Embed Size (px)
Citation preview

Regulation of epithelial cell migration and
macropinocytosis by septin GTPases
A Thesis
Submitted to the Faculty
of
Drexel University
by
Lee Dolat
in partial fulfillment of the
requirements for the degree
of
Doctor of Philosophy
June 2016

© Copyright 2015 Lee Dolat. All Rights Reserved.

ii
Acknowledgements
I thank my adviser, Dr Elias Spiliotis, for instilling in me a deep appreciation for cell
biology. Equally important, he provided me with countless opportunities to develop as a
scientist, explore my own ideas and interests, and accomplish everything I set out to do.
My labmates – Jon Bowen, Xiaobo Bai, Eva Karasmanis, and Jimmy Angelis -
have been exceptional colleagues and friends.
I thank my thesis committee for their critical comments on my work. Drs
Saunders, Stanford and Anandan also shared with me invaluable career advice, and I
am especially grateful for their support during the search for my next position.
Finally, I thank my family and friends for their help and support along the way.

iii
Table of Contents
List of Figures ................................................................................................................ v
List of Tables ............................................................................................................... vii
Thesis Abstract .......................................................................................................... viii
Chapter 1: Introduction ................................................................................................. 1
Septin structure, assembly and dynamics ................................................................... 1
Septins and the membrane skeleton ........................................................................... 6
Septins and the actin cytoskeleton .............................................................................. 8
Septins and the microtubule cytoskeleton ................................................................. 12
Chapter 2: Tissue-specific expression of septins in health and disease ................ 21
Septin expression and roles in embryogenesis ......................................................... 21
Cardiovascular system .............................................................................................. 23
Immune system ......................................................................................................... 25
Nervous system ........................................................................................................ 27
Reproductive system ................................................................................................. 31
Urinary and digestive system .................................................................................... 33
Respiratory system ................................................................................................... 35
Endocrine system...................................................................................................... 36
Integumentary system ............................................................................................... 37
Chapter 3: Septins in Cancer ...................................................................................... 40
Septins in hematological malignancies ...................................................................... 40
Altered septin expression in solid tumors .................................................................. 41
Septins in the tumor microenvironment ..................................................................... 44
Chapter 4: Septin overexpression: implications in tumor cell migration, trafficking and metabolism ........................................................................................................... 46
Septins functions in tumor cell migration ................................................................... 47
Septins in membrane traffic and metabolism ............................................................. 50
Chapter 5: Septins promote stress-fiber mediated maturation of focal adhesions and renal epithelial motility ........................................................................................ 57
Abstract ..................................................................................................................... 57
Introduction ............................................................................................................... 57
Materials and Methods .............................................................................................. 59

iv
Results and Discussion ............................................................................................. 68
Figures ...................................................................................................................... 74
Chapter 6: Septins 9 exhibits polymorphic binding to F-actin and inhibits myosin and cofilin activity ....................................................................................................... 88
Abstract ..................................................................................................................... 88
Introduction ............................................................................................................... 88
Materials and Methods .............................................................................................. 90
Results ...................................................................................................................... 96
Discussion............................................................................................................... 103
Figures .................................................................................................................... 106
Chapter 7: Septins promote macropinosome maturation and traffic to lysosomes by facilitating membrane fusion ............................................................................... 120
Abstract ................................................................................................................... 120
Introduction ............................................................................................................. 120
Materials and Methods ............................................................................................ 132
Results and Discussion ........................................................................................... 139
Figures .................................................................................................................... 140
Chapter 8: Conclusions and future directions ........................................................ 155
List of References ..................................................................................................... 162
Appendix .................................................................................................................... 190
Vita ............................................................................................................................. 192

v
List of Figures Septin filament assembly ............................................................................................... 16
Septins regulate membrane organization, shape and dynamics .................................... 18
Septins interact with the actin and microtubule cytoskeleton ......................................... 19
Septin functions in organ systems and their connection to human disease .................... 39
Modes of single tumor cell migration ............................................................................. 55
Macropinosome formation, maturation and traffic to endolysosomes ............................. 56
Septin filaments interface with RSF and TA stress fibers in the leading lamella of renal
epithelia ......................................................................................................................... 74
Septins regulate the organization of the lamellar actin network and are required for the
stabilization of nascent FAs ........................................................................................... 76
SEPT9 functions as an actin cross-linking protein in FA maturation .............................. 79
SEPT9 promotes the motility of renal epithelia during EMT ........................................... 81
Lamellar septin fibers are composed of SEPT2/6/7/9 .................................................... 83
Quantification of septin knockdown and overexpression in MDCK cells ......................... 85
Septin depletion does not affect the levels of pSer19 myosin II RLC; myosin II N93K
rescues FA phenotype in septin-depleted cells .............................................................. 86
SEPT9 bundles F-actin through the B-domain of its NTD ............................................ 106
The NTD and B-domain of SERPT9 bind to three sites on the surface of F-actin ........ 108
SEPT9 inhibits the activity and interaction of myosin with actin ................................... 109
SEPT9 reduces the extent of cofilin-driven F-actin depolymerization ........................... 111
Overall 3D reconstruction of F-actin decorated with the NTD suggest that sites of SEPT9
binding to the actin filament ......................................................................................... 113
Convergence of structural classes validates model-based crosscorrelation sorting ..... 114
Reference free 2D averages of occupied classes ........................................................ 115

vi
Comparison of the frequencies of structural classes upon cross-correlation sorting of F-
actin segments in the presence or absence of the A-domain of SEPT9 ....................... 116
Full length SEPT9 and SEPT9 B-domain do not significantly affect F-actin bundling over
concentration ranges that inhibit myosin activity .......................................................... 117
Cosedimentation assay of full length SEPT9 with F-actin ............................................ 118
Resolution determination ............................................................................................. 119
Septins localize to macropinocytic vacuoles and tubule-vesicular endosomes, and their
contact/fusion sites ...................................................................................................... 139
Septins associate preferentially with maturing macropinosomes in a PI(3,5)P2-
dependent manner ...................................................................................................... 141
Septin depletion decreases macropinocytic fusion events and hinders macropinosome
maturation and turnover .............................................................................................. 143
Septins are required for the lysosomal delivery of fluid phase cargo ............................ 145
Septins are required for fusion of macropinosomes/endosomes with lysosomes, and
promote fusion directly in an in vitro reconstitution assay ............................................ 147
PM-mCherry and SEPT2 localization with respect to Rab5 and Rab7 ......................... 149
SEPT2 knock-down increases macropinosome size and number, but does not disrupt
colocalization of macropinocytic cargo with Rab5 and Rab7 ........................................ 151
SEPT2 does not colocalize with Sec6/8 and Exo70 at the contact sites of
macropinosomes/endosomes ...................................................................................... 153

vii
List of Tables
Table 1.1 ....................................................................................................................................... 17
Table 2.1 ....................................................................................................................................... 38

viii
ABSTRACT
Regulation of epithelial cell motility and macropinocytosis
by septin GTPases
Lee Dolat
Supervisor: Elias T. Spiliotis, PhD
Epithelial cancers, or carcinomas, account for approximately 90% of all cancers.
Unregulated cellular proliferation drives tumor growth and expansion, while metastatic
behavior, or the motility of tumor cells to distant sites, underlies the majority of cancer-
related deaths. Thus, better understanding of epithelial cell biology will provide novel
anti-cancer therapeutics.
Septin GTPases are a novel component of the mammalian cytoskeleton. Human
septins are comprised of thirteen genes that are divided into four groups based on
sequence similarity. Septins bind and hydrolyze GTP in order to assemble into
heteropolymers and higher-order structures such as bundles, ring and patches. Septins
bind directly to the actin and microtubule cytoskeleton and cell membranes. Through
these interactions, septins are posited to regulate their organization and dynamics by
acting as scaffolds and diffusion barriers. Importantly, septins are frequently
overexpressed in carcinomas, but their role in tumorigenesis and metastasis is unknown.
In the first part of this thesis, I investigated how septins function in renal epithelial
cell migration. Epithelial cell migration is driven by the actin cytoskeleton, which
assembles into a network of stress fibers that exert contractile forces at focal adhesions
with the extracellular matrix (ECM). Here, I discovered that septin 9 (SEPT9) crosslinks
directly actin filaments and controls focal adhesions stability, which is essential for
productive epithelial migration. Importantly, I found that SEPT9 expression is

ix
upregulated during epithelial-to-mesenchymal transition (EMT), a critical step during
tumor cell invasion, and SEPT9 overexpression promotes renal carcinoma and renal
epithelial cell migration. These results are among the first evidence that septins are bona
fide actin crosslinking and suggest that septin overexpression may promote tumor cell
invasion.
In the second part of this thesis, I investigated the molecular interaction between
SEPT9 and actin filaments. I identified that a basic region in the SEPT9 N-terminus is
sufficient to crosslink actin filaments. In addition, SEPT9 binds to a common site on the
actin filament and interferes with the binding of the myosin motor domain and the actin-
severing protein cofilin. By inhibiting myosin binding and cofilin-mediated severing,
SEPT9 may protect nascent actin filaments from depolymerization. These results
provide the first evidence that septins regulate spatially the binding of actin-binding
proteins along the actin filament.
In the last part of this thesis, I identified a novel role of septins in epithelial
macropinocytosis, a form of clathrin-independent endocytosis that is frequently
upregulated in cancer and provides essential molecular intermediates for carbon
metabolism during tumor cell proliferation. Here, I found that septins localize to
macropinosome contact sites with endosomes in a phosphoinositide-specific manner.
Significantly, septins regulate the delivery of macropinocytic cargo to the lysosome by
promoting membrane fusion. These results provide the first evidence of a role for septins
in a clathrin-independent endocytic pathway and endocytic vesicle fusion.
Collectively, these findings advance our knowledge of septin cell biology and
provide new evidence for how septin overexpression may promote tumor growth and
metastasis through two different cellular pathways.


1
CHAPTER 1. Introduction
Similar to microtubules, actin microfilaments and intermediate filaments, septins
constitute a network of filamentous polymers, which are essential for the development
and physiological functions of eukaryotic organisms. Septin filaments associate with cell
membranes and the actin and microtubule cytoskeleton, exhibiting a structural and
functional interdependence that distinguishes them apart from the conventional
cytoskeleton. Septin filaments function as diffusion barriers and scaffolds that affect the
compartmentalization of cell membranes and the spatial organization and functions of
the microtubules and actin cytoskeleton (Caudron and Barral, 2009; Spiliotis and
Gladfelter, 2012).
Septins are guanosin-5’-triphosphate (GTP)-binding proteins that self assemble
into non-polar oligomers and polymers (Weirich et al., 2008; Zhang et al., 2000). Similar
to actin and microtubules, septin assembly is coupled to nucleotide-binding and
hydrolysis (Gasper et al., 2009). Resembling the hetero-polymeric nature of intermediate
filaments, septin filaments consist of various subunits, which in mammalian organisms
are encoded by 13-14 different genes with alternative splicing and translation initiation
sites that give rise to multiple septin isoforms (Caudron and Barral, 2009).
I. Septin structure, assembly and dynamics Classified under the P-loop superfamily of nucleotide-binding and hydrolyzing
proteins, which includes the Ras-like GTPases and the kinesin and myosin motors,
septins possess a highly conserved GTP-binding domain (G domain), which is
structurally characterized by α-helices and β-sheets that alternate between flexible loops
(Leipe et al., 2002). Septin G domains contain the G1 Walker A (GxxxxGKS/T), G3
Walker B (DxxG) and G4 (NKxD) motifs, which interact directly with atoms of the

2 guanine nucleotide. In addition to these signature motifs, the N- and C-termini of the
septin G domains are respectively characterized by a polybasic region, which binds
phosphoinositides (Casamayor and Snyder, 2003; Zhang et al., 1999), and a sequence
of variable length and unknown function termed the septin unique element (SUE)
(Versele and Thorner, 2005). Beyond these homology domains, septins differ
significantly in the length and sequence of their N- and C-terminal extensions, which
flank their G domains and consist of proline-rich and α-helical domains, respectively
(Luedeke et al., 2005).
Based on amino acid similarity, mammalian septin paralogs are categorized into
four groups: SEPT2, SEPT6, SEPT7 and SEPT9 (Dobbelaere and Barral, 2004;
Luedeke et al., 2005). The majority of septins fall under the SEPT2 group, which
includes SEPT1, SEPT2, SEPT4 and SEPT5, and the SEPT6 group, which consists of
SEPT6, SEPT8, SEPT10, SEPT11 and SEPT14 (Figure 1.1). Lacking a critical threonine
residue from their G domains, septins of the SEPT6 group are hydrolytically inactive and
always bound to GTP (Gulesserian et al., 2007; Sirajuddin et al., 2009). The remaining
septins are classified under the SEPT9 group, which comprises SEPT3, SEPT9 and
SEPT12, and the SEPT7 group, which contains SEPT7 isoforms (Figure 1.1). SEPT2,
SEPT7 and SEPT9 are nearly ubiquitously expressed, while SEPT1, SEPT3, SEPT12
and SEPT14 are tissue-specific . The remaining septins are expressed widely, but not
present in every tissue (Dolat et al., 2014a).
Septin polymerization is driven by the propensity of their G domains to homo-
and hetero-dimerize (Sirajuddin et al., 2007). In the presence of guanosine nucleotide,
septin G domains dimerize by interacting directly through their GTP-binding pockets (G
interface), which are oriented opposite to one another, or, alternatively, through their N-
and C-terminal helices (NC interface). Using the G and NC dimerization interfaces in

3 tandem, septin monomers assemble linearly into non-polar oligomers (Sirajuddin et al.,
2007) (Figure 1.1). In mammalian systems, the basic septin oligomer is a hetero-
octamer composed of septins from each of the SEPT2, SEPT6, SEPT7 and SEPT9
groups at a 2:2:2:2 stoichiometry (SEPT9-SEPT7-SEPT6-SEPT2-SEPT2-SEPT6-
SEPT7-SEPT9) (Kim et al., 2011; Sellin et al., 2011b). This mode of hetero-octameric
assembly is conserved between mammals and yeast (Bertin et al., 2008; Farkasovsky et
al., 2005), septin filaments could also consist of hetero-hexameric (SEPT7-SEPT6-
SEPT2-SEPT2-SEPT6-SEPT7) and –tetrameric (SEPT6-SEPT7-SEPT7-SEPT6) units
(Engidawork et al., 2003a; Kinoshita, 2003). In addition, “non-canonical” hetero-
oligomers that consist of multiple subunits of the same septin group may also exist
(Beites et al., 1999; Blaser et al., 2002; Hsu et al., 1998; Martinez et al., 2004; Shinoda
et al., 2010). Overall, septin hetero-oligomers could vary in size and composition
depending on cell and tissue type. Irrespective of subunit identity, presence of GDP and
thus GTP hydrolysis favors oligomerization by stabilizing the G interface, while GTP-
binding appears to destabilize the NC interface (Gulesserian et al., 2007). With the
exception of the SEPT6 group septins, which cannot hydrolyze GTP, oligomeric septins
are GDP-bound and septin polymers contain an excess of GDP relative to GTP
(Farkasovsky et al., 2005; Sirajuddin et al., 2007). Therefore, in contrast to microtubules
and actin microfilaments whose polymerization is favored by a non-hydrolyzed
nucleotide, septin polymerization is favored by GTP hydrolysis.
Septin hetero-octamers polymerize into higher order structures such as linear
and curved filaments, rings and gauze-like meshworks (Bertin et al., 2008; Garcia et al.,
2011; Kinoshita, 2002) (Figure 1.1). End-to-end binding of septin hetero-octamers
results in filaments, which are 5-10 nm wide and up to several microns long (Bertin et al.,
2008; Bertin et al., 2010; Kinoshita, 2002). Septin filaments pair with one another by

4 interacting laterally through the C- (coiled coil domains) and possibly N-terminal
extensions that stretch outwards from the plane of oligomerization (Bai et al., 2013;
Bertin et al., 2008; de Almeida Marques et al., 2012; John et al., 2007). Thus, septin
bundles form by lateral stacking of individual septin filaments and adaptor proteins
further enhance septin bundling (Sadian et al., 2013). Owing to the flexibility of the
SEPT2 NC interface, which behaves like a hinge, septin oligomers can bend within a 25-
30o angle of one another generating a curvature, which allows for the formation ring-like
structures with 0.5 – 0.7 µm diameter (Sirajuddin et al., 2007). Septin subunits that
enhance the flexibility of NC interface favor the formation of septin rings and post-
translational modifications can promote the assembly of a gauze-like meshwork of
orthogonally arranged filaments (Garcia et al., 2011). The filamentous pattern of septins
is further influenced by their association with membrane bilayers and the actin and
microtubule cytoskeleton. Giant unilamellar vesicle membranes enriched with
phosphatidylinositol-4,5-bisphosphate can promote the formation of tightly paired septin
filaments (Tanaka-Takiguchi et al., 2009) . Conversely, actin-templated assembly of
septins favors the formation of linear filaments as opposed to septin rings, which
become more prevalent upon actin depolymerization (Gulesserian et al., 2003;
Kinoshita, 2002). Similarly, the disk-like organization of septins in non-adherent cells
depends on microtubules (Engidawork et al., 2003b; Sellin et al., 2011a).
Septin filaments are less dynamic than microtubules and F-actin (Table 1.1).
Half-times of subunit exchange and turnover could vary from 10s of seconds for
cytoplasmic septin filaments in mammalian cells to 3 minutes for membrane-associated
septin polymers in yeast organisms (Dobbelaere et al., 2003; Engidawork and Lubec,
2003; Gulesserian et al., 2003; Hagiwara et al., 2011; Lubec and Engidawork, 2002).
Recent studies show that membrane-bound septin filaments grow by end-to-end

5 annealing (Bridges et al., 2014). Rod-shaped septin oligomers diffuse, collide and
anneal to the free ends of septin filaments, which do not exchange any subunits along
their length (Bridges et al., 2014). In contrast to membrane-bound septin filaments,
which incorporate new subunits after fragmentation, cytoplasmic septin filaments are
more dynamic, exchanging subunits along their length (Bridges et al., 2014; Engidawork
and Lubec, 2003; Gulesserian et al., 2003). Septin filament dynamics are affected by
post-translational modifications such as phosphorylation and acetylation, and vary
depending on the phase of the cell cycle (Hernandez-Rodriguez and Momany, 2012).
For example, in the budding yeast Saccharomyces cerevisiae, membrane-bound septin
filaments are dynamic in G1, but highly stable during the S, G2 and M phases of the cell
cycle (Dobbelaere et al., 2003). These changes in septin dynamics are mediated by
septin-dependent kinases and phosphatases that are recruited to septin filaments in the
beginning and end of the cell cycle, respectively (Dobbelaere et al., 2003).
The molecular mechanisms that regulate septin assembly and dynamics are not
fully understood. Recent advances indicate that regulation of GTP hydrolysis, septin
synthesis and degradation as well as post-translational modifications and protein-protein
interactions influence the filamentous organization and dynamics of septins. Septin
folding is mediated by the cytosolic chaperonin containing TCP-1 (CCT), which also
controls the folding of actin and tubulin, and septin degradation involves E3 ubiquitin
ligases including parkin, the von Hippel-Lindau (VHL) tumor suppressor and the Ring
Finger protein 8 (RNF8) (Chahwan et al., 2013; Craven et al., 2006a; Dekker et al.,
2008; Zhang et al., 2000). While these factors could regulate the availability of septin
subunits for polymerization, proteins that promote the GTPase activity of septins (e.g.,
Orc6) and cross-link septin filaments (e.g., Gic1) can directly enhance septin assembly
(Huijbregts et al., 2009; Sadian et al., 2013). In contrast, Cdc42, a signaling molecule of

6 the Rho family of GTPases, induces the unbundling and dissociation of septin filaments
(Sadian et al., 2013). Post-translational modifications such as phosphorylation,
acetylation and sumoylation further modulate septin assembly and dynamics, but many
of the details remain unknown (Hernandez-Rodriguez and Momany, 2012).
II. Septins and the membrane skeleton
Septin filaments assemble on the cytoplasmic leaflet of membrane bilayers
forming a meshwork that resembles the spectrin-actin membrane skeleton. In yeast
cells, septins form filamentous rings and gauze-like patches in actin-free areas of the cell
cortex (Engidawork et al., 2001a; Engidawork and Lubec, 2001), while in mammalian
cells septin filaments associate with the actin membrane skeleton (Hagiwara et al.,
2011). These membrane-bound septin filaments restrict protein diffusion and affect the
shape and rigidity of cell membranes (Caudron and Barral, 2009; Spiliotis and Gladfelter,
2012). Septins and are posited to interact with phosphoinositides, yet their specificity for
different phosphoinositides, which regulate the intracellular localization of membrane-
bound proteins, is not fully known.
In the budding yeast Saccharomyces cerevisiae, a membrane-bound septin ring
forms at the base of the emerging bud early in the cell cycle and evolves to a collar that
underlies the mother-bud neck (Engidawork et al., 2001a; Engidawork and Lubec, 2001).
Septin filaments are positioned between the plasma membrane and the smooth
endoplasmic reticulum (ER), impeding the lateral diffusion of cortical and ER membrane
proteins during polarized bud growth (Luedeke et al., 2005) (Figure 1.2 A). In mitosis,
splitting of the septin collar into two rings results in the formation of a membrane
compartment, which enables the accumulation of membrane-associated factors (e.g.,
vesicle tethering complexes) without diffusing into the mother and bud cell membranes

7 (Barral, 2000). During mammalian mitosis, a septin diffusion barrier is posited to similarly
limit the diffusion of inner leaflet membrane proteins across the equatorial plane of a
dividing cell (Schmidt and Nichols, 2004a).
Septins form membrane-bound rings at the base of primary and motile cilia and
the dendritic spines of neurons (Fliegauf et al., 2014; Gulesserian et al., 2001; Hu et al.,
2010; Tada et al., 2007). At the interface of the plasma and ciliary membrane, septins
control the localization of ciliary membrane proteins and thus, they are required for the
formation and maintenance of the primary cilium (Engidawork et al., 2001e; Hu et al.,
2010). Septins could similarly impede the lateral diffusion of post-synaptic proteins from
the dendritic spines to the shaft, aiding to the compartmentalization of synaptic receptors
and channels (Caudron and Barral, 2009). At the annulus of spermatozoa, septins form
a barrier between the principal and mid pieces of the sperm, and are required for the
proper localization of a protein that is essential for spermiogenesis (Engidawork et al.,
2001c; Ihara et al., 2005; Kissel et al., 2005). The mechanism by which septins restrict
lateral diffusion in cell membranes is unknown, but recent evidence indicates that
septins influence the organization of lipid domains and interact with the cytoplasmic tails
of membrane transporters (Hagiwara et al., 2011; Kinoshita et al., 2004; Sharma et al.,
2013b). Protein-lipid compartmentalization and domain identity could be further
reinforced by septin-mediated inhibition of vesicle delivery; septins interact with the
exocyst, a vesicle tethering complex, and components of the SNARE machinery of
vesicle fusion, and may introduce a physical barrier that interferes with vesicle delivery
to the plasma membrane (Engidawork et al., 2001b; Engidawork et al., 2001d;
Gulesserian et al., 2000; Yoo et al., 2001) (Figure 1.2 B). Hence, septins are an integral
component of the structure of cell membranes, contributing to the organization of

8 membrane domains by inhibiting selectively the lateral diffusion and delivery of
membrane proteins.
In addition to their diffusion barrier properties, membrane-bound septins affect
the shape and physical properties of lipid bilayers. Similar to the membrane-remodeling
functions of the Bin-Amphiphysin-Rvs (BAR)-domain proteins, septins have been shown
to induce the tubulation of spherical liposomes made of phosphatidylcholine and
phosphoinositides (Tanaka-Takiguchi et al., 2009) (Figure 1.2 C). These membrane
tubules have the diameter of septins rings (~0.5 µm) and are circumferentially braced by
septin filaments (Tanaka-Takiguchi et al., 2009). It is unknown, however, whether
septins affect membrane curvature and induce membrane tubules in vivo (Figure 1.2 D).
Conversely, several studies have shown that septin depletion decreases the rigidity and
stiffness of cell membranes (Mostowy et al., 2011; Tooley et al., 2009) (Figure 1.2 E). In
T lymphocytes, septins suppress membrane blebbing and protrusive activity is spatially
restricted to septin-free areas of the cell cortex (Gilden et al., 2012; Tooley et al., 2009).
Measurements of membrane elasticity and viscosity show a marked decrease in the
rigidity of cell membranes after septin depletion, which alters cell shape and morphology
(Mostowy et al., 2011). Thus, like the spectrin-actin membrane skeleton, septin filaments
contribute to the organization and mechanochemical properties of cell membranes.
III. Septins and the Actin Cytoskeleton
From yeast to mammals, septins are implicated in the organization and functions
of the actomyosin cytoskeleton. Septins colocalize with actin filaments and interact with
actin-binding proteins and signaling effectors that regulate the organization of the actin
cytoskeleton (Kinoshita, 2002; Mavrakis et al., 2014; Nagata and Inagaki, 2005) (Figure

9 1.3 A). Notably, septins interact directly with myosin II, regulating its localization and
activation in interphase and mitotic cells (Joo et al., 2007).
In the budding yeast S. cerevisiae, septins are essential for the recruitment of
proteins involved in actin cable assembly during bud growth (Figure 1.3 D). At the site of
bud formation, linear actin cables, which enable the polarized transport of vesicles, are
polymerized by the formins Bni1 and Bnr1 (Bretscher, 2013). In the absence of the
nonessential septin Shs1, Bnr1 is mislocalized and actin cables fail to assemble (Buttery
et al., 2012; Gao et al., 2010). The actin assembly activity of Bnr1 appears to depend on
Shs1 (Buttery et al., 2012) and the septin-interacting partner Bni5 is required for the
recruitment of myosin II, which drives actin cable retrograde flow (Nam, 2007). Thus,
septins scaffold and may activate the assembly of actin cables during bud growth.
Interestingly, mammalian septins scaffold the recruitment of cortactin, an activator of
Arp2/3, to actin patches in the axons of sensory neurons (Hu et al., 2012). Septin 6
(SEPT6) localizes to actin patches, which consist of branched actin filaments, and
SEPT6 depletion reduces the recruitment of cortactin to actin patches and their transition
to filopodia (Hu et al., 2012) (Figure 1.3 B and D). Reconstitution of branched actin
assembly in vitro and in the presence of Arp2/3 shows that SEPT6 localizes
preferentially to actin branch points (Hu et al., 2012). In agreement with this observation,
overexpression of SEPT6 increases the recruitment of cortactin to the lamellipodium, a
branched actin network at the leading edge of motile cells (Hu et al., 2012). Additional
septins (e.g., SEPT1, SEPT4) are enriched in the lamellipodia of squamous carcinoma
cells (Mizutani et al., 2013). Septins, therefore, have an evolutionarily conserved role in
scaffolding proteins involved in the formation of linear and branched actin filaments.
More recently, septins were found to cross-link and bundle actin filaments
directly, contributing to the assembly of contractile actin rings in Drosophila embryos

10 (Mavrakis et al., 2014). In developing Drosophila embryos, the blastoderm contains
thousands of nuclei, which become individual cells. Cellularization requires the assembly
of contractile rings of actomyosin filaments that furrow the plasma membrane resulting in
membrane canals that envelope the nuclei of the syncytial embryo. Embryos expressing
a septin mutant are characterized by a loss of circular actin filaments, which are less
bundled and contract at slower rates (Mavrakis et al., 2014). Drosophila septins bundle
actin filaments and copolymerization of actin with septins results in curved and circular
actin bundles (Mavrakis et al., 2014) (Figure 1.3 C and F). Mammalian septins also
bundle actin filaments in vitro and septin depletion results in loss of actin stress fibers
(Kinoshita, 2002; Kremer et al., 2007). These findings suggest that septins are actin-
binding and bundling proteins that regulate the organization of actin microfilaments.
However, the function of septins during actomyosin assembly is not fully understood.
In addition to their direct roles in actin organization, septins influence actin
organization and contractility by interacting with signaling factors and myosin II.
Assembly of stress fibers is under the control of Rho GTPases, which activate actin
nucleation factors and promote the contraction of myosin II (Chrzanowska-Wodnicka
and Burridge, 1996). Several studies have indicated that septins are involved in the Rho
signaling pathways that control stress fiber assembly. Septin 9 (SEPT9), which is
positioned at the terminal ends of septin hetero-octamers, interacts directly with a Rho
guanine exchange factor (GEF), termed the septin-associated Rho GEF (SA-Rho GEF),
and Rhotekin, a secondary effector of Rho signaling (Ito et al., 2005; Nagata and
Inagaki, 2005). Both SA-Rho GEF and Rhotekin regulate septin assembly and
localization to stress fibers. Interestingly, SEPT9 appears to negatively regulate Rho
activation by inhibiting GTP loading by the SA-Rho GEF (Nagata and Inagaki, 2005).
Septins also interact with Binders of Rho GTPases (Borgs), which are effectors of Cdc42

11 (Joberty et al., 2001). New studies show that Gic1, a yeast homologue of the human
Borg, binds and cross-links septin filaments in vitro in a Cdc42-dependent manner
(Sadian et al., 2013). Binding of the active Cdc42-GTP to Gic1 results in the dissociation
of Gic1 from septin filaments. The inactive Cdc42-GDP, however, also inhibits the
assembly of septin filaments by competing with Gic1 for binding to septins (Sadian et al.,
2013). In mammalian cells, Borgs have been shown to affect both septin and stress fiber
assembly (Joberty et al., 1999; Joberty et al., 2001).
While more work is needed to elucidate how the interactions between septins
and signaling molecules affect actin organization, structural and functional analysis of
the septin-myosin II interaction has provided a novel mechanism for the activation of the
non-muscle myosin II. The mammalian SEPT2 has been shown to interact directly with
the heavy chain of the non-muscle myosin II and yeast two-hybrid data indicate that
Myo1, the S. cerevisiae type II myosin, binds the sporulation-specific septin Spr3 (Drees
et al., 2001; Joo et al., 2007). Disruption of the SEPT2-myosin II interaction results in
loss of stress fibers in interphase cells and incomplete ingression of the cleavage furrow
during mitosis (Joo et al., 2007). These defects are accompanied by significant reduction
in the phosphorylation of the myosin light chain, which stimulates myosin II contractility.
Importantly, SEPT2 appears to scaffold the interaction of myosin II with the citron and
Rho-activated myosin kinases, which in the absence of septins dissociate from the
cleavage furrow (Joo et al., 2007). Thus, septins may control spatially and temporally the
localization and activation of myosin II. Importantly, this function might be evolutionarily
conserved as myosin activation and actin organization is also disrupted in Xenopus
embryos after Sept7 depletion, decreasing planar tension and protrusive activity during
the collective cell migration that drives body axis elongation (convergent extension)
(Shindo, 2014).

12
In summary, septins are integral components of the actomyosin cytoskeleton.
Recent studies have revealed that septins interact directly with actin filaments and
myosin II, but many of the molecular and functional details remain unknown. Future
studies will address the mechanisms by which septins affect the formation of linear and
branched actin filaments and myosin II-dependent contractility, and how septins
interface with signaling pathways that affect actin organization and contractility.
IV. Septins and the Microtubule Cytoskeleton
Septin filaments colocalize with microtubules in a diversity of eukaryotic
organisms and cells (Spiliotis, 2010). The extent of septin-microtubule colocalization
varies between septin paralogs and isoforms, and depends on cell type, phase of the
cell cycle and tubulin isoform expression and post-translational modifications. Recent
studies have shown that septins interact directly with microtubules, microtubule-
associated proteins (MAPs) and motors as well as enzymes that modify tubulin’s post-
translational modification. Thus, septins have emerged as novel regulators of
microtubule organization and dynamics, and may play a key role in the spatial regulation
of microtubule-dependent transport.
Mammalian SEPT2/6/7/9 complexes interact directly with microtubules through
their SEPT9, SEPT7 and SEPT6 subunits (Figure 1.3 I); SEPT2 does not bind
microtubules (Bai et al., 2013). It is unknown how SEPT6 and SEPT7 associate with
microtubules, but SEPT9 interacts with the acidic tails of βII-tubulin via its N-terminal
extension, which contains multiple repeats of the K/R-R/x-x-D/E motif (Bai et al., 2013).
Similar to the microtubule-binding sequence repeats of filamentous MAPs (e.g., tau,
MAP1), the SEPT9 repeat motifs consist of positively and negatively charged residues,
which interact electrostatically with one another and the acidic C-terminal tails of α-

13 tubulin (Bai et al., 2013). Thus, the N-termini of SEPT9 mediate septin-septin and septin-
microtubule interactions that cross-link microtubules into bundles. By forming
filamentous cross-bridges that bind microtubules in a staggered arrangement, septins
could increase the thickness and length of microtubule bundles as observed in vitro (Bai
et al., 2013). Consistent with this function, septins colocalize preferentially with
elongated microtubule bundles in cultured kidney epithelia (Bowen et al., 2011) (Figure
1.3 H). Moreover, expression of SEPT9 isoforms that lack N-terminal repeat motifs
reduces both microtubule bundling and septin-microtubule colocalization (Bai et al.,
2013). Hence, through the properties of SEPT9, mammalian septins function as bona
fide MAPs that facilitate the bundling of microtubules.
Like most MAPs, septins affect not only microtubule bundling, but also
microtubule dynamics. In kidney epithelia, SEPT2 knock-down increases microtubule
catastrophe and reduces the rate of microtubule growth, indicating that septins are
required for persistent microtubule growth (Bowen et al., 2011). Interestingly, loss of
SEPT2 also affects microtubule-microtubule interactions and the overall directionality of
microtubule growth (Bowen et al., 2011). In agreement with a septin requirement for
microtubule growth and bundling, SEPT9 knock-down decreased the density of the
microtubule network in mammary epithelia (Bai et al., 2013). Depletion of SEPT7,
however, has the opposite effect in HeLa cells, increasing microtubule density and
acetylation, a marker of microtubule stability (Kremer et al., 2005). A direct interaction
between SEPT2 and MAP4 indicates that cytoplasmic SEPT2/6/7 complexes inhibit
MAP4 from binding and bundling microtubules, but SEPT2/6/7 and MAP4 also compete
for binding to microtubules and a new study shows that SEPT7 mediates the interaction
of α-tubulin with the deacetylase HDAC6 (Ageta-Ishihara et al., 2013; Spiliotis et al.,
2008). Surprisingly, SEPT7 is required for α-tubulin deacetylation and SEPT7-depleted

14 neurons have increased levels of acetylated tubulin and reduced rates of microtubule
growth (Ageta-Ishihara et al., 2013). While the cause-and-effect relationship between
microtubule “stability” and acetylation is unclear, septins appear to affect microtubule
bundling, dynamics and post-translational modifications including acetylation and
polyglutamylation. Given that septins interact with α-tubulin, MAP4 and HDAC6 in a
paralog- and isoform-specific manner, it is possible that septins have distinctly different
roles, targeting different aspects of microtubule organization and dynamics.
Microtubule post-translational modifications and MAPs affect the binding and
processive movement of motors, generating a code for the spatial regulation of
intracellular transport (Verhey and Gaertig, 2007). Presence of septins on select
microtubule tracks (e.g., perinuclear microtubule bundles) raises the possibility that
septins regulate microtubule-dependent transport (Spiliotis et al., 2008). In kidney
epithelial cells, tubular vesicular carriers egress from the trans-Golgi network on septin-
coated microtubule tracks (Spiliotis et al., 2008). Microinjection of septin function-
blocking antibodies or SEPT2 depletion results in stalling of vesicle transport en route to
the plasma membrane (Spiliotis et al., 2008). Competition between SEPT2 and MAP4
for binding to microtubules suggests that septins could block the microtubule binding of
MAP4, which inhibits kinesin-dependent transport (Spiliotis et al., 2008). It is unknown
whether septins affect directly the binding and/or processive movement of microtubule
motors, but SEPT7 interacts directly with the mitotic motor CENP-E (kinesin 7) (Zhu et
al., 2008). Interestingly, septins are required for the proper localization of CENP-E and
consequently, for the stable capture and biorientation of the kinetochores of congressing
chromosomes (Spiliotis et al., 2005). Because SEPT7 interacts with the C-terminal tail of
CENP-E, which binds and inhibits the motor domain of CENP-E, SEPT7 could promote
the activation and motility of CENP-E by relieving its autoinhibition. Alternatively, septins

15 may scaffold the interaction of CENP-E with the Aurora A and B kinases or the protein
phosphatase 1 (PP1), which regulate CENP-E binding to microtubules; Aurora B has
been reported to interact directly with SEPT1 (Qi et al., 2005).

16
Figure 1.1. Septin filament assembly. Mammalian septins are classified in four groups
(SEPT2, SEPT6, SEPT7 and SEPT9) and characterized by highly conserved domains
such as a phosphoinositide-binding polybasic domain (PB), a GTP-binding domain, a
septin unique element (SUE) and C-terminal α-helical coiled-coil (CC) domains. Septins
form non-polar hetero-oligomeric complexes by dimerizing in tandem using their GTP-
binding domains, which interact with one another via their N- and C-terminal helices (NC
interface) or their GTP-binding pockets (G interface). GTP hydrolysis favors
oligomerization by stabilizing the G interface, but septins of the SEPT6 group do not
hydrolyze GTP. End-to-end assembly of septin hetero-oligomers results in the formation
of non-polar filaments, which interact laterally through the N- and C-terminal extensions
of their septin subunits. Owing to the flexibility of the NC interface, paired septin
polymers can bend into curved and ring-like filaments. In addition to septin bundles and
rings, membrane-associated septins form gauze-like structures, which consist of
orthogonally arranged filaments.
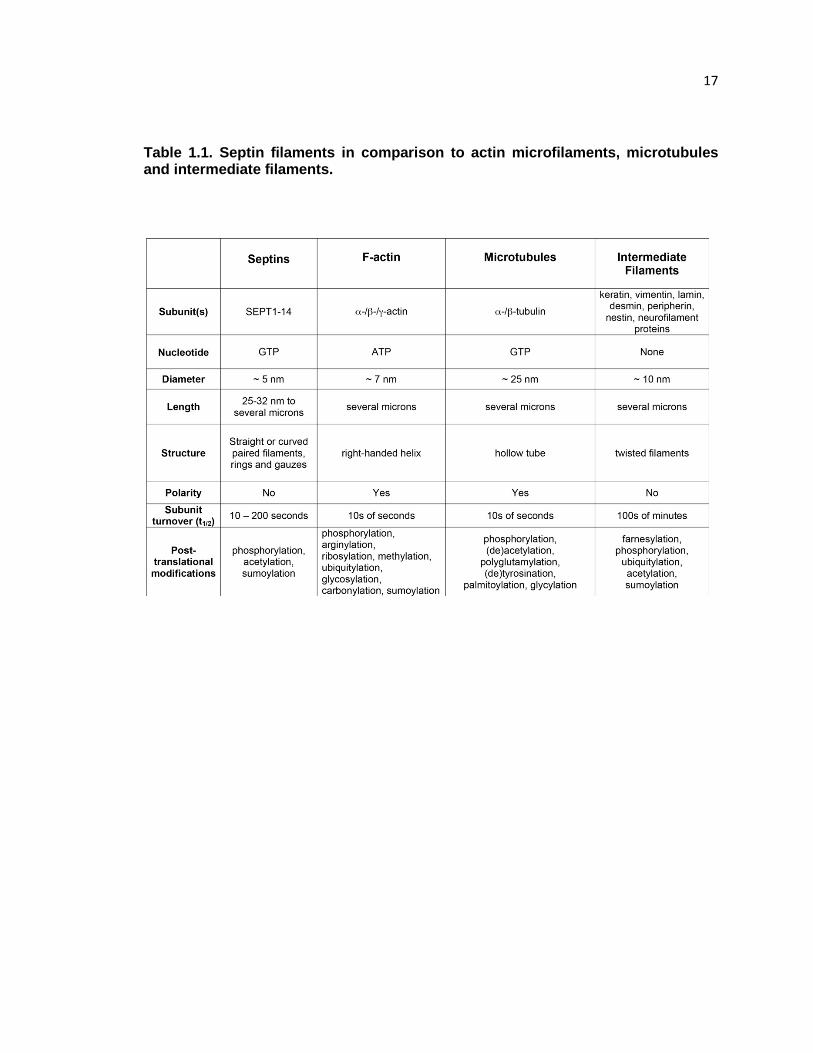
17 Table 1.1. Septin filaments in comparison to actin microfilaments, microtubules and intermediate filaments.
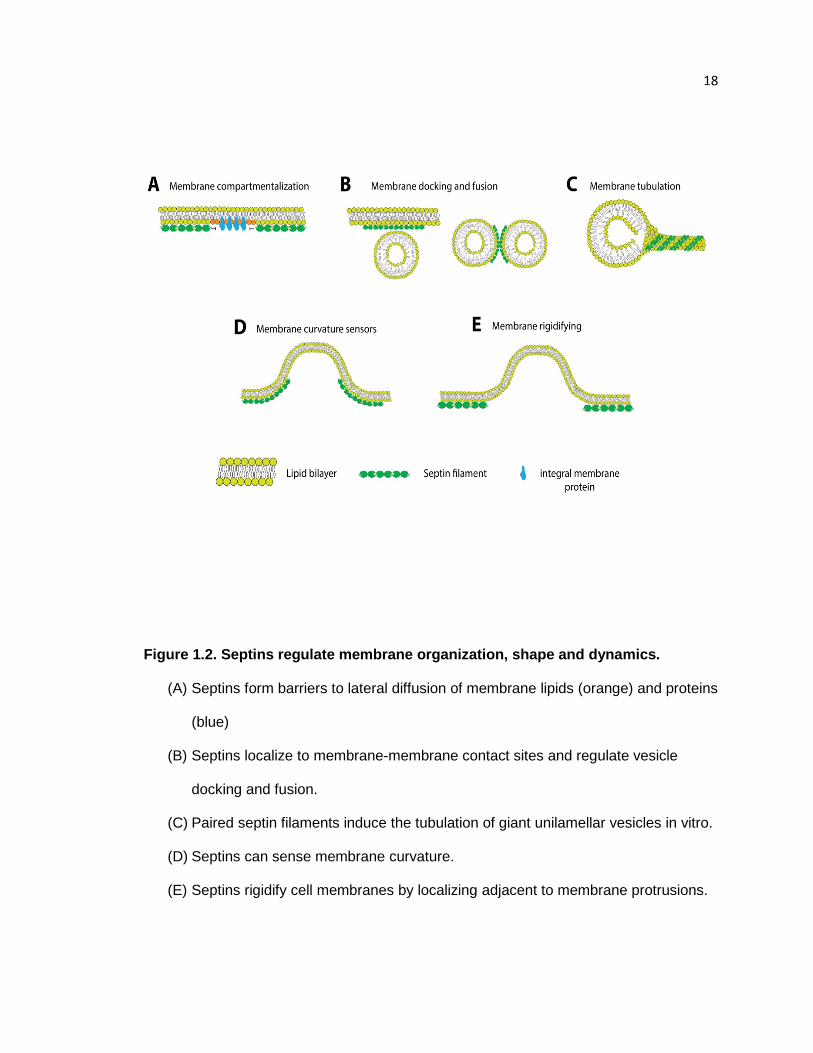
18
Figure 1.2. Septins regulate membrane organization, shape and dynamics.
(A) Septins form barriers to lateral diffusion of membrane lipids (orange) and proteins
(blue)
(B) Septins localize to membrane-membrane contact sites and regulate vesicle
docking and fusion.
(C) Paired septin filaments induce the tubulation of giant unilamellar vesicles in vitro.
(D) Septins can sense membrane curvature.
(E) Septins rigidify cell membranes by localizing adjacent to membrane protrusions.

19

20
Figure 1.3. Septins interact with the actin and microtubule cytoskeleton. (A – E)
Fluorescence images show purified recombinant septins tagged with the green
fluorescent protein decorating linear (A), branched (B) and circular (C) actin filaments.
Recent evidence indicates that septins crosslink actin filaments into curved and circular
bundles (Mavrakis et al, 2014) and scaffold the recruitment of cortactin, an actin
nucleation promoting factor, at Arp2/3-mediated branch points (Hu et al, 2012). In the
budding yeast Saccharomyces cerevisiae, septins are required for the localization and
activity of formins, which stimulate the formation of actin cables (D). In the axons of
sensory neurons, septins accumulate to actin patches (E), which consist of Arp2/3-
nucleated branched filaments, and in Drosophila embryos, septins are essential for the
assembly of the contractile rings of actomyosin (F), which drives membrane invagination
and furrow ingression during cellularization. Scale bars, 1 µm. (G – L) Fluorescent
images show colocalization of septin filaments with actin stress fibers (G) and
microtubules (H) in Madin-Darby canine kidney cells, and in vitro decoration of pre-
polymerized microtubules with purified recombinant SEPT7. Septins bundle
microtubules and inhibit their interaction with other microtubule-associated proteins (J).
Post-translational modifications (PTM) of tubulin such as acetylation and
polyglutamylation are influenced by septins, which can scaffold the interaction of tubulin
with the deacetylase HDAC6 (K). In addition, septins have been shown to guide spatially
microtubule-microtubule interactions and may influence the localization of microtubule
motors (L). Scale bars, 10 µm.

21
CHAPTER 2. Tissue-specific expression of septins in health and disease
Insights into the tissue expression and functions of mammalian septins have
emerged from DNA microarray analyses and septin knock-out mice. Serial analysis of
gene expression in a variety of human tissues shows unique expression of individual
septins (Table 2.1). SEPT2, SEPT7 and SEPT9 are ubiquitously expressed (Cao et al.,
2007; Connolly et al., 2011a; Hall et al., 2005a). While absent from some tissues, the
septins SEPT4, SEPT8, SEPT10 and SEPT11 are expressed widely (Cao et al., 2007;
Hall et al., 2005b). In contrast, expression of SEPT1, SEPT3, SEPT12 and SEPT14 is
limited to specific tissues. SEPT1 and SEPT3 are expressed in lymphoid and nervous
tissues, respectively (Hall et al., 2005a), and SEPT12 is a testis-specific septin (Steels et
al., 2007; Xue et al., 2004). SEPT14 was originally identified as a novel testis-specific
protein, but is also expressed in the nervous system (Peterson et al., 2007; Shinoda et
al., 2010). Little is known about how septin expression varies throughout development
and adult life, but a recent study showed that the levels of ten different septin proteins
change dramatically during brain development (Tsang et al., 2011).
I. Septin expression and roles in embryogenesis
To date, knockout mice have been generated for seven of the thirteen septins.
Genetic ablation of the ubiquitous Sept7, Sept9 and Sept11 resulted in embryonic
lethality. Sept7-/- mice were never born, arresting early in development possibly due to
mitotic failure (Kinoshita, 2008). Development of Sept11-/- mice ceased at day 11.5 in
utero and the mice died by day 13.5 (Roseler et al., 2011). Despite the development of a
healthy yolk sac, heartbeat and blood vessels, Sept9-/- embryos died by day 10 of
gestation, exhibiting mesenchymal tissue degeneration and extensive cell death

22 (Fuchtbauer et al., 2011). Surprisingly, loss of the neuron-specific Sept3 did not have
any discernible effects on the physiology and function of Sept3-/- mice (Tsang et al.,
2008). Functional redundancy between Sept3 and Sept9 or compensation of Sept3 loss
by Sept9 isoforms could account for the lack of phenotype. Similarly, Sept5-/- and Sept6-/-
mice were reported to be normal, but newer behavioral assays indicate changes in the
social interaction, rewarded goal approach and anxiety-related behavior of Sept5-/- mice
(Ono et al., 2005a; Suzuki et al., 2009). From all the septin knock-out mice, Sept4-/- are
the most well-studied and characterized. Male Sept4-/- mice are sterile due to a
morphologically altered and immotile sperm (Ihara et al., 2005; Kissel et al., 2005).
Neurologically, the cerebellum of Sept4-/- mice is mildly maldeveloped and hypo-
dopaminergic phenotypes such as an increase in the inhibition of the startling response
by weaker pre-stimuli have been reported (Kinoshita, 2008). Loss of Sept4 from mice
that express the alpha synuclein mutant A53T, which is common among familial forms of
Parkinson’s disease, increased amyloid deposition and neurodegenerative pathology,
leading to shorter lifespans (Ihara et al., 2007). Moreover, Sept4-/- mice are more
susceptible to tumor growth and liver fibrosis, and contain high levels of hematopoietic
and hair follicle stem cells (Fuchs et al., 2013; Garcia-Fernandez et al., 2010; Iwaisako
et al., 2008).
The embryonic lethality phenotype of the Sept7-/-, Sept9-/-, Sept11-/- mice
suggests that septins play an essential role in embryogenesis. It is unknown how septins
are involved in the embryonic development of mammals, but studies in model organisms
such as the fruit fly Drosophila melanogaster, the roundworm Caenorhabditis elegans
and the frog Xenopus lavis have shed some light on the developmental functions of
septins. In early Drosophila development, multiple rounds of mitotic nuclear divisions
lead to the formation of a multinucleated single cell embryo. The nuclei of this syncytial

23 blastoderm are partitioned into individual cells by a process termed cellularization. In
septin-deficient embryos, cellularization is incomplete resulting in multinucleated cells,
less imaginal discs (precursors of epithelial tissues) and larval death (Adam et al., 2000;
Neufeld and Rubin, 1994). In C. elegans, septins localize similarly to the cytokinetic
furrows, but are not essential for embryonic development (Nguyen et al., 2000). Septin-
depleted embryos rarely fail to complete cytokinesis, but the asymmetric geometry of
furrow ingression is disrupted and the contractile apparatus is less robust and more
prone to stochastic errors (Maddox et al., 2007). Post-embryonic development of C.
elegans tissues is more severely affected by septin mutations, which disrupt
gonadogenesis and the formation of a functional sensory and motor nervous system
(Finger et al., 2003; Nguyen et al., 2000). Recently, a study in Xenopus showed that
septins are involved in the planar cell polarity (PCP) signaling pathway, which directs the
collective cell movements of embryogenesis during gastrulation, axial elongation and
organogenesis. The PCP signaling protein Fritz, which interacts directly with Sept2, was
shown to control septin localization to the cortical membrane of gastrulating cells (Kim et
al., 2010). Moreover, Fritz and septins synergized toward the formation of cilia, which
are critical for the transduction of the Sonic Hedgehog signals that regulate
organogenesis (Kim et al., 2010). Thus, septins are essential components of the
molecular and cellular mechanisms that give rise to complex organ and tissue systems.
II. Cardiovascular System
The heart is the first organ to develop during embryogenesis. Terminal
differentiation of cardiomyocytes occurs near birth and is characterized by the cessation
of cell division and the development of contractile multinucleated cells. In mouse
embryonic cardiac cells, the levels of septin 2, 6, 7 and 9 levels are the highest in early

24 development and decrease at birth and adulthood (Ahuja et al., 2006). In embryonic
cardiomyocytes, septins localize to the cytokinetic ring and midbody of dividing cells and
are absent from sarcomeric actomyosin (Ahuja et al., 2006). Thus, septins could function
in early cardiac development by interacting with key components of the cytokinetic
machinery (e.g., myosin, anillin). Moreover, septin downregulation could trigger mitotic
arrest and the formation of terminally differentiated multinucleated cells. Interestingly,
SEPT5 is located within the 22qII locus that is commonly deleted in the DiGeorge/velo-
cardial-facial syndrome (DGS), which is characterized by cardial malformations and
lesions (Judith M. McKie, 1997). Studies in animal models suggest that 22qII
hemizygosity, which occurs in 90% of DGS cases, results in developmental impairment
of the right ventricle and outflow tract (Scambler, 2000). It is unknown if partial loss of
SEPT5 contributes to the pathology of DGS pathology, but decreased septin (SEPT8)
expression is also implicated in the toxic effects that anti-inflammatory drugs have on the
development of embryonic cardiomyocytes (Baek et al., 2010).
Septin expression and functions in the vascular network have been identified in
platelets, which are specialized secretory cells that regulate haemostasis, thrombosis
and injury repair. SEPT5 (CDCRel-1) localizes around platelet α-granules, storage
vesicles that contain growth factors and effector molecules involved in adhesion and clot
formation (Dent et al., 2002). Additionally, SEPT5 co-precipitates with syntaxin-4, a
component of the SNARE-SNAP complex that mediates vesicle fusion with the plasma
membrane (Dent et al., 2002). Platelets isolated from Sept5 null mice secrete serotonin
more readily upon stimulation, suggesting that Sept5 negatively regulates α-granule
fusion with the plasma membrane (Dent et al., 2002).
Abnormal SEPT5 expression has been linked to the Bernard-Soulier syndrome
(BSS), a rare autosomal recessive blood disorder characterized by excessive bleeding

25 and abnormal platelet morphology, count and secretion (Lopez et al., 1998). BSS
patients harbor mutations in the quaternary platelet glycoprotein (GP) Ibβ-IX receptor
subunit, which regulates platelet adhesion, activation and aggregation. A mouse model
lacking the IBβ gene recapitulates BSS pathology and isolated platelets contain
abnormally large α-granules and elevated levels of SEPT5 (Kato et al., 2004). More
recently, genotypic analysis of a juvenile patient with a rather severe case of BBS
showed homozygous deletions of the Ibβ and SEPT5 genes, which are positioned
directly next to one another (Bartsch et al., 2011; Zieger et al., 1997).
III. Immune System
The mammalian innate and adaptive immune systems encompass several
tissues and specialized cell types that defend against infectious agents. Despite that the
first mammalian septin (SEPT1) was cloned from lymphocytes twenty five years ago
(Carol., 1990), it was not until recently that researchers began to investigate the
immunological functions of septins.
Cells of the innate immunity (e.g. dendritic cells, macrophages) recognize and
engulf foreign bodies, and elicit a larger response through antigen presentation. SEPT10
was initially identified in dendritic cells, where it is abundantly expressed (Sui et al.,
2003). In contrast, SEPT10 is weakly expressed in the spleen, thymus and peripheral
blood leukocytes. In dendritic cells SEPT10 localizes to the cytoplasm and the nucleus,
and is transcriptionally upregulated upon stimulation with lipopolysaccharide (Sui et al.,
2003). Septins 2, 6, 8, 10 and 11 are abundantly expressed in macrophages, and
SEPT2 and SEPT11 localize to actin-based structures at the base and periphery of the
phagocytic cup (Huang et al., 2008). Disrupting septin assembly by expressing the
septin-binding domain of BORG3 or targeted knock down of SEPT2 or SEPT11 reduced

26 phagosome formation and uptake of IgG-coated beads (Huang et al., 2008). Further
studies are necessary to understand the precise functions of septins in dendritic and
macrophage cell biology.
T lymphocytes proliferate and mature in lymphoid tissues (e.g., thymus, spleen,
lymph nodes) and migrate to peripheral tissues, searching for antigen presenting cells.
Septins are essential for both T cell development and migration. During mouse T-cell
development, the transition from a double negative (CD4-CD8-) to a double positive
(CD4+CD8+) stage is accompanied by a 5- to 10-fold increase in Sept9 expression
(Lassen et al., 2013). Sept9 deletion affects T-cell development in the thymus leading to
an increase in stage 3 double negative cells (CD4-CD8-CD25+CD44-) at the expense of
stage 4 double negative cells (CD4-CD8-CD25-CD44-). Peripheral CD8+ and CD4+ T-
cells are reduced concomitantly and in vitro proliferation of Sept9-deleted CD8+ T-cells is
impaired; the effects of Sept9 deletion in vivo are more severe on CD8+ than CD4+ T-
cells (Lassen et al., 2013). These studies are the first to implicate septins in T-cell
development, but more work is needed to determine the mechanism by which SEPT9
regulates T-cell maturation and selection.
Lymphocytes migrate by forming a leading pseudopod and a trailing uropod, both
of which require polarized assembly of filamentous actin and contraction of the cortical
membrane. In T-cells, septins localize to the middle of the cell cortex and are distinctly
absent from the protruding pseudopod and uropod (Tooley et al., 2009). SEPT7-
depleted lymphocytes have elongated uropods and show poor persistent migration
despite maintaining normal levels of actin, microtubules and phosphorylated (active)
myosin (Tooley et al., 2009). In addition, SEPT7 depletion results in excessive blebbing
and reduces the rate of cortical retraction in a hydrostatic volume change assay, which
measures the ability of cells to expand and retract their cell membrane (Gilden et al.,

27 2012). In migrating lymphocytes, septins (SEPT6 and SEPT7) are absent from blebs
and filopodia, but are recruited to plasma membrane sites adjacent to these protrusive
structures (Gilden et al., 2012; Sellin et al., 2011a). Overall, the plasma membrane
organization and dynamics of migrating T-cells appear to depend on septins, which
affect persistent migration.
In non-immune cells, septins are essential for host defense against infectious
bacteria. The invasive bacterium Listeria monocytogenes enters mammalian cells
through the interaction of its surface proteins internalin A and B (InIA, InIB) with E-
cadherin and the Met receptor, respectively (Cossart, 2011). Targeted depletion of
SEPT2 inhibits bacterial invasion and impairs the InIB/Met signaling pathway (Mostowy
et al., 2009). Septins also thwart the intracellular mobility of the Shigella flexneri
bacterium, which propels itself through the cytoplasm by recruiting and activating the
Arp2/3 complex, which induces the polymerization of an actin tail (Egile et al., 1999).
Septin filaments are recruited to cytoplasmic Shigella and assemble cage-like structures
that inhibit bacterial mobility and facilitate the formation of an autophagosome for
bacterial degradation (Mostowy et al., 2010).
IV. Nervous system
Development of a functional nervous system involves the migration of embryonic
stem cells and neural precursors, and their differentiation into neurons and glial cells.
Neuronal morphogenesis requires the formation of axons, dendrites and synapses.
Recent studies have shown that septins play essential roles in neuronal migration,
axonal and dendritic arborization, and synaptic activity. In developing mouse embryos,
SEPT4 and SEPT14 are highly expressed in the cortical plate of the developing cortex,
which is formed by post-mitotic neurons that migrate tangentially from the subventricular

28 zone to the outer layers of the neocortex (Shinoda et al., 2010). In vivo knock down of
SEPT14 or SEPT4 expression and inhibition of their interaction perturb neuronal
migration to the cortical plate; neurons fail to extend leading processes and stall at the
ventricular and intermediate zones without reaching the cortical plate (Shinoda et al.,
2010). The sensory and motor neurons of the C. elegans septin mutants unc-59 and
unc-61 exhibit similar migration defects during larval development (Finger et al., 2003).
In cultured hippocampal neurons, septins are required for the formation of
dendritic spines (Cho et al., 2011; Tada et al., 2007; Xie et al., 2007). SEPT5/7/11
complexes localize to dendritic branch points and at the base of dendritic spines (Xie et
al., 2007). Targeted depletion of SEPT7, SEPT11 or SEPT6 reduces dendritic
arborization and alters the length, density and head morphology of dendritic spines (Cho
et al., 2011; Tada et al., 2007; Xie et al., 2007). In contrast, SEPT7 overexpression
increases dendritic protrusions and branchpoints (Tada et al., 2007; Xie et al., 2007).
Because dendritic spines are enriched with membrane proteins (e.g., neurotransmitter
receptors) that localize specifically at the post-synaptic cleft, septins are posited to form
a diffusion barrier at the base of dendritic spines blocking free diffusion of membrane
proteins in and out of the dendritic shaft (Caudron and Barral, 2009). In addition, septins
may also synergize with the actin and microtubule cytoskeleton for the formation of
dendritic spines.
Septin-mediated reorganization of the actin and microtubule cytoskeleton is
required for the collateral branching of axons. In sensory neurons isolated from chicken
dorsal root ganglia, SEPT6 and SEPT7 localize at the base of axonal filopodia and axon
branch points (Hu et al., 2012). This localization is reminiscent of septin complexes at
the base of dendritic spines, but in axons septins control the initiation and maturation of
collateral branches (Hu et al., 2012). SEPT6 enhances the transition of axonal patches

29 of F-actin to filopodia by enhancing the recruitment of the actin-binding protein cortactin,
an activator of Arp2/3-mediated actin polymerization (Hu et al., 2012). In contrast,
SEPT7 is involved in the entry of axonal microtubules into nascent filopodia, enabling
the formation of collateral branches (Hu et al., 2012). These regulatory roles of septins
are likely to influence not only the outgrowth and differentiation of neurites into axons
and dendrites, but also axon extension and growth cone motility. Indeed, septins have
been reported to affect axon length (Vega and Hsu, 2003), but more studies are needed
to understand their precise roles in axonal differentiation and growth.
Neurotransmission and organ innervation rely on the formation and function of
chemical synapses, which release and receive neurotransmitter molecules through their
pre- and post-synaptic clefts, respectively. Neurotransmitter secretion and uptake are
coupled to ion channels that sense and generate membrane potential. Initial studies
indicated that septins inhibit the release of synaptic vesicles (Beites et al., 2001), but
new evidence suggests that septins could also regulate the localization of ion channels
in membrane microdomains and their endocytosis from the cell membrane. The septins
3, 5, 6, 7 and 11 localize to the presynaptic cleft and their filamentous organization
coincides with the filament-like strands, which have been observed by electron
microscopy to connect synaptic vesicles with each other and the plasma membrane
(Hirokawa et al., 1989; Kinoshita et al., 2000; Tsang et al., 2011). SEPT5 and SEPT8
have been reported to interact with components of the molecular machinery of synaptic
vesicle fusion (Beites, 2005; Beites, 1999; Ito et al., 2009). SEPT5 and SEPT7 are
posited to inhibit vesicle fusion by inhibiting the formation of productive SNARE
complexes (Beites, 2005; Wasik et al., 2012). Interestingly, however, synaptic
transmission and morphology were not altered in Sept5-/- and Sept3-/- mice (Tsang et al.,
2008). Further analysis of the developing calyx of Held, which is a large glutamatergic

30 synapse of the auditory nervous system, showed that Sept5 filaments play a key role in
the positioning of pre-docked synaptic vesicles relative to the voltage gated calcium
channels (VGCCs) of the active zone (Yang et al., 2010). In Sept5-/- synapses, synaptic
vesicles are more tightly docked to the VGCCs. In addition, synaptic vesicles are more
readily and cooperatively released requiring a lower number of calcium channels to
trigger single fusion events (Yang et al., 2010). Thus, SEPT5 introduces a physical
barrier between synaptic vesicles and VGCCs regulating the efficiency of synaptic
transmission. A new study, however, suggests that SEPT5 and SEPT4 may modulate
synaptic activity by controlling the membrane organization and clustering of calcium and
possibly other ion channels (Sharma et al., 2013b). Although this hypothesis has yet to
be tested in neurons, SEPT4 is required for the organization of phosphatidyl-inositol-
(4,5)-bisphosphate-rich domains in the plasma membrane of HeLa cells and the
clustering of the calcium channel ORAI1, which triggers calcium entry and signaling
when calcium concentration is low in the endoplasmic reticulum (Sharma et al., 2013b).
Interestingly, the astrocyte glutamate transporter GLAST has been shown to interact
directly with SEPT2 (Kinoshita et al., 2004). SEPT2 mutations increase GLAST
internalization and decrease glutamate uptake (Kinoshita et al., 2004). Interaction of
septins (SEPT5, SEPT11) with dynamin, which mediates scission of vesicles from the
plasma membrane, raises the possibility that septins regulate endocytosis at synaptic
terminals, affecting the recycling of synaptic vesicles and ion transporters
(Maimaitiyiming et al., 2013; McNiven, 1998).
In summary, septins have fundamental roles in the development, activity and
connectivity of the nervous system. Thus, abnormalities in septin expression are likely to
compromise the function of the nervous system, contributing the pathology of
neurodevelopmental and neurodegenerative disorders. Hereditary neuralgic

31 amyotrophy, a disorder characterized by shoulder and arm pain and muscle atrophy, is
genetically linked to missense mutations and duplications in the N-terminal sequence of
SEPT9 (Chance and Windebank, 1996; Kuhlenbaumer et al., 2005). These
abnormalities may cause the axonal degeneration and focal demyelination observed in
the brachial plexus nerves of HNA patients (Ueda et al., 2010). In the maldeveloped
brain of fetuses with Down Syndrome, expression of SEPT6 and SEPT7 is markedly
reduced and SEPT4 might be hyper-phosphorylated due to over-expression of the
DYRK1A kinase (Sitz et al., 2008). Decreases in septin 7 and 11 expression were also
observed in Schizophrenia and bipolar disorders, and SEPT9 was abnormally
overexpressed in a mouse model of demyelinating neuropathy (Patzig et al., 2011;
Pennington et al., 2008). Histological analysis of brains from Alzheimer’s patients
identified SEPT1/2/4 in neurofibrillary tangles (Kinoshita et al., 1998), which are
intracellular aggregates of the hyper-phosphorylated tau protein. In addition, allelic
variations in SEPT3 isoforms have been observed in Alzheimer’s tissue samples
indicating a potential role or risk factor in AD pathogenesis (Takehashi et al., 2004). In
the brains of patients with Parkinson’s disease, SEPT4 is found in the Lewy body
aggregates of α-synuclein (Ihara et al., 2003). Interestingly, SEPT4 interacts with α-
synuclein directly, and loss of SEPT4 expression in the A53T familial Parkinson’s mouse
model increases amyloid-like aggregation and neurodegeneration (Ihara et al., 2007).
V. Reproductive system
Septins have been genetically linked to male infertility and ovarian cancer. The
importance of septins for sperm development was discovered by two independent
studies of SEPT4 knock-out mice. Male Sept4-/- mice are sterile and their sperm is
immotile and morphologically bent with an abnormal L-shape (Ihara et al., 2005; Kissel

32 et al., 2005). This phenotype correlates with Sept4 localization to the annulus, a cortical
ring separating the middle and principle pieces of spermatozoa. In Sept4-/- sperm, the
annular structure is abolished (Ihara et al., 2005; Kissel et al., 2005). Moreover, Sept4-/-
sperm mitochondria have abnormal size, cristiae and membrane morphology, and an
excess of cytoplasmic droplets is observed in the head and neck regions of the sperm
(Kissel et al., 2005). While these data suggest that Sept4 functions in mitochondrial
division and caspase-mediated removal of the cytoplasm during spermiogenesis, follow-
up studies explored the hypothesis that Sept4 maintains a cortical diffusion barrier at the
annulus. Hunnicutt and colleagues tracked the localization of the diffusing membrane
protein basigin, which localizes to the principle piece during spermatogenesis and shifts
to the middle piece during epididymal maturation (Kwitny et al., 2010). In the sperm of
Sept4-/- mice, basigin fails to localize to either the principle or middle pieces, indicating
that Sept4 is critical for maintaining the proper localization of sperm proteins through the
establishment of an annular diffusion barrier (Kwitny et al., 2010). Because a defective
annulus is common among patients with asthenospermia, screening for Sept4
localization is now used as a diagnostic tool for sperm malformation (Sugino et al.,
2008).
In addition to Sept4, septins 1, 6, 7 and 12 also localize to the annulus of the
sperm (Toure et al., 2011). Notably, Sept12 is expressed only in the testis (Steels et al.,
2007) and similar to the Sept4-/- mice, Sept12+/- chimeric mice are sterile and their sperm
is immotile with nuclear defects and distorted shape (Lin et al., 2009). In vitro fertilization
of mouse oocytes with Sept12+/- sperm has low success rates and fertilized oocytes do
not progress past the morula stage (Lin et al., 2011). Interestingly, genetic analysis of
infertile men revealed two missense SEPT12 mutations, T89M and D197N, which map
to the switch I and GTP recognition motifs of Sept12, respectively (Kuo et al., 2012).

33 Biochemical analysis showed that the SEPT12-T89M mutant had reduced rates of GTP
hydrolysis and SEPT12-D197N did not bind GTP (Kuo et al., 2012). Both mutants failed
to form filaments in tissue culture cells and SEPT12 was absent from the sperm annulus
of SEPT2-D197N patients (Kuo et al., 2012). Thus, mutations in the testis-specific
SEPT12 are genetically linked to male infertility.
Septin expression in ovarian tissue was first identified in Drosophila where sep1
localizes to follicle cells during embryonic development (Fares et al., 1995). The precise
function of septins in ovarian physiology is unknown. Misregulation of septin expression
has been identified in ovarian tumors and is discussed in the next chapter.
VI. Urinary and digestive systems
The tissue and organ surfaces of the urinary and digestive systems are
composed of epithelial cells. The epithelial plasma membrane is biochemically
differentiated into the apical and basolateral membrane domains. The directional
transport of water, ions and solutes depends on the proper localization of specific
channels (e.g., aquaporins, Na/K pump) and transporters (e.g., p-glycoprotein) to the
epithelial membrane domains. Establishment and maintenance of the polarized
distribution of these proteins is key for the development and function of organs such as
the kidney, intestine and lung.
In Madin-Darby canine kidney cells (MDCKs), a well-established model for
studying renal epithelial morphogenesis in 2D and 3D cultures, SEPT2 is essential for
the organization of the microtubule cytoskeleton and the transport of membrane vesicles
from the Golgi to the plasma membrane (Bowen et al., 2011). During the establishment
of columnar epithelial morphology, SEPT2 guides microtubule growth and microtubule-
microtubule interactions for the establishment of the subapical microtubule network.

34 Moreover, SEPT2 localization to a subset of perinuclear, microtubule bundles enables
the egress of Golgi-derived vesicles by hindering the binding of the microtubule-
associated protein 4 (MAP4), which impedes microtubule motor-driven transport .
Without proper microtubule architecture and post-Golgi membrane traffic, SEPT2-
depleted epithelia fail to assume a columnar shape and most apical and basolateral
membrane proteins accumulate in the cytoplasm. New studies suggest that septins are
also important for the two-dimensional expansion of the renal epithelium, which occurs
by planar division and asymmetric ingression of the cleavage furrow from the basal to
the apical membrane. In Drosophila epithelia, septins enable the closure of the
cytokinetic furrow by strengthening the cytokinetic actomyosin ring, which must
overcome the tensile forces exerted by the apical adherens junctions (Founounou et al.,
2013; Guillot and Lecuit, 2013).
Renal epithelia not only maintain water and ion homeostasis, but also monitor the
flow of fluid in the lumen of the nephron, which in turn can affect cell morphology and
proliferation. Fluid flow is sensed by the primary cilium, a microtubule-based antenna-
like organelle that projects from the epithelial apical membrane into the lumen of the
nephron (Pazour and Witman, 2003). Septins are integral and essential components of
the primary cilium (Chih et al., 2012; Ghossoub et al., 2013; Hu et al., 2010). In the inner
medullary collecting duct cells IMCD3, SEPT2 localizes to the base of the primary cilium
and is required for the formation of functional cilia (Hu et al., 2010). SEPT2 depletion
results in complete loss of primary cilia and the formation of shorter cilia, in which ciliary
membrane proteins diffuse freely into the surrounding plasma membrane (Hu et al.,
2010). Thus, SEPT2 is thought to be part of a membrane diffusion barrier, which
maintains the localization of ciliary membrane proteins. Recent studies, however,

35 indicate that SEPT2/7/9 complexes may also interact with axonemal microtubules
controlling the length of cilia (Ghossoub et al., 2013).
Podocytes are specialized epithelial cells that wrap around the capillaries of the
kidney’s glomeruli and respond to insulin. Podocytes prevent serum albumin and other
macromolecules from leaving the blood, while allowing water, salts and glucose to enter
the kidney lumen. Loss of podocyte function is prevalent in diabetic nephropathy, the
most common cause of renal failure (Pagtalunan et al., 1997). A new study shows that
SEPT7 colocalizes with the glucose transporter isoform 4 (GLUT4) storage vesicles and
interacts with nephrin, the CD2-associated protein CD2AP and the vesicle SNARE
protein VAMP2 (Wasik et al., 2012). SEPT7 knock-down increased the interaction of
VAMP2 with nephrin and syntaxin, and increased glucose uptake (Wasik et al., 2012).
Thus, SEPT7 could be involved in the regulation of glucose transport in the insulin-
sensitive podocytes.
VII. Respiratory System
Respiratory epithelia perform a number of key functions including gas exchange
and protection from particulate matter. In the lower respiratory tract, the multiciliated
bronchial epithelia protect the underlying tissue from noxious agents and push the
mucus toward the pharynx (Fahy and Dickey, 2010). Maintenance of the epithelial cell-
cell junctions is critical for limiting paracellular permeability, subepithelial inflammation
and injury (Frank, 2012). Physiological stresses in bronchial epithelia (e.g. particulate
matter) stimulate dynamic reorganization of the actin cytoskeleton at the cell cortex,
strengthening cell-cell junctions and restricting paracellular permeability (Youakim and
Ahdieh, 1999). In shear-stressed bronchial epithelia, SEPT2 is recruited to the apical
plasma membrane and interacts with the F-actin network more strongly (Sidhaye et al.,

36 2011). In SEPT2-depleted MDCK and bronchial cells challenged with shear stress or
particulate matter, cortical F-actin is reduced and low-molecular-weight dextran enters
into the basolateral space more readily (Sidhaye et al., 2011). Moreover, apical
application of EGF in SEPT2-depleted cells results in elevated activation of the
basolaterally-confined EGF receptor (Sidhaye et al., 2011). Taken together, these data
indicate that SEPT2 is critical for the maintenance of epithelial integrity during respiratory
stress.
VIII. Endocrine system
Tissues and cells of the endocrine system secrete hormones that regulate the
physiology and function of peripheral tissues and organs. In the pancreas, SEPT5 is
highly expressed in the islet of Langerhans, while its expression is markedly reduced
and absent from the acinar and ductal epithelia, respectively (Capurso et al., 2005). The
islet of Langerhans consists of α- and β-cells that secrete glucagon and insulin,
respectively, stimulating glucose release and uptake from the blood. The role of SEPT5
in exocytosis was tested in the HIT-T15 insulinoma cell line by assaying for the release
of exogenous human growth hormone (hGH) in the presence of wild-type or dominant-
negative SEPT5. Secretion of hGH was significantly reduced in cells expressing wild-
type SEPT5 indicating that SEPT5 inhibits exocytosis (Beites, 1999). In pancreatic β-
cells, SEPT2 interacts with EXO70, a component of the octameric exocyst complex that
mediates vesicle tethering to the plasma membrane (Rittmeyer et al., 2008). The
SEPT2-EXO70 complex localizes to the β-cell membrane and interacts directly with the
Rho GTPase effector, IQGAP1 (Rittmeyer et al., 2008). Interestingly, overexpression of
the SEPT2-EXO70 binding domain of IQGAP1 displaces SEPT2 from the plasma
membrane, and the constitutively-active form of CDC42 inhibits the interaction of

37 IQGAP1 with the SEPT2-EXO70 complex and blocks insulin secretion (Rittmeyer et al.,
2008). Although the precise role of septins in vesicle docking and fusion is unknown,
regulation of septin localization and function by IQGAP and Rho signaling might be
important for the secretion of insulin from pancreatic β-cells.
IX. Integumentary system
The integumentary system consists of the skin, hair follicles and sebaceous
glands. Collectively, these structures make up the largest organ system of the body
protecting all other organs from physical damage, infectious agents and water loss.
Regeneration of the epidermis after injury relies on skin and hair follicle stem cells,
whose growth and differentiation are tightly regulated so that tissue structure is
preserved without overgrowth. A recent study showed that the SEPT4 splice variant
ARTS (SEPT4_v2), is specifically expressed in hair follicle stem cells (HFSCs) (Fuchs et
al., 2013). Hair follicle bulges in mice lacking Sept4/ARTS are morphologically normal,
but contain twice as many CD34+ progenitor cells (Fuchs et al., 2013). Sept4/ARTS-
deficient CD34+ cells grow faster in culture without feeder cells and are highly-resistant
to pro-apoptotic stimuli, suggesting that SEPT4/ARTS restricts HFSC proliferation
(Fuchs et al., 2013). Remarkably, wound recovery in Sept4/ARTS-deficient mice is
extraordinarily fast. Sept4/ARTS has previously been shown to promote apoptosis by
targeting XIAP, an inhibitor of apoptosis, for degradation (Larisch et al., 2000). HFSCs
from mice deficient in both SEPT4/ARTS and XIAP showed reduced wound repair
(Fuchs et al., 2013) supporting the hypothesis that SEPT4/ARTS-mediated apoptosis
can limit stem cell growth.
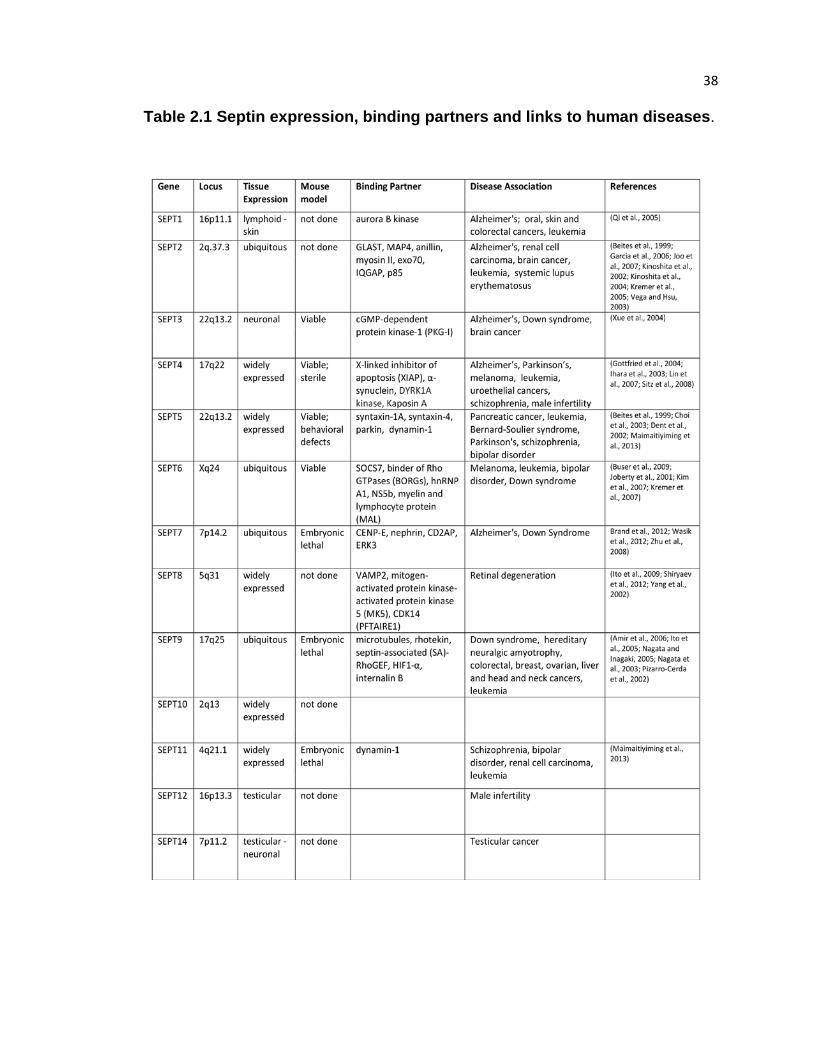
38 Table 2.1 Septin expression, binding partners and links to human diseases.

39
Figure 2.1 Septin functions in organ systems and their connection to human disease Schematic diagram depicts main organ systems of the human body and outlines organ-
and cell type-specific structures and processes that require septins. Color codes indicate
the intracellular involvement of septins, which includes septin functions in the
organization of actin (red), microtubules (green) and cell membranes (grey), vesicle
fusion (blue) and apoptosis (yellow). Human diseases are listed based on genetic,
histological, genomic and proteomic evidence that implicates septins in the pathology of
various organ disorders.

40
CHAPTER 3. Septins in cancer
Septin expression is frequently altered in cancer, including tumors of epithelial
origin and hematological malignancies. On the other hand, increased assembly of
septins with actin stress fibers in cancer-associated fibroblasts, resident cells of the
tumor microenvironment, was shown recently to promote ECM remodeling,
angiogenesis and tumor cell invasion in a mouse model of breast cancer (Calvo et al,
2015). Septins are also emerging biomarkers for urological and colon cancers, but their
role in tumorigenesis and metastasis is poorly understood.
Septins in hematological malignancies
Aberrant septin expression has been implicated in blood disorders including
acute myeloid leukemia (AML) and acute lymphoblastic leukemia (ALL) (Cerveira et al.,
2011). MLL is a histone methyltransferase that regulates homeobox (HOX) gene
expression during hematopoiesis (Muntean and Hess, 2012). Chromosomal
translocations of the mixed lineage leukemia (MLL) gene account for ~10% of the de
novo and therapy-induced leukemias (Muntean and Hess, 2012). Septins 2, 5, 6, 9 and
11 have all been identified as MLL fusion partners, accounting for the largest known
protein family associated with MLL rearrangements (Cerveira et al., 2011). The MLL-
septin chimera results from double-stranded intronic breaks in the MLL and septin
genes, producing in-frame fusions of the MLL N-terminus and almost the entire open-
reading frame of the septin (Cerveira et al., 2011). Ectopic expression of the MLL-
SEPT6 chimera has been shown to immortalize hematopoietic progenitor cells,
upregulating the expression of the Hox-7a and Hox-9a genes (Ono et al., 2005b).
Although MLL-SEPT6 expression in mice was not sufficient to induce leukemogenesis
(Ono et al., 2005a), mice transduced with an active FMS-like receptor tyrosine kinase 3,

41 which is frequently overexpressed in AML, required MLL-SEPT6 to stimulate
leukemogenesis (Ono et al., 2005b). MLL-SEPT6 expression also led to a lethal
myeloproliferative disease with long latency (Ono et al., 2005b).
Recent studies have shown that hematopoietic stem cell maturation and
malignancy are affected by the SEPT4 splice variant ARTS (apoptosis-related protein in
the TGF-beta signaling; SEPT4_v2) (Garcia-Fernandez et al., 2010). SEPT4/ARTS
binds to inhibitors of apoptosis proteins, initiating caspase activation and cell death ,
2004(Gottfried et al., 2004; Larisch et al., 2000). Sept4-deficient mice contain elevated
levels of hematopoietic stem cells, including progenitor and immature B-cells that are
resistant to pro-apoptotic stimuli (Garcia-Fernandez et al., 2010). Genetic crosses
between Sept4-/- and the leukemogenic Eµ-Myc mice showed that SEPT4 ablation
enhances lymphomagenesis and reduces lifetime by 50% (Garcia-Fernandez et al.,
2010). Significantly, ARTS expression is frequently altered in bone marrow lymphocytes
derived from patients with childhood ALL, the most common malignancy found in
children (Elhasid et al., 2004). Lymphocytes from ALL patients exhibit a significant
reduction in SEPT4/ARTS mRNA levels, and SEPT4/ARTS-deficient lymphocytes are
highly-resistant to apoptosis when treated with the chemotherapeutic agent, ara-C
(Elhasid et al., 2004). DNA methylation inhibitors can rescue SEPT4/ARTS expression
suggesting that aberrant methylation downregulates its expression during tumorigenesis
(Elhasid et al., 2004). Clinically, SEPT4/ARTS expression was rescued in patient
lymphocytes following therapy, indicating that SEPT4/ARTS expression suppresses
growth and possibly malignant transformation of hematopoietic cells.
Altered septin expression in solid tumors
Septins are overexpressed in renal cell carcinoma (RCC), which is an aggressive
and highly metastatic cancer of the kidney with poor response rates to therapeutics

42 (Craven et al., 2006b). SEPT2 and SEPT11 are upregulated in RCCs defective in the
von-Hippel Lindau (VHL) tumor suppressor, a ubiquitin E3 ligase that targets the
hypoxia-inducible factor 1 (HIF-1) transcription factor for degradation (Craven et al.,
2006a). It is unknown if SEPT2 and SEPT11 are transcriptionally regulated by HIF1 or
directly degraded by VHL. Moreover, it is unclear if and how septin upregulation
contributes to RCC growth and metastasis.
SEPT9 is over-expressed in hormonally regulated breast, prostate and ovarian
cancers and endometrial cancer. SEPT9 binds directly to HIF-1 and increases its
transcriptional activity in prostate cancer cells by inhibiting HIF-1 degradation. Septin 9
expression, however, is significantly altered in ovarian cancers. Initial studies in sporadic
ovarian tumor samples showed that the chromosomal locus 17q25.3, which includes the
SEPT9 gene, was partially deleted leading to allelic imbalances and loss of
heterozygosity (Kalikin et al., 2000; Russell et al., 2000). Subsequent studies revealed
that SEPT9 has a complex genomic organization encoding for up to 18 different mRNA
transcripts and 15 protein isoforms (McIlhatton et al., 2001). Analysis of SEPT9 mRNA
transcripts in benign, malignant and borderline, low-grade ovarian carcinomas showed
that the SEPT9_v1 and SEPT9_v4* transcripts were overexpressed in serous and
mucinous borderline tumors (Scott et al., 2006). The enhanced translational efficiency of
SEPT9_ v4* relative to SEPT9_v4 suggested that increased levels of SEPT9_i4 could
promote the malignant and metastatic potential of ovarian carcinomas (McDade et al.,
2007). Consistent with this possibility, over-expression of SEPT_i4 enhances cell motility
and resistance to anti-cancer drugs that target microtubules (Chacko et al., 2012).
Upregulation of SEPT9_i1 could also enhance tumor growth and angiogenesis by
slowing down the degradation of the c-Jun-N-terminal kinase (JNK) and the hypoxia-
inducible factor HIF1a (Amir et al., 2009; Gonzalez et al., 2009). While the cause of the

43 amplification of certain SEPT9 mRNA transcripts is not understood, epigenetic changes
such as the hypermethylation of alternative intragenic promoters could alter the relative
expression of mRNA transcripts (Connolly et al., 2011b).
An increase in the mRNA and protein levels of the Bradeion isoform of SEPT4
has also been detected in urine samples from patients with RCCs and transitional
bladder cancers (Bongiovanni et al., 2012; Tanaka et al., 2003). Quantification of SEPT4
Bradeion expression by immunochromatography and RT-PCR of urine samples has
been introduced for the diagnosis of urothelial cancers in Japan and Europe
(Bongiovanni et al., 2012; Tanaka et al., 2003). Abnormal expression of SEPT4 has also
been observed in colorectal cancer (CRC). While absent from patient-matched healthy
tissues, two SEPT4 isoforms (Bradeion α and β) were identified in colorectal tissue
isolated from human patients with mucinous carcinoma and rectal malignant melanoma
(Tanaka et al., 2001). Ribozyme-mediated cleavage of either or both isoforms arrests
CRC growth in the G2 phase of the cell cycle in vitro and prevents tumor growth in mice
injected with CRC cells (Tanaka et al., 2002). The SEPT4 isoforms exhibit cytoplasmic
and nuclear localization (Tanaka et al., 2001), but further studies are necessary to
elucidate their cellular functions and the molecular mechanisms that contribute to their
abnormal expression in CRC.
In squamous cell carcinoma (SCC) and malignant melanoma tissue sections,
SEPT1 is significantly upregulated (Mizutani et al., 2013). In SCC cells, SEPT1 and
SEPT5 coimmunoprecipitate and colocalize with SEPT4 in the lamellipodia, a dendritic
actin network at the leading edge of migrating cells (Mizutani et al., 2013). SEPT1-
depleted DJM-1 cells exhibit defective cell spreading, a process that depends on actin
polymerization and cell adhesion with the ECM. While absent from healthy epidermis,

44 SEPT9 is highly expressed in cultured SCC cells and coprecipitates with
SEPT7/10/11/14 (Mizutani et al., 2013).
Conversely, SEPT9 is highly expressed in normal colonic glandular and surface
epithelia, but is markedly reduced in adenomas and almost completely absent from
tumor tissues, indicating that SEPT9 levels decrease progressively during tumorigenesis
(Toth et al., 2011). Treatment of HT29 CRC cells with a demethylating agent results in
increased levels of SEPT9, suggesting that SEPT9 expression is downregulated by
gene methylation during CRC progression (Toth et al., 2011). Epigenetic modifications in
stem cell DNA within the colonic crypt are known to change with age and have been
identified in a subset of CRCs (Humphries and Wright, 2008; Taylor et al., 2003). More
specifically, hypermethylation of CpG islands (GC-rich regulatory sequences found in the
promoters of almost half of all human genes) are common in many cancers (Herman
and Baylin, 2003). Plasma DNA from CRC patients contains abnormally high levels of
methylated SEPT9 (Grutzmann et al., 2008). Significantly, SEPT9 is hypermethylated in
87% of early stage I and II colorectal tumors (Warren et al., 2011). These findings have
led to the development of the septin 9 test, which is a commercial non-invasive, blood-
based test that screens for the methylation of the SEPT9 gene as a diagnostic of early-
stage colon cancer.
Septins in the tumor microenvironment
Surrounding the tumor mass lies a complex environment referred to as the tumor
microenvironment (TME), whose organization and constituent cells become altered
during tumor growth and metastasis (Balkwill et al., 2012). Cells of the immune,
vasculature and lymphatic systems, along with resident cells of the ECM (e.g.

45 fibroblasts) can function in part to support tumor growth by promoting angiogenesis,
growth and motility.
In particular, cancer-associated fibroblasts (CAFs) are a subpopulation of
“activated” cells that reside within the tumor microenvironment that promote tumor cell
transformation, angiogenesis and invasion (Kalluri and Zeisberg, 2006). During tumor
progression, CAFs become highly contractile through the increased the assembly of
actomyosin stress fibers, which promotes ECM remodeling by protein deposition and
upregulating cell-ECM adhesions (Butcher et al., 2009). Furthermore, tumor-associated
macrophages (TAMs) also promote cell invasion by localizing to the invasive edge of
tumors where they sustain a paracrine signaling loop that stimulates cell motility
(Patsialou et al., 2009).
In normal mouse NIH3T3 fibroblasts, septins colocalize with subnuclear actin
stress fibers (Kinoshita et al., 2002). Septin knockdown results in stress fiber
disassembly, while pharmacological targeting of actin stress fibers induces septin
filament assembly (Kinoshita et al., 2002). Conversely, septins and actin are largely
cytoplasmic in fibroblasts derived from mouse mammary glands, but co-assemble into
actomyosin stress fibers in CAFs (Calvo et al., 2015). Septin depletion inhibits
actomyosin stress fiber assembly in CAFs and reduces their ability to promote tumor
growth in vivo (Calvo et al., 2015). Thus, septins and actin stress fibers show an
interdependent relationship, but whether septins bind directly to actin filaments and how
septins regulates the organization of the actin stress fiber network during cell migration
has remained unknown.

46
CHAPTER 4. Septin overexpression in tumor cell migration,
trafficking and metabolism
As discussed in the previous chapter, septin overexpression is frequently
reported in solid tumors of epithelial origin including renal, breast, ovarian and skin; yet,
how septins contribute to tumorigenesis and tumor metastasis at the molecular level is
unknown. I hypothesize that septin overexpression promotes tumor growth and invasion
through at least two distinct mechanisms: i) promoting the mesenchymal-like motility of
renal epithelia, and ii) regulating the intracellular trafficking of macropinocytic cargo.
Because septins are multi-functional proteins that interact directly with the
cytoskeleton and cell membranes, it is not surprising that these findings implicate septins
in different cellular processes. Furthermore, tumorigenesis is a complex process that
stems largely from changes in the genome, which can alter many different cellular
pathways (Hanahan and Weinberg, 2000). Tumor cells proliferate unchecked by evading
cell death signals and adapt to growing metabolic needs by changing the mechanisms
by which energy is produced in order to sustain proliferation (Hanahan and Weinberg,
2011). Through changes in cell polarity, cytoskeletal organization and the trafficking of
intracellular cargo, tumor cells can invade the surrounding ECM and migrate towards
tissues of the blood and lymph to initiate metastasis (Bravo-Cordero et al., 2012).
Septins functions in tumor cell migration
Epithelial tumor cell invasion is activated by the aberrant induction of epithelial-
to-mesenchymal transition (EMT), a developmental transcriptional program that
regulates the patterning of epithelial tissues (Thiery et al., 2009). During this process,
epithelia breakdown cell-cell contacts, upregulate the attachment and turnover of
adhesions with the ECM, and adopt a front-rear, mesenchymal-like polarity (Thiery et al.,

47 2009). Tumor cell invasion into the ECM is initiated by membrane protrusions, such as
invadopodia and pseudopodia, which are driven by the assembly of actin filaments
against the ventral cell membrane (Murphy and Courtneidge, 2011). Within these
protrusions, actin-binding proteins, cell-ECM receptors and ECM-degrading enzymes
are enriched (Linder, 2007).
Tumor cell invasion into the ECM is a dynamic process. Tumor cells adapt to
ECM organization and topography, which becomes stiffer and more crosslinked during
tumorigenesis, and can interconvert between different modes of motility (Figure 4.1): a)
mesenchymal-like; b) amoeboid; c) lobopodial (Friedl and Wolf, 2010; Petrie et al.,
2012). The mesenchymal-like motility of epithelia has been characterized extensively
(Figure 4.1). At the leading cell edge, actin polymerization by the Arp2/3 complex drives
the formation of protrusions termed lamellipodia, which make new contacts with the
ECM through integrin receptors (Parsons et al., 2010; Svitkina and Borisy, 1999). These
focal complexes mature into focal adhesions, which is induced under the tension and
contractile forces of a network of actin stress fibers. Radial actin stress fibers emanate
from growing focal adhesions and link up with the contractile transverse arcs in the
leading lamella, which transmits forces to focal adhesions to promote forward
translocation (Gardel et al., 2010). Assembly of this dynamic network involves proteins
that nucleate, bundle and cross-link actin filaments (Choi et al., 2008). Force
transduction is regulated by the myosin II regulatory light chain, which modulates the
ATPase activity and step rate of myosin II motor in response to ECM stiffness (Davis et
al., 2002; Schwartz, 2010). Thus, the force-generating mechanisms that drive cell
motility are in a direct crosstalk with the extracellular microenvironment. In contrast, bleb
and lobopodial-based cell migration rely upon the assembly of contractile actomyosin
fibers at the cell cortex, which induces the formation of membrane protrusions that help

48 tumor cells navigate through gaps in the ECM (Lammermann and Sixt, 2009; Petrie and
Yamada, 2012) (Figure 4.1). The conversion to amoeboid and lobopodial-based cell
motility correlates with the expression of actin-binding proteins and ECM-degrading
enzymes and ECM topography (Petrie and Yamada, 2012), but the mechanism by
which tumor cells interconvert between these motility modes is not fully known.
Importantly, invasive tumor cells frequently show elevated expression of actin-
binding proteins (Wang et al., 2004), which control the assembly and organization of the
actin cytoskeleton during cell migration (Parsons et al., 2010). Septins have been
reported previously to promote cell migration, but the mechanism has remained elusive.
In metastatic cancer cell lines, SEPT9 expression is elevated and essential for the
formation of actin-rich pseudopods and cell migration (Shankar et al., 2010). In
agreement with this study, overexpression of five different SEPT9 isoforms in the non-
invasive MCF-7 breast cancer cells promotes cell migration (Connolly et al., 2011b).
Thus, septins promote tumor cell migration, but how septins influence the organization of
the actin cytoskeleton at the molecular level during cell motility has been unclear.
Septins have been linked to the assembly of branched actin networks in epithelia
and branched actin patches and neurons. SEPT1 knockdown in squamous cell
carcinoma cells inhibits cell spreading, which is dependent upon the formation of the
lamellipodia by the Arp2/3 complex (Mizutani et al., 2013). Interestingly, SEPT6
overexpression promotes the recruitment of cortactin, an activator of the Arp2/3
complex, to actin patches in neurons and renal epithelial lamellipodia (Hu et al., 2012).
Experiments in vitro with purified actin filaments and the Arp2/3 complex show that
recombinant SEPT6 localizes to actin branch points (Hu et al., 2012), which correlates
with Arp2/3 localization. Despite these findings, how septins interact with and regulate
actin filament assembly remained unknown.

49
In the first part of this thesis, I present a novel role for septins in promoting the
mesenchymal-like motility of renal epithelia undergoing EMT by directly crosslinking
actin filaments and regulating focal adhesion stability. Septins localizes to transverse
actin arcs and their orthogonal junctions with radial stress fibers, which are anchored to
focal adhesions. By disrupting the actin stress fiber network, septin depletion reduces
focal adhesion stability and cell migration. I identified further that SEPT9 expression
specifically increases during EMT, suggesting that SEPT9 may be part of the EMT
program that promotes the mesenchymal-like motility of epithelia. Indeed, SEPT9
overexpression promotes the migration of renal carcinoma cells and the collective
migration of renal epithelia undergoing EMT in 3D collagen matrix. Conversely, SEPT9
depletion in single cells migrating in a 3D matrix inhibits motility and induces a transition
to a more rounded and protrusive shape, indicating that SEPT9 expression promotes
specifically the mesenchymal-like motility mode.
Because stress fiber assembly during mesenchymal motility is regulated by
plethora of actin-binding proteins, whose spatial and temporal recruitment is tightly
regulated, I investigated further the interaction between SEPT9 and actin filaments at the
molecular level. SEPT9 binds to three specific sites on the actin filament, two of which
are shared by other actin-binding proteins. Strikingly, SEPT9 interferes with the binding
of the myosin motor domain in its low-affinity state and the actin-severing protein cofilin,
which could stabilize and protect filaments from depolymerizing forces. Thus, I
hypothesize that SEPT9 protects actin filaments by regulating spatially the recruitment of
actin-binding proteins during filament assembly. In agreement with this hypothesis, I
investigated the assembly and stability of nascent actin stress fibers in live renal
epithelia. Indeed, septin depletion results in unstable radial actin stress fibers that
disassemble at a faster rate.

50
At the molecular level, septins operate collectively as hetero-polymers and
filaments. In renal epithelia, SEPT9 colocalizes and immunopreciptates with SEPT2,
SEPT6 and SEPT7, indicating that they co-assemble into complexes and filaments.
While SEPT6 and SEPT9 bind directly to actin, SEPT2 does not interact with actin
filaments, but it binds directly to myosin II and is posited to control the phosphorylation of
its regulatory light chain and myosin contraction (Joo et al., 2007). These differential
interactions may regulate spatially the binding and activation of myosin along the actin
filament and contraction of actomyosin stress fibers during cell motility. In the absence of
septins, myosin motor proteins may bind and contract uncrosslinked actin filaments,
which can induce filament buckling and disassembly. Conversely, actin filaments lacking
septins may contain more available binding sites for the actin-severing protein cofilin,
which is enriched in the leading cell edge. Thus, overexpression of septins in cancer
may reinforce the mesenchymal-like motility mode by controlling the interaction of actin-
binding proteins with actin filaments during stress fiber assembly and contraction.
Septins in membrane traffic and metabolism
Septins also associate with the microtubule cytoskeleton and cell membranes,
regulating the intracellular trafficking of membrane cargo (Spiliotis and Gladfelter, 2012).
Importantly, membrane traffic is an emerging regulator of tumor cell migration, growth
and metabolism (Jones et al., 2006). Intracellular vesicles are derived from organelles
and the plasma membrane via endocytosis. Through the recruitment of adaptor and
motor proteins, vesicles move along actin filaments and microtubules to distinct
subcellular compartments (Fu and Holzbaur, 2014). Intracellular vesicle dynamics are
controlled by the large family of Rab GTPases, which can recruit motor proteins (e.g.
dynein) and promote vesicle docking, tethering and fusion (Stenmark, 2009). Moreover,

51 intracellular vesicles can serve as signaling hubs that activate pathways to promote cell
motility and growth. Rab-dependent recycling of integrins to focal adhesions (Caswell et
al., 2008) and active members of the Rho family of GTPases (Palamidessi et al., 2008),
which regulate actin polymerization at the leading cell edge, promotes tumor cell motility
and endocytosis. Mutations present in human cancers that render Rab GTPases (e.g.
Rab35) constitutively active were identified recently to stimulate tumor cell proliferation
via aberrant trafficking of growth factor receptors to lysosomes and activation of the
PI3K/AKT/mTOR signaling pathway (Wheeler et al., 2015).
Septins regulate the intracellular delivery of golgi-derived to the plasma
membrane domains in renal epithelia, which is important for epithelial polarity and
morphogenesis (Spiliotis, 2008). Septin knockdown or inhibition with a function-blocking
antibody impedes the transport of plasma membrane proteins through the exocytic
pathways (Spiliotis, 2008). Septins colocalize with polyglutamylated microtubules and
regulate the organization and dynamics of the microtubule network (Bowen et al., 2011;
Spiliotis, 2008).
In contrast, little is known about the function of septins in endocytic pathways.
Recent evidence shows septins regulate epidermal growth factor receptor (EGFR)
surface levels and degradation (Diesenberg et al., 2015), which occurs through
endocytic recycling, and promote the formation of multivesciular bodies (Traikov et al.,
2014). Importantly, altered endocytosis is commonly observed in tumor cells.
Upregulation of the endocytic mechanisms that internalize adhesion proteins promote
the breakdown of cell-cell junctions (Yang et al., 2006), and increased macropinocytosis,
a form of clathrin-independent endocytosis, promotes tumor cell growth and possibly
migration (Commisso et al., 2013).

52
Macropinosomes are derived from plasma membrane ruffles, which fold back on
themselves forming large vesicles that can capture membrane proteins, extracellular
fluids and solutes (Lim and Gleeson, 2011). Following ruffle closure, macropinosomes
undergo a maturation process that involves changes in lipid composition, the recruitment
of Rab GTPases and fusion with late endosomes, which can recycle proteins back to the
plasma membrane, and lysosomes, which degrade proteins for catabolic pathways
(Bright et al., 2005; Swanson, 2008) (Figure 4.2). In motile fibroblasts, growth factor
stimulation induces the formation of macropinosomes that recycle integrins back to
nascent focal adhesions in the leading cell edge; and inhibition of macropinocytosis or
Rab-dependent integrin trafficking attenuates fibroblast migration (Gu et al., 2011). Thus,
macropinocytic trafficking of cell adhesion molecules may be necessary for tumor cell
invasion.
In addition, recent evidence also supports a role for macropinocytosis in tumor
cell growth via carbon metabolism (Commisso et al., 2013). Uncontrolled tumor cell
proliferation requires increased energy metabolism in order to sustain growth. Tumor
cells frequently bias energy production towards glycolysis and glutamine metabolism, or
glutaminolysis, which have emerged as hallmarks of tumor cell metabolism
(Deberardinis et al., 2008). Notably, the glycolytic and glutamine metabolic pathways are
frequently linked to cellular transformation by oncogenes, such as Ras GTPases (White,
2013). In addition, pancreatic tumor cells expressing a constitutively-active mutation in
the KRas oncogene show increased uptake of amino acids through macropinocytosis
and the delivery of glutamine to the central metabolic pathway (Commisso et al., 2013).
Strikingly, inhibition of macropinocytosis and subsequent glutamine metabolism reduces
tumor cell proliferation in vitro and tumor growth in vivo (Commisso et al., 2013).

53
In the last part of this thesis, I discovered a population of membrane-associated
septins that localize to macropinosomes and their contact sites with endosomes by
interacting directly with PI(3,5)P2, a low-abundant lipid that concentrates on late
endosome and lysosomes. Septin knockdown results in clusters of macropinosomes and
abrogates macropinosome fusion with lysosomes. Conversely, septin overexpression
enhances the trafficking of macropinocytic cargo to the lysosomes. Using an in vitro
vesicle fusion assay, I found that septins promote membrane fusion independently of the
actin and microtubule cytoskeleton.
Membrane-associated septins control the organization of plasma membrane
lipids and proteins (Spiliotis and Gladfelter, 2012). In addition, septins have been
reported to interact directly with exocytic SNAREs, whose clustering into membrane
domains is required for fusion. (Aoyagi et al., 2005). Based on this knowledge, septins
may promote vesicle fusion by controlling SNARE organization in membrane domains or
the dynamic assembly and disassembly of SNARE complexes during macropinosome
fusion with lysosomes. Through these potential mechanisms, septin overexpression may
promote tumor cell proliferation by promoting the trafficking of macropinocytic cargo to
the lysosome, which can supply molecular intermediates for the central metabolic
pathway.
In a similar manner, autophagy is a process by which stressed or starved cells
capture cellular material (e.g. organelles, proteins) into autophagosomes, which are
targeted to the lysosome for degradation (Kaur and Debnath, 2015; Levine and Klionsky,
2004). Autophagy has been linked to prolonged tumor cell survival, but also defects in
the autophagic machinery are associated with tumorigenesis (Mathew et al., 2007).
During microbial infection, septins target intracellular bacteria (e.g. Shigella) to
autophagosomes, which restricts their replication and reduces infection in vitro and in

54 vivo (Mostowy et al., 2010; Mostowy et al., 2013). Septins are posited to interact directly
with the autophagy machinery, which also targets autophagosomes to lysosomes by
interacting with lysosomal SNAREs (Liu et al., 2015; Mostowy et al., 2010). How septins
function in autophagy is not fully understood, and whether septin overexpression may
support autophagy-mediated cell survival in tumor cells is unknown. However, I
speculate that septins may have a wider role in targeting intracellular vesicles to
lysosomes.

55
Fig 4.1 Modes of single tumor cell migration. (A) Mesenchymal motility is characterized by the formation of focal adhesions and an
actin stress-fiber network containing radial stress fibers (SFs) and transverse arc
SFs that link up to promote focal adhesion growth.
(B) Amoeboid motility is characterized by the assembly of actomyosin fibers at the cell
cortex and the formation of protrusive blebs.
(C) Lobopodial motility is characterized by the assembly of an actomyosin network at the
cell cortex that connects with a network of intermediate filaments near the nucleus.
Through this cytoskeletal connectivity, the nucleus pressurizes the cell front in a
piston-like mechanism that promotes the extension of blunt membrane protrusions
termed lobopodia.

56
Figure 4.2 Macropinosome formation, maturation and traffic to endolysosomes. Plasma membrane ruffles fold back upon themselves to engulf soluble cargo (red) into
macropinosomes. Macropinosomes undergo a maturation process that is controlled by
changes in phosphoinositide lipid composition and the recruitment of Rab GTPases,
which collectively regulate macropinosome retrograde movement and fusion with late
endosomes and lysosomes.

57
CHAPTER 5. Septins promote stress fiber-mediated maturation of focal
adhesions and renal epithelial motility.
Abstract
Organogenesis and tumor metastasis involve the transformation of epithelia to
highly motile mesenchymal-like cells. Septins are filamentous G proteins, which are
over-expressed in metastatic carcinomas, but their functions in epithelial motility are
unknown. Here, we show that a novel network of septin filaments underlies the
organization of the transverse arc and radial (dorsal) stress fibers at the leading lamella
of migrating renal epithelia. Surprisingly, septin depletion resulted in smaller and more
transient and peripheral focal adhesions. This phenotype was accompanied by a highly
disorganized lamellar actin network and rescued by the actin bundling protein α-actinin-
1. We show that pre-assembled actin filaments are crosslinked directly by SEPT9,
whose expression is increased after induction of renal epithelial motility with the
hepatocyte growth factor. Significantly, SEPT9 over-expression enhanced renal cell
migration in 2D and 3D matrices, while SEPT9 knock-down decreased migration. These
results suggest that septins promote epithelial motility by reinforcing the cross-linking of
lamellar stress fibers and the stability of nascent focal adhesions.
Introduction
Embryonic patterning and organogenesis depend on the morphogenetic
movements of epithelial cells, which transition to migratory mesenchymal-like cells
(Thiery et al., 2009). In the developing kidney, extensive epithelial movements
accompany the formation, branching and tubulogenesis of the ureteric bud (Dressler,
2002; Zegers et al., 2003), and epithelial-to-mesenchymal transition (EMT) underlies

58 renal fibrosis and the metastasis of renal cell carcinomas (RCCs) (De Craene and Berx,
2013; Liu, 2010). EMT is characterized by disruption of cell-cell adhesions, cell
scattering and the enhancement of invasive cell motility with a front-rear polarity
(Guarino et al., 2007; Lim and Thiery, 2012).
Mesenchymal motility is driven by the actomyosin cytoskeleton and involves
adhesion to the ECM and its degradation (Bravo-Cordero et al., 2012; Friedl and Wolf,
2010). Nascent adhesions are stabilized and grow to focal adhesions (FAs) under the
tension exerted by a network of actin stress fibers (Geiger and Yamada, 2011; Schwartz,
2010). At the leading lamellae of migrating cells, forces are transduced to adhesions by
the radial (dorsal) stress fibers (RSFs), which extend dorsally from FAs and connect with
the transverse arc (TA), a contractile network of curved actin filaments (Burridge and
Wittchen, 2013; Gardel et al., 2010; Parsons et al., 2010). Actin nucleating and cross-
linking proteins and myosin II, whose activity is regulated by the phosphorylation of its
regulatory light chain (RLC), mediate the assembly, organization and contraction of actin
stress fibers (Burridge and Wittchen, 2013; Parsons et al., 2010). Actomyosin
organization and contractility are regulated by the small GTPases of the Ras
superfamily, which affect cell motility (Parri and Chiarugi, 2010; Raftopoulou and Hall,
2004).
Septins are evolutionarily and structurally related to the Ras GTPases (Leipe et
al., 2002), but unlike the monomeric small GTPases, septins form filamentous polymers
that consist of non-polar hetero-octameric complexes (Mostowy and Cossart, 2012;
Weirich et al., 2008) . Septins interact with the actomyosin cytoskeleton (Joo et al., 2007;
Kinoshita, 2002; Mavrakis et al., 2014) and are abnormally over-expressed in renal cell
carcinomas, but their precise function in stress fiber organization and mesenchymal-like

59 motility is unknown. Here, we show that in the leading edge of motile renal epithelia, a
novel network of septin filaments promotes cell motility by reinforcing the organization of
the lamellar stress fiber network and the stability of nascent FAs.
Materials and methods
Tissue culture and transfections.
MDCK II/G cells and the stable MDCK-SEPT2-YFP (Spiliotis et al., 2005), MDCK-
paxillin-GFP (Yamada and Nelson, 2007), MDCK-SEPT9-mCherry, MDCK-ABD-
ABP140-mCherry cell lines were cultured in low glucose DME with 1g/liter NaHCO3,
10% FBS (Cell Generation), and 1% PSK (penicillin, streptomycin, kanamycin) at 5%
CO2. The stable MDCK-SEPT9-mCherry and MDCK-ABP140-mCherry cells were
generated by standard selection with G418 sulfate (0.8 mg/mL; Corning) and stable
transfectants were cloned in 96-well plates by serial dilutions and levels of expression
were analyzed by western blotting. The renal cell carcinoma 786-O cell line was
purchased from ATCC and maintained in RPMI with 1 g/liter NaHCO3, 10% FBS, and
1% PSK. Coverslips were acid-washed with 1 M HCl for 4h at 50ºC and coated with 10-
20 µg/mL fibronectin (Invitrogen) for 2h at 37ºC or rat tail collagen. Cells were
transfected with 1.5 µg of plasmid or co-transfected with 1.5 µg plasmid and 1µg
shRNAs with Lipofectamine 2000 (Invitrogen). Prior to fixation or live-imaging, MDCK
cells were stimulated with 20 ng/mL recombinant HGF (Sigma) in DME supplemented
with 0.5% FBS for at least six hours. Live imaging studies were performed in phenol red-
free media containing 20 mM HEPES.
To generate polarized cysts, MDCK cells and cells that express stably SEPT9-
mCherry at low and high levels were cultured in collagen gels composed of 2mg/mL type
I collagen (Advanced Biomatrix) on transwell filters (0.4 µm pore; BD Biosciences) for
ten days. Polarized cysts were stimulated with 30 ng/mL HGF for three days (Fig. 4 C-E)

60 and with or without 50 µm FCF (Fig. 4E). To image and quantify single cell migration in
3D (Fig. 4 H and I), MDCK cells were transfected with control or SEPT9-targeting
shRNAs for 24h on tissue culture plates. Cells were dissociated by trypsinization and
cultured in 2 mg/mL type I collagen gels on 35 mm glass-bottom dishes (live imaging of
MDCK-ABP140-ABD-mCherry; Fig. 4 H) or transwell filters (morphometric analysis of
fixed MDCKs; Fig. 4I) and treated with 40 ng/mL HGF, 16 h prior to imaging.
Plasmids and shRNAs.
Canine SEPT2 was targeted with control (5’-TGA GTTCACACTAATGGTGGTCG-3’),
SEPT2 shRNA #1 (5’-CCTTAGACGTCGCAT TCATGAAA-3’) and SEPT2 shRNA #2 (5’-
GCAACTACAAGCCCGAAGAATAA-3’) (Bowen et al., 2011). To target human SEPT2,
the SEPT2 shRNA 5’-AAGGTGAATATTGTGCCTGTC-3’ was subcloned into the GFP-
expressing pG-SUPER vector. The SEPT9 shRNA construct was made by inserting the
SEPT9 shRNA 5’- GACCGGCTGGTGAACGAG AAGTT-3’ into mCherry-expressing
pSuper vector. The shRNA constructs against canine and human SEPT9 were made by
subcloning the shRNAs 5’-GACCGGCTGGTGAACGAGAAGTT-3’ and 5’-
GACCATCGAGATCAAGTCC-3’ into the mCherry-expressing pSuper and GFP-
expressing pGFP-V-RS vectors, respectively. ABP140-ABD-mCherry was generated by
annealing the primers 5’-ATCCTCCTCCTTG
CTGATGCTTCGAACTTCTTGATCAGGTC GGCCACGC CCATGGTGGCG-3’ (forward)
and 5’-AATTCGCCACCATGGGCGTGGCCGACCTGATCAAGAAGTCGAGAG
CATCAGCAAGGAGG-3’ (reverse), which correspond to the first 17 amino acids
(Lifeact) of ABP140, and inserting the sequence into the N1-mCherry vector (Invitrogen)
using the NcoI restriction site. SEPT9_mCherry was constructed by PCR amplification of
SEPT9_i1 from pET28a-SEPT9_i1 (Bai et al, 2013) using the primers 5’-

61 CGTAAGCTTGCATGAAGAAGT-3’ and 5’-GTAGTCGACCTACATCTCTGGGGC-3’ and
the product was ligated into the HindIII and SalI sites of the pmCherry-C1 vector. Mouse
SEPT2 was inserted into N1-YFP (Spiliotis et al, 2005). Avian paxillin was inserted into
C1-EGFP (West et al, 2001). Mouse myosin IIA-GFP, human α-actinin-1-GFP and
human myosin II RLC-DD-GFP (pEGFP-MRLC1 T18D, S19D) were purchased from
Addgene. The actin-binding domain of α-actinin-1 (amino acids 30-253) was deleted
from α-actinin-1-GFP (Roca-Cusachs et al., 2012). The myosin IIA-N93K-GFP mutant
was generated by site-directed mutagenesis using the primers 5’-
CTCACGTGCCTCAAGGAAGCTTCGGTG-3’ (forward) and 5’-CACCGAAGCTTCCTTG
AGGCACGTGAG-3’ (reverse).
Protein expression and purification.
SEPT2/6/7 was expressed by co-transforming E. coli BL21(DE3) cells with a bicistronic,
kanamycin-resistant pET-28a(+) plasmid that expresses SEPT6 and His-SEPT7 in
tandem, and an ampicillin resistant pET-15b(+) plasmid that expresses untagged SEPT2
(Bai et al., 2013). His-SEPT9_1 was expressed by transforming E. coli BL21(DE3) cells
with pET28a-SEPT9_i1 (Bai et al, 2013). Bacterial cultures of at least OD600 of 0.6 were
induced with 0.5 mM IPTG for 16h at 18C, centrifuged at 5,000 rpm for 5 m at 4ºC, and
resuspended in buffer containing 1% Triton X-100, 50 mM Tris, pH 8.0, 150 mM NaCl,
10% glycerol and 10 mM imidazole. Bacteria were lysed with a French pressure cell at
1,280 psi. Cell lysates were cleared using centrifugation (14,000 x g) for 30min at 4ºC
and the supernatants were passed through Ni-NTA beads by gravity flow. The columns
were washed with 10mL washing buffer (50mM Tris, pH 8.0, 300mM NaCl, 10%
glycerol, 25 mM imidazole) and proteins were eluted with elution buffer (50 mM Tris,
pH8.0, 300 mM NaCl, 10% glycerol, and 250 mM imidazole). Purification of SEP2/6/7

62 was verified by size exclusion chromatography using an AKTA FPLC system (GE
Healthcare). Purified proteins were dialyzed overnight in buffer containing 50mM Tris,
pH 8.0, 150 mM NaCl, and 10 mM imidazole.
Immunoprecipitations and immunoblots.
For immunoprecipitations, equal number of MDCK and SEPT2-YFP MDCK cells were
lysed with buffer containing 50 mM HEPES pH 7.4, 100 mM NaCl, 0.5% Triton X-100, 1
mM PMSF, and protease inhibitors (Millipore) and incubated with anti-GFP antibody
(3E6; Invitrogen) overnight at 4ºC. Protein A beads (Thermo Scientific) were incubated
for 4 h at 4ºC and were stripped with SDS buffer at 95ºC for 10min. Equal volumes were
resolved on a 10% SDS-PAGE gel and transferred to a nitrocellulose membrane using
100 V for 1 h at 4ºC. Membranes were blocked with buffer containing 5% milk and 1%
BSA for 1h at 25ºC. Primary antibodies targeting rabbit anti-SEPT2 (1:5000, Sigma),
rabbit anti-SEPT6 (1:5000, Santa Cruz Biotechnology, Inc.), rabbit anti-SEPT7 (1:8000,
ImmunoBiological Laboratories, Inc.) and rabbit anti-SEPT9 (1:2500, ITG Labs), and
anti- rabbit or mouse secondary antibodies (LiCor) conjugated to infrared dyes were
diluted in buffer containing 2% BSA, 0.1% Tween-20, and 0.025% sodium azide and
sequentially incubated on each membrane prior to scanning with an Odyssey Li-Cor
imaging system.
To test for changes in SEPT9 expression following HGF treatment, MDCK cells
were cultured on collagen-coated plates and stimulated with HGF for the indicated times.
Cell lysates of equal protein concentration were run on a 10% SDS-PAGE gel,
transferred to a nitrocellulose membrane and probed using primary antibodies targeting
rabbit anti-SEPT9 (PTG Labs), mouse anti-actin (1:8000, Sigma) and mouse anti-
GAPDH (1:8000, Abcam). The western blot shown in Fig. 4 A is representative of three

63 different experiments, which were reproduced with no limitations. To quantify SEPT9
overexpression in MDCK cells, untransfected cells and cells that express stably SEPT9-
mCherry at high levels were lysed, processed and blotted as described above (Fig. S2
D).
Immunofluorescence microscopy and quantifications.
Cells were simultaneously fixed and extracted with 4% PFA in warm PBS buffer
containing 0.1% Triton X-100 for 20min, or fixed with 4% PFA in cytoskeleton buffer (10
mM MES pH 6.1, 0.32 M sucrose, 138 mM KCl, 3 mM MgCl2, 2 mM EGTA) for 20 min
and extracted with 0.5% Triton X-100 for 10min (Fig. 3 D). Excess PFA was quenched
with 0.25% ammonium chloride. Prior to antibody incubation, cells were blocked with
PBS containing 2% BSA for 20 min. Primary antibodies to rabbit anti-SEPT2 (N5N; gift
from M. Kinoshita, Nagoya University, Nagoya, Japan), rabbit anti-SEPT6 (Tada et al.,
2007), rabbit anti-SEPT7 (IBL, Inc.), SEPT9 (ITG Labs), mouse anti-paxillin (BD, 349),
rabbit anti-pY118-paxillin (Millipore), rabbit anti-myosin-IIA (Sigma), mouse anti-pSer19-
MLC (Cell Signaling Technologies), and secondary donkey DyLight 488-, 594-, 647-
conjugated F(ab’)2 to mouse or rabbit IgG (Jackson ImmunoResearch Laboratories,
Inc.) were diluted in PBS with 2% BSA and 0.1% Triton X-100. F-actin was visualized
using phalloidin conjugated to Alexa-488 or -647 (Life Technologies). Coverslips were
mounted on slides using Vectashield (Vector Laboratories) or FluorSave (EMD) and
imaged using a confocal laser-scanning microscope (FluoView 1000; Olympus) with a
Plan Apocrhomat 60x/1.42NA objective. Optical sections (0.2 µm thick) were exported to
the Slidebook 5.0 (Intelligent Imaging Innovations), ImageJ or Volocity (Perkin Elmer)
softwares for processing and quantification. Kymographs were generated in Slidebook
by drawing a six pixel wide line down the center of a growing FA marked by paxillin or

64 ABP140-ABD. Super-resolution structured illumination microscopy was performed using
the DeltaVision OMX V4 (GE Healthcare) microscope with a 60x/1.42NA objective and
immersion oil with a refractive index of 1.514. A z-step size of 0.125 µm was used and
images were acquired with sCMOS pco.edge cameras (PCO) and reconstructed in the
softWoRx software.
FA sizes were quantified using masks of paxillin fluorescence intensity
segmentation; the area of individual masks were exported to excel and quantified. FA
distance from the cell edge was measured by converting the phalloidin-stained cell into a
binary image, which was subsequently inverted and transformed into a Euclidean
distance map (Schober et al., 2007). FAs were overlaid on the map and the distance
from the FA center to the cell edge was measured. To assess relative levels of pTyr118
paxillin to total paxillin, confocal images were obtained using identical laser settings
between control and septin-depleted samples. Individual FAs were masked using
intensity segmentation and the ratios were derived from the sum intensities.
Colocalization of septins with actin in the leading cell edge was quantified in
background subtracted images using the Mander’s Coefficient analysis in Slidebook.
Fluorescence intensity of actin as a function of the distance from the cell edge was
quantified from confocal images, which were acquired in during one sitting at identical
laser intensities. Protrusions at the leading cell edge were sampled 2-3 times using a 10
µm x 1 µm line scan drawn perpendicular from the cortex and were averaged and
plotted using custom software written in MATLAB (MathWorks). The number of RSFs
per micron was calculated by drawing a line parallel to the cell edge and counting the
number of radial fibers that intersected with the line. Fluorescence intensity ratios of
pSer19-myosin II light chain to myosin IIA heavy chain (Fig. S3) was quantified by

65 masking 5 µm segments of the TAs parallel to the leading cell edge. In each cell, the
sum intensities of at least six different segments were analyzed.
To visualize 3D cyst structure (Fig. 4 C), gels were briefly treated with
collagenase (Sigma) in PBS at 37ºC and fixed with 4% PFA in PBS with 0.5% Triton X-
100. Gels were incubated with phalloidin for 1 h under gentle agitation and mounted in
Vectashield. Optical sections (1.25 µm thick) were stacked as mass projections. The
number of extensions per cyst and the number of cells per extension were quantified
and plotted. Extensions were defined as protrusions from the main cyst body consisting
of three cells or more. To quantify single cell morphology in 3D (Fig. 4 I), gels were fixed,
stained and mounted using the same method but without the initial collagenase
treatment. The long and short axes were measured in Slidebook to derive the axial ratio
of each cell. To measure the velocity of cell movement in a 3D matrix (Fig. 4 H), time-
lapse optical sections were imported into the Volocity software, which identified, tracked
and quantified the movement of the cell centroid.
Time-lapse imaging.
All live imaging experiments were performed at 37ºC in DME lacking Phenol red
supplemented with 0.5% FBS, 1% PSK, and 20mM HEPES. Widefield microscopy was
performed with an inverted microscope (IX-81; Olympus) equipped with a motorized
stage (ProScanII; Prior), a camera (OrcaR2; Hamamatsu Photonics), a custom built
stage-top chamber and temperature controller (Air-Therm ATX; World Precision
Instruments) and the Slidebook 5.0 software. Fluorescent images were captured every
20s with a Plan Apocrhomat 60x/1.40 NA objective (Olympus). Spinning disk confocal
microscopy was performed at 37ºC using an inverted microscope (IX-71; Olympus)
equipped with a camera (ImagEM; Hamamatsu Photonics), an airstream incubator

66 (LiveCell; Pathology Devices), scan head (CSU10; Yokogawa), and the MetaMorph
software (Molecular Devices). Fluorescent images were captured every 20s with a 60x
1.49 NA objective. TIRF microscopy was performed at 37ºC with the DeltaVision OMX
V4 inverted microscope (GE Healthcare) equipped with 60x/1.49NA TIRF objective
(Olympus), motorized stage, sCMOS pco.edge cameras (PCO), stage-top incubator with
temperature controller and the softWoRx software. 3D cell migration experiments were
performed using a laser scanning confocal microscope (LSM700; Carl Zeiss) equipped
with a stage-top incubator and temperature controller (XL modules; Carl Zeiss), and the
Zen software (Carl Zeiss). One micron optical sections were acquired every two minutes
with a 40x C-Apochromat/1.2 NA water-objective (Carl Zeiss). Supplemental videos
were rendered using the Photoshop software (Adobe).
Quantification of FA dynamics.
FA dynamics were obtained using the stable MDCK-paxillin-GFP cell line and TIRF
microscopy; background subtraction and photobleach correction algorithms in the
Slidebook software were applied to normalize fluorescence intensities. Individual FA
lifetimes were quantified from the length of each adhesion signal in three cells using the
open-source Focal Adhesion Analysis Server (Berginski and Gomez, 2013). FA
assembly and disassembly rates were quantified as previously described (Webb et al.,
2004). In brief, individual FAs were masked in Slidebook and a semilogrithmic plot of
paxillin intensity as a function of time was generated. The rate constants were derived
from the slopes of 45 adhesions from the leading edge (three cells per condition).
Low speed co-sedimentation assay.
Human platelet actin (Cytoskeleton, Inc.) in G-buffer (5mM Tris-HCl, 0.2mM CaCl2) was
polymerized using F-buffer (20 mM HEPES, 100 mM KCl, 1 mM MgCl2) supplemented

67 with 0.5 mM ATP and 4 mM DTT for 1h at 25ºC to generate F-actin. 2 µM recombinant
SEPT2/6/7 and 4 µM recombinant his-tagged SEPT9 were incubated with 2 µM F-actin
for 1h at 25ºC and centrifuged atop a glycerol cushion (20 mM HEPES, 100 mM NaCl, 1
mM MgCl2, 10% glycerol) at 8,000 x g for 20min at 25ºC. Equal volumes of the
supernatant and pellet fractions were resolved using a 7.5% SDS-PAGE gel and stained
with Coomassie brilliant blue.
Electron Microscopy.
Skeletal striated muscle G-actin (2.5 μM) was polymerized in 10 mM HEPES buffer (pH
7.4), 100 mM KCl, 1 mM MgCl2, 0.5 mM DTT, and 0.5 mM ATP for 2 h. Actin filaments
were incubated with 14 μM His-SEPT6 or His-SEPT9 for 15-30 min at 4°C. To visualize
complexes of F-actin with septins, aliquots (10 µl) were applied to glow-discharged
carbon-coated grids (Electron Microscopy Sciences) and stained with 2% uranyl acetate.
The grids were examined in a JEOL 1200EXII transmission electron microscope (TEM)
(JEOL Inc, Peabody, MA) under regular-dose conditions at an accelerating voltage of 70
keV. The images shown in Fig. 3 F and G are representative of similar results obtained
from three independent experiments.
Transmigration Assays.
786-O cells were transfected with plasmids encoding for the indicated constructs for 48h,
trypsinized and plated on collagen-coated transwell filters (8 µm pore) for 12h. Filters
were fixed with 4% PFA in PBS and stained for nuclei with DAPI. Apical and basolateral
sections were imaged at 20x magnification using confocal microscopy. The number of
transfected cells in each section were quantified and plotted in excel.
Statistical Analyses
Data sets were analyzed using R and the Excel software. Normal distribution and equal
variance were assessed using the Shapiro and Levene tests, respectively. Data sets

68 with normal distribution and variance were analyzed using an unpaired student’s t-test.
Data sets that did not exhibit normal distributions or variance were further analyzed
using the Kruskal-Wallis non-parametric ANOVA. All values represent the mean +/-
SEM.
Results and Discussion
A network of septin filaments interfaces with RSF and TA stress fibers in the
leading lamella of renal epithelia.
To examine septin functions in the motility of renal epithelia, we used the MDCK
model of partial EMT, which allows the study of cell migration in 2D and 3D matrices
after stimulation with the hepatocyte growth factor (HGF) (Zegers et al., 2003).
Treatment of MDCKs with HGF disrupted cell-cell adhesions and induced scattering and
development of front-rear cell polarity as previously shown (de Rooij et al., 2005).
Resembling the stress fiber organization of migratory cells, the lamellae of fan-shaped
MDCK cells contained radial stress fibers that extended dorsally from FAs and
connected with transverse arc (TA) filaments (Fig. 1 A; arrows). Staining for endogenous
SEPT2, SEPT6, SEPT7 and SEPT9 revealed an extensive network of septin filaments
that localized at the interface of TA filaments and RSFs (Fig. 1 A, Fig. S1).
Colocalization and co-immunoprecipitation of SEPT2, SEPT6, SEPT7 and SEPT9 (Fig.
S2, A and B) indicated that these septin filaments consist of SEPT2/6/7/9 complexes,
which are the basic structural units of mammalian septin filaments (Kim et al., 2011;
Sellin et al., 2011b). Thus, we used SEPT2 as a marker for the lamellar network of
septin fibers, and SEPT2 and SEPT9 shRNAs were interchangeably applied to perturb
the localization and function of septins; knock-down of SEPT2 or SEPT9 affected the

69 expression and localization of both of these septins as well as SEPT6 and SEPT7 (Fig.
S2, C-D) as reported previously (Kim et al., 2011).
To determine the precise localization of septins with respect to the lamellar
network of stress fibers and FAs, we used confocal microscopy in combination with 3D
image reconstructions and super-resolution structured illumination microscopy (SIM).
Septins localized mainly on and between the curved actin bundles of the TA network and
on a subset of RSFs (Fig. 1 B-E; Video 1). Septins were present on the dorsal ends of
80% ± 2% RSFs (n = 72; 12 cells) that connected with the TA (Fig. 1, B and D). On the
quartile end of these RSFs, 1.9 ± 0.16 µm away from FAs (n = 58; 12 cells), septins
interdigitated with actin filaments as they flared from their FA-anchored bundle toward
the TA (Fig. 1, E; panel 1). Time-lapse microscopy showed that septins are recruited to
the distal ends of RSFs after the assembly of nascent FAs (Fig. 1 F and G). In live cells,
septins persisted on RSFs during their interactions with the TA, spanning segments of
both RSF and TA stress fibers (Fig. 1 H). Taken together, these data indicate that
septins localize at the interface of the contractile TA and RSFs, and thus, may be part of
the actomyosin machinery that drives the growth and turnover of nascent FAs in motile
renal epithelia.
Septins are required for the stabilization of nascent FAs and the organization of
the lamellar stress fiber network.
To test whether septins affect the formation and dynamics of FAs, HGF-treated
MDCK epithelia were depleted of SEPT2 or SEPT9. Septin knock-down with two
different SEPT2 shRNAs and a SEPT9 shRNA increased the number of FAs, which
were smaller and localized closer to the cell edge compared to control cells (Fig. 2 A-D).
Time-lapse imaging of paxillin-GFP showed that septin knock-down reduces the life time

70 of nascent FAs (Fig. 2E), which fail to grow significantly in size and do not move closer
to the cell body (Video 2). Analysis of the kinetics of paxillin-GFP fluorescence revealed
no differences in the rates of FA assembly and disassembly (Fig. 2 F), but septin
depletion abolished the stabilization phase of the FA life cycle (Fig. 2 G). Consistent with
lack of FA stabilization, which is characterized by dephosphorylation of paxillin’s Tyr118
(Zaidel-Bar et al., 2007), septin depletion increased the levels of pTyr118-paxillin relative
to total paxillin in MDCK and 786-0 RCC cells (Fig. 2 H-I).
Maturation of nascent FAs is mediated by stress fibers, which exert mechanical
forces and provide a template for the growth of cell-ECM adhesions (Burridge and
Wittchen, 2013; Gardel et al., 2010; Vicente-Manzanares et al., 2009). While myosin-
mediated tension is thought as the main trigger of FA maturation, recent work shows that
myosin II-dependent tension is necessary but not sufficient for FA maturation, which
requires actin-binding proteins that mediate the formation and maintenance of the
lamellar actin network (Choi et al., 2008; Oakes et al., 2012; Roca-Cusachs et al., 2013;
Stricker et al., 2013).
Next, we examined the organization and dynamics of the lamellar actin network
in septin-depleted cells. Strikingly, the lamellar actin meshwork was thin and devoid of
an organized TA (Fig. 2 J). F-actin was significantly reduced in the lamellar regions of
the cell (5-10 µm from the cell edge; Fig. 2 K), and the length and density of RSFs
decreased by ~50% (Fig. 2 L and M). In live SEPT2-depleted cells, RSFs were short-
lived, dissipating quickly after their formation, while in control cells the actin fluorescence
of RSFs persisted and increased over time (Fig. 2 N; Video 3). Taken together with the
localization of septins to the TA and distal ends of RSFs, these data suggest that septins

71 are required for the stabilization and maturation of nascent FAs by maintaining the
organization and possibly contractility of the lamellar stress fiber network.
Septins function as actin cross-linking proteins in FA maturation.
Because SEPT2 binds directly myosin II and this interaction is posited to facilitate the
activation of the myosin RLC (Joo et al., 2007), we sought to determine whether septins
affect FA maturation through their role in the organization of the lamellar actin network or
by a possible involvement in myosin-mediated tension. To distinguish between these
possibilities, we tested first whether septins affect the levels of activated myosin RLC on
the contractile TA, and second whether the actin bundling protein α-actinin-1 or the
constitutively active di-phosphomimetic myosin RLC-DD can rescue septin loss of
function. Analysis of the fluorescence intensity ratio of the phosphorylated pSer19
myosin RLC to total myosin IIA showed no difference between control and SEPT2-
depleted cells (Fig. S3 A and B). In agreement, RLC-DD did not rescue FA size and
number in SEPT2-depleted cells (Fig. 3 A-C). In contrast, α-actinin-1 restored the
number and size of FAs to near control levels (Fig. 3 A-C). This effect of α-actinin-1 was
also accompanied by a rescue of the lamellar actin network in septin-depleted cells,
which now contained a network of TA filaments and RSFs (Fig. 3 D). Notably, α-actinin-
1-∆ABD, which lacks its actin-binding domain, did not rescue FAs (Fig. 3 B and C), while
the actin-binding motor-dead myosin II-N93K did (Fig. S3 C-E). Thus, loss of septin
function in FA maturation is specifically rescued by the actin-binding and cross-linking
properties of α-actinin-1 and myosin IIA heavy chain.

72 Previous studies have reported that septin complexes comprising of SEPT2,
SEPT6 and SEPT7 do not bind actin filaments directly (Kinoshita et al., 2002), but new
work shows that copolymerization of actin with SEPT2/6/7 results in circular actin
filaments (Mavrakis et al., 2014). Because in the lamellar regions of motile MDCK
epithelia, septins interact with pre-assembled RSFs and TA filaments, we examined
whether septins have the ability to cross-link pre-polymerized actin filaments using low
speed actin sedimentation assays and negative stain EM. We found that SEPT9 directly
cross-links pre-polymerized actin filaments into bundles, while SEPT2/6/7 does not (Fig.
3 E-G). These results suggest that septins promote the maturation of nascent FAs by
cross-linking the lamellar stress fibers of motile epithelia.
SEPT9 is up-regulated in renal epithelia undergoing EMT and enhances renal cell
migration.
Given that septins are over-expressed in renal cell carcinomas and other cancers
(Connolly et al., 2011a; Craven et al., 2006a; Dolat et al., 2014a), we asked whether
SEPT9 is upregulated during renal EMT. Treatment of MDCK cells with HGF increased
the levels of SEPT9 relative to actin, while actin levels did not change (Fig. 4 A and B).
To test if increased SEPT9 expression promotes cell motility, we established stable
MDCK cell lines expressing SEPT9-mCherry at low endogenous-like and high levels of
expression, and cultured these cells in a 3D ECM, which enables the morphogenesis of
renal cysts. Treatment of the 3D MDCK cysts with HGF causes tubulogenesis, a process
that mimics the collective and invasive migration of metastatic tumors (Debnath and
Brugge, 2005). Over-expression of SEPT9 doubled the number of multicellular
extensions that protruded from the main cyst and the extensions were longer and
contained more cells (Fig. 4 C and D). In contrast, treatment of cysts with

73 forchlorfenuron (FCF), a septin-targeting compound (Hu et al., 2008), reduced
significantly the number of HGF-induced extensions (Fig. 4 E). Moreover, dissociated
single MDCK cells migrated slower in 3D and were less elongated and fibroblast-like
after SEPT9 depletion (Fig. 4 H and I; Video 4). To corroborate the effects of SEPT9
over-expression on renal cell motility, we performed a transmigration assay using the
786-0 renal carcinoma cells. SEPT9 over-expression increased the migration of 786-0
cells along a serum gradient, while SEPT9 knock-down decreased migration (Fig. 4 F
and G). These results, therefore, indicate that SEPT9 promotes the mesenchymal-like
migration of renal epithelia.
Previous studies have implicated septins in the amoeboid motility of T-cells and
neuronal migration (Shinoda et al., 2010; Tooley et al., 2009), which are mechanistically
different from the stress fiber-driven mode of mesenchymal migration. Septins have
been reported to colocalize with stress fibers in fibroblasts (Kinoshita et al., 1997;
Kremer et al., 2007; Schmidt and Nichols, 2004b), but this interaction has been
considered to be indirect (Joo et al., 2007; Kinoshita et al., 2002), and the precise
function(s) of septins in epithelial and mesenchymal motility are unknown (Chacko et al.,
2005; Connolly et al., 2011b). Here, we have found that septins are required for the
stabilization of nascent FAs. Our results indicate that septins cross-link stress fibers,
maintaining the organization of the lamellar actin network, and thereby, promoting the
transmission of forces from the contractile TA to FAs. This septin function relies on the
ability of SEPT9 to crosslink preassembled actin filaments. Thus, septin upregulation
might be part of the EMT program that retools the actin cytoskeleton for a migratory
mesenchymal-like phenotype.

74

75 Figure 5.1. Septin filaments interface with RSF and TA stress fibers in the leading
lamella of renal epithelia. (A) Confocal images of SEPT2 (green), actin stress fibers
(phalloidin; red) and FAs (paxillin; white) in HGF-treated (16 h) MDCK epithelia. White
arrows point to RSFs that connect with the TA. (B) 3D rendering of the lamellar region
outlined in panel A. Arrows point to SEPT2 fibers that localize on FA-anchored RSFs.
Scale bars, 1 µm. (C) Quantification of SEPT2 colocalization with F-actin (green) and
vice versa (red). (D) SIM images show ventral and dorsal optical sections from the
lamellar region of an HGF-treated MDCK cell. Arrowhead points to SEPT9 localization
on the dorsal segment of an RSF that connects with the TA. (E) SIM images show an
HGF-treated MDCK cell stained for F-actin (phalloidin; red), SEPT2 (green) and paxillin
(white). An FA-anchored RSF (1) and TA filaments (2) are shown in higher
magnification. Arrowheads point to SEPT2 localization on and between actin filaments.
(F) Still frames show FA formation (paxillin-mCherry; red) and subsequent recruitment of
SEPT2-YFP (green). (G) Total internal reflection fluorescence microscopy kymograph
shows recruitment of SEPT2-YFP (green) to a nascent RSF (ABP140-ABD-mCherry,
red). (H) Inverted monochrome images show frames from spinning disk confocal time-
lapse microscopy of HGF-treated MDCK cells expressing ABP140-ABD-mCherry and
SEPT2-YFP. A subset of TA filaments and an RSF were pseudo-colored red and super-
imposed onto the inverted monochrome image of the SEPT2-YFP channel. SEPT2-YFP
elements recruited to the junction of the RSF with the TA were pseudo-colored green.
Overlay image shows the pseudocolored stress fibers and SEPT2 outlined in the
inverted monochrome frames. Arrow points to the RSF end and its junction point with the
TA.

76

77 Figure 5.2. Septins regulate the organization of the lamellar actin network and are
required for the stabilization of nascent FAs. (A) Confocal images of MDCK cells,
which were transfected with mCherry-expressing plasmids that encode for control or
SEPT2 shRNAs, and stained for paxillin after treatment with HGF. Red dashed line
highlights the cell edge. (B and C) Graphs show FA size and number from MDCK cells
(n = 20) treated with control, SEPT2 and SEPT9 shRNAs. (D) Graph shows FA distance
from the cell edge of MDCK cells (n = 10) treated with control (n = 903 FAs), SEPT2 # 1
(n = 749 FAs), SEPT2 # 2 (n = 512 FAs) and SEPT9 shRNAs (n = 840 FAs). (E) Graph
shows FA lifetime in three different MDCK cells treated with control (n = 6,066 FAs) and
SEPT2 shRNAs (n = 10,528 FAs). (F) Graph shows the rates of FA (n = 45) assembly
and disassembly in cells treated with control and SEPT2 shRNAs. (G) Representative
profiles of the kinetics of paxillin-GFP fluorescence in MDCK cells treated with control
and SEPT2 shRNAs. (H) Confocal images show 786-O cells stained for total paxillin
(red) and phosphorylated pY118-paxillin (green). Insets show GFP expression from
plasmids encoding for control and SEPT2 shRNAs. (I) Bar graphs show the ratio of the
fluorescence intensity of pY118-paxillin to total paxillin in MDCK cells (n = 10) treated
with control (n = 740 FAs) and SEPT2 shRNAs (n = 962 FAs), and 786-O cells (n = 10)
treated with control (n = 757 FAs) and SEPT2 shRNAs (n = 602 FAs). (J) Confocal
images of control and SEPT2-depleted MDCK cells stained for F-actin (phalloidin; red)
and paxillin (white). Insets show GFP expression from plasmids encoding for control and
SEPT2 shRNAs. (K) Plot shows line scans of phalloidin fluorescence across the leading
edges of MDCK cells (n = 13) treated with control (n = 54 line scans) and SEPT2
shRNAs (n = 56 line scans). (L and M) Graphs show the mean number of RSFs per
lamellar length (L) and mean length of RSFs (M) in MDCK cells treated with control (n =
43) and SEPT2 shRNAs (n = 72). (N) Inverted monochrome frames show ABP140-ABD-

78 mCherry from total internal reflection fluorescence (TIRF) microscopy of MDCK cells
treated with control or SEPT2 shRNAs and HGF. Arrows point to RSFs that persist and
grow overtime in control cells, and stress fibers that dissipate in SEPT2-depleted cells.
79

80 Figure 5.3. SEPT9 functions as an actin cross-linking protein in FA maturation. (A)
Confocal images of paxillin-stained MDCK cells transfected with mCherry-expressing
plasmids that encode for control or SEPT2 shRNAs, and RLC-DD-GFP, α-actinin-GFP
or GFP vectors. Scale bars, 10 µm. (B and C) Graphs show FA size (B) and number per
µm2 (C) in control cells expressing GFP (n = 1,650 FAs; 25 cells), and SEPT2 knock-
down cells expressing GFP (n = 1,567 FAs; 24 cells), α-actinin-1-GFP (n = 1,556 FAs;
20 cells), RLC-DD-GFP (n = 1,962 FAs; 18 cells) and α-actinin-1-∆ABD-GFP (n = 1,962
FAs; 20 cells). (D) Confocal images of MDCK cells transfected with mCherry-expressing
plasmids encoding for control or SEPT2 shRNAs, and α-actinin-1-GFP or GFP vectors.
Cells were stained for F-actin (phalloidin). Scale bars, 10 µm. (E) Coomassie-stained gel
shows equal volumes of supernatant (S) and pellet (P) fractions from low speed
sedimentation of pre-polymerized actin filaments in the presence of recombinant
SEPT2/6/7 and SEPT9. (F-G) Negative stain EM images of actin filaments in the
absence and presence of recombinant SEPT6 or SEPT9. Outlined regions (F) are
shown in higher magnification in G. Scale bars, 0.5 µm.
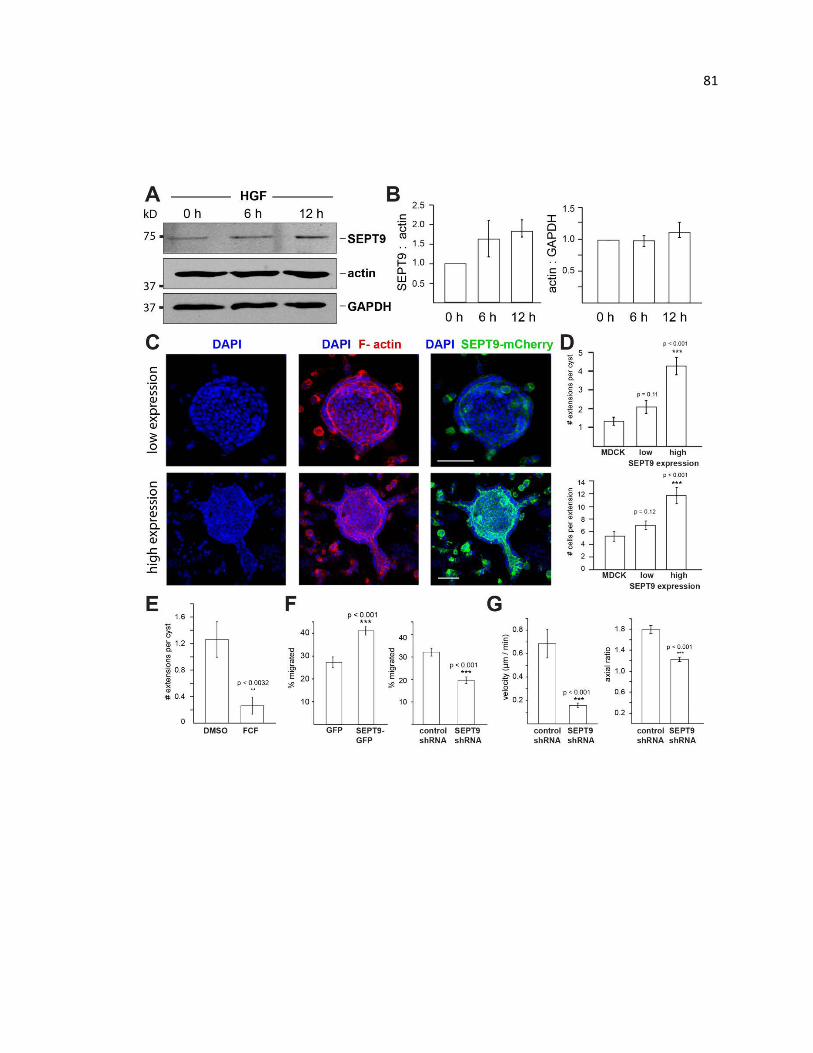
81

82 Figure 5.4. SEPT9 promotes the motility of renal epithelia during EMT. (A) Western
blot of HGF-treated MDCK cell lysates probed for SEPT9, actin and GAPDH. (B)
Graphs show the median, lowest and highest (error bars) ratio values of the SEPT9,
actin and GAPDH band intensities from three independent experiments. Values were
normalized to the protein band intensity ratios at 0 h of HGF treatment. (C) Maximum
intensity projections from 3D confocal microscopy images of HGF-treated renal cysts of
stable MDCK cells expressing SEPT9-mCherry (green) at low and high levels of
expression. 3D renal cysts were grown in collagen and stained for nuclei (DAPI; blue)
and F-actin (phalloidin; red). (D) Graphs show number of extensions per MDCK cyst (n =
23) and number of cells per extension from untransfected (n = 18), low- (n = 35) and
high-expressing (n = 51) MDCK cysts (n = 15). (E) Mean number of extensions per cyst
after treatment with DMSO (n = 23) and FCF (n = 19). (F and G) Graphs show the mean
percentage of transmigrated 786-0 cells expressing GFP and SEPT9-GFP (F), and
control and SEPT9 shRNAs from 20 different areas of a transwell filter. (H and I) Graphs
show the mean velocity (H; n = 16) and axial ratio of the long to short axes (I; n =31) of
MDCK cells migrating in a 3D matrix in the presence of HGF after transfection with
control or SEPT9 shRNAs.

83

84 Figure 5.5. Lamellar septin fibers are composed of SEPT2/6/7/9. (A-C) Confocal
images of MDCK cells stained for paxillin (white), F-actin (phalloidin; red) and
endogenous SEPT7, SEPT9 and SEPT6 (green). Outlined lamellar regions (dashed
rectangles) are shown in higher magnification. (D-F) Fluorescence images show MDCK
cells that expresses SEPT9-mCherry at endogenous-like levels after staining with
antibodies against SEPT2, SEPT6 and SEPT7. Scale bars, 10 µm. (G) Cell lysates from
a stable cell line that expresses SEPT2-YFP at subendogenous levels (see relative
intensity of SEPT2-YFP and SEPT2 bands in the “input” lanes) and untransfected
MDCKs were incubated with mouse anti-GFP antibody and immunoprecipitates were
blotted for rabbit antibodies against SEPT2, SEPT6, SEPT7 and SEPT9.
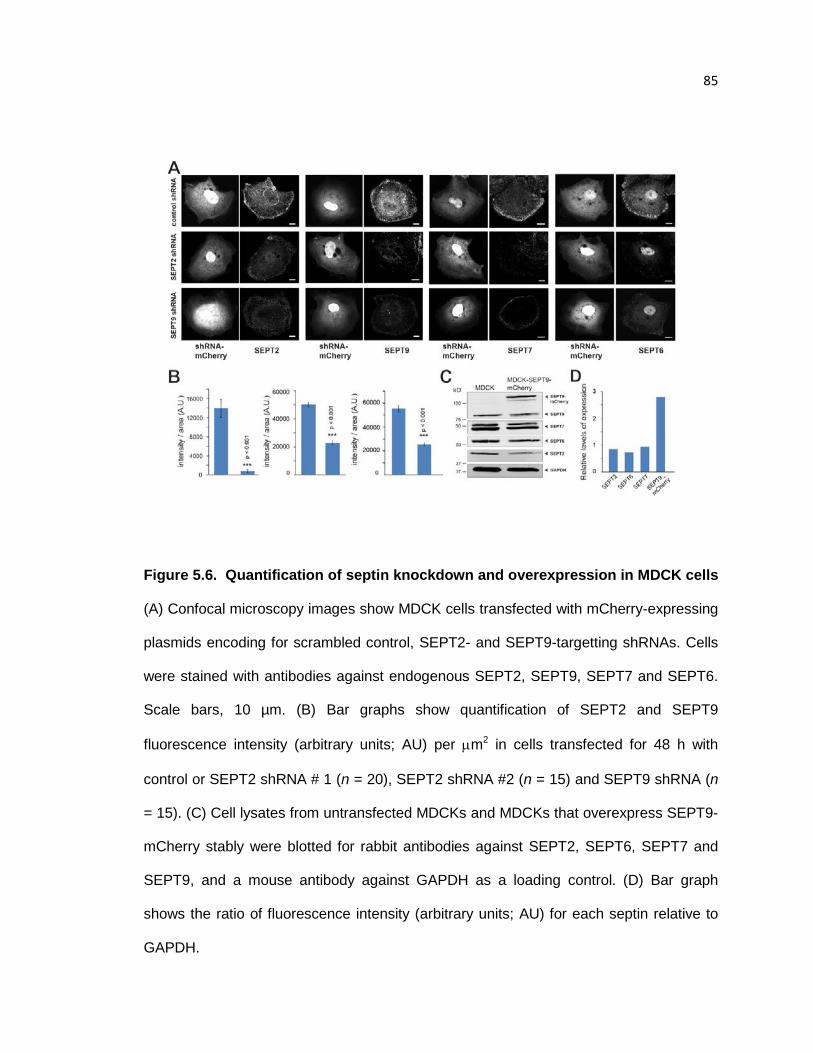
85
Figure 5.6. Quantification of septin knockdown and overexpression in MDCK cells
(A) Confocal microscopy images show MDCK cells transfected with mCherry-expressing
plasmids encoding for scrambled control, SEPT2- and SEPT9-targetting shRNAs. Cells
were stained with antibodies against endogenous SEPT2, SEPT9, SEPT7 and SEPT6.
Scale bars, 10 µm. (B) Bar graphs show quantification of SEPT2 and SEPT9
fluorescence intensity (arbitrary units; AU) per µm2 in cells transfected for 48 h with
control or SEPT2 shRNA # 1 (n = 20), SEPT2 shRNA #2 (n = 15) and SEPT9 shRNA (n
= 15). (C) Cell lysates from untransfected MDCKs and MDCKs that overexpress SEPT9-
mCherry stably were blotted for rabbit antibodies against SEPT2, SEPT6, SEPT7 and
SEPT9, and a mouse antibody against GAPDH as a loading control. (D) Bar graph
shows the ratio of fluorescence intensity (arbitrary units; AU) for each septin relative to
GAPDH.
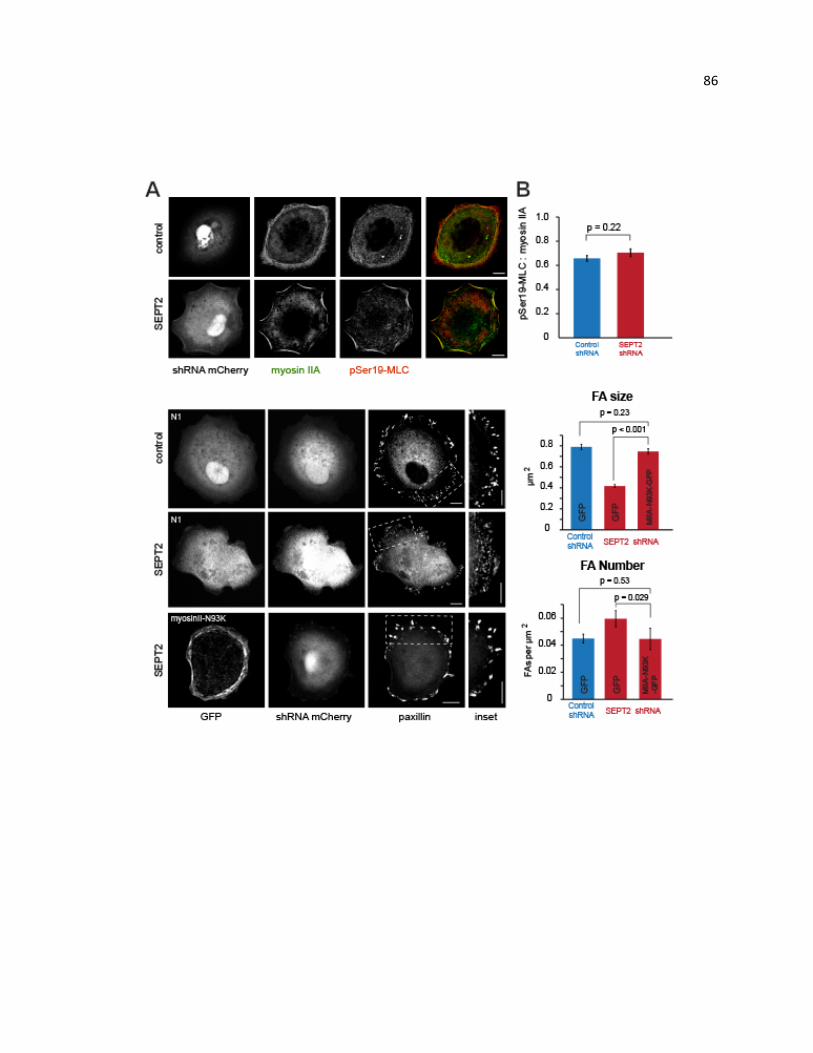
86

87 Figure 5.7. b. (A) Confocal microscopy images show MDCK cells treated SEPT2 and
control shRNAs and stained for myosin-IIA heavy chain (red) and phosphorylated Ser19-
myosin II regulatory light chain (green). Scale bars, 10 µm. (B) Bar graph shows
quantification of the ratio of pSer-myosin II light chain to myosin IIA heavy chain
fluorescence from 5 µm-long myosin filament segments, which were blindly selected
from the lamellar regions of cells treated with control (n = 93; 12 cells) and SEPT2 (n =
74; 11 cells) shRNAs. (C) Confocal microscopy images of paxillin-stained MDCK cells
transfected with mCherry-expressing plasmids that encode for control or SEPT2
shRNAs, and GFP or the GFP-tagged non-muscle myosin IIA heavy chain mutant N93K.
Scale bars, 10 µm. (D and E) Bar graph shows FA size (D) and number per µm2 (E) in
control cells (n = 16) expressing GFP (n = 1,722 FAs), and SEPT2 knock-down cells (n =
16) expressing GFP (n = 1,515 FAs) and myosin IIA N93K-GFP (n = 1,139 FAs).

88 CHAPTER 6. Septin 9 exhibits polymorphic binding to F-actin and inhibits
myosin and cofilin activity
Abstract
Septins are a highly conserved family of proteins in eukaryotes that is recognized
as a novel component of the cytoskeleton. Septin 9 (SEPT9) interacts directly with actin
filaments and functions as an actin stress fiber cross-linking protein that promotes the
maturation of nascent focal adhesions and cell migration. However, the molecular details
of how SEPT9 interacts with F-actin remain unknown. Here, we use electron microscopy
and image analysis to show that SEPT9 binds to F-actin in a highly polymorphic fashion.
We demonstrate that the basic domain (B-domain) of the N-terminal tail of SEPT9 is
responsible for actin cross-linking, while the GTP-binding domain (G-domain) does not
bundle F-actin. We show that the B-domain of SEPT9 binds to three sites on F-actin,
and the two of these sites overlap with the binding regions of myosin and cofilin. SEPT9
inhibits actin-dependent ATPase activity of myosin and competes with the weakly-bound
state of myosin for binding to F-actin. At the same time, SEPT9 significantly reduces the
extent of F-actin depolymerization by cofilin. Taken together, these data indicate that
SEPT9 protects actin filaments from depolymerization by cofilin and myosin, and
suggest a mechanism by which SEPT9 could maintain the integrity of growing and
contracting actin filaments.
Introduction
Septins were first discovered in the budding yeast as genes that are crucial for
cell division (Hartwell, 1971). Since the discovery of septins in yeast, proteins with
homologous sequences have been found in almost all eukaryotic cells. The number of
septin genes per organism is variable and ranges from two isoforms in Caenorhabditis

89 elegans to 13 isoforms in humans (Russell and Hall, 2011). Based on sequence
similarity, human septins are classified into four homology groups named after their
founding members: SEPT2, SEPT3, SEPT6 and SEPT7. In this manuscript we focus on
SEPT9 which belongs to the SEPT3 subgroup and has a unique N-terminal domain
(NTD) consisting of a basic domain (B-domain) and an acidic domain (A-domain) (Fig.
1a) (Bai et al., 2013). All septins have a GTP-binding domain (G-domain), which is
evolutionarily and structurally related to the Ras GTPases (Leipe et al., 2002). Unlike the
monomeric small GTPases, septins form polymers that consist of non-polar oligomeric
complexes(Bertin et al., 2008; Sirajuddin et al., 2007)
Septins have been recognized as the fourth component of the cytoskeleton
because of their filamentous appearance and their interdependence with actin filaments
and microtubules. The N-terminal domain of SEPT9 has been shown to bind and bundle
microtubules by interacting with the acidic C-terminal tails of β-tubulin (Bai et al., 2013).
Septins have been shown to bind actin filaments indirectly via anillin or the motor protein
myosin II(Joo et al., 2007; Kinoshita, 2002). Recent data, however, demonstrated that
Drosophila septin complexes (Sep1-Sep2-pnut) can directly interact with actin and
cross-link actin filaments into curved bundles, which are critical for the organization and
function of contractile actin rings during Drosophila embryonic cleavage (Mavrakis et al.,
2014). Moreover, we recently found that human SEPT9 directly binds and bundles F-
actin, and that cross-linking of actin filaments by SEPT9 promotes focal adhesion
maturation and epithelial cell motility (Dolat et al., 2014b).
Despite the new evidence that septins can directly interact with the actin
cytoskeleton, the molecular details of these interactions and how septins interact with
the surface of the actin filament remain unknown. Here we use electron microscopy,

90 image analysis and co-sedimentation assays to identify how SEPT9 interacts with F-
actin. Our data indicate that the B-domain of SEPT9 is responsible for its F-actin
bundling activity, while the G-domain of SEPT9 does not bundle actin filaments. We
demonstrate that SEPT9 binds to three sites on the surface of F-actin that are commonly
bound by other actin binding proteins. Importantly, two sites overlap with the regions of
the actin molecule involved in the binding of myosin and cofilin. As predicted by the
structural data, SEPT9 inhibits F-actin depolymerization by cofilin, which severs actin
filaments, and reduces the actin-dependent ATPase activity of myosin. Our results
suggest that SEPT9 could protect growing actin filaments from the combined
depolymerizing activity of cofilin and myosin during actin filament assembly and
contraction.
Materials and Methods
Protein expression and purification. G-Mg2+-actin was prepared as described (Orlova
and Egelman, 1995). SEPT9 constructs were prepared as follows. E. Coli BL21 (DE3)
cells were transformed with SEPT9_i1, SEPT9-NTD, SEPT9-G, SEPT9-A, or SEPT9-B
in kanamycin-resistant pET-28a (Bai et al., 2013). Plasmids expressing His-tagged
SEPT9-N (aa 1–283 of SEPT9_i1), SEPT9-B (aa 1–142 of SEPT9_i1), SEPT9-A (aa
143–283 of SEPT9_i1), SEPT9-G (aa 284–586 of SEPT9_i1) were made with the
QuikChange II Site-Directed Mutagenesis kit (Agilent Technologies) using the pET-
SEPT9_v1 and pET-SEPT9_v3 plasmids. Bacterial cultures of OD600 were induced with
0.5 mM IPTG for 16h at 18°C, collected by centrifugation and resuspended in lysis buffer
containing 50 mM Tris, pH 8.0, 1% Triton X-100, 150 mM NaCl, 10% glycerol, and 10
mM imidazole. Bacteria were lysed using a French pressure cell at 1,280 psi, and
lysates were cleared using centrifugation (14,000 g) for 30 min at 4°C. Columns

91 containing Ni-NTA beads were equilibrated with lysis buffer prior to passing
supernatants through by gravity flow. The beads were washed with buffer containing
50mM Tris, pH 8.0, 300 mM NaCl, 10% glycerol, and 10 mM imidazole, and proteins
were eluted with buffer containing 50mM Tris, pH 8.0, 300 mM NaCl, 10% glycerol, and
250 mM imidazole.
MyosinV-S1 heavy chain, 907 amino acid residues including 6IQ domains was
expressed in a baculovirus/Sf9 cell system. The proteins were purified using FLAG-
affinity chromatography and then concentrated and fractionated on a MonoQ ion-
exchange column with a linear gradient of 0.1−0.5 M NaCl in 10 mM MOPS, 0.1 mM
EGTA, 3 mM NaN3, and 1 mM DTT, pH 7.2, using HPLC. The concentration of myosinV-
S1 was determined from the A280 after correction for light scattering (A280−1.5A320).
The extinction coefficient was calculated to be 1.1 × 105 M-1 cm-1 from the number of
tyrosine and tryptophan residues and their molar extinction coefficients: ε = (n = 10)trp
× 5690 + (n = 42)tyr × 1280. All preparations were analyzed by SDS protein gel
electrophoresis and by active site titration with deac-aminoATP. Cofilin-1 was a
generous gift from Dr. Emil Reisler (UCLA).
Electron microscopy of negatively stained SEPT9 domains. G-actin was
polymerized for 1–2 hours in F-buffer (10mM HEPES, pH 7.4, 100mM KCl, 1mM MgCl2,
0.5mM ATP). For EM samples shown in Fig. 1c-g, 2 µM F-actin was incubated for 2 min
in tube with 34 µM NTD, B-domain, A-domain, or G-domain. Samples were blotted and
negatively stained with 2% (wt/vol) uranyl acetate. To obtain single actin filaments
decorated with SEPT9 domains was 2 µM F-actin was incubated for 1 min on carbon
coated glow discharged grids in a humidified chamber, gently blotted and incubated
again with 34 µM NTD, B-domain, A-domain, or G-domain. Samples were negatively

92 stained with 2% (wt/vol) uranyl acetate. A JEM-2100F (JEOL) Field Emission
Transmission Electron Microscope, equipped with 11-megapixel Gatan SC1000 ORIUS
CCD camera, was used at an accelerating voltage of 200 kV, and a nominal
magnification of 60,000x to record micrographs at a raster of 1.58 Å/pixel.
Electron microscopy of SEPT9 in presence of cofilin-1 and SEPT9. F-actin (2 µM)
was mixed with 10 µM of cofilin-1, or preincubated with 10 µM of SEPT9 before addition
of 10 µM of cofilin-1. Samples were left at room temperature and analyzed by TEM at 2
and 30 min time points.
Image analysis. The SPIDER software package (Galkin et al., 2001) was used for most
image processing, while the EMAN package (Galkin et al., 2003) was used to extract
filament images from micrographs. Images were corrected for the contrast transfer
function (CTF) using theoretical CTF obtained with the CTFIND software (Frank, 1984)
and decimated to 3.16 Å/pixel scale. From these CTF-corrected images segments (each
120 pixels long) of F-actin decorated with the NTD (n=8,193), B-domain (n=2,708), A-
domain (n=5,435) constructs were extracted. Segments of filament decorated with the
NTD were reconstructed using IHRSR approach (Ludtke et al., 1999). The resultant
reconstruction showed three additional densities bound to F-actin - one at the front
interface between SD1 and SD2 of two adjacent actin protomers, the other one at the
side of SD1, and one on the top of SD4 (Fig. S1). Three model volumes were created
by using pseudo-atomic model of F-actin (Mindell and Grigorieff, 2003) having model
density attached to the first site, second site, or third site. These volumes, along with a
naked F-actin volume, were projected onto 120 × 120-pixel images with an azimuthal
rotational increment of 4°, and the resultant 360 reference projections (4 × 90) were
cross-correlated with the 8,193 NTD-decorated actin filament segments. Each of the four

93 classes was reconstructed starting from the two independent starting models (Fig. S2).
The naked class (n=4,222) yielded a symmetry of 166.6°/27.3 Å, the mode1 class
(n=1,590) yielded a symmetry of 166.7°/27.2 Å, the mode 2 class (n=1,305) converged
to 166.1°/27.2 Å, while a set of images classified as mode 3 (n=1,079) reached a stable
solution having 167.0°/27.2 Å symmetry values. The same sorting procedure was
applied to actin filaments decorated with the B-domain. The naked segments extracted
from the filaments decorated with the B-domain (n=1,571) converged to a 166.6°/27.2 Å
solution, the mode1 class from that set (n=473) yielded 166.3°/27.1 Å, the mode 2 class
(n=332) converged to a symmetry of 166.1°/27.3 Å, while the mode 3 class (n=332)
yielded a symmetry of 166.5°/27.2 Å. Segments of filaments incubated with the A-
domain were mostly naked actin (n=4,935) with only 218 segments assigned to mode 1
class, 135 segments assigned to the mode 2 class, and 147 segments assigned to the
mode 3 class. Neither of these classes yielded reasonable 3D reconstruction (data not
shown). The naked class converged to the stable solution of 166.5°/27.1 Å. Fourier Shell
Correlation approach was used to determine the resolution of the 3D-reconstructions.
Each image set was split into two non-overlapping subsets, and these subsets were
reconstructed independently. Resultant volumes were used for resolution determination.
Using 0.5 criterion all maps yielded 27-28 Å resolution (Fig. S7). All the 3D-
reconstructions shown in the paper were filtered to the measured resolution. The
Chimera software (Egelman, 2000) was used to dock a protomer from the atomic model
of F-actin into the reconstruction manually. Figures 2, 3 and 4 were produced using the
Chimera software.
Actin activated steady-state ATPase experiments. Actin activated steady-state
experiments were done by the NADH coupled assay at 25 °C. 20 nM Myosin V-S16IQ
was mixed with 10 µM actin and increasing concentrations of SEPT9 A- domain , B-

94 domain or full length SEPT9 in a buffer containing 10 mM MOPS (pH 7.0), 2 mM MgCl2,
25 mM KCl, 0.15 mM EGTA, 2 mM ATP, 40 units/ml lactate dehydrogenase, 200
units/ml pyruvate kinase, 1 mM phosphoenolpyruvate, and 200 μM NADH (Fujii et al.,
2010). Changes in NADH absorption at 340 nm (€340 = 6220 M-1cm-1) were followed in
a Beckman DU640 spectrophotometer. The ATPase activity of blanks containing actin
but no myosin was subtracted from the actomyosin data. The data was fit to the equation
F (x) = Vmax/1+x/Ki .
Low-speed actin pelleting assays. G-actin in G buffer (5 mM Tris-HCl and 0.2 mM
CaCl2) was polymerized in buffer containing 20 mM Hepes, pH 7.4, 100 mM KCl, and 1
mM MgCl2 supplemented with 0.2 mM ATP and 4 mM DTT for 1 h at 25°C. In Figure 1, 8
μM of recombinant SEPT9-N, SEPT9-G, SEPT9-A, or SEPT9-B were incubated with 2
μM F-actin for 1 h at 25°C. In Supplemental Figure 6, increasing concentrations of full-
length SEPT9 or SEPT9-B were incubated with 2 μM F-actin for 1 h at 25°C. Reactions
were subsequently centrifuged on top of a glycerol cushion (20 mM Hepes, 100 mM
NaCl, 1 mM MgCl2, and 10% glycerol) at 8,000 g for 20 min at 25°C. Supernatant and
pellet fractions were collected in SDS sample buffer, and equal volumes of each fraction
were resolved with a 10% or 6-12% gradient SDS-PAGE gel and stained with
Coomassie brilliant blue. Protein band densities from scanned gels were quantified using
the Odyssey infrared scanning system (LI-COR Biosciences) and analyzed in excel. All
experiments were repeated 3 times.
High-speed actin pelleting assays. Competition between SEPT9 and the myosin V-S1
heavy chain for F-actin binding were performed in buffer containing 10mM MOPS, pH
7.0, 100 mM KCl, 2 mM MgCl2, 0.5 mM EGTA, and 2 mM DTT with 1.25 mM ATP or
without ATP. For experiments without ATP, the F-actin stock was dialyzed in F-buffer

95 without ATP. F-actin (2 μM) was pre-incubated with increasing concentrations of SEPT9
for 30 minutes at 25°C. Reactions were incubated with myosin V-S1 heavy chain (0.75 –
1 μM) for an additional 30 minutes at 25°C, placed on a glycerol cushion (10mM MOPS,
pH 7.0, 100 mM KCl, 2 mM MgCl2, 0.5 mM EGTA, 20% glycerol) and centrifuged at
200,000 g for 30 minutes at 21°C. Equal volumes of supernatant and pellet fractions
were collected in SDS sample buffer, and equal volumes of each fraction were resolved
using a 10% SDS-PAGE gel and stained with Coomassie brilliant blue. Protein band
densities were quantified using the Odyssey infrared scanning system and analyzed in
excel. Each experiment was repeated 3 times.
Co-sedimentation assays (Fig. 4) that tested for cofilin-mediated
depolymerization were performed by pre-incubation of aged F-actin with recombinant
SEPT9_i1 (5 μM) or 5 μM SEPT9 fragments (NTD, B-domain or A-domain) in buffer
containing 10 mM Hepes, pH 7.6, 100 mM KCl, 1 mM MgCl2, and 2 mM DTT for 30 min
at 25°C. Reactions were incubated with 5 μM recombinant human cofilin-1 for an
additional 30 min at 25°C, placed on a glycerol cushion (20 mM HEPES, pH 7.6, 100
mM NaCl, 1 mM MgCl2, and 10% glycerol) and centrifuged at 200,000 g for 30 min at
21°C in an ultracentrifuge (Optima TL100, Beckman Coulter). Supernatant and pellet
fractions were collected in SDS sample buffer, and equal volumes of each fraction were
resolved using a 8-15% gradient SDS-PAGE gel and stained with Coomassie brilliant
blue. Protein band densities from scanned gels were quantified using the Odyssey
infrared scanning system (LI-COR Biosciences) and analyzed in excel. Each experiment
was repeated 3 times.
High-speed pelleting assays between SEPT9 and F-actin (Fig. S5) were
performed by incubating full-length SEPT9 (1 μM) with increasing concentrations of F-

96 actin for 30 minutes at 25°C in buffer containing 20 mM Hepes, pH 7.6, 100 mM KCl,
and 1 mM MgCl2 supplemented with 0.2 mM ATP and 4 mM DTT. Reactions were
placed on a glycerol cushion (20 mM HEPES, pH 7.6, 100 mM NaCl, 1 mM MgCl2, and
20% glycerol) and centrifuged at 200,000 g for 30 min at 21°C. Supernatant and pellet
fractions were collected in SDS sample buffer, and equal volumes were resolved using a
10% SDS-PAGE gel. Gels were stained with Coomassie brilliant blue, and protein band
densities from scanned gels were quantified using the Odyssey infrared scanning
system. The fraction of SEPT9 protein in each pellet fraction was plotted against the
concentration of actin in GraphPad and fitted with a one-site specific binding curve to
determine the dissociation constant.
RESULTS
The B-domain of SEPT9 promotes F-actin bundling.
Using negative stain EM and low-speed pelleting assays, we sought to
understand how SEPT9 interacts with F-actin by expressing and purifying each of the
three SEPT9 domains; the domain composition of SEPT9 is shown in Fig. 1a. Full-length
SEPT9 (isoform 1) as well as the N-terminal domain (NTD) of SEPT9 effectively
assemble F-actin into bundles (Fig. 1c and d, respectively). Strikingly, the B-domain
alone promotes formation of ordered thick bundles (Fig. 1e), while the A-domain does
not possess any bundling activity (Fig. 1f). Similarly to the A-domain, the G-domain of
SEPT9 does not possess any bundling activity (Fig. 1g). Our results indicate that the B-
domain of SEPT9 is responsible for the SEPT9 F-actin bundling activity. To
independently corroborate these EM observations, we examined the ability of individual
SEPT9 domains to cross-link actin filaments using a low speed actin sedimentation

97 assay (Fig. 1h and i). In agreement with the EM data, the NTD (Fig. 1h) and B-domain
(Fig. 1i) of SEPT9 pelleted F-actin at low speed, while the G-domain (Fig. 1h) and the A-
domain (Fig. 1h) did not. Taken together, these data indicate that the B-domain of the
NTD of SEPT9 is responsible for the actin-bundling activity of SEPT9.
SEPT9 binds to three sites on the surface of F-actin.
To identify the binding site of SEPT9 on the surface of F-actin we performed a
3D-reconstruction of actin filaments decorated with the two SEPT9 actin bundling
fragments – the NTD and the B-domain (Fig. 2). In order to obtain single actin filaments
suitable for image analysis, the NTD or B-domain were incubated with actin filaments
that were pre-bound to a carbon film to reduce bundle formation. The overall 3D-
reconstruction of F-actin segments (each segment contained ~14 actin subunits)
decorated with the NTD of SEPT9 revealed three sites of interaction of the NTD with the
actin filament (Fig. S1). We used a modeling approach and cross-correlation sorting (see
Methods) to compute individual 3D-reconstructions for each of the three modes of
binding of the NTD to F-actin revealed in the overall reconstruction (Fig. 2a). Since
model-based sorting may be biased, we used two independent approaches to confirm
that our 3D-reconstructions reflected the intrinsic properties of the images. First, we
computed two independent 3D-reconstructions for each of the three structural modes
starting the IHRSR algorithm from a featureless solid cylinder, or from a 3D-
reconstruction of a different structural mode (Fig. S2). This approach proved that the
resultant EM map was independent from the starting model and hence all its features
reflected the intrinsic properties of the raw images (Fig. S2). Second, we calculated
reference free 2D-averages for each class of NTD-decorated F-actin segments to
confirm that all three 2D averages were different from each other and hence, the

98 differences in the 3D reconstructions reflected the differences in the raw images (Fig.
S3).
The NTD of SEPT9 binds to three distinct sites on the surface of F-actin. In mode
1 (Fig 2a, magenta arrows), the NTD binds to the side of the SD1/SD2 interface of the
actin molecule which includes actin residues 80-101, 125-130, and 357-360 (Fig. 2d,
shown in magenta). In mode 2 (Fig. 2a, red arrows), the NTD is bound to the front
portion of the SD1/2 interface, which involves residues 1-7, 21-30, 56-60, and 92-103
(Fig. 2d, shown in red). In mode 3 (Fig. 2a, cyan arrows), actin residues 223-238 and
248-251, which belong to the SD4 of F-actin (Fig. 2d, shown in cyan), are located at the
interface with the NTD.
We used the same set of references to determine how the B-domain of SEPT9
binds to F-actin (Fig. 2b). We found the B-domain is bound to the same three sites on
the surface of the actin molecule. We applied the same sorting procedure to F-actin
incubated with the A-domain of SEPT9 and compared the frequency of decorated and
naked actin segments between the three SEPT9 domains (Fig. 2c). In contrast to the
NTD and the B-domain, more than 90% of actin segments incubated with the A-domain
yielded a best match with naked F-actin. The remaining 10% generated an
uninterpretable 3D-reconstruction (data not shown). To exclude any possibility that the
A-domain binds to F-actin, we used the same set of references to run cross-correlation
sorting of pure F-actin segments (Fig. S4). Not surprisingly, the sorting showed a very
similar distribution of the pure F-actin segments between the four classes which
reflected the margin of error in the sorting procedure.
We did not find a predominant mode of NTD or B-domain binding to F-actin as
the frequencies of the three structural modes were quite similar (Fig. 2c). Because we

99 did not find any segments with all three sites occupied simultaneously, individual binding
modes of SEPT9 appear to be highly cooperative and mutually exclusive; note that full
length SEPT9 exhibited the same three modes of actin binding as the NTD and B-
domains (data not shown). It is therefore likely that if a SEPT9 molecule is bound to an
actin protomer in either mode 1, 2 or 3, adjacent SEPT9 molecules would also bind actin
protomers in the same mode. Such a cooperativity has been reported for many actin
binding protein and relies on cooperative structural transitions that can propagate along
the actin filament (Galkin et al., 2005; Galkin et al., 2010; Orlova and Egelman, 1997). In
addition to the structural cooperative transitions within the actin filament, the spatial
arrangement of the SEPT9 domains on the surface of F-actin could result in mutually
exclusive modes of SEPT9 binding to F-actin. Binding modes 1 and 2 have an
overlapping interface on the surface of F-actin (Fig. 2d, 80-101 helix in mode 1 and 92-
103 in mode 2), and modes 2 and 3 would clash spatially if present simultaneously
within the same actin filament. Taken together, our results indicate that SEPT9 interacts
with F-actin via the B-domain of its NTD in a polymorphic fashion. Importantly, this
interaction involves actin-binding sites, which are commonly bound by actin-binding
proteins including myosin and cofilin (Behrmann et al., 2012; Galkin et al., 2011).
SEPT9 inhibits actomyosin activity
The localization of myosin S1-fragment on the surface of F-actin in the absence
of ATP (rigor state) has been previously resolved (Behrmann et al., 2012). While the
position of myosin on the surface of the actin filament in the presence of ATP (weak
binding mode) has not been visualized, modeling studies based on the X-ray diffraction
from permeabilized muscles suggest that the N-terminus of F-actin is at the interface
with the weakly bound myosin head (Gu et al., 2002). Our data suggest that the position

100 of myosin in a rigor state would clash with the position of the NTD of SEPT9 in two of its
three binding modes (Fig. 3a-b, red arrows); the N-terminus of F-actin (denoted with red
asterisk in Fig. 3a-c) is also at the interface of NTD with F-actin. Thus, binding of SEPT9
to F-actin in modes 1 or 2 could interfere with the actin binding of myosin in both rigor
and weak states (Fig. 3a-b), while binding of SEPT9 to F-actin in mode 3 may not
interfere with the acto-myosin interaction (Fig. 3c).
To test these structural predictions, we first used an actin-activated steady-state
ATPase assay. In this assay, myosin ATP hydrolysis is coupled with its interaction with
F-actin. Thus, any protein that interferes with the actomyosin interaction will reduce the
ATPase activity of the myosin head. To evaluate the effect of SEPT9 on the actomyosin
cross-bridge cycle, we carried out the actin activated steady-state ATPase assay using
the NADH coupled method (Fig 3d). The S1 fragment of myosin V and F-actin were
mixed with increasing concentrations of full length SEPT9, or its individual B- and A-
domains domains (see Methods for details). The full length SEPT9 exhibited the largest
inhibition with an apparent Ki of 0.46 µM followed by the B-domain with a Ki of 2.44 µM.
The A-domain had almost no effect on myosin ATPase activity with an apparent Ki of
185 µM.
Since bundling would reduce the number of F-actin sites that are available for
myosin binding, we tested whether the reduction in myosin ATPase activity correlated
with an increase in F-actin bundling. Low-speed actin sedimentation assays showed that
both the full length SEPT9 and SEPT9 B-domain did not significantly affect F-actin
bundling over concentration ranges that caused a drastic reduction in myosin activity
(Fig. S5). Hence, the bundling activity of SEPT9 by itself could not account for full
inhibition of the acto-myosin ATPase activity. Taken together with our structural data that

101 predict a mutually exclusive binding of myosin and SEPT9 in modes 1 and 2 (Fig. 3a,b),
our results indicate that SEPT9 inhibits actin-dependent myosin ATPase activity by
competing with myosin for binding to the same sites of F-actin.
ATP hydrolysis and subsequent dissociation of the nucleotide from the myosin
head changes acto-myosin interaction from the initial weakly-bound state to a rigor-
bound state. This transition in the acto-myosin interaction is an essential part of myosin’s
ATPase cross-bridge cycle. To test how SEPT9 inhibits acto-myosin activity, we
performed competitive high-speed actin pelleting assays of myosin V S1-fragment with
the full length SEPT9 in the presence or absence of ATP, so we can distinguish between
the weakly-bound (ATP) and rigor-bound (no ATP) states of myosin. In the presence of
ATP, full length SEPT9 efficiently competed with the myosin S1-fragment for binding to
F-actin (Fig. 3e). Interestingly, in the absence of ATP (only ADP was present in solution)
SEPT9 co-sedimented with myosin S1-fragment (Fig. 3f). The affinity of the ADP-bound
myosin V to F-actin (~7.6 nM) is ~1000 fold higher than its affinity to F-actin in the
presence of ATP (~13 µM) (De La Cruz et al., 1999; Yengo et al., 2002). Taking into
account that the affinity of septins to F-actin is in the micromolar range(Mavrakis et al.,
2014) (Fig. S6), SEPT9 molecules that are bound to F-actin in modes 1 and 2 are likely
to be displaced by ADP-bound myosin, which has a high affinity for F-actin.
Cosedimentation of SEPT9 with rigor-bound myosin V suggests that SEPT9 is bound to
F-actin in mode 3 (Fig. 3c), which does not clash with the position of rigor-bound myosin.
In the presence of ATP, however, SEPT9 is likely to compete with myosin for the binding
modes 1 and 2, which account for ~70% of the binding of SEPT9 to F-actin (Fig. 2c), and
thereby, SEPT9 can inhibit the interaction of myosin with F-actin. To summarize, our
data indicate that SEPT9 inhibits the weak acto-myosin interaction through its B-domain,
while it can bind to F-actin in the presence of rigor-bound myosin.

102 SEPT9 reduces cofilin-driven F-actin depolymerization.
Superimposition of the 3D reconstructions of SEPT9-decorated F-actin in mode 1
(Fig. 4a, top) and mode 2 (Fig. 4a, bottom) revealed significant clashes between the
SEPT9 density (Fig. 4a, black arrows) and cofilin (Fig. 4a, shown as solid red surface).
Similar to our results with myosin, we hypothesized that SEPT9 may inhibit cofilin-actin
binding. Using electron microscopy and high-speed pelleting assays, we sought to test
whether SEPT9 affects cofilin-driven F-actin depolymerization.
EM imaging of actin filaments showed that within 2 min of incubation, cofilin
disrupts the meshwork of naked actin filaments, which are disintegrated into short
twisted filaments (Fig. 4b, 2 min). After 30 min, only a few cofilin-decorated actin
filaments remain (Fig. 4b, 30 min). In the presence of full length SEPT9, F-actin bundles
(Fig. 4c, 0 min) remain intact after 2 min of incubation with equimolar concentration of
cofilin-1 (Fig. 4c, 2 min). Even after 30 min of incubation with cofilin-1, F-actin bundles
are still present (Fig. 4c, 30 min). To further test whether SEPT9 protects against actin
depolymerization by cofilin, we performed high-speed sedimentation assays in the
presence of full length SEPT9 or its domains and cofilin-1 (Fig. 4,d-f). In our
experiments we used equimolar concentrations of SEPT9 and cofilin, because the
affinity of human ADF/cofilin to ADP-F-actin is similar to septins (Mavrakis et al., 2014) (Fig. S6).
After 30 min incubation of F-actin with cofilin-1, ~90% of actin is recovered in the
supernatant (Fig. 4d). Pre-incubation of F-actin with SEPT9 reduced the amount of actin
in the supernatant to ~30% (Fig. 4d,). The NTD (Fig. 4e) and the B-domain (Fig. 4f) of
SEPT9 also reduced the amount of depolymerized actin in the supernatant to ~50-60%
of total actin, while the A-domain had no effect (Fig. 4f). Thus, similar to the inhibition of

103 myosin binding to F-actin, SEPT9 appears to inhibit cofilin binding, and thereby, protect
actin filaments from cofilin-mediated depolymerization.
Discussion
Drosophila septins have been recently reported to directly bind and bundle F-
actin (Mavrakis et al., 2014). Mammalian septins (SEPT2/6/7 oligomers) have been
proposed to interact with F-actin indirectly via anillin and myosin II, but it was recently
shown that the mammalian SEPT9 can bind and bundle actin filaments (Dolat et al.,
2014b; Joo et al., 2007; Kinoshita et al., 2002). Our data show that the actin bundling
activity of SEPT9 resides in the NTD – a unique domain that is not present in other
septin paralogs. SEPT9 interacts with F-actin in a highly polymorphic fashion (Fig. 2a-b).
Such multiplicity in binding modes in not unique to SEPT9 and has been reported for
other actin binding proteins (ABPs) such as Arg/Abl kinase, drebrin, myosin binding
protein C, utrophin, and a giant muscle protein nebulin (Galkin et al., 2005; Galkin et al.,
2002; Grintsevich et al., 2010; Lukoyanova et al., 2002; Orlova et al., 2011) Since, the
N-terminal domain of SEPT9 does not possess any detectable amino acid identity with
these ABPs, it suggests that multiplicity of actin-binding modes is a common feature of
ABPs independently of their actin-binding sequences and domains.
Recently we showed that F-actin can exist in multiple structural states and that
the structural transitions in the mobile regions of the actin protomers are coupled (Galkin
et al., 2010). SEPT9 binds to the two mobile regions of actin molecule including
residues 1-7 of the N-terminus and residues 56-60 of the SD2 (Fig. 2d). It is plausible
that in the cell the structural mode of SEPT9 binding to F-actin is determined by the
structural state of the actin filament and the presence of other actin-bound ABPs. Our
results suggest that SEPT9 may switch from mode 1 and 2, where it interferes with the

104 actin-myosin interaction (Fig. 3a-b), to mode 3 where both rigor-bound myosin and
SEPT9 can simultaneously interact with F-actin. By switching between these structural
modes of binding, SEPT9 may alter acto-myosin interactions in migrating cells, where
tensile forces are tightly controlled and are linked to focal adhesion turnover (Parsons et
al., 2010; Peacock et al., 2007) and cell-ECM adhesion (Vicente-Manzanares et al.,
2007). Thus, SEPT9 may influence actomyosin activity at stress fibers and focal
adhesions.
SEPT9-dependent actin bundling activity is required for the maintenance of a
functional stress fiber network in the leading edge of migratory epithelial cells (Dolat et
al., 2014b). Interestingly, our results indicate that SEPT9 protects actin filaments from
depolymerization by cofilin. Actin cross-linking proteins have been shown to protect actin
filaments from depolymerization in vitro (Cano et al., 1992; Lebart et al., 2004; Zigmond
et al., 1992). In living cells, varying levels of expression of actin bundling proteins have
been shown to alter the rates of actin filaments turnover (Mukhina et al., 2007; Tilney et
al., 2003) and myosin has been shown to regulate the turnover of actin bundles in
neuronal growth cones (Ishikawa et al., 2003; Medeiros et al., 2006).
In addition, recent studies show that actin depolymerization by ADF/cofilin is
differentially regulated by actin motors and actin-cross-linking proteins (Schmoller et al.,
2011). While myosin II enhances the disintegration of F-actin bundles, cross-linking
proteins inhibit the disassembly of actin bundles by cofilin (Schmoller et al., 2011).
Notably, cross-linking proteins such as fascin may affect the severing activity of cofilin by
reducing the flexibility of bundled F-actin, and thereby, limit the torsional twist that cofilin
triggers on F-actin (Breitsprecher et al., 2011). Because the efficiency of cofilin severing
is the highest between decorated and non-decorated segments of F-actin, the severing

105 activity of cofilin will be the highest on more rigid bundled actin filaments, which do not
allow for a uniform, cooperative binding that is enabled by a torsional twist (Breitsprecher
et al., 2011). Thus, depending on the ABP composition of F-actin bundles and their
flexibility, cross-linking proteins such as fascin may either increase or decrease the
activity of cofilin (Breitsprecher et al., 2011; Elkhatib et al., 2014)
Our results indicate that SEPT9 inhibits both myosin and cofilin interaction with
F-actin, and hence may work as a protective factor for growing actin bundles. Given that
both myosin (Juanes-Garcia et al., 2016)and cofilin (Bernstein and Bamburg, 2010) are
involved in the regulation of actin organization and contractility in a diversity of cellular
processes and structures, our results suggest that the ubiquitously expressed SEPT9
(Dolat et al., 2014a) may be a key player in the regulation of F-actin organization and
function.
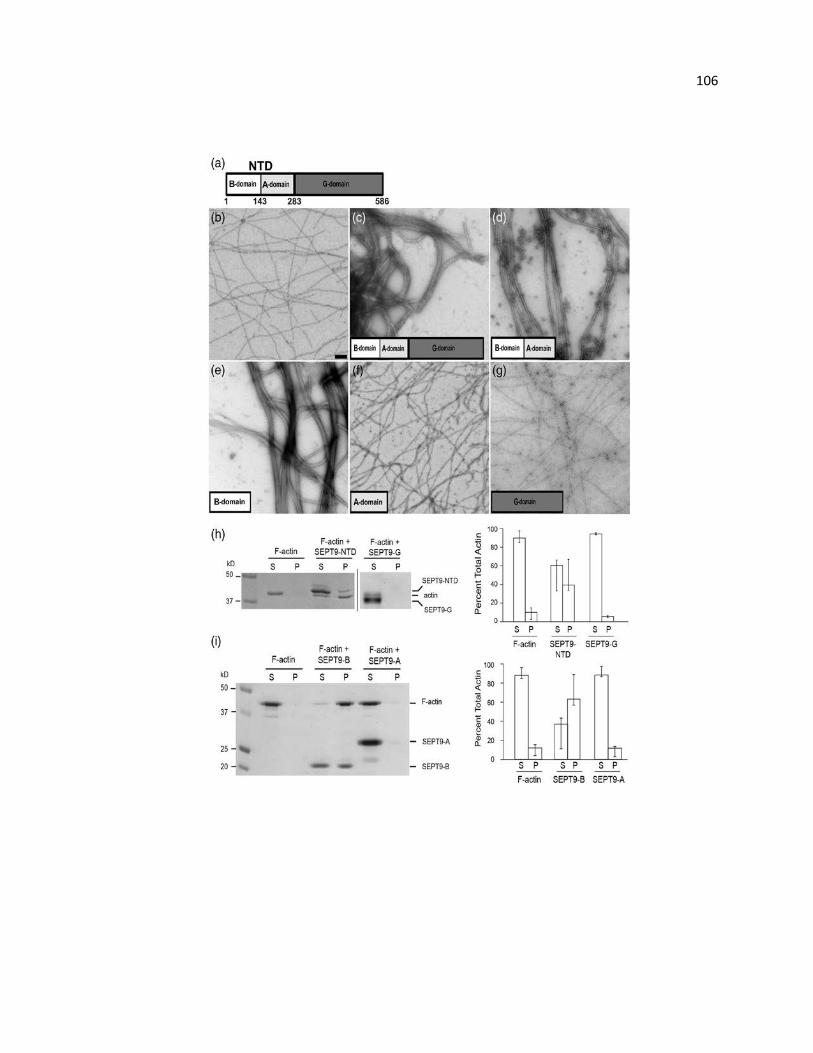
106

107 Figure 6.1. SEPT9 bundles F-actin through the B-domain of its NTD. (a) Schematic
representation of SEPT9 domains. In addition to the GTP-binding domain (G-domain)
found in all septin paralogs, SEPT9 has a unique N-terminal domain (NTD) consisting of
a basic domain (B-domain) and an acidic domain (A-domain). (b) Pure F-actin, scale
bar: 1 nm. (c) Full-length SEPT9 efficiently bundles actin filaments within 2 min of
incubation. (d) NTD consisting of the B- and A-domains packs F-actin into tight bundles.
(e) The B-domain bundles F-actin into tight bundles morphologically similar to those
shown in (d). (f) The A-domain does not bind or bundle F-actin. (g) The G-domain does
not bundle F-actin. (h and i) Low-speed sedimentation of F-actin in the presence of
SEPT9 domains. Coomassie-stained gels with the corresponding bar graphs show that
F-actin is recovered in the pellet in the presence of the NTD [(h), SEPT9-NTD] and the
B-domain [(i), SEPT9-B]. Error bars correspond to the maximum and minimum values
from three independent experiments. In the presence of the G-domain [(h), SEPT9-G]
and the A-domain [(i), SEPT9-A], most of the actin is recovered in the supernatant.
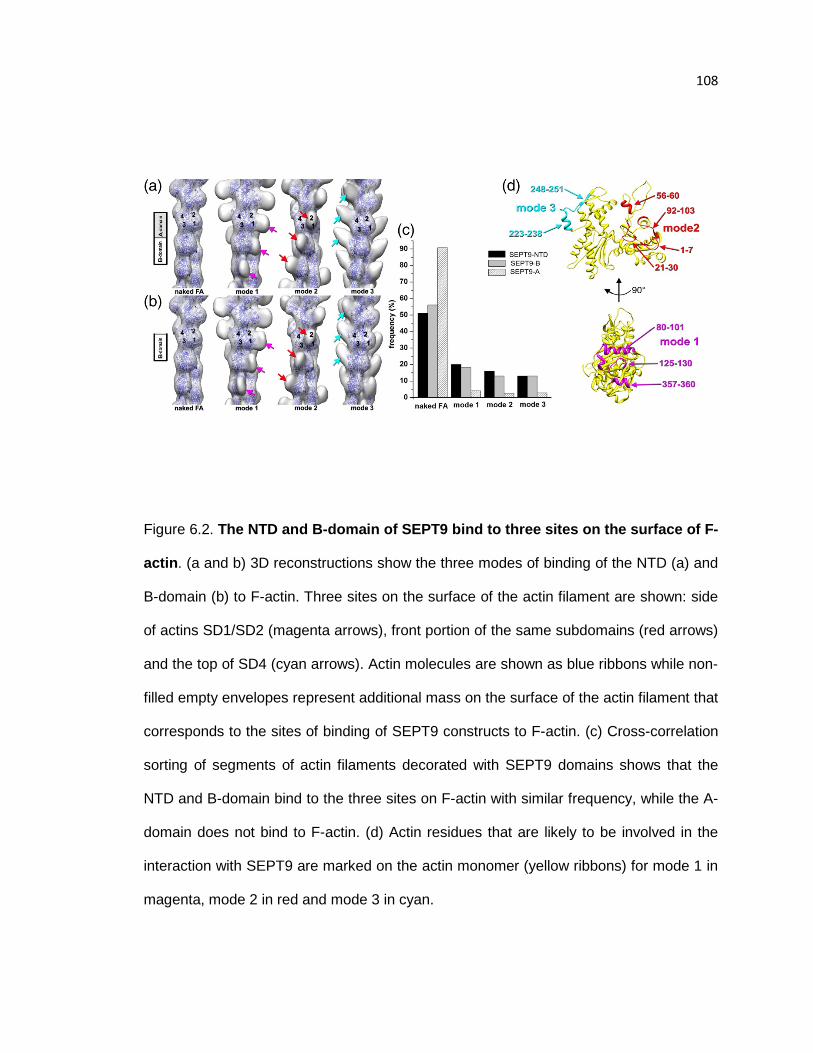
108
Figure 6.2. The NTD and B-domain of SEPT9 bind to three sites on the surface of F-
actin. (a and b) 3D reconstructions show the three modes of binding of the NTD (a) and
B-domain (b) to F-actin. Three sites on the surface of the actin filament are shown: side
of actins SD1/SD2 (magenta arrows), front portion of the same subdomains (red arrows)
and the top of SD4 (cyan arrows). Actin molecules are shown as blue ribbons while non-
filled empty envelopes represent additional mass on the surface of the actin filament that
corresponds to the sites of binding of SEPT9 constructs to F-actin. (c) Cross-correlation
sorting of segments of actin filaments decorated with SEPT9 domains shows that the
NTD and B-domain bind to the three sites on F-actin with similar frequency, while the A-
domain does not bind to F-actin. (d) Actin residues that are likely to be involved in the
interaction with SEPT9 are marked on the actin monomer (yellow ribbons) for mode 1 in
magenta, mode 2 in red and mode 3 in cyan.
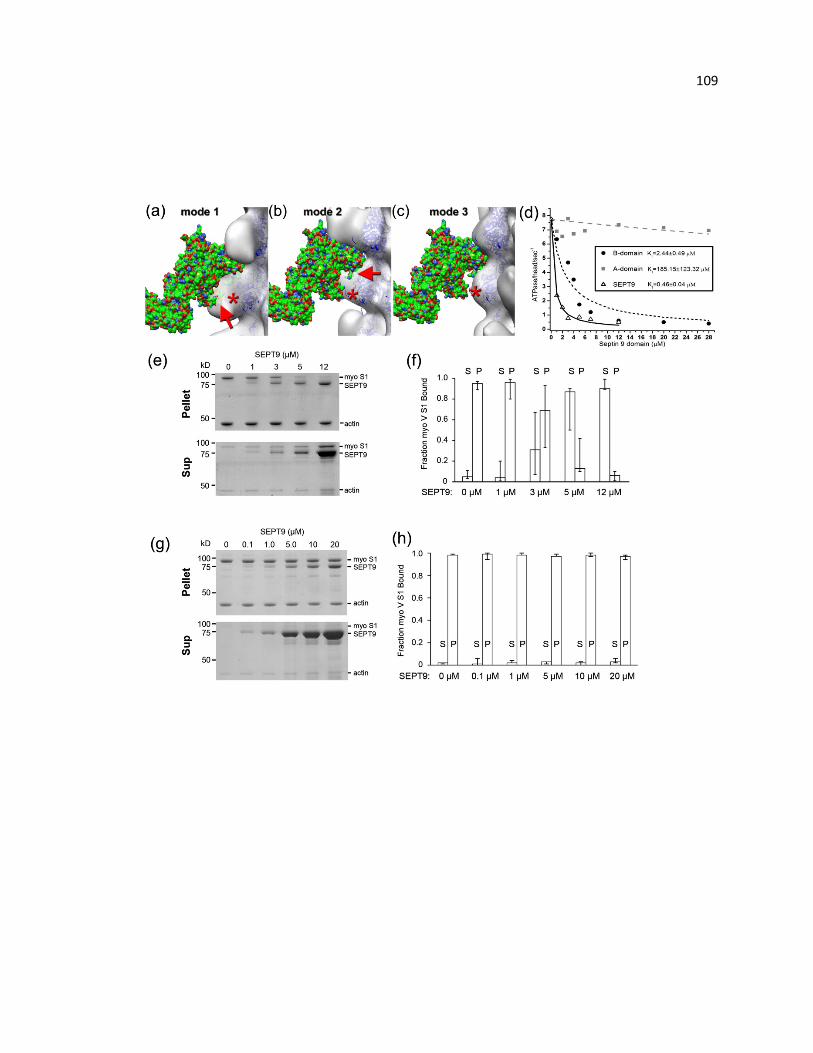
109

110 Figure 6.3. SEPT9 inhibits the activity and interaction of myosin with actin. (a–c) An
atomic model of the actomyosin complex in rigor state [14] was docked onto the 3D
reconstructions of the three modes of binding of the SEPT9 NTD to F-actin (Fig. 2a)
using the best fit of the actin molecules. Actin molecules are shown as blue ribbons and
myosin head is depicted as solid surface. The clashes between the myosin head and the
NTD density (red arrows) in modes 1 and 2 are marked with red arrows (a and b). There
are no such clashes in mode 3 (c). The position of the N-terminus of F-actin that resides
at the interface between the NTD and F-actin is denoted with red asterisks. (d) Plot
shows the ATPase activity of the S1 fragment of myosin V in the absence and presence
of full-length SEPT9 and the A- and B-domains of SEPT9. As predicted by the structural
data, full-length SEPT9 and the B-domain inhibit the ATPase activity of myosin, while the
A-domain does not have a significant effect. (e) Coomassie-stained gels show the pellet
(P; top) and supernatant (S; bottom) fractions from a competitive high-speed actin
pelleting assay in the presence of ATP (1.25 μM). F-Actin (2 μM) was incubated with
increasing concentrations of recombinant full-length SEPT9 and the myosin S1 fragment
was added at a constant concentration (0.75 μM). (f) Bar graphs show the fraction of
total S1 fragment in the pellet and supernatant. Error bars correspond to the minimum
and maximum values obtained from three independent experiments. (g) Coomassie-
stained gels show the pellet (P; top) and supernatant (S; bottom) fractions from a
competitive high-speed actin pelleting assay in the absence of ATP. F-Actin (2 μM) was
incubated with increasing concentrations of recombinant full-length SEPT9 and the
myosin S1 fragment was added at a constant concentration (1 μM). (f) Bar graphs show
the fraction of total S1 fragment in the pellet and supernatant. Error bars correspond to
the minimum and maximum values obtained from three independent experiments.
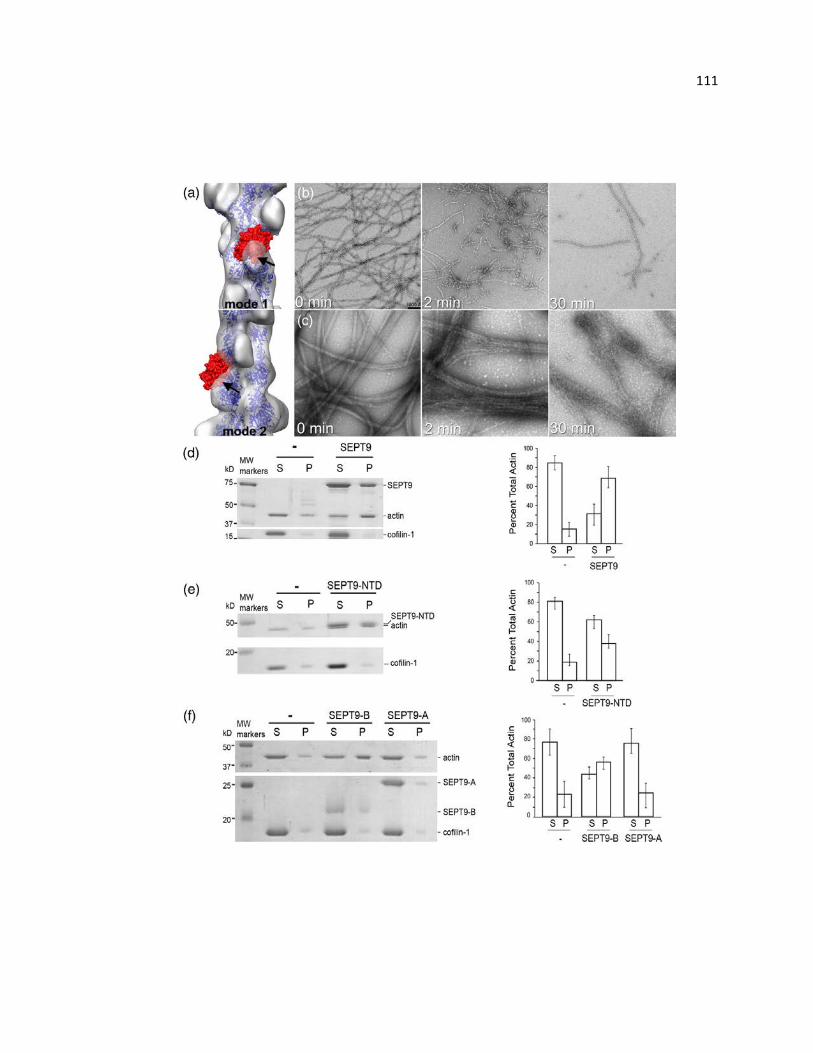
111

112 Figure 6.4. SEPT9 reduces the extent of cofilin-driven F-actin depolymerization. (a)
Atomic model of actin–cofilin complex [15] docked into the 3D reconstructions of modes
1 and 2 (transparent gray surfaces) shows a clash between the cofilin molecule (red
surface) and SEPT9 density (marked with black arrows). (b) Long F-actin filaments
formed after 2 h of polymerization (b; 0 min) are disrupted into short segments within
2 min of incubation with cofilin-1 (b; 2 min); after a 30-min incubation with cofilin (b;
30 min), only few filaments that are heavily decorated with cofilin remain. (c) F-actin
forms tight bundles in the presence of SEPT9 (c; 0 min). After 2 min of incubation with
cofilin-1, SEPT9-induced F-actin bundles still remain intact (c; 2 min), and even after
30 min of incubation, F-actin bundles still persist (c; 30 min). (d–f) High-speed
sedimentation assay of F-actin in the presence of SEPT9 domains and cofilin-1. Actual
gels with the corresponding bar graphs (d–f) show that, in the presence of cofilin-1, most
of the actin is found in the supernatant (d), while in the presence of full-size SEPT9 (d),
NTD (e) or the B-domain (f), more than half of the actin is recovered in the pellet. In the
presence of the A-domain, most of the actin remains in the supernatant (f).

113
Figure 6.5. Overall 3D reconstruction of F-actin decorated with the NTD suggest
the sites of SEPT9 binding to the actin filament. Segments of actin filaments
decorated with the NTD of SEPT9 were reconstructed using IHRSR approach starting
from a solid cylinder. The resultant reconstruction possessed three additional densities
on the surface of F-actin. The first one is at the front interface between SD1 and SD2 of
the two adjacent actin protomers (red arrow). The second density is at the side of SD1
(magenta arrow), while the third one is on the top of SD4 (cyan arrow)
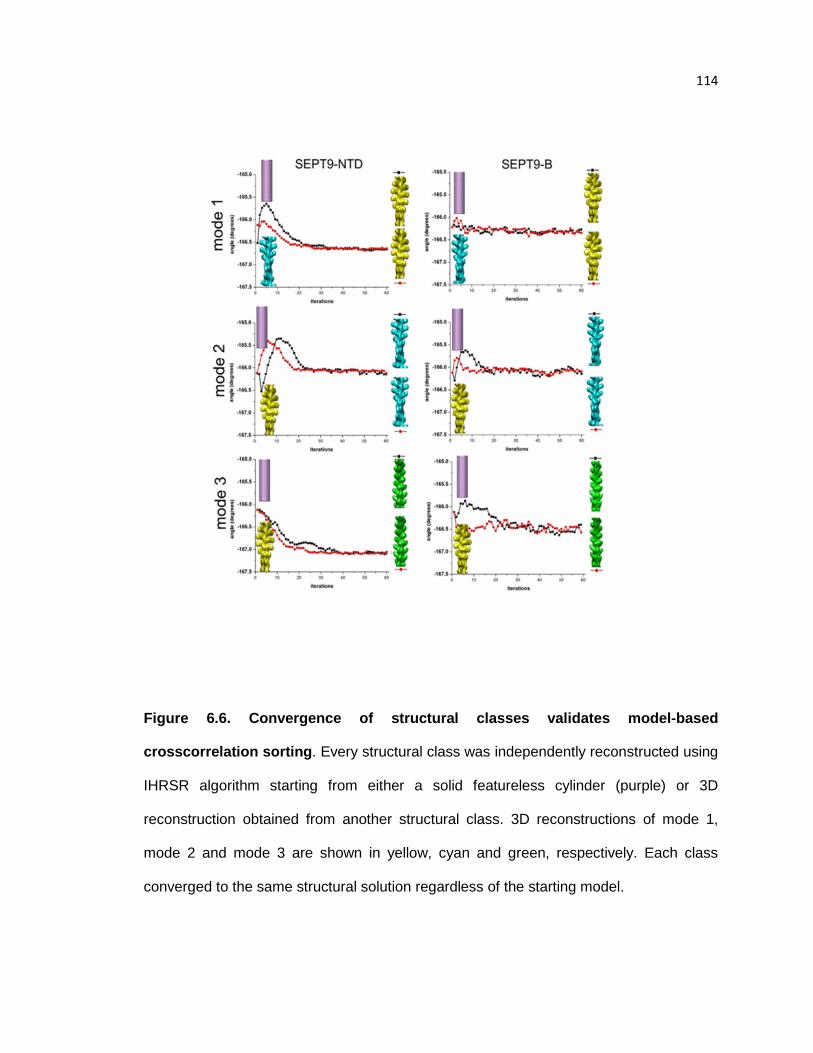
114
Figure 6.6. Convergence of structural classes validates model-based
crosscorrelation sorting. Every structural class was independently reconstructed using
IHRSR algorithm starting from either a solid featureless cylinder (purple) or 3D
reconstruction obtained from another structural class. 3D reconstructions of mode 1,
mode 2 and mode 3 are shown in yellow, cyan and green, respectively. Each class
converged to the same structural solution regardless of the starting model.

115
Figure 6.7. Reference free 2D averages of occupied classes. SPIDER1 AP SR
routine was used to generate reference free 2D averages of the three structural modes
recovered in the NTDdecorated actin filaments. Red arrows indicate visible differences
in the SEPT9-NTD positions.
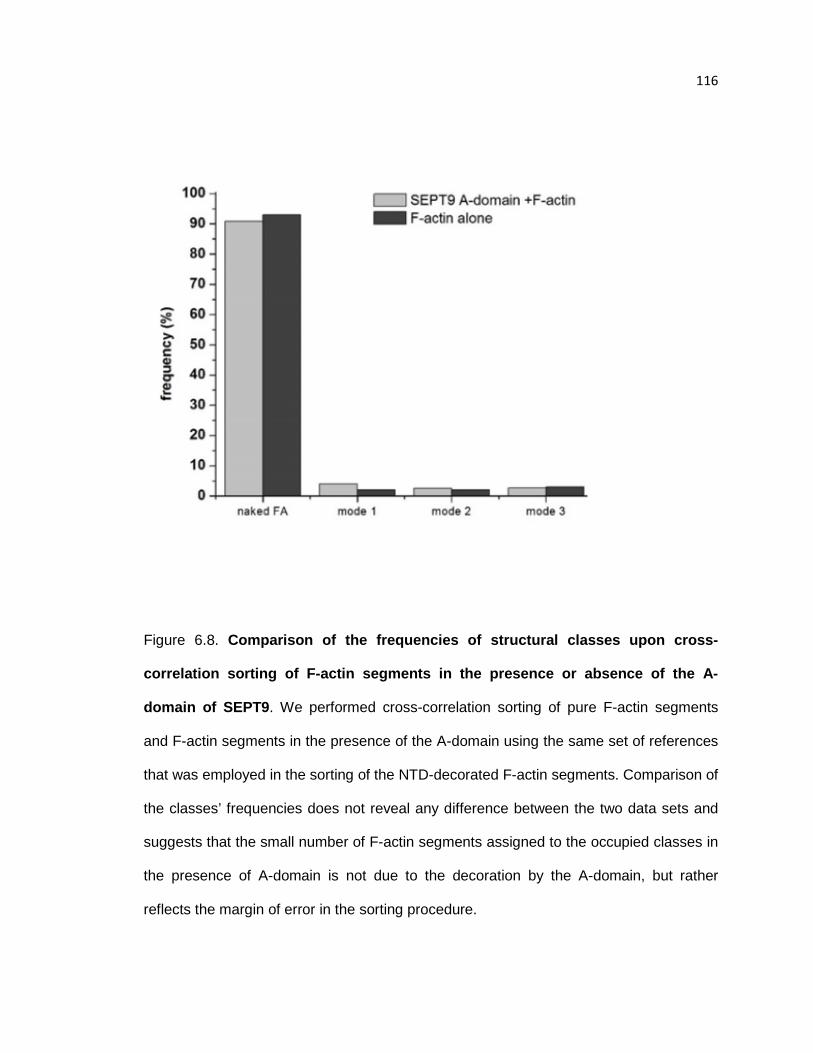
116
Figure 6.8. Comparison of the frequencies of structural classes upon cross-
correlation sorting of F-actin segments in the presence or absence of the A-
domain of SEPT9. We performed cross-correlation sorting of pure F-actin segments
and F-actin segments in the presence of the A-domain using the same set of references
that was employed in the sorting of the NTD-decorated F-actin segments. Comparison of
the classes’ frequencies does not reveal any difference between the two data sets and
suggests that the small number of F-actin segments assigned to the occupied classes in
the presence of A-domain is not due to the decoration by the A-domain, but rather
reflects the margin of error in the sorting procedure.
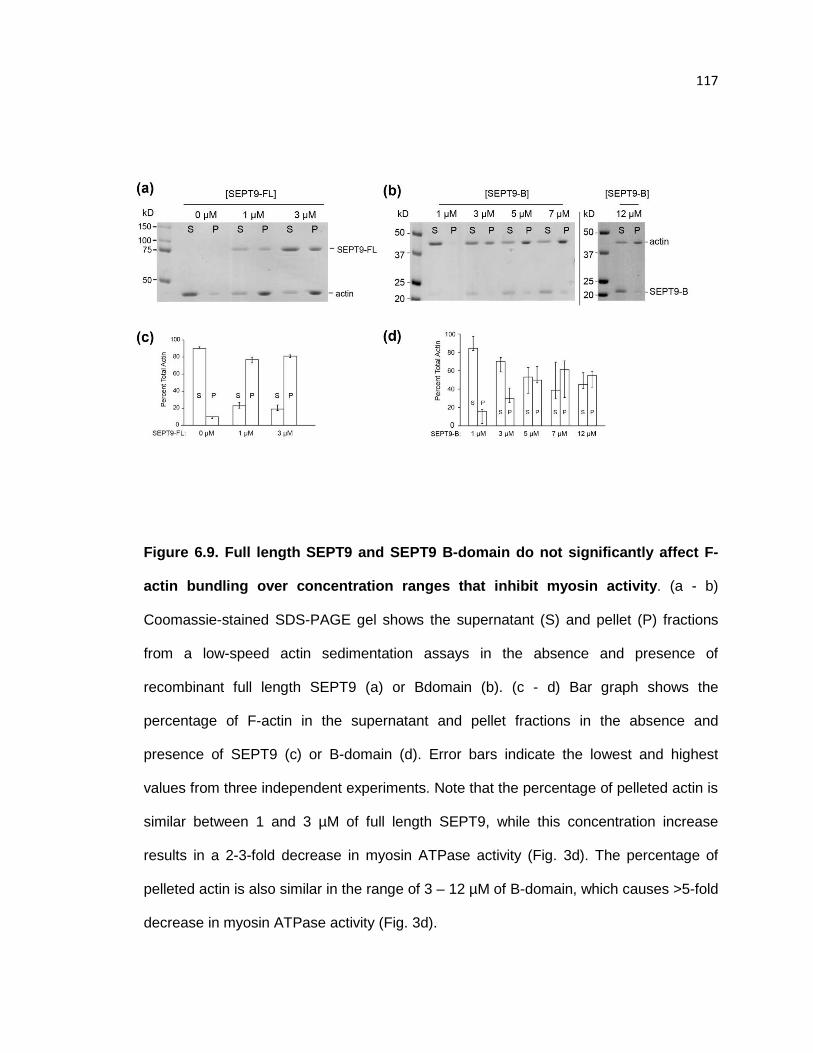
117
Figure 6.9. Full length SEPT9 and SEPT9 B-domain do not significantly affect F-
actin bundling over concentration ranges that inhibit myosin activity. (a - b)
Coomassie-stained SDS-PAGE gel shows the supernatant (S) and pellet (P) fractions
from a low-speed actin sedimentation assays in the absence and presence of
recombinant full length SEPT9 (a) or Bdomain (b). (c - d) Bar graph shows the
percentage of F-actin in the supernatant and pellet fractions in the absence and
presence of SEPT9 (c) or B-domain (d). Error bars indicate the lowest and highest
values from three independent experiments. Note that the percentage of pelleted actin is
similar between 1 and 3 µM of full length SEPT9, while this concentration increase
results in a 2-3-fold decrease in myosin ATPase activity (Fig. 3d). The percentage of
pelleted actin is also similar in the range of 3 – 12 µM of B-domain, which causes >5-fold
decrease in myosin ATPase activity (Fig. 3d).

118
Figure 6.10. Cosedimentation assay of full length SEPT9 with F-actin. (a)
Coomassie-stained gels show the pellet and supernatant fractions (equal volumes) from
a representative high-speed sedimentation assay. SEPT9 (1 µM) was incubated with
increasing concentrations of F-actin for 30 min at room temperature and was centrifuged
at 200,000 g. (b) Binding curve shows the fraction of F-actin-bound SEPT9 as a function
of increasing concentrations of F-actin. Data were fitted in GraphPad with one site
specific binding curve and an estimated Kd of 2.23 ± 0.60 µM was derived based on
three independent experiments.

119
Figure 6.11. Resolution determination. Each structural class was split in two non-
overlapping sets and the IHRSR procedure was used on each set. The correlation
between the corresponding Fourier shells of the two resultant volumes was calculated
for each data set.

120 CHAPTER 7. Septins promote macropinosome maturation and traffic to the
lysosome by facilitating membrane fusion
Abstract
Macropinocytosis, the internalization of extracellular fluid and material by plasma
membrane ruffles, is critical for antigen presentation, cell metabolism and signaling.
Macropinosomes mature through homotypic and heterotypic fusion with endosomes,
and ultimately merge with lysosomes. The molecular underpinnings of this clathrin-
independent endocytic pathway are largely unknown. Here, we show that the
filamentous septin GTPases associate preferentially with maturing macropinosomes in a
phosphatidylinositol 3,5-bisphosphate-dependent manner, and localize to their
contact/fusion sites with macropinosomes/endosomes. Septin knock-down results in
large clusters of hemifused macropinosomes, which persist longer and exhibit less
fusion events. Septin depletion and over-expression down-regulate and enhance,
respectively, the delivery of fluid-phase cargo to lysosomes, without affecting Rab5 and
Rab7 recruitment to macropinosomes/endosomes. In vitro reconstitution assays show
that fusion of macropinosomes/endosomes is abrogated by septin immunodepletion and
function-blocking antibodies, and is induced by recombinant septins in the absence of
cytosol and polymerized actin. Thus, septins regulate fluid phase cargo traffic to
lysosomes by promoting macropinosome maturation and fusion with
endosomes/lysosomes.
Introduction
Macropinocytosis is a clathrin-independent pathway of endocytosis that
originates from the closure of plasma membrane ruffles into large vesicles, which

121 internalize extracellular fluid and particles (Kerr and Teasdale, 2009; Swanson, 2008).
Macropinocytosis is upregulated by growth factors and oncogenes, which stimulate the
uptake of nutrients for cell proliferation and growth (White, 2013). Many bacterial and
viral pathogens invade cells by hijacking macropinocytosis, which is critical for the
antigen presentation (de Carvalho et al., 2015; Lim and Gleeson, 2011; Mercer and
Helenius, 2012). By modulating the internalization of plasma membrane receptors and
adhesion molecules, macropinocytosis is also implicated in cell signaling, adhesion and
motility (Bryant et al., 2007; Gu et al., 2011; Schmees et al., 2012).
Macropinosomes traffic to lysosomes while undergoing a process of maturation,
which is characterized by size reduction concomitant with membrane tubulation and
fusion with other macropinosomes and early/late endosomes (Bright et al., 2005;
Racoosin and Swanson, 1993). Macropinosome maturation is accompanied by changes
in phoshoinositide and Rab GTPase content (Levin et al., 2015). Nascent
macropinosomes become enriched with phopshatidylinositol 3-phosphate (PI3P) and
Rab5 (Feliciano et al., 2011; Yoshida et al., 2009), while maturing macropinosomes
contain phopshatidylinositol 3,5-biphosphate (PI(3,5)P2) and Rab7 (Kerr et al., 2006;
Kerr et al., 2010). Despite our knowledge of these transitions, the molecular basis and
regulation of the membrane fusion events that underlie macropinosome maturation and
merging with the lysosome remain unknown.
Septins are filamentous heteromeric GTPases that associate with cell
membranes and the cytoskeleton (Fung et al., 2014; Mostowy and Cossart, 2012).
Plasma membrane septins are essential for the maintenance of diffusion barriers and
modulate exocytosis (Bridges and Gladfelter, 2015; Caudron and Barral, 2009). Much
less is known about how septins function in the endomembrane system of organelles
and their communication. Septin mRNAs and proteins associate with endosomes

122 (Baumann et al., 2014; Baust et al., 2008) and are implicated in multivesicular body
formation (Traikov et al., 2014). However, how septins function in endocytic membrane
traffic is not known.
Materials and methods
Plasmids
Mouse SEPT2 was inserted into N1-YFP (Spiliotis et al., 2005). Mouse SEPT2
was inserted into N1-GFP (Bowen et al., 2011). PM-mCherry was constructed by
inserting the membrane-binding domain of lyn kinase in pcDNA 3.1-mCherry (Benjamin
et al., 2010). Rab5A was inserted into C1-GFP using the HindIII and SalI restriction
sites, and Rab7A was inserted into C1-GFP using the Xho1 and KpnI restriction sites.
Rab5B-mCherry and Rab7A-mCherry were purchased from addgene. Rat SEPT7v2 and
human SEPT6v3 were inserted into the pET-28a(+) vector, which expresses in tandem
His-SEPT7 and an untagged SEPT6 (Bai et al., 2013). Mouse SEPT2 was inserted into
pET-15b and expresses an untagged SEPT2 (Bai et al., 2013).
For experiments using shRNAs, the non-targeting control (5′-
TGAGTTCACACTAATGGTGGTCG-3′) was inserted in pG-SUPER-GFP (Bowen et al.,
2011) and pSUPER-mCherry (Dolat et al., 2014b). To target canine SEPT2, the shRNA
#1 (5′-CCTTAGACGTCGCATTCATGAAA-3′) was inserted in pSUPER-mCherry (Dolat
et al., 2014b) and SEPT2 shRNA #2 (5′-GCAACTACAAGCCCGAAGAATAA-3′) was
inserted in pG-SUPER-GFP. Human SEPT2 was targeted using the SEPT2 shRNA 5′-
AAGGTGAATATTGTGCCTGTC-3′, which was inserted in pG-SUPER-GFP (Bowen et
al., 2011). For siRNA experiments (Fig. 5), the SEPT2 siRNA oligonucleotides
(5’AAGGUGAAUAUUGUGCCUGUC-3) and mock GFP siRNA oligonucleotides (5’-

123 GGCUACGUCCAGGAGCGCACC-3’) were purchased from Dharmacon Research
(LaFayette, CO).
Tissue culture and transfections
MDCK II/G cells and the stable cell lines MDCK-SEPT2-YFP (CMV promoter;
N1-YFP) (Spiliotis et al., 2005) and MDCK-PM-mCherry (CMV promoter; pcDNA3.1)
were cultured in low glucose DME with 1 g/L NaHCO3, 10% FBS (Cell Generation), and
1% PSK (penicillin, streptomycin and kanamycin) at 37 ˚C with 5% CO2. HT1080 cells
(CCL-121) were purchased from ATCC and cultured in high glucose DME with 1 g/L
NaHCO3, 10% FBS (Cell Generation), and 1% PSK (penicillin, streptomycin and
kanamycin) at 37 ˚C with 5% CO2. Cells were plated on No. 1.5 glass coverslips (VWR),
which were pre-coated with 10 µg/mL type I collagen (PurCol; BioMatrix) in 0.1% acetic
acid for five minutes, and subsequently were dried and rinsed twice with media before
cell plating. Cells were transfected with 1.5 µg plasmid or co-transfected with 1.0 µg of
one plasmid and 1.5 µg of the second plasmid using lipofectamine 2000 (Invitrogen).
Dextran uptake and pulse-chase assays
Cells were cultured in low-serum media (0.5% FBS) overnight and switched to
media with 10% FBS to stimulate macropinocytosis for the duration of incubations and
pulse-chase treatments with fluorescent dextrans. Pulse-chase experiments were
performed by incubating cells with media containing 10% FBS and 0.2 mg/mL FITC- or
TR-dextran (10,000 MW, ThermoFisher Scientific) for 5 min and then washing three
times and incubating cells in dextran-free media for the indicated times. In Fig. 2C, cells
were incubated with 800 nM YM201636 (SelleckChem, Inc.) or vehicle control (DMSO)

124 for 2 h in low-serum media and then stimulated with media containing 10% FBS and 0.2
mg/mL TR-dextran with YM201636 or DMSO for 10 min.
Immunofluorescence microscopy
Cells were fixed with warm (37 ˚C) 2% paraformaldehyde (PFA) in PBS
(phosphate-buffered saline) and simultaneously permeabilized and blocked with 2%
BSA (bovine serum albumin; Sigma) and 0.1% saponin (Calbiochem) for 30 min. In Fig.
1A, cells were extracted prior to fixation by incubating for 30 s with warm (37 ˚C)
cytoskeleton buffer (CB) containing 10 mM MES (pH 6.1), 138 mM KCl, 3 mM MgCl2, 2
mM EGTA, 320 mM sucrose and 0.1% Triton X-100. Cells were subsequently rinsed
twice with CB and fixed with 2% PFA in CB.
Rabbit antibodies against SEPT2 (N5N; a gift from M. Kinoshita, Nagoya
University, Nagoya, Japan) and Rab5 (Stressgen), and mouse antibodies against Rab7A
(Abcam), LAMP1 (H4A3, Developmental Studies Hybridoma Bank; University of Iowa),
Sec6 (Exoc3; 10C2), Sec8 (Exoc4; 9H5), Exo70 (Exoc7; 13F3), gifts from C. Yeaman
(University of Iowa), were diluted in PBS containing 2% BSA and 0.1% saponin.
Secondary donkey Alexa Fluor 488-, AlexaFluor 594- or Alexa Fluor 647-conjugated
F(ab’) to mouse or rabbit IgG (Jackson ImmunoResearch Laboratories, Inc.) were
diluted in PBS containing 2% BSA and 0.1% saponin. Cells were stained for F-actin
using phalloidin conjugated to Alex Fluor 555 (ActiStain; Cytoskeleton, Inc.). Coverslips
were mounted on coverslides using Vectashield (Vector Laboratories).
Slides were imaged using a confocal laser-scanning microscope (FluoView 1000;
Olympus) equipped with three Hamamatsu R7862 photomultiplier tubes (PTMs) and
multi-line Argon ion (458 nm, 488 nm, 515 nm), Helium-Neon (543 nm) and diode 401
nm and 635 nm lasers. Images were acquired using 60x/1.42 NA oil objective and the

125 FV10-ASW 02.01 software. 3D rendering of confocal stacks was performed in the
Volocity software (Perkin Elmer).
Time-lapse imaging
All time-lapse microscopy experiments were performed at 37˚C using DME
without Phenol red supplemented with 10% FBS, 1% PSK and 20 mM Hepes. In Fig. 1
and 3A,, PM-mCherry dynamics were imaged with 3D deconvolution wide-field
microscopy using an inverted microscope (DeltaVision OMX v4; GE Healthcare)
equipped with a motorized stage, stage-tope incubator with temperature controller, and
three pco.edge sCMOS cameras (custom version). 3D stacks (1 – 2 µm; 0.125 nm
slices) were acquired every 10-20s using a 60x/1.49 NA objective lens (Olympus) and
were deconvolved in the softWoRx 6.1.1 software. In Fig. 3B-D and 3H, macropinosome
dynamics were imaged using an inverted microscope (IX81; Olympus) equipped with a
motorized stage (ProScanII; Prior), an OrcaR2 CCD camera (Hamamatsu Photonics), a
custom-built stage-top chamber and temperature controller (Air-Therm ATX; World
Precision Instruments) and the Slidebook 6.0 software. Fluorescent images were
acquired every 5 s for 8 min with a Plan-Apochromat 60x/1.40 NA objective. Time-lapse
videos were rendered in Photoshop (Adobe).
Protein expression and purification
Recombinant SEPT2/6/7 was expressed by co-transforming Escherichia coli
BL21(DE3) cells with a kanamycin-resistant, bicistronic pET-28a(+) that expresses His-
SEPT7 and SEPT6, and an ampicillin-resistant pET-15b(+) plasmid that expresses an
untagged SEPT2. Bacterial cultures were grown to 0.6 OD and induced with 0.5 mM
IPTG for 16 hours at 18 ˚C. Cultures were centrifuged (4,000 g) for 10 min at 4 ˚C, lysed

126 in buffer containing 50 mM Tris pH 7.5, 150 mM NaCl, 1 mM PMSF, 1 mM DTT, 10 mM
imidazole, and 10% glycerol using a French pressure cell at 1,280 psi. Cell lysates were
centrifuged (14,000 g) for 30 min at 4 ˚C and passed through a 0.45 µm filter before
applying to a gravity flow column with Ni-NTA beads. The columns were washed with
buffer containing 50 mM Tris pH 8.0, 300 mM NaCl, 1 mM DTT, 20 mM imidazole, and
10% glycerol, and SEPT2/6/7 complexes were eluted using buffer containing 50 mM Tris
pH 8.0, 150 mM NaCl, 10% glycerol, and 250 mM imidazole. Protein preps were
dialyzed overnight in dialysis buffer (50 mM Tris, pH 8.0, 150 mM NaCl, 1 mM DTT, and
10% glycerol).
Liposome flotation assays
All lipids were purchased from Avanti Polar Lipids, except PI(4,5)P2 diC8
(Echelon Biosciences; p-4508). L-α-phosphatidylcholine (840051) and L-α-
phosphatidylinositol (840042) were diluted in chloroform. PI(3,4,5)P3 (850156),
PI(3,5)P2 (850154), PI(3)P (850150), PI(4)P (850151), and PI(5)P (850152) were diluted
in a solution made of chloroform, methanol and water (20:9:1). PI(4,5)P2 was diluted in
water. To generate small unilamellar vesicles, lipids were mixed to a final concentration
of 75 %mol L-α-phosphatidylcholine, 20 %mol L-α-phosphatidylinositol, and 5 %mol of
each phosphoinositide. Lipid mixtures were dried completely under a nitrogen stream,
rehydrated in liposome buffer (50 mM Hepes pH 7.2, 50 mM KCl) for 30 min at 25 ˚C,
freeze/thawed five times, and passed through a mini-extruder using 0.1 µm pore filter at
least 12 times to isolate small unilamellar vesicles (SUVs). SUVs were stored at 4˚C and
used within 1-2 days.
To test for septin binding, 0.5 mM SUVs were mixed with 0.5 µM recombinant
SEPT2/6/7 in 50 µl liposome buffer containing 1 mM MgCl2 and incubated with rotation

127 at 25 ˚C for 1 h. Reactions were mixed with 200 µl liposome buffer with sucrose (75%)
and placed at the bottom of a polycarbonate tube (Beckman Coulter). Liposome buffer
(150 µl) with 25% sucrose was gently overlaid and on top of this layer, 50 µL of liposome
buffer was gently placed. Samples were centrifuged (220,000 g) in a SW60 Ti rotor
(Beckman Coulter) for 1 h at 20 ˚C. Equal volumes of the top and bottom fractions were
collected with a thin pipette tip, run on a 10% SDS-PAGE gel and stained with
Coomassie Brilliant Blue. Gels were scanned using the Odyssey imaging system (LI-
COR Biosciences).
In vitro fusion assay
Reconstitution of endosome fusion was performed in a cell-free fluorescence-
based assay as previously described (Barysch et al., 2010). MDCK cells were cultured
on 15 cm plates to 80% confluence, trypsinized and centrifuged (300 g) for 5 min. Cells
were resuspended in cold PBS containing 5 mg/mL BSA (PBS-BSA) and centrifuged
(250 g) for 5 min at 4˚C. The cell pellet was resuspended in cold internalization buffer
(0.2 mg/mL glucose in OPTI-MEM) and centrifuged (250 g) for 5 min at 4˚C. The
supernatant was discarded and the pellets were incubated at 37˚C for 5 min before
resuspending in warm internalization buffer with either FITC- or TR-dextran (0.5 mg/ml)
and incubating for an additional 5 min at 37 ˚C. Cells were transferred to ice and washed
three times with cold PBS-BSA and once with homogenization buffer (250 mM sucrose,
3 mM imidazole pH 7.4) while centrifuging (250 g) for 5 min at 4 ˚C in between washes.
The cell pellet was resuspended in 1 ml homogenization buffer containing protease
inhibitors (Calbiochem), passed through a ball-bearing homogenizer 20 times, and
centrifuged (1,200 g) for 15 min at 4 ˚C. The post-nuclear supernatant (PNS) was
recovered and protein concentration was determined using a Bradford assay (Thermo

128 Scientific). Small aliquots of the post-nuclear supernatants were flash frozen in liquid
nitrogen and stored in -80 ˚C. Canine kidney cytosol (gift from Dr. James Nelson) was
isolated as previously described (Davis et al., 1974). Canine kidney cortex was washed
in cold PBS, cut into 1 cm cubes and homogenized using a Waring blender in a PBS
containing 250 mM sucrose. The homogenate was filtered, centrifuged (100,000 g) to
remove cell membranes, and aliquots of the supernatant were drop flash frozen in liquid
nitrogen and stored at -80˚C.
Fusion reactions were assembled by mixing 5 µg canine kidney cytosol with 0.1
µg of post-nuclear extracts with FITC- and TR-dextran-labeled
macropinosomes/endosomes in homogenization buffer (50 µl) with protease inhibitors
and with or without 3.3 mM ATP. All reactions were incubated at 37 ˚C for 45 min. To
inhibit SEPT2 function, the cytosol was pre-incubated in homogenization buffer
containing 0.5 mg/ml rabbit anti-SEPT2 (N5N) or rabbit IgG (Sigma) for 1 h at 4 ˚C.
SEPT2 was immunodepleted from the cytosol as previous described (Pagano et al.,
2004). Cytosol was diluted to 1 mg/ml in buffer containing 20 mM Hepes pH 7.2, 90 mM
potassium acetate and 2 mM magnesium acetate with protease inhibitors and
subsequently incubated with 5 µg rabbit anti-SEPT2 or rabbit IgG overnight with rotation
at 4 ˚C. Protein A beads (Thermo Scientific) were blocked with 2% BSA overnight,
washed five times with homogenization buffer, and added to the cytosol for 3 hours at
4˚C. Samples were centrifuged (1,000 rpm) for 5 minutes at 4˚C and the supernatant
was collected. SEPT2 protein levels in the cytosol samples were assessed by western
blots.
To assess fusion microscopically, reactions (30 µl) were floated into chambers
created by immobilizing a glass coverslip on a slide with two strips of a double-side tape;
prior to mounting, coverslips were cleaned with 70% ethanol and flame-dried. Chambers

129 were washed with homogenization buffer prior to adding each reaction mix and sealing
with vacuum grease. Slides were centrifuged (300 g) upside-down for 10 min at 25 ˚C
using a swing-bucket rotor with microtiter adapters. Samples were imaged using an
inverted Zeiss AxioObserver Z1 wide-field microscope equipped with a Hamamatsu
Orca-R2 digital CCD camera, X-cite 120 metal halide lamp and a motorized z-drive.
Images were acquired using a 63X/1.4 NA objective and the Slidebook 6.0 software
(Intelligent Imaging Innovations).
Western blots
Cells were lysed in buffer containing 50 mM Hepes, pH 7.4, 100 mM NaCl, 0.5%
Triton X-100, 1 mM MgCl2, 1 mM PMSF and protease inhibitors. Samples were
centrifuged at 8,000 rpm for 10 min at 4˚C and the supernatants were collected. Equal
volumes were resolved with a 10% SDS-PAGE gel, transferred to a nitrocellulose
membrane at 100V for 1 h at 4˚C. Membranes were blocked with 5% dried milk (w/v) in
PBS containing 0.1% Tween-20. A rabbit antibody against SEPT2 (Sigma; 1:5000) and
a secondary anti-rabbit antibody (LiCor) conjugated to infrared dye was diluted in PBS
containing 2% BSA, 0.1% Tween-20, and 0.025% sodium azide, and sequentially
incubated on the membrane before scanning with the Odyssey imaging system (LI-
COR).
In Fig. 5, equal volumes of canine kidney cytosol samples were resolved on a
10% SDS-PAGE gel and transferred to a nitrocellulose membrane. Membranes were
blocked in 5% dried milk (w/v) in PBS containing 0.1% Tween-20. Rabbit antibody
against SEPT2 (Sigma; 1:5000), mouse anti-actin (Sigma, 1:5000), and anti-rabbit or –
mouse secondary antibodies (LiCor) conjugated to infrared dyes were diluted in PBS
containing 2% BSA, 0.1% Tween-20, and 0.025% sodium azide, and sequentially

130 incubated on each membrane before scanning with the Odyssey imaging system (LI-
COR).
Image analysis and quantifications
Image analysis was performed with the Slidebook 6.0 software (Intelligent Imaging
Innovations) unless otherwise noted. In Fig. 1B, the peripheral lamellar regions of cells
were masked by segmentation of F-actin fluorescence and within these regions, the sum
of SEPT2 fluorescence intensity per µm2 was quantified in each cell. In Fig. 2, the
percent of dextran particles with SEPT2 was calculated by creating dextran and SEPT2
masks using fluorescence intensity segmentations. SEPT2 and dextran masks were
subtracted to create a new artificial channel with masks that corresponds to all dextran
fluorescence that overlaps with SEPT2. The particles of this overlay channel were
automatically counted and their number was divided by the total number of dextran
particles to derive the percentage of dextran that colocalizes with SEPT2. The same
approach was used to derive the percentage of Rab5- and Rab7-mCherry compartments
with SEPT2 in peripheral lamellae (Fig. S1) and the percentage of dextran particles with
Rab5, Rab7 and LAMP1 (Fig. 3 and 4); LAMP1 background fluorescence was
subtracted by Gaussian smoothing. In Fig. 2B-D, time-lapse movies of PM-mCherry of
identical capture intervals (5 s) and duration (8 min) were analyzed after applying the 2-
D Laplacian of Gaussian function, which improves the clarity of membrane vacuoles and
tubules. The formation of nascent macropinosomes from membrane vesicles was
tracked manually and clusters of three or more macropinosomes per cell were scored
(Fig. 3B). Macropinosome lifetime was quantified by measuring the time that it took for a
nascent macropinosome to disappear or merge with another membrane vacuole or
tubule (Fig. 3C). The number of nascent macropinosomes that fused with another
membrane compartment was calculated as percentage of total macropinosomes

131 tracked. Number of fusion events per min was the inverse of the time that it took for a
nascent macropinosome to undergo a fusion event (Fig. 3D). In Fig. 3E, 2 µm-long line
masks were drawn along the hemifused membrane contact of two macropinocytic
vacuoles and along their non-contacting perimeters. The intensity profiles of each line
scan was exported and plotted in Excel. In Fig. 3F-G and S2, the number and size of
dextran particles were quantified in Image J (NIH) by generating masks of dextran
fluorescence intensity, which were gated by size (0.2 – 20 µm2) as previously done by
others (Wang et al., 2014). The rates of lysosomal delivery of dextran corresponded to
the slope of the lines derived from a least squares linear regression fit to the 5, 10 and
30 min points of the plot in Fig. 4C. In vitro fusion (Fig. 5) was quantified by generating
fluorescence segmentation masks that correspond to FITC- and TR-dextran-containing
macropinosomes/endosomes. The masks were subsequently subtracted from each
other to identify macropinosomes/endosomes with overlapping fluorescence. The
number of these new masks was automatically calculated and divided by the total
number of macropinosomes/endosomes, which was calculated by subtracting the
number of masks with FITC/TR overlap from the total number of FITC- and TR-labeled
masks.
Statistical analyses
All datasets were analyzed using the Shapiro and Levene tests, respectively, for
normality and equal variance in the R software, and an unpaired student’s t test used to
derive p values (95% confidence intervals) in the Excel software. All values represent
the mean ± SEM.

132 Results and Discussion
Septins associate preferentially with maturing macropinosomes in a PI(3,5)P2-
dependent manner
We previously identified a network of septins fibers in the leading lamellae of
migrating MDCK epithelia (Dolat et al., 2014b). This network is interwoven with actin
stress fibers, but it is reduced upon mild detergent extraction, indicating that a subset of
septins is membrane-bound (Fig. 1, A and B). Given that membrane ruffling activity is
high in cellular lamellae, we imaged the localization of septins in MDCK cells that stably
express PM-mCherry (Corbett-Nelson et al., 2006), a plasmalemmal marker that marks
membrane ruffles, macropinocytic vacuoles and vesicular-tubular endosomes (Fig. S1, A
and B). We stained for endogenous SEPT2 as a proxy for the lamellar network of septin
fibers, which consist of SEPT2/6/7/9 (Dolat et al., 2014b). SEPT2 was largely absent
from membrane ruffles, but localized at the contact sites and membranes of PM-
mCherry-labeled vacuoles (Fig. 1, C and D). Consistent with septin association with
macropinosomes, SEPT2 localized to the periphery and in between compartments that
contained PM-mCherry and dextran, a fluid phase cargo marker that enters cells by
macropinocytosis (Fig. 1E). Time-lapse microscopy of PM-mCherry and SEPT2-GFP
showed that SEPT2 accumulates at the fusion sites of macropinosomes (Fig. 1F, Video
1); SEPT2 was present on 87% ± 12% of macropinosome contact sites (n = 11 cells).
To examine whether SEPT2 associates with early or late stage macropinosomes,
we performed a pulse-chase experiment with fluorescent dextran. SEPT2 co-localization
with dextran peaked after 5-10 min of internalization and diminished after 20-30 min (Fig.
2A), when dextran accumulates in the lysosome (see Fig. 4C). Given that early
macropinosomes contain Rab5 while maturing macropinosomes become enriched with
Rab7 before fusing with lysosomes (Feliciano et al., 2011; Kerr et al., 2006; Kerr et al.,

133 2010), we analyzed SEPT2 localization in cells that expressed Rab5- and Rab7-
mCherry (Fig. S1, C-E). In peripheral lamellae, SEPT2 was present on 64% ± 4% of
Rab7-positive compartments compared to 21% ± 2% of Rab5 (Fig. S1E). SEPT2 was
also present on 72 ± 4% (n = 15 cells) of peripheral vacuoles labeled with the PI(3,5)P2-
binding ML1Nx2-GFP probe (Li et al., 2013), indicating that septins associate with
mature PI(3,5)P2-positive macropinosomes (Fig. 2B). Treatment of MDCK cells with
YM201636, an inhibitor of the FYVE finger containing phosphoinositide kinase (PIKfyve)
that converts PI(3)P to PI(3,5)P2 (Jefferies et al., 2008), reduced the intensity of the
lamellar septin network and SEPT2 localization on dextran-containing macropinosomes
(Fig. 2, C-E). Liposome floatation assays showed that SEPT2/6/7 associates
preferentially with PI(3,5)P2-containing membranes (Fig. 2, F and G). In contrast to
previous results with giant unilamellar vesicles (GUVs) (Tanaka-Takiguchi et al., 2009),
SEPT2/6/7 did not exhibit significant binding to PI(4,5)P2. Given that septin-lipid binding
is sensitive to membrane curvature (Bridges et al., 2016), this discrepancy may arise
from differences in the diameter of liposomes, which is 50-to-200-fold smaller than
GUVs. Our results nevertheless indicate that septins associate preferentially with mature
Rab7- and PI(3,5)P2-positive macropinosomes.
Septin depletion impedes the maturation and turnover of macropinosomes, and
reduces macropinosome fusion events
We sought to determine how septins affect macropinosome formation and
dynamics by imaging PM-mCherry in live MDCK cells, which were treated with control or
SEPT2 shRNAs that deplete SEPT2, but also SEPT6, 7 and 9 (Dolat et al., 2014b). In
control cells, membrane ruffles gave rise to multiple macropinosomes, which fused into
single vacuoles or dissipated within seconds of formation (Fig. 3A, Video 3). In SEPT2-

134 depleted cells, membrane ruffling and macropinosome formation were not affected.
Strikingly, however, SEPT2 knock-down resulted in clusters of numerous
macropinosomes (Fig. 3A, Video 3). Clusters of three or more macropinosomes were
present in four-fold more cells upon SEPT2 depletion and the average lifetime of
nascent macropinosomes increased by 1.7-fold (Fig. 3, B and C). Notably, several
macropinosomes appeared docked with their membranes adjoining laterally (Fig. 3E).
In agreement with a slower macropinosome turnover, dextran uptake assays
showed a significant increase in the number and size of dextran particles in SEPT2-
depleted cells (Fig. 3, F and G). Notably, this increase in number and size persisted after
30 and 60 min of dextran internalization, indicating a defect in the macropinocytic traffic
of dextran (Fig. 3, F and G). Similar results were obtained with a different SEPT2 shRNA
(Fig. S2, A and B) and in human fibrosarcoma HT1080 cells (Fig. S2, C and D), which
have intense membrane ruffling activity due to the expression of an oncogenic mutant of
N-ras (Paterson et al., 1987). Expression of an shRNA-resistant SEPT2-YFP rescued
the phenotype, reversing the number and size of macropinosomes to control levels (Fig.
S2B).
Because membrane tubulation and fusion events underlie the size reduction of
maturing macropinosomes, we analyzed macropinosome membrane dynamics in
MDCK-PM-mCherry cells. Septin depletion did not alter the percentage (~40%) of
macropinosomes that formed membrane tubules and had no effect on their detachment
(Fig. 3H, Video 4). Membrane fusion, however, was severely affected. The percentage
of nascent macropinosomes that fused with another macropinosome was reduced from
74% to 25% (n = 9 cells). Moreover, the frequency of fusion events between nascent
macropinosomes and tubulo-vesicular endosomes decreased markedly (Fig. 3D). Thus,

135 septins are required for the membrane fusion events underlying the shrinkage and
turnover of macropinosomes.
Septins regulate fluid phase cargo traffic and delivery to the lysosome
Given that macropinosomes transport fluid phase cargo from the plasma
membrane to the lysosome, we sought to determine whether septins affect the endocytic
trafficking and lysosomal delivery of extracellular dextran. Using a pulse-chase
approach, MDCK cells were pulse-labeled with fluorescent dextran and its localization
with respect to Rab5, Rab7 and the lysosomal associated membrane protein 1 (LAMP1)
was analyzed after 5, 10 and 30 min, respectively. Septin depletion did not affect dextran
colocalization with Rab5 and Rab7 (Fig. S2, E-H). However, the percentage of dextran
particles that colocalized with LAMP1 decreased by >60% and this defect was rescued
by shRNA-resistant SEPT2-YFP (Fig. 4, A and B). These data suggest that septins are
required for the delivery of dextran to lysosomes, but do not affect dextran traffic to
Rab5- and Rab7-positive endosomes or the recruitment of Rab5/Rab7 to dextran-
containing macropinosomes.
To further test whether septins regulate the lysosomal delivery of fluid phase
cargo, we analyzed kinetically the accumulation of dextran in lysosomes. Dextran
colocalization with LAMP1 was quantified after 5, 15 and 30 min of internalization under
conditions of septin depletion and over-expression. Between 5 and 30 min of
internalization, SEPT2 depletion decreased the rate of lysosomal delivery from 2.34% to
1.02% of total dextran per min (Fig. 4C). Notably, the percentage of dextran colocalizing
with LAMP1 in SEPT2-depleted cells did not change between 30 and 60 min of
internalization (45 ± 6% vs. 50 ± 4%), which was indicative of a block in the traffic of fluid
phase cargo.

136 Next, we tested the effects of SEPT2 over-expression using stable MDCK cell
lines that express SEPT2-YFP at low and high levels (Fig. 4D). In the cells that express
SEPT2-YFP at low levels, dextran delivery to the lysosome was similar to cells treated
with control shRNAs (Fig. 4E). High levels of SEPT2-YFP expression, however,
increased the efficiency of dextran delivery to the lysosome (Fig. 4E). Thus, lysosomal
delivery of fluid phase cargo is dependent on the expression levels of SEPT2.
Septins promote fusion of macropinosomes with endosomes/lysosomes.
To test whether septins regulate lysosomal delivery of fluid phase cargo by
directly modulating membrane fusion, we performed dual color dextran mixing assays in
live cells and in vitro using a reconstitution assay of endosomal fusion (Barysch et al.,
2010).
In MDCK cells, we tested how septin knock-down affects the delivery of FITC-
dextran into a cohort of endosomes/lysosomes that were pre-loaded with TexasRed
(TR)-dextran. After a 5 min pulse/15 min chase with TR-dextran, we performed 5 min
pulse/15 min chase with FITC-dextran to assess its macropinocytic traffic into TR-
dextran/LAMP1-positive lysosomes. Septin depletion decreased FITC-dextran entry into
lysosomes with TR-dextran by 50% (Fig. 5, A and B).
Next, we performed a cell-free assay of macropinosome/endosome fusion using
post-nuclear extracts from two MDCK cell populations, which were labeled separately
with TR- and FITC-dextran; fusion was induced with canine kidney cytosol and ATP and
assayed microscopically by quantifying the fraction of endosomes with both TR- and
FITC-dextran (Fig. 5, C and D). Addition of a SEPT2 function-blocking antibody or
immunodepletion of SEPT2 from the cytosol abrogated fusion (Fig. 5, C-F). Surprisingly,
recombinant SEPT2/6/7 and ATP induced fusion in the absence of cytosol (Fig. 5G). Of

137 note, addition of recombinant SEPT2/6/7 to the cytosol did not increase fusion, possibly
due to saturation by cytosolic septins. Cytochalasin D, an actin depolymerizing
compound, had no effect on the endosome fusion induced by SEPT2/6/7 in the absence
of cytosol and did not alter fusion in the presence of cytosol (Fig. 5, D and G). Similarly,
fusion was not altered by the microtubule depolymerizing drug nocodazole. Thus,
septins can directly induce membrane fusion independently of F-actin and microtubules.
Taken together, our results show that septins promote the membrane fusion
events that underlie the maturation of macropinosomes and the delivery of fluid phase
cargo to lysosomes. This is the first evidence for a direct role of septins in clathrin-
independent endocytosis and the fusion of endomembranes. In the context of
exocytosis, septins interact with SNARE proteins and the exocyst complex (Beites et al.,
1999; Hsu et al., 1998; Ito et al., 2009; Taniguchi et al., 2007). The SNARE and vesicle
tethering complexes that mediate the fusion events of the macropinocytic pathway are
largely unknown. The exocyst Sec6/8 complex localizes to the endosomes of MDCKs
(Oztan et al., 2007), but septins did not colocalize with Sec6/8 or Exo70 at the contact
sites of macropinosomes/endosomes (Fig. S3, C-E). Septin knock-down also did not
affect the localization of Vps39 (Fig. S3, F and G), a component of the HOPS vesicle-
tethering complex (Balderhaar and Ungermann, 2013). Because septin depletion
increases dramatically the fraction of docked macropinosomes/endosomes (Fig. 2),
septins appear to play a distinct role in membrane fusion without affecting membrane
tethering and/or docking. Notably, this septin role is coupled to PI(3,5)P2, a low
abundance phosphoinositide that is specifically involved in the fusion of mature
macropinosomes/endosomes with lysosomes (Kerr et al., 2006; McCartney et al., 2014).
Thus, similar to the evolutionarily related Rab GTPases, septin GTPases function as
phosphoinositide effectors that catalyze SNARE-mediated fusion. Future studies will

138 determine whether this function involves the stabilization of specific SNARE complexes
and the induction or sensing of membrane curvature, which promotes lipid mixing
(Martens and McMahon, 2008).
Oncogenic proteins of the Ras family of GTPases are long known to up-regulate
membrane ruffling and macropinocytosis, which in turn increases nutrient uptake to
support the metabolic needs of growing cancers (Kimmelman, 2015; White, 2013).
Moreover, lysosomal delivery of fluid phase cargo (e.g., amino acids) is critical for the
activation of signaling pathways that regulate cell growth and division (Commisso et al.,
2013; Settembre et al., 2013). Septins are over-expressed in many cancers, but their
functions in cancer are poorly understood (Connolly et al., 2011a; Dolat et al., 2014a).
Because SEPT2 alone does not induce fusion in vitro (Fig. 5G) and SEPT2-YFP over-
expression does not alter the expression levels of SEPT6/7 (data not shown), we posit
that over-expression of a single septin (e.g., SEPT2) may promote the formation of
membrane-bound SEPT2/6/7 polymers, which could functionally support the elevated
macropinocytic activity of cancer cells. Future studies may explore how this novel
function of septins might be therapeutically targetable.
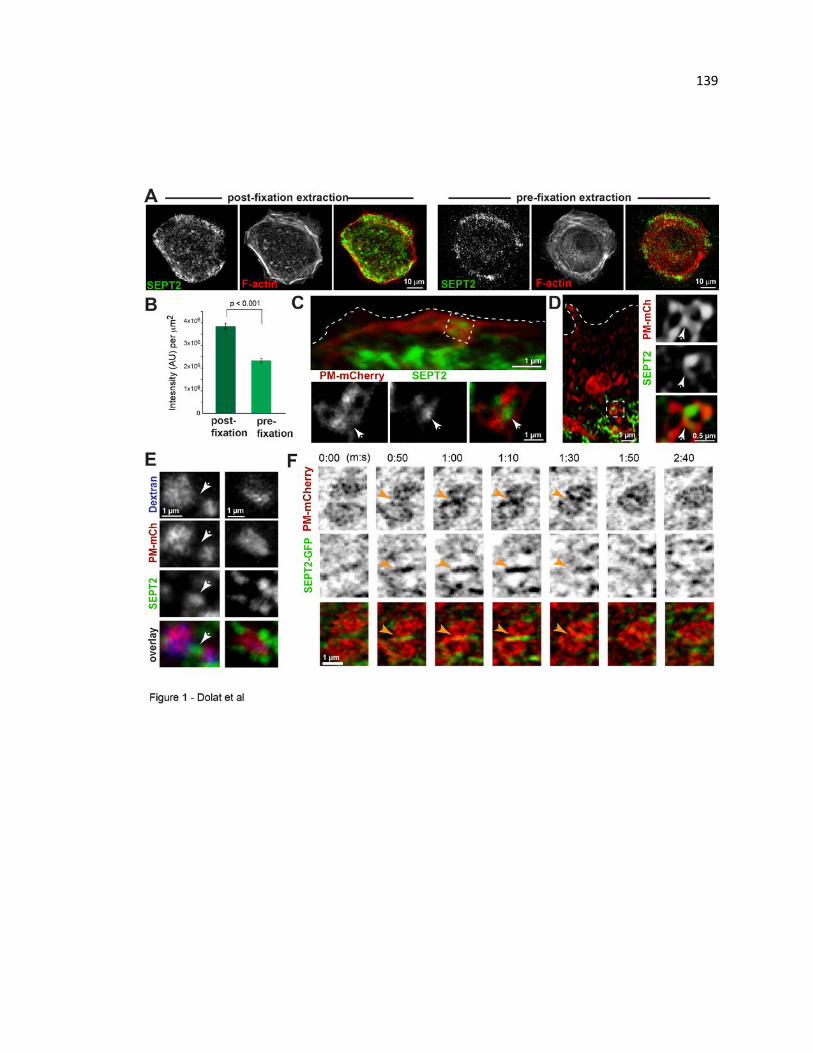
139

140 Figure 7.1. Septins localize to macropinocytic vacuoles and tubulo-vesicular
endosomes, and their contact/fusion sites. (A) Confocal images of MDCKs stained
for SEPT2 and F-actin with or without pre-extration with 0.1% Triton-X100 prior to
fixation. (B) Bar graph shows the sum intensity of SEPT2 per cell area (n = 15). (C-D)
3D-rendered confocal images of MDCK-PM-mCherry cells stained for endogenous
SEPT2. Arrows point to SEPT2 at the contact sites between macropinocytic vacuoles
(C) and tubulo-vesicular endosomes (D). (E) MDCK-PM-mCherry cells were incubated
with FITC-dextran and stained for SEPT2. Images show SEPT2 at the periphery and in
between (arrow) macropinosomes/endosomes. (F-G) MDCK-PM-mCherry cells were
transfected with SEPT2-GFP and imaged live with wide-field deconvolution microscopy.
Still frames show SEPT2 accumulation (arrows) at the site of fusion between two
macropinosomes (F) and the contacting membranes of a macropinosome and a tubular
endosome (G).

141

142 Figure 7.2. Septins associate preferentially with maturing macropinosomes in a
PI(3,5)P2-dependent manner. (A) MDCKs were pulsed with TR-dextran for 5 min,
chased for the indicated times, stained for SEPT2 and imaged with confocal microscopy.
Bar graph shows the percent of dextran-containing macropinosomes/endosomes with
SEPT2 (n = 12 cells). (B) Recombinant SEPT2/6/7 was mixed with liposomes of the
indicated phosphoinositides and centrifuged on a sucrose gradient. Coomassie-stained
gels show equal volumes from the top (liposome-bound SEPT2/6/7; B) and bottom
(unbound SEPT2/6/7; U) fractions. (C) Maximum intensity projections of confocal images
of MDCKs, which were incubated with DMSO (control) or YM201636 (800 nM) for 2 h,
and treated with TR-dextran for 10 min before fixing and staining for SEPT2. Insets show
outlined areas in higher magnification. (D) Bar graph shows the fraction of TR-dextran
puncta (>0.2 µm2) with SEPT2 (n = 20 cells).
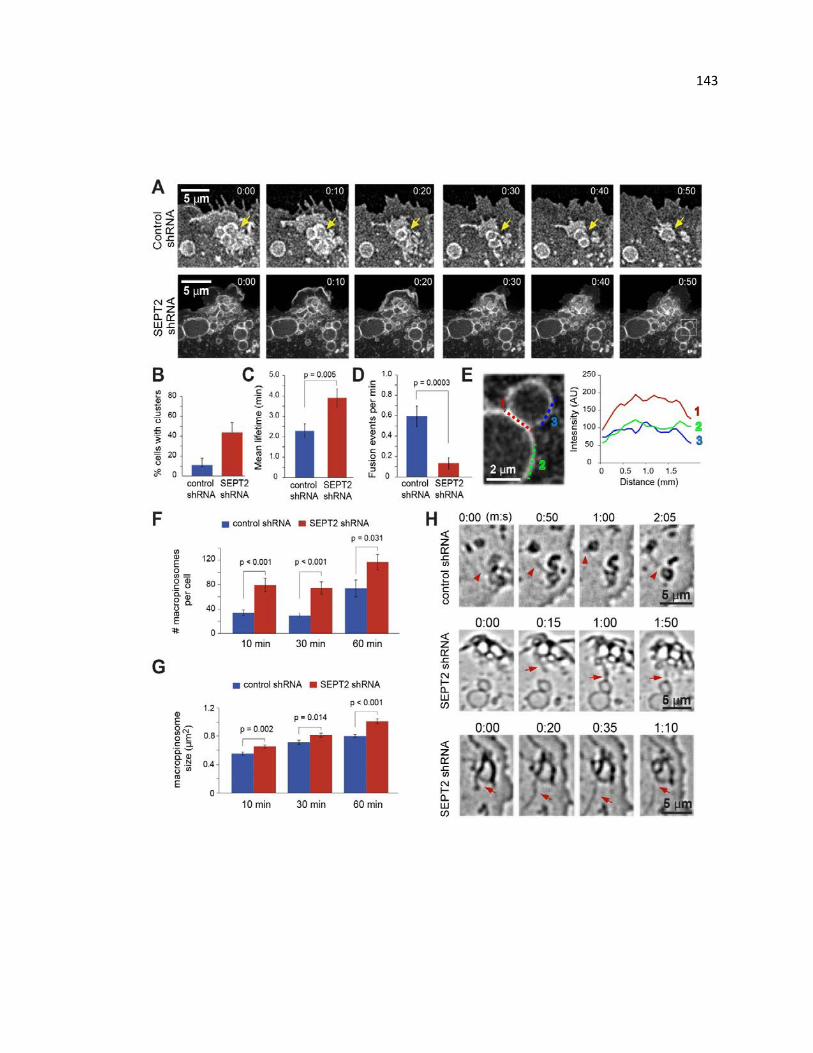
143

144 Figure 7.3. Septin depletion decreases macropinocytic fusion events and hinders
macropinosome maturation and turnover. (A) Time-lapse frames from widefield
deconvolution microscopy show the dynamics of PM-mCherry-labeled macropinosomes
in MDCK cells treated with control and SEPT2 shRNAs. (B) Control (n=41) and SEPT2-
depleted MDCK-PM-mCherry cells (n=31) were imaged live every 5 s for 8 min. Bar
graph shows percent cells with a cluster of three or more macropinosomes formed from
a lamellar ruffle. (C - D) Bar graphs show the lifetime (C) and fusion events (D) of
nascent macropinosomes (n = 24-27). (E) Quantification of PM-mCherry fluorescence
along the lateral hemi-fused contact (1) and non-contacting segments (2, 3) of two
macropinosomes from a SEPT2-depleted cell. (F-G) MDCKs were incubated with FITC-
dextran for the indicated times. Bar graphs show the number of FITC-dextran-containing
macropinosomes/endosomes (F) and their average size (G) per cell (n=18). (H) Still
frames show the formation and detachment of PM-mCherry-labeled membrane tubules
(arrows) in live MDCKs.
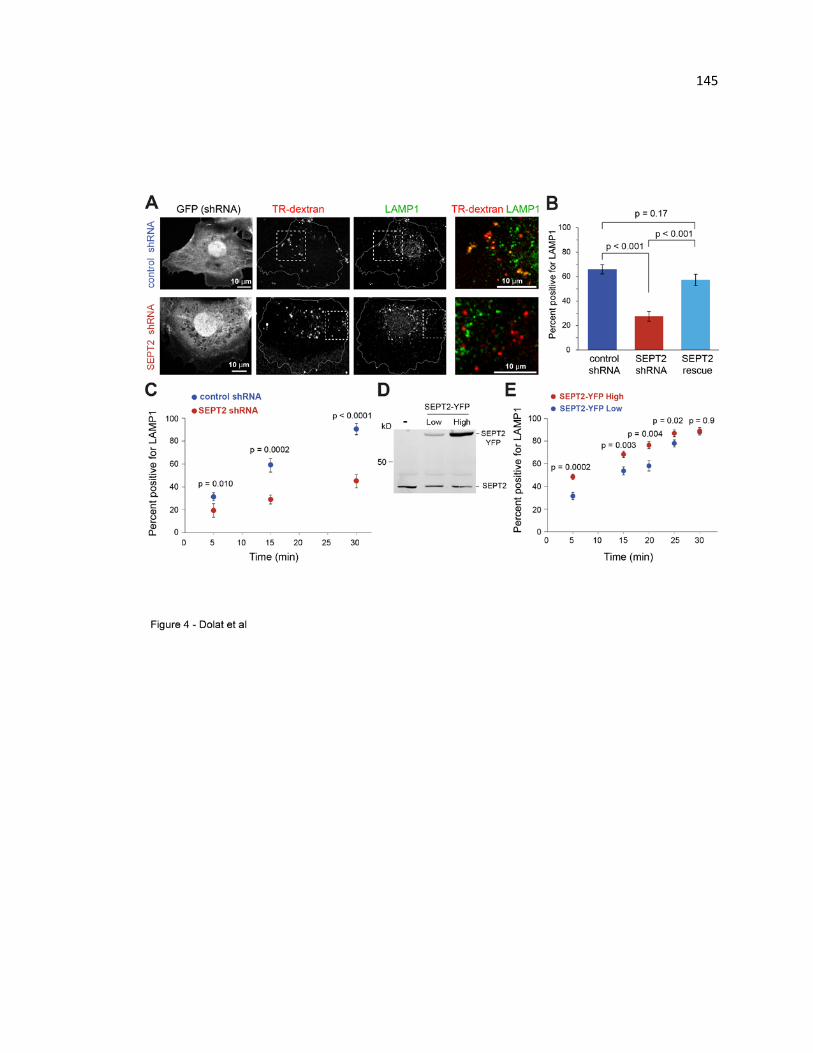
145

146 Figure 7.4. Septins regulate the lysosomal delivery of fluid phase cargo. (A)
MDCKs were transfected with GFP-expressing control or SEPT2 shRNAs for 48 h. After
a 5 min pulse/15 min chase with TR-dextran, cells were stained for LAMP1. Images
show maximum intensity projections of confocal image stacks. (B) Bar graph shows
percent of TR-dextran particles that colocalize with LAMP1 (n = 20 cells). (C) Plot shows
percent of TR-dextran with LAMP1 in control and SEPT2-depleted MDCKs (n=15), which
were pulsed with TR-dextran for 5 min and chased for the indicated times. (D) Lysates
from MDCKs that stably express SEPT2-YFP at low and high levels were analyzed by
SDS-PAGE and blotted for SEPT2. (E) Plot shows percent TR-dextran with LAMP1 in
MDCK cells (n=14) that express SEPT2-YFP at low and high levels. Cells were pulsed
with TR-dextran for 5 min and chased for the indicated times.
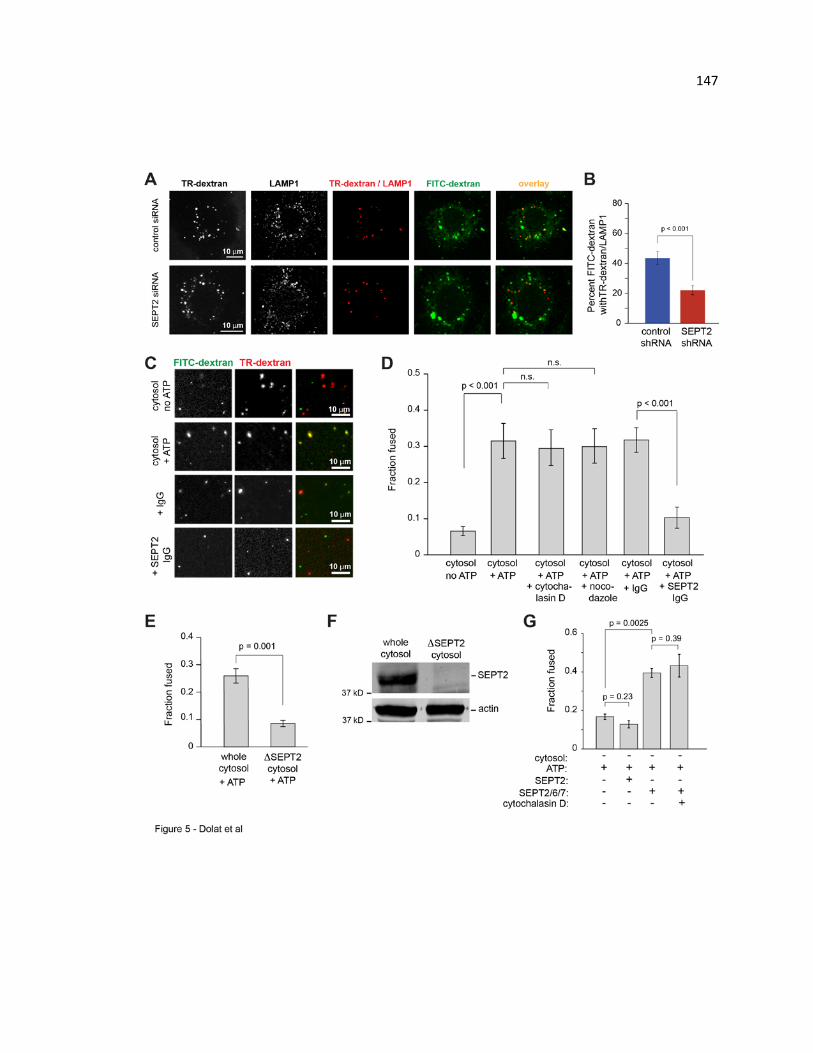
147

148 Figure 7.5. Septins are required for fusion of macropinosomes/endosomes with
lysosomes, and promote fusion directly in an in vitro reconstitution assay. (A)
MDCKs were treated with control or SEPT2 siRNA, and two rounds of 5 min pulse/15
min chase were performed with TR- and FITC-dextran, successively. Cells were stained
with anti-LAMP1 and imaged with confocal microscopy. Images show maximum intensity
projections of TR-dextran, LAMP1 and their areas of overlap (pseudo-colored in red) as
well as FITC-dextran (green), and its overlay with the pseudo TR-dextran/LAMP1
channel. (B) Bar graph shows percent TR-dextran/LAMP1 that contains FITC-dextran (n
= 18 cells). (C-D) MDCKs were separately incubated with TR- or FITC-labeled dextran
for 5 min, and post-nuclear supernatants (PNS) were mixed with canine kidney cytosol
with/without ATP, and with ATP plus cytochalasin D (cyto D; 10 µM), nocodazole (10
µM), control or anti-SEPT2 IgG. Images (C) show dextran-containing
macropinosomes/endosomes after incubation under the indicated conditions. Bar graph
(D) shows the fraction of fused macropinosomes/endosomes with both TR- and FITC-
labeled dextran (n = 15 images). (E) Bar graph shows the fraction of fused endosomes
after incubation of PNS with ATP and whole or SEPT2 immunodepleted (∆SEPT2)
cytosol (n = 15). (F) Gel shows equal volumes of whole and ∆SEPT2 cytosol blotted for
SEPT2 and actin. (G) Bar graphs show the fraction of fused endosomes after incubation
of PNS with/without recombinant SEPT2/6/7 in the presence/absence of 10 µM cyto D (n
= 15).

149

150 Figure 7.6. PM-mCherry and SEPT2 localization with respect to Rab5 and Rab7. (A-
B) MDCK-PM-mCherry cells were transfected with Rab5-GFP (A) and Rab7-GFP (B),
fixed and imaged by confocal microscopy. Images show 3D reconstructions from
confocal z-stacks. Outlined regions are shown in higher magnification. Arrows point to
vesicular endosomes with Rab5 or Rab7 and PM-mCherry. Arrowheads point to tubular
endosomes with Rab7 and PM-mCherry. (C-D) Confocal microscopy images show
MDCK cells that were transfected with Rab5 or Rab7-mCherry, fixed and stained for
endogenous SEPT2. Outlined regions are shown in higher magnification and 3D
reconstructions of the numbered endosomes (arrows) are shown in higher magnification.
(E) Bar graph shows the percentage of Rab5- and Rab7-mCherry-positive
compartments with SEPT2 (n = 17-18 cells).
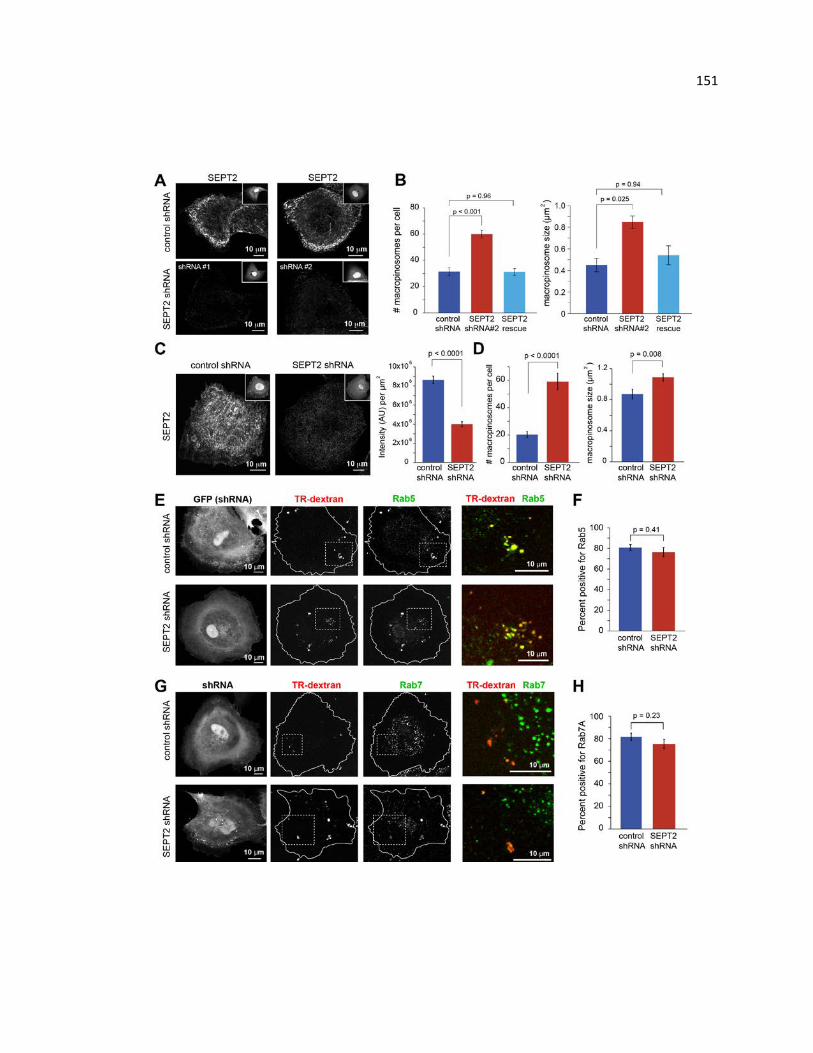
151

152 Figure 7.7. SEPT2 knock-down increases macropinosome size and number, but
does not disrupt colocalization of macropinocytic cargo with Rab5 and Rab7. (A)
MDCKs were transfected with plasmids that express GFP or mCherry and non-targeting
or SEPT2 shRNAs (GFP/shRNA #1, mCherry/shRNA #2) for 48 h. Cells were fixed and
stained for endogenous SEPT2. Images show maximum intensity projections of SEPT2
fluorescence from 3D confocal microscopy images; insets show GFP/mCherry
fluorescence. (B) MDCKs were treated with control or SEPT2 shRNA # 2, and incubated
with FITC-dextran for 10 min. Bar graphs show the number of FITC-dextran-containing
macropinosomes/endosomes (puncta >0.2 m2) and their average size per cell (n = 21
cells). (C) HT1080 cells were transfected with plasmids that express GFP and control or
SEPT2 shRNA, and stained for endogenous SEPT2. Images show maximum intensity
projections of SEPT2 and GFP (insets) fluorescence. Bar graph shows the quantification
of SEPT2 intensity per cell area (n = 20 cells). (D) Control and SEPT2-depleted HT1080
cells were incubated with TR-dextran for 10 min. Bar graphs show the number of TR-
dextran-containing macropinosomes/endosomes (puncta >0.2 m2) and their average
size per cell (n = 20 cells). (E – F) MDCKs were transfected with GFP-expressing control
or SEPT2 shRNAs for 48 h. Cells were pulsed with TR-dextran for 5 min and stained for
endogenous Rab5 (E) and Rab7 (F) after a 5 and 10 min chase, respectively. Images
show maximum intensity projections of confocal image stacks. Outlined regions with
merged fluorescence are shown in higher magnification. Bar graphs show percent of TR-
dextran particles that colocalize with Rab5 (E) and Rab7 (F) (n = 20 cells).
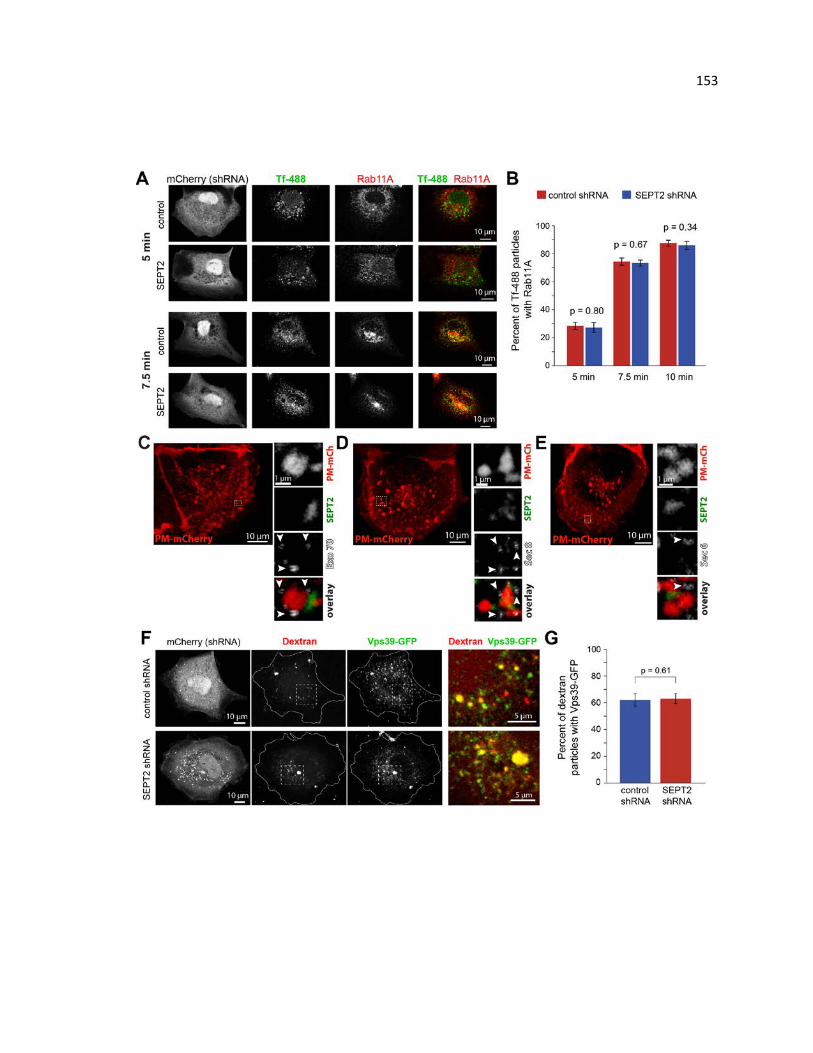
153

154 Figure 7.8. SEPT2 does not colocalize with Sec6/8 and Exo70 at the contact sites of macropinosomes/endosomes. (A – C) Images show 3D reconstructions of confocal
image stacks of MDCK-PM-mCherry cells stained for endogenous SEPT2 and Exo70
(A), Sec8 (B) or Sec6 (C). Outlined regions are shown in higher magnification (scale
bars, 1 µm). Green arrows point to SEPT2 at the sites of contact between PM-mCherry-
labeled macropinosomes/endosomes, and blue arrowheads point to the localization of
Exo70, Sec8 or Sec6.

155
CHAPTER 8. Conclusions and future directions
This dissertation focuses on the function of septin GTPases in epithelial cell
migration and macropinocytosis. While septins have been shown previously to associate
with the actin cytoskeleton and cell membranes, the molecular requirements have
remained poorly understood. Moreover, septins are frequently overexpressed in
epithelial cancers but the role of septins in tumor cell growth and metastasis is unknown.
In this dissertation, I uncovered a mechanism by which septins interact directly with the
actin cytoskeleton and promote epithelial migration and invasion. In addition, I
discovered that septins associate with macropinosomes in a PI(3,5)P2-dependent
manner and regulate the delivery of macropinocytic cargo to endolysosomes, a
molecular pathway that promotes tumor cell growth.
Septins in epithelial cell migration
Previous studies have shown that septins exhibit an interdependent relationship
with the actin cytoskeleton (Kinoshita et al., 1997), but whether septins bind directly to
actin was unknown. In the first part of this thesis, I discovered that septins localize to the
contractile transverse actin fibers and their points of connection with radial actin fibers.
Septin knock-down abolishes the actin stress fiber network by reducing the number of
radial fibers and thinning the transverse actin fibers. Significantly, I found that SEPT9
directly crosslinks actin filaments and interferes with the binding of the myosin motor
domain in its low-affinity state and cofilin, an actin-severing protein. Thus, septins are
bona fide actin cross-linking proteins that may protect nascent actin filaments from
depolymerizing forces, such as contractile myosin filaments and actin-severing proteins,
during epithelial cell migration.
Because septins are overexpressed frequently in cancers, I assayed for changes
in septin expression during epithelia-to-mesenchymal (EMT) transition, a critical step in

156 tumor cell invasion. I found a significant increase in SEPT9 expression and tested the
effects of SEPT9 overexpression on the motile properties of epithelia undergoing EMT.
Indeed, SEPT9 overexpression promotes epithelial cell migration in 2D and 3D
environments, while SEPT9 knockdown or pharmacological targeting of septins inhibits
cell migration. Taken together, my findings indicate that septins promote epithelial cell
migration by directly cross-linking actin filaments and regulating the assembly and
organization of the contractile actin stress fiber network.
Future studies for septins in actin filament assembly and organization
In the leading edge of motile epithelia, the actin stress fiber network is
characterized by the assembly radial actin fibers, which are anchored to focal adhesions,
and transverse arc stress fibers, which are crosslinked by contractile myosin II filaments.
Through the linkage of these two stress fiber networks, forces are transduced to focal
adhesions to promote forward translocation of the cell. My data indicate that septins are
essential for the assembly of a cohesive stress fiber network and may regulate force
transduction during cell migration. To test this hypothesis, the forces exerted upon focal
adhesions can be measured experimentally using atomic force microscopy and gel
contraction assays, which provide direct and indirect readouts, respectively.
Furthermore, radial actin fibers link up with transverse actin fibers in an
orthogonal manner. My preliminary studies in vitro show that recombinant SEPT9 can
crosslink and promote the orthogonal organization of purified actin filaments (Appendix
1.1). Actin filaments alone assemble into short linear filaments (Appendix 1.1B).
However, when co-polymerized with recombinant SEPT9, actin filaments appear thicker,
which suggests that they are crosslinked, and are arranged in a more branched-like
organization with orthogonal linkages between filaments (Appendix 1.1C).

157
More work is necessary to better understand how SEPT9 can promote this
geometric arrangement of actin filaments. Because SEPT9 can bind to three different
sites on the actin filament, it is possible that SEPT9 promotes orthogonal interactions
when bound to a specific site. High-resolution electron microscopy studies can reveal
the precise localization of SEPT9 on orthogonal actin filaments and how its position on
the actin filament may promote this geometry. Moreover, SEPT9 is a component of the
SEPT2-6-7-9 octamer and likely functions within the cell as part of a molecular complex,
but how SEPT9 binds to actin when in an octameric filament has not been determined.
The SEPT2-6-7 crystal structure shows a hinged region at the SEPT2 interface, which
provides flexibility to the filament (Sirajuddin et al., 2007), and could promote the
branched and orthogonal arrangement of actin filaments.
In addition, I showed that SEPT6 promotes the recruitment of cortactin to the
lamellipodium, a branched network of actin filaments (Hu et al., 2012). Co-
polymerization of actin with the recombinant SEPT2-6-7 complex results in curved and
circular actin filaments (Mavrakis et al., 2014). Interestingly, the Arp2/3 complex
preferentially initiates nascent filament assembly on curved actin filaments (Risca et al.,
2012). Thus, septins may promote branched actin assembly by favoring geometrically
the spatial localization of Arp2/3 activity. Moreover, yeast two-hybrid experiments
identified that SEPT6 interacts with the Arp2 of the Arp2/3 complex (Nakahira et al.,
2010), but we were unable to identify this interaction in cells or biochemically. Future
studies involving co-polymerization of actin with the Arp2/3 complex are necessary to
better understand the role of septins in branched actin assembly. Here, testing the
effects of recombinant septins in an actin pyrene assay, which provides a fluorescence
readout for actin polymerization, can reveal whether septins promote directly actin
polymerization.

158 Future studies for septins in integrin trafficking
The assembly of radial actin fibers is initiated by the engagement of integrin
receptors with ECM proteins (Parsons et al., 2010). Cytosolic tails of engaged, or
activated, integrin receptors recruit actin nucleation factors that polymerize actin
filaments and promote focal adhesion assembly and maturation. Thus far, two integrin
trafficking pathways have been identified: fast Rab4-dependent recycling and slow
Rab11-dependent recycling (Caswell and Norman, 2006). Significantly, the trafficking of
integrins is essential for tumor cell migration and invasion.
Based on previous studies and my preliminary data, I hypothesize that septins
may also have a role in integrin trafficking. Notably, Rab11 localizes to the golgi complex
and regulates the delivery of exocytic vesicles to the plasma membrane (Chen et al.,
1998). Septins localize to post-golgi microtubules and regulate the trafficking of
intracellular vesicles to the plasma membrane. Thus, septins may also promote the
trafficking of Rab11-dependent integrins to the membrane where they can engage with
ECM proteins. Alternatively, my preliminary studies show that septins colocalize with
juxtanuclear Rab4-positive vesicles (Appendix 1.2) in Cos7 cells, a renal fibroblast-like
cell line. Future studies investigating the role of septins in intracellular trafficking of
integrin receptors may unveil a second mechanism by which septins regulate cell
migration.
Septins in tumor cell migration and metastasis
In Chapter 5, I tested the effects of septin depletion on epithelial cell migration in
3D collagen matrices, which better recapitulates the physiological environment.
Interestingly, septin depletion transitions the cell to a more rounded and protrusive
shape and slows significantly cell motility, suggesting that septins may have a role in the
mechanism that regulates the mode of tumor cell motility. Future studies using

159 biomimetic cell-derived matrices derived from fibroblasts, which better mimic the tumor
microenvironment, will provide a better context for studying tumor cell migration. In these
matrices, tumor cells can transition between three different modes of migration:
mesenchymal-like, amoeboid and lobopodial. While the molecular pathways that
regulate the transition between these modes is poorly understood, septins may play a
role in the spatial organization of the actomyosin cytoskeleton, whose contractile activity
regulates focal adhesion dynamics during mesenchymal-like motility and the protrusive
behavior in amoeboid and lobopodial motility.
Ultimately, it will be necessary to test how septin expression affects tumor cell
motility in vivo using a mouse tumor model, which can be visualized with intravital
imaging, and whether pharmacological targeting of septins inhibits metastasis.
Collectively, these studies will report on the efficacy septin-targeting therapeutics in
cancer.
Septins in membrane fusion dynamics
From yeast to mammals, membrane-associated septins are posited to restrict the
lateral diffusion of membrane proteins and promote the compartmentalization of the
plasma membrane (Caudron and Barral, 2009). Septins also modulate exocytic events
at the plasma membrane (Tokhtaeva et al., 2015), but how septins function in endocytic
membrane traffic and dynamics is little understood. In Chapter 7, I show that septins
associate with macropinosomes in a PI(3,5)P2-dependent manner. Interestingly,
PI(3,5)P2 is a low-abundance lipid that localizes exclusively to late endosomes and
lysosomes and promotes macropinosome fusion (Kerr et al., 2010). In agreement with
this finding, septins colocalize with Rab7-positive late endosomes and control the
delivery of macropinocytic cargo to lysosomes. Moreover, reconstitution of vesicle fusion
in vitro shows that septin inhibition and immunodepletion blocks vesicle fusion, while the

160 addition of recombinant septins promotes vesicle fusion. Thus, septins are an essential
component of the molecular machinery that regulates membrane fusion.
Vesicle fusion dynamics
Membrane fusion relies on the coordinated recruitment of vesicle adaptor
proteins, which can recognize specific lipid domains, and the assembly and disassembly
of SNARE proteins between two vesicles. In addition, the microtubule and actin
cytoskeleton play intricate roles in localizing and docking intracellular vesicles,
respectively, to promote vesicle fusion.
In Chapter 7, I reconstituted vesicle fusion in vitro using a crude post-nuclear
supernatant that contained fluorescently-labeled endosomes, whose lipid composition
and constituent adaptor protein population was unknown. However, I was able to rule
out the role of the actin and microtubule cytoskeleton in this assay through
pharmacological inhibition. Thus, I hypothesize that septins function mechanistically at
the level of SNARE-mediated membrane fusion.
Septins were reported to interact directly with SNAREs at the plasma membrane
and inhibit exocytosis (Beites et al., 1999). However, the SNAREs constitute a large
family of proteins that localize to specific intracellular membrane compartments. The
SNAREs that promote macropinosome fusion with lysosomes are unknown, but
syntaxin-7 is important for lysosome fusion and syntaxin-17 regulates autophagosome
fusion with lysosomes (Itakura et al., 2012; Jahn and Scheller, 2006).
At a mechanistic level, septins may function at the level of SNARE assembly
and/or endosomal membrane organization, which in turn can control SNARE dynamics.
To address the first question, biochemical analyses of septin-SNARE interactions and
vesicle fusion assays using purified liposomes reconstituted with SNARE proteins will
provide a better readout for septin functions during membrane fusion. In this assay,

161 fluorescently-labeled liposomes containing donor and acceptor SNAREs are mixed
together and assayed microscopically. If septins promote membrane fusion through a
SNARE-dependent mechanism, the addition of recombinant septins could result in more
vesicle fusion by promoting SNARE assembly, which tethers vesicles together, or
SNARE disassembly, which drives membrane fusion (Jahn and Scheller, 2006).
Alternatively, septins may function at the level of membrane organization. By
compartmentalizing the plasma membrane, septins are required for the organization of
PI(4,5)P2-rich domains that organize the Orai1 calcium channel at ER-PM contact sites
(Sharma et al., 2013a). Moreover, septins were reported recently to bind preferentially to
highly-curved membranes, which are present on the plasma membrane and
endomembranes. Interestingly, both lipid domain organization and membrane curvature
can regulate the clustering of SNAREs into distinct membrane domains, which is
necessary for vesicle docking and fusion. High resolution microscopy, such as
superresoluation microscopy, which can provide a lateral resolution of ~ 20 – 100 nm,
and electron microscopy can report on the localization of septins with respect to
SNAREs on the endosomal membrane. In addition, testing the effects of septin depletion
on endolysosomal SNAREs will report on their assembly and clustering into discrete
domains on the endomembrane.
Taken together, these experiments could uncover the mechanism by which
septins regulate vesicle fusion, and whether septins have a more universal role in the
delivery of cargo from other endocytic pathways to the lysosome.

162
LIST OF REFERENCES Adam, J.C., J.R. Pringle, and M. Peifer. 2000. Evidence for functional differentiation among
Drosophila septins in cytokinesis and cellularization. Molecular biology of the cell. 11:3123-3135.
Ageta-Ishihara, N., T. Miyata, C. Ohshima, M. Watanabe, Y. Sato, Y. Hamamura, T. Higashiyama,
R. Mazitschek, H. Bito, and M. Kinoshita. 2013. Septins promote dendrite and axon development by negatively regulating microtubule stability via HDAC6-mediated deacetylation. Nat Commun. 4:2532.
Ahuja, P., E. Perriard, W. Trimble, J.-C. Perriard, and E. Ehler. 2006. Probing the role of septins in
cardiomyocytes. Experimental cell research. 312:1598-1609. Amir, S., R. Wang, J.W. Simons, and N.J. Mabjeesh. 2009. SEPT9_v1 up-regulates hypoxia-
inducible factor 1 by preventing its RACK1-mediated degradation. The Journal of biological chemistry. 284:11142-11151.
Aoyagi, K., T. Sugaya, M. Umeda, S. Yamamoto, S. Terakawa, and M. Takahashi. 2005. The
activation of exocytotic sites by the formation of phosphatidylinositol 4,5-bisphosphate microdomains at syntaxin clusters. The Journal of biological chemistry. 280:17346-17352.
Baek, S.M., J.S. Ahn, H.S. Noh, J. Park, S.S. Kang, and D.R. Kim. 2010. Proteomic analysis in
NSAIDs-treated primary cardiomyocytes. J Proteomics. 73:721-732. Bai, X., J.R. Bowen, T.K. Knox, K. Zhou, M. Pendziwiat, G. Kuhlenbaumer, C.V. Sindelar, and E.T.
Spiliotis. 2013. Novel septin 9 repeat motifs altered in neuralgic amyotrophy bind and bundle microtubules. The Journal of cell biology. 203:895-905.
Balderhaar, H.J., and C. Ungermann. 2013. CORVET and HOPS tethering complexes -
coordinators of endosome and lysosome fusion. Journal of cell science. 126:1307-1316. Balkwill, F.R., M. Capasso, and T. Hagemann. 2012. The tumor microenvironment at a glance.
Journal of cell science. 125:5591-5596. Barral, Y., Memall, V., Mooseker, M.S., and Synder, M. . 2000. Compartmentalization of the cell
cortex by septins is required for maintenance of cell polarity in yeast. Molecular Cell. 5:841-851.
Bartsch, I., K. Sandrock, F. Lanza, P. Nurden, I. Hainmann, A. Pavlova, A. Greinacher, U. Tacke, M.
Barth, A. Busse, J. Oldenburg, M. Bommer, B. Strahm, A. Superti-Furga, and B. Zieger. 2011. Deletion of human GP1BB and SEPT5 is associated with Bernard-Soulier syndrome, platelet secretion defect, polymicrogyria, and developmental delay. Thromb Haemost. 106:475-483.

163 Barysch, S.V., R. Jahn, and S.O. Rizzoli. 2010. A fluorescence-based in vitro assay for investigating
early endosome dynamics. Nat Protoc. 5:1127-1137. Baumann, S., J. Konig, J. Koepke, and M. Feldbrugge. 2014. Endosomal transport of septin mRNA
and protein indicates local translation on endosomes and is required for correct septin filamentation. EMBO Rep. 15:94-102.
Baust, T., M. Anitei, C. Czupalla, I. Parshyna, L. Bourel, C. Thiele, E. Krause, and B. Hoflack. 2008.
Protein networks supporting AP-3 function in targeting lysosomal membrane proteins. Molecular biology of the cell. 19:1942-1951.
Behrmann, E., M. Muller, P.A. Penczek, H.G. Mannherz, D.J. Manstein, and S. Raunser. 2012.
Structure of the rigor actin-tropomyosin-myosin complex. Cell. 150:327-338. Beites, C., Campbell, Kristen A., and WIlliam S. Trimble. 2005. The septin Sept5/CDCrel-1
competes with α-SNAP for binding to the SNARE complex. Biochemistry Journal. 385:347-353.
Beites, C.L., X.R. Peng, and W.S. Trimble. 2001. Expression and analysis of properties of septin
CDCrel-1 in exocytosis. Methods Enzymol. 329:499-510. Beites, C.L., H. Xie, R. Bowser, and W.S. Trimble. 1999. The septin CDCrel-1 binds syntaxin and
inhibits exocytosis. Nat Neurosci. 2:434-439. Beites, C.L., Xie, Hong., Bowswer, Robert and and William S. Trimble. 1999. The septin CDCrel-1
binds syntaxin and inhibits exocytosis. Nature Neuroscience. 2:434-439. Benjamin, J.M., A.V. Kwiatkowski, C. Yang, F. Korobova, S. Pokutta, T. Svitkina, W.I. Weis, and
W.J. Nelson. 2010. AlphaE-catenin regulates actin dynamics independently of cadherin-mediated cell-cell adhesion. The Journal of cell biology. 189:339-352.
Berginski, M.E., and S.M. Gomez. 2013. The Focal Adhesion Analysis Server: a web tool for
analyzing focal adhesion dynamics. F1000Res. 2:68. Bernstein, B.W., and J.R. Bamburg. 2010. ADF/cofilin: a functional node in cell biology. Trends in
cell biology. 20:187-195. Bertin, A., M.A. McMurray, P. Grob, S.S. Park, G. Garcia, 3rd, I. Patanwala, H.L. Ng, T. Alber, J.
Thorner, and E. Nogales. 2008. Saccharomyces cerevisiae septins: supramolecular organization of heterooligomers and the mechanism of filament assembly. Proceedings of the National Academy of Sciences of the United States of America. 105:8274-8279.
Bertin, A., M.A. McMurray, L. Thai, G. Garcia, 3rd, V. Votin, P. Grob, T. Allyn, J. Thorner, and E.
Nogales. 2010. Phosphatidylinositol-4,5-bisphosphate promotes budding yeast septin filament assembly and organization. Journal of molecular biology. 404:711-731.

164 Blaser, S., K. Jersch, I. Hainmann, D. Wunderle, A. Zgaga-Griesz, A. Busse, and B. Zieger. 2002.
Human septin-septin interaction: CDCrel-1 partners with KIAA0202. FEBS letters. 519:169-172.
Bongiovanni, L., F. Pirozzi, F. Guidi, M. Orsini, P. Chiurazzi, P.F. Bassi, and M. Racioppi. 2012.
Bradeion (SEPT4) as a urinary marker of transitional cell bladder cancer: a real-time polymerase chain reaction study of gene expression. J Urol. 187:2223-2227.
Bowen, J.R., D. Hwang, X. Bai, D. Roy, and E.T. Spiliotis. 2011. Septin GTPases spatially guide
microtubule organization and plus end dynamics in polarizing epithelia. The Journal of cell biology. 194:187-197.
Bravo-Cordero, J.J., L. Hodgson, and J. Condeelis. 2012. Directed cell invasion and migration
during metastasis. Current opinion in cell biology. 24:277-283. Breitsprecher, D., S.A. Koestler, I. Chizhov, M. Nemethova, J. Mueller, B.L. Goode, J.V. Small, K.
Rottner, and J. Faix. 2011. Cofilin cooperates with fascin to disassemble filopodial actin filaments. Journal of cell science. 124:3305-3318.
Bretscher, A. 2013. Deconstructing formin-dependent actin cable assembly. Proceedings of the
National Academy of Sciences of the United States of America. 110:18744-18745. Bridges, A.A., and A.S. Gladfelter. 2015. Septin Form and Function at the Cell Cortex. The Journal
of biological chemistry. 290:17173-17180. Bridges, A.A., M.S. Jentzsch, P.W. Oakes, P. Occhipinti, and A.S. Gladfelter. 2016. Micron-scale
plasma membrane curvature is recognized by the septin cytoskeleton. The Journal of cell biology. 213:23-32.
Bridges, A.A., H. Zhang, S.B. Mehta, P. Occhipinti, T. Tani, and A.S. Gladfelter. 2014. Septin
assemblies form by diffusion-driven annealing on membranes. Proceedings of the National Academy of Sciences of the United States of America. 111:2146-2151.
Bright, N.A., M.J. Gratian, and J.P. Luzio. 2005. Endocytic delivery to lysosomes mediated by
concurrent fusion and kissing events in living cells. Current biology : CB. 15:360-365. Bryant, D.M., M.C. Kerr, L.A. Hammond, S.R. Joseph, K.E. Mostov, R.D. Teasdale, and J.L. Stow.
2007. EGF induces macropinocytosis and SNX1-modulated recycling of E-cadherin. Journal of cell science. 120:1818-1828.
Burridge, K., and E.S. Wittchen. 2013. The tension mounts: stress fibers as force-generating
mechanotransducers. The Journal of cell biology. 200:9-19. Butcher, D.T., T. Alliston, and V.M. Weaver. 2009. A tense situation: forcing tumour progression.
Nature reviews. Cancer. 9:108-122.

165 Buttery, S.M., K. Kono, E. Stokasimov, and D. Pellman. 2012. Regulation of the formin Bnr1 by
septins anda MARK/Par1-family septin-associated kinase. Molecular biology of the cell. 23:4041-4053.
Calvo, F., R. Ranftl, S. Hooper, A.J. Farrugia, E. Moeendarbary, A. Bruckbauer, F. Batista, G.
Charras, and E. Sahai. 2015. Cdc42EP3/BORG2 and Septin Network Enables Mechano-transduction and the Emergence of Cancer-Associated Fibroblasts. Cell reports. 13:2699-2714.
Cano, M.L., L. Cassimeris, M. Fechheimer, and S.H. Zigmond. 1992. Mechanisms responsible for
F-actin stabilization after lysis of polymorphonuclear leukocytes. The Journal of cell biology. 116:1123-1134.
Cao, L., X. Ding, W. Yu, X. Yang, S. Shen, and L. Yu. 2007. Phylogenetic and evolutionary analysis
of the septin protein family in metazoan. FEBS letters. 581:5526-5532. Capurso, G., T. Crnogorac-Jurcevic, M. Milione, F. Panzuto, N. Campanini, S.E. Dowen, A. Di
Florio, C. Sette, C. Bordi, N.R. Lemoine, and G. Delle Fave. 2005. Peanut-like 1 (septin 5) gene expression in normal and neoplastic human endocrine pancreas. Neuroendocrinology. 81:311-321.
Carol., G., Michael W., and Tom St. John. 1990. Lymphocyte HEV adhesion variants differ in the
expression of multiple gene sequences. Gene. 95:279-284. Casamayor, A., and M. Snyder. 2003. Molecular dissection of a yeast septin: distinct domains are
required for septin interaction, localization, and function. Molecular and cellular biology. 23:2762-2777.
Caswell, P.T., M. Chan, A.J. Lindsay, M.W. McCaffrey, D. Boettiger, and J.C. Norman. 2008. Rab-
coupling protein coordinates recycling of alpha5beta1 integrin and EGFR1 to promote cell migration in 3D microenvironments. The Journal of cell biology. 183:143-155.
Caswell, P.T., and J.C. Norman. 2006. Integrin trafficking and the control of cell migration.
Traffic. 7:14-21. Caudron, F., and Y. Barral. 2009. Septins and the lateral compartmentalization of eukaryotic
membranes. Developmental cell. 16:493-506. Cerveira, N., S. Bizarro, and M.R. Teixeira. 2011. MLL-SEPTIN gene fusions in hematological
malignancies. Biological chemistry. 392:713-724. Chacko, A., S. McDade, S. Chanduloy, S. Church, R. Kennedy, J. Price, P. Hall, and S.E.H. Russell.
2012. Expression of the SEPT9_i4 isoform confers resistance to microtubule-interacting drugs. Cellular Oncology. 35:85-93.

166 Chacko, A.D., P.L. Hyland, S.S. McDade, P.W. Hamilton, S.H. Russell, and P.A. Hall. 2005.
SEPT9_v4 expression induces morphological change, increased motility and disturbed polarity. J Pathol. 206:458-465.
Chahwan, R., S. Gravel, T. Matsusaka, and S.P. Jackson. 2013. Dma/RNF8 proteins are
evolutionarily conserved E3 ubiquitin ligases that target septins. Cell Cycle. 12:1000-1008.
Chance, P.F., and A.J. Windebank. 1996. Hereditary neuralgic amyotrophy. Curr Opin Neurol.
9:343-347. Chen, W., Y. Feng, D. Chen, and A. Wandinger-Ness. 1998. Rab11 is required for trans-golgi
network-to-plasma membrane transport and a preferential target for GDP dissociation inhibitor. Molecular biology of the cell. 9:3241-3257.
Chih, B., P. Liu, Y. Chinn, C. Chalouni, L.G. Komuves, P.E. Hass, W. Sandoval, and A.S. Peterson.
2012. A ciliopathy complex at the transition zone protects the cilia as a privileged membrane domain. Nature cell biology. 14:61-72.
Cho, S.J., H. Lee, S. Dutta, J. Song, R. Walikonis, and I.S. Moon. 2011. Septin 6 regulates the
cytoarchitecture of neurons through localization at dendritic branch points and bases of protrusions. Mol Cells. 32:89-98.
Choi, C.K., M. Vicente-Manzanares, J. Zareno, L.A. Whitmore, A. Mogilner, and A.R. Horwitz.
2008. Actin and alpha-actinin orchestrate the assembly and maturation of nascent adhesions in a myosin II motor-independent manner. Nature cell biology. 10:1039-1050.
Chrzanowska-Wodnicka, M., and K. Burridge. 1996. Rho-stimulated contractility drives the
formation of stress fibers and focal adhesions. The Journal of cell biology. 133:1403-1415.
Commisso, C., S.M. Davidson, R.G. Soydaner-Azeloglu, S.J. Parker, J.J. Kamphorst, S. Hackett, E.
Grabocka, M. Nofal, J.A. Drebin, C.B. Thompson, J.D. Rabinowitz, C.M. Metallo, M.G. Vander Heiden, and D. Bar-Sagi. 2013. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature. 497:633-637.
Connolly, D., I. Abdesselam, P. Verdier-Pinard, and C. Montagna. 2011a. Septin roles in
tumorigenesis. Biological chemistry. 392:725-738. Connolly, D., Z. Yang, M. Castaldi, N. Simmons, M.H. Oktay, S. Coniglio, M.J. Fazzari, P. Verdier-
Pinard, and C. Montagna. 2011b. Septin 9 isoform expression, localization and epigenetic changes during human and mouse breast cancer progression. Breast Cancer Res. 13:R76.
Corbett-Nelson, E.F., D. Mason, J.G. Marshall, Y. Collette, and S. Grinstein. 2006. Signaling-
dependent immobilization of acylated proteins in the inner monolayer of the plasma membrane. The Journal of cell biology. 174:255-265.

167 Cossart, P. 2011. Illuminating the landscape of host-pathogen interactions with the bacterium
Listeria monocytogenes. Proceedings of the National Academy of Sciences of the United States of America. 108:19484-19491.
Craven, R.A., S. Hanrahan, N. Totty, P. Harnden, A.J. Stanley, E.R. Maher, A.L. Harris, W.S.
Trimble, P.J. Selby, and R.E. Banks. 2006a. Proteomic identification of a role for the von Hippel Lindau tumour suppressor in changes in the expression of mitochondrial proteins and septin 2 in renal cell carcinoma. Proteomics. 6:3880-3893.
Craven, R.A., A.J. Stanley, S. Hanrahan, J. Dods, R. Unwin, N. Totty, P. Harnden, I. Eardley, P.J.
Selby, and R.E. Banks. 2006b. Proteomic analysis of primary cell lines identifies protein changes present in renal cell carcinoma. Proteomics. 6:2853-2864.
Davis, J.S., C.L. Satorius, and N.D. Epstein. 2002. Kinetic effects of myosin regulatory light chain
phosphorylation on skeletal muscle contraction. Biophysical journal. 83:359-370. Davis, P.J., B.S. Handwerger, and F. Glaser. 1974. Physical properties of a dog liver and kidney
cytosol protein that binds thyroid hormone. The Journal of biological chemistry. 249:6208-6217.
de Almeida Marques, I., N.F. Valadares, W. Garcia, J.C. Damalio, J.N. Macedo, A.P. de Araujo,
C.A. Botello, J.M. Andreu, and R.C. Garratt. 2012. Septin C-terminal domain interactions: implications for filament stability and assembly. Cell Biochem Biophys. 62:317-328.
de Carvalho, T.M., E.S. Barrias, and W. de Souza. 2015. Macropinocytosis: a pathway to
protozoan infection. Front Physiol. 6:106. De Craene, B., and G. Berx. 2013. Regulatory networks defining EMT during cancer initiation and
progression. Nature reviews. Cancer. 13:97-110. De La Cruz, E.M., A.L. Wells, S.S. Rosenfeld, E.M. Ostap, and H.L. Sweeney. 1999. The kinetic
mechanism of myosin V. Proceedings of the National Academy of Sciences of the United States of America. 96:13726-13731.
de Rooij, J., A. Kerstens, G. Danuser, M.A. Schwartz, and C.M. Waterman-Storer. 2005. Integrin-
dependent actomyosin contraction regulates epithelial cell scattering. The Journal of cell biology. 171:153-164.
Deberardinis, R.J., N. Sayed, D. Ditsworth, and C.B. Thompson. 2008. Brick by brick: metabolism
and tumor cell growth. Current opinion in genetics & development. 18:54-61. Debnath, J., and J.S. Brugge. 2005. Modelling glandular epithelial cancers in three-dimensional
cultures. Nature reviews. Cancer. 5:675-688. Dekker, C., P.C. Stirling, E.A. McCormack, H. Filmore, A. Paul, R.L. Brost, M. Costanzo, C. Boone,
M.R. Leroux, and K.R. Willison. 2008. The interaction network of the chaperonin CCT. EMBO J. 27:1827-1839.

168 Dent, J., K. Kato, X.R. Peng, C. Martinez, M. Cattaneo, C. Poujol, P. Nurden, A. Nurden, W.S.
Trimble, and J. Ware. 2002. A prototypic platelet septin and its participation in secretion. Proceedings of the National Academy of Sciences of the United States of America. 99:3064-3069.
Diesenberg, K., M. Beerbaum, U. Fink, P. Schmieder, and M. Krauss. 2015. SEPT9 negatively
regulates ubiquitin-dependent downregulation of EGFR. Journal of cell science. 128:397-407.
Dobbelaere, J., and Y. Barral. 2004. Spatial coordination of cytokinetic events by
compartmentalization of the cell cortex. Science. 305:393-396. Dobbelaere, J., M.S. Gentry, R.L. Hallberg, and Y. Barral. 2003. Phosphorylation-dependent
regulation of septin dynamics during the cell cycle. Developmental cell. 4:345-357. Dolat, L., Q. Hu, and E.T. Spiliotis. 2014a. Septin functions in organ system physiology and
pathology. Biological chemistry. 395:123-141. Dolat, L., J.L. Hunyara, J.R. Bowen, E.P. Karasmanis, M. Elgawly, V.E. Galkin, and E.T. Spiliotis.
2014b. Septins promote stress fiber-mediated maturation of focal adhesions and renal epithelial motility. The Journal of cell biology. 207:225-235.
Drees, B.L., B. Sundin, E. Brazeau, J.P. Caviston, G.C. Chen, W. Guo, K.G. Kozminski, M.W. Lau, J.J.
Moskow, A. Tong, L.R. Schenkman, A. McKenzie, 3rd, P. Brennwald, M. Longtine, E. Bi, C. Chan, P. Novick, C. Boone, J.R. Pringle, T.N. Davis, S. Fields, and D.G. Drubin. 2001. A protein interaction map for cell polarity development. The Journal of cell biology. 154:549-571.
Dressler, G. 2002. Tubulogenesis in the developing mammalian kidney. Trends in cell biology.
12:390-395. Egelman, E.H. 2000. A robust algorithm for the reconstruction of helical filaments using single-
particle methods. Ultramicroscopy. 85:225-234. Egile, C., T.P. Loisel, V. Laurent, R. Li, D. Pantaloni, P.J. Sansonetti, and M.F. Carlier. 1999.
Activation of the CDC42 effector N-WASP by the Shigella flexneri IcsA protein promotes actin nucleation by Arp2/3 complex and bacterial actin-based motility. The Journal of cell biology. 146:1319-1332.
Elhasid, R., D. Sahar, A. Merling, Y. Zivony, A. Rotem, M. Ben-Arush, S. Izraeli, D. Bercovich, and
S. Larisch. 2004. Mitochondrial pro-apoptotic ARTS protein is lost in the majority of acute lymphoblastic leukemia patients. Oncogene. 23:5468-5475.
Elkhatib, N., M.B. Neu, C. Zensen, K.M. Schmoller, D. Louvard, A.R. Bausch, T. Betz, and D.M.
Vignjevic. 2014. Fascin plays a role in stress fiber organization and focal adhesion disassembly. Current biology : CB. 24:1492-1499.

169 Engidawork, E., N. Baiic, M. Fountoulakis, M. Dierssen, S. Greber-Platzer, and G. Lubec. 2001a.
Beta-amyloid precursor protein, ETS-2 and collagen alpha 1 (VI) chain precursor, encoded on chromosome 21, are not overexpressed in fetal Down syndrome: further evidence against gene dosage effect. J Neural Transm Suppl:335-346.
Engidawork, E., N. Balic, J.F. Juranville, M. Fountoulakis, M. Dierssen, and G. Lubec. 2001b.
Unaltered expression of Fas (CD95/APO-1), caspase-3, Bcl-2 and annexins in brains of fetal Down syndrome: evidence against increased apoptosis. J Neural Transm Suppl:149-162.
Engidawork, E., T. Gulesserian, M. Fountoulakis, and G. Lubec. 2003a. Aberrant protein
expression in cerebral cortex of fetus with Down syndrome. Neuroscience. 122:145-154. Engidawork, E., T. Gulesserian, M. Fountoulakis, and G. Lubec. 2003b. Expression of hypothetical
proteins in human fetal brain: increased expression of hypothetical protein 28.5kDa in Down syndrome, a clue for its tentative role. Mol Genet Metab. 78:295-301.
Engidawork, E., T. Gulesserian, R. Seidl, N. Cairns, and G. Lubec. 2001c. Expression of apoptosis
related proteins: RAIDD, ZIP kinase, Bim/BOD, p21, Bax, Bcl-2 and NF-kappaB in brains of patients with Down syndrome. J Neural Transm Suppl:181-192.
Engidawork, E., J.F. Juranville, M. Fountoulakis, M. Dierssen, and G. Lubec. 2001d. Selective
upregulation of the ubiquitin-proteasome proteolytic pathway proteins, proteasome zeta chain and isopeptidase T in fetal Down syndrome. J Neural Transm Suppl:117-130.
Engidawork, E., and G. Lubec. 2001. Protein expression in Down syndrome brain. Amino Acids.
21:331-361. Engidawork, E., and G. Lubec. 2003. Molecular changes in fetal Down syndrome brain. Journal of
neurochemistry. 84:895-904. Engidawork, E., J.C. Roberts, R. Hardmeier, R.J. Scheper, and G. Lubec. 2001e. Expression of the
multidrug resistance P glycoprotein (Pgp) and multidrug resistance associated protein (MRP1) in Down syndrome brains. J Neural Transm Suppl:35-45.
Fahy, J.V., and B.F. Dickey. 2010. Airway mucus function and dysfunction. N Engl J Med.
363:2233-2247. Fares, H., M. Peifer, and J.R. Pringle. 1995. Localization and possible functions of Drosophila
septins. Molecular biology of the cell. 6:1843-1859. Farkasovsky, M., P. Herter, B. Voss, and A. Wittinghofer. 2005. Nucleotide binding and filament
assembly of recombinant yeast septin complexes. Biological chemistry. 386:643-656. Feliciano, W.D., S. Yoshida, S.W. Straight, and J.A. Swanson. 2011. Coordination of the Rab5
cycle on macropinosomes. Traffic. 12:1911-1922.

170 Finger, F.P., K.R. Kopish, and J.G. White. 2003. A role for septins in cellular and axonal migration
in C. elegans. Dev Biol. 261:220-234. Fliegauf, M., A. Kahle, K. Haffner, and B. Zieger. 2014. Distinct localization of septin proteins to
ciliary sub-compartments in airway epithelial cells. Biological chemistry. 395:151-156. Founounou, N., N. Loyer, and R. Le Borgne. 2013. Septins Regulate the Contractility of the
Actomyosin Ring to Enable Adherens Junction Remodeling during Cytokinesis of Epithelial Cells. Developmental cell. 24:242-255.
Frank, J.A. 2012. Claudins and alveolar epithelial barrier function in the lung. Ann N Y Acad Sci.
1257:175-183. Frank, J.S., B.; Dowse, H. 1984. SPIDER-A modular software system for electron image
processing. Ultramicroscopy 6:343-358. Friedl, P., and K. Wolf. 2010. Plasticity of cell migration: a multiscale tuning model. The Journal
of cell biology. 188:11-19. Fu, M.M., and E.L. Holzbaur. 2014. Integrated regulation of motor-driven organelle transport by
scaffolding proteins. Trends in cell biology. 24:564-574. Fuchs, Y., S. Brown, T. Gorenc, J. Rodriguez, E. Fuchs, and H. Steller. 2013. Sept4/ARTS Regulates
Stem Cell Apoptosis and Skin Regeneration. Science. Fuchtbauer, A., L.B. Lassen, A.B. Jensen, J. Howard, S. Quiroga Ade, S. Warming, A.B. Sorensen,
F.S. Pedersen, and E.M. Fuchtbauer. 2011. Septin9 is involved in septin filament formation and cellular stability. Biological chemistry. 392:769-777.
Fujii, T., A.H. Iwane, T. Yanagida, and K. Namba. 2010. Direct visualization of secondary
structures of F-actin by electron cryomicroscopy. Nature. 467:724-728. Fung, K.Y., L. Dai, and W.S. Trimble. 2014. Cell and molecular biology of septins. Int Rev Cell Mol
Biol. 310:289-339. Galkin, V.E., A. Orlova, A.J. Koleske, and E.H. Egelman. 2005. The Arg non-receptor tyrosine
kinase modifies F-actin structure. Journal of molecular biology. 346:565-575. Galkin, V.E., A. Orlova, D.S. Kudryashov, A. Solodukhin, E. Reisler, G.F. Schroder, and E.H.
Egelman. 2011. Remodeling of actin filaments by ADF/cofilin proteins. Proceedings of the National Academy of Sciences of the United States of America. 108:20568-20572.
Galkin, V.E., A. Orlova, N. Lukoyanova, W. Wriggers, and E.H. Egelman. 2001. Actin
depolymerizing factor stabilizes an existing state of F-actin and can change the tilt of F-actin subunits. The Journal of cell biology. 153:75-86.

171 Galkin, V.E., A. Orlova, G.F. Schroder, and E.H. Egelman. 2010. Structural polymorphism in F-
actin. Nature structural & molecular biology. 17:1318-1323. Galkin, V.E., A. Orlova, M.S. VanLoock, I.N. Rybakova, J.M. Ervasti, and E.H. Egelman. 2002. The
utrophin actin-binding domain binds F-actin in two different modes: implications for the spectrin superfamily of proteins. The Journal of cell biology. 157:243-251.
Galkin, V.E., A. Orlova, M.S. VanLoock, A. Shvetsov, E. Reisler, and E.H. Egelman. 2003.
ADF/cofilin use an intrinsic mode of F-actin instability to disrupt actin filaments. The Journal of cell biology. 163:1057-1066.
Gao, L., W. Liu, and A. Bretscher. 2010. The yeast formin Bnr1p has two localization regions that
show spatially and temporally distinct association with septin structures. Molecular biology of the cell. 21:1253-1262.
Garcia-Fernandez, M., H. Kissel, S. Brown, T. Gorenc, A.J. Schile, S. Rafii, S. Larisch, and H. Steller.
2010. Sept4/ARTS is required for stem cell apoptosis and tumor suppression. Genes Dev. 24:2282-2293.
Garcia, G., 3rd, A. Bertin, Z. Li, Y. Song, M.A. McMurray, J. Thorner, and E. Nogales. 2011.
Subunit-dependent modulation of septin assembly: budding yeast septin Shs1 promotes ring and gauze formation. The Journal of cell biology. 195:993-1004.
Gardel, M.L., I.C. Schneider, Y. Aratyn-Schaus, and C.M. Waterman. 2010. Mechanical
integration of actin and adhesion dynamics in cell migration. Annu Rev Cell Dev Biol. 26:315-333.
Gasper, R., S. Meyer, K. Gotthardt, M. Sirajuddin, and A. Wittinghofer. 2009. It takes two to
tango: regulation of G proteins by dimerization. Nature reviews. Molecular cell biology. 10:423-429.
Geiger, B., and K.M. Yamada. 2011. Molecular architecture and function of matrix adhesions.
Cold Spring Harbor perspectives in biology. 3. Ghossoub, R., Q. Hu, M. Failler, M.-C. Rouyez, B. Spitzbarth, S. Mostowy, U. Wolfrum, S. Saunier,
P. Cossart, W. JamesNelson, and A. Benmerah. 2013. Septins 2, 7 and 9 and MAP4 colocalize along the axoneme in the primary cilium and control ciliary length. Journal of cell science. 126:2583-2594.
Gilden, J.K., S. Peck, Y.C. Chen, and M.F. Krummel. 2012. The septin cytoskeleton facilitates
membrane retraction during motility and blebbing. The Journal of cell biology. 196:103-114.
Gonzalez, M.E., O. Makarova, E.A. Peterson, L.M. Privette, and E.M. Petty. 2009. Up-regulation
of SEPT9_v1 stabilizes c-Jun-N-terminal kinase and contributes to its pro-proliferative activity in mammary epithelial cells. Cell Signal. 21:477-487.

172 Gottfried, Y., A. Rotem, R. Lotan, H. Steller, and S. Larisch. 2004. The mitochondrial ARTS protein
promotes apoptosis through targeting XIAP. EMBO J. 23:1627-1635. Grintsevich, E.E., V.E. Galkin, A. Orlova, A.J. Ytterberg, M.M. Mikati, D.S. Kudryashov, J.A. Loo,
E.H. Egelman, and E. Reisler. 2010. Mapping of drebrin binding site on F-actin. Journal of molecular biology. 398:542-554.
Grutzmann, R., B. Molnar, C. Pilarsky, J.K. Habermann, P.M. Schlag, H.D. Saeger, S. Miehlke, T.
Stolz, F. Model, U.J. Roblick, H.P. Bruch, R. Koch, V. Liebenberg, T. Devos, X. Song, R.H. Day, A.Z. Sledziewski, and C. Lofton-Day. 2008. Sensitive detection of colorectal cancer in peripheral blood by septin 9 DNA methylation assay. PloS one. 3:e3759.
Gu, J., S. Xu, and L.C. Yu. 2002. A model of cross-bridge attachment to actin in the A*M*ATP
state based on x-ray diffraction from permeabilized rabbit psoas muscle. Biophysical journal. 82:2123-2133.
Gu, Z., E.H. Noss, V.W. Hsu, and M.B. Brenner. 2011. Integrins traffic rapidly via circular dorsal
ruffles and macropinocytosis during stimulated cell migration. The Journal of cell biology. 193:61-70.
Guarino, M., B. Rubino, and G. Ballabio. 2007. The role of epithelial-mesenchymal transition in
cancer pathology. Pathology. 39:305-318. Guillot, C., and T. Lecuit. 2013. Adhesion Disengagement Uncouples Intrinsic and Extrinsic Forces
to Drive Cytokinesis in Epithelial Tissues. Developmental cell. 24:227-241. Gulesserian, T., E. Engidawork, N. Cairns, and G. Lubec. 2000. Increased protein levels of
serotonin transporter in frontal cortex of patients with Down syndrome. Neurosci Lett. 296:53-57.
Gulesserian, T., E. Engidawork, M. Fountoulakis, and G. Lubec. 2001. Antioxidant proteins in
fetal brain: superoxide dismutase-1 (SOD-1) protein is not overexpressed in fetal Down syndrome. J Neural Transm Suppl:71-84.
Gulesserian, T., E. Engidawork, M. Fountoulakis, and G. Lubec. 2003. Decreased brain levels of
Lupus La protein and increased U5 small ribonucleoprotein-specific 40 kDa protein in fetal Down syndrome. Cell Mol Biol (Noisy-le-grand). 49:733-738.
Gulesserian, T., E. Engidawork, M. Fountoulakis, and G. Lubec. 2007. Manifold decrease of sialic
acid synthase in fetal Down syndrome brain. Amino Acids. 32:141-144. Hagiwara, A., Y. Tanaka, R. Hikawa, N. Morone, A. Kusumi, H. Kimura, and M. Kinoshita. 2011.
Submembranous septins as relatively stable components of actin-based membrane skeleton. Cytoskeleton (Hoboken). 68:512-525.
Hall, P.A., K. Jung, K.J. Hillan, and S.E. Russell. 2005a. Expression profiling the human septin gene
family. J Pathol. 206:269-278.

173 Hall, P.A., K. Jung, K.J. Hillan, and S.E.H. Russell. 2005b. Expression profiling the human septin
gene family. J Pathol. 206:269-278. Hanahan, D., and R.A. Weinberg. 2000. The hallmarks of cancer. Cell. 100:57-70. Hanahan, D., and R.A. Weinberg. 2011. Hallmarks of cancer: the next generation. Cell. 144:646-
674. Hartwell, L.H. 1971. Genetic control of the cell division cycle in yeast. IV. Genes controlling bud
emergence and cytokinesis. Experimental cell research. 69:265-276. Herman, J.G., and S.B. Baylin. 2003. Gene Silencing in Cancer in Association with Promoter
Hypermethylation. New England Journal of Medicine. 349:2042-2054. Hernandez-Rodriguez, Y., and M. Momany. 2012. Posttranslational modifications and assembly
of septin heteropolymers and higher-order structures. Curr Opin Microbiol. 15:660-668. Hirokawa, N., K. Sobue, K. Kanda, A. Harada, and H. Yorifuji. 1989. The cytoskeletal architecture
of the presynaptic terminal and molecular structure of synapsin 1. The Journal of cell biology. 108:111-126.
Hsu, S.C., C.D. Hazuka, R. Roth, D.L. Foletti, J. Heuser, and R.H. Scheller. 1998. Subunit
composition, protein interactions, and structures of the mammalian brain sec6/8 complex and septin filaments. Neuron. 20:1111-1122.
Hu, J., X. Bai, J.R. Bowen, L. Dolat, F. Korobova, W. Yu, P.W. Baas, T. Svitkina, G. Gallo, and E.T.
Spiliotis. 2012. Septin-driven coordination of actin and microtubule remodeling regulates the collateral branching of axons. Current biology : CB. 22:1109-1115.
Hu, Q., L. Milenkovic, H. Jin, M.P. Scott, M.V. Nachury, E.T. Spiliotis, and W.J. Nelson. 2010. A
septin diffusion barrier at the base of the primary cilium maintains ciliary membrane protein distribution. Science. 329:436-439.
Hu, Q., W.J. Nelson, and E.T. Spiliotis. 2008. Forchlorfenuron alters mammalian septin assembly,
organization, and dynamics. The Journal of biological chemistry. 283:29563-29571. Huang, Y.W., M. Yan, R.F. Collins, J.E. Diciccio, S. Grinstein, and W.S. Trimble. 2008. Mammalian
septins are required for phagosome formation. Molecular biology of the cell. 19:1717-1726.
Huijbregts, R.P., A. Svitin, M.W. Stinnett, M.B. Renfrow, and I. Chesnokov. 2009. Drosophila Orc6
facilitates GTPase activity and filament formation of the septin complex. Molecular biology of the cell. 20:270-281.
Humphries, A., and N.A. Wright. 2008. Colonic crypt organization and tumorigenesis. Nature
reviews. Cancer. 8:415-424.

174 Ihara, M., A. Kinoshita, S. Yamada, H. Tanaka, A. Tanigaki, A. Kitano, M. Goto, K. Okubo, H.
Nishiyama, O. Ogawa, C. Takahashi, S. Itohara, Y. Nishimune, M. Noda, and M. Kinoshita. 2005. Cortical organization by the septin cytoskeleton is essential for structural and mechanical integrity of mammalian spermatozoa. Developmental cell. 8:343-352.
Ihara, M., H. Tomimoto, H. Kitayama, Y. Morioka, I. Akiguchi, H. Shibasaki, M. Noda, and M.
Kinoshita. 2003. Association of the cytoskeletal GTP-binding protein Sept4/H5 with cytoplasmic inclusions found in Parkinson's disease and other synucleinopathies. The Journal of biological chemistry. 278:24095-24102.
Ihara, M., N. Yamasaki, A. Hagiwara, A. Tanigaki, A. Kitano, R. Hikawa, H. Tomimoto, M. Noda,
M. Takanashi, H. Mori, N. Hattori, T. Miyakawa, and M. Kinoshita. 2007. Sept4, a component of presynaptic scaffold and Lewy bodies, is required for the suppression of alpha-synuclein neurotoxicity. Neuron. 53:519-533.
Ishikawa, R., T. Sakamoto, T. Ando, S. Higashi-Fujime, and K. Kohama. 2003. Polarized actin
bundles formed by human fascin-1: their sliding and disassembly on myosin II and myosin V in vitro. Journal of neurochemistry. 87:676-685.
Itakura, E., C. Kishi-Itakura, and N. Mizushima. 2012. The hairpin-type tail-anchored SNARE
syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell. 151:1256-1269.
Ito, H., K. Atsuzawa, R. Morishita, N. Usuda, K. Sudo, I. Iwamoto, K. Mizutani, R. Katoh-Semba, Y.
Nozawa, T. Asano, and K. Nagata. 2009. Sept8 controls the binding of vesicle-associated membrane protein 2 to synaptophysin. Journal of neurochemistry. 108:867-880.
Ito, H., I. Iwamoto, R. Morishita, Y. Nozawa, S. Narumiya, T. Asano, and K. Nagata. 2005. Possible
role of Rho/Rhotekin signaling in mammalian septin organization. Oncogene. 24:7064-7072.
Iwaisako, K., E. Hatano, K. Taura, A. Nakajima, M. Tada, S. Seo, N. Tamaki, F. Sato, I. Ikai, S.
Uemoto, and M. Kinoshita. 2008. Loss of Sept4 exacerbates liver fibrosis through the dysregulation of hepatic stellate cells. J Hepatol. 49:768-778.
Jahn, R., and R.H. Scheller. 2006. SNAREs--engines for membrane fusion. Nature reviews.
Molecular cell biology. 7:631-643. Jefferies, H.B., F.T. Cooke, P. Jat, C. Boucheron, T. Koizumi, M. Hayakawa, H. Kaizawa, T. Ohishi,
P. Workman, M.D. Waterfield, and P.J. Parker. 2008. A selective PIKfyve inhibitor blocks PtdIns(3,5)P(2) production and disrupts endomembrane transport and retroviral budding. EMBO Rep. 9:164-170.
Joberty, G., R.R. Perlungher, and I.G. Macara. 1999. The Borgs, a new family of Cdc42 and TC10
GTPase-interacting proteins. Molecular and cellular biology. 19:6585-6597.

175 Joberty, G., R.R. Perlungher, P.J. Sheffield, M. Kinoshita, M. Noda, T. Haystead, and I.G. Macara.
2001. Borg proteins control septin organization and are negatively regulated by Cdc42. Nature cell biology. 3:861-866.
John, C.M., R.K. Hite, C.S. Weirich, D.J. Fitzgerald, H. Jawhari, M. Faty, D. Schlapfer, R.
Kroschewski, F.K. Winkler, T. Walz, Y. Barral, and M.O. Steinmetz. 2007. The Caenorhabditis elegans septin complex is nonpolar. EMBO J. 26:3296-3307.
Jones, M.C., P.T. Caswell, and J.C. Norman. 2006. Endocytic recycling pathways: emerging
regulators of cell migration. Current opinion in cell biology. 18:549-557. Joo, E., M.C. Surka, and W.S. Trimble. 2007. Mammalian SEPT2 is required for scaffolding
nonmuscle myosin II and its kinases. Developmental cell. 13:677-690. Juanes-Garcia, A., C. Llorente-Gonzalez, and M. Vicente-Manzanares. 2016. Molecular control of
non-muscle myosin II assembly. Oncotarget. 7:5092-5093. Judith M. McKie, H.F.S., Emma Harvey, Ung-Jin Kim and Peter J. Scambler. 1997. A human gene
similar to Drosophila melanogaster peanut maps to the DiGeorge syndrome region of 22q11. Human Genetics. 101:6-12.
Kalikin, L.M., H.L. Sims, and E.M. Petty. 2000. Genomic and Expression Analyses of Alternatively
Spliced Transcripts of the MLL Septin-like Fusion Gene (MSF) That Map to a 17q25 Region of Loss in Breast and Ovarian Tumors. Genomics. 63:165-172.
Kalluri, R., and M. Zeisberg. 2006. Fibroblasts in cancer. Nature reviews. Cancer. 6:392-401. Kato, K., C. Martinez, S. Russell, P. Nurden, A. Nurden, S. Fiering, and J. Ware. 2004. Genetic
deletion of mouse platelet glycoprotein Ibbeta produces a Bernard-Soulier phenotype with increased alpha-granule size. Blood. 104:2339-2344.
Kaur, J., and J. Debnath. 2015. Autophagy at the crossroads of catabolism and anabolism. Nature
reviews. Molecular cell biology. 16:461-472. Kerr, M.C., M.R. Lindsay, R. Luetterforst, N. Hamilton, F. Simpson, R.G. Parton, P.A. Gleeson, and
R.D. Teasdale. 2006. Visualisation of macropinosome maturation by the recruitment of sorting nexins. Journal of cell science. 119:3967-3980.
Kerr, M.C., and R.D. Teasdale. 2009. Defining macropinocytosis. Traffic. 10:364-371. Kerr, M.C., J.T. Wang, N.A. Castro, N.A. Hamilton, L. Town, D.L. Brown, F.A. Meunier, N.F. Brown,
J.L. Stow, and R.D. Teasdale. 2010. Inhibition of the PtdIns(5) kinase PIKfyve disrupts intracellular replication of Salmonella. EMBO J. 29:1331-1347.
Kim, M.S., C.D. Froese, M.P. Estey, and W.S. Trimble. 2011. SEPT9 occupies the terminal
positions in septin octamers and mediates polymerization-dependent functions in abscission. The Journal of cell biology. 195:815-826.

176 Kim, S.K., A. Shindo, T.J. Park, E.C. Oh, S. Ghosh, R.S. Gray, R.A. Lewis, C.A. Johnson, T. Attie-
Bittach, N. Katsanis, and J.B. Wallingford. 2010. Planar cell polarity acts through septins to control collective cell movement and ciliogenesis. Science. 329:1337-1340.
Kimmelman, A.C. 2015. Metabolic Dependencies in RAS-Driven Cancers. Clin Cancer Res.
21:1828-1834. Kinoshita, A., M. Kinoshita, H. Akiyama, H. Tomimoto, I. Akiguchi, S. Kumar, M. Noda, and J.
Kimura. 1998. Identification of septins in neurofibrillary tangles in Alzheimer's disease. Am J Pathol. 153:1551-1560.
Kinoshita, A., M. Noda, and M. Kinoshita. 2000. Differential localization of septins in the mouse
brain. J Comp Neurol. 428:223-239. Kinoshita, M. 2003. Assembly of mammalian septins. J Biochem. 134:491-496. Kinoshita, M. 2008. Insight into septin functions from mouse models. John Wiley & Sons, Ltd. Kinoshita, M., C.M. Field, M.L. Coughlin, A.F. Straight, and T.J. Mitchison. 2002. Self- and actin-
templated assembly of Mammalian septins. Developmental cell. 3:791-802. Kinoshita, M., Field,C., Coughlin,M., Straight,A., and Mitchison, T. 2002. Self- and Actin-
Templated Assembly of Mammalian Septins. Developmental cell. 3:791–802. Kinoshita, M., S. Kumar, A. Mizoguchi, C. Ide, A. Kinoshita, T. Haraguchi, Y. Hiraoka, and M.
Noda. 1997. Nedd5, a mammalian septin, is a novel cytoskeletal component interacting with actin-based structures. Genes Dev. 11:1535-1547.
Kinoshita, N., K. Kimura, N. Matsumoto, M. Watanabe, M. Fukaya, and C. Ide. 2004. Mammalian
septin Sept2 modulates the activity of GLAST, a glutamate transporter in astrocytes. Genes Cells. 9:1-14.
Kissel, H., M.M. Georgescu, S. Larisch, K. Manova, G.R. Hunnicutt, and H. Steller. 2005. The Sept4
septin locus is required for sperm terminal differentiation in mice. Developmental cell. 8:353-364.
Kremer, B.E., L.A. Adang, and I.G. Macara. 2007. Septins regulate actin organization and cell-
cycle arrest through nuclear accumulation of NCK mediated by SOCS7. Cell. 130:837-850.
Kremer, B.E., T. Haystead, and I.G. Macara. 2005. Mammalian septins regulate microtubule
stability through interaction with the microtubule-binding protein MAP4. Molecular biology of the cell. 16:4648-4659.
Kuhlenbaumer, G., M.C. Hannibal, E. Nelis, A. Schirmacher, N. Verpoorten, J. Meuleman, G.D.
Watts, E. De Vriendt, P. Young, F. Stogbauer, H. Halfter, J. Irobi, D. Goossens, J. Del-Favero, B.G. Betz, H. Hor, G. Kurlemann, T.D. Bird, E. Airaksinen, T. Mononen, A.P.

177
Serradell, J.M. Prats, C. Van Broeckhoven, P. De Jonghe, V. Timmerman, E.B. Ringelstein, and P.F. Chance. 2005. Mutations in SEPT9 cause hereditary neuralgic amyotrophy. Nat Genet. 37:1044-1046.
Kuo, Y.C., Y.H. Lin, H.I. Chen, Y.Y. Wang, Y.W. Chiou, H.H. Lin, H.A. Pan, C.M. Wu, S.M. Su, C.C.
Hsu, and P.L. Kuo. 2012. SEPT12 mutations cause male infertility with defective sperm annulus. Hum Mutat. 33:710-719.
Kwitny, S., A.V. Klaus, and G.R. Hunnicutt. 2010. The annulus of the mouse sperm tail is required
to establish a membrane diffusion barrier that is engaged during the late steps of spermiogenesis. Biol Reprod. 82:669-678.
Lammermann, T., and M. Sixt. 2009. Mechanical modes of 'amoeboid' cell migration. Current
opinion in cell biology. 21:636-644. Larisch, S., Y. Yi, R. Lotan, H. Kerner, S. Eimerl, W. Tony Parks, Y. Gottfried, S. Birkey Reffey, M.P.
de Caestecker, D. Danielpour, N. Book-Melamed, R. Timberg, C.S. Duckett, R.J. Lechleider, H. Steller, J. Orly, S.J. Kim, and A.B. Roberts. 2000. A novel mitochondrial septin-like protein, ARTS, mediates apoptosis dependent on its P-loop motif. Nature cell biology. 2:915-921.
Lassen, L.B., A. Fuchtbauer, A. Schmitz, A.B. Sorensen, F.S. Pedersen, and E.M. Fuchtbauer. 2013.
Septin9 is involved in T-cell development and CD8(+) T-cell homeostasis. Cell Tissue Res. 352:695-705.
Lebart, M.C., F. Hubert, C. Boiteau, S. Venteo, C. Roustan, and Y. Benyamin. 2004. Biochemical
characterization of the L-plastin-actin interaction shows a resemblance with that of alpha-actinin and allows a distinction to be made between the two actin-binding domains of the molecule. Biochemistry. 43:2428-2437.
Leipe, D.D., Y.I. Wolf, E.V. Koonin, and L. Aravind. 2002. Classification and evolution of P-loop
GTPases and related ATPases. Journal of molecular biology. 317:41-72. Levin, R., S. Grinstein, and D. Schlam. 2015. Phosphoinositides in phagocytosis and
macropinocytosis. Biochim Biophys Acta. 1851:805-823. Levine, B., and D.J. Klionsky. 2004. Development by self-digestion: molecular mechanisms and
biological functions of autophagy. Developmental cell. 6:463-477. Li, X., X. Wang, X. Zhang, M. Zhao, W.L. Tsang, Y. Zhang, R.G. Yau, L.S. Weisman, and H. Xu. 2013.
Genetically encoded fluorescent probe to visualize intracellular phosphatidylinositol 3,5-bisphosphate localization and dynamics. Proceedings of the National Academy of Sciences of the United States of America. 110:21165-21170.
Lim, J., and J.P. Thiery. 2012. Epithelial-mesenchymal transitions: insights from development.
Development. 139:3471-3486.

178 Lim, J.P., and P.A. Gleeson. 2011. Macropinocytosis: an endocytic pathway for internalising large
gulps. Immunol Cell Biol. 89:836-843. Lin, Y.H., C.K. Chou, Y.C. Hung, I.S. Yu, H.A. Pan, S.W. Lin, and P.L. Kuo. 2011. SEPT12 deficiency
causes sperm nucleus damage and developmental arrest of preimplantation embryos. Fertil Steril. 95:363-365.
Lin, Y.H., Y.M. Lin, Y.Y. Wang, I.S. Yu, Y.W. Lin, Y.H. Wang, C.M. Wu, H.A. Pan, S.C. Chao, P.H. Yen,
S.W. Lin, and P.L. Kuo. 2009. The expression level of septin12 is critical for spermiogenesis. Am J Pathol. 174:1857-1868.
Linder, S. 2007. The matrix corroded: podosomes and invadopodia in extracellular matrix
degradation. Trends in cell biology. 17:107-117. Liu, R., X. Zhi, and Q. Zhong. 2015. ATG14 controls SNARE-mediated autophagosome fusion with
a lysosome. Autophagy. 11:847-849. Liu, Y. 2010. New insights into epithelial-mesenchymal transition in kidney fibrosis. J Am Soc
Nephrol. 21:212-222. Lopez, J.A., R.K. Andrews, V. Afshar-Kharghan, and M.C. Berndt. 1998. Bernard-Soulier
syndrome. Blood. 91:4397-4418. Lubec, G., and E. Engidawork. 2002. The brain in Down syndrome (TRISOMY 21). J Neurol.
249:1347-1356. Ludtke, S.J., P.R. Baldwin, and W. Chiu. 1999. EMAN: semiautomated software for high-
resolution single-particle reconstructions. Journal of structural biology. 128:82-97. Luedeke, C., S.B. Frei, I. Sbalzarini, H. Schwarz, A. Spang, and Y. Barral. 2005. Septin-dependent
compartmentalization of the endoplasmic reticulum during yeast polarized growth. The Journal of cell biology. 169:897-908.
Lukoyanova, N., M.S. VanLoock, A. Orlova, V.E. Galkin, K. Wang, and E.H. Egelman. 2002. Each
actin subunit has three nebulin binding sites: implications for steric blocking. Current biology : CB. 12:383-388.
Maddox, A.S., L. Lewellyn, A. Desai, and K. Oegema. 2007. Anillin and the septins promote
asymmetric ingression of the cytokinetic furrow. Developmental cell. 12:827-835. Maimaitiyiming, M., Y. Kobayashi, H. Kumanogoh, S. Nakamura, M. Morita, and S. Maekawa.
2013. Identification of dynamin as a septin-binding protein. Neurosci Lett. 534:322-326. Martens, S., and H.T. McMahon. 2008. Mechanisms of membrane fusion: disparate players and
common principles. Nature reviews. Molecular cell biology. 9:543-556.

179 Martinez, C., M.A. Sanjuan, J.A. Dent, L. Karlsson, and J. Ware. 2004. Human septin-septin
interactions as a prerequisite for targeting septin complexes in the cytosol. The Biochemical journal. 382:783-791.
Mathew, R., V. Karantza-Wadsworth, and E. White. 2007. Role of autophagy in cancer. Nature
reviews. Cancer. 7:961-967. Mavrakis, M., Y. Azou-Gros, F.-C. Tsai, J. Alvarado, A. Bertin, F. Iv, A. Kress, S. Brasselet, G.H.
Koenderink, and T. Lecuit. 2014. Septins promote F-actin ring formation by crosslinking actin filaments into curved bundles. Nature cell biology.
McCartney, A.J., Y. Zhang, and L.S. Weisman. 2014. Phosphatidylinositol 3,5-bisphosphate: low
abundance, high significance. Bioessays. 36:52-64. McDade, S.S., P.A. Hall, and S.E. Russell. 2007. Translational control of SEPT9 isoforms is
perturbed in disease. Hum Mol Genet. 16:742-752. McIlhatton, M.A., J.F. Burrows, P.G. Donaghy, S. Chanduloy, P.G. Johnston, and S.E. Russell.
2001. Genomic organization, complex splicing pattern and expression of a human septin gene on chromosome 17q25.3. Oncogene. 20:5930-5939.
McNiven, M.A. 1998. Dynamin: A Molecular Motor with Pinchase Action. Cell. 94:151-154. Medeiros, N.A., D.T. Burnette, and P. Forscher. 2006. Myosin II functions in actin-bundle
turnover in neuronal growth cones. Nature cell biology. 8:215-226. Mercer, J., and A. Helenius. 2012. Gulping rather than sipping: macropinocytosis as a way of
virus entry. Curr Opin Microbiol. 15:490-499. Mindell, J.A., and N. Grigorieff. 2003. Accurate determination of local defocus and specimen tilt
in electron microscopy. Journal of structural biology. 142:334-347. Mizutani, Y., H. Ito, I. Iwamoto, R. Morishita, H. Kanoh, M. Seishima, and K. Nagata. 2013.
Possible role of a septin, SEPT1, in spreading in squamous cell carcinoma DJM-1 cells. Biological chemistry. 394:281-290.
Mostowy, S., M. Bonazzi, M.A. Hamon, T.N. Tham, A. Mallet, M. Lelek, E. Gouin, C. Demangel, R.
Brosch, C. Zimmer, A. Sartori, M. Kinoshita, M. Lecuit, and P. Cossart. 2010. Entrapment of intracytosolic bacteria by septin cage-like structures. Cell Host Microbe. 8:433-444.
Mostowy, S., L. Boucontet, M.J. Mazon Moya, A. Sirianni, P. Boudinot, M. Hollinshead, P.
Cossart, P. Herbomel, J.P. Levraud, and E. Colucci-Guyon. 2013. The zebrafish as a new model for the in vivo study of Shigella flexneri interaction with phagocytes and bacterial autophagy. PLoS pathogens. 9:e1003588.
Mostowy, S., and P. Cossart. 2012. Septins: the fourth component of the cytoskeleton. Nature
reviews. Molecular cell biology. 13:183-194.

180 Mostowy, S., A. Danckaert, T.N. Tham, C. Machu, S. Guadagnini, J. Pizarro-Cerda, and P. Cossart.
2009. Septin 11 restricts InlB-mediated invasion by Listeria. The Journal of biological chemistry. 284:11613-11621.
Mostowy, S., S. Janel, C. Forestier, C. Roduit, S. Kasas, J. Pizarro-Cerda, P. Cossart, and F. Lafont.
2011. A role for septins in the interaction between the Listeria monocytogenes INVASION PROTEIN InlB and the Met receptor. Biophysical journal. 100:1949-1959.
Mukhina, S., Y.L. Wang, and M. Murata-Hori. 2007. Alpha-actinin is required for tightly regulated
remodeling of the actin cortical network during cytokinesis. Developmental cell. 13:554-565.
Muntean, A.G., and J.L. Hess. 2012. The pathogenesis of mixed-lineage leukemia. Annu Rev
Pathol. 7:283-301. Murphy, D.A., and S.A. Courtneidge. 2011. The 'ins' and 'outs' of podosomes and invadopodia:
characteristics, formation and function. Nature reviews. Molecular cell biology. 12:413-426.
Nagata, K., and M. Inagaki. 2005. Cytoskeletal modification of Rho guanine nucleotide exchange
factor activity: identification of a Rho guanine nucleotide exchange factor as a binding partner for Sept9b, a mammalian septin. Oncogene. 24:65-76.
Nakahira, M., J.N. Macedo, T.V. Seraphim, N. Cavalcante, T.A. Souza, J.C. Damalio, L.F. Reyes,
E.M. Assmann, M.R. Alborghetti, R.C. Garratt, A.P. Araujo, N.I. Zanchin, J.A. Barbosa, and J. Kobarg. 2010. A draft of the human septin interactome. PloS one. 5:e13799.
Nam, S.C., Sun, H., Kang, S.H., Joo, J.Y., Lee, S.J., Chung, Y.B., and Song, S. 2007. Phosphorylation-
Dependent Septin Interaction of Bni5 is Important for Cytokinesis. The Journal of Microbiology. 45:227-233.
Neufeld, T.P., and G.M. Rubin. 1994. The Drosophila peanut gene is required for cytokinesis and
encodes a protein similar to yeast putative bud neck filament proteins. Cell. 77:371-379. Nguyen, T.Q., H. Sawa, H. Okano, and J.G. White. 2000. The C. elegans septin genes, unc-59 and
unc-61, are required for normal postembryonic cytokineses and morphogenesis but have no essential function in embryogenesis. Journal of cell science. 113 Pt 21:3825-3837.
Oakes, P.W., Y. Beckham, J. Stricker, and M.L. Gardel. 2012. Tension is required but not
sufficient for focal adhesion maturation without a stress fiber template. The Journal of cell biology. 196:363-374.
Ono, R., M. Ihara, H. Nakajima, K. Ozaki, Y. Kataoka-Fujiwara, T. Taki, K. Nagata, M. Inagaki, N.
Yoshida, T. Kitamura, Y. Hayashi, M. Kinoshita, and T. Nosaka. 2005a. Disruption of Sept6, a fusion partner gene of MLL, does not affect ontogeny, leukemogenesis induced

181
by MLL-SEPT6, or phenotype induced by the loss of Sept4. Molecular and cellular biology. 25:10965-10978.
Ono, R., H. Nakajima, K. Ozaki, H. Kumagai, T. Kawashima, T. Taki, T. Kitamura, Y. Hayashi, and T.
Nosaka. 2005b. Dimerization of MLL fusion proteins and FLT3 activation synergize to induce multiple-lineage leukemogenesis. Journal of Clinical Investigation. 115:919-929.
Orlova, A., and E.H. Egelman. 1995. Structural dynamics of F-actin: I. Changes in the C terminus.
Journal of molecular biology. 245:582-597. Orlova, A., and E.H. Egelman. 1997. Cooperative rigor binding of myosin to actin is a function of
F-actin structure. Journal of molecular biology. 265:469-474. Orlova, A., V.E. Galkin, C.M. Jeffries, E.H. Egelman, and J. Trewhella. 2011. The N-terminal
domains of myosin binding protein C can bind polymorphically to F-actin. Journal of molecular biology. 412:379-386.
Oztan, A., M. Silvis, O.A. Weisz, N.A. Bradbury, S.C. Hsu, J.R. Goldenring, C. Yeaman, and G.
Apodaca. 2007. Exocyst requirement for endocytic traffic directed toward the apical and basolateral poles of polarized MDCK cells. Molecular biology of the cell. 18:3978-3992.
Pagano, A., P. Crottet, C. Prescianotto-Baschong, and M. Spiess. 2004. In vitro formation of
recycling vesicles from endosomes requires adaptor protein-1/clathrin and is regulated by rab4 and the connector rabaptin-5. Molecular biology of the cell. 15:4990-5000.
Pagtalunan, M.E., P.L. Miller, S. Jumping-Eagle, R.G. Nelson, B.D. Myers, H.G. Rennke, N.S.
Coplon, L. Sun, and T.W. Meyer. 1997. Podocyte loss and progressive glomerular injury in type II diabetes. J Clin Invest. 99:342-348.
Palamidessi, A., E. Frittoli, M. Garre, M. Faretta, M. Mione, I. Testa, A. Diaspro, L. Lanzetti, G.
Scita, and P.P. Di Fiore. 2008. Endocytic trafficking of Rac is required for the spatial restriction of signaling in cell migration. Cell. 134:135-147.
Parri, M., and P. Chiarugi. 2010. Rac and Rho GTPases in cancer cell motility control. Cell
Commun Signal. 8:23. Parsons, J.T., A.R. Horwitz, and M.A. Schwartz. 2010. Cell adhesion: integrating cytoskeletal
dynamics and cellular tension. Nature reviews. Molecular cell biology. 11:633-643. Paterson, H., B. Reeves, R. Brown, A. Hall, M. Furth, J. Bos, P. Jones, and C. Marshall. 1987.
Activated N-ras controls the transformed phenotype of HT1080 human fibrosarcoma cells. Cell. 51:803-812.
Patsialou, A., J. Wyckoff, Y. Wang, S. Goswami, E.R. Stanley, and J.S. Condeelis. 2009. Invasion of
human breast cancer cells in vivo requires both paracrine and autocrine loops involving the colony-stimulating factor-1 receptor. Cancer research. 69:9498-9506.

182 Patzig, J., O. Jahn, S. Tenzer, S.P. Wichert, P. de Monasterio-Schrader, S. Rosfa, J. Kuharev, K.
Yan, I. Bormuth, J. Bremer, A. Aguzzi, F. Orfaniotou, D. Hesse, M.H. Schwab, W. Mobius, K.A. Nave, and H.B. Werner. 2011. Quantitative and integrative proteome analysis of peripheral nerve myelin identifies novel myelin proteins and candidate neuropathy loci. J Neurosci. 31:16369-16386.
Pazour, G.J., and G.B. Witman. 2003. The vertebrate primary cilium is a sensory organelle.
Current opinion in cell biology. 15:105-110. Peacock, J.G., A.L. Miller, W.D. Bradley, O.C. Rodriguez, D.J. Webb, and A.J. Koleske. 2007. The
Abl-related gene tyrosine kinase acts through p190RhoGAP to inhibit actomyosin contractility and regulate focal adhesion dynamics upon adhesion to fibronectin. Molecular biology of the cell. 18:3860-3872.
Pennington, K., C.L. Beasley, P. Dicker, A. Fagan, J. English, C.M. Pariante, R. Wait, M.J. Dunn,
and D.R. Cotter. 2008. Prominent synaptic and metabolic abnormalities revealed by proteomic analysis of the dorsolateral prefrontal cortex in schizophrenia and bipolar disorder. Mol Psychiatry. 13:1102-1117.
Peterson, E.A., L.M. Kalikin, J.D. Steels, M.P. Estey, W.S. Trimble, and E.M. Petty. 2007.
Characterization of a SEPT9 interacting protein, SEPT14, a novel testis-specific septin. Mamm Genome. 18:796-807.
Petrie, R.J., N. Gavara, R.S. Chadwick, and K.M. Yamada. 2012. Nonpolarized signaling reveals
two distinct modes of 3D cell migration. The Journal of cell biology. 197:439-455. Petrie, R.J., and K.M. Yamada. 2012. At the leading edge of three-dimensional cell migration.
Journal of cell science. 125:5917-5926. Qi, M., W. Yu, S. Liu, H. Jia, L. Tang, M. Shen, X. Yan, H. Saiyin, Q. Lang, B. Wan, S. Zhao, and L.
Yu. 2005. Septin1, a new interaction partner for human serine/threonine kinase aurora-B. Biochem Biophys Res Commun. 336:994-1000.
Racoosin, E.L., and J.A. Swanson. 1993. Macropinosome maturation and fusion with tubular
lysosomes in macrophages. The Journal of cell biology. 121:1011-1020. Raftopoulou, M., and A. Hall. 2004. Cell migration: Rho GTPases lead the way. Dev Biol. 265:23-
32. Risca, V.I., E.B. Wang, O. Chaudhuri, J.J. Chia, P.L. Geissler, and D.A. Fletcher. 2012. Actin
filament curvature biases branching direction. Proceedings of the National Academy of Sciences of the United States of America. 109:2913-2918.
Rittmeyer, E.N., S. Daniel, S.-C. Hsu, and M.A. Osman. 2008. A dual role for IQGAP1 in regulating
exocytosis. Journal of cell science. 121:391-403.

183 Roca-Cusachs, P., A. del Rio, E. Puklin-Faucher, N.C. Gauthier, N. Biais, and M.P. Sheetz. 2013.
Integrin-dependent force transmission to the extracellular matrix by alpha-actinin triggers adhesion maturation. Proceedings of the National Academy of Sciences of the United States of America. 110:E1361-1370.
Roseler, S., K. Sandrock, I. Bartsch, A. Busse, H. Omran, N.T. Loges, and B. Zieger. 2011. Lethal
phenotype of mice carrying a Sept11 null mutation. Biological chemistry. 392:779-781. Russell, S.E., and P.A. Hall. 2011. Septin genomics: a road less travelled. Biological chemistry.
392:763-767. Russell, S.E., M.A. McIlhatton, J.F. Burrows, P.G. Donaghy, S. Chanduloy, E.M. Petty, L.M. Kalikin,
S.W. Church, S. McIlroy, D.P. Harkin, G.W. Keilty, A.N. Cranston, J. Weissenbach, I. Hickey, and P.G. Johnston. 2000. Isolation and mapping of a human septin gene to a region on chromosome 17q, commonly deleted in sporadic epithelial ovarian tumors. Cancer research. 60:4729-4734.
Sadian, Y., C. Gatsogiannis, C. Patasi, O. Hofnagel, R.S. Goody, M. Farkasovsky, and S. Raunser.
2013. The role of Cdc42 and Gic1 in the regulation of septin filament formation and dissociation. Elife. 2:e01085.
Scambler, P.J. 2000. The 22q11 deletion syndromes. Hum Mol Genet. 9:2421-2426. Schmees, C., R. Villasenor, W. Zheng, H. Ma, M. Zerial, C.H. Heldin, and C. Hellberg. 2012.
Macropinocytosis of the PDGF beta-receptor promotes fibroblast transformation by H-RasG12V. Molecular biology of the cell. 23:2571-2582.
Schmidt, K., and B.J. Nichols. 2004a. A barrier to lateral diffusion in the cleavage furrow of
dividing mammalian cells. Current biology : CB. 14:1002-1006. Schmidt, K., and B.J. Nichols. 2004b. Functional interdependence between septin and actin
cytoskeleton. BMC Cell Biol. 5:43. Schmoller, K.M., C. Semmrich, and A.R. Bausch. 2011. Slow down of actin depolymerization by
cross-linking molecules. Journal of structural biology. 173:350-357. Schober, M., S. Raghavan, M. Nikolova, L. Polak, H.A. Pasolli, H.E. Beggs, L.F. Reichardt, and E.
Fuchs. 2007. Focal adhesion kinase modulates tension signaling to control actin and focal adhesion dynamics. The Journal of cell biology. 176:667-680.
Schwartz, M.A. 2010. Integrins and extracellular matrix in mechanotransduction. Cold Spring
Harbor perspectives in biology. 2:a005066. Scott, M., W.G. McCluggage, K.J. Hillan, P.A. Hall, and S.E.H. Russell. 2006. Altered patterns of
transcription of the septin gene, SEPT9, in ovarian tumorigenesis. International Journal of Cancer. 118:1325-1329.

184 Sellin, M.E., P. Holmfeldt, S. Stenmark, and M. Gullberg. 2011a. Microtubules support a disk-like
septin arrangement at the plasma membrane of mammalian cells. Molecular biology of the cell. 22:4588-4601.
Sellin, M.E., L. Sandblad, S. Stenmark, and M. Gullberg. 2011b. Deciphering the rules governing
assembly order of mammalian septin complexes. Molecular biology of the cell. 22:3152-3164.
Settembre, C., A. Fraldi, D.L. Medina, and A. Ballabio. 2013. Signals from the lysosome: a control
centre for cellular clearance and energy metabolism. Nature reviews. Molecular cell biology. 14:283-296.
Shankar, J., A. Messenberg, J. Chan, T.M. Underhill, L.J. Foster, and I.R. Nabi. 2010. Pseudopodial
actin dynamics control epithelial-mesenchymal transition in metastatic cancer cells. Cancer research. 70:3780-3790.
Sharma, S., A. Quintana, G.M. Findlay, M. Mettlen, B. Baust, M. Jain, R. Nilsson, A. Rao, and P.G.
Hogan. 2013a. An siRNA screen for NFAT activation identifies septins as coordinators of store-operated Ca2+ entry. Nature. 499:238-242.
Sharma, S., A. Quintana, G.M. Findlay, M. Mettlen, B. Baust, M. Jain, R. Nilsson, A. Rao, and P.G.
Hogan. 2013b. An siRNA screen for NFAT activation identifies septins as coordinators of store-operated Ca entry. Nature.
Shindo, A.a.W., J. B. 2014. PCP and Septins Compartmentalize Cortical Actomyosin to Direct
Collective Cell Movement. Science. 3443:649-652. Shinoda, T., H. Ito, K. Sudo, I. Iwamoto, R. Morishita, and K. Nagata. 2010. Septin 14 is involved
in cortical neuronal migration via interaction with Septin 4. Molecular biology of the cell. 21:1324-1334.
Sidhaye, V.K., E. Chau, P.N. Breysse, and L.S. King. 2011. Septin-2 mediates airway epithelial
barrier function in physiologic and pathologic conditions. Am J Respir Cell Mol Biol. 45:120-126.
Sirajuddin, M., M. Farkasovsky, F. Hauer, D. Kuhlmann, I.G. Macara, M. Weyand, H. Stark, and A.
Wittinghofer. 2007. Structural insight into filament formation by mammalian septins. Nature. 449:311-315.
Sirajuddin, M., M. Farkasovsky, E. Zent, and A. Wittinghofer. 2009. GTP-induced conformational
changes in septins and implications for function. Proceedings of the National Academy of Sciences of the United States of America. 106:16592-16597.
Sitz, J.H., K. Baumgartel, B. Hammerle, C. Papadopoulos, P. Hekerman, F.J. Tejedor, W. Becker,
and B. Lutz. 2008. The Down syndrome candidate dual-specificity tyrosine phosphorylation-regulated kinase 1A phosphorylates the neurodegeneration-related septin 4. Neuroscience. 157:596-605.

185 Spiliotis, E.T. 2010. Regulation of microtubule organization and functions by septin GTPases.
Cytoskeleton (Hoboken). 67:339-345. Spiliotis, E.T., and A.S. Gladfelter. 2012. Spatial guidance of cell asymmetry: septin GTPases show
the way. Traffic. 13:195-203. Spiliotis, E.T., S.J. Hunt, Q. Hu, M. Kinoshita, and W.J. Nelson. 2008. Epithelial polarity requires
septin coupling of vesicle transport to polyglutamylated microtubules. The Journal of cell biology. 180:295-303.
Spiliotis, E.T., Hunt, S. J., Hu, Q., Kinoshita, M., and Nelson, W. J. 2008. Epithelial polarity
requires septin coupling of vesicle transport to polyglutamylated microtubules. The Journal of cell biology. 180:295-303.
Spiliotis, E.T., M. Kinoshita, and W.J. Nelson. 2005. A mitotic septin scaffold required for
Mammalian chromosome congression and segregation. Science. 307:1781-1785. Steels, J.D., M.P. Estey, C.D. Froese, D. Reynaud, C. Pace-Asciak, and W.S. Trimble. 2007. Sept12
is a component of the mammalian sperm tail annulus. Cell Motil Cytoskeleton. 64:794-807.
Stricker, J., Y. Beckham, M.W. Davidson, and M.L. Gardel. 2013. Myosin II-mediated focal
adhesion maturation is tension insensitive. PloS one. 8:e70652. Sugino, Y., K. Ichioka, T. Soda, M. Ihara, M. Kinoshita, O. Ogawa, and H. Nishiyama. 2008. Septins
as Diagnostic Markers for a Subset of Human Asthenozoospermia. J Urol. 180:2706-2709.
Sui, L., W. Zhang, Q. Liu, T. Chen, N. Li, T. Wan, M. Yu, and X. Cao. 2003. Cloning and functional
characterization of human septin 10, a novel member of septin family cloned from dendritic cells. Biochemical and Biophysical Research Communications. 304:393-398.
Suzuki, G., K.M. Harper, T. Hiramoto, T. Sawamura, M. Lee, G. Kang, K. Tanigaki, M. Buell, M.A.
Geyer, W.S. Trimble, S. Agatsuma, and N. Hiroi. 2009. Sept5 deficiency exerts pleiotropic influence on affective behaviors and cognitive functions in mice. Hum Mol Genet. 18:1652-1660.
Svitkina, T.M., and G.G. Borisy. 1999. Arp2/3 complex and actin depolymerizing factor/cofilin in
dendritic organization and treadmilling of actin filament array in lamellipodia. The Journal of cell biology. 145:1009-1026.
Swanson, J.A. 2008. Shaping cups into phagosomes and macropinosomes. Nature reviews.
Molecular cell biology. 9:639-649. Tada, T., A. Simonetta, M. Batterton, M. Kinoshita, D. Edbauer, and M. Sheng. 2007. Role of
Septin cytoskeleton in spine morphogenesis and dendrite development in neurons. Current biology : CB. 17:1752-1758.

186 Takehashi, M., T. Alioto, T. Stedeford, A.S. Persad, M. Banasik, E. Masliah, S. Tanaka, and K.
Ueda. 2004. Septin 3 gene polymorphism in Alzheimer's disease. Gene Expr. 11:263-270. Tanaka-Takiguchi, Y., M. Kinoshita, and K. Takiguchi. 2009. Septin-mediated uniform bracing of
phospholipid membranes. Current biology : CB. 19:140-145. Tanaka, M., H. Kijima, J. Itoh, T. Matsuda, and T. Tanaka. 2002. Impaired expression of a human
septin family gene Bradeion inhibits the growth and tumorigenesis of colorectal cancer in vitro and in vivo. Cancer Gene Ther. 9:483-488.
Tanaka, M., T. Tanaka, H. Kijima, J. Itoh, T. Matsuda, S. Hori, and M. Yamamoto. 2001.
Characterization of Tissue- and Cell-Type-Specific Expression of a Novel Human Septin Family Gene, Bradeion. Biochemical and Biophysical Research Communications. 286:547-553.
Tanaka, M., T. Tanaka, S. Matsuzaki, Y. Seto, T. Matsuda, K. Komori, J. Itoh, H. Kijima, K. Tamai,
M. Shibayama, Y. Hashimoto, H. Nakazawa, and H. Toma. 2003. Rapid and quantitative detection of human septin family Bradeion as a practical diagnostic method of colorectal and urologic cancers. Med Sci Monit. 9:MT61-68.
Taniguchi, M., M. Taoka, M. Itakura, A. Asada, T. Saito, M. Kinoshita, M. Takahashi, T. Isobe, and
S. Hisanaga. 2007. Phosphorylation of adult type Sept5 (CDCrel-1) by cyclin-dependent kinase 5 inhibits interaction with syntaxin-1. The Journal of biological chemistry. 282:7869-7876.
Taylor, R.W., M.J. Barron, G.M. Borthwick, A. Gospel, P.F. Chinnery, D.C. Samuels, G.A. Taylor,
S.M. Plusa, S.J. Needham, L.C. Greaves, T.B.L. Kirkwood, and D.M. Turnbull. 2003. Mitochondrial DNA mutations in human colonic crypt stem cells. J Clin Invest. 112:1351-1360.
Thiery, J.P., H. Acloque, R.Y. Huang, and M.A. Nieto. 2009. Epithelial-mesenchymal transitions in
development and disease. Cell. 139:871-890. Tilney, L.G., P.S. Connelly, L. Ruggiero, K.A. Vranich, and G.M. Guild. 2003. Actin filament
turnover regulated by cross-linking accounts for the size, shape, location, and number of actin bundles in Drosophila bristles. Molecular biology of the cell. 14:3953-3966.
Tokhtaeva, E., J. Capri, E.A. Marcus, J.P. Whitelegge, V. Khuzakhmetova, E. Bukharaeva, N. Deiss-
Yehiely, L.A. Dada, G. Sachs, E. Fernandez-Salas, and O. Vagin. 2015. Septin dynamics are essential for exocytosis. The Journal of biological chemistry. 290:5280-5297.
Tooley, A.J., J. Gilden, J. Jacobelli, P. Beemiller, W.S. Trimble, M. Kinoshita, and M.F. Krummel.
2009. Amoeboid T lymphocytes require the septin cytoskeleton for cortical integrity and persistent motility. Nature cell biology. 11:17-26.
Toth, K., O. Galamb, S. Spisak, B. Wichmann, F. Sipos, G. Valcz, K. Leiszter, B. Molnar, and Z.
Tulassay. 2011. The influence of methylated septin 9 gene on RNA and protein level in colorectal cancer. Pathol Oncol Res. 17:503-509.

187 Toure, A., B. Rode, G.R. Hunnicutt, D. Escalier, and G. Gacon. 2011. Septins at the annulus of
mammalian sperm. Biological chemistry. 392:799-803. Traikov, S., C. Stange, T. Wassmer, P. Paul-Gilloteaux, J. Salamero, G. Raposo, and B. Hoflack.
2014. Septin6 and Septin7 GTP binding proteins regulate AP-3- and ESCRT-dependent multivesicular body biogenesis. PloS one. 9:e109372.
Tsang, C.W., M.P. Estey, J.E. DiCiccio, H. Xie, D. Patterson, and W.S. Trimble. 2011.
Characterization of presynaptic septin complexes in mammalian hippocampal neurons. Biological chemistry. 392:739-749.
Tsang, C.W., M. Fedchyshyn, J. Harrison, H. Xie, J. Xue, P.J. Robinson, L.Y. Wang, and W.S.
Trimble. 2008. Superfluous role of mammalian septins 3 and 5 in neuronal development and synaptic transmission. Molecular and cellular biology. 28:7012-7029.
Ueda, M., N. Kawamura, T. Tateishi, N. Sakae, K. Motomura, Y. Ohyagi, and J.I. Kira. 2010.
Phenotypic spectrum of hereditary neuralgic amyotrophy caused by the SEPT9 R88W mutation. J Neurol Neurosurg Psychiatry. 81:94-96.
Vega, I.E., and S.C. Hsu. 2003. The septin protein Nedd5 associates with both the exocyst
complex and microtubules and disruption of its GTPase activity promotes aberrant neurite sprouting in PC12 cells. Neuroreport. 14:31-37.
Verhey, K.J., and J. Gaertig. 2007. The tubulin code. Cell Cycle. 6:2152-2160. Versele, M., and J. Thorner. 2005. Some assembly required: yeast septins provide the instruction
manual. Trends in cell biology. 15:414-424. Vicente-Manzanares, M., X. Ma, R.S. Adelstein, and A.R. Horwitz. 2009. Non-muscle myosin II
takes centre stage in cell adhesion and migration. Nature reviews. Molecular cell biology. 10:778-790.
Vicente-Manzanares, M., J. Zareno, L. Whitmore, C.K. Choi, and A.F. Horwitz. 2007. Regulation of
protrusion, adhesion dynamics, and polarity by myosins IIA and IIB in migrating cells. The Journal of cell biology. 176:573-580.
Wang, J.T., R.D. Teasdale, and D. Liebl. 2014. Macropinosome quantitation assay. MethodsX.
1:36-41. Wang, W., S. Goswami, K. Lapidus, A.L. Wells, J.B. Wyckoff, E. Sahai, R.H. Singer, J.E. Segall, and
J.S. Condeelis. 2004. Identification and testing of a gene expression signature of invasive carcinoma cells within primary mammary tumors. Cancer research. 64:8585-8594.
Warren, J.D., W. Xiong, A.M. Bunker, C.P. Vaughn, L.V. Furtado, W.L. Roberts, J.C. Fang, W.S.
Samowitz, and K.A. Heichman. 2011. Septin 9 methylated DNA is a sensitive and specific blood test for colorectal cancer. BMC Med. 9:133.

188 Wasik, A.A., Z. Polianskyte-Prause, M.-Q. Dong, A.S. Shaw, J.R. Yates, M.G. Farquhar, and S.
Lehtonen. 2012. Septin 7 forms a complex with CD2AP and nephrin and regulates glucose transporter trafficking. Molecular biology of the cell. 23:3370-3379.
Webb, D.J., K. Donais, L.A. Whitmore, S.M. Thomas, C.E. Turner, J.T. Parsons, and A.F. Horwitz.
2004. FAK-Src signalling through paxillin, ERK and MLCK regulates adhesion disassembly. Nature cell biology. 6:154-161.
Weirich, C.S., J.P. Erzberger, and Y. Barral. 2008. The septin family of GTPases: architecture and
dynamics. Nature reviews. Molecular cell biology. 9:478-489. Wheeler, D.B., R. Zoncu, D.E. Root, D.M. Sabatini, and C.L. Sawyers. 2015. Identification of an
oncogenic RAB protein. Science. 350:211-217. White, E. 2013. Exploiting the bad eating habits of Ras-driven cancers. Genes Dev. 27:2065-2071. Xie, Y., J.P. Vessey, A. Konecna, R. Dahm, P. Macchi, and M.A. Kiebler. 2007. The GTP-binding
protein Septin 7 is critical for dendrite branching and dendritic-spine morphology. Current biology : CB. 17:1746-1751.
Xue, J., C.W. Tsang, W.P. Gai, C.S. Malladi, W.S. Trimble, J.A. Rostas, and P.J. Robinson. 2004.
Septin 3 (G-septin) is a developmentally regulated phosphoprotein enriched in presynaptic nerve terminals. Journal of neurochemistry. 91:579-590.
Yamada, S., and W.J. Nelson. 2007. Localized zones of Rho and Rac activities drive initiation and
expansion of epithelial cell-cell adhesion. The Journal of cell biology. 178:517-527. Yang, J.Y., C.S. Zong, W. Xia, Y. Wei, M. Ali-Seyed, Z. Li, K. Broglio, D.A. Berry, and M.C. Hung.
2006. MDM2 promotes cell motility and invasiveness by regulating E-cadherin degradation. Molecular and cellular biology. 26:7269-7282.
Yang, Y.M., M.J. Fedchyshyn, G. Grande, J. Aitoubah, C.W. Tsang, H. Xie, C.A. Ackerley, W.S.
Trimble, and L.Y. Wang. 2010. Septins regulate developmental switching from microdomain to nanodomain coupling of Ca(2+) influx to neurotransmitter release at a central synapse. Neuron. 67:100-115.
Yengo, C.M., E.M. De la Cruz, D. Safer, E.M. Ostap, and H.L. Sweeney. 2002. Kinetic
characterization of the weak binding states of myosin V. Biochemistry. 41:8508-8517. Yoo, B.C., R. Vlkolinsky, E. Engidawork, N. Cairns, M. Fountoulakis, and G. Lubec. 2001.
Differential expression of molecular chaperones in brain of patients with Down syndrome. Electrophoresis. 22:1233-1241.
Yoshida, S., A.D. Hoppe, N. Araki, and J.A. Swanson. 2009. Sequential signaling in plasma-
membrane domains during macropinosome formation in macrophages. Journal of cell science. 122:3250-3261.

189 Youakim, A., and M. Ahdieh. 1999. Interferon-γ decreases barrier function in T84 cells by
reducing ZO-1 levels and disrupting apical actin. American Journal of Physiology - Gastrointestinal and Liver Physiology. 276:G1279-G1288.
Zaidel-Bar, R., R. Milo, Z. Kam, and B. Geiger. 2007. A paxillin tyrosine phosphorylation switch
regulates the assembly and form of cell-matrix adhesions. Journal of cell science. 120:137-148.
Zegers, M.M., L.E. O'Brien, W. Yu, A. Datta, and K.E. Mostov. 2003. Epithelial polarity and
tubulogenesis in vitro. Trends in cell biology. 13:169-176. Zhang, J., C. Kong, H. Xie, P.S. McPherson, S. Grinstein, and W.S. Trimble. 1999.
Phosphatidylinositol polyphosphate binding to the mammalian septin H5 is modulated by GTP. Current biology : CB. 9:1458-1467.
Zhang, Y., J. Gao, K.K. Chung, H. Huang, V.L. Dawson, and T.M. Dawson. 2000. Parkin functions
as an E2-dependent ubiquitin- protein ligase and promotes the degradation of the synaptic vesicle-associated protein, CDCrel-1. Proceedings of the National Academy of Sciences of the United States of America. 97:13354-13359.
Zhu, M., F. Wang, F. Yan, P.Y. Yao, J. Du, X. Gao, X. Wang, Q. Wu, T. Ward, J. Li, S. Kioko, R. Hu,
W. Xie, X. Ding, and X. Yao. 2008. Septin 7 interacts with centromere-associated protein E and is required for its kinetochore localization. The Journal of biological chemistry. 283:18916-18925.
Zieger, B., Y. Hashimoto, and J. Ware. 1997. Alternative expression of platelet glycoprotein
Ib(beta) mRNA from an adjacent 5' gene with an imperfect polyadenylation signal sequence. J Clin Invest. 99:520-525.
Zigmond, S.H., R. Furukawa, and M. Fechheimer. 1992. Inhibition of actin filament
depolymerization by the Dictyostelium 30,000-D actin-bundling protein. The Journal of cell biology. 119:559-567.
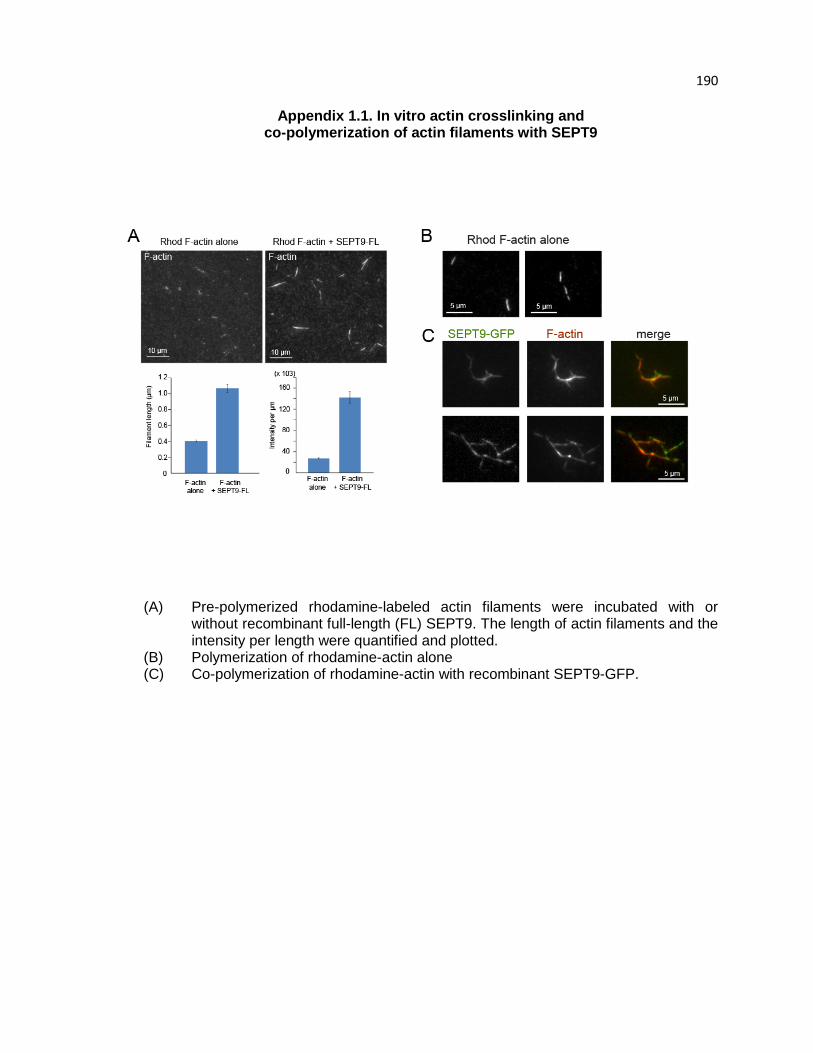
190
Appendix 1.1. In vitro actin crosslinking and co-polymerization of actin filaments with SEPT9
(A) Pre-polymerized rhodamine-labeled actin filaments were incubated with or
without recombinant full-length (FL) SEPT9. The length of actin filaments and the intensity per length were quantified and plotted.
(B) Polymerization of rhodamine-actin alone (C) Co-polymerization of rhodamine-actin with recombinant SEPT9-GFP.

191
Appendix 1.2. SEPT9 colocalization with Rab4
Cos7 cells were fixed and stained for SEPT9 (green) and Rab4 (red). SEPT9 colocalizes with Rab4-positive vesicles. Bottom panel shows higher magnification of the region of interest (ROI).

192
Vita Lee Dolat was born in Ledyard, CT, U.S.A on July 2, 1984. After receiving his Bachelors
of Science in Molecular and Cell Biology from the University of Connecticut, Lee worked
as a research technician at 454 Life Sciences, a high-throughput DNA sequencing
company, and Massachusetts General Hospital/Harvard Medical School. Lee conducted
his PhD studies in the laboratory of Elias T. Spiliotis, PhD in the Department of Biology
at Drexel University from 2010-2016.
Articles:
1) Lee Dolat and Elias T. Spiliotis. Septins promote macropinosome maturation and traffic to the lysosome by facilitating membrane fusion. In revision.
2) Elias T. Spiliotis and Lee Dolat. Priming for destruction: septins at the crossroads of mitochondrial fission and bacterial autophagy. EMBO Reports 17: 1-3, 2016.
3) Clayton Smith, Lee Dolat, Eva Forgacs, Elias T. Spiliotis and Vitold E. Galkin. Septin 9 exhibits polymorphic binding to F-actin and inhibits myosin and cofilin activity. Under Review in Journal of Molecular Biology 427: 3273 – 3284, 2015.
4) Lee Dolat, John L. Hunyara, Jonathan R. Bowen, Eva P. Karasmanis, Maha Elgawly, Vitold E. Galkin and Elias T. Spiliotis. Septins promote stress fiber-mediated focal adhesion maturation and renal epithelial motility. The Journal of Cell Biology 207: 225-235, 2014.
5) Lee Dolat, Qicong Hu, Elias T. Spiliotis. Septin functions in organ system physiology and pathology. Biological Chemistry 395:123-141, 2014.
6) Jianli Hu, Xiaobo Bai, Jonathan R. Bowen, Lee Dolat, Farida Korobova, Wenqian Yu, Peter W. Baas, Tatyana Svitkina, Gianluca Gallo, Elias T. Spiliotis. Septin-driven coordination of actin and microtubule remodeling regulates the collateral branching of axons. Current Biology 22: 1109-1115, 2012
Book Chapters:
1) Elias T. Spiliotis, Eva P. Karasmanis, Lee Dolat. Live-cell imaging of septins with the actin and microtubule cytoskeleton (2016). Methods in Cell Biology. In press.
2) Elias T. Spiliotis and Lee Dolat. Septins: cytoskeletal filaments with structural and regulatory functions (2016). Encyclopedia of Cell Biology. Elsevier. 2015.
Honors: Ruth L Kirschstein NRSA F31 pre-doctoral fellowship. NCI. NIH 2014 – 2016 President. Biology Graduate Student Association. 2014-2015 Teaching Assistant of the Year. University-wide nomination. 2011-2012






























