Embed Size (px)
Citation preview

THE JOURNAL OF BIOLOGICAL CHEMISTRY Vol. 258, No. 18, Issue of September 25, pp. 11174-11184,1983 Printed m U.S.A.
Characterization of Strand Displacement Synthesis Catalyzed by Bacteriophage T7 DNA Polymerase*
(Received for publication, March 21, 1983)
Robert L. LechnerS, Michael J. Englerg, and Charles C. Richardson From the Department of Biological Chemistry, Haruard Medical School, Boston, Massachusetts 021 15
The DNA polymerase induced after infection of Escherichia coli by bacteriophage T7 can exist in two forms. One distinguishing property of Form I, the elim- ination of nicks in double-stranded DNA templates, strongly suggests that this form of the polymerase cat- alyzes limited DNA synthesis at nicks, resulting in displacement of the downstream strand. In this paper, we document this reaction by a detailed characteriza- tion of the DNA product.
DNA synthesis on circular, duplex DNA templates containing a single site-specific nick results in circular molecules bearing duplex branches. Analysis of newly synthesized DNA excised from the product shows that the majority of the branches are less than 500 base pairs in length and that they arise from a limited number of sites. The branches have fully base-paired termini but are attached by two noncomplementary DNA strands that have a combined length of less than 30 nucleotides. The product molecules are topologi- cally constrained as a result of the duplex branch. DNA sequence analysis has provided an unequivocal struc- ture of one such product molecule. We conclude that strand displacement synthesis catalyzed by Form I of T7 DNA polymerase is terminated by a template- switching reaction. We propose two distinct models for template-switching that we call primer relocation and rotational strand exchange.
Strand displacement synthesis catalyzed by Form I of T7 DNA polymerase effectively converts T7 DNA circles that are held together by hydrogen bonds in their 160-nucleotide-long terminal redundancy to T7- length linear molecules. We suggest that strand dis- placement synthesis catalyzed by T7 DNA polymerase is essential in vivo to the processing of a T7 DNA concatemer to mature T7 genomes.
In the previous paper, we reported the existence of two forms of purified T7 DNA polymerase (1). Although no struc- tural differences have been found for the two forms, they can
* This investigation was supported by United States Public Health Service Grant AI-06045 and Grant NP-1L from the American Cancer Society, Inc. This is Paper 24 in a series entitled “Replication of Bacteriophage T7 Deoxyribonucleic Acid.” The previous paper is Ref. 1. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “aduertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
$Recipient of National Institutes of Health Fellowship 5 F32
3 Recipient of National Institutes of Health Fellowship 5 F32 AI05803. Present address, Department of Biochemistry and Molecu- lar Biology, University of Texas Medical School a t Houston, Houston, TX.
GM07288-02.
be differentiated on the basis of their activities. Form I has low specific activities of both single- and double-stranded DNA exonuclease activities relative to Form 11. Equally pro- nounced is the lack of stimulation by T7 gene 4 protein of DNA synthesis catalyzed by Form I1 on nicked,’ duplex templates; gene 4 protein stimulates DNA synthesis catalyzed by Form I manyfold on such a template. In addition, evidence was presented that Form I can use a 3’-hydroxyl group a t a nick as a primer to polymerize nucleotides, a reaction that leads to displacement of a 5”terminated strand as synthesis proceeds along the duplex. In this paper, we characterize the strand displacement synthesis catalyzed by Form I of T7 DNA polymerase.
The measured differences between the two forms suggest that Form I1 is most appropriate for catalyzing reactions that take place at the replication fork. Form I1 will not polymerize nucleotides starting at nicks, it has a high level of a proof- reading 3’ to 5’ exonuclease activity, and, as shown in the fourth paper of this series (2), DNA synthesis at gaps’ in DNA is limited to the filling of the gap, leaving a nick in the duplex.
On the other hand, strand displacement reactions may be relevant to other processes of DNA metabolism such as DNA maturation and recombination. For example, concatemers of T7 DNA are composed of a series of directly repeating mon- omer units, each of which contains the sequence of an entire T7 genome minus one of the 160-base pair terminal repeats (3). Watson (4) has suggested a model for the maturation of T7 concatemers in which an endonuclease makes a nick in each strand at the 5’ ends of each terminal repeat to provide primers for DNA polymerase. Mature T7 DNA molecules are then generated by DNA synthesis coupled to strand displace- ment. Here we show that the strand displacement reaction of Form I can effect such an event.
In addition to providing information on the physiological significance of Form I of T7 DNA polymerase, the studies described here provide additional insight into the mechanisms leading to strand displacement. We show that the end product of the strand displacement reaction is, as is also the case for Escherichia coli DNA polymerase I ( 5 ) , a duplex branch. Furthermore, the use of templates containing nicks at a unique sequence has made possible a detailed analysis of these branches, including the determination of the nucleotide se- quence of one. Our results suggest that a duplex branch can arise as a result of either primer relocation or a rotational strand exchange.
__-___
A nick in DNA is defined as a single phosphodiester bond inter- ruption that contains a 5’-phosphoryl and a 3‘-hydroxyl end group. A gap in DNA is defined as an internally located length of single- stranded DNA in a duplex DNA molecule.
11174
by guest on June 13, 2018http://w
ww
.jbc.org/D
ownloaded from

Strand Displacement Synthesis by T7 DNA Polymerase 11175
EXPERIMENTAL PROCEDURES
Materials
Bacterial Strains and Bacteriophage-Wild type bacteriophage, T7 phage, and E. coli carrying plasmid p A R l l l (6) were obtained from F. W. Studier (Brookhaven National Laboratories). E. coli carrying plasmid pBR322 (7) was obtained from W. Gilbert (Harvard Univer- sity). The plasmid pMB9 was constructed by Rodriguez et al. (8).
Enzymes-Form I and Form I1 of bacteriophage T7 DNA poly- merase were purified as described in the previous paper (1). The preparations used in these studies had specific activities of 5,800 and 10,300 units/mg, respectively. E. coli exonuclease I11 and restriction endonucleases HindIII and Hinfl were from New England Biolahs. Restriction endonuclease EcoRI was from two sources: New England Biolabs and R. Kolodner (Harvard University). T4 polynucleotide kinase was purified as previously described (9) or obtained from New England Biolabs. T4 DNA ligase was purified from E. coli E1150 as previously described (10). Pancreatic DNase was from Worthington. S1 nuclease was from Bethesda Research Laboratories.
Other Materials-Acrylamide, N,N'-methylene bisacrylamide and N,N,N',N'-tetramethylethylenediamine were from Bio-Rad. Agarose B (Type I) was obtained from Sigma Chemical Co. Ultrapure sucrose was from Schwarz/Mann. Formamide (99%) was obtained from MCB. DEAE-Sephadex A-25, Sephadex G-75 and Sephadex G-100 were obtained from Pharmacia. [w3'P]dATP and [a-32P]dTTP (500-1000 Ci/mmol) and [y3*P]dATP (7000 Ci/mmol) were purchased from New England Nuclear and were further purified by chromatography on DEAE-Sephadex A-25 (11).
Methods
DNA Preparations-Hydrogen-bonded T7 DNA circles were pre- pared by a method similar to that originally described by Ritchie et al. (12). The redundant sequence a t each end of the genome was exposed by partial digestion with E. coli exonuclease 111, and the resulting molecules were placed under conditions favorable to an- nealing of the complementary single-stranded ends. Our detailed procedure was as follows. T7 DNA was obtained by the method previously described (3) and incubated with T 7 DNA ligase to elimi- nate any nicks. The reaction mixture (60 p1) contained 20 pg of T7 DNA, 74 p~ ATP, 50 mM Tris-HC1 (pH 7.8), 6 mM MgC12, 6 mM dithiothreitol, and 0.01 unit of T7 DNA ligase. Incubation was for 20 min a t 30 "C. The 3"OH ends were then digested with E. coli exonuclease I11 until approximately 5% of the DNA was rendered acid soluble as measured by the acid solubility of radioactivity in an identical reaction mixture containing T7 (3H]DNA. The reaction mixture was added to 20 pl of a solution containing 0.4 M NaCl and 0.09 M Tris-HCI (pH 7.8) and 10 p1 of E. coli exonuclease I11 (58 units). After incubation for 20 min at 23 "C, the reaction was stopped by the addition of 18 pl of 0.1 M EDTA (pH 8). The DNA was diluted with 3.6 ml of a solution containing 0.3 M NaC1.0.03 M sodium citrate, and then incubated a t 65 "C for 40 min. Finally, the annealed DNA was dialyzed extensively a t 4 "C against 10 mM Tris-HC1 (pH 7.9) and 1 mM EDTA.
Supercoiled pMB9, pAR111, and monomeric pBR322 plasmid DNAs were prepared as previously described (14). Dimeric pBR322 plasmid DNA was prepared from cells which had been selected and grown on tetracycline-containing media (7), and an additional CsCl equilibrium centrifugation step was included after removing ethidium bromide.
Nicked circular DNA was prepared by digesting supercoiled DNA in the presence of ethidium bromide with either pancreatic DNase or the restriction endonuclease EcoRI. In Method A, plasmid pMB9 DNA was nicked with pancreatic DNase by a modification of the procedure of Greenfield et al. (15) as previously described (16). Plasmid pBR322 DNA was nicked by a slightly different procedure. The reaction mixture (1.7 ml) contained 97 pg of pBR322 DNA, 50 mM Tris-HC1 (pH 8.0), 50 mM NaC1, 10 mM MgC12, 350 pg/ml ethidium bromide, and 120 units of pancreatic DNase (17). After incubation a t 30 "C for 15 min, the reaction was stopped by the addition of 0.21 ml of 0.4 M EDTA (pH 8) and 0.2 ml of 10% Sarkosyl. Ethidium bromide was removed by ion exchange on a Dowex AG 50W-X8 column. The DNA was extracted twice with phenol and dialyzed exhaustively against 10 mM Tris-HC1 (pH 8), 10 mM NaC1, 1 mM EDTA. Analysis of denatured DNA by electron microscopy indicated that more than 80% of the molecules contained a single nick. In Method B, dimeric plasmid pBR322 was nicked with the restriction enzyme EcoRI as follows. The reaction mixture (10 ml)
contained 100 mM Tris-HC1 (pH 7.5), 50 mM NaC1,5 mM MgCl,, 100 pg/ml bovine serum albumin, 1 mg of pBR322 DNA, 125 pg/ml ethidium bromide, and 1000 units of EcoRI. After incubation at 20 "C for 180 min, the reaction mixture was transferred to 0 "C, and EDTA and Sarkosyl were added to a concentration of 7.5 mM and 0.7%, respectively. Ethidium bromide was removed by ion exchange on a Dowex AG 50W-X8 column equilibrated with 50 mM Tris-HCI (8.0), 7.5 mM EDTA. Fractions containing DNA were pooled recovery of DNA was greater than 95%. DNA was extracted twice with buffer- washed phenol (pH 8) and dialyzed against 1 liter of 10 mM Tris-HCI (pH 8.0), 1 mM EDTA, changed four times over a 48-h period. Analysis of the product by electrophoresis in agarose gels indicated that greater than 90% of the DNA was nicked. Electrophoresis of the heat-denatured product revealed equal quantities of single-stranded circular and linear DNA, indicating that most molecules contained only one nick.
Gel Electrophoresis-Agarose gel electrophoresis of circular plas- mid DNA was performed a t 4 "C in horizontal slabs. The gels con- tained from 0.6 to 1% agarose in electrophoresis buffer (90 mM Tris base, 90 mM boric acid, 2.5 mM disodium EDTA). Ethidium bromide (0.06 pg/ml) was present during analysis of closed circular DNA. The samples (10 to 25 11) contained 7% sucrose, 0.01% bromphenol blue, and 30 mM EDTA. Electrophoresis was carried out overnight a t voltage gradients of 2.5 to 5 V/cm. After electrophoresis, gels were stained in electrophoresis buffer containing 0.5 pg/ml ethidium bro- mide. The DNA bands were illuminated (Corning No. 9863 filter) by fluorescent Westinghouse FS-20 sunlamps and photographed through a Kodak Wratten No. 23A filter using Polaroid No. 55 or 57 film.
Acrylamide gels were cast in the same buffer as agarose gels except for the addition of 25% glycerol. Eight per cent vertical slabs (1.5 mm thick) were composed of a 20:l ratio of acrylamide to N,N'-methylene bisacrylamide. To prepare gels, 100 ml of acrylamide solution were mixed with ammonium persulfate (final concentration, 0.67%), de- gassed, and then mixed with 20 p1 of N,N,N',N'-tetramethylethyl- enediamine. Bands were visualized by autoradiography of a wet gel using Kodak X-Omat AR film.
Isolation and Sequencing of Snapback DNA-The nucleotide se- quence of the snapback DNA in the band labeled GGATG in Fig. 6 was determined as follows. EcoRI-nicked pBR322 DNA (10 pg) was incubated with Form I of T7 DNA polymerase (2 .5 units) in a 0.4-ml reaction mixture containing 50 mM Tris-HCI (pH 7.5), 10 mM MgC12, 10 mM 2-mercaptoethanol, and 150 p~ of each dNTP. After incuba- tion for 45 min at 30 "C, EDTA was added to a final concentration of 10 mM, and the DNA was extracted with phenol followed by chloroform/isoamyl alcohol (24:l). The extracted DNA solution was then concentrated by vacuum evaporation and passed over a Sepha- dex G-100 column. The DNA in the pooled void volume of the column eluate was digested to completion with endonuclease HindIII which cleaves 29 bases distal to the EcoRI site. The DNA was then treated with alkaline phosphatase (0.18 unit) for 45 min a t 65 "C, precipitated with ethanol, and then phosphorylated using T4 polynucleotide ki- nase (40 units) and [y"P]dATP (0.5 mCi, 7000 Ci/mmol) a t 37 "C for 1 h. The reaction was heated in a boiling water bath for 2 min and chilled, sucrose and bromphenol blue were added, and the reac- tion was electrophoresed on a neutral acrylamide gel at room temper- ature for 9 h a t 20 V/cm. The appropriate hand was identified by autoradiography and the DNA was electroeluted from the gel for DNA sequencing by the method of Maxam and Gilbert (18).
Electron Microscopy-DNA samples were mounted for electron microscopy by the formamide-cytochrome c method of Davis et al. (19). In the experiments shown in Table I and Fig. 2, the spreading solution (60 pl) contained either 14.5 pl of a reaction mixture after exonuclease I11 treatment (see Fig. 2) or 10 p1 containing 9 ng of pBR322 DNA (either panhandle or supercoiled) in 40 mM Tris-HC1 (pH 7.8), 5 mM sodium acetate, 1 mM EDTA, brought to 40% formamide, 50 pg/ml cytochrome c, 0.1 M Tris-HCI (pH 8.6), 10 mM EDTA, and 3 pg/ml ethidium bromide. The hypophase was 10 mM Tris-HCl (pH 8.6) containing 10% formamide and 3 pg/ml ethidium bromide. Micrographs were taken on a Zeiss EM10 electron micro- scope.
RESULTS
Identification and Isolation of the Product of DNA Synthesis at Nicks
In the preceding paper (l), we proposed that Form I of T7 DNA polymerase could catalyze the polymerization of nucleo-
by guest on June 13, 2018http://w
ww
.jbc.org/D
ownloaded from

11176 Strand Displacement Synthesis by T7 DNA Polymeraye
tides using the 3’-hydroxyl group at a nick as a primer. We interpreted the ability of Form I to destroy a nick in DNA as a substrate for DNA ligase as indicating that this form of the enzyme can catalyze such a reaction since this reaction was dependent upon the addition of dNTPs, thus providing evi- dence for the displacement of the 5’ terminus at a nick from its normal double helical conformation. Form 11, on the other hand, does not catalyze such a reaction and thus resembles T4 DNA polymerase, a well documented example of a poly- merase that does not catalyze strand displacement synthesis (5). As shown in the previous paper and as will be documented more quantitatively below, the amount of DNA synthesis catalyzed by Form I a t nicks is very low. However, even very limited strand displacement synthesis on singly nicked, cir- cular templates markedly changes their physical properties. We have taken advantage of this observation to separate the product of strand displacement synthesis from nicked tem- plates in order to study its structure and properties.
Detection of the Product by Gel Electrophoresis in the Pres- ence of Ethidium Bromide-The topological constraint in covalently closed circular DNA molecules prevents a change in the number of rotations of one strand about the other. Thus, ethidium bromide causes a positive shift in the super- helical winding number of these DNA molecules since it unwinds the DNA helix upon intercalation. I t has been shown that at subsaturating concentrations of ethidium bromide the relative electrophoretic mobilities of covalently closed circular SV40 DNA molecules having different numbers of superheli- cal turns before binding ethidium bromide are largely depend- ent upon the number of superhelical turns that result after binding of ethidium bromide (20). In the experiments de- scribed here, the ethidium bromide concentration (0.06 pg/ ml) causes binding of less than 0.01 mol of dye/mol of nucleo- tide to nicked circular DNA and relaxed covalently closed DNA and less than 0.10 mol of dye/mol of nucleotide to negatively supercoiled DNA of less than 50 superhelical turns (21). Under these conditions, as shown in Fig. 1, negatively supercoiled pMB9 DNA migrates considerably faster than circular pMB9 DNA containing a single nick (lane I ) . Relaxed covalently closed pMB9 DNA, prepared by incubation of nicked pMB9 DNA with DNA ligase, migrates faster than supercoiled DNA (lane 2).
When the nicked pMB9 DNA is used as a template for Form I of T 7 DNA polymerase, one product of the DNA synthesis reaction can be identified by its mobility during electrophoresis on agarose gels in the presence of ethidium bromide. This product is the band labeled X in lane 3 of Fig. 1. It migrates slightly slower than relaxed, covalently closed circles, but faster than naturally occurring, negatively super- coiled DNA. The sample shown in lane 3 was incubated with DNA ligase prior to electrophoresis. Therefore, all of the nicked DNA must have been used as template-primer by Form I of T 7 DNA polymerase since no DNA is observed at the position of relaxed covalently closed molecules, the product of ligase sealing of nicked circles. The product of DNA syn- thesis by Form I is produced in the same amounts even if DNA ligase and ATP are present at the onset of the reaction (data not shown).
Form I1 of T 7 DNA polymerase, as expected from the results presented in the previous paper (l), does not produce a product that can be identified by gel electrophoresis. After incubation with Form 11, the nicked pMB9 DNA is largely converted to covalently closed circles by incubation with DNA ligase (Fig. 1, lane 4 ) ; no species X is observed. Although not shown in Fig. 1, in the absence of ligase, no unique DNA product is observed and the addition of an equal number of units of Form I1 to a reaction mixture containing Form I did
1 2 3 4
NM- i h
of DNA synthesis by Form I of T7 DNA polymerase. Each FIG. 1. Agarose gel electrophoretic analysis of the product
reaction mixture (10 P I ) contained 0.1 fig of nicked pMB9 DNA, 40 mM Tris-HCI (pH 7.5), 20 mM MgCI2, 20 mM 2-mercaptoethanol, 300 p~ each of dCTP, dATP, dCTP, and dTTP, and 150 PM ATP. After the mixtures were incubated at 30 “C for 50 min in the presence of T7 DNA polymerase (0.024 unit), T7 DNA ligase (0.003 unit) was added to reaction mixtures 2, 3, and 4, and incubation continued for 30 additional min. Each reaction mixture was electrophoresed through a 0.8% agarose gel containing 0.06 pg/ml of ethidium bro- mide. Staining and photography were as described under “Experi- mental Procedures.” Lane 1, no DNA polymerase or DNA ligase; fane 2, T7 DNA ligase but no prior incubation with DNA polymerase; fane 3, Form I of T7 DNA polymerase followed by incubation with DNA ligase; fane 4, Form I1 of T7 DNA polymerase followed by incubation with T7 DNA ligase. The expected locations of several DNA species are indicated as follows: ND, nicked, circular dimer; NM, nicked, circular monomer; LM, linear monomer; RD, relaxed, covalently closed, circular dimer; SM, negatively supercoiled, circular monomer; RM, relaxed, covalently closed, circular monomer. The product of synthesis in the reaction catalyzed by Form I of T7 DNA polymerase is marked X .
not greatly reduce the production of species X . Electron Microscopic Analysis of the Product-The proper-
ties of the product of synthesis catalyzed by Form I on nicked circles suggested that i t was topologically constrained, yet it seemed unlikely that the molecule could be covalently closed since it could be formed in the absence of DNA ligase. In order to further characterize the product, the DNA labeled X in Fig. 1 was eluted from a gel and examined by electron microscopy after removal of ethidium bromide. All of the molecules observed were circular duplexes, and approximately 40% had a single branch arising from the circle; the lengths of the branches were too short for accurate measurement but most were less than 5% of the length of the 4362-bp2 pBR322 molecule. Four examples of typical branched circular mole- cules are shown in Fig. 2A. We refer to the branch as a “panhandle.”
In order to confirm that the product molecules isolated from the gel are indeed topologically constrained, the DNA was also prepared for electron microscopy in the presence of an amount of ethidium bromide known to be sufficient to
The abbreviation used is: bp, base pair.
by guest on June 13, 2018http://w
ww
.jbc.org/D
ownloaded from

Strand Displacement Synthesis by T7 DNA Polymerase 11177
create positive supercoiling of covalently closed, circular DNA molecules (22). Of the molecules observed, 76% appeared to be supercoiled molecules as shown in Fig. 2B. These topolog- ically constrained, circular molecules are not covalently closed since the constraint is removed by incubation with E. coli exonuclease 111. Exonuclease I11 is specific for double- stranded DNA and hydrolyzes phosphodiester bonds sequen- tially from the 3' end (23). After limited treatment with exonuclease III,83% of the product DNA molecules appeared relaxed when analyzed by electron microscopy with ethidium bromide (Fig. 2C, Table I). Incubation with exonuclease I11 was without effect on the covalently closed supercoiled pBR322 DNA molecules.
These results, taken together, show that the product mole- cules arising from DNA synthesis at a nick on a circular DNA molecule are not covalently closed. We conclude that the DNA circles are topologically closed by virtue of having a double-stranded branch. This branch, which we call a pan- handle, connects the 3' and 5' termini of the interrupted DNA strands in these molecules, thereby establishing a con- stant topological winding number (21).
FIG. 2. Electron microscopic analysis of the product of DNA synthesis catalyzed by Form I of T7 DNA polymerase. pBR322 DNA (0.6 pg) containing approximately 1 nick/molecule was incu- bated in a 60-pl reaction mixture containing 40 mM Tris-HC1 (pH 7.5). 20 mM MgC12, 20 mM 2-mercaptoethanol, 300 p M each of dGTP, M T P , dCTP, and dTTP, and 0.15 unit of Form I of T7 DNA polymerase. After incubation at 30 "C for 50 min, electrophoresis of the products of the reaction was carried out in a 0.8% agarose gel containing ethidium bromide (see "Experimental Procedures"). The product with the highest rate of migration on an agarose gel (the DNA corresponding to band X in Fig. 1) was electroeluted from the gel and dialyzed extensively to remove ethidium bromide (Sample A ) . Sufficie'nt ethidium bromide (3 pg/ml) to produce positive supertwist- ing was added to a portion of the DNA (Sample B). A third portion of the dialyzed DNA was incubated with exonuclease I11 to yield approximately 5% acid-soluble material (Sample C). The reaction mixture (13 pl) contained 6 ng of DNA, 5 mM MgC12, 10 mM 2- mercaptoethanol, and 0.013 unit of E. coli exonuclease 111. Incubation was at 37 "C for 30 min. The reaction was stopped by the addition of 1.5 pl of 0.4 M EDTA and the DNA was prepared for electron microscopy in the presence of ethidium bromide. A , DNA product in the absence of ethidium bromide; B, DNA product in the presence of ethidium bromide; C, exonuclease 111-treated DNA product in the presence of ethidium bromide.
TABLE I Release of topological constraint of branched circular product by
treatment with E. coli exonuclease III Singly nicked pBR322 DNA was incubated with all four dNTPs
and Form I of T7 DNA polymerase. Closed circular DNA product was purified by gel electrophoresis and incubated with E. coli exonu- clease I11 under conditions sufficient to cause approximately 200 nucleotides to be hydrolyzed per circular DNA molecule, as inferred from an incubation with 3H-labeled T7 DNA. The experimental reaction and control, which contained supercoiled pBR322 DNA instead of branched product, were mixed with 3 pg/ml ethidium bromide and mounted for electron microscopy. Circular DNA mole- cules were scored as either twisted or untwisted, and the data were normalized to 100%. Examples of twisted and untwisted branched circular molecules are shown in Fig. 2. Samples of a t least 100 molecules were counted to obtain these percentages.
DNA Nk!i:t- with exonu- Conversion" Incubation
clease I11 % twisted %
Branched product 76 13 83 pBR322 supercoil 88 87 1
Per cent conversion = 100 X ( F , - Fo) / ( l - Fo), where FI is the fraction of linear molecules after treatment with T7 DNA polymerase and Fo is the fraction of linear molecules before treatment.
3 DNA
a b c snapbock
FIG. 3. Schematic representation of the analysis of the product of DNA synthesis catalyzed by Form I of T7 DNA polymerase at nicks. A , pBR3222 DNA singly nicked with EcoRI serves as a primer-template for Form I of T7 DNA polymerase. DNA synthesis along the circular template strand gives rise to DNA having the sequence abc. Template switching allows the polymerase to copy the displaced single strand to give rise to a molecule bearing a duplex branch (panhandle), the newly synthesized strand having the se- quence c'b'a'. B, incubation of the product molecules with EcoRI eliminates the covalent attachment of newly synthesized DNA to the primer template. C, subsequent denaturation by heat or alkali disrupts the hydrogen bonding and releases the newly synthesized DNA. The self-complementary sequences in the newly synthesized DNA allow the DNA to renature rapidly to form a hairpin structure (snapback DNA).
Analysis of the Structure of the Newly Synthesized DNA The most likely structure that would account for the prop-
erties of the product synthesized by Form I of T7 DNA polymerase is a duplex branch on the circular template (Fig. 3, Structure A ) . Such a branch may arise by strand displace- ment during DNA synthesis from the nick followed by strand switching and polymerization on the displaced single strand. In order to characterize the product further, we have taken
by guest on June 13, 2018http://w
ww
.jbc.org/D
ownloaded from
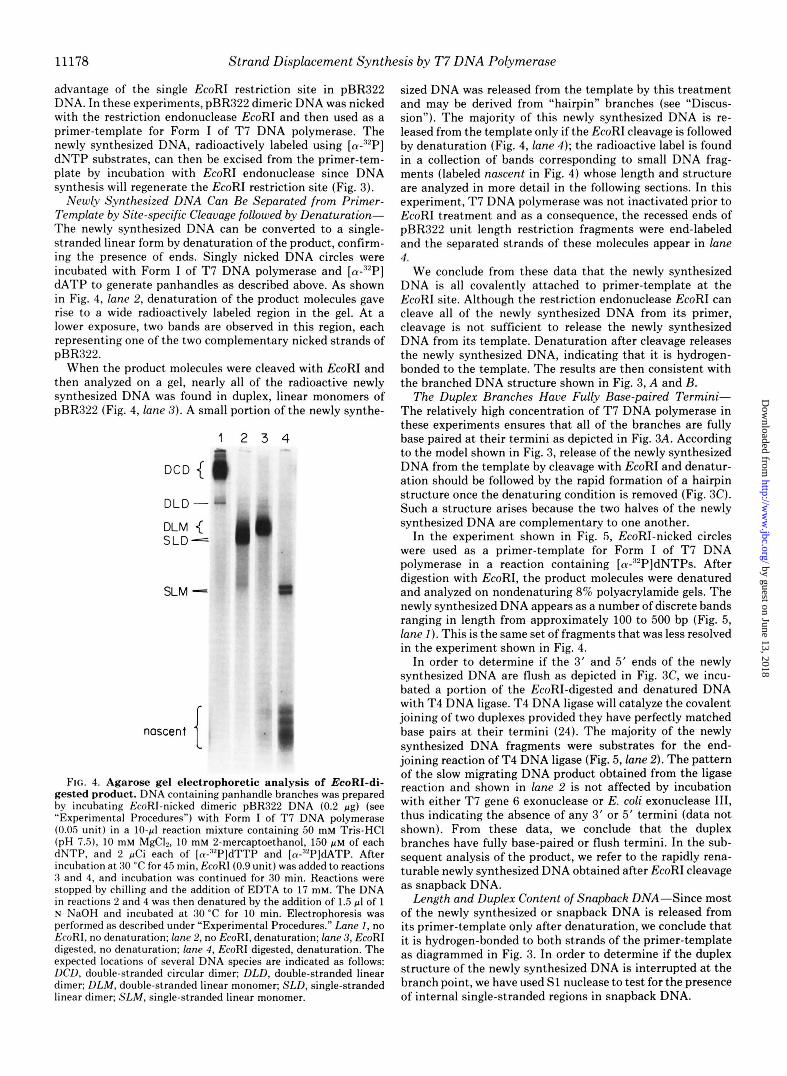
11178 Strand Displacement Synthesis by T7 DNA Polymerase
advantage of the single EcoRI restriction site in pBR322 DNA. In these experiments, pBR322 dimeric DNA was nicked with the restriction endonuclease EcoRI and then used as a primer-template for Form I of T 7 DNA polymerase. The newly synthesized DNA, radioactively labeled using [cY-’~P] dNTP substrates, can then be excised from the primer-tem- plate by incubation with EcoRI endonuclease since DNA synthesis will regenerate the EcoRI restriction site (Fig. 3).
Newly Synthesized DNA Can Be Separated from Primer- Template by Site-specific Cleavage followed by Denaturation- The newly synthesized DNA can be converted to a single- stranded linear form by denaturation of the product, confirm- ing the presence of ends. Singly nicked DNA circles were incubated with Form I of T 7 DNA polymerase and [a-“P] dATP to generate panhandles as described above. As shown in Fig. 4, lane 2, denaturation of the product molecules gave rise to a wide radioactively labeled region in the gel. At a lower exposure, two bands are observed in this region, each representing one of the two complementary nicked strands of pBR322.
When the product molecules were cleaved with EcoRI and then analyzed on a gel, nearly all of the radioactive newly synthesized DNA was found in duplex, linear monomers of pBR322 (Fig. 4, lane 3) . A small portion of the newly synthe-
1 2 3 4
DCD
DLD
DLM SLD -
SLM *
nascent
c I
FIG. 4. Agarose gel electrophoretic analysis of EcoRI-di- gested product. DNA containing panhandle branches was prepared by incubating EcoRI-nicked dimeric pBR322 DNA (0.2 pg) (see “Experimental Procedures”) with Form I of T7 DNA polymerase (0.05 unit) in a 10-pl reaction mixture containing 50 mM Tris-HCI (pH 7.5), 10 mM MgCI,, 10 mM 2-mercaptoethanol, 150 pM of each dNTP, and 2 pCi each of [a-‘*P]dTTP and [a-”PldATP. After incubation at 30 “C for 45 min, EcoRI (0.9 unit) was added to reactions 3 and 4, and incubation was continued for 30 min. Reactions were stopped by chilling and the addition of EDTA to 17 mM. The DNA in reactions 2 and 4 was then denatured by the addition of 1.5 pl of 1 N NaOH and incubated at 30 “C for 10 min. Electrophoresis was performed as described under “Experimental Procedures.” Lane 1 , no EcoRI, no denaturation; lane 2, no EcoRI, denaturation; lane 3, EcoRI digested, no denaturation; lane 4, EcoRI digested, denaturation. The expected locations of several DNA species are indicated as follows: DCD, double-stranded circular dimer; DLD, double-stranded linear dimer; DLM, double-stranded linear monomer; SLD, single-stranded linear dimer; SLM, single-stranded linear monomer.
sized DNA was released from the template by this treatment and may be derived from “hairpin” branches (see “Discus- sion”). The majority of this newly synthesized DNA is re- leased from the template only if the EcoRI cleavage is followed by denaturation (Fig. 4, lane 4); the radioactive label is found in a collection of bands corresponding to small DNA frag- ments (labeled nascent in Fig. 4) whose length and structure are analyzed in more detail in the following sections. In this experiment, T 7 DNA polymerase was not inactivated prior to EcoRI treatment and as a consequence, the recessed ends of pBR322 unit length restriction fragments were end-labeled and the separated strands of these molecules appear in lane 4.
We conclude from these data that the newly synthesized DNA is all covalently attached to primer-template at the EcoRI site. Although the restriction endonuclease EcoRI can cleave all of the newly synthesized DNA from its primer, cleavage is not sufficient to release the newly synthesized DNA from its template. Denaturation after cleavage releases the newly synthesized DNA, indicating that it is hydrogen- bonded to the template. The results are then consistent with the branched DNA structure shown in Fig. 3, A and B.
The Duplex Branches Have Fully Base-paired Termini- The relatively high concentration of T 7 DNA polymerase in these experiments ensures that all of the branches are fully base paired at their termini as depicted in Fig. 3A. According to the model shown in Fig. 3, release of the newly synthesized DNA from the template by cleavage with EcoRI and denatur- ation should be followed by the rapid formation of a hairpin structure once the denaturing condition is removed (Fig. 3C). Such a structure arises because the two halves of the newly synthesized DNA are complementary to one another.
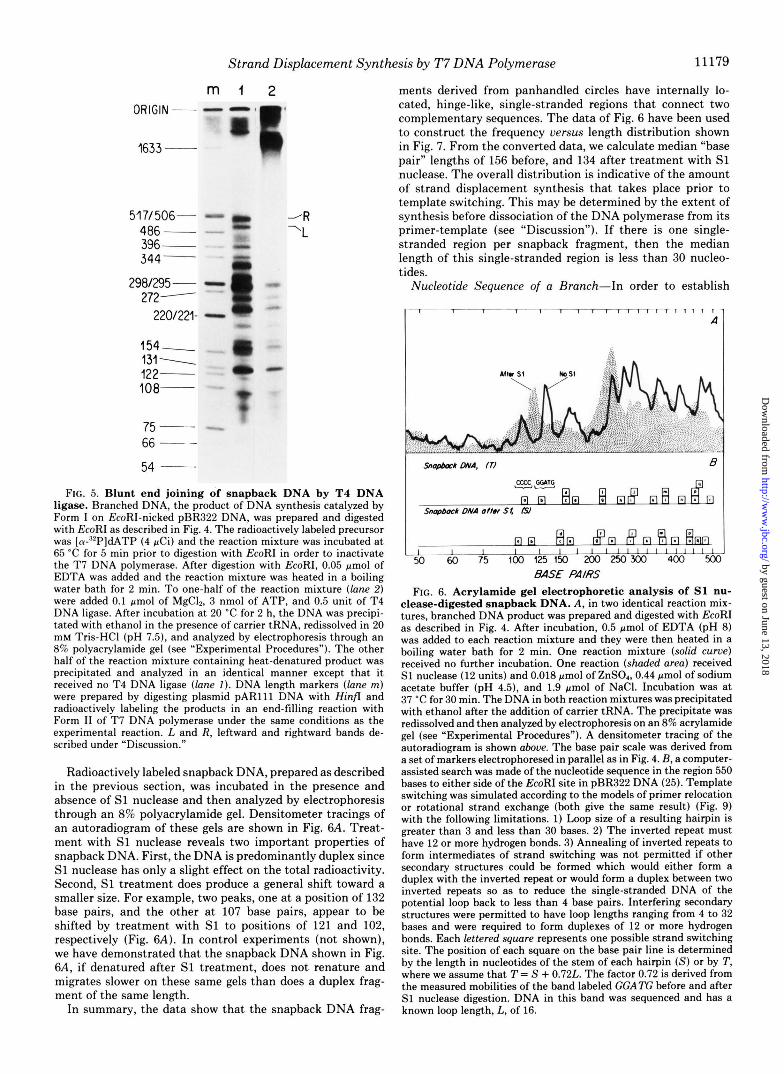
In the experiment shown in Fig. 5, EcoRI-nicked circles were used as a primer-template for Form I of T 7 DNA polymerase in a reaction containing [a-”PIdNTPs. After digestion with EcoRI, the product molecules were denatured and analyzed on nondenaturing 8% polyacrylamide gels. The newly synthesized DNA appears as a number of discrete bands ranging in length from approximately 100 to 500 bp (Fig. 5, lane 1 ) . This is the same set of fragments that was less resolved in the experiment shown in Fig. 4.
In order to determine if the 3’ and 5’ ends of the newly synthesized DNA are flush as depicted in Fig. 3C, we incu- bated a portion of the EcoRI-digested and denatured DNA with T4 DNA ligase. T4 DNA ligase will catalyze the covalent joining of two duplexes provided they have perfectly matched base pairs at their termini (24). The majority of the newly synthesized DNA fragments were substrates for the end- joining reaction of T4 DNA ligase (Fig. 5, lane 2). The pattern of the slow migrating DNA product obtained from the ligase reaction and shown in lane 2 is not affected by incubation with either T 7 gene 6 exonuclease or E. coli exonuclease 111, thus indicating the absence of any 3’ or 5’ termini (data not shown). From these data, we conclude that the duplex branches have fully base-paired or flush termini. In the sub- sequent analysis of the product, we refer to the rapidly rena- turable newly synthesized DNA obtained after EcoRI cleavage as snapback DNA.
Length and Duplex Content of Snapback DNA-Since most of the newly synthesized or snapback DNA is released from its primer-template only after denaturation, we conclude that it is hydrogen-bonded to both strands of the primer-template as diagrammed in Fig. 3. In order to determine if the duplex structure of the newly synthesized DNA is interrupted at the branch point, we have used S1 nuclease to test for the presence of internal single-stranded regions in snapback DNA.
by guest on June 13, 2018http://w
ww
.jbc.org/D
ownloaded from
Strand Displacement Synthesis by T7 DNA Polymerase 11179
m i 2 ORIGIN - a
1633 - w
517/506- -R 486 - ’L 396 - 344 -
2981295- - 2201221 -
272-
154- 131 1
122- 108-
75 ”
66 - -
54 -
L
FIG. 5. Blunt end joining of snapback DNA by T4 DNA ligase. Branched DNA, the product of DNA synthesis catalyzed by Form I on EcoRI-nicked pBR322 DNA, was prepared and digested with EcoRI as described in Fig. 4. The radioactively labeled precursor was [a-“PldATP (4 pCi) and the reaction mixture was incubated at 65 “C for 5 min prior to digestion with EcoRI in order to inactivate the T7 DNA polymerase. After digestion with EcoRI, 0.05 pmol of EDTA was added and the reaction mixture was heated in a boiling water bath for 2 min. To one-half of the reaction mixture ( l a n e 2) were added 0.1 pmol of MgC12, 3 nmol of ATP, and 0.5 unit of T4 DNA ligase. After incubation at 20 “C for 2 h, the DNA was precipi- tated with ethanol in the presence of carrier tRNA, redissolved in 20 mM Tris-HCI (pH 7.5), and analyzed by electrophoresis through an 8% polyacrylamide gel (see “Experimental Procedures”). The other half of the reaction mixture containing heat-denatured product was precipitated and analyzed in an identical manner except that it received no T4 DNA ligase (lane I ) . DNA length markers ( l a n e m) were prepared by digesting plasmid PAR111 DNA with Hinfr and radioactively labeling the products in an end-filling reaction with Form I1 of T7 DNA polymerase under the same conditions as the experimental reaction. L and R, leftward and rightward bands de- scribed under “Discussion.”
Radioactively labeled snapback DNA, prepared as described in the previous section, was incubated in the presence and absence of S1 nuclease and then analyzed by electrophoresis through an 8% polyacrylamide gel. Densitometer tracings of an autoradiogram of these gels are shown in Fig. 6A. Treat- ment with S1 nuclease reveals two important properties of snapback DNA. First, the DNA is predominantly duplex since S1 nuclease has only a slight effect on the total radioactivity. Second, S1 treatment does produce a general shift toward a smaller size. For example, two peaks, one at a position of 132 base pairs, and the other at 107 base pairs, appear to be shifted by treatment with S1 to positions of 121 and 102, respectively (Fig. 6A). In control experiments (not shown), we have demonstrated that the snapback DNA shown in Fig. 6A, if denatured after S1 treatment, does not renature and migrates slower on these same gels than does a duplex frag- ment of the same length.
In summary, the data show that the snapback DNA frag-
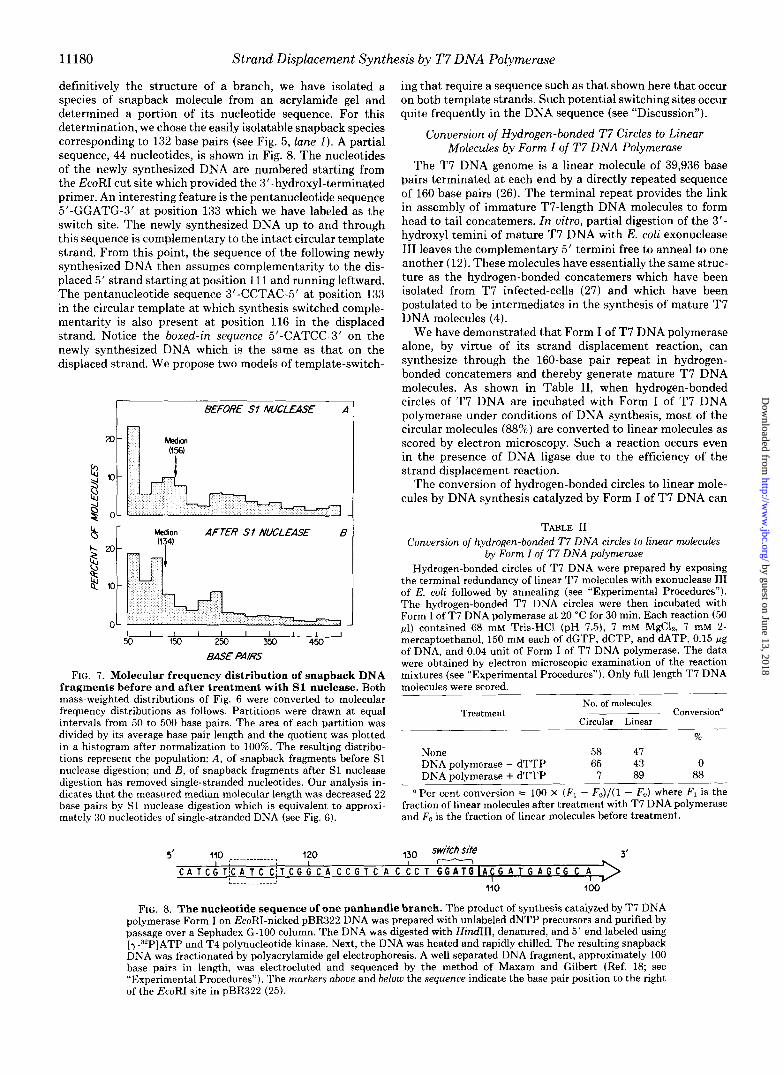
ments derived from panhandled circles have internally lo- cated, hinge-like, single-stranded regions that connect two complementary sequences. The data of Fig. 6 have been used to construct the frequency versus length distribution shown in Fig. 7. From the converted data, we calculate median “base pair” lengths of 156 before, and 134 after treatment with S1 nuclease. The overall distribution is indicative of the amount of strand displacement synthesis that takes place prior to template switching. This may be determined by the extent of synthesis before dissociation of the DNA polymerase from its primer-template (see “Discussion”). If there is one single- stranded region per snapback fragment, then the median length of this single-stranded region is less than 30 nucleo- tides.
Nucleotide Sequence of a Branch-In order to establish
9 - h DNd. IN I9
I I I I I I I 1 1 1 1 1 1 1 1 1 L &I $0 75 1 0 0 125 1 5 0 200 250300 400 5oc
BAS€ PAIRS FIG. 6. Acrylamide gel electrophoretic analysis of S1 nu-
clease-digested snapback DNA. A, in two identical reaction mix- tures, branched DNA product was prepared and digested with EcoRI as described in Fig. 4. After incubation, 0.5 pmol of EDTA (pH 8) was added to each reaction mixture and they were then heated in a boiling water bath for 2 min. One reaction mixture (solid curue) received no further incubation. One reaction (shaded area) received S1 nuclease (12 units) and 0.018 pmol of ZnSO,, 0.44 pmol of sodium acetate buffer (pH 4.5), and 1.9 pmol of NaCI. Incubation was at 37 “C for 30 min. The DNA in both reaction mixtures was precipitated with ethanol after the addition of carrier tRNA. The precipitate was redissolved and then analyzed by electrophoresis on an 8% acrylamide gel (see “Experimental Procedures”). A densitometer tracing of the autoradiogram is shown above. The base pair scale was derived from a set of markers electrophoresed in parallel as in Fig. 4. B, a computer- assisted search was made of the nucleotide sequence in the region 550 bases to either side of the EcoRI site in pBR322 DNA (25). Template switching was simulated according to the models of primer relocation or rotational strand exchange (both give the same result) (Fig. 9) with the following limitations. 1) Loop size of a resulting hairpin is greater than 3 and less than 30 bases. 2) The inverted repeat must have 12 or more hydrogen bonds. 3) Annealing of inverted repeats to form intermediates of strand switching was not permitted if other secondary structures could be formed which would either form a duplex with the inverted repeat or would form a duplex between two inverted repeats so as to reduce the single-stranded DNA of the potential loop back to less than 4 base pairs. Interfering secondary structures were permitted to have loop lengths ranging from 4 to 32 bases and were required to form duplexes of 12 or more hydrogen bonds. Each lettered sgwre represents one possible strand switching site. The position of each square on the base pair line is determined by the length in nucleotides of the stem of each hairpin (S) or by T, where we assume that T = S + 0.72L. The factor 0.72 is derived from the measured mobilities of the band labeled GGATG before and after S1 nuclease digestion. DNA in this band was sequenced and has a known loop length, L, of 16.
by guest on June 13, 2018http://w
ww
.jbc.org/D
ownloaded from
11180 Strand Displacement Synthesis by T7 DNA Polymerase
definitively the structure of a branch, we have isolated a species of snapback molecule from an acrylamide gel and determined a portion of its nucleotide sequence. For this determination, we chose the easily isolatable snapback species corresponding to 132 base pairs (see Fig. 5, lune I). A partial sequence, 44 nucleotides, is shown in Fig. 8. The nucleotides of the newly synthesized DNA are numbered starting from the EcoRI cut site which provided the 3”hydroxyl-terminated primer. An interesting feature is the pentanucleotide sequence 5’-GGATG-3’ at position 133 which we have labeled as the switch site. The newly synthesized DNA up to and through this sequence is complementary to the intact circular template strand. From this point, the sequence of the following newly synthesized DNA then assumes complementarity to the dis- placed 5’ strand starting at position 111 and running leftward. The pentanucleotide sequence 3’-CCTAC-5’ at position 133 in the circular template at which synthesis switched comple- mentarity is also present a t position 116 in the displaced strand. Notice the boxed-in sequence 5’-CATCC-3’ on the newly synthesized DNA which is the same as that on the displaced strand. We propose two models of template-switch-
I BEFORE Sf NUCLEASE A I
I 1 1 1 I I l l l I 50 150 250 350 450
BASE PAIRS
FIG. 7. Molecular frequency distribution of snapback DNA fragments before and after treatment with S1 nuclease. Both mass-weighted distributions of Fig. 6 were converted to molecular frequency distributions as follows. Partitions were drawn a t equal intervals from 50 to 500 base pairs. The area of each partition was divided by its average base pair length and the quotient was plotted in a histogram after normalization to 100%. The resulting distribu- tions represent the population: A, of snapback fragments before S1 nuclease digestion; and B, of snapback fragments after S1 nuclease digestion has removed single-stranded nucleotides. Our analysis in- dicates that the measured median molecular length was decreased 22 base pairs by S1 nuclease digestion which is equivalent to approxi- mately 30 nucleotides of single-stranded DNA (see Fig. 6).
ing that require a sequence such as that shown here that occur on both template strands. Such potential switching sites occur quite frequently in the DNA sequence (see “Discussion”).
Conversion of Hydrogen-bonded T7 Circles to Linear Molecules by Form I of T7 DNA Polymerase
The T7 DNA genome is a linear molecule of 39,936 base pairs terminated at each end by a directly repeated sequence of 160 base pairs (26). The terminal repeat provides the link in assembly of immature T7-length DNA molecules to form head to tail concatemers. In uitro, partial digestion of the 3‘- hydroxyl temini of mature T7 DNA with E. coli exonuclease I11 leaves the complementary 5’ termini free to anneal to one another (12). These molecules have essentially the same struc- ture as the hydrogen-bonded concatemers which have been isolated from T7 infected-cells (27) and which have been postulated to be intermediates in the synthesis of mature T7 DNA molecules (4).
We have demonstrated that Form I of T7 DNA polymerase alone, by virtue of its strand displacement reaction, can synthesize through the 160-base pair repeat in hydrogen- bonded concatemers and thereby generate mature T7 DNA molecules. As shown in Table 11, when hydrogen-bonded circles of T7 DNA are incubated with Form I of T7 DNA polymerase under conditions of DNA synthesis, most of the circular molecules (88%) are converted to linear molecules as scored by electron microscopy. Such a reaction occurs even in the presence of DNA ligase due to the efficiency of the strand displacement reaction.
The conversion of hydrogen-bonded circles to linear mole- cules by DNA synthesis catalyzed by Form I of T7 DNA can
TABLE I 1 Conversion of hydrogen-bonded T7 DNA circles to linear molecules
by Form I of T7 DNA polymerase Hydrogen-bonded circles of T7 DNA were prepared by exposing
the terminal redundancy of linear T 7 molecules with exonuclease 111 of E. coli followed by annealing (see “Experimental Procedures”). The hydrogen-bonded T7 DNA circles were then incubated with Form I of T7 DNA polymerase a t 20 “C for 30 min. Each reaction (50 @I) contained 68 mM Tris-HCl (pH 7.5), 7 mM MgCl,, 7 mM 2- mercaptoethanol, 150 mM each of dGTP, dCTP, and UTP, 0.15 Fg of DNA, and 0.04 unit of Form I of T7 DNA polymerase. The data were obtained by electron microscopic examination of the reaction mixtures (see “Experimental Procedures”). Only full length T 7 DNA molecules were scored.
No. of molecules
Circular Linear Treatment Conversion“
%
None 58 47 DNA polymerase - dTTP 65 43 0 DNA polymerase + dTTP 7 89 88
“Per cent conversion = 100 x (F1 - Fo)/(l - Fo) where FI is the fraction of linear molecules after treatment with T7 DNA polymerase and Fo is the fraction of linear molecules before treatment.
,?o swifcb site - 3’ > L ”””””. 2 A
110 I00
FIG. 8. The nucleotide sequence of one panhandle branch. The product of synthesis catalyzed by T7 DNA polymerase Form I on EcoRI-nicked pBR322 DNA was prepared with unlabeled dNTP precursors and purified by passage over a Sephadex G-100 column. The DNA was digested with HindIII, denatured, and 5’ end labeled using [y-32P]ATP and T4 polynucleotide kinase. Next, the DNA was heated and rapidly chilled. The resulting snapback DNA was fractionated by polyacrylamide gel electrophoresis. A well separated DNA fragment, approximately 100 base pairs in length, was electroeluted and sequenced by the method of Maxam and Gilbert (Ref. 18; see “Experimental Procedures”). The markers above and below the sequence indicate the base pair position to the right of the EcoRI site in pBR322 (25).
by guest on June 13, 2018http://w
ww
.jbc.org/D
ownloaded from

Strand Displacement Synthesis by T7 D N A Polymerase 11 181
be described as occurring in two steps. First, the polymerase extends the 3”hydroxyl terminus generated by exonuclease I11 treatment, using the single-stranded gap as template. When polymerization reaches the terminal redundancy, the enzyme encounters duplex template. Form I of T7 DNA polymerase continues DNA synthesis, displaces the 5’-ter- minated strand, and converts the circle to a linear form. Since the DNA polymerase does not specifically stop when it reaches the region of duplex template (i.e. creates a nick), DNA ligation does not compete with the conversion.
DISCUSSION
Strand Displacement-We have shown that Form I of T7 DNA polymerase catalyzes strand displacement synthesis on nicked, duplex DNA templates, a reaction that is character- istic of other, but not all, DNA polymerases. For example, DNA polymerase I of E. coli catalyzes strand displacement synthesis at nicks, but only after its 5’ to 3‘ exonuclease activity has removed 20 to 50 nucleotides (5). The 5’ to 3’ hydrolytic activity is not, however, a prerequisite for strand displacement since elimination of the exonuclease activity does not prevent this reaction ( 5 ) . Bacteriophage T5 DNA polymerase also catalyzes strand displacement synthesis (28), but it incorporates far more nucleotides (approximately 10,00O/nick) than does Form I of T 7 DNA polymerase. On the other hand, DNA polymerases I1 and 111 of E. coli, in the absence of other proteins, are limited to gap filling as is the T4 DNA polymerase ( 5 ) .
The mechanistic features required for strand displacement to occur during synthesis catalyzed by a DNA polymerase are not known. However, T7 DNA polymerase offers a unique opportunity to study this reaction since the enzyme can exist in two forms, only one of which can catalyze strand displace- ment synthesis. Since the secondary structure of duplex DNA in solution is dynamic (29), it does not appear unreasonable to assume that either form of T 7 DNA polymerase would have a propensity to synthesize DNA from a nick. Instead of forcing open the helix during polymerization, the polymerase might wait for thermally induced hydrogen bond breakage and/or base pair unstacking. The high rate of branch migra- tion gives credence to this idea (30).
As shown in the following paper (31), Form I1 of T7 DNA polymerase cannot catalyze strand displacement synthesis even if presented with a preformed displaced strand. If strand displacement synthesis can occur without actively forcing open the DNA duplex, then it seems likely that Form I1 has a mechanism by which it prevents this reaction. In this regard, its double-stranded 3’ to 5’ DNA exonuclease activity could be an important part of a mechanism designed to prevent strand displacement. By the same logic, Form I of T7 DNA polymerase, whose exonuclease activity has been subdued, would be capable of catalyzing strand displacement. Both the single- and double-stranded DNA exonuclease activities of Form I1 are at least 30-fold higher than those of Form I. As yet we do not know what chemical change converts Form I1 to Form I. Relevant to this topic is the fact that the gene 5 protein holds the active site of the 3’ to 5’ single-stranded DNA-specific exonuclease (32,33). We do not know, however, whether the double-stranded DNA-specific exonuclease, which apparently involves both subunits, shares part of its active site with the single-stranded DNA exonuclease.
Template Switching-The fact that duplex branches are the final product of strand displacement synthesis implies that a switch from the original template strand to the complemen- tary displaced strand has occurred. The duplex branches found with Form I of T7 DNA polymerase are similar to,
albeit shorter than, those found in the product synthesized by E. coli DNA polymerase I (5) and T5 DNA polymerase (28). The studies we have described here with T 7 DNA polymerase provide two new facts regarding the mechanism of template switching. First, from the nucleotide sequence, we find that a short stretch of single-stranded DNA exists at the branch point. This observation is not compatible with the model suggested by Kornberg (34) in which the polymerase trans- gresses from one strand to another without a contortion of the DNA. Second, the point at which synthesis switches from one strand to another is not random. Rather, it occurs at a site where a sequence on the original template strand is repeated on the displaced strand.
Our data are consonant with either of two alternative models for template switching which we shall call primer relocation and rotational strand exchange. Essential to both mechanisms depicted in Fig. 9 is the presence of a pair of short inverted repeat sequences separated by a variable num- ber of nucleotides. The number of nucleotides between the inverted repeats determines the length of the single-stranded region at the branch point. The molecular mechanism by which strand switching occurs is different between the two models. In the case of primer relocation, the sequence of one inverted repeat at the 3‘ end of the growing strand anneals to the complementary sequence of the other inverted repeat on the displaced strand. In rotational strand exchange, the ap- propriate DNA intermediate arises by annealing of the se- quences of the two inverted repeats within the displaced strand alone.
The individual steps involved in strand switching in the primer relocation model (Fig. 9) are as follows. 1) DNA polymerase binds to the 3”hydroxyl terminus at a nick and initiates the polymerization of nucleotides. 2) DNA polymer- ase proceeds through the two inverted repeats, but dissociates from the growing strand after copying the second inverted repeat sequence. It should be noted that T7 DNA polymerase, in the absence of other replication proteins, is not highly processive and we assume that the dissociation event occurs randomly during synthesis. 3) Branch migration occurs, un- wrapping the growing strand. 4) The inverted repeat at the 3’ end of the now single-stranded growing strand anneals to the complementary sequence on the 5”displaced strand to form a panhandle. Alternatively, it could anneal to the same se- quence on its own strand to form a hairpin branch. 5) Finally, the reannealed 3”hydroxyl end of the growing strand is used as a primer for T7 DNA polymerase where it completes the duplex branch. In Fig. lOA, we have depicted the structure of the final intermediate of the primer relocation reaction using the sequence of the branch determined in this study.
Template switching by rotational strand exchange is also shown schematically in Fig. 9. This mechanism involves the following steps, some of which are shared by the previous model. 1) As before, DNA polymerase initiates polymerization from the 3’-hydroxyl terminus of the nick. 2) After proceeding through the second inverted repeat, the polymerase disso- ciates from the growing strand. 3) At this point, the nucleotide sequences of both inverted repeats on the displaced strand have been exposed and can anneal to form a hairpin-like structure. 4) Rotation of the helical arms may make possible the alignment of the growing 3”hydroxyl-terminated strand with the displaced strand at the point of the inverted repeat annealing. 5) Finally, the 3’-hydroxyl end of the growing strand is extended, using the displaced strand as template to create a crossed strand exchange between two homologous duplex DNA segments. Such an intermediate structure is depicted in Fig. 10B using the nucleotide sequence of the branch determined in this study. In the vicinity of the cross-
by guest on June 13, 2018http://w
ww
.jbc.org/D
ownloaded from

11182 Strand Displacement Synthesis by T7 D N A Polymerase
nick
m+dNTP’s
ROTATIONAL STRAND EXCHANGE
or
panhandle
A 8 #A’
FIG. 9. Schematic representation of mechanisms of switching. As discussed in the text, thermally induced opening of the DNA helix may facilitate strand displacement synthesis at a nick. T7 DNA polymerase Form I cycles on and off the DNA, incorporating one or more nucleotides per cycle. Primer relocation (pathway on the left) involves the formation of two DNA intermediates. The first intermediate, which has two branches protruding from the same site, is formed by rewinding of the original displaced strand with concomitant unwinding of the 3’ end of the newly synthesized strand. The second intermediate is formed by annealing the terminal nucleotide sequence of the 3’ end of the newly synthesized DNA strand to any single-stranded complementary sequence. If annealing occurs to the 5”terminated displaced strand, DNA polymerase could use it as a primer to synthesize a stable panhandle. Alternatively, the second intermediate could be formed by reannealing the 3”terminal nucleotide seqeuence to a complementary sequence located internally on the same strand. This structure, if used as a primer by T7 DNA polymerase Form I, would give rise to a stable hairpin structure. Rotational strand exchange (pathway on the right) involves self-annealing of the original displaced strand between the sequence located at the replication fork and any complementary sequence closer to the 5’ end. The duplex in the intermediate thus formed can stack with the unreplicated duplex by a rotational movement which simultaneously brings the displaced strand into a position adjacent to the hydrogen-bonded 3’ end. Finally, the polymerase rebinds and extends the 3’ end using the displaced strand as a template, a step which requires transversal of the strand crossover point. This gives rise to a fully hydrogen-bonded bihelical DNA structure with a cross-strand exchange (37) that is topologically equivalent to the panhandle derived by primer relocation (see Fig. 10 for an example). m, double-stranded DNA; II1IIulllfl, single-stranded DNA, , a DNA polymerase molecule.
over point, this structure is essentially identical with the classic recombination intermediated proposed by Holliday (36). Studies with space-filling models have indicated that strands crossing from one double helix to the other can exist in a fully base-paired configuration (37). While this does not mean that they exist in a suitable configuration for DNA polymerase primer-template activity, the alignment of primer and template is reasonable.
In considering either model for strand switching, it is inter- esting to consider the role of the 3‘ to 5’ single- and double- stranded DNA exonuclease activities of Form I of T 7 D N A polymerase. Although these two activities are present in rel- atively low amounts in Form I, they probably contribute to either primer relocation or rotational strand exchange by removing any unpaired 3’-terminal nucleotides, thus provid- ing DNA polymerase with a suitable hydrogen-bonded primer. As a result, greater flexibility in dissociation of the polymerase
is possible. Studies with E. coli DNA polymerase I (38) and T4 DNA polymerase (39) have shown that such unpaired termini are removed quantitatively by the 3’ t o 5’ exonu- cleases of these enzymes even in the presence of dNTPs.
Both models of template switching invoke the use of in- verted repeat sequences in formally the same way. One can therefore predict, using either model, the location of all pos- sible branches. Electrophoretic analysis of the newly synthe- sized DNA revealed the presence of a limited set of discrete bands corresponding to branches of discrete lengths (Fig. 5). To demonstrate that all of these bands could have arisen through a DNA intermediate involving an inverted repeat, we have made a computer-assisted search of the nucleotide se- quence of pBR322 (25) 550 bases on either side of the EcoRI site. Template switching was simulated according to simple annealing rules which could apply to both primer relocation and rotational strand exchange. Our analysis, based upon a
by guest on June 13, 2018http://w
ww
.jbc.org/D
ownloaded from

Strand Displacement Synthesis by T7 DNA Polymerase 11183
A
new simnd
- di3phcod shond
B
IMr strm7d 120 loo ,’
circuior tm~io@ sirond
di3piaced strond
FIG. 10. Two topological conformers of a panhandle branch. The DNA sequence determined for one branch (see Fig. 8) is displayed as a panhandle in two topologically equivalent states. A, this diagram represents the structure expected to result after DNA synthesis from the relocated primer 5’-GGATG-3’ (see Fig. 9). Note that the circular template strand can be fully base paired to the complementary DNA present on either the new strand or the originally displaced strand (nucleotides 129 to 132). However, for the purpose of display, it is shown unbonded in this region. B, this diagram represents the struc- ture expected to result after extension of the 3”OH end over the cross-connection in the DNA intermediate proposed in the rotational strand exchange scheme (see Fig. 9). Note that this structure can he converted to the structure in A by “rotary diffusion” (35) of the cross- connection point, a distance of five nucleotides to the left.
requirement for an inverted repeat sequence capable of form- ing a duplex containing a t least 12 hydrogen bonds and separated by less than 30 bases (see Fig. 6 ) , indicated 42 possible switchover sites which could give rise to branches with total lengths of 500 base pairs or less. This number of branches is more than sufficient to be consistent with our data since we see approximately 15 prominent bands ranging in length from 50 to 500 bp (Fig. 5). Other factors would certainly affect the abundance of specific branches. For ex- ample, if one assumes that competition with other sequences could slow annealing of the inverted repeats (see Fig. 6B) , then the number of possible branches predicted is reduced to 18. Similarly, it seems likely that secondary structure would inhibit branch migration and that this in turn would reduce the probability of specific annealing events. Most of the 18 predicted branches can be tentatively identified with peaks in the gel tracing of Fig. 6 (compare A and B) . Each predicted branch has been marked by a lettered square on the base pair length scale of the gel tracings according to the length of snapback DNA which would result after EcoRI cleavage and denaturation. Some of the predicted snapback fragments are closely spaced, making their identification less certain, but it is compelling that spacing between predicted groups of frag- ments, or individual ones, is closely reflected by the peaks of the gel tracings in Fig. 6A. The DNA branch that was se- quenced, and which arose as a consequence of the existence of the inverted repeat GGATG, is well separated from other predicted panhandle fragments. Also well separated is the
branch DNA which is predicted to have arisen from annealing of the inverted repeat of CCCC (Fig. 6B, square a). In fact, it is the only one predicted to have a duplex stem shorter than 115 bp with a loop length of less than 30 bases even if competitive annealing is not invoked as a limitation to our search. This predicted species matches well with the first prominent band seen in the autoradiograms (Fig. 6A). Finally, we must mention that, as indicated by the number-length distribution (Fig. 7), the short snapback fragments are a numerically larger fraction of the population, a result that is consistent with both models. It is possible that some of the small molecular weight peaks observed in the region between 50 and 100 nucleotides are the result of strand switching at sites having less than 12 hydrogen bonds. Although these sites would form less stable intermediates, they would, according to the model, form duplexes more often than those located further from the EcoRI site.
There are only two heavily labeled bands in the region from 450 to 500 base pairs ( L and R in Fig. 5). Band L (leftward) could have arisen from relocation of either or both of two four-base inverted repeats, GGCG and CCCG, located at positions 457 and 460, respectively. Similarly, band R (right- ward) could have arisen from the repeat of GGCG a t position 506. It is interesting to note that, although there are some bands corresponding to molecules of lengths greater than 500 base pairs, none of these approaches the intensity of these two bands. Inspection of the sequence of pBR322 reveals that this higher molecular weight region is rich in GC base pairs. We suggest that Form I of T7 DNA polymerase makes slower progress through GC-rich duplex because of the unforced nature of its strand-displacing action. This effect, coupled with a low level of single strand-specific 3’ to 5’ exonuclease activity, could increase the frequency of strand switching in, or just preceding, a GC-rich region.
Role of Form Z in Viuo-Clearly, Form I1 of T7 DNA polymerase is an appropriate enzyme for most of the polym- erization reactions occurring on both leading and lagging strands. In fact, under proper conditions, it polymerizes nu- cleotides faster than Form I and with higher fidelity (1, 31). It does not displace the 5’ end of Okazaki fragments, allowing proper sealing by DNA ligase ( 2 ) . On the other hand, there is no reason at present to conclude that Form I does not have a function in uiuo. Both forms of the enzyme exhibit high fidelity (1) and, more important, some steps in T7 DNA replication would be best served by a polymerase that cata- lyzes limited strand displacement synthesis such as Form I. For example, a single-stranded tail such as that generated by the strand displacement reaction catalyzed by Form I could serve as an intermediate in recombination. Furthermore, from the DNA sequence of concatemers, it is almost certain that a strand displacement reaction such as the one proposed by Watson (4) must occur in order to generate the second copy of the terminal repeat sequence found a t each end of a mature T7 genome. In fact, we demonstrated here that, in vitro, intramolecular (circular) hydrogen-bonded T7 DNA whose structure mimics that of a concatemer can be converted to a unit-length linear T 7 DNA molecule by the strand displace- ment activity of Form I of T7 DNA polymerase. However, it is important to emphasize that we have not analyzed the detailed structure of the ends of the unit-length DNA gener- ated in this reaction. Nucleotide sequence analysis of the ends of the T7 genome reveals the presence of a t least seven inverted repeats that could potentially give rise to template switching prior to the completion of the strand displacement reaction. Clearly, further studies are necessary to investigate the effects of DNA-binding proteins or other replication pro- teins on the rate of strand displacement and template strand
by guest on June 13, 2018http://w
ww
.jbc.org/D
ownloaded from

11184 Strand Displacement Synthesis by T7 D N A Polymerase
switching. If both forms of T7 DNA polymerase do have physiological
roles, it is likely that there are mechanisms to regulate, or minimize, the action of Form I at nicks and a t gaps on the lagging strand side of the replication fork since strand dis- placement would be counterproductive. Two general mecha- nisms come to mind. First, the conversion of Form I1 to Form I might be timed to occur late in infection, when the matura- tion of concatemers occurs. Second, Form I1 may be converted to Form I only at the site where the 'activity of Form I is required. If this type of mechanism existed in the extreme, Form I would never be released as such from the DNA. If this were the case, the preparations of Form I could be considered artifactual even though its activity and basic structure are physiologically important.
Even if Form I does not play an essential role in T7 DNA metabolism, the presence of a small proportion of Form I in the T7-infected cell could give rise to a number of infrequent alterations, i.e. mutations in the viral DNA. Template switch- ing catalyzed by Form I could facilitate both frame shift and base substitution mutations (40). Deletions could arise as a consequence of polymerization across the base of a hairpin in a manner similar to that of template switching. The latter mechanism could, if fact, be a general one to give rise to new combinations of gene structure in both prokaryotic and eu- karyotic cells.
Acknowledgments-We thank Carl Fuller and Stanley Tabor for helpful discussions, assistance with the computer, and DNA sequenc- ing.
REFERENCES 1. Engler, M. J., Lechner, R. L., and Richardson, C. C. (1983) J.
2. Engler, M. J., and Richardson, C. C. (1983) J. Biol. Chem. 2 5 8 ,
3. Dunn, J. J., and Studier, F. W. (1981) J. Mol. Biol. 148,303-330 4. Watson, J. D. (1972) Nature New Biol. 239, 197-201 5. Masamune, Y., and Richardson, C. C. (1971) J. Biol. Chem. 246,
6. Romano, L. J., Tamanoi, F., and Richardson, C. C. (1981) Proc. Natl. Acad. Sci. U. S. A . 78,4107-4111
7. Bolivar, F., Rodriguez, R. L., Green, P. J., Betlach, M. C., Hey- neker, H. L., and Boyer, H. L. (1977) Gene 2,95-113
8. Rodriguez, R. L., Bolivar, F., Goodman, H. M., Boyer, H. W., and Betlach, M. (1976) in Molecular Mechanisms in the Control of Gene Expression (Nierlich, D. D., Rutter, W. J., and Fox, C. F., eds) Vol. 5, pp. 471-477, Academic Press, New York
9. Richardson, C. C. (1965) Proc. Natl. Acad. Sci. U. S. A. 5 4 , 158-
Biol. Chem. 2 5 8 , 11165-11173
11197-11205
2692-2701
10.
11. 12.
13. 14.
15.
16.
17.
18.
19.
20. 21.
22.
23.
24.
25.
26. 27.
28.
29.
30.
31.
32.
33.
34.
35. 36. 37. 38.
39. 165 40.
Davis, R. W., Botstein, D., and Roth, J. R. (1980) Aduanced Bacterial Genetics, p. 196, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY
Wu, R. (1970) J. Mol. Biol. 5 1 , 501-521 Ritchie, D. A., Thomas, C. A., Jr., MacHattie, L. A., and Wensink,
P. C. (1967) J. Mol. Biol. 2 3 , 365-376 Richardson, C. C. (1966) J. Mol. Biol. 15,49-61 Campbell, J. L., Richardson, C. C., and Studier, F. W. (1978)
Proc. Natl. Acad. Sci. U. S. A. 7 5 , 2276-2280 Greenfield, L., Simpson, L., and Kaplan, D. (1975) Biochim.
Biophys. Acta 407,365-375 Kolodner, R., Masamune, Y., LeClerc, J. E., and Richardson, C.
C. (1978) J. Biol. Chem. 253, 566-573 Weiss, B., Live, T. R., and Richardson, C. C. (1968) J. Biol. Chem.
Maxam, A. M., and Gilbert, W. (1980) Methods Enzymol. 6 5 ,
Davis, R., Simon, M., and Davidson, N. (1971) Methods Enzymol.
Keller, W. (1975) Proc. Natl. Acad. Sci. U. S. A . 7 2 , 4876-4880 Bauer, W., and Vinograd, J . (1974) in Basic Principles in Nucleic
Acid Chemistry (T'so, P. 0. P., ed) Vol. 11, pp. 262-305, Aca- demic Press, New York
Sebring, E. D., Garon, C. F., and Salzman, N. P. (1974) J. Mol. Biol. 90,371-379
Richardson, C. C., Lehman, I. R., and Kornberg, A. (1964) J . Biol. Chem. 239,251-258
Sgaramella, V., van de Sande, J. H., and Khorana, H. G. (1970) Proc. Natl. Acad. Sci. U. S. A . 67, 1468-1475
Sutcliffe, J. G. (1979) Cold Spring Harbor Symp. Quant. Biol. 43,
Dunn, J . J., and Studier, F. W. (1983) J. Mol. Biol., in press Schlegel, R. A,, and Thomas, C. A., Jr. (1972) J. Mol. Biol. 6 8 ,
Fujimura, R. K., and Roop, B. C. (1976) J. Biol. Chem. 2 5 1 ,
McConnell, B., and von Hippel, P. H. (1970) J. Mol. Biol. 50,
Radding, C. M., Beattie, K. L., Holloman, W. K., and Wiegand,
Lechner, R. L., and Richardson, C. C. (1983) J. Biol. Chem. 258,
Adler, S., and Modrich, P. (1979) J. Biol. Chem. 2 5 4 , 11605-
Hori. K.. Mark. D. F., and Richardson, C. C. (1979) J. Biol. C h ~ m .
243,4530-4542
499-560
21,413-428
77-90
319-345
2168-2175
297-316
R. C. (1977) J . Mol. Biol. 116, 825-839
11185-11196
11604
254, i1598-i1604' - . . . . . . .
Kornberg, A. (1980) DNA Replication, pp. 153-54, W. H. Freeman & Co., San Francisco
Meselson, M. (1972) J. Mol. Biol. 71, 795-798 Holliday, R. (1964) Genet. Res. 5 , 282-303 Sigal, N., and Alberts, B. (1972) J. Mol. Biol. 7 1 , 789-793 Brutlag, D., and Kornberg, A. (1972) J. Biol. Chem. 247, 241-
Englund, P. T. (1971) J. Biol. Chem. 2 4 6 , 5684-5687 Ripley, L. S. (1982) Proc. Natl. Acad. Sci. U. S. A. 79,4128-4132
248
by guest on June 13, 2018http://w
ww
.jbc.org/D
ownloaded from

R L Lechner, M J Engler and C C RichardsonDNA polymerase.
Characterization of strand displacement synthesis catalyzed by bacteriophage T7
1983, 258:11174-11184.J. Biol. Chem.
http://www.jbc.org/content/258/18/11174Access the most updated version of this article at
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
http://www.jbc.org/content/258/18/11174.full.html#ref-list-1
This article cites 0 references, 0 of which can be accessed free at
by guest on June 13, 2018http://w
ww
.jbc.org/D
ownloaded from





























