Embed Size (px)
Citation preview

RAB GTPASE-ACTIVATING PROTEINS
AT THE GOLGI:ENDOSOME INTERFACE
A DISSERTATION
SUBMITTED TO THE DEPARTMENT OF BIOCHEMISTRY
AND THE COMMITTEE ON GRADUATE STUDIES
OF STANFORD UNIVERSITY
IN PARTIAL FULFILLMENT OF THE REQUIREMENTS
FOR THE DEGREE OF
DOCTOR OF PHILOSOPHY
Ryan Michael Nottingham
May 2010

http://creativecommons.org/licenses/by-nc/3.0/us/
This dissertation is online at: http://purl.stanford.edu/dv301zj4923
© 2010 by Ryan Michael Nottingham. All Rights Reserved.
Re-distributed by Stanford University under license with the author.
This work is licensed under a Creative Commons Attribution-Noncommercial 3.0 United States License.
ii

I certify that I have read this dissertation and that, in my opinion, it is fully adequatein scope and quality as a dissertation for the degree of Doctor of Philosophy.
Suzanne Pfeffer, Primary Adviser
I certify that I have read this dissertation and that, in my opinion, it is fully adequatein scope and quality as a dissertation for the degree of Doctor of Philosophy.
Gilbert Chu
I certify that I have read this dissertation and that, in my opinion, it is fully adequatein scope and quality as a dissertation for the degree of Doctor of Philosophy.
Pehr Harbury
Approved for the Stanford University Committee on Graduate Studies.
Patricia J. Gumport, Vice Provost Graduate Education
This signature page was generated electronically upon submission of this dissertation in electronic format. An original signed hard copy of the signature page is on file inUniversity Archives.
iii

iv
ABSTRACT
Rab GTPases are master regulators of membrane trafficking in eukaryotic
cells. With GTP bound, they regulate trafficking by recruiting effectors to
specific membrane-bound compartments. Rab proteins are themselves
regulated by additional factors that mediate their proper localization as well as
their bound nucleotide state. GTPase-activating proteins (GAPs) stimulate a
Rabʼs intrinsic rate of GTP hydrolysis, thus inactivating the Rab by converting
bound GTP to GDP. Regulation of Rab proteins links the formation and
breakdown of sequential, Rab-regulated membrane domains in the secretory
and endocytic pathways. The first chapter introduces the function of
RabGTPases in membrane trafficking and the role of GAPs in regulating Rab
function.
The second chapter of this thesis presents the characterization of a novel
RabGAP, RUTBC1. This protein was identified through a yeast two hybrid
screen for RabGAPs that interact with Rab9, the key regulator of mannose 6-
phosphate receptor (MPR) recycling from late endosomes to the trans Golgi
network. RUTBC1 binds Rab9 in vitro and in cells through interactions with its
N-terminus. Overexpression of RUTBC1 only slightly disrupts MPR trafficking
and RUTBC1 does not function as a GAP for Rab9. An in vitro biochemical
screen of 32 mammalian Rab GTPases revealed that RUTBC1 has GAP
activity toward Rab33b and Rab32 that is catalyzed by its conserved TBC

v
domain. These data suggest that RUTBC1 might function to link inactivation of
these Rabs in relation to a Rab9 microdomain, in support of the existence of a
Rab cascade at the interface between the Golgi apparatus and endosomes.
The third chapter describes the functional role of RUTBC1 in cultured cells.
Depletion of RUTBC1 unexpectedly leads to concomitant depletion of
Atg16L1. Atg16L1 has an established and essential role in macroautophagy,
a highly conserved cellular recycling process. Overexpression of Atg16L1
causes the formation of large puncta in the cytoplasm, which are also labeled
by endogenous RUTBC1 and may represent autophagosomes. Atg16L1 is a
known Rab33b effector, suggesting that Rab33b, RUTBC1 and Atg16L1
function together to regulate autophagosome formation.
The fourth chapter describes another TBC-domain containing protein,
RUTBC2. This protein is highly related to RUTBC1 and also binds specifically
to Rab9. In vitro biochemical screening for RUTBC2ʼs Rab substrates showed
that RUTBC2 had highest GAP activity toward Rab34 and Rab36, two very
similar Rabs thought to play a role in secretion. The difference in substrate
specificity between RUTBC1 and RUTBC2 further exemplifies the highly
complex integration of diverse membrane trafficking pathways in mammalian
cells.

vi
ACKNOWLEDGEMENTS
Above all, I would like to thank my research advisor, Suzanne Pfeffer. During
my time at Stanford, I have learned so much from her: not only how to think
about science, but just as importantly, how to communicate the results. Most
of all though, I appreciate the independence she afforded me and moreover,
the patience that resulted from it. As a mentor, she helped me to realize that
nothing is impossible despite the obstacles and adversity one often
encounters in research.
Secondly, I would like to thank the other members of my committee, Gil Chu
and Pehr Harbury, for their advice and constructive criticism throughout my
time. Always helpful, I wished I had sought their advice more often than I did.
The Biochemistry Department deserves credit as well for making research
here easier through the great atmosphere maintained by the students and
postdocs. My collaborators in this work also deserve special mention: Francis
Barr and his laboratory as well as David Lambright and his laboratory. Without
their help, this thesis would have taken unimaginably longer. I also want to
thank my undergraduate advisor, Dorothy Shippen, for taking a chance on a
directionless undergrad and introducing me to the life of research.
Next, I want to express my thanks to the past and present members of the
Pfeffer Lab. What a ride! I am glad that I have met each of you – it is hard to

vii
imagine going through the ups and downs of graduate school with any other
group of people. I especially want to thank Maïka Deffieu for all of her helpful
discussions about autophagy (and for being a great friend!). I also especially
want to thank the postdocs who were here when I joined the lab (Pfeffer One!):
Dikran Aivazian, Leo Serrano, Ian Ganley, Sridevi Khambhanpaty and Monica
Calero. They made the lab a fun and joyful place…it was “beyond dreams.”
To my fellow Pfeffer Lab graduate students – I have never enjoyed discussing
science, music, religion and other esoterica more. A big thanks to Garret
Hayes, Eric Espinosa, Peter Lee and Frank Brown for being great friends as
well as great colleagues. Garret and Peter put up with me as bay mates and
were always great sounding boards for ideas about anything. Eric and Frank
were essentially my bay mates – I spent as much time in B455 as in B457 –
and showed me that “you, too, can get out of bed in the morning!”
Finally, I express my love and thanks to the people who have listened to whiny
phone calls and e-mails, trekked out to visit me, moved me half-way across
the country and never ever stopped being my biggest boosters and
supporters. Thank you to my friends in Texas and California. To my parents,
Mike and Patty, my brothers Dean and Sean, and my sister Erin, I love you
and none of this would have been possible without you.

viii
TABLE OF CONTENTS
1) Introduction 1
Rab GTPases 2
Rab Localization and Microdomains 5
The Rab Cycle 9
Rab GTPase-Activating Proteins 15
GAPs and GEFs: Defining Boundaries 28
References 34
Table 54
2) RUTBC1: a novel Rab9 effector that activates GTP hydrolysis by
Rab33B and Rab32 58
Abstract 59
Introduction 60
Methods 63
Results 70
Discussion 77
References 82
Figure Legends 88
Figures 91

ix
3) Interaction of RUTBC1, a Rab33B GAP, with the Rab33B effector,
Atg16L1 97
Abstract 98
Introduction 99
Methods 103
Results 106
Discussion 109
References 111
Figure Legends 114
Figures 116
4) Characterization of Rab substrates and binding
partners of RUTBC2 122
Abstract 123
Introduction 124
Methods 127
Results 133
Discussion 138
References 141
Figure Legends 145
Figures 148
5) Summary and Future Perspectives 153

x
LIST OF TABLES
Introduction
Table I. Summary of mammalian Rab GTPase-activating proteins 54

xi
LIST OF FIGURES
Chapter 2
Figure 1: RUTBC1 interacts with Rab9 91
Figure 2: RUTBC1 is an effector of Rab9 92
Figure 3: RUTBC1 binds to, but is not a GAP for Rab9 in cells 93
Figure 4: RUTBC1 TBC domain has GAP activity toward 94
Rab33B and Rab32 in vitro
Figure 5: RUTBC1 TBC domain stimulates GTP hydrolysis 95
Chapter 3
Figure 1: Domain architecture of RUTBC1 and Atg16L1 116
Figure 2: RUTBC1 solubilizes Atg16L1 from the Golgi 117
Figure 3: RUTBC1 and Atg16L1 interact in cells 118
Figure 4: RUTBC1 localizes to Atg16L1-positive puncta 120
Chapter 4
Figure 1: RUTBC2 interacts with Rab9 148
Figure 2: RUTBC2 is an effector of Rab9 149
Figure 3: RUTBC2 binds to, but is not a GAP for Rab9 in cells 150
Figure 4: RUTBC2 TBC domain has GAP activity toward 151
Rab36 and Rab34 in vitro
Figure 5: RUTBC2 is stably associated with membranes 152
in SK-N-SH cells

1
INTRODUCTION
Eukaryotic cells contain membrane bound compartments that segregate
different biochemical functions from each other and from those occurring in the
cytoplasm. Organelles are covered with different sets of proteins and lipids
that distinguish different compartments. The maintenance of individual
compartment identity and the transfer of protein and membrane between
compartments occur through molecular events collectively termed membrane
trafficking.
The molecules that provide specificity for each trafficking event, as well as
those that remodel membrane during vesicle budding and fusion, have been
investigated actively over the last thirty years. A fundamental question in the
field is how these organelles remain distinct despite the constant flux of
membrane and protein trafficked throughout the cell. This chapter focuses on
the master regulators of trafficking events, the Rab GTPases. I will discuss the
roles played by Rab proteins and their regulators, with emphasis on Rab
GTPase-activating proteins (RabGAPs), in giving compartments specific
identity through formation, maintenance and breakdown of membrane
microdomains.

2
Rab GTPases
Rab GTPases are members of the Ras-like GTPase superfamily – a large
group of proteins of approximately 25kDa that derive their function from their
ability to both bind and hydrolyze GTP. These so-called ʻGʼ proteins act as
switches governed by the identity of the bound nucleotide; this is a
consequence of a conformational change between active GTP- and inactive
GDP-bound states (Colicelli, 2004). Exchange of GDP for GTP leads to
changes in two regions of G proteins, called switch I and switch II, that form
hydrogen bond contacts with the phosphate groups of GTP. Hydrolysis of GTP
leads to the loss of these hydrogen bonds and a subsequent conformational
change back to the GDP-bound structure. This difference in conformation
allows “effector” proteins that bind small GTPases in their active state to
discriminate between the GTP- and GDP-bound forms and thus regulate
cellular processes. The superfamily is divided into five major subfamilies
including Ras, Rho, Ran, Arf and Rab. The last two subfamilies are critically
important in the regulation of membrane trafficking. Arf proteins (and the
related Arls) play a role in coat recruitment to vesicle budding sites, notably at
the Golgi and plasma membrane as well as in cytoskeletal dynamics
(Gillingham and Munro, 2007). Rab proteins, the largest subfamily, participate
in all steps of membrane trafficking including vesicle formation, motility,
tethering and docking and fusion (Segev, 2001; Zerial and McBride, 2001;
Stenmark, 2009).

3
The first members of the Rab family were discovered in S. cerevisiae. Sec4
was first identified in a screen for secretion mutants (Novick et al., 1980).
Yeast blocked in secretion become more dense than normal cells due to the
accumulation of dense secretory vesicles and other membranes. This property
allowed Schekman and coworkers to select for mutant cells by density
centrifugation. They discovered twenty-three complementation groups of
secretion mutants, many of which are well studied today. Before the product of
the SEC4 gene was identified, another gene named YPT1 was characterized
as an open reading frame between the tubulin and actin genes that had high
homology to the Ras oncogenes (Gallwitz et al., 1983). Both SEC4 and YPT1
are essential genes and YPT1 could not rescue a double deletion of
RAS1/RAS2 in yeast, suggesting that Ras-like GTPases performed a diverse
set of functions (Salminen and Novick, 1987; Schmitt et al., 1986). Soon after,
the SEC4 gene product was discovered to have homology to Ras and it was
even more closely related to YPT1; its overexpression could also suppress the
phenotypes of many of the late acting SEC mutants (Salminen and Novick,
1987). A ypt1 conditional-lethal mutant showed defects in protein secretion
(but at an earlier step than SEC4) and improper membrane growth (Segev et
al., 1988). Studies of these two proteins indicated that a novel family of
GTPases controlled membrane dynamics in cells. Later work in mammalian
cells identified four additional genes in a screen of cDNAs from rat brain with
oligonucleotides specific for Ras-like proteins (Touchot et al., 1987). This is

4
the genesis of the term Rab for “Ras-like GTPase from rat brain.” These genes
were quickly determined to have homology to YPT1 and SEC4 (Zahouri et al.,
1989). The remarkable discovery that mouse Rab1 could functionally replace
Ypt1 in yeast suggested that secretion (and likely other membrane trafficking
events) were regulated by conserved machineries (Haubruck et al., 1989).
This was consistent with contemporary findings showing homology between
the SEC18 gene product and NSF (N-ethylmaleimide sensitive factor), an
AAA+ ATPase required for vesicle fusion (Wilson et al., 1989). From eleven
members in yeast to over seventy members in mammalian cells, the Rab
subfamily is the largest group in the Ras-like GTPase superfamily (Pereira-
Leal and Seabra, 2001).
Rabs are composed of two different domains: a G domain that binds and
hydrolyzes guanine nucleotides and a C-terminal, so-called “hypervariable”
domain. The G domain is a typical nucleotide binding fold consisting of a
series of beta strands surrounded by alpha helices and all five conserved G
protein motifs are present in Rabs (Bourne et al., 1991, Colicelli, 2004). The
hypervariable domain is unique to the Rab subfamily of Ras-like small
GTPases. It is thought to have an irregular structure (Ostermeier and Brunger,
1999) and it plays a role in Rab localization (Chavrier et al., 1991; Aivazian et
al., 2006). Rab proteins associate with membranes by the addition of at least
one, but usually two, C-terminal cysteine-linked geranylgeranyl groups

5
(Khosravi-Far et al., 1991). Mutation of both cysteine residues abolishes Rab
membrane association and interferes with their functions in cells (Molenaar et
al., 1988; Walworth et al., 1989). Stable membrane association is thus
essential for Rabs to function as membrane organizers.
Rab Localization and Microdomains
One of the most striking discoveries in membrane trafficking was that each
organelle contains a specific set of Rab proteins (Chavrier et al., 1990). Zerial
and co-workers isolated eleven cDNA clones from Madin-Derby canine kidney
cells that had high homology to SEC4 and YPT1. Immunofluorescence and
electron microscopy showed that three of these Rabs were differentially
localized to organelles throughout the cell: e.g., Rab2 appeared to be on a
compartment intermediate between the endoplasmic reticulum (ER) and Golgi
apparatus, while Rab5 was localized to the plasma membrane and structures
in the cytoplasm (Chavrier et al. 1990). With over 70 Rabs in humans and at
least 40 expressed in one cell type alone (Nguyen et al., 2009), Rabs provide
both specificity and irreversibility to the organization of the diverse array of
membrane trafficking events.
The determinants of Rab localization are rather complex. Experiments using
chimeras of Rab5 and Rab7 led to the initial conclusion that the hypervariable
domain itself was sufficient to localize a Rab (Chavrier et al., 1991). Rab5

6
bearing the C-terminus of Rab7 seemed sufficient to re-localize Rab5 from
early endosomes to late endosomes. The C-termini of both Rab5 and Rab7
were also able to shift Rab2, normally on the Golgi, to early or late
endosomes, respectively (Chavrier et al., 1991). Similar experiments using
chimeras of Rab9, Rab5 and Rab1 revealed a more elaborate mechanism for
Rab localization. Pfeffer and co-workers showed that Rab localization was
dependent on effector binding and not simply the hypervariable domain
(Aivazian et al., 2006). They showed that re-localization of Rab5 bearing the
Rab9 hypervariable domain was dependent on the Rab9 effector TIP47 (Tail
Interacting Protein of 47kDa). Mutations in TIP47 that abrogate Rab9 binding
also failed to re-localize chimeric Rab5 (Aivazian et al., 2006). Thus, Rab
localization is dependent on both prenylation and effector binding to recruit
Rabs to the correct membrane. In several cases, the hypervariable domain is
important for effector binding, which may explain Chavrier et al.ʼs original
findings.
Rabs further define subdomains on organelles called microdomains (Zerial
and McBride, 2001). They provide identity to the membrane they are localized
to by specifically concentrating their effector proteins at these sites (Pfeffer,
2001). Effector proteins can include integral membrane proteins or soluble
proteins recruited to membranes by the Rab; moreover Rabs recruit lipid
modifying enzymes such as phosphoinositide kinases (Christoforidis et al.

7
1999a), further adding specificity by enriching for particular lipids. As an
example, Rab9 helps to define a microdomain on late endosomes separate
from Rab7 (Lombardi et al., 1993; Barbero et al., 2002). Rab7 functions in the
conversion of early endosomes into late endosomes and their eventual
maturation into lysosomes (Zhang et al., 2009). Mannose 6-phosphate
receptors (MPRs) deliver newly synthesized acid hydrolases to pre-lysosomal
compartments (Ghosh et al., 2003). There, the hydrolases dissociate from the
receptor and eventually localize to the lysosome, while MPRs must be
recycled back to the trans Golgi network (TGN) for another round of delivery.
Rab9 is essential for this recycling step and facilitates retrieval of MPRs
through its effector, TIP47. TIP47 binds to the cytoplasmic domains of MPRs
(Diaz and Pfeffer, 1998) The affinity of TIP47 for MPR cytoplasmic domains is
~1µM (Krise et al., 2000). In the presence of Rab9, the affinity of TIP47 for
MPR cytoplasmic tail is increased to ~300nM (Carroll et al. 2001). Thus, active
Rab9 works with its effector, TIP47, to segregate MPRs into a domain on late
endosomes that prevents them from being sorted to the lysosome.
Perhaps an even more elaborate example of Rab-mediated microdomain
formation comes from studies of Rab5. Rab5 and Rab4 are both found on
early endosomes (van der Sluijs et al., 1991; Bucci et al. 1992) but in distinct
domains. Expression of fluorescently tagged Rab5 and Rab4 showed that
endocytosed transferrin first entered a Rab5 domain on early endosomes and

8
then entered a Rab4 domain (Sönnichsen et al., 2000). This is consistent with
the function of both Rabs: Rab4 regulates recycling of transferrin receptor
back to the plasma membrane (van der Sluijs et al., 1991) while Rab5
functions in homotypic fusion of endosomes and clathrin-coated vesicles
through interaction with at least twenty different effectors (Christoforidis et al.,
1999b). While distinct, the Rab4 and Rab5 are coupled to each other. Each
Rab has a diverse set of effectors while effectors themselves generally only
bind to one or a few Rabs. Rabaptin-5 and Rabenosyn 5 bind both Rab5 and
Rab4 through distinct regions and might function to link these microdomains
together on the surface of early endosomes (Vitale et al., 1998; de Renzis et
al., 2002). Additional examples of “microdomain tethers” are likely to exist,
particularly in the Golgi stack. In a small number of cases where multiple Rabs
bind to a particular effector (Hayes et al., 2009; Sinka et al., 2008), these
interactions may be a dynamic but regulated way of keeping certain
microdomains linked in order to form some higher order structure or function,
e.g. an intact Golgi ribbon.
Rab5 microdomains seem to assemble cooperatively (Zerial and McBride,
2001). Rab5 binds to Rabaptin-5, which in turn, also forms a complex with
Rabex-5, a guanine nucleotide exchange factor or GEF (Stenmark et al., 1995;
Horiuchi et al., 1997). Thus, Rab activation is coupled to effector binding: in
this manner, a feedback loop exists where increasing amounts of active Rab5

9
exist on endosomes. Rab5 then recruits Early Endosome Antigen 1 (EEA1), a
tethering factor required for endosome fusion that also has a FYVE domain (a
binding domain for phosphatidylinositol-3-phosphate (PIP3)) and both Rab5
binding and PIP3 binding are required to recruit EEA1 to endosomes
(Christoforidis et al., 1999b; Stenmark et al., 1996; Simonsen et al., 1998).
Remarkably, Rab5 also binds Vps34, a phosphoinositide kinase that
generates PIP3 on early endosomes (Christoforidis et al., 1999a). Working
together with its effectors, Rab5 catalyzes the formation of this specialized
membrane domain. On other early endosomes, Rab5 also catalyzes a distinct
domain that contains APPL but not EEA1 (Miaczynska et al., 2004). What
causes microdomains to not simply diffuse apart in the plane of the
membrane? One model posits that oligomerization of effectors coupled with
lipid enrichment may stabilize a microdomain until GTP hydrolysis initiates its
dismantling (Rybin et al., 1996; Zerial and McBride, 2001).
The Rab Cycle
Key to proper microdomain formation, and therefore membrane trafficking, is
the regulation of active Rab localization. Rab localization is intimately tied to
two different cycles that generally correlate with each other: a cycle of
nucleotide binding and a cycle of membrane association. Cytosolic Rabs are
exclusively GDP-bound, and are activated after delivery to the membrane.
Membranes can contain both GTP- and GDP-bound Rabs, but only GDP Rabs

10
can be removed from membranes. These cycles provide multiple points of
regulation to ensure well organized membrane trafficking.
Newly synthesized, GDP-bearing Rabs are bound by Rab Escort Protein or
REP (Andres et al., 1993). REP presents the new Rab to the RabGGTase
prenylating enzyme, which utilizes only the REP:Rab complex as substrate
(Andres et al., 1993). RabGGTase has little affinity for Rab proteins on their
own and requires the presence of REP for efficient prenylation (Alexandrov et
al., 1999). The REP:Rab complex then dissociates from RabGGTase; after
prenylation, a prenyl group-triggered conformational change in REP disrupts
the REP:RabGGTase binding site (Rak et al., 2004). Another protein, GDP-
dissociation inhibitor or GDI, also delivers prenylated Rabs to membranes but
has the added function of being able to extract Rab proteins from membranes
as well (Sasaki et al., 1990, Araki et al., 1990). GDI binds only to prenylated
Rabs that are also bound to GDP (Shapiro and Pfeffer, 1995; Rak et al.,
2003). Both REP and GDI bind to the switch I and switch II regions of Rab
proteins, which explains the requirement for GDP-bound Rabs as well as the
name GDI. GDI is able to extract GDP-bound Rabs from membranes while
REP is poor at this function. This is explained by the binding specificities for
prenylated and unprenylated Rabs. REP has high affinity for the Rab itself
since it can bind either form of the Rab, while GDI has high affinity specifically
for prenylated Rabs. Thus, REP likely cannot overcome the energy barrier

11
required to extract the prenyl groups from the membrane, even though it
seems to be able to keep the prenyl groups from aggregating before
membrane delivery (Goody et al., 2005). Interestingly, the REP binding site
for RabGGTase has the highest sequence and structural homology to GDI
(Pylypenko et al., 2003). The difference in binding arises from the presence of
two REP residues, a phenylalanine and an arginine, found in REP that are
absent from GDI (Alory and Balch, 2000).
GDI:Rab complexes contain all information necessary to deliver Rabs to the
correct membrane, and nucleotide exchange occurs after association with
membranes (Soldati et al., 1994; Ullrich et al., 1994). Because of GDIʼs high
affinity for prenylated Rabs as well as the inability of some GEFs to use
GDI:Rab complexes as substrates, an enzymatic activity was hypothesized to
exist that catalyzed the release of Rabs from GDI (“GDF”; Dirac-Svejstrup et
al., 1997). Pfeffer and co-workers used reconstituted GDI:prenyl-Rab
complexes as substrates to probe purified late endosome membranes for this
activity. Rabs in complex with GDI do not exchange nucleotide (Sasaki et al.,
1993). Displacement of GDI would permit nucleotide exchange at either the
intrinsic or GEF-stimulated rate. Protease sensitive GDF activity was detected
in membranes. Intriguingly, this crude GDF was able to dissociate endosomal
Rab:GDI complexes but not those associated with the secretory pathway
(Dirac-Svejstrup et al., 1997). Yip3/PRA1 is the only mammalian protein

12
identified to date that possesses GDF activity and it too, is specific for
endosomal Rabs (Sivars et al., 2003). Yip3/PRA1 belongs to a conserved
family of integral membrane proteins from yeast to humans that interact with
Rabs, have distinct localizations, and at least one member has low affinity for
GDI (Yang et al., 1998; Abdul-Ghani et al., 2001; Hutt et al., 2000). These
properties make Yip/PRA proteins excellent candidates to be GDFs.
GDI displacement and subsequent insertion of the prenyl groups into
membranes are still poorly understood processes. After membrane
attachment, Rabs undergo nucleotide exchange, replacing GDP with GTP
(Soldati et al., 1994). This step is necessary to prevent solubilization of the
newly-delivered Rab by GDI. In vitro, GDP dissociation is the rate-limiting step
of the nucleotide cycle of GTPases. Rabs also have slow, intrinsic rates of
GDP dissociation, and so another enzymatic activity called a guanine
nucleotide exchange factor (GEF) is required to catalyze this process. It has
recently been proposed that GEF activity alone might be sufficient for GDI
displacement (Schoebel et al., 2009; Suh et al., 2010). This model is based on
studies of the Leishmania DrrA/SidM protein that was identified initially as a
protein containing both GDF and GEF activity (Ingmundson et al., 2007;
Machner and Isberg, 2007). DrrA is a potent GEF for Rab1 with a Kd of ~2pM
for nucleotide-free Rab1, which led the authors to argue that GDI displacement
is due to GEF activity alone (Schoebel et al 2009; Suh et al 2010). Because

13
pathogens probably have evolved highly efficient infection processes, it is
unclear if endogenous GEFs can apparently displace GDI as DrrA can.
Indeed, Transport protein particle complex I (TRAPPI), an endogenous GEF
for Ypt1 in yeast, has a much weaker Kd for nucleotide-free Ypt1 (~200nM),
so they may be unable to drive a GDF reaction (Chin et al., 2009).
The basic mechanism for GEF-catalyzed nucleotide exchange is very similar
among all Ras-like GTPases (Itzen et al., 2007). GEFs disrupt the nucleotide
binding site by driving out the phosphate groups of GDP (Bos et al., 2007).
This causes a conformational change in the switch I region that is incompatible
with nucleotide binding. GEF dissociates when GTP binds. GEFs do not have
specificity for one nucleotide over the other; nucleotide exchange is driven by
the almost 10-fold higher cellular levels of GTP over GDP (Goody and
Hoffman-Goody, 2002). This allosteric competition between nucleotide and
GEF binding is near universal among the Ras-like GTPase superfamily (Bos et
al., 2007). Currently, RabGEFs are poorly characterized: unlike other
regulatory proteins there is no common RabGEF domain. Vps9 domain-
containing proteins catalyze nucleotide exchange on Rab5 subfamily members
through a helical bundle (Delprato et al., 2004). Sec2 utilizes a coiled-coil
domain to catalyze nucleotide exchange on Sec4 (Dong et al., 2007). TRAPPI
consists of multiple subunits, most of which are required for GEF activity on
Ypt1 (Wang et al., 2000; Cai et al., 2008). In this case, multiple subunits form

14
the interface for Rab binding. Other work has shown that addition of three
further subunits changes both the localization and substrate specificity of
TRAPP. This complex is then called TRAPPII and possesses GEF activity on
Ypt31/32 (Jones et al., 2000; Morozova et al., 2006).
At this part of the cycle, Rabs are both membrane associated and bound to
GTP. This is the fully active state of the Rab and the state to which numerous
and diverse effector proteins bind. Since the membrane associated, GTP-
bound Rab gives a portion of membrane specific identity, inactivation of Rab
proteins is of critical importance to coordination and maintenance of organelle
function. Rabs that are mis-localized would recruit effectors to the wrong
compartment leading to establishment of “ectopic” microdomains. Rabs are
inactivated by hydrolysis of GTP to GDP and inorganic phosphate. Like all
GTPases, Rabs possess a slow, intrinsic rate of hydrolysis (Colicelli, 2004).
This presents another opportunity for regulation. Cells have evolved GTPase-
activating proteins (GAPs) to rapidly terminate active signaling. GAPs can
stimulate the intrinsic rate of hydrolysis by several orders of magnitude (Bos et
al., 2007). The discovery of RabGAPs as well as their proposed biochemical
mechanism of stimulation will be discussed below.
At the end of the cycle Rabs are again bound to GDP; effectors have much
lower affinity for their cognate, GDP-Rabs and the microdomain begins to

15
break down. Some Rabs may be simply re-activated by GEFs still present in
the microdomain; others may be extracted by GDI into the cytosol before a
GEF can act. GDI then recycles the Rab back to its donor compartment for re-
delivery to membranes. This recycling step is not essential for some Rab-
mediated events -- Ypt1 and Sec4 modified at their C-termini with permanent
membrane anchors were only slightly less efficient at growth and secretion
(Ossig et al., 1995). Presumably the lowered efficiency reflected the need to
synthesize new Rab proteins, as the permanently membrane anchored Rabs
might diffuse throughout the membrane system or eventually be degraded.
Rab GTPase-Activating Proteins
GAPs specific for Rab proteins were first identified in yeast (Strom et al.,
1993). Gallwitz and co-workers overexpressed a library of 2500 multi-copy
plasmids and a single transformant encoded a GTPase-activating activity that
showed highest activity toward Ypt6 (the yeast homolog of Rab6). The clone
was isolated and its identity was confirmed by expression and purification from
E. coli. This protein was termed Gyp6 (GAP for Ypt6). Gyp6 is not an essential
gene and deletion of Gyp6 produced no readily identifiable phenotypes. (Strom
et al., 1993). In vitro, Gyp6 was rather specific in substrate utilization; only
Ypt7 was comparable to Ypt6. Intriguingly, in gyp6∆ cells, Ypt7GAP activity
could still be detected, suggesting that each Rab might have its own GAP,
which could help to organize membrane trafficking events (Strom et al., 1993).

16
Eight yeast RabGAPs were identified: Gyp7 with activity on Ypt7 (Vollmer and
Gallwitz, 1995), Gyp1 with activity on Ypt1 and Sec4 (Du et al., 1998; Vollmer
et al., 1999), Gyp2 and Gyp3 that had broad substrate specificity (Albert and
Gallwitz, 1999), Gyp4 with activity on Sec4, Ypt6, and Ypt7 (Albert and
Gallwitz, 2000), and Gyp5 and Gyp8 which show preferred activation of Ypt1
(De Antoni et al., 2002).
These discoveries were aided by the fact that unlike RabGEFs, RabGAPs
have homology to each other. The Gyp proteins were originally thought to be
structurally unrelated, but sequence alignments proved otherwise. As the
amount of annotated sequences became available, it was noted that Gyp
proteins share a common domain (Neuwald, 1997). This domain was named
TBC for Tre2/Bub2/Cdc16. Tre2 is an oncogene that is formed by the fusion
of a TBC domain and a ubiquitin specific protease domain (Nakamura et al.,
1992). Bub2 and Cdc16 are homologous proteins in bakerʼs and fission yeast,
respectively, that are parts of a two-component GAP that regulates spindle
assembly and septum formation during mitosis through the Ras-like GTPase,
Tem1/Spg1 (Wang et al., 2000; Furge et al., 1998).
The TBC domain has six conserved motifs; the first three are nearly
universally conserved in all members. These “fingerprint” motifs are RxxxW in
motif A; IxxDxxR in motif B; and YxQ in motif C (Neuwald, 1997). Truncation

17
analyses of Gyp1 and Gyp7 confirmed functional homology in the putative
catalytic domain and identified conserved arginine residues in motifs A and B
that are critical for GAP activity (Albert and et al., 1999). This led Gallwitz and
co-workers to conclude that RabGAPs catalyze GTP hydrolysis by a
mechanism similar to GAPs for other Ras-like GTPases.
The crystal structure of Gyp1 showed that the TBC domain consists of 16
alpha helices (Rak et al., 2000). Surprisingly, the overall fold of the Gyp1 TBC
domain showed no similarity to that of other Ras-like GTPase family GAPs,
despite their content of alpha helices (Rak et al., 2000; Scheffzek et al., 1997;
Rittinger et al., 1997). The TBC domain adopts a “V” like shape, where
sequence motif A is in the core of the protein while motifs B and C are located
in the groove inside the “V” (Rak et al., 2000). The key arginine in motif A is
thought to contribute to overall fold stability rather than catalysis, while motifs
B and C define a putative Rab binding site. All RabGAPs have relatively low
affinities for their substrates, analogous to other GAP families (Bos et al.,
2007). This low affinity might be overcome in cells by recruitment of GAPs to
membranes. Gyp1, for example, localizes to the Golgi at steady-state, while
TBC1D20 even has a transmembrane domain (Du and Novick, 2001; Haas et
al., 2007; Sklan et al., 2007).

18
GTPase activation by most Ras-related GAPs involves an arginine “finger”
provided in trans by the GAP and a conserved glutamine provided in cis by the
GTPase. This conserved glutamine mediates GTP hydrolysis by coordination
of a water molecule for nucleophilic attack on the gamma phosphate, both in
the context of the GTPase alone and in complex with its GAP (Wittinghofer et
al., 1997). Binding of the GAP is thought to order the switch II region in order
to favor this alignment. This leads to a shift of negative charge from the
gamma to the beta phosphate; this charge distribution is closer to GDP than to
GTP (Allin et al., 2001). The accumulating negative charge is stabilized by the
guanidinium group of the arginine. This charge compensation by arginine is
thought to reduce the activation energy for breaking the beta-gamma
phosphoanhydride bond (Kotting et al., 2006). It is also thought to promote
the formation of a dissociative transition state with a penta-coordinated
phosphate group (Scheffzek et al., 1998). Co-crystals of Ras:RasGAP and
Rho:RhoGAP revealed that both GTPase:GAP pairs use this mechanism,
despite the absence of primary sequence conservation (Scheffzek et al., 1997;
Rittinger et al., 1997). This basic mechanism has many variations among
different family members of Ras-like GTPases: RanGAP uses an asparagine
to stabilize the glutamine orientation in Ran, while RapGAP uses an
asparagine, but this residue is thought to replace the canonical glutamine in
the active site as Rap lacks the conserved glutamine (Seewald et al., 2002;
Daumke et al., 2004). Sec23, the GAP for Sar1 (part of the COPII coat), also

19
uses an arginine finger but helps to align a histidine in place of the glutamine
in the Sar1 active site (Bi et al., 2002).
The co-crystal structure of Gyp1 in complex with human Rab33b revealed a
new variation in GAP mechanism: both the arginine and the glutamine
residues are provided by the GAP in trans (Pan et al., 2006). The GAPʼs
catalytic B and C motifs that contain the essential arginine and glutamine
residues, respectively, form a loop that extends into the Rab nucleotide-
binding pocket (Pan et al., 2006). The structure also confirmed that the Rab
does bind the GAP in the V-shaped groove via multiple α-helices from the
GAP (Pan et al., 2006). These helices interact primarily with both switch
regions and the P-loop, which helps explain why GAPs interact preferentially
with GTP-bound Rabs and suggests a possible mechanism for substrate
selectivity. Highly variable surfaces can exist in the switch regions due to the
varying conformations of three invariant hydrophobic residues (Merithew et al.,
2001). GAPs (as well as effectors) may use this surface to distinguish
between Rab substrates (Pereira-Leal and Seabra, 2000). This structural
work also helped to explain observations from numerous groups that GAP
activities in cytosol also stimulated the GTPase activity of Rab “Q-L” mutants
that lack the conserved G3 glutamine residue.

20
Puzzlingly, many purified, truncated yeast RabGAPs showed broad substrate
reactivity in vitro (Albert and Gallwitz, 1999; Will and Gallwitz, 2001). These
authors used truncated forms initially because of technical difficulties in
producing recombinant full-length protein. Rabs regulate specific trafficking
events and promiscuous GAPs might disrupt membrane identity. Data from
mammalian cells suggest that this apparent promiscuity is due to deleted
regions being necessary for discriminating between Rab substrates. Barr and
co-workers demonstrated that GAPCenA truncations decreased substrate
specificity -- full-length GAPCenA stimulated Rab4 exclusively (Fuchs et al.,
2007). Strict specificity has also been observed for other mammalian GAPs
including TBC1D30, which can even discriminate between closely related
Rab8 isoforms (Yoshimura et al., 2007).
Why do both Bub2 and Cdc16 require adaptors to function as GAPs for
Tem1/Spg1? Both contain the first three “fingerprint” motifs but neither has
the last three alpha helices (14-16) (Rak et al., 2000). Helix 15 truncations of
Gyp1 and Gyp7 lack GAP activity (Albert et al., 1999). Perhaps in a ternary
complex, the adaptor proteins provide the last three helices to complete the
TBC domain fold (Rak et al., 2000). Alternatively, this adaptor might change
the conformation of Tem1/Spg1 to one that promotes stimulation by
Bub2/Cdc16 (Geymonat et al., 2002). Crystal structures of Bub2/Cdc16
complexed with Tem1/Spg1 and their adaptors will resolve this puzzle.

21
While the biochemistry of RabGAPs has been studied extensively, their
functions in cells remain somewhat elusive. For Rab proteins, the functional
ramifications of GTP hydrolysis were initially somewhat controversial. In 1988,
Henry Bourne proposed that the secretory GTPases would not function like
Ras, where GTP hydrolysis attenuates a signal but does not affect protein
function. Instead, he proposed that they would function like elongation factors
where a cycle of nucleotide binding and hydrolysis was essential to ensure the
continued growth of the nascent polypeptide chain (Bourne, 1988). He
reasoned that secretion (and other transport events) might use nucleotide
hydrolysis to ensure irreversibility of a trafficking event.
Studies of Ras have greatly aided functional biochemical studies of Rab
proteins. Mutation of the conserved G3 motif glutamine of Ras-like GTPases
(often to leucine but also alanine) creates a protein deficient in hydrolysis and
thus predicted to be constitutively active (Polakis and McCormick, 1993). The
analogous mutants in Rabs have been used both in cells and in reconstituted
systems to probe requirements for nucleotide binding and hydrolysis in
different trafficking events.
Early cellular studies with Sec4Q71L and Rab5Q79L, mutants shown to be
deficient in GTP hydrolysis in vitro, gave somewhat conflicting results.

22
Secretion of invertase in Sec4 mutant cells was almost 3-fold slower at 13°C
compared to normal cells, and Sec4Q71L could not rescue other late acting
SEC mutants but instead generated synthetic lethal combinations (Walworth et
al., 1992). These mutant cells also accumulated secretory vesicles like the
original Sec4 temperature sensitive mutation (Novick and Schekman, 1980).
This suggested that the G3 motif Q to L mutant was a loss of function
mutation. In contrast, Rab5Q79L stimulated membrane fusion as shown by
the presence of enlarged early endosomes as well as increased fusion in a
cell-free endosome fusion assay (Stenmark et al., 1994). Compounding these
findings was the observation that GAP activity purified from lysates was able
to stimulate both wild-type and mutant Rab proteins. In addition, while GTPγS
was known to inhibit trafficking when added to cell free systems (Melançon et
al., 1987), it was unclear if this effect was due to Rab inhibition or other G
proteins involved in these events.
Zerial and co-workers took advantage of an analogous mutation in EF-Tu that
switches the nucleotide specificity of GTPases to xanthosine-5ʼ-triphosphate
(XTP) (Hwang and Miller, 1987). Using Rab5 mutated to bind XTP, Zerial and
co-workers discovered that Rab5 cycles between XDP and XTP on
membranes and that XTPγS does not inhibit in vitro endosome fusion (Rybin
et al., 1996). Studies of Ypt1Q67L corroborated these findings as this mutant
failed to block secretion (Richardson et al., 1998). This led to the hypothesis

23
that GTPase activity was needed simply for recycling of Rab proteins back to
the donor membranes, rather than playing an essential role in fusion, contrary
to the model of Bourne (Rybin et al., 1996; Richardson et al., 1998).
Termination of the active Rab signal would be key to keeping a target
membrane distinct from a donor membrane. Continual buildup of donor
microdomains at a target would otherwise lead to mis-sorting of receptors and
other cargo proteins within cells.
Deletion of RabGAPs in yeast revealed no obvious phenotypes, even when
putative GAPs for essential Rabs like Ypt1 and Sec4 were deleted. It is likely
that essential Rabs (such as Rab1/Ypt1) have multiple, dedicated GAPs. In
yeast, deletion of Gyp1, Gyp5 or Gyp8 alone (all GAPs with high in vitro
activity to Ypt1) did not yield any noticeable phenotype (De Antoni et al.,
2002). Synthetic, cold-sensitive growth phenotypes were observed in strains
harboring both the Ypt1Q67L mutant and various double deletions of Gyp1,
Gyp5 and Gyp8. The GAPs all have different cellular localizations, further
suggesting that GAP activity might be a way of preventing “stray” Rabs from
forming mis-localized microdomains at an incorrect membrane location.
In contrast with yeast, very little is known about RabGAP counterparts in
mammalian cells. The first mammalian RabGAP activity identified was
specific for Rab3A, an important regulator of exocytosis and synaptic vesicle

24
fusion (Burstein et al., 1991; Fukui et al., 1997). Interestingly, this protein
does not contain a TBC domain but does appear to catalyze hydrolysis by an
arginine finger mechanism, similar to Ras- and RhoGAPs (Clabecq et al.,
2000). GAPCenA was the first mammalian protein cloned containing a TBC
domain. This protein was originally discovered by a yeast two hybrid
interaction with Rab6Q72L via the GAPʼs C-terminus (a region with high
probability of coiled-coil structure). GAPCenA appeared to have GAP activity
on Rab6 and Rab4, the former requiring both prenylation of the Rab as well as
the coiled coil region C-terminal to the TBC domain (Cuif et al., 1999).
Approximately forty predicted GAPs for Rab GTPases in humans have been
identified by TBC domain sequence alignments (Bernards, 2003, Fuchs et al.,
2007; Table I). The key arginine in motif B is highly conserved but is not
absolute. For example, the arginine in Tre-2/USP6 is shifted one position
toward the N-terminus while TBC1D3 lacks this arginine. Interestingly,
TBC1D7 does not even have motif B, suggesting that it might completely lack
GAP activity altogether. Database searches also revealed that most
mammalian TBC domain proteins have multiple domains (Bernards, 2003).
These additional domains are very diverse and include lipid binding domains
such as the pleckstrin homology (PH) domain and glucosyltransferase/Rab-
like GTPase activators/myotubularins (GRAM) domains. Other domains
include phosphotyrosine-binding (PTB) domains and Src-homology-3 (SH3)

25
domains that are often involved in receptor tyrosine kinase signaling. Protein-
protein interaction domains such as coiled-coil domains and
RPIP8/Unc14/NESCA (RUN) domains are also sometimes found coupled with
TBC domains. Thus, RabGAPs may function as integrators of signals across
different trafficking events and perhaps even between different GTPase
families.
Recently, several efforts have been made to match mammalian RabGAPs with
their cognate Rabs partners. Initial attempts used yeast two hybrid screens
involving both mutant Rabs and GAPs to identify substrates with mixed
results. Through this approach, Barr and co-workers discovered a novel GAP
for Rab5 called RUTBC3/RabGAP-5. Overexpression of RUTBC3 blocked
uptake of both transferrin and epidermal growth factor (EGF), two known
Rab5-dependent processes (Haas et al., 2005). Fukuda and co-workers used
the same method to screen all Rabs against all RabGAPs and found many
GAPs that interacted with Rabs but most did not. Moreover, they found that
GAP activity did not seem to correlate with Rab binding in their screen (Itoh et
al., 2006).
Another method pioneered by the Barr laboratory, Rab inactivation screening,
has proven to be very successful at not only matching GAPs with their
substrate Rabs but also in identifying Rabs involved in fundamental cellular

26
processes. This method is based on the model that GAPs regulate the lifetime
of active Rabs. One prediction of this model is that overexpression of a GAP
in cells should lead to lower amounts of GTP-bound Rab. This in turn would
lead to a loss of microdomain integrity as evidenced by eventual loss of
effectors (and the Rabs themselves through GDI) from membranes. This loss
of microdomains would eventually lead to blocks in various transport steps.
These phenotypes would only be observed when overexpressing wild-type
GAPs and not their catalytically-inactive mutants, and would be predicted to be
yield phenotypes similar to that seen upon specific depletion of the substrate
Rabs.
The first example of this approach was used to probe which Rabs are required
for uptake of EGF and Shiga toxin (Stx; Fuchs et al., 2007). These ligands are
both internalized into small punctate structures within twenty minutes. The two
proteins then diverge in their trafficking pathways: STx is present in the Golgi
at sixty minutes post uptake, while EGF remains in endosomal structures.
Remarkably, Barr and co-workers discovered RabGAPs that differentially
regulate the uptake of these ligands (Fuchs et al., 2007). Overexpression of
RUTBC3/RabGAP-5 blocked EGF uptake but did not prevent STx from
trafficking to the Golgi. On the other hand, RN-tre, which previous work
suggested was involved in EGF signaling as a GAP for Rab5 (Lanzetti et al.,
2000), had no effect on EGF uptake, but did block STx trafficking to the Golgi.

27
These data suggested that these pathways are regulated by different Rab
GTPases. In vitro GAP assays confirmed that RUTBC3 was indeed a GAP for
Rab5, while RN-tre was a GAP for Rab43. Depletion of these Rabs produced
the same phenotypes as overexpression of their GAPs, confirming the
prediction of the model. Importantly, this work also highlighted possible
caveats of using so-called, constitutively active Rabs as probes in cells.
Rab5Q79L is known to block STx trafficking to the Golgi, although RUTBC3
overexpression does not block this process. Rab5Q79L causes enlarged
early endosomes that in the case of STx are less efficient in their transport
(Fuchs et al., 2007). Thus, constitutively active mutant phenotypes may not
reveal which Rabs are most important for a specific process but instead, might
shed light on the itinerary of a particular cargo. Stx passes through a Rab5
early endosome, but relies more heavily on Rab43 for its Golgi delivery.
Rab inactivation screening has also been used to probe Rab requirements in
maintaining Golgi structure, ciliagenesis, immunological synapse formation,
and melanosome aggregation, as well as for the identification of TBC domain
proteins that have Rab3 GAP activity. (Haas et al., 2007; Yoshimura et al.,
2007; Patino-Lopez et al., 2008; Itoh and Fukuda, 2006; Ishibashi et al., 2009).
In each of these cases, RabGAPs helped to define distinct requirements for
Rab GTPase function in a particular trafficking pathway. Perhaps most
striking was the discovery that only two Rab GAPs disrupted both Golgi

28
structure and protein secretion (Haas et al., 2007). Multiple GAPs caused
disruption of Golgi morphology in HeLa and hTERT-RPE1 cells, but only two
caused this phenotype in both cell types. These GAPs were TBC1D20, an
ER-localized GAP for Rab1 (Haas et al., 2007; Sklan et al., 2007) and RN-tre,
the above-mentioned GAP for Rab43. Overexpression of TBC1D20 caused
complete loss of Golgi structure, confirming the role of Rab1 in Golgi
biogenesis.
Do GAPs work alone in breaking down microdomains? Recent evidence
suggests that another layer of regulation exists above GAPs. In yeast, the
ability of Gyp7 to inactivate Ypt7 was shown to be commensurate with the
activity of Yck3, a kinase, (Brett et al., 2008). Yck3 helps complete
inactivation of Ypt7 by phosphorylating the HOPS tethering complex, which is
also a GEF for Ypt7; this phosphorylation is blocked by Ypt7-GTP. Perhaps
there are many different regulatory modes for GAPs in cells. Cells may have
evolved elaborate mechanisms to ensure tight regulation of highly conserved
Rabs. These results further highlight the complex integration of membrane
trafficking pathways in cells.
GAPs and GEFs: Defining Boundaries
Formation of Rab microdomains must be spatially and temporally regulated for
proper membrane transport. Coupling of different microdomains together on

29
the same organelle probably helps to maintain organelle identityand can
ensure vectorial flow of proteins and membranes. GAPs, as well as GEFs,
could provide many opportunities for regulation of these processes.
Current evidence supports a so-called Rab cascade model for microdomain
integration in which Rabs are activated and inactivated in sequential order to
coordinate vectorial flow of cargo. The first evidence in support of this model
came from the discovery that a late-Golgi localized Rab, Ypt32, recruits Sec2,
the GEF for the subsequent acting Sec4 onto secretory vesicles (Ortiz et al.,
2002).
The yeast secretory pathway has provided a large amount of evidence in
support of this model. The TRAPPI complex is a GEF for Ypt1, which functions
in the early secretory pathway between the ER and Golgi, and in intra-Golgi
transport (Wang et al., 2000). By binding Sec23, the TRAPP complex also
functions as a tether at the Golgi, for incoming COPII-coated vesicles (Cai et
al., 2007). This couples Ypt1 activation with cargo exit from the ER. With the
addition of two subunits, the specificity of the TRAPP complex (now TRAPPII)
may be switched to that of Ypt31/32, suggesting that there is a boundary at
the Golgi between active Ypt1 and active Ypt31/32 (Morozova et al., 2006 but
cf. Cai et al., 2008). This boundary would also require a way to inactivate Ypt1.
Satisfyingly, Novick and co-workers recently showed that Ypt32 is responsible

30
for recruiting Gyp1 to the Golgi, providing a mechanism for keeping early
Golgi, Ypt1 domains distinct from late Golgi, defined by Ypt31/32 (Rivera-
Molina and Novick, 2009). The cascade continues by recruitment of Sec2 onto
nascent secretory vesicles by Ypt32. This links Ypt32 to activation of Sec4,
the last Rab in the yeast secretory pathway. Activated Sec4 then binds Sec15,
a component of the large Exocyst tethering complex (Guo et al., 1999). Sec15
also associates with Sec2, leading to an intriguing mechanism by which a
Ypt32-regulated microdomain can be converted into a Sec4-regulated
microdomain (Medkova et al., 2006). Perhaps Sec15 displaces Ypt32 from
Sec2, thus incorporating a Sec4 GEF into a Sec4 microdomain to keep Sec4
active for tethering to the plasma membrane at the same time allowing access
for a Sec4-recruited Ypt32 specific GAP (Novick et al., 2006; Markgraf et al.,
2007).
There is also evidence for Rab cascades in mammalian cells. On endosomes,
Rab5 recruits the class C/VPS HOPS complex, which catalyzes nucleotide
exchange for the subsequently acting Rab7 (Rink et al., 2005). Using live cell
video microscopy, Zerial and co-workers showed that appearance of Rab7 is
followed by loss of Rab5, and that the ratio between the two Rabs controls the
identity of the compartment. This Rab conversion governs the maturation of
early endosomes into late endosomes (Rink et al., 2005). Presumably, a yet-
to-be identified GAP (perhaps recruited by Rab7) also inactivates Rab5 and

31
triggers its release from these membranes. It seems highly likely that similar
GEF/GAP recruitment will help other Rabs in all compartments of eukaryotic
cells to carry out their distinct functions. This mechanism provides for tight
control of the amount of active Rabs in cells.
Given the essential nature of Ypt1/Rab1 in yeast, the observation that
depletion of the mammalian Rab1 GAP, TBC1D20, caused no apparent defect
in Golgi structure or secretion of VSV-G protein from the ER to the Golgi is
somewhat puzzling (Haas et al., 2007). It is possible that depletion was not
adequate or that another GAP may substitute for TBC1D20 function upon its
depletion. On the other hand, this result is consistent with studies in yeast that
GTP hydrolysis is not required for Ypt1 function (Richardson et al., 1998), but
it also highlights differences in how various GAPs themselves may function.
Depletion of RUTBC3 leads to enlarged early endosomes (like Rab5Q79L),
and also re-localized Rab5 to the Golgi, similar to the localization of dominant
negative Rab5 (Haas et al., 2005). Why is there an apparent difference
between TBC1D20 depletion and RUTBC3 depletion and as a corollary, why
do Rab1 and Rab5 then function differently? This might be due to difference
in the site of GAP action relative to GEF action. If a GEF and GAP for a Rab
are both present at the donor compartment, this might be a way of

32
proofreading a process like cargo collection. If they are segregated from each
other at donor and acceptor compartments, it is likelier that GAP depletion will
show a phenotype.
Also unclear is how GAPs “poach” Rabs from a microdomain in the first place.
Rab proteins on membranes exist in complexes with their cognate effectors,
which likely help to exclude other Rabs (and their effectors) from a given
region of membrane (Zerial and McBride, 2001). These effector networks
would be able to block access to the Rab from its GAP. Most characterized
Rab GAPs have relatively high KM values for their substrates, likely reflecting a
combination of low binding affinity and high catalytic rate (Pan et al., 2006). In
contrast, most Rab effectors bind with higher affinity and could compete with
GAPs for Rab binding. If a GAP is actively recruited to a microdomain, the
network of reversible Rab–effector interactions would still allow a GAP (even
with poor affinity) to eventually break down that microdomain in the absence of
Rab activation. Thus, competition between GEF and GAP activities is likely
pervasive throughout all membrane trafficking pathways and could regulate
the formation and breakdown of Rab microdomains.
In summary, the ordered delivery of proteins and lipids to their correct
compartments is important for a vast array of cellular functions. Initially the
goal of this thesis was to find nucleotide cycle regulators of the Rab9 GTPase.

33
Our laboratory is interested in the function of Rab9, which is required for the
recycling of mannose 6-phosphate receptors from late endosomes to the trans
Golgi network. We wanted to know the molecular requirements for Rab9
microdomain segregation and how the consequences of segregation is
integrated into other trafficking pathways in the cell. In this thesis, I present an
investigation into the role of GAPs in regulating Rab9-dependent trafficking
events. A novel GAP, RUN- and TBC domain-containing 1 (RUTBC1) was
identified as a Rab9 effector. Its role in classical, Rab9-mediated trafficking
and its in vitro substrate specificity were investigated. Surprisingly, we
discovered a novel link from Rab9, through RUTBC1, to the autophagic
pathway, a highly-conserved but specialized trafficking process. Another
novel GAP, RUTBC2 was also identified as a Rab9 effector. Though highly
related, these proteins appear to have different substrate specificities in vitro,
suggesting that these proteins may integrate Rab9-mediated trafficking into
the broader context of other trafficking pathways.

34
References Abdul-Ghani et al. PRA isoforms are targeted to distinct membrane
compartments. J Biol Chem (2001) vol. 276 (9) pp. 6225-33.
Aivazian et al. TIP47 is a key effector for Rab9 localization. J Cell Biol (2006)
vol. 173 (6) pp. 917-26.
Albert and Gallwitz. Msb4p, a protein involved in Cdc42p-dependent
organization of the actin cytoskeleton, is a Ypt/Rab-specific GAP. Biol Chem
(2000) vol. 381 (5-6) pp. 453-6.
Albert and Gallwitz. Two new members of a family of Ypt/Rab GTPase
activating proteins. Promiscuity of substrate recognition. J Biol Chem (1999)
vol. 274 (47) pp. 33186-9.
Albert et al. Identification of the catalytic domains and their functionally critical
arginine residues of two yeast GTPase-activating proteins specific for Ypt/Rab
transport GTPases. EMBO J (1999) vol. 18 (19) pp. 5216-25.
Alexandrov et al. Characterization of the ternary complex between Rab7, REP-
1 and Rab geranylgeranyl transferase. Eur J Biochem (1999) vol. 265 (1) pp.
160-70.
Allin et al. Monitoring the GAP catalyzed H-Ras GTPase reaction at atomic
resolution in real time. Proc Natl Acad Sci USA (2001) vol. 98 (14) pp. 7754-9.
Alory and Balch. Molecular basis for Rab prenylation. J Cell Biol (2000) vol.
150 (1) pp. 89-103.

35
Andres et al. cDNA cloning of component A of Rab geranylgeranyl transferase
and demonstration of its role as a Rab escort protein. Cell (1993) vol. 73 (6)
pp. 1091-9.
Araki et al. Regulation of reversible binding of smg p25A, a ras p21-like GTP-
binding protein, to synaptic plasma membranes and vesicles by its specific
regulatory protein, GDP dissociation inhibitor. J Biol Chem (1990) vol. 265 (22)
pp. 13007-15.
Barbero et al. Visualization of Rab9-mediated vesicle transport from
endosomes to the trans-Golgi in living cells. J Cell Biol (2002) vol. 156 (3) pp.
511-8.
Bernards. GAPs galore! A survey of putative Ras superfamily GTPase
activating proteins in man and Drosophila. Biochim Biophys Acta (2003) vol.
1603 (2) pp. 47-82.
Bi et al. Structure of the Sec23/24-Sar1 pre-budding complex of the COPII
vesicle coat. Nature (2002) vol. 419 (6904) pp. 271-7.
Bos et al. GEFs and GAPs: critical elements in the control of small G proteins.
Cell (2007) vol. 129 (5) pp. 865-77.
Bourne et al. The GTPase superfamily: conserved structure and molecular
mechanism. Nature (1991) vol. 349 (6305) pp. 117-27.
Bourne. Do GTPases direct membrane traffic in secretion?. Cell (1988) vol. 53
(5) pp. 669-71.

36
Brett et al. Efficient termination of vacuolar Rab GTPase signaling requires
coordinated action by a GAP and a protein kinase. J Cell Biol (2008) vol. 182
(6) pp. 1141-51.
Bucci et al. The small GTPase rab5 functions as a regulatory factor in the
early endocytic pathway. Cell (1992) vol. 70 (5) pp. 715-28.
Burstein et al. Regulation of the GTPase activity of the ras-like protein
p25rab3A. Evidence for a rab3A-specific GAP. J Biol Chem (1991) vol. 266 (5)
pp. 2689-92.
Cai et al. The structural basis for activation of the Rab Ypt1p by the TRAPP
membrane-tethering complexes. Cell (2008) vol. 133 (7) pp. 1202-13.
Cai et al. TRAPPI tethers COPII vesicles by binding the coat subunit Sec23.
Nature (2007) vol. 445 (7130) pp. 941-4.
Carroll et al. Role of Rab9 GTPase in facilitating receptor recruitment by
TIP47. Science (2001) vol. 292 (5520) pp. 1373-6.
Chavrier et al. Hypervariable C-terminal domain of rab proteins acts as a
targeting signal. Nature (1991) vol. 353 (6346) pp. 769-72.
Chavrier et al. Localization of low molecular weight GTP binding proteins to
exocytic and endocytic compartments. Cell (1990) vol. 62 (2) pp. 317-29.
Chin et al. Kinetic analysis of the guanine nucleotide exchange activity of
TRAPP, a multimeric Ypt1p exchange factor. J Mol Biol (2009) vol. 389 (2) pp.
275-88.

37
Christoforidis et al. Phosphatidylinositol-3-OH kinases are Rab5 effectors. Nat
Cell Biol (1999) vol. 1 (4) pp. 249-52.
Christoforidis et al. The Rab5 effector EEA1 is a core component of endosome
docking. Nature (1999) vol. 397 (6720) pp. 621-5.
Clabecq et al. Biochemical characterization of Rab3-GTPase-activating protein
reveals a mechanism similar to that of Ras-GAP. J Biol Chem (2000) vol. 275
(41) pp. 31786-91.
Colicelli. Human RAS superfamily proteins and related GTPases. Sci STKE
(2004) vol. 2004 (250) pp. RE13.
Cuif et al. Characterization of GAPCenA, a GTPase activating protein for
Rab6, part of which associates with the centrosome. EMBO J (1999) vol. 18
(7) pp. 1772-82.
Dabbeekeh et al. The EVI5 TBC domain provides the GTPase-activating
protein motif for RAB11. Oncogene (2007) vol. 26 (19) pp. 2804-8.
Daumke et al. The GTPase-activating protein Rap1GAP uses a catalytic
asparagine. Nature (2004) vol. 429 (6988) pp. 197-201.
De Antoni et al. Significance of GTP hydrolysis in Ypt1p-regulated
endoplasmic reticulum to Golgi transport revealed by the analysis of two novel
Ypt1-GAPs. J Biol Chem (2002) vol. 277 (43) pp. 41023-31.

38
de Renzis et al. Divalent Rab effectors regulate the sub-compartmental
organization and sorting of early endosomes. Nat Cell Biol (2002) vol. 4 (2) pp.
124-33.
de Yebra et al. Reduced KIAA0471 mRNA expression in Alzheimer's patients:
a new candidate gene product linked to the disease?. Hum Mol Genet (2004)
vol. 13 (21) pp. 2607-12.
Delprato et al. Structure, exchange determinants, and family-wide rab
specificity of the tandem helical bundle and Vps9 domains of Rabex-5. Cell
(2004) vol. 118 (5) pp. 607-17.
Díaz and Pfeffer. TIP47: a cargo selection device for mannose 6-phosphate
receptor trafficking. Cell (1998) vol. 93 (3) pp. 433-43.
Dirac-Svejstrup et al. Identification of a GDI displacement factor that releases
endosomal Rab GTPases from Rab-GDI. EMBO J (1997) vol. 16 (3) pp. 465-
72.
Dong et al. A catalytic coiled coil: structural insights into the activation of the
Rab GTPase Sec4p by Sec2p. Mol Cell (2007) vol. 25 (3) pp. 455-62.
Du and Novick. Yeast rab GTPase-activating protein Gyp1p localizes to the
Golgi apparatus and is a negative regulator of Ypt1p. Mol Biol Cell (2001) vol.
12 (5) pp. 1215-26.
Du et al. Identification of a Sec4p GTPase-activating protein (GAP) as a novel
member of a Rab GAP family. J Biol Chem (1998) vol. 273 (6) pp. 3253-6.

39
Frasa et al. Armus Is a Rac1 Effector that Inactivates Rab7 and Regulates E-
Cadherin Degradation. Curr Biol (2010) vol. 20 (3) pp. 198-208.
Fuchs et al. Specific Rab GTPase-activating proteins define the Shiga toxin
and epidermal growth factor uptake pathways. J Cell Biol (2007) vol. 177 (6)
pp. 1133-43.
Fukui et al. Isolation and characterization of a GTPase activating protein
specific for the Rab3 subfamily of small G proteins. J Biol Chem (1997) vol.
272 (8) pp. 4655-8.
Furge et al. Byr4 and Cdc16 form a two-component GTPase-activating protein
for the Spg1 GTPase that controls septation in fission yeast. Curr Biol (1998)
vol. 8 (17) pp. 947-54.
Gallwitz et al. A yeast gene encoding a protein homologous to the human c-
has/bas proto-oncogene product. Nature (1983) vol. 306 (5944) pp. 704-7.
Geymonat et al. Control of mitotic exit in budding yeast. In vitro regulation of
Tem1 GTPase by Bub2 and Bfa1. J Biol Chem (2002) vol. 277 (32) pp. 28439-
45.
Ghosh et al. Mannose 6-phosphate receptors: new twists in the tale. Nat Rev
Mol Cell Biol (2003) vol. 4 (3) pp. 202-12.
Gillingham and Munro. The small G proteins of the Arf family and their
regulators. Annu Rev Cell Dev Biol (2007) vol. 23 pp. 579-611.

40
Goody et al. The structural and mechanistic basis for recycling of Rab proteins
between membrane compartments. Cell Mol Life Sci (2005) vol. 62 (15) pp.
1657-70.
Goody and Hofmann-Goody. Exchange factors, effectors, GAPs and motor
proteins: common thermodynamic and kinetic principles for different functions.
Eur Biophys J (2002) vol. 31 (4) pp. 268-74.
Guo et al. The exocyst is an effector for Sec4p, targeting secretory vesicles to
sites of exocytosis. EMBO J (1999) vol. 18 (4) pp. 1071-80.
Haas et al. Analysis of GTPase-activating proteins: Rab1 and Rab43 are key
Rabs required to maintain a functional Golgi complex in human cells. J Cell Sci
(2007) vol. 120 (Pt 17) pp. 2997-3010.
Haas et al. A GTPase-activating protein controls Rab5 function in endocytic
trafficking. Nat Cell Biol (2005) vol. 7 (9) pp. 887-93.
Hanono et al. EPI64 regulates microvillar subdomains and structure. J Cell
Biol (2006) vol. 175 (5) pp. 803-13.
Haubruck et al. The ras-related mouse ypt1 protein can functionally replace
the YPT1 gene product in yeast. EMBO J (1989) vol. 8 (5) pp. 1427-32.
Hayes et al. Multiple Rab GTPase binding sites in GCC185 suggest a model
for vesicle tethering at the trans-Golgi. Mol Biol Cell (2009) vol. 20 (1) pp. 209-
17.

41
Horiuchi et al. A novel Rab5 GDP/GTP exchange factor complexed to
Rabaptin-5 links nucleotide exchange to effector recruitment and function. Cell
(1997) vol. 90 (6) pp. 1149-59.
Hutt et al. PRA1 inhibits the extraction of membrane-bound rab GTPase by
GDI1. J Biol Chem (2000) vol. 275 (24) pp. 18511-9.
Hwang and Miller. A mutation that alters the nucleotide specificity of elongation
factor Tu, a GTP regulatory protein. J Biol Chem (1987) vol. 262 (27) pp.
13081-5.
Ingmundson et al. Legionella pneumophila proteins that regulate Rab1
membrane cycling. Nature (2007) vol. 450 (7168) pp. 365-9.
Ishibashi et al. Identification and characterization of a novel Tre-2/Bub2/Cdc16
(TBC) protein that possesses Rab3A-GAP activity. Genes Cells (2009) vol. 14
(1) pp. 41-52.
Itoh and Fukuda. Identification of EPI64 as a GTPase-activating protein
specific for Rab27A. J Biol Chem (2006) vol. 281 (42) pp. 31823-31.
Itoh et al. Screening for target Rabs of TBC (Tre-2/Bub2/Cdc16) domain-
containing proteins based on their Rab-binding activity. Genes Cells (2006)
vol. 11 (9) pp. 1023-37.
Itzen et al. Sec2 is a highly efficient exchange factor for the Rab protein Sec4.
J Mol Biol (2007) vol. 365 (5) pp. 1359-67.

42
Jones et al. The TRAPP complex is a nucleotide exchanger for Ypt1 and
Ypt31/32. Mol Biol Cell (2000) vol. 11 (12) pp. 4403-11.
Kane et al. A method to identify serine kinase substrates. Akt phosphorylates
a novel adipocyte protein with a Rab GTPase-activating protein (GAP) domain.
J Biol Chem (2002) vol. 277 (25) pp. 22115-8.
Kanno et al. Comprehensive Screening for Novel Rab-Binding Proteins by
GST Pull-Down Assay Using 60 Different Mammalian Rabs. Traffic (2010)
epub.
Khosravi-Far et al. Isoprenoid modification of rab proteins terminating in CC or
CXC motifs. Proc Natl Acad Sci USA (1991) vol. 88 (14) pp. 6264-8.
Kötting et al. A phosphoryl transfer intermediate in the GTPase reaction of Ras
in complex with its GTPase-activating protein. Proc Natl Acad Sci USA (2006)
vol. 103 (38) pp. 13911-6.
Krise et al. Quantitative analysis of TIP47-receptor cytoplasmic domain
interactions: implications for endosome-to-trans Golgi network trafficking. J
Biol Chem (2000) vol. 275 (33) pp. 25188-93.
Lan et al. Novel rab GAP-like protein, CIP85, interacts with connexin43 and
induces its degradation. Biochemistry (2005) vol. 44 (7) pp. 2385-96.
Lanzetti et al. The Eps8 protein coordinates EGF receptor signalling through
Rac and trafficking through Rab5. Nature (2000) vol. 408 (6810) pp. 374-7.

43
Lombardi et al. Rab9 functions in transport between late endosomes and the
trans Golgi network. EMBO J (1993) vol. 12 (2) pp. 677-82.
Luo et al. Identification of a novel nurr1-interacting protein. J Neurosci (2008)
vol. 28 (37) pp. 9277-86.
Machner and Isberg. A bifunctional bacterial protein links GDI displacement to
Rab1 activation. Science (2007) vol. 318 (5852) pp. 974-7.
Markgraf et al. Rab cascades and tethering factors in the endomembrane
system. FEBS Lett (2007) vol. 581 (11) pp. 2125-30.
Martinu et al. The TBC (Tre-2/Bub2/Cdc16) domain protein TRE17 regulates
plasma membrane-endosomal trafficking through activation of Arf6. Mol Cell
Biol (2004) vol. 24 (22) pp. 9752-62.
Medkova et al. The rab exchange factor Sec2p reversibly associates with the
exocyst. Mol Biol Cell (2006) vol. 17 (6) pp. 2757-69.
Melançon et al. Involvement of GTP-binding "G" proteins in transport through
the Golgi stack. Cell (1987) vol. 51 (6) pp. 1053-62.
Merithew et al. Structural plasticity of an invariant hydrophobic triad in the
switch regions of Rab GTPases is a determinant of effector recognition. J Biol
Chem (2001) vol. 276 (17) pp. 13982-8.
Miaczynska et al. APPL proteins link Rab5 to nuclear signal transduction via
an endosomal compartment. Cell (2004) vol. 116 (3) pp. 445-56.

44
Mîinea et al. AS160, the Akt substrate regulating GLUT4 translocation, has a
functional Rab GTPase-activating protein domain. Biochem J (2005) vol. 391
(Pt 1) pp. 87-93.
Molenaar et al. A carboxyl-terminal cysteine residue is required for palmitic
acid binding and biological activity of the ras-related yeast YPT1 protein.
EMBO J (1988) vol. 7 (4) pp. 971-6.
Morozova et al. TRAPPII subunits are required for the specificity switch of a
Ypt-Rab GEF. Nat Cell Biol (2006) vol. 8 (11) pp. 1263-9.
Nakamura et al. A novel transcriptional unit of the tre oncogene widely
expressed in human cancer cells. Oncogene (1992) vol. 7 (4) pp. 733-41.
Neuwald. A shared domain between a spindle assembly checkpoint protein
and Ypt/Rab-specific GTPase-activators. Trends Biochem Sci (1997) vol. 22
(7) pp. 243-4.
Nguyen et al. Analysis of the eukaryotic prenylome by isoprenoid affinity
tagging. Nat Chem Biol (2009) vol. 5 (4) pp. 227-35.
Novick et al. Interactions between Rabs, tethers, SNAREs and their regulators
in exocytosis. Biochem Soc Trans (2006) vol. 34 (Pt 5) pp. 683-6.
Novick et al. Identification of 23 complementation groups required for post-
translational events in the yeast secretory pathway. Cell (1980) vol. 21 (1) pp.
205-15.

45
Ortiz et al. Ypt32 recruits the Sec4p guanine nucleotide exchange factor,
Sec2p, to secretory vesicles; evidence for a Rab cascade in yeast. J Cell Biol
(2002) vol. 157 (6) pp. 1005-15.
Ossig et al. Functionality and specific membrane localization of transport
GTPases carrying C-terminal membrane anchors of synaptobrevin-like
proteins. EMBO J (1995) vol. 14 (15) pp. 3645-53.
Ostermeier and Brunger. Structural basis of Rab effector specificity: crystal
structure of the small G protein Rab3A complexed with the effector domain of
rabphilin-3A. Cell (1999) vol. 96 (3) pp. 363-74.
Pan et al. TBC-domain GAPs for Rab GTPases accelerate GTP hydrolysis by
a dual-finger mechanism. Nature (2006) vol. 442 (7100) pp. 303-6.
Patino-Lopez et al. Rab35 and its GAP EPI64C in T cells regulate receptor
recycling and immunological synapse formation. J Biol Chem (2008) vol. 283
(26) pp. 18323-30.
Peck et al. Insulin-stimulated phosphorylation of the Rab GTPase-activating
protein TBC1D1 regulates GLUT4 translocation. J Biol Chem (2009) vol. 284
(44) pp. 30016-23.
Pei et al. PRC17, a novel oncogene encoding a Rab GTPase-activating
protein, is amplified in prostate cancer. Cancer Res (2002) vol. 62 (19) pp.
5420-4.
Pereira-Leal and Seabra. Evolution of the Rab family of small GTP-binding
proteins. J Mol Biol (2001) vol. 313 (4) pp. 889-901.

46
Pereira-Leal and Seabra. The mammalian Rab family of small GTPases:
definition of family and subfamily sequence motifs suggests a mechanism for
functional specificity in the Ras superfamily. J Mol Biol (2000) vol. 301 (4) pp.
1077-87.
Pfeffer. Rab GTPases: specifying and deciphering organelle identity and
function. Trends Cell Biol (2001) vol. 11 (12) pp. 487-91.
Polakis and McCormick. Structural requirements for the interaction of p21ras
with GAP, exchange factors, and its biological effector target. J Biol Chem
(1993) vol. 268 (13) pp. 9157-60.
Pylypenko et al. Structure of Rab escort protein-1 in complex with Rab
geranylgeranyltransferase. Mol Cell (2003) vol. 11 (2) pp. 483-94.
Rak et al. Structure of the Rab7:REP-1 complex: insights into the mechanism
of Rab prenylation and choroideremia disease. Cell (2004) vol. 117 (6) pp.
749-60.
Rak et al. Structure of Rab GDP-dissociation inhibitor in complex with
prenylated YPT1 GTPase. Science (2003) vol. 302 (5645) pp. 646-50.
Rak et al. Crystal structure of the GAP domain of Gyp1p: first insights into
interaction with Ypt/Rab proteins. EMBO J (2000) vol. 19 (19) pp. 5105-13.
Richardson et al. GTP hydrolysis is not important for Ypt1 GTPase function in
vesicular transport. Mol Cell Biol (1998) vol. 18 (2) pp. 827-38.

47
Rink et al. Rab conversion as a mechanism of progression from early to late
endosomes. Cell (2005) vol. 122 (5) pp. 735-49.
Rittinger et al. Structure at 1.65 A of RhoA and its GTPase-activating protein in
complex with a transition-state analogue. Nature (1997) vol. 389 (6652) pp.
758-62.
Rivera-Molina and Novick. A Rab GAP cascade defines the boundary between
two Rab GTPases on the secretory pathway. Proc Natl Acad Sci USA (2009)
vol. 106 (34) pp. 14408-13.
Roach et al. Substrate specificity and effect on GLUT4 translocation of the
Rab GTPase-activating protein Tbc1d1. Biochem J (2007) vol. 403 (2) pp.
353-8.
Rybin et al. GTPase activity of Rab5 acts as a timer for endocytic membrane
fusion. Nature (1996) vol. 383 (6597) pp. 266-9.
Salminen and Novick. A ras-like protein is required for a post-Golgi event in
yeast secretion. Cell (1987) vol. 49 (4) pp. 527-38.
Sasaki et al. Purification and characterization from bovine brain cytosol of a
protein that inhibits the dissociation of GDP from and the subsequent binding
of GTP to smg p25A, a ras p21-like GTP-binding protein. J Biol Chem (1990)
vol. 265 (4) pp. 2333-7.
Sato et al. Activation of an oncogenic TBC1D7 (TBC1 domain family, member
7) protein in pulmonary carcinogenesis. Genes Chromosomes Cancer (2010)
vol. 49 (4) pp. 353-67.

48
Scheffzek et al. GTPase-activating proteins: helping hands to complement an
active site. Trends Biochem Sci (1998) vol. 23 (7) pp. 257-62.
Scheffzek et al. The Ras-RasGAP complex: structural basis for GTPase
activation and its loss in oncogenic Ras mutants. Science (1997) vol. 277
(5324) pp. 333-8.
Schmitt et al. The ras-related YPT1 gene product in yeast: a GTP-binding
protein that might be involved in microtubule organization. Cell (1986) vol. 47
(3) pp. 401-12.
Schoebel et al. RabGDI displacement by DrrA from Legionella is a
consequence of its guanine nucleotide exchange activity. Mol Cell (2009) vol.
36 (6) pp. 1060-72.
Seaman et al. Membrane recruitment of the cargo-selective retromer
subcomplex is catalysed by the small GTPase Rab7 and inhibited by the Rab-
GAP TBC1D5. J Cell Sci (2009) vol. 122 (Pt 14) pp. 2371-82.
Seewald et al. RanGAP mediates GTP hydrolysis without an arginine finger.
Nature (2002) vol. 415 (6872) pp. 662-6.
Segev. Ypt and Rab GTPases: insight into functions through novel
interactions. Curr Opin Cell Biol (2001) vol. 13 (4) pp. 500-11.
Segev et al. The yeast GTP-binding YPT1 protein and a mammalian
counterpart are associated with the secretion machinery. Cell (1988) vol. 52
(6) pp. 915-24.

49
Shapiro and Pfeffer. Quantitative analysis of the interactions between prenyl
Rab9, GDP dissociation inhibitor-alpha, and guanine nucleotides. J Biol Chem
(1995) vol. 270 (19) pp. 11085-90.
Simonsen et al. EEA1 links PI(3)K function to Rab5 regulation of endosome
fusion. Nature (1998) vol. 394 (6692) pp. 494-8.
Sinka et al. Golgi coiled-coil proteins contain multiple binding sites for Rab
family G proteins. J Cell Biol (2008) vol. 183 (4) pp. 607-15.
Sivars et al. Yip3 catalyses the dissociation of endosomal Rab-GDI
complexes. Nature (2003) vol. 425 (6960) pp. 856-9.
Sklan et al. TBC1D20 is a Rab1 GTPase-activating protein that mediates
hepatitis C virus replication. J Biol Chem (2007) vol. 282 (50) pp. 36354-61.
Sklan et al. A Rab-GAP TBC domain protein binds hepatitis C virus NS5A and
mediates viral replication. J Virol (2007) vol. 81 (20) pp. 11096-105.
Soldati et al. Membrane targeting of the small GTPase Rab9 is accompanied
by nucleotide exchange. Nature (1994) vol. 369 (6475) pp. 76-8.
Sönnichsen et al. Distinct membrane domains on endosomes in the recycling
pathway visualized by multicolor imaging of Rab4, Rab5, and Rab11. J Cell
Biol (2000) vol. 149 (4) pp. 901-14.
Stenmark. Rab GTPases as coordinators of vesicle traffic. Nat Rev Mol Cell
Biol (2009) vol. 10 (8) pp. 513-25.

50
Stenmark et al. Endosomal localization of the autoantigen EEA1 is mediated
by a zinc-binding FYVE finger. J Biol Chem (1996) vol. 271 (39) pp. 24048-54.
Stenmark et al. Rabaptin-5 is a direct effector of the small GTPase Rab5 in
endocytic membrane fusion. Cell (1995) vol. 83 (3) pp. 423-32.
Stenmark et al. Inhibition of rab5 GTPase activity stimulates membrane fusion
in endocytosis. EMBO J (1994) vol. 13 (6) pp. 1287-96.
Stone et al. TBC1D1 is a candidate for a severe obesity gene and evidence for
a gene/gene interaction in obesity predisposition. Hum Mol Genet (2006) vol.
15 (18) pp. 2709-20.
Strom et al. A yeast GTPase-activating protein that interacts specifically with a
member of the Ypt/Rab family. Nature (1993) vol. 361 (6414) pp. 736-9.
Suh et al. Structural insights into the dual nucleotide exchange and GDI
displacement activity of SidM/DrrA. EMBO J (2010) vol. 29 (2) pp. 496-504.
Tempel et al. First crystallographic models of human TBC domains in the
context of a family-wide structural analysis. Proteins (2008) vol. 71 (1) pp. 497-
502.
Touchot et al. Four additional members of the ras gene superfamily isolated by
an oligonucleotide strategy: molecular cloning of YPT-related cDNAs from a
rat brain library. Proc Natl Acad Sci USA (1987) vol. 84 (23) pp. 8210-4.

51
Ullrich et al. Membrane association of Rab5 mediated by GDP-dissociation
inhibitor and accompanied by GDP/GTP exchange. Nature (1994) vol. 368
(6467) pp. 157-60.
Van Der Sluijs et al. The small GTP-binding protein rab4 is associated with
early endosomes. Proc Natl Acad Sci USA (1991) vol. 88 (14) pp. 6313-7.
Vitale et al. Distinct Rab-binding domains mediate the interaction of Rabaptin-
5 with GTP-bound Rab4 and Rab5. EMBO J (1998) vol. 17 (7) pp. 1941-51.
Vollmer et al. Primary structure and biochemical characterization of yeast
GTPase-activating proteins with substrate preference for the transport
GTPase Ypt7p. Eur J Biochem (1999) vol. 260 (1) pp. 284-90.
Vollmer and Gallwitz. High expression cloning, purification, and assay of Ypt-
GTPase-activating proteins. Meth Enzymol (1995) vol. 257 pp. 118-28.
Wainszelbaum et al. The hominoid-specific oncogene TBC1D3 activates Ras
and modulates epidermal growth factor receptor signaling and trafficking. J
Biol Chem (2008) vol. 283 (19) pp. 13233-42.
Walworth et al. Hydrolysis of GTP by Sec4 protein plays an important role in
vesicular transport and is stimulated by a GTPase-activating protein in
Saccharomyces cerevisiae. Mol Cell Biol (1992) vol. 12 (5) pp. 2017-28.
Wang et al. The Bfa1/Bub2 GAP complex comprises a universal checkpoint
required to prevent mitotic exit. Curr Biol (2000) vol. 10 (21) pp. 1379-82.

52
Wang et al. TRAPP stimulates guanine nucleotide exchange on Ypt1p. J Cell
Biol (2000) vol. 151 (2) pp. 289-96.
Will and Gallwitz. Biochemical characterization of Gyp6p, a Ypt/Rab-specific
GTPase-activating protein from yeast. J Biol Chem (2001) vol. 276 (15) pp.
12135-9.
Wilson et al. A fusion protein required for vesicle-mediated transport in both
mammalian cells and yeast. Nature (1989) vol. 339 (6223) pp. 355-9.
Wittinghofer et al. The interaction of Ras with GTPase-activating proteins.
FEBS Lett (1997) vol. 410 (1) pp. 63-7.
Wolfman and Macara. A cytosolic protein catalyzes the release of GDP from
p21ras. Science (1990) vol. 248 (4951) pp. 67-9.
Yang et al. Specific binding to a novel and essential Golgi membrane protein
(Yip1p) functionally links the transport GTPases Ypt1p and Ypt31p. EMBO J
(1998) vol. 17 (17) pp. 4954-63.
Yoshimura et al. Functional dissection of Rab GTPases involved in primary
cilium formation. J Cell Biol (2007) vol. 178 (3) pp. 363-9.
Zahraoui et al. The human Rab genes encode a family of GTP-binding
proteins related to yeast YPT1 and SEC4 products involved in secretion. J Biol
Chem (1989) vol. 264 (21) pp. 12394-401.
Zerial and McBride. Rab proteins as membrane organizers. Nat Rev Mol Cell
Biol (2001) vol. 2 (2) pp. 107-17.

53
Zhang et al. Rab7: roles in membrane trafficking and disease. Biosci Rep
(2009) vol. 29 (3) pp. 193-209.
Zhang et al. TBC domain family, member 15 is a novel mammalian Rab
GTPase-activating protein with substrate preference for Rab7. Biochem
Biophys Res Commun (2005) vol. 335 (1) pp. 154-61.

54
Table I. Summary of mammalian Rab GTPase-activating proteins
TBC Protein
Aliases Reported substrates
Other domains
Function/Remarks References
EVI5 Rab11, -35
CC Regulates Shiga toxin uptake
Dabbeekeh et al., 2007; Fuchs et al., 2007
EVI5L Rab23, -10
CC Regulates primary cilia formation
Yoshimura et al., 2007; Itoh et al., 2006
RN-tre Rab5, -41 EGF receptor downregulation; Golgi biogenesis, regulates Shiga toxin uptake
Lanzetti et al., 2000; Haas et al., 2005; Fuchs et al., 2007
RUTBC1 SGSM2 Rab33b, -32
RUN Binds Rab9; interacts with Atg16L1
This study
RUTBC2 SGSM1 Rab34, -36
RUN Binds Nurr1 transcription factor
This study; Luo et al., 2008
RUTBC3 RabGAP-5, MAP, CIP85
Rab5 RUN; SH3
Regulation of endocytosis, binds tumor suppressor Merlin, gap junctions
Haas et al., 2005; Lee et al., 2004; Lan et al., 2005
TBC1D1 Rab8a, -10, -14
PTB Akt substrate; regulates insulin-stimulated GLUT4 receptor trafficking; genetically associated with obesity
Roach et al., 2004; Peck et al., 2009; Stone et al., 2006
TBC1D2A Armus Rab7 PH; CC Rac1 effector and regulator of E-cadherin degradation
Frasa et al., 2010
TBC1D2B KRAB; CC
Binds Rab22 Kanno et al., 2010

55
TBC Protein
Aliases Reported substrates
Other domains
Function/Remarks References
TBC1D3 PRC17 Hominoid specific; oncogene, activator of Ras?
Pei et al., 2002; Wainszelbaum et al., 2008
TBC1D4 AS160 Rab2a, -8a, -10, -14
PTB Akt substrate; regulates insulin-stimulated GLUT4 receptor trafficking
Kane et al., 2002; Mîlnea et al., 2005
TBC1D5 Rab7 Binds to retromer coat
Seaman et al., 2009
TBC1D6 GRTP1
TBC1D7 Rab17 Regulates primary cilia formation, oncogene
Yoshimura et al., 2007; Sato et al., 2010
TBC1D8A Vrp GRAM; CC
TBC1D8B GRAM
TBC1D9A GRAM; EF
TBC1D9B GRAM; EF
TBC1D10A EPI64 Rab27A, -35
Regulates microvillar structure; regulates Shiga toxin uptake
Itoh and Fukuda, 2006; Hanono et al., 2006; Fuchs et al., 2007
TBC1D10B Rab35, -3a
CC Regulates Shiga toxin uptake
Fuchs et al., 2007; Ishibashi et al., 2010
TBC1D10C Rab35 Regulates Shiga toxin uptake
Fuchs et al., 2007

56
TBC Protein
Aliases Reported substrates
Other domains
Function/Remarks References
TBC1D11 GAPCenA, RABGAP1
Rab4, -6 PTB; CC Controversial: has GAP activity on Rab6 but does not block Shiga toxin trafficking like Rab6 depletion; binds Rab36
Fuchs et al., 2007; Cuif et al., 1999; Kanno et al., 2010
TBC1D12
TBC1D13
TBC1D14 CC Crystal structure of TBC domain solved
Tempel et al., 2008
TBC1D15 Rab7 Binds Rab5A/B/C Zhang et al., 2005; Itoh et al., 2006
TBC1D16
TBC1D17 Rab21, -35
Regulates Shiga toxin uptake
Fuchs et al., 2007
TBC1D18 RabGAP1L Rab22 PTB Expression reduced in Alzheimer's patients
Itoh et al., 2006; de Yebra et al., 2004
TBC1D19
TBC1D20 Rab1 TM Regulates Golgi biogenesis; binds Hepatitis C NS5A protein
Haas et al., 2007; Sklan et al., 2007a; Sklan et al., 2007b
TBC1D21
TBC1D22A Crystal structure of TBC domain solved
Tempel et al., 2008
TBC1D22B
TBC1D23

57
TBC Protein
Aliases Reported substrates
Other domains
Function/Remarks References
TBC1D24 TLDc
TBC1D25 OATL1 Rab2a Itoh et al., 2006
TBC1D26
TBC1D27
TBC1D28
TBC1D29
TBC1D30 XM_037557 Rab8a Regulates primary cilia formation
Yoshimura et al., 2007
TBCKL PK; RHOD
USP6 Tre-2, TRE17
UCH Oncogene; founding member of TBC domain family, regulates plasma membrane to endomsome trafficking through Arf6 activation
Neuwald, 1997; Nakamura et al., 2002; Martinu et al., 2004
Domain Abbreviations:
CC - coiled-coli, RUN - RPIP8/Unc-14/NESCA, SH3 - Src homology domain 3,
PTB - Phosphotyrosine binding, PH - Pleckstrin homology, KRAB - Kruppel-
associated box, GRAM - Glucosyltransferases/Rab GTPase
Activators/Myotubularins, EF - EF hand, TM - transmembrane, TLDc - domain
in TBC and LysM containing proteins, PK - protein kinase, RHOD - rhodanese
homology domain, UCH - ubiquitin carboxy-terminal hydrolase

58
CHAPTER 2
RUTBC1: A NOVEL RAB9 EFFECTOR THAT
ACTIVATES GTP HYDROLYSIS BY RAB33B AND RAB32
(Manuscript in preparation)
Ryan M. Nottingham, Ian G. Ganley, Francis A. Barr,
David G. Lambright and Suzanne R. Pfeffer
Contributions: RMN contributed Fig. 1b, 2, 3a-c, 4 and 5. IGG contributed Fig.
3d. FAB contributed Fig. 1a and DGL contributed plasmids and reagents.
RMN and SRP conceived the project and wrote the paper.

59
ABSTRACT
Rab GTPases regulate all steps of membrane trafficking. In cells, their cycling
between active, GTP-bound states and inactive, GDP-bound states is
regulated by the action of antagonistic enzymatic activities called guanine
nucleotide exchange factors and GTPase-activating proteins (GAPs). The
substrates for most RabGAPs are unknown and the potential for cross talk
between different membrane trafficking pathways remains uncharted territory.
Rab9 and its effectors regulate recycling of mannose 6-phosphate receptors
from late endosomes to the trans Golgi network. We show here that RUTBC1
is a TBC domain-containing protein that binds to Rab9 specifically both in vitro
and in cultured cells but is not a GAP for Rab9. Biochemical screening of
RUTBC1ʼs Rab protein substrates revealed highest GAP activity toward
Rab33B and Rab32. These data support a model in which RUTBC1 is
recruited onto Rab33B positive microdomains adjacent to Rab9 microdomains.
This cross-talk between Rab domains suggests the existence of a Rab
cascade between endosomes and the Golgi.

60
INTRODUCTION
Spatial and temporal regulation are important for organizing and coordinating
movement of cargo throughout the secretory and endocytic pathways. Rab
proteins, small Ras-like GTPases, regulate all steps of intracellular trafficking
including cargo selection, vesicle motility along cytoskeletal elements,
tethering of vesicles near their targets and finally fusion of these vesicles with
target membranes (Stenmark, 2009). Rabs accomplish this regulation by
recruiting so-called effector proteins to create specific membrane
microdomains that help to identify different compartments (Zerial and McBride,
2001).
Rab proteins go through cycles of activation (i.e. nucleotide binding) that occur
against a cycle of membrane association. Active, GTP-bound Rabs bind so-
called effector proteins such as adaptors for coat or motor proteins, or
tethering factors that are the molecular machinery for each trafficking step.
Effectors display lower affinity for GDP-bound Rabs, thus favoring membrane
dissociation of effector proteins A GDP-bound Rab then becomes a substrate
for extraction from the membrane into the cytoplasm by a protein called GDI
(Pfeffer and Aivazian, 2004). GDI then recycles Rab proteins back to the
donor compartment for another round of membrane association.

61
In cells, the identity of the bound nucleotide is determined by the opposing
activities of two sets of enzymes: guanine nucleotide exchange factors
(GEFs), which catalyze the exchange of bound GDP for GTP, and GTPase-
activating proteins (GAPs), which catalytically accelerate a Rab proteinʼs slow,
intrinsic GTP hydrolysis rate.
Recently, the model of Rab cascades has been used to describe the linking of
one transport step to another to assure activation and inactivation of Rabs that
lie along the same transport pathway. In yeast, Ypt32p, a late Golgi Rab has
been found to be at the center of a far-ranging cascade in the secretory
pathway, not only recruiting Sec2p, the exchange factor for the next Rab in the
pathway (Sec4p; Ortiz et al. 2002) but also Gyp1p, the GAP for the previous
Rab in the pathway (Ypt1p; Rivera-Molina and Novick, 2009). Other examples
of Rab cascades have been found in mammalian cells including endosomal
maturation through Rab conversion from Rab5-positive early endosomes to
Rab7-positive late endosomes (Rink et al., 2005). It is reasonable to assume
that this satisfying model of Rab cascades will apply more generally
throughout trafficking pathways in each cell type.
Eukaryotes possess a family of proteins that contains the highly conserved
Tre2/Bub2/Cdc16 or TBC domain that has been shown to possess GAP
activity specifically on the Rab subfamily of GTPases (Neuwald, 1997, Strom

62
et al. 1993). TBC domains are often found in proteins that also contain
several other types of domains, suggesting a great amount of integration
between signaling pathways (Bernards, 2003). The human genome encodes
over 40 different TBC domain-containing proteins but only a few cognate pairs
of Rabs and GAPs have been functionally determined. Thus, much remains to
be learned about the functions of RabGAPs in cells: presumably, to
deconstruct established microdomains and form boundaries to focus Rab
microdomains, which would prevent mixing of different functional membrane
units.
The Rab9 GTPase is required for the recycling of mannose 6-phosphate
receptors from late endosomes to the trans Golgi network (Lombardi et al.,
1993; Barbero et al., 2002). It also plays a role in lysosome biogenesis
(Riederer et al., 1994) and late endosome morphology (Ganley et al, 2004). In
this study we analyze the role of a novel effector of Rab9 called RUTBC1. We
show here that RUTBC1 is a multi-domain protein that contains a TBC domain
and is not a GAP for Rab9 in cells but for other Rab GTPases.

63
METHODS
Yeast Two-Hybrid Analysis
Yeast two-hybrid analysis was carried out as described previously (Fuchs et
al., 2007). Briefly, 56 mutant Rab proteins deficient for GTP hydrolysis (Q to
A) were cloned into pGBT9 bait vector (Clontech). RUTBC1 and RUTBC2
were amplified from cDNA libraries and were cloned into pACT2 prey vector
(Clontech); growth on selective media indicated an interaction between a Rab
and RUTBC1 or RUTBC2.
Plasmids
For mammalian expression, N-terminally 3xmyc-tagged RUTBC1 was
obtained by amplification from a cDNA library and ligation into a modified
version of pCDNA3.1(+) (Invitrogen) (Fuchs et al., 2007). This construct
encodes the shorter of two isoforms found in GenBank. GFP-RUTBC1 was
constructed by amplification of this isoform by PCR and ligated into pEGFP-C1
(Clontech). RUTBC1-N and RUTBC1-C truncation constructs were amplified
by PCR and ligated into 3xmyc-pCDNA3.1(+). RUTBC1-RUN was created by
addition of a stop codon directly after the RUN domain by site-directed
mutagenesis using QuikChange (Stratagene). Predicted RUTBC1 GAP-dead
mutant (R803A) was also created using QuikChange.

64
For bacterial expression, RUTBC1-C was ligated into pET28a (Novagen) in
frame with the N-terminal His-tag. His-RUTBC1-C R803A was created by site-
directed mutagenesis. Plasmid encoding GST-Rab9A was previously
described (Aivazian et al, 2006). GST-Rab9B was amplified by PCR from
pET14-Rab9B (Hayes et al., 2009) and ligated into pGEX-4T-1 (GE
Healthcare). GST-Rab6A was amplified by PCR from His-Rab6A (Burguete et
al., 2008) and ligated into pGEX-4T-1. His-Rab33Q72A was described by
Hayes et al., 2009; His-Rab33 wild-type was obtained by site-directed
mutagenesis of this construct.
Plasmids of Rab proteins for biochemical screening of GAP activity were
previously described (Pan et al., 2006). Phosphate Binding Protein (PBP)
from E. coli was amplifed by PCR from bacteria and cloned into modified
pET15. His-PBP A197C was constructed by site-directed mutagenesis.
Protein Expression and Purification
His-RUTBC1-C was transformed into Rosetta2 (DE3) cells (Novagen) and
grown at 37°C until OD600 = 0.5. The cells were induced with 0.4mM isopropyl
β-D-thiogalactoside (IPTG) and grown for an additional 4 hours at 22°C. Cell
pellets were resuspended in lysis buffer (25mM HEPES, pH 7.4, 300mM NaCl,
50mM imidazole) supplemented with 1mM PMSF and lysed by two passes at
20,000 psi through an EmulsiFlex-C5 apparatus (Avestin). Cleared lysates

65
(20,000 rpm for 45 min at 4°C in a JA-20 rotor; Beckman Coulter) were
incubated with Ni-NTA (Qiagen) for 1 hour at 4°C. The resin was then washed
with lysis buffer and eluted with 25mM HEPES, pH7.4, 300mM NaCl and
250mM imidazole. Fractions containing RUTBC1-C were pooled and
concentrated using an Amicon Ultra concentrator (Millipore). The sample was
dialyzed to remove imidazole and then brought to 10%(v/v) glycerol. The
sample was aliquoted, snap frozen in liquid nitrogen and stored at -80°C. His-
RUTBC1-C R803A was purified using the same procedure.
His-Rab33B wild-type was transformed into Rosetta2 (DE3) cells and grown at
37°C until OD600 = 0.6. The cells were induced with 0.4mM IPTG and grown
for an additional 3.5 hours at 37°C. Cell pellets were resuspended in lysis
buffer (50mM MES, pH 6.5, 8mM MgCl2, 2mM EDTA, 0.5mM DTT and 10µM
GDP) supplemented with 1mM PMSF and lysed by two passes at 20,000 psi
through an EmulsiFlex-C5 apparatus. Cleared lysates (20,000 rpm for 45 min
at 4°C in a JA-20 rotor) were loaded onto an 30mL SP-Sepharose column (GE
Healthcare) and then eluted with a 10CV gradient of 0-500mM NaCl.
Fractions containing Rab33B were pooled and brought to 50% ammounim
sulfate. Precipitated protein was collected by centrifugation and resuspended
in S100 buffer (64mM Tris-HCl, pH 8.0, 100mM NaCl, 8mM MgCl2, 2mM
EDTA, 0.2µM DTT, 10µM GDP) and gel filtered by FPLC on a 16/60 Superdex
75 column (GE Healthcare) into S100 buffer. Fractions containing Rab33B

66
were pooled and concentrated using an Amicon Ultra concentrator. The
sample then brought to 10%(v/v) glycerol, aliquoted and snap frozen in liquid
nitrogen and stored at -80°C. His-Rab33B Q72A was purified using the same
procedure.
GST-Rab9A expression and purification were previously described (Aivazian
et al., 2006) and GST-Rab9B and GST-Rab6A were purified by the same
procedure. Expression and purification of Rab proteins for the GAP screen
were previously described (Pan et al., 2006). His-PBP A197C was purified
and labeled according to the method in Shutes and Der (2005).
Antibodies
Mouse monoclonal anti-myc (9E10), mouse monoclonal anti-Rab9A, mouse
monoclonal anti-CI-MPR (2G11) and rabbit anti-CI-MPR, were all previously
described (Ganley et al., 2008). Rabbit anti-GFP antibody was from
Invitrogen; mouse anti-GFP antibody was from Roche. Rabbit anti-Rab2
antibodies were from Santa Cruz Biotechnology. HRP-conjugated goat anti-
mouse and goat anti-rabbit secondary antibodies as well as protein-A-HRP
were from Bio-Rad.

67
Binding assays
Constructs encoding 3xmyc-RUTBC1 or 3xmyc-RUTBC1 truncations were
translated in vitro using a TNT Quick Coupled Transcription/Translation
System (Promega) following the manufacturerʼs protocol. GST-tagged Rabs
were loaded with GTPγS or GDP as described (Aivazian et al., 2006) and
mixed with TNT lysate for 1.5 hours at 25ºC in binding buffer (25mM HEPES-
NaOH, pH7.4, 150mM NaCl, 5mM MgCl2, 1mM DTT, 0.1mM GTPγS).
RUTBC1 constructs bound to GST-Rabs were isolated using glutathione-
Sepharose, washed in binding buffer (with 400mM NaCl) and then eluted by
addition of 25mM glutathione and analyzed by immunoblot.
Cell Culture and Transfections
HeLa, COS-1 and HEK293T cells were obtained from American Type Culture
Collection and cultured at 37°C and 5% CO2 in Dulbecco's modified Eagle's
medium supplemented with 7.5% fetal calf serum, 100U penicillin and
100µg/mL streptomycin. For overexpression studies, all cells were transfected
using Fugene 6 (Roche). Cells were harvested either 24 or 48 hours after
transfected as indicated.
Immunopreciptations, Protein Turnover and Lysosomal Enzyme Secretion
For immunoprecipitations, cells were transfected with indicated plasmids for
24 hours. Cells were then lysed in 50mM Tris-HCl, pH 7.4, 150mM NaCl, 1mM

68
MgCl2 and 1% Triton X-100 supplemented with Complete EDTA-free Protease
Inhibitor Cocktail (Roche) and spun for 15 minutes at full speed in a microfuge
at 4°C. Supernatants were pre-cleared using Protein-A agarose (Roche) for 15
minutes at room temperature and then immunoprecipitated with indicated
antibodies for 1.5 hours at room temperature. Immune complexes were
isolated on Protein-A agarose for 30 minutes at room temperature. The resin
was then washed 3 times with lysis buffer and once with PBS. The beads
were resuspended in 2X sample buffer and boiled and analyzed by
immunoblot. Protein turnover and lysosomal enzyme secretion assays were
performed as described previously (Ganley et al., 2008).
GAP Assays
For the biochemical screen of GAP substrates, the procedure followed by Pan
et al., 2006 was generally used with the exception that phosphate released
during the reaction was bound by modified PBP (A197C) labeled at position
197 with N-[2-(1-maleimidyl)ethyl]-7-(diethylamino)coumarin-3-carboxamide
(MDCC). Phosphate binding to MDCC-PBP causes a conformational change
in the phosphate binding cleft that results in an increase in MDCC
fluorescence (Brune et al., 1994). Reactions were started by adding a solution
containing GAP, MgCl2 and MDCC-PBP to desalted, GTP-exchanged Rabs by
a Precision 2000 liquid handling system (Biotek). Rab GTPases were at 2µM
for all reactions while the concentration of His-RUTBC1-C was varied.

69
Phosphate production was monitored by following fluorescence signal
continuously in a TECAN Saphire microplate reader using an excitation of
425nm and an emission cutoff filter of 455nm. Other assays were performed
as follows: purified Rab GTPases were exchanged with GTP-γ-32P as in
Aivazian et al. for 10 minutes at 25°C and desalted on PD-10 or PD-Mini
columns to remove free nucleotide. Loading efficiency was assayed by filter
binding and specific activity calculated from inputs. Various concentrations of
Rab-GTP were incubated with His-RUTBC1-C at 25°C. Aliquots of the
reaction were removed at various time points and quenched by addition of a
solution of 5% Norit-A in 50mM phosphoric acid. The quenched samples were
spun to pellet the charcoal and half of the supernatants were analyzed by
liquid scintillation counting in BioSafe-II scintillation fluid (Research Products
International) using an LS-6500 liquid scintillation counter (Beckman-Coulter).

70
RESULTS
We study the role of Rab9 GTPase in intracellular membrane traffic. As a
starting point to find regulators of Rab9, we used a two-hybrid screen
consisting of all TBC domain-containing proteins in the human genome as
prey against a comprehensive library of hydrolysis-deficient Rab GTPases as
bait (Haas et al., 2005, Fuchs, et al., 2007). This screen identified a TBC
domain-containing protein, RUTBC1, as a potential partner of both Rab9A and
Rab9B (Figure 1A). It also interacted to a lesser extent with Rab2 and Rab3
isoforms.
RUTBC1, and the closely related RUTBC2, are conserved proteins that
contain an N-terminal RPIP8/UNC-14/NESCA (RUN) domain and C-terminal
TBC domain (Figure 1B). RUN domains are entirely alpha-helical domains
that have been shown to interact with members of the small, Ras-like GTPase
superfamily including Rab6 and Rap1/2 (Callebaut et al., 2001; Recacha et al.,
2009; Janoueix-Lerosey et al., 1998). No enzymatic activity has been found to
be associated with RUN domains, suggesting that they likely mediate protein-
protein interactions. RUTBC1ʼs catalytic domain is unique in that there is a
large insertion between the first two “fingerprint” A and B motifs (Figure 1B,
sequence). In the structural model for Rab and RabGAP interaction, the
analagous region of Gyp1 is situated away from the Rab:GAP binding
interface (Pan et al., 2006). Most of the dissimilarity between RUTBC1 and

71
RUTBC2 TBC domains is found in this insertion. According to the NCBI
Homologene database, there is only one RUTBC1/2 protein in C. elegans (tbc-
8), Drosophila (CG1905) and zebrafish (LOC794373) while vertebrates have
both proteins. Drosophila actually has two RUTBC-like proteins but they are
thought to have diverged within flies, independent of the divergence that
occurred in vertebrates (Yang et al., 2007). Another protein, RabGAP-5, also
contains a RUN and TBC domain but the domain structure is reversed (Haas
et al., 2005; Yang et al., 2007).
RUTBC1 is a Rab9 effector
To confirm the results of the two hybrid screen and to test the nature of the
binding interactions, we tested if Rab9 could bind RUTBC1 in vitro. Full-length
RUTBC1 was difficult to express in E. coli so we utilized an in vitro
transcription/translation (IVT) system and assayed binding by GST-affinity
chromatography. As seen in Figure 2A, GST-Rab9A, but not GST-Rab9B or
GST-Rab6A, bound to in vitro translated full-length RUTBC1, confirming the
specificity seen in the screen. Rab9A and Rab9B are highly similar proteins
that localize to different organelles at steady-state: Rab9A on late endosomes
(Lombardi et al., 1993) and Rab9B at the Golgi (Yoshimura et al., 2007 and
our unpublished observations). Rab9A and 9B are most divergent in their C-
terminal, hypervariable domains. This suggests that RUTBC1 may recognize
part of the Rab9A hypervariable domain for binding. Next, we tested if

72
RUTBC1 preferred either the GTP or GDP-bound form of Rab9. Using the
GST-binding assay described above, in vitro translated RUTBC1 was bound
approximately 10-fold more efficiently by GST-Rab9 when loaded with GTPγS
than with GDP (Figures 1B and 1C). Thus, RUTBC1 is a bona fide effector of
Rab9A ( referred to as Rab9 below).
We next investigated the location of the Rab9 binding site in RUTBC1. A
series of RUTBC1 truncations were generated (Figure 2C) and tested using
the GST-binding assay. GST-Rab9 bound to the N-terminal half of RUTBC1,
and not to the C-terminal half that contains the TBC domain (Figure 2D). This
suggests that Rab9 is likely not a substrate for the predicted GAP activity of
RUTBC1. Further truncation revealed that Rab9 did not seem to bind to a
construct composed of amino acids 1-185 that stopped right after the RUN
domain. A previous two-hybrid screen for GAP-Rab interactions failed to
detect Rab9 binding to either RUTBC1 or RUTBC2 (Itoh et al., 2006) using
constructs for both proteins containing only the TBC domain. Taken together,
these data suggest that Rab9 binds RUTBC1 in a region distinct from the RUN
or TBC domains in a guanine nucleotide dependent manner.
RUTBC1 interacts with Rab9 in cells
Rab9 regulates the recycling of mannose 6-phosphate receptors (MPRs) from
late endosomes to the trans Golgi network (TGN) (Lombardi et al., 1994,

73
Carroll et al., 2001). To explore whether Rab9 and RUTBC1 interact in cells,
HEK 293T cells were transfected with GFP-RUTBC1 or GFP as a control. As
shown in Figure 3A, endogenous Rab9 co-immunoprecipitated with GFP-
RUTBC1 and not GFP (Figure 3A). Rab2, which showed interaction with
RUTBC1 by two hybrid was not detected in the immunoprecipitates. This
experiment confirms that RUTBC1 can interact with Rab9 in living cells.
We characterized this interaction further by investigating the effects of
overexpressed, exogenous RUTBC1 on MPR recycling. When Rab9 function
is disturbed in cells by overexpression of a dominant negative mutant Rab9
(Riederer et al., 1994) or by depletion of its effectors (Reddy et al., 2006;
Espinosa et al., 2009), MPRs are mis-sorted to the lysosome. We reasoned
that if RUTBC1 were a GAP for Rab9, the phenotype of RUTBC1
overexpression should be similar to that of a GDP-preferring Rab9 mutant or
Rab9 depletion. When RUTBC1 was overexpressed in COS-1 cells, MPRs
levels at steady state were decreased by ~35% (Figure 3B). This observation
led us to investigate the turnover rate of MPR in HeLa cells overexpressing
RUTBC1; under these conditions, it was turned over more rapidly (Figure 3C),
consistent with lower levels of MPRs at steady state. This suggested that
some amount of MPR was being mis-sorted to lysosomes, consistent with a
block in Rab9 function. As a possible GAP for Rab9, the predicted GAP-
deficient mutant (R803A) of RUTBC1 should not have the same effect upon

74
exogenous expression. In a functional test of MPR trafficking, we measured
the amount of hexosaminidase activity secreted into the media by cells
transfected with RUTBC1. Hexosaminidase is usually sorted to the lysosome
but when MPR levels are deficient in the TGN due to mis-sorting, the
hydrolase is secreted. In 293T cells transfected with RUTBC1, slightly higher
hexosaminidase activity was detected in the media than in control cells (Figure
3D). Cells transfected with RUTBC1 R803A showed a similar effect. Taken
together, perturbation of MPR trafficking seen in our experiments is likely due
to titration of Rab9 by the overexpression of RUTBC1 rather than the GAP
activity encoded by RUTBC1ʼs catalytic domain.
RUTBC1 has narrow substrate specificity
RUTBC1 binds Rab9 in the region between the RUN and TBC domains. and
does not appear to function as a Rab9-GAP. This suggested that Rab9 might
be part of a Rab cascade, in which Rab9 may bind a GAP that inactivates a
prior acting Rab GTPase. In this case, discovering the substrates of RUTBC1
would provide insight into the significance of Rab9-RUTBC1 interaction and
the identity of a prior acting Rab candidate protein.
Thirty-two different mammalian Rab GTPases were screened in vitro, under
single turnover conditions, as substrates for RUTBC1 using purified His-
tagged RUTBC1-C (Figure 2C) containing the TBC domain. Figure 4

75
summarizes these results by comparing observed second order rate constants
for GAP-catalyzed hydrolysis to the rate constants for each Rab proteins
intrinsic hydrolysis rate. The TBC domain of RUTBC1 had the highest activity
against Rab33B and Rab32, while no activity was detected against Rab9,
Rab2 or Rab3 proteins.
Further characterization of the kinetic parameters of the TBC domain found
that RUTBC1 has similar activity on Rab33B and Rab32. These Rabs were
mixed with increasing concentrations of RUTBC1-C and the data obtained
under pseudo first order conditions were simultaneously fit to the integrated
pseudo first order Michaelis-Menten equation. Apparent second order rate
constants from this fit were 2980 M-1s-1 for Rab33B and 1930 M-1s-1 for Rab32
(Figure 5A). Classical analysis of Rab33B GTP hydrolysis stimulated by
RUTBC1-C yielded a kcat of 12 min-1, which is approximately 5000-fold higher
than Rab33Bʼs intrinsic rate (0.0019 min-1; Figure 5B). We also determined a
KM of ~150µM. This value for KM is likely a lower estimate because of difficulty
preparing Rab33B-GTP substrate at amounts high enough to show complete
saturation. Catalytic efficiency of RUTBC1-C for Rab33B was calculated to be
only 1.8-fold lower (1680 M-1s-1), showing good agreement between the two
methods.

76
In the co-crystal of Gyp1p and Rab33B, Pan et al. (2006) suggested that
RabGAPs catalyze GTP hydrolysis by a dual-finger mechanism where both a
catalytic arginine and glutamine are supplied by the GAP. This model predicts
that RabGAPs will still be able to stimulate so-called constitutively active Rabs
that harbor a glutamine to alanine mutation in their G3 motifs. As shown in
Figure 5C (left panel) RUTBC1-C can efficiently stimulate GTP hydrolysis of
Rab33BQ92A. The dual finger mechanism also predicts that mutation of the
conserved arginine in the B motif should abrogate GAP activity. RUTBC1-C
R803A does not stimulate Rab33B hydrolysis above the intrinsic rate (Figure
5C, right).

77
DISCUSSION
Rab9 plays an essential role in the recycling of mannose 6-phosphate
receptors from late endosomes to the TGN, and it plays a role in lysosome
biogenesis and late endosome morphology. Regulators of the Rab9
nucleotide binding cycle are currently unknown although it is likely that both a
GEF and GAP for Rab9 exist in cells. Here we have shown that a predicted
RabGAP protein, RUTBC1, is a novel Rab9 effector that seems to bind
specifically to Rab9A and not Rab9B. Rab9A binds at a site in the linker
region between the RUN and TBC domains, and the proteins can interact in
cells. Despite specific binding, RUTBC1 does not possess GAP activity on
Rab9 but instead displays GAP activity on Rab33B and Rab32. This implies
that Rab33B or Rab32 act in a pathway that feeds into Rab9, and Rab9
binding to RUTBC1 helps to clear these Rabs from a Rab9A membrane
microdomain
Currently there are no known direct links between Rab9-mediated trafficking
and either Rab33B- or Rab32-regulated events. Rab33B is a ubiquitously
expressed Rab localized to the medial Golgi (Zheng et al., 1998).
Overexpression of its GTPase-deficient form (Rab33BQ92L) re-localizes
resident Golgi enzymes, like N-acetylglucosamine transferase I to the
endoplasmic reticulum (ER) (Valsdottir et al., 2001). Overexpression of its
GDP-preferring form (Rab33BT47N) blocks the re-localization of Golgi resident

78
enzymes to the ER in cells induced by expression of a dominant negative
mutant of Sar1 (Valsdottir et al., 2001). Rab33B function is somehow related
to that of Rab6 in a COPI independent retrograde trafficking pathway from the
Golgi to ER (Jiang and Storrie, 2005). Depletion of Rab33B from cells using
siRNA impairs Shiga-like toxin B trafficking from the Golgi to the ER (Starr et
al., 2010). Interestingly, it also rescues the dispersed Golgi phenotype
observed upon depletion of ZW10 (human homologue of yeast Dsl1p) or Cog3
(human homologue of yeast Sec34), two distinct tethering complexes involved
in Golgi to ER retrograde traffic. Depletion of Rab33B alone has no effect on
Golgi morphology (Starr et al., 2010). These data suggest that Rab33B might
regulate flux of material through the medial Golgi cisterna.
Depletion of tethers often leads to transport vesicle accumulation. In Cog3
depleted cells, the vesiculated Golgi contains both matrix proteins like GM130
and resident proteins like glycosyl transferases (Zolov and Lupashin, 2005). If
these are indeed bona fide transport intermediates, Rab33B might be involved
in the formation of these structures, i.e. in vesicle formation. This is similar to
role of Rab9 may play on late endosomes by collecting MPRs into nascent
vesicle buds by establishment of a Rab9-TIP47-MPR microdomain (Carroll et
al., 2001).

79
Other TBC domain containing proteins are predicted to be GAPs for Rab33B.
TBC1D22A and TBC1D22B are the mammalian homologues of yeast Gyp1p.
Gyp1p is an extensively studied TBC domain protein that has GAP activity on
Ypt1p in vitro and in cells. Gyp1p is localized to the Golgi and can potently
stimulate Rab33B in vitro (Pan et al., 2006). Overexpression of TBC1D22 in
HeLa cells leads to disruption of the Golgi and this phenotype is dependent on
its GAP activity (Haas et al., 2007). This argues against TBC1D22 being a
GAP for Rab33B in cells, as depletion of Rab33B leads to no change in Golgi
morphology, at least at the level of light microscopy (Starr et al., 2010).
Little is known about specific Rab33B effectors. Affinity chromatography using
GST-Rab33BQ92L showed that GM130, Rabaptin-5, and Rabex-5 all
appeared to be binding partners for Rab33B (Valsdottir et al., 2001). GM130 is
a known Rab1 effector while Rababptin-5 and Rabex-5 are well characterized
interacting partners of Rab5 (Moyer et al., 2001; Horiuchi et al. 1997). A
recent study reported that Atg16L1, an protein essential for autophagy in yeast
and mammals, is a specific Rab33B effector (Itoh et al., 2008). Autophagy is
the process by which cells recycle cytoplasm and organelles by enclosing
them in a unique double membrane structure called an autophagosome. This
structure then fuses with the lysosome to begin degradation of the contents
(Glick et al., 2010).

80
Rab32 has been reported to have divergent roles in different cell types.
Originally characterized as an A-kinase anchoring protein, mutants of Rab32
were reported to alter mitochrondrial distribution (Alto et al., 2002), but this
finding does not appear to be common to all cell types (Hirota and Tanaka,
2009; our unpublished observations). In mouse melanocytes, Rab32 controls
post-Golgi trafficking of melanogenic enzymes to melanosomes, a lysosome-
related organelle. The protein seems to have a redundant role in this pathway
with the closely related Rab38 (Wasmeier et al., 2006). In the cht mouse that
is deficient in Rab38, pigment defects are relatively mild; however, cht
melanocytes depleted of Rab32 show severe pigmentation defects. Rab32
and Rab38 share a common effector, VARP (Vps9-domain and Ankryin
Repeat Protein), which is also necessary for proper melanogenic enzyme
trafficking and is a GEF for Rab21 (Tamura et al., 2009). Rab32 is also
thought to function as an AKAP in Xenopus melanophores, regulating
aggregation and dispersion of melanosomes in response to hormones (Park et
al., 2007). RUTBC1 specifically showed GAP activity to Rab32 and not
Rab38, suggesting that Rab32 and Rab38 are not entirely redundant and that
their functions might diverge in different cell types. Intriguingly, a recent report
also linked Rab32 to autophagy; overexpression of GDP-preferring mutants of
Rab32 blocked basal autophagy (Hirota and Tanaka, 2009).

81
Taken together, these data suggest the existence of a Rab cascade involving
cross-talk between the Golgi and late endosomes. According to the Rab
cascade model, Rab9 would recruit (or activate) a GAP for the Rab that acts
before it in a trafficking pathway. Rab33Bʼs role in retrograde trafficking to the
ER seems less likely to be regulated by Rab9 than its putative role in
autophagy, as key regulators of autophagy like Atg9 are known to cycle
between Golgi and late endosomes (Young et al., 2006). Indeed, Rab9 has
even been suggested to play a role in an alternative autophagy mechanism
that is independent of the Atg5/Atg12/Atg16L1 complex (Nishida et al., 2009).
Rab9ʼs role vis-à-vis Rab33B and Rab32 is an exciting avenue for additional
experimentation, and experiments to examine the role of RUTBC1 in
autophagy and other trafficking pathways are currently in progress.

82
REFERENCES Aivazian et al. TIP47 is a key effector for Rab9 localization. J Cell Biol (2006)
vol. 173 (6) pp. 917-26.
Alto et al. Rab32 is an A-kinase anchoring protein and participates in
mitochondrial dynamics. J Cell Biol (2002) vol. 158 (4) pp. 659-68.
Barbero et al. Visualization of Rab9-mediated vesicle transport from
endosomes to the trans-Golgi in living cells. J Cell Biol (2002) vol. 156 (3) pp.
511-8.
Bernards. GAPs galore! A survey of putative Ras superfamily GTPase
activating proteins in man and Drosophila. Biochim Biophys Acta (2003) vol.
1603 (2) pp. 47-82.
Brune et al. Direct, real-time measurement of rapid inorganic phosphate
release using a novel fluorescent probe and its application to actomyosin
subfragment 1 ATPase. Biochemistry (1994) vol. 33 (27) pp. 8262-71.
Burguete et al. Rab and Arl GTPase family members cooperate in the
localization of the golgin GCC185. Cell (2008) vol. 132 (2) pp. 286-98.
Callebaut et al. RUN domains: a new family of domains involved in Ras-like
GTPase signaling. Trends Biochem Sci (2001) vol. 26 (2) pp. 79-83.
Espinosa et al. RhoBTB3: a Rho GTPase-family ATPase required for
endosome to Golgi transport. Cell (2009) vol. 137 (5) pp. 938-48.

83
Fuchs et al. Specific Rab GTPase-activating proteins define the Shiga toxin
and epidermal growth factor uptake pathways. J Cell Biol (2007) vol. 177 (6)
pp. 1133-43.
Ganley et al. A syntaxin 10-SNARE complex distinguishes two distinct
transport routes from endosomes to the trans-Golgi in human cells. J Cell Biol
(2008) vol. 180 (1) pp. 159-72.
Ganley et al. Rab9 GTPase regulates late endosome size and requires
effector interaction for its stability. Mol Biol Cell (2004) vol. 15 (12) pp. 5420-
30.
Glick et al. Autophagy: cellular and molecular mechanisms. The Journal of
pathology (2010) pp.
Haas et al. Analysis of GTPase-activating proteins: Rab1 and Rab43 are key
Rabs required to maintain a functional Golgi complex in human cells. J Cell Sci
(2007) vol. 120 (Pt 17) pp. 2997-3010.
Haas et al. A GTPase-activating protein controls Rab5 function in endocytic
trafficking. Nat Cell Biol (2005) vol. 7 (9) pp. 887-93.
Hayes et al. Multiple Rab GTPase binding sites in GCC185 suggest a model
for vesicle tethering at the trans-Golgi. Mol Biol Cell (2009) vol. 20 (1) pp. 209-
17.
Hirota and Tanaka. A small GTPase, human Rab32, is required for the
formation of autophagic vacuoles under basal conditions. Cell Mol Life Sci
(2009) vol. 66 (17) pp. 2913-32.

84
Horiuchi et al. A novel Rab5 GDP/GTP exchange factor complexed to
Rabaptin-5 links nucleotide exchange to effector recruitment and function. Cell
(1997) vol. 90 (6) pp. 1149-59.
Itoh et al. Golgi-resident small GTPase Rab33B interacts with Atg16L and
modulates autophagosome formation. Mol Biol Cell (2008) vol. 19 (7) pp.
2916-25.
Itoh et al. Screening for target Rabs of TBC (Tre-2/Bub2/Cdc16) domain-
containing proteins based on their Rab-binding activity. Genes Cells (2006)
vol. 11 (9) pp. 1023-37.
Jiang and Storrie. Cisternal rab proteins regulate Golgi apparatus redistribution
in response to hypotonic stress. Mol Biol Cell (2005) vol. 16 (5) pp. 2586-96.
Lombardi et al. Rab9 functions in transport between late endosomes and the
trans Golgi network. EMBO J (1993) vol. 12 (2) pp. 677-82.
Moyer et al. Rab1 interaction with a GM130 effector complex regulates COPII
vesicle cis--Golgi tethering. Traffic (2001) vol. 2 (4) pp. 268-76.
Neuwald. A shared domain between a spindle assembly checkpoint protein
and Ypt/Rab-specific GTPase-activators. Trends Biochem Sci (1997) vol. 22
(7) pp. 243-4.
Nishida et al. Discovery of Atg5/Atg7-independent alternative
macroautophagy. Nature (2009) vol. 461 (7264) pp. 654-8.

85
Ortiz et al. Ypt32 recruits the Sec4p guanine nucleotide exchange factor,
Sec2p, to secretory vesicles; evidence for a Rab cascade in yeast. J Cell Biol
(2002) vol. 157 (6) pp. 1005-15.
Pan et al. TBC-domain GAPs for Rab GTPases accelerate GTP hydrolysis by
a dual-finger mechanism. Nature (2006) vol. 442 (7100) pp. 303-6.
Park et al. Rab32 regulates melanosome transport in Xenopus melanophores
by protein kinase a recruitment. Curr Biol (2007) vol. 17 (23) pp. 2030-4.
Pfeffer and Aivazian. Targeting Rab GTPases to distinct membrane
compartments. Nat Rev Mol Cell Biol (2004) vol. 5 (11) pp. 886-96.
Recacha et al. Structural basis for recruitment of Rab6-interacting protein 1 to
Golgi via a RUN domain. Structure (2009) vol. 17 (1) pp. 21-30.
Reddy et al. A functional role for the GCC185 golgin in mannose 6-phosphate
receptor recycling. Mol Biol Cell (2006) vol. 17 (10) pp. 4353-63.
Riederer et al. Lysosome biogenesis requires Rab9 function and receptor
recycling from endosomes to the trans-Golgi network. J Cell Biol (1994) vol.
125 (3) pp. 573-82.
Rink et al. Rab conversion as a mechanism of progression from early to late
endosomes. Cell (2005) vol. 122 (5) pp. 735-49.
Rivera-Molina and Novick. A Rab GAP cascade defines the boundary between
two Rab GTPases on the secretory pathway. Proc Natl Acad Sci USA (2009)
vol. 106 (34) pp. 14408-13.

86
Shutes and Der. Real-time in vitro measurement of GTP hydrolysis. Methods
(2005) vol. 37 (2) pp. 183-89.
Starr et al. Rab33B and Rab6 Are Functionally Overlapping Regulators of
Golgi Homeostasis and Trafficking. Traffic (2010) pp.
Stenmark. Rab GTPases as coordinators of vesicle traffic. Nat Rev Mol Cell
Biol (2009) vol. 10 (8) pp. 513-25.
Strom et al. A yeast GTPase-activating protein that interacts specifically with a
member of the Ypt/Rab family. Nature (1993) vol. 361 (6414) pp. 736-9.
Tamura et al. Varp is a novel Rab32/38-binding protein that regulates Tyrp1
trafficking in melanocytes. Mol Biol Cell (2009) vol. 20 (12) pp. 2900-8.
Valsdottir et al. Identification of rabaptin-5, rabex-5, and GM130 as putative
effectors of rab33B, a regulator of retrograde traffic between the Golgi
apparatus and ER. FEBS Lett (2001) vol. 508 (2) pp. 201-9.
Wasmeier et al. Rab38 and Rab32 control post-Golgi trafficking of
melanogenic enzymes. J Cell Biol (2006) vol. 175 (2) pp. 271-81.
Yang et al. Identification of three novel proteins (SGSM1, 2, 3) which modulate
small G protein (RAP and RAB)-mediated signaling pathway. Genomics
(2007) vol. 90 (2) pp. 249-60.
Yoshimura et al. Functional dissection of Rab GTPases involved in primary
cilium formation. J Cell Biol (2007) vol. 178 (3) pp. 363-9.

87
Young et al. Starvation and ULK1-dependent cycling of mammalian Atg9
between the TGN and endosomes. J Cell Sci (2006) vol. 119 (Pt 18) pp. 3888-
900.
Zerial and McBride. Rab proteins as membrane organizers. Nat Rev Mol Cell
Biol (2001) vol. 2 (2) pp. 107-17.
Zheng et al. A novel Rab GTPase, Rab33B, is ubiquitously expressed and
localized to the medial Golgi cisternae. J Cell Sci (1998) vol. 111 ( Pt 8) pp.
1061-9.
Zolov and Lupashin. Cog3p depletion blocks vesicle-mediated Golgi
retrograde trafficking in HeLa cells. J Cell Biol (2005) vol. 168 (5) pp. 747-59.

88
FIGURE LEGENDS
Figure 1: RUTBC1 interacts with Rab9. (A) RUTBC1 was screened for
binding against a comprehensive library of 56 human Rab GTPases. Growth
after streaking on selective media indicates an interaction between RUTBC1
and a Rab GTPase. (B) Diagram of RUTBC1 domain structure. The RUN
domain is shown in blue and the TBC domain is shown in red. The first three
conserved motifs of the TBC domain are shown. Conserved residues are in
bold and the predicted catalytic arginine and glutamine are shown in red. The
extended insert between motif A and motif B of RUTBC1 is compared to the
same regions of human RabGAP-5 (RUTBC3) and S. cerevisiae Gyp1p.
Figure 2: RUTBC1 is an effector of Rab9. (A) In vitro translated (IVT)
3xmyc-RUTBC1 (IVT-3xmyc-RUTBC1) proteins were incubated with various
bacterially purified GST-tagged Rabs pre-loaded with GTPγS. Bound material
was eluted with glutathione and half of the eluate was analyzed by immunoblot
using anti-myc antibodies. Rabs were detected by Ponceau S staining. (B)
IVT-3xmyc-RUTBC1 was incubated with GST-Rab9A pre-loaded with either
GTPγS or GDP and analyzed as in A. (C) Quantitation of RUTBC1 nucleotide
preference shown in B. Error bars represent SEM from two independent
experiments. (D) Diagram of 3xmyc-tagged RUTBC1 constructs used in

89
binding assays. (E) IVT-3xmyc-RUTBC1 truncation constructs were incubated
with GST-Rab9A and analyzed as in A. “Input” indicates 1% of the IVT
reaction used in the binding assay.
Figure 3: RUTBC1 binds to, but is not a GAP for Rab9 in cells. (A)
HEK293T cells were transfected with GFP-RUTBC1 or GFP for 24 hrs and
immunoprecipitated with anti-GFP antibodies followed by immunoblotting for
with specific antibodies to different Rab proteins. “Input” represents 2% of the
lysate subjected to immunoprecipitation. (B) Left panel - Quantitation of COS-
1 cells were transfected with 3xmyc-RUTBC1 for 48 hours and extracts were
immunoblotted with anti-CI-MPR anitbodies. Right panel - Quantitation of CI-
MPR half-life measured from pulse/chase analysis of HeLa cells transfected
with 3xmyc-RUTBC1 for 48 hours. Extracts were immunoprecipitated with
anti-CI-MPR and exposed to phosphor screen. (C) HEK293T cells transfected
with 3xmyc-RUTBC1 wild type or R803A for 24 hours were assayed for
secreted and intracellular hexosaminidase activity. Error bars in all panels
represent SEM from at least two independent experiments.
Figure 4: RUTBC1 TBC domain has GAP activity toward Rab33B and
Rab32 in vitro. Purified, mammalian Rab GTPases (32) were pre-loaded with
GTP for one hour at room temperature and then desalted to remove free
nucleotide. MDCC-PBP, MgCl2 and varying concentrations of purified His-

90
RUTBC1-C were added to start the reaction. Phosphate release was
monitored continuously by microplate fluorimeter (see Materials and Methods).
Catalytic efficiency (kcat/KM) relative to the intrinsic rate constant (kintr) for GTP
hydrolysis was determined. Plots represent the mean from duplicate wells.
Figure 5: RUTBC1 TBC domain stimulates GTP hydrolysis. (A) GTP
hydrolysis by Rab33B (left) and Rab32 (right) in the presence of increasing
concentrations of RUTBC1-C. Values indicate the concentration of RUTBC1-
C in µM. Values below graphs represent catalytic efficiency extracted from a
simultaneous fit of the data to the integrated pseudo first order Michaelis-
Menten equation for each Rab substrate. (B) Velocity of RUTBC1-C catalyzed
reaction in the presence of increasing concentrations of Rab33B. (C) Left
panel - GTP hydrolysis by Rab33BQ92A in the presence of wild-type
RUTBC1-C. Right panel - GTP hydrolysis by Rab33B in the presence of
RUTBC1-C or the R803A mutant.

91
Figure 1

92
Figure 2

93
Figure 3

94
Figure 4

95
Figure 5

96
Figure 5, continued

97
CHAPTER 3
INTERACTION OF RUTBC1, A RAB33B GAP,
WITH THE RAB33B-EFFECTOR, ATG16L1
(Manuscript in preparation)
Ryan M. Nottingham and Suzanne R. Pfeffer
Contributions: RMN contributed Fig. 1-4.
RMN and SRP conceived the project and wrote the paper

98
ABSTRACT
Macroautophagy is a highly-conserved pathway that degrades bulk
cytoplasmic constituents including proteins, organelles and microorganisms.
This material is sequestered in a double membrane autophagosome.
Autophagosomes fuse with both early and late endosomes and lysosomes
leading to the degradation of the enclosed material. Two ubiquitin-like
conjugation systems are required for formation and expansion of the isolation
membrane. Atg16L1 forms a complex with Atg5-Atg12, which is required for
autophagosome formation in vivo. Currently, Atg16L1 is the only identified
effector of Golgi-localized Rab33B. Here we show that RUTBC1, a GTPase-
activating protein (GAP) for Rab33B, also interacts with Atg16L1. Depletion of
RUTBC1 from cells causes a concomitant depletion of Atg16L1, consistent
with their presence in a complex in cells. These proteins interact in cells, and
overexpression of both proteins causes them to co-localize to large
cytoplasmic puncta under nutrient rich conditions. Taken together, these data
suggest a model whereby RUTBC1 binds to Atg16L1, recruiting RUTBC1 to a
Rab33B microdomain.

99
INTRODUCTION
Macroautophagy is a highly-conserved pathway that degrades bulk
cytoplasmic constituents including proteins, organelles and microorganisms
and also plays a role in various disease states (Mizushima et al., 2008).
Macroautophagy involves building a membrane-bound structure called an
autophagosome around this material and then fusing this structure with
lysosomal compartments (Yorimitsu and Klionsky, 2005; Klionsky, 2007; Glick
et al., 2010). Macroautophagy is a non-selective process ; other kinds of
autophagy can be selective substrates, including mitochondria through
mitophagy, peroxisomes through pexophagy, chaperone-mediated autophagy,
and the cytoplasm-to-vacuole (Cvt) pathway).
Macroautophagy (simply called autophagy for the remainder of the chapter) is
unique among membrane trafficking pathways because the autophagosome is
bound by a double membrane whereas regular phagocytosis and conventional
trafficking involve only one bilayer. Autophagy occurs at basal levels in
vegetative cells but can be induced by different signaling pathways in
response to various stimuli such as starvation or inhibitors of master signaling
kinases such as mTOR. Autophagy can be suppressed by reagents that
induce lysosomal dysfunction, such as chloroquine, ammonium chloride or
protease inhibitors (Mizushima et al., 2010).

100
The current model of autophagosome formation involves a unique structure
termed the pre-autophagosomal structure in yeast and the isolation membrane
or phagophore in mammalian cells. Initiation of autophagosome formation is
regulated by the Atg1 complex (Levine and Klionsky, 2004; Young et al.,
2006). Autophagosome formation also requires phosphatidylinositol-3-
phosphate, which is generated by a particular PI3K complex consisting of
Vps34, p150, Beclin1 and Atg14L (Itakura et al., 2008). Atg9, the only known
integral membrane protein required for autophagosome formation, cycles
between the Golgi and Rab7- and Rab9-positive late endosomes, suggesting
the involvement of endocytic transport machinery in regulating
autophagosome formation (Young et al., 2006). Indeed, autophagosomes
most likely merge with both early and late endosomes in addition to lysosomes
(Eskelinen, 2005).
Two ubiquitin-like conjugation systems are required for formation and
expansion of the phagophore in yeast and mammals (Yorimitsu and Klionsky,
2005). The first system catalyzes the conjugation of Atg12 to Atg5. The Atg5-
Atg12 conjugate is required for the proper function of the second conjugation
system, which covalently attaches Atg8/LC3 to phosphatidylethanolamine
(PE). The Atg5-Atg12 conjugate, a cysteine protease Atg4, the E1-like
enzyme Atg7, the E2-like enzyme Atg3 and PE-containing liposomes are

101
sufficient for LC3 conjugation. In cells, another factor is required called Atg16
(Kuma et al., 2002).
Atg16 forms a complex with Atg5-Atg12 (called the Atg16 complex) and this
complex is thought to function as an E3-like enzyme, directing LC3-PE to the
correct membrane location (Fujita et al., 2008). This is consistent with the
observation that the Atg16 complex is observed on the isolation membranes
before LC3-PE. However, the Atg16 complex is not present on completed
autophagosomes, suggesting it is released before fusion of the expanding
double membrane or quickly thereafter. This has led some to propose that the
Atg5-Atg12:Atg16 complex might act as a type of coat complex similar to
those seen in conventional membrane trafficking.
Currently, Atg16L1 (mammalian Atg16) is the only identified effector of
Rab33B, a Golgi-localized small GTPase that has a role in retrograde
trafficking from the Golgi to the endoplasmic reticulum (Itoh et al., 2008;
Valsdottir et al., 2001). Overexpression of GTP-bound Rab33B leads to an
increase of the lipidated form of LC3, suggesting that Rab33B has a role in the
formation and expansion of autophagosomes through its interaction with
Atg16L1 (Itoh et al., 2008).

102
Autophagy is a highly conserved process, but many mammalian autophagy
proteins have additional domains absent from their yeast counterparts. In
mammalian cells, Atg16L1 forms a complex with Atg5-Atg12 just as in yeast
(Mizushima et al., 2003). Yeast Atg16 has an N-terminal Atg5 binding domain
and a C-terminal coiled-coil domain that is required for dimerization of the
complex. In contrast, mammalian Atg16L1 also has an array of seven tandem
WD40 domain repeats at its C-terminus (Fig. 1; Mizushima et al., 2003).
These repeats are not required for proper canonical autophagy in mammalian
cells (Fujita et al., 2009).
Here we show that RUTBC1, a GTPase-activating protein (GAP) for Rab33B,
also interacts with Atg16L1. Depletion of RUTBC1 from cells causes a
concomitant depletion of Atg16L1, consistent with their presence in a complex
in cells. These proteins interact in cells, and overexpression of both proteins
causes them to co-localize to large cytoplasmic puncta under nutrient rich
conditions. Taken together, these data suggest a model whereby RUTBC1
binds to Atg16L1, recruiting RUTBC1 to a Rab33B microdomain.

103
METHODS
Plasmids
For mammalian expression, N-terminally 3xmyc-tagged RUTBC1 was
obtained by amplification from a cDNA library and ligation into a modified
version of pCDNA3.1(+) (Invitrogen) (Fuchs et al., 2007). This construct
encodes the shorter of two isoforms found in GenBank. GFP-RUTBC1 was
constructed by amplification of this isoform by PCR and ligated into pEGFP-C1
(Clontech). The predicted RUTBC1 GAP-deficient mutant (R803A) was also
created using QuikChange (Stratagene). GFP-Rab33B was constructed by
amplification of Rab33B from His-Rab33BQ92A (Hayes et al., 2009) and
mutated back to wild-type using Quikchange. Myc-Atg16L1, encoding
Atg16L1 with an N-terminal, triple myc tag was a kind gift from Dr. Ramnik
Xavier.
Antibodies
Mouse monoclonal anti-myc (9E10) was previously described (Ganley et al.,
2008). Rabbit anti-GFP antibody was from Invitrogen; mouse anti-GFP
antibody was from Roche. Antibodies to RUTBC1 were produced in rabbits
(Josman, LLC) using the purified His-RUTBC1-C as antigen. Affinity
purification was carried out as described previously (Ganley et al., 2004).
Rabbit anti-Atg16L1 was from Abcam. HRP-conjugated goat anti-mouse and

104
goat anti-rabbit secondary antibodies as well as protein-A-HRP were from Bio-
Rad. Alexa fluor-conjugated secondary antibodies were from Invitrogen.
Cell Culture and Transfections
HeLa and COS-1 cells were obtained from American Type Culture Collection
and cultured at 37°C and 5% CO2 in Dulbecco's modified Eagle's medium
supplemented with 7.5% fetal calf serum, 100U penicillin and 100µg/mL
streptomycin. For over-expression studies, all cells were transfected using
Fugene 6 (Roche). For siRNA transfections, HeLa cells were transfected
using DharmaFECT1 (Thermo Dharmacon). Cells were harvested for
immunblot analysis or processed for immunofluorescence at 24, 48 or 72
hours post transfection as indicated. The targeting sequence for RUTBC1
siRNA was 5ʼ-GGACGCAGTCAAAGAGAAA-3ʼ.
Immunopreciptations
For immunoprecipitations, cells were transfected with indicated plasmids for
24 hours. Cells were then lysed in 50mM Tris-HCl, pH 7.4, 150mM NaCl,
1mM MgCl2 and 1% Triton X-100 supplemented with Complete EDTA-free
Protease Inhibitor Cocktail (Roche) and spun for 15 minutes at full speed in a
microfuge at 4°C. Supernatants were pre-cleared using Protein-A agarose
(Roche) for 15 minutes at room temperature and then immunoprecipitated with
indicated antibodies for 1.5 hours at room temperature. Immune complexes

105
were isolated on Protein-A agarose for 30 minutes at room temperature. The
resin was then washed 3 times with lysis buffer and once with PBS. The
beads were resuspended in 2X sample buffer and boiled and analyzed by
immunoblot.
Immunofluorescence Microscopy
Cells were transfected while attached to 22x22mm coverslips in a six-well
plate. After the indicated incubation time, cells were washed twice in
phosphate-buffered saline (PBS) and fixed for 20 min in 3.7% formaldehyde in
200 mM HEPES, pH 7.4. To quench fixation, cells were washed twice and
incubated for 15 min in DMEM and 10 mM HEPES, pH 7.4. Cells were
permeabilized and blocked for 5 min with 0.2% Triton X-100 in PBS, followed
by two washes and a 15-min incubation with 1% BSA in PBS. Cells were
incubated with primary antibody, diluted 1:500 (anti-Atg16L1) or undiluted
(anti-myc culture supernatant) in BSA/PBS for 30 min. This was followed by
three 5-min washes and a 30-min incubation in secondary antibody diluted
1:1000 in BSA/PBS. Micrographs were acquired using a microscope
(Axiophot2, Zeiss) fitted with a 63x/numerical aperture (NA) 1.25 Zeiss
NeoFluar objective lens and a charge-coupled device camera (Orca-R2;
Hamamatsu) and controlled by Zeiss Axiophot software. Pictures were
analyzed using Adobe Photoshop software.

106
RESULTS
We have shown previously that RUTBC1 is a Rab9 effector that displays GAP
activity on Rab33B and Rab32 in vitro. In order to characterize the functional
significance of RUTBC1's GAP activity, we investigated the ability of RUTBC1
to act as a GAP for these Rabs in cells. GAPs regulate the lifetime of active,
GTP-bound Rabs. Overexpression of a GAP in cells should decrease the
amount of GTP-bound Rab and thus lead to a shift in the equilibrium between
a Rab and its effectors. Effector proteins bind preferentially to GTP-bound
Rabs; this allows effectors to act as biosensors of the amount of GTP-bound
Rabs in cells. One predicted consequence of GAP overexpression would be
solubilization of membrane-localized effectors due to conversion of their
cognate Rab-GTP substrate(s) to RabGDP. Presumably, this would also lead
to solubilization of the Rab itself by subsequent GDI-mediated extraction.
Overexpression of GFP-Rab33B in HeLa cells recruited endogenous Atg16L1
to the Golgi apparatus (Fig. 2A) consistent with previous findings in NIH3T3
cells (Itoh et al., 2008). Since Atg16L1 is a direct effector of Rab33B,
overexpression of RUTBC1 might disrupt the Golgi-associated Atg16L1. As
shown in Figure 2B, overexpression of myc-tagged RUTBC1 dissociated
Atg16L1 from the Golgi. This suggests that RUTBC1 does indeed function as
a GAP for Rab33B in cells.

107
We next investigated the consequences of depleting RUTBC1 in cultured cells.
According to the GenBank database, the RUTBC1 gene encodes two isoforms
that differ by the inclusion of one exon in the linker region between the RUN
and TBC domains (Fig. 1). Our siRNA was designed to target the 5ʼ end of
the RUTBC1 ORF to deplete both isoforms. Amplification of RUTBC1 from a
HeLa cDNA library indicated that only isoform 2 (the shorter isoform) appeared
to be expressed in these cells (data not shown). HeLa cells were mock
transfected or transfected with RUTBC1 siRNA and further incubated for 72
hours. As shown in Fig. 3A and B, RUTBC1 was efficiently silenced upon
siRNA treatment (~95%). Levels of Cog3, a subunit of the COG tethering
complex, were unchanged and used as a loading control. To our surprise,
Atg16L1 levels also decreased (Fig. 3A). The Atg16L1 gene encodes three
splice variants. The alpha and beta isoforms are both expressed in HeLa cells
(REF). The larger beta isoform was depleted approximately 40%, while the
alpha isoform was depleted by almost 70% (Fig. 3B). All three isoforms can
form the oligomeric Atg16L1 complex, thus the functional significance of the
different isoforms remains unclear.
Concomitant depletion of Atg16L1 with RUTBC1 strongly suggests that these
two proteins interact in cells. We tested this directly by co-
immunoprecipitation analysis. Myc-Atg16L1 co-immunoprecipitated with GFP-
RUTBC1 and not with GFP (Fig. 3C). Furthermore, myc-Atg16L1 was also

108
immunoprecipitated with GFP-RUTBC1 R803A, a GAP-deficient mutant. In
the reverse experiment, GFP-RUTBC1 was co-immunoprecipitated with myc-
Atg16L1 (data not shown). Thus, RUTBC1 interacts with Atg16L1, and this
interaction is independent of its GAP activity.
The immunoprecipitation results led us to investigate whether RUTBC1 and
Atg16L1 colocalize in cells. Atg16L1 is usually cytosolic, but upon stimulation
of autophagy, it is recruited to the phagophore. Overexpression of Atg16L1
caused the formation of large puncta throughout the cytoplasm (Fig. 4A,
middle cell). GFP-RUTBC1 also co-localizes to these puncta, the size of
which seemed to correlate with RUTBC1 expression levels (Fig4A, compare
upper and lower panel). Interestingly, GFP-Rab33B also co-localized to
puncta labeled by Atg16L1 (Fig. 4B). These puncta were somewhat larger in
size than the RUTBC1-positive structures. It is currently unclear if RUTBC1-
positive structures are the equivalent to those labeled by Rab33B.
Alternatively, both kinds of structures could represent intermediates in
maturation of a compartment.

109
DISCUSSION
Here we show that RUTBC1 and a Rab33B-effector, Atg16L1, can interact in
cells. RUTBC1 displays GAP activity on Rab33B in vitro. It also appears to
have GAP activity in cells as shown by its ability to solubilize Rab33B-recruited
Atg16L1 from the Golgi. In co-immunoprecipitation experiments using
Atg16L1 as a biosensor for the level of Rab33B-GTP in cells, we showed that
both wild-type and the GAP-deficient mutant of RUTBC1 decreased the
amount of Rab33B co-immunoprecipitating with Atg16L1 (data not shown).
RUTBC1 may be recruited to a Rab33B microdomain containing Atg16L1,
displacing Atg16L1 from Rab33B in a sort of GAP “invasion” of a microdomain,
perhaps by indirectly competing for the Rab33B binding site on Atg16L1. This
interaction would also lead to inactivation of Rab33B by stimulation of GTP
hydrolysis. This model could be tested in vitro by adding wild-type and GAP-
deficient RUTBC1 to Rab33B-loaded liposomes bearing Atg16L1. Both
proteins would release Atg16L1 from membranes, but only addition of wild-
type RUTBC1 would lead to extraction of Rab33B from membranes by
addition of GDI. Alternatively, we could use Golgi enriched fractions from cells
expressing tagged Atg16L1 and Rab33B proteins.
The observation that depletion of RUTBC1 also leads to partial co-depletion of
Atg16L1 suggests that RUTBC1 is required for the stability of Atg16L1.
Indeed, the two proteins interact in cells as shown by co-immunoprecipitation

110
experiments. This could be through formation of a stable complex or by
RUTBC1-mediated regulation of Atg16L1 degradation.
Most Atg16L1 is found in a high molecular weight complex composed of the
Atg5-Atg12 conjugate and Atg16L1 (~800kDa; Mizushima et al., 2003). That
work also showed that a protein of 144kDa was also present in purified
Atg16L1 complex from mammalian cells but its identity was not reported.
RUTBC1 has a predicted molecular weight of ~118kDa and apparent
molecular weight of ~125kDa by SDS-PAGE. Gel filtration of bovine brain
cytosol revealed that RUTBC1 is in a large complex (~450kDa; data not
shown). Future experiments will characterize the composition of this complex.
Finally, Atg16L1 appears to be degraded by the ubiquitin proteasome system,
at least when overexpressed (Fujita et al., 2009). How RUTBC1 might
influence Atg16L1 stability will be another area for future work.

111
REFERENCES
Eskelinen. Maturation of autophagic vacuoles in Mammalian cells. Autophagy
(2005) vol. 1 (1) pp. 1-10.
Fuchs et al. Specific Rab GTPase-activating proteins define the Shiga toxin
and epidermal growth factor uptake pathways. J Cell Biol (2007) vol. 177 (6)
pp. 1133-43.
Fujita et al. Differential involvement of Atg16L1 in Crohn disease and
canonical autophagy: analysis of the organization of the Atg16L1 complex in
fibroblasts. J Biol Chem (2009) vol. 284 (47) pp. 32602-9.
Fujita et al. The Atg16L complex specifies the site of LC3 lipidation for
membrane biogenesis in autophagy. Mol Biol Cell (2008) vol. 19 (5) pp. 2092-
100.
Ganley et al. A syntaxin 10-SNARE complex distinguishes two distinct
transport routes from endosomes to the trans-Golgi in human cells. J Cell Biol
(2008) vol. 180 (1) pp. 159-72.
Ganley et al. Rab9 GTPase regulates late endosome size and requires
effector interaction for its stability. Mol Biol Cell (2004) vol. 15 (12) pp. 5420-
30.
Glick et al. Autophagy: cellular and molecular mechanisms. The Journal of
pathology (2010) pp.

112
Hayes et al. Multiple Rab GTPase binding sites in GCC185 suggest a model
for vesicle tethering at the trans-Golgi. Mol Biol Cell (2009) vol. 20 (1) pp. 209-
17.
Itakura et al. Beclin 1 forms two distinct phosphatidylinositol 3-kinase
complexes with mammalian Atg14 and UVRAG. Mol Biol Cell (2008) vol. 19
(12) pp. 5360-72.
Itoh et al. Golgi-resident small GTPase Rab33B interacts with Atg16L and
modulates autophagosome formation. Mol Biol Cell (2008) vol. 19 (7) pp.
2916-25.
Klionsky. Autophagy: from phenomenology to molecular understanding in less
than a decade. Nat Rev Mol Cell Biol (2007) vol. 8 (11) pp. 931-7.
Kuma et al. Formation of the approximately 350-kDa Apg12-Apg5.Apg16
multimeric complex, mediated by Apg16 oligomerization, is essential for
autophagy in yeast. J Biol Chem (2002) vol. 277 (21) pp. 18619-25.
Levine and Klionsky. Development by self-digestion: molecular mechanisms
and biological functions of autophagy. Dev Cell (2004) vol. 6 (4) pp. 463-77.
Mizushima et al. Methods in Mammalian Autophagy Research. Cell (2010) vol.
140 (3) pp. 313-326.
Mizushima et al. Autophagy fights disease through cellular self-digestion.
Nature (2008) vol. 451 (7182) pp. 1069-75.

113
Mizushima et al. Mouse Apg16L, a novel WD-repeat protein, targets to the
autophagic isolation membrane with the Apg12-Apg5 conjugate. J Cell Sci
(2003) vol. 116 (Pt 9) pp. 1679-88.
Valsdottir et al. Identification of rabaptin-5, rabex-5, and GM130 as putative
effectors of rab33b, a regulator of retrograde traffic between the Golgi
apparatus and ER. FEBS Lett (2001) vol. 508 (2) pp. 201-9.
Yorimitsu and Klionsky. Autophagy: molecular machinery for self-eating. Cell
Death Differ (2005) vol. 12 Suppl 2 pp. 1542-52.

114
FIGURE LEGENDS
Figure 1: Domain architecture of RUTBC1 and Atg16L1. RUTBC1
contains an N-terminal RUN domain (RPIP8/Unc-14/NESCA) and a C-terminal
TBC domain (Tre2/Bub2/Cdc16). Atg16L1 contains an N-terminal Atg5-
binding domain (BD) and a middle coiled-coil domain (CC). Mammalian
Atg16L1 also has a C-terminal WD40 repeat domain.
Figure 2: RUTBC1 solubilizes Atg16L1 from the Golgi. HeLa cells were
transfected with GFP-Rab33B and myc-RUTBC1 for 48 hours. Cells were
stained with rabbit anti-Atg16L1 antibodies and mouse anti-myc antibodies
followed by goat anti-rabbit-Alexa568 and goat anti-mouse-Alexa647
secondary antibodies. Scale bars represents 5µm.
Figure 3: RUTBC1 and Atg16L1 interact in cells. (A) HeLa cells were either
mock transfected or transfected with siRNA targeted to RUTBC1 and
incubated for 72 hours. Extracts were blotted with affinity purified RUTBC1
antibodies and anti-Atg16L1 antibodies. Cog3 was used as a loading control.
Asterisks indicate non-specific bands. (B) Quantitation of data presented in A.
Error bars represent standard deviation. (C) COS-1 cells transfected with
GFP-RUTBC1, GFP-RUTBC1 R803A and myc-Atg16L1 as indicated for 24
hours. Extracts were immunoprecipitated with anti-GFP antibodies and

115
immune complexes were analyzed by immunoblot. Immunoprecipitated GFP-
RUTBC1 and GFP were detected by Ponceau S stain.
Figure 4: RUTBC1 localizes to Atg16L1-positive puncta. (A) HeLa cells
were transfected with GFP-Rab33B and myc-Atg16L1 for 24 hours. Cells
were stained with mouse anti-myc antibodies and Alexa 594-conjugated goat
anti-mouse secondary antibodies. (B) HeLa cells were transfected with myc-
Atg16L1 and GFP-RUTBC1 for 24 hours. Cells were stained as in A. Scale
bars represents 5µm.

116
Figure 1

117
Figure 2

118
Figure 3

119
Figure 3 continued

120
Figure 4

121
Figure 4 continued

122
CHAPTER 4
CHARACTERIZATION OF RAB SUBSTRATES
AND BINDING PARTNERS OF RUTBC2
(Manuscript in preparation)
Ryan M. Nottingham, Ian G. Ganley, Francis A. Barr,
David G. Lambright and Suzanne R. Pfeffer
Contributions: RMN contributed Fig. 1b, 2, 3a-c, 4 and 5. IGG contributed
Fig.3d. FAB contributed Fig. 1a and plasmids. DGL contributed plasmids and
reagents. RMN and SRP conceived the project and wrote the paper

123
ABSTRACT
Rab GTPases regulate vesicle budding, motility, docking and fusion. In cells,
their cycling between active, GTP-bound states and inactive, GDP-bound
states is regulated by the action of opposing enzymes called guanine
nucleotide exchange factors and GTPase-activating proteins (GAPs). The
substrates for most RabGAPs are unknown and the potential for cross talk
between different membrane trafficking pathways remains uncharted territory.
Rab9 and its effectors regulate recycling of mannose 6-phosphate receptors
from late endosomes to the trans Golgi network. We show here that RUTBC2
is a TBC domain-containing protein that binds to Rab9 specifically both in vitro
and in cultured cells but is not a GAP for Rab9. Biochemical screening of
RUTBC1ʼs Rab protein substrates revealed highest GAP activity toward
Rab34 and Rab36, both Golgi-localized Rabs. These data suggest a model in
which RUTBC2 somehow links Rab9 function to Rab34/36 function at the
Golgi. This cross-talk between Rab domains suggests the existence of a Rab
cascade between endosomes and the Golgi.

124
INTRODUCTION
Rab GTPases are master regulators of membrane trafficking. They catalyze
the formation of functional membrane microdomains by recruiting effectors to
membranes while in their GTP-bound states. These effectors help Rabs
regulate every step of a trafficking event.
RUTBC2 is a putative Rab GTPase-activating protein (GAP) conserved in C.
elegans, Drosophila and vertebrates. The primary sequence reveals the
presence of two domains, an N-terminal RPIP8/Unc-14/NESCA (RUN) domain
and a C-terminal Tre-2/Bub2/Cdc16 (TBC) domain. RUN domains are thought
to function as protein-protein interaction domains. The TBC domain
possesses RabGAP activity and is conserved from yeast to man. There are
approximately 70 Rabs encoded by the human genome and likely ~40
expressed in any given cell. There are at least 40 human RabGAPs,
suggesting that RabGAPs are highly specific for their Rab protein substrates.
Little is known about either the biochemistry or cell biology of RUTBC2. It was
first detected in purified lysosomal membranes (Ausseil et al., 2006). The
original goal of that work was to find the causative agent of
mucopolysaccharidosis IIIC, a disease with defective lysosomal enzyme
activity. Later, Shimizu and co-workers analyzed the tissue distribution of both
the human and mouse transcripts by Northern blot. They found that RUTBC2

125
has a more restricted expression pattern than the highly related RUTBC1 or
RUTBC3 and that RUTBC2 was highly enriched in mouse brain (Yang et al.,
2007). Furthermore, RUTBC2 localized at steady-state to the trans Golgi
network in mouse neuroblastoma cells (Yang et al., 2007). RUTBC2 co-
immunoprecipitates with Rap1a/b and Rap2a/b via a region just C-terminal to
the RUN domain and with multiple Rab GTPases (Yang et al., 2007). This
suggests a link between Rap- and Rab-mediated signaling in cells but the
current Rab substrates of the RUTBC2 TBC domain are unknown.
Only one interacting partner of RUTBC2 has been identified. Nurr1 is an
orphan nuclear receptor required for the development of dopaminergic
neurons in the mouse brain. Co-expression of RUTBC2 and Nurr1 increased
transcription of endogenous tyrosine hydroxylase, part of the dopamine
synthesis pathway (Luo et al., 2008). Moreover, suppression of RUTBC2 by
siRNA led to decreased cell division and decreased expression of the
dopamine transporter, a Nurr1 target gene (Luo et al., 2008). This has led to
speculation that RUTBC2 may be important in the pathogenesis of Parkinsonʼs
disease, which is known to be influenced by Nurr1 (Federoff, 2009).
Rab9 GTPase is required for the recycling of mannose 6-phosphate receptors
from late endosomes to the trans Golgi network (Lombardi et al., 1993;
Barbero et al., 2002). It also plays a role in lysosome biogenesis (Riederer et

126
al., 1994) and late endosome morphology (Ganley et al, 2004). In this study
we report that RUTBC2 is a specific effector of Rab9. Rab9 binds RUTBC2 in
the linker region between the RUN and TBC domains. The RUTBC2 TBC
domain does not possess GAP activity on Rab9 but instead shows GAP
activity toward highly-related Rab34 and Rab36 proteins.

127
METHODS
Yeast Two-Hybrid Analysis
Yeast two-hybrid analysis was carried out as described previously (Fuchs et
al., 2007). Briefly, 56 mutant Rab proteins deficient for GTP hydrolysis (Q to
A) were cloned into pGBT9 bait vector (Clontech). RUTBC2 was amplified
from cDNA libraries and was cloned into pACT2 prey vector (Clontech); growth
on selective media indicated an interaction between a Rab and RUTBC2.
Plasmids
For mammalian expression, N-terminally 3Xmyc-tagged RUTBC2 was
obtained by amplification from a cDNA library and ligation into a modified
version of pCDNA3.1(+) (Invitrogen) (Fuchs et al., 2007). This construct
encodes the shortest of four isoforms found in GenBank and is missing the
first 25 amino acids at the N-terminus. GFP-RUTBC2 was constructed by
amplification of this isoform with the addition of the missing N-terminus by
PCR and ligated into pEGFP-C3 (Clontech). The predicted RUTBC2 GAP-
inactive mutant (R829A) was created using QuikChange.
For bacterial expression, RUTBC2-C was ligated into pET28a (Novagen) in
frame with the N-terminal His-tag. A plasmid encoding GST-RUTBC2 was
constructed by ligation of RUTBC2 into pGEX-6P-1 (GE Healthcare).
Untagged Rab9CLLL and GST-Rab9A were previously described (Aivazian et

128
al, 2006). GST-Rab9B was amplified by PCR from pET14-Rab9B (Hayes et
al., 2009) and ligated into pGEX-4T-1. GST-Rab6A was amplified by PCR
from His-Rab6A (Burguete et al., 2008) and ligated into pGEX-4T-1. GST-
C110 and GST-C123 were previously described (Reddy et al., 2006). His-p40
was previously described (Diaz et al., 1997). HisGFP was made by amplifying
GFP from pEGFP-C1 (Clontech) and ligation into pET14b (Novagen).
Rab proteins tested for GAP activity were previously described (Pan et al.,
2006). Phosphate Binding Protein (PBP) from E. coli was amplified by PCR
from bacteria and cloned into modified pET15. His-PBP A197C was
constructed by site-directed mutagenesis.
Protein Expression and Purification
His-RUTBC2-C was transformed into Rosetta2 (DE3) cells (Novagen) and
grown at 37°C until OD600 = 0.5. The cells were induced with 0.4mM isopropyl
β-D-thiogalactoside (IPTG) and grown for an additional 4 hours at 22°C. Cell
pellets were resuspended in lysis buffer (25mM HEPES, pH 7.4, 300mM NaCl,
50mM imidazole) supplemented with 1mM PMSF and lysed by two passes at
20,000 psi through an EmulsiFlex-C5 apparatus (Avestin). Cleared lysates
(20,000 rpm for 45 min at 4°C in a JA-20 rotor; Beckman Coulter) were
incubated with Ni-NTA (Qiagen) for 1 hour at 4°C. The resin was then washed
with lysis buffer and eluted with 25mM HEPES, pH7.4, 300mM NaCl and

129
250mM imidazole. Fractions containing RUTBC2-C were pooled and
concentrated using an Amicon Ultra concentrator (Millipore). The sample was
dialyzed to remove imidazole and then brought to 10%(v/v) glycerol. The
sample was aliquoted, snap frozen in liquid nitrogen and stored at -80°C.
GST-RUTBC2 was transformed into Rosetta2 (DE3) cells and grown at 37°C
until OD600 = 0.6. The cells were induced with 100µM IPTG and grown for an
additional 4.5 hours at 30°C. Cell pellets were resuspended in lysis buffer
(50mM HEPES, pH 7.4, 250mM NaCl, 1mM DTT, 1mM EDTA) supplemented
with 1mM PMSF and lysed by two passes at 20,000 psi through an
EmulsiFlex-C5. Cleared lysates (19,000 rpm for 30 min at 4°C in a JA-20
rotor; Beckman Coulter) were incubated with Glutathione-Sepharose FastFlow
(GE Healthcare) for 1.5 hours at 4°C. The resin was then washed with 25
column volumes lysis buffer and eluted with 50mM Tris-HCl, pH8.0, 250mM
NaCl and 20mM glutathione. Fractions containing RUTBC2-C were pooled
and concentrated using an Amicon Ultra concentrator (Millipore). The sample
was dialyzed to remove glutathione and brought to 10%(v/v) glycerol, snap
frozen in liquid nitrogen and stored at -80°C.
Untagged Rab9CLLL and GST-Rab9A expression and purification were
previously described (Aivazian et al., 2006) and GST-Rab9B and GST-Rab6A
were purified by the same procedure. His-p40 purification was previously
described (Diaz et al., 1997). His-GFP was purified using the Qiaexpressionist

130
Kit (Qiagen) according to the manufacturer. Expression and purification of
GST-C110 and GST-C123 were previously described (Reddy et al., 2006).
Expression and purification of Rab proteins for the GAP screen were
previously described (Pan et al., 2006). His-PBP A197C was purified and
labeled according to the method described (Shutes and Der, 2005).
Antibodies
Mouse monoclonal anti-myc (9E10), mouse monoclonal anti-Rab9A, mouse
monoclonal anti-CI-MPR (2G11) and rabbit anti-CI-MPR, were all previously
described (Ganley et al., 2008). Rabbit anti-GFP antibody was from
Invitrogen; mouse anti-GFP antibody was from Roche. Rabbit anti-RUTBC2
was purchased from Sigma. HRP-conjugated goat anti-mouse and goat anti-
rabbit secondary antibodies as well as protein-A-HRP were from Bio-Rad.
Binding assays
Constructs encoding 3xmyc-RUTBC2 were translated in vitro using a TNT
Quick Coupled Transcription/Translation System (Promega) following the
manufacturerʼs protocol. GST-tagged Rabs were loaded with GTPγS or GDP
as described (Aivazian et al., 2006) and mixed with TNT lysate for 1.5 hours at
25ºC in binding buffer (25mM HEPES-NaOH, pH7.4, 150mM NaCl, 5mM
MgCl2, 1mM DTT, 0.1mM GTPγS). RUTBC1 constructs bound to GST-Rabs
were isolated using glutathione-Sepharose, washed in binding buffer (with

131
400mM NaCl) and then eluted by addition of 25mM glutathione and analyzed
by immunoblot. Binding assays using purified proteins were as described
(Hayes et al., 2009).
Cell Culture and Transfections
HeLa, HEK293T, SK-N-SH and Vero cells were obtained from American Type
Culture Collection and cultured at 37°C and 5% CO2 in Dulbecco's modified
Eagle's medium supplemented with 7.5% fetal calf serum, 100U penicillin and
100µg/mL streptomycin. For overexpression studies, all cells were transfected
using Fugene 6 (Roche). Cells were harvested either 24 or 48 hours after
transfected as indicated. For siRNA treatment, HEK293T cells were
transfected using Lipodectamine 2000 (Invitrogen).
Immunopreciptation, Protein Turnover, Lysosomal Enzyme Secretion and
Fractionation
For immunoprecipitation, cells were transfected with indicated plasmids for 24
hours. Cells were then lysed in 50mM Tris-HCl, pH 7.4, 150mM NaCl, 1mM
MgCl2 and 1% Triton X-100 supplemented with Complete EDTA-free Protease
Inhibitor Cocktail (Roche) and spun for 15 minutes at full speed in a microfuge
at 4°C. Supernatants were pre-cleared using Protein-A agarose (Roche) for 15
minutes at room temperature and then immunoprecipitated with indicated
antibodies for 1.5 hours at room temperature. Immune complexes were

132
isolated on Protein-A agarose for 30 minutes at room temperature. The resin
was then washed 3 times with lysis buffer and once with PBS. Beads were
resuspended in 2X sample buffer boiled and analyzed by immunoblot. Protein
turnover and lysosomal enzyme secretion assays were performed as
described (Ganley et al., 2008). Cell fractionation was as previously described
(Ganley et al., 2004) except SK-N-SH cells were swollen in 10mM HEPES
pH7.4 for 5 minutes.
GAP Assays
For the biochemical screen of GAP substrates, the procedure followed by Pan
et al. (2006) was generally used with the exception that phosphate released
during the reaction was bound by modified PBP (A197C) labeled at position
197 with N-[2-(1-maleimidyl)ethyl]-7-(diethylamino)coumarin-3-carboxamide
(MDCC). Phosphate binding to MDCC-PBP causes a conformational change
in the phosphate binding cleft that results in an increase in MDCC
fluorescence (Brune et al., 1994). Reactions were started by adding a solution
containing GAP, MgCl2 and MDCC-PBP to desalted, GTP-exchanged Rabs by
a Precision 2000 liquid handling system (Biotek). Rab GTPases were at 2µM
for all reactions while the concentration of His-RUTBC2-C was varied.
Phosphate production was monitored by following fluorescence signal
continuously in a TECAN Saphire microplate reader using an excitation of
425nm and an emission cutoff filter of 455nm.

133
RESULTS
We are interested in the function and localization of the Rab9 GTPase. As a
starting point to find regulators of Rab9, we used a two-hybrid screen
consisting of all TBC domain-containing proteins in the human genome as
prey against a comprehensive library of hydrolysis-deficient Rab GTPases as
bait (Haas et al., 2005; Fuchs, et al., 2007). This screen revealed that a TBC
domain-containing protein, RUTBC2, interacted with both Rab9A and 9B
(Figure 1A). RUTBC2 also interacted to a lesser extent with Rab3 isoforms.
Like the closely-related RUTBC1, the TBC domain of RUTBC2 has a large
insertion between the first two “fingerprint” A and B motifs (Figure 1B,
sequence). In the structural model for Rab and RabGAP interaction, the
analogous region of Gyp1 is situated away from the Rab:GAP binding
interface (Pan et al., 2006). Most of the dissimilarity between RUTBC1 and
RUTBC2 in the TBC domain is found in this insertion. According to the NCBI
Homologene database, there is only one RUTBC1/2 protein in C. elegans (tbc-
8), Drosophila (CG1905) and zebrafish (LOC794373) while vertebrates have
both proteins.
RUTBC2 is a Rab9 effector
To confirm the results of the screen and to test the nature of the interactions,
we tested if Rab9 could bind RUTBC2 in vitro. Full-length RUTBC2 was

134
produced both in an in vitro transcription/translation system as well as in E.
coli, and assayed for binding to immobilized GST-Rabs. As shown in Figure
2A, GST-Rab9A but not GST-Rab9B or GST-Rab6A could bind in vitro
translated, full-length RUTBC2. These results seem contradictory to the
screen results but stronger Rab9B binding in two hybrid assays has been
observed before (Hayes et al., 2009). Next, we asked if RUTBC2 preferred
either the GTP or GDP-bound form of Rab9. Using the GST-binding assay
described above, in vitro translated RUTBC2 was bound much more efficiently
by GST-Rab9 when loaded with GTPγS than with GDP (Fig. 2B). Thus,
RUTBC2 is a bona fide effector of Rab9A (called Rab9 from now on).
We next investigated the location of the Rab9 binding site in RUTBC2. Using
purified proteins, we found that full-length GST-RUTBC2 bound as well to
Rab9 as the positive control, the GST-tagged, C-terminal 110 amino acids of
GCC185 (Reddy et al., 2006) (Fig. 2D, left panel). There was little Rab9
binding to the negative control, the GST-tagged, C-terminal 123 amino acids of
Golgin245. Rab9 bound poorly to the TBC domain alone (His-tagged
RUTBC2-C) but very well to a known Rab9 effector, His-p40 (Fig. 2D, right
panel). This suggests that the Rab9 binding site is likely located in the N-
terminus. An earlier two-hybrid screen for GAP-Rab interactions failed to
detect Rab9 binding to RUTBC2 (Itoh et al., 2006). This was likely due to the

135
use of constructs for both proteins that contained only the TBC domain.
Taken together, these data suggest that Rab9 binds directly to RUTBC2 in its
N-terminal region.
RUTBC1 interacts with Rab9 in cells
Rab9 regulates the recycling of mannose 6-phosphate receptors (MPRs) from
late endosomes to the trans Golgi network (TGN) (Lombardi et al., 1993;
Riederer et al., 1994). To ask whether Rab9 and RUTBC2 interact in cells,
HEK 293T cells were transfected with GFP-RUTBC2 or GFP as a control.
Endogenous Rab9 was immunoprecipitated with GFP-RUTBC2 and not GFP
(Figure 3A). Thus, RUTBC2 can interact with Rab9 in living cells.
We further characterized this interaction by investigating the effects of
exogenously expressed RUTBC2 on MPR recycling. When Rab9 function is
disturbed in cells by overexpression of a dominant negative mutant Rab9
(Riederer et al., 1994) or by depletion of its effectors (Reddy et al., 2006;
Espinosa et al., 2009), MPRs are mis-sorted to the lysosome. We reasoned
that if RUTBC2 had GAP activity on Rab9, the phenotype of RUTBC2
overexpression should be similar to that of a GDP-preferring Rab9 mutant or
Rab9 depletion. When RUTBC2 was overexpressed in HeLa cells, MPR
levels at steady state were decreased by ~40% (Figure 3B). This observation
led us to investigate the turnover rate of MPR in HeLa cells overexpressing

136
RUTBC1 and found it to be essentially unchanged (Figure 3C). This
suggested that though some amount of MPR was being mis-sorted to
lysosomes, there was likely an increase in MPR translation. As a possible
GAP for Rab9, the predicted GAP-deficient mutant (R829A) of RUTBC2
should not have the same effect when overexpressed.
In a functional test of MPR trafficking, we measured the amount of
hexosaminidase activity secreted into the media by cells transfected with
RUTBC2. Hexosaminidase is usually sorted to the lysosome but when MPR
levels are deficient in the TGN due to missorting events, the hydrolase is
secreted. In 293T cells transfected with RUTBC2, slightly higher
hexosaminidase activity was detected in the media than in control cells (Figure
3D). Cells transfected with RUTBC2 R829A showed a similar effect. Taken
together, perturbation of MPR trafficking is likely due to titration of Rab9 by the
overexpression of RUTBC2 rather than the GAP activity of this protein.
RUTBC2 has narrow substrate specificity
Discovering the Rab substrates of RUTBC2 should provide insight into the
significance of Rab9-RUTBC2 interaction. Thirty-two different mammalian
Rab GTPases were screened under single turnover conditions as substrates
for RUTBC2 using purified, His-tagged RUTBC2-C (Figure 2C) that contains
the catalytic TBC domain. Figure 4 summarizes these results by comparing
observed second order rate constants for GAP-catalyzed hydrolysis to the rate

137
constants for intrinsic hydrolysis by each Rab protein. The TBC domain of
RUTBC2 had the highest activity against Rab36 and Rab34 while no activity
was detected against Rab9 or Rab3. Both Rab36 and Rab34 are localized to
the Golgi (Wang and Hong, 2002; Chen et al., 2009).
RUTBC2 is enriched in neural cells
Analysis of different cell lines revealed that RUTBC2 is present in different
amounts in HEK293T, Vero and SK-N-SH cells (Fig 5A). SK-N-SH cells are a
human neuroblastoma cell line that might be similar to mouse Neuro2a
neuroblastoma cells used by Shimizu and co-workers in earlier RUTBC2
studies (Yang et al., 2007). RUTBC2 seems to be highly expressed in these
cells compared to kidney cells, consistent with that report. RUTBC2 could be
efficiently depleted by siRNA treatment of 293T cells (Fig. 5B). Quantitation
revealed an approximate ~90% level of depletion. RUTBC2 also stably
associated with membranes (Fig. 5C). Fractionation of SK-N-SH cells showed
that most of RUTBC2 was cytosolic; nevertheless, a small pool of RUTBC2
was stably associated with membranes. This is also consistent with the
steady-state localization observed previously (Yang et al., 2007).
Unfortunately, our antibody could not be used for immunofluorescence
microscopy to determine the specific membranes with which RUTBC2
associates. Together, these data suggest a model where RUTBC2 in some
way links late endosome and Golgi trafficking pathways.

138
DISCUSSION
Here we have shown that a predicted RabGAP protein, RUTBC2, is a novel
Rab9 effector. RUTBC2 does not display GAP activity on Rab9 but instead
has GAP activity for Rab36 and Rab34. RUTBC2 is present in many cell
types but is enriched in cells of neural origin. A small pool of RUTBC2 also
stably associates with membranes. Although Rab9B appeared to interact with
RUTBC2 in a two hybrid screen, only purified Rab9A bound directly to
RUTBC2. Binding to either Rab would be consistent with the observation that
murine RUTBC2 localizes to the trans Golgi network (Yang et al., 2007).
Rab9B is known to be on the Golgi while Rab9A regulates the transport of
MPRs from late endosomes to the TGN. It is not yet clear precisely where
Rab9A and RUTBC2 interact.
Currently there are no known direct links between Rab9-mediated trafficking
and either Rab36- or Rab34-regulated events. However, both Rab36 and
Rab34 are thought to mediate the localization of lysosomes (Wang and Hong,
2002; Chen et al., 2009), despite the fact that both Rabs localize to the Golgi.
GTP-restricted Rab34 or Rab36 cause LAMP1-positive lysosomes (and not
MPR-positive structures) to cluster in the perinuclear area (Wang and Hong,
2002; Chen et al., 2009; Goldenberg et al., 2007). This phenomenon is
mediated by RILP, an effector of both Rab34 and Rab36 as well as Rab7, a
Rab known to play a role in lysosomal biogenesis. A mutation in the switch I

139
region of Rab34 (K82Q) that blocks association with RILP fails to redistribute
lysosomes (Wang and Hong, 2002). Little else is known about Rab36 other
than it is deleted in many cases of malignant, rhabdoid tumors (Mori et al.,
1999).
Rab34 has also been suggested to play additional role in constitutive
secretion. It binds to hmunc-13, a diacylglycerol binding protein (Speight and
Silverman, 2005). Depletion of Rab34 and hmunc-13 from HeLa cells blocks
temperature sensitive Vesicular stomatitis virus glycoprotein (VSV-G) arrival at
the plasma membrane (Goldenberg et al., 2007; Goldenberg and Silverman,
2009). Brefeldin A treatment revealed that blocked VSV-G returned to the
endoplasmic reticulum, suggesting that Rab34 depletion causes a block in
intra-Golgi transport rather than exit from the trans Golgi network (Goldenberg
et al., 2007). A previous screen for the ability of overexpressed RabGAPs to
block VSV-G secretion in multiple cell types showed that RUTBC2 had no
effect on this process (Haas et al., 2007). This suggests that RUTBC2 may
only have activity on Rab36 in living cells.
There is also no known connection between Nurr1 and either Rab34 or Rab36.
One attractive, but highly speculative connection might be that either of these
Rabs are required for proper trafficking of the dopamine transporter. RUTBC2
might be able to integrate signals from the Golgi apparatus in regard to the

140
status of secretion to the Nurr1 transcriptional response. Interestingly, Rab34
has been shown to be a direct target of Hedgehog signaling (Vokes et al.,
2007). Hedgehog signaling helps mediate the specification of distinct cell
identities in the ventral neural tube through a Gli-mediated transcriptional
network, including dopaminergic neurons (Hynes et al., 2005). Experiments
elucidating the connections, if any, between these diverse signaling and
trafficking pathways could have potential impact on the pathogenesis and
treatment of Parkinson’s disease.

141
REFERENCES Aivazian et al. TIP47 is a key effector for Rab9 localization. J Cell Biol (2006)
vol. 173 (6) pp. 917-26.
Ausseil et al. An acetylated 120-kDa lysosomal transmembrane protein is
absent from mucopolysaccharidosis IIIC fibroblasts: a candidate molecule for
MPS IIIC. Mol Genet Metab (2006) vol. 87 (1) pp. 22-31.
Barbero et al. Visualization of Rab9-mediated vesicle transport from
endosomes to the trans-Golgi in living cells. J Cell Biol (2002) vol. 156 (3) pp.
511-8.
Brune et al. Direct, real-time measurement of rapid inorganic phosphate
release using a novel fluorescent probe and its application to actomyosin
subfragment 1 ATPase. Biochemistry (1994) vol. 33 (27) pp. 8262-71.
Burguete et al. Rab and Arl GTPase family members cooperate in the
localization of the golgin GCC185. Cell (2008) vol. 132 (2) pp. 286-98.
Chen et al. Rab36 regulates the spatial distribution of late endosomes and
lysosomes through a similar mechanism to Rab34. Mol Membr Biol (2010) vol.
27 (1) pp. 24-31.
Díaz et al. A novel Rab9 effector required for endosome-to-TGN transport. J
Cell Biol (1997) vol. 138 (2) pp. 283-90.
Espinosa et al. RhoBTB3: a Rho GTPase-family ATPase required for
endosome to Golgi transport. Cell (2009) vol. 137 (5) pp. 938-48.

142
Federoff. Nur(R1)turing a notion on the etiopathogenesis of Parkinson's
disease. Neurotoxicity research (2009) vol. 16 (3) pp. 261-70.
Fuchs et al. Specific Rab GTPase-activating proteins define the Shiga toxin
and epidermal growth factor uptake pathways. J Cell Biol (2007) vol. 177 (6)
pp. 1133-43.
Ganley et al. A syntaxin 10-SNARE complex distinguishes two distinct
transport routes from endosomes to the trans-Golgi in human cells. J Cell Biol
(2008) vol. 180 (1) pp. 159-72.
Ganley et al. Rab9 GTPase regulates late endosome size and requires
effector interaction for its stability. Mol Biol Cell (2004) vol. 15 (12) pp. 5420-
30.
Goldenberg and Silverman. Rab34 and its effector munc13-2 constitute a new
pathway modulating protein secretion in the cellular response to
hyperglycemia. Am J Physiol, Cell Physiol (2009) vol. 297 (4) pp. C1053-8.
Goldenberg et al. Golgi-bound Rab34 is a novel member of the secretory
pathway. Mol Biol Cell (2007) vol. 18 (12) pp. 4762-71.
Haas et al. Analysis of GTPase-activating proteins: Rab1 and Rab43 are key
Rabs required to maintain a functional Golgi complex in human cells. J Cell Sci
(2007) vol. 120 (Pt 17) pp. 2997-3010.
Haas et al. A GTPase-activating protein controls Rab5 function in endocytic
trafficking. Nat Cell Biol (2005) vol. 7 (9) pp. 887-93.

143
Hayes et al. Multiple Rab GTPase binding sites in GCC185 suggest a model
for vesicle tethering at the trans-Golgi. Mol Biol Cell (2009) vol. 20 (1) pp. 209-
17.
Hynes et al. Induction of midbrain dopaminergic neurons by Sonic hedgehog.
Neuron (1995) vol. 15 (1) pp. 35-44.
Itoh et al. Screening for target Rabs of TBC (Tre-2/Bub2/Cdc16) domain-
containing proteins based on their Rab-binding activity. Genes Cells (2006)
vol. 11 (9) pp. 1023-37.
Lombardi et al. Rab9 functions in transport between late endosomes and the
trans Golgi network. EMBO J (1993) vol. 12 (2) pp. 677-82.
Luo et al. Identification of a novel nurr1-interacting protein. J Neurosci (2008)
vol. 28 (37) pp. 9277-86.
Mori et al. Cloning and characterization of a novel Rab-family gene, Rab36,
within the region at 22q11.2 that is homozygously deleted in malignant
rhabdoid tumors. Biochem Biophys Res Commun (1999) vol. 254 (3) pp. 594-
600.
Pan et al. TBC-domain GAPs for Rab GTPases accelerate GTP hydrolysis by
a dual-finger mechanism. Nature (2006) vol. 442 (7100) pp. 303-6.
Reddy et al. A functional role for the GCC185 golgin in mannose 6-phosphate
receptor recycling. Mol Biol Cell (2006) vol. 17 (10) pp. 4353-63.

144
Riederer et al. Lysosome biogenesis requires Rab9 function and receptor
recycling from endosomes to the trans-Golgi network. J Cell Biol (1994) vol.
125 (3) pp. 573-82.
Shutes and Der. Real-time in vitro measurement of GTP hydrolysis. Methods
(2005) vol. 37 (2) pp. 183-89.
Speight and Silverman. Diacylglycerol-activated Hmunc13 serves as an
effector of the GTPase Rab34. Traffic (2005) vol. 6 (10) pp. 858-65.
Vokes et al. Genomic characterization of Gli-activator targets in sonic
hedgehog-mediated neural patterning. Development (2007) vol. 134 (10) pp.
1977-89.
Wang and Hong. Interorganellar regulation of lysosome positioning by the
Golgi apparatus through Rab34 interaction with Rab-interacting lysosomal
protein. Mol Biol Cell (2002) vol. 13 (12) pp. 4317-32.
Yang et al. Identification of three novel proteins (SGSM1, 2, 3) which modulate
small G protein (RAP and RAB)-mediated signaling pathway. Genomics
(2007) vol. 90 (2) pp. 249-60.

145
FIGURE LEGENDS
Figure 1: RUTBC2 interacts with Rab9. (A) RUTBC2 was screened against
a library of 56 human Rab GTPases. Growth after streaking on selective
media indicates an interaction between RUTBC2 and a Rab GTPase. (B)
Diagram of RUTBC2 domain structure. The RUN domain is shown in blue; the
TBC domain is shown in red. The first three conserved motifs of the TBC
domain are indicated. Conserved residues are in bold and the predicted
catalytic arginine and glutamine are shown in red, respectively. The extended
insert between motif A and motif B of RUTBC2 is compared to the same
regions of human RUTBC1 and RabGAP-5 (RUTBC3) and S. cerevisiae
Gyp1p.
Figure 2: RUTBC2 is an effector of Rab9. (A) In vitro translated (IVT)
3xmyc-RUTBC2 proteins were incubated with various bacterially purified,
GST-tagged Rabs pre-loaded with GTPγS. Bound material was eluted by
glutathione and half of the eluate was analyzed by immunoblot using anti-myc
antibodies. Rabs were detected by Ponceau S staining. (B) IVT-3xmyc-
RUTBC2 was incubated with GST-Rab9A pre-loaded with either GTPγS or
GDP and analyzed as in A. (C) Diagram of 3xmyc-tagged RUTBC1 constructs
used in binding assays. (D) Left panel: purified GST-RUTBC2 and control
proteins were incubated with Rab9A loaded with GTPγ[35S] and immobilized
using glutathione-Sepharose. Bound Rab was detected by scintillation

146
counting. Right panel: His-RUTBC2-C and control proteins were incubated
with Rab9A loaded with GTPγ[35S] and isolated using Ni-NTA. “Input”
indicates 1% of the IVT reaction used in the binding assay.
Figure 3: RUTBC2 binds to, but is not a GAP for Rab9A in cells. (A)
HEK293T cells were transfected with GFP-RUTBC2 or GFP for 24 hrs and
RUTBC2 was immunoprecipitated with rabbit anti-GFP antibodies followed by
immunoblotting with specific antibodies to Rab9A and GFP. “Input” represents
2% of the lysate subjected to immunoprecipitation. (B) Left panel - COS-1 cells
were transfected with 3xmyc-RUTBC2 for 48 hours and extracts were
immunoblotted with anti-CI-MPR antibodies. Right panel - Quantitation of CI-
MPR half-life measured from pulse/chase analysis of HeLa cells transfected
with 3xmyc-RUTBC2 for 48 hours. Extracts were immunoprecipitated with
anti-CI-MPR antibodies and exposed to a phosphor screen. (C) HEK293T
cells transfected with 3xmyc-RUTBC2 wild type or R829A for 24 hours were
assayed for secreted and intracellular hexosaminidase activity. Error bars in
all panels represent SEM from at least two independent experiments.
Figure 4: RUTBC2 TBC domain has GAP activity toward Rab36 and
Rab34 in vitro. Purified, mammalian Rab GTPases (32) were pre-loaded with
GTP for one hour at room temperature and then desalted to remove free
nucleotide. MDCC-PBP, MgCl2 and varying concentrations of purified His-

147
RUTBC2-C were added to start the reaction. Phosphate release was
monitored continuously by microplate fluorimeter (see Materials and Methods).
Catalytic efficiency (kcat/KM) relative to the intrinsic rate constant (kintr) for GTP
hydrolysis was determined. Plots represent the mean from duplicate wells.
Figure 5: RUTBC2 is stably associated with membranes in SK-N-SH
cells. (A) Detergent extracts (100µg) of indicated cell lines were resolved by
SDS-PAGE and immunoblotted using anti-RUTBC2 antibodies. (B) HEK293T
cells were either mock transfected or transfected with siRNA targeting
RUTBC2 for 72 hours. Shown is quantitation of the band corresponding to
RUTBC2. (C) SK-N-SH cells were fractionated into crude membranes and
cytosol. Increasing volumes of each fraction were analyzed by immunoblot
using RUTBC2 specific antibodies.

148
Figure 1
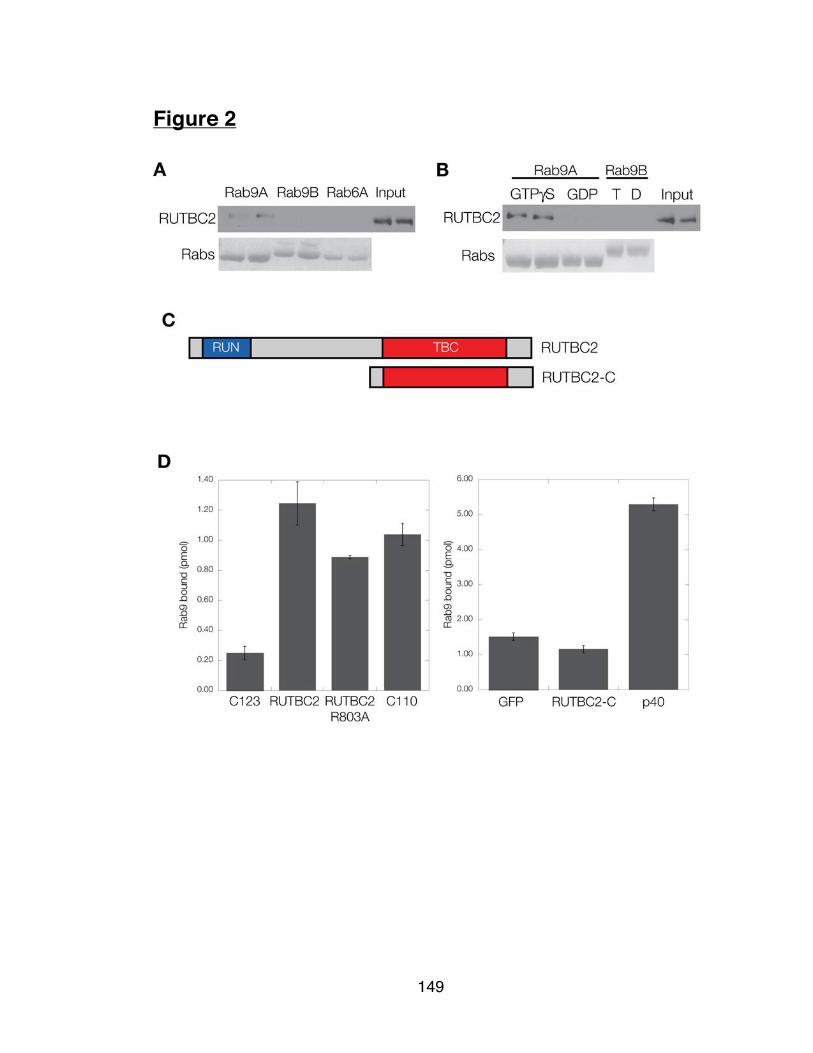
149
Figure 2

150
Figure 3

151
Figure 4

152
Figure 5

153
SUMMARY AND FUTURE PERSPECTIVES
Rab9 plays an essential role in the recycling of mannose 6-phosphate
receptors from late endosomes to the TGN, and it is also important for
lysosome biogenesis and late endosome morphology. Regulators of the Rab9
nucleotide binding cycle remain unknown, although it is likely that both a GEF
and GAP for Rab9 exist in cells. This thesis has added to our understanding
of Rab9 function in cells by providing new clues to links between different
trafficking pathways.
We have shown that two Rab GAP proteins, RUTBC1 and RUTBC2, are novel
Rab9 effectors that show preference for different Rab protein substrates for
their GAP activities. This implies that the substrate Rabs act in pathways that
feed into a Rab9-regulated pathway or organelle. Rab9 binding to RUTBC1 or
RUTBC2 might help to clear these Rabs from a Rab9 microdomain. It is now
imperative to investigate the significance of Rab9 interaction with these GAP
proteins. Does Rab9 simply localize these proteins transiently to late
endosomal membranes? Does Rab9 affect the catalytic activity of RUTBC1
and/or RUTBC2? Recent evidence suggests that binding of Rabs can
influence nucleotide hydrolysis activity directly (Rab9 and RhoBTB3: Espinosa
et al., 2009) or GEF activity (Rab11 and Rabin8: Knödler et al., 2010). The
latter case is especially germane to the proteins in this study because it
supports the Rab cascade model in a process (ciliagenesis) distinct from the

154
secretory pathway or the early-to-late endosome transition. It will thus be
important to test the ability of Rab9 to alter the catalytic activity of RUTBC1
and RUTBC2.
One of the more surprising findings in this thesis is the connection between
RUTBC1 and Atg16L1. Atg16L1 is required for conventional autophagy and
RUTBC1 depletion leads to concomitant depletion of Atg16L1. The most
important next goal must be to determine the effects of RUTBC1 depletion of
autophagosome formation and maturation. Rab9 does not have a
characterized role in autophagy but it would be reasonable to speculate that
mannose 6-phosphate receptor trafficking is important for autophagy, both for
maturation of the autophagosome into a late endosome-like structure and in its
canonical role delivering hydrolases to lysosomes.
Also of interest is our characterization of RUTBC2. Its enrichment in brain
tissue and connections to dopaminergic neuron biogenesis are important
justifications for further study. There is also no known connection between
Nurr1 (a reported binding partner for RUTBC2) and either Rab34 or Rab36.
One attractive, but highly speculative connection might be that either of these
Rabs are required for proper trafficking of the dopamine transporter. RUTBC2
might be able to integrate signals from the Golgi apparatus in regard to the
status of secretion to influence the Nurr1 transcriptional response.

155
Experiments elucidating the possible connections between these diverse
signaling and trafficking pathways could have potential impact on our
understanding of the fundamental pathogenesis of Parkinson’s disease.
References
Espinosa et al. RhoBTB3: a Rho GTPase-family ATPase required for
endosome to Golgi transport. Cell (2009) vol. 137 (5) pp. 938-48.
Knödler et al. Coordination of Rab8 and Rab11 in primary ciliogenesis.
Proceedings of the National Academy of Sciences (2010) vol. 107 (14) pp.
6346-51.





























