952). Możliwe jest także zastosowanie p-fluorofenyloalaniny, która, po
dodaniu do podłoża, powoduje spontaniczną haploidyzację szczepu diploidalnego (Lhoas,
1961). Jeśli szczep diploidalny uzyskano ze szczepów haploidalnych różniących się barwą
konidiów, po dodaniu p-fluorofenyloalaniny na szalkach zaobserwuje się różnobarwne
sektory.
W tej pracy diploidalność otrzymanego szczepu potwierdzono metodą hybrydyzacji
typu Southern. Zastosowanie tej metody było możliwe dzięki temu, że jeden ze szczepów
haploidalnych, użytych do otrzymania diploida, miał mutację uaZ11, polegającą na
wzajemnej translokacji fragmentów chromosomów I i VIII.
W niniejszej pracy wykorzystano metodę dysrupcji, zaproponowaną przez
(Chaveroche i wsp., 2000). Pozwala ona na dysrupcję dużych fragmentów genomu A.
nidulans. W metodzie tej wykorzystuje się bank genomowy tego grzyba, utworzony na
wektorze kosmidowym pWE15. Jest to metoda dwuetapowa (Rys. 11). Pierwszy etap polega
na dysrupcji genu na kosmidzie w bakteriach. W tym celu kosmid z badanym genem
wprowadza się do szczepu E. coli KS272 z plazmidem pKOBEG. Poieważ na kosmidzie
znajdują się geny warunkujące oporność na ampicylinę i kanamycynę, a plazmid pKOBEG
warunkuje oporność na chloramfenikol, transformanty selekcjonuje się na podłożu z tymi
trzema antybiotykami w 30°C (pKOBEG jest replikonem wrażliwym na wysoką
temperaturę). Plazmid pKOBEG nie tylko warunkuje oporność na chloramfenikol, ale także
74

DYSKUSJA
75
zawiera trzy geny operonu Redαβγ faga λ pod kontrolą promotora pBAD, indukowanego
arabinozą. Geny tego operonu kodują białka wydajnego systemu rekombinacji
homologicznej. Fakt ten umożliwia zastosowanie kaset dysrupcyjnych (zeo/pyrG),
uzyskiwanych w wyniku reakcji PCR. Taka kaseta ma na końcach 5’ i 3’ sekwencje
homologiczne (ok. 50 bp) do regionów DNA położonych powyżej i poniżej miejsca w
genomie A. nidulans, które ma ulec dysrupcji. Kaseta zawiera też geny umożliwiające jej
późniejszą selekcję zarówno w komórkach prokariotycznych (gen Sh ble – warunkujący
oporność na zeocynę), jak i w komórkach A. nidulans (gen pyrG z A. fumigatus). Do bakterii
KS272 z plazmidem pKOBEG i wtransformowanym uprzednio kosmidem, hodowanych na
podłożu z arabinozą, wprowadza się przygotowaną wcześniej kasetę dysrupcyjną. Następnie
na podłożu selekcyjnym: LB+Amp+Km+Cm+Zeo w 30°C poszukuje się transformantów, u
których nastąpiła rekombinacja homologiczna pomiędzy kasetą dysrupcyjną a kosmidem,
prowadząca do dysrupcji badanego genu na kosmidzie. Po transformacji przeprowadza się
gubienie plazmidu pKOBEG, hodując bakterie na podłożu bez chloramfenikolu w 42°C. By
sprawdzić czy dysrupcja genu rzeczywiście zaszła, kosmid z dysrupcją bada się metodą
analizy restrykcyjnej, PCR, hybrydyzacji typu Southern lub sekwencjonując region ulegający
dysrupcji.
Drugi etap polega na transformacji protoplastów A. nidulans kolistą lub
zlinearyzowaną formą kosmidu, w którym zaszła dysrupcja badanego genu. Po transformacji
kosmid z dysrupcją ulega rekombinacji homologicznej z chromosomem A. nidulans.
Następstwem tego procesu jest dysrupcja badanego genu na chromosomie. Selekcja
rekombinantów jest możliwa dzięki temu, że obecny na kasecie dysrupcyjnej gen pyrG z A.
fumigatus komplementuje mutacje w genie pyrG z A. nidulans. Szczep A. nidulans z
dysrupcją jest więc zdolny do wzrostu na podłożu bez uracylu i urydyny.
Dzięki obecności na kosmidzie dużego regionu homologicznego do sekwencji
genomu A. nidulans (obszar homologii może mieć do kilkudziesięciu tysięcy par zasad),
wydajność rekombinacji homologicznej i dysrupcji jest bardzo wysoka. Literatura podaje, że
stosując opisywaną metodę, stwierdza się wystąpienie dysrupcji badanego genu w 30 – 60%
stransformowanych komórek A. nidulans, podczas gdy użycie innych metod pozwala na
dysrupcję genu w 5 – 10% transformantów (Chaveroche i wsp., 2000).
Komercyjnie dostępne są banki genomowe A. nidulans, utworzone na dwóch
wektorach kosmidowych: pWE15 i LORIST2 (Brody i wsp., 1991; Prade i wsp., 1997).
Okazuje się jednak, że do omawianej metody dysrupcji nadają się tylko kosmidy, pochodzące

DYSKUSJA
76
od wektora pWE15, ponieważ po użyciu kosmidów pochodzących od wektora LORIST2, nie
udaje się uzyskać pozytywnych rezultatów dysrupcji genów (Chaveroche i wsp., 2000).
W niniejszej pracy potwierdzono hybrydyzacyjnie, że na kosmidzie W02D09,
pochodzącym od wektora pWE15, znajduje się fragment genomu A. nidulans z genem mcmA.
Na podstawie analizy restrykcyjnej kosmidu W02D09 in vitro oraz komputerowej analizy
restrykcyjnej sekwencji kontigu 1.159, pobranej z bazy genomowej A. nidulans Broad
Institute, oszacowano wielkość kosmidu na ok. 48 kb, a wielkość wstawki na tym kosmidzie
na ok. 39 kb. Wstawka obejmuje region 12-13 kb powyżej sekwencji kodującej (ORF) genu
mcmA oraz 25-26 kb poniżej tej sekwencji (Rys. 22). Dzięki obecności na kosmidzie tak
długich fragmentów homologicznych do genomu A. nidulans, otaczających gen mcmA, proces
rekombinacji pomiędzy kosmidem a chromosomem i towarzysząca mu dysrupcja genu mcmA
powinny zachodzić z wysoką wydajnością.
Zgodnie z informacjami dostępnymi w literaturze, metoda dysrupcji zaproponowana
przez Chaveroche i wsp. pozwala na wydajną dysrupcję genów u grzybów nitkowatych,
dlatego też tej właśnie metody zdecydowano się użyć do dysrupcji genu mcmA. Stosując tę
metodę, napotkano jednak pewne trudności, które opisano poniżej.
Transformacja szczepu KS272 (pKOBEG) kosmidem zachodziła z bardzo niską
wydajnoscią (ok. 102 stransformowanych komórek / μg DNA kosmidu). Pomimo że bakterie
po transformacji selekcjonowano na podłożu z trzema antybiotykami (LB+Amp+Km+Cm), u
większości potencjalnych transformantów analiza restrykcyjna wykazała, że zamiast kosmidu
W02D09, obecny jest fragment DNA o wielkości 5-6 kb, niewiadomego pochodzenia (Rys.
23). Spośród 17 potencjalnych transformantów, których DNA poddano analizie restrykcyjnej,
jedynie u dwóch wynik analizy był zgodny z oczekiwaniami i wskazywał na obecność
kosmidu W02D09, mimo że wszystkie transformanty wykazywały oporność na ampicylinę,
kanamycynę i chloramfenikol. U wszystkich też analiza restrykcyjna potwierdziła obecność
plazmidu pKOBEG. Ponieważ wiadomo, że duże replikony trudniej utrzymują się w
bakteriach, można przypuszczać, że wspomniany fragment o wielkości 5-6 kb powstaje z
kosmidu w wyniku rekombinacji, i że najprawdopodobniej niesie on oporność na dwa
antybiotyki: ampicylinę i kanamycynę. Co ciekawe, fragment 5-6 kb występuje w większości
stransformowanych komórek (13 / 17), lecz nie ma go w tych transformantach, które
zawierają kosmid W02D09 (Rys. 23).
Po wyselekcjonowaniu szczepu KS272 (pKOBEG + W02D09) i potwierdzeniu, że
zawiera on zarówno plazmid pKOBEG, jak i nieprzerekombinowany kosmid W02D09,
podjęto próbę transformacji tego szczepu kasetą dysrupcyjną zeo/pyrG. Wydajność

DYSKUSJA
77
transformacji, połączonej z rekombinacją homologiczną, wyniosła ok. 102 – 103
stransformowanych komórek / μg DNA kasety zeo/pyrG. Wynik, wykonanej wcześniej,
kontrolnej transformacji szczepu KS272 (pKOBEG + W02D09) plazmidem pBR322,
wskazywał na wydajność transformacji rzędu 103 – 104 stransformowanych komórek / μg
DNA plazmidu pBR322. Gdyby założyć, że transformacja kasetą zeo/pyrG zachodzi z
podobną wydajnością, oznaczałoby to, że w ok. 10% komórek stransformowanych kasetą
nastąpiła rekombinacja homologiczna, prowadząca do dysrupcji genu mcmA. Tak wysoki
odsetek rekombinantów wydaje się jednak mało prawdopodobny. Kosmid W02D09 posiada
miejsce startu replikacji ColE1, a plazmid pBR322 ma miejsce startu replikacji z plazmidu
pMB1. W literaturze dostępne są informacje, zgodnie z którymi oba replikony mogą być
wzajemnie niekompatybilne (Sutcliffe, 1979), co może być przyczyną niskiej wydajności
transformacji kontrolnej szczepu KS272 (pKOBEG + W02D09) plazmidem pBR322.
Plazmid pCDA21, będący matrycą w reakcji PCR, prowadzącej do uzyskania kasety
dysrupcyjnej zeo/pyrG, posiada miejsce startu replikacji R6Kγ. Zgodnie z informacjami
zawartymi w literaturze, replikony tego typu mogą się powielać jedynie w szczepie E. coli
BW19610 (Chaveroche i wsp., 2000). Mimo to uzyskano wyniki (niezamieszczone w
niniejszej pracy) wskazujące, że plazmid pCDA21 może się replikować także w szczepie
KS272 (pKOBEG + W02D09). W przypadku, gdy kasety zeo/pyrG nie oczyszczono przed
stransformowaniem tego szczepu, uzyskiwano transformanty, które zgodnie z oczekiwaniami,
były oporne na 4 antybiotyki: ampicylinę, kanamycynę, chloramfenikol i zeocynę. Nie
następowała w nich jednak dysrupcja genu mcmA, a analiza restrykcyjna wskazywała na
obecność w transformantach całego plazmidu pCDA21. Aby tego uniknąć, konieczne okazało
się oczyszczanie kasety zeo/pyrG przed transformacją poprzez izolację właściwego fragmentu
DNA z żelu agarozowego.
Po transformacji kasetą zeo/pyrG uzyskano ok. 100 transformantów / 200 ng DNA
kasety. Wydajność rekombinacji homologicznej pomiędzy kasetą zeo/pyrG a kosmidem
W02D09 oszacowano na ok. 10%, lecz prawdopodobnie jest ona niższa (niekompatybilność
replikonów, patrz wyżej). Wydajność gubienia plazmidu pKOBEG wynosiła 100%.
Analizując DNA kosmidowe z 15 klonów, u których spodziewano się dysrupcji genu mcmA,
w 5 przypadkach stwierdzono niewłaściwy wzór restrykcyjny (brakowało na przykład
fragmentu o wielkości 1,9 kb, odpowiadającego genowi pyrG na kasecie zeo/pyrG). U
pozostałych 10 klonów analiza restrykcyjna dała wynik zgodny z oczekiwanym, wskazujący
na wystąpienie dysrupcji genu mcmA na kosmidzie (Rys. 28). Potwierdzenie dysrupcji genu
mcmA metodą hybrydyzacji typu Southern było możliwe jedynie dla trzech klonów (2, 3, 7),

DYSKUSJA
78
co było związane z problemami, na jakie natknięto się podczas izolacji DNA kosmidowego ze
szczepu KS272 (W02D09::zeo/pyrG). Konieczne okazało się izolowanie kosmidów przy
użyciu zestawu komercyjnego do izolacji DNA plazmidowego, ponieważ po zastosowaniu
standardowej metody lizy alkalicznej, DNA kosmidów ulegało degradacji.
Na koniec warto zauważyć, że po transformacji szczepu KS272 (pKOBEG) kosmidem
W02D09 wyraźnie spada tempo wzrostu bakterii. Obecność kosmidu wpływa też negatywnie
na ich żywotność.
Doświadczenia wykonane w ramach niniejszej pracy zaowocowały uzyskaniem
diploidalnego szczepu A. nidulans oraz dysrupcją genu mcmA na kosmidzie W02D09.
Kolejny etap doświadczeń będzie obejmował transformację kosmidem z dysrupcją
haploidalnego i diploidalnego szczepu A. nidulans, a następnie analizę fenotypu
transformantów i stwierdzenie, czy dysrupcja genu mcmA będzie miała wpływ na regulację
katabolizmu argininy. Jeśli przypuszczenia dotyczące podobieństwa funkcji białek Mcm1p i
MCMA okażą się słuszne, można oczekiwać negatywnych efektów fenotypowych dysrupcji
genu mcmA u A. nidulans. Możliwe też, że dysrupcja tego genu w szczepie haploidalnym
będzie letalna.
II. Analiza wpływu mutacji mcmAI70A na regulację katabolizmu argininy u
A. nidulans
W promotorach genów katabolizmu argininy (agaA i otaA) u A. nidulans stwierdzono
obecność sekwencji analogicznych do drożdżowych UASarg. Sekwencji występującej w
promotorze genu otaA nadano nazwę AnUASarg (Dzikowska i wsp., 2003), a sekwencję
znalezioną w promotorze genu agaA nazwano ABS (Borsuk i wsp., 1999).
Porównanie sekwencji UASarg, AnUASarg i ABS umożliwiło znalezienie
charakterystycznych motywów o dużej homologii do sekwencji wiążących białka z rodziny
MADS (Rys. 35). W obrębie każdej drożdżowej sekwencji UASarg stwierdzono występowanie
dwóch takich konserwowanych motywów, nazwanych „arginine-box”, do których wiążą się
drożdżowe białka MADS: Mcm1p i ArgRIp (Messenguy i Dubois, 2000). W sekwencji
AnUASarg również występują dwa potencjalne motywy „arginine-box”, rozdzielone
(podobnie jak u drożdży) regionem bogatym w pary G-C. Ponadto, krótkie sekwencje
bezpośrednio poprzedzające motywy „arginine-box” są silnie konserwowane u obu
organizmów. Drożdżowe sekwencje „arginine-box” są poprzedzone trzema konserwowanymi

DYSKUSJA
79
nukleotydami, niezbędnymi dla właściwej funkcji białka Mcm1p, a nukleotyd tymidynowy
(T), znajdujący się w pozycji -2, ma kluczowe znaczenie w procesie zaginania cząsteczki
DNA (Acton i wsp., 1997). U A. nidulans w obrębie AnUASarg stwierdzono obecność pięciu
konserwowanych nukleotydów (ACCT
nukleotydami, niezbędnymi dla właściwej funkcji białka Mcm1p, a nukleotyd tymidynowy
(T), znajdujący się w pozycji -2, ma kluczowe znaczenie w procesie zaginania cząsteczki
DNA (Acton i wsp., 1997). U A. nidulans w obrębie AnUAS stwierdzono obecność pięciu
konserwowanych nukleotydów (ACCarg
TG), poprzedzających sekwencje „arginine-box”.
Podobnie jak u drożdży, w pozycji -2 występuje nukleotyd tymidynowy (T) (Rys. 35)
(Dzikowska i wsp., 2003).
T TMiejsce wiązania SRF TATA TATA T A T A TAAT AAT Miejsce wiązania Mcm1p NN NN CTTA CAR1 -215 GTG TTT CAR2 -220 CCT TTG ARG3 -110 TCA TGT ARG5,6 -57 ATA TTT
A A CC GG CC GG
A T A CC G CC G
G CTTA G
CTGCTAATGG -171 CACTTAGCGG
CTGTTAACGG -149 CCCTTAGCGG
CCTCTAAAGG -81 CCTTTA--AG
CCACTAAAGG -115 CCATTA--GG
agaA (-410) GGCAGCCGTCTT---TCTCCTCGACCTG TAAAAGCAG
otaA (-183) CACAG-CGTCATCGAACGGGTCGACCTG TG
Duże podobieństwo sekwencji UASarg i AnUASarg spowodowało, że podjęto próbę
ustalenia, czy sekwencja AnUASarg spełnia u A. nidulans podobną funkcję, jak UASarg u
drożdży. W tym celu w szczepie dzikim A. nidulans wykonano dwie różne delecje w obrębie
sekwencji AnUASarg. Usunięte regiony składały się z 7 (mutant TΔ3) lub 25 nukleotydów
TACTTAGGGGAAAGCCGCATTCCC GTCTTAGCGATG
CACTTAGAGGAATTGTACGGGTAC----ACC CACTTAGCGGTA
G CGC GAC
Region delecji w obrębie sekwencji ABS w promotorze genu agaA
(4L-abs) (3P-abs)
TΔ3 TΔ2
Rys. 35. Porównanie sekwencji DNA, do których wiążą się różne białka z rodziny MADS. Sekwencje zestawiono w następującej kolejności: sekwencja najwyższej zgodności motywu SRE, wiążącego SRF; sekwencja najwyższej zgodności motywu P, wiążącego Mcm1p; sekwencje „arginine-box”, występujące w promotorach genów katabolizmu (CAR1 i CAR2) i biosyntezy (ARG3 i ARG5,6) argininy u drożdży; fragmenty sekwencji dwóch promotorów genów agaA (ABS) i otaA (AnUASarg) u A. nidulans. Niebieską strzałką zaznaczono region delecji w obrębie sekwencji ABS, a niebieskimi literami oznaczono mutacje punktowe w obrębie tej sekwencji (szczegóły w tekście) (Grzelak, 2003). Kolorem różowym zaznaczono potencjalną sekwencję TATA. Niebieskie linie wyznaczają dwa regiony delecji w obrębie sekwencji AnUASarg (TΔ3 i TΔ2) (Dzikowska i wsp., 2003). Czarnymi strzałkami zaznaczono 12-nukleotydowe powtórzenia w sekwencji promotora genu otaA. Kolorem zielonym wyróżniono konserwowane nukleotydy tymidynowe w pozycji -2 (szczegóły w tekście). Rys. wg (Dzikowska i wsp., 2003; Grzelak, 2003).

DYSKUSJA
80
(mutant TΔ2) (Rys. 35). Obie delecje powodowały, że, in vivo, w obecności argininy nie
następowała aktywacja ekspresji genu otaA. Wprowadzenie tych samych delecji do szczepu z
mutacją arcAd47, w którym wytwarzane jest białko ARCA o właściwościach
superaktywatora, również uniemożliwiało aktywację ekspresji genu otaA (Dzikowska i wsp.,
2003). Wynik ten dowodzi, że sekwencja AnUASarg bierze udział w zależnej od argininy
regulacji ekspresji genu otaA, a także świadczy o tym, że w obrębie sekwencji AnUASarg
znajduje się miejsce wiązania aktywatora ARCA.
Promotor genu agaA ma długość ok. 1450 bp, obejmuje więc znacznie większy obszar
niż promotor genu otaA. Na obszarze tym stwierdzono występowanie pięciu miejsc startu
transkrypcji. Analiza sekwencji promotora genu agaA pozwoliła na znalezienie dwóch
odrębnych regionów, które nazwano górnym i dolnym promotorem. Górny promotor jest
położony w odległości 1440-900 bp, a dolny w odległości 680-160 bp, licząc od miejsca
startu translacji. W dolnym promotorze występuje sekwencja ABS (ang. ARCA binding site),
do której prawdopodobnie wiąże się białko ARCA (Borsuk i wsp., 1999).
W celu ustalenia roli, jaką odgrywa sekwencja ABS, wykonano delecję 70
nukleotydów w jej obrębie (Rys. 35) (Grzelak, 2003). Szczep z delecją charakteryzuje się
brakiem zdolności do wzrostu na pożywce z argininą jako jedynym źródłem azotu. W
szczepie tym nie obserwuje się też indukcji arginazy przez argininę. Co więcej, w
zmutowanym szczepie aktywność właściwa arginazy na poziomie podstawowym (bez
indukcji) jest o połowę niższa niż w szczepie kontrolnym. Taki wynik sugeruje, że sekwencja
ABS uczestniczy w zależnej od argininy regulacji ekspresji genu agaA. Ponadto sekwencja ta
może być zaangażowana w proces utrzymania na właściwym poziomie aktywności arginazy
w warunkach nieindukcyjnych. Warto zwrócić uwagę, że w usuniętym regionie ABS znajduje
się potencjalna sekwencja TATA. Obserwowane efekty delecji ABS mogą więc częściowo
wynikać z obniżonego poziomu transkrypcji genu agaA (Grzelak, 2003).
Inne doświadczenie polegało na wprowadzeniu dwóch mutacji w obrębie sekwencji
ABS. Jedna z mutacji (4L-abs) polegała na zamianie 4 reszt aminokwasowych, a druga (3P-
abs) na zamianie 3 reszt (Rys. 35). Genem agaA ze zmienionym promotorem
stransformowano dwa szczepy A. nidulans, z których jeden miał dziką kopię genu arcA, a
drugi zmutowany gen arcAd47. Efekt fenotypowy obu mutacji był podobny i polegał na
spadku aktywności właściwej arginazy o 30 – 60%, zarówno na poziomie podstawowym, jak
i po indukcji argininą, niezależnie od tego czy w komórce występowała dzika czy zmutowana
forma białka ARCA (Grzelak, 2003). Wyniki te potwierdzają, że ekspresja genu agaA zależy
od sekwencji ABS.

DYSKUSJA
81
Wzajemne podobieństwo sekwencji ABS i AnUASarg sugeruje, że ekspresja genów
agaA i otaA może być regulowana w taki sam sposób, tzn. przy udziale czynnika ARCA i co
najmniej jednego białka MADS.
Wzajemne podobieństwo sekwencji ABS i AnUASarg sugeruje, że ekspresja genów
agaA i otaA może być regulowana w taki sam sposób, tzn. przy udziale czynnika ARCA i co
najmniej jednego białka MADS.
helisa α Gly
region hydrofobowy miejsce substytucji
aminokwasowej I70A
Rys. 36. Przyrównanie sekwencji aminokwasowych domen typu MADS, pochodzących z kilku białek, należących do podrodziny MADS I. W przyrównaniu wykorzystano sekwencje aminokwasowe domen MADS następujących białek: Mcm1p (S. cerevisiae) (Ammerer, 1990); MCMA (A. nidulans = Emericella nidulans) (Dzikowska i wsp., 2005); ArgRIp (S. cerevisiae) (Dubois i wsp., 1987); SRF (Homo sapiens) (Norman i wsp., 1988). Liczby w nawiasach oznaczają pozycje pierwszych podanych reszt aminokwasowych w pełnej sekwencji aminokwasowej poszczególnych białek. Strzałką zaznaczono miejsce mutacji mcmAI70A, będącej przedmiotem badań w niniejszej pracy.
Jak wyżej wspomniano, delecja genu MCM1 jest letalna dla drożdży. Śmiertelne są też
mutacje punktowe, powodujące substytucje niektórych reszt aminokwasowych w domenie
MADS białka Mcm1p (Rys. 6) (Acton i wsp., 1997, 2000; Mead i wsp., 2002). Jest możliwe,
że zamiana aminokwasów, tworzących domenę MADS w potencjalnym białku MCMA u A.
nidulans, również może mieć negatywny wpływ na fenotyp. Szczep mcmAI70A, otrzymany od
Prof. T. Kobayashi z Uniwersytetu Nagoya w Japonii, posiada mutację, powodującą
substytucję I70A w obrębie domeny MADS białka MCMA. Zamieniona reszta izoleucyny
(I70) jest odpowiednikiem waliny (V34) w białku Mcm1p (Rys. 36). Walina (V34) kontaktuje
się z DNA oraz uczestniczy w procesie zaginania cząsteczki kwasu nukleinowego przez
czynnik Mcm1p. Bierze też udział w oddziaływaniu Mcm1p-Ste12 (Rys. 6, Rys. 7)
(Messenguy i Dubois, 2003). Stwierdzono, że mutacja V34A w białku Mcm1p wywołuje
znaczny spadek aktywności, objawiający się m.in. zmniejszeniem zdolności do aktywacji
transkrypcji genów in vivo. Efekt ten obserwuje się, jeżeli czynnik Mcm1p wiąże się z DNA
pojedynczo, a także wtedy, gdy oddziałuje z DNA w kompleksie z innymi białkami.
Szczególnie silny spadek zdolności do aktywacji (50-krotny) zaobserwowano dla kompleksu
Mcm1p-Ste12. Wydaje się, że taki spadek aktywności może być związany z poważnymi
zaburzeniami procesu zaginania cząsteczki kwasu nukleinowego przez czynnik Mcm1p, które
Jak wyżej wspomniano, delecja genu MCM1 jest letalna dla drożdży. Śmiertelne są też
mutacje punktowe, powodujące substytucje niektórych reszt aminokwasowych w domenie
MADS białka Mcm1p (Rys. 6) (Acton i wsp., 1997, 2000; Mead i wsp., 2002). Jest możliwe,
że zamiana aminokwasów, tworzących domenę MADS w potencjalnym białku MCMA u A.
nidulans, również może mieć negatywny wpływ na fenotyp. Szczep mcmAI70A, otrzymany od
Prof. T. Kobayashi z Uniwersytetu Nagoya w Japonii, posiada mutację, powodującą
substytucję I70A w obrębie domeny MADS białka MCMA. Zamieniona reszta izoleucyny
(I70) jest odpowiednikiem waliny (V34) w białku Mcm1p (Rys. 36). Walina (V34) kontaktuje
się z DNA oraz uczestniczy w procesie zaginania cząsteczki kwasu nukleinowego przez
czynnik Mcm1p. Bierze też udział w oddziaływaniu Mcm1p-Ste12 (Rys. 6, Rys. 7)
(Messenguy i Dubois, 2003). Stwierdzono, że mutacja V34A w białku Mcm1p wywołuje
znaczny spadek aktywności, objawiający się m.in. zmniejszeniem zdolności do aktywacji
transkrypcji genów in vivo. Efekt ten obserwuje się, jeżeli czynnik Mcm1p wiąże się z DNA
pojedynczo, a także wtedy, gdy oddziałuje z DNA w kompleksie z innymi białkami.
Szczególnie silny spadek zdolności do aktywacji (50-krotny) zaobserwowano dla kompleksu
Mcm1p-Ste12. Wydaje się, że taki spadek aktywności może być związany z poważnymi
zaburzeniami procesu zaginania cząsteczki kwasu nukleinowego przez czynnik Mcm1p, które

DYSKUSJA
82
również są efektem mutacji V34A. Mutacja ta powoduje ponadto nieznaczne (2,7-krotne)
zmniejszenie powinowactwa białka Mcm1p do DNA (Mead i wsp., 2002).
W literaturze nie znaleziono informacji na temat wpływu mutacji V34A na aktywność
kompleksu Mcm1p-ArgRp. Wydaje się jednak, że może ona w znacznym stopniu utrudniać
formowanie tego kompleksu, gdyż do jego powstania niezbędne jest zagięcie cząsteczki DNA
(Messenguy i Dubois, 2000).
Szczep A. nidulans z mutacją mcmAI70A został stworzony głównie w celu badania
wpływu tej mutacji na procesy płciowe u A. nidulans (T. Kobayashi, inf. ustna) (kompleks
Mcm1p-Ste12 powstaje u drożdży pod wpływem feromonów). Ponieważ nie wiadomo
również, czy mutacja mcmAI70A wpływa na regulację katabolizmu argininy, w niniejszej pracy
podjęto próbę wyjaśnienia tej kwestii.
Badając aktywność właściwą arginazy w szczepie z mutacją mcmAI70A, stwierdzono,
że jest ona niższa o 40 – 50% niż w szczepie dzikim (D6B-11), zarówno na poziomie
podstawowym, jak i po indukcji argininą. Z kolei różnice w aktywności właściwej OTA w
obu szczepach były w większości nieistotne statystycznie. Jedynie w warunkach hodowli na
podłożu z argininą jako jedynym źródłem azotu (–N+Arg) aktywność właściwa OTA w
szczepie mcmAI70A była o ok. 15% niższa niż w szczepie dzikim. Na podstawie uzyskanych
wyników można stwierdzić, że mutacja mcmAI70A ma silniejszy wpływ na regulację ekspresji
genu agaA.
Wiadomo, że drożdżowa mutacja V34A w białku Mcm1p objawia się m.in. 2,7-
krotnym zmniejszeniem powinowactwa tego białka do DNA (Mead i wsp., 2002). Jest
możliwe, że analogiczna mutacja I70A w białku MCMA u A. nidulans również wywołuje
podobny efekt. Sekwencja AnUASarg w genie otaA w większym stopniu przypomina
drożdżową sekwencję UASarg, niż sekwencja ABS w genie agaA, można więc założyć, że
białko MCMA będzie się silniej wiązało z promotorem genu otaA. W takim przypadku
mutacje zmniejszające powinowactwo białka MCMA do DNA powinny bardziej negatywnie
wpływać na regulację ekspresji genu agaA i tym zapewne można tłumaczyć silniejszy wpływ
mutacji mcmAI70A na aktywność właściwą arginazy.
Podsumowując, głównym celem tej pracy było poznanie funkcji białka MCMA u A.
nidulans, i mimo że nie udało się otrzymać dysrupcji genu mcmA w komórkach grzyba, to
jednak, dzięki analizie enzymatycznej mutanta mcmAI70A, ustalono, że białko MCMA
uczestniczy w regulacji katabolizmu argininy. Ponadto stwierdzono, że następstwem mutacji
mcmAI70A są zaburzenia konidiowania i spadek tempa wzrostu grzyba na różnych podłożach
minimalnych. Mutacja ta wpływa też negatywnie na wykorzystanie różnych źródeł azotu

DYSKUSJA
83
(Rys. 34). Zmutowany szczep wyraźnie gorzej konidiuje na podłożach, w których jedynym
źródłem azotu jest arginina, ornityna lub azotan. Z kolei na podłożu z proliną, jako jedynym
źródłem azotu, szczep mcmAI70A konidiuje podobnie jak szczep dziki (D6B-11), hodowany
na podłożu z azotanem. Na podłożu kompletnym (CM) szczep z mutacją rośnie normalnie,
choć nadal wytwarza znacznie mniej konidiów w porównaniu ze szczepem dzikim.
Przedstawione wyniki pokazują, że mutacja mcmAI70A u A. nidulans ma charakter
plejotropowy, podobnie jak, wspomniana wyżej, mutacja V34A w drożdżowym białku
Mcm1p, która również wywołuje liczne efekty fenotypowe, w tym także spadek tempa
wzrostu (Acton i wsp., 2000). Jest to kolejny dowód na to, że białka Mcm1p i MCMA mogą
pełnić w komórkach podobne funkcje.

LITERATURA
LITERATURA
1. Acton, T.B., Mead J., Steiner A.M., Vershon A.K. (2000). Scanning mutagenesis of Mcm1: residues required for DNA binding, DNA bending, and transcriptional activation by a MADS-box protein. Mol. Cell Biol., Volume: 20, pp. 1-11.
2. Acton, T.B., Zhong H., Vershon A.K. (1997). DNA-binding specificity of Mcm1: operator mutations that alter DNA-bending and transcriptional activities by a MADS box protein. Mol. Cell Biol., Volume: 17, pp. 1881-1889.
3. Albrecht, A.M., Vogel, H.J. (1964). Acetylornithine delta-transaminase. Partial purification and repression behavior. J Biol Chem., Jun;239:1872-6.
4. Althoefer, H., Schleiffer A., Wassmann K., Nordheim A., Ammerer G. (1995). Mcm1 is required to coordinate G2-specific transcription in Saccharomyces cerevisiae. Mol. Cell Biol., Volume: 15, pp. 5917-5928.
5. Alvarez-Buylla,E.R., Pelaz,S., Liljegren,S.J., Gold,S.E., Burgeff,C., Ditta,G.S., Ribas de Pouplana,L., Martinez-Castilla,L. and Yanofsky,M.F. (2000). An ancestral MADS-box gene duplication occurred before the divergence of plants and animals. Proc. Natl Acad. Sci. USA, 97, 5328–5333.
6. Amar, N., F. Messenguy, M. El Bakkoury, and E. Dubois (2000). ArgRIIp, a component of the ArgR-Mcm1 complex involved in the control of arginine metabolism in Saccharomyces cerevisiae, is the sensor of arginine. Mol. Cell. Biol. 20:2087-2097.
7. Ammerer G. (1990). Identification, purification, and cloning of a polypeptide (PRTF/GRM) that binds to mating-specific promoter elements in yeast. Genes Dev. 4:299-312.
8. Arabidopsis Genome Initiative. (2000). Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408:796-815.
9. Bartnik, E., Weglenski, P. (1973). Ammonium and glucose repression of the arginine catabolic enzymes in Aspergillus nidulans. Mol. Gen. Genet. 126, 75–84.
10. Bartnik, E., Weglenski, P. (1974). Regulation of arginine catabolism in Aspergillus nidulans. Nature, 250, 590–592.
11. Béchet, J., Grenson M., Wiame J.M. (1970). Mutations affecting the repressibility of arginine biosynthetic enzymes in Saccharomyces cerevisiae Eur. J. Biochem., Volume: 12, pp. 31-39
12. Bercy, J., E. Dubois, F. Messenguy (1987). Regulation of arginine metabolism in Saccharomyces cerevisiae: expression of the three ARGR regulatory genes and cellular localization of their products. Gene, 55 277–285.
13. Boros, J., Lim, F.-L., Darieva, Z., Pic-Taylor, A., Harman, R., Morgan, B. A., & Sharrocks, A. D. (2003). Molecular determinants of the cell-cycle regulated Mcm1p-Fkh2p transcription factor complex. Nucl. Acids. Res. 31 (9): 2279-2288.
14. Borsuk, P., Dzikowska, A., Empel, J., Grzelak, A., Grzeskowiak, R.,Weglenski, P. (1999). Structure of arginase coding gene and its transcript in Aspergillus nidulans. Acta Biochim. Pol. 46, 391–403.
15. Bowman, J.L., Meyerowitz, E.M. (1991). Genetic control of pattern formation during flower development in Arabidopsis. Symp. Soc. Exp. Biol. 45, 89-115.
16. Bowman, J.L., Smyth, D.R., Meyerowitz, E.M. (1991). Genetic interactions among floral homeotic genes of Arabidopsis. Development, 112, 1-20.
17. Bradford, M. M. (1976). A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem., May 7;72:248-54.
18. Brody, H., J Griffith, AJ Cuticchia, J Arnold and WE Timberlake (1991). Chromosome-specific recombinant DNA libraries from the fungus Aspergillus nidulans. Nucl. Acids Res. Vol 19, Issue 11 3105-3109.
19. Brown, T. A. (2001). Genomy, PWN, Warszawa. 20. Bruhn, L., Hwang-Shum J.J., Sprague G.F. Jr. (1992). The N-terminal 96 residues of MCM1, a
regulator of cell type-specific genes in Saccharomyces cerevisiae, are sufficient for DNA binding, transcription activation, and interaction with alpha 1 Mol. Cell Biol., Volume: 12, , pp. 3563-3572.
21. Carr, E.A., Mead J. and Vershon A.K. (2004). α1-induced DNA bending is required for transcriptional activation by the Mcm1/ α1 complex. Nucleic Acids Res., Vol. 32, pp. 2298-2305.
22. Chai, J., Tarnawski, A. S. (2002). Serum response factor: discovery, biochemistry, biological roles and implications for tissue injury healing. Journal of Physiology and Pharmacology 53 (2): 147-157.
84

LITERATURA
85
23. Chang, V.K., Donato J.J, Chan C.S., and Tye B.K. (2004). Mcm1 Promotes Replication Initiation by Binding Specific Elements at Replication Origins. Mol. Cel. Biol., Vol. 24, pp. 6514-6524.
24. Chang, V.K., Fitch M.J., Donato J.J., Christensen T.W., Merchant A.M., Tye B.K. (2003). Mcm1 binds replication origins. J. Biol. Chem., Volume: 278, pp. 6093-6100.
25. Chaveroche, M.-K., Ghigo, J.-M. and d’Enfert, C. (2000). A rapid method for efficient gene replacement in the filamentous fungus Aspergillus nidulans. Nucl. Acids Res. Vol. 28, No. 22 e97.
26. Coffman, J. A., R. Rai, T. Cunningham, V. Svetloy, T. G. Cooper (1996). Gat1p, a GATA family protein whose production is sensitive to nitrogen catabolite repression, participates in transcriptional activation of nitrogen-catabolic genes in Saccharomyces cerevisiae. Mol. Cell. Biol. 16 847–858.
27. Courchesne, W. E., B. Magasanik (1988). Regulation of nitrogen assimilation in Saccharomyces cerevisiae: roles of the URE2 and GLN3 genes. J. Bacteriol. 170 708–713.
28. Crabeel, M., M. DeRijcke, S. Seneca, H. Heimberg, I. Pfeiffer, A. Matisova (1995). Further definition of the sequence and position requirements of the arginine control element that mediates repression and induction by arginine in Saccharomyces cerevisiae. Yeast, 11 1367–1380.
29. Cybis, J., Piotrowska, M., and Weglenski, P. (1972). Genetics control of arginine pathways in Aspergillus nidulans. Common regulation of anabolism and catabolism. Mol Gen Genet 205: 170-175.
30. Cybis, J., Weglenski, P. (1972). Arginase induction in Aspergillus nidulans. The appearance and decay of the coding capacity of messenger. Eur J Biochem., Oct;30(2):262-8.
31. Davies, B. & Schwarz-Sommer, Z. (1994). Control of floral organ identity by homeotic MADS-box transcription factors, in Results and Problems in Cell Differentiation (Nover, L. ed.) vol. 20, pp. 235-258, Springer-Verlag, Berlin Heidelberg.
32. Davis, R.H. (1986). Compartmental and regulatory mechanisms in the arginine pathways of Neurospora crassa and Saccharomyces cerevisiae. Microbiol. Rev. 50: 280 – 313.
33. de Nadal E, Casadome L, Posas F. (2003). Targeting the MEF2-like transcription factor Smp1 by the stress-activated Hog1 mitogen-activated protein kinase. Mol Cell Biol. Jan;23(1):229-37.
34. DeRijcke, M., S. Seneca, B. Punyammalee, N. Glansdorff, M. Crabeel (1992). Characterization of the DNA target site for the yeast ARGR regulatory complex, a sequence able to mediate repression or induction by arginine. Mol. Cell. Biol. 12 68–81.
35. Dolan, J. W. & Fields, S. (1991). Cell-type-specific transcription in yeast. Biochem. Biophys. Acta 1088, 155-169.
36. Dubois, E. & Messenguy, F. (1991). In vitro studies of the binding of the ARGR proteins to the ARG5,6 promoter. Mol. Cell. Biol. 11, 2162-2168.
37. Dubois, E., F. Messenguy (1994). Pleiotropic function of ArgRIIIp (Arg82p), one of the regulators of arginine metabolism in Saccharomyces cerevisiae. Role in expression of cell-type-specific genes. Mol. Gen. Genet. 243 315–324.
38. Dubois, E., F. Messenguy (1997). Integration of the multiple controls regulating the expression of the arginase gene CAR1 of Saccharomyces cerevisiae in response to differentnitrogen signals: role of Gln3p, ArgRp-Mcm1p, and Ume6p. Mol. Gen. Genet. 253 568–580.
39. Dubois, E., J. Bercy, F. Messenguy (1987). Characterization of two genes, ARGRI and ARGRIII required for specific regulation of arginine metabolism in yeast. Mol. Gen. Genet. 207 142–148.
40. Dubois, E., Scherens B., Vierendeels F., Ho M.M., Messenguy F., Shears S.B. (2002). In Saccharomyces cerevisiae, the inositol polyphosphate kinase activity of Kcs1p is required for resistance to salt stress, cell wall integrity, and vacuolar morphogenesis. J. Biol. Chem., Volume: 277, pp. 23755-23763.
41. Dzikowska, A., Kacprzak, M., Tomecki, R., Koper, M., Scazzocchio, C., and Weglenski, P. (2003). Specific induction and carbon/nitrogen repression of arginine catabolism gene of Aspergillus nidulans – functional in vivo analysis of the otaA promoter. Fungal Genet Biol, 38: 175-186.
42. Dzikowska, A., Swianiewicz, M., Talarczyk, A., Wisniewska, M., Goras, M., Scazzocchio, C., Weglenski, P. (1999). Cloning, characterisation and regulation of the ornithine transaminase (otaA) gene of Aspergillus nidulans. Curr. Genet. 35, 118–126.
43. Dzikowska,A., Walczuk,B., Weglenski,P., Endo,Y., Kato,M. and Kobayashi,T. (2005). mcmA, the putative regulatory gene from MADS-box family. GenBank Accession Number: AY957455.
44. El Alami, M., Messenguy F., Scherens B., Dubois E. (2003). Arg82p is a bifunctional protein whose inositol polyphosphate kinase activity is essential for nitrogen and PHO gene expression but not for Mcm1p chaperoning in yeast. Mol. Microbiol., Volume: 49, pp. 457-468.
45. El Bakkoury, M., Dubois E., Messenguy F. (2000). Recruitment of the yeast MADS-box proteins, ArgRI and Mcm1 by the pleiotropic factor ArgRIII is required for their stability. Mol. Microbiol., Volume: 35, pp. 15-31.

LITERATURA
86
46. Elble, R., Tye B.K. (1992). Chromosome loss, hyperrecombination, and cell cycle arrest in a yeast mcm1 mutant. Mol. Biol. Cell, Volume: 3, pp. 971-980.
47. Empel, J., Sitkiewicz, I., Andrukiewicz, A., Lasocki, K., Borsuk, P., and Weglenski, P. (2001). arcA, the regulatory gene of the arginine catabolic pathway of Aspergillus nidulans. Mol Genet Genom 266: 591-597.
48. Errede, B. (1993). MCM1 binds to a transcriptional control element in Ty1. Mol. Cell Biol., Volume: 13, pp. 57-62.
49. Evans GA, Wahl GM. (1987). Cosmid vectors for genomic walking and rapid restriction mapping. Methods Enzymol., 152:604-610.
50. Fujioka,T., Furukawa,K., Mizutani,O., Abe,K., Yamagata,Y. and Nakajima,T. (2003). Functional analysis of Aspergillus nidulans rlmA gene encoding a transcription factor involved in cell integrity signal transduction. GenBank Accession Number: AB110208.
51. Garcia R, Bermejo C, Grau C, Perez R, Rodriguez-Pena JM, Francois J, Nombela C, Arroyo J. ( 2004). The global transcriptional response to transient cell wall damage in Saccharomyces cerevisiae and its regulation by the cell integrity signaling pathway. J Biol Chem. Apr 9;279(15):15183-95.
52. Giaever G, et al. (2002). Functional profiling of the Saccharomyces cerevisiae genome. Nature, Jul 25;418(6896):387-91.
53. Greenberg ME, Ziff EB. (1984). Stimulation of 3T3 cells induces transcription of the c-fos proto-oncogene. Nature, 311, 433-8.
54. Griffin D.H. (1994), Fungal Physiology. Wiley-Liss, Inc., Publication.
55. Grzelak, A. (2003). Analiza funkcjonalna genów agaA i suDpro związanych z katabolizmem argininy u Aspergillus nidulans. Praca doktorska wykonana w Zakładzie Genetyki UW.
56. Hasebe, M., and J. A. Banks (1997). Evolution of MADS gene family in plants. Pp. 179-197 in K. Iwatsuki and P. H. Raven, eds. Evolution and diversification of land plants. Springer-Verlag, Tokyo.
57. Herskowitz, I., J. Rine, and J. Strathern (1992). Mating-type determination and mating-type interconversion in Saccharomyces cerevisiae. vol. 2. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
58. Hfagy FC, Roper JA. (1952). Deoxyribonucleic acid content of haploid and diploid Aspergillus conidia. Nature, Oct 25;170(4330):713-714.
59. Hinnebusch, A. G. (1992), in: The Molecular and Cellular Biology of Yeast: Gene Expression, Vol. 2, E. W Jones, J. R. Pringle, J. R. Broach (Eds.), Cold Spring Harbor Press, Cold Spring Harbor pp. 319–401.
60. Hollenhorst, P.C., Bose M.E., Mielke M.R., Muller U., Fox C.A. (2000). Forkhead genes in transcriptional silencing, cell morphology and the cell cycle. Overlapping and distinct functions for FKH1 and FKH2 in Saccharomyces cerevisiae. Genetics, Volume: 154, pp. 1533-1548.
61. Hollenhorst, P.C., Pietz G., Fox C.A. (2001). Mechanisms controlling differential promoter-occupancy by the yeast forkhead proteins Fkh1p and Fkh2p: implications for regulating the cell cycle and differentiation. Genes Dev., Volume: 15, pp. 2445-2456.
62. Jamai, A., Dubois E., Vershon A.K., Messenguy F. (2002). Swapping functional specificity of a MADS box protein: residues required for Arg80 regulation of arginine metabolism. Mol. Cell Biol., Volume: 22, pp. 5741-5752.
63. Johansen, B., Pedersen, L. B., Skipper, M., & Frederiksen, S. (2002). MADS-box gene evolution-structure and transcription patterns. Molecular Phylogenetics and Evolution 23: 458-480.
64. Kerppola, T. K. & Curran, T. (1997). The transcription activation domains of Fos and Jun induce DNA bending through electrostatic interactions. EMBO J. 16, 2907-2916.
65. Koranda, M., Schleiffer A., Endler L., Ammerer G. (2000). Forkhead-like transcription factors recruit Ndd1 to the chromatin of G2/M-specific promoters. Nature, Volume: 406, pp. 94-98.
66. Kumar, R., Reynolds D.M., Shevchenko A., Goldstone S.D., Dalton S. (2000). Forkhead transcription factors, Fkh1p and Fkh2p, collaborate with Mcm1p to control transcription required for M-phase. Curr. Biol., Volume: 10, pp. 896-906.
67. Kuo, M.H., Nadeau E.T., Grayhack E.J. (1997). Multiple phosphorylated forms of the Saccharomyces cerevisiae Mcm1 protein include an isoform induced in response to high salt concentrations. Mol. Cell Biol., Volume: 17, pp. 819-832.
68. Lenouvel F., Nikolaev I., Felenbok B. (1997). In vitro recognition of specific DNA targets by AlcR, a zinc binuclear cluster activator different from the other proteins of this class. J Biol Chem 272:15521-15526.
69. Lhoas P. (1961) Mitotic haploidization by treatment of Aspergillus niger diploids with para-fluorophenylalanine. Nature, May 20;190:744.

LITERATURA
87
70. Lim FL, Hayes A, West AG, Pic-Taylor A, Darieva Z, Morgan BA, Oliver SG, Sharrocks AD. (2003). Mcm1p-induced DNA bending regulates the formation of ternary transcription factor complexes. Mol Cell Biol. Jan;23(2):450-61.
71. Lydall, D., Ammerer G., Nasmyth K. (1991). A new role for MCM1 in yeast: cell cycle regulation of SW15 transcription. Genes Dev., Volume: 5, pp. 2405-2419.
72. Maher, M., Cong F., Kindelberger D., Nasmyth K., Dalton S. (1995). Cell cycle-regulated transcription of the CLB2 gene is dependent on Mcm1 and a ternary complex factor. Mol. Cell Biol., Volume: 15, pp. 3129-3137.
73. Mai, B., Miles S., Breeden L.L. (2002). Characterization of the ECB binding complex responsible for the M/G(1)-specific transcription of CLN3 and SWI4. Mol. Cell Biol., Volume: 22, pp. 430-441.
74. Martin, J. E, Miano, J. M., Hustad, C. M., Copeland, N. G., Jenkins, N. A. & Olson, E. N. (1994). A Mef2 gene that generates a muscle-specific isoform via alternative mRNA splicing. Mol. Cell. Biol. 14, 1647-1656.
75. McDermott, J. C, Cardoso, M. C, Yu, Y.-T., Andres, V., Leifer, D., Krainc, D., Lipton, S. A. & Nadal-Ginard, B. (1993). hMEF2C gene encodes skeletal muscle- and brain-specific transcription factors. Mol. Cell. Biol. 13, 2564-2577.
76. McInerny, C.J., Partridge J.F., Mikesell G.E., Creemer D.P., Breeden L.L. (1997). A novel Mcm1-dependent element in the SWI4, CLN3, CDC6, and CDC47 promoters activates M/G1-specific transcription. Genes Dev., Volume: 11, pp. 1277-1288.
77. Mead, J., Bruning A.R., Gill M.K., Steiner A.M., Acton T.B., Vershon A.K. (2002). Interactions of the Mcm1 MADS box protein with cofactors that regulate mating in yeast. Mol. Cell Biol., Volume: 22, pp. 4607-4621.
78. Messenguy F., Dubois E. (2003). Role of MADS box proteins and their cofactors in combinatorial control of gene expression and cell development. Gene, Vol: 316, Complete, October 16, pp: 1-21.
79. Messenguy, F., Dubois E. (1993). Genetic evidence for a role for MCM1 in the regulation of arginine metabolism in Saccharomyces cerevisiae. Mol. Cell Biol., Volume: 13, pp. 2586-2592.
80. Messenguy, F., Dubois E. (2000). Regulation of arginine metabolism in Saccharomyces cerevisiae: a network of specific and pleiotropic proteins in response to multiple environmental signals. Food Technol. Biotechnol., Volume: 38, pp. 277-285.
81. Messenguy, F., E. Dubois, C. Boonchird (1991). Determination of the DNA-binding sequences of ARGR proteins to arginine anabolic and catabolic promoters. Mol. Cell. Biol. 11 2852–2863.
82. Messenguy, F.,Vierendeels F., Scherens B., and Dubois E. (2000). In Saccharomyces cerevisiae, expression of arginine catabolic genes CAR1 and CAR2 in response to exogenous nitrogen availability is mediated by the Ume6 (CargRI) – Sin3 (CargRII) – Rpd3 (CargRIII) complex. J. Bacteriol. Jun;182(11):3158-64.
83. Metcalf, W. W., W. Jiang, and B. L. Wanner (1994). Use of the rep technique for allele replacement to construct new Escherichia coli hosts for maintenance of R6K gamma origin plasmids at different copy numbers. Gene, 138:1-7.
84. Mueller, C.G., Nordheim A. (1991). A protein domain conserved between yeast MCM1 and human SRF directs ternary complex formation. EMBO J., Volume: 10, pp. 4219-4229.
85. Norman, C., Runswick, M., Pollock, R. & Treisman, R, (1988). Isolation and properties of cDNA clones encoding SRF, a transcription factor that binds to the c-fos serum response element. Cell 55, 989-1003.
86. Oehlen, L.J., McKinney J.D., Cross F.R. (1996). Ste12 and Mcm1 regulate cell cycle-dependent transcription of FAR1. Mol. Cell Biol., Volume: 16, pp. 2830-2837.
87. Oestreicher N, Scazzocchio C. (1993a). Sequence, regulation, and mutational analysis of the gene encoding urate oxidase in Aspergillus nidulans. J Biol Chem., Nov 5;268(31):23382-23389.
88. Oestreicher N, Sealy-Lewis H M, Scazzocchio C. (1993b). Characterisation, cloning and integrative properties of the gene encoding urate oxidase in Aspergillus nidulans. Gene, 132:185–192.
89. Parenicová L., de Folter S., Kieffer M., Horner DS., Favalli C., Busscher J., Cook HE., Ingram RM., Kater MM., Davies B., Angenent GC. and Colombo L. (2003). Molecular and Phylogenetic Analyses of the Complete MADS-Box Transcription Factor Family in Arabidopsis. New Openings to the MADS World. The Plant Cell, Vol. 15, 1538-1551.
90. Park H.-D., Scott S., Rai R., Dorrington R., Cooper T. G. (1999). Synergistic operation of the CAR2 (Ornithine transaminase) promoter elements in Saccharomyces cerevisiae. J. Bacteriol. 181 7052–7064.
91. Passmore, S., Maine G.T., Elble R., Christ C., Tye B.K. (1988). Saccharomyces cerevisiae protein involved in plasmid maintenance is necessary for mating of MAT alpha cells. J. Mol. Biol., Volume: 204, pp. 593-606.

LITERATURA
88
92. Pelaz, S., Ditta, G.S., Baumann, E., Wisman, E., Yanofsky, M.F. (2000). B and C floral organ identity functions require SEPALLATA MADS-box genes. Nature, 405, 200-203.
93. Pellegrini, L., Tan, S. & Richmond, T. J. (1995). Structure of serum response factor core bound to DNA. Nature, 376, 490- 498.
94. Pic, A., Lim F.L., Ross S.J., Veal E.A., Johnson A.L., Sultan M.R., West A.G., Johnston L.H., Sharrocks A.D., Morgan B.A. (2000). The forkhead protein Fkh2 is a component of the yeast cell cycle transcription factor SFF. EMBO J., Volume: 19, pp. 3750-3761.
95. Pollock, R., Treisman, R. (1990). A sensitive method for the determination of protein-DNA binding specificities. Nucleic. Acids Res. 18, 6197-6204.
96. Pollock, R., Treisman, R. (1991). Human SRF-related proteins: DNA-binding properties and potential regulatory targets. Genes Dev., Volume: 5, pp. 2327-2341.
97. Prade, R.A., James Griffith, Krys Kochut, Jonathan Arnold and William E. Timberlake (1997). In vitro reconstruction of the Aspergillus (= Emericella) nidulans genome. Proc. Natl. Acad. Sci. USA Vol. 94, pp. 14564-14569.
98. Pramila, T., Miles S., GuhaThakurta D., Jemiolo D., Breeden L.L. (2002). Conserved homeodomain proteins interact with MADS box protein Mcm1 to restrict ECB-dependent transcription to the M/G1 phase of the cell cycle. Genes Dev., Volume: 16, pp. 3034-3045.
99. Primig, M., Winkler H., Ammerer G. (1991). The DNA binding and oligomerization domain of MCM1 is sufficient for its interaction with other regulatory proteins. EMBO J., Volume: 10, pp. 4209-4218.
100. Qiu, H.F., Dubois E., Broen P., Messenguy F. (1990). Functional analysis of ARGRI and ARGRIII regulatory proteins involved in the regulation of arginine metabolism in Saccharomyces cerevisiae. Mol. Gen. Genet., Volume: 222, pp. 192-200.
101. Riechmann, J. L. & Meyerowitz, E. M. (1997). MADS domain proteins in plant development. J. Biol. Chem. 378, 1079-1101.
102. Rollins BJ., Stiles CD. (1989). Serum-inducible genes. Adv Cancer Res, 53, l-32.
103. Sambrook, J., Fritsch, E.F., Maniatis, T. (1989). Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, wyd. II, New York.
104. Schjerling, P., Holmberg, S. (1996). Comparative amino acid sequence analysis of C6 zinc cluster family of transcriptional regulators. Nucleic Acids Res. 24, 4599–4607.
105. Schwarz-Sommer, Z., Hue, I., Huijser, P., Flor, P.J., Hansen, R., Tetens, F., Lonnig, W.E., Saedler, H., Sommer, H. (1992). Characterization of the Antirrhinum floral homeotic MADS-box gene deficiens: evidence for DNA binding and autoregulation of its persistent expression throughout flower development. EMBO J. 11, 251-263.
106. Sharrocks, A. D., Gille. H. & Shaw, P. E. (1993). Identification of amino acids essential for DNA binding and dimerization in p67SRF: Implications for a novel DNA-binding motif. Mol. Cell. Biol. 13, 123-132.
107. Shore P., Sharrocks A.. D. (1995). The MADS-box family of transcription factors. Eur. J. Biochem. 229 1-13.
108. Smart, W. C., J. A. Coffman, T. G. Cooper (1996). Combinatorial regulation of the Saccharomyces cerevisiae CAR1 (arginase) promoter in response to multiple environmental signals. Mol. Cell. Biol. 16 5876–5887.
109. Sprague, G. F. (1990). Combinatorial associations of regulatory proteins and the control of cell type in yeast. Adv. Genet, 27, 33-62.
110. Stanbrough, M., D. W. Rowen, B. Magasanik (1995). Role of the GATA factors Gln3p and Nil1p of Saccharomyces cerevisiae in the expression of nitrogen-regulated genes. Proc. Natl. Acad. Sci. USA, 92 9450–9454.
111. Steger, D.J., Haswell E.S., Miller A.L., Wente S.R., O'Shea E.K. (2003). Regulation of chromatin remodeling by inositol polyphosphates. Science, Volume: 299, pp. 114-116.
112. Sutcliffe J.G. (1979). Complete nucleotide sequence of the Escherichia coli plasmid pBR322. Cold Spring Harb Symp Quant Biol., 43 Pt 1:77-90.
113. Tan, S., G. Ammerer, and T. J. Richmond (1988). Interactions of purified transcription factors: binding of yeast MATα1 and PRTF to cell type-specific, upstream activating sequences. EMBO J. 7:4255-4264.

LITERATURA
89
114. Tan, S., Richmond T.J. (1998). Crystal structure of the yeast MATalpha2/MCM1/DNA ternary complex. Nature, Volume: 391, pp. 660-666.
115. Theissen, G., J. T. Kim, and H. Saedler (1996). Classification and phylogeny of the MADS-box multigene family suggest defined roles of MADS-box gene subfamilies in the morphological evolution of eukaryotes. J. Mol. Evol. 43:484-516.
116. Todd, R.B., Andrianopoulos, A. (1997). Evolution of a fungal regulatory gene family: the Zn(II)2Cys6 binuclear cluster DNA-binding motif. Fungal Genet. Biol. 21, 388–405.
117. Treisman, R. & Ammerer, G. (1992). The SRF and MCM1 transcription factors. Curr. Opin. Genet. Dev. 2, 221-226.
118. Treisman, R. (1990). The SRE: a growth factor responsive transcriptional regulator. Semin. Cancer Biol. I, 47 – 58.
119. Wahl GM, Lewis KA, Ruiz JC, Rothenberg B, Zhao J, Evans GA. (1987). Cosmid vectors for rapid genomic walking, restriction mapping, and gene transfer. Proc Natl Acad Sci U S A. Apr;84(8):2160-2164.
120. Walczuk, B. (2005). I. Identyfikacja mutacji w allelu otaA1 genu kodującego transaminazę ornitynową Aspergillus nidulans. II. Identyfikacja genów kodujących białka zawierające domenę MADS u Aspergillus nidulans. Praca magisterska wykonana w Zakładzie Genetyki UW.
121. Weidner, G., d’Enfert, C. Koch, A., Mol, PC., Brakhage, AA. (1998). Development of a homologous transformation system for the human pathogenic fungus Aspergillus fumigatus based on the pyrG gene encoding orotidine 5’-monophosphate decarboxylase. Curr Genet, 33: 378–385.
122. West, A. G., & Sharrocks, A. D. (1999). MADS-box transcription factors adopt alternative mechanisms for bending DNA. J. Mol. Biol. 286, 1311-1323.
123. West, A.G., Shore P., Sharrocks A.D. (1997). DNA binding by MADS-box transcription factors: a molecular mechanism for differential DNA bending. Mol. Cell Biol., Volume: 17, pp. 2876-2887.
124. Wynne, J. & Treisman, R. (1992). SRF and MCM1 have related but distinct DNA binding specificities. Nucleic. Acids Res, 20, 3297-3303.
125. Yoon, S., Govind C.K., Qiu H., Kim S., Dong J. and Hinnebusch A.G. (2004). Recruitment of the ArgR/Mcm1p repressor is stimulated by the activator Gcn4p: A self-checking activation mechanism. PNAS, vol. 101, pp. 11713-11718.
126. York, J.D., Odom A.R., Murphy R., Ives E.B., Wente S.R. (1999). A phospholipase C-dependent inositol polyphosphate kinase pathway required for efficient messenger RNA export. Science, Volume: 285, pp. 96-100.
127. Yu, G., Fassler J.S. (1993). SPT13 (GAL11) of Saccharomyces cerevisiae negatively regulates activity of the MCM1 transcription factor in Ty1 elements. Mol. Cell Biol., Volume: 13, pp. 63-71.
128. Zhu, G., Spellman P.T., Volpe T., Brown P.O., Botstein D., Davis T.N., Futcher B. (2000). Two yeast forkhead genes regulate the cell cycle and pseudohyphal growth. Nature, Volume: 406, pp. 90-94.





























