Embed Size (px)
Citation preview

i
Lời cam doan
Tôi xin cam đoan: Luận văn này là công trình nghiên cứu của tôi, được
thực hiện dưới sự hướng dẫn khoa học của TS. Chu Xuân Quang.
Các số liệu, những kết luận nghiên cứu được trình bày trong luận văn
này trung thực và không trùng lặp với các đề tài khác. Học viên cũng xin cam
đoan rằng mọi sự giúp đỡ cho việc thực hiện luận văn này đã được cảm ơn và
các thông tin trích dẫn trong luận văn đã được chỉ rõ nguồn gốc.
Tôi xin chịu trách nhiệm về mọi vấn đề liên quan đến nội dung của đề
tài này.
Tác giả luận văn
Nguyễn Thị Xuân Thu

ii
Lời cảm ơn
Luận văn này được hoàn thành tại Trung tâm Kiểm định - Viện Vật liệu
xây dựng - Bộ Xây dựng và Trung tâm Công nghệ Vật liệu - Viện Ứng dụng
Công nghệ - Bộ Khoa học và Công nghệ. Trong quá trình nghiên cứu, em đã
nhận được nhiều sự giúp đỡ quý báu của các thầy cô, các đồng nghiệp, bạn bè
và gia đình.
Với lòng kính trọng và biết ơn sâu sắc nhất, em xin gửi lời cảm ơn tới
TS. Chu Xuân Quang - người thầy tâm huyết hướng dẫn khoa học, truyền cho
em tri thức cũng như chỉ bảo, động viên, giúp đỡ, khích lệ và tạo mọi điều
kiện tốt nhất để em hoàn thành luận văn này.
Em xin chân thành cảm ơn các thầy cô tại Học viện Khoa học và công
nghệ, Viện Hàn lâm khoa học và công nghệ Việt Nam; tập thể anh chị em
trong Trung tâm Kiểm định, Viện Vật liệu xây dựng, Bộ Xây dựng và Trung
tâm Công nghệ Vật liệu - Viện Ứng dụng Công nghệ - Bộ Khoa học và Công
nghệ đã giúp đỡ em trong quá trình thực nghiệm cũng như đóng góp nhiều ý
kiến quý báu về chuyên môn trong việc thực hiện và hoàn thiện luận văn.
Dù đã rất cố gắng, song do thời gian và kiến thức về đề tài chưa được
sâu rộng nên luận văn chắc chắn không tránh khỏi những thiếu sót và hạn chế.
Kính mong nhận được sự chia sẻ và những ý kiến đóng góp quý báu của các
thầy giáo, cô giáo, các bạn bè đồng nghiệp.
Một lần nữa em xin chân thành cảm ơn!
Tác giả luận văn
Nguyễn Thị Xuân Thu

iii
Danh mục các ký hiệu và chữ viết tắt
BOD Nhu cầu oxy sinh hóa
COD Nhu cầu oxy hóa học
MBBR Công nghệ xử lý nước thải bằng giá thể lơ lửng
tầng lưu động (Moving Bed BioReactor).
MLSS Hàm lượng chất rắn lơ lửng trong bùn lỏng
N Chất nitơ
NTSH Nước thải sinh hoạt
P Chất photpho
SS Chất rắn lơ lửng
TCVN Tiêu chuẩn Việt Nam
VSV Vi sinh vật

iv
Danh mục các bảng
Bảng 1.1. Tiêu chuẩn nước thải của một số loại cơ sở dịch vụ và công trình
công cộng ........................................................................................................ 10
Bảng 1.2. Tải trọng chất thải trung bình một ngày tính theo đầu người ......... 11
Bảng 1.3. Phân loại mức độ ô nhiễm theo thành phần hóa học điển hình của
nước thải sinh hoạt .......................................................................................... 12
Bảng 3.1. Sự phụ thuộc độ hấp thụ quang vào nồng độ +
4NH ......................... 44
Bảng 3.2. Độ hấp thụ quang của dung dịch 4NH 0,05 mg/L ......................... 45
Bảng 3.3. Sự phụ thuộc độ hấp thụ quang vào nồng độ +
4NH ......................... 45
Bảng 3.4. Độ hấp thụ quang của dung dịch 4NH theo tiêu chuẩn SEMWW
4500 C ............................................................................................................. 46
Bảng 3.5. Sự phụ thuộc độ hấp thụ quang vào nồng độ 2NO ......................... 46
Bảng 3.6. Độ hấp thụ quang của dung dịch 2NO theo TCVN 6178:1996 ...... 47
Bảng 3.7. Sự phụ thuộc độ hấp thụ quang vào nồng độ 3NO ......................... 48
Bảng 3.8. Độ hấp thụ quang của dung dịch 3NO theo TCVN 6180:1996 ...... 48
Bảng 3.9. Sự phụ thuộc độ hấp thụ quang vào nồng độ 3NO ......................... 49
Bảng 3.10. Độ hấp thụ quang của dung dịch 3NO 0,3 mg/L .......................... 49
Bảng 3.11. Sự phụ thuộc độ hấp thụ quang vào nồng độ 3
4PO ....................... 50
Bảng 3.12. Độ hấp thụ quang của dung dịch 3
4PO 0,02 mg/100 ml ............... 51
Bảng 3.13. Sự phụ thuộc độ hấp thụ quang vào hàm lượng nguyên tố P ....... 51
Bảng 3.14. Độ hấp thụ quang của dung dịch có hàm lượng P 1,0 mg/L ........ 52
Bảng 3.15. Thông số đánh giá độ thu hồi mẫu ............................................... 53
Bảng 3.16. Phương pháp phân tích theo TCVN 6179-1:1996 và
SEMWW4500 C ............................................................................................. 58

v
Bảng 3.17. Phương pháp phân tích theo TCVN 6180-1:1996 và
SEMWW4500 B ............................................................................................. 58
Bảng 3.18. Phương pháp phân tích theo TCVN 6202:2008 và SEMWW4500-P 59
Bảng 3.19. Ảnh hưởng của thời gian sục khí tới hiệu quả xử lý nitơ ............. 61
Bảng 3.20. Đánh giá hiệu quả xử lý N của hệ thống ...................................... 62
Bảng 3.21. Ảnh hưởng của thời gian sục khí tới hiệu quả xử lý P ................. 62
Bảng 3.22. Thông số các chất ô nhiễm trong nước thải giả lập ...................... 65
Bảng 3.23. Hàm lượng nitrat, nitrit đầu vào và đầu ra ................................... 70

vi
Danh mục các hình vẽ, đồ thị
Hình 1.1. Sơ đồ hệ thống xử lý nước bằng công nghệ MBBR ......................... 4
Hình 1.2. Mô hình công nghệ MBBR dạng hiếu khí và thiếu khí .................... 5
Hình 1.3. Lớp biofilm dính bám trên bề mặt giá thể ........................................ 7
Hình 3.1. Đường chuẩn xác định 4NH theo TCVN 6179-1: 1996 ................. 44
Hình 3.2. Đường chuẩn xác định 4NH theo tiêu chuẩn SEMWW4500 C ...... 45
Hình 3.3. Đường chuẩn xác định 2NO theo TCVN 6178:1996 ...................... 47
Hình 3.4. Đường chuẩn xác định 3NO theo tiêu chuẩn TCVN 6180:1996 .... 48
Hình 3.5. Đường chuẩn xác định 3NO theo tiêu chuẩn SEMWW 4500 B ..... 49
Hình 3.6. Đường chuẩn xác định hàm lượng Photphat theo TCVN 6202:2008 50
Hình 3.7. Đường chuẩn xác định Photphat theo tiêu chuẩn SMEWW 4500 - P 52
Hình 3.8. Hình ảnh của bùn hoạt tính bám trên giá thể vi sinh ...................... 63
Hình 3.9. Sự phát triển của bùn hoạt tính ....................................................... 64
Hình 3.10. Hiệu quả xử lý photphat ................................................................ 66
Hình 3.11. Hiệu quả xử lý amoni .................................................................... 67
Hình 3.12. Hiệu quả xử lý nitrat và nitrit ........................................................ 67
Hình 3.13. Hiệu quả xử lý photpho trong nước thải thực tế ........................... 68
Hình 3.14. Hiệu quả xử lý amoni trong nước thải thực tế .............................. 69

vii
MỤC LỤC
MỞ ĐẦU ........................................................................................................... 1
CHƯƠNG I. TỔNG QUAN NGHIÊN CỨU ................................................... 4
1.1. GIỚI THIỆU CHUNG VỀ CÔNG NGHỆ MBBR TRONG XỬ LÝ
NƯỚC THẢI ....................................................................................................... 4
1.1.1. Sơ đồ và mô hình xử lý bằng công nghệ MBBR ........................... 4
1.1.2. Các yếu tố ảnh hưởng đến quá trình xử lý bằng công nghệ MBBR 5
1.1.3. Ưu, nhược điểm của công nghệ MBBR ......................................... 8
1.2. TỔNG QUAN NƯỚC THẢI SINH HOẠT ............................................. 9
1.2.1. Nguồn gốc nước thải sinh hoạt ....................................................... 9
1.2.2. Đặc tính nước thải sinh hoạt ......................................................... 10
1.2.3. Tác động của nước thải sinh hoạt đến môi trường và sức khỏe con
người ....................................................................................................... 13
1.3. CÁC PHƯƠNG PHÁP XÁC ĐỊNH NỒNG ĐỘ CHẤT Ô NHIỄM
TRONG NƯỚC THẢI ..................................................................................... 14
1.3.1. Các phương pháp xác định nồng độ Photphat .............................. 14
1.3.2. Các phương pháp xác định nồng độ amoni .................................. 15
1.3.3. Các phương pháp xác định nồng độ nitrat .................................... 18
1.3.4. Các phương pháp xác định nồng độ nitrit .................................... 21
1.4. NGUYÊN TẮC XÁC ĐỊNH GIỚI HẠN PHÁT HIỆN VÀ GIỚI HẠN
ĐỊNH LƯỢNG .................................................................................................. 23
1.5. ĐÁNH GIÁ ĐỘ ĐÚNG CỦA PHƯƠNG PHÁP XÁC ĐỊNH 4NH .
2NO . 3NO . 3
4PO THÔNG QUA HIỆU SUẤT THU HỒI ............................. 25
CHƯƠNG 2. THỰC NGHIỆM ...................................................................... 31
2.1. NỘI DUNG NGHIÊN CỨU .................................................................... 31
2.2. PHƯƠNG PHÁP NGHIÊN CỨU ........................................................... 32

viii
2.2.1. Phương pháp lấy mẫu ................................................................... 32
2.2.2. Phương pháp bảo quản mẫu ......................................................... 32
2.2.3. Phương pháp phân tích mẫu ......................................................... 33
2.3. DỤNG CỤ, THIẾT BỊ .............................................................................. 33
2.4. PHƯƠNG PHÁP TRẮC QUANG XÁC ĐỊNH NH4+ BẰNG THUỐC
THỬ THYMOL - TCVN 6179-1 : 1996 ........................................................ 34
2.4.1 Chuẩn bị hóa chất, thuốc thử ......................................................... 34
2.4.2. Yếu tố ảnh hưởng ......................................................................... 35
2.4.3. Quy trình phân tích ....................................................................... 35
2.5. XÁC ĐỊNH AMONIAC (NH4+) TRONG NƯỚC– PHƯƠNG PHÁP
LÊN MÀU TRỰC TIẾP VỚI THUỐC THỬ NESSLER ............................ 35
2.5.1. Chuẩn bị hóa chất, thuốc thử ........................................................ 35
2.5.2. Yếu tố ảnh hưởng ......................................................................... 36
2.5.3. Quy trình phân tích ....................................................................... 36
2.6. XÁC ĐỊNH NITRIT - PHƯƠNG PHÁP ĐO MÀU VỚI THUỐC
THỬ GRIESS .................................................................................................... 36
2.6.1. Chuẩn bị hóa chất, thuốc thử ........................................................ 36
2.6.2. Yếu tố ảnh hưởng ......................................................................... 37
2.6.3. Quy trình phân tích ....................................................................... 37
2.7. XÁC ĐỊNH NITRAT TRONG NƯỚC – PHƯƠNG PHÁP ĐO MÀU
VỚI THUỐC THỬ AXIT SUNFOSALIXYLIC .......................................... 37
2.7.1. Chuẩn bị hóa chất, thuốc thử ........................................................ 37
2.7.2. Yếu tố ảnh hưởng ......................................................................... 38
2.7.3. Quy trình phân tích ....................................................................... 38
2.8. XÁC ĐỊNH NITRAT TRONG NƯỚC - PHƯƠNG PHÁP ĐO
QUANG PHỔ TIA UV VÀ DẪN XUẤT THỨ HAI................................... 39

ix
2.8.1. Chuẩn bị hóa chất, thuốc thử ........................................................ 39
2.8.2. Yếu tố ảnh hưởng ......................................................................... 39
2.8.3. Quy trình phân tích ....................................................................... 39
2.9. XÁC ĐỊNH PHOTPHAT (PO43-) TRONG NƯỚC - PHƯƠNG PHÁP
XANH MOLYBDEN ....................................................................................... 40
2.9.1. Chuẩn bị hóa chất, thuốc thử ........................................................ 40
2.9.2. Yếu tố ảnh hưởng ......................................................................... 41
2.9.3. Quy trình phân tích ....................................................................... 41
2.10. XÁC ĐỊNH PHOTPHAT (PO43-) TRONG NƯỚC - PHƯƠNG
PHÁP ĐO PHỔ HẤP THỤ PHÂN TỬ AXIT VANADOMOLYBDO
PHOSPHORIC .................................................................................................. 41
2.10.1. Chuẩn bị hóa chất, thuốc thử ...................................................... 41
2.10.2. Yếu tố ảnh hưởng ....................................................................... 42
2.10.3. Quy trình phân tích ..................................................................... 42
2.11. ẢNH HƯỞNG CỦA MỘT SỐ ĐIỀU KIỆN VẬN HÀNH ĐẾN
HIỆU SUẤT XỬ LÝ ........................................................................................ 42
2.11.1. Khảo sát quá trình phát triển của bùn hoạt tính .......................... 43
2.11.2. Khảo sát thời gian sục khí .......................................................... 43
2.11.3. Đánh giá hiệu quả xử lý hệ MBBR ............................................ 43
CHƯƠNG 3. KẾT QUẢ VÀ THẢO LUẬN .................................................. 44
3.1. XÁC ĐỊNH GIÁ TRỊ SỬ DỤNG CỦA PHƯƠNG PHÁP PHÂN
TÍCH ................................................................................................................... 44
3.1.1. Xây dựng đường chuẩn, tính toán LOD, LOQ của các phương
pháp xác định 4NH , 2NO , 3NO , 3
4PO ...................................................... 44
3.1.2. Đánh giá độ đúng của phương pháp xác định 4NH . 2NO . 3NO .
3
4PO thông qua hiệu suất thu hồi ............................................................. 52

x
3.1.3. So sánh hai phương pháp đánh giá độ chính xác
................................................................................................................ 57
3.2 PHÂN TÍCH MẪU THỰC TẾ ................................................................. 60
3.2.1. Lựa chọn phương pháp phân tích ................................................. 60
3.2.2 Kết quả phân tích mẫu thực tế ....................................................... 61
3.3. ĐÁNH GIÁ ẢNH HƯỞNG CỦA MỘT SỐ ĐIỀU KIỆN VẬN HÀNH
............................................................................................................................. 63
3.3.1. Quá trình phát triển của bùn hoạt tính .......................................... 63
3.3.2. Ảnh hưởng của thời gian sục khí tới hiệu suất xử lý ................... 64
3.3.3. Đánh giá hiệu quả xử lý hệ MBBR .............................................. 68
CHƯƠNG 4. KẾT LUẬN VÀ KIẾN NGHỊ .................................................. 71
4.1. KẾT LUẬN ................................................................................................ 71
4.2. KIẾN NGHỊ ............................................................................................... 72
TÀI LIỆU THAM KHẢO ............................................................................... 74

1
MỞ ĐẦU
1. Lý do chọn đề tài
Trong những năm gần đây, tình trạng ô nhiễm môi trường do nước thải
sinh hoạt (NTSH) đang diễn ra nghiêm trọng ở khắp nơi trên cả nước, đặc biệt
là tại các thành phố lớn, các trung tâm thương mại, các khu vui chơi giải trí.
Trong nước thải chứa hàm lượng lớn các chất nitơ (N), photpho (P) là nguyên
nhân gây ra các hiện tượng phú dưỡng và độc tính khi thải ra môi trường. Do
đó cần phải loại bỏ những chất này trong nước thải để làm giảm tác hại của
chúng đến môi trường. Hiện nay, hệ thống xử lý nước thải tại các cơ sở cũng
như các nhà máy xử lý nước thải đang đối mặt với vấn đề phải mở rộng quy
mô do sự tăng lên đáng kể của lưu lượng nước thải và tải trọng chất ô
nhiễm.... Tuy nhiên việc mở rộng quy mô rất khó khăn và có thể ảnh hưởng
đến quy hoạch của các địa bàn dân cư, do đó, tính khả thi không cao. Các
bước cải tiến kỹ thuật trong xử lý nước thải, cũng như ứng dụng các loại vật
liệu mới xử lý môi trường giúp giải quyết được các nhược điểm của các
phương pháp xử lý cũ, nâng cao chất lượng đầu ra và giảm chi phí quá trình
vận hành hệ thống xử lý. Để tăng hiệu quả xử lý đối với các nguồn thải thì
việc ứng dụng các thiết bị xử lý sinh học sử dụng giá thể vi sinh được coi là
giải pháp và hướng đi phù hợp do chúng làm tăng được nồng độ vi sinh trong
bể xử lý. Từ đó làm tăng hiệu quả xử lý trên cùng một thể tích và tiết kiệm
được mặt bằng xây dựng của hệ thống.
Một trong những công nghệ mới hiện nay đang được nghiên cứu và áp
dụng là công nghệ xử lý nước thải bằng giá thể lơ lửng tầng lưu động MBBR
(Moving Bed BioReactor - MBBR). Đây là một dạng của quá trình xử lý
nước thải bằng bùn hoạt tính, kết hợp giữa các điều kiện thuận lợi của quá
trình xử lý bùn hoạt tính và bể lọc sinh học. Trong quá trình MBBR, vi sinh
vật (VSV) phát triển và bám dính trên giá thể chuyển động trong chất lỏng
của bể xử lý. Ưu điểm của giải pháp này là hiệu suất xử lý cao và ổn định,
thời gian lưu bùn lâu, lượng bùn cần xử lý ít, chi phí vận hành không cao do
tốn ít năng lượng. Việc sử dụng giá thể vi sinh có trọng lượng, hình dạng phù
hợp, có khả năng chuyển động trong bể xử lý đã được nghiên cứu và ứng

2
dụng ở nhiều quốc gia có ngành công nghiệp môi trường phát triển. Ở nước
ta hiện nay, việc sản xuất và ứng dụng các loại vật liệu đệm vi sinh này đối
với các hệ thống xử lý nước thải như NTSH, nước thải bệnh viện, .v.v. đang
dần trở nên khá phổ biến. Nhiều nghiên cứu ứng dụng đệm vi sinh tại các
Viện nghiên cứu và các trường Đại học đã được triển khai. Tuy nhiên, hầu
hết các loại giá thể vi sinh đang được sử dụng tại Việt Nam khá đa dạng về
chủng loại, chủ yếu được nhập khẩu từ Trung Quốc, Đài Loan và của một số
ít cơ sở sản xuất nhựa trong nước. Thực tế, khả năng dính bám của VSV trên
các loại giá thể này còn hạn chế. Năm 2014, Trung tâm Công nghệ Vật liệu
đã nghiên cứu chế tạo được sản phẩm giá thể đệm vi sinh từ nhựa PE bằng
phương pháp ép phun và đưa vào hệ xử lý MBBR. Hiện nay, Trung tâm
Công nghệ Vật liệu đang tiếp tục triển khai hoàn thiện quy trình công nghệ,
nghiên cứu cải thiện tính chất bề mặt của giá thể vi sinh nhằm tăng hiệu quả
hình thành lớp vi sinh bám dính; qua đó tăng hiệu quả xử lý nước thải.
Bên cạnh việc đánh giá các chỉ tiêu cơ - lý của giá thể vi sinh như độ
bền uốn, độ bền kéo, độ bền va đập, độ cứng trên các thiết bị đo chuyên dụng,
việc đánh giá hiệu quả xử lý nước thải theo các tiêu chí, chỉ tiêu ô nhiễm cũng
rất quan trọng để có thể chứng minh được mức độ cải thiện của quá trình chế
tạo. Trong quá trình phân tích hàm lượng các chất chứa P và N nảy sinh các
vấn đề khi hàm lượng các chất trong nước thải đầu vào có hàm lượng lớn
nhưng nhờ hiệu quả xử lí cao nên nước thải đầu ra hàm lượng các hợp chất
chứa N và P có hàm lượng thấp hơn rất nhiều. Từ đó đặt ra vấn đề với cùng
một phương pháp phân tích thì có đảm bảo kết quả phân tích cho độ chính xác
cao không? Do vậy, chúng tôi đề xuất đề tài luận văn: “Phân tích hàm lượng
photphat và một số hợp chất của nitơ trong hệ xử lý nước thải sử dụng giá thể
vi sinh chuyển động (MBBR)”. Nội dung thử nghiệm và đánh giá hiệu quả sử
dụng của giá thể vi sinh đã chế tạo được trong hệ xử lý nước thải ở quy mô
phòng thí nghiệm là nghiên cứu có tính tiệm cận thực tế, tạo cơ sở chắc chắn
và cung cấp dữ liệu tin cậy cho việc ứng dụng giá thể vi sinh ở quy mô lớn
hơn. Do vậy, việc hoàn thiện quy trình phân tích và đánh giá chính xác được
sự thay đổi nồng độ của những thành phần chính cần xử lý trong nước thải là
cần thiết.

3
2. Mục tiêu nghiên cứu của đề tài
Phân tích hàm lượng photphat và một số hợp chất amoni, nitrat, nitrit
trong mẫu nước thải giả lập, mẫu nước thải đầu vào và đầu ra của hệ thống xử
lý nước thải bằng giá thể vi sinh chuyển động.
Đánh giá được hiệu quả xử lý photphat và một số hợp chất của nitơ
trong hệ xử lý nước thải sử dụng giá thể vi sinh chuyển động.
3. Đối tượng nghiên cứu
Hàm lượng photphat và một số hợp chất của nitơ trong mẫu nước thải
của hệ xử lý nước thải sử dụng giá thể vi sinh chuyển động (MBBR).
4. Phạm vi nghiên cứu
Đánh giá được hiệu quả xử lý photphat và một số hợp chất của nitơ
trong hệ xử lý nước thải sử dụng giá thể vi sinh chuyển động.
5. Ý nghĩa khoa học và thực tiễn của đề tài
Thử nghiệm và đánh giá hiệu quả sử dụng của giá thể vi sinh đã chế tạo
được trong hệ xử lý nước thải ở quy mô phòng thí nghiệm.
Cung cấp dữ liệu tin cậy cho việc ứng dụng giá thể vi sinh ở quy mô
lớn hơn.
Hoàn thiện quy trình phân tích và đánh giá chính xác được sự thay đổi
nồng độ của những thành phần chính cần xử lý trong nước thải.

4
CHƯƠNG I. TỔNG QUAN NGHIÊN CỨU
1.1. GIỚI THIỆU CHUNG VỀ CÔNG NGHỆ MBBR TRONG XỬ LÝ
NƯỚC THẢI
1.1.1. Sơ đồ và mô hình xử lý bằng công nghệ MBBR
Hệ màng sinh học (biofilm) trong xử lý nước thải hiện nay đang là xu
hướng gia tăng nhanh chóng bởi khả năng loại bỏ các chất ô nhiễm trong
nước thải với hiệu quả cao cũng như giảm thiểu các chi phí nhân công, giá
thành. Công nghệ xử lý sinh học với giá thể lơ lửng MBBR là một dạng của
quá trình xử lý nước thải bằng bùn hoạt tính bởi lớp màng sinh học (biofilm).
Trong quá trình sử dụng MBBR, lớp màng biofilm phát triển trên giá thể lơ
lửng trong lớp chất lỏng của bể phản ứng [1-2].
Hình 1.1. Sơ đồ nguyên lý hệ thống xử lý nước bằng công nghệ MBBR
MBBR có thể được thiết kế cho các cơ sở mới để loại bỏ nhu cầu oxy
sinh hóa/nhu cầu oxy hóa học (BOD/COD) hoặc loại bỏ nitơ, photpho từ các
dòng nước thải. Hiện tại các nhà máy áp dụng công nghệ bùn hoạt tính có thể
được nâng cấp để có thể khử nitơ và photpho hoặc BOD/COD ở lưu lượng
lớn. Các vi khuẩn nuôi cấy tăng trưởng nhờ phân hủy các chất hữu cơ hòa tan,
từng bước trưởng thành trong môi trường đó.
Công nghệ MBBR là công nghệ kết hợp giữa các điều kiện thuận lợi của
quá trình xử lý bùn hoạt tính hiếu khí và bể lọc sinh học. Bể MBBR hoạt động
giống như quá trình xử lý bùn hoạt tính hiếu khí trong toàn bộ thể tích bể. Đây

5
là quá trình xử lý bằng lớp màng biofilm với sinh khối phát triển trên giá mang
mà những giá mang này lại di chuyển tự do trong bể phản ứng và được giữ bên
trong bể phản ứng được đặt ở cửa ra của bể. Bể MBBR không cần quá trình
tuần hoàn bùn giống như các phương pháp xử lý bằng màng biofilm khác, vì
vậy nó tạo điều kiện thuận lợi cho quá trình xử lý bằng phương pháp bùn hoạt
tính trong bể, bởi vì sinh khối ngày càng được tạo ra trong quá trình xử lý. Bể
MBBR gồm 2 loại: bể hiếu khí và bể thiếu khí [2-5].
Bể hiếu khí Bể thiếu khí
Hình 1.2. Mô hình công nghệ MBBR dạng hiếu khí và thiếu khí
Trong bể hiếu khí sự chuyển động của các giá thể được tạo thành do sự
khuếch tán của những bọt khí có kích thước trung bình từ máy thổi. Trong khí
đó ở bể thiếu khí thì quá trình này được tạo ra bởi sự xáo trộn của các giá thể
trong bể bằng cánh khuấy. Hầu hết các bể MBBR được thiết kế ở dạng hiếu
khí có lớp lưới chắn ở cửa ra, ngày nay người ta thường thiết kế lớp lưới chắn
có dạng hình trụ đặt thẳng đứng hay nằm ngang.
1.1.2. Các yếu tố ảnh hưởng đến quá trình xử lý bằng công nghệ
MBBR
1.1.2.1. Giá thể
Trong công nghệ MBBR các giá thể chuyển động cùng với lớp màng
biofilm phát triển và bám trên bề mặt nhằm làm tăng sự tiếp xúc giữa VSV và
nước thải, từ đó gia tăng sinh khối làm quá trình phân hủy sinh học diễn ra
nhanh chóng với hiệu suất xử lý cao [1- 5]. Giá thể được thiết kế sao cho diện
tích bề mặt hiệu dụng là lớn nhất để lớp màng biofim dính bám trên bề mặt
của giá thể và tạo điều kiện tối ưu cho hoạt động của VSV khi những giá thể

6
này lơ lửng trong nước. Tất cả các vật liệu giá thể có tỷ trọng nhẹ hơn so với
tỷ trọng của nước, tuy nhiên mỗi loại giá thể có tỷ trọng khác nhau. Giá thể có
nhiều hình dạng khác nhau, thông thường các giá thể có hình trụ đứng, bên
trong và bề mặt ngoài có nhiều khe để tăng diện tích bề mặt.
Diện tích bề mặt tiếp xúc của các giá thể phổ biến nằm trong khoảng
120 – 950 m2/m
3. Đối với công nghệ MBBR, việc giữ cố định sinh khối trên
vật liệu đệm có ảnh hưởng lớn đến khả năng xử lý nước của hệ. Thêm vào đó,
việc duy trì tỷ lệ sinh khối cao hoạt động trong hệ là yếu tố cực kỳ quan trọng
để đánh giá các hệ màng lọc sinh học nói chung và hệ MBBR nói riêng. Do
đó, vật liệu chế tạo đệm vi sinh đóng vai trò chất mang là một yếu tố vô cùng
quan trọng trong quá trình xử lý nước thải bằng công nghệ MBBR.
1.1.2.2. Độ xáo trộn
Yếu tố khác có ảnh hưởng đến hiệu suất là dòng chảy và điều kiện xáo
trộn trong bể xử lý. Độ xáo trộn thích hợp là điều kiện lý tưởng đối với hiệu
suất của hệ thống. Lớp màng biofilm hình thành trên giá thể rất mỏng, phân
tán và vận chuyển cơ chất và oxy đến bề mặt biofilm. Vì vậy, lớp màng
biofilm dày và mịn không được mong đợi đối với hệ thống. Độ xáo trộn thích
hợp có tác dụng loại bỏ những sinh khối dư và duy trì độ dày thích hợp cho
biofilm. Độ dày của biofilm nhỏ hơn 100 m đối với việc xử lý cơ chất luôn
được ưu tiên.
Độ xáo trộn thích hợp giúp duy trì vận tốc dòng chảy cần thiết cho hiệu
suất quá trình. Độ xáo trộn cao sẽ tách sinh khối ra khỏi giá mang và chính vì
vậy sẽ làm giảm hiệu suất của quá trình xử lý. Thêm vào đó, sự va chạm và sự
ma sát của giá thể trong bể phản ứng làm cho biofilm tách rời khỏi bề mặt
phía ngoài của giá thể Kaldnes (giá mang được sử dụng thực nghiệm). Vì điều
này, giá mang MBBR được cung cấp với các rìa bên ngoài để bảo vệ sự hao
hụt của biofilm và đẩy mạnh sự phát triển của biofilm. Diện tích bề mặt của
các rìa bên ngoài không được tính vào diện tích thực tế của biofilm. Diện tích
trung bình hiệu quả của giá mang MBBR được báo cáo là khoảng 70% tổng
diện tích bề mặt để màng biofilm dính bám vào giá thể ở phía bên ngoài ít
hơn của giá mang [1-8].

7
Theo nghiên cứu của S. Winogradsly (1980), sau khi quan sát dưới
kính hiển vi lớp màng lọc trong bể lọc sinh học nhỏ giọt, đã tìm thấy rất nhiều
vi khuẩn Zoogleal, các vi khuẩn hình que, vi khuẩn hình sợi, nấm sợi,
protozoa và một số động vật bậc cao.
Một trong những nghiên cứu nhằm ước lượng các loại khuẩn trong hệ
thống lọc sinh học nhỏ giọt được tiến hành bởi M. Hotchkiss năm 1923. Kết
quả là đã tìm thấy nhiều loại vi khuẩn khác nhau ở độ sâu khác nhau trong bể
lọc. Các nhóm vi khuẩn bao gồm: vi khuẩn khử nitrat, sunfat tạo thành từ
protein, phân hủy ambumin, khử sunfat, oxi hóa sunfit được tạo thành từ các
protein nhiều nhất ở độ sâu 0,3m và giảm dần qua lớp lọc; vi khuẩn khử
sunfat hiện diện nhiều ở bề mặt và vi khuẩn oxi hóa sunfua có nhiều nhất ở độ
sâu 1,6m; các dạng vi khuẩn nitrit gia tăng theo độ sâu và có số lượng lớn hơn
các dạng vi khuẩn nitrat [7-17].
Hình 1.3. Lớp biofilm dính bám trên bề mặt giá thể
1.1.2.3. Tải trọng thể tích
Vì không thể xác định chính xác diện tích thực được bao bọc bởi
biofilm trên bề mặt của giá mang, người ta đưa ra hiệu suất quá trình theo thể
tích bể phản ứng thay vì diện tích bề mặt giá thể.
Nếu chỉ xử lý thứ cấp, hiệu quả tải tương đương 4-5 kg BOD7 /m3.ngày
đến 12-15 kg BOD7/m3.ngày ở mức 67% giá mang được lấp đầy (cung cấp
335m2 diện tích bề mặt giá thể trên m
3 thể tích bể phản ứng) [4, 10, 12].

8
1.1.3. Ưu, nhược điểm của công nghệ MBBR
1.1.3.1. Ưu điểm
Công nghệ MBBR với các ưu điểm nổi bật như: tăng cường chuyển
động để thúc đẩy tốc độ chuyển khối, tích lũy vi sinh cao nhờ sử dụng vật liệu
mang xốp và diện tích bề mặt lớn đang là công nghệ được ứng dụng nhiều
cho xử lý nước thải. So với phương pháp xử lý sinh học truyền thống bằng
bùn vi sinh hoạt tính, VSV được phân bố khá đồng đều trong thể tích của khối
phản ứng, công nghệ MBBR cho phép tăng đáng kể mật độ sinh khối trên một
đơn vị thể tích khối phản ứng. So với kỹ thuật lọc tầng t nh ngoài đặc điểm
tích lũy mật độ vi sinh cao, công nghệ MBBR thúc đẩy quá trình chuyển khối
nhờ chuyển động vật liệu mang trong môi trường phản ứng. So với kỹ thuật
tầng lưu thể, công nghệ MBBR cũng tích lũy mật độ vi sinh cao do sử dụng
vật liệu mang có diện tích bề mặt lớn (10.000 m2/m
3), tuy kém hơn về mặt
chuyển động (chuyển khối ngoài), nhưng bù lại vận hành đơn giản, không đòi
hỏi trình độ tự động hóa cao như khi sử dụng kỹ thuật tầng lưu thể. Chính vì
những ưu điểm nổi trội của công nghệ MBBR, hơn hẳn so với các kỹ thuật
lọc sinh học khác nên lựa chọn công nghệ này để giải quyết vấn đề xử lý nước
thải [1-5].
Có thể tổng quát các ưu điểm của công nghệ MBBR như sau:
- Tiết kiệm không gian (thể tích, diện tích) trạm xử lý hơn so với các
công nghệ truyền thống khác, giảm chi phí hoạt động, tự động, dễ vận hành
và bảo trì.
- Đạt hiệu quả kể cả trong nước thải có tỉ lệ BOD, COD cao. Hiệu suất
xử lý BOD > 90%.
- Xử lí nitơ, photphat trong nước thải: NH3 - N: 98 - 99%, TN: 80 -
85%, TP: 70 - 75%.
- Đáp ứng nhiều mức độ công suất.
- Có thể hoạt động ở nhiệt độ môi trường hạ thấp (gần 50C).
- Vật liệu làm giá thể: bền, nhỏ gọn, dễ sử dụng.

9
- Cách vận hành đơn giản, gần giống như quá trình bùn hoạt tính thông
thường.
- Dễ dàng nâng cấp, thích hợp cho việc cải tạo hệ thống cũ.
- Ổn định theo biến tải.
- Phát sinh bùn ít.
1.1.3.2. Nhược điểm
Có thể xảy ra quá trình nổi bùn phía sau hệ MBBR theo chu kỳ thay
màng sinh học dẫn đến hiệu quả lắng giảm.
1.2. TỔNG QUAN NƯỚC THẢI SINH HOẠT
1.2.1. Nguồn gốc nước thải sinh hoạt
NTSH là lượng nước được thải bỏ sau khi sử dụng cho các mục đích
sinh hoạt của con người: tắm, giặt giũ, tẩy rửa, vệ sinh cá nhân,... Chúng
thường được thải ra từ các căn hộ, cơ quan, trường học, bệnh viện, chợ và các
công trình công cộng khác. NTSH chiếm khoảng 50% nước thải đô thị. NTSH
của một khu dân cư phụ thuộc vào dân số, vào tiêu chuẩn cấp nước và đặc
điểm của hệ thống thoát nước.
Tiêu chuẩn cấp nước sinh hoạt cho một khu dân cư phụ thuộc vào khả
năng cung cấp nước của các nhà máy nước hay các trạm cấp nước hiện có.
Các trung tâm đô thị thường có tiêu chuẩn cấp nước cao hơn so với các vùng
ngoại thành và nông thôn, do đó lượng NTSH tính trên đầu người cũng có sự
khác biệt giữa thành thị và nông thôn. NTSH ở các trung tâm đô thị thường
thoát bằng hệ thống thoát nước dẫn ra các sông rạch, còn các vùng ngoại
thành và nông thôn do không có hệ thống thoát nước nên nước thải thường
được tiêu thoát tự nhiên vào các ao hồ hoặc thoát bằng biện pháp tự thấm.
Tiêu chuẩn NTSH của các khu dân cư đô thị thường là 100 - 200
L/người.ngày đêm (đối với các nước đang phát triển) và từ 150 - 500
L/người.ngày đêm (đối với các nước phát triển). Ở nước ta hiện nay, tiêu
chuẩn cấp nước dao động từ 120 – 180 L/người.ngày đêm. Ngoài ra, lượng

10
nước thải khu dân cư còn phụ thuộc vào điều kiện trang thiết bị vệ sinh nhà ở,
đặc điểm khí hậu thời tiết tập quán sinh hoạt của người dân.
Lượng NTSH tại các cơ sở dịch vụ, công trình công cộng phụ thuộc
vào loại công trình, chức năng và số người tham gia phục vụ trong đó. Tiêu
chuẩn thải nước của một số loại cơ sở dịch vụ và công trình công cộng được
liệt kê trong Bảng 1.1
Bảng 1.1. Tiêu chuẩn nước thải của một số loại cơ sở dịch
vụ và công trình công cộng
Công trình Đơn vị tính Lưu lượng (lít/ngày)
Nhà ga, sân bay Hành khách 7,5 - 15
Khách sạn
Khách 152 - 212
Nhân viên phục vụ 30 - 45
Nhà ăn Người ăn 7,5 - 15
Siêu thị Người làm việc 26 - 50
Bệnh viện
Giường bệnh 473 - 980
Nhân viên phục vụ 19 - 56
Trường đại học Sinh viên 56 - 113
Bể bơi Người tắm 19 - 45
Khu triển lãm, giải trí Người tham quan 15 - 30
1.2.2. Đặc tính nước thải sinh hoạt
NTSH khi chưa bị phân hủy có màu nâu, chứa nhiều cặn lơ lửng và
chưa bốc mùi khó chịu. Trong NTSH có chứa các chất rắn lơ lửng như phân
người và động vật, xác một số động vật chết, các mảnh vụn của thức ăn, dầu,
mỡ, băng gạc vệ sinh, gỗ, nhựa vụn, vỏ trái cây, và các phế thải khác sau khi
phục vụ cho ăn uống, sinh hoạt của con người thải ra môi trường nước. Dưới

11
điều kiện môi trường nhất định, vi khuẩn tự nhiêu có trong nước và đất tấn
công vào các chất thải gây ra các phản ứng sinh hóa làm biến đổi tính chất
của nước thải. Nước thải sẽ chuyển dần từ màu nâu sang màu đen và bốc mùi
khó chịu.
NTSH luôn có một số hợp chất chứa nitơ, photpho. Mỗi một người,
hàng năm có thể thải ra trung bình 4 kg N và 0,4 kg P trong nước tiểu và 0,55
kg N và 0,18 kg P trong phân. Tải trọng chất thải trung bình một ngày tính
theo đầu người được liệt kê trong Bảng 1.2.
Bảng 1.2. Tải trọng chất thải trung bình một ngày tính theo đầu người
Các chất Tổng chất thải,
g/người.ngày
Chất thải hữu cơ,
g/người.ngày
Chất thải vô cơ,
g/người.ngày
Tổng lượng chất thải 190 110 80
Các chất tan 100 50 50
Các chất không tan 90 60 30
Chất lắng 60 40 20
Chất không lắng 30 20 10
NTSH chiếm khoảng 65 – 80% lượng nước được cấp cho sinh hoạt.
NTSH thường chứa những tạp chất khác nhau. Các thành phần này bao gồm
52% chất hữu cơ, 48% các chất vô cơ. Đặc điểm cơ bản của NTSH là hàm
lượng cao các chất hữu cơ dễ phân hủy sinh học (như hydratcacbon, protein,
mỡ), chất dinh dưỡng (nitơ, photphat), chất rắn và mùi [6]. Trong NTSH còn
chứa nhiều loài sinh vật gây bệnh và các độc tố của chúng. Phần lớn các virut,
vi khuẩn gây bệnh tả, vi khuẩn gây bệnh lỵ, vi khuẩn gây bệnh thương hàn,...
Ngoài ra, nó thường chứa các thành phần dinh dưỡng rất cao. Nhiều trường
hợp, lượng chất dinh dưỡng này vượt quá nhu cầu phát triển của VSV dùng
trong xử lý bằng phương pháp sinh học. Mức độ ô nhiễm theo thành phần hóa
học điển hình của NTSH được phân loại như trong Bảng 1.3.
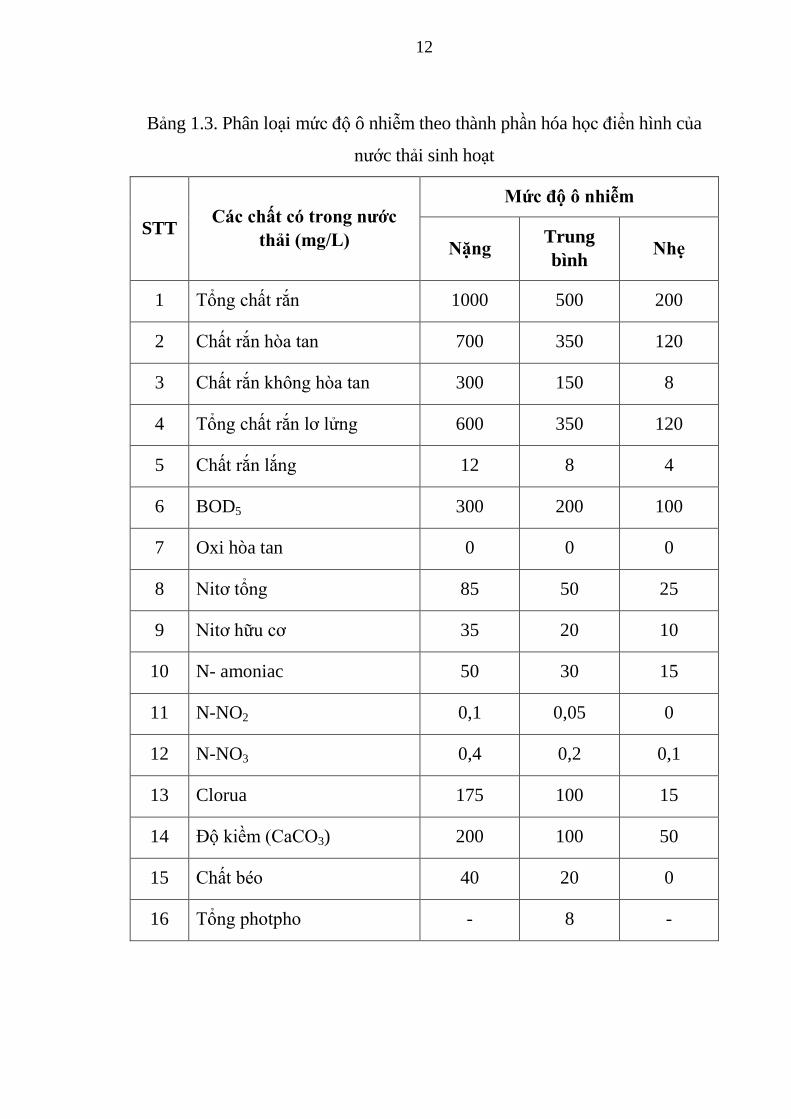
12
Bảng 1.3. Phân loại mức độ ô nhiễm theo thành phần hóa học điển hình của
nước thải sinh hoạt
STT Các chất có trong nước
thải (mg/L)
Mức độ ô nhiễm
Nặng Trung
bình Nhẹ
1 Tổng chất rắn 1000 500 200
2 Chất rắn hòa tan 700 350 120
3 Chất rắn không hòa tan 300 150 8
4 Tổng chất rắn lơ lửng 600 350 120
5 Chất rắn lắng 12 8 4
6 BOD5 300 200 100
7 Oxi hòa tan 0 0 0
8 Nitơ tổng 85 50 25
9 Nitơ hữu cơ 35 20 10
10 N- amoniac 50 30 15
11 N-NO2 0,1 0,05 0
12 N-NO3 0,4 0,2 0,1
13 Clorua 175 100 15
14 Độ kiềm (CaCO3) 200 100 50
15 Chất béo 40 20 0
16 Tổng photpho - 8 -

13
Như vậy, các chỉ tiêu chất lượng NTSH có các khoảng giá trị điển hình
khá lớn: COD - 500 mg/L, BOD5 - 250 mg/L, SS - 220 mg/L, photpho - 8
mg/L, nitơ amoni và nitơ hữu cơ - 40mg/L, pH - 6,8.
1.2.3. Tác động của nước thải sinh hoạt đến môi trường và sức
khỏe con người
NTSH từ các khu dân cư chưa được xử lý hoặc xử lý chưa đạt tiêu
chuẩn xả thải ra nguồn tiếp nhận sẽ gây ô nhiễm cho các khu vực đó. Mặc dù
các nguồn tiếp nhận vẫn có khả năng tự làm sạch song NTSH có các thành
phần vượt qua tiêu chuẩn thải cho phép, lưu lượng, hàm lượng chất ô nhiễm
ngày càng tăng, nên phần lớn chúng đã vượt qua khả năng tự làm sạch của
nguồn tiếp nhận. NTSH thải ra các nguồn tiếp nhận trước hết làm thay đổi lưu
lượng dòng chảy. Do lưu lượng NTSH hàng ngày tương đối cao khi thải vào
nguồn tiếp nhận làm tăng lưu lượng của nguồn và làm thay đổi đặc trưng
cũng như tính chất nguồn tiếp nhận. Các thông số ô nhiễm trong NTSH khá
cao cộng với thời gian dài làm thay đổi tính chất, thành phần nguồn tiếp nhận
làm cho nước vượt qua tiêu chuẩn cho phép.
NTSH là nguồn nước chứa nhiều loại vi khuẩn, VSV có hại, khi lượng
nước thải này không được xử lý thải vào môi trường sẽ là nguồn gây bệnh cho
con người, động vật và làm thay đổi tính chất, đặc trưng nguồn tiếp nhận ảnh
hưởng đến nuôi trồng đánh bắt thủy hải sản. Trong NTSH còn chứa một số
nguyên tố kim loại độc hại, chất độc hóa học sẽ ảnh hưởng trực tiếp hoặc gián
tiếp đến con người động thực vật. Ngoài ra, NTSH chứa lượng dầu mỡ lớn,
gây ra mùi và làm ngăn cách khuếch tán oxy trên bề mặt nước thải.
Cặn lắng chứa phần lớn là chất hữu cơ nên dễ bị oxi hóa sinh hóa làm
oxy hòa tan trong nước bị giảm. Trong lớp cặn lắng phía dưới diễn ra quá
trình lên men sinh ra các loại khí như: CH4, H2S, … thoát ra, xâm nhập vào
nước và không khí, gây mùi, làm nổi váng bọt trên bề mặt. Cặn lắng còn thay
đổi tiết diện dòng xả, thay đổi đáy sông hồ, cản trở dòng chảy. Nồng độ oxi
hòa tan trong sông hồ phía hạ lưu dòng chảy bị thay đổi do tiêu thụ oxi vào
quá trình oxi hóa sinh hóa. Nó ảnh hướng xấu đến sự ổn định của hệ sinh thái
trong hồ. Các nguyên tố dinh dưỡng có trong nước thải như: N, P, K và các
chất khoáng khác khi vào nước sẽ được phù du, thực vật nhất là tảo lam tiêu

14
thụ tạo nên sinh khối trong quá trình quang hợp. Sự phát triển đột ngột của tảo
lam trong nguồn nước giàu dinh dưỡng làm cho nước có mùi và độ màu tăng
lên. Hiện tượng này gọi là hiện tượng phú dưỡng.
1.3. CÁC PHƯƠNG PHÁP XÁC ĐỊNH NỒNG ĐỘ CHẤT Ô NHIỄM
TRONG NƯỚC THẢI
1.3.1. Các phương pháp xác định nồng độ Photphat
Photpho tồn tại ở dạng octophotphat ( 2 3
2 4 4 4 3 4H PO ,HPO ,PO ,H PO ) hay
poliphotphat Na3(PO3)6 và photphat hữu cơ.
Octophotphat có thể xác định bằng phương pháp so màu với thuốc thử
là amoni molipdat và thiếc clorua, còn poliphotphat và photphat hữu cơ cần
chuyển hóa thành octophotphat qua phản ứng với axit sau đó xác bằng
phương pháp so màu.
Lượng photpho tồn tại dưới dạng octophotphat có thể xác định thông
qua các phương pháp trọng lượng khi hàm lượng photpho lớn, phương pháp
thể tích khi nồng độ photphat lớn hơn 50 mg/L, phương pháp so màu khi xác
định hàm lượng photpho trong nước và nước thải.
a. Phương pháp Amino Acid và axit ascorbic
Phospho hoạt tính được xác định cơ bản theo hai bước với phương
pháp Ascorbic Acid (thang thấp) hoặc Amino Acid (thang cao). Bước đầu tiên
liên quan đến phản ứng của orthophosphate với molybdate trong dung dịch
axit, tạo thành phức hợp phosphomolybdate màu vàng. Phức hợp
phosphomolybdate sau đó được khử bởi amino acid hoặc ascorbic acid, tạo
nên màu xanh molybdenum đặc trưng.
b. Phương pháp dùng amoni molipdat
Phản ứng giữa ion octophosphat và một dung dịch axit chứa molipdat
và ion antimon tạo ra phức chất antimon phosphomolipdat. Khử phức chất
bằng axit ascobic tạo thành phức chất molipden màu xanh đậm. Đo độ hấp thụ
của phức chất để xác định nồng độ octophosphat [20]. Xác định polyphosphat
và một số hợp chất phospho hữu cơ bằng cách thủy phân chúng với axit

15
sulfuric để chuyển thành dạng octophosphat phản ứng với molipdat. Một số
hợp chất phospho hữu cơ được chuyển thành octophosphat bằng vô cơ hóa với
pesulfat. Nếu cần xử lý cẩn thận thì vô cơ hóa với axit nitric-axit sulfuric.
c. Phương pháp Molybdovanadate
Phospho hoạt tính kết hợp với molybdate trong môi trường axit để tạo
thành phức hợp phosphomolybdate. Vanadi, chứa trong chất thử
Molybdovanadate, phản ứng với phức để tạo thành axit
vanadomolybdophosphoric [21]. Cường độ của màu vàng tạo thành tỷ lệ
thuận với nồng độ phospho phản ứng.
1.3.2. Các phương pháp xác định nồng độ amoni
Trong nước và nước thải để xác định amoni có thể sử dụng một trong
ba phương pháp khác nhau:
- Tách amoni bằng cách chưng cất dung dịch chứa nó trong môi trường
kiềm, sau đó chuẩn độ bằng axit.
- Xác định điện thế sử dụng các điện cực nhạy ion
- Xác định bằng phương pháp trắc quang (so màu)
Để lựa chọn phương pháp thì nồng độ amoni và các chất gây ảnh
hưởng là những yếu tố chính cần xem xét. Mặt khác, cách lấy mẫu và xử lý
bảo quản mẫu cũng quyết định đến độ chính xác của phương pháp xác định
nitơ - amoni. Các mẫu phân tích ngay hoặc sớm hầu hết đều cho kết quả chính
xác. Nếu các mẫu được phân tích trong vòng 24h sau khi lấy mẫu, thì không
phải axit hóa mẫu và phải bảo quản lạnh ở nhiệt độ 4ºC. Trong ba phương
pháp xác định amoni kể trên, phương pháp chuẩn độ thể tích luôn phải có
bước chưng cất còn hai phương pháp sau thì có thể phải chưng cất và cũng có
thể không.
a. Phương pháp thể tích
Phương pháp áp dụng trong khoảng nồng độ từ 5mg đến 100mg N-
amoni/L.
Mẫu được đệm hóa bằng đệm borat ở pH = 9,5 để làm giảm sự thủy
phân xianat và các hợp chất hữu cơ chứa nitơ. Sau đó sử dụng bình Kjeldahl

16
để cất và hấp thụ amoniac vào dung dịch axit boric. Xác định amoni trong
phần cất bằng cách chuẩn độ với axit HCl 0,02 M hoặc với dung dịch HCl 0,1
M khi mẫu có hàm lượng amoni cao[22].
b. Phương pháp điện cực chọn lọc
Đây là phương pháp có khả năng xác định nitơ trong một khoảng rộng
với hai phương thức: sử dụng điện cực chọn lọc – ứng dụng trong khoảng
nồng độ từ 0,03 ÷ 1400 mg NH3-N/l và điện cực chọn lọc có sử dụng phương
pháp thêm xác định trong khoảng 2,5-30 mg NH3-N/l. Phương pháp này có
thể sử dụng để xác định amoni trong nhiều đối tượng như nước uống, nước
mặt, nước thải đô thị và nước thải công nghiệp.
Mẫu được đệm hóa bằng đệm borat ở pH = 9,5 và chưng cất và hấp thụ
vào 50 ml dung dịch H2SO4 0,04 N. Điện cực chọn lọc amoni sử dụng một
màng thấm kị khí để tách dung dịch mẫu khỏi một dung dịch NH4Cl trong
điện cực. Amoni hòa tan (NH3 và 4NH ) được chuyển thành NH3 nhờ tăng pH
đến 11 bằng một dung dịch kiềm mạnh. NH3 sẽ khuếch tán qua màng và làm
thay đổi pH. Sự thay đổi pH này được đo bằng một điện cực pH.
c. Phương pháp trắc quang
Trong các phương pháp trắc quang khác nhau để xác định amoni
phương pháp sử dụng thường xuyên nhất là phương pháp theo tiêu chuẩn
TCVN 6179-1:1996 hay phương pháp Nesler hoặc phương pháp phenat.
* Phương pháp thuốc thử thymol
Đo quang phổ ở bước sóng khoảng 655 nm của hợp chất màu xanh
được tạo bởi phản ứng của amoni với salixylat và ion hypoclorit có sự tham
gia của natri nitrosopentaxyano sắt (III) taxyano sắt (III) (natri nitroprusiat).
Các ion hypoclorit được tạo trong situ bằng cốc thuỷ phân kiềm của N,
N/dicloro-1,3,5-triazin2,4,6(1H,3H,5H)trion, muối natri (natri
diclorosoxyanurat). Phản ứng của cioramin với natri salixylat xảy ra ở độ pH
12.6 có sự tham gia của natri nitroprusiat. Bất kỳ chất cloramin nào có mặt
trong mẫu thử cũng đều được xác định. Natri xitrat có trong thuốc thử để cản
sự nhiễu do các cation, đặc biệt là canxi và magie [23].

17
* Phương pháp Nessler
Phương pháp sử dụng thuốc thử Nessler (K2HgI4) trong môi trường
kiềm được thêm vào một dung dịch muối amoni loãng, khi đó amoni sẽ nhanh
chóng phản ứng với thuốc thử tạo phức có màu vàng đến nâu sẫm. Màu tạo ra
giữa thuốc thử Nessler và amoni có cực đại hấp thụ quang ở bước sóng 420 –
500 nm tùy thuộc vào nồng độ amoni trong mẫu.[24]
Thuốc thử Nessler được sử dụng để xác định amoni trong dung dịch
amoni rất loãng và trong nước tự nhiên.
* Phương pháp phenat
Phương pháp dựa trên phản ứng tạo phức màu xanh đậm indophenol –
giữa amoni, phenol và hypoclorit. Phản ứng được xúc tác bởi muối Mn(II)
hoặc natri nitroprusit. Phương pháp đo màu ở bước sóng 640 nm.
Ngoài các phương pháp chủ yếu trên, các phương pháp phân tích dòng
chảy (FIA), sắc ký ion (IC), phương pháp enzym, phân tích dòng liên tục
(sequential injection analysis-SIA) và sắc ký khí – khối phổ (GC/MS) cũng
được sử dụng để xác định amoni trong nước.
d. Phương pháp trao đổi ion
Amoni trong dung dịch có thể xác định bằng cách cho dung dịch đi qua
cột trao đổi cation dạng H . Ion 4NH thay thế H trên nhựa trao đổi ion đẩy
H đi vào dung dịch, ta chuẩn độ H sẽ xác định được 4NH .[25]
3 4 3 4R SO NH R SO NH H (1.10)
e. Phương pháp phân tích dòng chảy sử dụng detector quang
Đo quang hợp chất màu xanh tạo ra bởi phản ứng giữa amoni với ion
salixilat và hypoclorit khi có mặt natri nitrosopentaxyanoferat(III) -
natrinitropussiat ở bước sóng khoảng 650nm. Ion hypoclorit được sinh ra thủy
phân 1,3 – diclo – 5 sodio – 1,3,5 – triazinanetrion (natri dicloisoxyanurat)
trong kiềm, và phản ứng với amoni tạo ra cloramin. Cloramin phản ứng với
natri salixilat ở pH 12,6 khi có mặt nitropussiat. Mọi cloramin có trong mẫu
đều được định lượng [23].

18
Natri xitrat được thêm vào để che một cation đặc biệt là 2Ca và 2Mg
(mọi phản ứng đều được thực hiện tự động nhờ kỹ thuật dòng chảy) chất màu
được đo bằng một máy trắc phổ dòng tự động. Hai cấu hình hệ thống đo được
quy định. Một hệ thống có khối thẩm tách và thích hợp để xác định nồng độ
amoni tính theo nitơ đến 50mg/l. Cấu hình không có khối thẩm tách và thích
hợp để xác định nồng độ amoni tính theo nitơ đến 0,5mg/l.
1.3.3. Các phương pháp xác định nồng độ nitrat
a. Phương pháp chuẩn độ
Người ta có thể xác định nitrat theo phương pháp này dựa trên phản
ứng khử 3NO về các trạng thái oxi hoá thấp hơn bằng các chất khử thích hợp.
Sau đó tiến hành phép chuẩn độ (có thể sử dụng chuẩn độ trực tiếp hay chuẩn
độ ngược).
Với phép chuẩn độ ngược thì một lượng chính xác dung dịch chuẩn
2Fe được cho dư so với lượng cần thiết vào dung dịch mẫu. Sau đó lượng dư
2Fe được chuẩn độ bằng dung dịch 2
2 7Cr O với chất chỉ thị là ferroin. Các
phản ứng xảy ra như sau:
2 3
3 2NO 3Fe 4H NO 3Fe 2H O (1.11)
2 2 3 3
2 7 26Fe Cr O 14H 6Fe 2Cr 7H O (1.12)
Phản ứng giữa 2Fe và 3NO xảy ra nhanh hơn khi đung nóng dung dịch
và có mặt của lượng dư axit H2SO4 65%. Phương pháp đơn giản, dễ thực hiện.
Cho phép xác định lượng 3NO với nồng độ cao 10-3M đến 10
-4M. Tuy nhiên,
do NO sinh ra phản ứng với oxi không khí tạo thành các chất có khả năng bị
khử hay bị oxi hoá bởi 2Fe nên trong quá trình phản ứng và chuẩn độ phải
được tiến hành trong môi trường khí CO2. Điều này được thực hiện bằng cách
thêm một lượng nhỏ NaHCO3 trước khi đun nóng và chuẩn độ. Phương pháp
này có thể xác định cả lượng nhỏ và lượng lớn 3NO trong mẫu phân tích [25].
b. Phương pháp trắc quang
* Phản ứng với thuốc thử axit phenoldisulfonic

19
Trong môi trường axit sunfuric đậm đặc, nitrat tham gia phản ứng với
axit phenoldisulfonic tạo thành phức chất không màu nitrophenoldisulfonic. Ở
môi trường bazơ mạnh phức này có màu vàng bền trong vòng 15-20 phút và
được đo bằng quang phổ kế ở bước sóng λ= 410 nm [26].
(1.13)
* Phản ứng với thuốc thử natri salixylat
Trong môi trường axit sulfuric đậm đặc, nitrat tham gia phản ứng với
natri salixylat tạo thành phức màu p-nitrosalixylat natri hoặc sản phẩm có thể
là o-nitrosalixylat natri. Ở môi trường bazơ mạnh phức này có màu vàng và
được đo bằng máy đo quang tại bước sóng λ = 410nm.
Phương trình phản ứng
(1.14)

20
Trong môi trường kiềm, phức chất phân ly thành ion gốc axit làm phân
tử trở nên phân cực. Vì vậy, các electron hóa trị chuyển động hỗn loạn hơn
nên phức chất có cường độ màu tăng và hấp thụ ánh sáng ở bước sóng dài.
Dung dịch phức màu bền trong vòng 10 - 15 phút [26].
Ưu điểm của hai phương pháp này là có độ nhạy cao, sử dụng đơn giản.
Bên cạnh đó chúng cũng có nhiều nhược điểm, phải loại trừ các ion cản do phải
cô, rất mất thời gian (để xác đinh được 3NO trong nước mất khoảng 5 - 6 giờ).
* Phản ứng với thuốc thử diphenylamin
Nitrat phản ứng với diphenylamine trong môi trường axit sunfuric đậm
đặc, sản phẩm tạo ra là muối có màu tím xanh. Phức màu ổn định trong vòng
2-3 giờ. Cường độ của màu tỷ lệ với nồng dộ nitrat trong nước. Tốc độ ôxi
hoá diphenilamin bằng nitrit lớn gấp nhiều lần so với oxi hoá bằng nitrat. Cụ
thể nếu oxi hoá bằng nitrit thì màu cực đại xuất hiện sau khoảng 15 phút,
trong khi đó nếu oxi hóa bằng nitrat thì màu phát triển trong vòng vài giờ. Vì
thế không nên tiến hành so màu quá sớm khi màu tím xanh do oxi hoá bằng
nitrat chưa ổn định. Nếu so màu quá sớm thì có thể xác định được nitrit là
chính [27]. Ngoài ra diphenylamin còn bị oxi hóa bởi các ion 4MnO , 2
2 7Cr O ,
[Fe(CN)6]3-
, 3ClO , 3BrO ... Các chất khử mạnh 2S , 3SO , 2
2 3S O ... bị oxi hóa
bởi hỗn hợp axit nitric và axit sunfuric đều gây cản trở cho việc định lượng
3NO .
c. Phương pháp cực phổ
Trong môi trường chất điện li với sự có mặt ion 3La hay 2Ba , ion
3NO cho sóng cực phổ tại thế từ -1,1 đến -1,4 V.
Để xác định nitrat người ta thường dùng sóng xúc tác urani. Trong môi
trường tạo phức nền Na2CO3 0,1M thì 2
2UO chỉ cho một sóng định lượng có
E1/2= 0,9 -1,1V phụ thuộc vào nồng độ 3NO .
Ngoài ra nitrat còn xác định trên điện cực giọt thuỷ ngân (DME) với
dung dịch nền là H2SO4 đặc : phenol = 4:1. Thế bán sóng của nitro phenol tạo
thành ở - 0,2 V. Giới hạn xác định 3NO theo phương pháp này là 5ppm.

21
Phương pháp này không bị ảnh hưởng bởi ion 2NO nên có độ chính xác
cao [21].
d. Phương pháp sắc kí trao đổi ion
Cơ sở của IEC là sự cạnh tranh các nhóm tích điện trái dấu trên chất
trao đổi giữa ion 3NO và ion 2NO chứa trong pha động gồm dung dịch mẫu
phân tích, đệm Lithium borate gluconate và dung môi acetonnitrile tại pH =
6,5. Pha động sẽ tương tác với pha t nh là cột sắc ký trao đổi ion Waters IC-
PacTM Anion HC 150 x 4,6 mm column. Hệ thống sắc kí làm việc với tốc độ
dòng là 1ml/phút với detecter là máy đo quang UV-VIS tại bước sóng 205nm.
Dung tích mẫu là 40 m [28].
Dựa vào thời gian lưu của ion 3NO và ion 2NO trong dung dịch chuẩn
ta có thể xác định được đỉnh nitrat/nitrit trong mẫu cần phân tích trong tập
hợp các chất mà sắc ký trao đổi ion tách ở 2 pic tương ứng.
Phương pháp khá hiệu quả, độ chọn lọc cao, ứng dụng rộng rãi. Sử
dụng lượng mẫu nhỏ. Tuy nhiên, hệ thống máy móc đắt, cần loại trừ màu của
dung dịch phân tích.
1.3.4. Các phương pháp xác định nồng độ nitrit
a. Phương pháp phân tích thể tích
Nguyên tắc: Nitrit được xác định dựa trên phản ứng oxi hóa nitrit thành
nitrat sử dụng tác nhân là KMnO4. Dung dịch chuẩn độ nhận biết thông qua
màu hồng của KMnO4. Phương trình chuẩn độ:
2
4 2 3 22MnO 5NO 6H 2Mn 5NO 3H O (1.15)
Trong môi trường axit ion 2NO bị phân hủy thành NO và NO2 theo
phương trình:
2 2 2 2NO H HNO NO NO H O (1.16)
Do đó cần phải đảo ngược thứ tự phản ứng (nhỏ từ từ dung dịch 2NO
vào dung dịch 4MnO trong môi trường axit.

22
Phương pháp này có độ nhạy thấp và tính chọn lọc kém nên thường
được dùng để xác định lại nồng độ dung dịch chuẩn gốc.
b. Phương pháp phân tích trọng lượng
Nguyên tắc: Nitrit có thể tạo thành muối khó tan với 2,4 –diamino-6
oxypyidin và 2,4 - nino – nitrozo – oxypyridin. Sấy khô muối ở nhiệt độ 120
đến 140 ºC rồi cân lấy khối lượng của muối. Ngoài ra chúng ta có thể xác
định nitrit gián tiếp bằng cách dựa trên phản ứng:
3HNO2 + AgBrO3 → AgBr + 3HNO3 (1.17)
Lọc và rửa kết tủa bằng dung dịch H2SO4 (1:4) và sấy ở nhiệt độ 85 ºC
đến 90 ºC rồi đem cân. Từ lượng AgBr ta có thể tính được 2NO .
c. Phương pháp trắc quang
* Thuốc thử Griess
Thuốc thử Griess là hỗn hợp axit sunfanilic và α- naphtylamin hòa tan
trong axit axetic 10%. Đầu tiên ion nitrit phản ứng với axit sunfanilic tạo
thành muối điazo:
(1.18)
Sau đó muối này phản ứng với α-naphtylamin tạo thành hợp chất azo
có màu hồng:
(1.19)
Độ hấp thụ quang được đo ở bước sóng 520 nm. Phản ứng thường được
tiến hành ở pH khoảng 1,7 – 3 và ở khoảng nhiệt độ là 0 – 50C. Nhiệt độ càng
cao phản ứng xảy ra càng nhanh nhưng lại dễ dàng bị phân huỷ thành các hợp
chất khác. Phương pháp có độ chọn lọc cao, khi có một lượng rất lớn (thường

23
gấp 100 lần) cloamin, clo, thiosunfat, natrithyophotphat và 3Fe thì sai số của
phương pháp là 10% [29].
Ngoài thuốc thử Griess, người ta còn có thể sử dụng dẫn xuất của Griss
như hỗn hợp thuốc thử 4 - amino benzene sunfonamit (NH2C6H4SO2NH2) và
N-(1-naphtyl) - 1,2 diaminoetan hidroclorua (C10H7NH-CH2-CH2-NH2.HCl).
Khi sử dụng hỗn hợp thuốc thử này có màu tím hồng ở pH = 1,9 ± 0,1 và cực
đại hấp thụ ở 540 nm [27, 29, 31].
* Thuốc thử axit barbituric
Nitrit phản ứng với axit barbituric trong môi trường axit tạo ra violuric
(dẫn xuất nitrosoaxit), trong nước có màu tím. Độ hấp thụ quang được đo ở
bước sóng 310 nm, khoảng tuyến tính 0,00 - 3,22 ppm. Hệ số hấp thụ mol
phân tử là 15330 ± 259,7 (95%). Phương pháp này áp dụng thành công để xác
định nitrit trong nước tự nhiên. Giới hạn định lượng là 1,66 g 2NO trong
100ml dung dịch làm việc tương ứng với lượng tối thiểu 9,5 ppb 2NO trong
mẫu nước. Nếu nồng độ nitrit thấp (3,0 g 2NO /L mẫu) thì ta sử dụng phương
pháp pha loãng mẫu với RSD thấp hơn 0,5 % [32].
Phương trình phản ứng:
(1.20)
1.4. NGUYÊN TẮC XÁC ĐỊNH GIỚI HẠN PHÁT HIỆN VÀ GIỚI HẠN
ĐỊNH LƯỢNG
Theo IUPAC, giới hạn phát hiện là nồng độ nhỏ nhất ( Lx ) của chất
phân tích tạo ra được một tín hiệu có thể phân biệt một cách tin cậy so với tín
hiệu trắng (hay tín hiệu nền). Giá trị của Lx được cho bởi phương trình :
Lx = i ib bx + k. S

24
Trong đó ibx là giá trị trung bình của nồng độ chất trong mẫu trắng,
ibS
là độ lệch chuẩn của tín hiệu chất phân tích trong mẫu trắng, k là đại lượng số
học được chọn theo độ tin cậy mong muốn. Với độ tin cậy 99% và bậc tự do
là 8 (tiến hành n=9 số thí nghiệm lặp lại) thì k 3 . Có hai cách tính LOD :
- Cách 1 : Tiến hành thí nghiệm để lập phương trình đường chuẩn. từ
đó xác định Sy (độ lệch chuẩn của tín hiệu y trên đường chuẩn) và chấp nhận
Sd = Sy. Như vậy LOD là nồng độ của chất phân tích cho tín hiệu 3Sy. Từ
phương trình đường chuẩn tính được nồng độ của chất phân tích. Cách này có
thể tiến hành nhanh và không tốn thời gian nhưng không thật chính xác vì đã
chấp nhận sự phụ thuộc của tín hiệu vào nồng độ mà thông thường mỗi
khoảng nồng độ có một hệ số góc khác nhau và đường chuẩn lập ra thường
trong khoảng nồng độ cách xa với LOD.
- Cách 2 : Tiến hành n thí nghiệm xác định nồng độ mẫu trắng thu được
các giá trị ybi (i =1÷n). Từ đó tính iby
và Sb
ttheo công thức :
iby = (∑ybi)/n; Sb=(∑(ybi-iby )
2/(n-1))
1/2
Từ iby thay vào phương trình đường chuẩn tính được
ibx thì :
LOD = xL = blank3.S A
B
Chúng tôi tiến hành đo lặp lại 9 lần tín hiệu của dung dịch nồng độ nhỏ
với các điều kiện ghi đo như lập đường chuẩn, chấp nhận sự sai khác độ lệch
chuẩn giữa dung dịch đo và mẫu trắng là không đáng kể. Vì bỏ qua A trong
phương trình đường chuẩn nên giới hạn phát hiện (LOD) và giới hạn định
lượng (LOQ) được xác định theo công thức 3σ và 10σ như sau :
LOD = ib
'
3.S
B; LOQ = ib
'
10.S
B
Với ibS là độ lệch chuẩn của tin hiệu mẫu đo sau 9 lần đo lặp lại. B
’ là
hệ số góc của phương trình hồi quy tính lại.

25
1.5. ĐÁNH GIÁ ĐỘ ĐÚNG CỦA PHƯƠNG PHÁP XÁC ĐỊNH 4NH .
2NO . 3NO . 3
4PO THÔNG QUA HIỆU SUẤT THU HỒI
Muốn xác định độ đúng cần phải tìm được giá trị đúng, có nhiều cách
khác nhau để xác định độ đúng bao gồm việc so sánh kết quả thực nghiệm với
kết quả thực hiện bởi một phương pháp đối chiếu hoặc sử dụng mẫu đã biết
nồng độ (mẫu kiểm tra hoặc mẫu chuẩn được chứng nhận) và phương pháp
xác định độ thu hồi (độ tìm lại).
Cách 1: So sánh với phương pháp chuẩn/đối chiếu
Phân tích mẫu chuẩn hoặc mẫu thử, thực hiện 10 lần bằng phương pháp
khảo sát và bằng một phương pháp đối chiếu. Phương pháp đối chiếu tốt nhất
là phương pháp tiêu chuẩn của các tổ chức có uy tín, nếu không phương pháp
đối chiếu là phương pháp đã qua thẩm định cho kết quả tin cậy trong dải đo
đang thực hiện. Tính toán các kết quả trung bình và độ lặp lại (hệ số biến
thiên) của hai phương pháp.
Đánh giá độ tương đồng về độ chụm của hai phương pháp bằng cách so
sánh phương sai S2 của hai phương pháp đó, dùng tiêu chuẩn F (Fisher) và so
sánh hai trị giá trung bình bằng tiêu chuẩn t (Student). Việc bố trí các thí
nghiệm phải được thực hiện theo phương pháp tham chiếu một cách nghiêm
ngặt và các phép đo phải được tiến hành dưới điều kiện lặp lại.
- So sánh hai phương sai (chuẩn F – Fisher)
Chuẩn F dùng để so sánh độ lặp lại của hai tập số liệu hoặc hai phương
pháp khác nhau. Với tập số liệu nhỏ, tính toán giá trị Ftn (F thực nghiệm) theo
công thức sau đây và so sánh với giá trị Fc (F tra bảng).
2
1
2tn
2
1SF
S
Trong đó:
+ Ftn là F thực nghiệm
+ S12 , S2
2 : Các phương sai của hai phương pháp với quy ước S1
2 > S2
2

26
Nếu: Ftn ≤ Fc (α, k1, k2): Hai phương pháp có độ lặp lại (độ chụm)
giống nhau.
Nếu: Ftn > Fc (α, k1, k2): Hai phương pháp có độ lặp lại khác nhau. trong
trường hợp này nếu độ lặp lại của phương pháp thử nghiệm khác với phương
pháp chuẩn thì cần xem xét thêm về độ lặp lại như đã mô tả trong phần trên.
Trong đó: Fc (α, k1, k2): Giá trị F tra bảng, với: k1, k2: Bậc tự do (k1 = n1
- 1; k2 = n2 - 1); n1, n2: Số lần làm thực nghiệm của hai phương pháp; α: Mức
ý ngh a (significance level), thường lấy α = 0,05 (tương ứng với độ tin cậy
(confidence level) 95%)
- So sánh hai giá trị trung bình (chuẩn t – Student)
Chuẩn t được dùng để so sánh xem có sự khác nhau giữa giá trị thực
nghiệm và giá trị thực hay không. Phương pháp này được ứng dụng hoặc để
so sánh kết quả thực nghiệm với giá trị chuẩn trong mẫu kiểm tra (xem thêm
cách 2) hoặc để so sánh kết quả của phương pháp phân tích với phương pháp
đối chiếu.
Trước khi so sánh hai giá trị trung bình cần so sánh hai phương sai. Với
số lần phân tích nhỏ hơn 30, khi hai phương sai có sự đồng nhất, tính độ lệch
chuẩn chung và giá trị ttn (t thực nghiệm) theo công thức sau đây và so sánh
với giá trị tc (t tra bảng):
2 22 1 1 2 2c
1 2
(n 1)S (n 1)SS
n n 1
1 2
tn
2
c
1 2
1 1
x xt
Sn n
Trong đó:
+ ttn : Giá trị t thực nghiệm
+ tc(α, k): Giá trị t tra bảng mức ý ngh a α, bậc tự do k

27
+ n1, n2: Số lần thí nghiệm lần lượt của phương pháp thử nghiệm và
phương pháp đối chiếu
+ 2
1S , 2
2S : Phương sai lần lượt của phương pháp thử nghiệm và phương
pháp đối chiếu
+ 1x , 2x : Giá trị trung bình lần lượt của phương pháp thử nghiệm và
phương pháp đối chiếu
Nếu ttn ≤ tc(α. k): Không có sự khác nhau về kết quả của hai phương pháp.
Nếu ttn > tc(α. k): Có sự khác nhau về kết quả của hai phương pháp,
phương pháp thử nghiệm mắc sai số hệ thống.
Trong trường hợp hai phương sai không đồng nhất (khác nhau có ý
ngh a). tính giá trị ttn và bậc tự do k theo các công thức sau và so sánh như trên.
1 2
tn 2 2
1 2
1 2
x xt
S Sn n
2
2 2
1 2
2 21 2
1 2k 2
2 21 2
1 2
1 1
S S
n n
S Sn n
n n
Cách 2: Sử dụng vật liệu chuẩn (Reference material)
Vật liệu chuẩn (còn gọi là mẫu chuẩn) là mẫu phân tích có hàm lượng
chất phân tích đã được xác định trước và là đúng. Có nhiều cấp vật liệu chuẩn
khác nhau. trong đó cao nhất là CRM (certified reference material – vật liệu
chuẩn được chứng nhận) được cung cấp bởi các tổ chức có uy tín trên thế giới.
Các mẫu CRM luôn có kết quả kèm theo khoảng dao động, do đó khi
phân tích mẫu CRM có thể đánh giá được độ đúng dựa vào khoảng dao động
cho phép (ví dụ: Mẫu thịt chỉ tiêu clenbuterol là 1 ng/g ± 0,05 ng/g, nếu kết
quả phân tích được trong khoảng từ 0,95-1.05 thì đạt).

28
Nếu không có các mẫu CRM có thể sử dụng các mẫu kiểm tra
(QCQuality Control) đã biết nồng độ. Phòng thử nghiệm có thể tự chuẩn bị
các loại mẫu này, hoặc sử dụng các mẫu thực có hàm lượng đã biết hoặc sử
dụng các mẫu lưu từ chương trình so sánh liên phòng thử nghiệm. Trong
trường hợp khác phòng thử nghiệm có thể sử dụng các mẫu thêm chuẩn để
đánh giá độ đúng, nội dung này sẽ được mô tả cụ thể trong cách 3 dưới đây.
Nhiều tổ chức có uy tín như USFDA, EURACHEM. ICH... quy định
tính độ chệch (bias) để xác định độ đúng như sau:
_
x100
Trong đó:
+ ∆: Độ chệch (bias), %
+ x :Giá trị trung bình của kết quả thử nghiệm
+ μ: Giá trị thực hoặc giá trị được chấp nhận là đúng USFDA quy định
độ chệch của các phương pháp xác định dư lượng phải không được lớn hơn
15% và không lớn hơn 20% tại LOQ.
Có thể sử dụng chuẩn t để đánh giá kết quả như sau:
Phân tích mẫu chuẩn lặp lại 10 lần (tối thiểu 6 lần), tính giá trị trung
bình và độ lệch chuẩn. từ đó tính giá trị ttn theo công thức sau đây và so sánh
với tc (p) :
tn 2
x
S
n
t
Trong đó:
+ ttn: Giá trị t thực nghiệm
+ tc(α. k): Giá trị t tra bảng với mức ý ngh a 0,05 (xem phụ lục 1) và
bậc tự do k = n - 1.
+ µ: Giá trị thực hoặc giá trị được chấp nhận (tham chiếu)

29
+ x : Giá trị trung bình của phương pháp thử nghiệm
+ S2 : Phương sai của phương pháp thử nghiệm, n: Số lần thí nghiệm
Nếu ttn ≤ tc : Không có sự khác nhau về kết quả của giá trị trung bình so
với giá trị tham chiếu ở mức ý ngh a α, tức là phương pháp có độ đúng đạt
yêu cầu.
Nếu ttn > tc : Có sự khác nhau về kết quả của phương pháp thử nghiệm
so với kết quả tham chiếu ở mức ý ngh a α, phương pháp thử nghiệm mắc sai
số hệ thống.
Cách 3: Xác định độ thu hồi
Các phương pháp tính độ đúng theo cách 1 hay cách 2 đều gặp những
khó khăn nhất định. Trong nhiều trường hợp không thể tìm hoặc áp dụng một
phương pháp tiêu chuẩn để so sánh kết quả, cũng như không thể dễ dàng có
được các mẫu chuẩn hoặc mẫu chuẩn được chứng nhận phù hợp với phương
pháp. Việc xác định độ đúng do đó có thể thực hiện thông qua xác định độ thu
hồi (còn gọi là độ tìm lại) của phương pháp.
Thêm một lượng chất chuẩn xác định vào mẫu thử hoặc mẫu trắng.
phân tích các mẫu thêm chuẩn đó, làm lặp lại tối thiểu bốn lần bằng phương
pháp khảo sát. tính độ thu hồi theo công thức sau đây:
- Đối với mẫu thử:
m c m
c
R% 100C CC
- Đối với mẫu trắng:
tt
c
R% 100CC
Trong đó: R%: Độ thu hồi. %
Cm+c: Nồng độ chất phân tích trong mẫu thêm chuẩn
Cm: Nồng độ chất phân tích trong mẫu thử
Cc: Nồng độ chuẩn thêm (lý thuyết)

30
Ctt: Nồng độ chất phân tích trong mẫu trắng thêm chuẩn
Sau đó tính độ thu hồi chung là trung bình của độ thu hồi các lần làm
lặp lại.
Thêm chất chuẩn ở ba mức nồng độ là mức thấp, trung bình và cao
trong khoảng nồng độ làm việc. Theo quy định của hội đồng châu Âu đối với
các chỉ tiêu an toàn (các chỉ tiêu thuộc nhóm độc có quy định giới hạn cho
phép, ví dụ tồn dư hormon, kháng sinh, hóa chất bảo vệ thực vật...) thêm
chuẩn vào mẫu trắng ở ba mức nồng độ tại 0,5 lần, 1 lần và 2 lần giới hạn cho
phép (MRL).
Hội đồng châu Âu cũng quy định đối với các mẫu phân tích hàng ngày
(routine) các chỉ tiêu thuộc cùng nhóm (ví dụ: hóa chất bảo vệ thực vật nhóm
clo hữu cơ) cần kiểm soát chất lượng bằng cách phân tích mẫu thêm chuẩn tối
thiểu 10% số lượng chất, các chất khác cần thay phiên kiểm tra với tần suất
tối đa 1 năm/lần cho từng chất.

31
CHƯƠNG 2. THỰC NGHIỆM
2.1. NỘI DUNG NGHIÊN CỨU
Bản luận văn này thực hiện nghiên cứu khảo sát hàm lượng
orthophosphat và một số hợp chất của nitơ gồm amoni, nitrit, nitrat trong hệ
xử lý nước thải sử dụng giá thể vi sinh chuyển động.
Mẫu nước thải sử dụng trong quá trình thí nghiệm được lấy từ bể mô
phỏng hệ thống MBBR. Nước thải sử dụng trong quá trình thí nghiệm có hàm
lượng các chất ô nhiễm COD = 500 mg/L; N = 16,67 mg/L; P = 3,33 mg/L,
giá trị pH trong khoảng 7,5 – 8,5.
- Hàm lượng bùn hoạt tính trong bể MLSS = 3000 mg/L.
- Thực nghiệm được tiến hành trong bể 5 lít
- Lấy 500 mL nước thải ban đầu, đây là mẫu nước thải đầu vào không
chứa bùn. Kí hiệu mẫu: NT
- Trộn đều lượng nước thải với bùn bằng máy sục khí, hàm lượng bùn
trong bể 3000 mg/L. Sau khi bùn và nước thải trộn đều với nhau, lấy 500 mL
mẫu, thu được lượng nước thải đầu vào chứa bùn, Kí hiệu mẫu NT00.
- Sục khí liên tục, cứ sau 2 giờ, 4 giờ, 6 giờ, 8 giờ, 24 giờ lấy 500 mL
mẫu vào các chai lần lượt được các mẫu NT02, NT04,NT06, NT08, NT24. (Các
mẫu đều chứa bùn).
- Sau khi sục khí 24 tiếng, tắt máy sục khí, cho lắng 2 giờ. Sau đó lấy
500 mL nước trong phía trên (không chứa bùn), kí hiệu mẫu NTSL.
Tiến hành lấy mẫu để phân tích các chỉ tiêu phốt phát, amoni, nitrat,
nitrit. Sau đó, tiến hành thí nghiệm với thời gian sục khí 8 giờ đối với nước
thải thực tế. Tiến hành thí nghiệm trong 30 ngày lấy mẫu theo tần suất 5
ngày/lần. Các nội dung thực nghiệm bao gồm:
- Bước 1: Khảo sát quy trình phân tích các thông số amoni, nitrit, nitrat,
phosphat cho mẫu nước thải trước và sau xử lí bằng phương pháp quang phổ
hấp thụ phân tử theo các tiêu chuẩn trong và ngoài nước. Tiến hành phân tích
mẫu thực tế theo các quy trình đã xây dựng dựa trên các tiêu chuẩn hiện hành.

32
Đưa ra ưu nhược điểm của các phương pháp để có sự lựa chọn phương pháp
cho phù hợp với tình hình thực tế.
- Bước 2: Đánh giá và so sánh số liệu theo 2 phương pháp phân tích
khác nhau, đưa ra khuyến cáo cho quá trình phân tích mẫu thực tế trên cơ sở
các yếu tố về độ nhạy, khoảng tuyến tính, thời gian phân tích mẫu để có sự
lựa chọn phương pháp tối ưu trong quá trình kiểm tra mẫu thực tế.
- Bước 3: Trên cơ sở số liệu phân tích, đánh giá ảnh hưởng của một số
điều kiện vận hành và hiệu quả hệ xử lí nước thải sử dụng giá thể vi sinh
chuyển động.
2.2. PHƯƠNG PHÁP NGHIÊN CỨU
2.2.1. Phương pháp lấy mẫu
Thực hiện lấy mẫu theo TCVN 5994:1995. Các mẫu được lấy tổ hợp
theo tần suất. Mẫu nước được lấy vào các chai PE, thể tích 500 ml. Trước khi
lấy mẫu, chai cần được rửa kỹ 2 - 3 lần bằng chính nước cần lấy. Điều cần lưu
ý là chai để lấy mẫu không sử dụng để đựng các chất lỏng khác.
2.2.2. Phương pháp bảo quản mẫu
Thực hiện bảo quản mẫu theo TCVN 5993-1995:
- Bảo quản mẫu để xác định Amoni: Phải xác định ngay lượng amoniac
trong mẫu lấy. Nếu không làm được phải cố định mẫu bằng 2 - 4ml Clorofooc
cho 1 lit nước và để ở 40C. Mẫu bảo quản không quá 1 tuần.
- Bảo quản mẫu để xác định Nitrit: Phải xác định ngay lượng nitrit
trong mẫu lấy. Nếu không làm được phải cố định mẫu bằng 2 - 4ml Clorofooc
cho 1 lit nước và để ở 40C. Bảo quản mẫu không quá 4 ngày.
- Bảo quản mẫu để xác định Nitrat: Mẫu để xác định nitrat phải được phân
tích ngay. Nếu không được bảo quản trong điều kiện đặc biệt thì không được để
quá 4 giờ. Ở nhiệt độ 40C giữ được 1 ngày. Nếu cho vào mỗi lít nước từ 2 - 4ml
Clorofooc hoặc 1ml H2SO4 đặc (d = 1,84) mẫu nước sẽ bền trong 4 ngày.
- Bảo quản mẫu để xác định Photphat: Mẫu để xác định photphat phải
được phân tích ngay, không được để quá 24 giờ.

33
2.2.3. Phương pháp phân tích mẫu
Mẫu phải được chưng cất hoặc lọc qua giấy lọc 0,45 m trước khi đưa
vào phân tích. Dựa vào các tiêu chuẩn trong và ngoài nước đang lưu hành
chúng tôi tiến hành xây dựng các quy trình phân tích cho phù hợp với các
mẫu nước thải trong quá trình xử lí. Các phương pháp được sử dụng để xác
định các thông số amoni, nitrit, nitrat và phosphat bao gồm:
- Xác định amoni theo Phương pháp quang phổ hấp thụ phân tử bằng
thuốc thử thymol - TCVN 6179-1:1996.
- Xác định Amoniac (NH4+) trong nước - Phương pháp lên màu trực
tiếp với thuốc thử Nessler.
- Xác định nitrit theo phương pháp quang phổ hấp thụ phân tử với
thuốc thử Griess - TCVN 6178:1996; Tiêu chuẩn SMEWW 4500 NO2- B.
- Xác định nitrat theo phương pháp quang phổ hấp thụ phân tử dùng
axit sunfosalixylic - TCVN 6180:1996.
- Xác định nitrat theo phương pháp đo quang phổ tia UV và dẫn xuất
thứ hai - Tiêu chuẩn SMEWW 4500 NO3- B.
- Xác định phospho theo phương pháp đo phổ dùng amoni molipdat –
TCVN 6202:2008.
- Xác định phospho theo phương pháp đo phổ hấp thụ phân tử với axit
Vanadomolybdo phosphoric - Tiêu chuẩn SMEWW 4500 - P C.
2.3. DỤNG CỤ, THIẾT BỊ
- Bếp điện
- Bát cô mẫu
- Bình định mức các loại 1000ml, 100 ml, 50 ml, 25 ml, phễu lọc, pipet
các loại.
- Bình nón, cốc thủy tinh, buret.

34
- Thiết bị chưng cất: bình thủy tinh chịu nhiệt có dung tích từ 800 ml
đến 2000 ml được gắn vào bình ngưng thẳng đứng để đầu ra cón thể chìm
dưới bề mặt của dung dịch axit nhận
- Máy đo pH.
- Máy quang phổ hấp thụ phân tử model Jasco V-730.
2.4. PHƯƠNG PHÁP TRẮC QUANG XÁC ĐỊNH NH4+ BẰNG THUỐC
THỬ THYMOL - TCVN 6179-1 : 1996
2.4.1 Chuẩn bị hóa chất, thuốc thử
- Thuốc thử màu: Hoà tan 130 g ± 1 g natri salixylat (C7H6O3Na) và
130 g g ± 1 g trinatri xytrat ngậm hai phân tử nước ((C6H5O7Na3.2H2O) trong
nước vào bình định mức 1000 ml. Thêm một lượng nước đủ để cho tổng thể
tích chất lỏng bằng khoảng 950 ml và sau đó thêm 0,970 g ± 0,005 g natri
nitrosopentaxyano sắt (III) 2 phân tử nước [natri nitroprusiat,
{Fe(CN)5NO}Na2.2H2O} vào dung dịch. Hoà tan chất rắn trong dung dịch,
sau đó pha loãng bằng nước tới vạch. Bảo quản trong lọ thuỷ tinh màu hổ
phách, thuốc thử này bền ít nhất trong hai tuần.
- Dung dịch natri diclorosoxyanurat: Hoà tan 32,0 g ± 0,1 g natri
hydroxit trong 500 ml ± 50 ml nước. Làm nguội dung dịch đến nhiệt độ trong
phòng và thêm 2,00 g ± 0,02 g natri diclorosoxyanurat 2 phân tử nước
(C2N3O3Cl2Na.2H2O) vào dung dịch. Hoà tan chất rắn và chuyển toàn bộ dung
dịch sang bình định mức dung tích 1000 ml. thêm nước tới vạch. Bảo quản
trong lọ thuỷ tinh màu hổ phách, thuốc thử này ổn định ít nhất trong hai tuần.
- Nitơ dạng amoni dung dịch chuẩn rN = 1000 mg/L: Hoà tan 3,819 g ±
0.004 g amoni clorua (đã được sấy khô ở 1050C ít nhất 2 giờ) vào khoảng 800
ml nước trong bình định mức dung tích 1000 ml. Pha loãng đến vạch mức
bằng nước. 1 ml dung dịch chuẩn này chứa 1 mg nitơ amoni. Bảo quản trong
lọ thuỷ tinh nút kín, dung dịch bền ít nhất trong một tháng.
- Dung dịch rửa: Hoà tan 100 g ± 2 g kali hydroxyt trong 100 ml nước
± 2 ml nước. Làm nguội dung dịch và thêm vào 900 ml ± 50 ml etanol 95%
(v/v). Bảo quản trong lọ polyetylen.

35
2.4.2. Yếu tố ảnh hưởng
Các chất gây nhiễu đáng kể thường gặp là nhiễu do anilin và
atanolamin, và thể hiện nhiễu nói chung là từ các amin bậc 1. Tuy nhiên, các
chất như vậy thường ít gặp trong các mẫu nước với các nồng độ bình thường.
Tính axit và tính kiềm mạnh sẽ gây nhiễu bằng việc tạo ra các hợp chất
hấp thụ, sự có mặt của bất kỳ chất nào gây nên việc khử các ion hypolcorit,
mặc dù các trường hợp này thường không chắc chắn xảy ra trong hầu hết các
mẫu nước. Đồng thời nhiễu do sự kết tủa của magiê xuất hiện khi khả năng
tạo phức của xytrat trong thuốc thử bị vượt quá mức. Vì nguyên nhân này
việc chưng cất sơ bộ mẫu thử là cần thiết.
2.4.3. Quy trình phân tích
Dùng pipet lấy phần mẫu thử lớn nhất là 50 mL vào bình định mức 100
ml. Thêm 4 ml thuốc thử màu và lắc k , sau đó thêm 4 ml dung dịch Natri
dicloisoxyanurat dihidrat và lại lắc k , định mức tới vạch.
Lắc k bình và đặt vào tủ ấm, giữ nhiệt độ 25 °C.
Sau ít nhất 60 phút, lấy bình ra khỏi tủ ấm và đo độ hấp thụ của dung
dịch tại bước sóng 655 nm.
2.5. XÁC ĐỊNH AMONIAC (NH4+) TRONG NƯỚC– PHƯƠNG PHÁP
LÊN MÀU TRỰC TIẾP VỚI THUỐC THỬ NESSLER
2.5.1. Chuẩn bị hóa chất, thuốc thử
- Dung dịch chỉ thị hỗn hợp: hòa tan 200 mg chỉ thị methyl đỏ trong
100 ml cồn 95% hoặc isopropyl alcohol. Hòa tan 100 mg xanh methylen trong
100 ml cồn hoặc isopropyl alcohol. Sau đó trộn hỗn hợp đó với nhau, hỗn hợp
được chuẩn bị hàng tháng.
- Thuốc thử Nessler được chuẩn bị trước 2 ngày: Hòa tan 100 g HgI2 và
70 g KI trong 100 ml nước. Hòa lẫn 224g KOH trong 700ml nước đựng trong
bình định mức 1000ml để nguội đến nhiệt độ phòng. Thêm từ từ dung dịch
thủy ngân (II) iodua/ Kaliiodua vào dung dịch KOH trong khi vẫn khuấy liên
tục. Pha loãng bằng nước đến vạch và trộn đều để yên ít nhất 2 ngày trước khi
sử dụng.

36
- Dung dịch chỉ thị axit boric: Hòa tan 20 g H3BO3 trong nước thêm 10
ml dung dịch chỉ thị hỗn hợp và pha loãng thành 1L.
- Dung dịch chuẩn axit H2SO4 0,02N.
- Dung dịch NaOH 6N: Cân 24g NaOH rồi hòa tan với một ít nước cất,
cho thêm 0,5ml Butanol, định mức thành 100ml.
- Dung dịch Kẽm sunfat 5%: Cân 5g ZnSO4, cho hòa tan vào trong
nước cất và định mức thành 100ml.
2.5.2. Yếu tố ảnh hưởng
Ion Ca2+
, Mg2+
(nước cứng), trong môi trường bazơ mạnh các ion này
sẽ tạo thành các hydroxide ở dạng keo, làm cho dung dịch bị vẩn đục cản trở
quá trình so màu. Các ion sắt, độ cứng cao của nước gây cảm trở phản ứng.
Độ đục và clo dư cũng gây cản trở phản ứng.
Các ion sắt, canxi, magie,... trong nước gây cản trở phản ứng nên cần
phải loại bỏ bằng cách chưng cất hoặc che bằng dung dịch Segnet hay dung
dịch EDTA. Nước đục xử lý bằng dung dịch ZnSO4 5%. Clo dư trong nước
được loại trừ bằng dung dịch Natrithiosunfat 5%.
2.5.3. Quy trình phân tích
- Xử lý mẫu: Với mẫu không chưng cất. Thêm 1mL ZnSO4 vào 100 ml
mẫu, thêm 0,1 đến 0,5 mL NaOH 6N để pH đạt 10,5. Trộn đều, lắc, để 5 đến
10 phút, cặn lắng xuống, lọc lấy phần nước trong.
- Phát triển màu: Lấy 50 mL mẫu vào bình định mức 100 ml thêm 1 giọt
dung dịch EDTA sau đó thêm 2 mL thuốc thử Nessler. Đợi 10 - 30 phút, so màu
trên máy với bước sóng 400 - 425 nm.
2.6. XÁC ĐỊNH NITRIT - PHƯƠNG PHÁP ĐO MÀU VỚI THUỐC
THỬ GRIESS [33]
2.6.1. Chuẩn bị hóa chất, thuốc thử
- Axit octhophosphoric, dung dịch 15 mol/L (r = 1.70 g/ml).
- Axit octhophosphoric, dung dịch 1,5 mol/L. Dùng pipet lấy 25 ml axit
octhophosphoric 15 mol/L vào 150 ml ± 25 ml nước. Khuấy đều và làm nguội

37
tới nhiệt độ trong phòng. Chuyển dung dịch sang bình định mức dung tích
250 ml và pha loãng với nước tới vạch. Bảo quản trong lọ thuỷ tinh màu hổ
phách, dung dịch bền ít nhất trong vòng 6 tháng.
- Thuốc thử màu: Hoà tan 40,0 g ± 0,5 g 4-aminobenzen sufonamid
(NH2C6H4SO2NH2) trong hỗn hợp của 100 ml ± 1 ml axit octophotphoric 15
mol/L và 500 ml ± 50 ml nước trong cốc thuỷ tinh có mỏ.
- Hoà tan 2,00 g ± 0,02 g N (1 naphtyl) 1.2 diamonietan dihidroclorua
(C10H7- NH-CH2-CH2-NH2- 2HCl) trong dung dịch tạo thành. Chuyển sang
bình định mức dung dịch 1000 ml và pha loãng với nước tới vạch. Lắc đều.
Bảo quản trong lọ thuỷ tinh màu hổ phách, dung dịch bền trong vòng 1 tháng
nếu giữ ở nhiệt độ từ 20C đến 5
0C.
- Dung dịch chuẩn nitrit, rN = 100 mg/L: Hoà tan 0,4922 g ± 0,0002 g
natri nitrit (sấy khô ở nhiệt độ 1050C trong thời gian ít nhất là 2 giờ) trong
khoảng 750 ml nước. Chuyển toàn bộ dung dịch sang bình định mức dung tích
1000 ml và pha loãng với nước tới vạch. Bảo quản trong lọ thuỷ tinh màu nâu
có nút kín ở nhiệt độ từ 20C đến 5
0C. Dung dịch này bền ít nhất là một tháng.
2.6.2. Yếu tố ảnh hưởng
Một số các chất thường gặp trong các mẫu nước đã được thí nghiệm về
khả năng gây nhiễu. Từ những chất đã qua thử, chỉ có cloramin, clo, thiosufat,
natri polyphotphat và sắt (III) là gây nhiễu một cách đáng kể.
2.6.3. Quy trình phân tích
Dùng pipet chuyển phần mẫu thử được lấy vào bình định mức dung tích
100 mL. Dùng pipet thêm 2 mL thuốc thử màu, lắc đều và pha loãng với nước
tới vạch. Sau 20 phút, đo độ hấp thu của của dung dịch ở bước sóng có độ hấp
thụ ở bước sóng 540 nm.
2.7. XÁC ĐỊNH NITRAT TRONG NƯỚC – PHƯƠNG PHÁP ĐO MÀU
VỚI THUỐC THỬ AXIT SUNFOSALIXYLIC
2.7.1. Chuẩn bị hóa chất, thuốc thử
- Axit sunfuric, H2SO418 mol/l, r = 1,84 g/ml.

38
- Axit axetic băng, CH3COOH 17 mol/L, r = 1,05 g/ml.
- Dung dịch kiềm, rNaOH = 200 g/L, r[CH2-N(CH2COOH)CH2-
COONa)]2.2H2O = 50 g/L: Hòa tan cẩn thận 200 g ± 2 g natri hidroxit dạng
hạt trong 800 ml nước. Thêm 50 g ± 0,5 g dinatri dihidro
etylendinitrilotetraaxetat ngậm 2 phân tử nước (EDTANa).{[CH2-
N(CH2COOH)CH2-COONa)]2.2H2O} và hòa tan. Để nguội đến nhiệt độ
phòng và thêm nước tới 1 L trong bình đong. Bảo quản trong chai polyetylen.
Thuốc thử này có thể bền trong thời gian dài.
- Dung dịch natri nitrua, rNaN3 = 0,5 g/L: Hòa tan cẩn thận 0,05 g ±
0,005 g natri nitrua trong khoảng 90 ml nước và pha loãng tới 100 ml bằng
nước trong bình đong. Bảo quản trong chai thủy tinh. Thuốc thử này có thể
bền trong thời gian dài.
- Dung dịch natri salixylat, rHO.C6H4.COONa = 10 g/L : Hòa tan 1 g ±
0,1 g natri salixylat (HO-C6H4-COONa) trong 100 ml ± 1 ml nước. Bảo quản
dung dịch trong chai thủy tinh hoặc chai polyetylen. Chuẩn bị dung dịch mới
trong ngày làm thí nghiệm.
- Dung dịch chuẩn gốc nitrat, rN = 1000 mg/L: Hòa tan 7,215 ± 0,001 g
kali nitrat (KNO3) (trước đó đã sấy khô ở 1050C ít nhất là 2 giờ) trong
khoảng 750 ml nước. Chuyển toàn bộ lượng đó sang bình định mức dung tích
1 L và thêm nước cho tới vạch. Bảo quản dung dịch trong chai thủy tinh
không quá 2 tháng.
2.7.2. Yếu tố ảnh hưởng
Các chất gây nhiễu tiềm tàng chính là clorua, octophotphat, magie và
mangan (II). Loại bỏ ion Cl- bằng dung dịch Ag2SO4. Loại bỏ chất hữu cơ hàm
lượng cao bằng cách cho kết tủa với Al(OH)3. Nitrit ở nồng độ cao gây sai số.
2.7.3. Quy trình phân tích
Dùng pipet hút dung dịch mẫu thử vào bát bay hơi sạch. Thêm 0,5 ml
dung dịch natri nitrua, và 0,2 ml axit axetic. Để yên ít nhất 5 phút và sau đó
để bay hơi hỗn hợp cho đến khô trong nồi cách thủy đang sôi. Thêm 1ml dung

39
dịch natri salixylat, trộn đều và cho bay hơi hỗn hợp đến khô lần nữa. Lấy bát
ra khỏi nồi cách thủy và để nguội bát đến nhiệt độ phòng.
Thêm 1 ml axit sunfuric và hòa tan cặn trên bát bằng cách lắc nhẹ. Để
hỗn hợp lắng trong 10 phút. Sau đó thêm 10 ml nước, tiếp theo là 10 ml dung
dịch kiềm.
Chuyển hỗn hợp sang bình định mức dung tích 25 ml nhưng không đổ
đến vạch. Đặt bình này vào nồi cách thủy ở 250C trong 10 phút ± 2 phút. Sau
đó lấy bình ra và thêm nước cho tới vạch. Đo độ hấp thụ của dung dịch ở
bước sóng 430 nm.
2.8. XÁC ĐỊNH NITRAT TRONG NƯỚC - PHƯƠNG PHÁP ĐO
QUANG PHỔ TIA UV VÀ DẪN XUẤT THỨ HAI [34]
2.8.1. Chuẩn bị hóa chất, thuốc thử
- Axit HCl 1M.
- Dung dịch chuẩn gốc nitrat, rN = 1000 mg/L: Hòa tan 7,215 ± 0,001 g
kali nitrat (KNO3) (trước đó đã sấy khô ở 1050C ít nhất là 2 giờ) trong
khoảng 750 ml nước. Chuyển toàn bộ lượng đó sang bình định mức dung tích
1 L và thêm nước cho tới vạch. Bảo quản dung dịch trong chai thủy tinh
không quá 2 tháng.
2.8.2. Yếu tố ảnh hưởng
- Chất hữu cơ hòa tan, chất hoạt động bề mặt, NO2-
và Cr6+
gây ảnh
hưởng đến phép xác định. Đối với mẫu đục phải được lọc hoặc làm mẫu đối
chứng. Một vài ion vô cơ không có trong nước tự nhiên như clorit, clorat cũng
gây ảnh hưởng.
- Axit hóa mẫu bằng dung dịch HCl 1M để loại bỏ ảnh hưởng của
hydroxit hay cacbonat. Ion clorua không ảnh hưởng đến phép đo này.
2.8.3. Quy trình phân tích
Lấy 50 mL mẫu (lọc và pha loãng nếu cần) vào bình định mức 100 ml.
Thêm 2 mL dung dịch HCl 1N và nước tới vạch định mức. Đo độ hấp thụ tại
hai bước sóng 220 nm và 275 nm. Tại bước sóng 220 nnm cho phép hấp thụ

40
NO3-
và các chất hữu cơ hòa tan, tại bước sóng 275 nm thì không hấp thụ
NO3-. Như vậy hiệu số tại hai bước sóng sẽ tính toán được nồng độ NO
3-.
2.9. XÁC ĐỊNH PHOTPHAT (PO43-
) TRONG NƯỚC - PHƯƠNG PHÁP
XANH MOLYBDEN
2.9.1. Chuẩn bị hóa chất, thuốc thử
- Dung dịch H2SO4 9M; H2SO4 4,5 M; H2SO4 2 M.
- Dung dịch NaOH 2 M.
- Dung dịch axit ascobic, r = 100g/L: Hòa tan 10 g ± 0,5 g axit ascobic
(C6H8O6) trong 100 ml ± 5 ml nước.
- Molipdat trong axit, Dung dịch I: Hòa tan 13 g ± 0,5 g amoni
heptamolipdat ngậm bốn nước [(NH4)6Mo7O24.4H2O)] trong 100 ml ± 5 ml
nước. Hòa tan 0,35 g ± 0,05 g antimon kali tartrat ngậm 1/2 nước
[K(SbO)C4H4O8.1/2 H2O] trong 100 ml ± 5 ml nước. Cho dung dịch molipdat
vào 300 ml ± 5 ml dung dịch axit sulfuric (4.1.1), khuấy liên tục. Thêm dung
dịch tartrat và trộn đều. Thuốc thử này ổn định ít nhất trong hai tháng nếu
được giữ trong bình thủy tinh màu nâu.
- Molipdat trong axit, dung dịch II: Hòa cẩn thận 230 ml ± 0,5 ml dung
dịch axit sulfuric trong 70 ml ± 5 ml nước, làm nguội. Hòa tan 13 g ± 0,5 g
amoni heptamolipdat ngậm bốn nước [(NH4)6Mo7O24.4H2O)] trong 100 ml ±
5 ml nước. Thêm dung dịch axit và trộn đều. Hòa tan 0,35 g ± 0,05 g antimon
kali tartrat ngậm 1/2 nước [K(SbO)C4H4O8.1/2 H2O] trong 100 ml ± 5 ml
nước. Thêm dung dịch axit – molipdat và trộn đều.
- Dung dịch natri thiosulphat pentahydrat, r = 12,0 g/L: Hòa tan 1,20 g
± 0,05 g natri thiosulphat ngậm năm nước (Na2S2O3.5H2O) trong 100 ml ± 5
ml nước. Thêm 0,05 g ± 0,005 g natri cacbonat (Na2CO3) làm chất bảo quản.
- Dung dịch tiêu chuẩn gốc octophosphat: Hòa 0,7165g KH2PO4 đã sấy
ở 1050C trong 2 giờ vào bình định mức 1000ml. Thêm nước cất đến vạch, lắc
đều. Thêm 2ml clorofrom để bảo quản. 1ml dung dịch này chứa 0,5mg PO43-

41
2.9.2. Yếu tố ảnh hưởng
Axit salisilix, ion Fe2+
, các chất hữu cơ cản trở phép xác định. Loại bỏ
các chất cản trở đó bằng cách tách riêng chúng nhờ các dung môi hữu cơ, sau
đó dùng Kalipecmanganat để oxy hóa. Dung môi chiết tốt nhất là butyl axetat.
Axit molipdic tạo phức với photpho, asen và silic tạo thành photphomolipdic,
asenomolipdic và silicomolipdic tương ứng. Do vậy phải loại bỏ ảnh hưởng
của asen và silic. Đối với silic ở nồng độ100 mg/L và Asen có nồng độ 0,2
mg/L không gây cản trở.
2.9.3. Quy trình phân tích
Dùng pipet lấy lượng mẫu thử đã định Vs vào bình định mức dung tích
100 ml và pha loãng với nước tới 40 ml, nếu cần. Thêm vào mỗi bình 1 ml
dung dịch axit ascorbic tiếp theo là 2 ml dung dịch axit molipdat. Thêm nước
tới vạch và lắc kỹ.
Nếu mẫu thử chứa asenat thì phải khử bằng thiosulphat trong môi
trường axit thành asenit. Việc khử được định lượng cho asenat đến nồng độ ít
nhất là 2 mg As/lit, được trình bày như sau:
Dùng pipet chuyển một lượng mẫu thử vào bình định mức 100 ml.
Thêm 0,4 ml dung dịch axit sulfuric 4,5M, 1 ml dung dịch axit ascobic 100 g/
Lvà 1 ml dung dịch thiosulphat 12 g/L khuấy và để quá trình khử kéo dài 10
phút ± 1 phút. Thêm 2 ml dung dịch axit molipdat II. Thêm nước tới vạch,
khuấy đều. Đo độ hấp thụ của mỗi dung dịch bằng máy đo phổ sau 10 phút và
30 phút ở bước sóng 820 nm.
2.10. XÁC ĐỊNH PHOTPHAT (PO43-
) TRONG NƯỚC - PHƯƠNG
PHÁP ĐO PHỔ HẤP THỤ PHÂN TỬ AXIT VANADOMOLYBDO
PHOSPHORIC
2.10.1. Chuẩn bị hóa chất, thuốc thử
- HCl.
- H2SO4.
- HNO3.

42
- Amoni heptamolipdat ngậm bốn nước (NH4)6Mo7O24.4H2O).
- NH4VO3.
- Thuốc thử vanadi-Molybdat:
Dung dịch A: Hòa tan 25 g (NH4)6Mo7O24.4H2O) trong 300ml nước.
Dung dịch B: Hòa tan 1,25 g NH4VO3 trong 300 ml nước đun đến sôi
sau đó để nguội thêm 330 ml HCl đặc.
Đổ dung dịch A và dung dịch B và trộn đều và định mức thành 1000 ml.
- Dung dịch tiêu chuẩn gốc octophosphat: Hòa 0,7165g KH2PO4 đã sấy
ở 1050C trong 2 giờ vào bình định mức 1000ml. Thêm nước cất đến vạch, lắc
đều. Thêm 2ml clorofrom để bảo quản. 1ml dung dịch này chứa 0,5mg PO43-
.
2.10.2. Yếu tố ảnh hưởng
Các ion Ca2+
, Mn2+
, Fe3+
có khả năng tạo kết tủa với các dạng của ion
photphat, các ion có màu có khả năng hấp thụ màu tốt ở gần bước sóng 470 nm
cũng có khả năng làm tăng mật độ quang khi định lượng phophat. Ion asenat là
một ion đồng dạng với photphat có thể ảnh hưởng đến quá trình tạo phức màu.
Silica làm tăng độ hấp thụ, fluoride, thorium, bismuth, sulfide, thiosulfate,
thiocyanate hoặc molybdate dư thừa gây ảnh hưởng đến phép xác định.
2.10.3. Quy trình phân tích
Dùng pipet hút 50 ml dung dịch mẫu thử vào bình định mức 100 ml.
Nếu như mẫu có pH lớn hơn 10, thêm vào mẫu 1 giọt chỉ thị phenolphtalein,
sau đó thêm dung dịch HCl vào để làm mất màu đỏ của mẫu. Thêm 10 ml
Vanadat molipdat sau đó định mức tới vạch bằng nước cất.
Sau 30 phút hoặc nhiều hơn đo mẫu tại bước sóng 400 – 490 nm.
Đối với mẫu trắng lấy 35 mL nước cất, thêm 10 mL Vanadat molipdat
sau đó định mức tới vạch bằng nước cất.
2.11. ẢNH HƯỞNG CỦA MỘT SỐ ĐIỀU KIỆN VẬN HÀNH ĐẾN
HIỆU SUẤT XỬ LÝ

43
2.11.1. Khảo sát quá trình phát triển của bùn hoạt tính
Tiến hành nuôi cấy bùn hoạt tính trên giá thể vi sinh vật trong thời gian
100 ngày và quan sát sự phát triển và bám dính. Giá thể vi sinh sử dụng trong
quá trình nuôi cấy là loại giá thể đã được biến tính bề mặt
Định lượng hàm lượng bùn hoạt tính bám trên giá thể đệm vi sinh và
xây dựng đồ thị hàm lượng theo thời gian .
2.11.2. Khảo sát thời gian sục khí
Chọn hàm lượng bùn hoạt tính tối ưu sau khi thực hiện theo mục 2.11.1.
Sử dụng nước thải giả lập để khảo sát.
Các thí nghiệm được thực hiện với thời gian sục khí kéo dài 24 giờ sau
đó ngừng sục khí, để nước lắng trong 2 giờ. Các khoảng thời gian sau khi sục
khí được 2 giờ, 4 giờ, 6 giờ, 8 giờ và 24 giờ, tiến hành lấy mẫu để phân tích
các chỉ tiêu photphat, amoni, nitrat, nitrit. Từ đó xác định được thời gian bể
xử lý các chất ô nhiễm đạt yêu cầu đặt ra.
2.11.3. Đánh giá hiệu quả xử lý hệ MBBR
Sử dụng các điều kiện tối ưu đã tìm được, tính toán hiệu suất xử lý theo
thời gian (tối đa 30 ngày). Chọn thời gian xử lý tối thiểu khi hàm lượng các
chất đạt quy chuẩn Việt Nam QCVN 14:2008/BTNMT

44
CHƯƠNG 3. KẾT QUẢ VÀ THẢO LUẬN
3.1. XÁC ĐỊNH GIÁ TRỊ SỬ DỤNG CỦA PHƯƠNG PHÁP PHÂN TÍCH
3.1.1. Xây dựng đường chuẩn, tính toán LOD, LOQ của các
phương pháp xác định 4NH , 2NO , 3NO , 3
4PO
3.1.1.1 Phương pháp xác định +
4NH
a. Theo TCVN 6179-1: 1996
Số liệu thực nghiệm thu được bảng 3.1:
Bảng 3.1. Sự phụ thuộc độ hấp thụ quang vào nồng độ +
4NH
Nồng độ amoni
chuẩn (mg/L) 0,04 0,08 0,20 0,40 0,60 0,80
Abs 0,0519 0,0738 0,1647 0,3281 0,4678 0,6057
Dựa theo bảng số liệu 3.1, sử dụng phần mềm excel để dựng đường
tuyến tính, ta được hình 3.1 sau:
Hình 3.1. Đường chuẩn xác định 4NH theo TCVN 6179-1: 1996
Giới hạn phát hiện và giới hạn định lượng 4NH : Tiến hành đo độ hấp
thụ quang theo TCVN 6179-1: 1996 tại bước sóng 700 nm của dung dịch
4NH 0,05 mg/L thu được số liệu theo bảng 3.2:
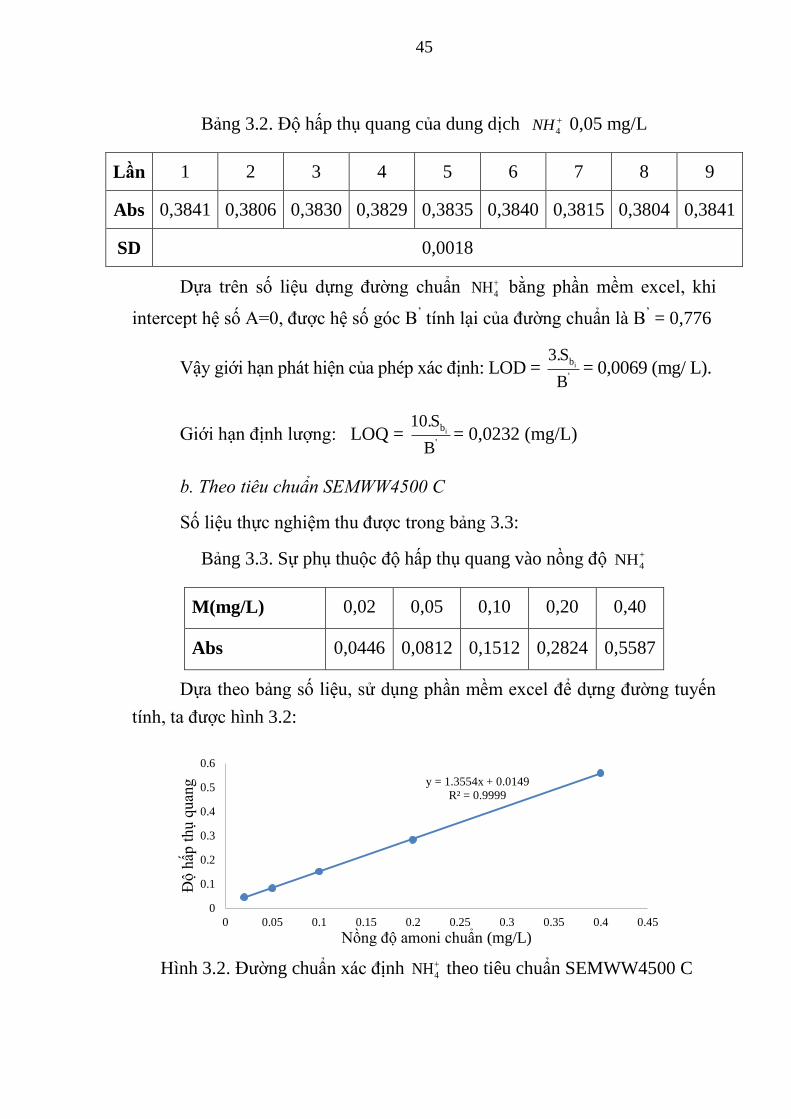
45
Bảng 3.2. Độ hấp thụ quang của dung dịch 4NH 0,05 mg/L
Lần 1 2 3 4 5 6 7 8 9
Abs 0,3841 0,3806 0,3830 0,3829 0,3835 0,3840 0,3815 0,3804 0,3841
SD 0,0018
Dựa trên số liệu dựng đường chuẩn 4NH bằng phần mềm excel, khi
intercept hệ số A=0, được hệ số góc B’ tính lại của đường chuẩn là B
’ = 0,776
Vậy giới hạn phát hiện của phép xác định: LOD = ib
'
3.S
B= 0,0069 (mg/ L).
Giới hạn định lượng: LOQ = ib
'
10.S
B= 0,0232 (mg/L)
b. Theo tiêu chuẩn SEMWW4500 C
Số liệu thực nghiệm thu được trong bảng 3.3:
Bảng 3.3. Sự phụ thuộc độ hấp thụ quang vào nồng độ +
4NH
M(mg/L) 0,02 0,05 0,10 0,20 0,40
Abs 0,0446 0,0812 0,1512 0,2824 0,5587
Dựa theo bảng số liệu, sử dụng phần mềm excel để dựng đường tuyến
tính, ta được hình 3.2:
Hình 3.2. Đường chuẩn xác định 4NH theo tiêu chuẩn SEMWW4500 C
y = 1.3554x + 0.0149
R² = 0.9999
0
0.1
0.2
0.3
0.4
0.5
0.6
0 0.05 0.1 0.15 0.2 0.25 0.3 0.35 0.4 0.45
Độ h
ấp thụ q
uan
g
Nồng độ amoni chuẩn (mg/L)

46
Giới hạn phát hiện và giới hạn định lượng 4NH : Tiến hành đo độ hấp
thụ quang theo tiêu chuẩn SEMWW4500 C đo ở bước sóng 420 nm của dung
dịch 4NH 0,03 mg/L.
Bảng 3.4. Độ hấp thụ quang của dung dịch 4NH theo tiêu chuẩn SEMWW 4500 C
Lần 1 2 3 4 5 6 7 8 9
Abs 0,0502 0,0485 0,0498 0,0533 0,0480 0,0537 0,0552 0,0545 0,0538
SD 0,0028
Dựa trên số liệu dựng đường chuẩn 4NH bằng phần mềm excel, khi
intercept hệ số A=0, được hệ số góc B’ tính lại của đường chuẩn là B
’ = 1,4093.
Vậy giới hạn phát hiện của phép xác định : LOD = ib
'
3.S
B= 0,0059 (mg/L).
Giới hạn định lượng: LOQ = ib
'
10.S
B= 0,0199 (mg/L)
3.1.1.2 Phương pháp xác định 2NO
a. Theo TCVN 6178:1996 ‘
Số liệu thực nghiệm thu được trong bảng 3.5:
Bảng 3.5. Sự phụ thuộc độ hấp thụ quang vào nồng độ 2NO
M(mg/ L) 0,01 0,03 0,04 0,05 0,10 0,20
Abs 0,0330 0,1008 0,1384 0,1763 0,3338 0,6629
Dựa theo bảng số liệu, sử dụng phần mềm excel để dựng đường tuyến
tính, ta được hình 3.3:

47
Hình 3.3. Đường chuẩn xác định 2NO theo TCVN 6178:1996
Giới hạn phát hiện và giới hạn định lượng 2NO : Tiến hành đo độ hấp
thụ quang theo tiêu chuẩn SEMWW4500 C đo ở bước sóng 540 nm của dung
dịch 2NO 0,02 mg/L.
Bảng 3.6. Độ hấp thụ quang của dung dịch 2NO theo TCVN 6178:1996
Lần 1 2 3 4 5 6 7 8 9
Abs 0,0652 0,0603 0,0670 0,0562 0,0575 0,0661 0,0583 0,0621 0,0674
SD 0,0043
Dựa trên số liệu dựng đường chuẩn 2NO bằng phần mềm excel, khi
intercept hệ số A=0, được hệ số góc B’ tính lại của đường chuẩn là B
’ = 3,3333.
Vậy giới hạn phát hiện của phép xác định : LOD = ib
'
3.S
B= 0,0039 (mg/L).
Giới hạn định lượng: LOQ = ib
'
10.S
B= 0,0130 (mg/L)
3.1.1.3 Phương pháp xác định 3NO
a. Theo tiêu chuẩn TCVN 6180:1996
Số liệu thực nghiệm thu được theo bảng 3.7:
y = 3.2982x + 0.0045 R² = 0.9997
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0 0.05 0.1 0.15 0.2 0.25
Độ h
ấp thụ q
uan
g
Nồng độ nitrit chuẩn (mg/ L)
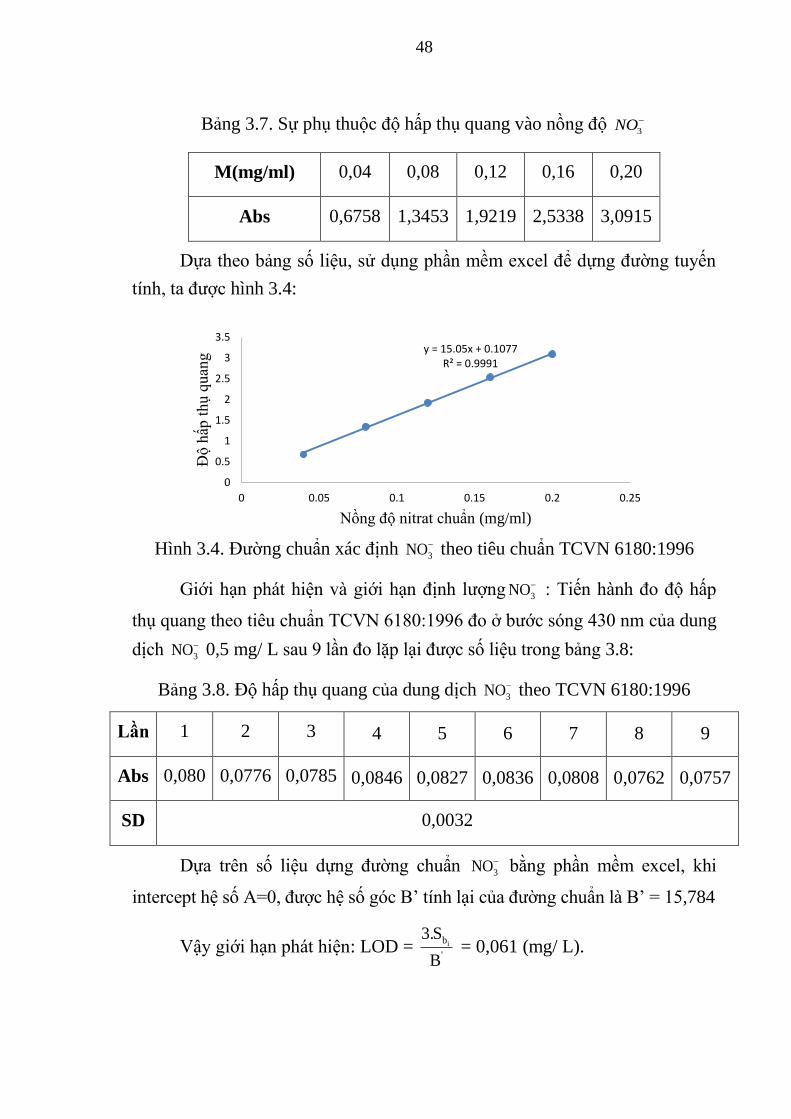
48
Bảng 3.7. Sự phụ thuộc độ hấp thụ quang vào nồng độ 3NO
M(mg/ml) 0,04 0,08 0,12 0,16 0,20
Abs 0,6758 1,3453 1,9219 2,5338 3,0915
Dựa theo bảng số liệu, sử dụng phần mềm excel để dựng đường tuyến
tính, ta được hình 3.4:
Hình 3.4. Đường chuẩn xác định 3NO theo tiêu chuẩn TCVN 6180:1996
Giới hạn phát hiện và giới hạn định lượng 3NO : Tiến hành đo độ hấp
thụ quang theo tiêu chuẩn TCVN 6180:1996 đo ở bước sóng 430 nm của dung
dịch 3NO 0,5 mg/ L sau 9 lần đo lặp lại được số liệu trong bảng 3.8:
Bảng 3.8. Độ hấp thụ quang của dung dịch 3NO theo TCVN 6180:1996
Lần 1 2 3 4 5 6 7 8 9
Abs 0,080 0,0776 0,0785 0,0846 0,0827 0,0836 0,0808 0,0762 0,0757
SD 0,0032
Dựa trên số liệu dựng đường chuẩn 3NO bằng phần mềm excel, khi
intercept hệ số A=0, được hệ số góc B’ tính lại của đường chuẩn là B’ = 15,784
Vậy giới hạn phát hiện: LOD = ib
'
3.S
B = 0,061 (mg/ L).
y = 15.05x + 0.1077 R² = 0.9991
0
0.5
1
1.5
2
2.5
3
3.5
0 0.05 0.1 0.15 0.2 0.25
Độ h
ấp thụ q
uan
g
Nồng độ nitrat chuẩn (mg/ml)
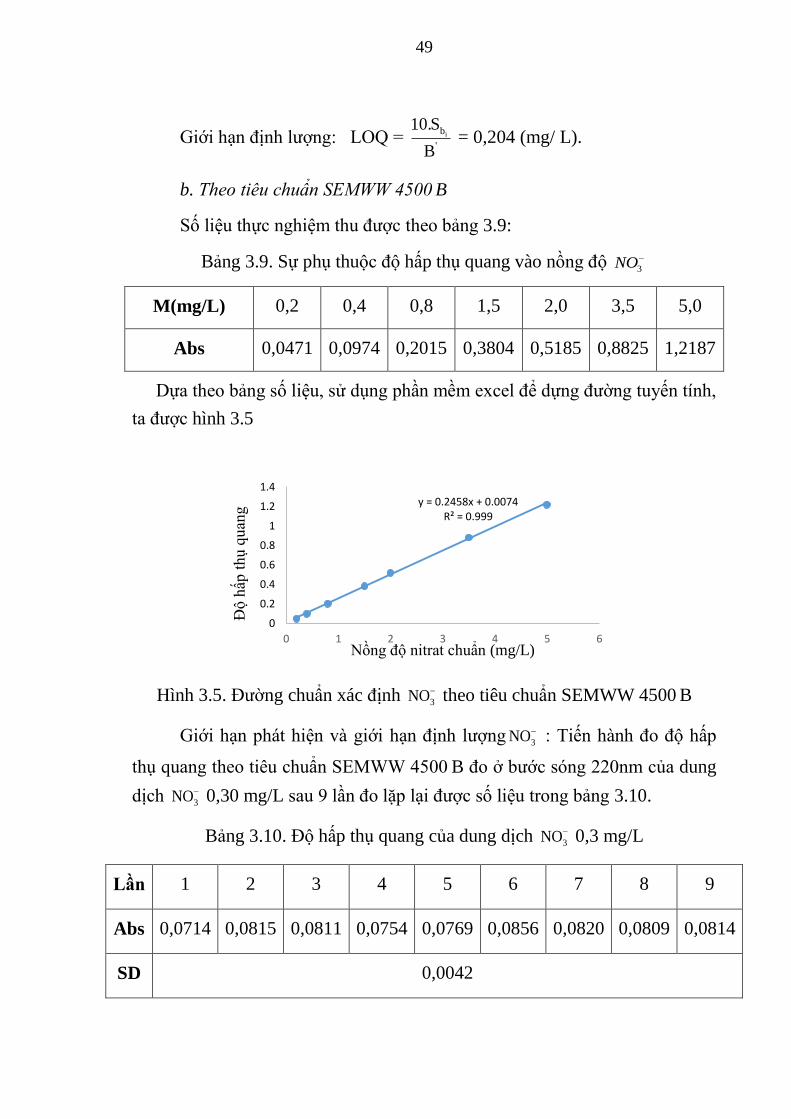
49
Giới hạn định lượng: LOQ = ib
'
10.S
B = 0,204 (mg/ L).
b. Theo tiêu chuẩn SEMWW 4500 B
Số liệu thực nghiệm thu được theo bảng 3.9:
Bảng 3.9. Sự phụ thuộc độ hấp thụ quang vào nồng độ 3NO
M(mg/L) 0,2 0,4 0,8 1,5 2,0 3,5 5,0
Abs 0,0471 0,0974 0,2015 0,3804 0,5185 0,8825 1,2187
Dựa theo bảng số liệu, sử dụng phần mềm excel để dựng đường tuyến tính,
ta được hình 3.5
Hình 3.5. Đường chuẩn xác định 3NO theo tiêu chuẩn SEMWW 4500 B
Giới hạn phát hiện và giới hạn định lượng 3NO : Tiến hành đo độ hấp
thụ quang theo tiêu chuẩn SEMWW 4500 B đo ở bước sóng 220nm của dung
dịch 3NO 0,30 mg/L sau 9 lần đo lặp lại được số liệu trong bảng 3.10.
Bảng 3.10. Độ hấp thụ quang của dung dịch 3NO 0,3 mg/L
Lần 1 2 3 4 5 6 7 8 9
Abs 0,0714 0,0815 0,0811 0,0754 0,0769 0,0856 0,0820 0,0809 0,0814
SD 0,0042
y = 0.2458x + 0.0074 R² = 0.999
0
0.2
0.4
0.6
0.8
1
1.2
1.4
0 1 2 3 4 5 6
Độ h
ấp thụ q
uan
g
Nồng độ nitrat chuẩn (mg/L)

50
Dựa trên số liệu dựng đường chuẩn 3NO bằng phần mềm excel, khi
intercept hệ số A=0, được hệ số góc B’ tính lại của đường chuẩn là B’ = 0,2481.
Vậy giới hạn phát hiện: LOD = ib
'
3.S
B = 0,0514 (mg/L).
Giới hạn định lượng: LOQ = ib
'
10.S
B= 0,1714 (mg/L).
3.1.1.4 Đường chuẩn xác định 3
4PO
a. Theo TCVN 6202:2008
Số liệu thực nghiệm thu được theo bảng 3.11:
Bảng 3.11. Sự phụ thuộc độ hấp thụ quang vào nồng độ 3
4PO
M(mg/100ml) 0,016 0,024 0,032 0,04 0,08 0,12
Abs 0,0948 0,1346 0,1645 0,1994 0,3580 0,5224
Dựa theo bảng số liệu, sử dụng phần mềm excel để dựng đường tuyến tính,
ta được hình 3.6:
Hình 3.6. Đường chuẩn xác định hàm lượng Photphat theo TCVN 6202:2008
Giới hạn phát hiện 3
4PO : Chúng tôi đo độ hấp thụ quang theo TCVN
6202:2008 đo tại bước sóng 725 nm của dung dịch 3
4PO 0,02 mg/100 ml thu
được số liệu theo bảng 3.12:
y = 4.0691x + 0.034 R² = 0.9997
0
0.1
0.2
0.3
0.4
0.5
0.6
0 0.02 0.04 0.06 0.08 0.1 0.12 0.14
Độ h
ấp thụ q
uan
g
Nồng độ photphat chuẩn (mg/ 100ml)

51
Bảng 3.12. Độ hấp thụ quang của dung dịch 3
4PO 0,02 mg/100 ml
Lần 1 2 3 4 5 6 7 8 9
Abs 0,1153 0,1154 0,1150 0,1156 0,1148 0,1149 0,1156 0,1153 0,1151
SD 0,0003
Dựa trên số liệu dựng đường chuẩn 3
4PO bằng phần mềm excel, khi
intercept hệ số A=0, được hệ số góc B’ tính lại của đường chuẩn là B’ = 4,5067
Vậy giới hạn phát hiện: LOD = ib
'
3.S
B = 0,0041 (mg/100 ml)
Giới hạn định lượng: LOQ = ib
'
10.S
B= 0,0136 (mg/100 ml)
b. Theo tiêu chuẩn SMEWW 4500 – P
Đo độ hấp thụ quang của dãy dung dịch chuẩn chứa 3
4PO tại bước sóng
cực đại 470nm theo tiêu chuẩn SMEWW 4500 – P được số liệu như trình bày
trong bảng 3.13.
Bảng 3.13. Sự phụ thuộc độ hấp thụ quang vào hàm lượng nguyên tố P
Hàm lượng
PO43-
(mg/L) 1 2 4 6.5 8 10 15
Abs 0,0150 0,0353 0,0698 0,1168 0,1455 0,1764 0,2668
Dựa theo bảng số liệu, sử dụng phần mềm excel để dựng đường tuyến
tính, ta được hình 3.7:
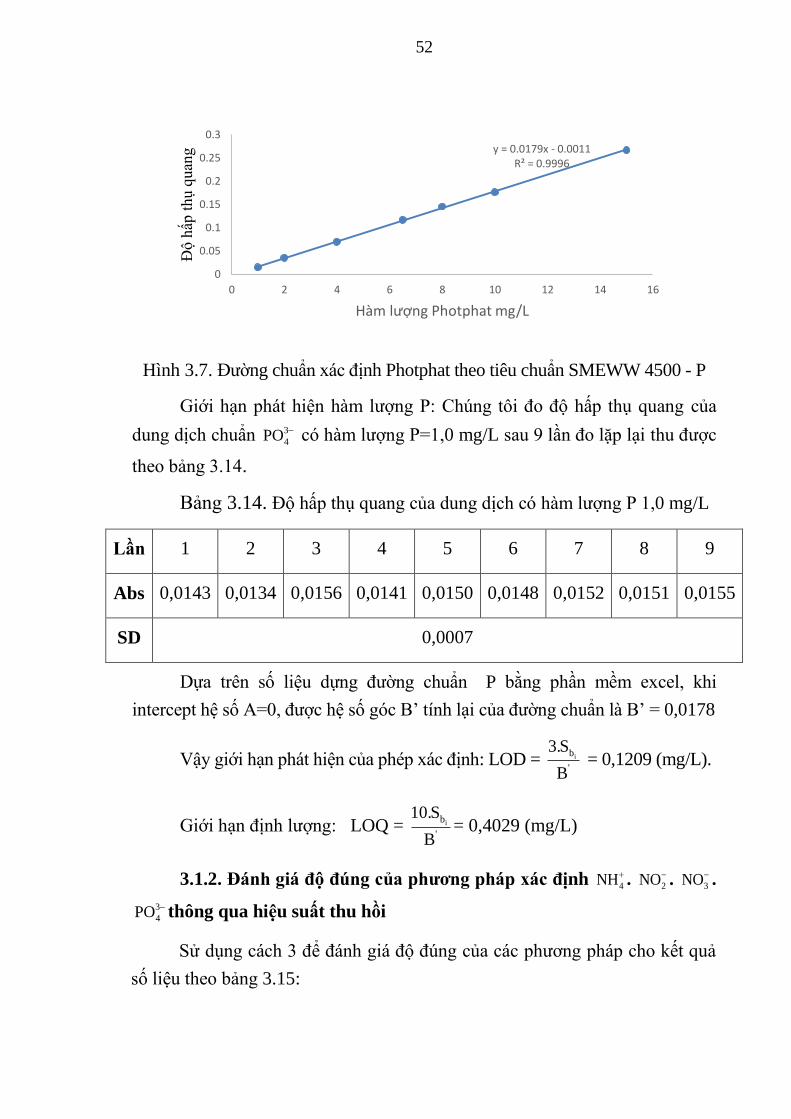
52
Hình 3.7. Đường chuẩn xác định Photphat theo tiêu chuẩn SMEWW 4500 - P
Giới hạn phát hiện hàm lượng P: Chúng tôi đo độ hấp thụ quang của
dung dịch chuẩn 3
4PO có hàm lượng P=1,0 mg/L sau 9 lần đo lặp lại thu được
theo bảng 3.14.
Bảng 3.14. Độ hấp thụ quang của dung dịch có hàm lượng P 1,0 mg/L
Lần 1 2 3 4 5 6 7 8 9
Abs 0,0143 0,0134 0,0156 0,0141 0,0150 0,0148 0,0152 0,0151 0,0155
SD 0,0007
Dựa trên số liệu dựng đường chuẩn P bằng phần mềm excel, khi
intercept hệ số A=0, được hệ số góc B’ tính lại của đường chuẩn là B’ = 0,0178
Vậy giới hạn phát hiện của phép xác định: LOD = ib
'
3.S
B = 0,1209 (mg/L).
Giới hạn định lượng: LOQ = ib
'
10.S
B= 0,4029 (mg/L)
3.1.2. Đánh giá độ đúng của phương pháp xác định 4NH . 2NO . 3NO .
3
4PO thông qua hiệu suất thu hồi
Sử dụng cách 3 để đánh giá độ đúng của các phương pháp cho kết quả
số liệu theo bảng 3.15:
y = 0.0179x - 0.0011 R² = 0.9996
0
0.05
0.1
0.15
0.2
0.25
0.3
0 2 4 6 8 10 12 14 16
Độ h
ấp thụ q
uan
g
Hàm lượng Photphat mg/L

53
Bảng 3.15. Thông số đánh giá độ thu hồi mẫu
STT Chỉ
tiêu
Tiêu
chuẩn
C chất
chuẩn
thêm ban
đầu
(mg/L)
Nồng độ
đo được
và suy ra
từ đường
chuẩn
(mg/L)
Độ
thu
hồi
(%)
Độ
thu
hồi
trung
bình
(%)
RSD%
1
4NH
TCVN
6179-1: 1996
0,04
0,0390 97,50
98,06 2,68
0,0395 98,75
0,0385 96,25
0,0399 99,75
0,2
0,1847 92,35
93,54 2,41
0,1850 92,50
0,1904 95,20
0,1882 94,10
0,8
0,7905 98,81
99,14 0,90 0,7950 99,38
0,7890 98,63
0,7980 99,75
2 4NH
SEMWW4500
0,02
0,0190 95,00
96,00 2,66
0,0194 97,00
0,0189 94,50
0,0195 97,50

54
C
0,1
0,0955 95,50
97,30 3,66
0,0956 95,60
0,0986 98,60
0,0995 99,50
0,4
0,3951 98,78
98,89 1,63 0,3905 97,63
0,3987 99,68
0,3980 99,50
3 2NO TCVN
6178:1996
0,01
0,0095 95,00
95.75 3.09
0,0098 98,00
0,0094 94,00
0,0096 96,00
0,04
0,0390 97,50
98.56 1.66 0,0395 98,75
0,0399 99,75
0,0393 98,25
0,2
0,1955 97,75
98,95 1,50 0,1980 99,00
0,1986 99,30
0,1995 99,75
4 3NO TCVN 0,04 0,0391 97,75 98,31 1,78
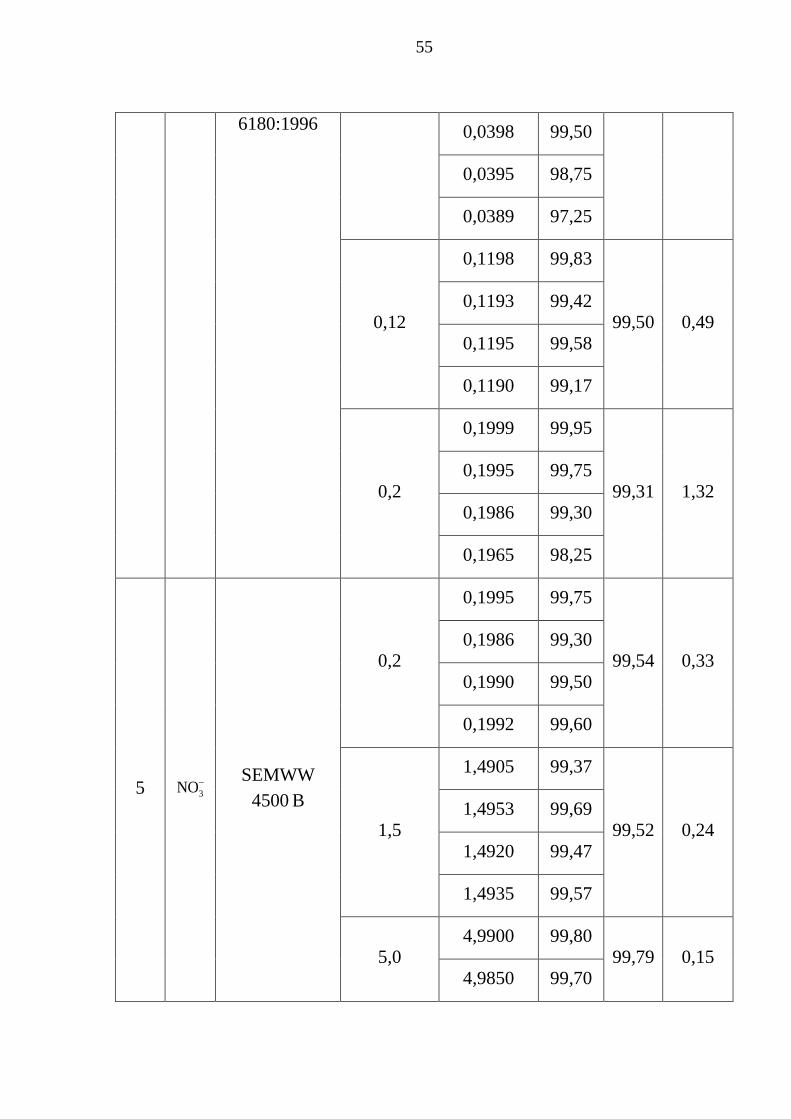
55
6180:1996 0,0398 99,50
0,0395 98,75
0,0389 97,25
0,12
0,1198 99,83
99,50 0,49 0,1193 99,42
0,1195 99,58
0,1190 99,17
0,2
0,1999 99,95
99,31 1,32
0,1995 99,75
0,1986 99,30
0,1965 98,25
5 3NO SEMWW
4500 B
0,2
0,1995 99,75
99,54 0,33 0,1986 99,30
0,1990 99,50
0,1992 99,60
1,5
1,4905 99,37
99,52 0,24
1,4953 99,69
1,4920 99,47
1,4935 99,57
5,0 4,9900 99,80
99,79 0,15
4,9850 99,70
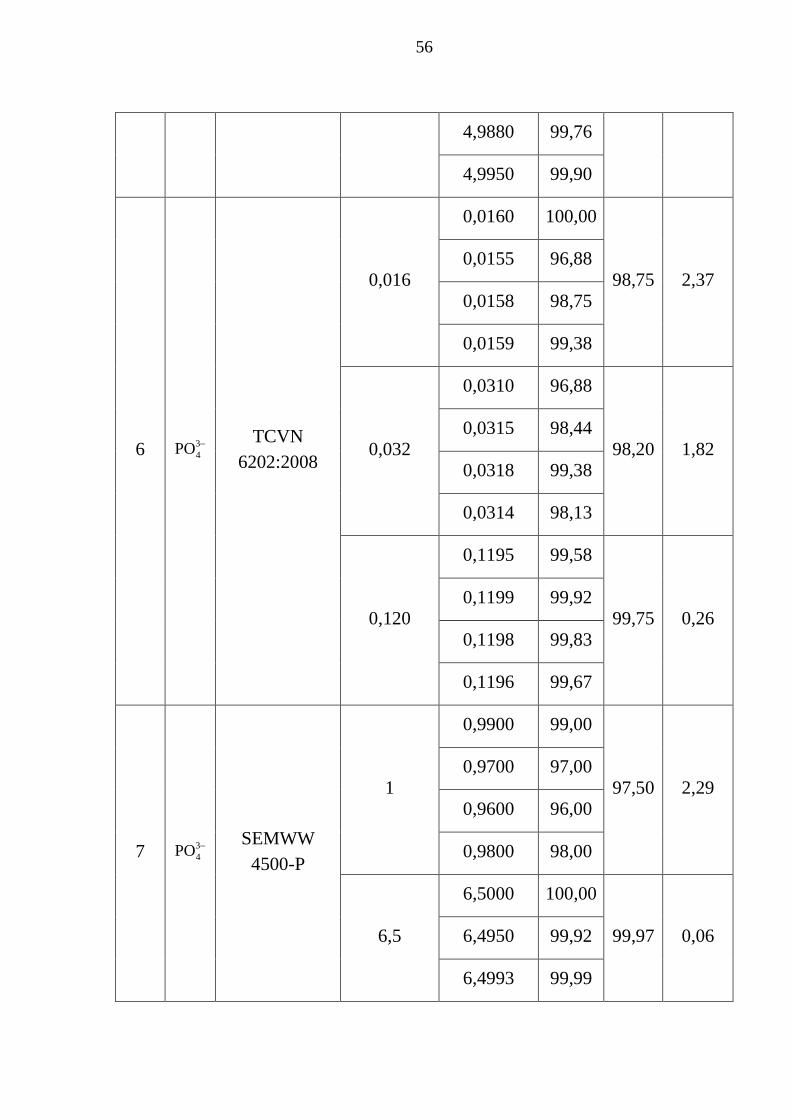
56
4,9880 99,76
4,9950 99,90
6 3
4PO TCVN
6202:2008
0,016
0,0160 100,00
98,75 2,37
0,0155 96,88
0,0158 98,75
0,0159 99,38
0,032
0,0310 96,88
98,20 1,82
0,0315 98,44
0,0318 99,38
0,0314 98,13
0,120
0,1195 99,58
99,75 0,26 0,1199 99,92
0,1198 99,83
0,1196 99,67
7 3
4PO SEMWW
4500-P
1
0,9900 99,00
97,50 2,29
0,9700 97,00
0,9600 96,00
0,9800 98,00
6,5
6,5000 100,00
99,97 0,06 6,4950 99,92
6,4993 99,99

57
6,4990 99,98
15
14,9900 99,93
99,80 0,21
14,9500 99,67
14,9800 99,87
14,9600 99,73
Kết luận: Tất cả các phương pháp phân tích đều đạt yêu cầu về độ thu
hồi theo AOAC
3.1.3. So sánh hai phương pháp đánh giá độ chính xác
So sánh độ chính xác của hai phương pháp định lượng khác nhau người
ta thường sử dụng kiểm định thống kê với phép so sánh hai phương sai sử
dụng chuẩn Fisher (F-test).
- Giả thiết thống kê:
H0: Hai phương sai đồng nhất.
H1: Hai phương sai không đồng nhất.
- Giá trị thống kê:
2
1tn 2
2
SF =
S với 2 2
1 2S >S ; f1=n1-1, f2 = n2-1
- Biện luận:
Nếu Ftn < Flt (f1, f2): Chấp nhận giả thiết H0.
Nếu Ftn > Flt (f1, f2): Bác bỏ giả thiết H0.
Sử dụng phần mềm Excel để xử lý số liệu theo các bước chính như sau:
Bước 1: Nhập số liệu vào bảng tính. Với mỗi phương pháp phân tích
một chất làm lặp lại 6 lần của cùng 1 mẫu, tổng 12 số liệu
Bước 2: Áp dụng “F-Test Two – Sample for Variances”: Chọn lệnh
Tools/Data Analysis; Chọn chương trình F-Test Two-Sample for Variances;
Trên hộp thoại F-Test Two-Sample for Variances ấn định thông số Label:
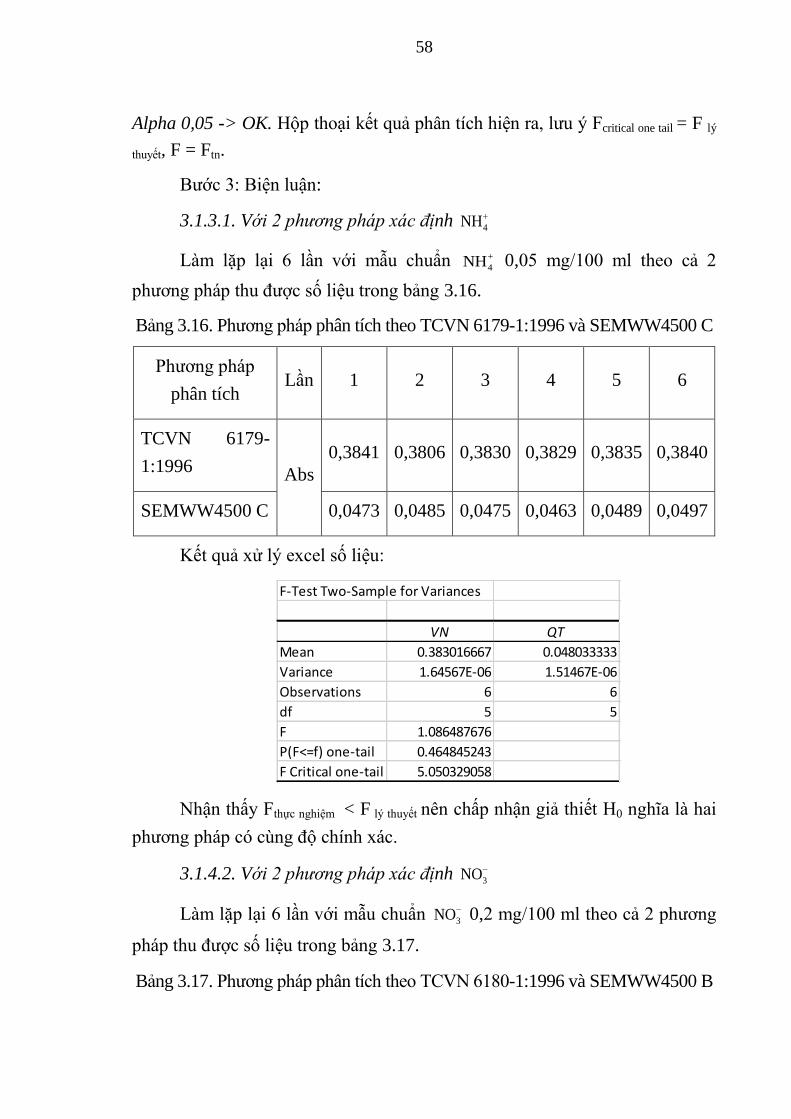
58
Alpha 0,05 -> OK. Hộp thoại kết quả phân tích hiện ra, lưu ý Fcritical one tail = F lý
thuyết, F = Ftn.
Bước 3: Biện luận:
3.1.3.1. Với 2 phương pháp xác định 4NH
Làm lặp lại 6 lần với mẫu chuẩn +
4NH 0,05 mg/100 ml theo cả 2
phương pháp thu được số liệu trong bảng 3.16.
Bảng 3.16. Phương pháp phân tích theo TCVN 6179-1:1996 và SEMWW4500 C
Phương pháp
phân tích Lần 1 2 3 4 5 6
TCVN 6179-
1:1996 Abs
0,3841 0,3806 0,3830 0,3829 0,3835 0,3840
SEMWW4500 C 0,0473 0,0485 0,0475 0,0463 0,0489 0,0497
Kết quả xử lý excel số liệu:
Nhận thấy Fthực nghiệm < F lý thuyết nên chấp nhận giả thiết H0 ngh a là hai
phương pháp có cùng độ chính xác.
3.1.4.2. Với 2 phương pháp xác định 3NO
Làm lặp lại 6 lần với mẫu chuẩn 3NO 0,2 mg/100 ml theo cả 2 phương
pháp thu được số liệu trong bảng 3.17.
Bảng 3.17. Phương pháp phân tích theo TCVN 6180-1:1996 và SEMWW4500 B
F-Test Two-Sample for Variances
VN QT
Mean 0.383016667 0.048033333
Variance 1.64567E-06 1.51467E-06
Observations 6 6
df 5 5
F 1.086487676
P(F<=f) one-tail 0.464845243
F Critical one-tail 5.050329058
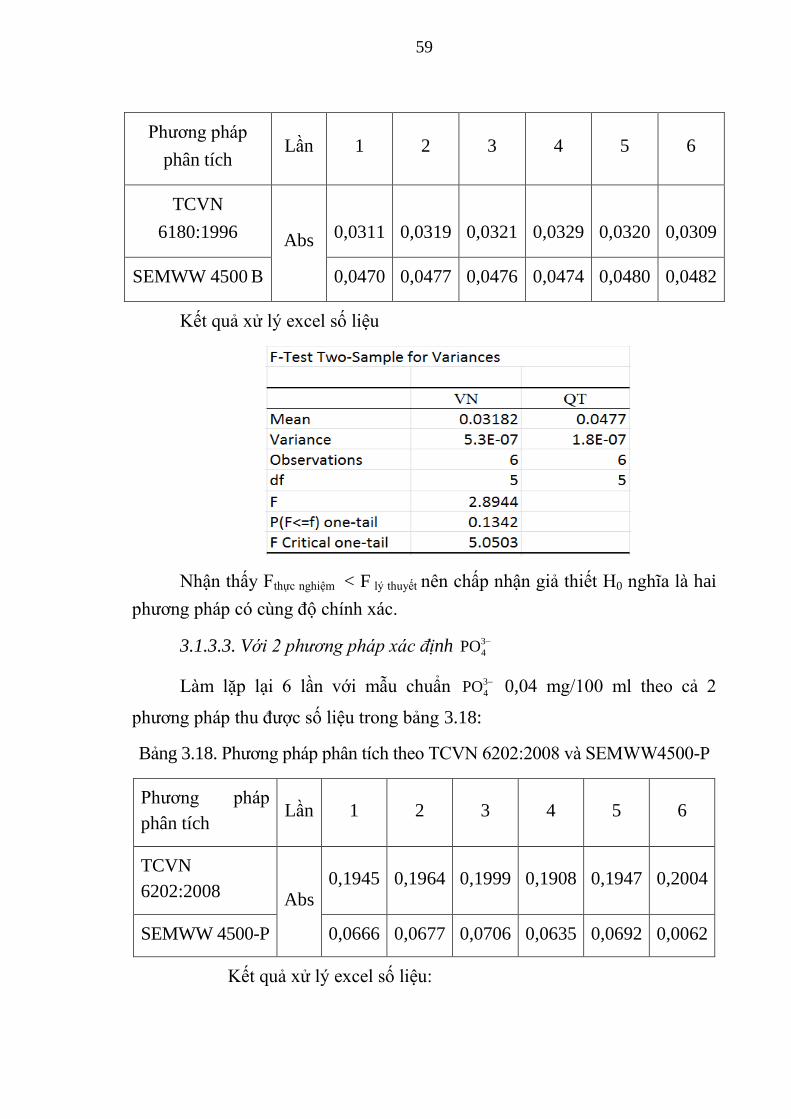
59
Phương pháp
phân tích Lần 1 2 3 4 5 6
TCVN
6180:1996 Abs 0,0311 0,0319 0,0321 0,0329 0,0320 0,0309
SEMWW 4500 B 0,0470 0,0477 0,0476 0,0474 0,0480 0,0482
Kết quả xử lý excel số liệu
Nhận thấy Fthực nghiệm < F lý thuyết nên chấp nhận giả thiết H0 ngh a là hai
phương pháp có cùng độ chính xác.
3.1.3.3. Với 2 phương pháp xác định 3
4PO
Làm lặp lại 6 lần với mẫu chuẩn 3
4PO 0,04 mg/100 ml theo cả 2
phương pháp thu được số liệu trong bảng 3.18:
Bảng 3.18. Phương pháp phân tích theo TCVN 6202:2008 và SEMWW4500-P
Phương pháp
phân tích Lần 1 2 3 4 5 6
TCVN
6202:2008 Abs 0,1945 0,1964 0,1999 0,1908 0,1947 0,2004
SEMWW 4500-P 0,0666 0,0677 0,0706 0,0635 0,0692 0,0062
Kết quả xử lý excel số liệu:

60
Nhận thấy Fthực nghiệm < F lý thuyết nên chấp nhận giả thiết H0 ngh a là hai
phương pháp có cùng độ chính xác.
3.2 PHÂN TÍCH MẪU THỰC TẾ
3.2.1. Lựa chọn phương pháp phân tích
Qua quá trình xác nhận giá trị sử dụng của phương pháp và so sánh độ
chính xác của hai phương pháp cũng như dựa trên các yếu tố ảnh hưởng. Cho
thấy với các thông số như amoni, nitrit (tiêu chuẩn việt nam và tiêu chuẩn
nước ngoài là giống nhau), phot phat và nitrat thì hai phương pháp trong và
ngoài nước có độ chính xác như nhau. Tuy nhiên với mẫu nước thải thực tế
trong hệ thống MBBR thì các thông số trong mẫu cho thấy với thông số
amoni trong nước thải đầu vào có hàm lượng rất lớn (28,3 mg/L) trong khi
mẫu nước thải đầu ra của hệ thống rất nhỏ (4,1 mg/L). Do đó, với mẫu nước
thải đầu vào chúng ta nên sử dụng phương pháp so màu bằng thuốc thử
thylmo để xác định hàm lượng amoni để tránh hiện tượng amoni bị kết tủa khi
sử dụng phương pháp lên màu trực tiếp với thuốc thử nesler.Với mẫu nước
thải đầu ra hàm lượng amoni nhỏ chúng tôi kiến nghị sử dụng phương pháp
lên màu trực tiếp với thuốc thử Nessler là phương pháp có độ nhạy cao hóa
chất đơn giản dễ sử dụng thời gian phân tích nhanh.
Đối với thông số nitrit thì các phương pháp trong và ngoài nước là như
nhau nên sử dụng phương pháp tiêu chuẩn TCVN 6178 :1996 để phân tích.
Thông số nitrat trong mẫu nước thải đầu vào lại có hàm lượng thấp trong khi
đó hàm lượng nitrat trong mẫu nước thải đầu ra hàm lượng cao hơn rất
nhiều.Vì vậy chúng tôi kiến nghị áp dụng phân tích mẫu nước thải đầu vào sử
F-Test Two-Sample for Variances
0.1945 0.0666
Mean 0.19644 0.055434
Variance 0.000015623 0.000765492
Observations 5 5
df 4 4
F 0.020409102
P(F<=f) one-tail 0.001184106
F Critical one-tail 0.156537812

61
dụng phương pháp đo quang phổ tia UV và dẫn xuất thứ hai - Tiêu chuẩn
SMEWW 4500 NO3- B là phương pháp có độ nhạy và độ chính xác cao. Với
mẫu nước thải đầu ra hàm lượng nitrat lớn nên sử dụng phương pháp quang
phổ hấp thụ phân tử dùng axit sunfosalixylic - TCVN 6180:1996.
Hàm lượng phot phat của mẫu nước thải đầu vào cũng cao hơn rất
nhiều so với hàm lượng phot phat của mẫu nước thải đầu ra. Trong khi đó với
phương pháp đo phổ dùng amoni molipdat – TCVN 6202:2008 có độ nhạy
cao phù hợp với việc xác định hàm lượng ở nước thải đầu ra. Xác định
phospho theo phương pháp đo phổ hấp thụ phân tử với axit Vanadomolybdo
phosphoric - Tiêu chuẩn SMEWW 4500 –P C lại rất phù hợp với việc xác
định Phot phat ở hàm lượng cao. Phương pháp này dễ dàng thao tác với hóa
chất đơn giản cùng thời gian phân tích nhanh.
3.2.2 Kết quả phân tích mẫu thực tế
Bảng 3.19. Ảnh hưởng của thời gian sục khí tới hiệu quả xử lý nitơ
Thời gian sục khí
(giờ)
Hàm lượng amoni
(mg/L)
Hiệu suất (%)
0 50,1 0
2 36,2 27,74
4 23,7 52,69
6 10 80,04
8 8,6 82,83
24 2,76 94,49
Sau lắng 2 giờ 2,73 94,55

62
Bảng 3.20. Đánh giá hiệu quả xử lý N của hệ thống
Ngày NH4+ (mg/L) NO3
- (mg/L) NO2
- (mg/L)
Đầu vào Đầu ra Đầu vào Đầu ra Đầu vào Đầu ra
5 40 12,4 0,1 12,5 0,02 2,1
10 35,2 11,6 0,05 10,3 0,04 2,15
15 37 11,2 0,07 6,5 0,03 2,01
20 39,2 5,4 0,12 5,4 0,1 1,15
25 32,2 4,5 0,11 5,2 0,12 1,1
30 28,3 4,1 0,1 5,1 0,08 1,08
Bảng 3.21. Ảnh hưởng của thời gian sục khí tới hiệu quả xử lý P
Thời gian sục khí
(giờ)
Hàm lượng P
(mg/L)
Hiệu suất (%)
0 20,3 0
2 17,6 13,30
4 15,1 25,62
6 11,4 43,84
8 9,5 53.,20
24 4,2 79,31
Sau lắng 2 giờ 4,1 79,80

63
3.3. ĐÁNH GIÁ ẢNH HƯỞNG CỦA MỘT SỐ ĐIỀU KIỆN VẬN HÀNH
3.3.1. Quá trình phát triển của bùn hoạt tính
Quá trình nuôi cấy bùn hoạt tính trên giá thể vi sinh thực hiện trong
thời gian 100 ngày. Hình ảnh bùn hoạt tính bám trên giá thể vi sinh thể hiện
trên Hình 3.8.
Hình 3.8. Hình ảnh của bùn hoạt tính bám trên giá thể vi sinh
Chú thích:
- (1) Hình ảnh giá thể vi sinh ban đầu
- (2); (3); (4); (5); (6); (7): Hình ảnh giá thể vi sinh sau 8 ngày, 25
ngày, 50 ngày, 65 ngày, 80 ngày, 100 ngày nuôi cấy.
Nhận thấy, sau 8 ngày đã có hiện tượng VSV bám trên giá thể, chủ yếu
tại bề mặt phía trong và các rãnh của giá thể đệm vi sinh. Tới ngày 25 của quá
trình thử nghiệm, VSV bám trên giá thể vi sinh với mật độ dày đặc hơn (hình
3.1), tuy nhiên, trong khoảng thời gian này vẫn chỉ tập trung tại các rãnh của
giá thể. Tới ngày thứ 50, VSV đã hình thành một lớp màng vi sinh trên giá
thể, VSV bám đồng đều trên bề mặt của giá thể đệm vi sinh. Từ ngày thứ 50
tới ngày thứ 65, VSV phát triển dày đặc hơn trên giá thể, hình thành một lớp
xốp, có chiều dày có thể nhìn thấy được. Đây là giai đoạn VSV phát triển
1 2 3
4 5
6 7

64
mạnh nhất. Từ ngày 80 tới ngày thứ 100, VSV vẫn tiếp tục sinh trưởng và
phát triển trên giá thể, tuy nhiên VSV phát triển chậm hơn, hàm lượng bùn
hoạt tính quan sát được trên giá thể không có sự khác biệt nhiều.
Hàm lượng bùn hoạt tính bám trên giá thể đệm vi sinh được thể hiện
trên Hình 3.9. Giá thể vi sinh sử dụng trong quá trình nuôi cấy là loại giá thể
đã được biến tính bề mặt nhằm tăng hiệu quả của quá trình VSV bám dính.
Hình 3.9. Sự phát triển của bùn hoạt tính
Sau 100 ngày nuôi cấy bùn hoạt tính trên giá thể vi sinh, kết quả cho
thấy, hàm lượng VSV bám trên giá thể vi sinh là 0,1228 gMLSS/g vật liệu
(tương đương với hàm lượng MLSS là 2200 mg/L). Trong khoảng thời gian
từ ngày thứ nhất tới ngày thứ 50, tốc độ tăng trưởng của VSV khá chậm, hàm
lượng bùn hoạt tính trong bể chỉ đạt 560 mg/L. Từ ngày 50 tới ngày thứ 100,
hàm lượng bùn hoạt tính tăng từ 560 lên 2200 mg/L, trong đó tốc độ VSV
bám dính trên giá thể vi sinh cao nhất trong khoảng thời gian từ ngày 60 tới
ngày thứ 85 của quá trình thử nghiệm.
3.3.2. Ảnh hưởng của thời gian sục khí tới hiệu suất xử lý
Bể phản ứng MBBR với hàm lượng bùn hoạt tính trong bể là 2200
mg/L. Thí nghiệm sử dụng nước thải nhân tạo (giả lập) được pha giả lập trong
phòng thí nghiệm, có tính chất tương tự như đối với nước thải thực tế. Ưu
điểm của nước thải nhân tạo là có thể cố định được hàm lượng các chất ô
0
500
1000
1500
2000
2500
0 20 40 60 80 100
ML
SS
(m
g/L
)
Thời gian (ngày)

65
nhiễm đầu vào, không bị biến động mạnh giống như nước thải thực tế, có thể
đánh giá được hiệu quả xử lý dễ dàng hơn (hình 3.2).
Thí nghiệm được thực hiện với thời gian sục khí kéo dài 24 giờ sau đó
ngừng sục khí, để nước lắng trong 2 giờ. Các khoảng thời gian sau khi sục khí
được 2 giờ, 4 giờ, 6 giờ, 8 giờ và 24 giờ, tiến hành lấy mẫu để phân tích các
chỉ tiêu photphat, amoni, nitrat, nitrit. Từ đó xác định được thời gian bể xử lý
các chất ô nhiễm đạt yêu cầu đặt ra.
Đặc trưng nguồn NTSH sử dụng trong quá trình nghiên cứu được thể
hiện trên Bảng 3.22:
Bảng 3.22. Thông số các chất ô nhiễm trong nước thải giả lập
STT Các chỉ tiêu Đơn vị Thông số
đầu vào
QCVN
14:2008/BTNMT- B
1 pH 8 5 - 9
3 COD mg/L 420 -
3 4N(NH ) mg N/L 50 10
4 3
4P(PO ) mg P/L 20 10
5 3NO mg/L 0,1 50
6 2NO mg/L 0,1 -
3.2.2.1. Ảnh hưởng của thời gian sục khí tới hiệu quả xử lý photphat
Hiệu quả xử lý photphat theo thời gian thể hiện trên hình 3.10
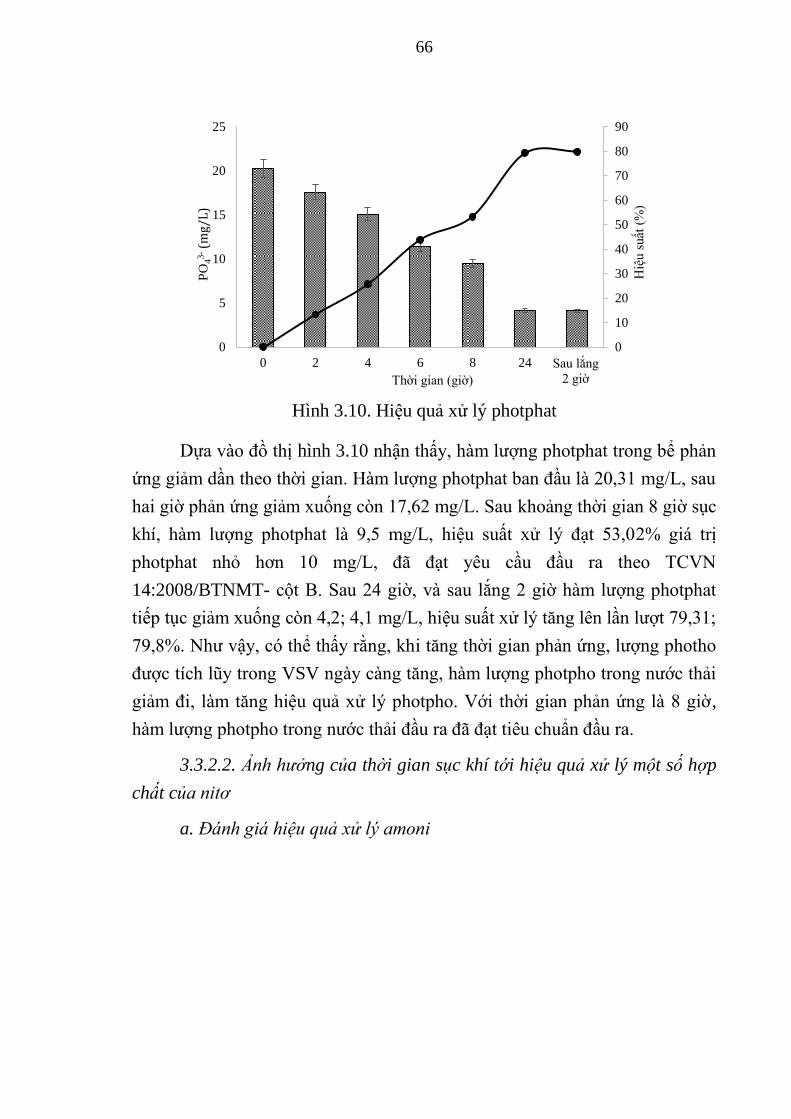
66
Hình 3.10. Hiệu quả xử lý photphat
Dựa vào đồ thị hình 3.10 nhận thấy, hàm lượng photphat trong bể phản
ứng giảm dần theo thời gian. Hàm lượng photphat ban đầu là 20,31 mg/L, sau
hai giờ phản ứng giảm xuống còn 17,62 mg/L. Sau khoảng thời gian 8 giờ sục
khí, hàm lượng photphat là 9,5 mg/L, hiệu suất xử lý đạt 53,02% giá trị
photphat nhỏ hơn 10 mg/L, đã đạt yêu cầu đầu ra theo TCVN
14:2008/BTNMT- cột B. Sau 24 giờ, và sau lắng 2 giờ hàm lượng photphat
tiếp tục giảm xuống còn 4,2; 4,1 mg/L, hiệu suất xử lý tăng lên lần lượt 79,31;
79,8%. Như vậy, có thể thấy rằng, khi tăng thời gian phản ứng, lượng photho
được tích lũy trong VSV ngày càng tăng, hàm lượng photpho trong nước thải
giảm đi, làm tăng hiệu quả xử lý photpho. Với thời gian phản ứng là 8 giờ,
hàm lượng photpho trong nước thải đầu ra đã đạt tiêu chuẩn đầu ra.
3.3.2.2. Ảnh hưởng của thời gian sục khí tới hiệu quả xử lý một số hợp
chất của nitơ
a. Đánh giá hiệu quả xử lý amoni
0
10
20
30
40
50
60
70
80
90
0
5
10
15
20
25
0 2 4 6 8 24 Sau lắng
2 giờ
Hiệ
u s
uất
(%
)
PO
43
- (m
g/L
)
Thời gian (giờ)

67
Hình 3.11. Hiệu quả xử lý amoni
Hàm lượng amoni ban đầu trong nước thải có giá trị 50,1 mg/L. Sau 6
giờ sục khí, hàm lượng amoni trong bể chỉ còn 10 mg/L, đạt tiêu chuẩn đầu
ra. Hàm lượng amoni tiếp tục giảm sau 8 giờ tới 24 giờ sục khí. Hiệu suất xử
lý amoni khá cao, có giá trị 80,02% sau 6 giờ; sau 24 giờ tăng lên 94,49%.
Sau lắng 2 giờ, hàm lượng amoni không khác nhiều, gần như không giảm so
với 24 giờ sục khí.
b. Đánh giá hiệu quả xử lý nitrat, nitrit
Hình 3.12. Hiệu quả xử lý nitrat và nitrit
Quá trình oxi hóa amoni thành nitrat làm giảm hàm lượng amoni trong
nước thải đồng thời làm tăng hàm lượng nitrat, nitrit trong nước thải. Hàm
lượng nitrat, nitrit ban đầu hầu như không có trong nước thải khoảng 0,1
mg/L. Dựa vào đồ thị hình 3.5 có thể thấy, lượng amoni bị oxi hóa chủ yếu
tạo thành nitrat trong khi oxi hóa rất ít thành nitrit. Sau 24 giờ sục khí, hàm
0
20
40
60
80
100
120
0
10
20
30
40
50
60
0 2 4 6 8 24 Sau lắng 2 giờ
Hiệ
u s
uất
(%
)
NH
4+ (
mg/L
)
Thời gian (giờ)
0
10
20
30
40
50
0 2 4 6 8 24 Sau
lắng
Nồng đ
ộ (
mg/
L)
Thời gian (giờ)
Nitrat Nitrit

68
lượng nitrat, nitrit trong nước thải là 45,2; 3,35 mg/L. Sau quá trình lắng, diễn
ra quá trình thiếu khí khử nitrat và nitrit tạo thành N2, quá trình xử lý nitơ
trong nước thải hoàn thành. Sau 2 giờ lắng, hàm lượng nitrat đã giảm từ 45,2
xuống còn 8,5 mg/L, nitrit từ 3,35 xuống 0,54 mg/L. Như vậy, với thời gian
lắng là 2 giờ, hàm lượng nitrat, nitrit đã được khử hiệu quả.
Nhận xét chung: Như vậy có thể thấy, hệ MBBR trong xử lý NTSH
nhân tạo mang lại hiệu quả xử lý cao. Sau 8 giờ phản ứng hàm lượng photpho
nhỏ hơn 10 mg/L đạt tiêu chuẩn đầu ra, chỉ sau 6 giờ hàm lượng amoni cũng
đạt tiêu chuẩn. Như vậy, có thể thấy, thời gian lưu thích hợp để xử lý hiệu quả
photpho và nitơ là 8 giờ.
3.3.3. Đánh giá hiệu quả xử lý hệ MBBR
Tiến hành thí nghiệm với thời gian sục khí 8 giờ đối với nước thải thực
tế. Đặc trưng của nước thải thực tế tương tự như đối với nước thải nhân tạo.
Tuy nhiên, nước thải thực tế dao động trong khoảng rộng. Cụ thể hàm lượng
amoni dao động từ 28 - 40 mg/L, hàm lượng photphat 12 - 18 mg/L. Thí
nghiệm được tiến hành trong khoảng thời gian 30 ngày.
3.3.3.1. Hiệu quả xử lý photpho
Hình 3.13. Hiệu quả xử lý photpho trong nước thải thực tế
Dựa vào đồ thị hình 3.13 và kết quả phân tích, có thể nhận thấy trong
khoảng thời gian đầu của quá trình vận hành, VSV trong giai đoạn thích nghi
với môi trường mới, hiệu quả xử lý trong 15 ngày đầu còn chưa cao, hàm
lượng photphat đầu ra mặc dù đã đạt tiêu chuẩn cho phép, hiệu suất xử lý vẫn
0
20
40
60
80
100
0
5
10
15
20
5 10 15 20 25 30
Hiệ
u s
uất
(%
)
Nồng đ
ộ (
mg/L
)
Thời gian (ngày)
HPO4 HPO4 HPO4

69
thấp từ 34,92 - 46,81%. Tới ngày thứ 20, hiệu quả xử lý photpho đã tăng, hàm
lượng photpho đầu ra đạt giá trị 3,48 mg/L. Tới ngày thứ 25 và 30, hiệu quả
xử lý photpho đã ổn định và giá trị photpho đầu ra 2,32; 2,61; hiệu suất xử lý
photphat đạt 82,81 - 84,18%.
3.3.3.2. Hiệu quả xử lý một số hợp chất nitơ
Hình 3.14. Hiệu quả xử lý amoni trong nước thải thực tế
Tương tự với quá trình xử lý photphat, trong giai đoạn đầu của quá
trình xử lý, VSV thích nghi với môi trường mới nên hiệu quả xử lý chưa cao,
hàm lượng amoni đầu ra chưa đạt tiêu chuẩn. Tới ngày thứ 15, hàm lượng
amoni đầu ra có giá trị 11,2 mg/L. Khi VSV đã thích nghi được với môi
trường, hiệu quả xử lý cao và ổn định hơn. Ngày thứ 20, hàm lượng amoni
đầu ra chỉ còn 5,4 mg/L. Hiệu quả xử lý amoni lớn hơn 85%. Như vậy sau 20
ngày, hiệu suất xử lý amoni cao, hàm lượng amoni đầu ra đạt QCVN
14:2008/BTNMT, cột B.
Một trong những đặc điểm của phương pháp sinh học hiếu khí bám
dính là có thể chịu được tải lượng ô nhiễm biến động mạnh, hàm lượng amoni
trong bể biến động khá mạnh từ 28 - 40 mg/L, tuy nhiên hệ vẫn xử lý amoni
rất hiệu quả, đảm bảo chất lượng nước đầu ra.
3.3.3.3. Hiệu quả xử lý nitrat, nitrit
Giá trị đầu vào và đầu ra của hàm lượng nitrat, nitrit được thể hiện trên
Bảng 3.23.
0
10
20
30
40
50
60
70
80
90
100
0
5
10
15
20
25
30
35
40
45
5 10 15 20 25 30
Hiệ
u s
uất
(%
)
Nồng đ
ộ (
mg/L
)
Thời gian (ngày)
HNH4 HNH4 HNH4
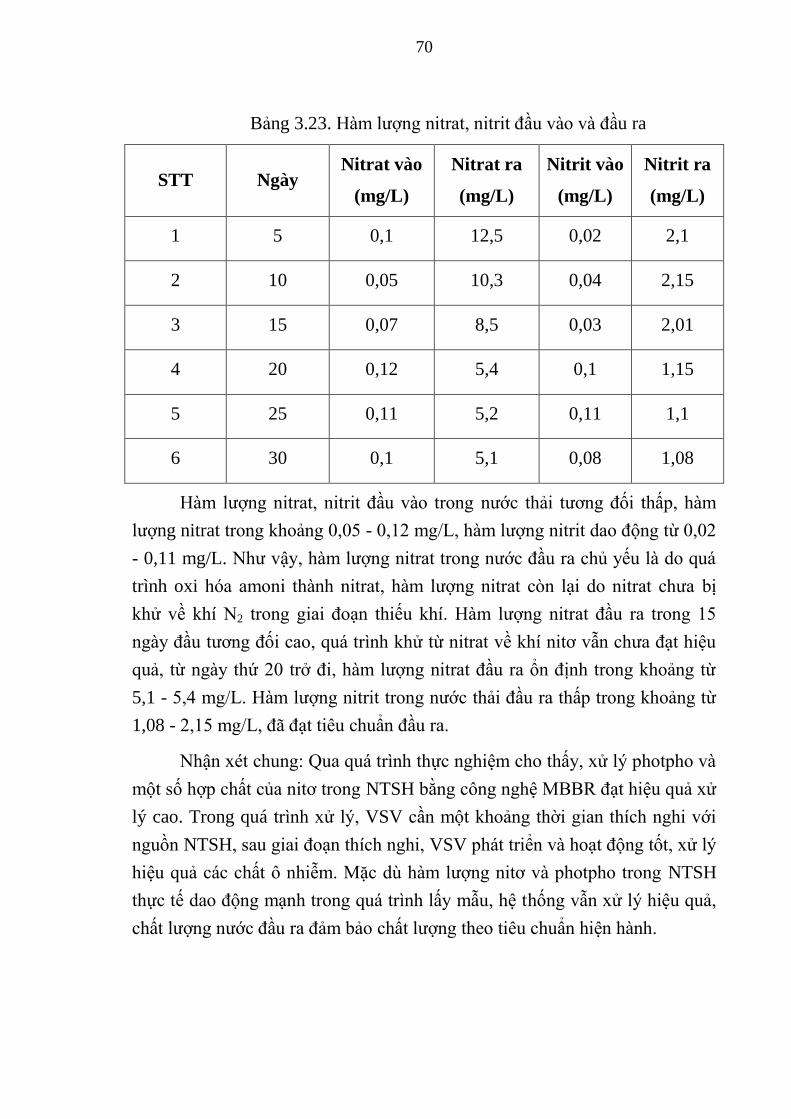
70
Bảng 3.23. Hàm lượng nitrat, nitrit đầu vào và đầu ra
STT Ngày Nitrat vào
(mg/L)
Nitrat ra
(mg/L)
Nitrit vào
(mg/L)
Nitrit ra
(mg/L)
1 5 0,1 12,5 0,02 2,1
2 10 0,05 10,3 0,04 2,15
3 15 0,07 8,5 0,03 2,01
4 20 0,12 5,4 0,1 1,15
5 25 0,11 5,2 0,11 1,1
6 30 0,1 5,1 0,08 1,08
Hàm lượng nitrat, nitrit đầu vào trong nước thải tương đối thấp, hàm
lượng nitrat trong khoảng 0,05 - 0,12 mg/L, hàm lượng nitrit dao động từ 0,02
- 0,11 mg/L. Như vậy, hàm lượng nitrat trong nước đầu ra chủ yếu là do quá
trình oxi hóa amoni thành nitrat, hàm lượng nitrat còn lại do nitrat chưa bị
khử về khí N2 trong giai đoạn thiếu khí. Hàm lượng nitrat đầu ra trong 15
ngày đầu tương đối cao, quá trình khử từ nitrat về khí nitơ vẫn chưa đạt hiệu
quả, từ ngày thứ 20 trở đi, hàm lượng nitrat đầu ra ổn định trong khoảng từ
5,1 - 5,4 mg/L. Hàm lượng nitrit trong nước thải đầu ra thấp trong khoảng từ
1,08 - 2,15 mg/L, đã đạt tiêu chuẩn đầu ra.
Nhận xét chung: Qua quá trình thực nghiệm cho thấy, xử lý photpho và
một số hợp chất của nitơ trong NTSH bằng công nghệ MBBR đạt hiệu quả xử
lý cao. Trong quá trình xử lý, VSV cần một khoảng thời gian thích nghi với
nguồn NTSH, sau giai đoạn thích nghi, VSV phát triển và hoạt động tốt, xử lý
hiệu quả các chất ô nhiễm. Mặc dù hàm lượng nitơ và photpho trong NTSH
thực tế dao động mạnh trong quá trình lấy mẫu, hệ thống vẫn xử lý hiệu quả,
chất lượng nước đầu ra đảm bảo chất lượng theo tiêu chuẩn hiện hành.

71
CHƯƠNG 4. KẾT LUẬN VÀ KIẾN NGHỊ
4.1. KẾT LUẬN
Qua quá trình xác nhận giá trị sử dụng của các phương pháp phân tích
theo các quy trình phân tích của các tiêu chuẩn trong và ngoài nước cho thấy
các thông số được phân tích theo 2 tiêu chuẩn có cùng độ chính xác. Nhưng
hệ thống MBBR với khả năng xử lí nước thải có hiệu suất cao nên hàm lượng
các chất trong nước thải đầu vào và đầu ra của hệ thống có nhiều sự khác biệt.
Vì vậy, quá trình phân tích mẫu nước luôn cần lựa chọn phương pháp phân
tích sao cho phù hợp với hàm lượng chất có trong mẫu. Qua quá trình khảo
sát nhóm đề tài đã đề xuất lựa chọn các phương pháp như sau:
- Phương pháp so màu bằng thuốc thử thylmo để xác định hàm lượng
amoni trong nước thải đầu vào.
- Phương pháp lên màu trực tiếp với thuốc thử Nessler để xác định hàm
lượng amoni trong nước thải đầu ra.
- Phương pháp tiêu chuẩn TCVN 6178 :1996 để phân tích hàm lượng
nitrit.
- Phương pháp đo quang phổ tia UV và dẫn xuất thứ hai - Tiêu chuẩn
SMEWW 4500 NO3- B để xác định hàm lượng nitrat trong nước thải đầu vào.
- Phương pháp quang phổ hấp thụ phân tử dùng axit sunfosalixylic -
TCVN 6180:1996 để xác định hàm lượng nitrat trong mẫu nước thải đầu ra.
- Phương pháp đo phổ dùng amoni molipdat – TCVN 6202:2008 phù
hợp với việc xác định hàm lượng phot phat trong nước thải đầu ra.
- Phương pháp đo phổ hấp thụ phân tử với axit Vanadomolybdo
phosphoric - Tiêu chuẩn SMEWW 4500 –P C thích hợp để xác định hàm
lượng phot phat trong nước thải đầu vào.
Qua các kết quả phân tích thực nghiệm cho thấy, hiệu quả xử lý
photphat và một số hợp chất của nitơ như amoni, nitrat, nitrit của công nghệ
MBBR đạt hiệu quả xử lý cao, đáp ứng yêu cầu đầu ra theo cột B, Quy chuẩn
kỹ thuật Quốc gia đối với nước thải sinh hoạt QCVN 14:2008/BTNMT. Cụ

72
thể, sau 30 ngày của quá trình xử lý, hiệu quả xử lý photphat đã ổn định và
hàm lượng photphat đầu ra 2,61mg/L; hiệu suất xử lý photphat đạt 84,18%.
Đối với các hợp chất của nitơ, hàm lượng amoni (NH4+), nitrat (NO3
-), nitrit
(NO2-) đầu ra lần lượt là 5,4; 5,1; 1,08 mg/L đều đạt yêu cầu đầu ra.
Công nghệ MBBR được đánh giá là một trong những công nghệ xử lý
nước thải tiên tiến hiện nay. Với nước thải sinh hoạt, sau khi xử lý đạt tiêu
chuẩn cột B, theo QCVN14: 2008/BTNMT. Công nghệ MBBR là một hệ
thống nhỏ gọn, việc ghép các module nhỏ thành một khối lớn nhằm tăng hay
giảm công suất, tăng hiệu quả xử lý được thực hiện dễ dàng, điều này cũng
tương đương với khả năng thích nghi trong điều kiện có mức độ dao động lưu
lượng lớn.
Xét về khía cạnh kinh tế, đầu tư vào công nghệ MBBR là một kế hoạch
hợp lý, có thể từng bước triển khai theo từng đợt xây dựng, theo từng quy mô
giai đoạn. Do rất nhỏ gọn nên cụm xử lý dùng MBBR có thể đặt tại bất kì đâu
trong tầng hầm của các tòa nhà, tại một góc nhỏ của khu đô thị, nhiều nghiên
cứu khác cũng cho thấy MBBR hầu như không có mùi, loại bỏ vi khuẩn và
đảm bảo yêu cầu trước khi xả ra bên ngoài môi trường.
Còn nếu nhìn nhận dưới góc độ môi trường, MBBR là công nghệ thân
thiện với môi trường, đem lại hiệu quả xã hội to lớn. Không những trả lại môi
trường nguồn nước chất lượng cao, không còn độc hại hay mầm bệnh từ các vi
khuẩn, virus, giảm được hiện tượng mùi phát tán ra môi trường không khí.
MBBR rất phù hợp với cụm xử lý phân tán do nhỏ gọn không chiếm
nhiều diện tích, quản lý vận hành dễ dàng, có thể quản lý thành từng cụm.
Chính vì ưu điểm này, MBBR đang được ứng dụng tại nhiều nước trên thế giới.
4.2. KIẾN NGHỊ
Công nghệ MBBR ứng dụng xử lý nước thải cùng với giá thể vi sinh từ
nhựa nhiệt dẻo có khả năng mở rộng ứng dụng từ quy mô phòng thí nghiệm
tới các cơ sở từ quy mô nhỏ tới lớn. Chính vì vậy, nếu tiếp tục có những đầu
tư về kinh phí sẽ tiến tới hoàn thiện công nghệ một cách đồng bộ và đưa vào
ứng dụng thực tế ở mức độ cao hơn.

73
Nước thải sau khi xử lý bằng công nghệ MBBR có chất lượng rất tốt,
không còn cặn lắng, vi khuẩn gây bệnh do đó có thể khuyến khích tái sử dụng
thải cho các mục đích công cộng để tăng lợi ích và hiệu quả đầu tư.
Công nghệ MBBR dù đã được áp dụng tại nhiều nước trên thế giới
nhưng cũng cần có nhiều nghiên cứu trong điều kiện của Việt Nam để công
nghệ MBBR phát huy được tối đa hiệu quả xử lý.

74
TÀI LIỆU THAM KHẢO
1. Borkar R.P, Gulhane M.L , and Kotangale A.J (2013). Moving Bed
Biofilm Reactor – A New Perspective in Wastewater Treatment. IOSR
Journal Of Environmental Science, Toxicology And Food Technology
(IOSR-JESTFT) 6 (6): 15-21.
2. Ankit B. Pinjarkar, Rushikesh D. Jagtap, Chaitanya K. Solanke, Hitesh
H. Mehta (2017), The Moving Bed Biofilm Reactor (MBBR).
International Advanced Research Journal in Science, Engineering and
Technology 4(3): 63 – 66.
3. Ødegaard H (2006). Innovations in wastewater treatment: the moving
bed biofilm process. Water Sci Technol. 53: 17-33.
4. Pal Shailesh R , Dr. Dipak S. Vyas , Arti N Pamnani (2016). Study the
efficiency of moving bed bio-film reactor (mbbr) for dairy wastewater
treatment. IJARIIE-ISSN(O)-2395-4396: 899 – 905.
5. Jamal Ali Kawan, Hassimi Abu Hasan, Fatihah Suja`, (2016) Othman
Jaafar, Rakmi Abd-Rahman, A review on sewage treatment and
polishing using moving bed bioreactor, Journal of Engineering Science
and Technology, 11(8), 1098-1120.
6. Pal Shailesh R , Dr. Dipak S. Vyas , Arti N Pamnani (2016). Study the
efficiency of moving bed bio-film reactor (mbbr) for dairy wastewater
treatment. IJARIIE-ISSN(O)-2395-4396: 899 – 905.
7. J. J. Marques, R. R. Souza, C. S. Souza1 and I. C. C. Rocha (2008),
Attached biomass growth and substrate utilization rate in a moving bed
biofilm reactor, Brazilian Journal of Chemical Engineering, 665 – 670.
8. Nguyễn Hoàng Như (2012), Nghiên cứu ứng dụng công nghệ MBBR để
xử lý nước thải sản xuất bia, Luận văn Thạc s , trường Đại học Bách
Khoa, Đại học Quốc gia Thành phố Hồ Chí Minh.
9. Majid Kermani, Bijan Bina, Hossein Movahedian, Mohammad Mehdi
Amin, Mahnaz Nikaeen (2009). Biological phosphorus and nitrogen

75
removal from wastewater using moving bed biofilm process. Iranian
Journal of biotechnology 7 (1): 19 – 27.
10. Wang XJ, Xia SQ, Chen L, Zhao JF, Renault NJ, Chovelon JM (2006).
Nutrients removal from municipal wastewater by chemical precipitation
in a moving bed biofilm reactor. Process Biochem. 41: 824-828.
11. Hooshyari B, Azimi A, Mehrdadi N (2009). Kinetic analysis of enhanced
biological phosphorus removal in a hybrid integrated fixed film
activated sludge process. Int J Environ Sci Tech. 6: 149-158.
12. Chen S, Sun D, Chung JS (2008), Simultaneous removal of COD and
ammonium from landfill leachate using an anaerobic–aerobic moving
bed biofilm reactor system. Waste Manage. 28:339-346.
13. Aygun, A., Nas, B., Berktay, A., (2008), Influence of high organic
loading rates on COD removal and sludge production in moving bed
biofilm reactor. Environ. Eng. Sci. 25, 1311–1316
14. Shore, J.L., M’Coy, W.S., Gunsch, C.K., Deshusses, M.A., (2012),
Application of a moving bed biofilm reactor for tertiary ammonia
treatment in high temperature industrial wastewater. Bioresour.
Technol. 112, 51–60.
15. Zhang, S., Wang, Y., He, W., Wu, M., Xing, M., Yang, J., Gao, N., Yin,
D., (2013), Responses of biofilm characteristics to variations in
temperature and NH+-N loading in a moving-bed biofilm reactor
treating micro-polluted raw water. Bioresour. Technol. 131, 365–373.
16. Zhuang, H., Han, H., Jia, S., Zhao, Q., Hou, B., (2014), Advanced
treatment of biologically pretreated coal gasification wastewater using a
novel anoxic moving bed biofilm reactor (ANMBBR)–biological aerated
filter (BAF) system. Bioresour. Technol. 157, 223–230.
17. Rafiei, B., Naeimpoor, F., Mohammadi, T., (2014), Bio-film and bio-
entrapped hybrid membrane bioreactors in wastewater treatment:
comparison of membrane fouling and removal efficiency. Desalination
337, 16–22.

76
18. Đồng Kim Loan, Trần Hồng Côn, Phạm Ngọc Hồ (2007), Quan trắc và
phân tích môi trường, Trường Đại học Khoa học Tự nhiên, Đại học
Quốc gia Hà nội.
19. Nguyễn Văn Ri, Tạ Thị Thảo (2013), Thực Tập Hóa phân tích- Phần 1:
Các phương pháp phân tích hóa học, Khoa Hóa học, Đại học Khoa học
Tự nhiên, Đại học Quốc gia Hà Nội.
20. Tiêu chuẩn quốc gia (2008). Vi-TCVN6202-2008, Chất lượng nước -xác
định photpho - phương pháp đo phổ dùng Amoni molipdat.
21. SMEWW 4500-P.C:2017, Vanadomolybdophosphoric Acid Colorimetric
Method.
22. Trần Thị Lý (2010), Xác định nitrat, nitrit trong một số mẫu nước mặt và
nước ngầm xung quanh khu vực nhà máy đạm Bắc Giang bằng phương
pháp trắc quan và phương pháp phân tích dòng chảy, luận văn thạc sỹ
Hóa học - Đại học Sư phạm Thái Nguyên.
23. Tiêu chuẩn quốc gia (1996). Vi-TCVN6179-1:1996, Xác định amoni
bằng phương pháp trắc phổ thao tác bằng tay.
24. Trần Tứ Hiếu (2003), Phân tích trắc quang, Nhà xuất bản Đại học Quốc
gia Hà Nội.
25. Trần Tứ Hiếu (2000), Hóa học phân tích, Nhà xuất bản Đại học Quốc
gia Hà Nội.
26. Zanardi E, Dazzi G, Madarena G, Chizzolini R. (2002), Comparative
study on nitrite and nitrate ions determination, Ann.Fac.Medic.Vet.di
Parma, vol XXII, p.79-86.
27. Timmer-TenHoor(1974), Sulfide interaction on colorimetric nitrite
determination, Marine Chemistry vol 2 (2), p. 149-151.
28. S.Marten, J. Harms, Wissenschaftliche Geratebau Dr. Ing.H. Knauer
GmbH (2000), Determination of nitrite and nitrate in fruit juies by UV
detection, KNAUER - ASI - Advanced Scientific Instruments.

77
29. Norwitz, P. N Keliher (1984), Spectrophotometric determination of
nitrite with composite reagents containing sulphanilamide, sulphanilic
acid or 4-nitroaniline as the diozotisable aromatic amine and N-(1-
naphthyl) ethylene diamine as coupling agent, Analyst, Vol. 109, pp
1281-1286.
30. Adnan Aydın, Ozgen Ercan, Sulin Tascioglu (2005), A novel method for
the spectrophotometric determination of nitrite in water, Talanta 66
1181-1186.
31. Fostr Dee Snell and Leslie S. Ettre (1972), Encyclopedia of industrial
chemiscal analysic, Vol 16, Interscience Publishers.
32. Rafiei, B., Naeimpoor, F., Mohammadi, T., (2014), Bio-film and bio-
entrapped hybrid membrane bioreactors in wastewater treatment:
comparison of membrane fouling and removal efficiency, Desalination
337, 16-22.
33. SMEWW 4500-NO2-.B:2017, Colorimetric Method.
34. SMEWW 4500-NO3-.B:2017, Ultraviolet Spectrophotometric Screening
Method.





























