Embed Size (px)
Citation preview

427
Drug discovery is a prolonged process that uses a variety oftools from diverse fields. To accelerate the process, a number ofbiotechnologies, including genomics, proteomics and a numberof cellular and organismic methodologies, have beendeveloped. Proteomics development faces interdisciplinarychallenges, including both the traditional (biology andchemistry) and the emerging (high-throughput automation andbioinformatics). Emergent technologies include two-dimensionalgel electrophoresis, mass spectrometry, protein arrays, isotope-encoding, two-hybrid systems, information technologyand activity-based assays. These technologies, as part of thearsenal of proteomics techniques, are advancing the utility ofproteomics in the drug-discovery process.
AddressesActivX Biosciences, Inc., 11025 North Torrey Pines Road, Suite 120,La Jolla, CA 92037, USA*e-mail: [email protected]
Current Opinion in Chemical Biology 2002, 6:427–433
1367-5931/02/$ — see front matter© 2002 Elsevier Science Ltd. All rights reserved.
Published online 6 June 2002
Abbreviations2DGE two-dimensional gel electrophoresisABP activity-based probeESI electrospray ionizationICAT isotope-coded affinity taggingMALDI matrix-assisted laser desorption ionizationMudPIT multidimensional protein identification technologyPCR polymerase chain reaction
IntroductionThe drug-discovery process involves many phases, includingtarget identification, lead identification, small-moleculeoptimization, and pre-clinical/clinical development. Efficiencyin this process relies on timely knowledge of biologicalcause-and-effect in the course of disease and treatment,which ultimately rests on knowledge of protein functionand regulation. One of the key steps, target identification,has been fostered through applied genomics, primarilybecause both high-throughput methods and tools that allownucleic acid amplification have enabled large-scale profilingof expressed genes [1]. However, analysis of the informationproduced by genomics, when measured against comparableinformation regarding protein expression, has led to theconclusion that message abundance fails to correlate withprotein quantity [2]. Further, post-translational processessuch as protein modifications or protein degradation remainunaccounted for in genomic analysis [3–5]. Because bothcell function and its biochemical regulation depend on protein activity, and because the correlation between message level and protein activity is low, the measurementof expression has proven to be inadequate. Consequently,the development of drug-discovery technologies has begun
to shift from genomics to proteomics. This shift hasoccurred not only in target discovery but also in many otherareas of the process, including patient treatment and care[6]. This review focuses on the burgeoning field of proteomics as it applies to drug discovery, which relies uponthe determination of cellular function and regulationthrough large-scale measurement of protein function and interaction.
Proteomics techniquesProteomics, as a scientific field, is defined as the study ofthe protein products of the genome, and their interactionsand functions. Similarly, the proteins expressed at a giventime in a given environment constitute a proteome [7].From a technology viewpoint, traditional proteomicsinvolves separation of proteins in a proteome, coupled to ameans of identification. Until recently, the tools of choicewere two-dimensional gel electrophoresis (2DGE) for separation, and mass spectrometry (MS) for protein identification. However, 2DGE is limited because it failsto detect proteins at the extremes of separation either bysize or by isoelectric point, and because it is insufficientlysensitive for low-abundance proteins [8]. From the perspective of drug discovery, 2DGE fails in two importantways. First, 2DGE is ineffective for the separation of membrane proteins, which represent nearly 50% of importantdrug targets [9]. Second, low-abundance proteins areunder-represented in a 2DGE analysis, yet often representkey sites of biological regulation. Specifically, it has beenestimated that more than 50% of proteins in cells are of lowabundance [10,11•]. Therefore, although 2DGE is powerful,researchers wishing to apply proteomics to drug discoverymust seek innovative ways to measure both protein abundance and activity.
Proteomics presents researchers with a formidable challenge for a number of reasons. First, protein levels varywidely with both cell type and environment [12•]. Second,unlike genomics, which can amplifybenefits from theamplification of single genes using the polymerase chainreaction (PCR), protein science has no comparable ampli-fication method [13]. Third, proteomics is complicated bythe fact that the absolute quantity of protein is of limitedinterest to drug discovery, because protein activities arehighly regulated post-translationally [5]. Therefore, proteinscan be abundant, yet possess little activity. Finally, becauseproteins interact functionally in vivo, protein–protein andprotein–small-molecule interactions need to be evaluatedin processes of interest [14].
For drug discovery, the ideal proteomics method would beone that is:
1. Sensitive enough to detect low-abundance proteins.
Proteomics in drug discoveryJonathan Burbaum* and Gabriela M Tobal

2. Able to detect activity over in addition to abundance.
3. Able to detect protein–protein and protein–small-molecule interactions.
4. Easily implemented and performed quickly.
Research in proteomics seeks to satisfy all, or some, ofthese conditions by developing new methods to understand
428 Next-generation therapeutics
Table 1
Properties of various proteomics techniques.
Analyticaltechnique 2DGE MudPIT Protein chips 2-Hybrid systems ICAT ABPs
Measure-ment
Polypeptide chain size
Polypeptide chain size
Surface affinity Protein interaction
Relative abundance
Catalytic activity
potentialIsoelectric point Active site peptide
sequence
Abundance Polypeptide chain size
Identity MS MS MS DNA MS MSestablishedby:
Applications Target selection Target selection Target selection Target selection Target selection Target selectionin drug
Protein express- ion profile
Profiling for diagnostics
Drug screening Drug screening Profiling for diagnostics
Drug screening and pan selectivity
discovery
Profiling fordiagnostics
Profiling for diagnostics
Detection of protein– protein and protein–drug interactions
Pros Can detect 1000s of proteins at once
Can detect 1000s of proteins at once
Can detect 1000s of proteins at once
Can detect potential protein interactions
Circumvents the proteome coverage problems of 2DGE
Circumvents the proteome coverage problems of2DGE
Circumvents the Circumvents the Is easily Is easily Can detect 1000s of proteome coverage problems of
proteome coverage problems of
automated automated proteins at once
2DGE 2DGE Is easily automated
Is easily automated Results in cloned genes for all
Detects protein activity,rather than protein
proteins abundance interrogated
Cons Cannot detect proteins that are very small, large, acidic or basic, poorly soluble and of low abundance
Does not detect abundance, activity, or interactions
At a proteomic scale, will require the cloning of 100s of 1000s of proteins
False negatives and positives
Does not detectpost-translation-al modifications or interactions
Probes needed for all protein families, hence proteomic-scale coverage difficult to ascertain
Difficult to automate
Does not detect interactions in physiologically relevant situations
Limited to proteins localized in the nucleus

protein function and interactions in a biological context.Recent technological advances in this area include developments in separation and identification technologies(i.e. MS, protein-chip technologies, and phage display),bioinformatics, and technologies that detect protein interactions and activities (i.e. activity-based assays, andtwo-hybrid assays) (Table 1).
Mass spectrometryAdvances in MS have allowed the rapid sequencing of proteins [15]. In particular, techniques that enable thetransfer and charging of large molecules such as proteinsand peptides peptides, as well as transfer into a gaseousphase (e.g. electrospray ionization [ESI] and matrix-assistedlaser desorption ionization [MALDI]), have allowed proteins to be analyzed by MS [16••]. ES ionizationESIproduces a fine spray of charged particles through acharged needle, whereas MALDI involves crystallizing thesample of interest within a matrix that can be vaporizedquickly using a laser pulse [15]. Two general MS methodsare employed for protein identification using MS. Thefirst, peptide-mass fingerprinting, compares the pattern ofmolecular weights of peptides generated from a proteolyticdigestion to theoretical fingerprints derived from proteindatabases. The second method, tandem mass spectrometry(MSn), selects peptides of interest and uses a secondaryfragmentation process to determine the peptide sequence,which is then identified using protein sequence databases.Further technology improvements, including ion-sourceminiaturization (for sensitivity) and detectors (for massaccuracy), have greatly expanded the method [17•]. Thecentral importance of MS in proteomics is attributable tothe large amounts of structural data that the method cancreate at great speed, and is demonstrated by the fact thatmost separation and detection methods rely on MS for protein identification.
BioinformaticsAs a method of analysis, MS is vital to proteomicsresearchers because it identifies their data, playing thesame role as nucleotide sequencing in genomics.Interpreting the roles of the identified proteins, however,is equally important. Bioinformatics provides importanttools that systematize the data produced by genomics andproteomics to enable computer-aided data interpretation.These technologies have increased the speed and thoroughness of data analysis to a degree that would otherwise be impossible. Bioinformatics methods not onlycatalog experiments, but also provide algorithms for dataanalysis and comparisons in numerous contexts, includingprotein and gene identification, protein structure–functionrelationship predictions, and functional connectionsbetween proteins [18–20]. This type of organization andanalysis is crucial to all areas of the drug-discovery processfrom the identification of novel drug targets, in whichinformatics technology enables the mining of DNA andprotein sequences databases for analysis of similarities, toscreening of active compounds in silico by virtual screening
and/or docking of compound collections. Further along inthe drug-development process, informatics enables thefacile optimization of leads in drug design, and the selectionof pre-clinical candidates [19]. The need for such automatedanalyses of large data sets has been met by growth in bothcomputing power and systematic databases [15].
Microfabrication and miniaturizationMicrofabrication has also played an important role in functional the development of proteomics technology.Miniaturization has two advantages. First, it providesminiaturized instruments that can improve the developmentand automation of current techniques through reduction ofsample amount, increased sample numberthroughput, andincreased sensitivity. Second, it provides miniaturized‘containers’ to segregate and identify samples. In thebroadest sense, such microfabricated containers encompassboth protein-array technology (discussed later in thisreview) and lab-on-a-chip technology. The latter refers toenclosed fluidics devices that create channels and reactionchambers on substrates such as glass. Although in proteomics,microfabrication of analytical devices is an emerging field,it has made important progress towards developing lab-on-a-chip MS technologies, protein separation techniques,and is the basis of protein-array technologies [21••]. In thefuture, proteomics will use many techniques that wereestablished on a macroscopic level, and become stream-lined by microfabricated instrumentation.
Types of information from proteomicsThe methods for producing proteomic data can be dividedinto two major categories: the ‘classical’ approach thatstrives to catalogue all proteins, and the ‘functional’approach that seeks to classify proteins to be studied by properties such as activity or affinity. The classicalapproach tends to provide information about identity and,in some cases, abundance. The functional approach providesidentity and abundance, as well as information about protein function and, in some cases, protein interactions.
Proteomics in drug discovery Burbaum and Tobal 429
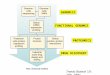
Figure 1
The proteomics information pyramid describes the escalatingcomplexity of different types of information collected usingproteomics techniques.
Identity
Abundance
Activity
InteractionsFunctional
Classical
Current Opinion in Chemical Biology

Both approaches provide valuable information that can beintegrated into a pyramid of proteomics information(Figure 1), with the classical approach providing the baseof the pyramid. The most basic, necessary informationabout a proteome is the identity of the comprised proteins.The next level of information is abundance. The classicalapproach tends to concentrate on detecting those twofoundation layers of the proteomics information pyramid.The next two layers of the pyramid involve informationproduced by more ‘functional’ techniques that either measure the activity of the proteins directly or interrogate theinteractions of proteins, or both. Although activity, the nextlayer in the pyramid, may be related to abundance, pro-teins are often post-translationally regulated. Therefore,functional techniques that clarify the relationship betweenabundance and activity, and determine how active proteinsare in biologically relevant circumstances, are important inunderstanding the complexities of the proteome. The nextlayer of the pyramid involves information produced by techniques that determine protein interactions. This information is the most complex and difficult to acquire, asproteins may interact with many different proteins undervarious circumstances. Information from all areas of theproteomics information pyramid is important for a well-rounded proteomics approach to drug discovery.
For classical proteome analysis, a chemical modificationstrategy called isotope-coded affinity tagging (ICAT) hasbeen developed to catalogue and quantify all the proteinsin a proteome [22•]. ICAT uses a reagent with three components: a reactive group to covalently bind aminoacids (e.g. cysteines), an isotopically light or heavy linker,and an affinity tag (e.g. biotin). The isotopic differencespermit protein abundance comparisons between two samples.In this process, samples for comparison are alkylated with either ‘light’ or ‘heavy’ reagent. The two samples arethen combined, undergo proteolytic digestion, and thelabeled peptides are isolated using the affinity tag. Thesample is then analyzed by LC/MS, with quantitation (notnormally a feature of MS methods) based on the ratio ofthe light and heavy signalsnondeuterated and deuteratedisotopes. This method is advantageous both for sensitivityand for throughput [8,14]. The information acquired fromthis method, as is common in classical methods, involvesidentification of proteins, and relative abundance [8].
Another improvement in classical proteome analysis is the2DLCMS method called multidimensional protein identi-fication technology (MudPIT) [23•,24]. In this method,the separation column in LC/MS is packed with two stationary phases (a strong cation exchange, and a reverse-phase adsorbent), and a solvent program separates thesample both by charge and hydrophobicity [23•]. Thismethod is employed directly on crude samples (without2DGE), and provides more complete coverage of proteomicspace [23•]. However, it requires the dedicated use of large amounts of instrument time and quantifies neither abundance nor activity [23•,24].
Methods that bridge both the classical approach and thefunctional approach use chemically reactive, activity-basedprobes (ABPs) to analyze the active protein component ofproteomes. Some ABPs bind proteins in a class-selectivemanner on the basis of features common to active species.To date, ABPs have been reported for the serine hydro-lases (a super-family that includes proteases, esterases,amidases, lipases and transacylases) [25••,26••], and thecysteine hydrolases [27,28]. Combinatorial methods havealso been applied to identify ABPs for several other families,including the aldehyde dehydrogenases, the carbonylreductases and the epoxide hydrolases [29••]. An advantageof such activity-based labeling methods is that the readoutis related to total protein activity such that the tight regulation of protein activity through post-translationalmodifications is accounted for [14].
Functional proteomic analysisPurely functional assays include protein arrays, phage-display methods and two-hybrid systems. These methodsuse technologies that are, in general, expensive to developon a proteomic scale, so have been more aggressively pursuedbyexplored by biotechnology companies. Currently, themethod being pursued by the most companies is proteinarray technology combined with MS. Companies such asCiphergen Biosystems and Phylos, among many others, aredeveloping a wide variety of protein arrays. There are twobasic designs for protein arrays. In the first, a surface isarrayed with a non-specific affinity substrate that binds asubset of the proteome. Ciphergen Biosystems’ arrays, forexample, interact with proteins using a number of suchweak affinity associations, followed by MS analysis [30,31].In the second type of design, a surface is arrayed with specific antibodies, proteins, peptides, nucleic acids orother small molecules to the surface of the chip, whichselect specific proteins [32•,33••]. If the associations aresufficiently specific, MS identification is not required.Variations on this design include immobilization methods,and native versus denatured capture. Phylos’ arrays, forinstance, attach proteins cotranslationally to their mRNAsand then use nucleic acid arrays to immobilize the fusion[34]. The advantages of array technologies are ease of useand throughput. Protein arrays provide the basis for anassay for the presence of specific proteins. In some cases,potential interactions between the proteins or small molecules on the chip and the proteins captured by the particular proteins are also revealed. However, such arraysare difficult to calibrate, because non-specific affinity-basedarrays are dominated by abundant proteins, rendering low-abundance proteins invisible [5]. Unlike nucleic-acid-basedarrays, the rules for protein–protein interactions are difficultto adjust and quantify. Specific affinity-based arrays areuntenable for classical proteomics because high coverage ofproteins would require hundreds of thousands of proteinsand small molecules to be isolated or synthesized [35•].
Two other assays that rely on an ability to segregate proteinsby affinity are phage display, employed by companies such
430 Next-generation therapeutics

as Dyax and Cambridge Antibody Technologies, and bar-coded nanoparticles, developed by SurroMed. Phagedisplay identifies interacting proteins by DNA sequencingof phage that bind to a selective surface of phage, whilebar-coded nanoparticles are created such that individualparticles cover a single small molecule, peptide or protein,and provide the same sort of encoding through a non-biological means. The barcodes identify the attached molecules [32•]. Like protein arrays and the two-hybridsystem, phage display and bar-coded nanoparticles cannotquantify activity, but can create protein profiles for different environments.
The two-hybrid system is a more traditional method thatattempts to decipher protein–protein interaction networks.In this method, first developed in yeast, one protein (or setof proteins) is genetically fused to a DNA-binding domain,while the other protein is fused to a transcription activationdomain. Both hybrids are expressed in a cell containingone or more reporter genes that are only activated whenthe proteins interact [36]. This method assumes that if twoproteins interact, such interaction is physiologically important and the two proteomes are involved in the sameprocesses [37,38••]. The advantages of this technology arethat it can be used in a high-throughput manner [39••], andthat it results in the immediate availability of the clonedgenes for any of the proteins involved [2,36]. There aremany disadvantages, however. For instance, the proteins ofinterest must interact when expressed as fusion proteins,which may result in false positives, or false negatives[35•,38••,40]. Further, the results are reported to be difficult to reproduce [38••,39••].
A novel type of array that interrogates both protein–proteininteractions and transcriptional regulation utilizes a processcalled reverse transfection [41•]. In this process, cDNA isarrayed on a glass slide, and cells that attach to the slide arelocally transfected with the cDNA. After fixing, theexpressed protein is visualized either by incubation withfluorescently-labeledfluorescently labeled antibodiesagainst the protein, or by fluorescent microscopy if the proteinof interest is fused to a fluorescent tag. This technique hasbeen used successfully in many ways, such as localizingexpressed proteins to the nucleus and cytoplasm, assessingactivation of signaling pathways, determining proteininteractions using two-hybrid approaches (see above), andinvestigating protein function by replacing missing function in knockout cell lines [41•]. The major limitationfor the use of this technique is the availability of completecDNAs for large-scale use.
Methods involving the ABPs previously mentioned arebeing used by ActivX Biosciences. In addition to providingthe ability to catalogue active proteins in a sample, theseactivity-based methods probe functional properties that arerelevant to drug discovery. For example, because the ABPsbind to active sites, they can help to evaluate the target specificity of potential drug candidates. In the presence of a
modifier of protein activity (e.g. a small-molecule lead), theABPs may label more slowly because the binding site isoccluded by the drug [26••]. Therefore, activity profiles inthe presence and absence of the drug can be compared.Further, the inhibition profiles of the drug target and ‘off-target’ proteins can be quantified without additional purification or characterization. Approximately 75% (orapproximately US $24 billion) of the total cost of drug development is attributed to failures in development [42,43].Consequently, the ability to screen for factors that could leadto failure in development, such as ‘off-target’ binding thatcan lead to toxicity is beneficial. Standard proteomics doesnot probe these factors directly, but only detects secondaryeffects of drug interactions. Activity-based methods, on theother hand, permit the direct visualization of primary drugtargets, as well as their downstream effectors (MP Patricelli,personal communication). Also, because activity-basedmethods have the ability to detect proteins based on a common chemical feature of the active site, they can be usedto detect novel proteins. Thus, methods developed byActivX Biosciences have the ability to measure relative activityrather than abundance, detect novel proteins, provide thesensitivity associated with fluorescence (the detection limit for this method is about 100 amol) [44••], and detect protein–drug interactions. Using this technique, proteinactivities can easily be profiled in different cellular environ-ments, both to identify potential drug targets, and tointerrogate drug–protein interactions and drug specificity.
ConclusionsBecause the challenges that face proteomics technologiesare far reaching, the development of proteomics will requirethe concurrent refinement of a number of techniques. It islikely that a combination of techniques will be necessary forthe generation and interpretation of data to help scientistsunderstand the processes involved in cell function and regulation. Thus, proteomics, particularly applied to drugdiscovery, is evolving toward an increasingly interdisciplinarypursuit that combines aspects of biology, chemistry, engineering and information science. The techniques beingdeveloped in proteomics are applicable to all areas of drugdiscovery, from target identification, to assessment of drugefficacy, both in pre-clinical (target selectivity) and clinicalsituations (patient response), to protein profiles for diagnostics.Future improvements in these technologies will continue to propel the quest for safer, more effective, and more cost-effective drugs.
AcknowledgementsWe acknowledge John Kozarich for critical reading of the manuscript, andMatt Patricelli for sharing unpublished results.
References and recommended readingPapers of particular interest, published within the annual period of review,have been highlighted as:
• of special interest••of outstanding interest
1. Nutall ME: Drug discovery and target validation. Cells TissuesOrgans 2001, 169:265-271.
Proteomics in drug discovery Burbaum and Tobal 431

2. Pandey A, Mann M: Proteomics to study genes and genomes.Nature 2000, 405:837-846.
3. Anderson L, Seilhamer J: A comparison of selected mRNA andprotein abundances in human liver. Electrophoresis 1997,18:533-537.
4. Mann M: Quantitative proteomics? Nat Biotechnol 1999,17:954-955.
5. Srinivas PR, Srivastava S, Hanash S, Wright GL Jr: Proteomics inearly detection of cancer. Clin Chem 2001, 47:1901-1911.
6. Simpson RJ, Dorow DS: Cancer proteomics: from signalingnetworks to tumor markers. Trends Biotechnol 2001, 19:S40-S48.
7. Dove A: Proteomics: translating genomics into products? NatBiotechnol 1999, 17:233-236.
8. Moseley MA: Current trends in differential expression proteomics: isotopically coded tags. Trends Biotechnol 2001,19:S10-S16.
9. Drews J: Drug discovery: a historical perspective. Science 2000,287:1960-1964.
10. Gygi SP, Corthals GL, Zhang Y, Rochon Y, Aebersold R: Evaluationof two-dimensional gel electrophoresis-based proteome analysistechnology. Proc Natl Acad Sci USA 2000, 97:9390-9395.
11. Issaq HJ: The role of separation science in proteomics research.• Electrophoresis 2001, 22:3629-3638.This is an excellent review of protein separation methods.
12. Bichsel VE, Liotta LA, Petricoin EF III: Cancer proteomics: from• biomarker discovery to signal pathway profiling. Cancer J 2001,
7:69-78.This is a good review covering the applications of current proteomics techniques in cancer research and therapeutic discovery.
13. Blackstock WP, Weir MP: Proteomics: quantitative and physicalmapping of cellular proteins. Trends Biotechnol 1999, 17:121-127.
14. Cravatt BF, Sorensen EJ: Chemical strategies for the globalanalysis of protein function. Curr Opin Chem Biol 2000,4:663-668.
15. Martin DB, Nelson PS: From genomics to proteomics:techniques and applications in cancer research. Trends Cell Biol2001, 11:S60-S65.
16. Mann M, Hendrickson RC, Pandey A: Analysis of proteins and•• proteomes by mass spectrometry. Annu Rev Biochem 2001,
70:437-473.This is an excellent review of mass spectrometry and its applications withregard to proteomics.
17. Papac DI, Shahrokh Z: Mass spectrometry innovations in drug• discovery and development. Pharm Res 2001, 18:131-145.This is an excellent review about the roles of mass spectrometry in drug discovery and development
18. Searls DB: Using bioinformatics in gene and drug discovery. DrugDiscov Today 2000, 5:135-143.
19. Bajorath J: Rational drug discovery revisited: interfacingexperimental programs with bio- and chemo-informatics. DrugDiscov Today 2001, 6:989-995.
20. Maggio ET, Ramnarayan K: Recent developments in computationalproteomics. Drug Discov Today 2001, 6:996-1004.
21. Figeys D, Pinto D: Proteomics on a chip: promising developments.•• Electrophoresis 2001, 22:208-216.This review highlights the importance of microfabrication and miniaturizationin proteomics technologies.
22. Gygi SP, Rist B, Gerber SA, Turecek F, Gelb MH, Aebersold R:• Quantitative analysis of complex protein mixtures using
isotope-coded affinity tags. Nat Biotechnol 1999, 17:994-999.The authors describe the ICAT method for detection of the relative abundance of proteins in complex mixtures. The results they describe correlate with known yeast metabolic function.
23. Washburn MP, Wolters D, Yates JR III: Large-scale analysis of the• yeast proteome by multidimensional protein identification
technology. Nat Biotechnol 2001, 19:242-247.Using MudPIT [24], the authors detected and identified 1484 proteins fromSaccharomyces cerevisiae.
24. Link AJ, Eng J, Schieltz DM, Carmack E, Mize GJ, Morris DR,Garvik BM, Yates JR III: Direct analysis of protein complexes using mass spectrometry. Nat Biotechnol 1999, 17:676-682.
25. Liu Y, Patricelli MP, Cravatt BF: Activity-based protein profiling: •• the serine hydrolases. Proc Natl Acad Sci USA 1999,
96:14694-14699.The authors describe the chemical synthesis and function of an ABP thatdetects the activity of the serine hydrolases.
26. Kidd D, Liu Y, Cravatt BF: Profiling serine hydrolase activities in•• complex proteomes. Biochemistry 2001, 40:4005-4015.The authors describe the use of ABPs for profiling serine hydrolase activitiesin complex proteomes, and describe the use of these probes for evaluatingtarget selectivity of serine hydrolase inhibitors.
27. Greenbaum D, Medzihradszky KF, Burlingame A, Bogyo M: Epoxide electrophiles as activity-dependent cysteine protease profiling and discovery tools. Chem Biol 2000,7:569-581.
28. Bogyo M, Verhelst S, Bellingard-Dubouchaud V, Toba S,Greenbaum D: Selective targeting of lysosomal cysteineproteases with radiolabeled electrophilic substrate analogs.Chem Biol 2000, 7:27-38.
29. Adam GC, Cravatt BF, Sorensen EJ: Profiling the specific reactivity•• of the proteome with non-directed activity-based probes. Chem
Biol 2001, 8:81-95.The authors describe a library of biotinylated sulfonate esters that can beused as ABPs to interrogate complex mixtures.
30. von Eggeling F, Junker K, Fiedle W, Wollscheid V, Durst M,Claussen U, Ernst G: Mass spectrometry meets chip technology:a new proteomic tool in cancer research? Electrophoresis 2001,22:2898-2902.
31. Boyle MD, Romer TG, Meeker AK, Sledjeski DD: Use of surface-enhanced laser desorption ionization protein chip system to analyze streptococcal exotoxin B activity secreted by Streptococcus pyogenes. J Microbiol Methods 2001, 46:87-97.
32. Zhou H, Roy S, Schulman H, Natan MJ: Solution and chip • arrays in protein profiling. Trends Biotechnol 2001,
19:S34-S39.The authors discuss array technologies, including solid-phase protein arrays,and solution arrays using self-encoded beads and nanoparticles.
33. Zhu H, Bilgin M, Bangham R, Hall D, Casamayor A, Bertone P, •• Lan N, Jansen R, Bidlingmaier S, Houfek T et al.: Global analysis of
protein activities using proteome chips. Science 2001,293:2101-2105.
The authors created a yeast proteome microarray, which was screened for particular protein and phospholipid interactions. They identified new calmodulin- and phospholipid-binding proteins, demonstrating that proteinfunction can be determined using this method.
34. Roberts RW, Szostak JW: RNA-peptide fusions for the in vitroselection of peptides and proteins. Proc Natl Acad Sci USA 1997,94:12297-12302.
35. Kodadek T: Protein microarrays: prospects and problems. Chem• Biol 2001, 8:105-115.This is an excellent review of protein microarrays.
36. Phizicky EM, Fields S: Protein–protein interactions: methods fordetection and analysis. Microbiol Rev 1995, 59:94-123.
37. Mrowka R, Patzak A, Herzel H: Is there a bias in proteomeresearch? Genome Res 2001, 11:1971-1973.
38. Ito T, Chiba T, Ozawa R, Yoshida M, Hattori M, Sakaki Y: •• A comprehensive two-hybrid analysis to explore the yeast
protein interactome. Proc Natl Acad Sci USA 2001, 98:4569-4574.
The authors use a previously described two-hybrid method to examine allpossible combinations between the ~6000 proteins of S. cerevisiae, andcompare their results with those of Uetz et al. [39•• ].
39. Uetz P, Giot L, Cagney G, Mansfield TA, Judson RS, Knight JR, •• Lockshon D, Narayan V, Srinivasan M, Pochart P et al.:
A comprehensive analysis of protein–protein interactions inSaccharomyces cerevisiae. Nature 2000, 403:623-627.
The authors describe and compare two large-scale methods of the yeasttwo-hybrid system in which they interrogate all possible combinationsbetween the ~6000 proteins of S. cerevisiae.
432 Next-generation therapeutics

40. MacBeath G, Schreiber SL: Printing proteins as microarrays forhigh-throughput function determination. Science 2000,289:1760-1763.
41. Blagoev B, Pandey A: Microarrays go live — new prospects for• proteomics. Trends Biochem Sci 2001, 26:639-641.The authors describe reverse transfection, a method in which full-lengthcDNAs arrayed on a glass slide are used to transfect mammalian cells forfunctional proteomics research.
42. Grass GM, Sinko PJ: Effect of diverse datasets on the predictivecapabilities of ADME models in drug discovery. Drug Discov Today2001, 6:S54-S61.
43. Pharmaceutical Research and Manufacturers of America (PhRMA):Pharmaceutical Industry Profile. Washington DC: PharmaceuticalResearch and Manufacturers of America (PhRMA); 2001:119.
44. Patricelli MP, Giang DK, Stamp LM, Burbaum JJ: Direct visualization•• of serine hydrolase activities in complex proteomes using
fluorescent active site-directed probes. Proteomics 2001,1:1067-1071.
The authors describe fluorescent-activity-directed probes for the serinehydrolase family of enzymes that have increased sensitivity and quantitativeaccuracy when compared with previously described ABPs, and provide aplatform for high-throughput proteome analysis.
Proteomics in drug discovery Burbaum and Tobal 433