Embed Size (px)
Citation preview

5/12/2018 Talasemia - slidepdf.com
http://slidepdf.com/reader/full/talasemia-55a35973b5eb1 1/39

5/12/2018 Talasemia - slidepdf.com
http://slidepdf.com/reader/full/talasemia-55a35973b5eb1 2/39
HEMOGLOBINAS
Algunos Datos
• 1 milímetro cúbico de sangre contiene aprox.5 millones de glóbulos rojos
• Cada eritrocito contiene aprox. 600 millonesde moléculas de hemoglobina• La hemoglobina es la proteína más
abundante de la naturaleza• La fracción proteica (globinas) permite ilustrar
una gran variedad de facetas de la Genética

5/12/2018 Talasemia - slidepdf.com
http://slidepdf.com/reader/full/talasemia-55a35973b5eb1 3/39
VARIANTES DE HEMOGLOBINAS
1. Variantes estructurales
2. Variantes que afectan a latasa de síntesis de lasglobinas

5/12/2018 Talasemia - slidepdf.com
http://slidepdf.com/reader/full/talasemia-55a35973b5eb1 4/39
VARIANTES DE HEMOGLOBINAS
1. Variantes estructurales
• Variantes por sustitución de aminoácidos.La mutación productora es una sustitución de
un nucleótido de un triplete por otronucleótido distinto. Es el grupo más numeroso(>600 variantes). Ej.: HbS (Anemiafalciforme), HbC, etc. Solo detectables si
cambia la carga neta de la proteína(electroforesis) o si se producen anomalíasfuncionales patentes (hemoglobinopatías).

5/12/2018 Talasemia - slidepdf.com
http://slidepdf.com/reader/full/talasemia-55a35973b5eb1 5/39
VARIANTES DE HEMOGLOBINAS
2. Variantes tasa de síntesis
TALASEMIAS
Variantes patológicas de hemoglobinascaracterizadas porque la síntesis de
globinas alfa o las beta se reduce en
mayor o menor medida.

5/12/2018 Talasemia - slidepdf.com
http://slidepdf.com/reader/full/talasemia-55a35973b5eb1 6/39
Hemoglobina
La Hemoglobina es una proteina cuya principal función es transportar el oxígeno a los tejidos.
Son tres los tipos de Hemoglobina en adultos
Hb A α 2β 2 96%
Hb A2 α 2δ 2 3%
Hb F α 2γ 2 1%
Alfa globinas: Cr16 (4genes)
Beta globinas: Cr11 (2 genes)

5/12/2018 Talasemia - slidepdf.com
http://slidepdf.com/reader/full/talasemia-55a35973b5eb1 7/39
Síntesis de Hemoglobinas a lo
largo del desarrolloA2b2: AA2d2: A2A2g2: F

5/12/2018 Talasemia - slidepdf.com
http://slidepdf.com/reader/full/talasemia-55a35973b5eb1 8/39
Talasemia
• La Talasemia es un desorden congénito hemolítico
causado por una deficiencia parcial o completa de
la síntesis de las cadenas alfa o beta de las globinasde la hemoglobina.
• Se manifiesta en una amplia gama de cuadros
clínicos que van desde la muerte intrauterina hastala microcitosis asintomática sin anemia.

5/12/2018 Talasemia - slidepdf.com
http://slidepdf.com/reader/full/talasemia-55a35973b5eb1 9/39
Síndromes talasémicos alfa-beta
Síntesis disminuida o ausente de cadenas de
globina
• Hemoglobinopatías muy frecuentes• Consecuencias:
– Descenso en la síntesis de Hb
– Desequilibrio de cadenas alfa/beta – Eritropoyesis compensadora
– Alteraciones en el espectro electroforético de la Hb

5/12/2018 Talasemia - slidepdf.com
http://slidepdf.com/reader/full/talasemia-55a35973b5eb1 10/39
Distribución geográfica
• La distribución mundial de las talasemias, coincide con ladel paludismo, siendo muy frecuente en los países de laCuenca del Mediterráneo, Norte de África, Oriente, India y
Sudeste Asiático.• La migración de las poblaciones ha extendido a las
talasemias a otros países europeos, América del Norte,Sudamérica y Australia.

5/12/2018 Talasemia - slidepdf.com
http://slidepdf.com/reader/full/talasemia-55a35973b5eb1 11/39
¿Cuando sospechar una
talasemia?
• En toda Anemia Hipocromica Microcítica
como hallazgo o en un paciente mayor deun año que no presenta ferropenia
• Antecedente Familiar de Portador talasémico

5/12/2018 Talasemia - slidepdf.com
http://slidepdf.com/reader/full/talasemia-55a35973b5eb1 12/39
LABORATORIO
• Hemograma, frotis
• Metabolismo del hierro
• Cuantificación de las Hemoglobinas
• Datos de hemólisis• Biología Molecular

5/12/2018 Talasemia - slidepdf.com
http://slidepdf.com/reader/full/talasemia-55a35973b5eb1 13/39
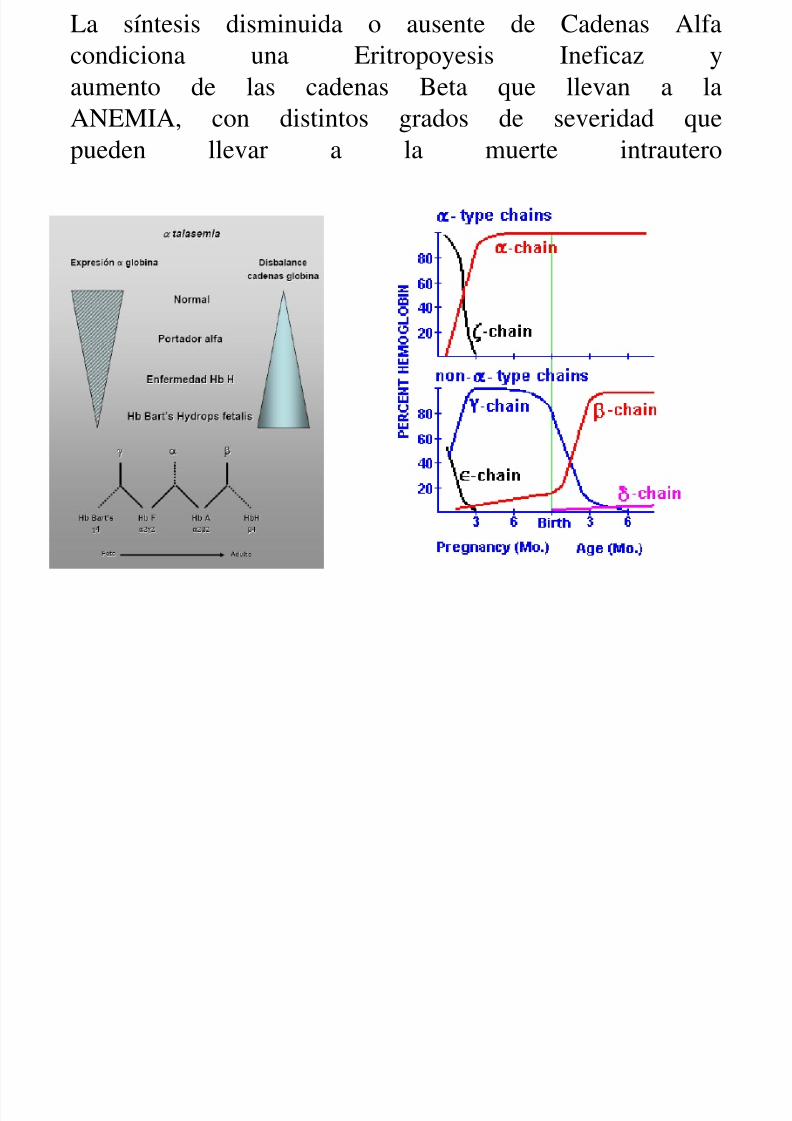
Alfa talasemia
• Es una alteracion del Cromosoma 16 – Síntesis disminuida o ausente de cadenas de α
globina• Exceso de cadenas gamma en el feto (Hb Bart; g4)
• Exceso de cadenas beta en el adulto (Hb H; b4)
• El grado de anemia dependerá de la afectación delos genes alfa y del exceso de cadenas gamma/beta
5/12/2018 Talasemia - slidepdf.com
http://slidepdf.com/reader/full/talasemia-55a35973b5eb1 14/39
La síntesis disminuida o ausente de Cadenas Alfacondiciona una Eritropoyesis Ineficaz yaumento de las cadenas Beta que llevan a laANEMIA, con distintos grados de severidad que
pueden llevar a la muerte intrautero

5/12/2018 Talasemia - slidepdf.com
http://slidepdf.com/reader/full/talasemia-55a35973b5eb1 15/39
Alfa talasemias
• Cada cromosoma 16 lleva 2 genes. Por ello, la cantidad de genes alfaheredados es de 4.
En las α-talasemias las mutaciones más frecuentemente halladas
son deleciones (-α), que pueden afectar: – un gen alfa – dos genes alfa en tandem – el cluster globínico α
• Menos frecuentemente las α-talasemias se deben a mutacionespuntuales (αT)
/ : Indica división de genes heredados de ambos padres:
/

5/12/2018 Talasemia - slidepdf.com
http://slidepdf.com/reader/full/talasemia-55a35973b5eb1 16/39
Clasificación y terminología de
alfa talasemia• Normal / • Portador silente - /
• Menor - /- --/
• Enfermedad Hb H --/-
• Hydrops fetalis --/--

5/12/2018 Talasemia - slidepdf.com
http://slidepdf.com/reader/full/talasemia-55a35973b5eb1 17/39

5/12/2018 Talasemia - slidepdf.com
http://slidepdf.com/reader/full/talasemia-55a35973b5eb1 18/39
Sobrevida eritrocitaria acortada Hemólisis intra – extravascular
Anemia severidad variable Palidez – Muerte intraúteroVisceromegalias
Orinas oscuras
Crisis de hiperhermólisis oaplásticas
Exacerbación por ingesta de drogas

5/12/2018 Talasemia - slidepdf.com
http://slidepdf.com/reader/full/talasemia-55a35973b5eb1 19/39
Laboratorio

5/12/2018 Talasemia - slidepdf.com
http://slidepdf.com/reader/full/talasemia-55a35973b5eb1 20/39
Manifestaciones clínicas• Portador silente
– Asintomático – No tratamiento (hierro / fólico / B12)
• Portador α Talasemia/Menor – Asintomático
– Anemia leve microcítica hipocrómica – Raro: anemia hemolítica (anemia, ictericia, esplenomegalia)
• Diagnóstico diferencial – Deficiencia de hierro – Anemia de los procesos crónicos
– Beta talasemia con Hb A2 normal• No suelen requerir tratamiento ni transfusiones.
– Ácido fólico
• ¡CONSEJO GENÉTICO!

5/12/2018 Talasemia - slidepdf.com
http://slidepdf.com/reader/full/talasemia-55a35973b5eb1 21/39
Enfermedad por Hb H
•Manifestaciones clínicas extremadamente variables•Asintomático•Anemia crónica•“Talasemia intermedia”

5/12/2018 Talasemia - slidepdf.com
http://slidepdf.com/reader/full/talasemia-55a35973b5eb1 22/39
Enfermedad por hemoglobina H• Palidez,
• Hepatomegalia (70%), Esplenomegalia (80%)
• Hiperesplenismo (10%). Excepcionalmente esplenectomía
• Crisis de hiperhemólisis y/o aplásticas, tras la ingesta de medicamentos• Litiasis vesicular, colecistitis
• Requerimiento transfusional esporádico
• Evaluación anual de la sobrecarga de hierro
• Ácido fólico; hierro y/o B12 sólo si se demuestra el déficit• RARAMENTE: TRASPLANTE ALOGÉNICO DE PROGENITORES
• CONSEJO GENÉTICO

5/12/2018 Talasemia - slidepdf.com
http://slidepdf.com/reader/full/talasemia-55a35973b5eb1 23/39
Estudio medular/sangre periférica

5/12/2018 Talasemia - slidepdf.com
http://slidepdf.com/reader/full/talasemia-55a35973b5eb1 24/39
Hydrops fetalis
• Ausencia de Hbs funcionales
• Anemia (Hb 2,5-10g/dl: Hb embrionarias, Portland 2γ2)• Hematopoyesis extramedular• Edema generalizado, ascitis, derrame pleural, pericárdico• Insuficiencia cardíaca• Muerte• 17% anomalías constitucionales
• Complicaciones maternas

5/12/2018 Talasemia - slidepdf.com
http://slidepdf.com/reader/full/talasemia-55a35973b5eb1 25/39
Beta Talasemia
• Es una alteracion del Cromosoma 11
• Síntesis o ausente de cadenas beta de globina+: Indica producción disminuida de la cadena de globina
por el gen (pero presente en menor medida)
b+ 0 :Indica ausencia de producción de globina por el gen :
b0

5/12/2018 Talasemia - slidepdf.com
http://slidepdf.com/reader/full/talasemia-55a35973b5eb1 26/39
Clasificación y terminología Beta
Talasemia• Normal b / b • Menor b / b0
b / b+ • Intermedia b+ / b+
• Mayor b0 / b0
b0 / b+

5/12/2018 Talasemia - slidepdf.com
http://slidepdf.com/reader/full/talasemia-55a35973b5eb1 27/39
Talasemia menor o rasgo beta
talasémico: transmisión genética

5/12/2018 Talasemia - slidepdf.com
http://slidepdf.com/reader/full/talasemia-55a35973b5eb1 28/39
Beta Talasemia Mayor:
Transmisión genética

5/12/2018 Talasemia - slidepdf.com
http://slidepdf.com/reader/full/talasemia-55a35973b5eb1 29/39
La sintesis disminuida o ausente de Cadenas Beta condicionauna Eritropoyesis Ineficaz yaumento de las cadenas alfa que llevan a la
ANEMIA, con distintos grados de severidad

5/12/2018 Talasemia - slidepdf.com
http://slidepdf.com/reader/full/talasemia-55a35973b5eb1 30/39
Rasgo beta talasémico/talasemia
minor• Recuento de hematíes alto
• Hb 11-13 g/dL
• VCM disminuido(<78 fl)
• ADE normal
• Anisopoiquilocitosis;dianocitos, punteado basófilo
• Niveles elevados de Hb A2(menor del 6%)
• Ligerahepatoesplenomegalia
• Astenia discreta
• Crisis esporádicas deictericia
• No tratamiento salvo ácidofólico

5/12/2018 Talasemia - slidepdf.com
http://slidepdf.com/reader/full/talasemia-55a35973b5eb1 31/39
Frotis de sangre periférica

5/12/2018 Talasemia - slidepdf.com
http://slidepdf.com/reader/full/talasemia-55a35973b5eb1 32/39
Beta talasemia intermedia
• Recuento de hematíes alto
• Hb 7-10 g/dL
• VCM disminuido(<65 fl)
• Niveles más elevados de HbA2 y de Hb F
• Moderadahepatoesplenomegalia
• Alteraciones óseas
• Sobrecarga de hierro -quelantes
• Pueden precisartransfusiones
BETA TALASEMIA MAYOR

5/12/2018 Talasemia - slidepdf.com
http://slidepdf.com/reader/full/talasemia-55a35973b5eb1 33/39
BETA TALASEMIA MAYOR O
ENFERMEDAD DE COOLEY

5/12/2018 Talasemia - slidepdf.com
http://slidepdf.com/reader/full/talasemia-55a35973b5eb1 34/39
Beta Talasemia Mayor
• ¿Cómo llegamos al Diagnostico?
• Paciente de 6 meses de vida que tiene
• Hemoglobina <6 g/dl• Retardo de crecimiento
• Palidez progresiva
• Inapetencia, irritabilidad, sudoración profusa• Infecciones repetitivas
• Distensión abdominal sin causa quirúrgica
5/12/2018 Talasemia - slidepdf.com
http://slidepdf.com/reader/full/talasemia-55a35973b5eb1 35/39
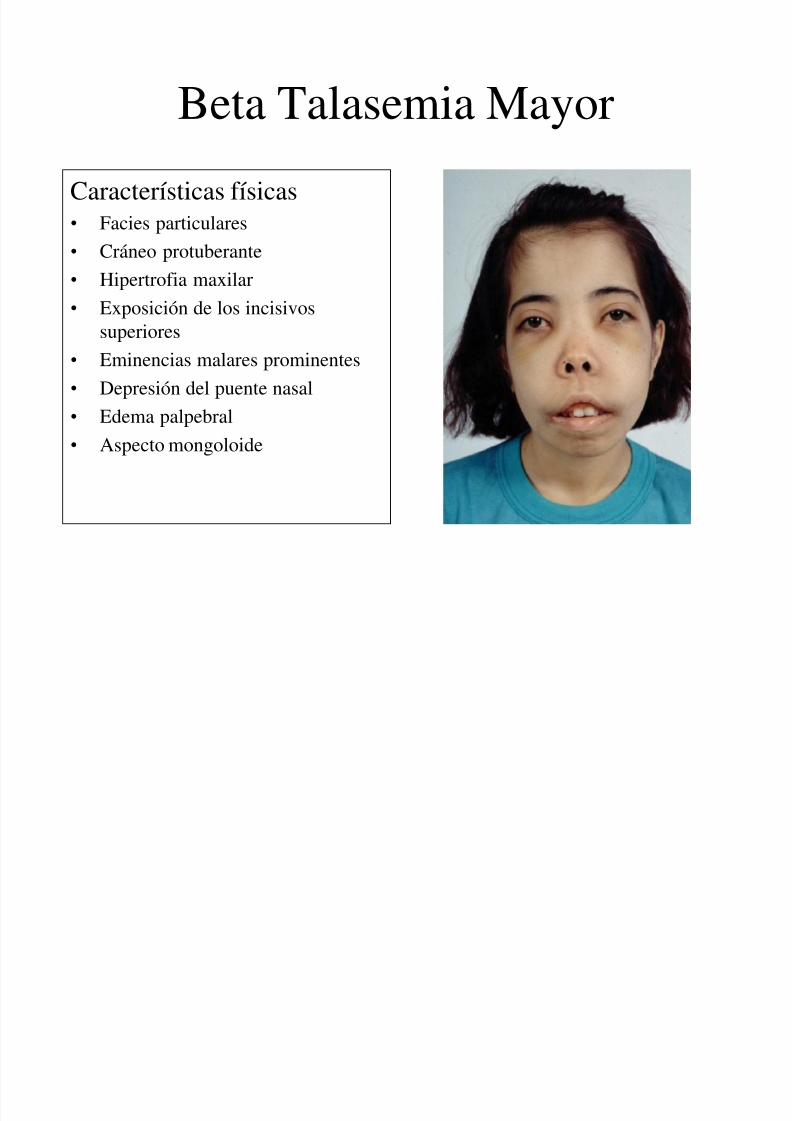
Beta Talasemia Mayor
Características físicas• Facies particulares
• Cráneo protuberante
• Hipertrofia maxilar• Exposición de los incisivos
superiores
• Eminencias malares prominentes
• Depresión del puente nasal
• Edema palpebral• Aspecto mongoloide

5/12/2018 Talasemia - slidepdf.com
http://slidepdf.com/reader/full/talasemia-55a35973b5eb1 36/39
Beta Talasemia Mayor
• Complicaciones• Retardo de crecimiento• Palidez, tinte ictérico• Musculatura pobre.
• Genu valgum• Pérdida de la grasa corporal• Extremidades delgadas• Distensión abdominal
(esplenomegalia)• Úlceras cutáneas
• Sobrecarga de hierro

5/12/2018 Talasemia - slidepdf.com
http://slidepdf.com/reader/full/talasemia-55a35973b5eb1 37/39
Beta talasemia mayor:
diagnóstico de laboratorio•Severa anisopoiquilocitosis•Hb < 7 g/dL•Hipocromía marcada•Microcitosis (VCM 50-70 fl)•Dianocitos•Eritroblastos circulantes;aumento de reticulocitos
•Hb A muy descendida o ausente•Gran predominio de HbF (60-95%)

5/12/2018 Talasemia - slidepdf.com
http://slidepdf.com/reader/full/talasemia-55a35973b5eb1 38/39
Beta Talasemia Mayor
Anomalías óseas:Aspecto de “cráneo en
cepillo” de la cortical por
expansión de la médula ósea

5/12/2018 Talasemia - slidepdf.com
http://slidepdf.com/reader/full/talasemia-55a35973b5eb1 39/39
Beta Talasemia Mayor:
Tratamiento• Es importante tener en cuenta que como encualquier patología el diagnostico precoz esimportante. En estas enfermedades es fundamental
el CONSEJO GENETICO.
• REGIMEN DE HIPERTRANSFUSION• ESPLENECTOMIA
• QUELANTES DEL HIERRO • TRASPLANTE ALOGÉNICO DEPROGENITORES HETOPOYÉTICOS





























